

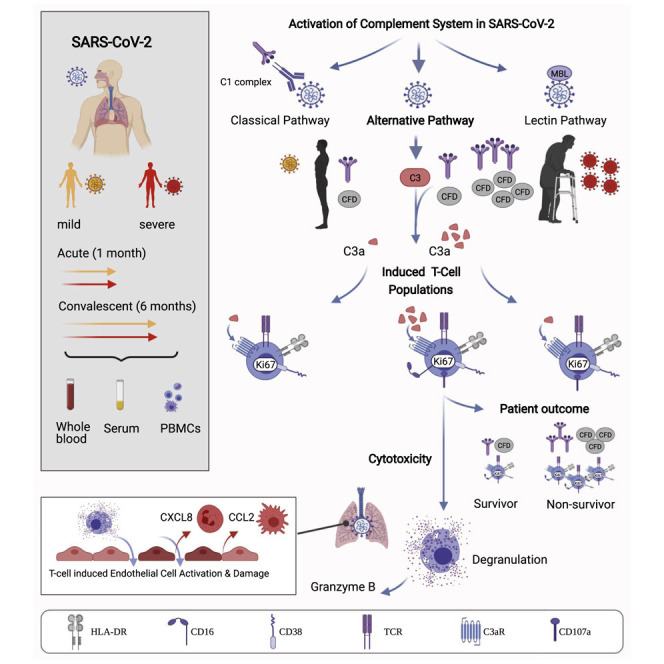
Abstract
Severe COVID-19 is linked to both dysfunctional immune response and unrestrained immunopathology, and it remains unclear whether T cells contribute to disease pathology. Here, we combined single-cell transcriptomics and single-cell proteomics with mechanistic studies to assess pathogenic T cell functions and inducing signals. We identified highly activated CD16+ T cells with increased cytotoxic functions in severe COVID-19. CD16 expression enabled immune-complex-mediated, T cell receptor-independent degranulation and cytotoxicity not found in other diseases. CD16+ T cells from COVID-19 patients promoted microvascular endothelial cell injury and release of neutrophil and monocyte chemoattractants. CD16+ T cell clones persisted beyond acute disease maintaining their cytotoxic phenotype. Increased generation of C3a in severe COVID-19 induced activated CD16+ cytotoxic T cells. Proportions of activated CD16+ T cells and plasma levels of complement proteins upstream of C3a were associated with fatal outcome of COVID-19, supporting a pathological role of exacerbated cytotoxicity and complement activation in COVID-19.
Keywords: COVID-19, T cells, complement, cytotoxicity, immunopathology
Graphical abstract
Generation of the C3a complement protein fragment by SARS-CoV-2 infection drives differentiation of a CD16-expressing T cell population that associates with severe COVID-19 disease outcomes.
Introduction
Severe acute respiratory distress syndrome coronavirus 2 (SARS-CoV-2) infection in humans causes a diverse spectrum of clinical manifestations, ranging from asymptomatic disease to acute respiratory distress syndrome (ARDS) and multi-organ failure (Miyazawa, 2020).
In addition to direct virus-induced injury to the respiratory system and other organs, increasing evidence suggests that the immune response evoked by SARS-CoV-2 infection contributes to the pathophysiology of Coronavirus disease (COVID-19), particularly during severe disease courses (Gustine and Jones, 2021; McKechnie and Blish, 2020; Vabret et al., 2020).
Both CD4+ T helper cells and CD8+ cytotoxic T lymphocytes (CTL) contribute to the control of respiratory viral infections. Consequently, SARS-COV-2-specific CD4+ and CD8+ T cells have been associated with milder COVID-19 (Jacob, 2020; Tan et al., 2021). Although this had been interpreted as a predominantly protective role of T cell responses (Rydyznski Moderbacher et al., 2020), complementary data do not unequivocally support this idea (Feng et al., 2020; Mathew et al., 2020; Peng et al., 2020; Thieme et al., 2020). The extent of SARS-CoV-2-specific T cell responses could not be directly tied to disease severity, with high T cell numbers not necessarily translating into mild COVID-19 (Le Bert et al., 2021). In fact, the number of SARS-CoV-2-specific CD4+ and CD8+ T cells were found to be comparable or even higher in COVID-19 patients displaying severe versus mild disease (Feng et al., 2020; Le Bert et al., 2021; Mathew et al., 2020; Peng et al., 2020; Thieme et al., 2020).
A higher state of T cell activation in all T cell compartments (CD4+, CD8+, double-negative) in patients progressing to severe COVID-19 was reported (Zenarruzabeitia et al., 2021).
Interstitial T cell infiltration is observed in pathological specimens of COVID-19 pneumonia along with macrophage accumulation in the alveolar space, and it has been hypothesized that infiltrating T cells also contribute to alveolar wall damage and endothelial cell injury known as lymphocytic endotheliitis (Miyazawa, 2020; Varga et al., 2020).
All this argues for a complex relationship between T cell immune responses and disease outcome during COVID-19 beyond a mere quantitative influence. It is likely that additional factors present in the microenvironment shape the quality of T cell responses and consequently impact pathology. Therefore, it is important to identify whether and which T cell subsets have a pathogenic role. Also, mechanisms by which potentially pathogenic T cells are induced need to be revealed, as studies on this matter are currently lacking (Yan et al., 2021).
Here, we combined single-cell proteomics and transcriptomics with mechanistic studies to reveal alterations in the T cell compartment, their upstream signals, and functional relevance, which explain important immunopathological features observed in severe COVID-19. Mass cytometry (cytometry by time of flight [CyTOF]) and single-cell RNA-seq (scRNA-seq) combined with VDJ-sequencing (VDJ-seq)-based T cell clonotype identification were used to determine COVID-19- and severity-specific alterations in the T cell compartment. In addition to the severity-independent formation of highly activated HLA-DRhiCD38hiCD137+Ki67+ T follicular helper (TFH)-like cells and CD8+ CTLs in COVID-19, we describe a C3a-driven induction of activated CD16 expressing cells in patients with severe COVID-19. These T cells display increased immune complex-mediated, TCR-independent cytotoxicity causing activation and release of chemokines by lung endothelial cells. This mechanism may contribute to the profound lung damage and endotheliitis observed in patients with severe COVID-19.
Results
Profound T cell activation and induction of CD16 expressing CD4+, CD8+ TCRab+, and TCRgd+ T cells in severe COVID-19
We performed CyTOF of whole blood samples from mild and severe COVID-19 patients during the acute and convalescent phase, alongside patients with other acute respiratory infections (Flu-like illness ), as well as patients chronically infected by human immunodeficiency virus (HIV) or hepatitis B (HBV) and controls (Figure 1 A). We previously reported that T cell numbers were reduced in acute COVID-19 compared with controls (Schulte-Schrepping et al., 2020). To further interrogate the T cell space, acquired T cells (CD45+CD3+CD19−CD15−) were pre-gated into CD4+ T helper cells (CD3+, CD8−TCRgd−), CD8+ CTLs (CD3+, CD8+TCRgd−), and TCRgd+ (CD3−, CD8−TCRgd+) cells. Unsupervised clustering analysis on samples from control, FLI, HIV, HBV, and acute COVID-19 using 29 surface antigens and the proliferation marker Ki67 partitioned the pre-gated T helper cells, CTLs, and TCRgd+ T cells into 19, 15, and 14 individual cell clusters, respectively (Figures 1B–1D). A significant proportion of cells from patients with mild or severe COVID-19 were clearly separated in Uniform Manifold Approximation and Projection for Dimension Reduction (UMAP) space from those of the other patient groups or controls (Figure 1B).
Figure 1.

Accumulation of HLA-DRhiCD38hi highly activated but also CD16 expressing CD4+ and CD8+ T cells in severe COVID-19
(A) Overview of the study cohort and methodological pipeline. Samples were collected from mild and severe COVID-19 patients during the acute and convalescent phase enrolled in Berlin (cohort 1), Bonn (cohort 2), or Aachen (cohort 3), patients suffering from other acute respiratory infections (FLI, being chronically infected HIV, or HBV, patients with non-infectious ARDS as well as controls. CyTOF and scRNA-seq combined with VDJ-seq-based T cell clonotype identification were used to determine COVID-19 as well as severity-specific alterations in the T cell compartment. The obtained results together with serum proteomics and in situ immunofluorescence data were used to develop hypotheses on their functional properties and inducing mechanisms, which were tested in ex vivo cultures. Detailed sample information included in all reported assays can be found in Table S1.
(B and C) UMAPs generated of CD4+ (left), CD8+ (middle), and TCRgd+ (right) T cells from CyTOF. Cells are colored according to donor (B) or cluster (C) origin. For visualization purposes, each UMAP shows 30,000 cells.
(D) Heatmap of CyTOF data (covering CD4+ (left panel), CD8+ (middle panel), and TCRgd+ (right panel) T cells. Z score standardized staining intensity of each marker (rows) per cluster (1–48, in columns, lower part). Clusters were grouped into metaclusters, as defined by the numbers 1–13 (in columns, upper part). Significance levels of differential cluster frequency for the following groups: controls (n = 9), FLI (n = 8), HIV (n = 6), HBV (n = 5), mild acute COVID-19 (n = 28), and severe acute COVID-19 (n = 35). Kruskall-Wallis test and post hoc Dunn’s multiple comparison test. All combinations where tested, only comparisons with healthy controls are shown.
(E) Box plots of CD4+ (7, 8, 18) and CD8+ (25, 26) T cell clusters determined by CyTOF generated from controls (n = 9), FLI (n = 8), HIV (n = 6), HBV (n = 5), mild acute COVID-19 (n = 20), and severe acute COVID-19 (n = 23) patient samples. Kruskall-Wallis test and post hoc Dunn’s multiple comparison test. KW∗: adjusted p value (Benjamini-Hochberg) of a Kruskal-Wallis test. All combinations where tested, only comparisons with healthy controls are shown (∗p < 0.1, ∗∗p < 0.01,∗∗∗p < 0.001,∗∗∗∗p < 0.0001).
Samples of COVID-19 patients collected during the first three weeks after symptom onset independent of the severity were characterized by increased proportions of CD4+ T cell cluster 7 (CD38hiHLA-DR+Ki67+ICOS+) compared with other infections or controls, whereas the abundance of cluster 18 (CD27−CD25+) was lower (Figures 1D and 1E). Cluster 7 T cells are characterized by high expression of activation markers such as HLA-DR, CD38, CD137, CD69, and Ki67. Furthermore, the T cells in this cluster express CXCR5, ICOS, and PD-1, resembling TFH-like cells. The increase of TFH-like cells was not seen for FLI, neither HIV, nor HBV. T helper cells in severe COVID-19 patients showed higher proportions of cluster 8 T cells (CD4+, highly activated natural killer T cell [NKT-like]), which in addition to the expression of CD38, HLA-DR, CD137, CD69, Ki67, and CXCR3, were characterized by high levels of CCR6 and CD16. Those T cells belong to the CD62L+CD45RO+ central memory metacluster, reflecting a more recent differentiation. In contrast, in patients suffering from chronic HIV or HBV infection we observed a tendency toward a higher proportion of cluster 19 (TIGIT+CD38+CD16+). T cells from this cluster were classified as terminally differentiated RA+ cells, expressed low levels of CD16, and showed no signs of recent activation.
This activation pattern was not restricted to the CD4+ T cell compartment and was also detectable in CD8+ CTLs and TCRgd+ T cells. Both mild and severe COVID-19 patients displayed increased proportions of cluster 25 T cells (CD8+CD38hiHLA-DR+Ki67+) in comparison with the other groups (Figures 1D and 1E). Severe COVID-19 was further characterized by increased abundance of cluster 26 T cells (CD8+, highly activated NKT-like), also expressing high levels of CCR6 and CD16. We observed that severity explained on average 96% of the variance in cluster frequencies, whereas age only 4% (Figure S1A), indicating that the increased proportions of activated CD16+ T cells are mainly due to disease severity and not to age.
Figure S1.

Weekly changes in CD4+ and CD8+ T cell cluster composition in mild versus severe COVID-19, related to Figure 1
(A) Percentage of variance in the frequency of activated CD16+ CyTOF clusters C8 and C26 explained by age and severity.
(B) Box plots of CD16+HLA-DR+ CD3+ T cells determined by flow cytometry (cohort 2) of samples from controls (n = 11) as well as mild COVID-19 acute (n = 5) and severe COVID-19 acute (n = 9) patients collected during the acute infection (for each donor the first sample available was selected) or samples collected during week two and three post-symptom onset only (right panel, control = 11, mild week 2 = 1, mild week 3+ = 4, severe week 2 = 4, severe week 3+ = 8). KW∗ shows the adjusted p value (Benjamini-Hochberg) of a Kruskal-Wallis test. The abundance of each cluster was compared between severity groups via adjusted Dunn’s post hoc test (Benjamini-Hochberg).
(C) UMAPs generated of CD4+ and CD8+ T cells from mass cytometry data of samples from COVID-19 patients collected during week one, two, and three post-symptom onset. Cells are colored according to (left) disease severity (yellow, mild COVID-19 acute phase; red, severe COVID-19 acute phase), and (right) patient ID.
(D) Box plots of CD4+ (7, 8, 18) and CD8+ (25, 26) T cell clusters determined by mass cytometry (whole blood, cohort 1) of samples from controls (n = 9), FLI (n = 8), HIV (n = 6), HBV (n = 5) as well as acute mild COVID-19 week 1, (n = 5), week 2 (n = 8), week 3+ (n = 11), and acute severe COVID-19 week 1 (n = 5), week 2 (n = 6), week 3+ (n = 18) patients collected during week 1, 2, and 3 post-symptom onset. KW∗ shows the adjusted p value (Benjamini-Hochberg) of a Kruskal-Wallis test. The abundance of each cluster was compared between severity groups via adjusted Dunn’s (Benjamini-Hochberg) for clusters with KW∗ < 0.1. All combinations where tested, only comparisons with healthy controls and within COVID-19 disease are shown (∗p < 0.1, ∗∗p < 0.01, ∗∗∗p < 0.001,∗∗∗∗p < 0.0001).
We confirmed the existence of a CD3+CD16+HLA-DR+ T cell population by flow cytometry in a second cohort of COVID-19 patients (Figure S1B).
Time-dependent analysis of the CD4+ and CD8+ T cell clusters revealed a trend to faster accumulation of the activated CD38hiHLA-DR+Ki67+-expressing clusters 7 and 25 during the first week after SARS-CoV-2 infection in severe versus mild COVID-19 (Figures S1C and S1D). In contrast, the highly activated CD16+ NKT-like CD4+ and CD8+ T clusters appeared only during the second week of infection.
Our findings concerning the identification of highly activated and proliferating T helper cells and CTLs after SARS-CoV-2 infection is in line with recent reports (Mathew et al., 2020; Rydyznski Moderbacher et al., 2020; Stephenson et al., 2021). Strikingly, we uncovered a unique activation and differentiation program in a subset of T cells across all three major T cell compartments (T helper cells, CTLs, and TCRgd+ T cells), characterized by CD16 and CCR6 expression in severe COVID-19. As the increase of highly activated CD16+ T cell clusters in severe COVID-19 versus control or mild COVID-19 samples was only significant for the CD4+ and CD8+ T cells and not the TCRgd+ T cells, we focused our subsequent analyzes on the former two compartments.
Single-cell transcriptomics reveal shift toward disproportionally high cytotoxic and degranulation potential of T cells in severe COVID-19
To obtain functional information on the COVID-19- and severity-specific T cell clusters, we performed scRNA-seq analysis of peripheral blood mononuclear cell (PBMC) samples as well as purified CD38 expressing T cells from acutely infected and convalescent mild and severe COVID-19, as well as FLI, HBV, and controls. To align the CyTOF and scRNA-seq T cell clusters, we applied a feature-based cluster annotation approach on the whole T cell space from PBMC and CD38+ T cell libraries (STAR Methods), which resulted in 17 clusters (Figures 2A and 2B). The proportion of T cells belonging to clusters 7, 8, and 10 was higher in COVID-19 patients compared with FLI or HBV patients as well as controls (Figures 2D and 2E). We observed other T cell clusters with FCGR3A expression (clusters 9, 11, 12, and 13). Of these, only cluster 9 transcribed SELL (CD62L) and displayed a central memory phenotype, whereas clusters 11, 12, and 13 T cells displayed a more advanced differentiation profile. Overall, cluster 7 contains TFH-like cells similar to the CyTOF cluster 7, whereas cluster 8, 9, and 10 most likely contain a mixture of highly activated and CD16+ NKT-like cells with similarities to the CyTOF clusters 8, 24, and 25. Next, we performed a Gene Ontology (GO) enrichment analysis across clusters 7, 8, 9, and 10, comparing mild and severe COVID-19 T cells (Figure 2F). Interestingly, whereas T cells from mild patients showed an enrichment for cellular responses to type I interferon and antiviral defense, we observed a specific enrichment of genes involved in degranulation in severe COVID-19 T cells. This selective enrichment was validated by a gene set enrichment analysis (GSEA) for genes belonging to the GO terms “response to type I interferon” and “defense response to virus,” which were enriched in mild, and for genes belonging to a cytotoxicity signature, which were enriched in severe COVID-19 T cells (all p_adj < 0.005, Figure 2G). Indeed, several genes that are known to promote degranulation (LAMP1 and STX11) or exert cytotoxic potential (PRF1, GZMB, GZMH, and GZMK) were increased in severe COVID-19 across clusters 7, 8, 9, and 10 T cells (Figures 2H and S2B) or the whole T cell space (Figure S2C). We validated these findings in scRNA-seq data from our second cohort (Figure S3 ). Here, clusters 6 and 7 contained CD4+ T cells transcribing ICOS, CD40LG, PDCD1, and CXCR5 and, thus, resembled cluster 7 T cells from cohort 1, although the expression level of CD38 and HLA-DR genes seemed to be lower. Clusters 10 and 13 from the second cohort resembled clusters 10 and 8 in cohort 1, respectively. Furthermore, GSEA similarly showed increased expression of genes mediating cytotoxicity in severe COVID-19 T cells across these clusters (Figures S3E and S3F).
Figure 2.

Single-cell transcriptomics of T cells during acute mild and severe COVID-19
(A) UMAP of T cell clusters from controls (n = 6), FLI (n = 8), HBV (n = 4), mild COVID-19 (n = 9), and severe COVID-19 (n = 10) patients.
(B) Heatmap showing the Z score standardized gene expression (rows) per T cell cluster (columns).
(C and D) UMAPs as shown in (A) with superimposed CD4, CD8A, FCGR3A, MKI67, CD38, and HLA-DRA expression (C), with cells colored according to disease group origin. For visualization purposes, cells were downsampled to 10,000 cells per disease group.
(E) Box plots of a selection of scRNA-seq T cell clusters whose abundances are higher in both mild and severe COVID-19 compared with other severity groups (the analyzed number of patients are specified in the legend of ). KW, KW∗: raw and adjusted p value (Benjamini-Hochberg) of a Kruskal-Wallis test, respectively.
(F) Bar plot indicating the negative log2-transformed adjusted p value (Benjamini-Hochberg) of the 20 most significant enriched pathways that are (top) upregulated in mild COVID-19 acute phase, compared with severe COVID-19 acute phase, (bottom) vice versa. Pseudobulk gene expression was calculated per sample among scRNA-seq T cell clusters 7, 8, 9, and 10.
(G) Enrichment plots from GSEA performed on the ranked gene list of the comparison severe versus mild COVID-19. The graph shows the mapping of the signature genes on the ranked gene list. The curve corresponds to the running sum of the weighted enrichment score (ES). The ranked gene list was calculated from the normalized pseudobulk expression data of severe and mild COVID-19 acute phase among scRNA-seq T cell clusters 7, 8, 9, and 10.
(H) Box plots of the average log2-transformed expression among T cell clusters 7, 8, 9, and 10 from mild (n = 9) and severe (n = 10) COVID-19 acute samples, for three genes included in the cytotoxicity signature (LAMP1, GZMB, and PRF1).
Figure S2.

scRNA-seq T cell clusters and their cytotoxic gene signature in samples from COVID-19 patients or patients with other infections, related to Figure 2
(A) Box plots of the percentage of cells in the remaining scRNA-seq T cell clusters generated from controls (n = 6), FLI (n = 8), HBV (n = 4), mild COVID-19 (n = 9), and severe COVID-19 (n = 10) patient samples of cohort 1. KW∗ shows the adjusted p value (Benjamini-Hochberg) of a Kruskal-Wallis test.
(B) Box plots of the average log2-transformed expression of all genes defining the cytotoxicity gene signature in cells belonging to clusters 7, 8, 9, and 10, generated from controls (n = 6), FLI (n = 8), HBV (n = 4), mild COVID-19 acute phase (n = 9), and severe COVID-19 acute (n = 10) patient samples of cohort 1.
