

Abstract
Although the individual structures and respiratory functions of cytochromes are well studied, the structural basis for their assembly, including transport of heme for attachment, are unknown. We describe cryo-EM structures of CcsBA, a bifunctional heme transporter and cytochrome c synthase. Models built from the cryo-EM densities show that CcsBA is trapped with heme in two conformations, herein termed the closed and open states. The closed state has heme located solely at a transmembrane (TM) site, with a large periplasmic domain oriented such that access of heme to the cytochrome acceptor is denied. The open conformation contains two heme moieties, one in the TM-heme site and another in external site (P-heme site). The presence of heme in the periplasmic site, at the base of a chamber, induces a large conformational shift which exposes the heme for reaction with apocytochrome c. Consistent with these structures, in vivo and in vitro cytochrome c synthase studies suggest a mechanism for transfer of the periplasmic heme to cytochrome.
INTRODUCTION
Heme is an essential cofactor for proteins like hemoglobins, which carry gases, and for cytochromes which are involved in cellular redox reactions. It is well understood that heme traverses bilayer membranes for assembly of heme proteins1–3. Heme proteins are ubiquitous throughout the tree of life and dysfunction in their biogenesis is associated with numerous pathologic disease states. For instance, defective heme transport is postulated as the basis in specific anemias4, abnormal erythropoiesis5,6, neurodegeneration7, and even obesity8. Cytochrome assembly factors are abnormal in certain diseases (eg. MLS syndrome9) as well as in mitochondrial-based pathologies10. Nevertheless, our mechanistic understanding of heme trafficking and cytochrome assembly are limited by a lack of structural information on the various putative transporters and assembly factors1,2. An inability to trap endogenous heme in purified transporter and assembly complexes, due to the transient nature of heme associations, hampers such efforts11.
Here, we study a bifunctional, integral membrane protein from Helicobacter hepaticus called CcsBA, that acts both as a heme transporter and cytochrome c synthase (Extended Data Fig. 1a). In bacteria (and chloroplasts), it is suggested that CcsBA exports heme from inside (cytoplasm) to outside (periplasm)12–14, where it facilitates its covalent attachment to two cysteines of cytochrome c (cyt c) in a conserved CXXCH motif (Extended Data Fig. 1b)15,16. Once attached, cyt c (with heme) is released from the synthase active site and folding ensues, with the histidine of CXXCH acting as a ligand to heme. Results from molecular genetics and spectroscopic studies have led to the hypothesis that the mechanism of heme export through CcsBA is via a transmembrane heme binding site (TM-heme site) to an external site close to the periplasm and the highly conserved tryptophan rich loop i.e. the WWD domain (P-heme site), with each site formed by a pair of histidine ligands (Extended Data Fig. 1a)14,17.
Here, we describe both cryo-EM structural analysis and in vitro and in vivo reconstitution studies of CcsBA to elucidate the structural and molecular basis for heme transport, attachment, and release. The two conformations of CcsBA shown here, represent the first near-atomic structures of a heme transporter with heme and an active cytochrome c synthase, providing unique mechanistic insights into both processes.
RESULTS
Biochemistry of CcsBA containing co-purified endogenous heme.
Recombinant, GST-fused CcsBA has been previously purified with heme and used to probe an internal heme binding site that requires two histidines (TM-His1 and 2) within the fourteen TMs of CcsBA (Extended Data Fig. 1a)14,17. An external heme binding site contains a pair of histidines in periplasmic loops (called P-His1 and 2), as well as the WWD domain. To date, the function of a large periplasmic region (Extended Data Fig. 1a), has remained a mystery. GST-CcsBA is active in vivo in recombinant E. coli, with periplasmic thioreduction proteins present to reduce cysteine thiols within the CXXCH motif18,19. Recently, in vitro reconstitution of the cyt c synthase activity of CcsBA has shown that no other factors are necessary for function20.
Although GST-CcsBA has been purified from recombinant E. coli, hexahistidine tagged CcsBA had previously shown toxicity and low yields14,17. We recently engineered a ccsBA vector with a hexahistidine tag (Supplementary Fig. 1a), with this construct yielding substantial levels of recombinant protein as well as high levels of trapped endogenous heme compared to the GST tagged version20. The recombinant C-terminal, hexahistidine-tagged CcsBA is active in vivo, attaching heme to the co-expressed reporter: Bordetella pertussis cyt c4 in E. coli (Fig. 1a, b). Only in the presence of CcsBA, is the heme covalently attached to the 24KD polypeptide, detected by a heme stain after denaturing SDS PAGE. As shown previously, some of the cyt c4 is degraded into a monoheme 12KD polypeptide that also has heme attached by CcsBA. Below we show that FPLC-purified CcsBA has in vitro cyt c synthase activity, thus we conclude that our structural studies reflect active CcsBA in its cycle of biological activities. Hexahistidine tagged CcsBA (here termed CcsBA) was solubilized in dodecyl-maltoside (DDM), and purified by cobalt affinity chromatography, then FPLC size exclusion chromatography (SEC). CcsBA co-elutes with substantial levels of bound heme upon FPLC SEC (Fig. 1c, heme at 415nm, protein at 280nm), eluting at 260 kDa (Supplementary Fig. 1b–c) with stoichiometric CcsB and CcsA polypeptides (Fig. 1d). Detergent/protein ratios indicate that 260 kDa is within the expected size of a DDM-solubilized 14 TM protein21,22 that is likely monomeric. Negative stain transmission electron microscopy images of FPLC-purified CcsBA resulted in well aligned 2D class averages (Supplementary Fig. 1d), as did initial single particle cryo-EM images obtained at 300kV (Fig. 1e), confirming the presence of monomeric complexes.
Fig. 1: CcsBA Cryo EM maps reveal two conformations.

a. GST:CcsBA and His-tagged variants were co-expressed with B. pertussis cyt c4 in E. coli Δccm. The ability of variants to mature cyt c4 was monitored by cell lysis, separation by SDS-PAGE, and heme stain. b. Quantitation of results in a with synthase function normalized to 100% of GST:CcsBA function. n=3 biologically independent experiments were performed. Data are presented as mean values +/− SD. c. FPLC elution profile of CcsBA on the Bio-Rad SEC 650. Absorbance at 280 nm shows total protein and absorbance at 415 nm is from heme. Collected fractions are demarcated by the dashed red lines. d. SDS-PAGE of purified CcsBA after purification by size exclusion chromatography. Sizes in parentheses are predicted sizes of the CcsBA protein complex (107kDa, undissociated) and the CcsA-His* (65.1kDa) and CcsB* (42.2kDa), which migrate faster on SDS PAGE than the predicted molecular weights, as shown previously14,17. e. FPLC-purified CcsBA was frozen and imaged using the Titan Krios cryo electron microscope. Two classes of particles were captured which we call the closed and open states. f. Front and top views of CcsBA cryo map highlighting the closed (blue) and open (red) conformations in the periplasmic domain. To better visualize heme locations, halos were added. The top down view shows the accessibility of the P-heme to the CXXCH substrate at the membrane surface. g. The periplasmic domain alone is shown in the closed (blue) and open (red) conformations. The open conformation dimensions are shown.
CcsBA has closed and open conformations based on heme traffic
FPLC-purified CcsBA was placed onto an EM grid, vitrified via plunge freezing into liquid ethane, then imaged on a 300kV Titan Krios G3 cryo-EM, resulting in the acquisition of 1.35 million single particles (Supplementary Table 1). Two conformations of CcsBA were resolved after multiple rounds of 2D and 3D classification. The iron centers density of TM-heme (green, Fig 1f) and P-heme (yellow, see Fig. 1f, CcsBA-open) become obvious as the protein and DDM densities were progressively thresholded, revealing in a conformational specific way, either single or double densities (Extended Data Fig. 2). The conformation with two hemes present has the large periplasmic domain open to the putative CXXCH substrate and is thus called CcsBA-open (Fig. 1f, red), unlike CcsBA-closed, which has a single heme present (Fig. 1f, blue). The dimensions of the periplasmic chamber in the open state are approximately 42 × 29 Å with a 22 Å wide “door” and a height of 17 Å (Fig. 1g, red). Thus, the P-heme present in the chamber should easily be accessible to the CXXCH substrate (approximately 10 × 12 × 8 Å dimension, subsequence in PDB 1j3s). A top-down view of the open state confirms this postulation (Fig. 1f, red). In the closed state however, the periplasmic domain occludes (caps) the position of the P-heme (Supplementary Fig. 2).
