

Abstract
Antibody-drug conjugates (ADCs) are cancer therapeutic agents comprised of an antibody, a linker and a small-molecule payload. ADCs use the specificity of the antibody to target the toxic payload to tumor cells. After intravenous administration, ADCs enter circulation, distribute to tumor tissues and bind to the tumor surface antigen. The antigen then undergoes endocytosis to internalize the ADC into tumor cells, where it is transported to lysosomes to release the payload. The released toxic payloads can induce apoptosis through DNA damage or microtubule inhibition and can kill surrounding cancer cells through the bystander effect. The first ADC drug was approved by the United States Food and Drug Administration (FDA) in 2000, but the following decade saw no new approved ADC drugs. From 2011 to 2018, four ADC drugs were approved while in 2019 and 2020 five more ADCs entered the market. This demonstrates an increasing trend for the clinical development of ADCs. This review summarizes the recent clinical research, with a specific focus on how the in vivo processing of ADCs influences their design. We aim to provide comprehensive information about current ADCs to facilitate future development.
Keywords: antibody-drug conjugates (ADCs), cancer therapy, clinical research, metabolism
1. Introduction
As early as the mid-20th century, doctors and scientists began to use cytotoxic chemicals to treat patients with advanced cancers (Miller et al., 2010). One of the earliest trials was to apply nitrogen mustard to treat patients with non-Hodgkin’s lymphoma (Conrad & Crosby, 1960). After treatment with nitrogen mustard, tumor regression occurred. Even though regression was short and incomplete, it encouraged researchers in this field at that time. In the following decades, alkylating agents (e.g. cyclophosphamide and cisplatin) and anti-metabolites (e.g. methotrexate and fluorouracil) were developed and utilized as cancer treatments (DeVita & Chu, 2008). In the late 20th century, the emergence of monoclonal antibodies (mAb) made it possible to develop more targeted anti-cancer drugs (Liu, 2014; Slamon, et al., 2001). In parallel, a growing number of tumor markers and tumor surface antigens were found and identified, providing targets for antibody therapy (Walts & Said, 1983). When combined to form antibody-drug conjugates (ADCs), the toxic compounds (payloads) provide the tumor cell killing effect, while antibodies provide targeted distribution (Latif et al., 1980).
After decades of time and effort, the first ADC drug, Mylotarg, was approved by the United States Food and Drug Administration (FDA) in 2000 (Norsworthy et al., 2018). It is used to treat patients with relapsed cluster of differentiation (CD) 33 positive acute myeloid leukemia (Zaro, 2015). Although Mylotarg was delisted in the United States for a period of time due to toxicity concerns, it is a milestone of ADC drug development and sets a precedent for ADC-based cancer therapy. In the following ten years, no new ADCs reached clinical approval. After 2011, several new ADC drugs were launched in quick succession. In 2011, Adcetris, targeting CD30, was approved for the treatment of Hodgkin’s lymphoma (Leal et al., 2015). In 2013, ado-trastuzumab emtansine (T-DM1) targeting human epidermal growth factor receptor 2 (HER2) was approved for the treatment of HER2-positive breast cancer (Reichert, 2014). In 2017 and 2018, Besponsa and Lumoxiti, respectively, which target CD22, were approved for use in the treatment of acute lymphoblastic leukemia (Kaplon & Reichert, 2018). By 2019, ADC development was on the rise evidenced by the approval of three ADC drugs within the year. Polivy, Padcev and Enhertu were approved by the FDA for treatment of B-cell lymphoma, urothelial tumor and HER2-positive breast cancer, respectively (Kaplon et al., 2020). In the past year, two more new ADC drugs gained approval, Trodelvy for triple-negative breast cancer and Blenrep for relapsed and refractory multiple myeloma (as shown in Table 1 and Figure 1).
Table 1.
ADC drugs approved by FDA (up to December 2020)
| ADC name | Brand name | Tumor target Antibody | Linker | Payload DAR | Indication | Pharmaceutical companies | Launch date | |
|---|---|---|---|---|---|---|---|---|
| 1 | Mylotarg | Gemtuzumab ozogamicin (GO) | CD33 Humanized IgG4 |
Cleavable hydrazone linker | Calicheamicin 3–5 |
CD33 positive acute myeloid leukemia | Pfizer | 5/20009/2017* |
| 2 | Adcetris | Brentuximab vedotin (SGN-35) | CD30 Chimeric IgG1 |
Cleavable maleimidocaproyl valine-citrulline linker | MMAE 4 |
Hodgkin’s lymphoma | Seattle Genetics/Takeda | 8/2011 |
| 3 | Kadcyla | Trastuzumab emtansine (T-DM1) | HER2 Humanized IgG1 |
Non-cleavable thioether linker | DM1 3–4 |
HER2 positive breast cancer | Roche | 2/2013 |
| 4 | Besponsa | Inotuzumab ozogamicin | CD22 Humanized IgG4 |
Cleavable hydrazone linker | Calicheamicin 6 |
B-cell acute lymphoblastic leukemia | Pfizer | 8/2017 |
| 5 | Lumoxiti | Moxetumomab pasudotox | CD22 Recombinant murine immunoglobulin variable domain |
Fusion protein (antibody and payload) | Pseudomonas exotoxin | Hairy cell leukemia | AstraZeneca | 9/2018 |
| 6 | Polivy | Polatuzumab vedotin | CD79b Fully humanized IgG1κ |
Cleavable maleimidocaproyl valine-citrulline linker | MMAE 3–4 |
Diffuse large B-cell lymphoma | Roche | 6/2019 |
| 7 | Padcev | Enfortumab vedotin | Nectin-4 Fully humanized IgG1κ |
Cleavable maleimidocaproyl valine-citrulline linker | MMAE 3–4 |
Urothelial carcinoma | Seattle Genetics/Astellas | 12/2019 |
| 8 | Enhertu | Fam-trastuzumab deruxtecan (DS-8201) | HER2 Fully humanized IgG1κ |
Cleavable maleimide tetrapeptide linker | DXd 8 |
HER2 positive breast cancer | AstraZeneca/Daiichi Sankyo | 12/2019 |
| 9 | Trodelvy | Sacituzumab govitecan | Trop-2 Humanized IgG1κ | Cleavable carbonate linker | SN-38 7–8 |
Triple-negative breast cancer | Immunomedics | 4/2020 |
| 10 | Blenrep | Belantamab mafodotin | BCMA Humanized IgG1κ |
Non-cleavable maleimidocaproyl linker | MMAF 4 |
Relapsed and refractory multiple myeloma | GlaxoSmithKline | 8/2020 |
Mylotarg was first approved in 2000, withdrawn in 2010 and relaunched in 2017.
Figure 1.
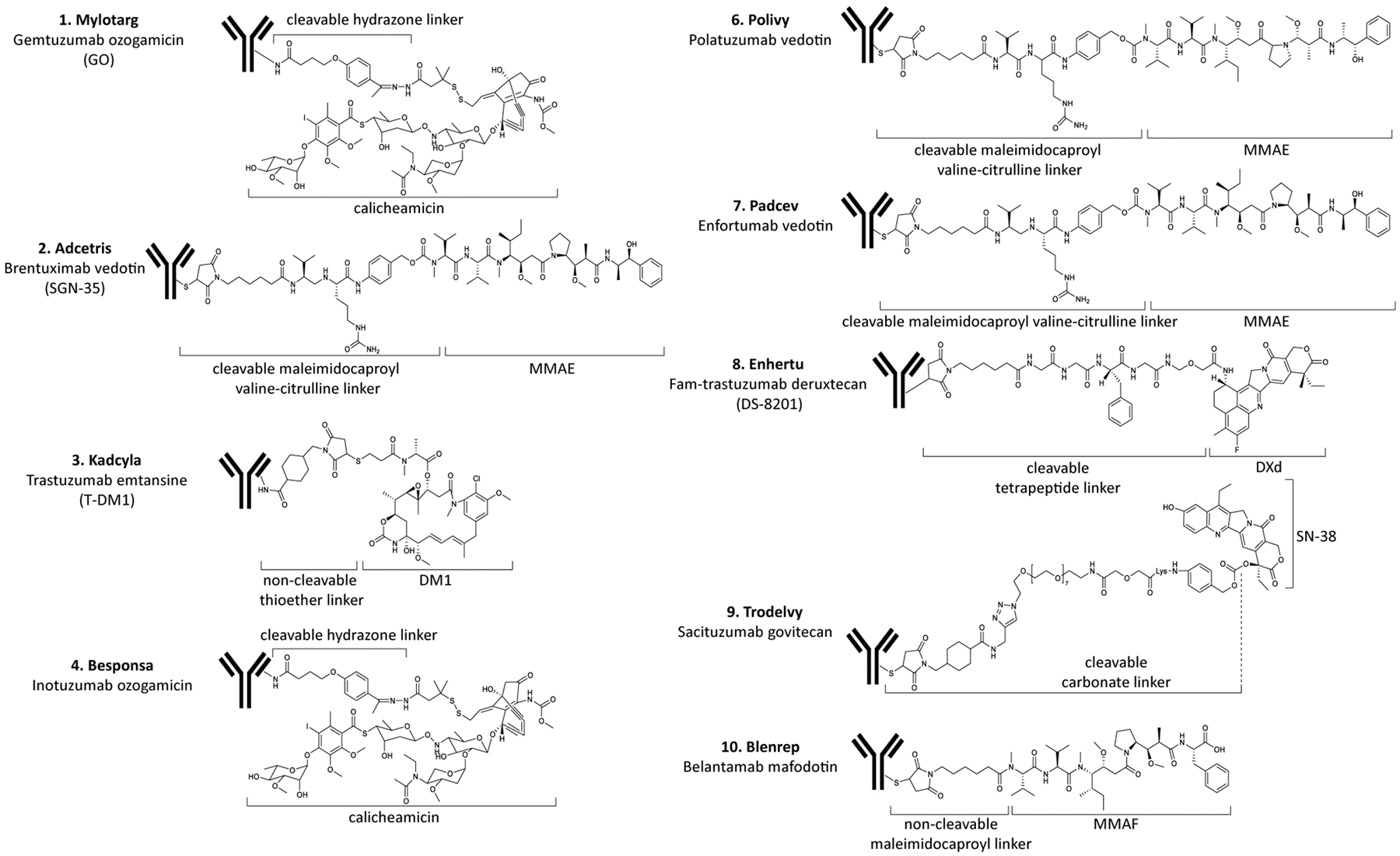
Structures of the approved ADC drugs listed in Table 1.
At the same time, pharmaceutical companies have made great efforts to overcome technical barriers related to ADCs, including plasma stability, payload dissociation, low blood retention time, minimal tumor penetration, decreased payload efficiency, immunogenicity, off-target toxicity, and drug resistance (Sassoon & Blanc, 2013). In the decade from 2010–2019, more than 60,000 research papers on ADCs were published, while only a few drugs entered the market. Behind the newly launched ADC drugs are a large number of clinical studies on ADCs that have been terminated due to safety or efficacy concerns (Tolcher, 2016). A better understanding of ADCs and their small molecule payloads would improve the likelihood of clinical success. Therefore, this review examines the mechanisms of ADC drugs according to their intended route of in vivo processing. It summarizes several clinical studies on ADCs over recent years according to different targeted tumor types in order to provide a framework for pre-clinical development of ADCs.
2. The structure of ADCs
An ADC is a complex formed by covalently coupling a small molecule drug (payload) with a monoclonal antibody through a linker. These three parts jointly define the overall physical and chemical properties, efficacy and possible problems of ADC drugs. The typical ADC design is shown in Figure 2.
Figure 2. Structure of ADCs.
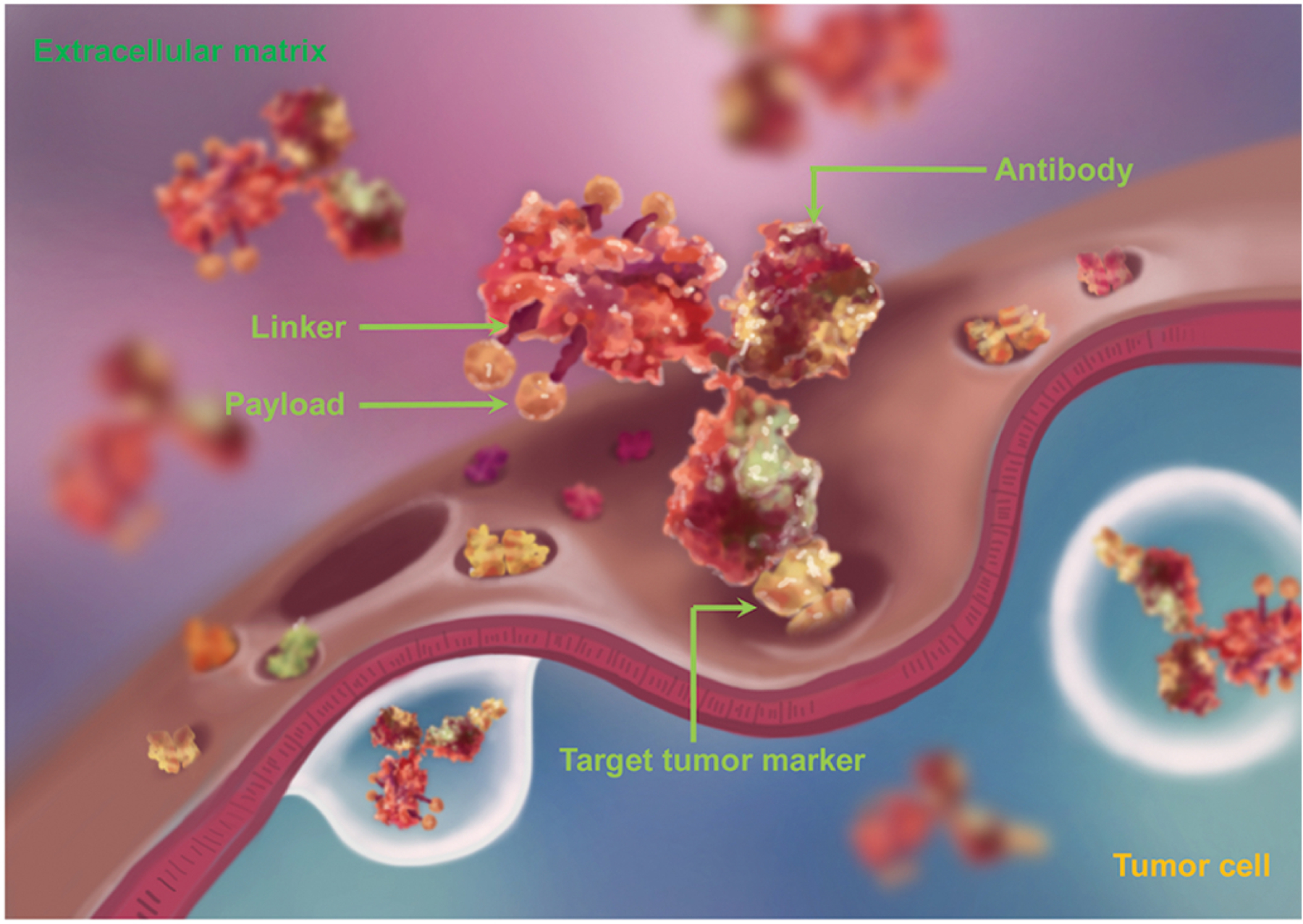
An ADC consists of three parts: an antibody, a linker and a payload (not to scale). The payloads are covalently coupled to the monoclonal antibody using linkers. This figure is based on a figure from www.adcreview.com.
2.1. Antibodies
Antibodies (Abs) are glycoproteins produced by plasma cells that can specifically bind to a corresponding antigen and generally consist of a variable antigen binding domain (Fab) and a constant domain (Fc) that binds immune cell receptors (Buss et al., 2012). The term immunoglobulins (Ig) refers more generally to Abs and all globulin proteins that have structural similarities to antibodies (Cushley & Owen, 1983). Antibodies can be divided into five categories: IgG, IgA, IgD, IgE and IgM (Kuroda et al., 2010). Among the five types, IgM is a pentamer and although it has high affinity, its molecular weight is too large and penetrability is poor (Shimizu et al., 2004). IgA has a dimer structure and a large molecular weight. IgD is sensitive to protease and is easy to degrade, so its half-life is short. IgE is very rare, only accounting for 0.002% of the total immunoglobulin in human serum. However, IgG can account for 75% ~ 85% of total immunoglobulin in human serum and has a molecular weight of about 150 kilodalton (kDa) (Smith, 1974). Because of its moderate molecular weight, high affinity, long half-life, strong penetrability and easy preparation, IgG has been the first choice in selection of ADC antibodies (Ritchie et al., 2015). There are four subtypes of IgG: IgG1, IgG2, IgG3 and IgG4. IgG1 is easy to prepare and has an intracellularly degradable hinge structure, therefore most ADCs are constructed with an IgG1 scaffold (Rees, 2015). IgG2 and IgG4 have weakly active Fc portions, so their ability to initiate antibody-dependent cellular cytotoxicity and complement-dependent cytotoxicity is weak (Steplewski et al., 1988; Armour et al., 1999), therefore IgG2 and IgG4-based ADCs can reduce side effects of non-target tissue aggregation caused by Fc segments (Aalberse et al., 2010). Some ADCs lacking Fc function have been FDA approved (e.g. Mylotarg and Besponsa) (Sau et al., 2017). Cytotoxic drugs are usually connected to the Fc or constant region, but not to the Fab region to avoid a negative impact on antigen detection and binding (as shown in Figure 2).
As navigation systems for ADC tumor distribution, antibodies used in ADCs need to have weak immunogenicity, high target specificity, high binding affinity, long half-life and good stability in blood circulation (Nejadmoghaddam, et al., 2019). The immunogenicity of antibodies and ADCs is one determinant of their circulation half-life (Hock et al., 2015). Early antibody therapies and ADC studies used mouse mAbs, which could induce a strong, acute immune response (Kuus-Reichel et al., 1994), while at present most ADCs use partially humanized or fully humanized antibodies (Lonberg, 2008). High specificity helps concentrate the cytotoxic agents on tumor sites, so as to achieve a targeted pharmacological effect. ADCs with low specificity are more likely to cause toxicity to normal tissues (Nejadmoghaddam et al., 2019). ADC antibodies should have high binding affinity and most ADCs have binding affinities in the range of 0.1 to 1.0 nM, also described as the equilibrium dissociation constant or Kd value, but few published data have solved the optimal binding affinity of ADCs. One theory on binding affinity (the binding site barrier theory) explains that the higher the antibody affinity and the higher the antigen density, the more of that antibody binds to the solid tumor surface rather than penetrates to the interior of the tumor tissue (Juweid, et al., 1992). One recent study prepared three ADCs that had different target binding affinities and found almost the same in vitro cytotoxicity among the three ADCs. The three ADCs had equivalent antitumor effects against small BxPC3 tumors. However, for large BxPC3 tumors, the high affinity ADC showed stronger anti-tumor activity relative to the low affinity ADC. In addition, immunofluorescence staining indicated that the high affinity ADC distributed throughout the whole tumor, whereas the low affinity ADC mainly localized close to tumor vessels, suggesting distribution and anti-tumor activity may depend on binding affinity (Tsumura et al., 2018). Kang et al. prepared an ‘acid-switched’ ADC targeting HER2. It had higher affinity at near neutral pH (in plasma and the extracellular microenvironment), but lower affinity at acidic pH (in endosome or lysosome), which allowed more cellular payload release. The antibody showed 250-fold weaker affinity intracellularly compared to outside cells. In xenograft tumor models, compared with T-MD1, the engineered ADC showed increased lysosomal delivery and higher therapeutic efficacy (Kang et al., 2019). In addition, for the same tumor surface antigen, antibodies of ADCs bound to different epitopes have different internalization rates (Rohrer, 2017). Fast internalization can improve efficiency of ADCs. Compared with small molecules, antibodies enter into tissues from plasma more slowly (Schlom et al., 1990). The use of antibodies as a targeting mechanism in ADCs demonstrates substantial benefit for localized delivery of cytotoxic agents.
2.2. Payloads
Payloads, also known as “cytotoxic molecules” or “warheads”, are important factors affecting the properties and activities of ADCs (Damelin, 2018). The mechanisms of cytotoxic molecules determine efficacy and adverse reactions of ADCs. At present, the commonly used cytotoxic molecules are nearly all natural-product based including microtubule inhibitors (such as auristatin, maytansinoids), DNA damaging agents (such as calicheamicin, duocarmycins, anthracyclines, pyrrolobenzodiazepine dimers) and DNA transcription inhibitors (amatoxin and quinoline alkaloid (SN-38)) (Yaghoubi et al., 2020; Theocharopoulos et al., 2020; Chen et al., 2017a; Gromek & Balunas, 2015). Among nine of the ten approved ADC drugs, six different small molecule payloads are used (shown in Table 1, Figure 1 and Figure 3). Three ADCs deliver monomethyl auristatin E (MMAE), two drugs deliver calicheamicin, and monomethyl auristatin F (MMAF), mertansine (DM1), SN-38 and DXd are also successfully applied. The toxins or toxin skeletons suitable as ADCs payloads (Chen et al., 2017a; Gromek & Balunas, 2015) must have the complex characteristics described in the following paragraphs.
Figure 3.
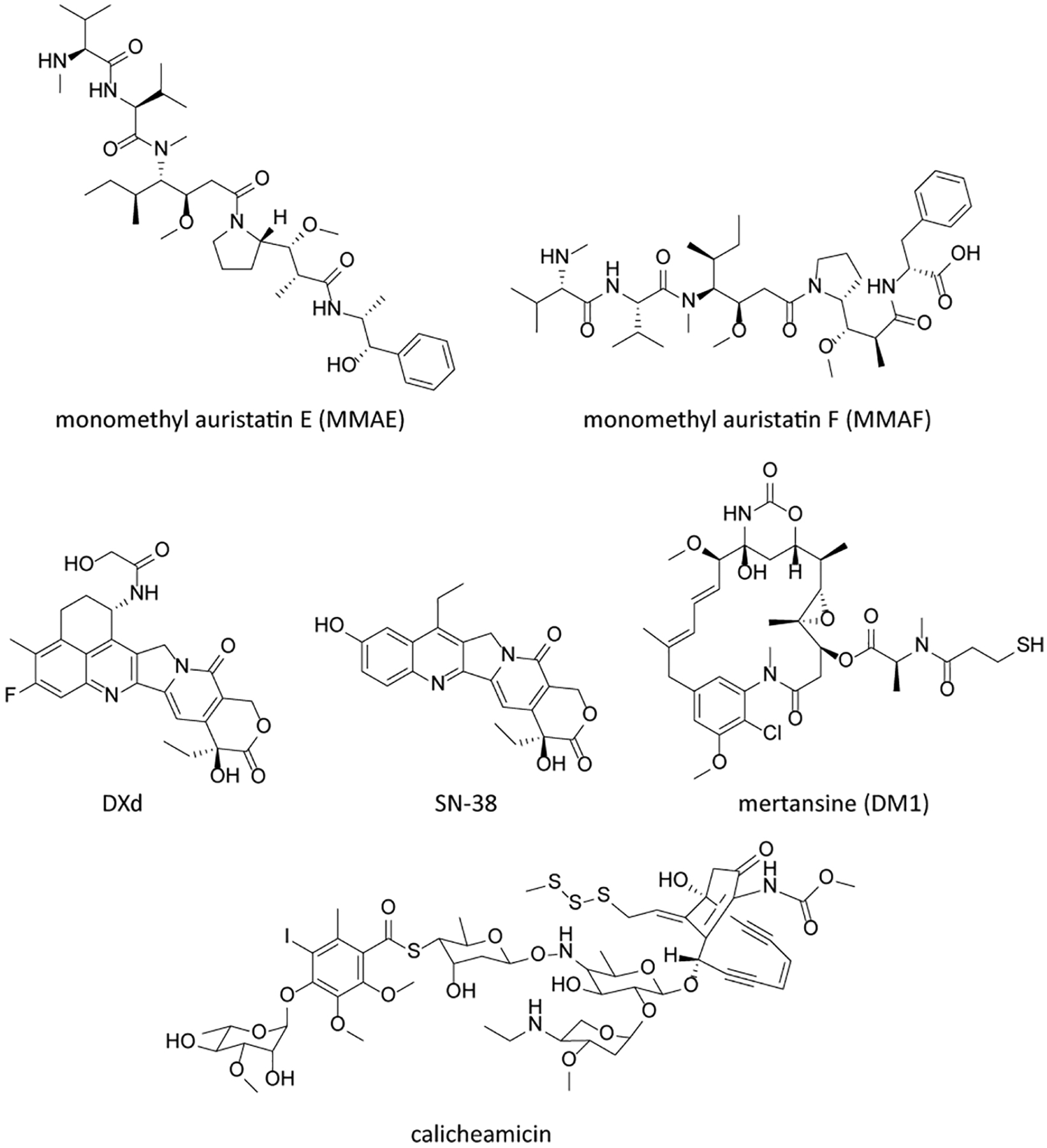
Chemical structures of clinical ADC payloads.
