

Summary
Resistance to platinum compounds is a major determinant of patient survival in high-grade serous ovarian cancer (HGSOC). To understand mechanisms of platinum resistance and identify potential therapeutic targets in resistant HGSOC, we generated a data resource composed of dynamic (±carboplatin) protein, post-translational modification, and RNA sequencing (RNA-seq) profiles from intra-patient cell line pairs derived from 3 HGSOC patients before and after acquiring platinum resistance. These profiles reveal extensive responses to carboplatin that differ between sensitive and resistant cells. Higher fatty acid oxidation (FAO) pathway expression is associated with platinum resistance, and both pharmacologic inhibition and CRISPR knockout of carnitine palmitoyltransferase 1A (CPT1A), which represents a rate limiting step of FAO, sensitize HGSOC cells to platinum. The results are further validated in patient-derived xenograft models, indicating that CPT1A is a candidate therapeutic target to overcome platinum resistance. All multiomic data can be queried via an intuitive gene-query user interface (https://sites.google.com/view/ptrc-cell-line).
Keywords: carboplatin, ovarian cancer, resistance, CPT1A, fatty acid oxidation, proteomic, oxidative phosphorylation, proteogenomic, reactive oxygen species
Graphical abstract
Highlights
-
•
Multi-omic profiles of platinum-resistant and sensitive ovarian cancer models
-
•
Significant alterations in multiomic profiles after carboplatin exposure
-
•
Oxidative phosphorylation and fatty acid oxidation (FAO) implicated in resistance
-
•
FAO/CPT1A may be a candidate druggable pathway to overcome platinum resistance
Huang et al. report extensive multiomic profiling of preclinical models of high-grade serous ovarian cancer and identify molecular features associated with resistance to standard-of-care, platinum-based chemotherapy. Functional data are presented, demonstrating that CPT1A is a candidate therapeutic target to overcome platinum resistance.
Introduction
Since the 1970s, platinum compounds have been widely used to treat malignancies, e.g., lung, ovarian, head and neck, testicular, bladder, and other cancers.4 Platinum compounds form covalent adducts on DNA, RNA, and proteins.5 Platinum reacts preferentially at the N7 position of guanine and adenine to form intra- and/or inter-strand crosslinks that disrupt DNA transactions (e.g., replication and transcription), leading to DNA strand breaks and cell death.6
Despite initial responses, most tumors develop platinum resistance, associated with poor survival.7 Platinum resistance is multifactorial, involving alterations in drug transporters, detoxification, removal of reactive oxygen species (ROS), DNA repair, oncogenes, metabolic reprogramming, and cell-death pathways.8, 9, 10, 11 Remarkably, despite >30 years of literature on platinum responses in human cancer,12,13 none of these findings is used clinically to stratify patients for platinum resistance or exploited therapeutically to treat platinum-resistant disease.
High-grade serous ovarian cancer (HGSOC) is the most common and lethal epithelial ovarian cancer (OC).14 Standard of care is surgical debulking coupled with platinum-based chemotherapy.15,16 HGSOCs are typically diagnosed at late stage, and tumor response to carboplatin-based chemotherapy is a major determinant of patient survival.17 Although ∼85% of HGSOCs are initially sensitive to platinum-based therapy, most become resistant. The remaining 15% of HGSOCs are refractory to platinum-based therapy at the time of diagnosis, showing no response or even growing through chemotherapy.18,19 Thus, understanding mechanisms of platinum resistance is an urgent clinical goal, both to identify predictive biomarkers of platinum response (to spare patients with resistant tumors futile platinum therapy) and to develop efficacious therapies for platinum-resistant disease.
Limited quantitative proteomic studies have focused on understanding platinum resistance. Li et al.20 identified 28 proteins associated with resistance using the OC COC1/DDP cell line. A 2013 study of genetically engineered mouse mammary tumors indicated upregulation of fatty acid synthesis and metabolism genes in the cisplatin-resistant mouse model.21 A 2017 study identified proteomic differences between cisplatin-sensitive (M019i) and resistant (M019iCis) HGSOC cells, and the results suggested that increased phosphorylation of sequestosome-1 (p62/SQSTM1) was associated with cisplatin resistance.22 In 2018, a study showed that phosphorylation of p38 mitogen-activated protein kinase (MAPK) was increased by carboplatin more markedly in the cisplatin-sensitive OC cell line A2780s than its derivative cisplatin-resistant A2780cp cells.23 A 2020 study identified 48 proteins differentially expressed between A2780 and A2780cp; the glycolysis enzyme Enolase-1 (ENO1) was significantly decreased in the cisplatin-resistant OC cells.24 Most recently, differentially expressed proteins were identified between platinum-resistant OC cell lines (TOV-112D, OVSAHO, and MDAH-2774) and their parental cells, and HSP90 was implicated as a central hub of these protein networks.25 To date, no multiomic profiling of the dynamic response of cancer cells to platinum has been reported.
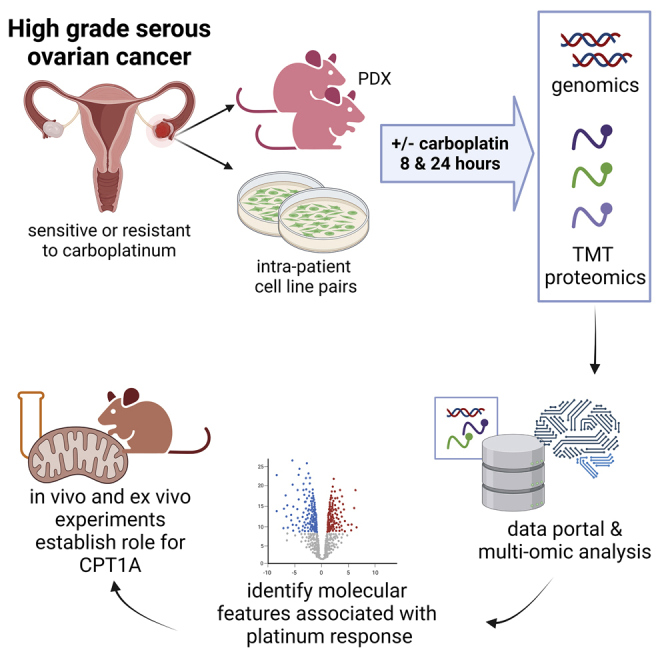
To study mechanisms underlying platinum resistance in HGSOC, we performed comprehensive, dynamic (±carboplatin) profiling of DNA, RNA, protein, and post-translational modifications (phosphorylation, ubiquitination, and acetylation) to identify the cellular networks that respond to platinum treatment and associate with platinum resistance in 3 HGSOC intra-patient cell line pairs (PEA1S/PEA2R, PEO1S/PEO4R, and PEO14S/PEO23R). The cell line pairs were derived from ascites or pleural effusions26 from 3 patients both before (PEA1S, PEO1S, and PEO14S) and after (PEA2R, PEO4R, and PEO23R) their tumors became clinically platinum resistant (i.e., in vivo).27,28 Unlike many HGSOC cell lines,29, 30, 31, 32 PEA1S/PEA2R, PEO1S/PEO4R, and PEO14S/PEO23R have been shown to recapitulate critical aspects of human HGSOCs.18,27,33,34 Genomic analyses revealed that the resistant lines were derived from pre-existing minor clones before chemotherapy, as opposed to a direct linear descent from sensitive cells in response to platinum challenge.27 PEO1S cells carry a germline mutation of BRCA2 (5193C > G (Y1655X)), and the paired PEO4R cells acquired cisplatin resistance by a secondary mutation that restores BRCA2 function.27,35 Follow-up studies indicated that additional factors (e.g., higher expression of HIF1A, MYC, EZH2, DNA-PK, etc.) also contribute to the platinum resistance in PEO4R cells,28,36, 37, 38, 39 and increased activities of HDAC4 and STAT1 may affect platinum responses in all 3 (PEA2R, PEO4R, and PEO23R) resistant cell lines.28 Finally, increased ROS levels and elevated production of interleukin-6 (IL-6) and IL-8 were also reported to be associated with platinum resistance in the PEA2R cell line.40
All molecular profiles in this current study were performed in complete process triplicates (i.e., full biological and technical replication) and can be readily explored via an online portal with an intuitive gene-query user interface (https://sites.google.com/view/ptrc-cell-line).
Results
Overview
PEA1S/PEA2R, PEO1S/PEO4R, and PEO14S/PEO23R intra-patient cell line pairs were exposed (or mock exposed) to 80 μM carboplatin for 8 or 24 h, after which RNA sequencing (RNA-seq) and liquid chromatography-tandem mass spectrometry (LC-MS/MS)-based proteomic profiling of the global (unmodified), phospho, pTyr-enriched, ubiquitinated, and acetylated proteomes was performed (Figure 1A). All experiments were performed in complete triplicates (i.e., biological and technical). Whole-genome sequencing (WGS) was performed for all 6 cell lines.
Figure 1.

Overview of experimental design and results
(A) Cells were treated with 80 μM carboplatin and harvested after 8 or 24 h of treatment. A control sample (“mock”) was treated with vehicle and harvested at 24 h. The experiment was repeated on 3 days (complete process triplicates). Nucleic acids were extracted for RNA-seq whole-genome sequencing (WGS). Protein lysates were generated and digested with trypsin, and global or sub-proteomes were isolated. Samples were TMT-labeled, pooled, and analyzed by LC-MS/MS.
(B) Regression analysis identifies protein and RNA features responsive to carboplatin exposure (Bonferroni-adjusted p < 0.05). Numbers are shown for significantly increased or decreased mRNAs, proteins, or PTMs in response to carboplatin at 8 h and 24 h.
(C) Phosphorylation of Ser1524 of BRCA1 and Ser343 and Ser615 of NBN. Gray line indicates average of 6 samples. ∗adjusted p < 0.05, ∗∗adjusted p < 0.0001, and NS, adjusted p > 0.05.
Proteomic profiling was performed using a tandem mass tag (TMT) isobaric labeling strategy41 for multiplexing (54 samples: 6 cell lines; 3 time points; and 3 complete process replicates) and relative quantification (Figure S1A). 1,503,465 peptides and 55,785 post-translational modifications (PTMs) were observed in the dataset, representing 11,120 proteins (global proteome), 35,357 phosphorylation sites (mapping to 7,073 proteins), 16,555 ubiquitinated sites (mapping to 4,141 proteins), 3,436 acetylated sites (mapping to 646 proteins), and 437 pTyr sites (mapping to 48 proteins; Figure S1B; Table S1). The proteomic results were reproducible. Based on the ratio quantification, across the triplicate complete (biological and technical) replicates, the median %CV ranges between 3.6% and 11.9% (Figure S1C). Additionally, based on the fragments per kilobase of transcript per million mapped reads (FPKM) values (log based), the median (and inter-quartile range) %CV for RNA was 17.8% (7%–39%; Figure S1C). All proteogenomic data can be readily explored via an online portal with an intuitive gene-query user interface (https://sites.google.com/view/ptrc-cell-line), including links to databases providing additional gene and pathway-level information.
Carboplatin induces robust responses
A linear-mixed-effects regression analysis was performed to identify protein and RNA features responsive to carboplatin exposure (i.e., combined analysis of both sensitive and resistant cells; Bonferroni-adjusted p < 0.05; Table S1). The global and phosphoproteome responses were much larger at 24 than at 8 h (Figure 1B). For example, after 8 h of carboplatin exposure, the expression levels of 530 proteins and 570 phosphopeptides were altered, although after 24 h of exposure, the expression levels of 2,158 proteins and 3,022 phosphopeptides were altered. In contrast, the ubiquitinated (2,498 and 2,351 peptides at 8 and 24 h, respectively) and acetylated (425 and 386 peptides at 8 and 24 h, respectively) proteomes, as well as the transcriptome (8,659 and 8,220 transcripts at 8 and 24 h, respectively), showed similar responsiveness at both times. PTMs showed a greater carboplatin responsiveness than the global proteome (Figure S1D). Although RNA expression showed positive correlation with protein expression at baseline (median Spearman correlation = 0.54; Figure S1E), the transcriptional response did not correlate with protein abundance changes in response to platinum exposure (Figure S1F), indicating significant regulation at the post-transcriptional level. Consistent with these findings, hierarchical clustering of the proteomic data is not driven by treatment status, whereas the RNA-seq-based data clusters were driven primarily by ±platinum exposure (Figure S2), perhaps reflecting the fact that the technologies enable detection of more platinum-responsive transcriptome features compared to the proteomes (Figure 1B; Table S1).
DNA damage response (DDR) pathways were upregulated following 24 h of carboplatin treatment in both sensitive and resistant cells, including increased activity of DNA damage checkpoints, DNA replication, replication stress response, and DNA repair, as observed in both the global and phosphoproteome (Table S2). Examples of DNA damage responses include time-dependent phosphorylation of Ser1524 of BRCA1 and Ser343 and Ser615 of NBN (Figure 1C), known targets of the ataxia telangiectasia mutated (ATM) kinase,42, 43, 44, 45 as well as increased ubiquitination of lys561 of FANCD2 and lys523 of FANCI (Figure S3A), indicating activation of the Fanconi anemia pathway.46 Furthermore, we observed increased ubiquitination of ribosomal proteins after carboplatin treatment (Table S2; Figure S3B), extending prior reports that doxorubicin and UV irradiation induce extensive ubiquitination of ribosomal proteins.47,48
Proteasome and spliceosome components showed upregulation (in the global and ubiquitin proteomes and the transcriptome) in response to carboplatin (Table S2). Elevated expression of proteasome proteins and their ubiquitination may be a consequence of carboplatin-induced oxidative stress,49 and spliceosome activity has been reported to be affected by DNA damage and regulated by ubiquitination.50, 51, 52 On the other hand, carboplatin exposure was associated with reduced expression of the cell adhesion and extracellular matrix (ECM) network at both RNA and protein levels (Table S2). Cisplatin has been reported to suppress the expression of ECM proteins (fibronectin, collagens, integrins, etc.) in kidney cells, which contributes to apoptosis and kidney injury in mice.53
To identify kinases responsive to carboplatin, we inferred kinase activity from substrate phosphorylation levels using single sample gene set enrichment analysis (ssGSEA)54 and performed a mixed-effect regression. At 8 h, in addition to ATR and ATM, we detected activation of p38-δ MAPK (MAPK13) and p38 MAPK-activated kinase MAPKAPK2 (MK2) (Figure 2A), consistent with previous reports of p38MAPK/MK2 stress-kinase-pathway-mediated cell cycle checkpoint’s being activated by ATM and ATR in response to DNA-damaging agents.55, 56, 57
Figure 2.

Phosphoproteomic signatures responsive to carboplatin
(A and B) Kinase activity inferred from phosphorylation of its substrates using ssGSEA at (A) 8 h and (B) 24 h of carboplatin exposure.
(C) Kinase activity inferred from phosphorylation of its substrates (red box) and phosphorylation of its activating sites (red circle). Gray circles indicate no activating phosphorylation is available. Black arrows indicate a direct phosphorylating kinase on the activating site, and dashed arrows indicate an indirect relationship.
(D) Log2 fold change of PTM-SEA phosphosite perturbation and pathway signature scores between 24 h of carboplatin exposure and mock-treated cells. The top 10 in each direction are shown. Dashed line indicates an FDR threshold of 0.05.
In line with our observation of larger phosphoproteome responses at 24 h, we detected more activated kinases at 24 versus 8 h, including the DDR checkpoint kinases, p38 and MAPK/JNK pathway members, as well as Cdks, protein kinase Cs (PKCs), PKD, PKA, CK2s, AMPK, and AKT1 (Figure 2B). These changes included the previously unreported activation of CK2, CDC7, and Ca/Calmodulin kinase 2 (CAMK2A) in the HGSOC cell lines in response to carboplatin (Table S3). We also detected downregulation of some kinase activities at 24 h, including HIPK2, PLK1, DYRK1A, CDK5, and ERKs.
To supplement the ssGSEA analysis, we also considered alteration of kinase activity based on phosphorylation of regulatory sites on kinase proteins. Of the 3,022 platinum-responsive phosphopeptides at 24 h, 157 mapped to a kinase. We found increased kinase-activating phosphorylation for PRKD1, MAPK14, ATR, and CHEK1, consistent with the ssGSEA analysis (Figure 2C, red rectangles and circles). Together, these methods identified a robust activation of DDR-related kinases that each phosphorylated the next kinase’s activating site (black arrows, Figure 2C). ATM and ATR have also been linked to indirect activation of AKT158 and MAPK1459 in DNA repair (dashed arrows).
To identify enrichment of phosphosite perturbation and pathway signatures (that can include increased and decreased sites), we performed PTM-SEA to calculate normalized enrichment scores for all samples. There was a significant enrichment of phosphosite signatures related to DNA-damaging ultraviolet and ionizing radiation in cells treated with carboplatin compared to mock-treated cells (Figures 2D and S3C). Additionally, Tie2, AGE/RAGE, and epidermal growth factor receptor (EGFR) pathway signatures were all increased in carboplatin-treated cells at 24 h (Figure 2D).
