

Summary
These preliminary data from an ongoing first-in-human phase 1/2, open-label study provide proof-of-concept that pluripotent stem cell-derived pancreatic endoderm cells (PEC-01) engrafted in type 1 diabetes patients become islet cells releasing insulin in a physiologically regulated fashion. In this study of 17 subjects aged 22-57 with type 1 diabetes, PEC-01 cells were implanted subcutaneously in VC-02 macroencapsulation devices, allowing for direct vascularization of the cells. Engraftment and insulin expression were observed in 63% of VC-02 units explanted from subjects at 3–12 months post-implant. Six of 17 subjects (35.3%) demonstrated positive C-peptide as early as 6 months post-implant. Most reported adverse events were related to surgical implant or explant procedures (27.9%) or to side-effects of immunosuppression (33.7%). Initial data suggest that pluripotent stem cells, which can be propagated to the desired biomass and differentiated into pancreatic islet-like tissue, may offer a scalable, renewable alternative to pancreatic islet transplants.
Graphical abstract
Highlights
-
•
Findings are shared for the first 17 participants in a phase 1/2 trial of VC-02
-
•
This investigational device was implanted into type 1 diabetes patients
-
•
VC-02 contains pluripotent stem cell-derived pancreatic endoderm cells
-
•
C-peptide levels and insulin expression correlate with engraftment in 63% of subjects
Shapiro et al. report preliminary proof-of-concept that in 17 people with type 1 diabetes, pancreatic endoderm cells in an investigational subcutaneous device (VC-02) achieved engraftment and insulin expression in 63% of units at 3–12 months post-implant. Pluripotent stem cells may be a scalable, renewable alternative to pancreatic islet transplants.
Introduction
Approximately 100 years since the discovery of insulin, type 1 diabetes (T1D) remains a life-altering and sometimes life-threatening diagnosis. While insulin is lifesaving, it fails to prevent progression of end-stage microvascular complications in many subjects, and imprecision with insulin delivery often results in hypoglycemia and glycemic lability. Modern insulin delivery systems include advances in insulin pumps, continuous glucose monitoring, and glucose-responsive systems, but these advances fail to prevent hypoglycemia, do not consistently enable subjects to achieve glycemic targets, are burdensome to wear for protracted periods, and can malfunction. Further, even with careful administration of exogenous insulin, long-term complications are common (Figures 1 and 2; Table 1).
Figure 1.

The two different types of units used in the present study
(Upper photo) Sentinels (VC-02-20), which are smaller in size (1.5 cm x 1 cm) and have fewer cells, are explanted and examined periodically to evaluate status of the dose-finding units. For size comparison a US quarter is shown.
(Lower photo) The larger dose-finding units (VC-02-300, 9 cm x 3 cm)) are used to develop the procedures and to assess the safety and efficacy of various quanta of cell doses.
Figure 2.

Trial Design of Cohort 2
Table 1.
Inclusion and exclusion criteria
| Inclusion criteria |
|---|
| Men and non-pregnant women |
| Diagnosis of T1D for a minimum of 5 years |
| Hypoglycemia unawareness (Clarke score) or significant glycemic lability |
| Stable diabetic treatment |
| Willingness to use a continuous glucose meter |
| Acceptable candidate for surgical implantation |
| Exclusion criteria |
| History of islet cell, kidney, and/or pancreas transplant |
| Six or more severe, unexplained hypoglycemic events within 6 months of enrollment |
| Uncontrolled or untreated thyroid disease or adrenal insufficiency |
| Diabetic complications such as severe kidney disease or renal dysfunction, proliferative retinopathy, foot ulcers, amputations, and/or severe peripheral neuropathy |
| Detectable stimulated serum C-peptide during the screening perioda |
C-peptide was assessed at two separate screening visits.
Cell-based therapy may ultimately offer a more effective alternative to injected insulin. Over the past 20 years, islet transplantation has provided important proof-of-concept that restoring internally regulated insulin and glucagon production is a safe and effective treatment option for selected subjects with difficult-to-control T1D. Islet cell transplantation with concomitant immunosuppression can lead to clinically relevant endogenous insulin secretion.1,2,3 A few islet allograft recipients have maintained insulin-independence for over 20 years duration, but most return over time to needing minimal-to-moderate exogenous insulin administration. The most durable benefit of islet transplantation has been elimination of severe hypoglycemia while normalizing hemoglobin A1c (HbA1c) in high-risk subjects.2,3
However, cadaveric islet cell transplants are not approved for routine clinical use in the US. Extracting islets from donor pancreas organs is a complex, highly skilled, and unreliable process not available in most medical centers. Moreover, the small potential supply of human organ donors is so markedly imbalanced with the high incidence and prevalence of diabetes that a therapy relying solely on organ donors would not be sustainable even if islet transplant therapy were to become standardized, approved, and reimbursed.
We set out to address the limitations of cadaveric islet transplantation by harnessing the potential of pluripotent stem cells (PSCs) to generate a renewable supply of cells for islet replacement.4 The distinct advantages of PSCs are that they can be differentiated into any cell type, including pancreatic islet endocrine cells, and can be propagated to the desired biomass prior to directed differentiation.5,6,7 Further, consistent and reproducible manufacturing can be performed as needed under clinically compliant current good manufacturing practices (cGMP) with robust quality control. Thus, human PSCs allow for a potentially unlimited, large-scale supply of replacement human islet-like cells.7,8,9
Extensive preclinical work has been performed on PSC-derived pancreatic endoderm cells, such as the PEC-01 cell population, including multipotent pancreatic progenitor cells and immature endocrine cells; these have proven to be robust in culture and handling and to mature in a predictable and consistent fashion into functional islets, including glucose-responsive, insulin-secreting β cells and glucagon-secreting α cells, without off-target effects, following implantation in rodents.7,10,11,12,13 Pancreatic endoderm cells have also been shown to function effectively in rodents when delivered subcutaneously in macroencapsulation devices.13,14,15,16
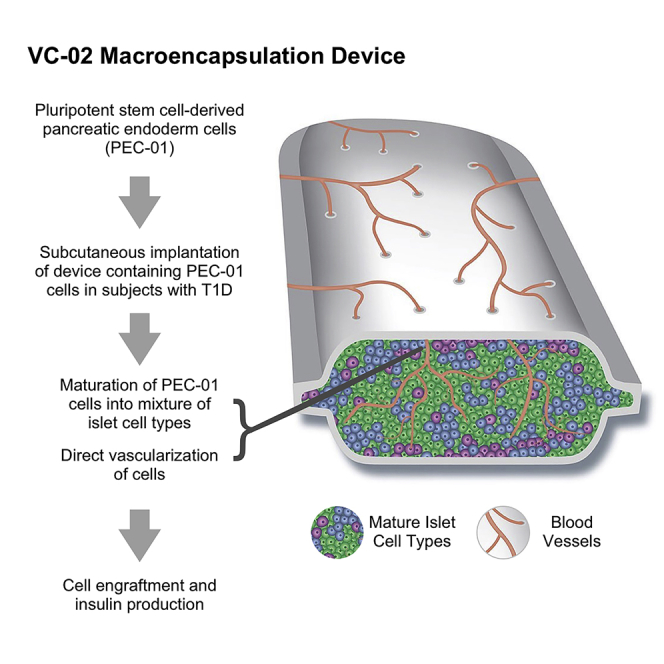
In the present study and accompanying paper,17 we show the success of an investigational biologic product capable of producing C-peptide when implanted successfully. C-peptide is produced by the pancreas as a byproduct of the formation of insulin but has a longer half-life in the body. These preliminary results are from an ongoing first-in-human, open-label phase 1/2 clinical trial where human PEC-01 pancreatic endoderm cells were implanted subcutaneously in subjects with T1D, utilizing an implantable delivery device that allows for direct vascularization of the cells. (Figures 1 and 2; Table 1) The device utilized here is conceptually and physically similar to that used in published preclinical studies as well as in another related clinical trial (NCT02239354, https://clinicaltrials.gov/ct2/show/NCT02239354; the “Encaptra’ immunoprotective macroencapsulation device, which includes a cell-impermeable membrane). However, unlike Encaptra, the device used in the present trial has ports engineered in the membrane to allow the subject’s vasculature to grow into the lumen and directly vascularize the engrafted cells. The intention of direct vascularization is to enhance cell survival through improved oxygenation and metabolic exchange, recognizing that the device in this form would not offer immunoprotection. The product candidate comprising PEC-01 delivered in an open device is termed VC-02 combination product (also known as “PEC-Direct”). Due to the lack of physical immunoprotection, trial subjects were given pharmacological immunosuppression to prevent allo- and auto-immune rejection. Subjects who became C-peptide positive are referred to as “responders” and subjects who remained C-peptide negative are referred to as “non-responders.”
