

Abstract
CD9, a member of the tetraspanin superfamily, has been implicated in regulating various physiological processes, including cell motility, adhesion, apoptosis and metastasis. Recently, interleukin-16 (IL-16), a pro-inflammatory cytokine released by normal airway and alveolar epithelial cells, has been implicated as a possible ligand for CD9 as an alternative receptor. In this study, using A549 cells as a model of human alveolar epithelium, CD9 expression was ablated using CRISPR/Cas technology. Decreased expression of CD9 mRNA and protein levels was confirmed through RT-qPCR and flow cytometry, respectively. Individual clones were generated that expressed high levels of CD9 (wild-type, WT) or significantly less CD9 (knockdown, KD). Both wild-type and knockdown A549 cell migration was quantified using a FluoroBloc transwell chemotaxis assay. Our results indicate that wild-type A549 cells migrated towards chemoattractants. Moreover, CD9 expression was required in this process since the CD9 knockdown cells had a significantly reduced migration towards growth serum and IL-16. These results support the migratory properties for CD9 in human lung cells and support the hypothesis that CD9 serves as an alternative receptor for IL-16.
Keywords: Interleukin-16, CRISPR/Cas technology, A549 cells, Migration, CD9, Cancer
1. Introduction
Interleukin 16 (IL-16) is a proinflammatory cytokine produced from peripheral blood mononuclear cells predominately T cells. IL-16 exists as both a pro-IL-16 protein with an N-terminal domain, and as a mature, C-terminal secreted protein. Secreted IL-16 is generated by a caspase-3-dependent cleavage of the precursor IL-16 protein. Secreted IL-16 has diverse immunoregulatory functions including inducing cell migration and lymphocyte activation [11].
Many IL-16 effects including migration and cell activation are mediated by the CD4 receptor. IL-16 was originally identified as a lymphocyte chemoattractant factor due to its migratory effects on CD4+ T cells [10]. Numerous cell types including mast cells, dendritic cells and epithelial cells express CD4 and, therefore, are responsive to this potent chemoattractant. IL-16 can also activate T-lymphocytes and inhibit HIV replication through a mechanism that requires IL-16/CD4-dependent activation of p56lck [13]. In CD4+ macrophages, IL-16 can activate the stress-activated protein kinase pathway, and can lead to the phosphorylation of c-Jun and p38 MAPK (mitogen-activated protein kinase), without inducing MAPK-family members ERK-1 and ERK-2 [5].
Although CD4 is thought to be the primary receptor for IL-16-induced chemotaxis [2], other studies have identified CD9 as a possible alternative receptor [7,8,12]. Previous studies have demonstrated that CD4−/− cells are responsive to IL-16 in terms of cytokine production, migration, and neural outgrowth [7,3]. Transient siRNA knockdown studies of CD9 in HMC-1 cells inhibited IL-16 binding and reduced intracellular calcium release [8]. A549 cells are known to express CD9 [6], but not CD4 [12], and are an excellent in vitro model in order to understand the inductive effects of IL-16 through CD9 receptor activation. The present study, using CRISPR/Cas technologies to genetically ablate the CD9 gene, provides clear support of CD9 as an alternate co-receptor for IL-16 and its migratory properties.
2. Materials and methods
2.1. Cell culture and reagents
A549 cells (human lung epithelial cell line) were purchased from the American Type Culture Collection (Manassas, VA). Cells were cultured at 37 °C in a 5% CO2 incubator in Dulbecco’s Modified Eagle Medium (DMEM) containing 10% fetal bovine serum (FBS) and antibiotics (Life Technologies, Grand Island, NY). The pSPCas9(BB)-2A-Puro (PX459) vector was obtained from Addgene (Cambridge, MA). Lipofectamine® 2000 and primer and probe sets were obtained from Life Technologies (Grand Island, NY). Anti-CD9, anti-CD4 antibodies and recombinant human IL-16 were obtained from BD Biosciences (San Jose, CA).
2.2. CRISPR/Cas knockout and transfection
A549 cells were cultured overnight as described without antibiotics and transfected the next day according to the manufacturer’s instructions with 2000 ng of plasmid [9]. Twenty-four hours post-transfection, cells were passed at 1:20 into fresh growth medium containing puromycin (1.5 μg/ml) for selection. Individual clones were selected 14 days after transfection.