Ultra-high toxicity
The intrinsic potency of a payload must be very high, because the accumulation of payloads in target cells is limited due to the low permeability of mAbs into tumor tissue, limited expression of tumor cell antigens, low cellular internalization and metabolic decomposition of ADCs in blood circulation (Ducry, 2013). Current payloads can kill tumor cells at sub-nM concentrations (Diamantis & Banerji, 2016). ADC payloads generally act on a small number of cell targets involved in basic cell survival processes, which ensures high cytotoxicity in genetically heterogeneous tumor tissues and prevent cancer cells from escaping through a drug resistance mechanism (Anderl et al., 2013).
Intracellular toxic targets
The target of a typical ADC payload is located within the cell and most ADCs need to enter tumor cells to release their payloads (Chen et al., 2017a). Many highly toxic agents from plants, animals and microorganisms are membrane targeted, blocking ion channels on neurons or causing coagulation disorders. Such toxins are not suitable for use as ADC payloads. At present, intracellular targets for ADCs are focused on nucleic acids or tubulin protein (Yaghoubi et al., 2020).
Payload characteristics
The molecular weight of payloads should be relatively small, so as to reduce the risk of an immunogenic reaction (Birrer et al., 2019). In addition, payloads should have appropriate solubility in a water-based buffer to facilitate antibody coupling. Payloads should contain a functional group suitable for coupling with an antibody through linkers in its structure (Birrer et al., 2019). The payload should also be stable in the low pH environment of the lysosomes. When non-cleavable linkers are used, the toxins should retain their cytotoxicity when degraded into linker residue-payload form, such as thiol derivative forms after breakage of disulfide bonds in tumor cells (Ponziani et al., 2020).
2.3. Linker
The third important component of ADCs is the linker, which functions as a connection between the antibody and payload. The linker needs to be stable in blood to keep the cytotoxic payload attached to the antibody, but once the ADC enters the tumor cell or is transported to lysosomes, the linker should quickly break down to release the payload (Filntisi et al., 2014). The linker impacts many important properties of ADCs, such as drug-to-antibody ratio or DAR value (the number of payloads attached to one antibody), payload release time, therapeutic index (TI) and pharmacokinetics/pharmacodynamics. The stability of the linker in blood is very important. Linkers with limited stability are prone to cause nonspecific cleavage, which leads to wider drug release and off-target toxicity (Excoffier et al., 2016). The stability of linkers used in initial ADCs was limited and the subsequent rapid release of payload led to poor therapeutic indices and side effects that were similar to non-conjugated chemotherapy (Excoffier et al., 2016). After application of new linkers, this situation has improved. Commonly used linkers include the valine-citrulline (VC) linker, N-succinimidyl 4-(2-pyridyldithio)butanoate (SPDB) linker, hydrazone linker, succinimidyl 4-(N-maleimidomethyl)cyclohexane-1-carboxylate (SMCC) linker, maleimidocaproyl (MC) linker, N-succinimidyl-4-(2-pyridyldithio)pentanoate (SPP) linker, thioether linker, tetrapeptide linker and carbonate linker (Tsuchikama & An, 2016; Yao et al., 2016) (Table 1, Figure 2 and Figure 4).
Figure 4.
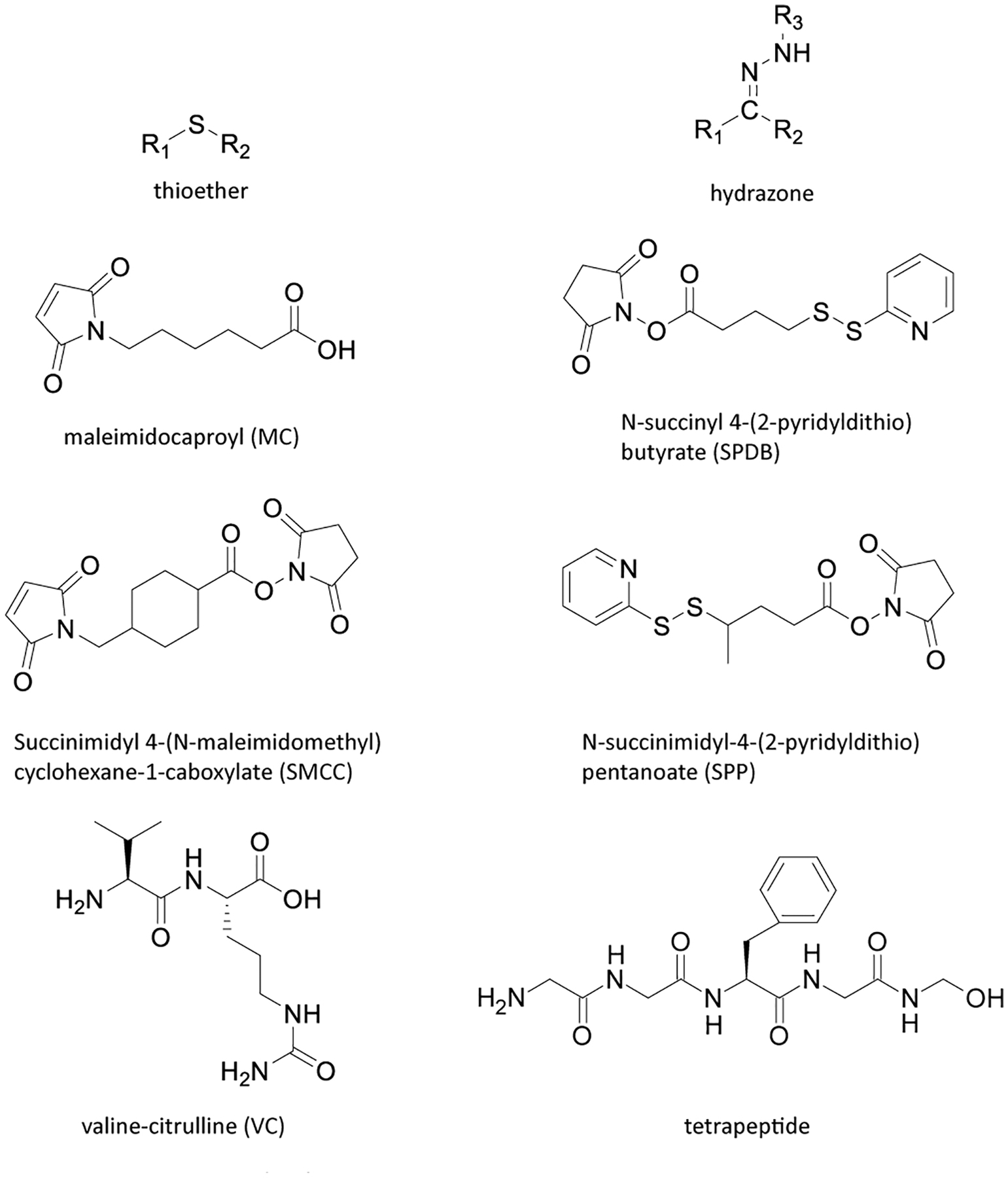
Chemical structures of commonly used ADC linkers.
Drug-to-antibody ratio (DAR)
The drug-to-antibody ratio describes the average number of drug molecules conjugated to the antibodies. In general, interchain disulfide bridges and surface-exposed lysine and cysteine residues are the most commonly used linker modification sites on antibodies (Dimasi et al., 2017). The hydroxyl on glycosylation groups and other residues on antibodies are rarely used as conjugation sites (Nolting, 2013). Most of the clinical ADCs adopt hetero-coupling technology and carefully control average modification to achieve a DAR value in range of 2 to 8, which often results in mixtures of ADCs with different DAR values (Su et al., 2018; Yao et al., 2016). At present, a number of site-specific linker methods (homologous conjunction technologies) are under investigation to control DAR value and prepare better stability and low aggregation ADCs (Panowski et al., 2014; Tsuchikama & An, 2016).
Based on the cleavage properties of linkers within tumor cells, linkers can be divided into two types: cleavable and non-cleavable (stable) (Tsuchikama & An, 2016). ADCs with cleavable linkers can release payloads quickly in tumor cells through linker cleavage, while ADCs with stable linkers release payloads only after entering the lysosomes of tumor cells where the antibody is fully degraded by proteases (Dan et al., 2018). Cleavable linkers have higher metabolism in circulation, which can contribute to off-target toxicity (Tumey & Han, 2017). The metabolites produced by the two linker types are also different. ADCs with cleavable linkers can release intact cytotoxic molecules, while ADCs with stable linkers release payloads that are linked to amino acids (Bargh et al., 2019). Generally, ADCs with stable linkers have longer half-lives and lower clearance rates than those with cleavable linkers (Tumey & Han, 2017). In addition, ADCs with cleavable linkers can release their payload even if the internalization process is not smooth (Bargh et al., 2019).
Cleavable linkers
Cleavable linkers can be cleaved by a number of mechanisms, including hydrolysis of acid labile bonds, enzymatic cleavage of amide or ester bonds, or reductive cleavage of disulfide bonds (Mueller et al., 1990). These mechanisms may operate in primary and late endosomes of tumor cells, and there are no strict requirements for lysosomes (Klussman et al., 2004). Cleavable linkers are sensitive to the intracellular environment. For example, acid cleavable linkers are usually stable in blood, but rapidly cleaved in the low pH lysosomal environment (such as Besponsa and Mylotarg). Disulfide linkers release payloads through intracellular glutathione (GSH) reduction reactions (Mueller et al., 1990). If the released payloads can cross tumor cell membranes, they can kill nearby cancer cells- this is referred to as the bystander effect (Bargh et al., 2019; Kovtun et al., 2006). For example, the cleavable ADC DS-8201a releases its membrane permeable payload DXd, which kills HER2-positive cells surrounding the targeted cancer cells, but not more distant cells. This is beneficial for treatment of HER2 heterogeneous tumors (Ogitani et al., 2016). However, cleavable linkers do not always enable the bystander effect, rather it depends on membrane-penetrability and charge properties of the released payload (Chari et al., 2014). The bystander effect is advantageous for tackling heterogeneous tumors and for penetrating deeper into solid tumors which are less accessible to the conjugate. At the same time there are disadvantages such as killing normal cells or immune cells nearby the intended target tumor cells (Chari et al., 2014).
Non-cleavable linkers
Non-cleavable linkers are composed of structures that have enzyme-resistant properties and are stable in blood (Filntisi et al., 2014). Representing non-cleavable linkers are thioether and MC linkers (Dorywalska et al., 2015). Their payloads can be released only after the ADCs are transported to lysosomes and broken down into amino acids (Lu et al., 2016). The advantages of stable linkers include minimal payload release outside of cells, thus reducing off-target toxicity and improving therapeutic index, but the disadvantages include lower release efficiency and the need for a good internalization process (McCombs & Owen, 2015; Gianolio et al., 2012). In addition, stable linkers do not achieve bystander effects because the metabolized payload form (lysine or cysteine residue-linker-payload form) possesses a charge, restricting its diffusion (Bargh et al., 2019).
3. Mechanism and in vivo processing to activate ADCs
3.1. First step - ADCs enter blood through IV injection and distribute to tissues
3.1.1. Tissue distribution of ADCs
ADCs are usually administered by intravenous (IV) injection to ensure rapid whole body distribution and avoid degradation (Dan et al., 2018). After injection, ADCs are initially distributed to organs with rich blood flow (Xu, 2015) (shown in Figure 5, step 1). Barrier systems of some organs and tissues, such as the blood-testis barrier and blood-brain barrier reduce tissue entry of ADCs (Pardridge, 2015). With time, ADCs gradually enter interstitial space of organs and tissues which decreases blood concentration and increases distribution volume (Boswell et al., 2011). During the process, the ADC reaches the tumor epitope and the antibody portion binds the target antigen (Schneider et al., 2017).
Figure 5. In vivo processing of a typical antibody drug conjugate.
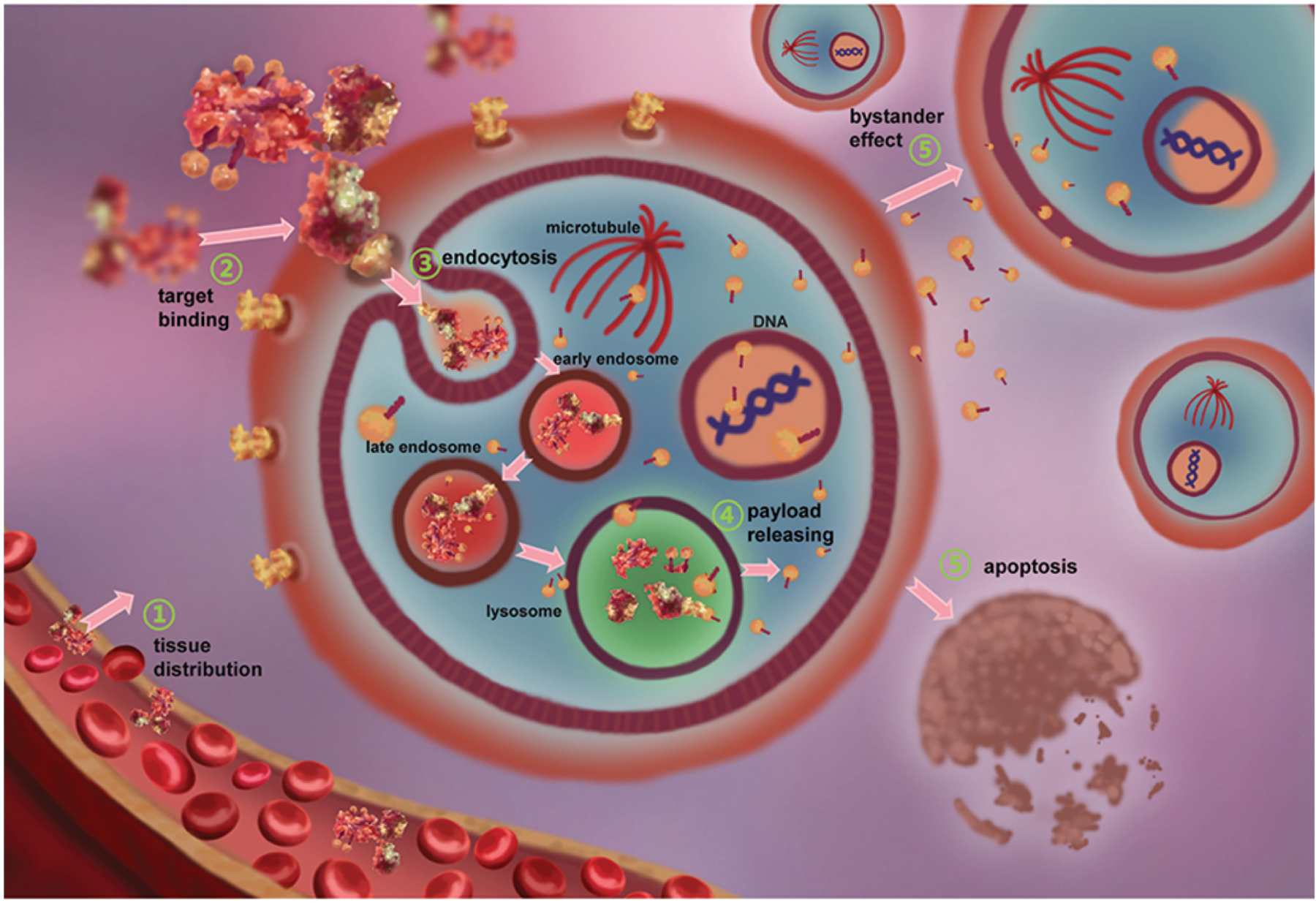
(1) The ADC enters blood circulation through IV injection and then distributes throughout the body over time. (2) The ADC binds to targeted antigen on tumor cell surfaces by antigen-antibody specific binding. (3) The ADC is internalized into tumor cells via receptor-mediated endocytosis and then is transported to the lysosome. (4) Within the lysosome, cytotoxic payloads are released after ADC proteolysis or linker split. (5) Cytotoxic payloads destroy the tumor cell and nearby tumor cells through the bystander effect. The figure is based on figures in (Birrer et al., 2019) and (Lambert & Berkenblit, 2018).
The distribution characteristics of ADCs are mainly determined by the antibody, which accounts for more than 95% of the whole ADC molecular weight (Chen & Zhan, 2019). ADCs show similar PK characteristics as antibodies, such as low clearance rate, long half-life, low volume of distribution, poor oral bioavailability, nonlinear distribution and elimination, and immunogenicity (Chen & Zhan, 2019). The molecular weight of IgG is about 150 kDa and its radius is about 15 nm (Bournazos & Ravetch, 2017). The half-lives of antibodies such as IgG can reach 21 days boosted by neonatal Fc receptor (FcRn) mediated IgG recycling (Pernille et al., 2017; Roopenian & Akilesh, 2007). The Fc segment of IgG can specifically bind FcRn of the major histocompatibility complex family to form an IgG-FcRn complex, which can be internalized into cells with high binding affinity under the acidic conditions of endosomes (Wang et al., 2014; Gurbaxani et al., 2013). Then the IgG-FcRn complex can be recycled back to the cell membrane surface and exposed to low alkaline pH, which decreases the affinity of the complex and releases the antibody into circulation (Maas & Cao, 2018). In this way, the half-life of IgG is significantly prolonged. In addition, when an ADC, as a biological macromolecule, enters the human body, it may stimulate humoral immunity and cause an anti-ADC immune response, thus accelerating the inactivation or elimination of the ADC and preventing its tissue distribution (Waldmann, 2019; Petkova et al., 2006).
The kinetics of ADCs in circulation are also affected by payloads and linkers, altering charge distribution and hydrophilicity of the antibody, thus changing the aggregation and sedimentation characteristics of the antibody in circulation (Kamath & Iyer, 2015). Due to the difference in the DAR and the payload distribution heterogeneity, ADCs show structure diversity, resulting in heterogeneous dissociation, which is different from simple antibody drugs. The actual DAR values of ADCs generally fluctuate in the range of 0 to 8 (the average value is generally 3 to 5), making PK studies more difficult (Tang et al., 2019).
3.1.2. Assessment of ADC in tissue distribution
Since an ADC is composed of an antibody, a linker and a payload, the three parts need to be considered as a whole to study absorption and distribution. At the same time, because ADCs may degrade in vivo, distribution and metabolism analysis of the three parts individually is also important, which can be used to examine the consequences of potential degradation of an ADC before it reaches its target (Kamath & Iyer, 2015).
Radioisotope labeling is a common method applied to study ADC distribution (Pimm et al., 1987). Radioisotopes, such as 3H, 131I and 89Zr, can be used to label antibodies and small molecule drugs at the same time or separately (Pimm et al., 1987). After IV administration, the distribution of whole ADCs or individual antibodies and payloads after release can be monitored in real time (Terwisscha et al., 2017). In blood or tumor tissue, antibodies and drug molecules labeled with different radioisotopes can be counted to determine the distribution and cleavage of ADCs. Simultaneously, an isotope-labeled ADC can be compared to an isotope-labeled antibody to study distribution differences caused by addition of the payload. Liquid scintillation counting of blood samples can be used to analyze drug distribution parameters after administration (Fand et al., 1986; Schmidt et al., 2002). In recent years, whole body quantitative autoradiography has also been applied to study distribution of ADCs (McEwen & Henson, 2015).
Before binding to the tumor surface antigen, linker cleavage and ADC metabolism could happen within circulation. This is one of the focal points in ADC development and clinical research. DAR variation impacts decomposition of ADCs (Roberts et al., 2013) as it determines drug loading and reflects safety and effectiveness of ADCs. For ADCs in blood circulation, DAR analysis mainly examines molar concentration changes in the ratio of total drug molecules and antibody molecules in ADCs with respect to administration time. The main analytical method is liquid chromatography-mass spectrometry (LC-MS), which has high sensitivity, resolution and accuracy (Mou et al., 2018). Moreover, the absolute molecular weight of ADC molecules with varying DAR values is different, so the DAR can be analyzed by electrospray ionization or matrix assisted laser desorption MS. The ADCs with different DAR values can be identified by individual mass charge ratio peaks on the mass spectrum, and then the relative contents (i.e. the distribution of DAR) can be obtained according to calculation of peak areas (Chen et al., 2016). The latest high-resolution mass analyzers, such as time of flight mass and orbital ion trap analyzer (orbitrap), combined with deconvolution calculation of intelligent software, make MS analysis of ADCs more convenient and accurate (Beck et al., 2019; Chen et al., 2016; Lanshoeft et al., 2016).
3.1.3. Problems of ADC tissue distribution and strategies for improvement
The biological distribution of ADCs enables their therapeutic effects (Lambert & Berkenblit, 2018). Very early clinical dosimetric studies of patients with radiolabeled antibodies showed that only ~0.1% of the antibody dose could be located in solid tumor mass 24 hours after infusion, regardless of tumor type or antibody target (Mach et al., 1980; Sedlacek et al., 1992). On the bright side, antibodies including ADCs as a high molecular weight agent can extravasate from leaky tumor vessels and sinusoidal vessels in the liver, bone marrow, or lymphoid organs, but not from other normal vessels, known as the enhanced permeability and retention (EPR) effect (Yasunaga, 2020). The EPR effect or enhanced vascular permeability can sustain an adequate supply of nutrients and oxygen for rapid tumor growth, which also provides accumulations of both endogenous and exogenous proteins larger than 60 kDa, benefiting macromolecule-based solid tumor therapy (Maeda et al., 2000). However, characteristics such as increased hydrostatic pressure and blockage of tumor lymphatic channels and blood vessels are reported to obstruct ADCs outside tumor tissues (Dahlgren & Lennernäs, 2020). Different from direct binding with hematological tumors, four steps occur for ADCs to bind solid tumors: reaching tumor blood vessels, exuding the blood vessel, distributing to tumor tissue and binding with the tumor target (Thurber et al., 2008). The best drug effect with limited cytotoxic molecules depends on tumor uptake of the ADC, tumor vascular density, membrane permeability and other rate limiting steps (Sapra et al., 2011). In addition, the affinity between the ADC and FcRn, the charge and physiochemical properties of the ADC, and the internalization of target will affect the distribution of ADCs (Maas & Cao, 2018).
In circulation, the toxicity and/or side effects caused by early payload release or ADC binding to normal tissue are problems that cannot be ignored (Bander, 2013). Non-target tissues and organs can capture ADCs, especially in tissues with large blood flow and strong phagocytic function (such as intestine and liver) (Maas & Cao, 2018). For example, liver cells phagocytize a number of ADC molecules and their rich metabolic enzymes decompose these ADCs where the released payloads then experience first and second phase metabolism resulting in drug inactivation (Tumey et al., 2015; Bumbaca et al., 2011).
Dispersion of ADCs in target-expressing normal cells can also lead to side effects or treatment failure (Bander, 2013). For example, treatment with epidermal growth factor receptor (EGFR)-targeted ADC (depatuxizumab mafodotin) for recurrent glioblastoma caused side effects including corneal abnormalities (Goss et al., 2018). When HER2-targeted ADC (trastuzumab emtansine (T-DM1)) was used for treatment of HER2-positive breast cancer the side effects of nodular regenerative hyperplasia and corneal abnormalities were observed (Lepelley et al., 2018; Tsuda et al., 2016). A folate receptor α-targeted ADC (mirvetuximab soravtansine) also showed corneal abnormalities following treatment of platinum-resistant ovarian cancer patients (Lepelley et al., 2018; Tsuda et al., 2016). Treating non-Hodgkin’s lymphoma patients with CD79b-targeted ADC (polatuzumab vedotin) resulted in 70% of patients showing peripheral neuropathy (Lu et al., 2017).
Additionally, off-target toxicity based on the Fc domain has been reported. An ADC antibody that lacked terminal galactose caused receptor mediated endocytosis by the mannose receptor (Gorovits & Krinos-Fiorotti, 2013). The mannose receptor is a lectin that specifically binds and internalizes antibodies with specific agalactosylated Fc-segments which has been found on the surface of endothelial cells and immune cells in hepatic and splenic sinusoids.
