

Abstract
Traditional experimental testing to identify endocrine disruptors that enhance estrogenic signaling relies on expensive and labor-intensive experiments. We sought to design a knowledge-based deep neural network (k-DNN) approach to reveal and organize public high-throughput screening data for compounds with nuclear estrogen receptor α and β (ERα and ERβ) binding potentials. The target activity was rodent uterotrophic bioactivity driven by ERα/ERβ activations. After training, the resultant network successfully inferred critical relationships among ERα/ERβ target bioassays, shown as weights of 6521 edges between 1071 neurons. The resultant network uses an adverse outcome pathway (AOP) framework to mimic the signaling pathway initiated by ERα and identify compounds that mimic endogenous estrogens (i.e., estrogen mimetics). The k-DNN can predict estrogen mimetics by activating neurons representing several events in the ERα/ERβ signaling pathway. Therefore, this virtual pathway model, starting from a compound’s chemistry initiating ERα activation and ending with rodent uterotrophic bioactivity, can efficiently and accurately prioritize new estrogen mimetics (AUC = 0.864–0.927). This k-DNN method is a potential universal computational toxicology strategy to utilize public high-throughput screening data to characterize hazards and prioritize potentially toxic compounds.
Keywords: estrogen receptor, uterotrophic activity, high-throughput screening data, deep learning, adverse outcome pathway, artificial intelligence
Graphical Abstract
INTRODUCTION
Estrogen receptors (ERs) play critical roles in cell differentiation,1 fertility,2,3 and morphogenesis.4 ERs are present in many forms within cells, including estrogen-related receptors, membrane-bound G-protein coupled ERs, and nuclear ERs α and β.5 The interaction of estrogen with ERα and ERβ represents a classical ligand-dependent transcriptional regulation paradigm.1,6 ERs are more promiscuous than other steroid receptors due to their sizeable binding pockets.7 Therefore, environmental compounds (e.g., pesticides and plasticizers) can act as estrogen mimetics by binding to and activating ERs. Estrogen mimetics can act as endocrine disruptors, resulting in diverse adverse outcomes such as breast, vaginal, and uterine cancers8-11 and congenital anomalies.11
Traditional experimental testing to identify endocrine disruptors relies on expensive and labor-intensive low-throughput experiments, such as those employed in the Environmental Protection Agency (EPA)’s Endocrine Disruptor Screening Program (EDSP).12 These traditional methods are not practical to assess the toxicity potentials of the massive amount of new compounds that need to undergo screening, especially before chemical synthesis.13 Computational modeling is a promising alternative method for toxicity evaluations, including identifying potential estrogen mimetics. Traditional computational methods based on machine learning approaches, such as quantitative structure–activity relationship (QSAR) modeling, have been applied to model various toxicities.14-16 However, these conventional models were purely based on chemical structure information, which causes flaws in predicting complicated toxicities, such as narrow chemical space coverage17 and model overfitting.18
Deep learning approaches emerged as an essential field of artificial intelligence (AI), particularly adept for handling big data.19-21 Deep learning efforts employ various neural network (NN) methods.22 Although the comparison between machine learning and deep learning methods has no clear conclusion,23-27 deep learning based on NNs attracted considerable attention in computational toxicology. For example, it showed certain advantages in previous toxicity modeling challenge projects.28-30 The success of NN methods in these tasks is attributed to their ability to learn complex, non-linear relationships within data through several layers. In these layers, individual neurons transform multiple inputs (consisting of individual weights and outputs from the previous layer) into a singular output to proceed to the next layer. In this way, individual neurons are trained to recognize individual portions of abstract, transformed data and approximate complex relationships between input data and a target variable. However, NNs often perform as “black boxes,” making the neurons and weights within the layers challenging to interpret.31 In 2007, the Organization for Economic Co-operation and Development (OECD) released guidelines regarding suitable validations of computational toxicology models.32 One requirement outlined in these guidelines is that computational toxicology models must be mechanistically explainable.32 The black-box nature of NNs causes critical concerns for using deep learning in computational toxicology, particularly for chemical risk assessments.
High-throughput screening (HTS) is widely used in toxicology and represents one of the current primary public data sources. One of the most prominent HTS initiatives in toxicology was the EPA Toxicity Forecaster (ToxCast) program.33,34 This program later progressed to the collaborative Toxicity in the 21st Century (Tox21) initiative among the EPA, Food and Drug Administration (FDA), National Center for Advancing Translational Sciences (NCATS), and National Toxicology Program (NTP).35-37 The EPA developed a mathematical model that utilizes data from 16 HTS bioassays among hundreds in the ToxCast and Tox21 programs that, by design, measure the early key events (KEs) of an adverse outcome pathway (AOP) initiated by ERα activation.38-40 This model predicts the apical outcome of in vivo rodent uterotrophic activity and is therefore predictive of the potential for a compound to cause diverse adverse outcomes associated with estrogenicity. In 2015, the EPA announced their acceptance of this model’s results as replacements for the EDSP Tier 1 testing battery for estrogenicity screening.41
A specific molecular feature initiates an AOP when it interacts with a biomolecule, such as a receptor42,43 A total of 3 of the 16 bioassays used in the EPA model represent the molecular initiating event (MIE) in this AOP by measuring compound binding to bovine, human, and mouse nuclear ERα.44-47 The MIE of an AOP triggers a cascade of measurable KEs at increasing biological system levels (i.e., cellular, tissue, and organ), ultimately leading to an adverse outcome at the organism level.42,43 The 13 remaining bioassays use various human cell lines to represent the KEs of this AOP: protein stabilization,48-50 DNA binding,50 transcriptional regulation,51-53 and cell proliferation.54,55 The existing computational models require new compounds of interest to undergo experimental testing using these 16 HTS bioassays, which are not always commercially available. The fast screening of millions of compounds is still not applicable based on this strategy. Furthermore, although a framework for the nuclear ER agonism pathway was defined, the existing studies did not describe the chemical fragment involved in the MIE (i.e., the toxicophore).
Computational modeling in toxicology has incorporated the data from HTS programs such as ToxCast and Tox21.20,56-59 Our previous studies successfully integrated HTS data for modeling complicated toxicity endpoints,60-62 including ER binding activities.63 In this study, a novel computational modeling framework was developed to
reveal a hidden ERα/ERβ agonism pathway from public data, including toxicophores and biological targets;
directly evaluate animal estrogen mimetics (i.e., rodent uterotrophic bioactivity) from existing HTS data and chemical structures; and
virtually simulate the toxicity pathway perturbation for each predicted toxic compound.
This study presents a new knowledge-based deep NN (k-DNN) method to mimic a toxicity pathway for ERα and ERβ agonists using a virtual AOP (vAOP) framework.43 A data set of 42 compounds with known in vivo rodent uterotrophic bioactivity was used to train this network.40 This vAOP framework accurately mimics the comprehensive effects of in vitro bioassays related to toxicity pathway KEs to predict in vivo outcomes by incorporating chemical fragments and hierarchically structured biological data during the training process. This computational framework shows great promise for developing future predictive models of complicated toxicity endpoints. This study highlights the applicability of using both advanced AI techniques, such as deep learning, and public HTS data to replace classic animal models.
METHODS
In Vivo Rodent Uterotrophic Data Set.
The in vivo data set used in this study was obtained from an extensive literature curation by the US EPA to identify a set of compounds with high-quality and guideline-like rodent uterotrophic bioassay results.40 For this study, compounds were only used for ER k-DNN training if they had reproducible and independently verified results in at least two separate studies.40 Compounds with conflicting in vivo test results from multiple studies or single in vivo test results unconfirmed by an independent study were excluded.39,40 In total, 43 compounds met these criteria. However, the chemical fragments used in the ER k-DNN cannot handle salts and are two-dimensional (i.e., cannot identify differences between stereoisomers). Therefore, the CASE Ultra v1.8.0.0 DataKurator tool was used to curate this data set before modeling. The heaviest organic fraction was retained from all salts. 17α-estradiol (CAS 57-91-0) and 17β-estradiol (CAS 50-28-2) were considered duplicate structures because the curation step removed the compounds’ stereochemistry. In this situation, 17β-estradiol was retained because of its use as the control in the ToxCast/Tox21 ERα/ERβ assays. The final data set used to train the network in this study contained 42 compounds (Table S1). Each of these compounds showed reproducible and independently verified in vivo rodent uterotrophic or non-uterotrophic activity.
In Vitro Bioassay Data Set.