2.3. RT-qPCR
Total RNA was isolated using the RNeasy Mini Kit (Qiagen). Reverse transcription of RNA was performed using the Multiscribe RT Kit (Life Technologies). Gene expression was measured using Taqman on demand probe sets. Relative fold change was calculated using the following formulas: ΔCT = CT (Actin)–CT (target gene) and , in which ΔCT1 represents the lowest CT value among control samples and ΔCT2 represents the value of a particular sample as previously described [1]. Twelve independent knockdown clones were quantified for CD9 mRNA using two technical replicates for each experiment (n = 2). Two independent CD9 knockdown clones were subsequently identified and used to quantify CD9 protein expression and chemotaxis.
2.4. Flow cytometry
CD9 and CD4 expression levels were quantified by standard flow cytometry. Briefly, cells were trypsinized, and 106 cells were blocked in 2% FBS with 0.1% sodium azide. Cells were stained with 5 μL of anti-human CD9 or anti-human CD4 antibody for 30 min. Fluorescence was quantified though a BD Accuri C6 Flow cytometer. Two independent knockout clones were quantified for membrane bound CD9 using three technical replicates for each experiment (n = 3).
2.5. Immunoblot analysis
Wild-type (WT) and knockdown (KD) CD9 cells were seeded into 6-well plates and starved for 24 h in 1% FBS/DMEM. Cells were then stimulated with 2.5 μg/ml IL-16 for 5, 15, 30, 60 and 120 min or 10 ng/ml IL-1β for 10 min as a positive control. Cell extracts were isolated using RIPA buffer followed by sonication. Proteins were resolved on SDS–PAGE gels and transferred onto PVDF membranes. Immunoblots were probed with either an anti-phospho-p38, an anti-phospho-SAPK/JNK, an anti-SAPK/JNK, or an anti-p38 rabbit polyclonal antibody (Cell Signaling Technology, Danvers, MA). Blots were probed with a goat anti-rabbit IgG HRP conjugated secondary (Santa Cruz Biotechnology, Santa Cruz, CA) and binding was visualized through chemiluminescence. An increased signal for p38 or JNK (phosphorylated or total) was defined as the ratio of p38 or JNK over Actin that had a ratio greater than two standard deviations above the unstimulated, negative control (i.e. background levels). Densitometry was quantified through Image J software (NIH). Two independent biological replicates were performed using two technical replicates for each experiment (n = 2).
2.6. Chemotaxis assay
Chemotaxis was quantified fluorescently using BD Falcon FluoroBlok 24-Multiwell Insert Plate with an 8.0-μm pore size and Calcien AM. Two independent KD clones were selected for migration analysis. Briefly, A549 cells were suspended in the apical chambers and chemoattractant (FBS or IL-16) was added to the basal chambers. Following overnight incubation, transwells were stained in a second 24-well plate containing 500 μL/well of 4 μg/mL Calcein AM in HBSS and incubated for 1 h at 37 °C. Fluorescence of invaded cells was read at wavelengths of 494/517 nm (Ex/Em) on a Tecan bottom-reading fluorescent plate reader. Percent migration was calculated as RFU (background corrected value) – RFU of negative control/RFU of negative control. Three independent biological replicates were performed using two or more technical replicates for each experiment (n = 2–5)
2.7. Statistical analysis
Data are presented as mean ± SEM. Analyses were done using GraphPad Prism 6.0 (San Diego, CA). A two-way ANOVA was used to compare four experimental groups (DMEM, 10% FBS, PBS and IL-16 as chemoattractants) with two independent variables (WT and KD cells). A t-test was used to compare two groups (WT and KD cells) with one independent variable (IL-16). Significance was noted at P < 0.05.
3. Results/discussion
3.1. CD9 expression
A549 cells expressed high levels of CD9 mRNA and protein (Fig. 1B, G and H). A549 cells did not express CD4 (Fig. 1D, E and F) as previously described [12]. In order to genetically delete CD9 in A549 cells, guide RNAs were engineered targeting either the first or second exon of the CD9 gene, ligated into the pSPCas9(BB)-2A-Puro vector, and transfected as previously described [9]. For wild-type A549 cells, real-time PCR amplification of CD9 mRNA was measured at approximately threshold cycle (CT) 24. In contrast, in putative CD9 knockdown cells (two independent clones) expression of CD9 mRNA was nearly undetectable (CT = 38). In addition, approximately 75% of wild-type A549 cells expressed CD9, whereas after genetic deletion less than 3% of A549 cells expressed CD9 on the cell surface (Fig. 1. These data indicated that CD9 had been successfully deleted from A549 cells using CRIPSR/Cas gene editing.
Fig. 1.