(C) Box plots of the average log2-transformed expression of all genes defining the cytotoxicity gene signature in cells belonging to all T cell clusters generated from controls (n = 6), FLI (n = 8), HBV (n = 4), mild COVID-19 (n = 9), and severe COVID-19 (n = 10) patient samples of cohort 1.
Figure S3.

scRNA-seq T cell cluster and their cytotoxic gene signature in samples from COVID-19 patients or controls of cohort 2, related to “scRNA-seq data analysis of Rhapsody data (cohort 2)” in STAR Methods
(A) UMAP of the T cell subset from the PBMC dataset of Schulte-Schrepping et al. (2020), including controls (n = 13), mild COVID-19 (n = 21) and severe COVID-19 (n = 29) patients.
(B) Heatmap of selected marker expression of the T cell subset from the PBMC dataset of Schulte-Schrepping et al. (2020), including controls (n = 13), mild COVID-19 (n = 21) and severe COVID-19 (n = 29) patients.
(C) UMAP of T cell clusters as shown in (A) with cells colored according to disease group origin: blue, controls (n = 13); yellow, mild COVID-19 acute phase (n = 21); red, severe COVID-19 acute phase (n = 29).
(D) Box and whisker (10–90 percentile) plots of a selection of cohort 2 scRNA-seq T cell clusters, generated from controls (n = 13), mild COVID-19 acute phase (n = 8) and severe COVID-19 acute phase (n = 9) patient samples. Selected clusters show a TFH phenotype (cluster 6 and 7) or display FCGR3A expression and increased frequency in severe COVID-19 (cluster 10 and 13) generated from controls (n = 13), mild COVID-19 acute phase (n = 8) and severe COVID-19 acute phase (n = 9) patient samples. KW∗ shows the adjusted p value (Benjamini-Hochberg) of a Kruskal-Wallis test. The abundance of each cluster was compared between severity groups via adjusted Dunn’s post hoc test (Benjamini-Hochberg). When multiple samples for the sample patients were available only the earliest sample was used for visualization and statistical testing. All combinations where tested, only comparisons with healthy controls are shown (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001).
(E) GSEA performed on the ranked gene list of the comparison severe versus mild COVID-19. The graph shows the mapping of the signature genes on the ranked gene list. The curve corresponds to the running sum of the weighted enrichment score (ES). The ranked gene list was calculated from the normalized pseudobulk expression data of severe and mild samples weeks 1 and 2 post symptoms onset across clusters 6, 7, 10, and 13.
(F) Dot plot of the expression of the genes included in the “cytotoxicity” and “response to type I interferon” signatures in control, mild COVID-19, and severe COVID-19 samples in weeks 1 and 2 post symptoms onset across clusters 6, 7, 10, and 13. The dots are colored by the scaled gene expression across the groups and the size is proportional to the ratio of cells expressing the specific gene.
Thus, scRNA-seq analysis of samples from two independent cohorts supported our finding of a subset of activated CD16+ T cells across the major T cell compartments in severe COVID-19 and identified an increase in cytotoxicity-associated transcriptional programs.
CD16-mediated degranulation of CD8+ T cells causes chemokine release by endothelial cells
CyTOF and scRNA-seq analyses identified two main T cell activation features: (1) formation of highly activated, proliferating TFH-like CD4+ cells and CXCR3 expressing CTLs independent of disease severity, and (2) activated CD16+ T cells specific for severe COVID-19. Because TFH cells promote B cell help (Crotty, 2014) and because we observed a trend for a faster formation of activated TFH-like cells in severe COVID-19 patient samples, we tested whether SARS-CoV-2-specific antibody responses were more pronounced in those patients. Serum concentrations of SARS-CoV-2-specific IgA, but particular IgG levels were higher in severe COVID-19 (Figure 3 A), which is in line with previous reports (Garcia-Beltran et al., 2021). Furthermore, maximal antibody levels determined during the second week post-symptom onset correlated positively with the cell proportions of the TFH-like CyTOF cluster 7 (Figure 3B), whereas the CD16+CD4+ T cell cluster 8 did not correlate with antibody levels (IgA: R2 = 0.11, p = 0.19; IgG: R2 = 0.18, p = 0.08; data not shown). Although severe COVID-19 patients developed a faster antibody response, all study cohort patients were reactive (optical density ratios > 1.1) at later time points.
Figure 3.

Increased degranulation and cytotoxic potential of T cells from severe COVID-19
(A) Box and whisker (5–95 percentile) plots of SARS-CoV-2-specific IgG and IgA antibody levels detected in serum samples from mild (n = 15) and severe (n = 17) COVID-19 patients collected between day 10 and 14 post symptom onset. Wilcoxon test ∗∗p < 0.01.
(B) Linear regression analysis of TFH cell proportions (CyTOF cluster 7) determined in samples collected from mild (n = 8) and severe (n = 11) COVID-19 (cohort 1) during day 5 and 14 post-symptom onset and SARS-CoV-2-specific IgG and IgA serum levels.
(C) Box and whisker (min − max) plots summarizing the intracellular granzyme B expression (unstimulated) of CD8+ T cells from PBMCs of mild (n = 21) and severe (n = 28) COVID-19 patients as well as controls (n = 21). Kruskal-Wallis & post hoc Dunn’s multiple comparison test ∗p < 0.05, ∗∗∗p < 0.001.
(D) Box and whisker (5–95 percentile) plots summarizing the degranulation capacity of CD8+ T cells from PBMCs of mild (n = 21) and severe (n = 28) COVID-19 patients as well as controls (n = 20) defined by their increase of cell surface CD107a expression upon stimulation with anti-CD16 antibody-coated or isotype-coated beads. Multiple Mann-Whitney test ∗p < 0.05, ∗∗∗p < 0.001.
(E) Scatter plot of the degranulation capacity from PBMCs of severe COVID-19 patients (n = 12) upon stimulation with spike-protein-coated beads pre-incubated with control serum, with spike-protein-coated beads pre-incubated with COVID-19 serum or with anti-CD16 antibody-coated beads. Kruskal-Wallis and post hoc Dunn’s multiple comparison test ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
(F) Enrichment of CD137+CD69+ cells in activated CD16− (CD38+CD16−) and activated CD16+ (CD38+CD16+) over the total CD8+ T cell compartment upon restimulation of PBMC samples from mild (n = 5) and severe (n = 7) COVID-19 patients with a SARS-CoV-2 peptide pool. Enrichment was calculated by dividing the proportions of CD137+CD69+ T cells in non-activated CD16− (CD38+CD16−) and activated CD16+ (CD38+CD16+) by the proportions of total CD8+ T cells. Friedmann test & post hoc Dunn’s multiple comparison test. ∗p < 0.05, ∗∗p < 0.01.
(G) Box and whisker (5–95 percentile) plots summarizing the normalized release of CXCL8 and CCL2 by primary lung endothelial cells co-cultured with CD8+ T cells enriched from PBMCs of mild (n = 6) and severe (n = 5) COVID-19 patients as well as non-infected controls (n = 5) upon stimulation with ConA or anti-CD16 antibody-coated beads. Wilcoxon test ∗p < 0.05.
(H) Endothelial cell resistance upon stimulation with ConA alone (n = 5) or additional co-culture with CD8+ T cells enriched from PBMCs of mild (n = 6) and severe (n = 5) COVID-19 patients as well as non-infected controls (n = 5). Kruskal-Wallis test ∗p < 0.05.
(I) Representative immunofluorescence staining of CD3 (green) and CD16 (red) in autopsy lung tissues of patients without lung pathology, with COVID-19, ARDS, or influenza pneumonia.
(J) Quantification of CD3/CD16 double-positive T lymphocytes per mm2 in the autopsy cohorts of deceased patients without lung pathology (n = 4) compared with COVID-19 (n = 13), ARDS (n = 8) and influenza pneumonia (n = 6). The COVID-19 cohort was separated into early stage (death after 7–14 days after first symptoms), mid stage (15–30 days after symptom onset), and late stage (>35 days). One-way ANOVA; ∗∗, p < 0.01.
Next, we investigated the functional properties related to the CD16+ CD4+ and CD8+ clusters. As indicated by scRNA-seq, samples from patients with severe COVID-19 contained significantly more Granzyme B expressing CD8+ T cells compared with controls (Figure 3C). Because CD16 is known to mediate antibody-mediated degranulation of NK cells (Moretta et al., 2008), we tested whether T cells from patients with severe COVID-19 display enhanced CD16-dependent degranulation potential. As a surrogate for immune complex-mediated stimulation, we assessed cell surface CD107a in PBMCs from mild or severe patients as well as controls after 6 h incubation with anti-CD16 antibody or isotype antibody-coated beads (Figure 3D). Stimulation with anti-CD16 elicited strong degranulation of CD8+ T cells from patients with severe COVID-19, compared with T cells from non-infected controls (Figure 3D). T cells from patients with mild COVID-19 showed intermediate degranulation potential. As a second approach to assessing immune-complex-mediated degranulation, we stimulated T cells with SARS-CoV-2 spike-protein-coated beads complexed with patient-derived serum. Serum from COVID-19 patients induced similar degranulation as anti-CD16 antibody-coated beads, which was not observed with serum from uninfected controls (Figure 3E).
We also investigated SARS-CoV-2 specificity of activated CD16+ T cells. PBMCs from mild and severe COVID-19 patients were restimulated with a SARS-CoV-2 peptide pool and the proportion of SARS-CoV-2-reactive CD8+CD38+CD16+ T cells (CD137+CD69+) was determined after 24 h. On average 5% of the T cells upregulated CD137 and CD69 expression. The level of SARS-CoV-2- specificity among CD38+CD16+ T cells was similar in patients with severe and mild COVID-19, yet the absolute number of activated CD16+ T cells was 4-fold higher in patients with severe disease. When comparing the frequencies of SARS-CoV-2 reactive T cells between the total CD8+, activated CD16− (CD38+CD16−) and activated CD16+ (CD38+CD16+) compartment, we detected a significant enrichment in activated CD16+ T cells but not activated CD16− T cells compared with total CD8+ T cells in samples of patients with severe COVID-19 (Figure 3F).
One of the most likely implications of CD16 engagement in severe COVID-19 is enhanced T cell degranulation during interaction with endothelial cells (Bagnato and Harari, 2015; Degauque et al., 2021). Indeed, based on findings from autopsies in severe COVID-19, T cell infiltration along with endothelial cell injury known as lymphocytic endotheliitis has been observed (Miyazawa, 2020; Varga et al., 2020). It is, therefore, conceivable that immune complex-mediated degranulation of CD16+ T cells contributes to endothelial injury. To test this hypothesis, we co-cultured primary lung microvascular endothelial cells with enriched non-naive CD8+ T cells isolated from patients with mild or severe COVID-19 or controls in the presence of anti-CD16 antibodies. Subsequently, we analyzed the release of inflammatory mediators (Figure 3G). Anti-CD16-triggered severe COVID-19 T cells elicited enhanced CXCL8 (IL-8) and CCL2 (MCP-1) release by co-cultured endothelial cells. Chemokines were produced by endothelial cells, as we did not observe chemokine release by anti-CD16-triggered T cells alone (below <0.4 pg/mL for CXCL8 and <1.9 pg/mL for CCL2). T cells from COVID-19 patients amplified Concanavalin A-induced loss of transendothelial electrical resistance, indicating endothelial barrier disruption, as compared with control T cells, but this effect was only significant for T cells from patients with severe COVID-19 (Figure 3H).
Complementing our findings in peripheral blood, we investigated the tissue localization of CD16+ T cells in the lungs of patients with COVID-19. We co-stained CD3 and CD16 in autopsy lung tissues from COVID-19 patients and from three different cohorts of non-COVID patients, i.e., patients without inflammatory or fibrotic lung disease (control), patients with non-infectious ARDS, and influenza pneumonia (either H1N1 or seasonal influenza) positive patients (FLU). In autopsy lung tissue from COVID-19 patients, we found an increased number of CD3+CD16+ T lymphocytes in comparison with lung tissue from the different control autopsy groups (Figures 3I and 3J). The pulmonary accumulation of CD3+/CD16+ cells was most prominent in autopsy samples from COVID-19 patients between 7 and 14 days and declined at late time points of death (Figure 3J).
We also made use of a published scRNA-seq dataset of bronchoalveolar lavage (BAL) samples from COVID-19 and non-COVID-19 pneumonia (Wauters et al., 2021) to investigate T cells and their phenotypes (STAR Methods; data and code availability). Whereas most T cell clusters contained CD38 and HLA-DRA transcribing cells, FCGR3A expression was roughly confined to clusters 3, 6, 10, and 11. The abundance of clusters 3, 10, and 11 was higher in COVID-19 samples. Cluster 10, showing the strongest enrichment in COVID-19 samples, was characterized by the highest FCGR3A expression. T cells belonging to this cluster transcribed nearly all genes of the cytotoxicity signature including LAMP1 (CD107A) at the highest level.
These findings support our hypothesis of an enhanced generation and local accumulation of CD16+, highly cytotoxic T cells in patients with severe COVID-19, which can induce activation and injury of lung endothelial cells. T-cell-induced release of chemoattractants CXCL8 and CCL2 can contribute to increased infiltration of neutrophils and monocytes in COVID-19 pneumonia.
FCGR3A expressing T cell clones induced during acute severe COVID-19 persist and maintain their increased cytotoxic potential
Knowing that CD16+ T cells from severe COVID-19 patients display enhanced cytotoxic properties potentially contributing to organ damage, we analyzed their persistence after clearance of the acute infection. We obtained VDJ sequence information in addition to the gene expression data of T cells from acute and convalescent samples of mild and severe COVID-19 as well as FLI, HBV, and controls, allowing us to study the fate of early expanded T cell clones during convalescence at months 3–8 post-symptom onset (Figures 4 A–4E). First, we analyzed whether the individual COVID-19 T cell clusters differ in their clonal enrichment. The FCGR3A expressing scRNA-seq clusters enriched in patients with COVID-19 (8, 9, and 10) showed a high level of clonal enrichment during acute COVID-19 infection, whereas it was rather low for cluster 7, containing CD4+ TFH-like cells (Figure 4A). Consequently, only clones belonging to clusters 8, 9, and 10, composed of mixed CD4/CD8A or CD8A/TCRgd T cell clusters, displayed a high degree of persistence with up to 50% of the TCR clones being recovered in convalescent samples (Figure 4B). Furthermore, clones expanded in the highly proliferating cluster 9 showed a higher persistence in severe COVID-19 patients (Figure 4B). Following the clone-specific VDJ sequences also allowed us to track their differentiation trajectory during convalescence (Figures 4C and 4D). T cells expanded during the acute infection belonging to clusters 7–10 dropped dramatically during convalescence (Figure 4D), the respective T cell clones evolved into T cells mainly identified in clusters 11 and 12, which retained FCGR3A expression (Figures 4C and 4D). GSEA further revealed the high cytotoxic potential of these clones, among clusters 6, 11, and 12 T cells of COVID-19 patients versus controls, with an evident difference even when comparing severe versus mild COVID-19 (p_adj < 0.01, Figure 4E).
Figure 4.

Time-dependent evolution and phenotype of T cell clones expanded during acute COVID-19
(A) Percentage of expanded and non-expanded T cell clones in clusters 7, 8, 9, and 10. A cell that has the same clonotype in more than 1 per 1,000 cells over all T cells per patient was considered as an expanded clone (controls, n = 6; FLI, n = 8; HBV, n = 4; mild COVID-19 acute, n = 9; severe COVID-19 acute, n = 10).
(B) Percentage of T cell clones from clusters 7, 8, 9, and 10 acute phase found in convalescent samples (mild COVID-19, n = 7; severe COVID-19, n = 6).
(C) Flow diagram representing the cluster trajectory of clones present in acute (left) and convalescent (right) COVID-19 (mild, n = 7; severe, n = 6).
(D) Percentage of cells in selected clusters for each COVID-19 sample (mild COVID-19, n = 7; severe COVID-19, n = 6).
(E) Enrichment plots from GSEA performed for the comparison (left) control versus convalescent COVID-19 (mild and severe), and (right) severe versus mild convalescent COVID-19.
(F) Box plots summarizing the percentage of cells belonging to the indicated CD4+ (7, 8, and 19) and CD8+ (25, 26, 29, and 33) CyTOF cluster in samples from mild and severe patients during the acute (mild n = 24, severe n = 29) and convalescent phase (mild n = 11, severe n = 9). KW∗: adjusted p value (Benjamini-Hochberg) of a Kruskal-Wallis test. All combinations were tested, only comparisons between the acute and convalescent phase within each COVID-19 severity group are shown (∗p<0.1, ∗∗p < 0.01, ∗∗∗p < 0.001).
Using the VDJ sequence information allowed us also to define whether the activated FCGR3A + clusters are mainly composed of NKT cells known to show innate immune cell functions (Krovi and Gapin, 2018). With the vast majority of iNKT cells expressing an identical TCRα chain (TRAV10-TRAJ18) paired to a restricted set of TCRβ chains (TRBV25), we determined the proportion of TRAV10-TRAJ18-TRBV25 pairing T cell clones across all T cell clusters. The median proportion of all T cells in our dataset expressing the TCR alpha-beta pair was 0.04%. We did not observe a major iNKT cell enrichment in the described COVID-19 T cell clusters 7–10, with the highest frequency of 0.03% in cluster 7.
Furthermore, we labeled cells from samples during COVID-19 convalescence with the CyTOF clusters previously found via k-nearest neighbor (Figures 4F and S4A). Similar to our scRNA-seq findings, we observed alterations in T cell cluster abundances during convalescence. For CD8+ T cells, we observed a strong increase in T cell proportions for cluster 29, which belongs to the effector memory meta cluster and is characterized by PD-1, TIGIT as well as CD137 expression. In line with the scRNA-seq data, proportions of highly activated (clusters 7 and 25) and CD16+ NKT-like (clusters 8 and 26) CD4+ and CD8+ clusters decreased in the convalescent phase. However, CD16+ T cells did not disappear. In the CD4+ T cell compartment, we observed an increase in the CD16+ late differentiated TEMRA cluster 19, which was more pronounced in severe COVID-19 patients. Similarly, proportions of T cells belonging to the CD8+ CD16+ TEMRA cluster 33 were higher in the convalescent phase.
Thus, CD16+ CD8+ T cells of severe COVID-19 patients persist during convalescence, adopt a more differentiated, CD62L− phenotype but maintain their high cytotoxic potential.
C3a promotes differentiation of CD16 expressing highly cytotoxic T cells
Because mortality and severe morbidity in COVID-19 disproportionately affect older individuals (Nanda et al., 2020), we investigated whether the formation of total CD16+ T cells is associated with increased age. We utilized published flow cytometry data (Kverneland et al., 2016), which revealed some CD16+ CD4+ and CD8+ T cells in controls, yet at very low proportions (Figure 5 A). Especially for CD8+ T cells, we detected significantly higher proportions of CD16+ cells in samples from older individuals, supporting our hypothesis of an age-dependent increase. Those are total CD16+ T cells, which cannot directly be compared with the proportions of activated CD16+ CyTOF clusters. It shows, however, that CD16+ T cells accumulate during aging.
Figure 5.

C3a promotes differentiation of CD16 expressing highly cytotoxic T cells
(A) Box and whisker (5–95 percentile) plots showing the proportions of CD16 expressing CD8+ and CD4+ T cells in whole blood samples of controls (n = 95) according to their age (<50 n = 59, >50 n = 36). Wilcoxon test ∗p < 0.05.
(B) Linear regression analysis of age and plasma CFD levels determined by mass spectrometry in samples from Messner et al. (2020).
(C) Box and whisker (5–95 percentile) plots summarizing the CFD plasma levels determined by mass spectrometry in samples collected from mild (week 2, n = 5; week 3, n = 7) and severe (week 2, n = 7; week 3, n = 17) COVID-19 patients during week two or three post-symptom onset. Wilcoxon test ∗p < 0.05. Longitudinal changes of plasma C3a and C5a concentrations in samples from mild (n = 12) and severe (n = 17) COVID-19 and FLI (n = 8). Kruskal-Wallis and Dunn’s multiple comparison test ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
(D) Scatter plots showing the differences in C3a binding capacity of non-naive CD4+ and CD8+ T cells enriched from PBMCs of mild (n = 3) or severe (n = 5) COVID-19 patients or controls (n = 6) determined by flow cytometry. Wilcoxon test ∗p < 0.05.
(E) Linear regression for the proportions of T cells belonging to CyTOF cluster 8 or 26 and plasma C3a levels in acute COVID-19 samples (n = 26).