Differences in carboplatin responses between platinum-sensitive versus resistant cells
We identified individual proteins, PTMs, and RNA features that show differences in response to carboplatin between sensitive and resistant cell lines by jointly analyzing all 3 pairs of cell lines (false discovery rate [FDR] < 0.05; STAR Methods). Differences in the platinum response were more robust after 24 h of exposure compared with 8 h. For example, 23 proteins, 67 phosphosites, 48 ubiquitin sites, and 2 acetylated sites displayed significant differential responses to carboplatin between sensitive and resistant cells after 24 h of exposure (Figure 3; Table S1), as compared to no individual proteins in the global and acetylated proteomes, and only 7 phosphosites and 1 ubiquitin site showing differential responses to carboplatin after 8 h exposure (FDR < 0.05; Figure 3; Table S1). 5 (ENC1, NKTR, PSMA3, SLC39A7, and VAPA) of the 23 proteins whose expression levels showed differential responses (sensitive versus resistant) to carboplatin at 24 h (Figure 3A) showed significantly larger average fold changes (FCs) (defined as the average ratio of protein abundances post-carboplatin treatment over mock treatment) in the sensitive cell lines compared to the resistant lines (FDR < 0.05; Table S1). All 5 proteins showed increased expression in the sensitive cell lines but decreased (or less increased) expression in the resistant cells post-platinum (Figure 3A). The remaining 18 proteins displayed either larger FCs in the resistant cell lines (FDR < 0.05; Table S1; Figure 3A) relative to the sensitive cell lines (8 proteins) or were more downregulated in the sensitive cell lines relative to the resistant cell lines (10 proteins) at 24 h post-carboplatin treatment. 9 of these 18 proteins have been previously reported to be associated with platinum resistance (Table S4A). For example, high expression levels of the small ubiquitin-binding domain (CUE)-containing protein (CUEDC2) contribute to cisplatin resistance through regulation of p38 MAPK signaling.60
Figure 3.

Differential responses to carboplatin treatment between sensitive and resistant cell lines (FDR < 0.05)
(A) Global proteins, (B) phosphosites, (C) ubiquitin sites, and (D) mRNAs. The values depicted by color represent log2 fold change (8 or 24 h and 0 h).
Twenty-eight phosphosites showed larger FCs in the sensitive cell lines (24 h; Table S1; Figure 3B) and are enriched for the Gene Ontology (GO) biological process of “mRNA processing” (FDR = 3.6 × 10−4). Thirty-nine phosphosites (representing 35 proteins) show larger FCs in phosphorylation in the resistant cell lines, and 6 of these proteins were previously reported to be associated with platinum resistance (Table S4B). For example, both Rb Ser249 and Ser807 can be phosphorylated by Cdks or p38 MAPK, two modifications that affect Rb activity and cell cycle progression and may alter platinum-induced cell cycle arrest.61, 62, 63 Additionally, phosphorylation of CREB1 at Ser271 by HIPK2 was previously reported to respond to DNA damage and promote survival.64,65
Of the 48 proteins showing differential ubiquitin response at 24 h (Figure 3C; Table S1), 13 ubiquitin sites (representing 12 proteins) showed larger FCs in the sensitive cell lines. The remaining 35 ubiquitin sites with larger FCs in resistant cell lines are enriched for the GO biological processes of “anion transport” (FDR = 5.1 × 10−6). 6 of the 31 proteins (with larger FCs in resistant cell lines) have been associated with platinum resistance (Table S4C), including the ubiquitination on K561 of FANCD2 and K523 of FANCI (Figures 3C and S3A).
For mRNA expression, 5 and 7 transcripts show significantly different responses to platinum (comparing sensitive and resistant cell lines) after 8 and 24 h of carboplatin exposure, respectively (Figure 3D), and these are not enriched for any biological process.
Similar analyses (Table S1) identified genes, proteins, and PTMs possibly showing different responses to carboplatin exposure within individual intra-patient pairs of sensitive and resistant cell lines, although these analyses were greatly limited due to the small sample size.
Baseline differences in sensitive versus resistant cells
After correcting for multiple hypothesis testing, no individual protein or post-translational modification expression level at baseline (i.e., mock treatment) was significantly associated with sensitivity to carboplatin, likely due to the underlying inter-patient heterogeneity66 and the multifactorial nature of resistance mechanisms for platinum compounds.67, 68, 69 However, baseline differences between sensitive and resistant cell lines were observed at the pathway level (Figure 4A; Table S2). For example, expression of the interferon alpha and gamma pathways were elevated in resistant cell lines (global proteome and RNA; Figures 4B and 4C). Conversely, the Kyoto Encyclopedia of Genes and Genomes (KEGG)_ribosome pathway members are expressed at higher levels in sensitive cell lines, consistently across the global proteome, RNA, and ubiquitin datasets (Figures 4D and 4E). Interestingly, dysregulated ribosome biogenesis in cancer is being considered as a potential therapeutic target in HGSOC.70,71
Figure 4.

Pathways showing baseline expression differences between sensitive and resistant cell lines
(A) Significant baseline differences observed between sensitive and resistant cell lines at the pathway level.
(B) Volcano plot showing higher expression of proteins in the interferon alpha pathway in the resistant cell lines.
(C) Volcano plot showing higher expression of proteins in the interferon gamma pathway in the resistant cell lines.
(D) Volcano plot showing higher expression of ribosomal proteins (RPs) in the sensitive cell lines.
(E) Volcano plot showing higher ubiquitination of RPs in the sensitive cell lines.
Baseline differences in protein complexes in sensitive versus resistant cells
We identified 1,729 protein complexes annotated in the CORUM database72 with at least two protein members observed in our global proteomic dataset. A subset of 33 of these complexes showed differential abundances between sensitive and resistant cell lines (FDR ≤ 0.05; Figure S4A; Table S5). Of these, 16 were expressed at a higher level in resistant cell lines, and 17 were expressed at a higher level in sensitive cell lines. Most of the protein complexes expressed at higher levels in the resistant cell lines are associated with DNA repair, for example, the MRE11A-RAD50-NBN-TRF2 and the ERCC1-ERCC4-MSH2 complexes. Consistent with their high expression in the resistant cell lines, ERCC1-XPF endonuclease and MSH2 were found to be required for the recombinational repair processing of the ICL induced by carboplatin.73,74
High expression of the MRN complex (MRE11–RAD50–NBS1) was previously reported to be associated with chemoresistance in human squamous cell carcinoma cells and gastric cancers.75,76 To test the hypothesis that higher expression of MRN complex components in PEA2R, PEO4R, and PEO23R contributes to their platinum resistance, we examined the ability of mirin (a small molecule inhibitor of MRN)77 to sensitize resistant cell lines to carboplatin. We found that mirin enhanced platinum lethality in both the sensitive and resistant cell lines, and resistant cell lines were no more sensitive to mirin than the sensitive cell lines (Figures S4B–S4D). In contrast, mirin did not sensitize a non-tumor fallopian tube control cell line FT478 to carboplatin treatment (Figure S4E).
Of the 17 protein complexes that were expressed at a higher level in sensitive cells (Figure S4A; Table S5), both the 40S and 60S cytoplasmic ribosome subunits are represented, consistent with our pathway analyses described above (Figure 4D). The remaining protein complexes are involved in various functions, including cell-cell and cell-extracellular matrix interactions, cytoskeleton remodeling, chromatin remodeling, cycle control, and glycosylation. Of note, changes in ECM complex expression in HGSOC cell lines derived from the same patient may reflect the change of orthometastatic capacity during disease progression.34
Integrated multiomic analysis identifies baseline differences between sensitive and resistant cells
To increase our statistical power to identify differences between sensitive and resistant cell lines, we performed WGS analysis and derived CNV profiles of the 6 cell lines (Table S6A). We observed significant heterogeneity in the CNV profiles of the cell lines, also noted in previous work.27 We performed an integrated multi-omics analysis to identify individual features that showed significant differences between resistant and sensitive cell lines consistently across all datasets, including copy number, mRNA transcript levels, and protein abundance (Table S6B). Specifically, we identified 9 genes with consistently higher expression across all 3 resistant cell lines and 11 genes with consistently higher expression across all 3 sensitive cell lines (combined CNA, RNA, and protein p < 0.05; Table S7; Figures S5A and S5B). The 9 genes showing increased expression in the resistant cells are enriched for OAS antiviral response (FDR = 3.6 × 10−7), including OAS1, OAS3, OASL, and TRAFD1, with the top hit being OAS3 (combined CNA, RNA, and protein p = 1.5 × 10−5), which encodes an enzyme that is induced by interferons and catalyzes the formation of 2′, 5′ oligomers of ATP.79 These oligos promote degradation of both viral and endogenous RNA as part of the cellular innate antiviral response.80 This result is consistent with our findings (Figures 4B and 4C) that expression of the interferon alpha and gamma pathways were elevated in resistant cell lines at baseline. Two additional genes displaying increases in CNV, mRNA, and protein expression in the resistant cells are MSLN and PPL (Figure S5A). MSLN encodes mesothelin, a membrane glycoprotein that is frequently overexpressed in malignancies, including HGSOC.81,82 PPL encodes periplakin, a component of the cornified envelope of keratinocytes, and acts as a localization signal in PKB/AKT-mediated signaling.83 Both genes have been reported to be associated with platinum resistance.84,85 The remaining genes, including the 11 genes elevated in sensitive cell lines, are not enriched in any pathways, nor have they been previously reported to be associated with platinum resistance.
Altered expression of proteins involved in fatty acid oxidation (FAO) and oxidative phosphorylation (OXPHOS) is associated with platinum resistance in HGSOC, both in vitro and in vivo
We observed baseline differences in expression of metabolic pathway proteins between sensitive and resistant cell lines (Figure 4A; Table S2). For example, proteins in the Hallmark “OXPHOS” (adjusted p = 2 × 10−9) and “adipogenesis pathways” (adjusted p = 9.5 × 10−5), as well as the Reactome “citric acid cycle TCA cycle” (adjusted p = 0.026), “fatty acid metabolism” (adjusted p = 0.016), and “lipid metabolism” pathways (adjusted p = 0.018), were expressed at higher levels in resistant cell lines (Table S2). In addition to these baseline differences, carboplatin exposure was accompanied by reduced acetylation of proteins in the OXPHOS and fatty acid metabolism pathways (Table S2). Notably, mitochondrial proteins are frequently acetylated, which in most cases negatively impacts their activities,86 and most acetylated proteins in mitochondria are involved in regulating energy metabolism, such as fatty acid metabolism and OXPHOS.86, 87, 88
To determine whether the metabolic signature associated with platinum resistance in our cell line data is relevant in vivo, we performed global proteomic profiling of 20 human-in-mouse patient-derived xenograft (PDX) models derived from patients with HGSOC (10 platinum-sensitive and 10 platinum-refractory).89 Consistent with the cell line results, we found that both the TCA cycle and FAO pathways are increased in the proteomic profiles of platinum refractory PDX-derived tumors compared with platinum-sensitive tumors (Figures S5C and S5D). We also found an association between elevated TCA cycle pathway activity and overall survival (p < 0.05) in a previously reported proteomic analysis of human OCs (Figure S5E; note, platinum response is a major determinant of survival for HGSOC patients.)17,90
Pharmacologic inhibitors of CPT1A sensitize platinum-resistant cell lines to carboplatin
Our observation of increased expression of FAO/OXPHOS pathway members in carboplatin-resistant cell lines and PDX models is consistent with prior reports that altered FAO/OXPHOS metabolism may be associated with platinum resistance in cancers.9, 10, 11,40,91, 92, 93, 94, 95 To determine whether increased FAO/OXPHOS metabolism plays a causal role in platinum resistance in our cell line models, we performed a series of functional studies. The rate-limiting step of FAO is catalyzed by carnitine palmitoyltransferase 1A (CPT1A), which shuttles long-chain fatty acids into mitochondria. Interestingly, CPT1A is overexpressed in a subset of HGSOCs and associated with shorter progression-free survival. CPT1A inactivation induces accumulation of OC cell lines in the G1 phase and inhibits tumorigenicity in severe combined immunodeficiency (SCID) mice.96 We found that CPT1A was expressed at higher levels in the resistant cell lines from 2 of 3 cell line pairs (PEO4R/PEO1S and PEA2R/PEA1S). Conversely, the mitochondrial acetyl-coenzyme A (CoA) carboxylase 2 (ACACB), a negative regulator of FAO,97 was expressed at relatively lower levels in the corresponding cell line pairs (Figure 5A). These observations are consistent with increased FAO/OXPHOS activities in PEA2R and PEO4R cells as compared to PEA1S and PEO1S, and, as would be expected with a high-OXPHOS status,40,98 we found that PEA2R and PEO4R cells exhibit higher ROS production than their paired sensitive cell lines, both at baseline and after challenge with carboplatin for 1 h (Figures S6A and S6B). The PEO23R/PEO14S pair showed opposite but consistent trend compared with the other 2 pairs, with the lower CPT1A level and higher ACACB level (Figure 5A), and a correspondingly lower ROS production in PEO23R compared to PEO14S (Figure S6C). Heterogeneity among cell lines from different patients is expected due to the underlying inter-patient heterogeneity66 and the multi-factorial nature of resistance mechanisms for platinum.67, 68, 69 Consistent with the CPT1A overexpression in resistant cell lines from 2 of the 3 pairs (PEA1S/PEA2R and PEO1S/PEO4R), a subset of platinum resistant and refractory HGSOC PDX models (Figure S5F) and human tumors (Figure S5G)90 show high expression of CPT1A. CPT1A overexpression has been associated with platinum resistance in HGSOCs.96
Figure 5.

CPT1A and ACACB expression levels are associated with carboplatin resistance in 2 of the 3 cell line pairs, and pharmacologic inhibition of CPT1A sensitizes cells to carboplatin
(A) CPT1A protein abundance is higher in PEA2R and PEO4R, although acetyl-CoA carboxylase 2 (ACACB) protein abundance is lower in PEA2R and PEO4R. Western blot data (see image and bar graphs) confirm the LC-MS/MS results.
(B–D) Perhexiline sensitizes HGSOC cell lines to carboplatin.
(E) Perhexiline sensitizes non-tumorigenic FT4 cells to carboplatin.
Data in (B)–(E) are an average of 3 biological repeats each with 3 technical repeats. p values (Student’s t test) are provided.
To test whether high FAO is required for platinum resistance, we examined whether the carboplatin sensitivity of the cell lines was affected by two CPT1A inhibitors, etomoxir (2[6(4-chlorophenoxy) hexyl] oxirane-2-carboxylate)99 and perhexiline (2-(2,2-dicyclohexylethyl) piperidine).100,101 Perhexiline is a more potent inhibitor of FAO than etomoxir, inhibiting not only CPT1A but also CPT2, which converts acyl-carnitine to acyl-CoA, the next step downstream of CPT1A.100,101 Interestingly, both perhexiline (Figures 5B–5D) and etomoxir (Figures S6D–S6F) sensitized all 6 HGSOC cell lines to platinum, and the combined effect of perhexiline and carboplatin was dramatic (Figures 5B–5D). The interaction between carboplatin and perhexiline was most significant for the PEO1S and PEO4R pair (Figure 5B), as the concentrations required to achieve >95% loss of viability were 2 μM perhexiline plus 20 μM carboplatin for PEO1S and 4 μM perhexiline plus 40 μM carboplatin for PEO4R as compared to 8 μM perhexiline plus 80 μM carboplatin for PEA1S/PEA2R and PEO14S/PEO23R pairs (Figures 5C and 5D). The non-tumor fallopian tube (FT4) control cell line78 was also sensitized to carboplatin by perhexiline (Figure 5E) and etomoxir (Figure S6G).
CPT1A is a determinant of platinum resistance in PEO4R cells
To confirm that the platinum-sensitizing effect of CPT1A inhibitors was not due to off-target effects,102, 103, 104 we knocked out CPT1A in PEO4R and PEO1S cells using CRISPR-Cas9. Individual clones were isolated by limiting dilution, deletions were confirmed by DNA sequencing, and loss of CPT1A protein was confirmed by western blotting (Figure 6A). Complete loss of CPT1A protein results in significantly increased sensitivity to carboplatin consistently across 6 independent PEO4R KO clones (Figures 6B and S7A). On the other hand, none of the 7 independent CPT1A KO clones isolated from the PEO1S cell line showed significant change in carboplatin sensitivity as compared to parental PEO1S cells (Figures 6B and S7B). Of note, clones C15, C52, and C86 with a deletion of 33 amino acids in the N-terminal region of CPT1A displayed no increase in sensitivity to carboplatin treatment (Figures 6B and S7A), suggesting that they may retain CPT1A activity. Of note, the N-terminal of CPT1A binds to malonyl-CoA and plays an inhibitory role on CPT1A activity.105 We further confirmed the increased carboplatin sensitivity in PEO4R CPT1A knockout (KO) clone C6 using a colony formation assay (Figures 6C and S7C).
Figure 6.

CPT1A is a determinant of platinum resistance both in vitro and in vivo
(A) Inventory of CPT1A knockout (KO) clones with western blot confirmation in selected clones.
(B) Sensitivity of CPT1A KO clones to carboplatin (cell viability assay, average of 3 biological repeats each with 3 technical repeats).
(C) Sensitivity of selected CPT1A KO clones to carboplatin (colony formation assay). One PEO4 KO clone (C6) and one PEO1 KO clone (B85) were tested along with WT controls (C5 and A3, respectively).
(D) Retroviral complementation of PEO4 CPT1A-KO clone C6 and WT control clone C5. Vec, vector control; WT, WT CPT1A; Mut, mutant CPT1A (G710E) (3 biological repeats each with 3 technical repeats; Student’s t test performed between WT and KO; p values provided in the graph).
(E) Western blot confirmation of the expression of CPT1A WT or G710E mutant in PEO4 cells.
(F) Combination efficacy of carboplatin + CPT1A inhibitors in the platinum refractory HGSOC PDX model PHO48. Tumor area was monitored weekly by transabdominal ultrasound. Change in tumor size over time is plotted as the statistical model estimated average of all animals at each time point for a given treatment, scaled relative to baseline estimate for that treatment. Shading indicates 95% confidence intervals. The p values are provided in the table.