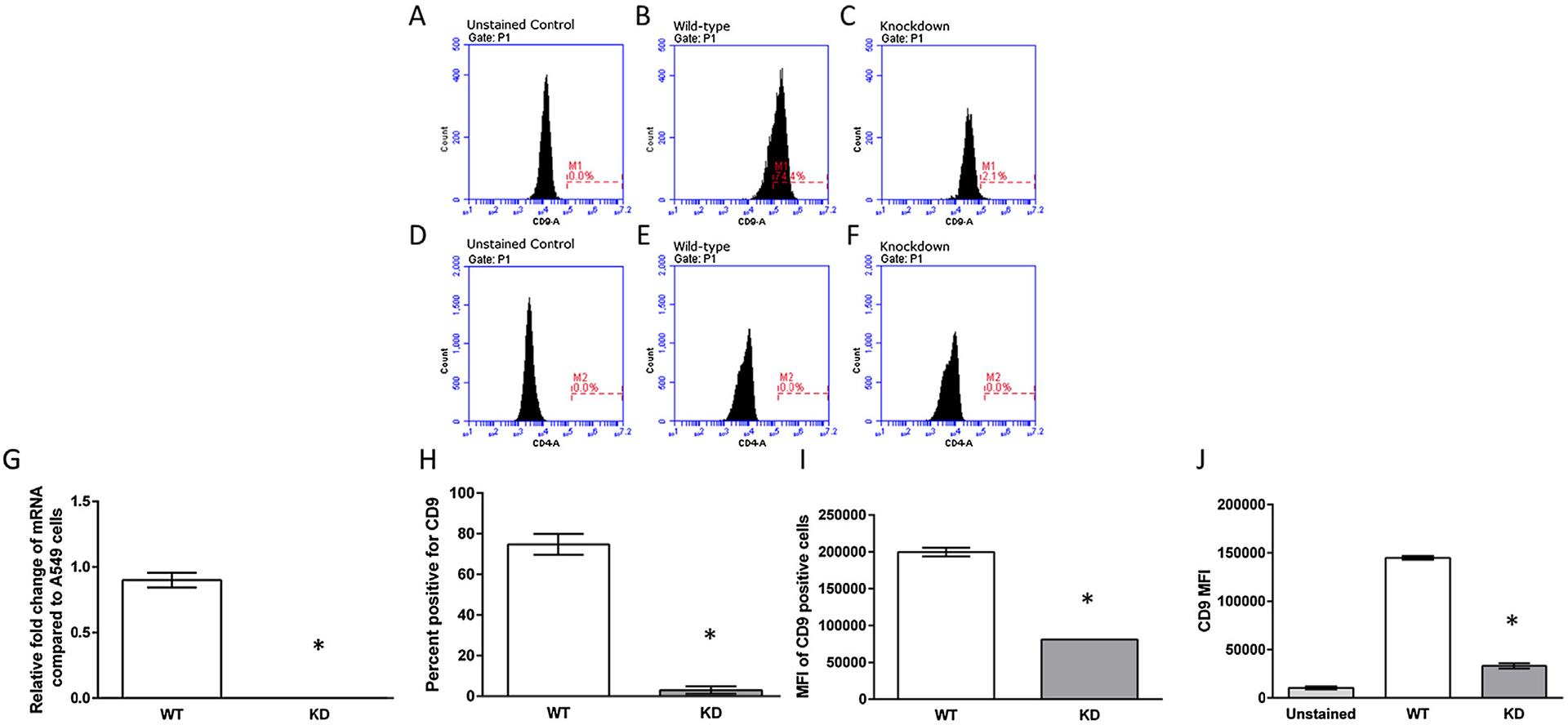
Characterization of CRISPR/Cas CD9 deletion clones. (A–C) Representative histograms of the WT and KD cells unstained or stained with an anti-human CD9 antibody. (D–F) Representative histograms of the WT and KD cells unstained or stained with an anti-human CD4 antibody. (G) Representative quantitative mRNA levels of knockout clones were compared to the parental A549 cells, which is indicated as an expression level of 0.1 mRNA expression levels of CD9 and Actin were quantified through RT-qPCR. Data presented are mean relative fold change ± SEM (n = 3). Asterisks indicate a significant difference between wild-type cells and knockdown cells (P < 0.05). (H and I) CD9 protein expression of CD9 knockdown clones. Cells were stained with an anti-CD9 antibody and the percent of cells that were CD9 positive (cells within the M1 gate) and the MFI of the gated M1 cells were quantified through flow cytometry. (J) MFI of all WT and KD cells quantified through flow cytometry. Data presented are mean relative fold change ± SEM (n = 3). Asterisks indicate a significant difference between wild-type cells and knockdown cells (P < 0.05).