(F) Box and whisker (5–95 percentile) plots showing the percentage of CD16 expressing CD4+ and CD8+ T cells upon stimulation of CD3+ T cells from controls with anti-CD3/CD28 antibodies and IL-2 in medium containing serum from mild (n = 19), severe (n = 19) COVID-19 patients or AB serum in the presence or absence of recombinant C3a (n = 8). Wilcoxon test ∗p < 0.05, ∗∗p < 0.01.
(G) Scatter plots showing the cell surface CD107a expression level of CD4+ and CD8+ T cells upon stimulation of CD3+ T cells from controls with anti-CD3/CD28 antibodies and IL-2 in medium containing serum from mild (n = 6), severe (n = 6) COVID-19 patients or AB serum (n = 6). Friedmann and Dunn’s multiple comparison test ∗p < 0.05.
(H) Scatter plots revealing the changes in the proportions of CD16 expressing CD4+ and CD8+ T cells upon stimulation of CD3+ T cells from controls with anti-CD3/CD28 antibodies and IL-2 in medium containing serum from mild or severe COVID-19 patients upon neutralization of C3a (n = 6). Wilcoxon test ∗p < 0.05.
A key feature of severe COVID-19 is increased complement generation and activation (Carvelli et al., 2020; Ma et al., 2021; Sinkovits et al., 2021). The complement system is an integral part of the innate immune defense but its effector functions also extend to an instruction of the adaptive immune system (Lubbers et al., 2017). In particular, complement enhances T cell activation as T cells are known to express receptors for C3a and C3b (Arbore et al., 2018; Hess and Kemper, 2016; West et al., 2018).
To corroborate a potential link between aging and complement activity, we analyzed the correlation of the complement component complement factor D (CFD) with age in the generation Scotland study (Figure 5B; Messner et al., 2020), which revealed a clear age-dependent increase of CFD. We then extended these findings to COVID-19 patients and showed that particularly severe COVID-19 at early time points was characterized by significantly elevated CFD plasma concentrations (Figure 5C, left panel).
CFD is a serine protease that catalyzes the formation of the active C3 convertase in the alternative pathway (Noris and Remuzzi, 2013). C3a plasma levels detected in severe COVID-19 patients during the first three weeks after symptom onset exceeded C3a levels detected in mild COVID-19 or other acute respiratory infections (Figure 5C, middle panel). C5a levels peaked in week three post-onset of symptoms but did not reach significance (Figure 5C, right panel). Both, CD4+ and CD8+ T cells from COVID-19 patients regardless of disease severity displayed higher C3a binding potential than cells from controls (Figure 5D). Finally, plasma C3a levels in COVID-19 patients measured at week 2 post-symptom onset correlated with proportions of COVID-19-specific activated CD16 expressing CD4+ and CD8+ CyTOF clusters (cluster 8+26) (Figure 5E).
Next, we stimulated enriched CD3+ cells from healthy unexposed controls with plate-bound anti-CD3/CD28 antibodies in the presence of recombinant IL-2 and serum from mild or severe COVID-19 patients or control serum. The addition of serum from severe COVID-19 patients resulted in a 10- and 20-fold increase of CD16+ T cells, which was higher compared with the increase observed when adding serum from mild COVID-19 patients (Figure 5F). Furthermore, the addition of recombinant C3a to cells stimulated in the presence of control serum enhanced the formation of CD16+ T cells (Figure 5F). The in vitro-generated T cells phenotypically and functionally resembled the T cells identified by CyTOF in severe COVID-19 patients displaying a higher degranulation potential (Figure 5G).
Finally, we tested whether C3a is responsible for the altered T cell differentiation potential of serum from patients with severe COVID-19. Neutralization of C3a reduced CD16+ T cells in most T cell differentiation cultures (Figure 5H).
In summary, complement split products, such as C3a, produced at high levels in severe COVID-19 generate an inflammatory milieu that promotes differentiation of CD16+, highly cytotoxic T cells.
High proportions of activated CD16+ T cells and plasma complement protein levels are associated with a worse outcome of COVID-19
We compared proportions of activated CD16+ T cells (STAR Methods) in patients who died from COVID-19 and those who survived. We observed significantly higher percentages of activated CD16+ TCRab+ cells among all CD4+ and CD8+ T cells in samples from severe COVID-19 patients who died (non-survivor) compared with those who survived (survivor) (Figure 6 A, right panel). Furthermore, proportions of CD3+CD16+HLA-DR+ T cells measured by multicolor flow cytometry (Figure S4B ) and activated FCGR3A expressing scRNA-seq clusters (Figure S4C) in samples from cohort 2 showed a trend to be higher in severe COVID-19 patients who deceased during follow up.
Figure 6.

Proportion of activated CD16+ T cells and plasma complement protein levels is associated with outcome in COVID-19
(A) Subgroup analysis of activated CD16+ T cell proportions according to survival of severe COVID-19 patients. Left panel, illustration summarizing the data analysis, data obtained from severe COVID-19 patients (WHO 5–7) were divided according to patient survival. Right panel, box plots of proportions of activated CD16+TCRab+ T cells determined by CyTOF (cohort 1) generated from controls (n = 9), FLI (n = 8), HIV (n = 6), HBV (n = 5), mild acute COVID-19 (n = 16), severe surviving acute COVID-19 (n = 8) and severe non-surviving acute COVID-19 (n = 7) (Wilcoxon test with Benjamini-Hochberg correction, ∗p < 0.1, ∗∗p < 0.01).
(B) Box plots showing plasma expression levels of complement proteins upstream of C3a generation determined by mass spectrometry, which were significantly different between samples from mild (n = 44) and severe (n = 66) COVID-19 patients of cohort 1 (Wilcoxon and post hoc Benjamini-Hochberg test).
(C) Subgroup analysis of plasma complement proteins according to worsening of WHO grade. Upper panel, illustration summarizing the data analysis, data obtained from samples of mild and severe COVID-19 samples were divided according to the subsequent WHO grade progression (delta WHO). Lower panel, box plots showing plasma expression levels of complement proteins, which are significantly different between samples from patients with non-worsening WHO grade (n = 91) and worsening WHO grade (n = 19) of mild or severe COVID-19 patients (cohort 1) (Wilcoxon and post hoc Benjamini-Hochberg test).
(D) Subgroup analysis of plasma complement expression levels according to survival of severe COVID-19 patients. Box plots showing plasma expression levels of complement proteins upstream of C3a generation, which are significantly different between samples from severe COVID-19 patients (WHO 5–7) who were divided according to patient survival (survivors, n = 48; non-survivors, n = 18) (Wilcoxon and post hoc Benjamini-Hochberg test).
Figure S4.

Assignment of CyTOF clusters to COVID-19 convalescent samples, related to Figure 4, survival data of cohort 2, related to Figure 6, and gating strategy of CyTOF data, related to Figure 1
(A) Exemplary graph visualizing the assignment of CD4+ T cells measured during the convalescent phase to CyTOF T cell clusters identified during acute COVID-19. UMAP generated with CD4+ T cells from mass cytometry data, coming from acute (non-COVID-19 and COVID-19) and convalescent COVID-19 samples. (Left) Cells from acute samples are colored according to the cell cluster origin (see legend), whereas cells from convalescent COVID-19 samples have not been assigned to a specific cluster (NA). (Middle) Cells from acute and convalescent COVID-19 samples are colored in red and black, respectively. (Right) Cells from acute and convalescent COVID-19 samples are colored according to the cell cluster, after assigning clusters to cells from convalescent COVID-19 samples via KNN approach.
(B) Box plots of CD16+HLA-DR+ CD3+ T cells determined by flow cytometry (cohort 2) of week 2 or 3+ samples from control (n = 11) mild (n = 5) or surviving (n = 3), non-surviving (n = 6) COVID-19 patients collected during the acute infection. The earliest sample (from week 2 on) was selected per patient in case of repeated measurements. The proportions of T cells between severity groups were compared using the Wilcoxon test with Benjamini-Hochberg correction.
(C) Proportions of activated FCGR3A+ T cells (from scRNA-seq clusters 10, 13, and 18, cohort 2) within the whole TCRab+ T cell space from control (n = 13) mild (n = 58) or surviving (n = 36), not-surviving (n = 6) severe COVID-19 patients collected during the acute infection. The earliest sample (from week 2 on) was selected per patient in case of repeated measurements. The proportions of T cells between severity groups were compared using the Wilcoxon test with Benjamini-Hochberg correction.
(D) Gating of CD3+CD45+CD19−CD15− T cells and the three T cell compartments for a representative CyTOF dataset of cohorts 1 prior to clustering, as shown in Figures 1E and S1C.
Next, we tested in a larger cohort whether plasma levels of complement proteins upstream of C3a generation are associated with patient disease course and outcome. Levels of positive regulators of the classical and alternative pathway, such as C1QA, C1QB, C1QC, C1R, and CFD, were higher in plasma samples of patients with severe compared with mild COVID-19 (Figure 6B). We also analyzed complement protein levels in relation to disease trajectory, specifically subsequent worsening of disease severity. C1R and CFD were elevated in samples from patients who showed a clinical deterioration, whereas the abundance of complement factor I (CFI), which inhibits the classical and lectin-dependent complement pathway, was lower in samples from patients with subsequent disease progression (Figure 6C). Finally, amounts of C1QA, C1QB, C1QC, and CFD were not only higher in severe COVID-19 but were also associated with fatal outcome (Figure 6D).
Altogether these data further support the pathological role of the complement system and of activated CD16+ T cells during severe COVID-19.
Discussion
Excessive T cell activation and altered phenotypes can contribute to infection-associated organ damage. Early after the first reports on immune profiles of COVID-19 patients (Sette and Crotty, 2021) discussions on their putative role in immune protection versus pathology started. In our study, we provide evidence that SARS-CoV-2 infection—in contrast to other acute and chronic infections—promotes the formation of highly activated and proliferating HLA-DR+CD38hiCD137+CD69+ T helper cells and CD8+ T cells independent of disease severity, although this response occurred faster in severe COVID-19 patients. More importantly, in severe COVID-19 patients, we detected differentiation of activated CD16+ T cells, which showed an increased immune complex-mediated cytotoxic potential and a potential to activate lung microvascular endothelial cells. Expanded clones within the CD16+ T cell compartment persisted and maintained their high cytotoxic potential. We identified C3a as an upstream signal for the differentiation of the altered activated T cell phenotype. Proportions of activated CD16+ T cells and plasma complement protein abundance levels were associated with worse outcomes of patients with severe COVID-19. Thus, SARS-CoV-2-triggered complement activation creates an inflammatory milieu that drives differentiation of T cells with high immunopathogenic potential.
A balanced T cell activation is decisive for the course of infection. The formation of CD8+ tissue-resident memory T cells (Trm) during primary infection is known to restrain viral spread upon secondary influenza infections. Yet, enhanced accumulation of Trm cells in an imbalanced environment such as during aging can support excessive inflammation leading to organ damage and impaired repair (Goplen et al., 2020). In COVID-19, large numbers of such Trm-like CD8+ T cells have been identified in the airways (Liao et al., 2020).
Blood samples acquired during the acute phase of severe COVID-19 contain high numbers of HLA-DR+CD38hiKi67+ in both CD4+ and CD8+ T cell compartments (Mathew et al., 2020; Rydyznski Moderbacher et al., 2020; Stephenson et al., 2021), a finding that we corroborated by CyTOF analysis. Severe COVID-19 patients showed a faster increase of the CD38hiHLA-DR+Ki67+ICOS+ TFH-like CyTOF cluster 7 proportions accompanied by an earlier antibody response (Figures 5A and S1B). We identified an elevated and activated T cell population expressing CD16 across the three major T cell compartments. Activated CD16+ T cells showed increased TCR-independent pathogenic potential.
The activated CD16+ CD4+ and CD8+ T cells enriched in severe COVID-19 expressed high levels of chemokine receptors such as CXCR3 and CCR6 (Figure 1D). This clearly distinguished them from the other CD16lo T cell clusters, e.g., cluster 31. CXCR3 and CCR6 might promote the migration of activated CD16+ T cells into the inflamed lungs (Oja et al., 2018; Shanmugasundaram et al., 2020). Pronounced CCR6 expression on T cells has been described in severe COVID-19 (Fenoglio et al., 2021; Tiwari-Heckler et al., 2021), which we can link here to unexpected phenotypic and functional properties. Immunofluorescence co-staining of CD3 and CD16 in lung samples of an autopsy cohort showed enrichment of CD3+CD16+ T cells in COVID-19 compared with influenza pneumonia or other causes of ARDS (Figures 3I and 3J). Although strong T cell activation is a feature of both severe COVID-19 and influenza pneumonia, specific differences have been described between both diseases (Youngs et al., 2021).
We found that approximately 5% of the activated CD16+ CD8+ T cells respond to stimulation with SARS-CoV-2 peptides. This is in line with the previously described positive correlation between ex vivo-determined HLA-DR+CD38hiKi67+CD8+ T cells and SARS-CoV-2-specific CD8+ T cells (Rydyznski Moderbacher et al., 2020). Activated CD16+ T cells show significant enrichment of SARS-CoV-2 specific T cells compared with activated CD16− T cells (Figure 5F). The remaining, non-responding activated CD16+ T cells may recognize other SARS-CoV-2 epitopes or may be driven by bystander activation and/or homeostatic proliferation (Bergamaschi et al., 2021; Mathew et al., 2020). This indicates that cognate T cell activation plays an important role in the generation of activated CD16+ T cells along with environmental signals in a complement split product-rich inflammatory milieu.
A very high proportion of T cells from acute COVID-19 and especially severe patients express cytotoxic molecules such as Perforin and Granzyme B (Shuwa et al., 2021). The increased cytotoxic profile persisted for up to six months and was associated with poorer recovery. The CD16+ T cells identified in severe COVID-19 did not only express higher levels of PRF1 and GZMB but also LAMP1 and STX11 (Figures 2H and S2B), which explains their increased general degranulation potential (Figures 3D and 3E; Spessott et al., 2017). So far, CD16+ T cells have been described mainly in patients with chronic infections or inflammation (Björkström et al., 2008; Clémenceau et al., 2011; Jacquemont et al., 2020). In these conditions, CD16+ T cells displayed a more differentiated phenotype like the one adopted during COVID-19 convalescence (Figures 4C, 4D and 4F). In acute COVID-19, we also identified elevated transcription of various granzyme genes including “Granzyme K” (Figure S2B). Interestingly, increased numbers of Granzyme K expressing effector memory T cells have been observed in blood samples of older individuals and these T cells were shown to augment cytokine and chemokine production by fibroblasts (Mogilenko et al., 2021). It was shown that extracellular Granzyme K proteolytically activates Protease-activated receptor-1 leading to increased release of IL-6 and CCL2 (MCP-1) by endothelial cells (Sharma et al., 2016). Particularly in severe COVID-19, we demonstrated that T cells also induce CCL2 and XCL8 by co-cultured primary lung endothelial cells upon anti-CD16 mediated degranulation (Figure 3G). This establishes a general link between the immune complex triggering of local CD16+ T cells and endothelial cell-mediated release of monocyte and neutrophil chemoattractants, a hallmark of severe COVID-19 (Rendeiro et al., 2021). Altogether, activated CD8+ T cells adopt antibody-dependent cellular cytotoxicity (ADCC) properties known for NK cells (Lee et al., 2021). ADCC can have protective but also disease exaggerating roles (Yu et al., 2021). The binding of antigen-antibody complexes to CD16 on activated T cells might, therefore, counteract anti-inflammatory immune complex clearing systems via complement receptor 1 (Fernandez-Arias et al., 2013; Kavai, 2008). Notably, patients suffering from severe COVID-19 have been reported to display high levels of spike-reactive IgG with significantly reduced Fc fucosylation. This change in the Fc glycosylation increases binding affinity to CD16, leading to increased CD16-mediated effector function (Ferrara et al., 2011; Hoepel et al., 2021; Vivier et al., 2008). As such, the distinct serological profile observed in severe COVID-19 with afucosylated, spike-directed IgG, and an inherently increased inflammatory capacity could further enhance the pathogenic potential of CD16+ T cells.
In a search for important environmental signals driving differentiation of activated CD16+ T cells, we detected a positive correlation between high serum C3a levels and proportions of CD16+ T cell clusters (Figure 5E). It has been reported that serum C3 hyperactivation is a risk factor for COVID-19 mortality (Sinkovits et al., 2021) and widespread complement activation by all three pathways and, thus, generation of C3a has also been described in patients with severe COVID-19 (Chouaki Benmansour et al., 2021; Defendi et al., 2021; Satyam et al., 2021). The disease-promoting activity of the complement system was observed for other coronaviruses, as SARS-CoV infection caused less systemic inflammatory response and lung injury in C3 knock-out as compared with wild-type mice (Gralinski et al., 2018). Furthermore, it has been shown that SARS-CoV-2 infection of lung epithelial cells induces transcription of complement genes leading to the generation of activated C3a (Yan et al., 2021). Signaling via complement receptors such as C3AR1 and cell-autonomous complosome in human T cells enhances induction of CD4+ Th1 responses and cytotoxic function of CD8+ T cells (Arbore et al., 2018). Here, we show that increased C3a generation in severe COVID-19 patients promotes differentiation of CD16+, highly cytotoxic CD4+ and CD8+ T cells (Figures 5F–5H). Interestingly, targeting distal complement effects by receptor blockade in a humanized preclinical model of SARS-CoV-2 infection prevented acute lung injury (Carvelli et al., 2020). Results from the first clinical trials on complement inhibition in COVID-19 also showed promising effects resulting in reduced inflammation and faster normalization of neutrophil and lymphocyte counts (Mastaglio et al., 2020; Mastellos et al., 2020; Polycarpou et al., 2020). In this context, C3 inhibition enabled a broader and better therapeutic potential as compared with C5 neutralization (Mastellos et al., 2020).
Limitations of the study
The limited number of convalescent samples did not allow us to perform correlations with patient recovery. Our major focus was to reveal immunopathogenic functions of severity-associated T cell populations during acute COVID-19 and to identify driving signals.
In this context, it would be of great interest to see whether application of the C3 inhibitor AMY-101 in COVID-19 patients with ARDS will ameliorate differentiation of CD16+, cytotoxic T cells and, thus, endothelial cell injury and ultimately improved patient outcome.
It is very likely that the role of complement activation in COVID-19 goes beyond the here-described mechanisms, which should be investigated in future studies. Furthermore, although C3a neutralization significantly reduced the CD16+ differentiation potential of severe COVID-19 serum, we cannot exclude that other pro-inflammatory mechanisms could induce this activated CD16+ T cell phenotype. Certainly, here also a more detailed comparative investigation of T cell responses between COVID-19 and influenza infection on a larger and more stratified cohort is needed.
Also, It will be interesting to investigate whether the described inflammatory circuit is also active in other immune pathologies for which complement activation and immune complex formation have been described.
Taken together, particularly severe COVID-19 leads to an elevated number of activated CD16+ T cells that link triggering of the complement cascade via TCR-independent cytotoxic T cell functionality to endothelial damage and patient survival. This functionally links the innate and the adaptive immune system with endothelial injury, which might constitute an important molecular axis explaining the vast spectrum of organ damage observed in COVID-19.
