

Graphical abstract
Keywords: Glycoside hydrolase, CAZymes, Rumen microbiome, Glycome profiling, Linkage analysis, Transcriptome, Differential gene expression, Nutrient utilization, Dietary polysaccharides
Abbreviations: AIR, alcohol insoluble residue; AB, arabinan; ADF, acid detergent fibre; AG, arabinogalactan; AGP, arabinogalactan protein; AO, ammonium oxalate; AX, arabinoxylan; BS, barley straw; CAZyme, carbohydrate active enzyme; CE, carbohydrate esterase; CH, chlorite; ELISA, enzyme-linked immunosorbent assay; DE, differentially expressed; FID, flame ionization detection GC, gas chromatography; GH, glycosyl hydrolase; HG, homogalacturonan; HPAEC-PAD, high performance anion exchange chromatography coupled with pulsed amperometric detection; HX, heteroxylan; mAbs, monoclonal antibodies; MS, mass spectrometry; NDF, neutral detergent fibre; PC, post-chlorite; PL, polysaccharide lyase; RG, rhamnogalacturonan; SC, sodium carbonate; TTIR, total tract indigestible residue; XG, xyloglucan
Abstract
There is a knowledge gap regarding the factors that impede the ruminal digestion of plant cell walls or if rumen microbiota possess the functional activities to overcome these constraints. Innovative experimental methods were adopted to provide a high-resolution understanding of plant cell wall chemistries, identify higher-order structures that resist microbial digestion, and determine how they interact with the functional activities of the rumen microbiota. We characterized the total tract indigestible residue (TTIR) from cattle fed a low-quality straw diet using two comparative glycomic approaches: ELISA-based glycome profiling and total cell wall glycosidic linkage analysis. We successfully detected numerous and diverse cell wall glycan epitopes in barley straw (BS) and TTIR and determined their relative abundance pre- and post-total tract digestion. Of these, xyloglucans and heteroxylans were of higher abundance in TTIR. To determine if the rumen microbiota can further saccharify the residual plant polysaccharides within TTIR, rumen microbiota from cattle fed a diet containing BS were incubated with BS and TTIR ex vivo in batch cultures. Transcripts coding for carbohydrate-active enzymes (CAZymes) were identified and characterized for their contribution to cell wall digestion based on glycomic analyses, comparative gene expression profiles, and associated CAZyme families. High-resolution phylogenetic fingerprinting of these sequences encoded CAZymes with activities predicted to cleave the primary linkages within heteroxylan and arabinan. This experimental platform provides unprecedented precision in the understanding of forage structure and digestibility, which can be extended to other feed-host systems and inform next-generation solutions to improve the performance of ruminants fed low-quality forages.
1. Introduction
Lignocellulosic biomass can be converted into a broad suite of fuels, chemicals, and biomaterials. The recalcitrance of cellulosic material and high cost of hydrolysis to simple sugar is a major barrier to the commercial viability of bioproducts developed from cellulosic biomass. With an ever-growing human population and emergence of more affluent societies, the global demand for food, meat and milk is projected to increase exponentially [1], [2]. Ruminant livestock are in a unique position to satisfy the growing demand for high-quality protein, as ruminants can produce milk and meat via the microbial fermentation of cellulose-rich forages, crop residues, and food by-products. In this light, the rumen microbiota represents an underexploited repository of carbohydrate-active enzymes (CAZymes) and microorganisms for applications in animal nutrition, biofuel, and bio-based chemical industries.
Globally, more than 73.9 million metric tons of crop residues are produced annually [3], [4]. Within Western Canada, barley straw (BS) residues are of increasing significance as they represent a significant by-product of barley production in the prairies, second only to that of wheat [3]. Although these residues could serve as feed for ruminants, usually less than half of the biomass they contain is digested, and total tract indigestible residues (TTIR) within feces represents underutilized nutritive material. Supplementing ruminant diets with exogenous enzymes has the potential to improve the utilization of crop residues for meat and milk production [5], [6], [7], [8]. However, current commercial enzyme products have been largely developed for biorefinery and bioprocessing applications and were not designed to function in the gastrointestinal tract of ruminants. Physiological conditions within the rumen are not favorable for some enzymes within cellulase mixtures [7], [8], with many of these products possessing enzyme activities that are redundant to those already produced by the rumen microbial community. Quantifying the digestibility of neutral detergent fibre (NDF) and acid detergent fibre (ADF) in ruminants provides only a cursory insight into the factors that limit plant cell wall digestion in the rumen. Previously, immunofluorescence microscopy coupled with cell wall specific monoclonal antibodies (mAbs) has proven a more informative method to investigate the polysaccharide composition of plant cell walls [9], [10]. In this light, an informed, tailored approach is still needed to investigate ruminal fibre digestion. Such an approach should be anchored in a higher understanding of cell wall recalcitrance to ruminal enzymatic digestion and identification of rate-limiting enzyme activities. Once rate-limiting enzymatic activities within the rumen are identified, CAZymes with synergistic activities could be used to enhance the digestion of fibre by rumen microbiota.
Here, we have developed a platform for coupling glycomic analyses to comparative metatranscriptomes of rumen microbiota cultivated ex vivo with BS and TTIR as substrates. We aimed to characterize abundant, recalcitrant cell wall structures and identify candidate enzyme activities with the potential to enhance the rumen digestion of BS. We identified indigestible cell wall moieties within TTIR and characterized the glycosidic linkages that contribute to recalcitrance within BS. Using a complementary metatranscriptomic approach and predictive bioinformatic methods, a suite of genes was selected for downstream production of recombinant enzymes that target recalcitrant residues as a means of overcoming constraints to the digestion of BS plant cell walls. Although this study focused on the digestion of BS, the experimental pipeline presented here holds high promise for the improvement of lignocellulosic feedstock from diverse plant sources in support of the sustainable conversion of lignocellulose biomass into industrial feedstock.
2. Results
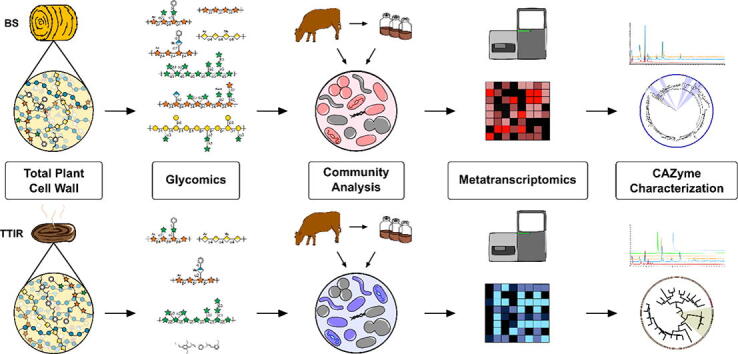
The evaluation of indigestible residues in BS and the identification of rumen enzymes active on recalcitrant polysaccharides requires a combinatorial glycomics and transcriptomics workflow (Fig. 1). In brief, cattle were fed BS-rich diets and the TTIR from cow fecal material were collected and washed, and the polysaccharides were purified as alcohol insoluble residue (AIR). ELISA-based glycome profiling and glycosidic linkage analyses were employed to determine the relative abundance of different cell wall components pre- and post-digestion. To evaluate the potential for further hydrolysis of TTIR, the rumen microbiota was harvested from cannulated cattle fed a BS-rich diet and incubated in vitro with BS and TTIR. Transcriptomic data of these microbial communities were analyzed to identify differentially expressed genes encoding CAZymes that actively contributed to the digestion of recalcitrant cell wall polysaccharides in TTIR. High-resolution phylogenetic fingerprinting and biochemical characterization of recombinant enzymes were adopted to determine the substrate specificity of selected differentially expressed transcripts. This multi-pronged and substrate-informed approach to CAZyme discovery tailored the selection of candidate rumen microbial genes to those with demonstrated specific activity against recalcitrant residual polysaccharides.
Fig. 1.

Experimental pipeline for the identification of rumen enzymes active on barley straw (BS) and total tract indigestible residue (TTIR). Barley straw is the predominant component in the cattle diet. The BS feed and total tract indigestible residue (TTIR) are subjected to chemical fractionation, releasing pectin, hemicellulose, and lignocellulose fractions. These fractions are then analyzed by comprehensive glycomics, including antibody-based glycome profiling and glycosidic linkage analysis, to determine total cell wall structure. Rumen microbes are grown ex vivo using BS and TTIR as the sole carbon source for transcriptomics and differential gene expression analysis. Transcriptomics informed by whole cell wall characterization provided evidence for the identification of rumen enzymes active against recalcitrant TTIR polysaccharides.
2.1. Glycome profiling of BS and total tract indigestible residue
The sequential extraction of cell wall material with ammonium oxalate, sodium carbonate, 1 M KOH, 4 M KOH, chlorite, and post-chlorite 4 M KOH, selectively enriched for various cell wall glycan fractions based on their embeddedness in the plant cell wall [11]. Ammonium oxalate and sodium carbonate fractions contain loosely bound pectic and arabinogalactan polysaccharides, while extraction with 1 M and 4 M KOH solubilized hemicelluloses (e.g., xyloglucans and xylans) and tightly bound pectic polysaccharides. Chlorite treatment released lignin and lignin-associated polysaccharides, while a subsequent post-chlorite extraction after delignification recovered remaining recalcitrant hemicellulose and pectin fractions embedded within crystalline cellulose. Chemical fractionation of BS and TTIR followed by enzyme-linked immunosorbent assay (ELISA) identified differences in the glycome profile of these substrates (Fig. 2, with detailed information on monoclonal antibodies and their epitopes available [12]). One major difference between BS and TTIR was the amount of carbohydrate recovered in the ammonium oxalate extracts of these two fractions; about 2.5-fold carbohydrate was recovered (mg recovered/gram of cell wall) in the ammonium oxalate extract from TTIR as compared with BS. Yet, with the exception of the de-esterified homogalacturonan (HG) epitopes (α-1,4 linked HG epitope (DP > 4)), which were enriched in TTIR ammonium oxalate extract as compared to that from BS (Fig. 2; box 1), there was very little antibody reactive material in the TTIR extract. This suggests that most of the carbohydrate extracted by ammonium oxalate was too low in molecular weight to bind to the ELISA plate, or to be recognized as full epitopes by the antibodies used in this study. In general, the ammonium oxalate and sodium carbonate fractions of TTIR had minimal amounts or lacked most xyloglucan (XG), xylan, type I rhamnogalacturonan (RG-I), and arabinogalactan (AG) moieties, suggesting that these carbohydrates were digested during passage through the ruminant digestive tract. In addition, there was a notable reduction in the AG-3 (Rha-(1,4)-GalA-(1,2)-(Rha-(1,4)-GalA-(Rha-(1,4)-GalA) and AG-4 epitopes and in the lignin-associated 4-O-methyl-D-glucuronosyl (MeGlcA)-substituted xylans (Fig. 2, Box 7) in TTIR compared with BS. In contrast, the somewhat loosely associated MeGlcA-substituted xylans present in the sodium carbonate extract appeared resistant to degradation during passage through the bovine digestive tract (Fig. 2, Box 2).
Fig. 2.