Results
The 17 clinical trial participants described here were negative for baseline C-peptide and received an identical immune suppression regimen. These 17 participants (9 male, 8 female) had ages ranging from 22 to 57 years and had had diabetes for 8 to 52 years. The subject population is further described in Table 2. Because the trial is still in progress, results from a subset of subjects are reported here. These results are from consecutive subjects in Cohort 2 of the ongoing trial and from their first 12 months of implant. Participation of some subjects was terminated early at around 12 months post-implant based upon a risk-benefit analysis, while others have reached longer time points, including 24 months post-implant; accordingly, more information is provided for these subjects.
Table 2.
Patient demographics upon enrollment
| Parameter (allowed; units) | Median (range) |
|---|---|
| Age (18-65 years) | 47 (22–57) |
| Gender (male/female) | N/A (9 M/8 F) |
| Weight (kg) | 79.5 (53.5–104) |
| Years T1D (>5 years) | 37 (8–52) |
| Insulin usage (units/day) | 36.5 (11.1–96.6) |
| Glucose AUC (ng/mL) | 940 (314a–1388) |
| HbA1c (< 10%) | 7.1 (5.9–9.0) |
| Hypoglycemia unawareness (Clarke score >4) | 5 (4–7) |
| Time in range % (70–180 mg/dL) | 60 (37–77) |
| Time above range % (>180 mg/dL) | 34 (19-61) |
| Time below range % (<70 mg/dL) | 5 (1–15) |
| Hypoglycemic events (patient-reported <70 mg/dL; 7-day total) | 2 (0–17) |
One subject’s glucose values were available for analysis only through t = 90 min for the 4 h MMTT.
Engraftment and insulin expression were observed in 63% of VC-02 units explanted from trial subjects at time periods from 3 to 12 months post-implant. Statistical analyses indicate that subjects who had units with substantial cell engraftment were more likely than those without this feature to have glucose- and/or mixed meal-responsive C-peptide measured in their blood at time periods from 6 to 24 months post-implant.
Safety measures
For the 17 subjects, in the initial 12 months post-implant, there were 297 adverse events (AEs) recorded (224 were mild, 59 moderate, 10 severe, and 4 life-threatening). Causality assessments by investigators concluded that 83 AEs (27.9%) were possibly related to the surgical procedures (implant or explant) and 100 AEs (33.7%) were possibly related to immunosuppression, and 6 AEs (2%) were possibly related to the investigational product. Seven of 10 AEs that were reported as severe in intensity (2.4%) were determined by the investigators to be possibly related to VC-02 and included device protrusion (3 events), post-operative pain (2 events), swelling at incision sites (1), and post-procedural numbness (1). In addition, of the 10 serious adverse events (SAEs) occurring in 6 subjects, 5 were severe hypoglycemia, and the other 5 were uterine leiomyoma, parvovirus B19 infection, liver abscess, colitis, and pleural effusion. None of these SAEs were considered to be related to VC-02 as determined by investigators after further evaluation and are consistent with the preclinical safety data.
Primary efficacy endpoint: C-peptide measurements
To have the greatest opportunity to detect functional engraftment of implanted combination products, stimulated human C-peptide response to a mixed-meal tolerance test (MMTT) prior to enrollment was used as a sensitive measure intended to reveal any residual endogenous β cell function. To be eligible for study inclusion, subjects had to demonstrate undetectable circulating C-peptide (below the limit of quantitation of < 0.1 ng/mL) prior to implant during the MMTT. As observed in preclinical studies, when implanting a progenitor cell population, MMTT and simplified meal challenges (sample collection at 0 min and at 60–90 min) performed in the initial few months following implantation continued to reveal no detectable circulating C-peptide at fasting or upon stimulation. Subsequently, a total of 6/17 subjects (35.3%) demonstrated stimulated C-peptide (>0.1 ng/mL) at varying time points as early as 6 months post implant. Table 3 provides sequential peak C-peptide measurements at multiple clinical visits, after glucose challenge through a MMTT. At 1–5 subsequent visits (to the visit where C-peptide was first detectable), a group of individual subjects showed positive C-peptide measurements during a MMTT. These C-peptide-positive subjects all presented with detectable engraftment as assessed by standard histology of hematoxylin and eosin-stained slides and expression of insulin as assessed by immunohistochemistry on explanted units, as shown in the two right-hand columns of Table 3. Graft-derived cell viability and insulin expression were lower among explants obtained from C-peptide negative subjects.
Table 3.