A pathway emerges for CcsBA-mediated heme transport
Through multiple rounds of 3D classification we resolved only two well-defined cryo-EM densities that corresponded to the open and closed states (Supplementary Fig. 3). No minor population that would have corresponded to a conformation with heme only in the P-heme site was identified. Therefore, this state, if present, is highly transient. This observation suggests a mechanism for the initiation of heme flux from the TM-heme site to the P-heme site that includes filling the TM-heme site whenever heme traffics to the P-heme site. We postulate that heme entering the CcsBA cytoplasmic vestibule displaces the TM-heme such that it moves towards the P-heme site (Fig. 2a). Both the closed and open conformations have TM-heme, liganded by the imidazoles of TM-His1 and 2 (Fig. 2b). Only small positional differences between the open and closed states exist in this region with respect to TMs, heme, or the vestibule leading to the TM-His site (Supplementary Table 2). In contrast, the region at the P-heme site shows major differences between the open and closed conformations. P-His1 and 2 are clearly defined in the open state, with each imidazole liganded to the P-heme (Fig. 2c). Unlike TM-His ligands, density from the P-His2 and loop is not resolved in the closed state (Extended Data Fig. 3), suggesting that the P-His2 loop is flexible in the closed state (see “active site” below). Because P-His1 and loop are resolved in both states, but P-His 2 only in the open state, we suggest that P-His 2 has a specific function that requires mobility, as described below.
Fig. 2: CcsBA transmembrane helices and its heme binding domains.
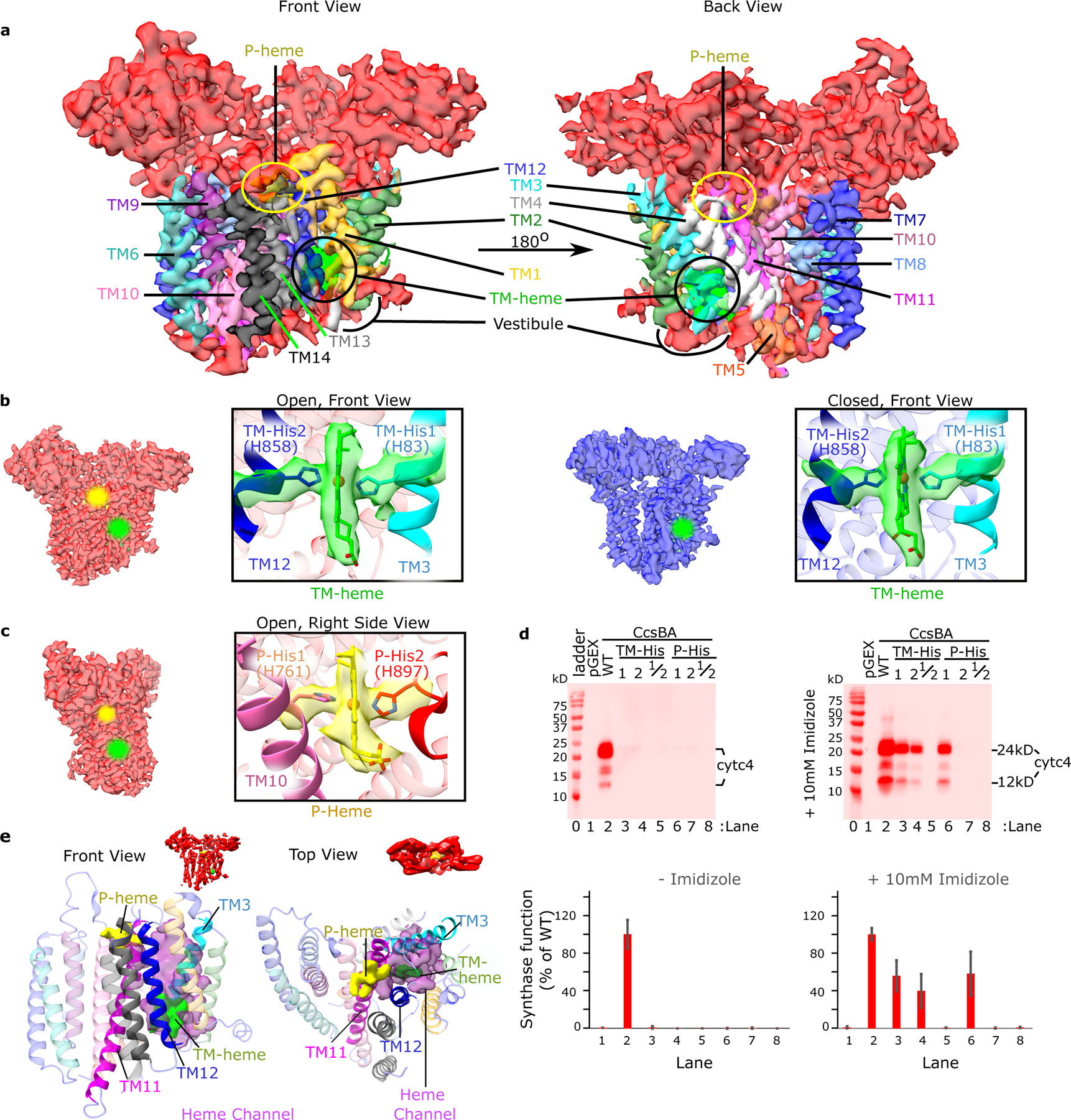
a. Front and back views of the open conformation of CcsBA cryo-EM map at 3.56 Å with labeled TMs, hemes, and the heme vestibule. b. CcsBA-open cryo-EM map (red) at 3.56 Å, next to the CcsBA-closed cryo-EM map (blue) at 4.14 Å. The boxed panels are a close-up view of the protein models docked into the electron density of the TM-heme (TMs 1 and 2 are hidden for better visualization of heme). c. A right side view rotated approximately 90° from that in b. The boxed panel is a close-up view of the protein-heme model of CcsBA-open docked into the electron density of P-Heme (TMs 8 and 9 are hidden to better visualize heme). To mark the heme locations, halos were added in the b and c cryo-EM densities. d. Representative heme stains of the reporter, cyt c4 in E. coli Δccm are shown and were used to assess the function of CcsBA His variants. Controls were pGEX vector (no CcsBA, lane 1) and wild-type CcsBA(WT, lane 2). Single and double His to Gly substitutions were made to produce variants at positions TM-His1, TM-His2, TM-His1/2, P-His1, P-His2, and P-His 1/2 that were nonfunctional. Results of chemical complementation of CcsBA His variants with 10mM imidazole (right panel). The bar graph (mean, n=3) and error bars (SD of the mean) are from representative data from 1 of 3 independent trials. e. For elucidation of the channel (magenta), P-heme (yellow) was modelled into the closed state based on the position of P-His 1 and the WWD domain. Then Pymol was used to find the channel cavity connecting TM-heme (green) to P-heme and exported to ChimeraX for display. The proposed channel lies between TM11 (magenta) and TM12 (blue).
We performed in vivo functional assays on TM-His and P-His variants (where His is replaced with Gly) with the co-expressed cyt c4 reporter, to determine if they attach heme to CXXCH motifs. As expected, and consistent with results obtained using GST-CcsBA, all four histidine variants were unable to attach heme to cyt c4 (Fig. 2d)14,17. We used an in vivo imidazole correction approach with the variants to probe for “cavities” that might be formed in each His/Gly variant. This approach draws from recombinant studies on the myoglobin distal His93Gly variant, whereby exogenous imidazole corrected for heme binding in growing E. coli by entering the apomyoglobin cavity (of His93Gly) and providing the axial ligand to allow for heme binding23. In the case of CcsBA, we assayed imidazole correction (10mM) using its in vivo function: heme attachment to cyt c4 (Fig. 2d). Chemical correction by exogenous imidazole would suggest that heme binding has been corrected and that a cavity for imidazole in the His/Gly variant is present. TM-His1, 2 and P-His1 variants were each corrected by imidazole but the P-His2 variant was not. Thus, the P-His2 residue and its loop do not form a cavity for imidazole correction; it is not equivalent to P-His1 liganding. Consistent with the lack of P-His2 density in the closed state, we suggest that P-His2 plays an active role in the reaction chamber for CXXCH heme attachment (see active site below).
We evaluated the CcsBA-open and CcsBA-closed densities for a possible heme “channel” between the TM-heme and P-heme sites. We identified a direct channel (magenta-colored cavity) between TM11 and TM12 that could accommodate heme as it moves from TM-His to P-His sites (Fig. 2e). The distance between hemes in the TM- and P-heme sites is approximately 17 Å in the open state (edge to edge of heme, Extended Data Fig. 2c). Because there is no access to the CXXCH acceptor in the closed state, it is likely that binding the acceptor would not induce transport of heme to the P-heme site. Thus, some other force(s) must stimulate heme transport without resulting in heme only in the P-heme site. As described above, we propose that heme binding in the vestibule displaces heme in the TM-heme site into the channel towards the P-heme site. Initially, P-His1, then P-His2, ligand this heme, in conjunction with key sidechains of the WWD domain for specific interactions. The vestibule heme then enters the TM-heme site (see Supplementary Video 1, video of heme flux).
The CcsBA active site reveals mechanisms of cyt c biogenesis.