Glycome profile of barley straw (BS) and total tract indigestible residue (TTIR). BS and TTIR samples were sequentially extracted with ammonium oxalate (AO), sodium carbonate (SC), 1 M (1 M) and 4 M (4 M) KOH, chlorite (CH), and post-chlorite 4 M KOH (PC). The total carbohydrate recovered per gram of cell wall is indicated by bar graph (top). Cell wall extracts were analyzed through an ELISA-based screen against a collection of plant cell wall glycan-directed monoclonal antibodies (mAbs), and results shown as a heat map, where each row reflects the binding of a single mAb against the different extracts. The colour of each element in the heat map represents the strength of the ELISA signal, as indicated (black = no signal, yellow = strong signal). The panel on the righthand side depicts the clade of mAbs specific to various plant cell wall glycans. Numbered white boxes represent major differences in mAb binding intensities between BS and TTIR samples. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
In the remainder of the glycome profiles, there appeared to be an increased abundance of particular epitopes in TTIR as compared to BS. For example, there was an increase in the relative abundance of Gal-XG epitopes in the 1 M and 4 M KOH extracts of TTIR compared to BS (Fig. 2, Boxes 3, 4), as well an increased enrichment of the Xylan-3 and 2-arabinosyl-substituted xylans (Fig. 2, Boxes 5, 6). Compared to BS, diverse epitopes of 6-linked β-galactan epitopes (common in RG-I and arabinogalactan-proteins (AGPs)) were enriched in 4 M KOH extract from TTIR (Fig. 2, Box 9), but these epitopes were less abundant in the 1 M KOH extract (Fig. 2, Box 8). There also was an enrichment in some RG-I epitopes in the 4 M KOH extract from TTIR (Fig. 2, Box 10) compared to the corresponding extract from BS. Lastly, there was an overall enrichment in the TTIR biomass for the most tightly bound glycans released in the 4 M KOH post-chlorite extraction compared to BS. It is important to note that chlorite and post-chlorite extracts represent the most recalcitrant cell wall fractions. The persistent presence of cross-linked (glucurono)arabinoxylan, xyloglucan, and cellulose-embedded pectins (galactans, RG-I, and AG-2) in the KOH, chlorite and post-chlorite extracts of TTIR is indicative of the recalcitrance of these fractions to microbial digestion within the digestive tract.
2.2. Glycosidic linkage analysis of BS and total tract indigestible residue
To further define the extent of plant cell wall digestibility by ruminant microbiota, we conducted glycosidic linkage analysis to identify and quantify the total glycosidic bonds found within BS and TTIR. This method provided structural insights into the total composition of the carbohydrate matrix (i.e., is not limited by epitope accessibility) and polymerization state (i.e., is not limited to polysaccharides). First, the relative abundance of individual monosaccharide linkages in these fractions was determined (Supplemental File 1), which enabled a comparison of the ratio of linkage abundance in BS to TTIR to be used as a reflection of overall digestibility (Fig. 3A). Several linkages within the pectin-rich fraction (EDTA + Na2CO3); 2,4-Xylp, t-GalAp, 4-GalAp, 3,6-Galp, 3-Araf, 5-Araf, 2,3,5-Araf, 4-Manp, and 3-Glcp were more readily digested. Linkages within hemicellulose fraction (4 M KOH), t-Araf, t-Xylp, 2-Xylp, 4-Xylp, 2,4-Xylp, 3,4-Xylp, t-GalAp, 4-GalAp, 4-Manp, and 3-Glcp, were also freely digested. As expected, the final recalcitrant cellulosic fraction exhibited the lowest level of digestibility for most linkages in all fractions (Fig. 3A). Notably, while lignin was not removed from the whole cell wall polysaccharide samples, it cannot be detected by the MS-based linkage analysis.
Fig. 3.

Digestibility of barley straw plant cell wall linkages. (A) Glycans were sampled from barley straw (BS) and total tract indigestible residue (TTIR) plant whole cell wall (de-starched alcohol insoluble residue, AIR) and isolated non-starch polysaccharides fractions (EDTA + Na2CO3, 4 M KOH, and cellulosic-residue). The relative abundance (molar %) of glycosidic linkages was determined by gas chromatography – mass spectrometry/flame ionization detection (GC–MS/FID) of partially methylated acetylated alditol (PMAA) derivatives. Heat maps were generated based on the calculated ratio of linkage abundance in the BS control over the digested TTIR (black = indigestible, bright green = most digestible). (B) Bar graphs were generated for the top 10 indigestible linkages within each plant cell wall fraction, representing the ratio of the highest average abundance calculated in TTIR over that of BS control. Linkages with standard deviations of greater than 50% were excluded as they were considered unreliable. (C) Linkage composition data for BS and TTIR were assigned to classes of polysaccharides according to Pettolino, et al. [13]. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
In order to evaluate the recalcitrance of BS to digestion by intestinal microbial communities and identify rate-limiting enzymatic activities, the ten most abundant indigestible linkages were calculated as the change in abundance ratio for TTIR over BS, and ranked highest to lowest (Fig. 3B). In the alcohol insoluble residue (AIR) of the whole cell wall, t-Xylp was found to be highly abundant (1.6 ± 0.6), whereas 3,4-Xylp (5.4 ± 0.4), t-Manp (2.4 ± 0.7), and 2-Araf (2.6 ± 0.9) were the most indigestible linkages within the EDTA + Na2CO3, 4 M KOH, and cellulosic residue fractions, respectively.
Polysaccharide composition was estimated by assigning the linkage composition data to classes of polysaccharides according to the method developed by Pettolino et al. [13] (Fig. 3C, Table 1, Supplemental File 1B). Differences in polysaccharide composition were observed between BS and TTIR samples. TTIR contained more arabinan, heteroxylan, and xyloglucan than BS in the AIR and final residual cellulosic fractions. The EDTA + Na2CO3 fraction of TTIR showed higher heteroxylan and xyloglucan content. AG-2 content within the TTIR 4 M KOH fraction was observed to be higher as compared to BS.
Table 1.
Prediction of polysaccharide composition (%) based on glycosidic linkage data.
| Polysaccharides1 | AIR |
EDTA + Na2CO3 |
4 M KOH |
Residue |
||||
|---|---|---|---|---|---|---|---|---|
| Barley | TTIR | Barley | TTIR | Barley | TTIR | Barley | TTIR | |
| Arabinan (AB) | 1.9 | 2.1 | 6.3 | 5.3 | 2.1 | 2.5 | 0.2 | 0.4 |
| Heteroxylan (HX) | 30.8 | 33.8 | 17.1 | 32.4 | 79.6 | 76.2 | 4.7 | 7.9 |
| Type I arabinogalactan (AG-I) | 1.2 | 1.5 | 2.9 | 3.7 | 0.3 | 0.4 | t.r. | 0.1 |
| Type II arabinogalactan (AG-II) | 2.2 | 2.2 | 12.1 | 11.6 | 2.9 | 4.3 | 0.3 | 0.6 |
| Xyloglucan (XG) | 5.1 | 5.5 | 5.5 | 8.2 | 9.0 | 9.8 | 2.3 | 3.0 |
| Mixed linkage glucan (MLG) | 3.3 | 1.9 | 23.7 | 21.0 | 4.2 | 4.8 | 1.2 | 1.4 |
| Cellulose | 49.7 | 47.2 | n.a. | n.a. | n.a. | n.a. | 84.4 | 78.3 |
| Homogalacturonan (HG) | 0.2 | 0.3 | 7.0 | 2.4 | 0.4 | 0.3 | t.r. | t.r. |
| Heteromannan (HM) | 0.9 | 0.8 | 9.6 | 5.7 | 0.5 | 0.5 | 0.5 | 0.8 |
| Unassigned (UA) | 4.8 | 4.7 | 15.8 | 9.8 | 1.1 | 1.1 | 6.1 | 7.4 |
2.3. RNA-Seq output and de novo assembly
RNA-Seq and differential gene expression analysis was conducted to study the metabolically active members of rumen microbial communities in vitro and to compare microbial CAZyme expression profiles of rumen microbes cultured on TTIR or BS. Rumen microbial samples were cultured in vitro in triplicate for each substrate (TTIR and BS), as reported previously [14], and metatranscriptomic analyses were performed. A total of 364,583,860 sequence reads were used for quantitative RNA-Seq analysis. The number of reads per sample ranged from 56.95 million to 66.20 million. The transcriptome was assembled de novo using the Trinity assembler [15] and led to 1,998,343 distinct transcripts with a median transcript length of 336 bases and N50 of 533 bases (Supplemental File 2).
2.4. Microbial taxonomic classification based on putative mRNA
RNA-Seq raw reads were submitted as input to the Kaiju [16], [17] web server for taxonomical assignment at the whole transcriptome level. Bacteria represented the majority (88%) of rumen microbial transcripts (Table 2), Firmicutes and Bacteroidetes, were found to be the most dominant phyla. At the family level, Lachnospiraceae (9%) Ruminococcaceae (7%), Spirochaetaceae (5%), Prevotellaceae (4%), Clostridiaceae (4%), Fibrobacteraceae (2%) were most abundant. Transcripts associated with Eukaryotes accounted for 8% of the total transcripts; Ascomycota, Basidiomycota, Neocallimastigomycota, and Intramacronucleata were found to be the major phyla within these samples (Table 2).
Table 2.
Taxonomic analyses of rumen microbiota transcriptome.
| Domain | Phylum | Family |
|---|---|---|
| Bacteria (88%) | Firmicutes (41%) | Lachnospiraceae (9%) |
| Ruminococcaceae (7%) | ||
| Clostridiaceae (4%) | ||
| Unclassified Clostridiales (4%) | ||
| Paenibacillaceae (1%) | ||
| Bacteroidetes (17%) | Prevotellaceae (4%) | |
| Bacteroidaceae (2%) | ||
| Unclassified Bacteroidales (2%) | ||
| Rikenellaceae (1%) | ||
| Porphyromonadaceae (1%) | ||
| Flavobacteriaceae (1%) | ||
| Actinobacteria (9%) | Streptomycetaceae (3%) | |
| Fibrobacteres (2%) | Fibrobacteraceae (2%) | |
| Proteobacteria (12%) | Pseudomonadaceae (0.6%) | |
| Spriochaetae (5%) | Spirochaetaceae (5%) | |
| Lentisphaerae (0.9%) | Unclassified Lentisphaerae (0.9%) | |
| Eukaryota (8%) | Opisthokonta (64%) | Ascomycota (32%) |
| Neocallimastigomycota (6%) | ||
| Basidiomycota (18%) | ||
| Alveolata (9%) | Intramacronucleata (2%) |
Note: Taxonomical affiliations of whole transcriptomic data was obtained from taxonomic analyses of mRNA by Kaiju [16]. Relative abundance (%) at the domain level were calculated as a percentage of total mRNA transcripts, whereas relative abundance at the phyla and family level are calculated as a percentage of mRNA transcripts within the affiliated domain. Only those phyla and families with relative abundance higher than 0.6% are reported.
2.5. CAZyme expression under in vitro batch culture
To investigate the functional activities of these data, the assembled transcripts were parsed through BLASTx and the CAZy database (v2017-07–20) [18]. Transcripts annotated as CAZymes belonged to 169 different families: 126 glycoside hydrolases (GH), 27 polysaccharide lyases (PL), and 16 carbohydrate esterases (CE) (Supplemental File 3). The overall transcriptome contained all the major GH families putatively involved in cellulose and hemicellulose digestion. Substrates for individual GH, PL, and CE families were assigned according to Couger, et al. [19]. A total of 13 GH families possibly involved in cellulose degradation were identified, collectively accounting for ∼18% of total CAZymes (Table 3, Supplemental File 4). Major cellulose-targeting GH families included GH5, GH9, GH45, GH48, and GH124 endoglucanases; GH6 and GH48 cellobiohydrolases; and GH1 and GH3 β-glucosidases. Similarly, transcripts implicated in hemicellulose degradation included: GH10, GH11, GH43, GH5, GH26, and GH8; debranching enzymes belonging to GH2, GH3, GH43, GH51, and GH95; and acetyl xylan esterases of the CE1 family. Out of 39 hemicellulose-targeting GH families, only 12 had a relative abundance greater than 1% (accounting for ∼34% of CAZyme expression). Twelve GH, 6 PL, and 3 CE families putatively involved in pectin degradation were identified, including polygalacturonases (GH28), rhamnosidases (GH78), galactanases (GH53), pectin lyases (PL1) and pectin acetylesterases (CE12). In addition, transcripts encoding enzymes that degrade starch (GH13, GH31, and GH77 families) were also identified. Similarly, several CBM families were identified (Supplemental File 4) whereby ∼68% putatively bind to cellulose or hemicellulose, with starch and chitin-binding CBMs also observed.