Summary of stimulated C-peptide and histology analysis of graft explants
| Pre-implant | Post-implant |
Histology (Number of insulin-positive graft cells per section) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Maximum stimulated [C-peptide] (ng/mL) | ||||||||||
| Subject | Screeninga | 6 mb | 9 mc | 12 mb | 15 mc | 18 mc | 21 mc | 24 mb | 3 m | 9-12 m |
| A-006 | C-peptide negative |
BQL | BQL | 0.3 | 0.2 | n/a | TBD | TBD | 202 | 25 |
| B-004 | BQL | BQL | 0.1 | BQL | 0.2 | TBD | TBD | 758 | 20 | |
| D-001 | 0.1 | BQL | 0.1 | TBD | TBD | TBD | TBD | 461 | 1419 | |
| E-002 | 0.1 | OUT | OUT | OUT | OUT | OUT | OUT | 56 | 387 | |
| E-006 | BQL | 0.1 | 0.2 | ND | 0.2 | 0.2 | 0.2 | 70 | 132 | |
| E-007 | BQL | 0.1 | 0.2 | 0.2 | 0.2 | TBD | TBD | 346 | 1123 | |
| p value Responders (N = 6) versus Non-Responders (N = 11) | 0.0348 | 0.0514 | ||||||||
| A-001 | C-peptide negative | BQL | BQL | BQL | OUT | OUT | OUT | OUT | 4 | 17 |
| A-002 | BQL | BQL | BQL | OUT | OUT | OUT | OUT | 18 | 43 | |
| A-003 | BQL | BQL | BQL | OUT | OUT | OUT | OUT | 2 | 41 | |
| A-004 | BQL | BQL | BQL | OUT | OUT | OUT | OUT | 5 | ND | |
| A-005 | BQL | BQL | BQL | OUT | OUT | OUT | OUT | 38 | 25 | |
| B-003 | BQL | BQL | BQL | OUT | OUT | OUT | OUT | 19 | 34 | |
| C-001 | BQL | BQL | BQL | OUT | OUT | OUT | OUT | 75 | 1 | |
| D-002 | BQL | BQL | BQL | OUT | OUT | OUT | OUT | 2230 | ND | |
| E-003 | BQL | BQL | BQL | OUT | OUT | OUT | OUT | 46 | 57 | |
| E-005 | BQL | BQL | BQL | OUT | OUT | OUT | OUT | 227 | 1 | |
| E-008 | BQL | BQL | BQL | OUT | OUT | OUT | OUT | 72 | 0 | |
Data from the 17 subjects in Cohort 2 are shown; the six subjects who exhibited de novo stimulated C-peptide measurements (peak measurement in ng/mL) are sorted to the top of the table.
Letters A-E in the subject number represent different clinical sites.
The same C-peptide ELISA analysis was performed for all samples. Histology of explanted units from these six subjects corresponds with the graft-derived C-peptide measurements shown in the two right-hand columns. Histology is based on the number of insulin-positive cells counted per graft section; KEY: BQL, below quantifiable limit; OUT, subject no longer in trial; ND, not determined; TBD, to be determined.
Data from the 17 subjects in Cohort 2 are shown; the 6 subjects who exhibited de novo stimulated C-peptide measurements (peak measurement in ng/mL) are sorted to the top of the table. The different letters in the subject identifier represent different clinical sites. The p value was determined by the Wilcoxon-Mann-Whitney test.
Glucose challenge test varied at different time points: simplified oral glucose challenge at screening;
4 h MMTT at pre-implant V2 and at 6-, 12-, and 24-month post-implant visits;
2 h MMTT at the 9-, 15-, 18-, and 21-month visits.
In 71% of the explant procedures (24/34 overall) performed at either 3 months or at 9-12 months, more than one sentinel unit was removed from the subject. When multiple units were removed, the histology scoring was determined by the average number of cells staining positive for insulin across multiple explanted units. There was a distinct subset of subjects showing circulating C-peptide, classified as responders (n = 6), who demonstrated glucose-responsive C-peptide (increase in C-peptide after increase in blood glucose) after an MMTT.17 Explants from these subjects contained greater numbers of graft-derived, insulin-staining cells. Conversely, explants from non-responders (n = 11), who did not show detectable C-peptide measurements, were observed to have fewer graft-derived β cells. Statistical analysis indicates significantly more graft cells staining positive for insulin at 3 months when comparing responders to non-responders (Table 3). Insulin content and secretion per β cell increased with implantation duration, suggesting that the accumulation of β cells and/or their maturation may contribute to the delayed appearance of detectable C-peptide.
Histology of explanted units
As represented in Figure 3, Figure 4, and Figures S1 and S2, at least one sentinel explanted between 3 and 12 months from 15/17 subjects in the trial showed detectable engraftment, with expression of insulin in at least 30 graft-derived cells per histological section shown via immunohistochemical staining. Importantly, explanted dose-finding units showed proportionally similar histological results to sentinels at the respective time points and within a subject.
Figure 3.

Histology of cross-sections of a sentinel explanted from “Subject D-001” with glucose-responsive C-peptide illustrates both the cellular and non-cellular regions of a mature graft
(A–C) Three serial cross-sections (B) of approximately 4 mm apart within the explant stained with hematoxylin and eosin (H&E) reveal cellular regions (purple, hematoxylin-stained, enlarged in C) and non-cellular regions (pink, eosin-stained, enlarged in A). The non-cellular regions consist of collagenous fibers corresponding to regions of infiltrating host myofibroblasts observed at earlier time points (see Figure S2).
(D–G) Panels (D)–(G) show immunohistochemistry of the cellular region identified in (C). Chromogranin A-staining shows the vast majority of the graft consists of endocrine cells (D). Staining for alpha smooth muscle actin (αSMA) identifies host-derived myofibroblasts (E), especially in the bottom left within the device lumen, and in larger blood vessels, but not or only weakly so smaller capillaries. Glucagon (brown) and insulin (magenta) staining account for the majority of graft-derived endocrine cells, glucagon being more abundant (F). Staining for CD34 identifies small capillaries as well as larger blood vessels (G). Note paucity of CD34 staining in the region devoid of graft-derived cells (bottom left within device lumen).
Counterstain in all IHC panels is hematoxylin. Scale bars, 2.5 mm (top center) and 250 μm (all other images).
Figure 4.

Histology of cross-sections from an explanted dose-finding unit at approximately 11 months from “Subject E-002”
Several panels illustrate the histological features routinely observed in the cellular regions of VC-02 grafts. Shown are “nearly adjacent” sections. Gray strips seen at the top and bottom of each image are the delivery device membranes.
(A) H&E staining reveals densely packed cells reminiscent of human endocrine islet morphologies, albeit without the macro-anatomical “islet” nature.
(B) Nearly all graft-derived cells are endodermal in nature as demonstrated by nuclear staining to the transcription factors FOXA2 and CDX2. The open structure lined with endodermal cells, exemplified in the lower left of each image, is not a prominent feature but observed occasionally in the cellular regions of the grafts.
(C) A subset of graft-derived cells coincident with insulin-expressing cells show nuclear immunoreactivity for NKX6-1, a transcription factor specific to β cells in mature human islets.
(D) The cellular regions are well vascularized with microcapillary structure, demonstrated with the endothelial cell marker CD34 (magenta). CD3 staining (brown) marks T cells, not abundant but observed occasionally outside the device lumen, but rarely inside the lumen among graft-derived cells.