The public in vitro bioassay hit calls for these 42 compounds were extracted from the EPA ToxCast/Tox21 summary files for invitroDBv3.264,65 (https://www.epa.gov/chemical-research/exploring-toxcast-data-downloadable-data). In total, the ToxCast and Tox21 programs tested these 42 compounds against 1241 bioassays. Besides 14 ERα/ERβ assays (Table 1), 43 additional bioassays were included in the network architecture (Table S2), totaling approximately 3 times the number of included ERα/ERβ assays. These additional bioassays each had the same ToxCast-defined biological process target as one of the 14 ERα/ERβ assays (Table 1): receptor binding, protein stabilization, regulation of gene expression, regulation of transcription factor activity, or cell proliferation. These biological process targets describe the high-level biological process measured by an assay’s readout. The additional bioassays were selected based on data availability for the 42 compounds in the in vivo data set, with preference given to nuclear receptor bioassays when possible. This effort resulted in a data set containing 42 training compounds with in vitro data from 57 ToxCast/Tox21 bioassays and in vivo guideline-like uterotrophic data (Table S1).
Table 1.
ToxCast and Tox21 ER Agonism Pathway-Related Assays Used in the vAOP Modela
| ID | assay name | assay source | gene name | time point (min) |
biological process target | assay design type | cell line | ER KE | vAOP model layer |
|---|---|---|---|---|---|---|---|---|---|
| E1 | NVS_NR_hER | NovaScreen | ESR1 | 1080 | receptor binding | radioligand binding | NA (cell-free) | ER binding | 2 |
| E2 | OT_ER_ERaERa_0480 | Odyssey Thera | ESR1 | 480 | protein stabilization | protein fragment complementation assay | HEK293T | receptor dimerization | 3 |
| E3 | OT_ER_ERaERa_1440 | Odyssey Thera | ESR1 | 1440 | protein stabilization | protein fragment complementation assay | HEK293T | receptor dimerization | 3 |
| E4 | OT_ER_ERaERb_0480 | Odyssey Thera | ESR1, ESR2 | 480 | protein stabilization | protein fragment complementation assay | HEK293T | receptor dimerization | 3 |
| E5 | OT_ER_ERaERb_1440 | Odyssey Thera | ESR1, ESR2 | 1440 | protein stabilization | protein fragment complementation assay | HEK293T | receptor dimerization | 3 |
| E6 | OT_ER_ERbERb_0480 | Odyssey Thera | ESR2 | 480 | protein stabilization | protein fragment complementation assay | HEK293T | receptor dimerization | 3 |
| E7 | OT_ER_ERbERb_1440 | Odyssey Thera | ESR2 | 1440 | protein stabilization | protein fragment complementation assay | HEK293T | receptor dimerization | 3 |
| E8 | OT_ERa_EREGFP_0120 | Odyssey Thera | ESR1 | 120 | regulation of gene expression | fluorescent protein induction | HeLa | DNA binding | 4 |
| E9 | OT_ERa_EREGFP_0480 | Odyssey Thera | ESR1 | 480 | regulation of gene expression | fluorescent protein induction | HeLa | DNA binding | 4 |
| E10 | ATG_ERa_TRANS_up | Attagene, Inc. | ESR1 | 1440 | regulation of transcription factor activity | mRNA induction | HepG2 | transcriptional activation | 5 |
| E11 | ATG_ERE_CIS_up | Attagene, Inc. | ESR1 | 1440 | regulation of transcription factor activity | mRNA induction | HepG2 | transcriptional activation | 5 |
| E12 | TOX21_ERa_BLA_Agonist_ratio | Tox21 | ESR1 | 1440 | egulation of transcription factor activity | β lactamase induction | HEK293T | transcriptional activation | 5 |
| E13 | TOX21_ERa_LUC_VM7_Agonist | Tox21 | ESR1 | 1320 | regulation of transcription factor activity | luciferase induction | VM7 | transcriptional activation | 5 |
| E14 | ACEA_ER_80hr | ACEA Biosciences, Inc. | ESR1 | 1920 | cell proliferation | real-time cell-growth kinetics | T47D | cell proliferation | 6 |
NA: not applicable.
All compounds tested by the ToxCast/Tox21 programs in each of the 57 bioassays were used as training data to develop QSAR models to fill missing data. These training data included distributed structure-searchable toxicity identifiers (DTXSIDs), preferred compound names, and hit calls (active, inactive, or inconclusive). Chemical structure information was retrieved from PubChem’s PUG-REST service using the DTXSIDs or preferred compound names as queries.66 Inconclusive results were removed from each data set before modeling. The RDKit implementation of functional connectivity fingerprints (FCFPs) and Molecular ACCess System (MACCS) fingerprints were used in QSAR model training.
External validation compounds were obtained from the Estrogen Receptor Activity Prediction Project (CERAPP), which had known estrogenic or non-estrogenic activity from low-throughput in vitro assays.67 The same data curation process, including removing overlapped compounds to the training set, resulted in a validation set containing 6286 new compounds (Table S4).
Chemical Fragments.
Two-dimensional binary FCFPs were generated for all compounds. FCFPs represent functional group information about substructures in a compound.68 The FCFP algorithm calculates this information by evaluating the environment surrounding a compound’s atoms using a specified bond radius (i.e., a modified version of the Morgan algorithm).68 FCFPs with a radius of 2 or 3 perform well for capturing a wide range of chemical features for modeling purposes,25,69 including for modeling ER activity.23 In this study, 1024 FCFPs were calculated for all compounds using a bond radius of 3 in Python v3.6.2 using the cheminformatics package RDKit v2017.09.1 (http://rdkit.org/). Our results showed that a bond radius of 3 could balance the representation of wide chemical diversity and the potential of losing information (Table S3).
Quantitative Structure–Activity Relationship Models.
The network designed in this study cannot deal with missing data. Therefore, it was necessary to fill in the data gaps caused by inconclusive experimental results or untested compound-bioassay pairs. This data imputation was accomplished using QSAR modeling, consistent with previous computational toxicology studies.60,62 A total of 6 algorithms were used to develop the QSAR models to fill in missing data for each of the 57 bioassays included in the network: Bernoulli Naïve Bayes (BNB), k-nearest neighbors (kNN), random forest (RF), support vector machines (SVM), single-endpoint DNNs, and multitask DNNs. In the network training set, 25 compounds required imputations across 9 assays. However, QSAR models were developed for all 57 bioassays to predict new compounds in the external prediction process. BNB, kNN, RF, and SVM were implemented using the Python machine-learning library scikit-learn v0.19.0 (http://scikit-learn.org/).70 Both DNN methods were implemented using the Python deep-learning library keras v2.1.2 (https://keras.io/) and Google Tensorflow v1.4.0 (https://tensorflow.org/ for GPU training and CPU for prediction) as a backend with parameters as previously described.71
Each algorithm was trained using two types of chemical descriptors: MACCS and FCFPs.68 Briefly, BNB models apply Bayes’ theorem to data sets with binary features such as chemical fingerprints by “naively” assuming that each feature is independent of all others.72 kNN models learn and predict a compound’s activity based on several (k) nearest neighbors’ properties using user-defined similarity measures.73 RF models produce an average of the outputs from a series of decision trees using a random selection of features and training set compounds.74 SVM models divide the descriptor space to distinguish active and inactive compounds.75 Both the single-endpoint and multitask DNNs used in this study consisted of an input layer containing chemical fingerprints and an output layer containing a prediction for the assay of interest. Between the input and output layers, each network had three additional layers such that every node in each layer had a weighted connection to every node in the preceding and next layers. Each network layer was composed of the same number of neurons (i.e., 167 neurons for networks using MACCS fingerprints and 1024 for networks using FCFPs). Descriptions of these algorithms’ hyper-parameters25 and their optimization during QSAR model training25,71 were described previously. Data imputation used the combination that yielded the highest AUC for each included bioassay.
Network Architecture.
The ER k-DNN was trained in Python v3.6.2 using Google Tensorflow v1.4.0. Neural networks begin with an input layer that contains information about the features of the data, such as chemical fingerprints. The input layer of the network described here consisted of 1024 FCFPs plus 3 known ERα/ERβ toxicophores [i.e., steroid and diethylstilbestrol (DES) scaffolds and the phenol group].76 Neural networks end with an output layer, which predicts the activity of interest. Here, the modeled activity was in vivo rodent uterotrophic bioactivity.22 The output layer of the ER k-DNN used a sigmoid activation function, which results in a value between 0 and 1 that represents a probability.77 The probability calculated represented the likelihood that a compound has in vivo rodent uterotrophic bioactivity. Five layers connect the ER k-DNN’s input and output layers, organized and ordered using an AOP framework. Each layer represented a higher level of biological organization than the last (Figure 3). These 5 layers contained 57 neurons in total, which each represented one in vitro bioassay included in the training set.
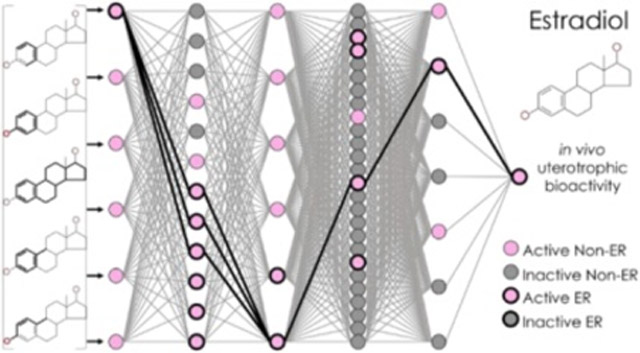
Figure 3.