Table 3.
Proportional expression of CAZy families involved in cell wall degradation.
| CAZy family | Relative Transcript Abundance |
|---|---|
| Cellulose | |
| GH3 | 7.0 |
| GH5 | 3.7 |
| GH9 | 1.9 |
| GH51 | 1.2 |
| GH94 | 1.2 |
| GH1 | 1.0 |
| GH8 | 0.4 |
| GH48 | 0.2 |
| GH45 | 0.2 |
| GH74 | 0.2 |
| GH6 | 0.1 |
| GH44 | 0.03 |
| GH124 | 0.02 |
| Hemicellulose | |
| GH3 | 7.0 |
| GH43 | 6.3 |
| GH2 | 5.5 |
| GH5 | 3.7 |
| CE1 | 2.6 |
| CE4 | 1.9 |
| GH16 | 1.5 |
| GH95 | 1.3 |
| GH10 | 1.3 |
| GH51 | 1.2 |
| GH36 | 1.2 |
| GH92 | 1.1 |
| Other GH (21) | 8.7 |
| Other CE (6) | 2.7 |
| Pectin | |
| GH28 | 1.7 |
| PL1 | 1.5 |
| GH78 | 1.4 |
| GH53 | 1.2 |
| CE12 | 0.9 |
| GH4 | 0.6 |
| GH4 | 0.6 |
| Other GH (8) | 2.9 |
| Other PL (5) | 0.8 |
| Other CE (2) | 0.6 |
| Starch | |
| GH13 | 7.9 |
| GH31 | 1.8 |
| GH77 | 0.9 |
| Other GH (8) | 2.7 |
Note: The relative abundance of transcripts is defined as the proportion of transcripts for a given family in the total of all CAZyme (GH, CE, and PL only) transcripts targeting each plant polysaccharide.
Transcripts predicted to be involved in the digestion of cellulose, hemicellulose, pectin and starch (GH + PL + CE) were taxonomically classified to gain insight into the predominant species involved in cell wall degradation. The majority of the cellulose-targeting transcripts were associated with the Firmicutes (42%; Ruminococcus, 9%, Lachnospiraceae, 5%), Bacteroidetes (23%; Prevotella, 7%, Bacteroides, 3%), and Fibrobacteres (4%; Fibrobacter 3%) (Fig. 4, Supplemental File 5). Fungi contributed ∼4% of transcripts that were predicted to target cellulose digestion (Fig. 4). Similarly, Firmicutes contributed ∼38% (Clostridium 6%, Ruminococcus 5%) and Bacteroidetes 26% (Prevotella 7%) to the pool of transcripts targeting hemicellulose (Fig. 4, Supplemental File 5). Likewise, transcripts for families putatively involved in dismantling pectin were extensively assigned to bacteria and primarily to Firmicutes (39%; Ruminococcus, 5%) and Bacteroidetes (24%; Prevotella, 6%). In contrast, starch-degrading transcripts mostly originated from Bacteroidetes (34%; with Prevotella contributing 11% of total starch digesting transcripts).
Fig. 4.

Taxonomic classification of transcripts involved in the digestion of key plant cell wall components. Taxonomic affiliation and relative abundance of transcripts putatively known to be involved in the digestion of cellulose, hemicellulose, pectin, and starch as assigned by Kaiju [16]. Relative abundance was defined as the proportion of transcripts from a given subphyla of the total CAZyme (GH, CE, and PL only) transcripts targeting individual plant polysaccharided.
2.6. Gene expression at CAZyme family level
Metatranscriptomic data was used to evaluate the contribution of specific CAZyme families to cell wall degradation. Thus, for those CAZyme family transcripts known to mediate the deconstruction of major cell wall polysaccharides, total expression levels of transcripts for the individual CAZyme family were calculated as a percentage of total expression levels for all CAZyme transcripts (Supplemental File 6). Endo-glucanase and β-glucosidase (GH5, GH8, and GH9) transcripts were higher in TTIR incubations (Supplemental File 6A), while BS incubations had a higher abundance of cellobiohydrolase (GH6, GH48) transcripts (Supplemental File 6B). Interestingly, both GH6 and GH48 families contributed towards the cellobiohydrolase pool for BS degradation, whereas GH48 was the only major family of cellobiohydrolases associated with TTIR. Similar transcript abundance was seen for xylan degradation families (GH10, GH11, GH43, and GH39) for TTIR and BS (Supplemental File 6D). Putative pectin-degrading GH families showed higher expression on BS with GH78 as the major CAZyme family, whereas GH28 and GH53 were the major pectin-associated GH families expressed with TTIR (Supplemental File 6F).
2.7. Differential gene expression at individual transcript level
Identifying transcripts that catalyze the cleavage of carbohydrate linkages that limit ruminal digestion could provide insight into improving the digestibility of BS. Therefore, we focused on differentially expressed transcripts (fold change > 1.5, FDR < 0.05, and p < 0.05 based on DEseq analysis) detected in cultures cultivated with either BS or TTIR. A total of 1,912 differentially expressed transcripts (1,245 BS-overexpressed and 667 TTIR-overexpressed, Supplemental File 7A) were identified and further analyzed for taxonomic affiliations. The most abundant TTIR-overexpressed transcripts (8.5% of total) were associated with Prevotella (Prevotella sp. tf2-5, Prevotella sp. tc2-28) and Ruminococcus particularly Ruminococcus flavefaciens (8%) (Fig. 5). While transcripts of fungal origin, such as Neocallimastix californiae (5.3%), Piromyces finnis (3.8%) and Aneromyces robustus (2.8%), also contributed considerably towards the pool of BS-overexpressed transcripts. A high proportion of BS-overexpressed transcripts was found to be associated with the methanogen Methanomicrobium mobile (4.4%). Around 8% of upregulated BS transcripts were associated with Lachnospiraceae G11. A high proportion of differentially expressed transcripts were unclassified in both samples (Fig. 5).
Fig. 5.

Taxonomic affiliation of differentially expressed transcripts. Differentially expressed (DE) transcripts from BS (red) and TTIR (blue) representing >1% of the total DE pool were included and taxonomically assigned using Kaiju [16]. Transcripts with a fold change of 1.5 or more, with an FDR < 0.05 and p < 0.05 as calculated by DEseq were considered DE. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Differential expression analyses of transcripts classified as CAZymes (via BLAST against the CAZy database) identified a total of 96 GH genes that were upregulated when rumen microbiota were cultured with TTIR, while in the presence of BS, 156 GH transcripts were upregulated (Table 4, Supplemental File 7A). In TTIR samples, 8 PL and 12 CE transcripts were upregulated, whereas 6 PL transcripts and 32 CE transcripts were upregulated with BS. To increase the resolution of functional annotation, the nucleotide sequences for differentially expressed transcripts were submitted to the recent version of the dbCAN2 server [20] and manually curated for CAZyme annotation (Supplemental File 7B).
Table 4.
Total number of differentially expressed CAZyme transcripts on BS and TTIR substrates.
| CAZy Class | Number of transcripts |
|
|---|---|---|
| BS | TTIR | |
| Glycoside hydrolase (GH) | 156 | 96 |
| Polysaccharide lyase (PL) | 6 | 8 |
| Carbohydrate esterase (CE) | 32 | 12 |
| Carbohydrate binding module (CBM) | 74 | 38 |
Note: Transcripts with a fold change of 1.5 or more, with an FDR < 0.05, and p < 0.05 as calculated by DEseq were considered differentially expressed.
Due to the high variation in expression levels across replicate batch cultures, no statistical difference between BS and TTIR culture was noted at the CAZyme family level for the total collection of differentially expressed transcripts within a given CAZy family, with the sole exception of CBM4 being significantly upregulated in TTIR and CBM36 in BS (Supplemental File 8). CBM4s may represent a useful family for CAZyme ‘fishing’ as they are most commonly associated with xylanases [21], β1,3-glucanases [22], or β1,4-glucanases [23]. To distinguish dominant individual differentially expressed transcripts, the transcriptional expression pattern between TTIR and BS cultures was studied for CAZy families of interest known to target XG, HX, and pectin (Fig. 6). Transcripts TR363754|c1_g3 (GH11), TR744494|c3_g1 (GH16), and TR463944|c0_g2 (CE15) accounted for a large proportion of the TTIR overexpressed transcripts. Whereas other transcripts, such as TR400632|c0_g1 (GH11), TR849235|c0_g1 (GH11), and TR300921|c2_g2 (GH43), were observed to be upregulated in BS cultures as compared to TTIR cultures.
Fig. 6.

Relative individual differentially expressed transcript expression levels for CAZy families of interest. The contribution levels of differentially expressed individual transcripts for GH, CE, and PL families of interest was calculated as the abundance (%) of the individual transcript relative to the expression levels of transcripts observed for those GH families listed, for each substrate (BS or TTIR), and a heat map was generated. Statistical significance was calculated by two-way ANOVA and indicated on the plot (**** p < 0.001).
2.8. Prediction of enzyme activities for rumen differentially expressed CAZyme transcripts
Heteroxylan and arabinan were identified as recalcitrant polysaccharides by glycome and linkage analyses, and therefore, differentially expressed transcripts from GH11, GH43, and CE15 (i.e., relevant polyspecific families targeting various glycosidic linkages within a given CAZy family) were selected for high-resolution functional prediction. For transcripts that did not encode the full-length enzyme, sequences were determined by comparison to the closest characterized gene using SACCHARIS [24]. Phylogenetic trees generated for biochemically characterized members of GH11 (Fig. 7A) indicated that TTIR overexpressed transcripts (TR363754|c1_g3, TR668965|c0_g1, TR196282|c0_g1, TR190685|c0_g1, and TR83509|c0_g1) partitioned into distantly related clades, as compared to those overexpressed in BS (TR476599|c0_g1, TR849235|c0_g1, TR327227|c0_g1, TR291133|c0_g1, TR400632|c0_g1, and TR1014827|c1_g1). All GH11 transcripts were predicted to function as endo-β-1,4-xylanases; the dominant enzymatic activity of this family. For GH43s (Fig. 7B), TTIR-overexpressed transcript TR712918|c0_g1 was classified as an endo-α-1,5-L-arabinanase, while BS-overexpressed transcripts TR400300|c0_g2 and TR451123|c0_g1 were predicted to be putative arabinofuranosidases. Both TR941931|c2_g1 and TR300921|c2_g2 were overexpressed in BS and were predicted to be endo-α-1,5-L-arabinanases. All three TTIR-overexpressed CE15 transcripts were classified as acetyl xylan esterases (Fig. 7C). Notably, a number of differentially expressed transcripts encoded for dockerin domains and CBMs. Additionally, transcripts in CAZy families GH5 and GH16 were also analyzed by SACCHARIS, based on the presence of XG, MLG, and AG-I found within BS and TTIR (Supplemental File 10). The transcript TR744494|c3_g1 was found to be highly homologous to a xylanase from GH16 subfamily 21.