(E) Many cells show glucagon (GCG) immunoreactivity.
(F) Many cells show insulin (INS) immunoreactivity in regions coincident with NKX6-1 expression. Scale bars, 250 μm. Counterstain is hematoxylin.
By 12 weeks post-implant, graft-derived cells were organized structurally into clusters primarily with endocrine cell morphology and expressing, in order of abundance, glucagon, insulin, or somatostatin (Figure S1). In total, chromogranin A-staining endocrine cells accounted for 66% of graft-derived cells. Host lymphocytes, exclusively T cells identified through CD3 expression with B cells excluded through the absence of CD22 staining, were sporadically observed around the external periphery of the units, with T cells rarely detectable among graft cells within the unit lumen. Host cells were fully integrated into the devices, with a thin fibrous tissue capsule established around the implant.
Blood vessels were observed in the tissue capsule and within the device lumen, indicating they traversed through ports in the membrane. Blood vessels were identified through staining with CD34 (Pusztaszeri et al., 2006),18 marking the luminal side vascular endothelial cells of larger vessels as well as capillaries, and with alpha smooth muscle actin (αSMA) staining larger vessels but only weakly detectible in capillaries (Figure 3 and Figure S2). Non-vascular host cells were also observed inside the unit lumen. By histological phenotype, many of the infiltrating cells of host origin were characterized as myofibroblasts associated with the wound healing response and also staining positive for αSMA (Figure 3 and Figure S2). While graft-derived cells were detected in the majority of explants, host-derived fibroblasts predominated within the lumen of nearly all explants, with graft-derived cells constituting on average just 40% of total cells among explants from responders and 26% in explants from non-responders. Within regions of the device lumen where host-derived fibroblasts were dominant, graft-derived cells were rare or absent (Figure S2). Host cells could be distinguished from graft cells in female subjects by their absence of Y chromosome-specific FISH, which positively marks the implanted cells of CyT49 male origin. Additional histological analyses are described in the accompanying manuscript by Ramzy et al.17
At one year, by design of the protocol, subjects without measurable C-peptide during the MMTT were withdrawn from the study, and all units explanted and immunosuppression discontinued after explantation. If the units had not engrafted, matured, and yielded C-peptide by this point, it was believed it would be highly unlikely they would acquire robust function subsequently. When balancing the declining possibility of graft function against the risks and demands of trial participation for the second year of the 17 subjects described herein (including immunosuppression), 11 were terminated soon before or at one year post-implant; these are the subjects noted “below the line’ in Table 3.
Characterization of units explanted at 9 months to 1 year after implant are shown in Figure 3 and Figure 4. Blood vessels were observed traversing the capsule, against the external aspect of the unit membranes, and within the lumen. A robust matrix of blood vessels was elaborated in the regions of the device containing graft cells of endocrine profile, but not in regions where graft-derived cells were rare. Based on CD34-staining quantitation, areas of the device lumen containing graft-derived cells had 10-fold higher blood vessel density compared to non-cellular areas of the device (Figure 3). Two major regions could be characterized inside the lumen: cellular and largely non-cellular. Units showed different proportions of these two regions. Figure 3 illustrates the two regions in “Subject D-001,” who showed glucose-responsive C-peptide at several time points. For “Subject E-002,” a dose-finding unit examined at approximately 1 year (11 months) appeared similar to findings with sentinels at the same time (Figure 4).
Secondary efficacy measures
When considering responders and non-responders, no statistically significant differences were observed comparing pre- and post-implant measures, including changes in glycemic parameters (time-below-range, time-in-range, time-above-range, HbA1C, insulin dosing; data shown in Table S1).
Discussion
Glucose-responsive insulin secretion as captured by de novo C-peptide production was observed in T1D subjects implanted with PSC-derived cells. Similar findings were subsequently noted, as described in the accompanying paper.17 This phenomenon has previously been described in animal models using PSC-derived pancreatic progenitors8,10,11,12,13,14,15,16 and in the somewhat analogous clinical context of cadaveric islet transplants with immunosuppression.1 However, this report is a clinical observation of the nexus, whereby a scalable source of cells (PSC-derived pancreatic progenitors) is used to generate implantable material that engrafts and matures into glucose-responsive insulin-secreting tissue. The progressive accumulation of functional, insulin-secreting cells occurs over a period of approximately 6–9 months from the time of implant. However, once a subject shows positive C-peptide, this appears to be durable, and correlates clearly with survival and differentiation of human insulin-producing cells over time. Although one subject at one time point appears to have dipped below the limit of quantitation, every subject who showed circulating C-peptide at some point also showed it upon a subsequent MMTT, and all still had detectable C-peptide upon their most recent MMTT. To further support these data, we now have evidence of higher levels of glucose-responsive C-peptide with clinically relevant glycemic improvements (including time-in-range, HbA1C, and insulin dosing) as seen in a subsequent subject in VC-02 from a later group of patients with a device with a different configuration.19
The previous related clinical study testing PSC-derived pancreatic endoderm cells (PEC-01 cells) for T1D in a fully encapsulating device (VC-01 trial NCT02239354 [https://clinicaltrials.gov/ct2/show/NCT02239354]) showed that (1) such a product candidate can acquire neovasculature and (2) PEC-01 cells mature to insulin-expressing β cells in human subjects with T1D.20 Neither of these observations was a foregone conclusion. T1D is associated with impaired wound healing, which might also manifest as an inability to generate neovasculature, particularly in the subcutaneous location. Although PEC-01 and other pancreatic progenitor cell populations are now well known to mature into functional human islet tissue in animals, including T1D models, it was possible the same might not hold true for implants in human T1D subjects. Both of these critical elements of the general approach—vascularization and maturation into functional endocrine islet tissue in subjects with T1D—were again demonstrated using the VC-02 paradigm.
Beyond the surgical implantation and explant procedural risks and side-effects of immunosuppression, which together accounted for the great majority of the mild-to-moderate adverse effects, the VC-02 product candidate in general appears to be safe and well-tolerated. Importantly, there were no safety concerns reported as a result of the biologic activity of VC-02 with respect to off-target growth; specifically, there were no teratomas seen within the units, nor was there evidence of metastatic growth observed outside of the containment devices. Moreover, we observed no HLA sensitization (based upon lack of clinically relevant, de novo reactivity to donor-specific antigens post-implant), no more frequent or severe hypoglycemia secondary to unregulated insulin or other hormonal secretion, and no other untoward effects. While immunosuppression is an additional challenge for a T1D subject to endure, for those experiencing hypoglycemia unawareness or glycemic lability, the risk-benefit balance could well tip in favor of using such a product, if this enables better glycemic control and fewer life-threatening severe hypoglycemic events. Despite potent systemic immune suppression, multiple surgical implantation sites, and the presence of foreign materials, the risk of local infection was exceedingly low, suggesting that this approach is well tolerated in subjects with T1D who are at risk for a poor healing response.