Consortia
Sascha S. Haenel, Mirja Mittermaier, Fridolin Steinbeis, Tilman Lingscheid, Bettina Temmesfeld-Wollbrück, Thomas Zoller, Daniel Grund, Christoph Ruwwe-Glösenkamp, Miriam S. Stegemann, Katrin M. Heim, Ralf H. Hübner, Bastian Opitz, Kai-Uwe Eckardt, Martin Möckel, Ulrike Bachmann, Felix Balzer, Claudia Spies, Marc Kastrup, Steffen Weber-Carstens, Frank Tacke, Christina Pley, Claudia Fink, Sarah Berger, Chantip Dang-Heine, Michael Hummel, Georg Schwanitz, Constanze Lüttke, Yinan Wu, Uwe D. Behrens, Maria Rönnefarth, Sein Schmidt, Christian Drosten, Martin Witzenrath, Alexander Krannich and Christof von Kalle for set up and realization of the study platform;
Linda Jürgens, Malte Kleinschmidt, Sophy Denker, Moritz Pfeiffer, Belén Millet Pascual-Leone, Luisa Mrziglod, Felix Machleidt, Sebastian Albus, Felix Bremer, Jan-Moritz Doehn, Tim Andermann, Carmen Garcia, Philipp Knape, Philipp M. Krause, Liron Lechtenberg, Yaosi Li, Panagiotis Pergantis, Till Jacobi, Teresa Ritter, Berna Yedikat, Lennart Pfannkuch, Christian Zobel, Ute Kellermann, Susanne Fieberg, Laure Bosquillon de Jarcy, Anne Wetzel, Christoph Tabeling, Markus C. Brack, Moritz Müller-Plathe, Jan M. Kruse, Daniel Zickler, Andreas Edel, Silke Leonhardt, Timur Özkan, Carola Misgeld, David Steindl, Marcel Wittenberg, Claas J. Steffen, Jan A. Graaw, Katharina Tielling, Ludwig Wiegank, Philipp Engelmann, Gottfried Lürzer, Victor Wegener, Stefan Angermair, Julia Heeschen, Moritz Weigeldt, Eike Wolter, Christoph Töpper, Anna Nothnagel, Sara Lange, Ralf F. Trauzeddel, Britta Stier, Roland Körner, Nils B. Müller, and Philipp Enghard for obtaining informed consent and biosamples;
Paula Stubbemann, Nadine Olk, Willi M. Koch, Alexandra Horn, Katrin K. Stoyanova, Saskia Zvorc, Yvonne Ahlgrimm, Wiebke Herud, Lucie Kretzler, Lil A. Meyer-Arndt, Linna Li, and Isabelle Wirsching for collection of clinical data;
Denise Treue∗, Dana Briesemeister∗ and Jenny Schlesinger∗, (∗Central Biobank Charité/BIH; ZeBanC), for biobanking of samples.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Purified NA/LE Mouse Anti-Human CD3 |
BD Pharmingen | Cat# 555329; LOT 829056K00781551; Clone UCHT1; RRID: AB_395736 |
| Purified NA/LE Mouse Anti-Human CD28 |
BD Pharmingen | Cat# 555725; LOT 8311929; Clone CD28.2; RRID: AB_396068 |
| Ultra LEAF purified anti-human C3a | Biolegend | Cat# 518106; LOT B307339; Clone K13/16; RRID: AB_2861044 |
| CD3_BV421 | Biolegend | Cat# 300434; LOT B312569; Clone UCHT1; RRID: AB_10897105 |
| HLA-DR_BV785 | Biolegend | Cat# 307642; LOT B324516; Clone L243; RRID: AB_2563461 |
| CD8_FITC | Biolegend | Cat# 301050; LOT B279939; Clone RPA-T8; RRID: AB_2888883 |
| CD38_PE-Cy7 | Biolegend | Cat# 356608; LOT B281408; Clone HB7; RRID: AB_2860985 |
| CD16_AF700 | Biolegend | Cat# 302026; LOT B231546; Clone 3G8; RRID: AB_2278418 |
| Ki67_eFluor506 | Invitrogen, Thermo Fisher Scientific | Cat# 69-5698-82; LOT 1915581; Clone SolA15; RRID: AB_2637482 |
| CD14 160Gd (RMO52) | Fluidigm | Cat# 3160006; RRID: AB_2661801 |
| CD28 purified (L293) | BD Bioscience | Cat# 348040; RRID:AB_400367, |
| CD69 162Dy (FN50) | Fluidigm | Cat# 3162001B; RRID: N/A |
| CD294 163Dy (BM16) | Fluidigm | Cat# 3163003B; RRID: AB_2810253 |
| RANKL APC Biotec | Miltenyi | Cat# 130-098-511; RRID: AB_2656691 |
| Anti-APC 163Dy | Fluidigm | Cat# 3163001B; RRID: AB_2687636 |
| CXCR5 164Dy (51505) | Fluidigm | Cat# 3164016B; RRID: AB_2687858 |
| Siglec 8 164Dy (7C9) | Fluidigm | Cat# 3164017B; RRID:AB_2892691 |
| CD34 166Er (581) | Fluidigm | Cat# 3166012B; RRID: AB_2756424 |
| CD38 167Er (HIT2) | Fluidigm | Cat# 3167001B; RRID: AB_2802110 |
| Ki67 168Er (Ki-67) | Fluidigm | Cat# 3168007B; RRID: AB_2800467 |
| CD25 169Tm (2A3) | Fluidigm | Cat# 3169003; RRID: AB_2661806 |
| CD24 169Tm (ML5) | Fluidigm | Cat# 3169004B; RRID: AB_2688021 |
| Lag3 purified (11C3C65) | Biolegend | Cat# 369302; RRID: AB_2616876 |
| RANK purified (80704) | R&D Systems | Cat# MAB683; RRID: AB_2205330 |
| CD161 purified (HP-3G10) | Biolegend | Cat# 339919; RRID: AB_2562836 |
| CD11b purified (ICRF44) | Biolegend | Cat# 301337; RRID: AB_2562811 |
| CD45RO purified (4G11) | DRFZ Berlin | N/A |
| CD44 purified (BJ18) | Biolegend | Cat# 338811; RRID: AB_2562835 |
| CD137 173Yb (4B4-1) | Fluidigm | Cat# 3173015B; RRID: N/A |
| PD-1 175Lu (EH12.2H7) | Fluidigm | Cat# 3175008; RRID: AB_2687629 |
| PD-L1 175Lu (29.E2A3) | Fluidigm | Cat# 3175017B; RRID: AB_2687638 |
| CD56 176Yb (NCAM16.2) | Fluidigm | Cat# 3176008; RRID: AB_2661813 |
| CD8A purified (GN11) | DRFZ Berlin | N/A |
| IgM purified (MHM-88) | Biolegend | Cat# 314502; RRID: AB_493003 |
| CD11c purified (Bu15) | Biolegend | Cat# 337221; RRID: AB_2562834 |
| B2M purified (2M2) | Biolegend | Cat# 316302; RRID: AB_492835 |
| CD16 209Bi (3G8) | Fluidigm | Cat# 3209002B; RRID: AB_2756431 |
| TotalSeq-C0251 anti-human Hashtag 1 | Biolegend | Cat# 394661; RRID: AB_2801031 |
| TotalSeq-C0252 anti-human Hashtag 2 | Biolegend | Cat# 394663; RRID: AB_2801032 |
| TotalSeq-C0253 anti-human Hashtag 3 | Biolegend | Cat# 394665; RRID: AB_2801033 |
| TotalSeq-C0254 anti-human Hashtag 4 | Biolegend | Cat# 394667; RRID: AB_2801034 |
| TotalSeq-C0255 anti-human Hashtag 5 | Biolegend | Cat# 394669; RRID: AB_2801035 |
| TotalSeq-C0256 anti-human Hashtag 6 | Biolegend | Cat# 394671; RRID: AB_2820042 |
| TotalSeq-C0257 anti-human Hashtag 7 | Biolegend | Cat# 394673; RRID: AB_2820043 |
| TotalSeq-C0258 anti-human Hashtag 8 | Biolegend | Cat# 394675; RRID: AB_2820044 |
| TotalSeq-C0259 anti-human Hashtag 9 | Biolegend | Cat# 394677; RRID: AB_2820045 |
| TotalSeq-C0260 anti-human Hashtag 10 | Biolegend | Cat# 394679; RRID: AB_2820046 |
| CD3_PE | Biolegend | Cat# 300441; clone UCHT1; RRID: AB_2562047 |
| CD3_BV711 | Biolegend | Cat# 300464; clone UCHT1; RRID: AB_2566036 |
| CD8_BV605 | Biolegend | Cat# 301040; clone RPA-T8; RRID: AB_2563185 |
| CD8_FITC | Biolegend | Cat# 301050; clone RPA-T8; RRID: AB_2562055 |
| CD8_VioGreen | Miltenyi Biotec | Cat# 130-110-684; clone REA734; RRID: AB_2659241 |
| CD4_BV421 | Biolegend | Cat# 317434; clone OKT4; RRID: AB_2562134 |
| CD4_APC/Fire 750 | Biolegend | Cat# 300560; clone RPA-T4; RRID: AB_2629693 |
| CD16_BV605 | Biolegend | Cat# 302040; clone 3G8; RRID: AB_2562990 |
| CD16_FITC | Biolegend | Cat# 980112; clone 3G8; RRID: AB_2876771 |
| CD16_Biotin 3G8 | Biolegend | Cat# 302004; clone 3G8; RRID: AB_314204 |
| CD45RA_PerCP/Cyanine 5.5 | Biolegend | Cat# 304122; clone HI100; RRID: AB_893357 |
| CCR7 _BV421 | Biolegend | Cat# 353208; clone G43H7; RRID AB_11203894 |
| Perforin_PE | Biolegend | Cat# 308106; clone dG9; RRID:AB_314704 |
| Perforin_PEcy7 | Biolegend | Cat# 308126; clone dG9; AB_2572049 |
| CD56_PEcy7 | BD Biosciences | Cat# 335820: clone NCAM16; RRID: N/A |
| IgG1_Biotin Isotype Ctrl |
Biolegend | Cat# 400104; clone MOPC21; RRID: AB_326427 |
| IFNy_BV785 | Biolegend | Cat# 502542; clone 4S.B3; RRID: AB_2563882 |
| Anti human/mouse Granzyme B_Pacific Blue | Biolegend | Cat# 515408; clone GB11; RRID: AB_2562196 |
| anti-human/mouse Granzyme B_APC/Fire 750 | Biolegend | Cat# 372210; clone QA16A02; RRID: AB_2728377 |
| Granzyme K_PerCP/Cyanine5.5 | Biolegend | Cat# 370514; clone GM26E7; RRID: AB_2632852 |
| CD107a_APC | Biolegend | Cat# 328620; clone H4A3; RRID: AB_1279055 |
| CD69 _APC | Biolegend | Cat# 310910; clone FN50; RRID:AB_314844 |
| OX40_PE | Biolegend | Cat# 350003; clone BER-ACT35; RRID:AB_10641708 |
| CD137_BV421 | Biolegend | Cat# 309820; clone 4B4-1; RRID:AB_2563830 |
| CD40L_FITC | Miltenyi Biotec | Cat# 130-113-606; clone 5C3; RRID:AB_2751141 |
| CCR7_AlexFluor 700 | Biolegend | Cat# 353244; clone G043H7; RRID:AB_2617001 |
| functional grade CD40 | Miltenyi Biotec | Cat# 130-094-133; clone HB14; RRID:AB_10839704 |
| Human TruStain FcX (Fc Receptor Blocking Solution) | Biolegend | Cat# 422302; RRID: AB_2818986 |
| Fc receptor blocking solution | Miltenyi Biotec | Cat# 130-059-901; RRID:AB_2892112 |
| Mouse anti-CD3 | BioRad | Cat# MCA1477; clone CD3-12; RRID: RRID:AB_321245 |
| Mouse anti-CD16 | Santa Cruz Biotechnology | Cat# sc-20052; clone DJ130c; RRID:AB_2890161 |
| Mouse anti-CD31 | Agilent Technologies | Cat# M0823; clone JC70A; RRID:AB_2114471) |
| Biological samples | ||
| PBMC | please refer to Table S1. “Cohort details” | N/A |
| Autopsy lung tissue samples | please refer to Table S1. “Cohort details” | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| RPMI 1640 | Pan Biotech | Cat# P04-18525 |
| RPMI 1640 | Gibco, Thermo Fisher Scientific | Cat# 21875034 |
| Human Serum, Type AB, male | PanBiotech | Cat# P30-2902; Lot#PO20920 |
| Human AB serum | Pan Biontech | Cat# P30-2901 |
| Fetal Calf Serum | Biochrom | Cat# S0115 |
| Fetal Calf Serum | Sigma-Aldrich | Cat# F7524; Lot# 0001638262 and 0001638308 |
| Sodium Pyruvate | Gibco, Thermo Fisher Scientific | Cat# 11360-070 |
| GlutaMAX | Gibco, Thermo Fisher Scientific | Cat# 35050-038 |
| HEPES | Sigma-Aldrich | Cat# H0887-100mL |
| MEM Non-essential amino Acid solution | Sigma-Aldrich | Cat# M7145-100mL |
| Pierce Universal Nuclease for Cell Lysis | Thermo Fisher Scientific | Cat# 88701 |
| DPBS | Gibco, Thermo Fisher Scientific | Cat# 14190-094 |
| Bovine Albumin Fraction V (BSA) | Serva | Cat# 11930 |
| MACS BSA Stock solution | Miltenyi Biotec | Cat# 130-091-376 |
| EDTA | Sigma | Cat# E5134-100G |
| IL-2 (Proleukin S) | Novartis | Cat# 02238131 |
| h-C3a | Almac | Cat# CN-91 |
| Beriglobin | CSL Behring | PZN 4616123 |
| Anti-Biotin MACSiBead Particles | Miltenyi Biotec | Cat# 130-092-357 |
| SARS-CoV-2 Spike Trimer (HEK)-Biotin | Miltenyi Biotec | Cat# 130-127-685 |
| PepTivator SARS-CoV-2 Select | Miltenyi Biotec | Cat# 130-127-309 |
| DAPI (4',6-Diamidino-2-Phenylindole, Dihydrochloride) | Thermo Fisher Scientific | Cat# D1306; RRID: AB_2629482 |
| SPRIselect | Beckmann Coulter | Cat# B23318 |
| 10% Tween 20 | BIO-RAD | Cat# 1662404 |
| Buffer EB | QIAGEN | Cat# 19086 |
| Ethanol, Absolute | Fisher Bioreagents | Cat# BP2818-500 |
| Glycerol, 85% | Merck | Cat# 1040941000 |
| Nuclease-Free Water | Invitrogen | Cat# AM9937 |
| Formalin solution 4%, phosphate buffered | Formafix | Cat# 01-1005 |
| Critical commercial assays | ||
| Pan T Cell Isolation Kit, human | Miltenyi Biotec | Cat# 130-096-535 |
| Zombie Red Fixable Viability Kit | Biolegend | Cat# 423109; RRID: N/A |
| Zombie UV Fixable Viability Kit | Biolegend | Cat# 423108; RRID: N/A |
| Foxp3 Staining buffer kit | Miltenyi Biotec | Cat# 130-093-142; RRID: N/A |
| BD Perm/Wash | BD Biosciences | Cat# 554723; RRID: AB_2869011 |
| BD Cytofix | BD Biosciences | Cat# 554655; RRID: AB_2869005 |
| Chromium Next GEM Chip G Single Cell Kit | 10x Genomics | Cat# 1000120 |
| Chromium Next GEM Single Cell 5′ Library and Gel Bead Kit v1.1 | 10x Genomics | Cat# 1000167 |
| Chromium Single Cell 5′ Library Construction Kit | 10x Genomics | Cat# 1000020 |
| Chromium Single Cell V(D)J Enrichment Kit, Human T Cell | 10x Genomics | Cat# 1000005 |
| Single Index Kit T Set A | 10x Genomics | Cat# 1000213 |
| Single Index Kit N Set A | 10x Genomics | Cat# 1000212 |
| Chromium Single Cell 5′ Feature Barcode Library Kit | 10x Genomics | Cat# 1000080 |
| Qubit 1X dsDNA HS Assay Kit | Thermo Fisher Scientific | Cat# Q33231 |
| High Sensitivity DNA Kit | Agilent Technologies | Cat# 5067-4626 |
| Maxwell 16 LEV RNA FFPE Purification Kit | Promega | Cat# AS1260 |
| TaqMan Exogenous Internal Positive Control Reagents Kit | Applied Biosystems | Cat# 4308323 |
| TaqMan Fast Virus 1-step master mix | Applied Biosystems | Cat# 4444434 |
| DAKO REAL PEROXIDASE-BLOCKING SOLUTION | Agilent Technologies | Cat#S2023 |
| DAKO REAL ANTIBODY DILUENT | Agilent Technologies | Cat# S2022 |
| EnV FLEX, High pH, (Link) | Agilent Technologies | Cat# K800021-2; RRID: N/A |
| EnVision FLEX/ HRP goat anti-mouse (ready-to-use) | Agilent Technologies | Cat# K8000; RRID:RRID: AB_2890017 |
| EnVision FLEX, High pH (Link), HRP. Rabbit/ Mouse | Agilent Technologies | Cat# DM802 |
| EnVision FLEX Target Retrieval Solution Low pH | Agilent Technologies | Cat# K8005 |
| Opal 7-Color Manual IHC Kit | Akoya Biosciences | Cat# NEL811001KT |
| Opal 690 | Akoya Biosciences | Cat# FP1497001KT |
| Opal 520 TSA Plus | Akoya Biosciences | Cat# FP1487001KT |
| Opal 620 TSA Plus | Akoya Biosciences | Cat# FP1495001KT |
| Spectral DAPI | Akoya Biosciences | Cat# FP1490 |
| Deposited data | ||
| scRNA-seq | this paper (cohort 1) | GEO: GSE175450; Zenodo: 10.5281/zenodo.5771937 |
| CyTOF | this paper (cohort 1) | FlowRepository: FR-FCM-Z4P5; Zenodo: 10.5281/zenodo.5771937 |
| Experimental models: Cell lines | ||
| Human pulmonary microvascular endothelial cells (HPMECs) | Promocell | Lot# 455Z003; Lot# 463Z013.1 |
| Blaer1 cells GFP-/- | Prof. Holger Heine, Forschungszentrum Borstel | N/A |
| Oligonucleotides | ||
| E_Sarbeco_R 5′ ATATTGCAGCAGTACGC ACACA 3′ |
metabion | N/A |
| E_Sarbeco_F 5′ ACAGGTACGTTAATAGTTA ATAGCGT 3′ |
metabion | N/A |
| E_Sarbeco_P1 5′ FAM-ACACTAGCCATCCT TACTGCGCTTCG-BHQ-2 3′ |
metabion | N/A |
| RdRp_SARSr-R 5′ CARATGTTAAASACA CTATTAGCATA 3′ |
metabion | N/A |
| RdRp_SARSr-F 5′ GTGARATGGTCAT GTGTGGCGG 3′ |
metabion | N/A |
| RdRp_SARSr-P1 5′ Cy5-CCAGGTGGWACRT CATCMGGTGATGC-BHQ-2 3′ |
Metabion | N/A |
| Software and algorithms | ||
| analysis_cytof.Rmd | this paper (cohort 1) | Zenodo: 10.5281/zenodo.5771937 |
| scRNAseq_analysis_1preprocessing.Rmd | this paper (cohort 1) | Zenodo: 10.5281/zenodo.5771937 |
| scRNAseq_analysis_2clustering.Rmd | this paper (cohort 1) | Zenodo: 10.5281/zenodo.5771937 |
| scRNAseq_analysis_3convalescent.Rmd | this paper (cohort 1) | Zenodo:10.5281/zenodo.5771937 |
| CellRanger | 10x genomics | v3.1.0 and v5.0.0 |
| Bcl2fastq2 | Illumina | v2.20 |
| STAR | (Dobin et al., 2013) | v2.6.1b |
| Cutadapt | (Martin, 2011) | v1.16 |
| Dropseq-tools | https://github.com/broadinstitute/Drop-seq/ | v2.0.0 |
| R | https://www.cran.r-project.org | v3.6.2; v4.0.3 |
| Seurat (R package) | (Butler et al., 2018; Hafemeister and Satija, 2019; Stuart et al., 2019) | v3.1.4; v3.1.2; v3.9.9. (CRAN) |
| Harmony (R package) | (Korsunsky et al., 2019) (https://github.com/immunogenomics/harmony) |
v1.0 |
| Destiny (R package) | (Angerer et al., 2016) | v 3.0.1 |
| ClusterProfiler (R package) | (Yu et al., 2012) | v3.10.1 (CRAN) |
| SingleR (R package) | (Aran et al., 2019) | v1.0.5 (Bioconductor) |
| DirichletReg (R package) | (Maier, 2014) | v0.6.3.1 (CRAN) |
| AUCell (R package) | (Aibar et al., 2017) | v1.6.1 (CRAN) |
| Cytobank | (Kotecha et al., 2010) https://www.cytobank.org | https://doi.org/10.1002/0471142956.cy1017s53 |
| SPADE (Cytobank) | (Qiu et al., 2011) | Cytobankis running a version of SPADE derived from v1.10.2 |
| flowCore (R package) | https://www.bioconductor.org/packages/release/bioc/html/flowCore.html | v1.48.1 (Bioconductor), 10.18129/B9.bioc.flowCore |
| CytoML (R package) | https://github.com/RGLab/CytoML | v1.8.1 (Bioconductor), 10.18129/B9.bioc.CytoML |
| CytofBatchAdjust (R package) | https://github.com/CUHIMSR/CytofBatchAdjust | (Schuyler et al., 2019) |
| uwot (R package) | https://cran.r-project.org/web/packages/uwot/index.html | v0.1.8 (CRAN) |
| ComplexHeatmap (R package) | (Gu et al., 2016) | v1.20.0 (Bioconductor) |
| lme4 (R package) | (Nowicka et al., 2017) | v1.1-21 (CRAN) |
| multcomp (R package) | (Hothorn et al., 2008) | v1.4-13 (CRAN) |
| lsmeans (R package) | (Lenth, 2016) | v2.30-0 (CRAN) |
| phenoptr (R package) | (Johnson, 2021) | v0.2.9 https://github.com/akoyabio/phenoptr/ |
| phenoptrReports (R package) | (Johnson, 2021) | v0.2.10 https://akoyabio.github.io/phenoptrReports |
| Prism (software) | https://www.graphpad.com | v9 |
| FlowJo | https://www.flowjo.com | v10.6.1 |
| Cytoscape | https://www.cytoscape.org | v3.7.1 (Shannon et al., 2003) |
| iRegulon | (Janky et al., 2014) | v1.3 |
| inForm | Akoya Biosciences | v2.4.8 |
| BioRad CFX Maestro 1.0 Version 4.0.2325.0418 | BioRad 2017 | 12004110 |
| ZEN 3.0 black edition | Carl Zeiss AG | v3.0 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to the lead contact, Birgit Sawitzki ( birgit.sawitzki@charite.de ).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Samples from patients with COVID-19 were collected within two cohort studies (Schulte-Schrepping et al., 2020) designed to allow deep molecular and immunological studies of COVID-19 in blood. This study was primarily designed to describe immunological deviations in COVID-19 patients without the intention of the development of new treatments or new diagnostics, and, therefore, sample size estimation was not included in the original study design.