To demonstrate that P-heme is attached to the cyt c (CXXCH) substrate, we used in vitro reconstitution of the cyt c synthase activity (Fig. 3a), which we recently developed for both the human cyt c synthase(HCCS) and CcsBA to reveal that no other protein factors are required for in vitro synthesis20. Here we reconstitute the FPLC-purified CcsBA and the CcsBA P-His1/2Gly variant. The assay is performed anaerobically, requiring DTT for reduction of the cyt c thiols and to maintain the heme of CcsBA in the reduced state, previously determined prerequisites for attachment24–26. We used a mammalian apocyt c (Equus caballus, Sigma, 89% identity to human) as a substrate that was chemically stripped of heme20,27. We and others have shown that human cyt c is recognized in vivo by bacterial cyt c synthesis systems (when a signal sequence is added to the cyt c to divert to the periplasm)28,29. Cyt c with covalently attached heme absorbs light at 550 nm while the b-hemes in CcsBA, when reduced, have a 560 nm maximum (Fig. 3a). Within minutes of initiating the reaction, a 550 nm peak is observed with FPLC-purified CcsBA and becomes prominent after three hours (Fig. 3b, left panel). We confirmed formation of the covalent attachment using heme stains after SDS-PAGE. We previously proposed that heme bound in the P-heme site (and adjoining WWD domain, Extended Data Fig. 1a), is covalently attached to the CXXCH motif but this has not been proven14,17. CcsBA P-His variants still possess heme in the TM-heme site (~70% compared to wild type CcsBA) so we tested whether heme is still able to be attached in vitro using the FPLC-purified CcsBA P-His1/2Gly variant. Since the P-His variant is unable to attach heme in vitro (Fig. 3b, right panel) we conclude that the P-heme site represents the active site.
Fig. 3: The Active Site of CcsBA with WWD Domain.

a. Diagram of in vitro reactions highlighting the spectra of heme in CcsBA initially (P-heme yellow, TM-heme green) and after reacting (TM-heme only) with apo cyt c to form holo cyt c. Cartoon similar to that used20 to show assay for cyt c synthase activity. b. UV vis spectra of in vitro reactions. CcsBA (left) or CcsBA mut (P-His 1/2 Gly) (right) were reacted with apocyt c as in a. Initial spectra (black) and final (3hr) spectra (red) show that CcsBA matures apo cyt c (550 nm peak), while CcsBA mut does not (560 nm, b-heme). c. Sequence alignment of the following WWD domains from the prokaryotic cyt c biogenesis pathways: CcsBA (System II), CcmC (System I), and CcmF (System I). Invariant and semiconserved residues are colored in grey, and variable residues are indicated with an “X.” CcsBA and CcmC residues that form a Cys/heme crosslink are indicated with an asterisk. Homologous residues of CcsBA and CcmC that crosslink are highlighted in yellow. Modified from ref.14. d. A top view of the open conformation of CcsBA with P-heme density (yellow), P-His2 Loop (pink), and WWD domain density (green). Labeled are the 2-vinyl and 4-vinyl of the heme, P-His2 (H897), and four highly conserved Trp residues within the WWD domain. e. Top view of the open conformation of CcsBA with P-heme density (yellow), and WWD Domain density (green). The density of P-His2 has been removed and replaced by the CXXCH (blue) that ligands the heme. The PDB model of the heme is docked into the map, and the CXXCH is placed as it was positioned by AutoDock Vina 1.5.651 (Supplementary Table 3). The 2-vinyl and 4-vinyl of the P-heme are labeled, due to their interaction with the cysteines of the CXXCH.
It has been speculated for many years that the WWD domain binds heme and somehow presents this heme for covalent attachment to its acceptor via vinyls 2 and/or 430. Three integral membrane proteins contain the WWD domain (Fig. 3c, Supplementary Fig. 4): CcsBA and CcmF (acceptor cyt c), and CcmC (acceptor CcmE)12,31. In the case of CcmF, the cyt c synthase is a CcmF/H complex and is part of a separate cyt c maturation pathway called System I32–34. Recently the crystal structure for CcmF of System I has been published (see Discussion). CcmC of System I transports heme by an unknown mechanism to the periplasmic heme chaperone CcmE, which then donates this heme to the CcmF/H cyt c synthase35. Each of the three WWD family members possesses the adjoining P-His heme ligands that bracket the WWD domain (Supplementary Fig. 4). We focus on the positions of the WWD domain and P-His ligands in the external site to further understand the mechanisms of cyt c synthase (i.e. covalent attachment to CXXCH and release of the holocyt c product).
In the open state, the WWD domain of CcsBA is compact and appears to interface with the edges of heme, acting in concert with the P-His2 loop to separate heme from the CXXCH substrate as it enters the chamber (Fig. 3d, e). Resolution of P-heme allows for identification of the vinyls and propionates, showing that the vinyls are accessible to the incoming CXXCH substrate (Fig 3d,e, Extended Data Fig. 4). We recently published heme/cysteine crosslinking experiments whereby cysteine substitutions of WWD residues in CcsBA and CcmC crosslink in vivo to vinyl groups on the heme (see Fig. 3c, yellow highlights)14,35. Each crosslinked residue (eg W828) in the structure is near a vinyl group of heme, supporting the active site structure revealed for the open state (Fig. 3d). Moreover, each crosslinked residue can now be assigned a likely vinyl group for the crosslink (ie vinyl 2 or 4). Densities at the active site include W828, W833, W837, W839 and P-His1 and 2 (H761, H897 respectively), suggesting that the compact WWD domain is involved in binding the edge of heme, presenting it to the incoming acceptor protein.
We propose a mechanism for CcsBA cyt c synthase activity, whereby a periplasmic reaction chamber forms upon entry of the heme substrate into the active site. This chamber is sufficiently large to accommodate the CXXCH motifs, which is the only motif necessary for recognition by CcsBA (unlike the unrelated human holocyt c synthase (HCCS), which requires the N-terminal alpha helix 120). Given the flexibility of the P-His2 loop, and that it is easily accessible in the chamber (Fig. 3d, e), we suggest that upon binding CXXCH, the histidine of CXXCH exchanges with P-His2. After computationally removing the P-His2 ligand, we docked the human cyt c CXXCH sequence (CSQCH) in the chamber (Fig. 3e; Supplementary Table 3). The histidine of CSQCH bound as the ligand to heme iron and each cysteine was positioned near a vinyl of heme. Since the first and second cysteines were docked near vinyl 2 and vinyl 4, respectively, the docking would stereochemically attach CXXCH to the heme. Once attached, the holocyt c must be released from the active site. Although requiring further study (e.g. trapping the CXXCH substrate at the active site), the WWD domain is well positioned to facilitate release, thus providing assistance for binding, attachment and release functions.
Release of holocyt c will result in the presence of only TM-heme, inducing a conformational change back to the closed state. Supplementary Video 1 shows how CXXCH enters through the chamber door, docks, attaches, and is released, causing CcsBA to transition back to the closed state. The chamber door is ideally suited to allow entry of unfolded apocyt c. As we have demonstrated using in vitro reconstitutions, many different peptides containing CXXCH are recognized with subsequent heme attachment20. Thus, an unfolded motif may be a major criterion for chamber entry.
Heme transport-mediated conversion from closed to open state
Conformational changes in the periplasmic domain during the conversion from CcsBA-closed to CcsBA-open include a large 19 Å shift of the two periplasm domains (PD1 and PD2) that open the chamber above the active site (Fig. 4). A large periplasmic region is present among all CcsBA (or in CcsB for organisms that encode separate ccsB and ccsA genes). The most conserved feature in this periplasmic region among all CcsBA proteins is represented by PD1, which is comprised of a Beta sheet structure (see “PD1” in Fig. 4a). However, it is largely the beta sheet secondary structure in PD1 that is conserved, with only a few residues that are completely conserved. We believe this is consistent with a capping function whereby PD1 occludes the CXXCH acceptor when in the closed state. Other potential roles for the PDs in the closed and opened states are presented in Discussion. The present density resolution (3.56 Å) does not allow for robust identification of individual residues that interact in the periplasmic region. However, analyses of the TMs in the closed and open conformations provide insight on the structural changes that drive chamber formation when the P-heme site is occupied (Supplementary Table 2, Fig. 4b-c). While most TMs show minor rearrangements from closed to open states, TMs 6 through 9 have shifts that range from 10.3 Å to 14.8 Å. We consider these two shifts, caused by P-heme liganding, likely to contribute to chamber opening. First, the periplasmic loop between TM9 and TM10 (TM9-10 loop) shifts 10 Å and forms new interactions with chamber residues in the open state (Fig. 4b). The TM9-10 loop is extended deep into the periplasmic domain in the closed state but is brought close to the membrane TMs in the open state (Fig. 4b). Note that the TM9-10 loop contains P-His1, which is positioned similarly in both conformations in the membrane, yet the remainder of the TM9-10 loop rearranges upon P-heme binding. We propose that the TM9 -TM10 loop helps control chamber opening. A second notable rearrangement is in TM6, which follows PD2 by shifting 11.1 Å (Fig. 4c). Interaction of PD2 with TM6 upon P-heme binding could induce conformational change in the periplasmic domain of CcsBA-open, resulting in the 19 Å movement that opens the chamber. Together, these shifts are likely responsible for the heme transport induced formation of the cyt c synthesis reaction chamber. Supplementary Videos 1 and 2 show the shifts in TMs 6–9 occurring simultaneously with chamber opening.