Fig. 7.

Prediction of enzyme activities by CAZyme fingerprinting. Translated protein sequences annotated by dbCAN2 [20] were used as query sequence inputs for SACCHARIS [24] and embedded into phylogenetic trees of characterized (A) GH11, (B) GH43, and (C) CE15 enzymes. Transcript sequences are highlighted with a coloured circle to represent whether they were highly expressed in in vitro with BS (red) or TTIR (blue) as the substrate. EC number and CAZy database annotated functions are colour-coded as indicated. CAZy subfamilies for GH43 are indicated as GH43_#. Domain boundaries are based on predictions by dbCAN [20] and InterProScan [73], with all schematics to scale and colour-coded for signal peptide (dark red), dockerin (teal), glycoside hydrolase (GH) (dark blue), carbohydrate esterase (CE) (light blue), carbohydrate binding module (CBM) (grey), or CBM-like (white). The polypeptide length for the open reading frame of each transcript is shown. Schematics of typical plant cell wall substrates, as identified by EC number, are shown with the targeted bond highlighted. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
2.9. Characterization of rumen differentially expressed xylanases
Two GH11 sequences, TTIR-induced WP_049962670.1 (the closest related sequence of TR363754|c1_g3) and BS-induced TR400632|c0_g1, were recombinantly expressed and purified, and characterized using heteroxylan and arabinoxylan substrates (Fig. 8A). Both enzymes demonstrated an endo-xylanase activity when analyzed by thin layer chromatography (Fig. 8B) and high-performance anion-exchange chromatography coupled with pulsed amperometric detection (HPAEC-PAD) (Fig. 8C). The product profiles suggested release of xylotriose (X3) by WP_049962670.1 from beechwood xylan whereas, TR40632|c0_g1 liberated xylose from beechwood xylan, and xylose and longer xylooligosaccharides (X4, X5, and X6) in higher abundance from oat spelt xylan (Fig. 8C). HPAEC-PAD traces lacked evidence of a xylohexaose (X6) product in the beechwood xylan digest by TR400632|c0_g1. Together, these HPAEC-PAD analyses suggest there are subtle differences in the specificities of these two enzymes. A large peak (∼22.5′) was also noticed for both enzymes on oat spelt and beechwood xylan, suggesting that these enzymes might accommodate or prefer branched substrates, such as those found in arabinoxylan. Therefore, WP_049962670.1 and TR400632|c0_g1 were used to digest rye and wheat arabinoxylan. Major products from these reactions eluted in proximity to 32-α-L-arabinofuranosyl-xylobiose (A3X) and 23,33-di-α-L-arabinofuranosyl-xylotriose (A2,3XX) standards, in addition generating small xylooligosaccharides (Fig. 8C, Supplemental File 11). No product formation was observed for digestion with BS, however xylobiose (X2) product was generated when both enzymes were incubated with TTIR (Supplemental File 11). The insoluble and complex nature of these samples may require a more comprehensive mixture of enzymes to expose xylan backbones for efficient hydrolysis by GH11 endo-xylanases.
Fig. 8.

Digestion of xylan and arabinoxylan substrates by differentially expressed CAZymes from barley straw and total tract indigestible residue rumen cultures. (A) Substrates used to detect GH11 activity: oat spelt and beechwood heteroxylan, and rye and wheat arabinoxylan. (B) Thin layer chromatography of GH11 substrates without (-) and with (+) GH11 enzymes, as indicated. (C) Soluble products of enzymatic digestion were analyzed by HPAEC-PAD using a gradient of 10–120 mM sodium acetate in a constant background of 30 mM NaOH. Xylan mono- and oligosaccharide standards were included as controls: xylose (X), xylobiose (X2), xylotriose (X3), xylotetraose (X4), xylopentaose (X5), and xylohexaose (X6), and/or a xylose ladder of X1-X6 forms. Arabinoxylan oligosaccharide standards are as follows: 32-α-L-arabinofuranosyl-xylobiose (A3X), 23-α-L-arabinofuranosyl-xylobiose (A2XX), 23,33-di-α-L-arabinofuranosyl-xylotriose (A2,3XX).
3. Discussion
Glycome profiling provided unprecedented insight into the architecture of BS and its residual fraction that resists digestion as it passes through the digestive tract of cattle. This approach detected diverse structural features present within plant cell wall fractions and polysaccharide classes (Fig. 2). For example, a comprehensive suite of more than 25 mAbs recognized different substructures within xyloglucan, including each of the major side-chains. Another set of approximately 30 mAbs enabled diverse xylan epitopes including Ara, GlcA and MeGlcA substitutions as well as various lengths of backbone residues [25] to be characterized (Fig. 2). This allowed us to identify recalcitrant polymers with high precision and to differentiate polysaccharides that were removed during digestion from those that were enriched in TTIR. For instance, while degradation of lignin by ruminal microorganisms is severely limited due to anaerobic conditions and short retention times [26], 4-O-MeGlcA xylan epitopes conjugated to lignin, as well as xylans loosely associated with the cell wall in BS, were removed during passage through the digestive tract. This suggests that lignin-associated heteroxylan moieties are not among those recalcitrant groups in BS (Fig. 2). Alternatively, 2-Ara-substituted xylans, as well as xylan epitopes recognized by the Xylan-2 and Xylan-3 groups of antibodies, were enriched in TTIR, suggesting that these epitopes directly contribute to recalcitrance (Fig. 2 boxes 5a and 6a).
The glycan composition of TTIR was not fully described by glycan profiling. For example, there were a significant fraction of glycans that were loosely associated with TTIR (AO fraction), yet with the exception of de-esterified homogalacturonan epitopes (DP > 4), there was limited antibody binding to this fraction. The lack of antibody binding is likely attributable to the fact that short-chain oligosaccharides released during digestion fail to bind to ELISA plates [10]. In addition, while the antibody collection used here recognizes a large and diverse set of plant cell wall glycan epitopes, there may be other epitopes native to BS or generated during passage through the ruminant digestive tract that are not recognized by the current antibody library. Therefore, to confirm glycome profiling results we quantified individual glycosidic linkages using partially methylated acetyl alditol derivatization, enabling most glycan linkages to be measured. The linkage data showed that TTIR was enriched in heteroxylan, xyloglucan, and arabinan when compared with BS (Fig. 3, Table 1), suggesting that these components were more recalcitrant than other components. Fractionation of the walls showed that the EDTA+ carbonate extract was enriched in heteroxylan and xyloglucan (Fig. 3, Table 1), which was in contrast to the results from glycome profiling, where the equivalent extract (ammonium oxalate) only showed enhanced homogalacturonan content (Fig. 2). This may reflect the composition of the large fraction of cell wall glycans that were loosely associated with TTIR, but not detected in the ELISAs glycome profiling. A consistent abundance of heteroxylan and xyloglucan within the whole cell wall and the EDTA/carbonate and KOH extracts of TTIR suggest that these cell wall components contribute to the recalcitrance of BS within the ruminant digestive tract. Apart from cell wall composition, linkage analysis also provided meaningful insight into the predominant bonds within identified recalcitrant polysaccharides. ELISA-based glycome profiling and glycosidic linkage analysis are two methods used extensively to study the structure of plant cell walls [9], [10], [27], however these methods have not be used previously to study fibre utilization and recalcitrance after passage through the ruminant digestive tract. The results presented in this study could prove to be a valuable resource for crop breeders to develop feedstocks that exhibits increased digestibility in ruminants.
Informed by glycomics and metatranscriptomics, differential expression analyses provided additional complementary approaches to identify candidate linkages to target for augmenting plant cell wall digestion. Incubation with TTIR provided a unique experimental approach that enabled the rumen microbial digestion of recalcitrant plant cell wall to be studied. The residual nutritional value and effective fermentation of TTIR was confirmed by total gas production using BS and TTIR as substrates for in vitro cultures (Supplemental File 9). These findings confirm that TTIR is a product of transit time (host physiology), fibre properties, and chemical and microbial digestive processes. Firmicutes (Lachnospiraceae, Ruminococcaceae, Clostridiaceae), Bacteroidetes (Prevotellaceae, Bacteroidaceae and unclassified Bacteroidiales), Actinobacteria, Fibrobacteres, Proteobacteria and Spirochaetae were found to be dominant bacterial phyla contributing to the transcriptome (Table 2; Fig. 5). These results are in agreement with previously reported metatranscriptomic analysis, such as Dai et al. [28]. However, there was variation in the abundance of taxonomic families as compared to previous literature [28], [29]. This variation can be attributed to the shorter incubation time (48 h), and absence of host effects in closed ex vivo batch culture systems as compared to in vivo animal-based studies. Shorter incubation times favor rapidly growing bacteria over slower growing fungi and protozoa within ruminal microbial communities. In contrast to the host environment where fermentation by-products like VFA and ammonia are absorbed across the intestinal epithelium, these products accumulate in closed batch culture systems, which can bias the fermentation towards certain types of microbial communities. Furthermore, host genetics have also been reported to influence the rumen microbial population [30], as a region on chromosome 6 in cattle has recently been shown to be associated with Actinobacteria, Euryarchaeota and Fibrobacteres densities [31]. Whole animal experiments cannot specifically be used to study the digestion of TTIR without establishing its nutritional value and direct utilization by rumen microbes, whereas in vitro batch cultures provided the control over experimental variables needed to study its digestion. Moreover, the metabolic activity and effective fermentation of recalcitrant TTIR by rumen microbiota was demonstrated in batch cultures (Supplemental File 9), validating the use of this method for unbiased enzyme discovery in the absence of host control and variability.
Gene expression analyses of rumen microbial communities cultured on BS and TTIR demonstrated substrate-specific modes of digestion, as evidenced by the expression levels of CAZy family transcripts. At the family level, higher expression of putative endo-glucanases GH5, GH8, and GH9 on TTIR indicated an abundance of metabolizable cellulose within TTIR, as confirmed in the linkage analyses. Furthermore, GH48 cellobiohydrolases were the dominant exo-glucanases expressed during the digestion of TTIR by rumen microbes. In contrast, GH6 and GH48 were the predominant families associated with the microbial digestion of BS (Supplemental File 6B). Both GH6 and GH48 are known to efficiently hydrolyze the crystalline regions of cellulose [28], [32]; cellobiohydrolases within the GH48 family act on the the reducing ends of cellulose chains, while GH6 carbohydrases act on non-reducing ends, generating cellobiose [33]. The high expression levels of these exo-glucanases suggest there is a greater abundance of amorphous cellulose at the reducing ends of micro-fibrils in TTIR as compared to BS. The level of expression of GH43 hemicellulases in both TTIR and BS was higher than other hemicellulose-targeting activities (Supplemental File 6E). Among the 501 bacteria in the Hungate catalogue, the GH43 family was reported as the most abundant hemicellulase [34], and both metagenomic [35], [36], [37] and metatranscriptomic [28], [29], [38], [39], [40] studies have suggested that members of the GH43 family are the principal hemicellulases within the rumen. The GH43 family encompasses a range of debranching enzymes, including arabinofuranosidases, xylanases, galactanases, arabinanases and β-xylosidases, which aid in the degradation of the arabinoxylan [41]. Glycomics revealed that in TTIR, arabinoxylan was enriched, highlighting its resistance to ruminal digestion. Additionally, the abundance of linkages consistent with branched polysaccharides like 3,4-Xylp, t-GlcAp, 2,3,4-Xylp, t-Araf in TTIR also confirms the high resistance of heteroxylan to degradation by intestinal microbiota (Fig. 3). Metatranscriptomic analyses reflected the efforts of the rumen microbiota to degrade this polysaccharide, with the GH10 family being the dominant family of xylanases (Supplemental File 6D). This family has been previously reported to digest heteroxylan and release xylotriose with a MeGlcA moiety at the non-reducing end, which in turn is hydrolyzed by members of the GH5 family of xylanases [42]. The GH5 xylanases are active against glucuronoxylan and MeGlcA substituents on the xylan backbone [42]. Pectin polysaccharides differed between BS and TTIR (Fig. 2, Fig. 3). Accordingly, expression levels of pectin-targeting CAZy families differed between rumen microbiota incubated with TTIR vs BS (Supplemental File 6F). Rumen microbial communities cultured on BS expressed more GH78 transcripts; a family that contains α-l-rhamnosidases that act on rhamnogalacturonan (RG-I) and arabinogalactan-protein (AGP) linkages [43]. In contrast, with TTIR there was a higher expression of GH28 transcripts, a family that encodes polygalacturonases and rhamnogalacturonases that hydrolyse the α-1,4 galacturonsyl linkages in homogalacturonan and RG-I respectively, and in GH53, which encode for β-1,4-galactanases [44].