To additionally confirm that CPT1A plays an important role in platinum resistance in PEO4R cells, we performed genetic complementation studies. We reintroduced either a wild-type or mutant CPT1A gene in CPT1A-KO clones in both the PEO4R and PEO1S cells and examined the effect on carboplatin sensitivity. As shown in Figure 6D, expression of retroviral-expressed human wild-type (WT) CPT1A protein in PEO4 CPT1A KO clone C6 restored resistance to carboplatin, although expression of a mutant CPT1A protein (G710E) that lacks carnitine palmitoyltransferase (CPTase) catalytic activity106 did not affect the carboplatin sensitivity of the PEO4 CPT1A-KO. This result not only supports the central role of CPT1A in carboplatin resistance in PEO4R cells but also demonstrates that the CPTase activity (and not the lysine succinyltransferase activity)106 of CPT1A is required for this role. As controls, retroviral vector expression in PEO4 CPT1A KO, all retroviral expression lines of PEO4 WT clone C5 (C5 WT+Vec, C5 WT+WT, and C5 WT+Mut) and PEO1 WT clone A3 (A3 WT+Vec, A3 WT+WT, and A3 WT+Mut), and PEO1 CPT1A KO clone B85 (B85 KO+Vec, B85 KO+T, and B85 KO+Mut) did not affect carboplatin sensitivity of PEO4 WT cells and PEO1 WT and CPT1A KO cells (Figures 6D and S7E). The successful restoration of WT or G710E mutant CPT1A protein expression in PEO4 and PEO1 CPT1A KO was validated by western blot (Figures 6E and S7F).
We noted differences in the level of sensitization to carboplatin between CPT1A-KO (Figures 6B and S7A) and the CPT1A inhibitors etomoxir (Figure S6D) and perhexiline (Figure 5B). Additionally, CPT1A KO did not increase the carboplatin sensitivity of PEO1S cells (Figure S7B), whereas the inhibitors sensitize both PEO4R and PEO1S (Figures S6D and 5B). To check whether there are compensatory increases of other isoforms of the CPT1 gene family members as well as CPT2 due to loss of the CPT1A gene (potentially contributing to platinum resistance), we assessed protein expression levels of CPT1B, CPT1C, and CPT2 by western blotting (Figure S7D). No significant compensatory increases in any of these 3 proteins were found in the absence of CPT1A, suggesting that loss of CPT1A is responsible for sensitizing PEO4R cells, although off-target and/or non-specific effects104,107 of the two inhibitors may also contribute to further sensitize PEO1S.
Carboplatin-induced ROS is associated with higher induction of DNA damage and apoptotic cell death in CPT1A-KO compared with CPT1A WT cells
Carboplatin induces ROS, leading to DNA damage and apoptosis.108,109 We found that basal level of ROS in PEO4 CPT1A-KO cells (untreated) was significantly higher compared to PEO4 WT (Figure 7A), suggesting that lack of CPT1A is associated with oxidative stress in PEO4 cells. As expected, carboplatin exposure for 24 h increased ROS production in both PEO4 WT and PEO4 CPT1A-KO cells in a concentration-dependent manner (Figure 7A). However, when cells were treated with carboplatin at 160 μM for 48 h, a significant drop of ROS below basal control was observed (Figure 7A), likely due to induction of NRF2 protein (Figure 7B), a transcription factor that mediates the antioxidant response.110 Consistent with this hypothesis, NRF2 target transcripts (e.g., NQO1, PRDX1, ME1, and PIR) were induced following carboplatin exposure (Figure 7C). NRF2 levels at baseline are not different between PEO4 WT and KO cells (Figure 7B). Although both PEO4 CPT1A-KO and PEO4 WT show similar NRF2 induction in response to carboplatin, apoptotic cell death is nonetheless significantly higher in the PEO4 CPT1A-KO compared to the PEO4 parental cell line, based on both increased annexin V binding (Figure 7D) and caspase-3 cleavage (Figure 7E). Furthermore, treatment with the ROS inhibitor N-acetyl-cysteine (NAC) rescued the apoptotic effects of carboplatin (Figure 7E), confirming that ROS plays a role in carboplatin-induced cell death. Interestingly, carboplatin-induced cell death in the PEO4 CPT1A-KO was associated with a significant increase in DNA damage that was not observed in PEO4 WT, as shown by the augmented level of γH2AX (Figure 7B). Taken together, our data indicate that CPT1A plays a critical role in regulating oxidative stress in PEO4 cells and that lack of CPT1A re-sensitizes cells to carboplatin by increasing DNA damage.
Figure 7.

Carboplatin-induced ROS is associated with higher induction of DNA damage and apoptotic cell death in CPT1A-KO versus CPT1A WT cells
(A) The basal level of ROS (detected with 2,7’–dichlorofluorescin diacetate [DCFDA] dye) was compared between PEO4 CPT1A-KO cells and the parental PEO4 cell line (n = 3; ∗p < 0.05). Relative changes in ROS production for both PEO4 WT and PEO4 CPT1A-KO cells upon carboplatin exposure were plotted for both 24 h and 48 h.
(B) Representative western blot showing the baseline and effects of carboplatin exposure on NRF2 and γH2AX expression (48 h). Quantification of NRF2 and γH2AX proteins was done using NIH ImageJ software and plotted as line graphs. Data are expressed as mean ± SEM (n = 3; ∗p < 0.05; ∗∗p < 0.001).
(C) RNA expression levels of NQO1, PRDX1, ME1, and PIR at 0 h and 8 h after carboplatin treatment were compared for PEO1S and PEO4R cells.
(D) Detection of carboplatin-induced apoptosis via Annexin V staining in PEO4-WT and PEO4-KO cells. Cells were treated with different concentrations of carboplatin for 48 h and incubated with AV-fluorescein isothiocyanate (FITC) and phosphatidylinositol (PI). Stained cells were analyzed by flow cytometry. Percentage of intact cells (AV−/PI−) and different stage apoptotic cells (AV+/PI−, AV+/PI+, and AV−/PI+) are presented. Data represent mean ± SEM (n = 3; ∗p < 0.001).
(E) Western blot showing the effects of carboplatin treatment (160 μM; 48 h) in PEO4-WT versus PEO4-KO on caspase-3 cleavage as an indicator of cell death via apoptosis. Data are expressed as mean ± SEM (n = 3; ∗p < 0.05; ∗∗p < 0.001).
Combining carboplatin with CPT1A inhibitors reduces tumor growth in vivo
We evaluated the preclinical efficacy of the combination of carboplatin and CPT1A inhibitors in an HGSOC PDX model (PH048). PH048 was generated from a patient diagnosed with HGSOC whose refractory tumor showed aggressive growth during adjuvant carboplatin and paclitaxel. Tumor-bearing mice were randomized into 6 groups for treatment: (1) control (saline); (2) etomoxir (40 mg/kg, intraperitoneally [i.p.] 5 days/week); (3) perhexiline (80 mg/kg, oral gavage 5 days/week); (4) carboplatin (51 mg/kg, i.p. weekly); (5) etomoxir + carboplatin; and (6) perhexiline + carboplatin. Doses were based on published literature in mice.111,112 Response to treatment was assessed by weekly transabdominal ultrasound, as described.89 The combination of carboplatin plus either etomoxir or perhexiline resulted in significantly greater tumor growth inhibition than the carboplatin monotherapy group (+etomoxir p = 0.0018; +perhexiline p = 0.0045; Figure 6F), consistent with our in vitro findings, indicating that CPT1A inhibitors may increase the therapeutic efficacy of carboplatin in HGSOC.
Discussion
Platinum compounds are widely used chemotherapy agents and are expected to remain in use, even in the era of precision medicine.113 Platinum resistance is a major determinant of survival, particularly in OCs, which are frequently diagnosed at late stage. Decades of literature demonstrate that resistance is multifactorial,12,13 and there has been no clinical translation of biomarkers to predict platinum response or treatments to overcome platinum resistance, and these remain unmet clinical needs. To date, no study using modern proteogenomic technologies has been undertaken to characterize cancer cell responses to platinum or to understand mechanisms of resistance.
In this study, we present a proteogenomic interrogation of the dynamic response of human cancer cells to carboplatin, focusing on intra-patient cell line pairs from HGSOC patients. This multiomic data resource is comprehensive and reproducible (Figures 1 and S1). All data in the resource are publicly available, and the results can be visualized via a searchable database with an intuitive gene-query user interface, including links to databases providing additional gene- and pathway-level information (https://sites.google.com/view/ptrc-cell-line).
Our data reveal that carboplatin induces robust responses in platinum-sensitive and resistant cells and uncover known and novel biology. Due to their therapeutic tractability,114, kinases are of particular interest. In addition to previously reported kinases responsive to platinum (e.g., ATM/ATR/CHEK1, CDKs, PKCs, MAPKs, AKT, and AMPK), we identified novel evidence of activation of CK2, CDC7, and CAMK2A in the HGSOC cell lines in response to carboplatin. It has been reported that the kinase activity of CAMK2A may be stimulated by platinum-induced elevation of intracellular calcium and ROS,115, 116, 117, 118 and activated CAMK2 hyper-phosphorylates downstream target molecules to stimulate ROS and induce cell death.118,119
Our results also identify platinum-induced activation of CK2, a pleiotropic kinase involved in a variety of cellular processes, including cell proliferation and apoptosis.120,121 Our observation that CK2 kinase activity is induced by carboplatin is consistent with reports that CK2 localizes to sites of DNA double-strand breaks,122 and its kinase activity toward p53 is activated by UV.123 CK2 has been proposed as a potential anti-cancer therapeutic target, and a CK2 inhibitor was shown to synergize with cisplatin in models of OC.124 The CK2 inhibitor CX4945 is currently in early-phase clinical trials for renal tumors and recurrent medulloblastoma (NCT03571438 and NCT03904862).
Our data also identify platinum-induced activation of CDC7, an essential S phase kinase that regulates DNA replication through phosphorylation of MCM proteins.125 How CDC7 is regulated upon genotoxic stress is controversial. Some reports suggest that CDC7 is downregulated, leading to inhibition of the late-origin firing.126,127 Others have reported that CDC7 activity is preserved upon DNA damage and required for checkpoint action and DNA damage tolerance during replication stress.128, 129, 130, 131 Our results indicate that CDC7 is activated by carboplatin treatment, in line with the latter reports. Indeed, CDC7 inhibitors have been found to enhance platinum cytotoxicity.132 A CDC7 inhibitor (LY3143921 hydrate) is currently being evaluated (NCT03096054) in patients with advanced solid tumors, including HGSOC.
We also find elevated expression of FAO/OXPHOS metabolic pathway proteins in platinum-resistant versus platinum-sensitive cell lines. OCs have a predilection to metastasize to the omentum,133 a hormonally and immunologically active fatty tissue in the peritoneal cavity.134 Elegant studies by Lengyel et al.133,135 and others have shown that metastatic OC cells in the omentum initiate lipolytic signals in adipocytes that result in the release of long-chain fatty acids that are taken up by OC cells through the CD36 receptor136 and used for energy production through β-oxidation. HGSOC cells depend on FAO to overcome anoikis during dissemination to metastatic sites in the peritoneal cavity or survive in ascites.137 FABP4, a lipid transport protein in adipocytes, is a critical regulator of lipid responses in ovarian cancer cells co-cultured with adipocytes135 and is a determinant of metastatic potential in OC.138 Furthermore, FABP4 expression may be a predictor of residual disease in HGSOC.139 CRISPR KO of FABP4 in HGSOC cells reduced metastatic tumor burden in mice, and an FABP4 inhibitor additionally increased the sensitivity of cancer cells toward carboplatin both in vitro and in vivo, suggesting possible therapeutic use to reduce omental metastasis and help sensitize OC cells to platinum.135
Our results complement and extend prior findings by demonstrating that CRISPR KO or pharmacologic inhibition of CPT1A, which catalyzes the rate limiting step in FAO, sensitizes HGSOC cells to carboplatin. Our findings extend prior work showing that reduced CPT1A expression is associated with platinum sensitivity in a collagen type XI alpha 1 (COL11A1)-dependent in vitro model of platinum resistance,140,141 in which culturing OC cells on COL11A1-coated plates confer cisplatin resistance by engaging α1β1 integrin and discoidin domain receptor 2 (DDR2) on ovarian cancer cells to induce inhibitor of apoptosis proteins and upregulate both fatty acid synthesis and oxidation. Moreover, our results extend these findings by demonstrating that the effect of CPT1A expression on platinum sensitivity can also be a cell autonomous trait associated with tumor cell upregulation of proteins involved in FAO and downregulation of proteins involved in fatty acid synthesis (e.g., FASN and ACLY; Table S1). The convergence of experimental evidence from these different models of platinum resistance (i.e., cell autonomous versus ECM dependent) on the association of increased FAO in resistant cells demonstrates that OC cells can use more than one mechanism (i.e., genetic or via ECM) to increase FAO and underscores the importance of FAO in platinum resistance. This has implications for therapeutic intervention, because molecules targeting the ECM to disrupt FAO-inducing signals may not work in tumors in which increased FAO is an intrinsic property of the cancer cell.
The mechanistic link between platinum resistance and CPT1A is not fully understood. CPT1A facilitates FAO, contributing to ATP and NADPH production.142,143 One hypothesis consistent with our data (Figure 7) and the literature is that lack of CPT1A could result in reduced levels of NADPH and thereby exacerbate the oxidative stress induced by platinum,144 leading to increased DNA single-strand and double-strand breaks.145,146 Repair of DNA damage is associated with substantial energetic requirements.147 For example, PARP-1 activation consumes large amounts of NAD+ and ATP.148 In CPT1A-KO cells, which have less ATP available,96,149 PARP-1-mediated repair of DNA strand breaks may become compromised. Consistent with this, CRISPR-mediated CPT1A KO was previously reported to reduce resistance to IR in breast cancer cells.150
Our in vitro and in vivo data implicate CPT1A as a potential therapeutic target in HGSOC. Inhibition of CPT1A has been proposed as a therapeutic target in AML and Burkitt’s lymphoma based on preclinical studies.151, 152, 153 Of note, perhexiline has been used in clinical trials targeting cardiac disease (e.g., NCT02862600, NCT00839228, NCT00845364, NCT00500552, and NCT00841139) and is or has been used in some countries to treat cardiac disease, including angina.154, 155, 156, 157, 158 These clinical experiences have shown that, with monitoring of drug levels and attention to cytochrome P-450 function,159 the drug can be given to patients safely,160 paving a potential path forward for repurposing CPT1A inhibitors in the adjuvant setting in clinical trials with cancer patients receiving platinum-based chemotherapies. In this respect, it is interesting to note that the combination of 4 μM perhexiline plus 40 μM carboplatin killed >95% of platinum-resistant PEO4R cells while the non-tumorigenic FT4 control cell line retained ∼30% viability (Figure 5E), suggesting a potential therapeutic window for preferentially killing cancer cells and minimizing toxicity.