Fig. 4: CcsBA-closed and -open structures which suggest a mechanism of periplasmic domain movement.
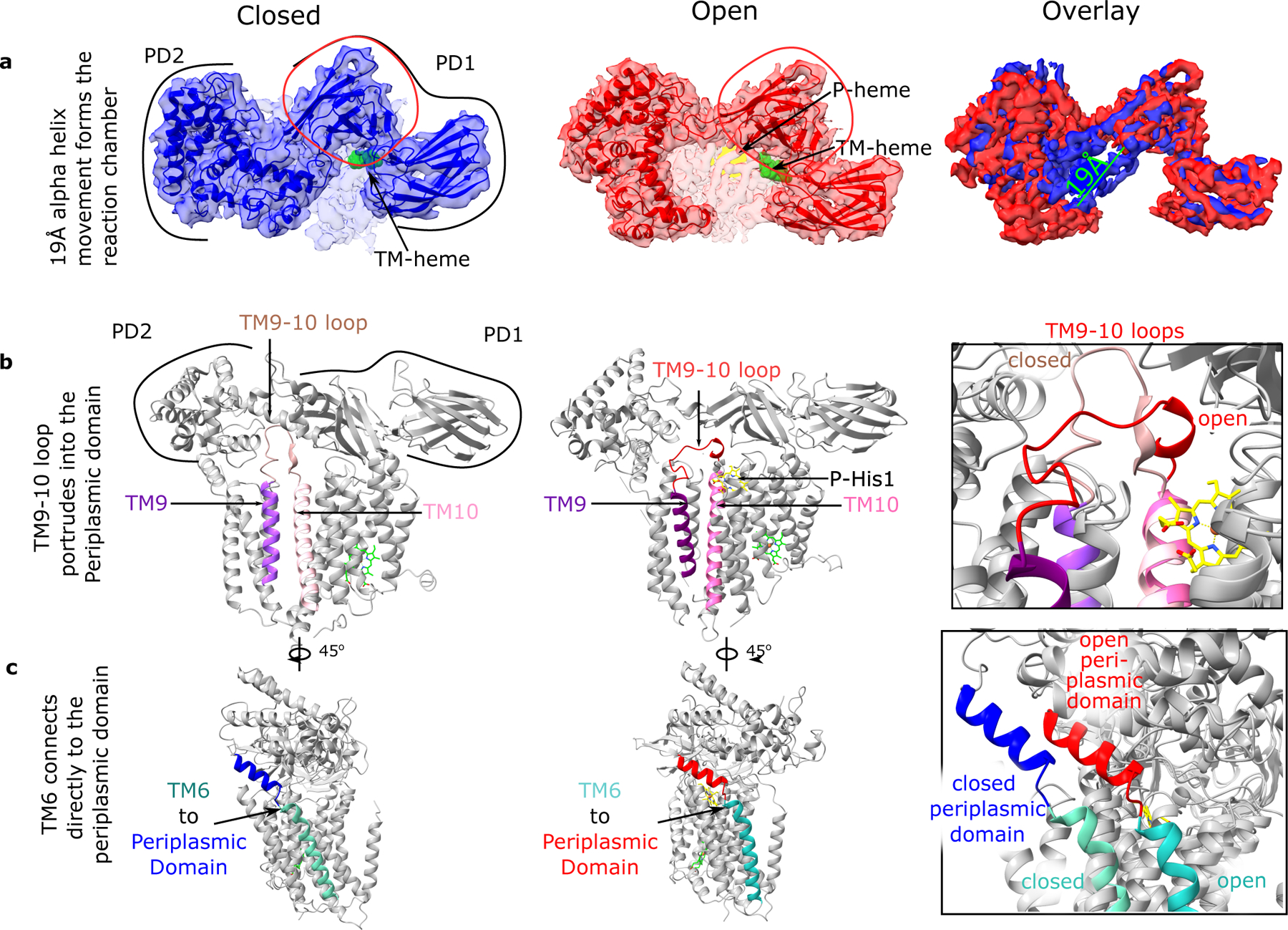
a. Top view of 4.14 Å CcsBA-closed and 3.56 Å CcsBA-open cryo-EM maps and the respective models and their overlay (rightmost panel). P-heme is yellow, and TM-heme is green. Measurement of the periplasmic alpha helices 19 Å movement to open the door to the active site is shown. The red circle encloses the most conserved set of residues in the periplasmic domain. b. Model of CcsBA-closed with the periplasmic domain backbone showing the TM9 to TM10 loop (brown) extending deeply into the periplasmic domain. In contrast, CcsBA-open has the TM9 to TM10 loop (red) extended near the membrane surface. The periplasmic domain was selectively made transparent to see the loops in each panel. c. Model of CcsBA-closed and -open showing the connection of TM6 to the periplasmic domain.
DISCUSSION
We report, to our knowledge, the first cryo-EM derived structures of a heme transporter with bound heme. Model fitting of CcsBA into the refined cryo-EM density yielded two conformations that highlight intermediates in the transport and cyt c synthesis cycles. Structures of two crystallized ABC transporters that import heme in Gram-negative pathogens have previously been published36–38. These require a periplasmic binding protein (PBP) that shuttles heme to the membrane components for release into the cytoplasm or inner leaflet, upon ATP hydrolysis. In these cases, an external crevice was postulated to bind heme that is attached to the PBP, suggesting a mechanism that is similar to other ABC importers. In CcsBA, we observe a clear path between TMs that could allow heme to travel from the TM-heme site to the external heme site. Since heme goes down its concentration gradient during export, we do not envision an energy source requirement for this process. We have not captured a conformation that has heme only in the external site, suggesting that heme entering a cytoplasmic vestibule beneath the TM-heme displaces the TM-heme towards the external site (P-heme site) and fills the TM-heme site. The very low levels and locations of free (labile) heme in the cell has recently been characterized using heme reporters, levels that are likely due to its toxicity39,40. Given heme’s amphipathic features and the CcsBA structures, heme could enter the CcsBA vestibule from the inner leaflet, from the cytoplasm, or directly from the terminal step in heme synthesis (eg. ferrochelatase, the enzyme that inserts reduced iron into protopophryin IX41–43.)
Notably, the CcsBA structures reveal that a periplasmic chamber forms in the open state as a consequence of heme transport to the external site. We suggest that the open conformation provides a reaction chamber for the stereochemical attachment of the external heme to the cyt c CXXCH motif. All cyt c open reading frames in bacteria possess a SEC-dependent signal sequence to direct the unfolded apoprotein to the periplasm. The CXXCH motifs could enter the chamber door as the motif exits the SEC complex44,45. In the chamber, the reduced P-heme is bound by two P-His ligands, shown to be essential in vivo and in vitro, and is directly accessible to the CXXCH acceptor, separated only by the P-His2 loop. We propose a stereospecific ligand exchange in which the histidine of CXXCH replaces P-His2, which is likely to be flexible based on the CcsBA-closed structure and imidazole correction results. Once the two thioether bonds form between the CXXCH thiols and heme vinyls, heme is released from the active site of CcsBA. The conserved WWD domain, including key tryptophans, is positioned on the edge of heme and could aid in binding, attachment, and release. Further analyses of CcsBA variants, both structurally and biochemically, will address the machinery of heme transport, conformational induction of a chamber, as well as mechanisms of the active site.
Upon attachment of P-heme to CXXCH and release of cyt c from the chamber, the CcsBA-closed state reforms. The molecular reason for the capping (closing) of the active site is unclear. Potentially, binding of the CXXCH substrate with no heme present (e.g. when heme levels are low) might be deleterious and/or provide feedback control. It has been suggested that the two pairs of histidines that bind transported heme could well be protective17,46. Therefore, capping might further protect the TM-heme from reactions with other substrates (e.g. oxidants) or perhaps prevent permeation of CcsBA to ions or protons when the P-heme site is unoccupied. It is also possible that the periplasmic domain is involved in the release function of the product. More studies will be needed to address these possibilities, guided by the structural features. The CcsBA family (also called ResBC or the System II cyt c biogenesis pathway) is widely distributed in the bacterial (and chloroplast) kingdom (Supplementary Fig. 5), examples include many Gram positives and negatives, mycobacteria, bacteroides, and cyanobacteria. Indeed, all of the key heme ligands (P-His and TM-His) discussed here are conserved, as well as the WWD domain. The structural and mechanistic insights reported here will be widely applicable, filling large gaps in the field.