Knowledge-based predictions from the NN. Predictions are shown for (A) estradiol, a uterotrophic training compound; (B) naringenin and (C) pregnenolone, estrogenic external validation compounds; and (D) tributyltin chloride, the false positive training compound. Each panel contains a diagram of the neurons and weighted edges of the NN’s inner layers, where pink neurons with a value of 1 represent active bioassay results, and gray neurons with a value of 0 represent inactive bioassay results. Neurons with heavy edges represent bioassays known to encompass KEs in the nuclear ER alpha and beta adverse outcome pathway. Edges with heavy edges represent connections between the activated neuron(s) in each layer.
Each of the 57 neurons was placed into one of the 5 layers based on the ToxCast-defined biological process target of the bioassay it represented (Tables 1 and S2). Layer 2 bioassays (n = 6) represented receptor binding, including binding to ERα, the first step of the signaling pathway (E1, A1–A5).44-47 Receptor dimerization assays (n = 12) were grouped into layer 3, with other bioassays having the “protein stabilization” biological process target (E2–E7, A6–A11). Layer 3’s bioassays included six assays measuring the formation of ERα and ERβ homo- and heterodimers triggered by ERα activation (i.e., the second step of the pathway).48-50 Layer 4 bioassays (n = 6) represented the third step of the pathway being modeled. This layer contained bioassays with the ToxCast-defined biological process target “regulation of gene expression,” including those measuring DNA binding of ERα and ERβ homo- and heterodimers (E8–E9, A12–15).50 Bioassays with the biological process target “regulation of transcription factor activity” (n = 26), including those measuring ER-mediated transcriptional activation triggered by the binding of ERα and ERβ dimers to DNA, were placed into layer 5 (E10–13, A16–37).51-53 The last layer contained bioassays representing cell proliferation (n = 7), including an ER-sensitive assay (E14, A38–43).54,55 These five center layers were densely connected. Each of the 57 neurons shared weighted connections with every neuron in the layers before and after it. Here, these weighted connections were initialized with a value of 0.10 so that the relative weights after training were interpretable. Then, they underwent optimization in the network training process.
Network Training and Cross-Validation.
The network’s edge weights were optimized during training using the stochastic gradient descent algorithm,78 with a starting learning rate of 0.01 and momentum of 0.9.79 The k-DNN was evaluated during training using binary cross-entropy, a standard evaluation method for classification tasks (eq 1).80 If the binary cross-entropy did not change after 50 consecutive iterations, the learning rate was automatically reduced by 90% until it reached a minimum allowed value of 0.00001.81 Optimization of the ER k-DNN ended upon 200 iterations with no binary cross-entropy improvement.82 Dropout was induced at the neurons that represented assays in which a given training compound was classified as inactive.83 Half of the neurons in each layer were also randomly removed via the dropout technique to increase the network’s ability to generalize to new compounds,83
| (1) |
where Z represents the calculated output, Y represents the known class, and m represents the number of training compounds.
The network’s predictive value was evaluated during training using a leave-one-out cross-validation procedure. Briefly, this procedure used all but one training set compound to train the network and predict the remaining compound. This procedure was repeated 42 times until all training set compounds were used for predictions one time. At the end of this procedure, the complete training set’s predictions were evaluated using the area under the receiver-operating curve (ROC) metric (AUC). The ER k-DNN from this study computed a probability that each compound will show in vivo rodent uterotrophic bioactivity. A compound was considered uterotrophic when its calculated probability exceeded a certain threshold. The ROC for model performance is a plot of the true-positive rate (TPR, eq 2) and false positive rate (FPR, eq 3) using various probability thresholds for distinguishing between uterotrophic and non-uterotrophic compounds.84 The AUC represents the ER k-DNN’s likelihood of correctly classifying compounds as uterotrophic or non-uterotrophic. An AUC value of 0.5 represents random classifications, and an AUC value of 1 represents 100% predictivity.
| (2) |
| (3) |
Network Analysis.
The output of the matrix multiplication between the input vector from the previous layer and the optimized edge weights after the dropout of neurons corresponding to inactive bioassay results was examined for each compound. The neuron or neurons with the highest value in a layer’s output vector were considered to be activated by this compound. The activated neurons with the associated bioassays and their connection edges for each predicted compound were recorded as a virtual pathway for analysis based on the embedded biological data.
RESULTS
In Vivo and In Vitro Training Data.
The quality of models developed within an AOP framework relies more on the number of pathway steps captured by the model than the abundance and diversity of training compounds.85 For this reason, highly mechanistic toxicity models typically have small training sets composed of extensively curated, high-quality experimental data. The ER k-DNN underwent training with compounds with known in vivo uterotrophic or non-uterotrophic bioactivity in rodents (n = 42) (Table S1). These compounds had reproducible and independently verified in vivo test results in two or more guideline-like in vivo rodent uterotrophic studies.39,40 Of the 42 compounds, 29 were uterotrophic and 13 were non-uterotrophic (Table S1).
The hit calls for these 42 compounds in hundreds of high-throughput bioassays were extracted from the EPA ToxCast/Tox21 database.64,65 Among these bioassays, 14 were included for network training because they represented KEs in the ERα/ERβ agonism pathway (Table 1). An additional 43 bioassays with the same ToxCast-defined “biological process target” as at least one ER bioassay were incorporated into the ER k-DNN (Figure 1A, Tables 1 and S2). These biological process targets describe the high-level biological activity (e.g., regulation of gene expression) represented by a bioassay’s measured readout. The additional ToxCast/Tox21 bioassays were selected based on the training compound data availability, with preference given to nuclear receptor bioassays when possible, to assess the network’s ability to discern data representing the ERα/ERβ pathway from data representing pathways with related biological functions. The in vitro bioassay profile for the training set compounds contained 797 active results, 1485 inactive results, and 112 untested compound–bioassay pairs. The included bioassays represented 31 targets (Figure 1A).
Figure 1.

Summary of the training data set. Distributions of the network’s included assays are shown by (A) target gene and (B) number of active and inactive compounds.
Missing data are common when using high-throughput bioassay data for modeling.21,56,59,86 QSAR approaches are not always reliable for complicated in vivo endpoints (e.g., uterotrophic bioactivity), but they perform well in predicting in vitro bioassay results with relatively simple mechanisms.21 Inhouse predictive QSAR model development approaches filled in the 112 missing activities before k-DNN training.71 The QSAR models were evaluated using the AUC metric. The AUC measures the likelihood of the QSAR model to distinguish active from inactive compounds correctly. Most QSAR models have AUC values between 0.5 and 1. An AUC of 0.5 represents performance equivalent to random classifications, and an AUC of 1 represents perfect performance. On average, the QSAR models developed for data imputation in this study had an AUC of about 0.72 (Figure S1). A total of 12 QSAR models were developed for each of the 57 bioassays for a total of 684 QSAR models. The data imputation process used a consensus model, which was the average of all predictions from individual models developed using the same descriptors. Consensus models typically represent the best individual predictivity for a given bioassay by leveraging the strengths of many algorithms.14,67,71,87,88 The final in vitro bioassay profile used for modeling contained 847 active results and 1547 inactive results (Figure 1B, Table S1). This imbalanced ratio reflects the nature of high-throughput bioassay data in toxicity testing, which usually contains more inactive results than active results.21,56,59,86
Network Design.
The ER k-DNN used chemical structure information, in vitro data, and target in vivo toxicity endpoint data for training (Figure 2). The ER k-DNN architecture mirrored the biological levels inherent in the AOP framework. As a result, the network could capture and simultaneously learn the complex sequence of interactions that usually link structural features (i.e., toxicophores) to different levels of biological responses and eventually cause in vivo adverse toxicity outcomes.43 Each potential toxicophore, in vitro hit call, and in vivo rodent uterotrophic bioactivity was represented by one neuron in the ER k-DNN. The input layer contained 1027 neurons representing potential toxicophores, and the output layer was a single neuron representing in vivo uterotrophic bioactivity. In vitro bioassays were organized between these layers based on the biological processes in the ERα/ERβ pathway: receptor binding, receptor dimerization, DNA binding, transcriptional activation, and cell proliferation (Figure 3). Every neuron received input from each neuron in the preceding layer and sent output to each neuron in the subsequent layer. The weight of connections underwent automatic optimization during training. The ER k-DNN contained 1084 neurons organized into 7 layers. A combination of a compound’s input data and the final optimized weights is a single score representing the probability that a compound will be uterotrophic in rodents.
Figure 2.

Workflow used in this study. The modeling workflow employed in this study consists of four main stages: generation of chemical fingerprints and retrieval of in vitro bioassay data for each compound in the training set, generation and implementation of QSAR models to fill data gaps, organization of in vitro data into biological levels of organization (MIE through adverse outcome), and network training. Pink cells and neurons with a value of 1 represent active bioassay results, and gray neurons and cells with a value of 0 represent inactive bioassay results.