3.2. Second step - ADCs bind to target antigens on tumor cell surfaces
3.2.1. Interactions of ADCs with tumor surface antigens
After sufficient time in blood circulation, ADCs can spread throughout the body and reach tumor tissue (Nejadmoghaddam et al., 2019). An ADC that reaches tumor tissue can bind to its target antigen on the surface of tumor cells using the antigen-binding site of the antibody (shown in Figure 5, step 2). The target antigen-ADC complex can then undergo receptor-mediated endocytosis, which will transfer the ADC into tumor cells for subsequent transport and processing (Ritchie et al., 2013). Typically, target tumor cells should have moderate antigen expression level on the cell surface (105 /cell) (Chari et al., 2014),
Selection of tumor target proteins is a good starting point for ADC design. It determines the type of tumor that the ADC will target, and can predict problems that may be encountered in clinical trials (Bander, 2013). In recent years, many targets have been evaluated in the development process of ADCs (De Stefano et al., 2018). Most of these targets are tumor surface antigens (Ishii, 1995). Tumor surface antigens can be divided into two types, tumor associated antigens (TAA) and tumor specific antigens (TSA) (Butterfield, 2015; Trail et al., 2003). Among them, TAAs are proteins with high expression in tumor cells, but low expression in normal cells (Li et al., 2010). TAAs are often growth factor receptors, carcinoembryonic antigens or leukocyte differentiation antigens, which can be used in clinical diagnosis and classification of tumors (Li et al., 2010). For example, HER2 is a growth factor receptor expressed on the cell membrane (Nemeth et al., 2017) with high expression in breast cancer, gastric cancer and other cancer cells. At present, the ADCs targeting HER2 are Fam-trastuzumab deruxtecan (Enhertu) and Ado trastuzumab emtansine (Kadcyla) (Meric-Bernstam et al., 2019). Alternatively, CD30 is a leukocyte differentiation antigen which is widely overexpressed on the surface of various types of Hodgkin’s lymphoma cells (Pierce & Mehta, 2017). Brentuximab vedotin (Adcetris) targets CD30 and is used to treat lymphoma (Pierce & Mehta, 2017).
TSAs only exist on the surface of tumor cells, but not normal cells, so they can be used as markers to distinguish tumor tissue from normal cells. TSAs are unique antigens and generated by mutations. The external environment of physical factors such as radiation, chemical factors such as carcinogens, and biological factors such as viruses can also induce the production of TSAs (Rammensee et al., 2002). When cells expressing TSAs die or tumor tissue expressing TSAs undergo necrosis, the TSAs will be released into the blood, which can be detected and identified (Stauss, 2003). TSAs have the potential to be used as an antibody target to develop corresponding ADC drugs. At present, most targets of ADCs in clinical trials are TAAs, but many ADCs targeting TSAs are undergoing clinical or preclinical studies (Chen & Zhan, 2019). Therefore, ADCs targeting TSAs may be approved in the future, which have potential to reduce the off-target side effects caused by ADCs.
3.2.2. Research on tumor marker detection
For clinical use of ADC drugs, patient stratification is very important (Goutsouliak et al., 2020). Some cancer patients can express tumor antigens corresponding to ADC targets, while some patients do not. For the latter group, other treatment schemes are necessary. For example, Trastuzumab-DM1 is approved for treatment of breast cancer patients. The target is HER2, but ~75% of breast cancer patients are HER2 negative (Nemeth et al., 2017). Detection of tumor markers in patients is particularly important. Tumor markers are substances produced by tumor cells and related to the formation and occurrence of tumors (Hong et al., 2018). These substances originally exist in the nucleus, cytoplasm, membrane, or cell fluid of tumor cells. With tumor development, expression of the markers is elevated.
Radioimmunoassay and enzyme-linked immunosorbent assay (ELISA) are traditional identification methods of tumor markers (Marsigliante et al., 1993). At present, automatic immunochemical analysis instruments are commonly used detectors, which can rapidly and accurately measure serum tumor markers (Bi et al., 2009). The detection mechanism is based on the antigen-antibody interaction. Generally, luciferase-labeled antibodies specifically bind tumor markers and emit light following addition of a luminescent agent (Webster et al., 1990). In detection of the chemical reactions, some chemical groups are oxidized to form excited states and emit photons of a certain wavelength while returning to the ground state. This is a highly sensitive analytical method combining chemical reaction with immune response, also known as photomultiplier technique (Min et al., 2018). An automated instrument makes the experimental process very simple; obtained blood samples can be directly added to the chemiluminescent immunoassay system for detection and subsequent output to analyze results (Bi et al., 2009).
The above detection methods are serological detection methods, which are generally used in preliminary identification (Holdenrieder, 2016). The detection of tumor markers in tumor tissue is more important for ADC treatment selection. Histological detection of tumor markers can show the quantity, distribution and uniformity of tumor marker expression in tumor tissues (Shi et al., 1991). In addition, the study of tumor antigen regulatory factors, such as tumor marker renewal rate and heterogeneity, are important elements for consideration (Bander, 2013).
3.2.3. Problems and strategies for ADC target selection
Specificity and expression level
Target specificity and expression level are the core of target selection in ADC design. High specificity means one antigen is highly expressed on tumor cells with corresponding low or non-existent expression on normal cells. An antigen with a high expression level means this antigen is present in much larger numbers on the tumor cell surface or tumor tissue when compared to expression levels of the same antigen on either normal cells or other tumor cell types (Bander, 2013). High specificity and high expression level of the antigen can increase ADC recruitment to tumor tissues and decrease delivery to normal tissues.. Most of the selected targets are those with high specificity and high expression levels in tumors and their metastases (Wang et al., 2011). Although uncommon, if a tumor antigen that is also expressed in normal tissue is selected as the target, the expression in normal tissue should be limited to a small region or the normal tissue should have regenerative ability to minimize toxicity of the ADC towards the healthy tissue (Ducry, 2013).
Internalization
Most current payloads have intracellular targets, thus internalization is particularly important for cytotoxic activity (Bander, 2013). As internalization is determined by the properties of the target antigen, target internalization should be understood before ADC preparation. Moreover, successful recycling or replenishment of cell surface antigen is also essential (Collins et al., 2019). A cell surface antigen that can be quickly replenished can accumulate more ADC into the cell, thus improving payload delivery and the possibility of tumor cell death (Ducry, 2013). However, antigens that are quickly replenished can also deplete ADCs from the tumor tissue surface and prevent interior tumor penetration.
Heterogeneity
Heterogeneity of targets can be found between tumor cells or between patients. With respect to breast cancer, HER2 positive breast cancer patients can benefit from HER2 targeted ADCs, but the remaining patients would not benefit from them. The heterogeneity within tumor cells means that for instance not all tumor cells in a given HER2 positive breast cancer patient express HER2 (Ducry, 2013). The larger the proportion of target-negative cells, the weaker the therapeutic effect of ADCs (Ducry, 2013). Accordingly, for tumor heterogeneity, combination treatment with two or more ADCs may be efficacious and worth investigating (Hamilton, 2015).
Tumor stroma as target
At present, most ADCs are designed to target tumor surface antigens, which is endocytosis dependent. However, ADCs targeting tumor matrix (which is endocytosis independent) are getting increasing attention (Joubert et al., 2017). Cancer-related microenvironments from different cancer types can share many common features, so this approach may eventually extend applications of ADC therapy beyond strict selection of antigen positive patients (Ahmed et al., 2008). Moreover, most solid tumors have abundant stroma that hinders the distribution of high molecular weight agents, which functions as a barrier that prevents ADCs from attacking cancer cells (Yasunaga et al., 2011). Stromal targeted ADCs can release payload after binding to tumor stromal cells through mechanisms like extracellular plasmin or other proteolytic cleavage (Matsumura, 2020; Gébleux et al., 2017). The released small molecule payload can readily enter into and kill cancer cells across the stromal obstacle.
3.3. Third step – ADCs enter tumor cells through receptor-mediated endocytosis
3.3.1. Endocytosis pathways of ADCs
After circulation in the blood, ADCs reach tumor tissues and bind to target antigens on the surface of tumor cells (Damelin, 2018). Upon forming an antigen-antibody complex, the complex will undergo receptor-mediated endocytosis to transport the ADC to lysosomes for payload release (Kalim et al., 2017) (shown in Figure 5, step 3). According to the mechanism of action, endocytosis of ADCs can be divided into four types: clathrin-mediated endocytosis, cell membrane cave-like invagination also known as caveolae-mediated endocytosis, macropinocytosis, and clathrin- and caveolin-independent endocytosis (Kalim et al., 2017). Usually, ADCs enter cells through the clathrin-mediated endocytosis pathway (Chalouni & Doll, 2018).
Clathrin-mediated endocytosis
The clathrin-mediated endocytosis pathway is the most important receptor-dependent endocytosis pathway for biomacromolecule internalization (Swan, 2013). The clathrin pathway exists in mammalian cells to intake nutrients, regulate cell surface receptor levels, and transmit cell-cell signaling (Kalim et al., 2017). There are four steps in clathrin-mediated endocytosis: assembly of clathrin coats, membrane invagination and coated pit maturation, constriction of pit and fission of vesicle, and uncoating of vesicles (Kaksonen & Roux, 2018). Clathrin (190 kDa) can coat the antigen-ADC complex to form a 100–150 nm diameter vesicle (Chalouni & Doll, 2018). After formation of the complex, clathrin and adaptor protein 2 will be recruited to cell membranes, encapsulating the antigen-ADC complex then invaginating into cells to form clathrin capsule vesicles (Chen & Zhan, 2019). Dynein, a GTPase, will be recruited to the junction site between the cell membrane and vesicle, and forms a ring to separate the vesicle from the cell membrane allowing the vesicle to enter the tumor cell (Chalouni & Doll, 2018). Subsequently, the vesicle will transform into early endosomes with a pH of 5.9 to 6.0. Early endosomes can either transport the antigen-ADC complex back to the cell surface, or transform into late endosomes and transport the complex to the lysosomes. Late endosomes (also known as polyvesicular bodies) with a lower pH value are gradually formed in the early endosome (Chen & Zhan, 2019). The late endosome then fuses with the lysosome to release the ADC (Kalim et al., 2017). From endocytosis to degradation, this process takes several hours (Thurber et al., 2008), during which the payload accumulates in the tumor cells.
Caveolin-mediated endocytosis
Like clathrin-mediated endocytosis, endocytosis mediated by caveolin is generally receptor-dependent (Sargiacomo et al., 1995). Caveolae are a lipid raft structure (55–80 nm diameter) with complex cholesterol and are formed by polymerization of caveolin (21 kDa) (Chalouni & Doll, 2018). Caveolae can mediate cave-like invagination to encapsulate the antigen-ADC complex for internalization, the content and volume of which is smaller than those of clathrin-mediated endocytosis (Chen & Zhan, 2019). Caveolae are dynamic endocytic carriers. Caveolae buds and fuses back to the plasma membrane within one second (Chalouni & Doll, 2018). This is a dynamic cycle of caveolae budding from the plasma membrane, some of which fuse with plasma membranes in an apparent futile cycle, whereas others transform into early endosomes and then return to the plasma membrane. Caveolin-1 was shown to promote T-DM1 internalization and enhance drug sensitivity (Chung et al., 2018). In addition, one study showed that caveolae-mediated endocytosis was a novel T-DM1 resistance mechanism, internalizing T-DM1 and then expelling it out of the cell (Sung, et al., 2018).
Macropinocytosis
Macropinocytosis is a spontaneous endocytosis pathway (Bauer et al., 2016). It can also be induced by the engagement of growth factor, chemokine, or toll-like receptors (Mitrenga et al., 1975). This pathway, like phagocytosis, is actin-mediated. Additionally, microtubules and microfilaments play important roles in membrane ruffling and macropinosome closure (Marques et al., 2017). After extending a certain length, the plasma membrane forms about 50–100 microvilli-like structures (about 0.5 μm to 5 μm), which can encapsulate the ADC (Chalouni & Doll, 2018). This vesicle is called the macropinosome. The existence time of macropinocytosis in the cell is short (about 5–20 minutes). This process transports encapsulated contents to lysosomes (Bauer et al., 2020; Bauer et al., 2016). For normal tissues, macropinocytosis can function in clearing apoptotic cells, participating in an immune response, mediating the invasion of some pathogens and renewing the cell membrane (Marques et al., 2017). Macropinocytosis provides an effective way for non-selective endocytosis of extracellular nutrients and liquid macromolecules. In tumor cells, mutated genes such as v-Src or K-Ras can enhance macropinocytosis. There are a large number of dead cells (70–80%) in the center of tumor tissues due to a lack of nutrient supply (Jayashankar & Edinger, 2020). Surrounding living tumor cells can absorb the biomacromolecules and nutrients from dead cells through macropinocytosis, greatly improving the utilization rate of cell resources (Jayashankar & Edinger, 2020).
3.3.2. Research methods for intracellular transport and localization of ADCs
For the evaluation of ADC endocytosis, the rate and intracellular localization are the most important aspects (Harper et al., 2013). Determining the mechanism of endocytosis can offer valuable information including the proportion of ADCs in the tumor environment that enter into tumor cells and the time necessary for ADC internalization of the target antigen or target cancer cells. These determinants can be used to select the best target cancer cells, target antigen and optimal ADC composition. Intracellular localization of ADCs can offer information such as the proportion of ADC excreted outside of tumor cells after endocytosis, the rate limiting step in lysosome transport, the drug release rate in tumor cells, and the stability of different linkers throughout transport.
Flow cytometry is often used as a method for determining endocytosis of ADCs (Marder et al., 1987). At 4°C ADCs can specifically bind to the cell surface target antigen, but will not be endocytosed. At 37°C, cells can mediate endocytosis to intake ADCs from the cell surface. Thus the endocytosis rate of an ADC can be calculated indirectly by comparing the fluorescence intensity changes of cell surface at 4°C with that after incubation at 37°C (Harper et al., 2013; Chen & Zhan, 2019).
The use of confocal microscopy allows visualization of ADCs after endocytosis (Harper et al., 2013). This technique uses different fluorophores to label the ADC, payload, cell membranes, and organelles including lysosome-associated membrane protein-1, clathrin heavy chain and caveolin-1 (Donaldson, 2015; Chen & Zhan, 2019). The localization of the ADC in the cells at different times is observed directly by a high-resolution confocal microscope (Kalim et al., 2017; Harper et al., 2013).
3.3.3. Problems and methods to improve ADC endocytosis efficiency
Most current ADCs in development and all marketed ADCs target tumor cell surface antigens and depend upon internalization to release payloads, which emphasizes the importance of studying ADC endocytosis and methods for improving endocytosis efficiency (Nejadmoghaddam et al., 2019). The endocytosis efficiency of an ADC is related to endocytosis characteristics of antigens, antibody binding sites on antigens and tumor cell type (Birrer et al., 2019). The antibody internalization rate, internalization time, recycling and intracellular drug accumulation are all key parameters to measure and estimate ADC endocytosis efficacy (Birrer et al., 2019; Muhammad et al., 2017). Durbin et. al. evaluated the endocytosis effectiveness of an ADC against EGFR-overexpressing cancer cell lines. They found that the same ADC (Ab033) has an internalization rate of 0.047/minute for A431 cells and 0.15/minute for H441 cells. In addition, they found up to 45% of internalized Ab033 returned to the cell surface. Fortunately, despite the recycling after 24 hours, about 5×106 free toxic drug molecules accumulated in a single tumor cell. Based on the same antibody, Ab033, they prepared ADCs with either cleavable or non-cleavable linkers. Ab033 ADC with a cleavable linker showed high payload accumulation compared with the non-cleavable linker Ab033 ADC (Durbin et al., 2018). This suggests that many different conditions occurring at the same time affect the endocytosis efficiency and therapeutic efficacy of ADCs.
The structure of the antibody can be modified to improve the endocytosis efficiency (Thakur et al., 2018). Bispecific antibodies (those that bind two targets) and bisepitope antibodies (those that bind two locations on one target) are two developing strategies to improve ADC endocytosis efficiency (Chiu & Gilliland, 2016).
Bispecific antibody
Bispecific antibodies can bind two target antigens at the same time (the applicable target tumor cells should express both of the two antigens on their surface). The antigen with high endocytosis efficiency can internalize the ADC regardless of a low endocytosis efficiency of the other antigen (normally a low endocytosis antigen is a tumor marker, while a high endocytosis antigen does not have to be a tumor antigen) (Pegram et al., 2020; Andreev et al., 2017). For example, when antibodies targeting the fast-internalized epithelial cell kinase A2 (EphA2) and non-internalized high expression activated leukocyte cell adhesion molecule (ALCAM) are combined to form a bispecific ADC, the number of bispecific antibodies entering tumor cells exceeds that of a single mAb or a mixture of two antibodies, showing a bispecific-dependent amplification effect (Lee et al., 2019). The anti-ALCAM binding domain provides tumor-specific binding sites, and the anti-EphA2 binding domain can promote endocytosis. A small amount of internalized antigen EphA2 induces a large number of internalized ALCAM. On this basis, the bispecific ADC was constructed. Results demonstrate the killing effect of the bispecific ADC on tumor cells in vitro and in vivo were better than that of a single specific ADC (Lee et al., 2019), though it is unclear whether this strategy might negatively impact tumor specificity.
Bisepitope antibody
Dual epitope (or bisepitope) ADCs do not target two kinds of antigens, but target different epitopes of one target through two Fab fragments of the antibody (Robert et al., 1999). At present, a dual epitope ADC targeting HER2, a poor internalization antigen after antibody binding, has undergone clinical studies (Pegram et al., 2020). Li et. al. prepared a bivalent biparatopic antibody targeting two non-overlapping epitopes on HER2 and subsequently prepared an ADC that coupled the bisepitope antibody and microtubule inhibitor. They found that the bisepitope antibody induced HER2 receptor clustering and brought robust internalization, lysosomal trafficking, and degradation. In vivo studies showed superior anti-tumor activity over T-DM1 (Li et al., 2019).
3.4. Fourth step – ADCs release payloads within target cells
3.4.1. Lysosomal treatment of ADCs
The late endosomes carrying ADCs fuse with lysosomes and transport ADCs for degradation, thereafter releasing the payload (Howard, 2017) (shown in Figure 5, step 4). Lysosomes maintain an acidic environment of pH 4.5 to 5.0 through continuous proton input via H+-ATPase (Chalouni & Doll, 2018). They are also rich in proteolytic enzymes, such as cathepsin and collagenase. When ADCs enter tumor cells, the antibody part of the ADC has completed its mission. Part of the antibody will be hydrolyzed by proteases in the cell and broken down into amino acids (Ritchie et al., 2013). From endocytosis to degradation, this process takes several hours (Thurber et al., 2008). During this time, the payload will be released and accumulate in the cytoplasm. The release mechanism is determined by the type of linker (Staudacher & Brown, 2017).
The first approved ADC, Mylotarg, uses hydrazone linker technology. Hydrazone linkers are chemically unstable linkers, which are joined to the lysine residues of antibodies and release the payload as a result of environmental differences between the plasma and cytoplasm (Lu et al., 2016). Hydrazone linkers break at acidic pH. In general, they are stable at the neutral pH (7.3 to 7.5) of blood, where their degradation rate is only ~6% in 24 hours. After entering tumor cells, ADCs in late endosomes (pH 5.0 to 6.5) and lysosomes (pH 4.5 to 5.0) rapidly release their payloads. The 24-hour degradation can be up to 97% (Nolting, 2013).
Disulfide bond-based linkers are also a type of chemically unstable linker (Lu et al., 2016). The disulfide bond is stable in plasma. After entering tumor cells, it contacts with the strong reductive environment of the cytoplasm and releases the payload. Disulfide linker reduction requires sulfhydryl cofactors, such as reductive glutathione (GSH), or disulfide isomerase (Nolting, 2013). On average, the concentration of GSH in cells ranges from 0.5 to 10 mmol/L, while in blood GSH is very low at about 5 μmol/L (Tsuchikama & An, 2016).
Enzyme catalyzed linkers are more stable than disulfide and hydrazone linkers (Tumey & Han, 2017). Peptide linkers are a typical enzyme-catalyzed linker, which can better control the release of the ADC payload. Due to the neutral pH of blood and endogenous inhibitors, the activity of protease in blood is very low, which ensures the stability of peptide linkers in blood (McCombs & Owen, 2015). The half-life of a peptide linker is generally 7–10 days (Nolting, 2013). After an ADC enters a tumor cell and is transported to the lysosome, its peptide linker will break down gradually by protease degradation and release the payload. In addition, the β-glucuronide linker is a type of enzyme-cleavable linker which is cleaved by β-glucosidase (Jeffrey et al., 2010). This enzyme is overexpressed in some types of tumors, and its activity is very low in the extracellular environment. An advantage of the β-glucuronide linker is its hydrophilicity, which can reduce ADC aggregation.
At present, the most common non-cleavable linker in ADCs is the thioether linker, which is obtained by reaction of maleimide and mercaptan (Lu et al., 2016). After degradation of the thioether linker in lysosomes, the lysine-containing payload is released (Walles et al., 2017). Generally, a thioether linker has a half-life of about 7 days, which is close to that of a cleavable linker (Alley et al., 2008). For example, the marketed ADC T-DM1 applies a non-cleavable thioether linker, designed to retain the payload inside the tumor cells (Lambert & Chari, 2014). Additionally, the marketed ADC Blenrep applies a non-cleavable maleimidocaproyl linker connecting belantamab and MMAF.
3.4.2. Research on linker technology
Research on linker technology mainly focuses on linker stability determination, optimization of old linkers, development of new linker strategies and mapping of coupling kinetic models (Nolting, 2013). The determination of ADC stability in vivo is key in evaluating the linker (Lu et al., 2016). The most important detection methods are ELISA and LC-MS (Chen et al., 2016). The lysine residues on the antibody surface contain amino groups, which can be used as coupling sites due to their strong nucleophilicity (Nolting, 2013). In the simplest coupling reaction, the active ester (O-succinimide or N-hydroxythiosuccinimide) can react directly with lysine residues to form stable amide bonds (Behrens et al., 2015). Other active esters such as hydroxybenzotriazole, fluorobenzene or nitrobenzene derivatives can also be used to form linkers (Yao et al., 2016). This coupling method is widely used for non-cleavable linkers.
Alternatively, a two-step coupling method can be used to couple antibodies and payloads (Chih et al., 2011). In this method, lysine residues of the antibody are modified, the linker is added, and then payloads are coupled to the linker (Gandhi et al., 2018). The second coupling reaction usually uses a sulfhydryl group in the payload (Chih et al., 2011). For example, T-DM1 is linked to the antibody by using SMCC as its linker (Wakankar et al., 2010). After preparation, DAR determination and structure characterization of the ADC are needed. The available detection methods of ADCs include UV-Vis spectroscopy, hydrophobic interaction chromatography (HIC) and LC-MS (D’Atri et al., 2018; Ouyang, 2013).
3.4.3. Problems and strategies related to linkers
There are, on average, 40 lysine sites on a typical antibody that can be used to couple payloads, giving ~106 forms of the ADC, theoretically (Chen & Zhan, 2019). IgG1 contains only four inter-chain disulfide bonds, which have higher reactivity than intra-chain disulfide bonds and can be used for modification (Badescu et al., 2014). After disulfide bond breaking, eight cysteine residues are produced as potential junction sites. However, the use of cleaved cysteine as a payload binding site will affect stability of the antibody (Dimasi et al., 2017). Some of the early ADCs were constructed using glycosyl groups on the antibody surface as junction sites, with the advantage of being able to connect large numbers of payloads (Tumey & Han, 2017). However, the disadvantage lies in the inability to control the junction site and DAR, leading to the junction site covering the antibody specific recognition sites. As a result, part of these ADCs cannot bind to the target antigen, or alternatively, the excessive connections can enhance the hydrophobicity of the ADC resulting in hydrophobic aggregation and precipitation (Panowski et al., 2014). Early development of Mylotarg used lysine on antibodies to couple payloads. Due to a relatively large amount of lysine on the antibody, the DAR is as high as 14, which makes inter-batch consistency and ADC diversity of Mylotarg difficult to control. At present, research has focused on reducing heterogeneity of ADCs (Sochaj et al., 2015; Behrens et al., 2015).
Site-specific coupling technology is used to couple a certain number of payload molecules to specific sites of the antibody, ensuring consistency of the prepared ADC, with the benefit of improving efficacy and reducing toxicity (Dan et al., 2018). Enzyme based coupling is an important example of this technology. Enzymatic modification of antibodies can produce completely homogeneous ADCs through enzyme-specific substrate catalysis. The commonly used enzymes are glycosyltransferase, glutamine transferase and β-galactosidase (Yamada & Ito, 2019). The Fc segment of all IgG subtypes has a pair of N-glycosylated aspartates (Asn297), which can be used for glycosyltransferase modification to introduce linker and payload (Kubizek et al., 2017). Specifically, β-N-acetylglucosaminidase is used to break oligosaccharides on N-glycosylated aspartate to expose active sites for modification (the original oligosaccharide has unfixed length and lacks a payload conjugating site). Then β-N-acetylglucosaminidase mutants are used to add oligosaccharides with azide-based payload binding sites. Following this addition, payloads can be attached onto the oligosaccharide by azides through biorthogonal reactions (Manabe & Yamaguchi, 2021). In one study the authors designed a β-galactosidase-cleavable linker for ADCs. A galactoside linker, monomethyl auristatin E (MMAE) and cysteine-reactive trastuzumab were conjugated to form an ADC. Evaluation of the ADC, both in vitro and in vivo, found superior therapeutic efficacy in mice compared to trastuzumab emtansine (Kolodych, et al., 2017).