As discussed above, parallels between the System II CcsBA complex and Ccm proteins (Supplementary Fig. 4) from System I have been proposed. Although the structure of an active cyt c synthase has not been published, very recently the crystal structure of CcmF47, a component of the CcmF/H cyt c synthase complex, has been determined. For the Ccm pathway (System I), the mechanism for delivery of substrate heme to the synthase is via a periplasmic heme chaperone, CcmE48,49. Brausemann et al show that CcmF presents a cavity in the outer leaflet region, where the WWD domain surrounds the putative heme (attached to CcmE as a covalent adduct)47. They postulate that the CcmE heme enters the cavity, not from above, but from the outer leaflet. Although a structure with CcmE heme in this active site will be needed to support this50, their study highlights some key mechanistic differences between CcmF/H and CcsBA. It has been known for over a decade that CcmF proteins contain another stable heme b that is liganded by TM-His1 and TM-His233, also previously proposed that this redox active b heme would reduce the incoming heme from CcmE (to Fe2+)12,33. Such reduction would eject the CcmE-heme adduct and prepare this substrate heme for attachment to CXXCH12. Recent work shows that the TM-heme is indeed liganded by TM His1 & 2, similar to that in CcsBA47. However, while CcsBA displays a transport channel between TM-heme and P-heme sites, the “channel” in CcmF is occluded by at least two tryptophans (W215 and W251), not present in CcsBA. This exciting result is consistent with two key mechanistic differences between CcmF/H and CcsBA: first, that CcmF does not transport its TM-heme and second, that the tryptophans could well participate in a reduction relay of the substrate heme (holoCcmE). Thus, CcmF/H is a heme reductase and cyt c synthase. The System II CcsBA is unique, it is a machine that carries out heme transport, CXXCH recognition and scanning, cyt c synthase attachment, and then release of the holocyt c47. Here we address the structural basis for how the CcsBA machine functions, revealing major dynamic switches that occur during the cycle.
MATERIALS AND METHODS
Bacterial growth conditions
Escherichia coli strains were grown in Luria-Bertani (LB; Difco) broth with selective antibiotics and inducing reagents as required. Antibiotic/induction concentrations: carbenicillin, 50 μg/ml; chloramphenicol, 20 μg/ml, isopropyl β-D-1-thiogalactopyranoside (IPTG; Gold Biotechnology), 1.0 mM or 0.1 mM; arabinose (alfa Aesar), 0.2% (wt/vol).
Construction of strains, plasmids, and primers
Cloning was performed using E. coli NEB-5α with the QuikChange II site-directed mutagenesis kit (Agilent Technologies) following the manufacturer’s instructions. Strains, plasmid, and primers are provided in Supplementary Table 4 and are available upon request.
Protein Purifications
GST*CcsBA:His and *CcsBA:His affinity purifications were performed as in17,20, with the following modifications. Resuspended membrane pellets were bound to Talon Metal Affinity Resin (Takara). Columns were washed by gravity flow in Resin Buffer (20 mM Tris pH 8, 100 mM NaCl) using 0, 2, and 5 mM imidazole, then eluted with 125 mM imidazole.
Heme Staining, Coomassie protein staining, and immunoblotting
Samples were prepared in loading dye at 1:1 (v/v) that did not contain reducing agents and were not boiled to maintain heme signals. Samples were separated by SDS-PAGE. Heme staining was performed by transfer to nitrocellulose and detection of heme signal using the SuperSignal Femto kit (Pierce) and imaged on a LI-COR odyssey Fc (LI-Cor Biosciences). Total protein was detected by staining SDS-PAGE gels with Coomassie stain.
UV-visible absorption spectroscopy
UV-visible absorption spectroscopy was obtained with a Shimadzu UV-1800 spectrophotometer. Spectra were recorded in the assay buffer and under anaerobic conditions. Heme quantification by Soret absorbance was performed with 50 μg of protein.
In vivo functional (heme attachment) assays.
Assays were performed as in14. Briefly, the indicated CcsBA construct was co-expressed with B. pertussis cytochrome c4:His (pRGK332) in E. coli C43Δccm::KanR. Starter cultures were grown overnight in LB with appropriate antibiotics at 37°C and back diluted 1:5 into 5ml of LB with appropriate antibiotics for 3 hours at 37°C and 200rpm. CcsBA and cytochrome c expression were induced with 0.1 mM IPTG and 0.2% arabinose for 3 hours at 37°C and 200rpm. Cells were collected by centrifugation at 5000 rpm for 10 minutes at 4°C and pellets were stored at −80°C. Cells were lysed in 200 μl B-PER reagent (Thermo Fisher Scientific) according to the manufacturer’s instructions. Protein concentration was determined by Nanodrop. 100 μg of cell lysis were separated by SDS-PAGE and analyzed by heme stain. Heme stains were quantified by ImageJ 1.53e52 or Image Studio Lite v5.2.
In vitro reconstitution of synthase function
In vitro reconstitutions were performed as in20. For anaerobic reactions, all reagents were equilibrated with N2 (95%) and H2 (5%) in a Coy anaerobic chamber for 30 min. 10 μM FPLC purified synthase (CcsBA) was combined with 20 μM apo equine heart cytochrome c (see20 for apo cyt c preparation). An initial spectrum was obtained. 5 mM DTT was added to initiate the reaction. Reactions were placed at 37 °C and spectra were taken at indicated time points.
CryoEM sample preparation
Purified *CcsBA:His was centrifuged at 10,000g for 15 minutes, and a Hamilton syringe was used to inject 240 μl of supernatant onto a Bio Rad NGC Quest 10 Plus Chromatography System with an ENrich SEC 650 column. Elution buffer (20 mM Tris pH 8, 100 mM NaCl, 125 mM imidazole, 0.03% DDM) was used to load the column (2 ml, 1 ml/min) and isolate the protein (26 ml, 1 ml/min). Absorbances were recorded (280 nm, 415 nm, 530 nm, 560 nm), and fractions of 0.3 – 1 mL were collected. Peak fractions were pooled and centrifuged in a 100K Sartorius concentrator at 5,000 rpm, 4 C. Protein concentration was determined via a Bradford Assay. Samples were transported on ice for vitrification and initial screening, prior to subsequent single particle cryo-EM data acquisition on a Titan Krios G3 cryo-EM operating at 300kV.
Negative stain Grid Preparation, Imaging and Processing
Purified CcsBA at a concentration of 0.01mg/ml was used for negative staining. Glow-discharged (1 min at 15 mA with GloQube glow-discharger) carbon coated 200 mesh copper grids were placed on a 10 μl drop of purified CcsBA and incubated for 1 min at room temperature. Post-incubation, the grids were washed serially with 5 ddH2O drops and stained with 0.75% uranyl formate for 2 min. Excess uranyl formate was blotted off using filter paper then the grids were air dried. Grids were imaged on a JEOL JEM-1400 TEM equipped with an AMT CCD camera operating at 120 kV at 60,000x nominal magnification resulting in a magnified pixel size of 3.08 Å. Using cisTEM53 28,777 particles were picked and used for 2D classification with 50 classes. 23 2D class average were selected totaling 11,741 particles and a used in a second round of 2D classification with 10 classes.
Cryo-EM Grid Preparation and Imaging
CcsBA cryo EM samples were prepared on quantifoil holey carbon grids (R2/2 300 mesh copper) and plunge frozen a Vitrobot Mark IV (ThermoFisher Scientific, Brno, CZ). Prior to plunge freezing the girds were plasma cleaned for 1 minute using a Gatan Solarus 950 (Gatan, Pleasanton, CA). The Vitrobot sample chamber was set to 4°C and 100 % humidity. 3 μL of purified CcsBA at a concentration of 2.8 mg/ml was applied to the plasma cleaned quantifoil grids and allowed to incubate for 20 seconds. Grids were then blotted for 2 seconds at a blot force of −1 and plunge frozen into liquid ethane. Vitrified samples were then imaged using a Cs-corrected Thermo Fisher Titan Krios G3 cryo-electron microscope (ThermoFisher Scientific, Eindhoven, NL) operating at an accelerating voltage of 300 kV equipped with a Gatan K2-Summit detector (Gatan, Pleasanton, CA) placed on a BioQuantum 968 GIF-quantum energy filter (Gatan, Pleasanton, CA) operating with a slit width of 20 eV. Data acquisition was automated using the EPU software (ThermoFisher Scientific, Brno, CZ) at a magnification of 105,000× which corresponds to a pixel size of 1.1 Å. Micrograph movies were recorded for 8s with 40 frames. This results in a frame rate of 0.2 sec per frame with a dose rate of 1.65 electrons per Å2 per frame (a total dose of 66 electrons per Å2). The defocus was varied between −1 to −2.5 μm.