Limitations of the study
One limitation of this study is that the limited number of patients represented in the study likely does not represent all the heterogeneous mechanisms underlying clinical resistance to platinum compounds.13 Future studies with more patients will be required to determine whether the findings in our study are generalizable to all HGSOC tumors or only specific subclasses. Additionally, our preclinical models do not capture the influence of the immune system and tumor microenvironment on platinum responsiveness. Considering recent observations that cytotoxic death of ovarian cells could stabilize PD-L1 or other negative immune regulatory receptors,161 a combinatorial strategy targeting immune-negative regulators could be required for the CPT1A inhibitors to be fully effective in patients. Finally, although we chose to focus functional studies on CPT1A, we hope this high-quality dataset will prove a valuable resource to the research community to stimulate additional studies to advance our understanding of platinum resistance.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| P-Tyr-1000 rabbit antibody | Cell Signaling Technology | Cat#8954S; RRID:AB_2687925 |
| diGly | Cell Signaling Technology | Cat#5562 |
| acetyl-lysine motif | Cell Signaling Technology | Cat#13416 |
| anti-rabbit IgG HRP-linked secondary antibody | Cell Signaling Technology | Cat#7074; RRID: AB_2099233 |
| recombinant anti-CPT1A antibody | Abcam | Cat#ab220789; RRID: AB_2847832 |
| recombinant anti-CPT1B antibody | Abcam | Cat#ab134135; RRID: AB_2847833 |
| recombinant anti-CPT2 antibody | Abcam | Cat#ab181114; RRID: AB_2687503 |
| CPT1C-specific antibody | Proteintech | Cat#12969-1-AP; RRID: AB_2084844 |
| GAPDH antibody | Cell Signaling Technology | Cat#5174; RRID: AB_10622025 |
| phospho-Histone H2A.X (Ser139) antibody | Cell Signaling Technology | Cat#9718; RRID: AB_2118009 |
| Caspase-3 antibody | Cell Signaling Technology | Cat#9662; RRID: AB_331439 |
| a-Actinin (D6F6) XP antibody | Cell Signaling Technology | Cat#6487; RRID: AB_11179206 |
| NRF2 antibody | Proteintech | Cat#16396-1-AP; RRID: AB_2782956 |
| histone H3 antibody | Proteintech | Cat#17168-1-AP; RRID :AB_2716755 |
| Biological samples | ||
| Patient-derived xenografts, platinum sensitive | Mayo Clinic, Rochester, MN | PH013, PH063, PH077, PH088, PH242, PH249, PH299, PH361, PH423, PH454 |
| Patient-derived xenografts, platinum refractory | Mayo Clinic, Rochester, MN | PH026, PH048, PH271, PH341, PH550, PH081, PH232, PH586, PH626, PH763 |
| Chemicals, peptides, and recombinant proteins | ||
| Carboplatin | APP Pharmaceuticals, Selleckchem | Cat#S1215 |
| Urea | Sigma-Aldrich | Cat#U0631 |
| Trizma base (Tris) | Sigma-Aldrich | Cat#T2694 |
| iodoacetamide (IAM) | Sigma-Aldrich | Cat#A3221 |
| EDTA | Sigma-Aldrich | #E7889 |
| EGTA | Sigma-Aldrich | #E0396 |
| Phosphatase Inhibitor Cocktail 2 | Sigma-Aldrich | #P5726 |
| Phosphatase Inhibitor Cocktail 3 | Sigma-Aldrich | #P0044 |
| Protease Inhibitor Cocktail | Sigma-Aldrich | #P8340 |
| phosphate buffered saline | Thermo Fisher Scientific | #BP-399-20 |
| tris(2-carboxyethyl)phosphine | Thermo Fisher Scientific | Cat#77720 |
| DTT | Sigma-Aldrich | Cat#11583786001 |
| EPPS | Sigma-Aldrich | Cat#E9502 |
| Lys-C | Wako | Cat#12505061 |
| trypsin | Promega | Cat#V5111 |
| TMT reagents | Thermo Fisher Scientific | Cat#90406 |
| 100 mg Sep-Pak solid-phase extraction column | Waters | Cat#WAT023590 |
| protein A-agarose beads | Sigma-Aldrich | Cat#11134515001 |
| Fe-NTA phosphopeptide enrichment kit | Thermo Fisher Scientific | Cat# A32992 |
| Protein A resin | Thermo Fisher Scientific | Cat#53142 |
| StageTip | Thermo Fisher Scientific | Cat#SP301 |
| Acetonitrile | Sigma-Aldrich | Cat#A955 |
| water | Sigma-Aldrich | Cat#W6 |
| ammonium bicarbonate | Sigma-Aldrich | CatA6141 |
| mirin | Selleckchem | Cat#S8096 |
| etomoxir | Sigma-Aldrich | Cat#E1905 |
| perhexiline | Sigma-Aldrich | Cat#SML0120 |
| N-Acetyl-L-cysteine | Sigma-Aldrich | Cat#1009005 |
| Carboplatin, clinical grade | Mayo Clinic Pharmacy | N/A |
| Etomoxir, clinical grade | Target Molecule Corporation | Targetmol T4535 |
| Perhexiline, clinical grade, obtained as Pexsig (perhexiline maleate tablet, 100mg) | Aspen Pharma Pty Ltd | N/A |
| Critical commercial assays | ||
| AllPrep DNA/RNA FFPE kit | QIAGEN | Cat#80234 |
| QIAmp DNA FFPE Tissue kit | QIAGEN | Cat#56404 |
| miRNeasy FFPE kit | QIAGEN | Cat#217504 |
| Illumina TruSeq stranded Total RNA sample preparation kit | Ilumina | Cat#20020597 |
| Kapa DNA Hyper prep reagents | Roche Diagnostics Corporation | Cat#KK8504 |
| Micro BCA assay | ThermoFisher Scientific | Cat#23235 |
| crystal violet assay kit | Abcam | Cat#ab232855 |
| Cellular Reactive Oxygen Species Detection Assay Kit | Abcam | Cat#ab186027 |
| Synthego’s Gene Knockout Kit V2 | Synthego | N/A |
| FITC Annexin V Apoptosis Detection Kit I | BD PharMingen | N/A |
| Deposited data | ||
| Cell line and PDX proteomics datasets (Global, phospho, acetyl, ubiquitin, pTyr-enriched proteomics) | This paper | PRIDE: PXD020764 |
| RNA sequencing data | This paper | GEO: GSE163152 |
| Genome sequencing data | This paper | SRA: PRJNA684350 |
| TCGA CPTAC Ovarian cancer data | Zhang et al.90 | https://proteomics.cancer.gov/data-portal |
| PTMsigDB v1.9 | Krug et al.162 | https://github.com/broadinstitute/ssGSEA2.0 |
| PhosphoSitePlus | Hornbeck et al.163 | https://www.phosphosite.org |
| HPRD v9.0 | Keshava Prasad et al.164 | http://hprd.org |
| MSigDB Canonical Gene Sets (C2 CP) | Liberzon et al.165 | https://www.gsea-msigdb.org/gsea/msigdb/index.jsp |
| CORUM | Giurgiu et al.72 | https://mips.helmholtz-muenchen.de/corum |
| Experimental models: cell lines | ||
| Human: PEO1 | Toshiyasu Taniguchi, Sigma-Aldrich | RRID: CVCL_2686 |
| Human: PEO4 | Toshiyasu Taniguchi, Sigma-Aldrich | RRID: CVCL_2690 |
| Human: PEA1 | Sigma-Aldrich | RRID: CVCL_2682 |
| Human: PEA2 | Sigma-Aldrich | RRID: CVCL_2683 |
| Human: PEO14 | Sigma-Aldrich | RRID: CVCL_2687 |
| Human: PEO23 | Sigma-Aldrich | RRID: CVCL_2689 |
| Human: FT-4 non-tumorigenic fallopian tube cell line (SV40 immortalized) | Drs. Anna Lokshin and Katherine Aird | N/A |
| HEK293T | Sigma-Aldrich | RRID: CVCL_0063 |
| Experimental models: organisms/strains | ||
| female SCID beige mice (C.B.-17/IcrHsd- Prkdcscid Lystbg) | ENVIGO | N/A |
| Oligonucleotides | ||
| BRCA2 primer: 5′-CTATTGAG ACTGTGGTGCCACCTAAG |
Thermo Fisher Scientific/Invitrogen | Custom oligos |
| BRCA2 primer: 5′-GCAGGGT GAAGAGCTAGTCACAAGTT |
Thermo Fisher Scientific/Invitrogen | Custom oligos |
| CPT1A guide #1: U∗C∗U∗GAUGAACUUCU UUUUCC + synthego modified EZ scaffold |
Synthego | N/A |
| CPT1A guide #2: G∗A∗G∗CUUCAUGGCU CAGCCGC + synthego modified EZ scaffold |
Synthego | N/A |
| CPT1A guide #3: G∗G∗C∗AGAAG CUCACCAAGCUG + synthego modified EZ scaffold |
Synthego | N/A |
| CPT1A PCR forward primer: 5′-CCT GATGATCATCTTGGGGCTC |
Thermo Fisher Scientific/Invitrogen | Custom oligos |
| CPT1A PCR reverse primer: 5′-CCT CCTATTAAGTAGGTCGCTGGC |
Thermo Fisher Scientific/Invitrogen | Custom oligos |
| CPT1A sequencing primer: 5′-TCT TTGTAGCGGTGGACAGGC |
Thermo Fisher Scientific/Invitrogen | Custom oligos |
| Recombinant DNA | ||
| CPT1A WT | Taro Hitosugi | Kurmi et al.106 |
| CPT1A G710E | Taro Hitosugi | Kurmi et al.106 |
| pLHCX vector | Taro Hitosugi | Kurmi et al.106 |
| EcoPac | Taro Hitosugi | Kurmi et al.106 |
| pAmphopac | Taro Hitosugi | Kurmi et al.106 |
| pVSVG | Taro Hitosugi | Kurmi et al.106 |
| Software and algorithms | ||
| ComBat | Johnson et al.166 | https://www.bioconductor.org/packages/release/bioc/html/sva.html |
| sumer | Savage et al.167 | https://github.com/bzhanglab/sumer |
| GSVA | Barbie et al.54 | https://bioconductor.org/packages/release/bioc/html/GSVA.html |
| PTM-SEA | Krug et al.162 | https://github.com/broadinstitute/ssGSEA2.0 |
| WebGestalt | Liao et al.168 | http://webgestalt.org |
| ImageJ | Schneider et al.169 | https://imagej.nih.gov/ij/ |
| Summit | Beckman Coulter | N/A |
| ComplexHeatmap | Gu et al.170 | https://www.bioconductor.org/packages/release/bioc/html/ComplexHeatmap.html |
| R | The R Foundation | https://www.r-project.org |
| SAS | SAS Institute Inc. | https://www.sas.com/en_us/home.geo.html |
| Sequest-based software pipeline | Huttlin et al.171 | https://gygi.hms.harvard.edu/software.html |
| Ascore | Rose et al.172 and Erickson et al.173 | https://gygi.hms.harvard.edu/software.html |
| BIC-seq | Xi et al.174 | https://github.com/ding-lab/BICSEQ2 |
| GISTIC | Mermel et al.175 | https://github.com/broadinstitute/gistic2 |
| wilcox.test | Bauer176 | Base R-package: stats |
| nlme | Laird and Ware177 | https://cran.r-project.org/web/packages/nlme/index.html |
| Other | ||
| Web portal for data visualization | This paper | https://sites.google.com/view/ptrc-cell-line |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Amanda Paulovich (apaulovi@fredhutch.org).
Materials availability
Multiple partial and complete CPT1A knockout clones in the background of PEO1s and PEO4R generated by CRISPR-Cas9 method as described in this study are available in Dr. Paulovich’s lab upon request.
Experimental model and subject details
Cell lines
PEA1S, PEA2R, PEO14S, and PEO24R cells were purchased from Sigma-Aldrich (European Collection of Authenticated Cell Cultures, PEA1 Sigma 10032306-1VL/ECACC 10032306; PEA2 Sigma 10032307-1VL/ECACC 10032307; PEO14 Sigma 10032311-1VL/ECACC 10032311; PEO23 Sigma 10032313-1VL/ECACC 10032313). PEO1S and PEO4R cells were provided by Toshiyasu Taniguchi (Fred Hutchinson Cancer Research Center) (for the multiomic profiles) and also separately purchased from Sigma-Aldrich (European Collection of Authenticated Cell Cultures, PEO1 Sigma 10032308-1VL/ECACC 10032308, PEO4 Sigma 10032309-1VL/ECACC 10032309) (for the in vitro functional studies). Cell lines were authenticated by STR profiling (University of Arizona Genetics Core). The status of the BRCA2 premature stop codon mutation (5193C > G) in PEO1S cells35 was confirmed by PCR amplifying this region of BRCA2 using oligonucleotide primers (5′-CTATTGAGACTGTGGTGCCACCTAAG and 5′-GCAGGGTGAAGAGCTAGTCACAAGTT) and sequencing the resulting PCR fragment using the same primers. There was no evidence of the previously reported reversion mutation that restores BRCA2 function in some PEO1S cultures178. Cells were cultured in RPMI1640 (Corning 10-040-CV) supplemented with L-glutamine and 10% fetal bovine serum (heat Inactivated FBS, Atlanta Biologicals S10250). Cultures were re-initiated from cryopreserved parental stocks every three months and tested for mycoplasma (MycoAlert, Lonza) every 6 months. FT-4 non-tumorigenic fallopian tube cell line (SV40 immortalized)78 was a gift from Drs. Anna Lokshin and Katherine Aird (University of Pittsburgh Medical Center) and was cultured in DMEM/F-12 with 10% FBS and 1% penicillin-streptomycin.
Patient-derived xenograft (PDX)
Fresh human tumor tissues from consenting patients with ovarian cancer were collected at the time of primary debulking surgery and coded with a patient heterotransplant (PH) number in accordance with the Mayo Clinic Institutional Review Board and the Health Insurance Portability and Accountability Act regulations. All animal procedures were approved by the Mayo Clinic Institutional Animal Care and Use Committee (IACUC). Tumors were established by IP injection into female SCID beige mice (C.B.-17/IcrHsd- Prkdcscid Lystbg; ENVIGO) as previously described89. Briefly, minced patient tumor in McCoy’s 5a medium was supplemented with rituximab (10 mg/kg, Genentech, Inc.) to prevent lymphoma development179 in ∼0.3 mL of total volume for each injection. After engraftment, PDX tumors were expanded into additional mice prior to cryogenic preservation for future experiments89. The minimal information standard for PDX models is provided in the following table:
| 048 | |
|---|---|
| Gender | F |
| Age | 60 |
| Diagnosis | Ovarian Cancer |
| Consent | Academic |
| Primary Tissue | Ovary |
| Collection Site | Primary |
| Specimen collected | Ovary |
| Histology | Serous |
| Grade | High |
| Stage | IIIC |
| Markers | N/A |
| Treatment | Naive |
| Mouse Strain | SCID-bg |
| Mouse Humanized | No |
| Preparation | Solid Tumor |
| Injection site | IP |
| Characterization | Histology |
| Negative murine/EBV | Yes |
| Passage | P7 |
Method details
Proteomics sample preparation and TMT labeling
For cell line proteomic analysis, cells were plated in 150-mM tissue culture plates and cultured for 48 hours, at which point they were approximately 50% confluent. The cultures were then treated with vehicle (water) or 80 μM carboplatin (APP Pharmaceuticals, Schaumburg, IL; dissolved in water at 10 mg/mL) for 8 or 24 hours. Cells were trypsinized with 2 mL of 0.25% trypsin (Corning 25-053-CI) at 37°C until cells were released from the plate. The released cells were washed twice with DPBS and lysed in freshly prepared lysis buffer (6 M urea, 25 mM Tris, pH 8.0, 1 mM EDTA, 1 mM EGTA, 1% Sigma Phosphatase Cocktail 2, 1% Sigma Phosphatase Cocktail 3, and 1% Sigma Protease Inhibitor Cocktail) at 4°C (1 mL lysis buffer per 5 × 107 cells). Lysates were sonicated (Virsonic 600 microprobe at full power) for 12 s, incubated on ice for 20 s, again sonicated for 12 s, and centrifuged at 21,000 g at 4°C for 10 min. Protein concentrations were determined using bicinchoninic acid (Pierce BCA Protein Assay Kit). Lysates were stored in the liquid phase of liquid nitrogen. Each cell line was grown and treated (or mock-treated) once each on three independent days, producing 3 biological replicates. Each biological replicate was independently processed and subjected to ‘omic analyses (representing technical replicates), and thus the dataset represents three independent complete process replicates for each cell line, time point and treatment condition.
For PDX tumor proteomic analysis, tumors (average mass 0.4 g, range 0.1-1.3 g) were harvested and briefly rinsed in ice-cold PBS to remove contaminating blood, transferred to a cryovial and then snap frozen in liquid nitrogen. Frozen tumors were cryo-pulverized with a Covaris cryoPREP CP02 Impactor, and protein was solubilized in 1 mL urea lysis buffer (6 M urea, 25 mM Tris, pH 8.0, 1 mM EDTA, 1 mM EGTA, 1% Sigma Phosphatase Cocktail 2, 1% Sigma Phosphatase Cocktail 3, and 1% Sigma Protease Inhibitor Cocktail). Samples were vortexed at maximum speed for 15 s, and lysates were transferred to 1.7 mL screw-top microfuge tubes. Samples were subjected to 3x 30 s of sonication using a Fisher Scientific 550 Sonic Dismembrator at 50% power in a cup horn probe filled with ice-cold water. Samples were then cleared by centrifugation (20,000 RCR, 10 minutes at 4°C) and transferred to cryovials (NUNC #363401) for storage in the vapor phase in an LN2 tank. Protein concentrations of the clarified lysates were measured by Micro BCA assay (ThermoFisher Scientific, Cat# 23235).
1 mg of protein from each cell line or PDX tumor lysate was reduced with 5 mM tris(2-carboxyethyl)phosphine (TCEP; ThermoFisher Scientific, Cat#77720) for 15 min at room temperature, alkylated with 10 mM iodoacetamide (Sigma-Aldrich, Cat#I1149) in the dark for 30 min and quenched with 10 mM DTT (Sigma-Aldrich, Cat#11583786001). Samples were chloroform−methanol precipitated180. ∼300 μg protein pellets were reconstituted in 200 mM EPPS (Sigma-Aldrich, Cat#E9502) at pH 8.5 and digested by Lys-C (Wako, Cat#12505061) overnight at a 1:50 protease-to-protein ratio and trypsin (Promega, Cat#V5111) for 6 hours at a 1:100 protease-to-protein ratio. For global proteome and phosphotyrosine-containing peptide analysis, a final volume of 30% acetonitrile was added together with TMT reagents (ThermoFisher Scientific, Cat#90406) at a 1:2 peptide-to-TMT ratio (w/w). Samples were mixed 1:1 across all TMT channels, desalted using a 100 mg Sep-Pak solid-phase extraction column (Waters, Cat#WAT023590) and dried in vacuum centrifugation. For other post-translational modification analysis, including phosphorylation, ubiquitination, and acetylation, modified peptides were enriched individually from protein digests and sequentially as the order given (Figure 1), then labeled with TMT reagent and pooled.
Enrichment of pTyr-containing peptides
Phosphotyrosine enrichment was performed with 2 mg labeled peptide mixture. P-Tyr-1000 rabbit antibody (Cell Signaling Technology, Cat#8954S) was coupled with protein A-agarose beads (Sigma-Aldrich, Cat#11134515001) overnight prior to immunoaffinity purification. Beads were washed two times with 1 mL cold PBS and then two times with 1 mL cold immunoaffinity purification (IAP) buffer. Labeled peptide mixture was resuspended in 1.4 mL IAP buffer, mixed with the beads and incubated on a rotator with gentle end-over-end rotation for 2 hours at 4°C. After centrifugation at 1500 g for 30 s and removal of supernatant, beads were further washed two times with 1 mL cold IAP buffer, followed by two washes with 1 mL ice-chilled PBS. Supernatant was removed and beads were transferred onto a 0.2 μm Ultrafree-MC Centrifugal Filter (Millipore). Enriched peptides were eluted with 75 μL 0.15% TFA, desalted using homemade StageTips181, and dried via vacuum centrifugation.
Enrichment of phosphopeptides with IMAC
The Pierce High-Select Fe-NTA phosphopeptide enrichment kit (ThermoFisher Scientific, Cat# A32992) was used to enrich phosphopeptides from 1 mg of each individual protein digest following the manufacturer’s protocol. The enriched modified peptides were desalted and labeled with TMT prior to basic-pH reverse-phase (BPRP) high-performance liquid chromatography (HPLC) fractionation.