In summary, the present study demonstrates, in a small number of human subjects with T1D, that PSC-derived pancreatic progenitor cells have the capacity to survive, engraft, differentiate, and mature into human islet-like cells when implanted subcutaneously. These cells demonstrate strong insulin staining and produce measurable glucose- and meal-responsive C-peptide production in peripheral blood. Induction and maintenance immunosuppression appear to be effective in preventing allogeneic and autoimmune destruction of the graft cells, at least in this subset of subjects. However, immune sensitization from non-compliance with immunotherapy could potentially lead to a decreased stem cell survival, leading to graft failure. Investigations are underway to access how to best promote graft vascularization and survival, reduce the infiltration of host-derived fibroblasts, and eliminate the need for immunosuppressive therapy.
Limitations of the study
Despite the measurable C-peptide observed in the responder subjects, no statistically significant measurable clinical benefit has yet been observed in this pilot study. This lack of therapeutic response is most likely attributable to insufficient numbers of engrafted PSC-derived pancreatic progenitor cells, i.e., insufficient β cell mass and the need for further optimization of the device and/or administration method. One of the goals of the trial is to understand how to effectively deliver the cells in a way that achieves maximal engraftment such that the dosing requirements can be determined for future and ongoing iterative trials. While approximately 30% of the subjects exhibited some regions of the device lumen with notable graft cells at 3 and 12 months post-implant, every graft explanted also contained substantial regions of non-cellular material, consistent with noted graft-to-graft variability. These regions are most likely collagen and other extracellular material produced by the infiltrating host myofibroblasts observed at earlier time points. The infiltration of myofibroblasts and their secretion of extracellular matrix may be an impediment for cellular engraftment, given that (1) large regions of extracellular matrix are not observed in fully encapsulated VC-01 explants (into which no host cells can enter), and (2) the non-cellular regions are observed closest to the ports where there is the greatest access of host cells to the lumen.
It is most likely that the appearance of glucose-responsive C-peptide in some of the subjects was related to the implanted cells and not to other manipulations, in particular the addition of an immunosuppressive regimen per se. While there is some suggestion that immunosuppression might dampen autoimmunity in T1D, and therefore create an opportunity for the endogenous β cell population to recover, attempts to demonstrate this principle clinically have not been successful. In the accompanying paper,17 subjects implanted with PEC-01 cells showed demonstrable C-peptide levels prior to explant with levels disappearing post-explant. This provides strong evidence that the observed C-peptide levels are derived from the graft cells and are not due to the stimulatory effect of the immunosuppression regimen.
Clinical islet recipients also represent an important control for this possibility, as we have not observed incremental increases in C-peptide production over time in failing grafts while identical maintenance immune suppression is continued in subjects with T1D (unpublished data). Moreover, the correlation observed in the current study between C-peptide detection and histological evidence of islet-like differentiation is further compelling evidence that the implanted cells are effectively stimulated by circulating glucose levels and release insulin in response (see Table 3).17
We do not have data on the number or percentage of ductal and acinar cells in the histological sections shown in the supplementary figures. However, the accompanying paper from Ramzy et al. 17 describes data that show the majority of graft cells to be endocrine, with a minority of ductal and very rare exocrine cells. Additional cellular analysis, including more detailed analysis of pancreatic cells and non-endocrine cells such as vascular cells and fibroblasts, would have been helpful, and will be addressed in future.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-alpha smooth muscle actin (ACTA) | Abcam | ab125044 |
| Mouse monoclonal anti-CD22 | Leica | PA0249 |
| Mouse monoclonal anti-CD3 | Leica | PA0553 |
| Mouse monoclonal anti-CD34 | Leica | PA0212 |
| Rabbit monoclonal anti-CDX2 | Abcam | ab76541 |
| Rabbit monoclonal anti-Chromogranin A | Abcam | ab68271 |
| Rabbit monoclonal anti-FOXA2 | Abcam | ab108422 |
| Mouse monoclonal anti-Glucagon | Sigma | G2654 |
| Rabbit monoclonal anti-Insulin | Cell Signaling Technology | 3014S |
| Goat polyclonal anti-NKX6-1 | LSBio | LS-C124275 |
| Critical commercial assays | ||
| C-Peptide | ADVIA Centaur and ADVIA Centaur XP systems. | 03649928 |
| Experimental models: Cell lines | ||
| Endoderm cells | Schulz et al., 2012 | PEC-01, pancreatic |
| Human embryonic stem cells | Ethically consented embryo, NIH Registration Number 0041 | CyT49 |
| Software and algorithms | ||
| Image analysis software | Definiens | Tissue Studio |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to the lead contact, Howard Foyt, MD, PhD, FACP (HFoyt@viacyte.com).
Materials availability
Reagents utilized in this study that are not commercially sold will be made available by the corresponding author and shared upon reasonable request (consideration will be given to limited quantity and availability of suitable alternatives). The proprietary device used in this study is still at investigational stages and cannot be provided to external parties.
Study design and subject details
The first-in-human Phase 1/2, ongoing open-label study called “A Safety, Tolerability, and Efficacy Study of VC-02™ Combination Product in Subjects with Type 1 Diabetes Mellitus and Hypoglycemia Unawareness” has been conducted at 7 centers in North America and recently expanded to include 1 center in Belgium (clinicaltrials.gov NCT03163511). Long-standing T1D subjects were selected based on confirmation of hypoglycemia unawareness and/or glycemic lability as part of stringent selection criteria (Table 1). All subjects signed the informed consent, and all study sites were approved by an Institutional Review Board. Subjects were both genders (9 male, 8 female), ages ranged from 22-57 years and had had diabetes for 8-52 years. A Clarke score 34 confirmed subjects qualifying for study participation on the basis of hypoglycemia unawareness (Clarke et al., 1995).1 The primary efficacy endpoint was the measurement of C-peptide in response to a Mixed-Meal Tolerance Test (MMTT) and simplified meal challenges (sample collection at 0 min and at 60 to 90 min) as a measure intended to indicate implanted progenitor cell function. Primary outcome measures were incidence of all AEs reported for subjects in the first cohort, and change in C-peptide from baseline for subjects in the second cohort.
Inclusion criteria specified men and non-pregnant women, diagnosis of T1D for a minimum of five years, hypoglycemia unawareness (Clarke score) or significant glycemic lability, stable diabetic treatment, willingness to use a continuous glucose meter, and being an acceptable candidate for surgical implantation. Exclusion criteria included history of islet cell, kidney, and/or pancreas transplant; 6 or more severe, unexplained hypoglycemic events within 6 months of enrollment; uncontrolled or untreated thyroid disease or adrenal insufficiency; diabetic complications such as severe kidney disease or renal dysfunction, proliferative retinopathy, foot ulcers, amputations, and/or severe peripheral neuropathy; or detectable stimulated serum C-peptide during the screening period (C-peptide was assessed at two separate screening visits) (Table 1).