Cryo-EM Image Processing
Processing of cryo-EM data was carried out using CryoSPARC v3.2.054. Motion correction of 8,676 raw image movies was performed using patch motion correction and CTF estimation was performed using the patch CTF estimation routine. Particles were initially picked using CryoSPARC’s blob picker using a minimum and maximum particle diameter of 120Å and 170Å respectively with a separation distance of 96Å. After refining the picks using the Inspect Pick routine,1,186,400 particles were extracted using a box size of 256 pixels. Three successive 2D classifications were used to exclude low-resolution particles resulting in a final selection of 147,346 particles. These particles served as the input of a three-class ab-Initio reconstruction job which resulted in two good initial densities representing the open and closed confirmation. All 147,346 particles selected from the 2D classification and the three initial densities (low passed to 30Å) from the ab initio reconstruction were used as inputs for a three-class heterogeneous refinement. This allowed the clear separation of the open and closed confirmations. Particles for each confirmation were subsequently used for independent homogenous refinement resulting in resolutions for the open and closed conformations of 4.89Å and 7.22Å respectively. Non-uniform refinement55 was used to further improve the alignment of each conformation, resulting in densities with resolutions of 3.83Å and 6.20Å for the open and closed conformations respectively.
These density maps were utilized to generate 2D templates for each conformation, which were subsequently used on the complete dataset to ensure all particles were picked correctly. 2D templates generated from each conformation were combined and used as the input for the template picker. Template picks were refined using the inspect particle picks routine resulting in a particle selection of 1,352,166 particles which were subsequently extracted using a 256-pixel box size. An initial 2D classification with 200 classes resulted in the selection of 68 good classes that represented 610,236 particles. These particles were used as the input for an additional 2D classification with the following parameters: 100 classes, maximum resolution 4Å, number of final full iterations 2, number of online-EM iteration 28, and batchsize per class 200. From this 2D classification 75 good classes were selected that represented 499,162 particles. To ensure removal of all non-contributing particles, a final 2D classification was run with the following parameters: 80 classes, Maximum resolution 3Å, number of final full iterations 2, number of online-EM iteration 25, and batchsize per class 200. 70 good classes were selected resulting in dataset size of 468,788 particles, which were subsequently used as the input for an ab initio reconstruction with four classes. Two classes clearly represented the open and closed conformations with the remaining two classes showing very little detail. All volumes from the ab initio reconstruction job were used as initial models for heterogeneous refinement with four classes. Again, both the open and closed confirmations represented only two of the four classes, with the open confirmation class containing 173,569 particles at a FSC resolution estimate of 6.12Å and the closed confirmation class containing 167,336 particles with a FSC resolution estimate of 6.95Å. The remaining two classes showed densities which contained little to no fine detail, thus the particles in these classes were discarded from moving forwards. The particle subsets representing the open and closed classes were used as the inputs for a second round of heterogeneous refinement. We repeated this routine twice more as we saw continued improvement in the fine structure of the open and closed confirmation density maps as well as in their estimated FSC resolution.
Post the final heterogeneous refinement step, the open confirmation contained 140,727 particles and the closed confirmation contained 117,488 particles with FSC resolution estimates of 5.02Å and 6.51Å respectively. Each particle subset was subsequently used as the input to separate homogeneous refinements which improved the alignment for both conformations with FSC resolution estimates of 4.30Å and 4.94Å for the open and closed conformations respectively. Since CcsBA is membrane protein, where particle alignment within the solvent ring can be challenging, we leveraged the newly released non-uniform refinement53 approach in Cryosparc for both conformations. Substantial improvement was seen in both the fine structure of the density maps with the open conformation having a final GSFSC resolution of 3.56A and the closed conformation having a final GSFSC resolution of 4.14A at a threshold of 0.143.
Model Building and Refinement
The open and closed conformations were modelled de novo into densities of the 3.56Å map (open) and 4.14Å map (closed). Initially, all helices and beta strands were placed into the map as to begin a poly-alanine model using COOT (WinCoot 0.8.9.2)56. Helices and strands were then connected by following the backbone in the density and placing residues, generating a full poly-alanine model. Identity of helices were confirmed by using known landmarks (eg. P-His, TM-His, WWD Domain, connections to periplasm) and following the trace of the density. Sequence identity was obtained by using the known landmarks in the structure as well as bulky residues visible in density, and the structure was mutated to the correct sequence in COOT. After mutating all residues to the correct sequence manually adjusting residues into their respective density using COOT was carried out. The structure was refined with Phenix 1.17.1 real space refine57 with 5 cycles of energy minimization and secondary structure restraints, as well as through simulation using ISOLDE 1.258. In the open density two heme cofactors and one phospholipid were modelled into the densities using COOT. In the closed density one heme cofactor and one phospholipid were modelled into the densities using COOT. The final step was manual refinement of density fit and geometrical errors using COOT. Model validation was performed using Molprobity 4.5.159. ChimeraX 1.160 and Pymol 2.0 were used to generate figures.
Not present in density in the open conformation were residues 345–385 and in the closed 344–383. These are likely disordered due to the natural proteolysis that occurs within this region (see Extended Data Fig. 1). Additionally, within the closed state density, residues 894–905 were not resolved. As described in the text, these residues represent the P-His2 loop and is likely disordered (see Extended Data Fig. 1). They are present in the open state density, due to stabilization from P-Heme, with the P-His2 imidazole acting as an axial ligand to P-heme. P-heme is not present in the closed conformation.
We compared the CcsBA Cryo-EM experimental structures (densities) with a trRosetta-generated theoretical model (Supplementary Fig. 6). It is clear that the trRosetta 1.13 model is a transition between the open and closed states, albeit closer to the closed conformation density. Only experimental structures have thus facilitated structural elucidation of the active open state (and active site) with P-heme, and the major conformational changes that occur during the transport cycle. Nevertheless, the trRosetta structure is consistent with the overall architecture of the fitted densities, including TMs.
Data Availability
Cryo-EM electron density maps of CcsBA-open and CcsBA-closed have been deposited in the Electron Microscopy Data Bank under accession numbers EMD-24941 and EMD-24942, respectively. Coordinates for atomic models of CcsBA-open and CcsBA-closed have been deposited in the Protein Data Bank under accession numbers 7S9Y and 7S9Z, respectively. Source data are provided with this paper.
Source data of gels and densitometry numbers used to calculate the error bars in Figures 1AB and 2D are available in the file Mendez_Figure1AB-SourceDataFile.xlsx and Mendez_Figure2D-SourceDataFile.xlsx.
Extended Data
Extended Data Fig. 1. Topological map of CcsBA and proposed method of apocytc heme attachment.
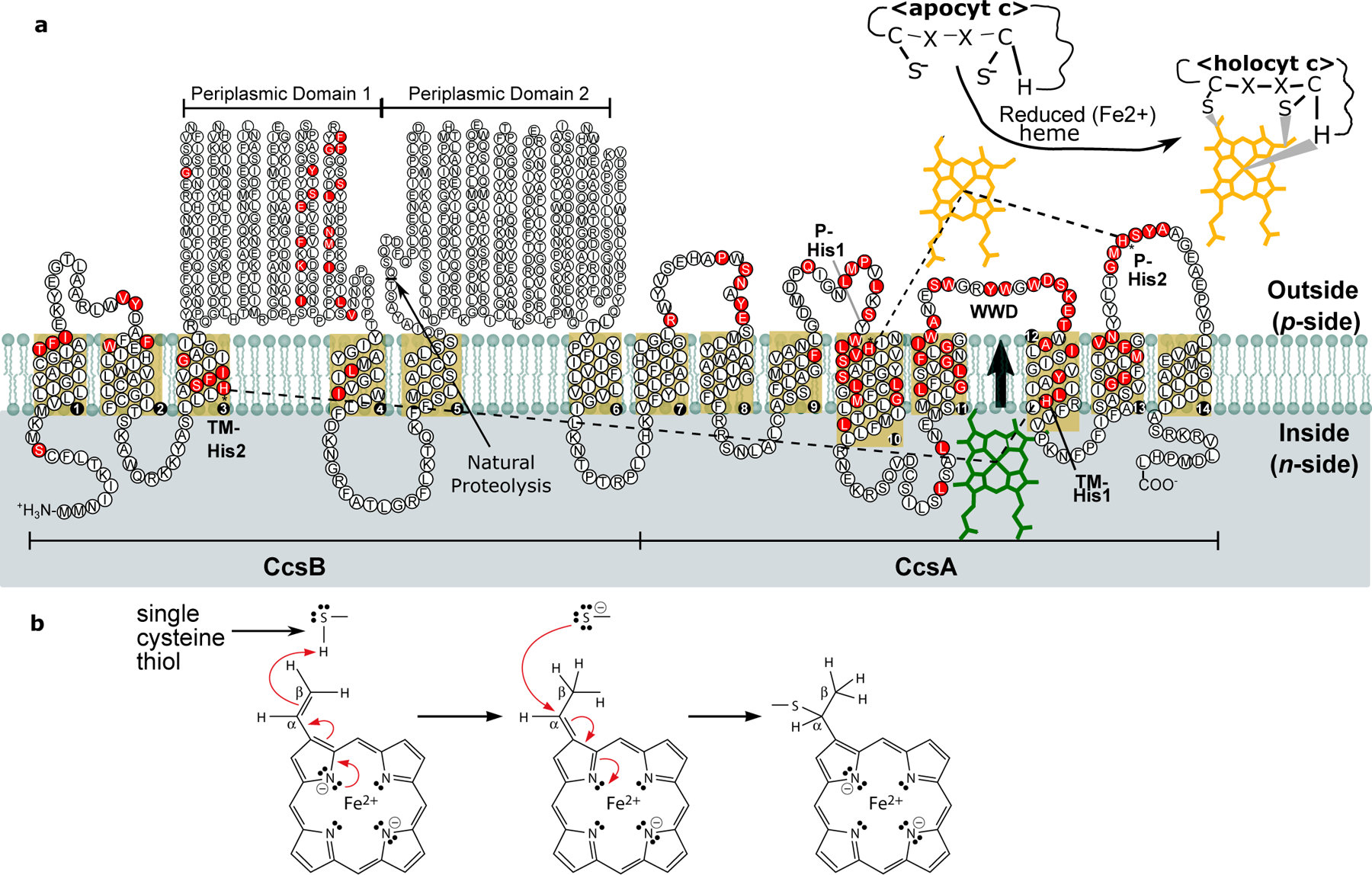
a. Schematic of H. hepaticus CcsBA topology catalyzing heme attachment to apocytc. CcsBA consists of fourteen transmembrane domains and two major periplasmic domains. Conserved features are shown: two conserved histidines in the transmembrane domain (TM-His1-H858, TM-His2-H83) and two conserved periplasmic histidines (P-His1-H897, P-His2-H761) which flank the heme-handling WWD domain. The WWD domain positions heme for attachment to the CXXCH motif in apocytochrome c to form holocytc. Heme enters through a vestibule and is liganded by the TM-His1 and TM-His2. Exact, conserved substitutions and semi-conserved substitutions are all colored in red (T-Coffee analysis1) derived from comparing organisms: M. tuberculosis, B. pertussis, Synechocystis, B. theta, B. subtilis, Wolinella, and H. hepaticus. b. Chemistry of thioether formation. Modified from3. Red arrows indicate two electron transfer.