Enrichment of diGly-containing peptides
The diGly-containing peptide enrichment was performed following a procedure published previously172. The diGly monoclonal antibody (Cell Signaling Technology, Cat#5562) (32 μg/IP) was coupled to Protein A Plus Ultralink resin (40 μL slurry/IP) (ThermoFisher Scientific, Cat#53142) overnight at 4°C prior to its chemical cross-linking reaction182. Dried peptides (1 mg for each sample) were resuspended in 1.4 mL of cold IAP buffer [50 mM MOPS (pH 7.2), 10 mM sodium phosphate and 50 mM NaCl] and centrifuged at maximum speed for 5 min at 4°C to remove any insoluble material. Supernatants (pH ∼7.2) were incubated with the antibody beads for 2 hours at 4°C with gentle end-over-end rotation. After centrifugation at 1000 g for 30 s, beads were washed three times with cold IAP buffer and twice with cold PBS. The diGly peptides were eluted twice with 75 μL 0.15% TFA, desalted using homemade StageTips181 and dried via vacuum centrifugation.
Enrichment of acetylated peptides
Acetylated peptides were enriched using PTMScan acetyl-lysine motif [Ac-K] kit (Cell Signaling Technology, Cat#13416). Antibody beads were washed with cold PBS and IAP buffer for three times each. Unbound flow-through fraction from diGly enrichment was loaded onto the beads and incubated for 2 hours at 4°C with gentle end-over-end rotation. After centrifugation at 1000 g for 30 s, beads were washed three times with cold IAP buffer and twice with cold PBS. Beads were then transferred onto a 0.2 μm Ultrafree-MC Centrifugal Filter (Millipore, Cat#UFC30LG25) and eluted twice with 75 μL 0.15% TFA. The enriched peptides were desalted using homemade StageTips181 and dried via vacuum centrifugation.
Basic-pH reverse-phase (BPRP) high-performance liquid chromatography (HPLC) fractionation
Peptides from the full proteome and IMAC-enrichment were resuspended in Buffer A (10 mM ammonium bicarbonate, 5% ACN, pH 8). Peptides from the full proteome were subjected to a 50 min linear gradient from 13% to 42% of Buffer B (10 mM ammonium bicarbonate, 90% ACN, pH 8) at a flow rate of 0.6 mL/min while IMAC-enriched peptides were subjected to a 50 min linear gradient from 5% to 32% B. 96 fractions were collected and consolidated into 12 samples in a checkerboard manner180. Fractions were vacuum-centrifuged until dry and desalted via StageTip (Thermo Scientific SP301) for LC-MS analysis.
Cell line liquid chromatography and tandem mass spectrometry analysis
Mass spectrometry data were collected using an Orbitrap Fusion mass spectrometer (ThermoFisher Scientific, Cat#IQLAAEGAAPFADBMBCX) coupled to a Proxeon EASY-nLC 1000 liquid chromatography (LC) pump (ThermoFisher Scientific, Cat# LC120). Peptides were separated on a 100 μm inner diameter microcapillary column packed with 30 cm of Accucore150 resin (2.6 μm, 150 Å, ThermoFisher Scientific). LC separation was achieved using a 3 h gradient of 7 to 30% acetonitrile in 0.125% formic acid at a flow rate of ∼550 nL/min. Each analysis used a synchronous precursor selection (SPS)-MS3-based TMT method to reduce reporter ion interference and resulting ratio compression183,184. The scan sequence began with an MS1 spectrum collected at 120,000 resolution with an AGC target of 400,000 and a max injection time of 50 ms. The ten most intense multiply charged ions (required z > 1) were selected for MS/MS. Monoisotopic precursor selection was enabled. Isolation width was set at 0.7 m/z. ITMS2 spectra were collected at turbo speed with an AGC of 20,000, max injection time of 120 ms and CID collision energy of 35%. For phosphorylation data acquisition, Multi-Stage Activation (MSA) was used in addition to the CID fragmentation185. Following acquisition of each MS2 spectrum, we collected an MS3 spectrum with the SPS-MS3 technology. Synchronous-precursor-selection (SPS) was enabled to include 10 MS2 fragment ions in the FTMS3 spectrum. For the FTMS3 scan, the Orbitrap was operated at 50,000 resolution with an AGC target of 100,000 and a max injection time of 150 ms and an HCD collision energy of 65%.
PDX liquid chromatography and tandem mass spectrometry analysis
Mass spectrometry data were collected using an Orbitrap Fusion Lumos mass spectrometer (ThermoFisher Scientific, Cat#IQLAAEGAAPFADBMBHQ) coupled to a Proxeon EASY-nLC 1200 liquid chromatography (LC) pump (ThermoFisher Scientific, Cat# LC140). Peptides were separated on a 100 μm inner diameter microcapillary column packed with 30 cm of Accucore150 resin (2.6 μm, 150 Å, ThermoFisher Scientific). LC separation was achieved using a 3 h gradient of 7 to 30% acetonitrile in 0.125% formic acid at a flow rate of ∼550 nL/min. Each analysis used an SPS-MS3-based TMT method to reduce reporter ion interference and resulting ratio compression183,184. The scan sequence began with an MS1 spectrum collected at 120,000 resolution with an AGC target of 200,000 and a max injection time of 50 ms. The ten most intense multiply charged ions (z > 1) were selected for MS/MS. Monoisotopic precursor selection was enabled. Isolation width was set at 0.5 m/z. ITMS2 spectra were collected at turbo speed with an AGC of 20,000, max injection time of 120 ms and CID collision energy of 35%. For full proteome analysis, since proteins in a PDX model can be either mouse- or human-origin making PDX tissue inherently more complex than human or mouse alone, a real-time search-based data acquisition method186 was utilized to only perform quantitative SPS-MS3 scans on precursors that were matched uniquely to a peptide of human origin on the fly in order to improve coverage of the human proteome which is of more interest compared to mouse proteins in the matrix. Only performing SPS-MS3 scans on human-unique peptides reduced subsequent data analysis complexity caused by interspecies interference. For the FTMS3 acquisition, the Orbitrap was operated at 50,000 resolution with an AGC target of 150,000, a max injection time of 300 ms, and an HCD collision energy of 65%. Synchronous-precursor-selection (SPS) was enabled to include up to 10 matched MS2 fragment ions in the FTMS3 spectrum. For phosphorylation data acquisition, Multi-Stage Activation (MSA) was used in addition to the CID fragmentation185 and a SPS-MS3 scan was collected following each MS2 scan.
Database search
Mass spectra were processed using a Sequest-based software pipeline171. Raw files were converted to mzXML format, and monoisotopic m/z measurements and charge state assignments were corrected. Spectra were searched against a database including all entries from the human UniProt database (February 04, 2014). This database was concatenated with one composed of all protein sequences in the reversed order as well as known common protein contaminants. Sequest searches were performed using a 50 ppm precursor ion tolerance, requiring trypsin protease specificity, while allowing up to two missed cleavages. The product ion tolerance was set to 0.9 Da. TMT tags on peptide N termini/lysine residues (+229.162932 Da) and carbamidomethylation of cysteine residues (+57.02146 Da) were set as static modifications while methionine oxidation (+15.99492 Da) was set as variable modifications. For each PTM analysis, phosphorylation on serine, threonine, and tyrosine (+79.966 Da), lysine ubiquitylation (+114.04293 Da), or lysine acetylation (−187.15237 Da) was included as variable modification. Peptide-spectrum matches (PSMs) were adjusted to a 1% false discovery rate (FDR)187. PSM filtering was performed using a linear discriminant analysis, as described previously171, while considering the following parameters: XCorr, ΔCn, missed cleavages, peptide length, charge state, and precursor mass accuracy. PSMs were identified, quantified, and collapsed to a 1% peptide false discovery rate (FDR) and then collapsed further to a final protein-level FDR of 1%. To quantify the confidence of each PTM site, we used a modified version of Ascore172,173, and only PTM sites with Ascore values > 13 (p < 0.05) were considered. Moreover, protein assembly was guided by principles of parsimony to produce the smallest set of proteins necessary to account for all observed peptides. Proteins and PTM sites were quantified by summing reporter ion counts across all matching PSMs. For TMT-based reporter ion quantitation, we found the closest matching centroid in a 0.003 Da window around the expected m/z of the TMT reporter ion and extracted the summed signal-to-noise (S/N) ratio for each TMT channel. MS3 spectra with TMT reporter summed signal-to-noise ratio less than 100, or a MS/MS isolation specificity less than 0.5 were excluded from quantification.
Data pre-processing: normalization and batch-correction (cell line and PDX proteomic data)
We first filtered out presumed contaminant proteins/PTM-sites (keratins) and the reverse database hits from proteomics, phospho-proteomics, and other PTM datasets. Then we take the ratio of the raw intensities to the intensity of the reference bridge sample in each TMT and convert the ratio to the logarithm scale. For the global proteomics data, after removing the reference bridge sample from every TMT, we perform global normalization to align the sample median and scale by median absolute deviation to remove any systematic variation across the samples. However, the global normalization is based on the assumption that the distribution of the features is roughly similar across the samples, except for a constant (such as, median). The inefficiency of the instruments to detect or quantify the weak signal of low-abundance peptides leads to the considerable amount of missing protein abundances, with the chance of missingness being higher for low abundance ions. Hence in such cases the assumptions of global normalization fail to hold. For the phospho-proteomics data and other PTMs, we observed the missing rate varies significantly across the patients and/or time-points, and hence we adopted “truncated” global normalization188 to avoid possible bias of considering overall mean/median. To calculate the sample median, we consider the top L ordered statistic of the feature intensities, where L was chosen to be 0.9∗n_min, n_min being the minimum of the number of features observed in each ordered set. After normalization we perform outlier truncation. Any intensity exceeding median ± 4∗IQR is truncated by median ± 4∗IQR. Next we filtered out the proteins/phospho-/PTM-sites with batch-level missing data-points (missing from all samples in a TMT plex). Finally, we applied batch correction on global, phospho and other PTMs normalized data to remove the technical difference (batch-effect) between different TMT 9-plex. We used an R tool: ComBat to remove batch-effect166.
Nucleic acid extractions
Cells were pelleted by centrifugation, and DNA and RNA were extracted using a protocol adapted from QIAGEN AllPrep DNA/RNA FFPE Kit (Cat# 80234), QIAamp DNA FFPE Tissue Kit (Cat# 56404) and miRNeasy FFPE kit (Cat# 217504). Briefly, cell pellets were resuspended in 240 μL Buffer PKD and 16 μL proteinase K (QIAGEN # 80234), lysed by vortexing, and centrifuged 20 min (15-30 min) at > 20,000 x g (room temperature). The supernatant was transferred to a new 1.5 mL centrifuge tube for RNA extraction, and the pellet was reserved for DNA extraction. The supernatant was incubated at 80°C for 15 min (on a thermal mixer at 300 rpm), and then centrifuged at 14,000 rpm for 2 min. The supernatant was transferred to a new 2.0 mL centrifuge tube and RNA was extracted using the miRNeasy FFPE kit (QIAGEN cat# 217504) according to the manufacturer’s instructions. The pellet containing the DNA was extracted using the QIAamp DNA FFPE Tissue Kit (QIAGEN cat# 56404) according to the manufacturer’s instructions. Both RNA and DNA kits utilize spin columns to wash and then elute nucleic acids.
RNaseq library preparation
Purified total RNA samples were evaluated for quality and quantity by Agilent Bioanalzyer using RNA 6000 Nano chip and reagents (Cat#5067-1511). Sequencing libraries were prepared using Illumina TruSeq stranded Total RNA sample preparation kit (Cat# 20020597) from 200ng of RNA according to the manufacturer’s protocol.
Whole genome library preparation
Purified gDNA was quantified by Qubit Fluorometer and sheared to 300bp using a Covaris M220. Sequencing libraries were prepared using Kapa DNA Hyper prep reagents (Cat# KK8504) from 100ng of DNA according to the manufacturer’s protocol.
Next-generation sequencing
The finished libraries were quantified by Qubit fluorometer, Agilent TapeStation 2200 D1000 screentape (Cat#5067-5582), and RT-qPCR using the Roche Kapa Biosystems library quantification kit (Cat#KK4854) according to the manufacturer’s protocols. Whole genome libraries for copy number analysis were sequenced with > 25M 75bp read pairs and RNaseq libraries were sequenced with > 50M 75bp read pairs by the Molecular Biology Core Facilities at Dana-Farber Cancer Institute.
Pre-processing for cell line RNA data
The RNA data had 25,435 genes. We first removed the transcripts that were missing from all 6 cell lines. We also removed 10 samples that were missing more than 50% of the features. Then we converted the counts to the logarithm scale and performed global normalization to align the sample median. We further removed the genes with missing counts from 50% of the samples.
Cell viability assay
The effect of carboplatin (S1215, Selleckchem US), mirin (S8096, Selleckchem US), etomoxir (E1905, Sigma-Aldrich), and perhexiline (SML0120, Sigma-Aldrich) alone and in combination on cell viability was evaluated using a crystal violet assay kit (ab232855, Abcam). Cells from 3 pairs of patient-derived cell lines and FT-4 cells were seeded into clear 96-well plates at a density of 7,500-10,000 cells per well in 100 μL of media and allowed to attach for 48 hours to form a monolayer. Cells were then exposed to a serial dilution of single treatment of carboplatin (5 μM to 320 μM), mirin (0.5 μM to 40 μM), etomoxir (1 μM to 160 μM), or perhexiline (0.1 μM to 8 μM) or as a combination therapy (carboplatin plus mirin, carboplatin plus etomoxir, or carboplatin plus perhexiline) for 72 hours. Following treatment, cells were washed with PBS, 40 μL of 1% crystal violet staining solution was added to each well, and plates were incubated for 20 min with shaking at room temperature. After incubation, staining solution was removed, and wells were washed 4 times with 200 μL PBS, 100 μL of solubilization solution (1% SDS) was added to each well, and plates were incubated for 30 min at room temperature. The absorbance (A) at 595 nm was determined using a Biotek 2 microplate reader (Biotek USA). Cell viability was determined by the following formula: Cell viability = A of treated cells/A of untreated cells.
Colony formation assay
PEO1S (WT), PEO1S (CPT1Ako, clone B12), PEO4R (WT), and PEO4R (CPT1Ako, clone C6) were plated at 1125 cells/plate for PEO1 cells and 750 cells/plate for PEO4 cells in 60-mm dishes containing normal growth medium. Cells were allowed to adhere for 48 hours, and exposed continuously to various carboplatin concentrations (0, 0.05, 0.1, 0.2, 0.4, and 1.0 μM for PEO1 cells, and 0, 0.1, 0.2, 0.4, 1.0, and 2.5 μM for PEO4 cells). Medium containing the appropriate concentration of carboplatin was changed every other day. Treatment lasted for 14-17 days, and cells were stained with 1% crystal violet staining solution (50 mL of 1% crystal violet staining solution was made from 5% crystal violet stock solution in ddH2O by adding 10 mL of 5%, 5 mL of methanol, and 35 mL of dd H2O). Pictures of the plates were taken, and images were scored for colony formation using ImageJ software169. All experiments were done in biological triplicates (each was seeded from a different plate of cells), and colony formation was counted independently by two people and the averages of the two counts were reported.
Reactive oxygen species assay
Reactive Oxygen Species (ROS) levels were measured for PEO1S, PEO4R, PEA1S, PEA2R, PEO14S, PEO23R, and FT4 cells using the Cellular Reactive Oxygen Species Detection Assay Kit (Red fluorescence, ab 186027, Abcam, USA). Briefly, 10,000 cells/well of each cell line were plated into Thermo Scientific Nunc MicroWell 96-well optical-bottom plates with polymer base (Cat#165305, Thermo Scientific, USA). Cells were allowed to attach for 48 hours. The manufacturer’s protocol was followed for the ROS assay, and time-resolved fluorescence was monitored at Ex/Em = 520/605 nm with bottom read mode on a SpectraMax M5 Multi-mode microplate reader (Molecular Devices, USA). ROS levels were measured for PEO4 WT and PEO4 KO using a flow cytometry-based assay. Single-cell suspensions were treated with 20 μM DCFDA and the fluorescence (Ex/Em = 485/535 nm) was measured. Flow cytometry was performed at the University of Illinois at Chicago RRC facility using CyAn flow cytometer (Beckman Coulter Inc., Fullerton, CA). All data were analyzed by Summit software (Beckman Coulter Inc., Fullerton, CA). N-Acetyl-L-cysteine (NAC) was purchased from Sigma (MO, USA), (#1009005).
CRISPR-Cas9 knockout of CPT1A gene in ovarian cancer cell lines
CPT1A gene was knocked out in PEO1 and PEO4 cell lines using Synthego’s Gene Knockout Kit V2 (Synthego, WA). Briefly, three guide sgRNAs (see below) were designed to target the first exon of the CPT1A gene to induce multiple concurrent double strand breaks by SpCas9 nuclease, followed by random non-homologous end joining to create a mixture of various length of nucleotide deletions in the target region.
CPT1A guide #1: U∗C∗U∗GAUGAACUUCUUUUUCC + synthego modified EZ scaffold
CPT1A guide #2: G∗A∗G∗CUUCAUGGCUCAGCCGC + synthego modified EZ scaffold
CPT1A guide #3: G∗G∗C∗AGAAGCUCACCAAGCUG + synthego modified EZ scaffold
Individual clones were isolated by limited dilution and expanded by multiple rounds of clonal expansion. Nucleotide deletions were confirmed by DNA sequencing of the PCR product using the following primers: 5′-CCTGATGATCATCTTGGGGCTC (PCR forward primer), 5′-CCTCCTATTAAGTAGGTCGCTGGC (PCR reverse primer) and 5′-TCTTTGTAGCGGTGGACAGGC (sequencing primer). The loss of CPT1A protein production was confirmed by western blotting as described below.