Taxonomic affiliations of differentially expressed transcripts suggested distinct origins for upregulated transcripts between BS and TTIR cultures (Fig. 5). The abundance of fungal transcripts within BS-overexpressed transcripts (Fig. 5) suggested Neocallimastix californiae, Piromyces finnis and Aneromyces robustus to be among the most metabolically active taxa, possibly suggesting synergistic interactions between fungal and bacterial species during the fermentation of BS. Within TTIR-overexpressed transcripts, Ruminococcus flavefaciens and Prevotella spp. (Prevotella sp. tf2-5, Prevoltella sp. tc2-28) were dominant taxa, and both are well-poised to efficiently degrade and utilize polysaccharides within TTIR. Ruminococcus flavefaciens is known to produce an efficient cellulosome system, while genomic studies of Prevotella sp. have reported complete polysaccharide utilization loci (PULs) that contribute to hemicellulose digestion [45]. Indeed, several GH and CBM families putatively involved in cellulose, hemicellulose, and pectin digestion were identified based on differential expressions (Supplemental File 8).
Based upon the glycomic analysis, differentially expressed transcripts were selected for candidate CAZyme families with a focus on activities important for digestion of plant cell wall polysaccharides (Fig. 6). No significant difference was seen between BS and TTIR in the relative contribution of GH5 and GH9 family transcripts (Fig. 6), suggesting that rumen microbes may lack specialized xyloglucanases for the utilization of XG, despite its abundance within TTIR. Consequently, a lack of xyloglucanases may limit the extent that BS is digested within the ruminant digestive tract, a constraint that may be overcome by adding exogenous xyloglucanases to BS diets.
Higher resolution predictive analysis based upon phylogenetic relationships of CAZymes was conducted in an attempt to postulate enzyme function and specificity for differentially expressed transcripts. We focused on the representative families; GH5, GH11, GH16, GH43, and CE15, which are known to be important for xyloglucan, heteroxylan, and arabinan saccharification (Fig. 7, Supplemental File 10). Although no target xyloglucanases were identified in rumen batch cultures, substrate-specific differential gene expression of CAZymes involved in heteroxylan and arabinan digestion was evident from phylogenetic trees of GH11, GH43 and CE15. This suggested that these transcripts may represent specialized xylanases and arabinanases induced within the rumen microbial community for the digestion of TTIR. In addition, a number of differentially expressed transcripts encoded dockerin domains and CBMs, highlighting that some of these CAZymes participate within highly evolved multimeric protein complexes that metabolize complex plant cell wall carbohydrates.
GH11 transcripts that were overexpressed in TTIR cultures clustered distantly from BS-overexpressed GH11 transcripts. Indeed, some of the early diverging sequences may be more aptly described as GH11-like based upon their relatedness to characterized GH11s (Fig. 7A). The partitioning of TTIR-overexpressed transcripts also suggests that changes in expression levels reflects changes in the predominant bacterial species that utilize heteroxylan in TTIR vs BS. The TTIR-overexpressed GH11 transcript TR83509|c0_g1 was observed to contain a unique arrangement with four tandem GH11-like domains (Fig. 7A), which we believe represents the first tetramodular xylanase architecture described. The phylogenetic analyses of the overexpressed GH43 transcripts revealed that both TTIR (TR712918|c0_g1) and BS (TR941931|c2_g1 and TR300921|c2_g2) clustered together with endo-α-1,5-l-arabinanases (Fig. 7B), possibly suggesting differential modes of arabinanase regulation in rumen microbiota cultured on BS vs TTIR. The recalcitrance of arabinan to microbial digestion was also reflected by linkage analysis and glycome profiling (Fig. 2, Fig. 3; Table 1). Transcripts TR400300|c0_g2 and TR451123|c0_g1, were overexpressed in incubations with BS and clustered with arabinofuranosidases, reflective of the cross-linked nature of heteroxylan in BS (Fig. 7B). The three overexpressed CE15 transcripts with TTIR partitioned with acetyl xylan esterases, activities also important for the degradation of heteroxylan.
Metatranscriptomic studies to date have been conducted mostly at the level of CAZy family, with a limited number of CAZy families delineated into subfamilies. This analysis often provides limited or no information about substrate specificity [28], [9], [38], [39], [40]. In this study, SACCHARIS analyses of differentially expressed transcripts and biochemical characterization of overexpressed recombinant xylanases in combination with glycome profiling and glycosidic linkage analyses, suggested inducible, substrate-specific CAZyme transcript expression. Further characterization of the putative activities of transcripts over expressed with TTIR, based on their homology with biochemically characterized CAZymes could provide insight into improving the digestion of this recalcitrant residue. Building on in silico predictions, in vitro enzyme characterization of statistically significant transcripts showed that the expression profiles of the rumen microbiota were consistent with the metabolism of these linkages within BS and TTIR (Fig. 8, Supplemental File 11). As RNA-Seq often results in truncated sequences, it is possible that many of the transcript polypeptides may contain auxiliary domains, such as CBMs, and dockerin and cohesin modules that potentiate the activity of CAZymes on complex substrates.
The BS-overexpressed transcripts, and their closely clustered CAZymes, represent important enzyme activities for effective BS digestion and may hold high intellectual value for application in industrial sectors, such as bioenergy and green chemistry. Previous studies have demonstrated that supplementing arabinofuranosidases and acetyl xylan esterases to a mixture of rumen enzymes enhanced the saccharification efficiency of plant cell wall carbohydrates [6], [14]. Thus, candidate CAZymes of rumen origin stand a better chance of success as enzyme additives as they are optimized for the complex physiological conditions within the rumen. Furthermore, the high transcriptional activity of Prevotella sp. and Ruminococcus flavefaciens within TTIR batch cultures highlights their potential as probiotics for the digestion of recalcitrant BS residues.
Conclusion: Analysis of cell wall glycan composition, glycosidic linkages, and the relative extractability of polysaccharides within TTIR and BS provided unprecedented insight into nutrient utilization by rumen microbes and helped to delineate the role of matrix polysaccharides in cell wall recalcitrance. Adopting TTIR as a substrate for rumen batch cultures and the application of next-generation sequencing to study gene expression by the rumen microbial community identified a number of CAZyme genes coding for enzymes with potential to enhance the hydrolysis of recalcitrant plant cell wall residues. Furthermore, incubation of rumen microbial communities on TTIR was proven to be a powerful approach to enzyme discovery. In this regard, characterization of the small oligosaccharide fraction associated with TTIR may allow targeted searches of other organisms for hydrolytic enzymes capable of digesting these oligosaccharides. Taken together, the results presented suggest that the cell wall layered structure hinders enzymatic accessibility to recalcitrant cell wall components like heteroxylan and xyloglucan. Considering the rumen is one of the most efficient biomass digestive systems known, these findings hold high value for advances in bioproduct and bioprocess development using lignocellulosic feedstocks. The integration of high-resolution glycomics with targeted metatranscriptomics analyses is an innovative experimental platform that can be extended to other feed-host systems and holds promise for defining rate-limiting reactions in the microbial ecosystems of digestive organs.
4. Material and methods
4.1. Glycome profiling of BS and total tract indigestible residue
Alcohol insoluble residue (AIR) from BS and TTIR was prepared according to Wood et al. [46]. Briefly, AIR samples were treated with 70% alcohol, chloroform–methanol (1:1 v/v), and acetone to remove contaminants like attached biomass, lipids, solubles, or any contaminant DNA, RNA or protein. AIR samples were de-starched by incubation with porcine pancreatic α-amylase in 10 mM Tris–maleate buffer (pH 6.9) following Pettolino et al. [13], then sequentially extracted using 50 mM AO (pH ∼ 5.0), 50 mM SC (pH ∼ 10.0) [with 0.5% (w/v) NaBH4], 1 M KOH [with 1% (w/v) NaBH4], 4 M KOH [with 1% (w/v) NaBH4], 207 mM SC solution (acidified by the addition of 150 μL glacial acetic acid), and 4 M KOH [with 1% (w/v) NaBH4] resulting in five different cell wall fractions designated AO, SC, 1 M KOH, 4 M KOH, CH, and PC, respectively [47]. These plant cell wall extracts were probed with a collection of 154 cell wall glycan-directed mAbs [48] using an ELISA, and binding intensities (measured as secondary antibody-conjugated peroxidase activity) from this experiment presented as a heat map as described previously [47].