During antibody preparation, unnatural amino acids can be added into the antibody through genetic engineering (Axup et al., 2012). The biggest advantage of introducing unnatural amino acids is that the prepared antibody can easily complete specified chemical reactions at these residues to connect a payload (Liang et al., 2019). The most common method is to change a stop codon into a coded codon (Axup et al., 2012). This method also requires synthetic tRNA and tRNA synthetase. Additionally, the strategy of converting amino acid residues in a non-antigen-recognition region of an antibody into cysteine by genetic engineering has also been reported (Dimasi et al., 2017). This method can be used to modify a disulfide bond in the antibody chain to couple payloads without destroying inter-chain disulfide bonds or affecting the stability of the antibody (Dimasi et al., 2017).
3.5. Fifth step – cytotoxic ADC payloads inhibit tumor cell growth
3.5.1. ADC payload mechanism of action and toxicity
Once ADCs are degraded in lysosomes, their payloads are released into the cytoplasm to engage their corresponding targets (Casi & Neri, 2012). Typically, for moderately potent payloads, accumulation over 106 molecules/cell will kill one tumor cell (Chari et al., 2014). The most common targets are DNA in the nucleus and microtubules in the cytoplasm. Drugs targeting nuclear DNA include adriamycin, duocarmycin and pyrrole benzodiazepines (PBDs) (Fu & Ho, 2018). These drugs embed into small grooves on the double helix structure of DNA, breaking the DNA apart and leading to cellular apoptosis (Fu & Ho, 2018). There are two kinds of ADC payloads targeting microtubules: auristatin derivatives (including MMAE and MMAF) and maytansine derivatives (including DM1 and DM4) (Akaiwa et al., 2020). More than 60% of ADCs in clinical trials employ microtubule inhibitors as their payloads (Fu & Ho, 2018). Compared to payloads that target DNA, these drugs better limit cell growth. They mainly target microtubules of rapidly dividing cells, blocking the cell cycle and leading to apoptosis. After degradation of the ADC, the small molecules released into the cytoplasm can cross cell membranes, enter adjacent cells and inhibit proliferation (Staudacher & Brown, 2017). The bystander effect is helpful to inhibit tumor cells with weak or no expression of TSA in heterogeneous tumors (Staudacher & Brown, 2017) (shown in Figure 5, step 5), though at the same time it could have negative consequences if neighboring normal cells are affected.
Tubulin inhibitors
Tubulin polymerization is essential for a variety of cellular processes, including intracellular transport, mitosis, and structural integrity maintenance (Chen et al., 2017a). Microtubules have at least five known binding sites, including vinca alkaloid, colchicine, paclitaxel, meticin and lauramide binding sites. Microtubule-targeting agents block separation of the mitotic spindle, resulting in unstable cytoskeleton structure, which induces cell cycle arrest in the G2/M phase leading to cell death. The structure of microtubules can be influenced by the binding of β-subunit or α-β heterodimer of tubulin (Chen et al., 2017a; Haider et al., 2019). Maytansines can bind to the extended microtubules and inhibit formation of microtubules by blocking the tubulin heterodimer formation (Steinmetz & Prota, 2018). Drugs with this mechanism are suitable for rapidly proliferating cells. Therefore, microtubule inhibitors are particularly cytotoxic to cancer cells, because cancer cells divide and grow faster than most normal cells (Steinmetz & Prota, 2018). This can also reduce the off-target killing of normal tissues and cells. However, early release of microtubule inhibitors can kill some rapidly dividing normal cells, resulting in adverse side effects (Gébleux et al., 2015). In addition, static and undifferentiated cancer cells, such as cancer stem cells or tumor initiating cells, are less sensitive to microtubule inhibitors, thus avoiding killing and producing drug resistance (Takebe et al., 2015). It should be noted that most mouse xenotransplantation models contain tumors that grow much faster than normal human tumors (Morton et al., 2016). Therefore, drugs that exhibit strong tumor killing activity in animal experiments may not necessarily show the same efficacy in human clinical trials.
Maytansines are cytotoxins similar in structure to rifamycin, geldanamycin and cladosporin, which have a common nineteen membered macrolide structure (Elliott & Fried, 1976; Erickson et al., 2006). Maytansine and its derivatives are potent inhibitors of microtubule assembly. They can induce mitotic arrest of poisoned cells by binding to tubulin at the vinblastine binding site, therefore, the mechanism of maytansines is similar to that of vinblastine (Huang et al., 1985). At low concentrations, microtubule dynamic instability and cell migration are inhibited; at higher concentrations, microtubule assembly and cell division are inhibited. These effects result in antiproliferative activity during mitosis (Su et al., 2018). The ED50 (effective dose) of maytansine ranges from 10−5 μg/ml to 10−4 μg/ml (Cassady et al., 2004). Premature release of maytansines can lead to side effects such as neurotoxicity, gastrointestinal toxicity, weakness, nausea, vomiting and diarrhea (Kowalczyk et al., 2017).
DNA damaging agents
DNA damaging agents have a long history of application to cancer chemotherapy (Hartley & Hochhauser, 2012). These molecules exert their cytotoxic role by binding to the DNA double helix. According to their mechanisms of action against DNA, they are divided into four types: double strand break agents (such as doxorubicin), alkylating agents (such as PBDs), intercalating agents (such as calicheamicins) and crosslinking agents (such as cisplatin) (Hartley & Hochhauser, 2012). DNA damage agents have two advantages as ADC payloads relative to microtubule targeting agents: (1) DNA damaging agents (pM IC50 values) have more potent cytotoxicity than microtubule targeting agents (nM IC50 value), which enables ADCs to kill cancer cells at lower doses, including cancer cells with low expression of target antigens; (2) DNA damaging agents kill tumor cells regardless of their cell cycle stages (dividing or non-dividing cells) (Bander, 2013). This can produce cytotoxic activity against less proliferative cancer cells, such as cancer stem cells (Takebe et al., 2015). In addition, calicheamicins can insert into the minor DNA groove and induce cell apoptosis. The cytotoxicity of calicheamicin is more than 100 times that of standard chemotherapy drugs (Ricart & D., 2011). Among the ADC drugs currently on the market, Mylotarg and Besponsa both use calicheamicin as their payload.
3.5.2. Metabolism studies of ADCs
Payloads released from ADCs in the lysosome kill tumor cells. Research in this area mainly focuses on the metabolism of the ADC, the toxicity mechanism of the payload, and the adverse reactions of the ADC (Sarrut et al., 2016). As an ADC enters circulation, payloads are slowly released, and the average DAR value will decrease with time until reaching zero (Hedrich et al., 2018). The change rate of DAR can reflect the release rate of payloads from the antibody. At the same time, the molar concentration of cytotoxic molecules in circulation will increase, which affects efficacy and safety. The metabolism of ADCs includes: (1) the study of small molecules and their metabolites and (2) the study of antibodies and their metabolites (Malik et al., 2017). These studies help uncover the toxicity mechanism and metabolic process of ADCs, to understand more about these molecules to optimize their design and achieve a better curative effect.
ELISA is a typical ligand binding assay for qualitative and quantitative analysis of antibody molecules in ADCs (Shah & Maghsoudlou, 2016) (Li et al., 2016). The ADC and its metabolism can be measured conveniently and accurately by this method. A downfall of this assay is that it only measures the ADC antibody, but not released payloads. Additionally, it is only suitable for the detection of known metabolites, not unknown metabolites (Li et al., 2016).
LC-MS can be used to detect small-molecule payloads. With the improvement of analytic methods and instrument sensitivity, LC-MS can be used to analyze the DAR of ADCs and the metabolism of toxic small molecules (Zhu et al., 2020). This method is quite beneficial especially because it is capable of identifying new metabolites (Huo et al., 2015). The connection sites and the number of connections of the payload on the ADC can be obtained (Yu et al., 2010).
3.5.3. Problems with ADC payloads
Effects of payloads on ADCs
Often times, cytotoxic small molecule drugs used as ADC payloads are hydrophobic in nature (Adair et al., 2012; Anderl et al., 2013). The hydrophobicity of the payload affects physical and chemical properties of the ADC (Lyon et al., 2015). Excessive hydrophobic cytotoxic molecules coupling to the ADC structure make ADCs easy to aggregate and become unstable. This is not only due to the hydrophobicity, but also because of changes in antibody secondary and tertiary structures and the resulting impact on conformation stability (Lyon et al., 2015). For example, the serum half-life of the ADC T-DM1 (3.94 days) is far shorter than its antibody trastuzumab (18.3 days) (Pegram et al., 2020). In addition, differences in conjugation sites and the DAR also affect pharmacokinetic (PK) properties of ADCs leading to ADC heterogeneity and characterization difficulties (Lyon et al., 2015). In contrast, hydrophilic payloads decrease ADC aggregation, but also decrease ADC membrane permeability and bystander effect. The off-target effects and metabolism of payloads directly impact the function of ADCs. Payloads that are substrates, inhibitors or inducers of metabolic enzymes such as cytochrome P450 isozymes or even transporters (such as P-glycoprotein) likely impact ADC metabolism as well (Lynch & Price, 2007; Shen et al., 2012).
Limit adverse reactions
One of the key research points of ADC development is to limit off-target toxicity. This toxicity of the ADC is mainly driven by prematurely released payload or payload released in normal cells (Mahalingaiah et al., 2019). In blood, the ADC linker may break (especially cleavable linker), which can lead to toxicity. ADCs collected by normal cells through nonselective endocytosis also produce toxicity (Su et al., 2018). In addition, some ADCs will bind non-tumor cells that also express the target antigen. Ocular toxicity has been reported in more than a dozen ADCs targeting a variety of antigens, most of which have limited expression in the eyes (Su et al., 2018). These adverse reactions include blurred vision, abnormal cornea, or dry eyes (Masters et al., 2018). Most of the ADCs involved contain MMAF or DM4 indicating that there is a relationship between these tubulin inhibitory payloads and the development of ocular side effects (Wolska-Washer & Robak, 2019). It is speculated that this effect, at least in part, is a result of damage to stem cells living in the limbus (Parrozzani et al., 2020). Compared with MMAF, ADCs containing MMAE have less ocular toxicity (Akaiwa et al., 2020). However, most MMAE based ADCs have toxic effects such as neutropenia and peripheral neuropathy (Masters et al., 2018). These are also common side effects of various microtubule inhibitors. As mature neurons do not actively divide, neuronal cell death is not related to mitotic blockade (Masters et al., 2018). In contrast, peripheral neuropathy is thought to be caused by damage to microtubule-dependent transport pathways. The development of vadastuximab talirine (SGN-CD33A) based on PBDs was suspended by the FDA due to its severe hepatotoxicity (Saber et al., 2019). In one study the authors carried out a meta-analysis of 70 publications to obtain clinical toxicity information of ADCs. In the analysis, anemia, neutropenia, and peripheral neuropathy were found for MMAE ADCs, where thrombocytopenia and hepatic toxicity were evident for DM1, and ocular toxicity followed MMAF (Masters et al., 2018). In another example, T-DM1 can bind to HER2 in hepatocyte cell surfaces, thus leading to hepatic toxicity (Pegram et al., 2020). Additionally, T-DM1 can internalize into megakaryocytes via a FcγRIIa-dependent manner, resulting in diminished megakaryocytes. Platelets are produced by megakaryocytes, so T-DM1 treatment can lead to thrombocytopenia (Pegram, et al., 2020). ADCs that extravasate into a normal lung have the potential to cause interstitial pneumonia as a severe adverse effect. EGFR-targeted therapy causes dermatitis in some patients, and while the mechanism is unknown, it is observed in both EGFR-high and -low tumors (Lenz, 2006). These adverse reactions are concluded as off-target toxicities and related to payloads. Overall, it is necessary to study new compounds as potential payloads for ADCs that reduce the risk of adverse reactions and off-target toxicity, while providing the same or better tumor cell killing.
4. Current ADC clinical landscape
Use of ADCs for tumor therapy is promising for the treatment of a number of cancer types. So far, at least ten ADC drugs have been approved by the FDA (shown in Table 1). Among them, six ADCs treat hematological malignancies. Compared with solid tumors, target antigens of hematological malignancies are more easily contacted by circulating ADCs. Lymphoma and breast cancer are two of the most researched cancers. There are three ADCs approved for breast cancer and another two for lymphoma. The antigens in these ten approved ADCs (e.g., CD33, CD30, CD22) are tumor cell surface molecules with highly restricted distribution patterns, which avoid ADCs targeting hematopoietic tissues and pluripotent stem cells, providing better specificity and tolerance.
The number of ADCs in clinical trials is increasing rapidly. At present, there are more than 280 ADC-related clinical trials at different stages studying about 138 different ADCs to treat roughly 60 kinds of cancer (https://clinicaltrials.gov). Pharmaceutical companies around the world have contributed to the vigorous development of ADCs in clinical research, which brings new hope to the fight against cancer. Table 2 summarizes some of the ADCs in clinical trials in the most recent years, focusing on tumor types, payloads, linker type, sponsors, and clinical status.
Table 2.
ADCs under clinic trials in the most recent years
| ADC name | Tumor target Antibody | Payload DAR | Linker type | Clinical status | ClinicalTrials Identifier Sponsor | Indication | Reference |
|---|---|---|---|---|---|---|---|
| Targeted cancer | |||||||
| 1. Multiple kinds of tumors | |||||||
| Trastuzumab deruxtecan | HER2 Humanized antibody |
Deruxtecan (camptothecin) | Cleavable | Phase I |
NCT02564900 Daiichi Sankyo Co, Ltd. |
Advanced breast and gastric or gastro-esophageal tumors | (Doi et al., 2017) |
| Lifastuzumab vedotin (LIFA) | NaPi2b (SLC34A2) Humanized IgG1 monoclonal antibody (MNIB2126A) |
MMAE | Cleavable VC linker | Phase I |
NCT01911598 Genentech, Inc. |
Non-small cell lung cancer or platinum-resistant ovarian cancer | (Gerber et al., 2020) |
| Anetumab ravtansine (BAY 94–9343) | Mesothelin Fully human IgG1 monoclonal antibody |
DM4 3–4 |
Cleavable disulfide linker | Phase I |
NCT01439152 Bayer HealthCare Pharmaceuticals |
Advanced or metastatic solid tumors | (Hassan et al., 2020) |
| PF-06664178 (RN927C) |
Trop-2 Humanized IgG1 |
Aur0101 (auristatin derivative) 2 |
Cleavable C-terminus of heavy chain via an enzymatic process | Phase I |
NCT02122146 Pfizer |
Advanced or metastatic solid tumors | (King et al., 2018) |
| IMGN853 Mirvetuximab soravtansine |
Folate receptor α | DM4 | Phase I |
NCT01609556 ImmunoGen, Inc. |
Solid tumors | (Moore et al., 2017a) | |
| ABT-414 | EGFR Humanized recombinant antibody |
MMAF | Non-cleavable MC linker | Phase I/II |
NCT01741727 AbbVie |
Advanced solid tumors | (Munasinghe et al., 2017) |
| Telisotuzumab Vedotin (ABBV399) |
c-Met Humanized monoclonal antibody |
MMAE | Cleavable VC linker | Phase I |
NCT02099058 AbbVie Inc. |
Advanced solid tumors | (Strickler et al., 2018) |
| PF-06647263 | EFNA4 | Calicheamicin | Phase I |
NCT02078752 Pfizer |
Advanced solid tumors | (Garrido-Lag una et al., 2019) | |
| PF-06263507 | 5T4 Humanized IgG1 antibody |
MMAF 4 |
Cleavable MC linker | Phase I |
NCT01891669 Pfizer |
Advanced solid tumors | (Shapiro et al., 2017) |
| Depatuxizumab mafodotin (ABT-414) | EGFR | MMAF 3–4 |
Non-cleavable MC linker | Phase I/II |
NCT01800695 AbbVie |
Advanced solid tumors likely to overexpress EGFR |
(Goss et al., 2018) |
| Enfortumab vedotin (EV) | Nectin-4 Fully humanized IgG1κ monoclonal antibody |
MMAE 3–4 |
Cleavable maleimidocaproyl VC linker | Phase I |
NCT02091999 Astellas Pharma and Seattle Genetics |
Nectin-4-positive solid tumors | (Rosenberg et al., 2020) |
| Brentuximab vedotin | CD30 Chimeric IgG1 antibody |
MMAE | Cleavable | Phase II |
NCT01461538 Seattle Genetics, Inc. |
CD30-expressing solid tumors | (Sharman et al., 2019) |
| Aprutumab Ixadotin (BAY 1187982) | Fibroblast Growth Factor Receptor 2 (FGFR2) fully human monoclonal antibody | Auristatin W | Non-cleavable | Phase I |
NCT02368951 Bayer Healthcare |
Advanced FGFR2 positive solid tumors | (Kim et al., 2019) |
| 2. Hodgkin’s lymphoma | |||||||
| Brentuximab vedotin | CD30 Monoclonal antibody |
MMAE | Cleavable | Phase III |
NCT01712490 Millennium Pharmaceuticals and Seattle Genetics |
Stage III or IV Hodgkin’s lymphoma | (Suri et al., 2019) |
| Brentuximab vedotin | CD30 | MMAE | Phase II |
NCT01569204 Takeda Pharmaceuticals |
Advanced classical Hodgkin’s lymphoma | (Eichenauer et al., 2017) | |
| 3. Non-Hodgkin lymphoma | |||||||
| IMGN529 | CD37 Humanized monoclonal antibody |
DM1 | Phase I |
NCT01534715 ImmunoGen, Inc. |
Relapsed/refractory B-cell non-Hodgkin lymphoma | (Stathis et al., 2018) | |
| Pinatuzumab vedotin | CD22 Monoclonal antibody |
MMAE | Cleavable | Phase I |
NCT01209130 Genentech, Inc. |
Relapsed or refractory B-cell non-Hodgkin lymphoma | (Advani et al., 2017) |
| Coltuximab ravtansine (SAR3419) | CD19 Monoclonal antibody |
DM4 | Cleavable hindered disulfide bond | Phase II |
NCT01472887 Sanofi |
Relapsed/refractory diffuse large B-cell lymphoma | (Trnĕný et al., 2018) |
| Inotuzumab ozogamicin (InO) | CD22 Humanized antibody |
Calicheamicin | Cleavable | Phase III |
NCT01232556 Pfizer Inc. |
Relapsed/refractory aggressive B-cell non-Hodgkin lymphoma | (Dang et al., 2018) |
| SGN-CD70A | CD70 | PBD | Cleavable | Phase I |
NCT02216890 Genetics, Inc. |
CD70-positive diffuse large B cell lymphoma and mantle cell lymphoma | (Phillips et al., 2019) |
| Brentuximab vedotin | CD30 | MMAE | Phase II |
NCT01421667 Genetics, Inc. |
Diffuse large B-cell lymphoma | (Bartlett et al., 2017) | |
| Brentuximab vedotin | CD30 | MMAE | Phase I/II |
NCT02581631 Bristol-Myers Squibb and Seattle Genetics |
Relapsed/refractory primary mediastinal large B-cell lymphoma | (Zinzani et al., 2019) | |
| 4. Lung cancers | |||||||
| Rovalpituzumab tesirine (SC16LD6.5) | DLL3 Humanized IgG1 monoclonal antibody |
SC-DR002 | Cleavable | Phase I |
NCT01901653 Stemcentrx Inc. |
Small-cell lung cancer | (Rudin et al., 2017) |
| Sacituzumab govitecan | Trop-2 Humanized antibody |
SN-38 | Phase I |
NCT01631552 Immunomedics, Inc. |
Small-cell lung cancer | (Gray et al., 2017) | |
| Lorvotuzumab mertansine (IMGN901) |
CD56 Humanized monoclonal antibody |
DM1 | Cleavable disulfide linker | Phase I/II | NCT01237678 | Small-cell lung cancer | (Socinski et al., 2017) |
| Sacituzumab govitecan | Trop-2 Humanized antibody |
SN-38 | Phase I/II | NCT01631552 | Advanced non-small-cell lung cancer | (Heist et al., 2017) | |
| Trastuzumab emtansine (T-DM1) | HER2 | DM1 | Non-cleavable thioether linker | Phase II | HER2-positive non-small cell lung cancer | (Hotta et al., 2018) | |
| Rovalpituzumab tesirine | DLL3 Humanized IgG1 monoclonal antibody |
PBD | Cleavable | Phase I |
NCT03086239 AbbVie Inc. |
Advanced, recurrent small cell lung cancer | (Udagawa et al., 2019) |
| Ado-Trastuzumab emtansine (T-DM1) | HER2 | DM1 | Non-cleavable thioether linker | Phase II |
NCT02675829 Genentech |
HER2-mutant lung cancer | (Li et al., 2018) |
| 5. Cancers in esophagus, stomach or intestine | |||||||
| Trastuzumab deruxtecan | HER2 Fully humanized IgG1κ monoclonal antibody |
DXd 8 |
Non-cleavable maleimide tetrapeptide linker | Phase II |
NCT03329690 Daiichi Sankyo, Inc. and AstraZeneca |
HER2-positive gastric cancer | (Shitara et al., 2020) |
| TAK-264 (MLN0264) | Guanylyl cyclase C Human mAb | MMAE | Cleavable | Phase II |
NCT02202759 Millennium Pharmaceuticals Inc. |
Metastatic or recurrent adenocarcinoma of the stomach or gastroesophageal junction | (Almhanna et al., 2017a) |
| Ado-Trastuzumab Emtansine (T-DM1) | HER2 Humanized monoclonal antibody | DM1 | Non-cleavable thioether linker | Phase I |
NCT01641939 Roche Ltd, and Genentech, Inc. |
HER2-positive advanced gastric cancer | (Chen et al., 2017b) |
| 6. Pancreatic adenocarcinoma | |||||||
| LMB-100 | Mesothelin (MSLN) Humanized Fab | Pseudomonas exotoxin A (PE) | Phase I/II |
NCT02810418 Bayer AG, Aduro BioTech, and Morphotek Inc. |