Subjects were enrolled into Cohort 1 to establish the initial safety profile of the product candidate. A total of 30 additional subjects were then implanted to initiate evaluation of product efficacy in Cohort 2 by the time of the manuscript data cut. Of these subjects, 19 had reached a minimum of 9-12 months of study participation (or had withdrawn from the study). Preliminary results from 17 of 19 subjects in Cohort 2 of the trial, who were confirmed as C-peptide-negative prior to implant, and received the same immunodepletion regimen, are reported here. Of the 2 subjects not included in these results, 1 had measurable C-peptide prior to implant; the other received basiliximab as opposed to ATG for immunodepletion. These two subjects are excluded from these analyses to describe results from a more uniform cohort (Table 1).
Manufacturing test article
PEC-01 cells were manufactured from CyT49 PSCs as described and cryopreserved to facilitate lot release prior to combination product manufacture.8 PEC-01 is a cell population largely comprised of pancreatic progenitor and immature endocrine cells manufactured via directed differentiation of hESC. The CyT49 hESC line was derived by ViaCyte from an ethically-consented embryo in a process compliant with governing regulations and is the starting material for cell product manufacture. It is the only cell line being handled in the GMP manufacturing facility, and the cell line identity is assured via the quality process, batch records, and labeling. Tiered Master and Working Cell Banks have been qualified. PEC-01 is manufactured during a 28-day process involving expansion of hESC-, aggregation into suspension culture, directed differentiation to highly enriched pancreatic endoderm cells followed by harvesting and cryopreservation. Quality control testing and release of a PEC-01 lot are performed in the 2 to 3 weeks after each lot is cryopreserved.
The delivery devices were comprised of a multi-layer ‘sandwich’ including (1) a semi-permeable membrane made of expanded polytetrafluoroethylene with engineered portals, and (2) an external polyester mesh to provide rigidity.1 PEC-01 cells were thawed and cultured for 3 days to re-establish robust viability, formulated in enriched media, and loaded into devices with media via access tubing, which was subsequently sealed and trimmed before final aseptic packaging of the VC-02 units for shipment and implant.
Implanted VC-02 units
Implanted units were either ‘sentinel’ units or ‘dose-finding’ units (Figure 1). Both were implanted in one surgery session. Units were explanted at various time points post-implant as described here.
The dose-finding units (300 μL capacity = VC-02-300) are larger units, approximately 3cm x 9cm ‘footprint’ surface area x 1mm thick and are intended to represent quanta of cell dosing. Results from these units help determine how many will eventually be needed for a potentially therapeutic dose. Although there are multiple considerations with cell therapy dosing, especially in this context (progenitors terminally differentiate into endocrine cells post-implant with potential to proliferate, and proliferation may be constrained by the physical device limits), each dose-finding unit is loaded with approximately 75 million PEC-01 cells. Based upon preclinical studies completed at ViaCyte, each unit has the potential to deliver approximately 250,000 islet equivalents upon maturity.
Sentinel units (20 μL capacity = VC-02-20) allow serial ‘sampling’ via explant for characterization at various time points post-implant. Sentinels are small delivery devices with a negligible dose of cells intended to not significantly affect outcomes upon periodic removal, but sufficient to reflect the histological characteristics of the larger units. The ‘footprint’ surface area (X-Y dimensions) of the two unit sizes are different, but the microenvironment (Z-dimension) defining the distance of cells within the device from the subject’s tissue, is analogous. Sentinels are approximately 1.5cm x 1cm in area x 1mm thick, and each is loaded with approximately 6 million PEC-01 cells. As described in ‘Results,’ we found that observations from sentinels did indeed reflect the characteristics of the dose-finding units.
Each subject was implanted with up to 4 dose-finding units in the deep subcutaneous tissues of the anterior and lateral abdominal wall, and up to 6 of the sentinel units placed in the volar forearm or abdominal wall. Sentinels were explanted sequentially for histology at various time points with any and all remaining sentinel units explanted at study completion (up to 24 months). As this is an open-label clinical trial with iterative incorporation of learnings, actual numbers and locations of units varied from subject to subject, depending upon information available at time of implant.
VC-02 units include engineered membrane portals to allow host cells to pass freely into the unit and for the subject’s vasculature to grow into the graft. The portals traverse the entire thickness of the membrane and are large enough bore to allow meaningfully-sized diameter vasculature to ingress, develop, and mature (Figure 1).
Cohort 2
This cohort evaluates the ability to achieve engraftment and demonstrate efficacy while further evaluating the safety of the product (the objective of Cohort 1). Subjects in Cohort 2 have remained implanted for up to 2 years. While sentinels are explanted at various time points, the larger dose-finding units are intended to remain implanted for the full duration of subject participation. Subjects in Cohort 2 received up to 10 units total in 1 surgical implantation session, of which up to 4 were VC-02-300 units and the remainder, sentinels (Figure 2).
Methods details
Mixed meal tolerance test (MMTT)
A 4 h MMTT was conducted during the screening period to establish the pre-implant basal and stimulated C-peptide and glucose levels of each subject, and at specific study visits post-implant in accordance with the primary endpoint. At multiple interim study visits post-implant, C-peptide levels were instead assessed using 2 h MMTTs to limit subject burden. The MMTT could only be started if the subject had been fasting for a minimum of 8 h. Prior to initiating the MMTT, the subject’s fasting glucose was required to be within 70-200 mg/dL to begin. If it was not within range after repeat testing, the subject was requested to return to the site within visit window to repeat the MMTT. During the MMTT, subjects were allowed to continue exogenous basal insulin; however, short-acting insulin was held for 4 h before starting the test. A 4 h MMTT was also performed at month 12.
Blood glucose monitoring
Continuous glucose monitoring (CGM) data were acquired through use of study-provisioned Dexcom G4 or G5 m. CGM data analysis was based on the most recent 14-day period prior to each study visit.
C-peptide ELISA
Serum samples taken during MMTTs were sent to a central lab for measurement of C-peptide concentration (ADVIA Centaur® assay from Siemens). The lower limit of quantitation for this assay is 0.1 ng/mL (reference range 0.8-4.4 ng/mL).
Immunosuppressive regimens
To protect implanted cells from allo- and auto-immunity, immunosuppression was used similar to current practice in islet transplantation.1 2 All 17 subjects received depletional induction immunosuppression with anti-thymocyte globulin (ATG: up to 6 mg/kg i.v. over 5 days), tumor necrosis factor-alpha blockade with etanercept (50 mg i.v. pre-implant followed by 25 mg subcutaneous x 3 doses out to 10 days post-implant) followed by maintenance tacrolimus (target 10-12 ng/mL for the first 3 months; 7-9 ng/mL thereafter) and mycophenolate mofetil (up to 2 g per day as tolerated). Tacrolimus levels during the 12 months in responders and non-responders are illustrated in Table S1 (shown as TAC level in the final line of each table).