Network Training and Hidden Pathway Identification.
The ER k-DNN developed in this study was evaluated using predictions on the training set compounds with the fully trained network and using a leave-one-out cross-validation procedure during network training. Cross-validation procedures are reliable ways to evaluate a model’s predictivity.32,89 Briefly, the leave-one-out cross-validation procedure used 41 compounds for training and left out the remaining compound for prediction. This procedure was repeated 42 times, leaving each training compound out for prediction once. The fully trained ER k-DNN had a training AUC of 0.956, predicting the uterotrophic or non-uterotrophic bioactivity of 38 out of 42 compounds correctly (Table 2). The k-DNN’s leave-one-out cross-validation AUC was 0.867 (Figure 4).
Table 2.
Training Set Performance of the k-DNNa
| performance metric | value |
|---|---|
| AUC | 0.956 |
| true positives | 26 |
| true negatives | 12 |
| false positives | 1 |
| false negatives | 3 |
| unique neuron patterns activated by predicted toxic compounds | 5 |
| true positives activating only ERα and ERβ neurons | 23 |
AUC: area under the ROC.
Figure 4.

ROCs from leave-one-out cross-validation. The solid line at y = x represents performance equivalent to assigning uterotrophic vs non-uterotrophic designations to compounds randomly. The solid curve represents the ROC for the k-DNN. The dashed line represents the ROC for the first control network, which was trained using 100 permutations of the in vitro bioassay and in vivo rodent uterotrophic training data. The dotted line represents the ROC for the second control network, which was trained without any bioassays known to encompass KEs in the nuclear ERα and ERβ AOP.
Unlike traditional “black-box” NNs, this k-DNN approach effectively captured chemical-in vitro–in vivo relationships (i.e., from initial events of compound-receptor bindings to KEs measured in vitro and eventually to in vivo organ-level outcomes) and virtually identified an ERα/ERβ pathway for each prediction. ERα/ERβ toxicophores and KEs embedded in the network were identifiable by examining the neurons of relevant bioassays activated during the prediction of toxic compounds. A total of 27 training compounds were predicted as uterotrophic, including 26 true positives and 1 false positive (Table 2). Among these predicted uterotrophic compounds, 23 activated only neurons representing the relevant ERα/ERβ pathway. For example, when predicting estradiol (CAS 50-28-2), an endogenous uterotrophic compound, seven neurons were activated among the ER k-DNN’s five center layers. The computed probability that this compound would have uterotrophic bioactivity was 1 (Figure 3A). This prediction activated neurons that represent ERα binding, dimerization (two ERα/ERβ bioassays and an ERβ/ERβ bioassay), DNA binding, transcriptional activation, and cell proliferation. This neuron pattern, including the neurons and their connections, represents a virtual pathway affected by this biologically active compound when showing in vivo rodent uterotrophic bioactivity.
Examining the information of the first two layers of the ER k-DNN can reveal relevant chemical fragments as toxicophores of the ERα/ERβ pathway. Weighted edges tuned during the training process (n = 6162) connect the ER k-DNN’s first and second layers. The most heavily weighted edge between a chemical fragment’s neuron and a receptor binding bioassay neuron indicates the most influenced receptor binding bioassay by that fragment. The second layer of the ER k-DNN contained six neurons representing binding to various steroidal nuclear receptors (Figure 5). A total of 3 toxicophores critical for ERα/ERβ binding were in the first layer: the phenol group and the steroid and DES scaffolds (contained in 25, 9, and 1 training compounds, respectively).76 Each of these toxicophores had six edges connecting to the second layer (i.e., connecting to each neuron in the second layer). After training, the highest-weighted edge from each of these three toxicophores connected with the ERα binding bioassay neuron, rather than the other five neurons representing androgen, glucocorticoid, and progesterone receptor (PR) binding. The difference between the ERα edge weight and the other nuclear receptor edge weights is more pronounced for the steroid scaffold and phenol group than for the DES scaffold, consistent with the higher occurrence of these two fragments among training set compounds. Therefore, the ER k-DNN captured several toxicophores that activate ERα (i.e., the MIE of the relevant AOP) by inferring relationships between three toxicophores and a human ERα binding bioassay (Figure 5). This receptor activation represents a critical step by initiating the pathway and determining relevant compounds’ resulting estrogenicity.
Figure 5.

Analysis of three chemical fragments known to bind to nuclear ERα and ERβ: the steroid scaffold, phenol group, and DES scaffold. Each column of the heatmap represents one chemical fragment and shows the weights of edges connecting the neuron associated with that fragment and each of the neurons in the network’s receptor binding layer: human ERα; chimpanzee, human, and rat AR; the human glucocorticoid receptor; and human PR. Each column is normalized such that the highest weighted edge leaving the fragment’s neuron is shown as the darkest color, and the lowest weighted edge is the lightest color.
The ER k-DNN’s robustness was ensured by comparing the results to two control networks. First, a permutation test was performed by constructing an NN with randomized in vitro bioassay and in vivo rodent uterotrophic activities (control #1).90 The randomized biological data were used as input data to train the control network. This process was repeated with 100 randomizations of the biological data. During these 100 leave-one-out cross-validation procedures, control #1 showed an AUC of 0.492 ± 0.086, which is close to a random prediction result. This performance was dramatically lower than the ER k-DNN’s cross-validation AUC of 0.867 (Figure 4). For the 29 compounds predicted to be uterotrophic by control #1, 28 unique neuron sequences were activated, indicating overfitting rather than learning from the available biological data. Compared to this, the ER k-DNN predicted 29 compounds as estrogenic by activating 14 unique sequences.
A second control NN was constructed with the same inner structure as the k-DNN but with useful data (i.e., the ERα/ERβ bioassay data) removed (control #2). Control #2’s cross-validation AUC decreased from 0.867 to 0.594 (Figure 4). During cross-validation, control #2 predicted 14 training set compounds to be uterotrophic. These 14 compounds activated 10 unique neuron sequences. The decreased network performance and an increased number of activated sequences in the control networks indicate no clear relationship between a set of randomly selected bioassays and the control networks’ predictions. The ER k-DNN consistently activated the neuron sequences, including ERα/ERβ bioassays for uterotrophic training compounds, whereas the two control networks randomly and erratically activated neurons. Therefore, the ER k-DNN made predictions based on accurately identified toxicological mechanisms rather than a black-box network with thousands of arbitrarily tuned parameters with no biological basis.
External Predictions.
Due to a lack of high-quality uterotrophic compounds outside of the training set, the ER k-DNN was used to predict a data set obtained from the Collaborative Estrogen Receptor Activity Prediction Project (CERAPP).67 This external validation data set contained compounds with available estrogenic versus non-estrogenic classification data from low-throughput in vitro ER agonism screening. Compounds existing in the training set were removed from this data set, resulting in 6286 new compounds that were unknown to the trained ER k-DNN but tested in various numbers of ToxCast/Tox21 bioassays (Table S4). This data set was used to test the k-DNN’s predictivity in the external validation process. Like the training compounds, missing ToxCast/Tox21 HTS bioassay results were filled using QSAR models for all new compounds. Compounds tested in the ToxCast/Tox21 programs were further excluded to evaluate external compounds that required QSAR predictions for all biological data embedded in the network’s center layers. This curation process resulted in a second, smaller data set of 368 new compounds for external validation purposes (Table S4).
Figure 6 shows the ROCs of external validations. When considering the entire validation set (n = 6286), the AUC value was 0.747. The AUC value was similar when considering the second smaller validation set (n = 368), equaling 0.759. Because the k-DNN strongly depends on mechanistic biological data representing pathway steps rather than on a large and chemically diverse training set, its predictions are reliable when making predictions that are biologically consistent with the virtual pathways. In this study, predicted estrogenic compounds are most reliable if they activate neurons representing steps in the ERα/ERβ pathway in at least three network layers. Predicted non-estrogenic compounds are most reliable if they activate neurons representing events in the ERα/ERβ pathway in less than three network layers. When considering external compounds that meet these criteria for reliability, the AUC values improved from 0.743 to 0.864 for the first validation set (n = 693) and from 0.803 to 0.927 for the second external validation set (n = 99). These criteria can be used as an applicability domain in future studies.
Figure 6.

ROCs from external validation. The top panel shows the ROC for the complete external validation set. The bottom panel shows the ROC for a subset of the external validation set containing only compounds experimentally untested in the HTS assays embedded in the network. The solid line at y = x in each panel represents performance equivalent to assigning estrogenic vs non-estrogenic designations to compounds randomly. In each panel, the solid curve represents the ROC for the complete data set using no mechanism-based filter. The dashed curve represents the ROC for a smaller subset that meets the mechanism-based criteria. Compounds that meet the mechanism-based criteria include predicted estrogens with at least three and predicted non-estrogens with no more than two network layers containing activated neurons related to nuclear ERα and ERβ signaling.