3.2. Cell activation
To determine whether IL-16 could activate A549 cells in the presence or absence of CD9, WT and KD cells were stimulated with either IL-1β for 10 min or IL-16 for up to two hours. IL-1β has been shown to lead to the phosphorylation of p-38 and JNK in human A549 cells [4]. No phosphorylated p-38 or p-JNK was detected with IL-16 stimulation; however, IL-1β was able to phosphorylate p-38 and p-JNK (Fig. 2A). Moreover, the levels of total p-38 and JNK were not different between the WT and KD cells and did not change with IL-16 stimulation. These data indicate that acute IL-16 stimulation does not lead to the activation of a stress response in A549 cells.
Fig. 2.
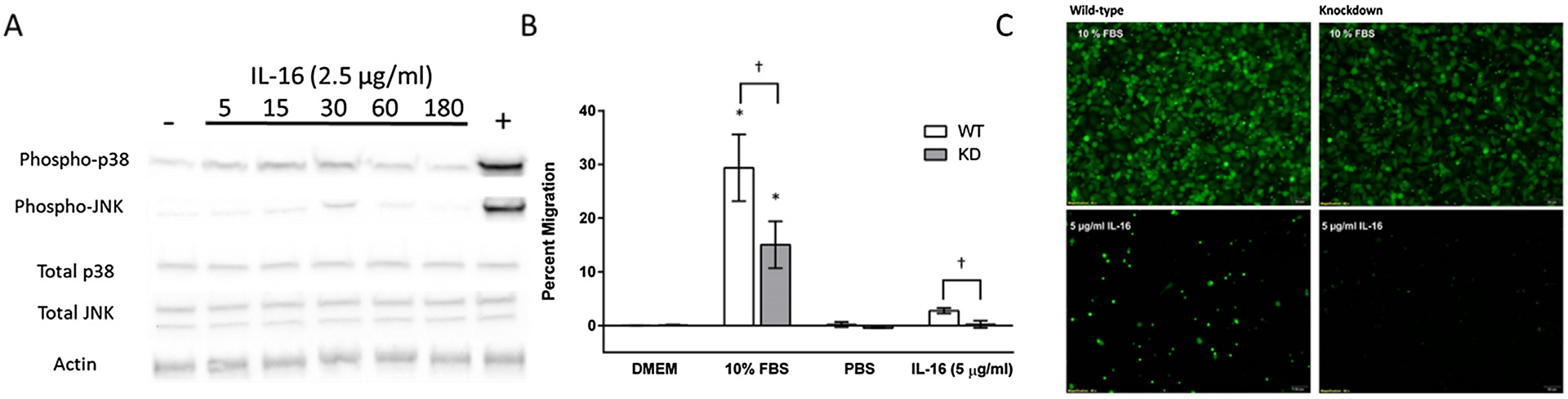
Cell activation and migration in the presence of IL-16. (A) Representative western blots from wild-type cells, which were incubated in the presence of IL-16 or IL-1β (positive control) and then probed for phosphorylated JNK and p38, total JNK and p38, and Actin. (B) Wild-type and knockout cells (5*106 cells/transwell) were incubated in the apical chamber and chemoattractants were added to the basal chambers for 20 h. Cells were stained 4 μg/mL Calcein AM and incubated for 1 h at 37 °C, 5% CO2. Fluorescence was read at wavelengths of 494/517 nm (Ex/Em). Data presented are mean relative fold change ± SEM (n = 5–9). (C) To ensure that fluorescence was correlated with the number of migrating cells, images from each transwells were obtained through fluorescence microscopy. Asterisk indicate a significant difference between control cells and cells exposed to 10% FBS (positive control) (P < 0.05). Daggers indicate a significant difference between wild-type cells and knockdown cells exposed to 10% FBS or IL-16 (P < 0.05).
3.3. Migration
Chemotaxis was quantified in both WT and KD cells in the presence of IL-16 or 10% FBS. Migration was quantified through a fluorescent migration assay using FluoroBlok transwells as described in the Materials and Methods. Both WT and KD cells significantly increased the percent of migrating cells in response to FBS compared to unstimulated controls (Fig. 2B and C). However, there was a significant difference in the percent of migrating cells between the WT and KD cells in response to 10% FBS. Without CD9 expression, knockdown cells had a significant decrease in migration compared to the wild-type (29% compared to 15% migration) in response to FBS. In addition, in the presence of IL-16 (5 μg/ml), wild-type cells had a significant increase in migration compared to the KD cells (2.8% compared to 0.2% migration). These data strongly support the necessary role of CD9 in cell migration and the hypothesis that IL-16 is a chemoattractant that can bind CD9 leading to actin reorganization and cell migration.