Retrovirus production, retroviral infection, and stable cell line selection
Retroviral human CPT1A WT and G710E constructs were kindly provided by Taro Hitosugi from Mayo Clinic. These plasmids were pLHCX-hygro- Gateway destination vector-based and were constructed as previously described144. Each of the pLHCX vector plasmid (RV), CPT1A WT (RW), and CPT1A G710E (RM) plasmid was co-transfected with packaging plasmids (EcoPac, pAmphopac, pVSVG) into HEK293T (Sigma-Aldrich) cells using lipofectamine 2000. Retrovirus was harvested 48 h after transfection, filtered w/ 0.45 μm filter, and 8 μg/ml final concentration of polybrene was added. Retroviral infection of the PEO1 WT (A3) and PEO1-CPT1A KO clones (B85), PEO4 WT (C5) and PEO4-CPT1A KO clones (C6) was conducted with freshly harvested retrovirus. Infected cell lines were selected in 25 μg/ml hygromycin for 3 weeks to obtain stable cell lines. Stable expression of the CPT1A WT and G710E mutant protein was verified by western blot of cell lysates as described below.
Cell apoptosis assay
Cell apoptosis was determined by using a BD PharMingen FITC Annexin V Apoptosis Detection Kit I. Briefly, cells were harvested and washed with PBS twice. The pellets were resuspended in 1x Binding Buffer and incubated for 15 minutes at room temperature with 3 μL of FITC Annexin V and 3 μL of Propidium Iodide in the dark. Then 400 μL of 1x Binding Buffer was added to each tube prior to analysis. Samples were analyzed with a flow cytometer (LSR Fortessa with HTS, BD Biosciences, NJ, USA) and Summit (Beckman Coulter Inc.; Fullerton, CA).
Western blot and protein lysate preparation
Protein was extracted from cell pellets using freshly prepared ice-cold urea lysis buffer (containing 6 M urea (Sigma, U0631), 25 mM Tris (pH 8.0) (Sigma, T2194), 1 mM EDTA (Sigma, E7889), 1 mM EGTA (Sigma, E0396), 1% phosphatase inhibitor cocktail 2 (Sigma, P5726), 1% phosphatase inhibitor cocktail 3 (Sigma, P0044), and 1% protease inhibitor cocktail (Sigma, P3840)). Lysis buffer was added directly to cell pellets (1 mL of lysis buffer per 5 × 107 cells, or a minimum of 0.1 mL of lysis buffer for < 5 × 106 cells), followed by two rounds of sonication (using a cup horn probe (Fisher Scientific, 550 Sonic Dismembrator) filled with ice water (30 s at 50% power), separated by a 10 s incubation on ice. The lysates were vortexed at maximum power for 15 s and centrifuged at 20,000 g for 10 min at 4°C to pellet the debris. The cleared lysate was then transferred to a fresh pre-cooled microcentrifuge tube and stored at −80°C. The protein concentration of the lysate was determined using the Pierce Micro BCA Protein Assay (Thermo, 23235).
Cell lysates were prepared for gel electrophoresis by diluting to a protein concentration of 1.2 ug/uL using 4X NuPAGE LDS Sample Buffer (Thermo, NP0007), 10X NuPAGE Sample Reducing Agent (Thermo, NP0009) and PBS (where the final concentration of the LDS Sample Buffer and Reducing Agent are 1X). Samples were heated at 98°C for 5 min, spun down (20,000 g for 10 s at room temperature), and 30 μg of total protein was loaded per well onto a polyacrylamide gel (NuPAGE 3%–8% Tris-Acetate Gels (Thermo, EA0375BOX) in 1X Tris-Acetate SDS Running Buffer (Thermo, LA0041) for CPT1A, CPT1B, CPT1C and CPT2 western blots; NuPAGE 4%–12% Bis-Tris Gels (Thermo, EA0321BOX) in 1X MES SDS Running Buffer (Thermo, NP0002) for GAPDH western blots). Proteins were then transferred to a membrane (Thermo, LC2001) using a traditional wet transfer with NuPAGE Transfer Buffer (Thermo, NP0006) and the XCell II Blot Module (Thermo, EI9051). Membranes were washed with 1X TBS (Cell Signaling, 12498S) for 5 min, blocked with 5% nonfat dried milk (Cell Signaling, 9999S) for 1 hour at room temperature, incubated with an antibody (dilution and solution according to manufacturer’s instructions) overnight at 4°C, washed three times with 1X TBST (Cell Signaling, 9997S), incubated with an anti-rabbit IgG HRP-linked secondary antibody (Cell Signaling, 7074) for 1 hour at room temperature, washed three times with 1X TBST and then incubated with a working solution of SuperSignal West Pico PLUS Chemiluminescent Substrate (Thermo, 34580) for 5 min at room temperature. Chemiluminescence on the membrane was detected using Bio-Rad’s ChemiDoc XRS+ Imaging System. Novex Sharp Pre-stained Protein Standard (Thermo Fisher Scientific, Cat # LC5800) was used in the molecular marker lane in western blots, with 80kDa and 40kDa MW marker band shown in related western. The following antibodies were obtained from commercial resources: recombinant anti-CPT1A antibody (Abcam, ab220789); recombinant anti-CPT1B antibody (Abcam, ab134135); recombinant anti-CPT2 antibody (Abcam, ab181114); CPT1C-specific antibody (Proteintech, Rosemont, IL, USA, 12969-1-AP); GAPDH antibody (Cell Signaling, 5174), phospho-Histone H2A.X (Ser139) antibody (Cell Signaling antibodies, 9718), Caspase-3 antibody (Cell Signaling antibodies, 9662), a-Actinin (D6F6) XP antibody (Cell Signaling antibodies, 6487), NRF2 antibody (Proteintech, 16396-1-AP), and histone H3 antibody (Cell Signaling Technologies, Inc. Boston, MA, USA, 17168-1-AP).
PDX drug treatment
For in vivo drug studies, PDX tumors were injected IP into SCID beige mice. When tumors reached a minimum threshold of 0.3-0.5 cm2 by cross-sectional area on ultrasound imaging, animals were randomized into one of six groups: (i) saline control, (ii) carboplatin (51 mg/kg, IP weekly), (iii) etomoxir (40 mg/kg, IP 5 days/week), (iv) perhexiline (80mg /kg, oral gavage 5days /week), (v) carboplatin + etomoxir, or (vi) carboplatin + perhexiline for up to 9 weeks. The clinical grade reagents were used for animal experiments as required by IACUC. Carboplatin was purchased from Mayo Clinic pharmacy. Etomoxir was obtained from Target Molecule Corporation (Targetmol T4535). Perhexiline was obtained as Pexsig (perhexiline maleate tablet, 100mg) from Aspen Pharma Pty Ltd (NSW, Australia). Both combination groups (v and vi) were treated at the same dose and schedule as the monotherapy groups. Ultrasound measurements of tumor size were taken weekly. Mice were removed from the study if predetermined moribund criteria were met: tumors ≥ 10% of animal body weight (estimated by ultrasound based on experience and IACUC guidance), weight loss ≥ 20%, or body condition score ≤ 5189, animal weight loss ≥ 20% of baseline, inability to ambulate, inability to reach for food and/or water, skin ulceration from tumor burden, or a body condition score of ≤ 5189.
Quantification and statistical analysis
Mixed effect model for association tests
We performed a mixed effect linear regression model to test for the association of the individual gene/protein/PTM with platinum response. Specifically, we considered the following set of models for testing the three hypotheses discussed in the main text:
Protein abundance ∼sen/res_0 + patient_0|cellline_0 … (1)
Protein abundance ∼sen/res + time_8hr + time_24hr + patient|cellline … (2)
Protein abundance ∼sen/res + time_8hr + time_24hr + sen/res∗time_8hr + Sen/Res∗time_24hr … (3)
For identifying the genes/proteins/PTMs associated with a baseline difference between sensitive and resistant cell lines, we tested for the regression coefficient of the factor sen/res_0 (considering only samples at baseline) in model 1. We also added the random effect of patients nested within cell lines (patient_0|cellline_0) in the model to take into account the subject level variation.
For identifying the markers associated with platinum response at 8 hours and 24 hours, we tested the coefficients of time_8hr and time_24hr respectively in model 2. We also added the random effect of patients within the cell lines.
Finally, for identifying the markers with different platinum responses between sensitive and resistant cell lines, we tested for the coefficient of the interaction effects sen/res∗time_8hr and Sen/Res∗time_24hr at the two time points, in model 3; taking into account the random variation of patients within the cell lines as in the previous two models.
Pathway enrichment analysis using Wilcoxon test
Pathway enrichment analysis was conducted to characterize the baseline difference between sensitive and resistant cell lines, the overall platinum effect on the cells, and the platinum response differences between resistant and sensitive cell lines at two time points, based on results from association tests (described in the previous section). Gene set enrichment was conducted across a collection of gene sets from MSigDB’s Canonical database that includes: KEGG, Biocarta, Reactome, PID, and from MSigDB’s Hallmark collection. These collections were downloaded from https://www.gsea-msigdb.org/gsea/msigdb/index.jsp 165,190,191. We performed the Wilcoxon test to compare the distribution of signed p values (obtained from mixed-effect model-based regression analysis) of the genes within the pathways to the remaining genes in the dataset. Gene sets with < 5 or > 300 member genes were excluded. To help identify pathways distinctly associated with platinum response and to consolidate redundant pathway results, Sumer software was utilized167.
Over-representation analysis
Genes, proteins, or proteins containing PTMs that demonstrated larger fold changes in the sensitive cells versus the resistant cells (and vice versa) in response to platinum were submitted to WebGestalt168 for over-representation enrichment analysis of Gene Ontology Biological Process terms. The reference list were genes or proteins quantified in the same omics type. Terms were considered significant with a Benjamini-Hochberg corrected p value < 0.05.
Kinase activity analysis
Kinase activity analysis was performed using single sample Gene Set Enrichment Analysis (ssGSEA) implemented in the GSVA R package54. Phosphorylation site data were combined by average in each sample if multiple peptides contained the site. Substrates of kinases were collected from PhosphoSitePlus (version June 2017), SwissProt (version June 2017), and HPRD (v9.0) and were converted to a 13-mer motif (+/− 6 amino acids surrounding the phosphorylated site)163,164,192. Kinases were required to have at least 5 substrates in the data. Differences between carboplatin-treated and mock-treated cells were calculated using the mixed effect linear regression model.
Enzyme activity from substrate phosphorylation
Phosphorylation site 13-mer motifs on kinases and phosphatases that were significantly altered (adjusted p < 0.05) were annotated with regulatory information downloaded from PhosphoSitePlus (version November 2019). Sites with an ‘on function’ of ‘enzymatic activity, induced’, were considered activating sites. Sites with an ‘on function’ of ‘activity, inhibited’ or ‘enzymatic activity, inhibited’ were considered inhibitory sites. Sites without known regulatory information were excluded from further analysis.
Phosphosite signature enrichment analysis
PTM-SEA162 was used to calculate normalized enrichment scores for the perturbation and pathway phosphosite signature sets from PTMsigDB v1.9 for all samples. The file with human flanking identifiers was modified to contain 13-mers instead of 15-mers. PTM-SEA was performed using default parameters, with the exception of requiring at least 5 phosphosites in the data. Differences between carboplatin-treated and mock-treated cells were calculated using the mixed effect linear regression model.
Pathway activity score calculation
Pathway activity scores for individual PDX and human tumor proteomic data were calculated using the GSVA method in the GSVA R package. The gene sets were the same as described in ‘Pathway enrichment analysis using Wilcoxon test’ and default parameters were used with the requirement of a minimum of 5 overlapping genes. Scores were compared using Student’s t test.
Protein complex analysis
In this analysis, the goal is to identify protein complexes showing differential expression levels between the sensitive and resistant cell lines. Considering the CORUM protein complex database72, we identified 1729 protein complexes with at least two protein members observed in our cell line global proteomic data. Then for each protein complex, we tested its association with the sensitive/resistant status by applying the regularized Hoteling T2 test193, a multivariate two-sample test, on the global abundances of proteins in the complex from the 3 sensitive and 3 resistant cell lines. Note, the average abundances across biological replicates of each cell line at the base line (i.e., mock treatment) was used as the input of the statistical tests. In addition, the regularized Hotelling T2 test was used to better accommodate the small sample size in the analysis. In the end, we obtained the adjusted p values after accounting for multiple hypotheses testing. For significantly differentially expressed protein complexes, the up/downregulation direction in the sensitive compared to the resistant cell lines were annotated based on the mean abundance differences between two cell line groups across all proteins in the complex. In Table S5, we also reported the proteins in each complex that are marginally significant (p < 0.05) based on the univariate association test using mixed effects regression models.
Copy number variation (CNV) analysis
We performed data analysis based on 10x whole genome sequencing copy number variation data to identify any difference in copy number between three sensitive and 3 resistant cell lines. DNA copy number segmentation and amplification/deflection calls were made using BIC-seq and GISTIC based on the WGS. There were 24,579 genomic segments, many of which had identical copy numbers across all 6 cell lines. We first collapsed the segments with identical copy numbers, and this gave us 477 segments. We then performed a univariate association test using paired t test to test if there is mean difference in copy number between sensitive and resistant cell lines. We report the p value, log 2 (fold change) and adjusted p value (FDR) for genes in corresponding segments in Table S6. We do not see any individual gene passing an FDR cut-off of 0.1. We then combined the p values of the univariate association test analysis at baseline based on protein and RNA with the p values of the CNV univariate association analysis using Fisher’s method and also obtained the adjusted p value (reported in Table S6).
PDX drug response
PDX growth curves were analyzed by repeated-measures implemented via linear mixed effects models194. The dependent variable was ultrasound tumor area on the natural log scale. Independent variables were day, treatment arm, day by treatment interaction, and day squared, where the day variable was centered. The functional form of the mean model was chosen based on Akaike information criterion (AIC), Bayesian information criterion (BIC), plots of predicted trajectories and residual plots assuming independent observations. The form of the mean model was then held fixed and plausible covariance structures estimated via restricted maximum likelihood estimation; AIC and BIC indicated that a spatial (power) covariance structure was a good fit. Treatment arms were compared via two degree of freedom coincident curve hypothesis tests.
Statistical software
All analyses were performed via R (R foundation for statistical computing, Vienna, Austria 2017)) and SAS software (copyright 2016, SAS Institute Inc., Cary, NC, USA) unless otherwise noted. Heatmaps were generated using the ComplexHeatmap package170 in R.
Additional resources
All processed proteogenomic data are presented via an online portal with an intuitive gene-query user interface (https://sites.google.com/view/ptrc-cell-line). The web portal provides visualizations of the proteogenomic data and pathway analysis.
Acknowledgments
We thank Anna Lokshin and Katherine Aird (University of Pittsburgh) and Toshiyasu Taniguchi (Fred Hutchinson Cancer Research Center) for the FT-4 and the PEO1S and PEO4R cell lines, respectively. We thank Taro Hitosugi (Mayo Clinic) for retroviral human CPT1A constructs. This work was done with funding from the National Cancer Institute’s Clinical Proteomic Tumor Analysis Consortium and supported by National Institutes of Health grants U01CA214114 (A.G.P. and M.J.B.), R50CA211499 (J.R.W.), U24CA210993 (P.W.), U24CA210954 (B.Z.), P50CA136393 (S.H.K), and S10OD028685 (A.G.P); U.S. Department of Defense (DOD) grants W81XWH-20-1-046 (S.G.) and W81XWH-16-2-0038 (M.J.B.); and generous support from the Aven Foundation (A.G.P.) and the Christl Burgess Memorial Fund for ovarian cancer research (S.G.).
Author contributions
Conceptualization, A.N.H., L.M.K., S.J.W., S.H.K., P.W., S.G., M.J.B., and A.G.P.; methodology, S.P.G., L.M.K., S.J.W., S.H.K., B.Z., P.W., S.G., M.J.B., and A.G.P.; software, S.C., S.R.S., C.L., A.C., and P.W.; validation, H.W., R.G.I., J.J.K., Q.Y., and S.P.G.; formal analysis, S.C., S.R.S., C.L., A.C., and P.W.; investigation, H.W., R.G.I., J.J.K., X.H., N.E., D.A.D., C.J.H., U.J.V., Z.J.S., Q.Y., S.P.G., and Z.T.H.; resources, H.W., R.G.I., J.J.K., X.H., Z.J.S., T.D.L., Q.Y., S.P.G., Z.T.H., L.M.K., S.J.W., S.H.K., and M.J.B.; data curation, S.C., S.R.S., R.G.I., J.J.K., C.L., A.C., and P.W.; writing – original draft, D.H., S.C., H.W., S.R.S., R.G.I., J.J.K., Q.Y., L.M.K., S.J.W., S.H.K., P.W., M.J.B., and A.G.P.; writing – review & editing, D.H., S.C., H.W., S.R.S., R.G.I., J.J.K., J.R.W., A.N.H., T.D.L., L.M.K., S.J.W., S.H.K., S.G., P.W., M.J.B., and A.G.P.; visualization, D.H., S.C., H.W., S.R.S., S.G., P.W., M.J.B., and A.G.P.; supervision, P.W., M.J.B., and A.G.P.; project administration, J.R.W., T.D.L., L.M.K., S.J.W., S.H.K., B.Z., P.W., M.J.B., and A.G.P.; funding acquisition, M.J.B. and A.G.P.
Declaration of interests
M.J.B. has participated in advisory boards for Clovis, Astra Zeneca, and GSK-Tesaro. A.N.H. has a financial interest in the company Seattle Genetics.
Published: December 21, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2021.100471.
Supplemental information
Differential proteogenomic features were identified that associated with 3 hypotheses, as follows. Hypothesis 1: Platinum induces changes in both sensitive and resistant cell lines (i.e., a combined analysis of all cell lines). Hypothesis 2: Sensitive and Resistant cell lines differ at baseline (mock). Hypothesis 3: Platinum-induced changes differ between Sensitive and Resistant cell lines. On the “Assn test Summary” worksheet, the number of significant proteins, PTMs or RNAs under each hypothesis are shown in each cell, with hyperlinks to the results in other worksheets. In the result tabs, for Hypothesis 1, “FC_time_8 ” is 8Hr / 0Hr fold change for all cell lines (both sensitive and resistant). “FC_time_24 ” is 24Hr / 0Hr fold change. (FC > 1 indicates upregulation after treatment; FC < 1 indicates downregulation.) For Hypothesis 3, “Dir_int_8” or “Dir_int_24” are directions whether the fold changes over baseline (FC = 8hr/0hr or 24hr/0hr) is larger or smaller in sensitive cell lines as compared to resistant cell lines. “p-value” and “FDR” are the Wilcoxon statistic values. A single sample overview of proteins and posttranslational modifications that were both identified and quantified is provided in the Data Summary worksheet.