Because the network inferred connections among chemical and biological data during training, a critical mechanistic context is available to justify a new compound’s prediction. For example, naringenin (CAS 480-41-1) was an estrogenic external validation compound. The ER k-DNN correctly predicted this compound as having estrogenic potential with a probability of 1. When predicting this compound, the neuronal activation pattern shows the predicted bioassay responses that resulted in this estrogenic result (Figure 3B). Naringenin contains a similar scaffold to DES (CAS 56-53-1), a classic ER agonist. When predicting naringenin, the neurons activated in the center layers represented KEs of the ERα/ERβ pathway. The prediction of this compound activated neurons representing ERα binding, ER dimerization (two ERα/ERβ bioassays and an ERβ/ERβ bioassay), DNA binding, transcriptional activation, and cell proliferation. This explicit ERα/ERβ pathway neuron activation provides a biological justification for this estrogenic prediction.
DISCUSSION
With computational toxicology’s progress into a big data era, developing methods to use the available data for mechanism-driven chemical toxicity predictions efficiently is a central challenge.56,58,59,91 Many existing models have flaws in meeting the international guidelines for computational toxicity predictions to be accurate and allow for mechanism-based interpretation.32 Although some computational models that successfully incorporate biological data exist for several toxicity endpoints, including ER binding, these models are usually limited by presenting black-box-type predictions or requiring experimental data.38,39,63,71 The k-DNN approach developed in this study addresses these challenges by employing an approach that can make interpretable end-to-end predictions (Figure 2). The k-DNN approach described here combines chemical and biological public data into an explainable AOP context for toxicity predictions using the network architecture inherent to NNs (Figure 3). This network successfully inferred connections among potential toxicophores (Figure 5), a set of in vitro HTS bioassays (Table S2), and in vivo uterotrophic bioassay results (Figures 3A and 4, Table S1).
The ER k-DNN can also make correct predictions for compounds that may not show active responses for all KEs in the pathway. For example, 4-tert-butylphenol (CAS 98-54-4) is a uterotrophic training compound. This compound was experimentally inactive in the ERα binding bioassay but active in bioassays relating to the remaining four KEs in the pathway (i.e., ERβ/ERβ dimerization, DNA binding, transcriptional activation, and cell proliferation). The ER k-DNN activated an androgen receptor (AR) binding bioassay in the first layer and an ERβ/ERβ dimerization bioassay in the second layer. The availability of non-ERα/ERβ edges allowed this compound’s activation of the rest of the pathway and, therefore, a correct estrogenic prediction. This behavior shows similar utility in predicting external test compounds with predicted inactive results in some ERα/ERβ bioassays. Pregnenolone (CAS 145-13-1) is an estrogenic external test compound. This compound was also inactive in the ERα binding bioassay. However, it did show activity in bioassays representing the other KEs in the ERα/ERβ pathway. The learned behavior of the ER k-DNN to take advantage of other connections likely to be positive for estrogenic compounds ultimately allows for a correct prediction of this external compound explained by a biological mechanism (Figure 3C).
The false-positive training compound was tributyltin chloride (CAS 1461-22-9). This compound did not show rodent uterotrophic bioactivity. However, the ER k-DNN predicted it as uterotrophic with a probability of 0.5003, slightly above the threshold of 0.5. As shown by the virtual pathway, one neuron of an ERα/ERβ–related bioassay was activated, representing DNA binding (E9, Figure 3D). However, the activated neurons in other layers did not represent ERα/ERβ-related bioassays. One neuron represented an antioxidant response element bioassay. The remaining three bioassays represent other nuclear receptor pathways (i.e., androgen, farsenoid X, and constitutive androstane). This virtual pathway resulted in a weakly positive prediction because some training compounds showing in vivo uterotrophic bioactivity coincidentally showed activity in these non-ERα/ERβ assays (Table S1), increasing these edge weights.
Using the k-DNN approach, users can exclude false positives like tributyltin chloride by manually reviewing the biological information embedded into the network. Alternatively, users can lift the threshold for distinguishing between predicted uterotrophic and non-uterotrophic compounds (e.g., increasing the threshold from 0.5 to 0.6). By allowing for the critical evaluation of positive results according to a rational screening of mechanistic evidence, the k-DNN approach represents a significant advancement over traditional black-box computational approaches for chemical toxicity predictions. Such advancements are critical in pursuing the acceptance of computational models by regulatory bodies such as the OECD.32 Using the k-DNN to generate probabilities with supporting mechanistic data allows toxicologists to make more informed decisions about further experimental testing.
The k-DNN approach developed in this study aims to learn which toxicophores and bioassays targeted the appropriate pathway steps without manual specifications. This capability highlights the practical applications of the approach for other complex toxicity modeling studies that need to reveal obscure toxicity pathways. Within the developed framework, available bioassay data for high-quality data sets representing apical outcomes can be organized based on their biological levels. The k-DNN training process can then elucidate new pathways by linking relevant toxicophores and bioassays during the model training process. Therefore, this approach is expected to identify new toxicity pathways when experimental data representing steps at higher levels of biological organization are available for the training process. The abundance and specificity of the identified toxicophores will also increase with more animal data available for network training. These mechanistically related toxicophores and bioassay combinations are then available to reduce, refine, and eventually replace animal testing in chemical safety assessments through weight-of-evidence frameworks utilizing AOPs, such as Integrated Approaches to Testing and Assessment (IATA).92,93 Ultimately, this approach can provide a strategy for the efficient utilization of both rapidly growing biological data and computational methods to pave the way for the regulatory acceptance of non-animal methods in toxicity assessments.
Supplementary Material
ACKNOWLEDGMENTS
H.L.C., D.P.R., and H.Z. were partially supported by the National Institute of Environmental Health Sciences (NIEHS) grants (R01ES031080 and R15ES023148) and an ExxonMobil Research Grant for Rutgers University. L.M.A. was partially supported by NIEHS grant R01ES029275 and National Center for Advancing Translational Sciences grant UL1TR003017. L.M.A. and H.Z. were partially supported by NIEHS grant P30ES005022.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.est.1c02656.
Five-fold cross-validation scores of quantitative structure–activity relationship (QSAR) models used for the imputation of missing bioassay data (PDF)
In vivo rodent uterotrophic bioassay data; additional Toxicity Forecaster (ToxCast) and toxicity in the 21st century (Tox21) assays included in the model; training set performance with varying bond radiuses used for generating FCFPs; and Collaborative Estrogen Receptor Activity Prediction Project (CERAPP) external validation sets (XLSX)
The authors declare no competing financial interest.
Contributor Information
Heather L. Ciallella, Center for Computational and Integrative Biology, Rutgers University Camden, Camden, New Jersey 08103, United States
Daniel P. Russo, Center for Computational and Integrative Biology, Rutgers University Camden, Camden, New Jersey 08103, United States; Department of Chemistry, Rutgers University Camden, Camden, New Jersey 08102, United States
Lauren M. Aleksunes, Department of Pharmacology and Toxicology, Ernest Mario School of Pharmacy, Rutgers University, Piscataway, New Jersey 08854, United States
Fabian A. Grimm, ExxonMobil Biomedical Sciences, Inc., Annandale, New Jersey 08801, United States
Hao Zhu, Center for Computational and Integrative Biology, Rutgers University Camden, Camden, New Jersey 08103, United States; Department of Chemistry, Rutgers University Camden, Camden, New Jersey 08102, United States.