Advanced pancreatic adenocarcinoma | (Alewine et al., 2020) | |
| TAK-264 (MLN0264) | Guanylyl cyclase C Human monoclonal antibody | MMAE | Cleavable | Phase II |
NCT02202785 Millennium Pharmaceuticals, Inc. |
Advanced or metastatic pancreatic adenocarcinoma | (Almhanna et al., 2017b) |
| 7. Colorectal cancer | |||||||
| Labetuzumab Govitecan (IMMU-130) | Carcinoembryonic antigen-related cell adhesion molecule 5 (CEACAM5) (CD66e) Humanized antibody |
SN-38 (camptothecin) | Phase I/II |
NCT01605318 Immunomedics. Inc. |
Refractory or relapsing metastatic colorectal cancer | (Dotan et al., 2017) | |
| 8 Breast cancer | |||||||
| Trastuzumab deruxtecan (T-DXd) | HER2 Humanized antibody |
Exatecan derivative (a topoisomerase I inhibitor) | Cleavable peptide linker | Phase Ib |
NCT02564900 Daiichi Sankyo. Inc. |
HER2-low-expressing advanced breast cancer | (Modi et al., 2020) |
| Sacituzumab govitecan (IMMU-132) | Trop-2 | SN-38 | Phase I/II |
NCT01631552 David M. Goldenberg |
Metastatic triple-negative breast cancer | (Bardia et al., 2017) | |
| Trastuzumab emtansine | HER2 Humanized monoclonal antibody |
DM1 | Phase III |
NCT00829166 Genentech. |
HER2-positive advanced breast cancer | (Diéras et al., 2017) | |
| 9. Ovarian cancer | |||||||
| Mirvetuximab soravtansine (IMGN853) | Folate receptor α Humanized antibody | DM4 | Cleavable disulfide linker | Phase Ib |
NCT02606305 ImmunoGen, Inc. |
Sensitive ovarian cancer (in combination with carboplatin); FRα-positive, platinum-resistant ovarian cancer (in combination with bevacizumab) |
(Moore et al., 2018a; O’Malley et al., 2020) |
| Mirvetuximab soravtansine (IMGN853) | Folate receptor α Humanized antibody | DM4 | Cleavable | Phase I |
NCT01609556 ImmunoGen. |
Relapsed epithelial ovarian cancer | (Martin et al., 2017) |
| Lifastuzumab vedotin (DNIB0600A) | NaPi2b Humanized IgG1 monoclonal antibody |
MMAE | Phase II |
NCT01991210 Genentech, Inc. |
Platinum-resistant ovarian cancer | (Banerjee et al., 2018) | |
| Mirvetuximab Soravtansine IMGN853 | Folate receptor α Humanized monoclonal antibody | DM4 | Cleavable | Phase I |
NCT01609556 ImmunoGen. |
Platinum-resistant ovarian, fallopian tube, or primary peritoneal cancer | (Moore et al., 2017b) |
| Mirvetuximab soravtansine (IMGN853) | Folate receptor α Humanized monoclonal antibody | DM4 3–4 |
Cleavable disulfide linker | Phase III | NCT02631876 | Platinum-resistant ovarian cancer | (Moore et al., 2018b) |
| 10. Cervical cancer | |||||||
| Tisotumab Vedotin | Tissue factor (TF) Fully human monoclonal antibody |
MMAE | Protease-cleavable linker | Phase I/II |
NCT02001623 Genmab A/S |
Recurrent or metastatic cervical cancer | (Hong et al., 2020) |
| 11. Renal cell carcinoma | |||||||
| AMG 172 | CD27L Fully human IgG1κ monoclonal antibody |
DM1 5 |
Non-cleavable 4-[N-maleimidomethyl] cyclohexane-1-carb oxylate conjugated to lysine residues | Phase I |
NCT01497821 Amgen Inc. |
Relapsed/refractory renal cell carcinoma | |
| AGS-16M8F and AGS-16C3F | ENPP3 Fully human IgG2a antibodies |
MMAF | Non-cleavable | Phase I |
NCT01114230 NCT01672775 Agensys, Inc. |
Advanced refractory renal cell carcinomas | (Thompson et al., 2018) |
| SGN-CD70A | CD70 | PBD | Cleavable | Phase I |
NCT02216890 Genetics, Inc. |
CD70-positive metastatic renal cell carcinoma | (Pal et al., 2019) |
| 12. Prostate cancer | |||||||
| PSMA ADC | Prostate-specific membrane antigen (PSMA) Fully human IgG1 monoclonal antibody |
MMAE 4 |
Cleavable VC linker | Phase I |
NCT01414283 Progenics Pharmaceuticals, Inc. |
Chemotherapy-refractory prostate cancer | (Petrylak et al., 2019) |
| DSTP3086S | Six-transmembrane epithelial antigen (STEAP1) Humanized IgG1 monoclonal antibody |
MMAE | Cleavable | Phase I |
NCT01283373 Genentech, Inc. |
Metastatic castration-resistant prostate cancer | (Danila et al., 2019) |
| ASG-5ME | SLC44A4 Fully human IgG2κ monoclonal antibody |
MMAE | Cleavable MC-VC linker | Phase I |
NCT01228760 NIH/NCI Cancer Center |
Metastatic castration-resistant prostate cancer | (McHugh et al., 2019) |
| PSMA ADC | PSMA Fully human IgG1 monoclonal antibody |
MMAE | Cleavable VC linker | Phase II |
NCT01695044 Progenics Pharmaceuticals, Inc |
Progressive metastatic castration-resistant prostate cancer | (Petrylak et al., 2020) |
| 13. Epithelial cancers | |||||||
| Sacituzumab govitecan (IMMU-132) | Trop-2 Humanized antibody |
SN-38 (camptothecin) | Phase I/II |
NCT01631552 Immunomedics, Inc. |
Diverse epithelial cancers | (Ocean et al., 2017) | |
| 14. Melanoma | |||||||
| Glembatumumab vedotin | Glycoprotein NMB Fully human IgG2 monoclonal antibody | MMAE | Cleavable VC linker | Phase II |
NCT02302339 Celldex Therapeutics, Inc. |
Advanced melanoma | (Ott et al., 2019) |
| 15. Myeloma | |||||||
| AMG 224 | BCMA Human IgG1 antibody |
DM1 | Non-cleavable | Phase I |
NCT02561962 Amgen Inc. |
Relapsed or refractory multiple myeloma | (Lee et al., 2021) |
| DFRF4539A | FcRH5 Humanized IgG1 monoclonal antibody |
MMAE | Cleavable MC-VC-PABC linker | Phase I |
NCT01432353 Genentech, Inc. |
Relapsed or refractory multiple myeloma | (Lee et al., 2021) |
| GSK2857916 | B-cell Maturation antigen Humanized IgG1 monoclonal antibody |
MMAF | Non-cleavable Protease-resistant MC linker |
Phase I |
NCT02064387 Glaxo SmithKline. |
Relapsed or refractory multiple myeloma (BMA117159) | (Trudel et al., 2018; Trudel, et al., 2019) |
| Lorvotuzumab Mertansine (IMGN901) |
CD-56 | DM1 | Cleavable disulfide linker | Phase I | NCT00346255 | Relapsed and/or Refractory CD-56-positive multiple myeloma |
(Ailawadhi et al., 2019) |
| 16. Osteosarcoma | |||||||
| Glembatumumab vedotin (CDX-011) | Glycoprotein non-metastatic B Fully human IgG2 monoclonal antibody | MMAE | Phase II | AOST1521 NCT02487979 |
Recurrent osteosarcoma | (Kopp et al., 2019) | |
| 17. Leukemia | |||||||
| Camidanlumab tesirine |
CD25 Humanized antibody |
PBD dimer (SG3199) | Cleavable Cathepsin-valine-alanine linker | Phase I |
NCT02588092 Therapeutics SA |
Acute myeloid leukemia or acute lymphoblastic leukemia | (Goldberg et al., 2020) |
| Vadastuximab talirine (SGN-CD33A) | CD33 Monoclonal antibody |
PBD dimer | Cleavable MC-VC linker | Phase I |
NCT01902329 Genetics, Inc. |
CD33-positive acute myeloid leukemia | (Stein et al., 2018) |
| Azacitidine and Gemtuzumab ozogamicin (GO) | CD33 Humanized IgG4 antibody |
Calicheamicin | Phase I/II |
NCT00766116 Celgene, Inc. |
Relapsed acute myeloid leukemia | (Medeiros et al., 2018) | |
| 18. Glioma | |||||||
| AMG 595 | EGFRvIII Fully human monoclonal antibody |
DM1 3–4 |
Phase I |
NCT01475006 Amgen Inc. |
Recurrent malignant glioma expressing EGFRvIII | (Rosenthal et al., 2019) | |
| Depatuxizumab mafodotin (ABT-414) | EGFR monoclonal antibody | MMAF | Non-cleavable MC linker | Phase I |
NCT01800695 AbbVie |
EGFR-amplified recurrent glioblastoma | (van den Bent et al., 2017) |
| ABT-414 | EGFR Humanized recombinant IgG1κ monoclonal antibody |
MMAF | Non-cleavable MC linker | Phase I |
NCT02573324 NCT02343406 AbbVie |
Glioblastoma | (Reardon et al., 2017) |
Of the clinically used agents, Mylotarg was first approved by the FDA in May 2000. In 2010, Pfizer withdrew Mylotarg from the market due to the side effect of veno-occlusive disease causing the early death of patients (Cohen et al., 2002; Norsworthy et al., 2018). In 2014, analysis of data from five randomized clinical trials found that low-dose Mylotarg improved patient survival compared with supportive treatment. Based on these data, in September 2017, the FDA again approved it for treatment of adult patients with newly diagnosed acute myeloid leukemia expressing CD33 antigen (Norsworthy et al., 2018).
Adcetris was initially approved for Hodgkin’s lymphoma (HL) and systemic anaplastic large cell lymphoma (sALCL) in 2011 (Scott, 2017). Additionally, it was approved as a first-line treatment in November 2018 for treating adult patients with CD30 positive peripheral T-cell lymphoma (PTCL) (NCT01777152, NCT01578499 and NCT01100502). Around HL, sALCL and PTCL, Adcetris has obtained six treatment indications, demonstrating a rapid indication expansion (van der Weyden et al., 2019).
Kadcyla was approved for use in treating HER2-positive breast cancer in 2013. Currently, it is the only ADC approved as a monotherapy drug in 104 countries (including the United States and EU Member States) for HER2-positive metastatic breast cancer patients who have previously received Herceptin and/or paclitaxel chemotherapy (Lambert & Chari, 2014; Phillips et al., 2016) (NCT00829166).
Besponsa is the second ADC from Pfizer. Launched in 2017, it targets CD22 for the treatment of relapsed/refractory B-cell precursor acute lymphoblastic leukemia (NCT01564784). Besponsa uses the same acid-unstable hydrazone linker as Mylotarg. Lumoxiti (Moxetumomab pasudotox) was approved in September 2018 for the treatment of patients with hairy cell leukemia. Eighty percent of patients (64) achieved complete remission of tumors over 180 days after treatment (NCT01829711). The most common adverse reactions were tissue edema and renal failure (Kreitman & Arons, 2018).
Polivy (polatuzumab vedotin) was approved in June 2019 for patients with B-cell lymphoma who had received at least two other prior treatments (Deeks, 2019). It was approved because of good therapeutic outcome in clinical trial (NCT02257567). Padcev (enfortumab vedotin) was approved in December 2019 for treatment of locally advanced or metastatic urothelial carcinoma (Hanna, 2020). In a clinical trial involving 125 patients, Padcev brought 44% objective response rate (ORR), 12 complete responses and 7.6 months in median duration response contributing to its approval (NCT03219333). The most common side effects were hyperglycemia, peripheral neuropathy and eye diseases. Enhertu (trastuzumab deruxtecan) was approved in December 2019 for patients with HER2-positive breast cancer who had received at least two other previous treatments (Keam, 2020). In the clinical trial, 60.3% ORR reflected significant tumor regression, and median relapse free time of 14.8 months supporting approval (NCT03248492). The common adverse reactions of Enhertu were neutropenia and left ventricular dysfunction. Severe adverse reactions included interstitial lung disease and embryo-fetal toxicity (Keam, 2020).
Trodelvy (sacituzumab govitecan) was approved by the FDA in April 2020 for patients with advanced triple negative breast cancer who had received at least two other prior treatments. In the clinical study (NCT01631552), 33.3% ORR and a median relapse free time of 7.7 months. The most common adverse reactions include nausea and vomiting, neutropenia, diarrhea, and fatigue. Blenrep (bellantamab mafodotin) was approved by the FDA in August 2020 for the treatment of recurrent multiple myeloma. In its clinical study (NCT03525678), good outcomes were observed with 31% ORR and 73% patients showing no recurrence for more than six months. The most common adverse reactions include corneal disease (corneal epithelial changes on ophthalmic examination), vision loss and nausea (Kaplon et al., 2020). These clinical studies and patents outline successful applications of ADCs and meanwhile suggest some drawbacks of current ADCs.
5. Conclusions and future directions
ADCs are considered to be exciting and promising treatments for cancer (Tsuchikama & An, 2016). The number of ADCs registered in clinical trials continues to grow. This is closely related to the expansion of ADC antibody target ranges, optimization of linker technologies and innovation of new payloads (Damelin, 2018). The research of ADCs is undergoing a transition period. The old conjugation methods are giving way to newer point-specific modification methods, lower toxicity payloads are giving way to super-high toxicity payloads, mouse derived antibodies are giving way to fully humanized antibodies, and so on (Bander, 2013). A better understanding of all aspects of in vivo processing of ADCs and lessons from 20 years of clinical experience are vital to developing more effective ADCs (Damelin, 2018; Tolcher, 2016). With the approval of three ADCs in 2019, they have attracted worldwide attention. In fact, we are in the era of an ADC boom, as a large number of new and emerging ADCs are under development or in clinical trials (Theocharopoulos et al., 2020; Tolcher, 2016). It is worth noting that the addition of ADCs in clinics, clinical trials and research environments not only reflects the growing interest and confidence of doctors and pharmaceutical companies in this field, but also highlights the value and benefits that ADCs provide to cancer patients (Howard, 2017). Currently, ten ADCs have demonstrated effectiveness as therapeutic agents and gained approval by the FDA for treatment of cancers. (Table 1). In the future, more ADC drugs are expected to be approved for marketing to treat additional types of cancer (such as those in Table 2).
ADCs have the distinct advantage of transporting cytotoxic drugs into cancer cells specifically reducing the toxicity of these drugs to healthy tissues (Anderl et al., 2013; Sedlacek et al., 1992). Earlier studies tried a number of natural toxins as payloads for ADCs (Walts & Said, 1983). However, the cytotoxic activity of these toxins was not strong enough and lacked effectiveness in both animal and clinical trials. In the past 20 years, great efforts have been devoted to the development of super toxins, especially those for intracellular targets (Anderl et al., 2013). At present, many super toxins and their derivatives have been developed. These usually target microtubules or DNA. Tubulin inhibitors, such as auristatin and maytansine, are widely used in ADC development and selectively targets rapidly dividing cancer cells and induces less damage to non-dividing healthy cells (Akaiwa et al., 2020; Saber et al., 2019). DNA damaging agents, such as calicheamicins and PBDs, can induce apoptosis in all cells, including non-dividing cancer cells (Saber et al., 2019). However, despite the progress made in payload effectiveness, alternative types of payload skeletons for ADCs has not increased, and most cytotoxic agents are derivatives of former core skeletons (Yaghoubi et al., 2020). Therefore, in the next generation of ADCs, it is necessary to develop new scaffolds of high-efficiency payloads with diversified targets, mechanisms of action, and decreased side effects (Masters et al., 2018). We expect complex natural product libraries will continue to be an excellent source of novel cytotoxic molecules for use as ADC payloads.
A central difficulty within ADC development lies in the linker technology. The traditional linker technique usually couples drugs through an antibody disulfide bond utilizing cysteine or lysine residues (Tsuchikama & An, 2016). The approximately 20 potential joinable lysine residues and randomly conjugation lead to production of millions of different ADC forms (Bargh et al., 2019; Birrer et al., 2019; Tsuchikama & An, 2016). The PK properties of the heterogeneous ADCs in vivo vary greatly and as a result, industrial production faces great challenges to ensure sample consistency between batches (Tsuchikama & An, 2016). This heterogeneity in ADCs is an obstacle in gaining approval by the FDA (Birrer et al., 2019). Therefore, many companies producing ADCs are developing site-specific linker technology (White et al., 2019). Site-specific linker technology can ensure a fixed number of drug molecules coupled to specific sites of the antibody, which can ensure drug homogeneity and batch production stability to a large extent (Panowski et al., 2014). In addition, it can accurately control DAR values at 2 to 4 (Behrens et al., 2015). Presently, site-specific ADC technologies mainly rely on engineering modification of cysteine, modification of unnatural amino acids, selenocysteine replacement or site-specific enzyme catalysis (Nolting, 2013; Panowski et al., 2014). Looking toward the future, innovative and low-cost methods for coupling cytotoxicity payloads to antibodies will undoubtedly attract attention.
What are the current challenges facing the field of ADC development? Although it has been more than 35 years since the initial development of ADCs (Deguchi et al., 1986; Garnett et al., 1983), the FDA has only approved ten ADC drugs to date. Several ADCs have exhibited great potential in preclinical research, but did not perform well in clinical trials (Nejadmoghaddam et al., 2019). Toxicity and incomplete characterization data are the two main factors contributing to these failures (Hinrichs & Dixit, 2015). The nature of ADCs is more complex than mAbs, and requires additional data to support clinical trials (Lansita et al., 2015). This includes 1) complete ADC structure data: different characterizations of ADCs between batches (MS, peptide map, glycosylation, circular dichroism, differential scanning calorimetry, etc.), DAR values, coupling site, cytotoxin distribution uniformity, etc.; 2) efficacy of ADC: antigen-binding affinity, time required for internalization, timing and localization of ADC in blood circulation, intracellular ADC entry ratio, effect of ADC in different cancers and effect of combination therapy with other drugs; 3) analysis of adverse reactions: causes and severity of side effects, toxicity to normal tissues with low expression of target antigen, adverse reactions caused by premature cleavage of linker (early release of toxin) and toxicity caused by non-specific endocytosis in non-tumor tissues (Masters et al., 2018; Roberts et al., 2013).
What are the possibilities for the future of ADC development? A large number of ADC technologies developed over the past decade have created a wide range of opportunities for designing ADCs specific to a given target (Nejadmoghaddam et al., 2019; White, et al., 2019). In combination with the approval of novel ADCs, new technologies may be able to shine in the coming years. These areas for development may include: 1) ADCs targeting tumor microenvironments. Several characteristics and targets in the tumor microenvironment are common regardless of tumor types, which can greatly broaden the therapeutic window of ADCs and improve their antitumor activity. 2) New forms of ADCs, such as X-drug conjugate (XDC, such as peptides and small molecules). Many publications in recent years reveal novel attempts on ADC-like peptide-drug conjugates, small molecule-drug conjugates, and nanoparticle-drug conjugates. 3) Recombinant DNA technology and other interdisciplinary and multidisciplinary methods that can bring low-cost site-specific payload coupling (Birrer et al., 2019; Chen & Zhan, 2019). 4) Payloads without direct cytotoxicity, such as those that may guide the immune system to target the cancerous cells. On the whole, decades of time has been spent on ADC development, thousands of papers on ADCs have been published and importantly ten ADCs have been approved up to now, all of which predict a prosperous future for ADCs.
Acknowledgments:
Financial support from the NIH (CA186935 and AI150869 to A.J.W.), the Herman Frasch Foundation for Chemical Research, Bank of America, N.A., Trustee (HF17 to A.J.W.) and the University of Connecticut Research Excellence Program (to A.J.W. and M.J.B.) is gratefully acknowledged.
Abbreviations
- Abs
Antibodies
- ADC
Antibody-drug conjugate
- ALCAM
Activated leukocyte cell adhesion molecule
- BCMA
B-cell maturation antigen
- CD
Cluster of differentiation
- DAR
Drug-to-antibody ratio
- DM1
Mertansine 1
- ED
Effective dose
- EGFR
Epidermal growth factor receptor
- ELISA
Enzyme linked immunosorbent assay
- EphA2
Epithelial cell kinase A2
- EPR
Enhanced permeability and retention
- Fab
Antigen binding fragment
- Fc
Constant domain fragment
- FcRn
Neonatal Fc receptor
- FDA
Food and Drug Administration
- GO
Gemtuzumab ozogamicin
- GSH
Glutathione
- HER2
Human epidermal growth factor receptor 2
- HIC
Hydrophobic interaction chromatography
- HL
Hodgkin’s lymphoma
- Ig
Immunoglobulin
- IV
Intravenous
- kDa
kilodalton
- LC
Liquid chromatography
- mAb
Monoclonal antibody
- MC
Maleimidocaproyl
- MMAE
Monomethyl auristatin E
- MMAF
Monomethyl auristatin F
- MS
Mass spectrometry
- ORR
objective response rate
- PBD
Pyrrole benzodiazepines
- PK
Pharmacokinetics
- PTCL
Peripheral T-cell lymphoma
- sALCL
systemic anaplastic large cell lymphoma
- SGN-35
Brentuximab vedotin
- SMCC
Succinimidyl 4-(N-maleimidomethyl)cyclohexane-1-carboxylate
- SPDB
N-succinimidyl 4-(2-pyridyldithio)butanoate
- SPP
N-succinimidyl-4-(2-pyridyldithio)pentanoate
- TAA
Tumor associated antigens
- T-DM1
Ado-trastuzumab emtansine
- TSA
Tumor specific antigens
- VC
Valine citrulline
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Statement: A.J.W. owns shares in Terpenoid Therapeutics, Inc. The current work did not involve the company. Y.J., M.A.S., X.H., and M.J.B. have no conflicts to declare.
Recommended readings: For additional background information on ADCs and their metabolism, the following publications are highly recommended: Chen S. Q. & Zhan J. B. (2019). Antibody-drug conjugates and cellular metabolic dynamics. Hangzhou, Zhejiang University Press. (ISBN: 9787308197281) and Ducry L. (2013). Antibody-Drug Conjugates. New Jersey, Humana Press. (ISBN: 9781627035415).