Number of significant pathways in each of the 3 hypotheses (described in the Table S1 legend) are shown in each cell, with hyperlink to results from the “Pathway test summary” worksheet. In the result worksheets, “size” is the number of genes observed in the data from the pathway. “p-value” and “FDR” are the Wilcoxon statistic values. “dir” is directions whether the expression of the pathways is up or down (8 or 24 hour compared to 0 hour for Hypothesis 1; sensitive compared to resistant for Hypothesis 2 and 3).
related to Figure 4 A) Gene level DNA copy number data derived from segmented CNV profiles based on WGS (See STAR Methods). For a given cell line and a given gene, its CNV number is set to be the log ratio measure of the CNV-segment the gene belongs to. For the column names: “Start” is the starting location on the chromosome for the CNV segment the gene belongs to; “End” is the end location; “PEA1.S” through “PEO23.R” are the CNV (log ratio measure) of genes for each of the 6 cell lines. B) Integrated multiomic association studies conducted on all three cell line pairs to identify differences between sensitive and resistant cell lines. For the column names: “Start” is the starting location on the chromosome for the CNV segment the gene belongs to; “End” is the end location. For a given gene, “CNV_FC” is the average CNV (log) fold change (FC) across the three pairs of cell lines, where the log FC of each pair of cell line is calculated as the differences between its log ratio CNV in the sensitive cell line and that in the paired resistant cell line; “CNV_p,” “CNV_FDR” are the p value and FDR of the paired test. Similarly, “prot_FC” or “rna_FC” are the mean of log2(sensitive/resistant) of the protein abundance or RNA in the paired tests between sensitive and resistant cell lines; “prot_p” and “rna_p” are p values of the corresponding paired tests; “prot_RNA_cnv_p” is the combined p value based on protein, RNA and CNV; “prot_RNA_cnv_FDR” is the combined FDR based on protein, RNA and CNV.
Data and code availability
All LC-MS/MS proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository1 with the dataset identifier PRIDE:PXD020764 (http://proteomecentral.proteomexchange.org/cgi/GetDataset?ID=PXD020764). All RNA sequencing data have been deposited to the National Center for Biotechnology Information Gene Expression Omnibus (GEO)2 with GEO Series accession GEO:GSE163152 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE163152). All whole genome sequencing data have been deposited to the National Center for Biotechnology Information Sequencing Read Archive (SRA)3 with the BioProject accession # SRA:PRJNA684350 (https://www.ncbi.nlm.nih.gov/sra/?term=PRJNA684350).
References
- 1.Vizcaíno J.A., Côté R.G., Csordas A., Dianes J.A., Fabregat A., Foster J.M., Griss J., Alpi E., Birim M., Contell J., et al. The PRoteomics IDEntifications (PRIDE) database and associated tools: status in 2013. Nucleic Acids Res. 2013;41:D1063–D1069. doi: 10.1093/nar/gks1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Edgar R., Domrachev M., Lash A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shumway M., Cochrane G., Sugawara H. Archiving next generation sequencing data. Nucleic Acids Res. 2010;38:D870–D871. doi: 10.1093/nar/gkp1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.DeVita V.T., Rosenberg S.A., Lawrence T.S. Eleventh Edition. Wolters Kluwer; 2018. DeVita, Hellman, and Rosenberg’s Cancer: Principles & Practice of Oncology (Cancer Principles and Practice of Oncology) [Google Scholar]
- 5.Pascoe J.M., Roberts J.J. Interactions between mammalian cell DNA and inorganic platinum compounds. I. DNA interstrand cross-linking and cytotoxic properties of platinum(II) compounds. Biochem. Pharmacol. 1974;23:1359–1365. doi: 10.1016/0006-2952(74)90355-4. [DOI] [PubMed] [Google Scholar]
- 6.Dasari S., Tchounwou P.B. Cisplatin in cancer therapy: molecular mechanisms of action. Eur. J. Pharmacol. 2014;740:364–378. doi: 10.1016/j.ejphar.2014.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Matsuo K., Lin Y.G., Roman L.D., Sood A.K. Overcoming platinum resistance in ovarian carcinoma. Expert Opin. Investig. Drugs. 2010;19:1339–1354. doi: 10.1517/13543784.2010.515585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Galluzzi L., Senovilla L., Vitale I., Michels J., Martins I., Kepp O., Castedo M., Kroemer G. Molecular mechanisms of cisplatin resistance. Oncogene. 2012;31:1869–1883. doi: 10.1038/onc.2011.384. [DOI] [PubMed] [Google Scholar]
- 9.Cocetta V., Ragazzi E., Montopoli M. Mitochondrial involvement in cisplatin resistance. Int. J. Mol. Sci. 2019;20:3384. doi: 10.3390/ijms20143384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Montopoli M., Bellanda M., Lonardoni F., Ragazzi E., Dorigo P., Froldi G., Mammi S., Caparrotta L. “Metabolic reprogramming” in ovarian cancer cells resistant to cisplatin. Curr. Cancer Drug Targets. 2011;11:226–235. doi: 10.2174/156800911794328501. [DOI] [PubMed] [Google Scholar]
- 11.Morandi A., Indraccolo S. Linking metabolic reprogramming to therapy resistance in cancer. Biochim. Biophys. Acta Rev. Cancer. 2017;1868:1–6. doi: 10.1016/j.bbcan.2016.12.004. [DOI] [PubMed] [Google Scholar]
- 12.Muggia F. Platinum compounds 30 years after the introduction of cisplatin: implications for the treatment of ovarian cancer. Gynecol. Oncol. 2009;112:275–281. doi: 10.1016/j.ygyno.2008.09.034. [DOI] [PubMed] [Google Scholar]
- 13.Huang D., Savage S.R., Calinawan A.P., Lin C., Zhang B., Wang P., Starr T.K., Birrer M.J., Paulovich A.G. A highly annotated database of genes associated with platinum resistance in cancer. Oncogene. 2021;40:6395–6405. doi: 10.1038/s41388-021-02055-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Torre L.A., Trabert B., DeSantis C.E., Miller K.D., Samimi G., Runowicz C.D., Gaudet M.M., Jemal A., Siegel R.L. Ovarian cancer statistics, 2018. CA Cancer J. Clin. 2018;68:284–296. doi: 10.3322/caac.21456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cannistra S.A. Cancer of the ovary. N. Engl. J. Med. 2004;351:2519–2529. doi: 10.1056/NEJMra041842. [DOI] [PubMed] [Google Scholar]
- 16.Matulonis U.A., Sood A.K., Fallowfield L., Howitt B.E., Sehouli J., Karlan B.Y. Ovarian cancer. Nat. Rev. Dis. Primers. 2016;2:16061. doi: 10.1038/nrdp.2016.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bast R.C., Jr., Hennessy B., Mills G.B. The biology of ovarian cancer: new opportunities for translation. Nat. Rev. Cancer. 2009;9:415–428. doi: 10.1038/nrc2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lisio M.A., Fu L., Goyeneche A., Gao Z.H., Telleria C. High-grade serous ovarian cancer: basic sciences, clinical and therapeutic standpoints. Int. J. Mol. Sci. 2019;20:952. doi: 10.3390/ijms20040952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu J., Matulonis U.A. New strategies in ovarian cancer: translating the molecular complexity of ovarian cancer into treatment advances. Clin. Cancer Res. 2014;20:5150–5156. doi: 10.1158/1078-0432.CCR-14-1312. [DOI] [PubMed] [Google Scholar]
- 20.Li S.-L., Ye F., Cai W.-J., Hu H.-D., Hu P., Ren H., Zhu F.-F., Zhang D.-Z. Quantitative proteome analysis of multidrug resistance in human ovarian cancer cell line. J. Cell. Biochem. 2010;109:625–633. doi: 10.1002/jcb.22413. [DOI] [PubMed] [Google Scholar]
- 21.Warmoes M., Jaspers J.E., Xu G., Sampadi B.K., Pham T.V., Knol J.C., Piersma S.R., Boven E., Jonkers J., Rottenberg S., Jimenez C.R. Proteomics of genetically engineered mouse mammary tumors identifies fatty acid metabolism members as potential predictive markers for cisplatin resistance. Mol. Cell. Proteomics. 2013;12:1319–1334. doi: 10.1074/mcp.M112.024182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nguyen E.V., Huhtinen K., Goo Y.A., Kaipio K., Andersson N., Rantanen V., Hynninen J., Lahesmaa R., Carpen O., Goodlett D.R. Hyper-phosphorylation of sequestosome-1 distinguishes resistance to cisplatin in patient derived high grade serous ovarian cancer cells. Mol. Cell. Proteomics. 2017;16:1377–1392. doi: 10.1074/mcp.M116.058321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Han X., Chen H., Zhou J., Steed H., Postovit L.M., Fu Y. Pharmacological inhibition of p38 MAPK by SB203580 increases resistance to carboplatin in A2780cp cells and promotes growth in primary ovarian cancer cells. Int. J. Mol. Sci. 2018;19:E2184. doi: 10.3390/ijms19082184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Santana-Rivera Y., Rabelo-Fernández R.J., Quiñones-Díaz B.I., Grafals-Ruíz N., Santiago-Sánchez G., Lozada-Delgado E.L., Echevarría-Vargas I.M., Apiz J., Soto D., Rosado A., et al. Reduced expression of enolase-1 correlates with high intracellular glucose levels and increased senescence in cisplatin-resistant ovarian cancer cells. Am. J. Transl. Res. 2020;12:1275–1292. [PMC free article] [PubMed] [Google Scholar]
- 25.Lombardi R., Sonego M., Pucci B., Addi L., Iannelli F., Capone F., Alfano L., Roca M.S., Milone M.R., Moccia T., et al. HSP90 identified by a proteomic approach as druggable target to reverse platinum resistance in ovarian cancer. Mol. Oncol. 2021;15:1005–1023. doi: 10.1002/1878-0261.12883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Langdon S.P., Lawrie S.S., Hay F.G., Hawkes M.M., McDonald A., Hayward I.P., Schol D.J., Hilgers J., Leonard R.C., Smyth J.F. Characterization and properties of nine human ovarian adenocarcinoma cell lines. Cancer Res. 1988;48:6166–6172. [PubMed] [Google Scholar]
- 27.Cooke S.L., Ng C.K., Melnyk N., Garcia M.J., Hardcastle T., Temple J., Langdon S., Huntsman D., Brenton J.D. Genomic analysis of genetic heterogeneity and evolution in high-grade serous ovarian carcinoma. Oncogene. 2010;29:4905–4913. doi: 10.1038/onc.2010.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stronach E.A., Alfraidi A., Rama N., Datler C., Studd J.B., Agarwal R., Guney T.G., Gourley C., Hennessy B.T., Mills G.B., et al. HDAC4-regulated STAT1 activation mediates platinum resistance in ovarian cancer. Cancer Res. 2011;71:4412–4422. doi: 10.1158/0008-5472.CAN-10-4111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gillet J.P., Varma S., Gottesman M.M. The clinical relevance of cancer cell lines. J. Natl. Cancer Inst. 2013;105:452–458. doi: 10.1093/jnci/djt007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Domcke S., Sinha R., Levine D.A., Sander C., Schultz N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat. Commun. 2013;4:2126. doi: 10.1038/ncomms3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jacob F., Nixdorf S., Hacker N.F., Heinzelmann-Schwarz V.A. Reliable in vitro studies require appropriate ovarian cancer cell lines. J. Ovarian Res. 2014;7:60. doi: 10.1186/1757-2215-7-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lengyel E., Burdette J.E., Kenny H.A., Matei D., Pilrose J., Haluska P., Nephew K.P., Hales D.B., Stack M.S. Epithelial ovarian cancer experimental models. Oncogene. 2014;33:3619–3633. doi: 10.1038/onc.2013.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Coscia F., Watters K.M., Curtis M., Eckert M.A., Chiang C.Y., Tyanova S., Montag A., Lastra R.R., Lengyel E., Mann M. Integrative proteomic profiling of ovarian cancer cell lines reveals precursor cell associated proteins and functional status. Nat. Commun. 2016;7:12645. doi: 10.1038/ncomms12645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goyeneche A., Lisio M.A., Fu L., Srinivasan R., Valdez Capuccino J., Gao Z.H., Telleria C. The capacity of high-grade serous ovarian cancer cells to form multicellular structures spontaneously along disease progression correlates with their orthotopic tumorigenicity in immunosuppressed mice. Cancers (Basel) 2020;12:E699. doi: 10.3390/cancers12030699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sakai W., Swisher E.M., Jacquemont C., Chandramohan K.V., Couch F.J., Langdon S.P., Wurz K., Higgins J., Villegas E., Taniguchi T. Functional restoration of BRCA2 protein by secondary BRCA2 mutations in BRCA2-mutated ovarian carcinoma. Cancer Res. 2009;69:6381–6386. doi: 10.1158/0008-5472.CAN-09-1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ai Z., Lu Y., Qiu S., Fan Z. Overcoming cisplatin resistance of ovarian cancer cells by targeting HIF-1-regulated cancer metabolism. Cancer Lett. 2016;373:36–44. doi: 10.1016/j.canlet.2016.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dai Y., Jin S., Li X., Wang D. The involvement of Bcl-2 family proteins in AKT-regulated cell survival in cisplatin resistant epithelial ovarian cancer. Oncotarget. 2017;8:1354–1368. doi: 10.18632/oncotarget.13817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li Z., Zhou W., Zhang Y., Sun W., Yung M.M.H., Sun J., Li J., Chen C.W., Li Z., Meng Y., et al. ERK regulates HIF1α-mediated platinum resistance by directly targeting PHD2 in ovarian cancer. Clin. Cancer Res. 2019;25:5947–5960. doi: 10.1158/1078-0432.CCR-18-4145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sun J., Cai X., Yung M.M., Zhou W., Li J., Zhang Y., Li Z., Liu S.S., Cheung A.N.Y., Ngan H.Y.S., et al. miR-137 mediates the functional link between c-Myc and EZH2 that regulates cisplatin resistance in ovarian cancer. Oncogene. 2019;38:564–580. doi: 10.1038/s41388-018-0459-x. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 40.Matassa D.S., Amoroso M.R., Lu H., Avolio R., Arzeni D., Procaccini C., Faicchia D., Maddalena F., Simeon V., Agliarulo I., et al. Oxidative metabolism drives inflammation-induced platinum resistance in human ovarian cancer. Cell Death Differ. 2016;23:1542–1554. doi: 10.1038/cdd.2016.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nusinow D.P., Szpyt J., Ghandi M., Rose C.M., McDonald E.R., 3rd, Kalocsay M., Jané-Valbuena J., Gelfand E., Schweppe D.K., Jedrychowski M., et al. Quantitative proteomics of the Cancer Cell Line Encyclopedia. Cell. 2020;180:387–402.e16. doi: 10.1016/j.cell.2019.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fabbro M., Savage K., Hobson K., Deans A.J., Powell S.N., McArthur G.A., Khanna K.K. BRCA1-BARD1 complexes are required for p53Ser-15 phosphorylation and a G1/S arrest following ionizing radiation-induced DNA damage. J. Biol. Chem. 2004;279:31251–31258. doi: 10.1074/jbc.M405372200. [DOI] [PubMed] [Google Scholar]
- 43.Okada S., Ouchi T. Cell cycle differences in DNA damage-induced BRCA1 phosphorylation affect its subcellular localization. J. Biol. Chem. 2003;278:2015–2020. doi: 10.1074/jbc.M208685200. [DOI] [PubMed] [Google Scholar]
- 44.Olson E., Nievera C.J., Lee A.Y., Chen L., Wu X. The Mre11-Rad50-Nbs1 complex acts both upstream and downstream of ataxia telangiectasia mutated and Rad3-related protein (ATR) to regulate the S-phase checkpoint following UV treatment. J. Biol. Chem. 2007;282:22939–22952. doi: 10.1074/jbc.M702162200. [DOI] [PubMed] [Google Scholar]
- 45.Wu X., Ranganathan V., Weisman D.S., Heine W.F., Ciccone D.N., O’Neill T.B., Crick K.E., Pierce K.A., Lane W.S., Rathbun G., et al. ATM phosphorylation of Nijmegen breakage syndrome protein is required in a DNA damage response. Nature. 2000;405:477–482. doi: 10.1038/35013089. [DOI] [PubMed] [Google Scholar]