Extended Data Fig. 2. Raising the threshold for CcsBA-open and CcsBA-closed reveals two and one dense regions, respectively.

a. Front view of the cryo-EM map of CcsBA-open (red, 3.56 Å) with TM-heme (green) and P-heme (yellow). Density thresholding reveals the two densest regions that indicate the iron in heme. b. Cryo-EM map of the 4.14 Å closed conformation (blue) with TM-heme (green). Filtering the electron density reveals a single dense particle which is assigned to iron in heme. c. The heme to heme distances, the vertical, horizontal and then the edge to edge distances, were measured edge to edge using Autodock Vina.
Extended Data Fig. 3. The P-His2 loop is likely disordered in closed conformation.

a. Top view of the 3.56 Å open conformation (red) with TM-heme (green), TM13 (black), and P-His2 loop (orange). A lower resolution version of the open conformation was used to allow for comparisons with the closed conformation. b. Top view of the 4.14 Å closed conformation (blue) viewed from the same angle with TM-heme, and TM13. There is no density going into TM13 in the closed conformation, so there is no orange colored P-His2 density in the closed conformation.
Extended Data Fig. 4. Thioether attachment of CXXCH to the P-Heme.

a. CXXCH is attached to heme with the His liganding the iron in heme, and cytc Cys sulfur groups forming thioether attachments to the 2- and 4-vinyl. Modified from35. b. The 3.56 Å CcsBA open conformation (red) zoomed in to the active site, with P-Heme and PDB heme (both yellow) and WWD Domain (green). The PDB of CXXCH (teal) was docked into the map by Autodock Vina61(Supplementary Table 3). The positions of the 2-and 4-vinyl sites of thioether attachment are labeled.
Supplementary Material
Heme flux from the TM-heme site to the P-heme site is accompanied by conformational change. The movie starts with the closed conformation: one heme (green) in the TM-heme site, the transmembrane domain of the protein (red), the periplasmic region of the protein (blue), both TM-His (red), and both P-His (yellow). A new heme comes from the bottom of the protein into the vestibule where it bumps the current TM-heme into the heme channel as the movie zooms to show this action. The movie then shifts to a top view where conformational change happens: the upper heme finishes moving into the P-His site and is liganded by the P-His.
Movie of the CXXCH entry, thioether attachment, release of P-Heme into the chamber, and conformational change. Movie starts with a side view (TM6 and TM7 side) of the open conformation of CcsBA with Transmembrane Domain (red), Periplasmic Domain (blue), P-His (yellow), P-Heme (green) and CXXCH (dark green). The CXXCH moves into the door of the chamber. The CXXCH then moves into the chamber and replaces the P-His2(H897) and the H of the CXXCH ligands the heme. The CXXCH attaches to the 2- and 4-vinyl. Following this attachment, the view zooms out to show the release of the P-heme and ends with conformational change back to the closed conformation.
ACKNOWLEDGEMENTS
This work was funded by the National Institutes of Health (R01 GM47909 to RGK). MJR and JAJF are supported by the Washington University Center for Cellular Imaging (WUCCI), which is funded in part by Washington University School of Medicine, The Children’s Discovery Institute of Washington University and St. Louis Children’s Hospital (CDI-CORE-2015-505 and CDI-CORE-2019-813), the Foundation for Barnes-Jewish Hospital (3770). JAJF is also supported by a Chan Zuckerberg Initiative Imaging Scientist award (2020-225726). We thank Joshua Jarodsky and Ernest Burgie for critically reading the manuscript and providing insightful feedback.
Footnotes
COMPETING INTERESTS STATEMENT The authors declare no competing interests.
REFERENCES
- 1.Donegan RK, Moore CM, Hanna DA & Reddi AR Handling heme: The mechanisms underlying the movement of heme within and between cells. Free Radic. Biol. Med 133, 88–100 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reddi AR & Hamza I Heme Mobilization in Animals: A Metallolipid’s Journey. Acc. Chem. Res 49, 1104–1110 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pitt JN & Kaeberlein M Inter-organ regulation of haem homeostasis. Nat. Cell Biol 19, 756–758 (2017). [DOI] [PubMed] [Google Scholar]
- 4.Mercurio S et al. Alteration of heme metabolism in a cellular model of Diamond–Blackfan anemia. Eur. J. Haematol 96, 367–374 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Quigley JG et al. Identification of a Human Heme Exporter that Is Essential for Erythropoiesis. Cell 118, 757–766 (2004). [DOI] [PubMed] [Google Scholar]
- 6.Rajagopal A et al. Haem homeostasis is regulated by the conserved and concerted functions of HRG-1 proteins. Nature 453, 1127–1131 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chiabrando D et al. Mutations in the Heme Exporter FLVCR1 Cause Sensory Neurodegeneration with Loss of Pain Perception. PLoS Genet. 12, e1006461 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Galmozzi A et al. PGRMC2 is an intracellular haem chaperone critical for adipocyte function. Nature 576, 138–142 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Prepeluh N et al. A mosaic form of microphthalmia with linear skin defects. BMC Pediatr. 18, 254 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schiffmann LM et al. Mitochondrial respiration controls neoangiogenesis during wound healing and tumour growth. Nat. Commun 11, 3653 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ponka P, Sheftel AD, English AM, Scott Bohle D & Garcia-Santos D Do Mammalian Cells Really Need to Export and Import Heme? Trends Biochem. Sci 42, 395–406 (2017). [DOI] [PubMed] [Google Scholar]
- 12.Kranz RG, Richard-Fogal C, Taylor J-S & Frawley ER Cytochrome c biogenesis: mechanisms for covalent modifications and trafficking of heme and for heme-iron redox control. Microbiol. Mol. Biol. Rev 73, 510–528 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Simon J & Hederstedt L Composition and function of cytochrome c biogenesis System II. FEBS J. 278, 4179–4188 (2011). [DOI] [PubMed] [Google Scholar]
- 14.Sutherland MC et al. Structure-Function Analysis of the Bifunctional CcsBA Heme Exporter and Cytochrome c Synthetase. mBio 9, e02134–18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xie Z et al. Genetic analysis of chloroplast c-type cytochrome assembly in Chlamydomonas reinhardtii: One chloroplast locus and at least four nuclear loci are required for heme attachment. Genetics 148, 681–692 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Feissner RE et al. Recombinant cytochromes c biogenesis systems I and II and analysis of haem delivery pathways in Escherichia coli. Mol. Microbiol 60, 563–577 (2006). [DOI] [PubMed] [Google Scholar]
- 17.Frawley ER & Kranz RG CcsBA is a cytochrome c synthetase that also functions in heme transport. Proc. Natl. Acad. Sci 106, 10201–10206 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beckett CS et al. Four genes are required for the system II cytochrome c biogenesis pathway in Bordetella pertussis, a unique bacterial model. Mol. Microbiol 38, 465–481 (2000). [DOI] [PubMed] [Google Scholar]
- 19.Schiött T, von Wachenfeldt C & Hederstedt L Identification and characterization of the ccdA gene, required for cytochrome c synthesis in Bacillus subtilis. J. Bacteriol 179, 1962–1973 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sutherland MC et al. In vitro reconstitution reveals major differences between human and bacterial cytochrome c synthases. eLife (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chaptal V et al. Quantification of Detergents Complexed with Membrane Proteins. Sci. Rep 7, 41751 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ilgü H et al. Variation of the detergent-binding capacity and phospholipid content of membrane proteins when purified in different detergents. Biophys. J 106, 1660–1670 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barrick D Replacement of the proximal ligand of sperm whale myoglobin with free imidazole in the mutant His-93-->Gly. Biochemistry 33, 6546–6554 (1994). [DOI] [PubMed] [Google Scholar]