4.2. Linkage analysis of BS and total tract indigestible residue (TTIR)
AIRs were prepared as above and further fractionated based on the literature [49], [50], [51], [52] with modifications. Briefly, the cell walls (∼55 mg) were treated with 10 mL of 0.25 M sodium borodeuteride (NaBD4, 99 atom % D, Alfa Aesar) deionized water solution at 4 °C for 24 h, followed by quenching the excess reductant by dropwise addition of 10% (v/v) acetic acid and adjusting pH to neutral. The suspensions were then centrifuged (3000 × g, 0.5 h), and resulting supernatants were pooled with three deionized water washes of the pellets (with centrifugation between each wash), and a pooled solution (Solution 1) was stored at 4 °C. The pellets were then subjected to sequential extraction of polysaccharides using 15 mL of each of the following solutions: 50 mM EDTA (pH 6.5), 50 mM Na2CO3 (containing 25 mM NaBD4), and 4 M KOH (containing 25 mM NaBD4) over 24 h at room temperature with gentle magnetic stirring. After extraction, soluble fractions were separated from residues by centrifugation (3000 × g, 0.5 h), followed by neutralizing the supernatants, washing the residues three times with deionized water (with centrifugation between washes), and pooling all washes into corresponding fractions. The EDTA and Na2CO3 extracts and the aforementioned Solution 1 were pooled (designated FEDTA+Na2CO3). The 4 M KOH fraction and the final residue left after strong alkaline extraction were designated F4MKOH and FResidue, respectively. All fractions were dialyzed against deionized water and lyophilized. For linkage analysis of the whole cell walls and the isolated fractions, uronic acids in the samples (∼2 mg) were first converted to their 6,6-dideuterio neutral sugars using carbodiimide activation at pH 4.75 followed by NaBD4 reduction at pH 7.0. Samples were then dialysized against deionized water, and freeze-dried. The carboxyl-reduced samples were converted to their partially methylated alditol acetate (PMAA) derivatives by permethylation with iodomethane and sodium hydroxide in dimethyl sulfoxide [53], [54] with 2 M trifluoroacetic acid at 121 °C for 1.5 h, reduction with NaBD4, and peracetylation with acetic anhydride [13]. Deutero-methylation using iodomethane-d3 (≥99.5 atom % D, Sigma) was applied to the whole cell wall and F4MKOH fractions in order to identify and quantify the linkages from endogenously O-methylated sugars (e.g., 4-O-methyl glucuronic acids) [55], [56], [57]. The PMAAs were then subjected to gas chromatography-mass spectrometry (GC–MS) and gas chromatography-flame ionization detection (GC-FID) analyses on an Agilent 7890A-5977B GC–MS/FID system (Agilent Technologies, Santa Clara, CA). The PMAAs were separated using a medium polarity Supelco SP-2380 capillary column (30 m × 0.25 mm × 0.20 μm, Sigma-Aldrich) with a constant column outlet helium flow rate of 0.8 mL/min. Sample solutions were injected at an inlet temperature of 250 °C with a split ratio of 10:1. The oven temperature was programmed to start at 120 °C (hold 4 min) followed by increasing at 8 °C/min to 175 °C, 0.5 °C/min to 183 °C (hold 8 min), 0.5 °C/min to 195 °C, 4 °C/min to 210 °C, and 20 °C/min to 255 °C (hold 8 min). The transfer line temperature was kept at 280 °C. The mass spectrometer was operated in electron ionization (EI), full-scan mode (ionization energy, 70 eV; source temperature, 230 °C; quad temperature, 150 °C). The FID detector was operated at 300 °C (H2 flow, 30 mL/min; air flow, 400 mL/min; N2 makeup flow, 25 mL/min). The PMAAs were then identified by comparing their MS fragmentation patterns with those of reference derivatives and the literature [58], and quantified based on the previously reported FID response factors calculated using the effective carbon number concept [59]. Glycosidic linkages were assigned to relevant cell wall polysaccharides, and the relative compositions of each type of polysaccharides were then estimated by summing up corresponding linkage compositions [13], [60]. Six separate experiments were conducted for the whole cell walls and the F4MKOH fractions, of which half were used for deuterio-methylation analyses and the other half for methylation analyses. The FResidue fractions were subjected to six separate experiments (methylation analysis only). Three separate methylation analyses were conducted for the FEDTA+Na2CO3 fractions.
4.3. Preparation of TTIR
All animal procedures and protocols used in this study were reviewed and approved by the Lethbridge Research and Development Center Animal Care Committee (ACC number 1501) in accordance with the guidelines of Canadian Council on Animal Care (CCAC, 2009). An overview of the experimental setup is graphically represented in Fig. 1.
In vitro batch culture was used to culture rumen microbes on BS and TTIR as substrates, as described previously [14]. TTIR was obtained from faecal samples collected from Angus × Hereford cross beef heifers (six) fed BS-based diets containing 70:30 forage-to-concentrate on a dry matter (DM) basis [61]. A high proportion of barley straw was used in the feed to ensure sufficient indigestible material was generated for downstream analyses. Collected faecal samples were pooled, extensively washed (6–7 times) with 50 mM citrate buffer (3 L) and treated with 70% alcohol and acetone to remove soluble particles as discussed above. Given the high digestibility of the concentrate, it was assumed that the majority of TTIR originated from BS. The fecal residue obtained was dried for 72 h in an oven at 40 °C and defined as TTIR for use in batch culture experiments. BS was dried at 40 °C and ground to pass through a 1 mm screen.
4.4. Batch culture experiments
In vitro batch cultures were set up in 3 replicate serum vials. BS and TTIR were weighed (0.7 g/bag) into acetone-washed and pre-weighted filter bags (F57 ANKOM bag, Ankom Technology Corp., Macedon, NY) that were then heat sealed. Individual bags were then placed into 125 mL serum vials. Rumen fluid from 4 to 5 different sites within rumen was collected from four ruminally cannulated Angus × Hereford heifers fed 50% grass hay, 30% BS, 15% corn dried distillers grains plus solubles and 5% mineral/vitamin supplement (DM basis) [14]. Grass hay, BS and distillers grains were included in the diets as fibre sources in an effort to increase the potential diversity of microorganisms involved in plant cell wall digestion. Collected rumen fluid was strained through cheesecloth and equal volumes from each cow were combined and anaerobically maintained under a stream of O2-free CO2. The inoculum was prepared by mixing rumen fluid 1:4 with mineral buffer [14]. Inoculum (65 mL) was transferred to each vial under a stream of O2-free CO2. Vials were sealed with rubber stoppers and placed in a rotary shaker (120 rpm) in an incubator at 39 °C. Gas pressure in each vial was measured at 3, 6, 12, 24 and 48 h of incubation by inserting a 23 gauge (0.6 mm) needle attached to a pressure transducer (model PX4200-015GI, Omega Engineering, Inc., Laval, QC, Canada) and used to estimate gas production according to Romero-Pérez and Beauchemin (2018) [62]: Gas volume = 4.7047 × (gas pressure) + 0.0512 × (gas pressure2).
4.5. RNA extraction and sequencing
Total RNA from the solid bound ruminal microbes were extracted as described previously [63]. Briefly, the solid contents within nylon bags from in to vitro batch cultures were each recovered upon completion of 48 h of incubation and processed individually without pooling replicates together. The recovered solid content was manually ground to a fine powder using a mortar and pestle for 5 min in liquid nitrogen. Ground samples (∼200 mg) were placed in 2 mL microfuge tubes, 1.5 mL of TRIzol reagent was added and incubated at room temperature for 5 min, and the RNA was extracted using the acid guanidinium-phenol–chloroform (AGPC) method [64]. Total RNA was further purified with a MEGAclear kit according to manufacturer instructions (Applied Biosystems/Ambion). The RNA concentration and integrity were estimated using an Agilent 2100 bioanalyzer (Agilent Technologies, Mississauga, Ontario, Canada) and RNA 6000 Nano kit (Agilent Technologies) according to the manufacturer recommendations. RNA sequencing was conducted on rRNA depleted library (KAPA rRNA-depleted (bacteria) library preparation kit (KAPA Biosystems, Wilmington, MA, USA)) using the HiSeq 4000 PE100 platform at McGill University-Genome Quebec Innovation center.
4.6. Data analysis, bioinformatics, and CAZyme phylogenetic analyses
RNA-Seq raw reads were submitted as input to the Kaiju [16] webserver for taxonomic assignment at the whole transcriptome level. Kaiju was run in greedy mode with a minimum match length of 11 and a minimum match score of 55, with 5 mismatches allowed. The results of the taxonomic classification with Kaiju were visualized with Krona [17]. The raw sequenced reads were further processed using MUGQIC RNA-Seq de novo assembly and differential analysis pipeline [65]. Raw sequenced reads were trimmed and clipped for sequencing adapters, low quality and short sequences were filtered using Trimmomatic [66], normalized and assembled using Trinity normalization utility inspired by the Diginorm algorithm [67]. The sequenced reads were assembled by the de novo transcriptomic assembly program Trinity. Transcripts from Trinity were aligned against the uniprot_sprot.trinotate_v2.0 protein database using the BLASTx program. The Trinotate suite was used for homology searches to known sequence data (BLAST+/SwissProt/Uniref90), protein domain identification (HMMER/PFAM), protein signal peptides and transmembrane domain prediction (signal/tmHMM), and for comparison to currently curated annotation databases (EMBL Uniprot eggnog/GO Pathways databases). The derived functional annotation data were integrated into an SQLite database to prepare the annotation report. To identify putative carbohydrate active enzymes, all sequences were profiled against the CAZy database on 2017–07-20 [18]. Gene abundance was estimated using RSEM and DE transcripts between substrates were identified using DEseq [68] and edgeR [69] package from Bioconductor.
For DE transcript functional analyses, DE transcript nucleotide sequences were re-submitted to the dbCAN2 meta server [20], manually curated, and categorized by CAZy family. The amino acid sequences from the dbCAN-annotated CAZymes were submitted as input to SACCHARIS for phylogenetic analyses [24]. Sequences and accession numbers of characterized GH11, GH43, and CE15 enzymes were extracted from the CAZy database [18], and ProtTest [70] was used for best-fit model selection using the sequence alignment. FastTree [71] was used to generate the phylogenetic trees which were then annotated using iTOL [72].
4.7. Transcript CAZyme gene synthesis and expression in E. coli
Transcript targets TR400632|c0_g1 and TR363754|c1_g3 were selected based on their statistically significant overexpression in BS and TTIR cultures, respectively. TR363754|c1_g3 did not cover the entire GH11 domain as identified by dbCAN2 [20] and InterProScan [73], and a search with BLASTx revealed the next nearest homolog to be GenBank accession no. WP_049962670.1 from Ruminococcus sp. HUN007. Nucleotide sequences for TR400632|c0_g1 and WP_049962670.1 were analyzed by dbCAN2 [20] and InterProScan [73], and modelled using Phyre2 [74]. Phyre2 models were manually curated with PyMOL (Schrödinger, Inc), and GH11 domain amino acid sequence boundaries were chosen based on these analyses; TR400632|c0_g131-248, and WP_049962670.131-250. GH11 nucleotide sequences were codon optimized for expression in E. coli and gene synthesized including flanking NdeI and XhoI restriction sites at the 5′ and 3′ ends, respectively, and a stop codon at the 3′ end (BioBasic Inc.). Genes were ordered synthesized into the pET28a vector for recombinant protein expression with a C-terminal His6-tag for downstream protein purification. Protein construct vectors were transformed into E. coli BL21 Tuner (DE3) cells (Novagen). Cells were grown in LB Miller broth containing 50 μg mL−1 kanamycin at 37 °C to an OD600 nm of 0.6, when protein expression was induced by the addition of IPTG to a final concentration of 1 mM. The cell culture was incubated at 18 °C for 16 h prior to being harvested by centrifugation at 6500 × g for 20 min at 4 °C. Cell pellets were stored at −20 °C until needed.
4.8. Recombinant protein purification
The cell pellet from 1 L of bacterial culture was thawed and resuspended in 50 mL of lysis buffer (20 mM Tris pH 8.0, 500 mM NaCl, 0.1 mg mL−1 lysozyme). Cells were homogenized by sonication for 2 min of 1 s intervals of medium intensity sonic pulses at a power setting of 4.5 (Heat Systems Ultrasonics Model W-225 and probe). Cellular debris was removed by centrifugation at 17,500 × g for 45 min at 4 °C. The filtrate was loaded onto Ni-NTA resin and purified by immobilized metal affinity chromatography; recombinant protein was eluted by an increasing gradient 0–500 mM imidazole in 20 mM Tris pH 8.0 and 500 mM NaCl. Protein was concentrated by centrifugation with 10 kDa cut-off Amicon Ultra centrifugal concentrators (Millipore Sigma). His6-tagged protein was further purified using a HiLoad 16/60 Superdex 200 prep-grade size exclusion column (GE Healthcare) in 20 mM Tris pH 8, 500 mM NaCl, 2% glycerol. Pure protein fractions were pooled and concentrated. Protein purification was monitored throughout by SDS-PAGE. All recombinant proteins were used freshly prepared for downstream enzyme activity assays.