Cohort 1/Berlin cohort
This study includes a subset of patients enrolled between March 2 and April 30 2021 in the Pa-COVID-19 study, a prospective observational cohort study assessing pathophysiology and clinical characteristics of patients with COVID-19 at Charité Universitätsmedizin Berlin (Kurth et al., 2020). The study was approved by the Institutional Review board of Charité (EA2/066/20). Written informed consent was provided by all patients or legal representatives for participation in the study. The patient population included in all analyses of cohort 1 consists of 43 control donors (samples collected in 2019 before SARS-CoV-2 outbreak or collected in the fall of 2020 who did not have a SARS-CoV-2 infection), 8 patients presenting with flu-like illness but tested SARS-CoV-2-negative, 6 patients with chronic HIV infection, 5 patients with chronic HBV infection as well as 35 mild and 42 severe COVID-19 patients during the acute or convalescent phase (Figures 1A and 1B; Table S1). Information on age, sex, medication, and comorbidities is listed in Table S1. All COVID-19 patients were tested positive for SARS-CoV-2 RNA in nasopharyngeal swabs and allocated to mild (WHO 2-4, mild+moderate) or severe (WHO 5-8, severe+critical) disease according to the WHO ordinal scale for clinical improvement
Cohort 2/Bonn cohort
This study was approved by the Institutional Review board of the University Hospital Bonn (073/19 and 134/20). After providing written informed consent, 14 control donors and 22 COVID-19 patients were included in the study. In patients who were not able to consent at the time of study enrollment, consent was obtained after recovery. Information on age, sex, medication, and comorbidities are provided in Table S1. COVID-19 patients who tested positive for SARS-CoV-2 RNA in nasopharyngeal swabs were recruited at the Medical Clinic I of the University Hospital Bonn between March 30 and June 17, 2020 and allocated to mild (WHO 2-4, mild+moderate, n = 9) or severe (WHO 5-8, severe+critical, n = 13) disease according to the WHO ordinal scale for clinical improvement. Controls in cohort 2 were collected from healthy people or from otherwise hospitalized patients with a wide range of diseases and comorbidities including chronic inflammatory immune responses. These individuals were either tested negative for SARS-CoV-2, serologically negative or had no indication for acute COVID-19 disease based on clinical or laboratory parameters.
Cohort 3/Aachen cohort
The study was approved by the local ethics committee (EK 304/20, EK 119/20, and EK 092/20). Informed consent for all autopsies was obtained from the legal representatives of the deceased patients. We consecutively included 14 autopsies of COVID-19-positive patients from April 2020 to January 2021. Each patient had a positive clinical PCR test for upper or lower respiratory SARS-CoV-2 before autopsy. Autopsies were performed in two steps according to a modified standard protocol to minimize staff exposure (developed as part of the German Registry for COVID-19 Autopsies – DeRegCOVID [Boor et al., 2021]). As a control, 18 lung tissues from three cohorts of non-COVID autopsies in an anonymized manner were included: four samples without inflammatory or fibrotic lung disease (“normal”) from 2009 to 2015, six samples from influenza patients from 2009 to 2018, and eight samples from 2010 to 2019 from patients with acute respiratory distress syndrome (“ARDS”) without acute inflammation of the selected ARDS samples (see Table S1 for cohort details).
Method details
Antibodies used for mass cytometry (cohort 1)
All anti-human antibodies pre-conjugated to metal isotopes were obtained from Fluidigm Corporation (San Francisco, USA). All remaining antibodies were obtained from the indicated companies as purified antibodies and in-house conjugation was done using the MaxPar X8 labeling kit (Fluidigm, USA). Antibodies are listed in the key resources table.
Sample processing, antigen staining, and data analysis of mass cytometry-based immune cell profiling (cohort 1)
Sample processing, cell staining and acquisition was done as previously described (Schulte-Schrepping et al., 2020). T cells were identified based on the expression of CD3 and CD45, but exclusion of CD19+ and CD15+ cells. Pregating on CD8-TCRgd-, CD8+TCRgd- and TCRgd+ cells was used to define CD4+, CD8+ and TCRgd+ T cells, respectively (Figure S4D). As expected, samples from HIV patients showed an inverted CD4/CD8 ratio, whereas the proportions of all three main T cell compartments were similar between all other patient groups (mild and severe COVID-19, FLI, HBV) and controls (median levels: T helper cells 63.5-71.5%, CTLs 27.1-34.8%, TCRgd+ 0.9-2.9%). Each T cell compartment was then clustered based on the expression of 30 markers: CD62L, CD45RO, CD28, CD27, CD226, ICOS, PD-1, LAG3, TIGIT, CD96, CD25, CD56, HLA-DR, CD38, CD137, CD69, Ki67, CXCR3, CXCR5, CCR6, CRTH2, KLRB1, KLRG1, KLRF1, CD95, CD10, CD16, CD34, CD123, and CD11cThe batch-normalized CyTOF values (described in Schulte-Schrepping et al., [2020]) were first transformed with the inverse hyperbolic sine function (asinh) and then z-score normalized per marker across all samples and all cells. We then clustered each T cell compartment excluding convalescent COVID-19 samples using Phenograph (Levine et al., 2015) with 30 nearest neighbors (k = 30). We originally found 50 clusters, which were annotated based on the average z-score transformed CyTOF expression across T cells of the markers in each cluster. Four clusters were merged in pairs, as each pair slightly differed in the expression of only one marker (CD95) resulting in 48 clusters. Cells from the convalescent samples were classified into the clusters previously found, via k-nearest neighbor (in Euclidean distance) using the knn function from “class” R package. (Figure S4A). UMAPs were calculated across all acute and convalescent samples based on the 30 markers used for clustering, using the R package “uwot” (arXiv:1802.03426 ), n_neighbors = 30, spread = 1, min_dist = 0.5, based on Euclidean distance).
The frequency of each cluster was calculated as the percentage of cells in each cluster for each patient and for each T cell compartment. Statistical testing for the difference in the frequency of each cluster across severity groups was calculated with the adjusted Dunn's post-hoc test (Benjamini-Hochberg) for clusters with significant Kruskal-Wallis test (adjusted p-value (Benjamini-Hochberg) < 0.1, adjustment was done across all clusters in each T cell compartment). For the non-weekly analysis, we considered the first sample per patient when multiple samples were available. Similarly, for the weekly analysis, only the first sample per patient per week was included, and the repeated samples in the same week were excluded from the analysis.
To test whether the age of the patients in cohort 1 was confounding the abundance of the activated CD16+ T cell CyTOF clusters C8 and C26, we did a multivariate model for the frequencies of these clusters, with severity and age as predictors using the R function lm (stats package, version 4.0.3). In this analysis, we tested the cluster frequencies based on age and severity for healthy controls and acute COVID-19 patients selected for the non-weekly analysis (Figures S1C and S1D).
For the survival analysis (Figure 6A), we selected mild and severe COVID-19, samples acquired between 7 and 28 days post-symptom onset. In case of repeated measurements per patient, the first sample was selected. The proportions of T cells between severity groups were compared using the Wilcoxon test with Benjamini-Hochberg correction. To define an objective way to select the sum of all activated CD16+ TCRab+ clusters of our CyTOF dataset (cohort 1), we computed an activation value for each cluster in the TCRab+ space, defined as the mean of the average z-scored expression of activation markers (CD25, HLA-DR, CD38, CD137, CD69, and Ki67). Then, clusters with an activation value higher than the average, and with an average z-scored CD16 expression higher than 1, were considered activated CD16+ TCRab+ T cell clusters (CyTOF cluster 8, 13, 26).
Blood processing and data analysis for multi-color flow cytometry-COVID-19 cohort (cohort 2)
Whole blood was prepared by treatment of 1ml peripheral blood with 10ml of RBC lysis buffer (Biolegend, USA). After one wash in DPBS, cells were directly processed for scRNA-seq (BD Rhapsody) or multi-color flow cytometry (MCFC). After RBC lysis, cells were washed with DPBS and from each sample 1-2 million cells were stained for flow cytometric analysis (Table S3). Antibody staining was performed in DPBS with the addition of BD Horizon Brilliant Stain Buffer (Becton Dickinson, USA) for 30min at 4°C as described before (Schulte-Schrepping et al., 2020). To remove dead cells from the analysis, a staining for dead cells was included (LIVE/DEAD Fixable Yellow Dead Cell Stain Kit; 1:1000 – Thermo Scientific, USA). To prevent any possible contamination of the operator, after staining, the samples were fixed for 5 minutes with 4% PFA. Flow cytometry analysis was performed on a BD Symphony instrument (Becton Dickinson, USA) configured with 5 lasers (UV, violet, blue, yellow-green, red). Flow cytometry data analysis was performed with FlowJo V10.7.1, CD16/HLA-DR double-positive cells were gated from the total T lymphocytes (living/CD45+/CD66b-/CD19-/CD3+), STAR Methods, data and code availability reports the detailed gating strategy and representative plots. With this analysis we confirmed the existence of a CD3+CD16+HLA-DR+ T cell population.
Age-dependent control cohorts
We used our flow cytometry datasets (fcs files) described previously (Kverneland et al., 2016) to report the proportions of CD16 expressing CD4+ and CD8+ T cells. Briefly, whole blood samples collected from healthy controls spanning an age range between 20 and 84 years with equal distribution of females and males in each 10-year age bin were stained using the ONE Study antibody panels as previously described (Sawitzki et al., 2020; Streitz et al., 2013) and acquired on a 10-Color Navios Flow Cytometer (Beckman Coulter, USA). For the here reported results data from panel 1 were used. The flow cytometry data analysis was performed in Kaluza version 1.2 (Beckman Coulter, USA, Figure S5A, gating strategy and representative plots).
Figure S5.

Representative plots of gating strategy, related to Figure 5A
(A) Gating of CD3+CD4+CD16+ and CD3+CD8+CD16+ T cells, as shown in Figure 5A (age-dependent accumulation of CD16+ T cells in controls).
Isolation of blood cells for scRNA-seq (cohort 1)
Human peripheral blood mononuclear cells (PBMCs) were isolated from heparinized whole blood by density gradient centrifugation over Pancoll (density: 1,077g /ml, PAN-Biotech, Germany). Subsequently, the cells were counted, frozen and stored in liquid nitrogen.
On the day of the experiment, the frozen PBMCs were thawed in pre-warmed thawing medium (RPMI 1640, Gibco; 2% FCS, Sigma; 0,01% Pierce Universal Nuclease, Thermo Fisher, USA).
10x Genomics Chromium single-cell RNA-seq (cohort 1)
Approximately 2-3 x105 PBMCs were resuspended in staining buffer (DPBS, Gibco; 0,5% BSA, Miltenyi Biotec, Germany; 2 mM EDTA, Gibco, Thermo Fisher Scientific, USA) and hashtagged with 0.5 μg Total-Seq-C™ Hashtag antibodies for 30 min at 4°C. After the incubation, PBMCs were washed three times. Subsequently, PBMC were counted and up to four different samples were pooled equally. The PBMCs were washed three times, resuspended in DPBS, filtered through a 40 μm mesh (Flowmi™ Cell Strainer, Merck, Germany) and counted using the C-Chip hemocytometer (NanoEntek, South Korea). The cell suspension was super-loaded with 40000 - 50,000 cells per lane, in the Chromium™ Controller for partitioning single-cells into nanoliter-scale Gel Bead-In-Emulsions (GEMs).
The remaining PBMCs were subjected to flow-cytometric sorting based on DAPI, CD3 (clone UCHT1), CD4 (clone RPA-T4), CD8 (RPA-T8) and CD38 (clone HB7) antibody staining and simultaneously hashtagged as described above. We did not include CD45RA or CD45RO as we did not want to exclude activated naive T cells from our analysis. Live (DAPI-) CD3+CD4+CD38+, as well as CD3+CD8+CD38+ cells, were sorted using the FACS Aria II (BD Biosciences, USA). Afterwards, CD4+CD38+ and CD8+ CD38+ T cells from each donor were pooled equally and CD38+ T cells from up to four samples were pooled to equal proportions. The resulting cell pool was resuspended in DPBS, filtered through a 40 μm mesh (Flowmi™ Cell Strainer, Merck, Germany) and counted using the C-Chip hemocytometer (NanoEntek, South Korea). The cell suspension was super-loaded with 40,000 - 50,000 cells per lane, in the Chromium™ Controller for partitioning single-cells into nanoliter-scale Gel Bead-In-Emulsions (GEMs).
The Chromium Next GEM Single Cell 5′ Kit v1.1 was used for reverse transcription, cDNA amplification and library construction of the gene expression libraries (10x Genomics, USA). For additional VDJ and hashtag libraries the Chromium Single Cell V(D)J Enrichment Kit, Human T Cell (10x Genomics, USA) and the Chromium Single Cell 5’ Feature Barcode Library Kit (10x Genomics, USA) were used, respectively. All libraries were prepared following the detailed protocols provided by 10x Genomics, quantified by Qubit Flex Fluorometer (Thermo Fisher, USA) and quality was checked using 2100 Bioanalyzer with High Sensitivity DNA kit (Agilent, USA). Sequencing was performed in paired-end mode with a S1 and S2 flow cell using NovaSeq 6000 sequencer (Illumina, USA).
BD Rhapsody single-cell RNA-seq (cohort 2)
For the analysis of the T lymphocytes compartment of COVID-19 patients, the PBMC dataset from Schulte-Schrepping et al. (Schulte-Schrepping et al., 2020) was used. The dataset was provided as a Seurat object of the PBMCs samples from the original study including all pre-processing and filtering criteria as described in the original manuscript. The data can be downloaded from FASTGenomics [ FASTGenomics: https://beta.fastgenomics.org/p/schulte-schrepping_covid19 ] or directly explored using the web platform.
Data pre-processing of 10x Genomics Chromium scRNA-seq data (cohort 1)
The Cell Ranger Software Suite (Version 5.0.0) was used to process raw sequencing data, using the cellranger multi workflow and the GRCh38 references for gene expression and VDJ data and totalSeq antibody barcode sequences for feature barcoding data. Multiplexed samples from multiple donors were demultiplexed by detecting SNPs using CellSNP-lite (Huang and Huang, 2021).
ScRNA-seq data analysis of 10x Chromium data (cohort 1)
Data quality control
For each sample, the number of RNA counts per cell was mean-normalized, and cells with a mean-normalized RNA count lower than 0.45 and higher than 5 were excluded from the analysis, as well as cells with a percentage of mitochondrial reads higher than 10%. The filtered data were then log-normalized and scaled using the Seurat package.
Definition of the T lymphocyte space
After filtering, gene expression data from PBMCs and enriched CD38+ T cells were log-normalized, merged, and scaled. We merged different libraries using the FindIntegrationAnchors and IntegrateData functions of Seurat with reduction.use='rpca'. We then performed PCA based on a subset of genes that exhibited high cell-to-cell variation (FindVariableFeatures Seurat function), constructed a KNN graph based on the Euclidean distance in PCA space considering the first 15 PCs (FindNeighbors Seurat function), and clustered the cells with the Louvain algorithm implemented in FindClusters Seurat function, with a resolution of 0.5.
Clusters from merged CD38+ T cell and PBMC libraries that contained mainly cells from PBMC libraries were excluded from the T cell space. T cell clusters were then confirmed by their high expression of CD3D, CD3E, and CD3G genes. At least 80% of the cells in the selected T cell clusters had a V(D)J sequence, whereas excluded clusters had a maximum of 10% of cells with a V(D)J sequence.
T cell clusters
After selecting T cells from PBMCs and enriched CD38+ T cells, we performed PCA based on a subset of genes that exhibited high cell-to-cell variation (FindVariableFeatures Seurat function). We then constructed a KNN graph based on the Euclidean distance in PCA space considering the first 15 PCs (FindNeighbors Seurat function) and clustered the cells with the Louvain algorithm implemented in FindClusters Seurat function, with a resolution of 0.5. UMAP was computed with the first 15 PCs.
Cluster annotation and statistical test
To align the CyTOF and scRNA-seq T cell clusters, we applied a feature-based cluster annotation approach. The selected feature list contained genes for all markers analyzed in CyTOF measurements plus additional genes such as T cell state-specific transcription markers (data and code availability). The specific name was assigned according to the Z-score standardized gene expression level of a particular set of genes (e.g., MKI67 ++/FCGR3A ++) or well-established cellular states (e.g. CD4 naive). To annotate the clusters, we generated a heatmap (Figure 2B) to visualize the expression of selected genes that were also selected as variable features (in the previous step). We found 20 clusters, and some of them displayed nearly identical phenotypes and, thus, were merged, ending up with 17 clusters (Figures 2A and 2B). T cell clusters were composed of i) solely CD4 expressing T cell clusters (clusters 1 to 7), ii) clusters of CD4, CD8A/B & TRGC2 expressing T cells (clusters 8 & 9), iii) clusters containing CD8A/B & TRGC2 expressing T cells (clusters 10 to 13 and 16) and iv) clusters of solely CD8 transcribing T cells (clusters 14 & 15). Cluster 8-10 were characterized by highly activated T cells with high transcription of CD38 and HLA-DR genes (Figures 2B and 2C). Cluster 7 showed the typical features of activated TFH cells, as in addition to CD38 and HLA-DR genes, they transcribed ICOS, CD40LG, PDCD1 (PD-1) and CXCR5 (Figure 2B). Cluster 8 T cells transcribed the highest MKI67 (Ki67) levels, indicative of the highest proliferative potential (Figures 2B and 2C). In addition, they expressed FCGR3A (CD16A), which was even more pronounced for cluster 10 T cells (Figures 2B and 2C).
The complete processing, dimensionality reduction, and clustering were performed with the Seurat package.
The frequency of each cluster was calculated as the percentage of cells in each cluster for each patient. Statistical testing for the difference in the frequency of each cluster was calculated with adjusted Dunn's post-hoc test (Benjamini-Hochberg) for clusters with significant Kruskal-Wallis test (adjusted p-value (Benjamini-Hochberg) < 0.05, depicted as KW∗ in e.g.: Figures 2E and S2A).
Gene Ontology (GO) Enrichment Analysis
For the identification of differentially expressed genes between disease groups, we used pseudobulk gene expression, i.e., the sum of the raw counts from all cells in each patient among selected clusters of interest. The pseudobulk samples were then normalized according to the DESeq2 pipeline ((Love et al., 2014), v. 1.30.0). For further enrichment analysis, we selected differentially expressed genes with high counts (“baseMean” > 100) and p-value lower than 0.05. Differentially expressed genes were identified with the R package “DESeq2” v. 1.30.1 and GO enrichment analysis was performed with the R package “enrichR” v. 3.0.
Gene Set Enrichment Analysis (GSEA)
The log2-fold change of differentially expressed genes (with high counts, i.e., “baseMean” > 100) from DESeq2 was used to define the ranked gene list used for GSEA. We tested three signatures (listed in Table S2): (1) RESPONSE TO TYPE I INTERFERON (GO:0034340), DEFENSE RESPONSE TO VIRUS (GO:0051607), and cytotoxicity. Gene lists (1) and (2) were obtained from the Molecular Signatures Database (MSigDB) (Subramanian et al., 2005), and for the cytotoxicity signature we used 17 cytotoxicity-associated genes taken from T_CELL_MEDIATED_CYTOTOXICITY (GO:0001913) and (Zhang et al., 2020). GSEA was performed with the R package “fgsea” v. 1.16.0 with 1000 permutations for statistical testing.
scRNA-seq data analysis of Rhapsody data (cohort 2)
Definition of the T lymphocytes space
From the complete landscape of immune cells in the PBMCs dataset, according to the labels of the original manuscript (Schulte-Schrepping et al., 2020), CD4+ T cells, CD8+ T cells, Activated T cells, Prol. cells and NK cells were selected for downstream analysis. The subset was scaled, centered, and regressed against the number of detected transcripts per cell. PCA was calculated on the top 2000 most variable genes using the vst method integrated into Seurat’s FindVariableFeatures function and UMAP dimensionality reduction was performed. In the newly defined cellular space, we identified unwanted cells (non-T cells) according to the expression of lineage markers. With this approach, we removed NK cells and a small proportion of monocytes found to contaminate the original lymphocytes cluster. We further optimized the selection of the T cells space by removing cells that overlap with a recent study on NK cells, which utilizes the same dataset (Krämer et al., 2021). The cleaned T cell space was once more scaled and PCA, as well as UMAP dimensionality reduction, were computed as described above. Cell clusters were calculated on the first 20 principal components (PCs) using Louvain clustering with a resolution of 1. All analysis steps were performed with Seurat [(Hao et al., 2021), v. 3.9.9.9032].