References
- Aalberse RC, Stapel SO, Schuurman J, & Rispens T (2010). Immunoglobulin G4: an odd antibody. Clinical & Experimental Allergy, 39, 469–477. [DOI] [PubMed] [Google Scholar]
- Adair JR, Howard PW, Hartley JA, Williams DG, & Chester KA (2012). Antibody-drug conjugates - a perfect synergy. Expert Opinion On Biological Therapy, 12, 1191–1206. [DOI] [PubMed] [Google Scholar]
- Advani RH, Lebovic D, Chen A, Brunvand M, Goy A, Chang JE, … Cheson BD (2017). Phase I study of the anti-CD22 antibody-drug conjugate Pinatuzumab Vedotin with/without Rituximab in patients with relapsed/refractory B-cell non-Hodgkin lymphoma. Clinical Cancer Research, 23, 1167–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed F, Steele JC, Herbert JM, Steven NM, & Bicknell R (2008). Tumor stroma as a target in cancer. Curr Cancer Drug Targets, 8, 447–453. [DOI] [PubMed] [Google Scholar]
- Ailawadhi S, Kelly KR, Vescio RA, Jagannath S, Wolf J, Gharibo M, … Chanan-Khan A (2019). A phase I study to assess the safety and pharmacokinetics of single-agent Lorvotuzumab Mertansine (IMGN901) in patients with relapsed and/or refractory CD-56-positive multiple myeloma. Clin Lymphoma Myeloma Leuk, 19, 29–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akaiwa M, Dugal-Tessier J, & Mendelsohn BA (2020). Antibody-drug conjugate payloads: study of auristatin derivatives. Chem Pharm Bull (Tokyo), 68, 201–211. [DOI] [PubMed] [Google Scholar]
- Alewine C, Ahmad M, Peer CJ, Hu ZI, Lee MJ, Yuno A, … Pastan I (2020). Phase I/II study of the Mesothelin-targeted immunotoxin LMB-100 with Nab-Paclitaxel for patients with advanced pancreatic adenocarcinoma. Clinical Cancer Research, 26, 828–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alley SC, Benjamin DR, Jeffrey SC, Okeley NM, Meyer DL, Sanderson RJ, … Senter PD (2008). Contribution of linker stability to the activities of anticancer immunoconjugates. Bioconjug Chem, 19, 759–765. [DOI] [PubMed] [Google Scholar]
- Almhanna K, Miron ML, Wright D, Gracian AC, Hubner RA, Van Laethem JL, … Kalebic T (2017a). P hase II study of antibody-drug conjugate, TAK-264 (MLN0264) in previously treated patients with advanced or metastatic pancreatic adenocarcinoma expressing guanylyl cyclase C. Invest New Drugs, 35, 235–241. [DOI] [PubMed] [Google Scholar]
- Almhanna K, Wright D, Mercade TM, Van Laethem JL, Gracian AC, Guillen-Ponce C, … Kalebic T (2017b). A phase II study of antibody-drug conjugate, TAK-264 (MLN0264) in previously treated patients with advanced or metastatic pancreatic adenocarcinoma expressing guanylyl cyclase C. Invest New Drugs, 35, 634–641. [DOI] [PubMed] [Google Scholar]
- Anderl J, Faulstich H, Hechler T, & Kulke M (2013). Antibody-drug conjugate payloads. Methods in Molecular Biology, 1045, 51–70. [DOI] [PubMed] [Google Scholar]
- Andreev J, Thambi N, Perez BA, Delfino F, Martin J, Kelly MP, … Thurston G (2017). Bispecific Antibodies and Antibody-Drug Conjugates (ADCs) Bridging HER2 and Prolactin Receptor Improve Efficacy of HER2 ADCs. Molecular Cancer Therapeutics, 16, 681–693. [DOI] [PubMed] [Google Scholar]
- Armour KL, Clark MR, Hadley AG, & Williamson LM (1999). Recombinant human IgG molecules lacking Fc receptor I binding and monocyte triggering activities. European Journal of Immunology, 29, 2613–2624. [DOI] [PubMed] [Google Scholar]
- Axup JY, Bajjuri KM, Ritland M, Hutchins BM, Kim CH, Kazane SA, … Schultz PG (2012). Synthesis of site-specific antibody-drug conjugates using unnatural amino acids. Proc Natl Acad Sci U S A, 109, 16101–16106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badescu G, Bryant P, Bird M, Henseleit K, Swierkosz J, Parekh V, … Godwin A (2014). Bridging disulfides for stable and defined antibody drug conjugates. Bioconjug Chem, 25, 1124–1136. [DOI] [PubMed] [Google Scholar]
- Bander NH (2013). Antibody-drug conjugate target selection: critical factors.. New Jersey, Humana Press. [DOI] [PubMed] [Google Scholar]
- Banerjee S, Oza AM, Birrer MJ, Hamilton EP, Hasan J, Leary A, … Liu JF (2018). Anti-NaPi2b antibody-drug conjugate lifastuzumab vedotin (DNIB0600A) compared with pegylated liposomal doxorubicin in patients with platinum-resistant ovarian cancer in a randomized, open-label, phase II study. Annals of Oncology, 29, 917–923. [DOI] [PubMed] [Google Scholar]
- Bardia A, Mayer IA, Diamond JR, Moroose RL, Isakoff SJ, Starodub AN, … Vahdat LT (2017). Efficacy and safety of anti-Trop-2 antibody drug conjugate Sacituzumab Govitecan (IMMU-132) in heavily pretreated patients with metastatic triple-negative breast cancer. Journal of Clinical Oncology, 35, 2141–2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bargh JD, Isidro-Llobet A, Parker JS, & Spring DR (2019). Cleavable linkers in antibody-drug conjugates. Chemical Society Reviews, 48, 4361–4374. [DOI] [PubMed] [Google Scholar]
- Bartlett NL, Smith MR, Siddiqi T, Advani RH, O’Connor OA, Sharman JP, … Jacobsen ED (2017). Brentuximab vedotin activity in diffuse large B-cell lymphoma with CD30 undetectable by visual assessment of conventional immunohistochemistry. Leuk Lymphoma, 58, 1607–1616. [DOI] [PubMed] [Google Scholar]
- Bauer A, Frascaroli G, & Walther P (2020). Megapinosome: Morphological description of a novel organelle. Journal of Structural Biology, 210, 107505–107505. [DOI] [PubMed] [Google Scholar]
- Bauer A, Subramanian N, Villinger C, Frascaroli G, Mertens T, & Walther P (2016). Megapinocytosis: a novel endocytic pathway. Histochemistry and Cell Biology, 145, 617–627. [DOI] [PubMed] [Google Scholar]
- Beck A, D’Atri V, Ehkirch A, Fekete S, Hernandez-Alba O, Gahoual R, … Cianférani S (2019). Cutting-edge multi-level analytical and structural characterization of antibody-drug conjugates: present and future. Expert Rev Proteomics, 16, 337–362. [DOI] [PubMed] [Google Scholar]
- Behrens CR, Ha EH, Chinn LL, Bowers S, Probst G, Fitch-Bruhns M, … Jackson DY (2015). Antibody-drug conjugates (ADCs) derived from interchain cysteine cross-linking demonstrate improved homogeneity and other pharmacological properties over conventional heterogeneous ADCs. Mol Pharm, 12, 3986–3998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi S, Zhou H, & Zhang S (2009). Multilayers enzyme-coated carbon nanotubes as biolabel for ultrasensitive chemiluminescence immunoassay of cancer biomarker. Biosensors & Bioelectronics, 24, 2961–2966. [DOI] [PubMed] [Google Scholar]
- Birrer MJ, Moore KN, Betella I, & Bates RC (2019). Antibody-drug conjugate-based therapeutics: state of the science. J Natl Cancer Inst, 111, 538–549. [DOI] [PubMed] [Google Scholar]
- Boswell CA, Mundo EE, Zhang C, Bumbaca D, Valle NR, Kozak KR, … Lin K (2011). Impact of drug conjugation on pharmacokinetics and tissue distribution of anti-STEAP1 antibody-drug conjugates in rats. Bioconjug Chem, 22, 1994–2004. [DOI] [PubMed] [Google Scholar]
- Bournazos S & Ravetch JV (2017). Diversification of IgG effector functions. International Immunology, 29, 303–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bumbaca D, Wong A, Drake E, Reyes AN, Lin BC, Stephan JP, … Dennis MS (2011). Highly specific off-target binding identified and eliminated during the humanization of an antibody against FGF receptor 4. MAbs, 3, 376–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buss NA, Henderson SJ, Mcfarlane M, Shenton JM, & de Haan L (2012). Monoclonal antibody therapeutics: history and future. Current Opinion in Pharmacology, 12, 615–622. [DOI] [PubMed] [Google Scholar]
- Butler JE (2000). Enzyme-linked immunosorbent assay. Journal of immunoassay, 21, 165–209. [DOI] [PubMed] [Google Scholar]
- Butterfield LH (2015). Cancer vaccines. British Medical Journal, 350, 988–988. [Google Scholar]
- Casi G & Neri D (2012). Antibody-drug conjugates: Basic concepts, examples and future perspectives. Journal of Controlled Release, 161, 422–428. [DOI] [PubMed] [Google Scholar]
- Cassady JM, Chan KK, Floss HG, & Leistner E (2004). Recent developments in the maytansinoid antitumor agents. Chem Pharm Bull (Tokyo), 52, 1–26. [DOI] [PubMed] [Google Scholar]
- Chalouni C & Doll S (2018). Fate of antibody-drug conjugates in cancer cells. J Exp Clin Cancer Res, 37, 20–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chari RV, Miller ML, & Widdison WC (2014). Antibody-drug conjugates: an emerging concept in cancer therapy. Angew Chem Int Ed Engl. 53, 3796–827. [DOI] [PubMed] [Google Scholar]
- Chen H, Lin Z, Arnst KE, Miller DD, & Li W (2017a). Tubulin inhibitor-based antibody-drug conjugates for cancer therapy. Molecules, 8, 22–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SC, Kagedal M, Gao Y, Wang B, Harle-Yge ML, Girish S, … Li C (2017b). Population pharmacokinetics of trastuzumab emtansine in previously treated patients with HER2-positive advanced gastric cancer (AGC). Cancer Chemother Pharmacol, 80, 1147–1159. [DOI] [PubMed] [Google Scholar]
- Chen SQ & Zhan JB (2019). Antibody-drug conjugates and cellular metabolic dynamics. Hangzhou, Zhejiang University Press. [Google Scholar]
- Chen T, Chen Y, Stella C, Medley CD, & Zhang K (2016). Antibody-drug conjugate characterization by chromatographic and electrophoretic techniques. Journal of chromatography. B, Analytical technologies in the biomedical and life sciences, 1032, 39–50. [DOI] [PubMed] [Google Scholar]
- Chih HW, Gikanga B, Yang Y, & Zhang B (2011). Identification of amino acid residues responsible for the release of free drug from an antibody-drug conjugate utilizing lysine-succinimidyl ester chemistry. J Pharm Sci, 100, 2518–2525. [DOI] [PubMed] [Google Scholar]
- Chiu ML & Gilliland GL (2016). Engineering antibody therapeutics. Curr Opin Struct Biol, 38, 163–173. [DOI] [PubMed] [Google Scholar]
- Chari RV, Miller ML, & Widdison WC (2014). Antibody-drug conjugates: an emerging concept in cancer therapy. Angew Chem Int Ed Engl. 15, 3796–3827. [DOI] [PubMed] [Google Scholar]
- Chung YC, Chang CM, Wei WC, Chang TW, Chang KJ, & Chao WT (2018). Metformin-induced caveolin-1 expression promotes T-DM1 drug efficacy in breast cancer cells. Sci Rep, 8, 3930–3935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen AD, Luger SM, Sickles C, Mangan PA, Porter DL, Schuster SJ, … Stadtmauer EA (2002). Gemtuzumab ozogamicin (Mylotarg) monotherapy for relapsed AML after hematopoietic stem cell transplant: efficacy and incidence of hepatic veno-occlusive disease. Bone Marrow Transplant, 30, 23–28. [DOI] [PubMed] [Google Scholar]
- Collins DM, Bossenmaier B, Kollmorgen G, & Niederfellner G (2019). Acquired resistance to antibody-drug conjugates. Cancers (Basel), 11, 394–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad MJ & Crosby WH (1960). Massive nitrogen mustard therapy in Hodgkin’s disease with protection of bone marrow by tourniquets. Blood, 16, 1089–1103. [PubMed] [Google Scholar]
- Cushley W & Owen MJ (1983). Structural and genetic similarities between immunoglobulins and class-1 histocompatibility antigens. Immunology Today, 4, 88–92. [DOI] [PubMed] [Google Scholar]
- Dahlgren D & Lennernäs H (2020). Antibody-drug conjugates and targeted treatment strategies for hepatocellular carcinoma: a drug-delivery perspective. Molecules, 25, 2861–2867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damelin M (2018). Innovations for Next-Generation Antibody-Drug Conjugates. New Jersey, Humana Press. [Google Scholar]
- Dan N, Setua S, Kashyap VK, Khan S, Jaggi M, Yallapu MM, … Chauhan SC (2018). Antibody-Drug Conjugates for Cancer Therapy: Chemistry to Clinical Implications. Pharmaceuticals (Basel), 11, 32–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang NH, Ogura M, Castaigne S, Fayad LE, Jerkeman M, Radford J, … Ando K (2018). Randomized, phase 3 trial of inotuzumab ozogamicin plus rituximab versus chemotherapy plus rituximab for relapsed/refractory aggressive B-cell non-Hodgkin lymphoma. Br J Haematol, 182, 583–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danila DC, Szmulewitz RZ, Vaishampayan U, Higano CS, Baron AD, Gilbert HN, … Scher HI (2019). Phase I Study of DSTP3086S, an Antibody-Drug Conjugate Targeting Six-Transmembrane Epithelial Antigen of Prostate 1, in Metastatic Castration-Resistant Prostate Cancer. Journal of Clinical Oncology, 37, 3518–3527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Atri V, Fekete S, Stoll D, Lauber M, Beck A, & Guillarme D (2018). Characterization of an antibody-drug conjugate by hydrophilic interaction chromatography coupled to mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci, 1080, 37–41. [DOI] [PubMed] [Google Scholar]
- De Stefano F, Chacon E, Turcios L, Marti F, & Gedaly R (2018). Novel biomarkers in hepatocellular carcinoma. Dig Liver Dis, 50, 1115–1123. [DOI] [PubMed] [Google Scholar]
- Deeks ED (2019). Polatuzumab Vedotin: First Global Approval. Drugs, 79, 1467–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deguchi T, Chu TM, Leong SS, Horoszewicz JS, & Lee CL (1986). Effect of methotrexate-monoclonal anti-prostatic acid phosphatase antibody conjugate on human prostate tumor. Cancer Research, 46, 3751–3755. [PubMed] [Google Scholar]
- DeVita VT & Chu E (2008). A History of Cancer Chemotherapy. Cancer Research, 68, 8643–8653. [DOI] [PubMed] [Google Scholar]
- Diamantis N & Banerji U (2016). Antibody-drug conjugates-an emerging class of cancer treatment. British Journal of Cancer, 4, 362–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diéras V, Miles D, Verma S, Pegram M, Welslau M, Baselga J, … Gianni L (2017). Trastuzumab emtansine versus capecitabine plus lapatinib in patients with previously treated HER2-positive advanced breast cancer (EMILIA): a descriptive analysis of final overall survival results from a randomised, open-label, phase 3 trial. Lancet Oncology, 18, 732–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimasi N, Fleming R, Zhong H, Bezabeh B, Kinneer K, Christie RJ, … Gao C (2017). Efficient Preparation of Site-Specific Antibody-Drug Conjugates Using Cysteine Insertion. Mol Pharm, 14, 1501–1516. [DOI] [PubMed] [Google Scholar]
- Doi T, Shitara K, Naito Y, Shimomura A, Fujiwara Y, Yonemori K, … Tamura K (2017). Safety, pharmacokinetics, and antitumour activity of trastuzumab deruxtecan (DS-8201), a HER2-targeting antibody-drug conjugate, in patients with advanced breast and gastric or gastro-oesophageal tumours: a phase 1 dose-escalation study. Lancet Oncology, 18, 1512–1522. [DOI] [PubMed] [Google Scholar]
- Donaldson JG (2015). Immunofluorescence Staining. Curr Protoc Cell Biol, 69, 3–4. [DOI] [PubMed] [Google Scholar]
- Dorywalska M, Strop P, Melton-Witt JA, Hasa-Moreno A, Farias SE, Galindo CM, … Rajpal A (2015). Site-Dependent Degradation of a Non-Cleavable Auristatin-Based Linker-Payload in Rodent Plasma and Its Effect on ADC Efficacy. PLoS One, 10, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dotan E, Cohen SJ, Starodub AN, Lieu CH, Messersmith WA, Simpson PS, … Berlin JD (2017). Phase I/II Trial of Labetuzumab Govitecan (Anti-CEACAM5/SN-38 Antibody-Drug Conjugate) in Patients With Refractory or Relapsing Metastatic Colorectal Cancer. Journal of Clinical Oncology, 35, 3338–3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducry L (2013). Antibody-Drug Conjugates. New Jersey, Humana Press. [Google Scholar]
- Durbin KR, Phipps C, & Liao X (2018). Mechanistic Modeling of Antibody-Drug Conjugate Internalization at the Cellular Level Reveals Inefficient Processing Steps. Molecular Cancer Therapeutics, 17, 1341–1351. [DOI] [PubMed] [Google Scholar]
- Eichenauer DA, Plütschow A, Kreissl S, Sökler M, Hellmuth JC, Meissner J, … Borchmann P (2017). Incorporation of brentuximab vedotin into first-line treatment of advanced classical Hodgkin’s lymphoma: final analysis of a phase 2 randomised trial by the German Hodgkin Study Group. Lancet Oncology, 18, 1680–1687. [DOI] [PubMed] [Google Scholar]
- Elliott WJ & Fried J (1976). Maytansinoids. Synthesis of a fragment of known absolute configuration involving chiral centers C-6 and C-7. Journal of Organic Chemistry, 41, 2469–2475. [DOI] [PubMed] [Google Scholar]
- Erickson HK, Park PU, Widdison WC, Kovtun YV, Garrett LM, Hoffman K, … Blättler WA (2006). Antibody-maytansinoid conjugates are activated in targeted cancer cells by lysosomal degradation and linker-dependent intracellular processing. Cancer Research, 66, 4426–4433. [DOI] [PubMed] [Google Scholar]
- Excoffier M, Janin-Bussat MC, Beau-Larvor C, Troncy L, Corvaia N, Beck A, … Klinguer-Hamour C (2016). A new anti-human Fc method to capture and analyze ADCs for characterization of drug distribution and the drug-to-antibody ratio in serum from pre-clinical species. J Chromatogr B Analyt Technol Biomed Life Sci, 1032, 149–154. [DOI] [PubMed] [Google Scholar]
- Fand I, Sharkey RM, McNally WP, Brill AB, Som P, Yamamoto K, … Goldenberg DM (1986). Quantitative whole-body autoradiography of radiolabeled antibody distribution in a xenografted human cancer model. Cancer Research, 46, 271–277. [PubMed] [Google Scholar]
- Filntisi A, Vlachakis D, Matsopoulos GK, & Kossida S (2014). Computational Construction of Antibody–Drug Conjugates Using Surface Lysines as the Antibody Conjugation Site and a Non-cleavable Linker. Cancer Informatics, 13, 179–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn AA, Pedley RB, Green AJ, Dearling JL, El-Emir E, Boxer GM, … Begent RH (2003). The nonuniformity of antibody distribution in the kidney and its influence on dosimetry. Radiation Research, 159, 182–189. [DOI] [PubMed] [Google Scholar]
- Fu Y & Ho M (2018). DNA damaging agent-based antibody-drug conjugates for cancer therapy. Antib Ther, 1, 33–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi AV, Arlotta KJ, Chen HN, Owen SC, & Carpenter JF (2018). Biophysical Properties and Heating-Induced Aggregation of Lysine-Conjugated Antibody-Drug Conjugates. Journal of Pharmaceutical Sciences, 107, 1858–1869. [DOI] [PubMed] [Google Scholar]
- Garnett MC, Embleton MJ, Jacobs E, & Baldwin RW (1983). Preparation and properties of a drug-carrier-antibody conjugate showing selective antibody-directed cytotoxicity in vitro. International Journal of Cancer, 31, 661–670. [DOI] [PubMed] [Google Scholar]
- Garrido-Laguna I, Krop I, Burris HR, Hamilton E, Braiteh F, Weise AM, … Hong DS (2019). First-in-human, phase I study of PF-06647263, an anti-EFNA4 calicheamicin antibody-drug conjugate, in patients with advanced solid tumors. International Journal of Cancer, 145, 1798–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gébleux R, Wulhfard S, Casi G, & Neri D (2015). Antibody format and drug release rate determine the therapeutic activity of noninternalizing antibody-drug conjugates. Molecular Cancer Therapeutics, 14, 2606–2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gébleux R, Stringhini M, Casanova R, Soltermann A, & Neri D (2017). Non-internalizing antibody-drug conjugates display potent anti-cancer activity upon proteolytic release of monomethyl auristatin E in the subendothelial extracellular matrix. International Journal of Cancer, 140, 1670–1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber DE, Infante JR, Gordon MS, Goldberg SB, Martín M, Felip E, … Burris HR (2020). Phase Ia Study of Anti-NaPi2b Antibody-Drug Conjugate Lifastuzumab Vedotin DNIB0600A in Patients with Non-Small Cell Lung Cancer and Platinum-Resistant Ovarian Cancer. Clinical Cancer Research, 26, 364–372. [DOI] [PubMed] [Google Scholar]
- Gianolio DA, Rouleau C, Bauta WE, Lovett D, Cantrell WJ, Recio AR, … Teicher BA (2012). Targeting HER2-positive cancer with dolastatin 15 derivatives conjugated to trastuzumab, novel antibody-drug conjugates. Cancer Chemother Pharmacol, 70, 439–449. [DOI] [PubMed] [Google Scholar]
- Goldberg AD, Atallah E, Rizzieri D, Walter RB, Chung KY, Spira A, … Solh M (2020). Camidanlumab tesirine, an antibody-drug conjugate, in relapsed/refractory CD25-positive acute myeloid leukemia or acute lymphoblastic leukemia: A phase I study. Leuk Res, 95, 106385–106407. [DOI] [PubMed] [Google Scholar]
- Gorovits B & Krinos-Fiorotti C (2013). Proposed mechanism of off-target toxicity for antibody-drug conjugates driven by mannose receptor uptake. Cancer Immunol Immunother, 62, 217–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goss GD, Vokes EE, Gordon MS, Gandhi L, Papadopoulos KP, Rasco DW, … Tolcher AW (2018). Efficacy and safety results of depatuxizumab mafodotin (ABT-414) in patients with advanced solid tumors likely to overexpress epidermal growth factor receptor. Cancer, 124, 2174–2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goutsouliak K, Veeraraghavan J, Sethunath V, De Angelis C, Osborne CK, Rimawi MF, … Schiff R (2020). Towards personalized treatment for early stage HER2-positive breast cancer. Nature Reviews Clinical Oncology, 17, 233–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray JE, Heist RS, Starodub AN, Camidge DR, Kio EA, Masters GA, … Goldenberg DM (2017). Therapy of Small Cell Lung Cancer (SCLC) with a Topoisomerase-I-inhibiting Antibody-Drug Conjugate (ADC) Targeting Trop-2, Sacituzumab Govitecan. Clinical Cancer Research, 23, 5711–5719. [DOI] [PubMed] [Google Scholar]
- Gromek SM & Balunas MJ (2015). Natural products as exquisitely potent cytotoxic payloads for antibody- drug conjugates. Current Topics in Medicinal Chemistry, 14, 2822–2834. [DOI] [PubMed] [Google Scholar]
- Gurbaxani B, Dostalek M, & Gardner I (2013). Are endosomal trafficking parameters better targets for improving mAb pharmacokinetics than FcRn binding affinity? Molecular Immunology, 56, 660–674. [DOI] [PubMed] [Google Scholar]
- Haider K, Rahaman S, Yar MS, & Kamal A (2019). Tubulin inhibitors as novel anticancer agents: an overview on patents (2013–2018). Expert Opinion On Therapeutic Patents, 29, 623–641. [DOI] [PubMed] [Google Scholar]
- Hamilton GS (2015). Antibody-drug conjugates for cancer therapy: The technological and regulatory challenges of developing drug-biologic hybrids. Biologicals, 43, 318–332. [DOI] [PubMed] [Google Scholar]
- Hanna KS (2020). Enfortumab vedotin to treat urothelial carcinoma. Drugs Today (Barc), 56, 329–335. [DOI] [PubMed] [Google Scholar]
- Harper J, Mao S, Strout P, & Kamal A (2013). Selecting an optimal antibody for antibody-drug conjugate therapy: internalization and intracellular localization. Methods Mol Biol, 1045, 41–49. [DOI] [PubMed] [Google Scholar]
- Hartley JA & Hochhauser D (2012). Small molecule drugs - optimizing DNA damaging agent-based therapeutics. Current Opinion in Pharmacology, 12, 398–402. [DOI] [PubMed] [Google Scholar]
- Hassan R, Blumenschein GJ, Moore KN, Santin AD, Kindler HL, Nemunaitis JJ, … Bendell JC (2020). First-in-Human, Multicenter, Phase I Dose-Escalation and Expansion Study of Anti-Mesothelin Antibody-Drug Conjugate Anetumab Ravtansine in Advanced or Metastatic Solid Tumors. Journal of Clinical Oncology, 38, 1824–1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedrich WD, Fandy TE, Ashour HM, Wang H, & Hassan HE (2018). Antibody-Drug Conjugates: Pharmacokinetic/Pharmacodynamic Modeling, Preclinical Characterization, Clinical Studies, and Lessons Learned. Clinical Pharmacokinetics, 57, 687–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heist RS, Guarino MJ, Masters G, Purcell WT, Starodub AN, Horn L, … Camidge DR (2017). Therapy of Advanced Non-Small-Cell Lung Cancer With an SN-38-Anti-Trop-2 Drug Conjugate, Sacituzumab Govitecan. Journal of Clinical Oncology, 35, 2790–2797. [DOI] [PubMed] [Google Scholar]