4.9. Thin layer chromatography
Purified GH11 enzymes were screened for activity on heteroxylan and arabinoxylan substrates: oat spelt xylan (Fluka), beechwood xylan (Megazyme), rye arabinoxylan (Megazyme), wheat arabinoxylan (Megazyme). Reactions (200 μL) contained 1 μM enzyme and 10 mg mL−1 substrate in 20 mM Tris pH 8.0 and incubated at room temperature for 64 h with mild shaking to maximize accessibility of insoluble substrates. After incubation, the samples were heat treated at 100 °C to denature the enzyme and terminate the reaction. Samples were then shortly centrifuged at 8000 × g to pellet denatured protein and undigested insoluble substrate particles from the product. Digested samples were spotted (total 6 μL; spotted 3 times with 2 μL each) onto thin layer chromatography plates (Silica Gel 60; EMD Millipore). The samples were dried between multiple rounds of spotting. Appropriate mono- and oligosaccharide standards were included as controls (6 μL of 1 mM concentration); xylose (X), xylobiose (X2), xylotriose (X3), xylotetraose (X4), xylopentaose (X5), and xylohexaose (X6). The samples were resolved using a mobile phase of 2:1:1 butanol:acetic acid:H2O, and dried prior to visualization with an orcinol solution (70:3, acetic acid:sulfuric acid with 1% orcinol) and heating at 100 °C for 3–5 min.
4.10. High-performance anion-exchange chromatography with pulsed amperometric detection (HPAEC-PAD)
HPAEC-PAD was performed with a Dionex ICS-3000 chromatography system (Thermo Scientific) equipped with an autosampler as well as a pulsed amperometric (PAD) detector. Taken from the soluble fraction of enzyme digests as subjected to thin layer chromatography, 10 μL of aqueous samples were injected onto an analytical (3 × 150 mm) CarboPac PA20 column (Thermo Scientific) and eluted at a 0.5 mL min−1 flow rate with a sodium acetate gradient (0 to 40 min, 10 to 120 mM) in a constant background of 30 mM NaOH. The elution was monitored with a PAD detector (standard quadratic waveform). Xylan mono- and oligosaccharide standards were included as a xylose ladder of X1-X6 forms. Arabinoxylan oligosaccharide standards were also used: 32-α-L-arabinofuranosyl-xylobiose (A3X), 23– α-L-arabinofuranosyl-xylobiose (A2XX), 23,33-di-α-L-arabinofuranosyl-xylotriose (A2,3XX).
5. Ethics approval and consent to participate
All animal procedures and protocols used in this study were reviewed and approved by the Lethbridge Research and Development Center Animal Care Committee (ACC number 1501) in accordance with the guidelines of Canadian Council on Animal Care (CCAC, 2009).
6. Consent for publication
No consent was required for this publication.
7. Availability of data and material
Sequenced transcripts have been deposited into the NCBI SRA database BioProject accession number: PRJNA673210.
Funding
This work was funded by Alberta Agriculture and Forestry, Canada: project ID 2018F182R, and Agriculture and Agri-Food Canada: project ID: J-001589. Glycome profiling of biomass samples was supported by the Center for Bioenergy Innovation (Oak Ridge National Laboratory), US Department of Energy (DOE) Bioenergy Research Center supported by the Office of Biological and Environmental Research in the DOE Office of Science.
Author contributions
XX, AKB, DRJ, SV, and MGH performed and analyzed data for cell wall characterization. AKB, ROP performed in vitro batch culture, RNA extraction, and transcriptome sequence analysis. KEL performed recombinant enzyme production and characterization. KEL, LK, and MRMM contributed towards data processing, statistical support, and graphical representation of data. AKB, DWA, and TAM funded and conceived of the study. AKB, KEL, DWA, and TAM wrote the manuscript and all authors reviewed and approved the manuscript.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
The authors thank the Lethbridge Research and Development Centre (AAFC) staff for their technical and animal care assistance.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.csbj.2021.12.009.
Contributor Information
D. Wade Abbott, Email: wade.abbott@agr.gc.ca.
Tim A. McAllister, Email: tim.mcallister@agr.gc.ca.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Alexandratos N, Bruinsma J World agriculture towards 2030/2050: the 2012 revision. In: ESA Working paper No. 12-03. Rome. FAO. 2012. http://www.fao.org/3/a-ap106e.pdf. Accessed 26 Oct 2020.
- 2.Elam TE Projections of global meat production through 2050. 2006. https://docplayer.net/21349988-Projections-of-global-meat-production-through-2050.html. Accessed 26 Oct 2020.
- 3.Sokhansanj S., Mani S., Stumborg M., Samson R., Fenton J. Production and distribution of cereal straw on the Canadian prairies. Can Biosys Eng. 2006;48:3.39-3.46. [Google Scholar]
- 4.Kim S., Dale B.E. Global potential bioethanol production from wasted crops and crop residues. Biomass Bioenerg. 2004;26(4):361–375. [Google Scholar]
- 5.Hatfield R.D., Ralph J., Grabber J.H. Cell wall structural foundations: molecular basis for Improving forage digestibilities. Crop Sci. 1999;39(1):27–37. [Google Scholar]
- 6.Badhan A., Wang Y., Gruninger R., Patton D., Powlowski J., Tsang A., et al. Formulation of enzyme blends to maximize the hydrolysis of alkaline peroxide pretreated alfalfa hay and barley straw by rumen enzymes and commercial cellulases. BMC Biotechnol. 2014;14(1) doi: 10.1186/1472-6750-14-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ribeiro GO, Gruninger RJ. Badhan A, McAllister TA (2016) Mining the rumen for fibrolytic feed enzymes. Anim Front 6: 20-26.
- 8.Meale SJ, Beauchemin KA, Hristov AN, Chaves AV, McAllister TA (2014) Board-invited review: Opportunities and challenges in using exogenous enzymes to improve ruminant production. J Anim Sci 92: 427-442. [DOI] [PubMed]
- 9.Sun X.Z., Joblin K.N., Andrew I.G., Hoskin S.O., Harris P.J. Degradation of forage chicory by ruminal fibrolytic bacteria. J Appl Microbiol. 2008;105(5):1286–1297. doi: 10.1111/j.1365-2672.2008.03861.x. [DOI] [PubMed] [Google Scholar]
- 10.Pattathil S., Avci U., Zhang T., Cardenas C.L., Hahn M.G. Immunological approaches to biomass characterization and utilization. Front Bioeng Biotechnol. 2015;3:173. doi: 10.3389/fbioe.2015.00173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pattathil S., Hahn M.G., Dale B.E., Chundawat S.P.S. Insights into plant cell wall structure, architecture, and integrity using glycome profiling of native and AFEXTM-pre-treated biomass. J Exp Bot. 2015;66(14):4279–4294. doi: 10.1093/jxb/erv107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Plant cell wall monoclonal antibody database. http://glycomics.ccrc.uga.edu/wall2/antibodies/antibodyHome.html. Accessed 11 May 2021.
- 13.Pettolino F.A., Walsh C., Fincher G.B., Bacic A. Determining the polysaccharide composition of plant cell walls. Nat Protoc. 2012;7(9):1590–1607. doi: 10.1038/nprot.2012.081. [DOI] [PubMed] [Google Scholar]
- 14.Ribeiro G.O., Badhan A., Huang J.L., Beauchemin K.A., Yang W.Z., et al. New recombinant fibrolytic enzymes for improved in-vitro ruminal fiber degradability of barley straw. J Anim Sci. 2018;96:3928–3942. doi: 10.1093/jas/sky251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grabherr M.G., Haas B.J., Yassour M., Levin J.Z., Thompson D.A., Amit I., et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29(7):644–652. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Menzel P., Ng K., Krogh A. Fast and sensitive taxonomic classification for metagenomics with Kaiju. Nat Commun. 2016;7:11257. doi: 10.1038/ncomms11257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ondov B.D., Bergman N.H., Phillippy A.M. Interactive metagenomic visualization in a Web browser. BMC Bioinf. 2011;12:385. doi: 10.1186/1471-2105-12-385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lombard V., Golaconda Ramulu H., Drula E., Coutinho P.M., Henrissat B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014;42(D1):D490–D495. doi: 10.1093/nar/gkt1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Couger M.B., Youssef N.H., Struchtemeyer C.G., Liggenstoffer A.S., Elshahed M.E. Transcriptomic analysis of lignocellulosic biomass degradation by the anaerobic fungal isolate Orpinomyces sp. strain C1A. Biotechnol Biofuels. 2015;8:208. doi: 10.1186/s13068-015-0390-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang H, Yohe T, Huang L, Entwistle S, Wu P, et al. (2018) dbCAN2: a meta server for automated carbohydrate-active enzyme annotation. Nucl Acids Res 46: W95–W101. [DOI] [PMC free article] [PubMed]
- 21.Hachem M.A., Karlsson E.N., Bartonek-roxå E., Raghothama S., Simpson P.J., Gilbert H.J., et al. Carbohydrate-binding modules from a thermostable Rhodothermus marinus xylanase: cloning, expression and binding studies. Biochem J. 2000;345(1):53–60. [PMC free article] [PubMed] [Google Scholar]
- 22.Boraston A.B., Warren R.A., Kilburn D.G. beta-1,3-Glucan binding by a thermostable carbohydrate-binding module from Thermotoga maritima. Biochemistry. 2001;40(48):14679–14685. doi: 10.1021/bi015760g. [DOI] [PubMed] [Google Scholar]
- 23.Tomme P., Creagh A.L., Kilburn D.G., Haynes C.A. Interaction of polysaccharides with the N-terminal cellulose-binding domain of Cellulomonas fimi CenC. 1. Binding specificity and calorimetric analysis. Biochemistry. 1996;35(44):13885–13894. doi: 10.1021/bi961185i. [DOI] [PubMed] [Google Scholar]
- 24.Jones D.R., Thomas D., Alger N., Ghavidel A., Inglis G.D., Abbott D.W. SACCHARIS: an automated pipeline to streamline discovery of carbohydrate active enzyme activities within polyspecific families and de novo sequence datasets. Biotechnol Biofuels. 2018;11(1) doi: 10.1186/s13068-018-1027-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ruprecht C., Bartetzko M.P., Senf D., Dallabernadina P., Boos I., Andersen M.C.F., et al. A synthetic glycan microarray enables epitope mapping of plant cell wall glycan-directed antibodies. Plant Physiol. 2017;175(3):1094–1104. doi: 10.1104/pp.17.00737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hatfield R.D., Jung H.J.G., Broderick G., Jenkins T.C. In: Forages: The science of grassland agriculture. Barnes R.F., Nelson K.J., Collins M.M., editors. Blackwell publishing; Ames, IA: 2007. Nutritional chemistry of forages; pp. 467–485. [Google Scholar]