REFERENCES
- (1).Hall JM; Couse JF; Korach KS The Multifaceted Mechanisms of Estradiol and Estrogen Receptor Signaling. J. Biol. Chem 2001, 276, 36869–36872. [DOI] [PubMed] [Google Scholar]
- (2).Eddy EM; Washburn TF; Bunch DO; Goulding EH; Gladen BC; Lubahn DB; Korach KS Targeted Disruption of the Estrogen Receptor Gene in Male Mice Causes Alteration of Spermatogenesis and Infertility. Endocrinology 1996, 137, 4796–4805. [DOI] [PubMed] [Google Scholar]
- (3).Lubahn DB; Moyer JS; Golding TS; Couse JF; Korach KS; Smithies O Alteration of Reproductive Function but Not Prenatal Sexual Development after Insertional Disruption of the Mouse Estrogen Receptor Gene. Proc. Natl. Acad. Sci. U.S.A 1993, 90, 11162–11166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Heldring N; Pike A; Andersson S; Matthews J; Cheng G; Hartman J; Tujague M; Ström A; Treuter E; Warner M; Gustafsson J-Å Estrogen Receptors: How Do They Signal and What Are Their Targets. Physiol. Rev 2007, 87, 905–931. [DOI] [PubMed] [Google Scholar]
- (5).Prossnitz ER; Arterburn JB International Union of Basic and Clinical Pharmacology. XCVII. G Protein-Coupled Estrogen Receptor and Its Pharmacologic Modulators. Pharmacol. Rev 2015, 67, 505–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).De Coster S; van Larebeke N Endocrine-Disrupting Chemicals: Associated Disorders and Mechanisms of Action. J. Environ. Public Health 2012, 2012, 713696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Brzozowski AM; Pike ACW; Dauter Z; Hubbard RE; Bonn T; Engström O; Öhman L; Greene GL; Gustafsson J-Å; Carlquist M Molecular Basis of Agonism and Antagonism in the Oestrogen Receptor. Nature 1997, 389, 753–758. [DOI] [PubMed] [Google Scholar]
- (8).Ciocca DR; Fanelli MA Estrogen Receptors and Cell Proliferation in Breast Cancer. Trends Endocrinol. Metab 1997, 8, 313–321. [DOI] [PubMed] [Google Scholar]
- (9).Felty Q; Roy D Estrogen, Mitochondria, and Growth of Cancer and Non-Cancer Cells. J. Carcinog 2005, 4, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Doisneau-Sixou SF; Sergio CM; Carroll JS; Hui R; Musgrove EA; Sutherland RL Estrogen and Antiestrogen Regulation of Cell Cycle Progression in Breast Cancer Cells. Endocr.-Relat. Cancer 2003, 10, 179–186. [DOI] [PubMed] [Google Scholar]
- (11).Birnbaum LS; Fenton SE Cancer and Developmental Exposure to Endocrine Disruptors. Environ. Health Perspect 2003, 111, 389–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Fenner-Crisp PA; Maciorowski AF; Timm GE The Endocrine Disruptor Screening Program Developed by the U.S. Environmental Protection Agency. Ecotoxicology 2000, 9, 85–91. [Google Scholar]
- (13).Meigs L; Smirnova L; Rovida C; Leist M; Hartung T Animal Testing and Its Alternatives – the Most Important Omics Is Economics. ALTEX 2018, 35, 275–305. [DOI] [PubMed] [Google Scholar]
- (14).Solimeo R; Zhang J; Kim M; Sedykh A; Zhu H Predicting Chemical Ocular Toxicity Using a Combinatorial QSAR Approach. Chem. Res. Toxicol 2012, 25, 2763–2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Zhu H; Ye L; Richard A; Golbraikh A; Wright FA; Rusyn I; Tropsha A A Novel Two-Step Hierarchical Quantitative Structure-Activity Relationship Modeling Work Flow for Predicting Acute Toxicity of Chemicals in Rodents. Environ. Health Perspect 2009, 117, 1257–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Zhu H; Tropsha A; Fourches D; Varnek A; Papa E; Gramatica P; Öberg T; Dao P; Cherkasov A; Tetko IV Combinatorial QSAR Modeling of Chemical Toxicants Tested against Tetrahymena Pyriformis. J. Chem. Inf. Model 2008, 48, 766–784. [DOI] [PubMed] [Google Scholar]
- (17).Stouch TR; Kenyon JR; Johnson SR; Chen X-Q; Doweyko A; Li Y Silico ADME/Tox: Why Models Fail. J. Comput.-Aided Mol Des 2003, 17, 83–92. [DOI] [PubMed] [Google Scholar]
- (18).Dearden JC; Cronin MTD; Kaiser KLE How Not to Develop a Quantitative Structure-Activity or Structure-Property Relationship (QSAR/QSPR). SAR QSAR Environ. Res 2009, 20, 241–266. [DOI] [PubMed] [Google Scholar]
- (19).Najafabadi MM; Villanustre F; Khoshgoftaar TM; Seliya N; Wald R; Muharemagic E Deep Learning Applications and Challenges in Big Data Analytics. J. Big Data 2015, 2, 1. [Google Scholar]
- (20).Zhang L; Tan J; Han D; Zhu H From Machine Learning to Deep Learning: Progress in Machine Intelligence for Rational Drug Discovery. Drug Discovery Today 2017, 22, 1680–1685. [DOI] [PubMed] [Google Scholar]
- (21).Zhu H Big Data and Artificial Intelligence Modeling for Drug Discovery. Annu. Rev. Pharmacol. Toxicol 2020, 60, 573–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Schmidhuber J Deep Learning in Neural Networks: An Overview. Neural Network. 2015, 61, 85–117. [DOI] [PubMed] [Google Scholar]
- (23).Russo DP; Zorn KM; Clark AM; Zhu H; Ekins S Comparing Multiple Machine Learning Algorithms and Metrics for Estrogen Receptor Binding Prediction. Mol. Pharm 2018, 15, 4361–4370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Zhou Y; Cahya S; Combs SA; Nicolaou CA; Wang J; Desai PV; Shen J Exploring Tunable Hyperparameters for Deep Neural Networks with Industrial ADME Data Sets. J. Chem. Inf. Model 2019, 59, 1005–1016. [DOI] [PubMed] [Google Scholar]
- (25).Korotcov A; Tkachenko V; Russo DP; Ekins S Comparison of Deep Learning with Multiple Machine Learning Methods and Metrics Using Diverse Drug Discovery Data Sets. Mol. Pharm 2017, 14, 4462–4475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Koutsoukas A; Monaghan KJ; Li X; Huan J Deep-Learning: Investigating Deep Neural Networks Hyper-Parameters and Comparison of Performance to Shallow Methods for Modeling Bioactivity Data. J. Cheminf. 2017, 9, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Byvatov E; Fechner U; Sadowski J; Schneider G Comparison of Support Vector Machine and Artificial Neural Network Systems for Drug/Nondrug Classification. J. Chem. Inf. Comput. Sci 2003, 43, 1882–1889. [DOI] [PubMed] [Google Scholar]
- (28).Mayr A; Klambauer G; Unterthiner T; Hochreiter S DeepTox: Toxicity Prediction Using Deep Learning. Front. Environ. Sci 2015, 3, 80. [Google Scholar]
- (29).Huang R; Xia M; Nguyen D-T; Zhao T; Sakamuru S; Zhao J; Shahane SA; Rossoshek A; Simeonov A Tox21Challenge to Build Predictive Models of Nuclear Receptor and Stress Response Pathways as Mediated by Exposure to Environmental Chemicals and Drugs. Front. Environ. Sci 2016, 3, 85. [Google Scholar]
- (30).Kleinstreuer NC; Karmaus AL; Mansouri K; Allen DG; Fitzpatrick JM; Patlewicz G Predictive Models for Acute Oral Systemic Toxicity: A Workshop to Bridge the Gap from Research to Regulation. Comput. Toxicol 2018, 8, 21–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Castelvecchi D Can We Open the Black Box of AI? Nature 2016, 538, 20–23. [DOI] [PubMed] [Google Scholar]
- (32).Organisation for Economic Co-operation and Development. Guidance Document on the Validation of (Quantitative)Structure-Activity Relationship [(Q)SAR] Models. OECD Environment Health and Safety Publications. Series on Testing and Assessment, 2007; Vol. 69, pp 1–154. [Google Scholar]
- (33).Dix DJ; Houck KA; Martin MT; Richard AM; Setzer RW; Kavlock RJ The ToxCast Program for Prioritizing Toxicity Testing of Environmental Chemicals. Toxicol. Sci 2007, 95, 5–12. [DOI] [PubMed] [Google Scholar]