Histological analyses
Formalin-fixed and paraffin embedded sections of explants were stained on a Leica BOND-III immunohistochemistry instrument according to the manufacturer’s suggestions, using the following antibodies to detect alpha smooth muscle actin (ACTA2, Abcam ab125044); CD22 (Leica PA0249); CD3 (Leica PA0553); CD34 (Leica PA0212); CDX2 (Abcam ab76541); chromogranin A (Abcam ab68271); FOXA2 (Abcam ab108422); glucagon (Sigma G2654); insulin (Cell Signaling Technology 3014S); NKX6-1 (LSBio LS-C124275). Quantitation of insulin-staining cells and CD34-staining density determinations were made with Tissue Studio image analysis software from Definiens.
Quantification and statistical analysis
Statistical analysis
For the histology analysis and the secondary efficacy measures, including changes in glycemic parameters (time-below-range, time-in-range, time-above-range, HbA1C, insulin dosing), the median was used as the measure of central tendency, as the data were not normally distributed, and the non-parametric Wilcoxon Two-Sample test, also known as the Wilcoxon-Mann-Whitney test, was used, with the Kruskal-Wallis p value used for determining statistical significance. Results of the statistical analysis of histology are shown in Table 3.
The data for these 17 subjects were analyzed using SAS 9.4. There were 6 responders and 11 non-responders included in the data analysis. Data from Month 3 and Months 9 or 12 were included in the analysis. If there were multiple observations for a subject at a time point, then the average of those observations was included in the analysis. Normality was assessed using the Shapiro-Wilk test, and visually inspected using Q-Q plots. Both the test and the plots indicated the data violated normality assumptions (i.e., the data were not normally distributed) for the variables that were analyzed. Hence, the comparison of the responders and non-responders was performed using the Wilcoxon Two-Sample test, also known as the Wilcoxon-Mann-Whitney test. A significance level of α = 0.05 was used to determine statistical significance.
Additional resources
Clinical trial registry description: URL https://clinicaltrials.gov/ct2/show/NCT03163511
Funding
Funding sources for this work included ViaCyte, the California Institute for Regenerative Medicine, the JDRF, and the Stem Cell Network of Canada.
Acknowledgments
This work was supported in part by grants from CIRM (CLIN2-09672), JDRF (IDDP), and the Stem Cell Network of Canada. The authors thank Khaled Dajani, Peter A Senior, Kathleen Dungan, and Shumei Meng for their hard work on the trial.
Author contributions
A.M.J.S.: investigation, project administration, writing-original draft and writing-review & editing. D.T.: investigation, project administration, writing-review & editing. T.W.D.: investigation, project administration, writing-review & editing. M.D.B.: investigation, project administration, writing-review & editing. W.H.: investigation, project administration, writing-review & editing. J.P.: investigation, project administration, writing-review & editing. J.W.: investigation (lead surgeon), conceptualization, writing-review & editing. M.D.: conceptualization and methodology, writing-review & editing. R.M.W.: conceptualization and methodology, writing-original draft and writing-review & editing. E.P.B.: supervision, conceptualization and project administration, writing-original draft and writing-review & editing. M.S.J.: project administration, conceptualization and methodology, writing-review & editing. E.J.K.: lead scientist, conceptualization and methodology, writing-review & editing. K.A.D.: supervision, conceptualization and project administration, writing-review & editing. H.L.F.: supervision (medical director), conceptualization and project administration, writing-original draft and writing-review & editing.
Declaration of interests
M.D., R.M.W., E.J.K., E.P.B., K.A.D., M.J., and H.L.F. are employees of ViaCyte and may have equity interest in ViaCyte, Inc., a privately held company. J.S. and J.W. are consultants for ViaCyte and may have equity interest in ViaCyte. ViaCyte has 850 global and US patents. The remaining authors declare no conflicts of interest. Funding sources for this work included the California Institute for Regenerative Medicine, JDRF, and the Stem Cell Network of Canada. These groups were not involved in the research, the writing, or the decision to submit the manuscript for publication. Authors have not been paid by a pharmaceutical company or other organization to write this article.
Published: December 2, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2021.100466.
Supplemental information
C-peptide positive and c-peptide negative subjects, related to the STAR Methods
Data and code availability
Data: Individual deidentified participant data collected and presented in this publication will be made available by reasonable request to the corresponding author. Complete study data including detailed study protocol will be made available by the clinical trial sponsor after trial completion, currently scheduled for 2023. Composite data will be released by the sponsor to clinicaltrials.gov within 12 months of study completion.
Individual deidentified participant data that underlie the results reported in this article alongside the detailed study protocol will be made available indefinitely to researchers who provide a methodologically sound proposal to the clinical trial sponsor.
Code: There is no unique code to report.
General Statement: Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request.
References
- 1.Shapiro A.M., Lakey J.R., Ryan E.A., Korbutt G.S., Toth E., Warnock G.L., Kneteman N.M., Rajotte R.V. Islet transplantation in seven patients with type 1 diabetes mellitus using a glucocorticoid-free immunosuppressive regimen. N. Engl. J. Med. 2000;343:230–238. doi: 10.1056/NEJM200007273430401. [DOI] [PubMed] [Google Scholar]
- 2.Hering B.J., Clarke W.R., Bridges N.D., Eggerman T.L., Alejandro R., Bellin M.D., Chaloner K., Czarniecki C.W., Goldstein J.S., Hunsicker L.G., et al. Clinical Islet Transplantation Consortium Phase 3 Trial of Transplantation of Human Islets in Type 1 Diabetes Complicated by Severe Hypoglycemia. Diabetes Care. 2016;39:1230–1240. doi: 10.2337/dc15-1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lablanche S., Vantyghem M.C., Kessler L., Wojtusciszyn A., Borot S., Thivolet C., Girerd S., Bosco D., Bosson J.L., Colin C., et al. TRIMECO trial investigators Islet transplantation versus insulin therapy in patients with type 1 diabetes with severe hypoglycaemia or poorly controlled glycaemia after kidney transplantation (TRIMECO): a multicentre, randomised controlled trial. Lancet Diabetes Endocrinol. 2018;6:527–537. doi: 10.1016/S2213-8587(18)30078-0. [DOI] [PubMed] [Google Scholar]