T cell cluster annotation
The 19 cell clusters were manually annotated according to a set of T cell hallmark genes (Figures 2B and S3B). Cell cluster names were assigned according to the expression levels of particular sets of genes (e.g., MKI67 ++/FCGR3A ++) or well-established cellular states (e.g., CD4 naive)
Gene Set Enrichment Analysis (GSEA)
For the analysis of the differential regulation of specific pathways in moderate or severe COVID-19, GSEA was performed on pseudobulk samples. In this analysis, we included only samples from weeks 1 and 2 post-symptom onset. A pseudobulk sample, defined as the sum of the raw counts from all cells in each group, was generated for moderate and severe patients. The pseudobulk samples were then normalized according to the DEseq2 pipeline ((Love et al., 2014), v. 1.30.0). Log2-transformed fold change of each gene was calculated to define the ranked gene list used for GSEA. GSEA was performed with the fgsea package with 1000 permutations for statistical testing.
Data visualization
All the graphical visualization of the data was performed in R with the ggplot2 package with the exception of the heatmaps, which were displayed using the pheatmap library
Box plots:
Box plots are calculated in the style of Tukey, shortly the center of the box represents the median of the values, the hinges the 25th and 75th percentile and the whiskers are extended no further than the 1.5 ∗ IQR (interquartile range). When multiple samples from the same donors were present in the same group only the earliest sample was used for visualization and statistical testing.
Heatmap:
The heatmap shows the mean value of the scaled expression of each gene in each cluster.
Dot plot:
The dot plot of the signature genes shown in Figure S3F was calculated according to the Dotplot Seurat function scaling the expression values by gene.
Statistical testing
To calculate the statistical significance of the changes observed in the frequency of selected T cell clusters we first performed a Kruskal-Wallis rank sum test with Benjamini-Hochberg correction for multiple testing when more than one test was performed. For the clusters showing statistically significant results, we performed a pairwise Dunn post-hoc test with Benjamini-Hochberg correction for multiple testing. An adjusted p-value < 0.05 was considered significant and annotated above the box plot as follow: < 0.05 ∗, < 0.01 ∗∗, < 0.001 ∗∗∗.
Analysis of lung autopsy samples (cohort 3)
RNA isolation and RT-PCR
COVID-19 autopsy lung tissue samples were screened for the presence of morphological criteria consistent with diffuse alveolar damage and best tissue preservation. Three samples of each case were tested for SARS-CoV-2 RNA with RT-PCR as previously reported (von Stillfried et al., 2021; Wong et al., 2021).
Briefly, using a Maxwell® 16 LEV RNA FFPE Purification Kit (Promega GmbH, Walldorf, Germany), we extracted RNA from formalin-fixed, paraffin-embedded (FFPE) lung tissue blocks on the Maxwell® 16 IVD instrument (Promega GmbH) according to the manufacturer's instructions. RNA eluates were processed immediately or stored at -80°C for further processing. We used the TaqMan™ Fast 1-Step Master Mix for the qualitative detection of the E gene and the RdRp gene as a second target with primer and probe sets labeled with fluorescent reporters and quencher dyes. Internal Positive Control reagents were used as internal PCR controls. RT-PCR was performed according to a previous publication (Remmelink et al., 2020). The primer and probe sequences used are listed in the key resources table. Results were analyzed using BioRad CFX Maestro software. The E gene was used to screen for SARS-CoV-2 RNA and validated with the detection of the RdRp gene as a second target. Samples with RT-PCR Ct (cycle threshold) values >40 for E and RdRp genes were considered negative. Samples were interpreted particularly as positive if Ct values for the E gene only were ≤40, and if additionally, the Ct value for the RdRp gene was ≤40. Finally, the sample from each case with the lowest Ct value for SARS-CoV-2 E gene RNA (indicating the highest RNA quantity) was selected for immunofluorescent staining.
Immunofluorescence
FFPE blocks of the samples with the lowest Ct value of SARS-CoV-2 E gene RNA were cut to 1 μm thin sections, deparaffinized, rehydrated and washed in phosphate-buffered saline (PBS). To analyze lymphocyte subtypes, we stained tissue sections using our pre-established protocol as follows: slides underwent antigen retrieval in low pH (citrate) buffer using the PT-Link module (Agilent, Santa Clara, USA). After fixation in 4% formaldehyde for 10 min, slides were washed, and blocking was performed with peroxidase blocking solution followed by 30 min incubation with antibody diluent. Immunofluorescence multiplex staining was performed with the Opal 7-Color Manual IHC Kit. The slides were incubated for 1h with primary antibodies CD3 (clone CD3-12, Bio Rad) and CD16 (clone DJ130c, Santa Cruz Biotechnology) as well as CD31 (clone JC70A, Agilent Technologies), followed by incubation with EnVision FLEX HRP and visualized with Opal 520 TSA Plus (Akoya Biosciences), Opal 690 (Akoya Biosciences) and Opal 620 TSA Plus (Akoya Biosciences), respectively. The nuclei were counterstained using Spectral DAPI.
Analysis of Immunofluorescence
We scanned immunofluorescence slides using the VECTRA automated quantitative pathology imaging system (Akoya Biosciences, Marlborough, USA) and generated ten multispectral images per slide (each multispectral image had the size 334μm∗250μm). After deploying automated cell detection using the InForm software, we trained an in-built cell phenotyping algorithm to detect CD3+/CD16+ cells in our cohorts. From each cohort, multispectral images of one patient were used for training and manually annotated using the above-mentioned phenotype. This algorithm was then deployed on all samples to detect the above-mentioned phenotypes. Measurement outputs of the inForm-Software were analyzed using the phenoptr and phenoptrReports packages in R version 4.0.3. Our final readout was the number of CD3/CD16 double-positive cells per mm2 within each cohort. Representative immunofluorescence pictures were obtained with a Zeiss Axio Imager 2 microscope using 40x objective and image analysis software ZEN 3.0 black edition.
Statistical testing
To calculate the statistical significance of changes observed in CD3/CD16 double-positive cells we performed a one-way ANOVA with Bonferroni’s multiple comparison test for all columns. A p-value < 0.05 was considered significant and annotated above the column as follow: <0.05 ∗, <0.01 ∗∗, <0.001 ∗∗∗.
Detection of SARS-CoV-2-specific IgG and IgA antibodies
Detection of IgG and IgA to the S1 domain of the SARS-CoV-2 spike (S) protein were assessed by anti-SARS-CoV-2 S1 IgG ELISAs (Euroimmun AG, Germany), as described elsewhere (Schlickeiser et al., 2020). Serum samples were tested at a 1:101 dilution and optical density (OD) ratios were calculated by dividing the OD at 450 nm by the OD of a calibrator sample tested within each run. Therefore, the calculated OD ratios can be used as a relative measure for the concentration of IgA and IgG antibodies in the tested sample. For IgG and IgA, OD ratios above 1.1 were considered to be reactive. Statistical analysis and data visualization was done in Prism9.
Ex vivo functional analyses of T cells
Degranulation assay and C3a binding assay
Frozen PBMC were thawed using Benzonase-containing wash buffer (RPMI, 2% FCS, Pierce Universal Nuclease, 250U/mL) seeded at 0.25x106/well and rested overnight in a humidified incubator. Subsequent to overnight rest, PBMC were washed and resuspended in complete medium (RPMI1640 (Gibco, Thermo Fisher Scientific, USA), 2mM(1% V/V) GlutaMAX (Gibco, Thermo Fisher Scientific, USA), 10mM (1% V/V) HEPES (Sigma-Aldrich, USA), 10mM (1%v/v) Sodium Pyruvate (Gibco, Thermo Fisher Scientific, USA), 10% v/v heat-inactivated Fetal Calf Serum (Sigma-Aldrich, USA), 1%(V/V) Penicillin/Streptomycin (Bio&Cell, Germany), 1%(V/V) MEM Non-essential amino Acid solution (Sigma-Aldrich, USA). Cells were stimulated with MACSiBeads (Miltenyi Biotec, Germany) coated with Biotin conjugated CD16 antibody (clone 3G8, Biolegend, USA), the corresponding isotype control (clone MOPC21, Biolegend, USA) or MACSiBead-recombinant Spike protein-Serum-immune complexes at a ratio of 1:10 (cell to particle). Beads were loaded with indicated antibodies according to the protocol provided by the manufacturer. Immune complex generation is described below.
PBMC were cultured in a humidified incubator (37°C, 5% C02) for 6h in the presence of 1x Brefeldin A (eBioscienceTM, Thermo Fisher Scientific, USA) and 1x Monensin (Biolegend, USA). LAMP1 directed antibody (CD107a-APC, clone H4A3, final dilution 1:40) was added to the culture for the whole incubation period. Subsequent to 6h incubation cells were subjected to surface and intracellular staining. Fc receptor-mediated unspecific binding of antibodies was blocked by preincubation with human TruStain FcX Fc receptor blocking solution (Biolegend, USA) for 10 min at 4°C. Cells were stained for CD3 BV711 (clone UCHT1, Biolegend), CD4 BV421 (clone OKT4, Biolegend), CD8 FITC (clone RPA-T8, Biolegend), CD16 BV605 (clone 3G8, Biolegend), CD38-PEcy7 (clone HB7, Biolegend), HLA-DR BV785 (clone L243, Biolegend) for 30 min at 4 C°. The fixable viability dye (ZombieRed, Biolegend) was incorporated in the surface staining mix. Surface antibody staining was performed in DPBS (Gibco, Thermo Fisher Scientific, USA), 2mM EDTA (Sigma-Aldrich, USA, 0,5%BSA Miltenyi Biotec, Germany). Cells were fixed for 20 min at 4°C with BD Cytofix/Cytoperm solution (BD Biosciences, USA) and intracellularly stained for 30 min at 4°C for Granzyme B APC/Fire 750 (clone QA16A02, Biolegend, USA), Perforin PE (clone dG9, Biolegend), Granzyme K PerCP/Cyanine5.5 (clone GM26E7, Biolegend) or TNFα PerCP/Cyanine5.5 (clone Mab11, Biolegend). Intracellular staining was performed in 1x Perm/Wash permeabilization buffer (BD Biosciences, USA). The antibody panel overview is provided in Table S4. Data analysis was performed in FlowJo™version 10.6.2 (BD Life Sciences, USA, Figure S6A includes detailed gating strategy and representative plots), statistical analysis and data visualization was done in Prism9.
Figure S6.

Representative plots of gating strategy, related to Figures 3C–3E
(A) Gating of GZMB expression and degranulation/CD107a expression of CD8+ T cells, also discriminationg between CD8low and CD8high cells, as shown in Figures 3C–3E. Especially the CD8high expressing T cells from patients with severe COVID-19 are characterized by increased GZMB expression and degranulation.
Flow sorted non-naive T cells (CD3+, CD45RA+/-, CCR7- CD4+ or CD8+), were rested overnight in complete medium and washed in cold staining buffer (DPBS 2mM EDTA, 0,5% BSA). Prior to surface marker staining, non-specific, Fc receptor-mediated staining was blocked by 10 min pre-incubation at 4°C with human TruStainFcX (Biolegend, USA) blocking solution or Fc receptor blocking solution (Miltenyi Biotec, Germany). Surface marker staining (CD3 PE(clone UCHT1), CD8 VioGreen (clone REA734), CD16 BV605 (clone 3G8), CD38 PECy7 (clone HB7), HLA-DR BV785 (cloneL243), Fixable Live dead dye (Zombie Red)) was performed for 30 min at 4°C. Binding of complement split product hC3a was tested by incubation of cells for 60 min at 4°C with 50nM synthetic human C3a labelled with AF647 (Almac, UK) subsequent to fixation and permeabilization with BD Cytofix/Cytoperm™ (BD Biosciences, USA) solution. Staining with C3a-AF647 was performed in 1x Perm/Wash permeabilization buffer (BD Biosciences, USA). The antibody panel overview is provided in Table S5. Data analysis was performed in FlowJo™ version 10.6.2 (BD Life Sciences, USA, STAR Methods, data and code availability includes detailed gating strategy and representative plots), statistical analysis and data visualization was done in Prism9.
Serum immune complex generation
Anti-Biotin MACSiBeads (Miltenyi Biotec, order# 130-092-357) were incubated with recombinant SARS-CoV-2 Spike Trimer (HEK)-Biotin (Miltenyi Biotec, order # 130-127-685) at 30μg recombinant protein per 1x108 beads at 4°C with repeated resuspension. Subsequent to washing in PBS, spike coated beads were incubated with human serum derived from a COVID19 patient or serum from healthy male donors (Pan Biotech, order# P30-2902, lot#P041801) at 4°C. Serum was removed by repeated washing in complete medium and MACSiBead-rSpike-Serum immune complexes were added at a 1:10 cell to bead ratio.
Detection of SARS-CoV-2-specific T cells
Frozen PBMC were thawed using Benzonase-containing wash buffer (RPMI, 2% FCS, Pierce Universal Nuclease, 250U/mL) seeded in 96 round well plate and rested for 3-4h in complete medium (RPMI1640 (Gibco, Thermo Fisher Scientific, USA), 2mM(1% V/V) GlutaMAX (Gibco, Thermo Fisher Scientific, USA), 10mM (1% V/V) HEPES (Sigma-Aldrich, USA), 10mM (1%v/v) Sodium Pyruvate (Gibco, Thermo Fisher Scientific, USA), 10% v/v heat-inactivated human AB type Serum (PanBiotech, Germany), 1%(V/V) Penicillin/Streptomycin (Bio&Cell, Germany), 1%(V/V) MEM Non-essential amino Acid solution (Sigma-Aldrich, USA) in a humidified incubator at 37°C. Prior to stimulation, PBMC were preincubated with CD40 blocking antibody for 15min (Miltenyi Biotec, order# 130-094-133, 0.5μg/mL). Peptide pool PepTivator SARS-CoV-2 Select (Miltenyi Biotec, order# 130-127-309) was added to cultures at a concentration of 1μg/mL. After 24h stimulation, cells were washed and subsequently stained with CD3 BV711 (clone UCHT-1,Biolegend), CD4 APC/Fire 750 (clone RPTA-4, Biolegend, CD8 VioGreen (clone REA734, Miltenyi Biotec), OX40 PE ( clone BER-ACT35, Biolegend), HLA-DR BV785 (clone L243, Biolegend), CD137 BV421 (clone 4B4-1, Biolegend), CD38-PEcy7 (clone HB7, Biolegend), CD69 APC (clone FN50, Biolegend), CD40L FITC (clone 5C3, Miltenyi Biotec), CD16 BV605 (clone 3G8, Biolegend), CD45RA PerCP-Cy5.5 (clone HI100, Biolegend), CCR7 Alexa Fluor 700 (clone G043H7, Biolegend) for 30 min at 4°C. The fixable viability dye (ZombieRed, Biolegend) and FC receptor block human TruStainFcX (Biolegend, USA) were incorporated in the surface staining mix. Finally, cells are fixed for 15 min at 4°C in 2% PFA in PBS. Surface antibody staining and washing steps are performed in DPBS (Gibco, Thermo Fisher Scientific, USA), 2mM EDTA (Sigma-Aldrich, USA) 0.5%BSA (Miltenyi Biotec, Germany). The antibody panel overview is provided in Table S6. Data analysis was performed in FlowJo™ version 10.6.2 (BD Life Sciences, USA, Figure S7A), statistical analysis and data visualization was done in Prism9.
Figure S7.

Representative plots of gating strategy, related to Figure 3F
(A) Gating of CD137+CD69+ of CD8+CD16+CD38+ T cells, also discriminationg between CD8low and CD8high cells, as shown in Figure 3F. Both, CD8low & CD8high expressing T cells from patients with severe COVID-19 contain high proportions of CD16+CD38+ cells.
Endothelial-T cell-co-cultures
Human pulmonary microvascular endothelial cells (HPMECs, Promocell, Germany, passage 4-8) were plated on 96-well plates (96W10idf PET, Applied Biophysics Inc., USA) and grown to confluency for 48-72 h. Cell impedance was quantified by electric cell impedance sensing (ECIS® Z-Theta Applied Biophysics Inc., USA) at 4000Hz every 60 sec. For co-cultivation experiments, media was replaced by Opti-MEM™ (Gibco, Thermo Fisher Scientific, USA). After 1 h stabilization phase, Convanacalin A (10 μM) from Canavalia ensiformis (Jack bean, Sigma-Aldrich Chemie GmbH, Munich, Germany, anti-CD16 beads (5 beads / T cell), and 20,000 flow-sorted, non-naive CD8+ T cells were added, and cell barrier integrity was monitored continuously for 24 h. Resistance was normalized for each individual well to the baseline before treatment. Statistical analysis and data visualization was done in Prism9.
T cell differentiation cultures and C3a neutralization
Frozen PBMC were thawed, washed twice with RPMI 1640 medium containing 2 % FBS and 0,02 % nuclease and afterwards resuspended in MACS buffer (DPBS with 0.5 % BSA, 2 mM EDTA) followed by enrichment of CD3+ cells using the human Pan T cell enrichment kit, (Miltenyi Biotec, Germany) according to the manufacturer’s protocol. CD3+ cells were activated in round-bottom wells (20000 cells/96 well) with 5 μg/ml plate-bound anti-human CD3, soluble 1 μg/ml anti-human CD28 and 20 IU/ml IL-2 (Proleukin) and the following additives: 1) 20 % non-inactivated serum (AB serum or serum from mild or severe COVID-19 patients), 2) 20 % heat-inactivated AB serum (30 min, 56 °C) +/- human recombinant C3a (20 nM), 3) 20 % non-inactivated serum from mild or severe COVID-19 patients +/- anti-human C3a (20 μg/ml). After 7 days of culture, cells were harvested and stained for flow cytometry using Zombie UV™ fixable viability kit (Biolegend, USA) for live/dead discrimination, Beriglobin® (CSL Behring, USA) for blocking of unspecific antibody binding (3,2 mg/ml) and CD3 BV421 (clone UCHT1), HLA-DR BV785 (clone L243), CD8 FITC (clone RPA-T8), CD38 PE-Cy7 (clone HB7) and CD16 AF700 (clone 3G8) for surface staining (all Biolegend, USA). Foxp3 staining buffer kit (Miltenyi Biotec, Germany) was used according to the manufacturer’s directions for intracellular staining of Ki67 eFluor506 (clone SolA15, Invitrogen, USA). Cells were acquired at a CytoFLEX LX flow cytometer and data analysis was done using Kaluza Analysis Software (both Beckman Coulter, USA, STAR Methods; data and code availability includes the detailed gating strategy).
T cells were differentiated as described above and rested overnight in a humidified incubator 37°C, 5% C02. 20,000 T cells were transferred to a 96 well round plate, washed 1x and resuspended in complete medium. Blaer1 GFP-/- cells were added to a ratio of 1: 5 (T cell: Blaer1 cell) after labelling with humanized monoclonal CD38-directed antibody (Daratumumab). To this end, Blaer1 cells were labelled for 30 min at 4°C with 50μg/mL Daratumumab. T cells and Blaer1 cells were incubated in a humidified incubator (37°C, 5% C02) for 6h in the presence of 1x Brefeldin A (eBioscienceTM, Thermo Fisher Scientific, USA) and 1x Monensin (Biolegend, USA). LAMP1-directed antibody (CD107a-APC, clone H4A3, final dilution 1:40) was added to the culture for the whole incubation period. Prior to 6h incubation, cells were centrifuged for 1 min at 350 rcf. Subsequently to 6h incubation, cells were subjected to surface and intracellular staining. Fc receptor-mediated unspecific binding of antibodies was blocked by preincubation with human TruStain FcX Fc receptor blocking solution (Biolegend, USA) for 10 min at 4°C. TrueStain FCX is furthermore added to the respective staining cocktails. Subsequently, cells were stained for CD3 PE (clone UCHT1, Biolegend), CD4 APC/Fire 750 (clone RPT-A4, Biolegend), CD8 BV605 (clone RPA-T8, Biolegend) and CD16 FITC (clone 3G8, Biolegend). The fixable viability dye (ZombieRed, Biolegend) was incorporated in the surface staining mix. Surface epitope staining was performed in DPBS (Gibco, Thermo Fisher Scientific, USA), 2mM EDTA (Sigma-Aldrich, USA, 0,5%BSA (Miltenyi Biotec, Germany). Cells were fixed for 20 min at 4°C with BD Cytofix/Cytoperm solution (BD Biosciences, USA) and intracellularly stained for 30 min at 4°C Perforin PEcy7 (clone dG9, Biolegend, USA), IFNγ BV785 (clone 4S.B3, Biolegend, USA) for 30 min at 4 C°. Intracellular staining was performed in 1x Perm/Wash permeabilization buffer (BD Biosciences, USA). Statistical analysis and data visualization was done in Prism9.