- 24.Barker PD et al. Transmutation of a heme protein. Proc. Natl. Acad. Sci 90, 6542–6546 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nicholson DW & Neupert W Import of cytochrome c into mitochondria: reduction of heme, mediated by NADH and flavin nucleotides, is obligatory for its covalent linkage to apocytochrome c. Proc. Natl. Acad. Sci 86, 4340–4344 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kranz R, Lill R, Goldman B, Bonnard G & Merchant S Molecular mechanisms of cytochrome c biogenesis: three distinct systems. Mol. Microbiol 29, 383–396 (1998). [DOI] [PubMed] [Google Scholar]
- 27.Babul J & Stellwagen E Participation of the protein ligands in the folding of cytochrome c. Biochemistry 11, 1195–1200 (1972). [DOI] [PubMed] [Google Scholar]
- 28.Kleingardner JG & Bren KL Comparing substrate specificity between cytochrome c maturation and cytochrome c heme lyase systems for cytochrome c biogenesis. Metallomics 3, 396 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Richard-Fogal CL, San Francisco B, Frawley ER & Kranz RG Thiol redox requirements and substrate specificities of recombinant cytochrome c assembly systems II and III. Biochim. Biophys. Acta 1817, 911–919 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Beckman DL, Trawick DR & Kranz RG Bacterial cytochromes c biogenesis. Genes Dev. 6, 268–283 (1992). [DOI] [PubMed] [Google Scholar]
- 31.Lee J-H, Harvat EM, Stevens JM, Ferguson SJ & Saier MH Evolutionary origins of members of a superfamily of integral membrane cytochrome c biogenesis proteins. Biochim. Biophys. Acta - Biomembr 1768, 2164–2181 (2007). [DOI] [PubMed] [Google Scholar]
- 32.Verissimo AF & Daldal F Cytochrome c biogenesis System I: an intricate process catalyzed by a maturase supercomplex? Biochim. Biophys. Acta 1837, 989–998 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Richard-Fogal CL et al. A conserved haem redox and trafficking pathway for cofactor attachment. EMBO J. 28, 2349–2359 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.San Francisco B, Sutherland MC & Kranz RG The CcmFH complex is the system I holocytochrome c synthetase: engineering cytochrome c maturation independent of CcmABCDE. Mol. Microbiol 91, 996–1008 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sutherland MC, Jarodsky JM, Ovchinnikov S, Baker D & Kranz RG Structurally Mapping Endogenous Heme in the CcmCDE Membrane Complex for Cytochrome c Biogenesis. J. Mol. Biol 430, 1065–1080 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Naoe Y et al. Crystal structure of bacterial haem importer complex in the inward-facing conformation. Nat. Commun 7, 13411 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Woo J-S, Zeltina A, Goetz BA & Locher KP X-ray structure of the Yersinia pestis heme transporter HmuUV. Nat. Struct. Mol. Biol 19, 1310–1315 (2012). [DOI] [PubMed] [Google Scholar]
- 38.Huang W & Wilks A Extracellular Heme Uptake and the Challenge of Bacterial Cell Membranes. Annu. Rev. Biochem 86, 799–823 (2017). [DOI] [PubMed] [Google Scholar]
- 39.Hanna DA et al. Heme dynamics and trafficking factors revealed by genetically encoded fluorescent heme sensors. Proc. Natl. Acad. Sci 113, 7539–7544 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yuan X et al. Regulation of intracellular heme trafficking revealed by subcellular reporters. Proc. Natl. Acad. Sci 113, E5144–E5152 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Medlock A, Swartz L, Dailey TA, Dailey HA & Lanzilotta WN Substrate interactions with human ferrochelatase. Proc. Natl. Acad. Sci. U. S. A 104, 1789–1793 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Medlock AE et al. Identification of the Mitochondrial Heme Metabolism Complex. PloS One 10, e0135896 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dailey HA et al. Ferrochelatase at the millennium: structures, mechanisms and [2Fe-2S] clusters. Cell. Mol. Life Sci. CMLS 57, 1909–1926 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Breyton C, Haase W, Rapoport TA, Kühlbrandt W & Collinson I Three-dimensional structure of the bacterial protein-translocation complex SecYEG. Nature 418, 662–665 (2002). [DOI] [PubMed] [Google Scholar]
- 45.Tsirigotaki A, De Geyter J, Šoštaric N, Economou A & Karamanou S Protein export through the bacterial Sec pathway. Nat. Rev. Microbiol 15, 21–36 (2017). [DOI] [PubMed] [Google Scholar]
- 46.Merchant SS His protects heme as it crosses the membrane. Proc. Natl. Acad. Sci 106, 10069–10070 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brausemann A, Zhang L, Ilcu L & Einsle O Architecture of the membrane-bound cytochrome c heme lyase CcmF. Nat. Chem. Biol 1–6 (2021) doi: 10.1038/s41589-021-00793-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.San Francisco B & Kranz RG Interaction of holoCcmE with CcmF in heme trafficking and cytochrome c biosynthesis. J. Mol. Biol 426, 570–585 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schulz H, Hennecke H & Thöny-Meyer L Prototype of a heme chaperone essential for cytochrome c maturation. Science 281, 1197–1200 (1998). [DOI] [PubMed] [Google Scholar]
- 50.Brown BL & Iverson TM Handling heme with care. Nat. Chem. Biol 1–2 (2021) doi: 10.1038/s41589-021-00821-7. [DOI] [PubMed] [Google Scholar]
- 51.Trott O & Olson AJ AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem 31, 455–461 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rasband WS ImageJ. US Natl. Inst. Health Bethesda MD Available at, http://imagej.nih.gov/ij/. (1997). [Google Scholar]
- 53.Grant T, Rohou A & Grigorieff N cisTEM, user-friendly software for single-particle image processing. eLife 7, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Punjani A, Rubinstein JL, Fleet DJ & Brubaker MA cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017). [DOI] [PubMed] [Google Scholar]
- 55.Punjani A, Zhang H & Fleet DJ Non-uniform refinement: adaptive regularization improves single-particle cryo-EM reconstruction. Nat. Methods 17, 1214–1221 (2020). [DOI] [PubMed] [Google Scholar]
- 56.Emsley P & Cowtan K Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr 60, 2126–2132 (2004). [DOI] [PubMed] [Google Scholar]
- 57.Liebschner D et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. Sect. Struct. Biol 75, 861–877 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Croll TI ISOLDE: a physically realistic environment for model building into low-resolution electron-density maps. Acta Crystallogr. Sect. Struct. Biol 74, 519–530 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Williams CJ et al. MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci. Publ. Protein Soc 27, 293–315 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pettersen EF et al. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. Publ. Protein Soc 30, 70–82 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Heme flux from the TM-heme site to the P-heme site is accompanied by conformational change. The movie starts with the closed conformation: one heme (green) in the TM-heme site, the transmembrane domain of the protein (red), the periplasmic region of the protein (blue), both TM-His (red), and both P-His (yellow). A new heme comes from the bottom of the protein into the vestibule where it bumps the current TM-heme into the heme channel as the movie zooms to show this action. The movie then shifts to a top view where conformational change happens: the upper heme finishes moving into the P-His site and is liganded by the P-His.
Movie of the CXXCH entry, thioether attachment, release of P-Heme into the chamber, and conformational change. Movie starts with a side view (TM6 and TM7 side) of the open conformation of CcsBA with Transmembrane Domain (red), Periplasmic Domain (blue), P-His (yellow), P-Heme (green) and CXXCH (dark green). The CXXCH moves into the door of the chamber. The CXXCH then moves into the chamber and replaces the P-His2(H897) and the H of the CXXCH ligands the heme. The CXXCH attaches to the 2- and 4-vinyl. Following this attachment, the view zooms out to show the release of the P-heme and ends with conformational change back to the closed conformation.
Data Availability Statement
Cryo-EM electron density maps of CcsBA-open and CcsBA-closed have been deposited in the Electron Microscopy Data Bank under accession numbers EMD-24941 and EMD-24942, respectively. Coordinates for atomic models of CcsBA-open and CcsBA-closed have been deposited in the Protein Data Bank under accession numbers 7S9Y and 7S9Z, respectively. Source data are provided with this paper.
Source data of gels and densitometry numbers used to calculate the error bars in Figures 1AB and 2D are available in the file Mendez_Figure1AB-SourceDataFile.xlsx and Mendez_Figure2D-SourceDataFile.xlsx.