- Hinrichs MJ & Dixit R (2015). Antibody Drug Conjugates: Nonclinical Safety Considerations. AAPS Journal, 17, 1055–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hock MB, Thudium KE, Carrasco-Triguero M, & Schwabe NF (2015). Immunogenicity of Antibody Drug Conjugates: Bioanalytical Methods and Monitoring Strategy for a Novel Therapeutic Modality. AAPS Journal, 17, 35–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holdenrieder S (2016). Biomarkers along the continuum of care in lung cancer. Scandinavian journal of clinical and laboratory investigation, 245, S40–S45. [DOI] [PubMed] [Google Scholar]
- Hong DS, Concin N, Vergote I, de Bono JS, Slomovitz BM, Drew Y, … Coleman RL (2020). Tisotumab Vedotin in Previously Treated Recurrent or Metastatic Cervical Cancer. Clinical Cancer Research, 26, 1220–1228. [DOI] [PubMed] [Google Scholar]
- Hong Y, Park C, Kim N, Cho J, Moon SU, Kim J, … Yoon S (2018). QSurface: fast identification of surface expression markers in cancers. BMC Systems Biology, 12, 17–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotta K, Aoe K, Kozuki T, Ohashi K, Ninomiya K, Ichihara E, … Kiura K (2018). A Phase II Study of Trastuzumab Emtansine in HER2-Positive Non-Small Cell Lung Cancer. Journal of Thoracic Oncology, 13, 273–279. [DOI] [PubMed] [Google Scholar]
- Howard PW (2017). Antibody-drug Conjugates (ADCs). Weinheim, Wiley-VCH Verlag GmbH & Co. KGaA. [Google Scholar]
- Huang AB, Lin CM, & Hamel E (1985). Maytansine inhibits nucleotide binding at the exchangeable site of tubulin. Biochemical & Biophysical Research Communications, 128, 1239–1246. [DOI] [PubMed] [Google Scholar]
- Huo H, Jia P, Zhang X, Zhang Z, Yang H, Zhang Q, … Zhang L (2015). Tentative identification of new metabolites of cnidilin by liquid chromatography-mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci, 995–996, 85–92. [DOI] [PubMed] [Google Scholar]
- Ishii M (1995). Limitation of clinical usefulness of tumor marker. Cancer & chemotherapy, 22, 1139–1145. [PubMed] [Google Scholar]
- Jayashankar V & Edinger AL (2020). Macropinocytosis confers resistance to therapies targeting cancer anabolism. Nature Communications, 11, 1121–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffrey SC, De Brabander J, Miyamoto J, & Senter PD (2010). Expanded Utility of the β-Glucuronide Linker: ADCs That Deliver Phenolic Cytotoxic Agents. ACS Medicinal Chemistry Letters, 1, 277–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joubert N, Denevault-Sabourin C, Bryden F, & Viaud-Massuard MC (2017). Towards antibody-drug conjugates and prodrug strategies with extracellular stimuli-responsive drug delivery in the tumor microenvironment for cancer therapy. European Journal of Medicinal Chemistry, 142, 393–415. [DOI] [PubMed] [Google Scholar]
- Juweid M, Neumann R, Paik C, Perez-Bacete MJ, Sato J, van Osdol W, … Weinstein JN (1992). Micropharmacology of monoclonal antibodies in solid tumors: direct experimental evidence for a binding site barrier. Cancer Research, 52, 5144–5153. [PubMed] [Google Scholar]
- Kaksonen M & Roux A (2018). Mechanisms of clathrin-mediated endocytosis. Nat Rev Mol Cell Biol, 19, 313–326. [DOI] [PubMed] [Google Scholar]
- Kalim M, Chen J, Wang S, Lin C, Ullah S, Liang K, … Zhan J (2017). Intracellular trafficking of new anticancer therapeutics: antibody-drug conjugates. Drug Des Devel Ther, 11, 2265–2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamath AV & Iyer S (2015). Preclinical Pharmacokinetic Considerations for the Development of Antibody Drug Conjugates. Pharm Res, 32, 3470–3479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JC, Sun W, Khare P, Karimi M, Wang X, Shen Y, … Ward ES (2019). Engineering a HER2-specific antibody–drug conjugate to increase lysosomal delivery and therapeutic efficacy. Nature Biotechnology, 37, 523–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplon H, Muralidharan M, Schneider Z, & Reichert JM (2020). Antibodies to watch in 2020. MAbs, 12, 170–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplon H & Reichert JM (2018). Antibodies to watch in 2018. MAbs, 10, 183–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keam SJ (2020). Trastuzumab Deruxtecan: First Approval. Drugs, 80, 501–508. [DOI] [PubMed] [Google Scholar]
- Kim SB, Meric-Bernstam F, Kalyan A, Babich A, Liu R, Tanigawa T, … Berlin J (2019). First-in-Human Phase I Study of Aprutumab Ixadotin, a Fibroblast Growth Factor Receptor 2 Antibody-Drug Conjugate (BAY 1187982) in Patients with Advanced Cancer. Targeted Oncology, 14, 591–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King GT, Eaton KD, Beagle BR, Zopf CJ, Wong GY, Krupka HI, … El-Khoueiry AB (2018). A phase 1, dose-escalation study of PF-06664178, an anti-Trop-2/Aur0101 antibody-drug conjugate in patients with advanced or metastatic solid tumors. Invest New Drugs, 36, 836–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klussman K, Mixan BJ, Cerveny CG, Meyer DL, Senter PD, & Wahl AF (2004). Secondary mAb--vcMMAE conjugates are highly sensitive reporters of antibody internalization via the lysosome pathway. Bioconjug Chem, 15, 765–773. [DOI] [PubMed] [Google Scholar]
- Kolodych S, Michel C, Delacroix S, Koniev O, Ehkirch A, Eberova J, … Wagner A (2017). Development and evaluation of β-galactosidase-sensitive antibody-drug conjugates. European Journal of Medicinal Chemistry, 142, 376–382. [DOI] [PubMed] [Google Scholar]
- Kopp LM, Malempati S, Krailo M, Gao Y, Buxton A, Weigel BJ, … Janeway KA (2019). Phase II trial of the glycoprotein non-metastatic B-targeted antibody-drug conjugate, glembatumumab vedotin (CDX-011), in recurrent osteosarcoma AOST1521: A report from the Children’s Oncology Group. European Journal of Cancer, 121, 177–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovtun YV, Audette CA, Ye Y, Xie H, Ruberti MF, Phinney SJ, … Goldmacher VS (2006). Antibody-drug conjugates designed to eradicate tumors with homogeneous and heterogeneous expression of the target antigen. Cancer Research, 66, 3214–3221. [DOI] [PubMed] [Google Scholar]
- Kowalczyk L, Bartsch R, Singer CF, & Farr A (2017). Adverse Events of Trastuzumab Emtansine (T-DM1) in the Treatment of HER2-Positive Breast Cancer Patients. Breast Care (Basel), 12, 401–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitman RJ & Arons E (2018). Update on hairy cell leukemia. Clin Adv Hematol Oncol, 16, 205–215. [PMC free article] [PubMed] [Google Scholar]
- Kubizek F, Eggenreich B, & Spadiut O (2017). Status Quo in Antibody-Drug Conjugates - Can Glyco- Enzymes Solve the Current Challenges? Protein Pept Lett, 24, 686–695. [DOI] [PubMed] [Google Scholar]
- Kuroda D, Shirai H, Kobori M, & Nakamura H (2010). Structural classification of CDR-H3 revisited: a lesson in antibody modeling. Proteins-structure Function & Bioinformatics, 73, 608–620. [DOI] [PubMed] [Google Scholar]
- Kuus-Reichel K, Grauer LS, Karavodin LM, Knott C, & Kay NE (1994). Will immunogenicity limit the use, efficacy, and future development of therapeutic monoclonal antibodies? Clin Diagn Lab Immunol, 1, 365–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert JM & Chari RV (2014). Ado-trastuzumab Emtansine (T-DM1): an antibody-drug conjugate (ADC) for HER2-positive breast cancer. Journal of Medicinal Chemistry, 57, 6949–6964. [DOI] [PubMed] [Google Scholar]
- Lambert JM & Berkenblit A (2018). Antibody-drug conjugates for cancer treatment. Annu Rev Med. 29, 191–207. [DOI] [PubMed] [Google Scholar]
- Lanshoeft C, Stutz G, Elbast W, Wolf T, Walles M, Stoeckli M, … Kretz O (2016). Analysis of small molecule antibody-drug conjugate catabolites in rat liver and tumor tissue by liquid extraction surface analysis micro-capillary liquid chromatography/tandem mass spectrometry. Rapid Commun Mass Spectrom, 30, 823–832. [DOI] [PubMed] [Google Scholar]
- Lansita JA, Burke JM, Apgar JF, & Mounho-Zamora B (2015). An Introduction to the Regulatory and Nonclinical Aspects of the Nonclinical Development of Antibody Drug Conjugates. Pharmaceutical Research, 32, 3584–3592. [DOI] [PubMed] [Google Scholar]
- Latif ZA, Lozzio BB, Wust CJ, Krauss S, & Lozzio CB (1980). Evaluation of drug-antibody conjugates in the treatment of human myelosarcomas transplanted in nude mice. Cancer, 45, 1326–1333. [DOI] [PubMed] [Google Scholar]
- Leal M, Sapra P, Hurvitz SA, Senter P, Wahl A, Schutten M, … Kabbarah O (2015). Antibody–drug conjugates: an emerging modality for the treatment of cancer. Annals of the New York Academy of Sciences, 1321, 41–54. [DOI] [PubMed] [Google Scholar]
- Lee HC, Raje NS, Landgren O, Upreti VV, Wang J, Avilion AA, … Spencer A (2021). Phase 1 study of the anti-BCMA antibody-drug conjugate AMG 224 in patients with relapsed/refractory multiple myeloma. Leukemia, 35, 255–258. [DOI] [PubMed] [Google Scholar]
- Lee NK, Su Y, Bidlingmaier S, & Liu B (2019). Manipulation of Cell-Type Selective Antibody Internalization by a Guide-Effector Bispecific Design. Molecular Cancer Therapeutics, 18, 1092–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenz HJ. (2006). Anti-EGFR mechanism of action: antitumor effect and underlying cause of adverse events. Oncology. 20, 5–13. [PubMed] [Google Scholar]
- Lepelley M, Allouchery M, Long J, Boucherle D, Ranchoup Y, Le Marc’Hadour F, … Sturm N (2018). Nodular Regenerative Hyperplasia Induced by Trastuzumab Emtansine: Role of Emtansine? Annals of Hepatology, 17, 1067–1071. [DOI] [PubMed] [Google Scholar]
- Li BT, Shen R, Buonocore D, Olah ZT, Ni A, Ginsberg MS, … Kris MG (2018). Ado-Trastuzumab Emtansine for Patients With HER2-Mutant Lung Cancers: Results From a Phase II Basket Trial. Journal of Clinical Oncology, 36, 2532–2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JY, Perry SR, Muniz-Medina V, Wang X, Wetzel LK, Rebelatto MC, … Coats SR (2019). A Biparatopic HER2-Targeting Antibody-Drug Conjugate Induces Tumor Regression in Primary Models Refractory to or Ineligible for HER2-Targeted Therapy. Cancer Cell, 35, 948–949. [DOI] [PubMed] [Google Scholar]
- Li XL, Chen XY, & Zhong DF (2016). Bioanalysis in the development of antibody-drug conjugates. Acta pharmaceutica Sinica, 51, 517–528. [PubMed] [Google Scholar]
- Li Y, Cozzi PJ, & Russell PJ (2010). Promising tumor-associated antigens for future prostate cancer therapy. Medicinal Research Reviews, 30, 67–101. [DOI] [PubMed] [Google Scholar]
- Liang XJ, Gong LY, Zhou F, Zhou DM, & Zhu JJ (2019). Pharmacological effects of site specific conjugated anti-human epidermal growth factor receptor 2-antibody drug conjugate using unnatural amino acid technology. Beijing Da Xue Xue Bao Yi Xue Ban, 51, 797–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JKH (2014). The history of monoclonal antibody development- Progress, remaining challenges and future innovations. Annals of Medicine and Surgery, 3, 113–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonberg N (2008). Human Monoclonal Antibodies from Transgenic Mice. Handbook of experimental pharmacology, 181, 69–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu D, Gillespie WR, Girish S, Agarwal P, Li C, Hirata J, … Jin JY (2017). Time-to-Event Analysis of Polatuzumab Vedotin-Induced Peripheral Neuropathy to Assist in the Comparison of Clinical Dosing Regimens. CPT Pharmacometrics Syst Pharmacol, 6, 401–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Jiang F, Lu A, & Zhang G (2016). Linkers Having a Crucial Role in Antibody-Drug Conjugates. International Journal of Molecular Sciences, 17, 561–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch T & Price A (2007). The effect of cytochrome P450 metabolism on drug response, interactions, and adverse effects. American Family Physician, 76, 391–396. [PubMed] [Google Scholar]
- Lyon RP, Bovee TD, Doronina SO, Burke PJ, Hunter JH, Neff-LaFord HD, … Senter PD (2015). Reducing hydrophobicity of homogeneous antibody-drug conjugates improves pharmacokinetics and therapeutic index. Nature Biotechnology, 33, 733–735. [DOI] [PubMed] [Google Scholar]
- Maas BM & Cao Y (2018). A minimal physiologically based pharmacokinetic model to investigate FcRn-mediated monoclonal antibody salvage: Effects of K(on), K(off), endosome trafficking, and animal species. MAbs, 10, 1322–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mach JP, Carrel S, Forni M, Ritschard J, Donath A, & Alberto P (1980). Tumor localization of radiolabeled antibodies against carcinoembryonic antigen in patient with carcinoma. New England Journal of Medicine, 303, 5–10. [DOI] [PubMed] [Google Scholar]
- Maeda H, Wu J, Sawa T, Matsumura Y, & Hori K (2000). Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. J Control Release. 65, 271–84. [DOI] [PubMed] [Google Scholar]
- Mahalingaiah PK, Ciurlionis R, Durbin KR, Yeager RL, Philip BK, Bawa B, … Van Vleet TR (2019). Potential mechanisms of target-independent uptake and toxicity of antibody-drug conjugates. Pharmacol Ther, 200, 110–125. [DOI] [PubMed] [Google Scholar]
- Malik P, Phipps C, Edginton A, & Blay J (2017). Pharmacokinetic Considerations for Antibody-Drug Conjugates against Cancer. Pharm Res, 34, 2579–2595. [DOI] [PubMed] [Google Scholar]
- Manabe S & Yamaguchi Y (2021). Antibody glycoengineering and homogeneous antibody-drug conjugate preparation. Chem Rec. Epub ahead of print.. [DOI] [PubMed] [Google Scholar]
- Marder P, Apelgren LD, & Bumol TF (1987). Comparative analysis of monoclonal antibody-drug conjugate binding by flow cytometry. Journal of Immunological Methods, 96, 165–170. [DOI] [PubMed] [Google Scholar]
- Marques PE, Grinstein S, & Freeman SA (2017). SnapShot:Macropinocytosis. Cell, 169, 766–767. [DOI] [PubMed] [Google Scholar]
- Marsigliante S, Muscella A, Ciardo V, Barker S, Leo G, Baker V, … Storelli C (1993). Enzyme-linked immunosorbent assay of HER-2/neu gene product (p185) in breast cancer: its correlation with sex steroid receptors, cathepsin D and histologic grades. Cancer Letters, 75, 195–206. [DOI] [PubMed] [Google Scholar]
- Martin LP, Konner JA, Moore KN, Seward SM, Matulonis UA, Perez RP, … Birrer MJ (2017). Characterization of folate receptor alpha (FRα) expression in archival tumor and biopsy samples from relapsed epithelial ovarian cancer patients: A phase I expansion study of the FRα-targeting antibody-drug conjugate mirvetuximab soravtansine. Gynecologic Oncology, 147, 402–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massard C, Soria JC, Krauss J, Gordon M, Lockhart AC, Rasmussen E, … Henary H (2019). First-in-human study to assess safety, tolerability, pharmacokinetics, and pharmacodynamics of the anti-CD27L antibody-drug conjugate AMG 172 in patients with relapsed/refractory renal cell carcinoma. Cancer Chemother Pharmacol, 83, 1057–1063. [DOI] [PubMed] [Google Scholar]
- Masters JC, Nickens DJ, Xuan D, Shazer RL, & Amantea M (2018). Clinical toxicity of antibody drug conjugates: a meta-analysis of payloads. Invest New Drugs, 36, 121–135. [DOI] [PubMed] [Google Scholar]
- Matsuda Y, Malinao MC, Robles V, Song J, Yamada K, & Mendelsohn BA (2020). Proof of site-specificity of antibody-drug conjugates produced by chemical conjugation technology: AJICAP first generation. J Chromatogr B Analyt Technol Biomed Life Sci, 1140, 121981. [DOI] [PubMed] [Google Scholar]
- Matulonis UA, Birrer MJ, O’Malley DM, Moore KN, Konner J, Gilbert L, … Kim SK (2019). Evaluation of Prophylactic Corticosteroid Eye Drop Use in the Management of Corneal Abnormalities Induced by the Antibody-Drug Conjugate Mirvetuximab Soravtansine. Clinical Cancer Research, 25, 1727–1736. [DOI] [PubMed] [Google Scholar]
- Matsumura Y (2020). Cancer stromal targeting therapy to overcome the pitfall of EPR effect. Adv Drug Deliv Rev, 154–155, 142–150. [DOI] [PubMed] [Google Scholar]
- Mayor S, Parton RG, & Donaldson JG (2014). Clathrin-independent pathways of endocytosis. Cold Spring Harb Perspect Biol, 6, 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCombs JR & Owen SC (2015). Antibody drug conjugates: design and selection of linker, payload and conjugation chemistry. AAPS Journal, 17, 339–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwen A & Henson C (2015). Quantitative whole-body autoradiography: past, present and future. Bioanalysis, 7, 557–568. [DOI] [PubMed] [Google Scholar]
- McHugh D, Eisenberger M, Heath EI, Bruce J, Danila DC, Rathkopf DE, … Morris MJ (2019). A phase I study of the antibody drug conjugate ASG-5ME, an SLC44A4-targeting antibody carrying auristatin E, in metastatic castration-resistant prostate cancer. Invest New Drugs, 37, 1052–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medeiros BC, Tanaka TN, Balaian L, Bashey A, Guzdar A, Li H, … Ball ED (2018). A Phase I/II Trial of the Combination of Azacitidine and Gemtuzumab Ozogamicin for Treatment of Relapsed Acute Myeloid Leukemia. Clin Lymphoma Myeloma Leuk, 18, 346–352. [DOI] [PubMed] [Google Scholar]
- Meric-Bernstam F, Johnson AM, Dumbrava E, Raghav K, Balaji K, Bhatt M, … Piha-Paul SA (2019). Advances in HER2-Targeted Therapy: Novel Agents and Opportunities Beyond Breast and Gastric Cancer. Clinical Cancer Research, 25, 2033–2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller FP, Vandome AF, & Mcbrewster J (2010). History of Cancer Chemotherapy. Berkeley, Alphascript Publishing. [Google Scholar]
- Min X, Fu D, Zhang J, Zeng J, Weng Z, Chen W, … Xia N (2018). An automated microfluidic chemiluminescence immunoassay platform for quantitative detection of biomarkers. Biomedical Microdevices, 20, 1–9. [DOI] [PubMed] [Google Scholar]
- Mitrenga D, Arnold W, Müller O, & Mayersbach HV (1975). The fate of injected human IgG in the mouse liver: Uptake, immunological inactivation, and lysosomal reactions. Cell and Tissue Research, 156, 359–376. [DOI] [PubMed] [Google Scholar]
- Modi S, Park H, Murthy RK, Iwata H, Tamura K, Tsurutani J, … Takahashi S (2020). Antitumor Activity and Safety of Trastuzumab Deruxtecan in Patients With HER2-Low-Expressing Advanced Breast Cancer: Results From a Phase Ib Study. Journal of Clinical Oncology, 38, 1887–1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore KN, Borghaei H, O’Malley DM, Jeong W, Seward SM, Bauer TM, … Birrer MJ (2017a). Phase 1 dose-escalation study of mirvetuximab soravtansine (IMGN853), a folate receptor α-targeting antibody-drug conjugate, in patients with solid tumors. Cancer, 123, 3080–3087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore KN, Martin LP, O’Malley DM, Matulonis UA, Konner JA, Perez RP, … Birrer MJ (2017b). Safety and Activity of Mirvetuximab Soravtansine (IMGN853), a Folate Receptor Alpha-Targeting Antibody-Drug Conjugate, in Platinum-Resistant Ovarian, Fallopian Tube, or Primary Peritoneal Cancer: A Phase I Expansion Study. Journal of Clinical Oncology, 35, 1112–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore KN, O’Malley DM, Vergote I, Martin LP, Gonzalez-Martin A, Malek K, … Birrer MJ (2018a). Safety and activity findings from a phase 1b escalation study of mirvetuximab soravtansine, a folate receptor alpha (FRα)-targeting antibody-drug conjugate (ADC), in combination with carboplatin in patients with platinum-sensitive ovarian cancer. Gynecologic Oncology, 151, 46–52. [DOI] [PubMed] [Google Scholar]
- Moore KN, Vergote I, Oaknin A, Colombo N, Banerjee S, Oza A, … Birrer MJ (2018b). FORWARD I: a Phase III study of mirvetuximab soravtansine versus chemotherapy in platinum-resistant ovarian cancer. Future Oncology, 14, 1669–1678. [DOI] [PubMed] [Google Scholar]
- Morton JJ, Bird G, Refaeli Y, & Jimeno A (2016). Humanized Mouse Xenograft Models: Narrowing the Tumor-Microenvironment Gap. Cancer Research, 76, 6153–6158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mou S, Huang Y, & Rosenbaum AI (2018). ADME Considerations and Bioanalytical Strategies for Pharmacokinetic Assessments of Antibody-Drug Conjugates. Antibodies (Basel), 7, 41–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller BM, Wrasidlo WA, & Reisfeld RA (1990). Antibody conjugates with morpholinodoxorubicin and acid-cleavable linkers. Bioconjug Chem, 1, 325–330. [DOI] [PubMed] [Google Scholar]
- Muhammad K, Jie C, Wang S, Lin C, Saif U, Liang K, … Zhan JB (2017). Intracellular trafficking of new anticancer therapeutics: antibody–drug conjugates. Drug Design Development & Therapy, 11, 2265–2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munasinghe WP, Mittapalli RK, Li H, Hoffman DM, Holen KD, Menon RM, … Xiong H (2017). Evaluation of the effect of the EGFR antibody-drug conjugate ABT-414 on QT interval prolongation in patients with advanced solid tumors likely to over-express EGFR. Cancer Chemother Pharmacol, 79, 915–922. [DOI] [PubMed] [Google Scholar]
- Nejadmoghaddam MR, Minai-Tehrani A, Ghahremanzadeh R, Mahmoudi M, Dinarvand R, & Zarnani AH (2019). Antibody-Drug Conjugates: Possibilities and Challenges. Avicenna Journal of Medical Biotechnology, 11, 3–23. [PMC free article] [PubMed] [Google Scholar]
- Nemeth BT, Varga ZV, Wu WJ, & Pacher P (2017). Trastuzumab cardiotoxicity: from clinical trials to experimental studies. Br J Pharmacol, 174, 3727–3748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolting B (2013). Linker technologies for antibody-drug conjugates. Methods Mol Biol, 1045, 71–100. [DOI] [PubMed] [Google Scholar]
- Norsworthy KJ, Ko CW, Lee JE, Liu J, John CS, Przepiorka D, … Pazdur R (2018). FDA Approval Summary: Mylotarg for Treatment of Patients with Relapsed or Refractory CD33-Positive Acute Myeloid Leukemia. Oncologist, 23, 1103–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ocean AJ, Starodub AN, Bardia A, Vahdat LT, Isakoff SJ, Guarino M, … Goldenberg DM (2017). Sacituzumab govitecan (IMMU-132), an anti-Trop-2-SN-38 antibody-drug conjugate for the treatment of diverse epithelial cancers: Safety and pharmacokinetics. Cancer, 123, 3843–3854. [DOI] [PubMed] [Google Scholar]
- Ogitani Y, Hagihara K, Oitate M, Naito H, & Agatsuma T (2016). Bystander killing effect of DS-8201a, a novel anti-human epidermal growth factor receptor 2 antibody-drug conjugate, in tumors with human epidermal growth factor receptor 2 heterogeneity. Cancer Sci, 107, 1039–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Malley DM, Matulonis UA, Birrer MJ, Castro CM, Gilbert L, Vergote I, … Moore KN (2020). Phase Ib study of mirvetuximab soravtansine, a folate receptor alpha (FRα)-targeting antibody-drug conjugate (ADC), in combination with bevacizumab in patients with platinum-resistant ovarian cancer. Gynecologic Oncology, 157, 379–385. [DOI] [PubMed] [Google Scholar]
- Ott PA, Pavlick AC, Johnson DB, Hart LL, Infante JR, Luke JJ, … Hamid O (2019). A phase 2 study of glembatumumab vedotin, an antibody-drug conjugate targeting glycoprotein NMB, in patients with advanced melanoma. Cancer, 125, 1113–1123. [DOI] [PubMed] [Google Scholar]
- Ouyang J (2013). Drug-to-antibody ratio (DAR) and drug load distribution by hydrophobic interaction chromatography and reversed phase high-performance liquid chromatography. Methods Mol Biol, 1045, 275–283. [DOI] [PubMed] [Google Scholar]
- Pal SK, Forero-Torres A, Thompson JA, Morris JC, Chhabra S, Hoimes CJ, … Smith DC (2019). A phase 1 trial of SGN-CD70A in patients with CD70-positive, metastatic renal cell carcinoma. Cancer, 125, 1124–1132. [DOI] [PubMed] [Google Scholar]
- Panowski S, Bhakta S, Raab H, Polakis P, & Junutula JR (2014). Site-specific antibody drug conjugates for cancer therapy. MAbs, 6, 34–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardridge WM (2015). Blood-brain barrier drug delivery of IgG fusion proteins with a transferrin receptor monoclonal antibody. Expert Opin Drug Deliv, 12, 207–222. [DOI] [PubMed] [Google Scholar]