- 27.Pattathil S., Ingwers M.W., Victoriano O.L., Kandemkavil S., McGuire M.A., Teskey R.O., et al. Cell wall ultrastructure of stem wood, roots, and needles of a conifer varies in response to moisture availability. Front Plant Sci. 2016;7 doi: 10.3389/fpls.2016.00882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dai X., Tian Y., Li J., Su X., Wang X., et al. Metatranscriptomic analyses of plant cell call polysaccharide degradation by microorganisms in the cow rumen. Appl Environ Microbiol. 2015;81:1375–1386. doi: 10.1128/AEM.03682-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Comtet-Marre S., Parisot N., Lepercq P., Chaucheyras-Durand F., Mosoni P., Peyretaillade E., et al. Metatranscriptomics reveals the active bacterial and eukaryotic fibrolytic communities in the rumen of dairy cow fed a mixed diet. Front Micro. 2017;8 doi: 10.3389/fmicb.2017.00067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tapio I., Shingfield K.J., McKain N., Bonin A., Fischer D., Bayat A.R., et al. Oral Samples as non-invasive proxies for assessing the composition of the rumen microbial community. PLoS ONE. 2016;11(3):e0151220. doi: 10.1371/journal.pone.0151220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Golder H.M., Thomson J.M., Denman S.E., Mcsweeney C.S., Lean I.J. Genetic markers are associated with the ruminal microbiome and metabolome in grain and sugar challenged dairy heifers. Front Genet. 2018;9:62. doi: 10.3389/fgene.2018.00062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brunecky R., Alahuhta M., Xu Q.i., Donohoe B.S., Crowley M.F., Kataeva I.A., et al. Revealing nature’s cellulase diversity: the digestion mechanism of Caldicellulosiruptor bescii CelA. Science. 2013;342(6165):1513–1516. doi: 10.1126/science.1244273. [DOI] [PubMed] [Google Scholar]
- 33.Barr B.K., Hsieh Y.-L., Ganem B., Wilson D.B. Identification of two functionally different classes of exocellulases. Biochemistry. 1996;35(2):586–592. doi: 10.1021/bi9520388. [DOI] [PubMed] [Google Scholar]
- 34.Seshadri R., Leahy S.C., Attwood G.T., Teh K.H., Lambie S.C., Cookson A.L., et al. Cultivation and sequencing of rumen microbiome members from the Hungate1000 Collection. Nat Biotechnol. 2018;36(4):359–367. doi: 10.1038/nbt.4110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brulc J.M., Antonopoulos D.A., Berg-Miller M.E., Wilson M.K., Yannarell A.C., et al. Gene-centric metagenomics of the fibre-adherent bovine rumen microbiome reveals forage specific glycoside hydrolases. Proc Natl Acad Sci. 2009;106:1948–1953. doi: 10.1073/pnas.0806191105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dai X., Zhu Y., Luo Y., Song L., Liu D.i., Liu L.i., et al. Metagenomic insights into the fibrolytic microbiome in yak rumen. PLoS ONE. 2012;7(7):e40430. doi: 10.1371/journal.pone.0040430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pope P.B., Denman S.E., Jones M., Tringe S.G., Barry K., Malfatti S.A., et al. Adaptation to herbivory by the Tammar wallaby includes bacterial and glycoside hydrolase profiles different from other herbivores. Proc Natl Acad Sci. 2010;107(33):14793–14798. doi: 10.1073/pnas.1005297107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Qi M., Wang P., O'Toole N., Barboza P.S., Ungerfeld E., Leigh M.B., et al. Snapshot of the eukaryotic gene expression in muskoxen rumen—a metatranscriptomic approach. PLoS ONE. 2011;6(5):e20521. doi: 10.1371/journal.pone.0020521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shinkai T., Mitsumori M., Sofyan A., Kanamori H., Sasaki H., Katayose Y., et al. Comprehensive detection of bacterial carbohydrate-active enzyme coding genes expressed in cow rumen. Anim Sci J. 2016;87(11):1363–1370. doi: 10.1111/asj.12585. [DOI] [PubMed] [Google Scholar]
- 40.Li F., Guan L.L., McBain A.J. Metatranscriptomic profiling reveals linkages between the active rumen microbiome and feed efficiency in beef cattle. Appl Environ Microbiol. 2017;83(9) doi: 10.1128/AEM.00061-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mewis K., Lenfant N., Lombard V., Henrissat B., Nojiri H. Dividing the large glycoside hydrolase family 43 into subfamilies: a motivation for detailed enzyme characterization. Appl Environ Microbiol. 2016;82(6):1686–1692. doi: 10.1128/AEM.03453-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pollet A., Delcour J.A., Courtin C.M. Structural determinants of the substrate specificities of xylanase from different glycoside hydrolase families. Crit Rev Biotechnol. 2010;30:176–191. doi: 10.3109/07388551003645599. [DOI] [PubMed] [Google Scholar]
- 43.O'Neill E.C., Stevenson C.E.M., Paterson M.J., Rejzek M., Chauvin A.-L., Lawson D.M., et al. Crystal structure of a novel two domain GH78 family α-rhamnosidase from Klebsiella oxytoca with rhamnose bound. Proteins. 2015;83(9):1742–1749. doi: 10.1002/prot.24807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sprockett D.D., Piontkivska H., Blackwood C.B. Evolutionary analysis of glycosyl hydrolase family 28 (GH28) suggests lineage-specific expansions in necrotrophic fungal pathogens. Gene. 2011;479(1-2):29–36. doi: 10.1016/j.gene.2011.02.009. [DOI] [PubMed] [Google Scholar]
- 45.Terry S.A., Badhan A., Wang Y., Chaves A.V., McAllister T.A., Miglior F. Fibre digestion by rumen microbiota – a review of recent metagenomic and metatranscriptomic studies. Can J Ani Sci. 2019;99(4):678–692. [Google Scholar]
- 46.Wood J.A., Tan H.T., Collins H.M., Yap K., Khor S.F., et al. Genetic and environmental factors contribute to variation in cell wall composition in mature desi chickpea (Cicer arietinum L.) cotyledons. Plant Cell Environ. 2018;41:2195–2208. doi: 10.1111/pce.13196. [DOI] [PubMed] [Google Scholar]
- 47.Pattathil S., Avci U., Miller J.S., Hahn M.G. In: Biomass conversion. Methods in Molecular Biology (Methods and Protocols) Himmel M., editor. Humana Press; Totowa, NJ: 2012. Immunological approaches to plant cell wall and biomass characterization: Glycome profiling; p. 908. [DOI] [PubMed] [Google Scholar]
- 48.Pattathil S, Avci U, Baldwin D, Swennes AG, McGill JA, et al. (2010) A comprehensive toolkit of plant cell wall glycan-directed monoclonal antibodies. Plant Physiol 153: 514-525. [DOI] [PMC free article] [PubMed]
- 49.Hsieh Y.-Y., Chien C., Liao S.-S., Liao S.-F., Hung W.-T., Yang W.-B., et al. Structure and bioactivity of the polysaccharides in medicinal plant Dendrobium huoshanense. Bioorgan Med Chem. 2008;16(11):6054–6068. doi: 10.1016/j.bmc.2008.04.042. [DOI] [PubMed] [Google Scholar]
- 50.Li J., Wang D., Xing X., Cheng T.-J., Liang P.-H., Bulone V., et al. Structural analysis and biological activity of cell wall polysaccharides extracted from Panax ginseng marc. Int J Biol Macromol. 2019;135:29–37. doi: 10.1016/j.ijbiomac.2019.05.077. [DOI] [PubMed] [Google Scholar]
- 51.Bacic A., Moody S.F., Clarke A.E. Structural analysis of secreted root slime from maize (Zea mays L.) Plant Physiol. 1986;80(3):771–777. doi: 10.1104/pp.80.3.771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kim J.B., Carpita N.C. Esterification of maize cell wall pectins related to cell expansion. Plant Physiol. 1992;98:646–653. doi: 10.1104/pp.98.2.646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ciucanu I., Kerek F. A simple and rapid method for the permethylation of carbohydrates. Carbohyd Res. 1984;131(2):209–217. [Google Scholar]
- 54.Albersheim P., Nevins D.J., English P.D., Karr A. A method for the analysis of sugars in plant cell wall polysaccharides by gas liquid chromatography. Carbohyd Res. 1967;5(3):340–345. [Google Scholar]
- 55.Darvill A.G., McNeil M., Albersheim P. Structure of plant cell walls VIII. A new pectic polysaccharide. Plant Physiol. 1978;62(3):418–422. doi: 10.1104/pp.62.3.418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dell A, Khoo KH, Panic M, McDowell RA, Etienne A, et al. (1989) FAB-MS and ESI-MS of glycoproteins. In: Fukuda M, Kobata A, editor. Glycobiology, a practical approach. Oxford University Press. pp. 187-222.
- 57.Chiovitti A., Bacic A., Craik D.J., Kraft G.T., Liao M.L. A nearly idealized 6’-O-methylated iota-carrageenan from the Australian red alga Claviclonium ovatum (Acrotylaceae, Gigartinales) Carbohyd Res. 2004;339:1459–1466. doi: 10.1016/j.carres.2004.03.019. [DOI] [PubMed] [Google Scholar]
- 58.Carpita NC, Shea EM (1988) Linkage structure of carbohydrates by gas chromatography-mass spectrometry (GC-MS) of partially methylated alditol acetates. In: Biermann CJ, McGinnis GD, editors. Analysis of Carbohydrates by GLC and MS. CRC Press pp.157-216.
- 59.Sweet D.P., Shapiro R.H., Albersheim P. Quantitative analysis by various G.L.C. response-factor theories for partially methylated and partially ethylated alditol acetates. Carbohyd Res. 1975;40(2):217–225. [Google Scholar]
- 60.Sowinskia E.E., Gilbert S., Lamb E., Carpita N.C. Linkage structure of cell-wall polysaccharides from three duckweed species. Carbohyd Polym. 2019;223:115–119. doi: 10.1016/j.carbpol.2019.115119. [DOI] [PubMed] [Google Scholar]
- 61.Ribeiro G.O., Oss D.B., He Z., Gruninger R.J., Elekwachi C., Forster R.J., et al. Repeated inoculation of cattle rumen with bison rumen contents alters the rumen microbiome and improves nitrogen digestibility in cattle. Sci Rep. 2017;7(1) doi: 10.1038/s41598-017-01269-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Romero-Pérez A., Beauchemin K.A., Plaizier J. Estimating gas volume from headspace pressure in a batch culture system. Can J Anim Sci. 2018;98(3):593–596. [Google Scholar]
- 63.Wang P., Qi M., Barboza P., Leigh M.B., Ungerfeld E., Selinger L.B., et al. Isolation of high-quality total RNA from rumen anaerobic bacteria and fungi, and subsequent detection of glycoside hydrolases. Can J Microbiol. 2011;57(7):590–598. doi: 10.1139/w11-048. [DOI] [PubMed] [Google Scholar]
- 64.Chomczynski P., Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162(1):156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 65.Haas B.J., Papanicolaou A., Yassour M., Grabherr M., Blood P.D., Bowden J., et al. De novo transcript sequence reconstruction from RNA-Seq using the trinity platform for reference generation and analysis. Nat Protoc. 2013;8(8):1494–1512. doi: 10.1038/nprot.2013.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: A flexible trimmer for illumina sequence data. Bioinformatics 30: 2114–20. [DOI] [PMC free article] [PubMed]
- 67.Titus B., Howe C.A., Zhang Q., Pyrkosz A.B., Brom T.H. A reference-free algorithm for computational normalization of shotgun sequencing data. ArXiv.org. 2012 http://arxiv.org/abs/1203.4802 [Google Scholar]
- 68.Ander S., Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Robinson M.D., McCarthy D.J., Smyth G.K. EdgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Darriba D, Taboada GL, Doallo R, Posada D (2011) ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics 27: 1164–1165. [DOI] [PMC free article] [PubMed]
- 71.Price M.N., Dehal P.S., Arkin A.P., Poon A.F.Y. FastTree 2 – Approximately maximum-likelihood trees for large alignments. PLoS ONE. 2010;5(3):e9490. doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Letunic I, Bork P (2019) Interactive Tree Of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res 47: W256–W259. [DOI] [PMC free article] [PubMed]
- 73.Jones P., Binns D., Chang H.-Y., Fraser M., Li W., McAnulla C., et al. InterProScan 5: genome-scale protein function classification. Bioinformatics. 2014;30(9):1236–1240. doi: 10.1093/bioinformatics/btu031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kelley L.A., Mezulis S., Yates C.M., Wass M.N., Sternberg M.J.E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc. 2015;10(6):845–858. doi: 10.1038/nprot.2015.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequenced transcripts have been deposited into the NCBI SRA database BioProject accession number: PRJNA673210.