4. Conclusions
The present study confirms the role of CD9 in cell migration and supports the hypothesis that CD9 is an alternative receptor for IL-16. Our results demonstrate that CD9 is required in cell migration specifically towards IL-16 and this cellular effect is not mediated by either p-38 or p-JNK.
Acknowledgement
We would like to thank Rollin Leavitt for his technical assistance with this project.
Funding Sources
This project has been supported by the Faculty Development Grant for Traditional Research at Fort Lewis College (DB), Faculty Development Grant for Teaching, Innovation and Pedagogy at Fort Lewis College (DB and SF), and the Minority Access to Research Careers - Undergraduate Student Training in Academic Research Program (Grant 5T34GM092711-03) (DB).
Abbreviations:
- IL-16
interleukin-16
- CRISPR
clustered regularly interspaced short palindromic repeat
References
- [1].Blake DJ, Reese CM, Garcia M, Dahlmann EA, Dean A, Soluble extracellular Klotho decreases sensitivity to cigarette smoke induced cell death in human lung epithelial cells, Toxicol. In Vitro 29 (2015) 1647–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Cruikshank WW, Greenstein JL, Theodore AC, Center DM, Lymphocyte chemoattractant factor induces CD4-dependent intracytoplasmic signaling in lymphocytes, J. Immunol 146 (1991) 2928–2934. [PubMed] [Google Scholar]
- [3].Fenster CP, Chisnell HK, Fry CR, Fenster SD, The role of CD4-dependent signaling in interleukin-16 induced c-Fos expression and facilitation of neurite outgrowth in cerebellar granule neurons, Neurosci. Lett 485 (2010) 212–216. [DOI] [PubMed] [Google Scholar]
- [4].Jang BC, Lim KJ, Paik JH, Kwon YK, Shin SW, Kim SC, Jung TY, Kwon TK, Cho JW, Baek WK, Kim SP, Suh MH, Suh SI, Up-regulation of human beta-defensin 2 by interleukin-1beta in A549 cells: involvement of PI3K, PKC, p38 MAPK, JNK, and NF-kappaB, Biochem. Biophys. Res. Commun 320 (2004) 1026–1033. [DOI] [PubMed] [Google Scholar]
- [5].Krautwald S, IL-16 activates the SAPK signaling pathway in CD4+ macrophages, J. Immunol 160 (1998) 5874–5879. [PubMed] [Google Scholar]
- [6].Massin F, Rubinstein E, Faure GC, Martinet Y, Boucheix C, Béné MC, Tetraspan and beta-1 integrins expression pattern of the epithelial lung adenocarcinoma cell line A549 and its sensitivity to divalent cations, Cytometry B Clin. Cytom 60 (2004) 31–36. [DOI] [PubMed] [Google Scholar]
- [7].Mathy NL, Bannert N, Norley SG, Kurth R, Cutting edge: CD4 is not required for the functional activity of IL-16, J. Immunol 164 (2000) 4429–4432. [DOI] [PubMed] [Google Scholar]
- [8].Qi JC, Wang J, Mandadi S, Tanaka K, Roufogalis BD, Madigan MC, Lai K, Yan F, Chong BH, Stevens RL, Krilis SA, Human and mouse mast cells use the tetraspanin CD9 as an alternate interleukin-16 receptor, Blood 107 (2006) 135–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F, Genome engineering using the CRISPR-Cas9 system, Nat. Protoc 8 (2013) 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Rand TH, Cruikshank WW, Center DM, Weller PF, CD4-mediated stimulation of human eosinophils: lymphocyte chemoattractant factor and other CD4-binding ligands elicit eosinophil migration, J. Exp. Med 173 (1991) 1521–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Richmond J, Tuzova M, Cruikshank W, Center D, Regulation of cellular processes by interleukin-16 in homeostasis and cancer, J. Cell Physiol 229 (2014) 139–147. [DOI] [PubMed] [Google Scholar]
- [12].Yadav S, Shi Y, Wang H, IL-16 effects on A549 lung epithelial cells: dependence on CD9 as an IL-16 receptor? J. Immunotoxicol 7 (2010) 183–193. [DOI] [PubMed] [Google Scholar]
- [13].Yousefi S, Ma XZ, Singla R, Zhou YC, Sakac D, Bali M, Liu Y, Sahai BM, Branch DR, HIV-1 infection is facilitated in T cells by decreasing p56lck protein tyrosine kinase activity, Clin. Exp. Immunol 133 (2003) 78–90. [DOI] [PMC free article] [PubMed] [Google Scholar]