- Parrozzani R, Lombardi G, Midena E, Leonardi F, Londei D, Padovan M, … Frizziero L (2020). Corneal side effects induced by EGFR-inhibitor antibody-drug conjugate ABT-414 in patients with recurrent glioblastoma: a prospective clinical and confocal microscopy study. Therapeutic Advances in Medical Oncology, 12, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pegram MD, Miles D, Tsui CK, & Zong Y (2020). HER2-Overexpressing/Amplified Breast Cancer as a Testing Ground for Antibody-Drug Conjugate Drug Development in Solid Tumors. Clinical Cancer Research, 26, 775–786. [DOI] [PubMed] [Google Scholar]
- Pernille Foged, Jensen Angela, Schoch Vincent, … Tilman. (2017). A Two-pronged Binding Mechanism of IgG to the Neonatal Fc Receptor Controls Complex Stability and IgG Serum Half-life. Molecular & Cellular Proteomics Mcp, 16, 451–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petkova SB, Akilesh S, Sproule TJ, Christianson GJ, Khabbaz HA, Brown AC, … Roopenian ADC (2006). Enhanced half-life of genetically engineered human IgG1 antibodies in a humanized FcRn mouse model: potential application in humorally mediated autoimmune disease. International Immunology, 18, 1759–1769. [DOI] [PubMed] [Google Scholar]
- Petrylak DP, Kantoff P, Vogelzang NJ, Mega A, Fleming MT, Stephenson JJ, … Israel RJ (2019). Phase 1 study of PSMA ADC, an antibody-drug conjugate targeting prostate-specific membrane antigen, in chemotherapy-refractory prostate cancer. Prostate, 79, 604–613. [DOI] [PubMed] [Google Scholar]
- Petrylak DP, Vogelzang NJ, Chatta K, Fleming MT, Smith DC, Appleman LJ, … Israel RJ (2020). PSMA ADC monotherapy in patients with progressive metastatic castration-resistant prostate cancer following abiraterone and/or enzalutamide: Efficacy and safety in open-label single-arm phase 2 study. Prostate, 80, 99–108. [DOI] [PubMed] [Google Scholar]
- Phillips GDL, Haas SD, Girish S, & Guardino E (2016). ADCs Approved for Use: Trastuzumab Emtansine (Kadcyla®, T〥M1) in Patients with Previously Treated HER2 ㏄ ositive Metastatic Breast Cancer. New Jersey, John Wiley & Sons, Ltd. [Google Scholar]
- Phillips T, Barr PM, Park SI, Kolibaba K, Caimi PF, Chhabra S, … Smith DC (2019). A phase 1 trial of SGN-CD70A in patients with CD70-positive diffuse large B cell lymphoma and mantle cell lymphoma. Invest New Drugs, 37, 297–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce JM & Mehta A (2017). Diagnostic, prognostic and therapeutic role of CD30 in lymphoma. Expert Review of Hematology, 10, 29–37. [DOI] [PubMed] [Google Scholar]
- Pimm MV, Armitage NC, Ballantyne K, Baldwin RW, Perkins AC, Durrant LG, … Hardcastle JD (1987). Imaging of primary and metastatic colorectal carcinoma with monoclonal antibody 791T/36 and the therapeutic potential of antibody-drug conjugates. Cancer Detect Prev Suppl, 1, 249–262. [PubMed] [Google Scholar]
- Ponziani S, Vittorio GD, Pitari G, Cimini AM, & Giansanti F (2020). Antibody-Drug Conjugates: The New Frontier of Chemotherapy. International Journal of Molecular Sciences, 21, 5510–5536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rammensee HG, Weinschenk T, Gouttefangeas C, & Stevanović S (2002). Towards patient-specific tumor antigen selection for vaccination. Immunological Reviews, 188, 164–176. [DOI] [PubMed] [Google Scholar]
- Reardon DA, Lassman AB, van den Bent M, Kumthekar P, Merrell R, Scott AM, … Gan HK (2017). Efficacy and safety results of ABT-414 in combination with radiation and temozolomide in newly diagnosed glioblastoma. Neuro Oncol, 19, 965–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rees AR (2015). The Antibody Molecule From antitoxins to therapeutic antibodies. New York, Oxford University Press. [Google Scholar]
- Reichert JM (2014). Antibodies to watch in 2014. MAbs, 6, 5–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricart AD (2011). Antibody-Drug Conjugates of Calicheamicin Derivative: Gemtuzumab Ozogamicin and Inotuzumab Ozogamicin. Clinical Cancer Research, 17, 6417–6427. [DOI] [PubMed] [Google Scholar]
- Ritchie M, Bloom L, Carven G, & Sapra P (2015). Selecting an Optimal Antibody for Antibody- Drug Conjugate Therapy. Heidelberg, Springer International Publishing. [Google Scholar]
- Ritchie M, Tchistiakova L, & Scott N (2013). Implications of receptor-mediated endocytosis and intracellular trafficking dynamics in the development of antibody drug conjugates. MAbs, 5, 13–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert B, Dorvillius M, Buchegger F, Garambois V, Mani JC, Pugnières M, … Pèlegrin A (1999). Tumor targeting with newly designed biparatopic antibodies directed against two different epitopes of the carcinoembryonic antigen (CEA). International Journal of Cancer, 81, 285–291. [DOI] [PubMed] [Google Scholar]
- Roberts SA, Andrews PA, Blanset D, Flagella KM, Gorovits B, Lynch CM, … Warner G (2013). Considerations for the nonclinical safety evaluation of antibody drug conjugates for oncology. Regul Toxicol Pharmacol, 67, 382–391. [DOI] [PubMed] [Google Scholar]
- Rohrer T (2017). Manufacturing Concepts for Antibody-Drug Conjugates. New Jersey, John Wiley & Sons, Inc. [Google Scholar]
- Roopenian DC & Akilesh S (2007). FcRn: the neonatal Fc receptor comes of age. Nature Reviews Immunology, 7, 715–725. [DOI] [PubMed] [Google Scholar]
- Rosenberg J, Sridhar SS, Zhang J, Smith D, Ruether D, Flaig TW, … Petrylak DP (2020). EV-101: A Phase I Study of Single-Agent Enfortumab Vedotin in Patients With Nectin-4-Positive Solid Tumors, Including Metastatic Urothelial Carcinoma. Journal of Clinical Oncology, 38, 1041–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal M, Curry R, Reardon DA, Rasmussen E, Upreti VV, Damore MA, … Cloughesy T (2019). Safety, tolerability, and pharmacokinetics of anti-EGFRvIII antibody-drug conjugate AMG 595 in patients with recurrent malignant glioma expressing EGFRvIII. Cancer Chemother Pharmacol, 84, 327–336. [DOI] [PubMed] [Google Scholar]
- Rudin CM, Pietanza MC, Bauer TM, Ready N, Morgensztern D, Glisson BS, … Spigel DR (2017). Rovalpituzumab tesirine, a DLL3-targeted antibody-drug conjugate, in recurrent small-cell lung cancer: a first-in-human, first-in-class, open-label, phase 1 study. Lancet Oncology, 18, 42–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saber H, Simpson N, Ricks TK, & Leighton JK (2019). An FDA oncology analysis of toxicities associated with PBD-containing antibody-drug conjugates. Regul Toxicol Pharmacol, 107, 104429–104443. [DOI] [PubMed] [Google Scholar]
- Sapra P, Hooper AT, O’Donnell CJ, & Gerber HP (2011). Investigational antibody drug conjugates for solid tumors. Expert Opin Investig Drugs, 20, 1131–1149. [DOI] [PubMed] [Google Scholar]
- Sargiacomo M, Scherer PE, Tang Z, Kübler E, Song KS, Sanders MC, … Lisanti MP (1995). Oligomeric structure of caveolin: implications for caveolae membrane organization. Proc Natl Acad Sci U S A, 92, 9407–9411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarrut M, Corgier A, Fekete S, Guillarme D, Lascoux D, Janin-Bussat MC, … Heinisch S (2016). Analysis of antibody-drug conjugates by comprehensive on-line two-dimensional hydrophobic interaction chromatography x reversed phase liquid chromatography hyphenated to high resolution mass spectrometry. I - Optimization of separation conditions. J Chromatogr B Analyt Technol Biomed Life Sci, 1032, 103–111. [DOI] [PubMed] [Google Scholar]
- Sassoon I & Blanc V (2013). Antibody-drug conjugate (ADC) clinical pipeline: a review. Methods Mol Biol, 1045, 1–27. [DOI] [PubMed] [Google Scholar]
- Sau S, Alsaab HO, Kashaw SK, Tatiparti K, & Iyer AK (2017). Advances in antibody–drug conjugates: A new era of targeted cancer therapy. Drug Discovery Today, 22, 1547–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlom J, Hand PH, Greiner JW, Colcher D, & Raubitschek A (1990). Innovations that influence the pharmacology of monoclonal antibody guided tumor targeting. Cancer Research, 50, 820–823. [PubMed] [Google Scholar]
- Schmidt D, Asselineau B, B Ttger R, Klein H, Lebreton L, Neumann S, … Pichenot G (2002). Characterization of liquid scintillation detectors. Nuclear Inst & Methods in Physics Research A, 476, 186–189. [Google Scholar]
- Schneider JR, Carias AM, Bastian AR, Cianci GC, Kiser PF, Veazey RS, … Hope TJ (2017). Long-term direct visualization of passively transferred fluorophore-conjugated antibodies. Journal of Immunological Methods, 450, 66–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott LJ (2017). Brentuximab Vedotin: A Review in CD30-Positive Hodgkin Lymphoma. Drugs, 77, 435–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedlacek HH, Seemann G, Hoffmann D, Czech J, Lorenz P, Kolar C, … Bosslet K (1992). Antibodies as carriers of cytotoxicity. Basel, Karger. [Google Scholar]
- Shah K & Maghsoudlou P (2016). Enzyme-linked immunosorbent assay (ELISA): the basics. Br J Hosp Med (Lond), 77, 98–101. [DOI] [PubMed] [Google Scholar]
- Shapiro GI, Vaishampayan UN, LoRusso P, Barton J, Hua S, Reich SD, … Borghaei H (2017). First-in-human trial of an anti-5T4 antibody-monomethylauristatin conjugate, PF-06263507, in patients with advanced solid tumors. Invest New Drugs, 35, 315–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharman JP, Wheler JJ, Einhorn L, Dowlati A, Shapiro GI, Hilton J, … Jalal SI (2019). A phase 2, open-label study of brentuximab vedotin in patients with CD30-expressing solid tumors. Invest New Drugs, 37, 738–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen BQ, Bumbaca D, Saad O, Yue Q, Pastuskovas CV, Khojasteh SC, … Girish S (2012). Catabolic fate and pharmacokinetic characterization of trastuzumab emtansine (T-DM1): an emphasis on preclinical and clinical catabolism. Current Drug Metabolism, 13, 901–910. [DOI] [PubMed] [Google Scholar]
- Shi SR, Key ME, & Kalra KL (1991). Antigen retrieval in formalin-fixed, paraffin-embedded tissues: an enhancement method for immunohistochemical staining based on microwave oven heating of tissue sections. Journal of Histochemistry & Cytochemistry, 39, 741–748. [DOI] [PubMed] [Google Scholar]
- Shimizu T, Kozono Y, Kozono H, Oda M, & Azuma T (2004). Affinity maturation of secreted IgM pentamers on B cells. International Immunology, 5, 675–684. [DOI] [PubMed] [Google Scholar]
- Shitara K, Bang YJ, Iwasa S, Sugimoto N, Ryu MH, Sakai D, … Yamaguchi K (2020). Trastuzumab Deruxtecan in Previously Treated HER2-Positive Gastric Cancer. N Engl J Med, 382, 2419–2430. [DOI] [PubMed] [Google Scholar]
- Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, … Norton L (2001). Use of Chemotherapy plus a Monoclonal Antibody against HER2 for Metastatic Breast Cancer That Overexpresses HER2. New England Journal of Medicine, 344, 783–792. [DOI] [PubMed] [Google Scholar]
- Smith GP (1974). Antibody Diversity. (Book Reviews: The Variation and Adaptive Expression of Antibodies). Science, 183, 190–191. [Google Scholar]
- Sochaj AM, Świderska KW, & Otlewski J (2015). Current methods for the synthesis of homogeneous antibody-drug conjugates. Biotechnology Advances, 33, 775–784. [DOI] [PubMed] [Google Scholar]
- Socinski MA, Kaye FJ, Spigel DR, Kudrik FJ, Ponce S, Ellis PM, … Ares LP (2017). Phase 1/2 Study of the CD56-Targeting Antibody-Drug Conjugate Lorvotuzumab Mertansine (IMGN901) in Combination With Carboplatin/Etoposide in Small-Cell Lung Cancer Patients With Extensive-Stage Disease. Clinical Lung Cancer, 18, 68–76. [DOI] [PubMed] [Google Scholar]
- Stathis A, Flinn IW, Madan S, Maddocks K, Freedman A, Weitman S, … Lia PM (2018). Safety, tolerability, and preliminary activity of IMGN529, a CD37-targeted antibody-drug conjugate, in patients with relapsed or refractory B-cell non-Hodgkin lymphoma: a dose-escalation, phase I study. Invest New Drugs, 36, 869–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staudacher AH & Brown MP (2017). Antibody drug conjugates and bystander killing: is antigen-dependent internalisation required? Br J Cancer, 117, 1736–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stauss HJ, Kawakami Y, Parmiani G. (2003). Tumor antigens recognized by T cells and antibodies. Abingdon, Taylor & Francis Ltd. [Google Scholar]
- Stein EM, Walter RB, Erba HP, Fathi AT, Advani AS, Lancet JE, … Stein AS (2018). A phase 1 trial of vadastuximab talirine as monotherapy in patients with CD33-positive acute myeloid leukemia. Blood, 131, 387–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinmetz MO & Prota AE (2018). Microtubule-Targeting Agents: Strategies To Hijack the Cytoskeleton. Trends in Cell Biology, 28, 776–792. [DOI] [PubMed] [Google Scholar]
- Steplewski Z, Sun L, Shearman K, Ghrayeb J, Daddona P, & Koprowski H (1988). Biological activity of human-mouse IgG1, IgG2, IgG3, and IgG4 chimeric monoclonal antibodies with antitumor specificity. Proceedings of the National Academy of Sciences, 85, 4852–4856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart AK, Krishnan AY, Singhal S, Boccia RV, Patel MR, Niesvizky R, … Berdeja JG (2019). Phase I study of the anti-FcRH5 antibody-drug conjugate DFRF4539A in relapsed or refractory multiple myeloma. Blood Cancer Journal, 9, 2–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strickler JH, Weekes CD, Nemunaitis J, Ramanathan RK, Heist RS, Morgensztern D, … Kelly K (2018). First-in-Human Phase I, Dose-Escalation and -Expansion Study of Telisotuzumab Vedotin, an Antibody-Drug Conjugate Targeting c-Met, in Patients With Advanced Solid Tumors. Journal of Clinical Oncology, 36, 3298–3306. [DOI] [PubMed] [Google Scholar]
- Su D, Kozak K, Sadowsky J, Yu SF, Fourie-O Donohue A, Nelson C, … Ng C (2018). Modulating ADC payload metabolism by conjugation site and linker modification. Bioconjugate Chemistry, 4, 1155–1167. [DOI] [PubMed] [Google Scholar]
- Sung M, Tan X, Lu B, Golas J, Hosselet C, Wang F, … Loganzo F (2018). Caveolae-mediated endocytosis as a novel mechanism of resistance to Trastuzumab Emtansine (T-DM1). Molecular Cancer Therapeutics, 17, 243–253. [DOI] [PubMed] [Google Scholar]
- Suri A, Mould DR, Song G, Collins GP, Endres CJ, Gomez-Navarro J, … Venkatakrishnan K (2019). Population pharmacokinetic modeling and exposure-response assessment for the antibody-drug conjugate Brentuximab Vedotin in Hodgkin’s lymphoma in the phase III ECHELON-1 study. Clinical Pharmacology & Therapeutics, 106, 1268–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swan LE (2013). Initiation of clathrin-mediated endocytosis: all you need is two? Bioessays, 35, 425–429. [DOI] [PubMed] [Google Scholar]
- Takebe N, Miele L, Harris PJ, Jeong W, Bando H, Kahn M, … Ivy SP (2015). Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: clinical update. Nature Reviews Clinical Oncology, 12, 445–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang F, Shi W, & Huang W (2019). Homogeneous antibody-drug conjugates via glycoengineering. Methods Mol Biol, 2033, 221–238. [DOI] [PubMed] [Google Scholar]
- Terwisscha VSA, Ogasawara A, Pacheco G, Vanderbilt AN, Tinianow JN, Gupta N, … Williams SP (2017). Preclinical Efficacy of an Antibody-Drug Conjugate Targeting Mesothelin Correlates with Quantitative 89Zr-ImmunoPET. Molecular Cancer Therapeutics, 16, 134–142. [DOI] [PubMed] [Google Scholar]
- Thakur A, Huang M, & Lum LG (2018). Bispecific antibody based therapeutics: Strengths and challenges. Blood Reviews, 32, 339–347. [DOI] [PubMed] [Google Scholar]
- Theocharopoulos C, Lialios PP, Gogas H, & Ziogas DC (2020). An overview of antibody-drug conjugates in oncological practice. Therapeutic Advances in Medical Oncology, 12, 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JA, Motzer RJ, Molina AM, Choueiri TK, Heath EI, Redman BG, … Kollmannsberger CK (2018). Phase I Trials of Anti-ENPP3 Antibody-Drug Conjugates in Advanced Refractory Renal Cell Carcinomas. Clinical Cancer Research, 24, 4399–4406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurber GM, Schmidt MM, & Wittrup KD (2008). Factors determining antibody distribution in tumors. Trends in Pharmacological Sciences, 29, 57–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolcher AW (2016). Antibody drug conjugates: lessons from 20 years of clinical experience. Annals of Oncology, 27, 2168–2172. [DOI] [PubMed] [Google Scholar]
- Trail PA, King HD, & Dubowchik GM (2003). Monoclonal antibody drug immunoconjugates for targeted treatment of cancer. Cancer Immunol Immunother, 52, 328–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trnĕný M, Verhoef G, Dyer MJ, Ben YD, Patti C, Canales M, … Gianni AM (2018). A phase II multicenter study of the anti-CD19 antibody drug conjugate coltuximab ravtansine (SAR3419) in patients with relapsed or refractory diffuse large B-cell lymphoma previously treated with rituximab-based immunotherapy. Haematologica, 103, 1351–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trudel S, Lendvai N, Popat R, Voorhees PM, Reeves B, Libby EN, … Cohen AD (2018). Targeting B-cell maturation antigen with GSK2857916 antibody-drug conjugate in relapsed or refractory multiple myeloma (BMA117159): a dose escalation and expansion phase 1 trial. Lancet Oncology, 19, 1641–1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trudel S, Lendvai N, Popat R, Voorhees PM, Reeves B, Libby EN, … Cohen AD (2019). Antibody-drug conjugate, GSK2857916, in relapsed/refractory multiple myeloma: an update on safety and efficacy from dose expansion phase I study. Blood Cancer Journal, 9, 37–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchikama K & An Z (2016). Antibody-drug conjugates: recent advances in conjugation and linker chemistries. Protein & Cell, 9, 33–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuda M, Takano Y, Shigeyasu C, Imoto S, & Yamada M (2016). Abnormal corneal lesions induced by Trastuzumab Emtansine: an antibody-drug conjugate for breast cancer. Cornea, 35, 1378–1380. [DOI] [PubMed] [Google Scholar]
- Tsumura R, Manabe S, Takashima H, Koga Y, Yasunaga M, & Matsumura Y (2018). Influence of the dissociation rate constant on the intra-tumor distribution of antibody-drug conjugate against tissue factor. Journal of Controlled Release, 284, 49–56. [DOI] [PubMed] [Google Scholar]
- Tumey LN, Rago B, & Han X (2015). In vivo biotransformations of antibody-drug conjugates. Bioanalysis, 7, 1649–1664. [DOI] [PubMed] [Google Scholar]
- Tumey LN & Han S (2017). ADME considerations for the development of biopharmaceutical conjugates using cleavable linkers. Current Topics in Medicinal Chemistry, 17, 3444–3462. [DOI] [PubMed] [Google Scholar]
- Udagawa H, Akamatsu H, Tanaka K, Takeda M, Kanda S, Kirita K, … Okamoto I (2019). Phase I safety and pharmacokinetics study of rovalpituzumab tesirine in Japanese patients with advanced, recurrent small cell lung cancer. Lung Cancer, 135, 145–150. [DOI] [PubMed] [Google Scholar]
- van den Bent M, Gan HK, Lassman AB, Kumthekar P, Merrell R, Butowski N, … Reardon DA (2017). Efficacy of depatuxizumab mafodotin (ABT-414) monotherapy in patients with EGFR-amplified, recurrent glioblastoma: results from a multi-center, international study. Cancer Chemother Pharmacol, 80, 1209–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Weyden C, Dickinson M, Whisstock J, & Prince HM (2019). Brentuximab vedotin in T-cell lymphoma. Expert Review of Hematology, 12, 5–19. [DOI] [PubMed] [Google Scholar]
- Wakankar AA, Feeney MB, Rivera J, Chen Y, Kim M, Sharma VK, … Wang YJ (2010). Physicochemical stability of the antibody-drug conjugate Trastuzumab-DM1: changes due to modification and conjugation processes. Bioconjug Chem, 21, 1588–1595. [DOI] [PubMed] [Google Scholar]
- Waldmann H (2019). Human Monoclonal Antibodies: The Benefits of Humanization. Methods Mol Biol, 1904, 1–10. [DOI] [PubMed] [Google Scholar]
- Walles M, Connor A, & Hainzl D (2017). ADME and safety aspects of non-cleavable linkers in drug discovery and development. Current Topics in Medicinal Chemistry, 17, 3463–3475. [DOI] [PubMed] [Google Scholar]
- Walts AE & Said JW (1983). Specific tumor markers in diagnostic cytology. Immunoperoxidase studies of carcinoembryonic antigen, lysozyme and other tissue antigens in effusions, washes and aspirates. Acta Cytologica, 27, 408–416. [PubMed] [Google Scholar]
- Wang X, Ma D, Olson WC, & Heston WD (2011). In vitro and in vivo responses of advanced prostate tumors to PSMA ADC, an auristatin-conjugated antibody to prostate-specific membrane antigen. Molecular Cancer Therapeutics, 10, 1728–1739. [DOI] [PubMed] [Google Scholar]
- Wang Y, Tian Z, Thirumalai D, & Zhang X (2014). Neonatal Fc receptor (FcRn): a novel target for therapeutic antibodies and antibody engineering. Journal of Drug Targeting, 22, 269–278. [DOI] [PubMed] [Google Scholar]
- Webster HV, Bone AJ, Webster KA, & Wilkin TJ (1990). Comparison of an enzyme-linked immunosorbent assay (ELISA) with a radioimmunoassay (RIA) for the measurement of rat insulin. Journal of Immunological Methods, 134, 95–100. [DOI] [PubMed] [Google Scholar]
- White JB, Fleming R, Masterson L, Ruddle BT, Zhong H, Fazenbaker C, … Dimasi N (2019). Design and characterization of homogenous antibody-drug conjugates with a drug-to-antibody ratio of one prepared using an engineered antibody and a dual-maleimide pyrrolobenzodiazepine dimer. MAbs, 11, 500–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolska-Washer A & Robak T (2019). Safety and tolerability of antibody-drug conjugates in cancer. Drug Saf, 42, 295–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S (2015). Internalization, Trafficking, Intracellular Processing and Actions of Antibody-Drug Conjugates. Pharm Res, 32, 3577–3583. [DOI] [PubMed] [Google Scholar]
- Yaghoubi S, Karimi MH, Lotfinia M, Gharibi T, Mahi-Birjand M, Kavi E, … Abdollahpour-Alitappeh M (2020). Potential drugs used in the antibody-drug conjugate (ADC) architecture for cancer therapy. Journal of Cellular Physiology, 235, 31–64. [DOI] [PubMed] [Google Scholar]
- Yamada K & Ito Y (2019). Recent Chemical Approaches for Site-Specific Conjugation of Native Antibodies: Technologies toward Next-Generation Antibody-Drug Conjugates. Chembiochem, 20, 2729–2737. [DOI] [PubMed] [Google Scholar]
- Yao H, Jiang F, Lu A, & Zhang G (2016). Methods to Design and Synthesize Antibody-Drug Conjugates (ADCs). International Journal of Molecular Sciences, 17, 194–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasunaga M (2020). Antibody therapeutics and immunoregulation in cancer and autoimmune disease. Semin Cancer Biol. 64, 1–12. [DOI] [PubMed] [Google Scholar]
- Yasunaga M, Manabe S, & Matsumura Y (2011). New concept of cytotoxic immunoconjugate therapy targeting cancer-induced fibrin clots. Cancer Science, 102, 1396–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Qi-Tao, Zhi-Heng Huang, Zhou Yun-Li, … Qi-Ting. (2010). Mass spectrometry of maytansinoids-A study on the fragmentation mechanism and identification of synthetic analogs of maytansine. Acta Chimica Sinica, 4, 48–54. [Google Scholar]
- Zaro JL (2015). Mylotarg: Revisiting Its Clinical Potential Post-Withdrawal. In (Vol. 17, pp. 179–190). Cham, Springer International Publishing. [Google Scholar]
- Zhu X, Huo S, Xue C, An B, & Qu J (2020). Current LC-MS-based strategies for characterization and quantification of antibody-drug conjugates. Journal of Pharmaceutical Analysis, 10, 209–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinzani PL, Santoro A, Gritti G, Brice P, Barr PM, Kuruvilla J, … Moskowitz AJ (2019). Nivolumab combined with Brentuximab Vedotin for relapsed/refractory primary mediastinal large B-cell lymphoma: efficacy and safety from the Phase II checkmate 436 study. Journal of Clinical Oncology, 37, 3081–3089. [DOI] [PMC free article] [PubMed] [Google Scholar]