- (34).Judson RS; Houck KA; Kavlock RJ; Knudsen TB; Martin MT; Mortensen HM; Reif DM; Rotroff DM; Shah I; Richard AM; Dix DJ In Vitro Screening of Environmental Chemicals for Targeted Testing Prioritization: The ToxCast Project. Environ. Health Perspect 2010, 118, 485–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Attene-Ramos MS; Miller N; Huang R; Michael S; Itkin M; Kavlock RJ; Austin CP; Shinn P; Simeonov A; Tice RR; Xia M The Tox21 Robotic Platform for the Assessment of Environmental Chemicals–from Vision to Reality. Drug Discovery Today 2013, 18, 716–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Thomas R; Paules RS; Simeonov A; Fitzpatrick SC; Crofton KM; Casey WM; Mendrick DL The US Federal Tox21 Program: A Strategic and Operational Plan for Continued Leadership. ALTEX 2018, 35, 163–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Shukla SJ; Huang R; Austin CP; Xia M The Future of Toxicity Testing: A Focus on in Vitro Methods Using a Quantitative High-Throughput Screening Platform. Drug Discovery Today 2010, 15, 997–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Judson RS; Magpantay FM; Chickarmane V; Haskell C; Tania N; Taylor J; Xia M; Huang R; Rotroff DM; Filer DL; Houck KA; Martin MT; Sipes N; Richard AM; Mansouri K; Setzer RW; Knudsen TB; Crofton KM; Thomas RS Integrated Model of Chemical Perturbations of a Biological Pathway Using 18In VitroHigh-Throughput Screening Assays for the Estrogen Receptor. Toxicol. Sci 2015, 148, 137–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Browne P; Judson RS; Casey WM; Kleinstreuer NC; Thomas RS Screening Chemicals for Estrogen Receptor Bioactivity Using a Computational Model. Environ. Sci. Technol 2015, 49, 8804–8814. [DOI] [PubMed] [Google Scholar]
- (40).Kleinstreuer NC; Ceger PC; Allen DG; Strickland J; Chang X; Hamm JT; Casey WM A Curated Database of Rodent Uterotrophic Bioactivity. Environ. Health Perspect 2016, 124, 556–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).United States Environmental Protection Agency. Use of High Throughput Assays and Computational Tools; Endocrine Disruptor Screening Program; Notice of Availability and Opportunity for Comment. Fed. Regist 2015, 80, 35350–35355. [Google Scholar]
- (42).Ankley GT; Edwards SW The Adverse Outcome Pathway: A Multifaceted Framework Supporting 21st Century Toxicology. Curr. Opin. Toxicol 2018, 9, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Ankley GT; Bennett RS; Erickson RJ; Hoff DJ; Hornung MW; Johnson RD; Mount DR; Nichols JW; Russom CL; Schmieder PK; Serrrano JA; Tietge JE; Villeneuve DL Adverse Outcome Pathways: A Conceptual Framework to Support Ecotoxicology Research and Risk Assessment. Environ. Toxicol. Chem 2010, 29, 730–741. [DOI] [PubMed] [Google Scholar]
- (44).Haji M; Kato K.-i.; Nawata H; Ibayashi H Age-Related Changes in the Concentrations of Cytosol Receptors for Sex Steroid Hormones in the Hypothalamus and Pituitary Gland of the Rat. Brain Res. 1981, 204, 373–386. [DOI] [PubMed] [Google Scholar]
- (45).Knudsen TB; Houck KA; Sipes NS; Singh AV; Judson RS; Martin MT; Weissman A; Kleinstreuer NC; Mortensen HM; Reif DM; Rabinowitz JR; Setzer RW; Richard AM; Dix DJ; Kavlock RJ Activity profiles of 309 ToxCast chemicals evaluated across 292 biochemical targets. Toxicology 2011, 282, 1–15. [DOI] [PubMed] [Google Scholar]
- (46).O’Keefe JA; Handa RJ Transient Elevation of Estrogen Receptors in the Neonatal Rat Hippocampus. Dev. Brain Res 1990, 57, 119–127. [DOI] [PubMed] [Google Scholar]
- (47).Sipes NS; Martin MT; Kothiya P; Reif DM; Judson RS; Richard AM; Houck KA; Dix DJ; Kavlock RJ; Knudsen TB Profiling 976 ToxCast Chemicals across 331 Enzymatic and Receptor Signaling Assays. Chem. Res. Toxicol 2013, 26, 878–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).MacDonald ML; Lamerdin J; Owens S; Keon BH; Bilter GK; Shang Z; Huang Z; Yu H; Dias J; Minami T; Michnick SW; Westwick JK Identifying Off-Target Effects and Hidden Phenotypes of Drugs in Human Cells. Nat. Chem. Biol 2006, 2, 329–337. [DOI] [PubMed] [Google Scholar]
- (49).Yu H; West M; Keon BH; Bilter GK; Owens S; Lamerdin J; Westwick JK Measuring Drug Action in the Cellular Context Using Protein-Fragment Complementation Assays. Assay Drug Dev. Technol 2003, 1, 811–822. [DOI] [PubMed] [Google Scholar]
- (50).Stossi F; Bolt MJ; Ashcroft FJ; Lamerdin JE; Melnick JS; Powell RT; Dandekar RD; Mancini MG; Walker CL; Westwick JK; Mancini MA Defining Estrogenic Mechanisms of Bisphenol A Analogs through High Throughput Microscopy-Based Contextual Assays. Chem. Biol 2014, 21, 743–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Huang R; Xia M; Cho M-H; Sakamuru S; Shinn P; Houck KA; Dix DJ; Judson RS; Witt KL; Kavlock RJ; Tice RR; Austin CP Chemical Genomics Profiling of Environmental Chemical Modulation of Human Nuclear Receptors. Environ. Health Perspect 2011, 119, 1142–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Romanov S; Medvedev A; Gambarian M; Poltoratskaya N; Moeser M; Medvedeva L; Gambarian M; Diatchenko L; Makarov S Homogeneous Reporter System Enables Quantitative Functional Assessment of Multiple Transcription Factors. Nat. Methods 2008, 5, 253–260. [DOI] [PubMed] [Google Scholar]
- (53).Martin MT; Dix DJ; Judson RS; Kavlock RJ; Reif DM; Richard AM; Rotroff DM; Romanov S; Medvedev A; Poltoratskaya N; Gambarian M; Moeser M; Makarov SS; Houck KA Impact of Environmental Chemicals on Key Transcription Regulators and Correlation to Toxicity End Points within EPA’s ToxCast Program. Chem. Res. Toxicol 2010, 23, 578–590. [DOI] [PubMed] [Google Scholar]
- (54).Rotroff DM; Dix DJ; Houck KA; Kavlock RJ; Knudsen TB; Martin MT; Reif DM; Richard AM; Sipes NS; Abassi YA; Jin C; Stampfl M; Judson RS Real-Time Growth Kinetics Measuring Hormone Mimicry for ToxCast Chemicals in T-47D Human Ductal Carcinoma Cells. Chem. Res. Toxicol 2013, 26, 1097–1107. [DOI] [PubMed] [Google Scholar]
- (55).Xing JZ; Zhu L; Gabos S; Xie L Microelectronic Cell Sensor Assay for Detection of Cytotoxicity and Prediction of Acute Toxicity. Toxicol. In Vitro 2006, 20, 995–1004. [DOI] [PubMed] [Google Scholar]
- (56).Zhu H; Zhang J; Kim MT; Boison A; Sedykh A; Moran K Big Data in Chemical Toxicity Research: The Use of High-Throughput Screening Assays to Identify Potential Toxicants. Chem. Res. Toxicol 2014, 27, 1643–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Zhao L; Zhu H Big Data in Computational Toxicology: Challenges and Opportunities. In Computational Toxicology; Ekins S, Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, 2018, pp 291–312. DOI: 10.1002/9781119282594.ch11 [DOI] [Google Scholar]
- (58).Luechtefeld T; Rowlands C; Hartung T Big-Data and Machine Learning to Revamp Computational Toxicology and Its Use in Risk Assessment. Toxicol. Res 2018, 7, 732–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Ciallella HL; Zhu H Advancing Computational Toxicology in the Big Data ERa by Artificial Intelligence: Data-Driven and Mechanism-Driven Modeling for Chemical Toxicity. Chem. Res. Toxicol 2019, 32, 536–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Russo DP; Strickland J; Karmaus AL; Wang W; Shende S; Hartung T; Aleksunes LM; Zhu H Nonanimal Models for Acute Toxicity Evaluations: Applying Data-Driven Profiling and Read-Across. Environ. Health Perspect 2019, 127, 047001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Kim MT; Huang R; Sedykh A; Wang W; Xia M; Zhu H Mechanism Profiling of Hepatotoxicity Caused by Oxidative Stress Using Antioxidant Response Element Reporter Gene Assay Models and Big Data. Environ. Health Perspect 2016, 124, 634–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Zhao L; Russo DP; Wang W; Aleksunes LM; Zhu H Mechanism-Driven Read-Across of Chemical Hepatotoxicants Based on Chemical Structures and Biological Data. Toxicol. Sci 2020, 174, 178–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Ribay K; Kim MT; Wang W; Pinolini D; Zhu H Predictive Modeling of Estrogen Receptor Binding Agents Using Advanced Cheminformatics Tools and Massive Public Data. Front. Environ. Sci 2016, 4, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Judson R; Richard A; Dix D; Houck K; Elloumi F; Martin M; Cathey T; Transue TR; Spencer R; Wolf M ACToR - Aggregated Computational Toxicology Resource. Toxicol. Appl Pharmacol 2008, 233, 7–13. [DOI] [PubMed] [Google Scholar]