Quantification of cytokines, chemokines & complement split products
Interleukin-6 (IL-6), interleukin-8 (IL-8=CXCL8), tumor necrosis factor (TNF) and monocyte chemoattractant protein-1 (MCP-1=CCL2) levels of cell culture supernatants were determined using a Milliplex MAP human cytokine/chemokine magnetic bead panel kit (Merck Millipore, USA, HCYTOMAG-60k). The assay was performed overnight with half of the volume according to the manufacturer’s instructions. The median fluorescent intensities were quantified on a Bio-Plex 200 system (Bio-Rad Laboratories, Inc., USA) and the results were calculated using Bio-Plex- Manager 6.1 software (Bio-Rad Laboratories, Inc., USA).
Concentrations of the C3a and C5a activation products were measured in EDTA plasma from patients using commercially available enzyme-linked immunosorbent assays (ELISA) kits (HK354 (C3a) at a 1:1000 dilution, HK349 (C5a) at a 1:4 dilution, Hycult Biotech, the Netherlands) according to the manufacturer’s protocols.
Statistical analysis and data visualization was done in Prism9.
Plasma/serum proteomics
Proteomics was conducted using a recently developed platform technology for plasma and serum proteomics. (Demichev et al., 2021; Messner et al., 2020). In brief, plasma and serum samples were prepared for mass spectrometry through denaturation, reduction, alkylation, and tryptic digestion in 96 well plates using liquid handling robotics and cleaned using solid phase microextraction. The samples were separated using high-flow rate reversed-phase liquid chromatography with a 1290 Infinity II LC System (Agilent Technologies, USA), and data were acquired using a Triple TOF 6600 Hybrid mass spectrometer (Sciex Ltd, USA) running SWATH MS (Gillet et al., 2012). Data were processed using DIA-NN (Demichev et al., 2020), and post-normalized to control batch effects using information obtained from the repeated measurement of quality control samples.
Quantification and statistical analysis
The software used for statistical analysis is stated in the paragraphs describing the respective experimental procedures in the section “methods details.” For statistical details please refer to the figure legends and the respective experimental procedures in the section “methods details.”
The study was not blinded, and the sample size was calculated empirically prioritizing the inclusion of the highest number of COVID-19 samples and matching controls.
One ”n” represents the results of one experiment conducted with specimens of one individual at one time point. In the case of sequential analysis i.e., acute and convalescent, it refers to specimens from identical donors at two different time points.
Acknowledgments
We thank Katrin Vogt, Christine Appelt, Claudia Conrad, Anja Freiwald, Daniela Ludwig, and Vadim Farztdinov for technical support and Jonas Schulte-Schrepping, Elena de Dominico, Nico Reusch, Kristian Händler, Heidi Theis, Michael Kraut, and Kevin Baßler for generating the scRNA-seq dataset of cohort 2. In addition, we thank Franziska Scheibe and Il-Kang Na for providing human antibodies.
We also thank Desireé Kunkel and Jacqueline Keye from the BIHFlow and Mass cytometry Core facility for cell sorting, the BIH/MDC Genomics Platform for sequencing, and the Clinical Study Center (CSC) at the Berlin Institute of Health (BIH) and the Central Biobank of the BIH (ZeBanC) for ongoing support of the PA-COVID-19 Study.
We thank all members of the Pa-COVID-19 collaborative study group (Mendeley Data: https://doi.org/10.17632/9sw3trkv8p.3).
This work was supported by the German Research Foundation (DFG): SA1383/3-1 to B.S.; SFB-TR84 114933180 to L.E.S., S.B., P.G., S.H., and W.M.K. INST 37/1049-1, INST 216/981-1, INST 257/605-1, INST 269/768-1, INST 217/988-1, INST 217/577-1, and EXC2151-390873048 to J.L.S.; GRK 2168 – 272482170, ERA CVD 00160389 to J.L.S.; SFB 1454 – 432325352 to A.C.A. and J.L.S.; SFB TR57 and SPP1937 to J.N.; GRK2157 to A.-E.S.; ME 3644/5-1 to H.E.M.; RTG2424 to N.B.; SFB-TRR219 322900939, BO3755/13-1 Project-ID 454024652 to P.B.; the Berlin University Alliance (BUA) (PreEP-Corona grant to L.E.S. and V.M.C.); the Berlin Institute of Health (BIH) (to L.E.S., V.M.C., B.S., and W.M.K.); Helmholtz-Gemeinschaft Deutscher Forschungszentren, Germany (sparse2big to J.L.S.); EU projects SYSCID (733100 to J.L.S.); European Research Council Horizon 2020 (grant agreement no. 101001791 to P.B.); the DZIF, Germany (TTU 04.816 and 04.817 to J.N.); the Hector Foundation (M89 to J.N.); the EU projects ONE STUDY (260687), BIO-DrIM (305147) and INsTRuCT (860003) to B.S.; Federal Ministry of Education and Research FKZ 01KX2021 (COVIM to B.S., L.E.S., F.K., and V.M.C.; DEFEAT PANDEMIcs to P.B.; OrganoStrat to A.-E.S.); and STOP-FSGS-01GM1901A to P.B. and S.D.; the German Registry of COVID-19 Autopsies (www.DeRegCOVID.ukaachen.de), funded by the Federal Ministry of Health (ZMVI1-2520COR201 to P.B.) and through a resolution of the German Bundestag (Charité Corona Cross [CCC] and Charité Corona Cross 2.0 [CCC 2.0] to A.T.); the German Research Foundation (DFG; Project-IDs 322900939, 454024652, and 432698239 to P.B.; 432698239 to S.D.), the European Research Council (ERC; Consolidator Grant AIM.imaging.CKD, no. 101001791 to P.B.), and by the START-Program of the Faculty of Medicine of the RWTH Aachen University (grant numbers 125/17 to S.V.S. and 148/21 to R.D.B.); the Berlin Senate to German Rheumatism Research Centre (DRFZ); the Berlin Brandenburg School for regenerative Therapies (BSRT) to C.B.; the German Federal Ministry of Education and Research (BMBF) projects RECAST (01KI20337) to B.S., V.M.C., L.E.S., and M.R.; VARIPath (01KI2021) to V.M.C.; NUM COVIM (01KX2021) to L.E.S., V.M.C., F.K., J.L.S., J.N., and B.S.; RAPID to S.H.; SYMPATH to N.S. and W.M.K.; PROVID to S.H. and W.M.K.; ZissTrans (02NUK047E) to N.B.; National Research Node “Mass spectrometry in Systems Medicine” (MSCoresys) (031L0220A) to M.R. and N.B.; Diet-Body-Brain (DietBB) (01EA1809A) to J.L.S.; the UKRI/NIHR through the UK Coronavirus Immunology Consortium (UK-CIC), the Francis Crick Institute through the Cancer Research UK (FC001134), the UK Medical Research Council (FC001134), and the Wellcome Trust (FC001134 and IA 200829/Z/16/Z) to M.R.; a Charité 3R project (to B.S., S.H., and W.M.K.); and an intramural grant from the Department of Genomics & Immunoregulation at the LIMES Institute to A.C.A. The federal state of Bavaria (FOR-COVID to A.-E.S.).
We are grateful to the patients and donors volunteering to participate in this study making this research possible in the first place and wish for a speedy and full recovery. We are especially thankful to the next of kin of deceased COVID-19 patients who gave consent to the autopsy of their beloved ones.
Author contributions
Conceptualization: M.R., J.L.S., A.C.A., F.K., L.E.S., N.B., and B.S.; methodology: P.G., L.B., L.M., T.K., M. Streitz, V.M.C., P.B., S.D., W.M.K., and R.D.B; software/formal analysis: R.A.-G., L.B., V.D., I.G., S.K.A., L.E.S., V.M.C., P.B., and N.B.; investigation: P.G., L.B., S.B., L.M., T.K., C.I., L.P., C.G., M. Schneider, M. Streitz, J.S., H.-P.D., D.P., M.M., and P.B.; resources: L.J.L., P.T.-L., E.T.H., D.H., H.E.M., A.R.S., S.H., P.B., R.D.B., M.D., H.W., N.S., A.U., J.R., S.V.S., M.A.M., H.M.-R., J.N., C.M., C.T., and F.K.; data curation: D.B., E.W., M.L., and B.O.; writing–original draft: P.G., R.A.-G., L.B., S.B., L.M., T.K., M. Schneider, M.B., V.M.C., and B.S.; writing–review & editing: P.G., R.A.-G., L.B., M.R., A.T., R.E., I.L., W.M.K., C.M., J.L.S., A.C.A. C.T., F.K., L.E.S., N.B., and B.S.
Declaration of interests
V.M.C. is named together with Euroimmun GmbH on a patent application filed recently regarding SARS-CoV-2 diagnostics via antibody testing. A.R.S. and H.E.M. are listed as inventors on a patent application by the DRFZ Berlin in the field of mass cytometry.
Published: December 28, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.cell.2021.12.040.
Supplemental information
Data and code availability
Debarcoded fcs files of the CyTOF experiments of cohort 1 are deposited at FlowRepository under the Repository ID: FR-FCM-Z4P5. Raw count data of the scRNA-seq experiments of cohort 1 are deposited at GEO under the accession number GSE175450. R-scripts to analyse the CyTOF and single-cell data corresponding to cohort 1 will be available in Zenodo under the https://doi.org/10.5281/zenodo.5771937.
This includes supplementary tables, list of genes of interest selected for the clustering of scRNA-seq data (cohort 1), and also the reanalysis of a published scRNA-seq dataset (Wauters et al., 2021) of BAL samples from COVID-19 patients and patients with non-COVID-19 pneumonia, and detailed gating strategy and representative plots for Figures 5D, 5F–5H, and S1. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Aibar S., González-Blas C.B., Moerman T., Huynh-Thu V.A., Imrichova H., Hulselmans G., Rambow F., Marine J.-C., Geurts P., Aerts J., et al. SCENIC: single-cell regulatory network inference and clustering. Nat. Methods. 2017;14:1083–1086. doi: 10.1038/nmeth.4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angerer P., Haghverdi L., Büttner M., Theis F.J., Marr C., Buettner F. destiny: diffusion maps for large-scale single-cell data in R. Bioinformatics. 2016;32:1241–1243. doi: 10.1093/bioinformatics/btv715. [DOI] [PubMed] [Google Scholar]
- Aran D., Looney A.P., Liu L., Wu E., Fong V., Hsu A., Chak S., Naikawadi R.P., Wolters P.J., Abate A.R., et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat. Immunol. 2019;20:163–172. doi: 10.1038/s41590-018-0276-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbore G., West E.E., Rahman J., Le Friec G., Niyonzima N., Pirooznia M., Tunc I., Pavlidis P., Powell N., Li Y., et al. Complement receptor CD46 co-stimulates optimal human CD8+ T cell effector function via fatty acid metabolism. Nat. Commun. 2018;9:4186. doi: 10.1038/s41467-018-06706-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagnato G., Harari S. Cellular interactions in the pathogenesis of interstitial lung diseases. Eur. Respir. Rev. 2015;24:102–114. doi: 10.1183/09059180.00003214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergamaschi L., Mescia F., Turner L., Hanson A.L., Kotagiri P., Dunmore B.J., Ruffieux H., De Sa A., Huhn O., Morgan M.D., et al. Longitudinal analysis reveals that delayed bystander CD8+ T cell activation and early immune pathology distinguish severe COVID-19 from mild disease. Immunity. 2021;54:1257–1275.e8. doi: 10.1016/j.immuni.2021.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Björkström N.K., Gonzalez V.D., Malmberg K.-J., Falconer K., Alaeus A., Nowak G., Jorns C., Ericzon B.-G., Weiland O., Sandberg J.K., et al. Elevated numbers of Fc gamma RIIIA+ (CD16+) effector CD8 T cells with NK cell-like function in chronic hepatitis C virus infection. J. Immunol. 2008;181:4219–4228. doi: 10.4049/jimmunol.181.6.4219. [DOI] [PubMed] [Google Scholar]
- Boor P., Eichhorn P., Hartmann A., Lax S.F., Märkl B., Menter T., Skok K., Slotta-Huspenina J., von Stillfried S., Tzankov A., et al. Practical aspects of COVID-19 autopsies. Pathologe. 2021;42:197–207. doi: 10.1007/s00292-021-00925-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler A., Hoffman P., Smibert P., Papalexi E., Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018;36:411–420. doi: 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvelli J., Demaria O., Vély F., Batista L., Chouaki Benmansour N., Fares J., Carpentier S., Thibult M.-L., Morel A., Remark R., et al. Association of COVID-19 inflammation with activation of the C5a-C5aR1 axis. Nature. 2020;588:146–150. doi: 10.1038/s41586-020-2600-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chouaki Benmansour N., Carvelli J., Vivier E. Complement cascade in severe forms of COVID-19: recent advances in therapy. Eur. J. Immunol. 2021;51:1652–1659. doi: 10.1002/eji.202048959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clémenceau B., Vivien R., Debeaupuis E., Esbelin J., Biron C., Levy Y., Vié H. FcγRIIIa (CD16) induction on human T lymphocytes and CD16pos T-lymphocyte amplification. J. Immunother. 2011;34:542–549. doi: 10.1097/CJI.0b013e31822801d4. [DOI] [PubMed] [Google Scholar]
- Crotty S. T follicular helper cell differentiation, function, and roles in disease. Immunity. 2014;41:529–542. doi: 10.1016/j.immuni.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Defendi F., Leroy C., Epaulard O., Clavarino G., Vilotitch A., Le Marechal M., Jacob M.-C., Raskovalova T., Pernollet M., Le Gouellec A., et al. Complement alternative and mannose-binding lectin pathway activation is associated with COVID-19 mortality. Front. Immunol. 2021;12:742446. doi: 10.3389/fimmu.2021.742446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degauque N., Haziot A., Brouard S., Mooney N. Endothelial cell, myeloid, and adaptive immune responses in SARS-CoV-2 infection. FASEB J. 2021;35 doi: 10.1096/fj.202100024R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demichev V., Messner C.B., Vernardis S.I., Lilley K.S., Ralser M. DIA-NN: neural networks and interference correction enable deep proteome coverage in high throughput. Nat. Methods. 2020;17:41–44. doi: 10.1038/s41592-019-0638-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demichev V., Tober-Lau P., Lemke O., Nazarenko T., Thibeault C., Whitwell H., Röhl A., Freiwald A., Szyrwiel L., Ludwig D., et al. A time-resolved proteomic and prognostic map of COVID-19. Cell Syst. 2021;12:780–794.e7. doi: 10.1016/j.cels.2021.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Z., Diao B., Wang R., Wang G., Wang C., Tan Y., Liu L., Wang C., Liu Y., Liu Y., et al. The novel severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) directly decimates human spleens and lymph nodes. MedRxiv, MedRxiv. 2020 doi: 10.1101/2020.03.27.20045427. [DOI] [Google Scholar]
- Fenoglio D., Dentone C., Parodi A., Di Biagio A., Bozzano F., Vena A., Fabbi M., Ferrera F., Altosole T., Bruzzone B., et al. Characterization of T lymphocytes in severe COVID-19 patients. J. Med. Virol. 2021;93:5608–5613. doi: 10.1002/jmv.27037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Arias C., Lopez J.P., Hernandez-Perez J.N., Bautista-Ojeda M.D., Branch O., Rodriguez A. Malaria inhibits surface expression of complement receptor 1 in monocytes/macrophages, causing decreased immune complex internalization. J. Immunol. 2013;190:3363–3372. doi: 10.4049/jimmunol.1103812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrara C., Grau S., Jäger C., Sondermann P., Brünker P., Waldhauer I., Hennig M., Ruf A., Rufer A.C., Stihle M., et al. Unique carbohydrate-carbohydrate interactions are required for high affinity binding between FcgammaRIII and antibodies lacking core fucose. Proc. Natl. Acad. Sci. USA. 2011;108:12669–12674. doi: 10.1073/pnas.1108455108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Beltran W.F., Lam E.C., Astudillo M.G., Yang D., Miller T.E., Feldman J., Hauser B.M., Caradonna T.M., Clayton K.L., Nitido A.D., et al. COVID-19-neutralizing antibodies predict disease severity and survival. Cell. 2021;184:476–488.e11. doi: 10.1016/j.cell.2020.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillet L.C., Navarro P., Tate S., Röst H., Selevsek N., Reiter L., Bonner R., Aebersold R. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Mol. Cell. Proteomics. 2012;11 doi: 10.1074/mcp.O111.016717. O111.016717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goplen N.P., Wu Y., Son Y.M., Li C., Wang Z., Cheon I.S., Jiang L., Zhu B., Ayasoufi K., Chini E.N., et al. Tissue-resident CD8+ T cells drive age-associated chronic lung sequelae after viral pneumonia. Sci. Immunol. 2020;5 doi: 10.1126/sciimmunol.abc4557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gralinski L.E., Sheahan T.P., Morrison T.E., Menachery V.D., Jensen K., Leist S.R., Whitmore A., Heise M.T., Baric R.S. Complement activation contributes to severe acute respiratory syndrome coronavirus pathogenesis. mBio. 2018;9:e01753. doi: 10.1128/mBio.01753-18. e01718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z., Eils R., Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. 2016;32:2847–2849. doi: 10.1093/bioinformatics/btw313. [DOI] [PubMed] [Google Scholar]
- Gustine J.N., Jones D. Immunopathology of hyperinflammation in COVID-19. Am. J. Pathol. 2021;191:4–17. doi: 10.1016/j.ajpath.2020.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafemeister C., Satija R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 2019;20:296. doi: 10.1186/s13059-019-1874-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao Y., Hao S., Andersen-Nissen E., Mauck W.M., Zheng S., Butler A., Lee M.J., Wilk A.J., Darby C., Zager M., et al. Integrated analysis of multimodal single-cell data. Cell. 2021;184:3573–3587.e29. doi: 10.1016/j.cell.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess C., Kemper C. Complement-mediated regulation of metabolism and basic cellular processes. Immunity. 2016;45:240–254. doi: 10.1016/j.immuni.2016.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoepel W., Chen H.J., Geyer C.E., Allahverdiyeva S., Manz X.D., de Taeye S.W., Aman J., Mes L., Steenhuis M., Griffith G.R., et al. High titers and low fucosylation of early human anti-SARS-CoV-2 IgG promote inflammation by alveolar macrophages. Sci. Transl. Med. 2021;13 doi: 10.1126/scitranslmed.abf8654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hothorn T., Bretz F., Westfall P. Simultaneous inference in general parametric models. Biom. J. 2008;50:346–363. doi: 10.1002/bimj.200810425. [DOI] [PubMed] [Google Scholar]
- Huang X., Huang Y. Cellsnp-lite: an efficient tool for genotyping single cells. Bioinformatics. 2021;37:4569–4571. doi: 10.1093/bioinformatics/btab358. [DOI] [PubMed] [Google Scholar]
- Jacob C.O. On the genetics and immunopathogenesis of COVID-19. Clin. Immunol. 2020;220:108591. doi: 10.1016/j.clim.2020.108591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquemont L., Tilly G., Yap M., Doan-Ngoc T.-M., Danger R., Guérif P., Delbos F., Martinet B., Giral M., Foucher Y., et al. Terminally differentiated effector memory CD8+ T cells identify kidney transplant recipients at high risk of graft failure. J. Am. Soc. Nephrol. 2020;31:876–891. doi: 10.1681/ASN.2019080847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janky R., Verfaillie A., Imrichová H., Van de Sande B., Standaert L., Christiaens V., Hulselmans G., Herten K., Naval Sanchez M., Potier D., et al. iRegulon: from a gene list to a gene regulatory network using large motif and track collections. PLoS Comput. Biol. 2014;10 doi: 10.1371/journal.pcbi.1003731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson K.S. 2021. phenoptr: inForm Helper Functions. R package version 0.3.1.https://akoyabio.github.io/phenoptr/ [Google Scholar]
- Kavai M. Immune complex clearance by complement receptor type 1 in SLE. Autoimmun. Rev. 2008;8:160–164. doi: 10.1016/j.autrev.2008.06.002. [DOI] [PubMed] [Google Scholar]