- 4.Thomson J.A., Itskovitz-Eldor J., Shapiro S.S., Waknitz M.A., Swiergiel J.J., Marshall V.S., Jones J.M. Embryonic stem cell lines derived from human blastocysts. Science. 1998;282:1145–1147. doi: 10.1126/science.282.5391.1145. [DOI] [PubMed] [Google Scholar]
- 5.D’Amour K.A., Agulnick A.D., Eliazer S., Kelly O.G., Kroon E., Baetge E.E. Efficient differentiation of human embryonic stem cells to definitive endoderm. Nat. Biotechnol. 2005;23:1534–1541. doi: 10.1038/nbt1163. [DOI] [PubMed] [Google Scholar]
- 6.D’Amour K.A., Bang A.G., Eliazer S., Kelly O.G., Agulnick A.D., Smart N.G., Moorman M.A., Kroon E., Carpenter M.K., Baetge E.E. Production of pancreatic hormone-expressing endocrine cells from human embryonic stem cells. Nat. Biotechnol. 2006;24:1392–1401. doi: 10.1038/nbt1259. [DOI] [PubMed] [Google Scholar]
- 7.Schulz T.C., Young H.Y., Agulnick A.D., Babin M.J., Baetge E.E., Bang A.G., Bhoumik A., Cepa I., Cesario R.M., Haakmeester C., et al. A scalable system for production of functional pancreatic progenitors from human embryonic stem cells. PLoS ONE. 2012;7:e37004. doi: 10.1371/journal.pone.0037004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schulz T.C. Concise Review: Manufacturing of Pancreatic Endoderm Cells for Clinical Trials in Type 1 Diabetes. Stem Cells Transl. Med. 2015;4:927–931. doi: 10.5966/sctm.2015-0058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brandon E., Scott M., Zimmerman M., D’Amour K. In: Second Generation Cell and Gene-based Therapies: Biological Advances, Clinical Outcomes, and Strategies for Capitalisation. Vertès Alain A., Smith Devyn M., Qureshi Nasib, Dowden Nathan J., editors. Elsevier; 2020. Stem cell-derived islet replacement: Which cells & how to protect them from the immune system? [Google Scholar]
- 10.Kroon E., Martinson L.A., Kadoya K., Bang A.G., Kelly O.G., Eliazer S., Young H., Richardson M., Smart N.G., Cunningham J., et al. Pancreatic endoderm derived from human embryonic stem cells generates glucose-responsive insulin-secreting cells in vivo. Nat. Biotechnol. 2008;26:443–452. doi: 10.1038/nbt1393. [DOI] [PubMed] [Google Scholar]
- 11.Kelly O.G., Chan M.Y., Martinson L.A., Kadoya K., Ostertag T.M., Ross K.G., Richardson M., Carpenter M.K., D’Amour K.A., Kroon E., et al. Cell-surface markers for the isolation of pancreatic cell types derived from human embryonic stem cells. Nat. Biotechnol. 2011;29:750–756. doi: 10.1038/nbt.1931. [DOI] [PubMed] [Google Scholar]
- 12.Rezania A., Bruin J.E., Riedel M.J., Mojibian M., Asadi A., Xu J., Gauvin R., Narayan K., Karanu F., O’Neil J.J., et al. Maturation of human embryonic stem cell-derived pancreatic progenitors into functional islets capable of treating pre-existing diabetes in mice. Diabetes. 2012;61:2016–2029. doi: 10.2337/db11-1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Motté E., Szepessy E., Suenens K., Stangé G., Bomans M., Jacobs-Tulleneers-Thevissen D., Ling Z., Kroon E., Pipeleers D., Beta Cell Therapy Consortium EU-FP7 Composition and function of macroencapsulated human embryonic stem cell-derived implants: comparison with clinical human islet cell grafts. Am. J. Physiol. Endocrinol. Metab. 2014;307:E838–E846. doi: 10.1152/ajpendo.00219.2014. [DOI] [PubMed] [Google Scholar]
- 14.Agulnick A.D., Ambruzs D.M., Moorman M.A., Bhoumik A., Cesario R.M., Payne J.K., Kelly J.R., Haakmeester C., Srijemac R., Wilson A.Z., et al. Insulin-Producing Endocrine Cells Differentiated In Vitro From Human Embryonic Stem Cells Function in Macroencapsulation Devices In Vivo. Stem Cells Transl. Med. 2015;4:1214–1222. doi: 10.5966/sctm.2015-0079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bruin J.E., Rezania A., Xu J., Narayan K., Fox J.K., O’Neil J.J., Kieffer T.J. Maturation and function of human embryonic stem cell-derived pancreatic progenitors in macroencapsulation devices following transplant into mice. Diabetologia. 2013;56:1987–1998. doi: 10.1007/s00125-013-2955-4. [DOI] [PubMed] [Google Scholar]
- 16.Haller C., Piccand J., De Franceschi F., Ohi Y., Bhoumik A., Boss C., De Marchi U., Jacot G., Metairon S., Descombes P., et al. Macroencapsulated Human iPSC-Derived Pancreatic Progenitors Protect against STZ-Induced Hyperglycemia in Mice. Stem Cell Reports. 2019;12:787–800. doi: 10.1016/j.stemcr.2019.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ramzy A., Thompson D.M., Ward-Hartsonge K.A., Ivison S., Cook L., Garcia R.V., Loyal J., Kim P.T.W., Warnock G.L., Levings M.K., Kieffer T.J. Implanted pluripotent stem cell-derived pancreatic endoderm cells secrete glucose-responsive C-peptide in subjects with type 1 diabetes. Cell Stem Cell. 2021;28:2047–2061. doi: 10.1016/j.stem.2021.10.003. [DOI] [PubMed] [Google Scholar]
- 18.Pusztaszeri M.P., Seelentag W., Bosman F.T. Immunohistochemical expression of endothelial markers CD31, CD34, von Willebrand factor, and Fli-1 in normal human tissues. J. Histochem. Cytochem. 2006;54:385–395. doi: 10.1369/jhc.4A6514.2005. [DOI] [PubMed] [Google Scholar]
- 19.Keymeulen B., Jacobs-Tulleneers-Thevissen D., Sr., Kroon E.J., Jaiman M.S., Daniels M., Wang R., Pipeleers D., D’Amour K., Foyt H.L. 196-LB: Stem Cell–Derived Islet Replacement Therapy (VC-02) Demonstrates Production of C-Peptide in Patients with Type 1 Diabetes (T1D) and Hypoglycemia Unawareness. Diabetes. 2021 doi: 10.2337/db21-196-LB. [DOI] [Google Scholar]
- 20.Henry R.R., Pettus J.H., Wilensky J., Shapiro A.M., Senior P.A., Roep B.O., Wang R.S., Kroon E.J., Scott M.J., D’Amour K.A., Foyt H.L. Initial Clinical Evaluation of VC-01TM Combination Product: A Stem Cell-Derived Islet Replacement for Type 1 Diabetes (T1D) Diabetes. 2018 doi: 10.2337/db18-138-OR. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
C-peptide positive and c-peptide negative subjects, related to the STAR Methods
Data Availability Statement
Data: Individual deidentified participant data collected and presented in this publication will be made available by reasonable request to the corresponding author. Complete study data including detailed study protocol will be made available by the clinical trial sponsor after trial completion, currently scheduled for 2023. Composite data will be released by the sponsor to clinicaltrials.gov within 12 months of study completion.
Individual deidentified participant data that underlie the results reported in this article alongside the detailed study protocol will be made available indefinitely to researchers who provide a methodologically sound proposal to the clinical trial sponsor.
Code: There is no unique code to report.
General Statement: Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request.