- (65).Williams AJ; Grulke CM; Edwards J; McEachran AD; Mansouri K; Baker NC; Patlewicz G; Shah I; Wambaugh JF; Judson RS; Richard AM The CompTox Chemistry Dashboard: A Community Data Resource for Environmental Chemistry. J. Cheminf 2017, 9, 1–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Kim S; Thiessen PA; Bolton EE; Bryant SH PUG-SOAP and PUG-REST: Web Services for Programmatic Access to Chemical Information in PubChem. Nucleic Acids Res. 2015, 43, W605–W611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Mansouri K; Abdelaziz A; Rybacka A; Roncaglioni A; Tropsha A; Varnek A; Zakharov A; Worth A; Richard AM; Grulke CM; Trisciuzzi D; Fourches D; Horvath D; Benfenati E; Muratov E; Wedebye EB; Grisoni F; Mangiatordi GF; Incisivo GM; Hong H; Ng HW; Tetko IV; Balabin I; Kancherla J; Shen J; Burton J; Nicklaus M; Cassotti M; Nikolov NG; Nicolotti O; Andersson PL; Zang Q; Politi R; Beger RD; Todeschini R; Huang R; Farag S; Rosenberg SA; Slavov S; Hu X; Judson RS CERAPP: Collaborative Estrogen Receptor Activity Prediction Project. Environ. Health Perspect 2016, 124, 1023–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Rogers D; Hahn M Extended-Connectivity Fingerprints. J. Chem. Inf. Model 2010, 50, 742–754. [DOI] [PubMed] [Google Scholar]
- (69).O’Boyle NM; Sayle RA Comparing Structural Fingerprints Using a Literature-Based Similarity Benchmark. J. Cheminf 2016, 8, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Pedregosa F; Varoquaux G; Gramfort A; Michel V; Thirion B; Grisel O; Blondel M; Prettenhofer P; Weiss R; Dubourg V; Vanderplas J; Passos A; Cournapeau D; Brucher M; Perrot M; Duchesnay E Scikit-Learn: Machine Learning in Python. J. Mach. Learn. Res 2011, 12, 2825–2830. [Google Scholar]
- (71).Ciallella HL; Russo DP; Aleksunes LM; Grimm FA; Zhu H Predictive Modeling of Estrogen Receptor Agonism, Antagonism, and Binding Activities Using Machine- and Deep-Learning Approaches. Lab. Invest 2020, 101, 490–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Manning CD; Raghavan P; Schuetze H The Bernoulli Model. Introduction to Information Retrieval; Cambridge University Press, 2009; pp 234–265. [Google Scholar]
- (73).Cover T; Hart P Nearest Neighbor Pattern Classification. IEEE Trans. Inf. Theory 1967, 13, 21–27. [Google Scholar]
- (74).Breiman L Random Forests. Mach. Learn 2001, 45, 5–32. [Google Scholar]
- (75).Vapnik VN Methods of Pattern Recognition. The Nature of Statistical Learning Theory; Springer Science & Business Media: New York, 2000, pp 123–180. DOI: 10.1007/978-1-4757-3264-1_6 [DOI] [Google Scholar]
- (76).Hong H; Tong W; Fang H; Shi L; Xie Q; Wu J; Perkins R; Walker JD; Branham W; Sheehan DM Prediction of Estrogen Receptor Binding for 58,000 Chemicals Using an Integrated System of a Tree-Based Model with Structural Alerts. Environ. Health Perspect 2002, 110, 29–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Han J; Moraga C The Influence of the Sigmoid Function Parameters on the Speed of Backpropagation Learning. In From Natural to Artificial Neural Computation. IWANN 1995. Lecture Notes in Computer Science; Mira J, Sandoval F, Eds.; Springer: Berlin, Heidelberg, 1995; Vol. 930, pp 195–201. [Google Scholar]
- (78).Bottou L Large-Scale Machine Learning with Stochastic Gradient Descent. Proceedings of COMPSTAT’2010; Lechevallier Y, Saporta G, Eds.; Physica-Verlag Heidelberg: Paris, France, 2010; pp 177–186. [Google Scholar]
- (79).Sutskever I; Martens J; Dahl G; Hinton G On the Importance of Initialization and Momentum in Deep Learning. Proceedings of the 30th International Conference on Machine Learning; JMLR: W&CP, 2013; pp 1139–1147. [Google Scholar]
- (80).Ramos D; Franco-Pedroso J; Lozano-Diez A; Gonzalez-Rodriguez J Deconstructing Cross-Entropy for Probabilistic Binary Classifiers. Entropy 2018, 20().DOI: DOI: 10.3390/e20030208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Goodfellow I; Bengio Y; Courville A Challenges in Neural Network Optimization. Deep Learning; The MIT Press: Cambridge, MA, 2016; pp 279–290. [Google Scholar]
- (82).Li M; Soltanolkotabi M; Oymak S Gradient Descent with Early Stopping Is Provably Robust to Label Noise for Over-parameterized Neural Networks. Proceedings of the 23rd International Conference on Artificial Intelligence and Statistics (AISTATS) 2020; PMLR, 2020; pp 4313–4324. [Google Scholar]
- (83).Srivastava N; Hinton G; Krizhevsky A; Sutskever I; Salakhutdinov R Dropout A Simple Way to Prevent Neural Networks from Overfitting. J. Mach. Learn. Res 2014, 15, 1929–1958. [Google Scholar]
- (84).Fawcett T An Introduction to ROC Analysis. Pattern Recognit. Lett 2006, 27, 861–874. [Google Scholar]
- (85).Kostal J; Voutchkova-Kostal A Going All In: A Strategic Investment in In Silico Toxicology. Chem. Res. Toxicol 2020, 33, 880–888. [DOI] [PubMed] [Google Scholar]
- (86).Zhao L; Ciallella HL; Aleksunes LM; Zhu H Advancing Computer-Aided Drug Discovery (CADD) by Big Data and Data-Driven Machine Learning Modeling. Drug Discovery Today 2020, 25, 1624–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Mansouri K; Kleinstreuer N; Abdelaziz AM; Alberga D; Alves VM; Andersson PL; Andrade CH; Bai F; Balabin I; Ballabio D; Benfenati E; Bhhatarai B; Boyer S; Chen J; Consonni V; Farag S; Fourches D; García-Sosa AT; Gramatica P; Grisoni F; Grulke CM; Hong H; Horvath D; Hu X; Huang R; Jeliazkova N; Li J; Li X; Liu H; Manganelli S; Mangiatordi GF; Maran U; Marcou G; Martin T; Muratov E; Nguyen D-T; Nicolotti O; Nikolov NG; Norinder U; Papa E; Petitjean M; Piir G; Pogodin P; Poroikov V; Qiao X; Richard AM; Roncaglioni A; Ruiz P; Rupakheti C; Sakkiah S; Sangion A; Schramm K-W; Selvaraj C; Shah I; Sild S; Sun L; Taboureau O; Tang Y; Tetko IV; Todeschini R; Tong W; Trisciuzzi D; Tropsha A; Van Den Driessche G; Varnek A; Wang Z; Wedebye EB; Williams AJ; Xie H; Zakharov AV; Zheng Z; Judson RS CoMPARA: Collaborative Modeling Project for Androgen Receptor Activity. Environ. Health Perspect 2020, 128, 027002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Mansouri K; Karmaus A; Fitzpatrick J; Patlewicz G; Pradeep P; Alberga D; Alepee N; Allen TEH; Allen D; Alves VM; Andrade CH; Auernhammer TR; Ballabio D; Bell S; Benfenati E; Bhattacharya S; Bastos JV; Boyd S; Brown JB; Capuzzi SJ; Chushak Y; Ciallella H; Clark AM; Consonni V; Daga PR; Ekins S; Farag S; Fedorov M; Fourches D; Gadaleta D; Gao F; Hartung T; Hirn M; Karpov P; Korotcov A; Lavado GJ; Lawless M; Li X; Luechtefeld T; Lunghini F; Mangiatordi GF; Marcou G; Marsh D; Martin T; Mauri A; Muratov EN; Myatt GJ; Nguyen D; Nicolotti O; Note R; Pande P; Parks AK; Peryea T; Polash AH; Rallo R; Roncaglioni A; Rowlands C; Ruiz P; Russo DP; Sayed A; Sayre R; Sheils T; Siegel C; Tetko IV; Thomas D; Tkachenko V; Todeschini R; Toma C; Tripodi I CATMoS: Collaborative Acute Toxicity Modeling Suite. Environ. Health Perspect 2021, 129, 047013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Tropsha A; Gramatica P; Gombar VK The Importance of Being Earnest: Validation Is the Absolute Essential for Successful Application and Interpretation of QSPR Models. QSAR Comb. Sci 2003, 22, 69–77. [Google Scholar]
- (90).Rücker C; Rücker G; Meringer M Y-Randomization and Its Variants in QSPR/QSAR. J. Chem. Inf. Model 2007, 47, 2345–2357. [DOI] [PubMed] [Google Scholar]
- (91).Zhu H; Bouhifd M; Donley E; Egnash L; Kleinstreuer N; Kroese ED; Liu Z; Luechtefeld T; Palmer J; Pamies D; Shen J; Strauss V; Wu S; Hartung T Supporting Read-across Using Biological Data. ALTEX 2016, 33, 167–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Tollefsen KE; Scholz S; Cronin MT; Edwards SW; de Knecht J; Crofton K; Garcia-Reyero N; Hartung T; Worth A; Patlewicz G Applying Adverse Outcome Pathways (AOPs) to Support Integrated Approaches to Testing and Assessment (IATA). Regul. Toxicol. Pharmacol 2014, 70, 629–640. [DOI] [PubMed] [Google Scholar]
- (93).Organization for Economic Co-operation and Development. Guidance Document for the Use of Adverse Outcome Pathways in Developing Integrated Approaches to Testing and Assessment (IATA). Series on Testing and Assessment, 2016, No. 260. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.