- Korsunsky I., Millard N., Fan J., Slowikowski K., Zhang F., Wei K., Baglaenko Y., Brenner M., Loh P.-R., Raychaudhuri S. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods. 2019;16:1289–1296. doi: 10.1038/s41592-019-0619-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotecha N., Krutzik P.O., Irish J.M. Web-based analysis and publication of flow cytometry experiments. Curr. Protoc. Cytom. 2010;53 doi: 10.1002/0471142956.cy1017s53. 10.17.1–10.17.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krämer B., Knoll R., Bonaguro L., ToVinh M., Raabe J., Astaburuaga-García R., Schulte-Schrepping J., Kaiser K.M., Rieke G.J., Bischoff J., et al. Early IFN-α signatures and persistent dysfunction are distinguishing features of NK cells in severe COVID-19. Immunity. 2021;54:2650–2669.e14. doi: 10.1016/j.immuni.2021.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krovi S.H., Gapin L. Invariant natural killer T cell subsets-more than just developmental intermediates. Front. Immunol. 2018;9:1393. doi: 10.3389/fimmu.2018.01393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurth F., Roennefarth M., Thibeault C., Corman V.M., Müller-Redetzky H., Mittermaier M., Ruwwe-Glösenkamp C., Heim K.M., Krannich A., Zvorc S., et al. Studying the pathophysiology of coronavirus disease 2019: a protocol for the Berlin prospective COVID-19 patient cohort (Pa-COVID-19) Infection. 2020;48:619–626. doi: 10.1007/s15010-020-01464-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kverneland A.H., Streitz M., Geissler E., Hutchinson J., Vogt K., Boës D., Niemann N., Pedersen A.E., Schlickeiser S., Sawitzki B. Age and gender leucocytes variances and references values generated using the standardized one-study protocol. Cytometry A. 2016;89:543–564. doi: 10.1002/cyto.a.22855. [DOI] [PubMed] [Google Scholar]
- Le Bert N., Clapham H.E., Tan A.T., Chia W.N., Tham C.Y.L., Lim J.M., Kunasegaran K., Tan L.W.L., Dutertre C.-A., Shankar N., et al. Highly functional virus-specific cellular immune response in asymptomatic SARS-CoV-2 infection. J. Exp. Med. 2021;218 doi: 10.1084/jem.20202617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W.S., Selva K.J., Davis S.K., Wines B.D., Reynaldi A., Esterbauer R., Kelly H.G., Haycroft E.R., Tan H.-X., Juno J.A., et al. Decay of Fc-dependent antibody functions after mild to moderate COVID-19. Cell Rep. Med. 2021;2:100296. doi: 10.1016/j.xcrm.2021.100296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenth R.V. Least-squares means: the R package lsmeans. J. Stat. Softw. 2016;69:1–33. [Google Scholar]
- Levine J.H., Simonds E.F., Bendall S.C., Davis K.L., Amir E.-A.D., Tadmor M.D., Litvin O., Fienberg H.G., Jager A., Zunder E.R., et al. Data-driven phenotypic dissection of AML reveals progenitor-like cells that correlate with prognosis. Cell. 2015;162:184–197. doi: 10.1016/j.cell.2015.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao M., Liu Y., Yuan J., Wen Y., Xu G., Zhao J., Cheng L., Li J., Wang X., Wang F., et al. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat. Med. 2020;26:842–844. doi: 10.1038/s41591-020-0901-9. [DOI] [PubMed] [Google Scholar]
- Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubbers R., van Essen M.F., van Kooten C., Trouw L.A. Production of complement components by cells of the immune system. Clin. Exp. Immunol. 2017;188:183–194. doi: 10.1111/cei.12952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L., Sahu S.K., Cano M., Kuppuswamy V., Bajwa J., McPhatter J., Pine A., Meizlish M.L., Goshua G., Chang C.H., et al. Increased complement activation is a distinctive feature of severe SARS-CoV-2 infection. Sci. Immunol. 2021;6 doi: 10.1126/sciimmunol.abh2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier M. Institute for Statistics and Mathematics; Wirtschaftsuniversität Wien: 2014. DirichletReg: Dirichlet Regression for Compositional Data in R. [Google Scholar]
- Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011;17:10. [Google Scholar]
- Mastaglio S., Ruggeri A., Risitano A.M., Angelillo P., Yancopoulou D., Mastellos D.C., Huber-Lang M., Piemontese S., Assanelli A., Garlanda C., et al. The first case of COVID-19 treated with the complement C3 inhibitor AMY-101. Clin. Immunol. 2020;215:108450. doi: 10.1016/j.clim.2020.108450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastellos D.C., Pires da Silva B.G.P., Fonseca B.A.L., Fonseca N.P., Auxiliadora-Martins M., Mastaglio S., Ruggeri A., Sironi M., Radermacher P., Chrysanthopoulou A., et al. Complement C3 vs C5 inhibition in severe COVID-19: early clinical findings reveal differential biological efficacy. Clin. Immunol. 2020;220:108598. doi: 10.1016/j.clim.2020.108598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew D., Giles J.R., Baxter A.E., Oldridge D.A., Greenplate A.R., Wu J.E., Alanio C., Kuri-Cervantes L., Pampena M.B., D’Andrea K., et al. Deep immune profiling of COVID-19 patients reveals distinct immunotypes with therapeutic implications. Science. 2020;369 doi: 10.1126/science.abc8511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKechnie J.L., Blish C.A. The innate immune system: fighting on the front lines or fanning the flames of COVID-19? Cell Host Microbe. 2020;27:863–869. doi: 10.1016/j.chom.2020.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messner C.B., Demichev V., Wendisch D., Michalick L., White M., Freiwald A., Textoris-Taube K., Vernardis S.I., Egger A.-S., Kreidl M., et al. Ultra-high-throughput clinical proteomics reveals classifiers of COVID-19 infection. Cell Syst. 2020;11:11–24.e4. doi: 10.1016/j.cels.2020.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazawa M. Immunopathogenesis of SARS-CoV-2-induced pneumonia: lessons from influenza virus infection. Inflam. Regen. 2020;40:39. doi: 10.1186/s41232-020-00148-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogilenko D.A., Shpynov O., Andhey P.S., Arthur L., Swain A., Esaulova E., Brioschi S., Shchukina I., Kerndl M., Bambouskova M., et al. Comprehensive profiling of an aging immune system reveals clonal GZMK+ CD8+ T cells as conserved hallmark of inflammaging. Immunity. 2021;54:99–115.e12. doi: 10.1016/j.immuni.2020.11.005. [DOI] [PubMed] [Google Scholar]
- Moretta A., Marcenaro E., Parolini S., Ferlazzo G., Moretta L. NK cells at the interface between innate and adaptive immunity. Cell Death Differ. 2008;15:226–233. doi: 10.1038/sj.cdd.4402170. [DOI] [PubMed] [Google Scholar]
- Nanda A., Vura N.V.R.K., Gravenstein S. COVID-19 in older adults. Aging Clin. Exp. Res. 2020;32:1199–1202. doi: 10.1007/s40520-020-01581-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noris M., Remuzzi G. Overview of complement activation and regulation. Semin. Nephrol. 2013;33:479–492. doi: 10.1016/j.semnephrol.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowicka M., Krieg C., Crowell H.L., Weber L.M., Hartmann F.J., Guglietta S., Becher B., Levesque M.P., Robinson M.D. CyTOF workflow: differential discovery in high-throughput high-dimensional cytometry datasets. F1000Res. 2017;6:748. doi: 10.12688/f1000research.11622.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oja A.E., Piet B., Helbig C., Stark R., van der Zwan D., Blaauwgeers H., Remmerswaal E.B.M., Amsen D., Jonkers R.E., Moerland P.D., et al. Trigger-happy resident memory CD4+ T cells inhabit the human lungs. Mucosal Immunol. 2018;11:654–667. doi: 10.1038/mi.2017.94. [DOI] [PubMed] [Google Scholar]
- Peng Y., Mentzer A.J., Liu G., Yao X., Yin Z., Dong D., Dejnirattisai W., Rostron T., Supasa P., Liu C., et al. Broad and strong memory CD4+ and CD8+ T cells induced by SARS-CoV-2 in UK convalescent individuals following COVID-19. Nat. Immunol. 2020;21:1336–1345. doi: 10.1038/s41590-020-0782-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polycarpou A., Howard M., Farrar C.A., Greenlaw R., Fanelli G., Wallis R., Klavinskis L.S., Sacks S. Rationale for targeting complement in COVID-19. EMBO Mol. Med. 2020;12 doi: 10.15252/emmm.202012642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu P., Simonds E.F., Bendall S.C., Gibbs K.D., Bruggner R.V., Linderman M.D., Sachs K., Nolan G.P., Plevritis S.K. Extracting a cellular hierarchy from high-dimensional cytometry data with SPADE. Nat. Biotechnol. 2011;29:886–891. doi: 10.1038/nbt.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remmelink M., De Mendonça R., D’Haene N., De Clercq S., Verocq C., Lebrun L., Lavis P., Racu M.-L., Trépant A.-L., Maris C., et al. Unspecific post-mortem findings despite multiorgan viral spread in COVID-19 patients. Crit. Care. 2020;24:495. doi: 10.1186/s13054-020-03218-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rendeiro A.F., Ravichandran H., Bram Y., Chandar V., Kim J., Meydan C., Park J., Foox J., Hether T., Warren S., et al. The spatial landscape of lung pathology during COVID-19 progression. Nature. 2021;593:564–569. doi: 10.1038/s41586-021-03475-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rydyznski Moderbacher C., Ramirez S.I., Dan J.M., Grifoni A., Hastie K.M., Weiskopf D., Belanger S., Abbott R.K., Kim C., Choi J., et al. Antigen-specific adaptive immunity to SARS-CoV-2 in Acute COVID-19 and Associations with Age and Disease Severity. Cell. 2020;183:996–1012.e19. doi: 10.1016/j.cell.2020.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satyam A., Tsokos M.G., Brook O.R., Hecht J.L., Moulton V.R., Tsokos G.C. Activation of classical and alternative complement pathways in the pathogenesis of lung injury in COVID-19. Clin. Immunol. 2021;226:108716. doi: 10.1016/j.clim.2021.108716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawitzki B., Harden P.N., Reinke P., Moreau A., Hutchinson J.A., Game D.S., Tang Q., Guinan E.C., Battaglia M., Burlingham W.J., et al. Regulatory cell therapy in kidney transplantation (The ONE Study): a harmonised design and analysis of seven non-randomised, single-arm, phase 1/2A trials. Lancet. 2020;395:1627–1639. doi: 10.1016/S0140-6736(20)30167-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlickeiser S., Schwarz T., Steiner S., Wittke K., Al Besher N., Meyer O., Kalus U., Pruß A., Kurth F., Zoller T., et al. Disease severity, fever, age, and sex correlate With SARS-CoV-2 neutralizing antibody responses. Front. Immunol. 2020;11:628971. doi: 10.3389/fimmu.2020.628971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulte-Schrepping J., Reusch N., Paclik D., Baßler K., Schlickeiser S., Zhang B., Krämer B., Krammer T., Brumhard S., Bonaguro L., et al. Severe COVID-19 is marked by a dysregulated myeloid cell compartment. Cell. 2020;182:1419–1440.e23. doi: 10.1016/j.cell.2020.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuyler R.P., Jackson C., Garcia-Perez J.E., Baxter R.M., Ogolla S., Rochford R., Ghosh D., Rudra P., Hsieh E.W.Y. Minimizing batch effects in mass cytometry data. Front. Immunol. 2019;10:2367. doi: 10.3389/fimmu.2019.02367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sette A., Crotty S. Adaptive immunity to SARS-CoV-2 and COVID-19. Cell. 2021;184:861–880. doi: 10.1016/j.cell.2021.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanmugasundaram U., Bucsan A.N., Ganatra S.R., Ibegbu C., Quezada M., Blair R.V., Alvarez X., Velu V., Kaushal D., Rengarajan J. Pulmonary mycobacterium tuberculosis control associates with CXCR3- and CCR6-expressing antigen-specific Th1 and Th17 cell recruitment. JCI Insight. 2020;5 doi: 10.1172/jci.insight.137858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P., Markiel A., Ozier O., Baliga N.S., Wang J.T., Ramage D., Amin N., Schwikowski B., Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma M., Merkulova Y., Raithatha S., Parkinson L.G., Shen Y., Cooper D., Granville D.J. Extracellular Granzyme K mediates endothelial activation through the cleavage of protease-activated receptor-1. FEBS J. 2016;283:1734–1747. doi: 10.1111/febs.13699. [DOI] [PubMed] [Google Scholar]
- Shuwa H.A., Shaw T.N., Knight S.B., Wemyss K., McClure F.A., Pearmain L., Prise I., Jagger C., Morgan D.J., Khan S., et al. Alterations in T and B cell function persist in convalescent COVID-19 patients. Med (N. Y.) 2021;2:720–735.e4. doi: 10.1016/j.medj.2021.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinkovits G., Mező B., Réti M., Müller V., Iványi Z., Gál J., Gopcsa L., Reményi P., Szathmáry B., Lakatos B., et al. Complement overactivation and consumption predicts in-hospital mortality in SARS-CoV-2 infection. Front. Immunol. 2021;12:663187. doi: 10.3389/fimmu.2021.663187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spessott W.A., Sanmillan M.L., Kulkarni V.V., McCormick M.E., Giraudo C.G. Syntaxin 4 mediates endosome recycling for lytic granule exocytosis in cytotoxic T-lymphocytes. Traffic. 2017;18:442–452. doi: 10.1111/tra.12490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephenson E., Reynolds G., Botting R.A., Calero-Nieto F.J., Morgan M.D., Tuong Z.K., Bach K., Sungnak W., Worlock K.B., Yoshida M., et al. Single-cell multi-omics analysis of the immune response in COVID-19. Nat. Med. 2021;27:904–916. doi: 10.1038/s41591-021-01329-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streitz M., Miloud T., Kapinsky M., Reed M.R., Magari R., Geissler E.K., Hutchinson J.A., Vogt K., Schlickeiser S., Kverneland A.H., et al. Standardization of whole blood immune phenotype monitoring for clinical trials: panels and methods from the ONE study. Transplant. Res. 2013;2:17. doi: 10.1186/2047-1440-2-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M., Hao Y., Stoeckius M., Smibert P., Satija R. Comprehensive integration of single-cell data. Cell. 2019;177:1888–1902.e21. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan A.T., Linster M., Tan C.W., Le Bert N., Chia W.N., Kunasegaran K., Zhuang Y., Tham C.Y.L., Chia A., Smith G.J.D., et al. Early induction of functional SARS-CoV-2-specific T cells associates with rapid viral clearance and mild disease in COVID-19 patients. Cell Rep. 2021;34:108728. doi: 10.1016/j.celrep.2021.108728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thieme C.J., Anft M., Paniskaki K., Blazquez-Navarro A., Doevelaar A., Seibert F.S., Hoelzer B., Konik M.J., Berger M.M., Brenner T., et al. Robust T cell response toward spike, membrane, and nucleocapsid SARS-CoV-2 proteins is not associated with recovery in critical COVID-19 patients. Cell Rep. Med. 2020;1:100092. doi: 10.1016/j.xcrm.2020.100092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwari-Heckler S., Rauber C., Longhi M.S., Zörnig I., Schnitzler P., Jäger D., Giese T., Merle U. Dysregulated Host Response in Severe Acute Respiratory Syndrome Coronavirus 2-Induced Critical Illness. Open Forum Infect. Dis. 2021;8:ofab019. doi: 10.1093/ofid/ofab019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vabret N., Britton G.J., Gruber C., Hegde S., Kim J., Kuksin M., Levantovsky R., Malle L., Moreira A., Park M.D., et al. Immunology of COVID-19: current state of the science. Immunity. 2020;52:910–941. doi: 10.1016/j.immuni.2020.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varga Z., Flammer A.J., Steiger P., Haberecker M., Andermatt R., Zinkernagel A.S., Mehra M.R., Schuepbach R.A., Ruschitzka F., Moch H. Endothelial cell infection and endotheliitis in COVID-19. Lancet. 2020;395:1417–1418. doi: 10.1016/S0140-6736(20)30937-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivier E., Tomasello E., Baratin M., Walzer T., Ugolini S. Functions of natural killer cells. Nat. Immunol. 2008;9:503–510. doi: 10.1038/ni1582. [DOI] [PubMed] [Google Scholar]
- von Stillfried S., Villwock S., Bülow R.D., Djudjaj S., Buhl E.M., Maurer A., Ortiz-Brüchle N., Celec P., Klinkhammer B.M., Wong D.W.L., et al. SARS-CoV-2 RNA screening in routine pathology specimens. Microb. Biotechnol. 2021;14:1627–1641. doi: 10.1111/1751-7915.13828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wauters E., Van Mol P., Garg A.D., Jansen S., Van Herck Y., Vanderbeke L., Bassez A., Boeckx B., Malengier-Devlies B., Timmerman A., et al. Discriminating mild from critical COVID-19 by innate and adaptive immune single-cell profiling of bronchoalveolar lavages. Cell Res. 2021;31:272–290. doi: 10.1038/s41422-020-00455-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West E.E., Kolev M., Kemper C. Complement and the regulation of T cell responses. Annu. Rev. Immunol. 2018;36:309–338. doi: 10.1146/annurev-immunol-042617-053245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong D.W.L., Klinkhammer B.M., Djudjaj S., Villwock S., Timm M.C., Buhl E.M., Wucherpfennig S., Cacchi C., Braunschweig T., Knüchel-Clarke R., et al. Multisystemic cellular tropism of SARS-CoV-2 in autopsies of COVID-19 patients. Cells. 2021;10:1900. doi: 10.3390/cells10081900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan B., Freiwald T., Chauss D., Wang L., West E., Mirabelli C., Zhang C.J., Nichols E.-M., Malik N., Gregory R., et al. SARS-CoV-2 drives JAK1/2-dependent local complement hyperactivation. Sci. Immunol. 2021;6 doi: 10.1126/sciimmunol.abg0833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youngs J., Provine N.M., Lim N., Sharpe H.R., Amini A., Chen Y.-L., Luo J., Edmans M.D., Zacharopoulou P., Chen W., et al. Identification of immune correlates of fatal outcomes in critically ill COVID-19 patients. PLoS Pathog. 2021;17 doi: 10.1371/journal.ppat.1009804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G., Wang L.-G., Han Y., He Q.-Y. clusterProfiler: an R package for comparing biological themes among gene clusters. Omics. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y., Wang M., Zhang X., Li S., Lu Q., Zeng H., Hou H., Li H., Zhang M., Jiang F., et al. Antibody-dependent cellular cytotoxicity response to SARS-CoV-2 in COVID-19 patients. Signal Transduct. Target. Ther. 2021;6:346. doi: 10.1038/s41392-021-00759-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zenarruzabeitia O., Astarloa-Pando G., Terrén I., Orrantia A., Pérez-Garay R., Seijas-Betolaza I., Nieto-Arana J., Imaz-Ayo N., Pérez-Fernández S., Arana-Arri E., et al. T cell activation, highly armed cytotoxic cells and a shift in monocytes CD300 receptors expression is characteristic of patients with severe COVID-19. Front. Immunol. 2021;12:655934. doi: 10.3389/fimmu.2021.655934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.-Y., Wang X.-M., Xing X., Xu Z., Zhang C., Song J.-W., Fan X., Xia P., Fu J.-L., Wang S.-Y., et al. Single-cell landscape of immunological responses in patients with COVID-19. Nat. Immunol. 2020;21:1107–1118. doi: 10.1038/s41590-020-0762-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Debarcoded fcs files of the CyTOF experiments of cohort 1 are deposited at FlowRepository under the Repository ID: FR-FCM-Z4P5. Raw count data of the scRNA-seq experiments of cohort 1 are deposited at GEO under the accession number GSE175450. R-scripts to analyse the CyTOF and single-cell data corresponding to cohort 1 will be available in Zenodo under the https://doi.org/10.5281/zenodo.5771937.
This includes supplementary tables, list of genes of interest selected for the clustering of scRNA-seq data (cohort 1), and also the reanalysis of a published scRNA-seq dataset (Wauters et al., 2021) of BAL samples from COVID-19 patients and patients with non-COVID-19 pneumonia, and detailed gating strategy and representative plots for Figures 5D, 5F–5H, and S1. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.