- 46.Longerich S., Kwon Y., Tsai M.S., Hlaing A.S., Kupfer G.M., Sung P. Regulation of FANCD2 and FANCI monoubiquitination by their interaction and by DNA. Nucleic Acids Res. 2014;42:5657–5670. doi: 10.1093/nar/gku198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Boeing S., Williamson L., Encheva V., Gori I., Saunders R.E., Instrell R., Aygün O., Rodriguez-Martinez M., Weems J.C., Kelly G.P., et al. Multiomic analysis of the UV-induced DNA damage response. Cell Rep. 2016;15:1597–1610. doi: 10.1016/j.celrep.2016.04.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Halim V.A., García-Santisteban I., Warmerdam D.O., van den Broek B., Heck A.J.R., Mohammed S., Medema R.H. Doxorubicin-induced DNA damage causes extensive ubiquitination of ribosomal proteins associated with a decrease in protein translation. Mol. Cell. Proteomics. 2018;17:2297–2308. doi: 10.1074/mcp.RA118.000652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Aiken C.T., Kaake R.M., Wang X., Huang L. Oxidative stress-mediated regulation of proteasome complexes. Mol. Cell Proteomics. 2011;10 doi: 10.1074/mcp.M110.006924. R110.006924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bellare P., Small E.C., Huang X., Wohlschlegel J.A., Staley J.P., Sontheimer E.J. A role for ubiquitin in the spliceosome assembly pathway. Nat. Struct. Mol. Biol. 2008;15:444–451. doi: 10.1038/nsmb.1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lenzken S.C., Loffreda A., Barabino S.M. RNA splicing: a new player in the DNA damage response. Int. J. Cell Biol. 2013;2013:153634. doi: 10.1155/2013/153634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schmittgen T.D., Ju J.F., Danenberg K.D., Danenberg P.V. Inhibition of pre-mRNA splicing by cisplatin and platinum analogs. Int. J. Oncol. 2003;23:785–789. [PubMed] [Google Scholar]
- 53.Li M., Balamuthusamy S., Khan A.M., Maderdrut J.L., Simon E.E., Batuman V. Pituitary adenylate cyclase-activating polypeptide ameliorates cisplatin-induced acute kidney injury. Peptides. 2010;31:592–602. doi: 10.1016/j.peptides.2009.12.018. [DOI] [PubMed] [Google Scholar]
- 54.Barbie D.A., Tamayo P., Boehm J.S., Kim S.Y., Moody S.E., Dunn I.F., Schinzel A.C., Sandy P., Meylan E., Scholl C., et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009;462:108–112. doi: 10.1038/nature08460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Coulthard L.R., White D.E., Jones D.L., McDermott M.F., Burchill S.A. p38(MAPK): stress responses from molecular mechanisms to therapeutics. Trends Mol. Med. 2009;15:369–379. doi: 10.1016/j.molmed.2009.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hernández Losa J., Parada Cobo C., Guinea Viniegra J., Sánchez-Arevalo Lobo V.J., Ramón y Cajal S., Sánchez-Prieto R. Role of the p38 MAPK pathway in cisplatin-based therapy. Oncogene. 2003;22:3998–4006. doi: 10.1038/sj.onc.1206608. [DOI] [PubMed] [Google Scholar]
- 57.Reinhardt H.C., Aslanian A.S., Lees J.A., Yaffe M.B. p53-deficient cells rely on ATM- and ATR-mediated checkpoint signaling through the p38MAPK/MK2 pathway for survival after DNA damage. Cancer Cell. 2007;11:175–189. doi: 10.1016/j.ccr.2006.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xu N., Lao Y., Zhang Y., Gillespie D.A. Akt: a double-edged sword in cell proliferation and genome stability. J. Oncol. 2012;2012:951724. doi: 10.1155/2012/951724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhu K.Q., Zhang S.J. Involvement of ATM/ATR-p38 MAPK cascade in MNNG induced G1-S arrest. World J. Gastroenterol. 2003;9:2073–2077. doi: 10.3748/wjg.v9.i9.2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang A., Guo C., Sun Y., Lu L., Wang Y., Wang Q., Zhang Y., Zhang H., Wang L., Gu Y., Liu A. Overexpression of CUEDC2 predicts poor prognosis in ovarian serous carcinomas. J. Cancer. 2015;6:542–547. doi: 10.7150/jca.11420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chytil A., Waltner-Law M., West R., Friedman D., Aakre M., Barker D., Law B. Construction of a cyclin D1-Cdk2 fusion protein to model the biological functions of cyclin D1-Cdk2 complexes. J. Biol. Chem. 2004;279:47688–47698. doi: 10.1074/jbc.M405938200. [DOI] [PubMed] [Google Scholar]
- 62.Gubern A., Joaquin M., Marquès M., Maseres P., Garcia-Garcia J., Amat R., González-Nuñez D., Oliva B., Real F.X., de Nadal E., Posas F. The N-terminal phosphorylation of RB by p38 bypasses its inactivation by CDKs and prevents proliferation in cancer cells. Mol. Cell. 2016;64:25–36. doi: 10.1016/j.molcel.2016.08.015. [DOI] [PubMed] [Google Scholar]
- 63.Yang Q.E., Nagaoka S.I., Gwost I., Hunt P.A., Oatley J.M. Inactivation of retinoblastoma protein (Rb1) in the oocyte: evidence that dysregulated follicle growth drives ovarian teratoma formation in mice. PLoS Genet. 2015;11:e1005355. doi: 10.1371/journal.pgen.1005355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mayr B., Montminy M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat. Rev. Mol. Cell Biol. 2001;2:599–609. doi: 10.1038/35085068. [DOI] [PubMed] [Google Scholar]
- 65.Sakamoto K., Huang B.W., Iwasaki K., Hailemariam K., Ninomiya-Tsuji J., Tsuji Y. Regulation of genotoxic stress response by homeodomain-interacting protein kinase 2 through phosphorylation of cyclic AMP response element-binding protein at serine 271. Mol. Biol. Cell. 2010;21:2966–2974. doi: 10.1091/mbc.E10-01-0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Izar B., Tirosh I., Stover E.H., Wakiro I., Cuoco M.S., Alter I., Rodman C., Leeson R., Su M.J., Shah P., et al. A single-cell landscape of high-grade serous ovarian cancer. Nat. Med. 2020;26:1271–1279. doi: 10.1038/s41591-020-0926-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Catenacci D.V. Next-generation clinical trials: novel strategies to address the challenge of tumor molecular heterogeneity. Mol. Oncol. 2015;9:967–996. doi: 10.1016/j.molonc.2014.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sedletska Y., Giraud-Panis M.J., Malinge J.M. Cisplatin is a DNA-damaging antitumour compound triggering multifactorial biochemical responses in cancer cells: importance of apoptotic pathways. Curr. Med. Chem. Anticancer Agents. 2005;5:251–265. doi: 10.2174/1568011053765967. [DOI] [PubMed] [Google Scholar]
- 69.Siddik Z.H. Cisplatin: mode of cytotoxic action and molecular basis of resistance. Oncogene. 2003;22:7265–7279. doi: 10.1038/sj.onc.1206933. [DOI] [PubMed] [Google Scholar]
- 70.Yan S., Frank D., Son J., Hannan K.M., Hannan R.D., Chan K.T., Pearson R.B., Sanij E. The potential of targeting ribosome biogenesis in high-grade serous ovarian cancer. Int. J. Mol. Sci. 2017;18:E210. doi: 10.3390/ijms18010210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bursać S., Prodan Y., Pullen N., Bartek J., Volarević S. Dysregulated ribosome biogenesis reveals therapeutic liabilities in cancer. Trends Cancer. 2021;7:57–76. doi: 10.1016/j.trecan.2020.08.003. [DOI] [PubMed] [Google Scholar]
- 72.Giurgiu M., Reinhard J., Brauner B., Dunger-Kaltenbach I., Fobo G., Frishman G., Montrone C., Ruepp A. CORUM: the comprehensive resource of mammalian protein complexes-2019. Nucleic Acids Res. 2019;47(D1):D559–D563. doi: 10.1093/nar/gky973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhang N., Liu X., Li L., Legerski R. Double-strand breaks induce homologous recombinational repair of interstrand cross-links via cooperation of MSH2, ERCC1-XPF, REV3, and the Fanconi anemia pathway. DNA Repair (Amst.) 2007;6:1670–1678. doi: 10.1016/j.dnarep.2007.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bhagwat N., Olsen A.L., Wang A.T., Hanada K., Stuckert P., Kanaar R., D’Andrea A., Niedernhofer L.J., McHugh P.J. XPF-ERCC1 participates in the Fanconi anemia pathway of cross-link repair. Mol. Cell. Biol. 2009;29:6427–6437. doi: 10.1128/MCB.00086-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Abuzeid W.M., Jiang X., Shi G., Wang H., Paulson D., Araki K., Jungreis D., Carney J., O’Malley B.W., Jr., Li D. Molecular disruption of RAD50 sensitizes human tumor cells to cisplatin-based chemotherapy. J. Clin. Invest. 2009;119:1974–1985. doi: 10.1172/JCI33816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Altan B., Yokobori T., Ide M., Bai T., Yanoma T., Kimura A., Kogure N., Suzuki M., Bao P., Mochiki E., et al. High expression of MRE11-RAD50-NBS1 is associated with poor prognosis and chemoresistance in gastric cancer. Anticancer Res. 2016;36:5237–5247. doi: 10.21873/anticanres.11094. [DOI] [PubMed] [Google Scholar]
- 77.Dupré A., Boyer-Chatenet L., Sattler R.M., Modi A.P., Lee J.H., Nicolette M.L., Kopelovich L., Jasin M., Baer R., Paull T.T., Gautier J. A forward chemical genetic screen reveals an inhibitor of the Mre11-Rad50-Nbs1 complex. Nat. Chem. Biol. 2008;4:119–125. doi: 10.1038/nchembio.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Dahl E.S., Buj R., Leon K.E., Newell J.M., Imamura Y., Bitler B.G., Snyder N.W., Aird K.M. Targeting IDH1 as a prosenescent therapy in high-grade serous ovarian cancer. Mol. Cancer Res. 2019;17:1710–1720. doi: 10.1158/1541-7786.MCR-18-1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hovnanian A., Rebouillat D., Mattei M.G., Levy E.R., Marié I., Monaco A.P., Hovanessian A.G. The human 2′,5′-oligoadenylate synthetase locus is composed of three distinct genes clustered on chromosome 12q24.2 encoding the 100-, 69-, and 40-kDa forms. Genomics. 1998;52:267–277. doi: 10.1006/geno.1998.5443. [DOI] [PubMed] [Google Scholar]
- 80.Leisching G., Wiid I., Baker B. OAS1, 2, and 3: significance during active tuberculosis? J. Infect. Dis. 2018;217:1517–1521. doi: 10.1093/infdis/jiy084. [DOI] [PubMed] [Google Scholar]
- 81.Chang K., Pastan I. Molecular cloning of mesothelin, a differentiation antigen present on mesothelium, mesotheliomas, and ovarian cancers. Proc. Natl. Acad. Sci. USA. 1996;93:136–140. doi: 10.1073/pnas.93.1.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ghafoor A., Thomas A., Hassan R. Targeting mesothelin in ovarian cancer. Oncotarget. 2018;9:36050–36051. doi: 10.18632/oncotarget.26350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.van den Heuvel A.P., de Vries-Smits A.M., van Weeren P.C., Dijkers P.F., de Bruyn K.M., Riedl J.A., Burgering B.M. Binding of protein kinase B to the plakin family member periplakin. J. Cell Sci. 2002;115:3957–3966. doi: 10.1242/jcs.00069. [DOI] [PubMed] [Google Scholar]
- 84.Melaiu O., Stebbing J., Lombardo Y., Bracci E., Uehara N., Bonotti A., Cristaudo A., Foddis R., Mutti L., Barale R., et al. MSLN gene silencing has an anti-malignant effect on cell lines overexpressing mesothelin deriving from malignant pleural mesothelioma. PLoS ONE. 2014;9:e85935. doi: 10.1371/journal.pone.0085935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Suzuki A., Horiuchi A., Ashida T., Miyamoto T., Kashima H., Nikaido T., Konishi I., Shiozawa T. Cyclin A2 confers cisplatin resistance to endometrial carcinoma cells via up-regulation of an Akt-binding protein, periplakin. J. Cell. Mol. Med. 2010;14:2305–2317. doi: 10.1111/j.1582-4934.2009.00839.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Baeza J., Smallegan M.J., Denu J.M. Mechanisms and dynamics of protein acetylation in mitochondria. Trends Biochem. Sci. 2016;41:231–244. doi: 10.1016/j.tibs.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Guan K.L., Xiong Y. Regulation of intermediary metabolism by protein acetylation. Trends Biochem. Sci. 2011;36:108–116. doi: 10.1016/j.tibs.2010.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Parodi-Rullán R.M., Chapa-Dubocq X.R., Javadov S. Acetylation of mitochondrial proteins in the heart: the role of SIRT3. Front. Physiol. 2018;9:1094. doi: 10.3389/fphys.2018.01094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Weroha S.J., Becker M.A., Enderica-Gonzalez S., Harrington S.C., Oberg A.L., Maurer M.J., Perkins S.E., AlHilli M., Butler K.A., McKinstry S., et al. Tumorgrafts as in vivo surrogates for women with ovarian cancer. Clin. Cancer Res. 2014;20:1288–1297. doi: 10.1158/1078-0432.CCR-13-2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhang H., Liu T., Zhang Z., Payne S.H., Zhang B., McDermott J.E., Zhou J.Y., Petyuk V.A., Chen L., Ray D., et al. CPTAC Investigators Integrated proteogenomic characterization of human high-grade serous ovarian cancer. Cell. 2016;166:755–765. doi: 10.1016/j.cell.2016.05.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Aiderus A., Black M.A., Dunbier A.K. Fatty acid oxidation is associated with proliferation and prognosis in breast and other cancers. BMC Cancer. 2018;18:805. doi: 10.1186/s12885-018-4626-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Farge T., Saland E., de Toni F., Aroua N., Hosseini M., Perry R., Bosc C., Sugita M., Stuani L., Fraisse M., et al. Chemotherapy-resistant human acute myeloid leukemia cells are not enriched for leukemic stem cells but require oxidative metabolism. Cancer Discov. 2017;7:716–735. doi: 10.1158/2159-8290.CD-16-0441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ye H., Adane B., Khan N., Sullivan T., Minhajuddin M., Gasparetto M., Stevens B., Pei S., Balys M., Ashton J.M., et al. Leukemic stem cells evade chemotherapy by metabolic adaptation to an adipose tissue niche. Cell Stem Cell. 2016;19:23–37. doi: 10.1016/j.stem.2016.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Pastò A., Pagotto A., Pilotto G., De Paoli A., De Salvo G.L., Baldoni A., Nicoletto M.O., Ricci F., Damia G., Bellio C., et al. Resistance to glucose starvation as metabolic trait of platinum-resistant human epithelial ovarian cancer cells. Oncotarget. 2017;8:6433–6445. doi: 10.18632/oncotarget.14118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bauerschlag D.O., Maass N., Leonhardt P., Verburg F.A., Pecks U., Zeppernick F., Morgenroth A., Mottaghy F.M., Tolba R., Meinhold-Heerlein I., Bräutigam K. Fatty acid synthase overexpression: target for therapy and reversal of chemoresistance in ovarian cancer. J. Transl. Med. 2015;13:146. doi: 10.1186/s12967-015-0511-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Shao H., Mohamed E.M., Xu G.G., Waters M., Jing K., Ma Y., Zhang Y., Spiegel S., Idowu M.O., Fang X. Carnitine palmitoyltransferase 1A functions to repress FoxO transcription factors to allow cell cycle progression in ovarian cancer. Oncotarget. 2016;7:3832–3846. doi: 10.18632/oncotarget.6757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Currie E., Schulze A., Zechner R., Walther T.C., Farese R.V., Jr. Cellular fatty acid metabolism and cancer. Cell Metab. 2013;18:153–161. doi: 10.1016/j.cmet.2013.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Dar S., Chhina J., Mert I., Chitale D., Buekers T., Kaur H., Giri S., Munkarah A., Rattan R. Bioenergetic adaptations in chemoresistant ovarian cancer cells. Sci. Rep. 2017;7:8760. doi: 10.1038/s41598-017-09206-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kruszynska Y.T., Sherratt H.S. Glucose kinetics during acute and chronic treatment of rats with 2[6(4-chloro-phenoxy)hexyl]oxirane-2-carboxylate, etomoxir. Biochem. Pharmacol. 1987;36:3917–3921. doi: 10.1016/0006-2952(87)90458-8. [DOI] [PubMed] [Google Scholar]
- 100.Kennedy J.A., Kiosoglous A.J., Murphy G.A., Pelle M.A., Horowitz J.D. Effect of perhexiline and oxfenicine on myocardial function and metabolism during low-flow ischemia/reperfusion in the isolated rat heart. J. Cardiovasc. Pharmacol. 2000;36:794–801. doi: 10.1097/00005344-200012000-00016. [DOI] [PubMed] [Google Scholar]
- 101.Kennedy J.A., Unger S.A., Horowitz J.D. Inhibition of carnitine palmitoyltransferase-1 in rat heart and liver by perhexiline and amiodarone. Biochem. Pharmacol. 1996;52:273–280. doi: 10.1016/0006-2952(96)00204-3. [DOI] [PubMed] [Google Scholar]
- 102.Yao C.H., Liu G.Y., Wang R., Moon S.H., Gross R.W., Patti G.J. Identifying off-target effects of etomoxir reveals that carnitine palmitoyltransferase I is essential for cancer cell proliferation independent of β-oxidation. PLoS Biol. 2018;16:e2003782. doi: 10.1371/journal.pbio.2003782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Raud B., Roy D.G., Divakaruni A.S., Tarasenko T.N., Franke R., Ma E.H., Samborska B., Hsieh W.Y., Wong A.H., Stüve P., et al. Etomoxir actions on regulatory and memory T cells are independent of Cpt1a-mediated fatty acid oxidation. Cell Metab. 2018;28:504–515.e7. doi: 10.1016/j.cmet.2018.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Divakaruni A.S., Hsieh W.Y., Minarrieta L., Duong T.N., Kim K.K.O., Desousa B.R., Andreyev A.Y., Bowman C.E., Caradonna K., Dranka B.P., et al. Etomoxir inhibits macrophage polarization by disrupting CoA homeostasis. Cell Metab. 2018;28:490–503.e7. doi: 10.1016/j.cmet.2018.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Rao J.N., Warren G.Z.L., Estolt-Povedano S., Zammit V.A., Ulmer T.S. An environment-dependent structural switch underlies the regulation of carnitine palmitoyltransferase 1A. J. Biol. Chem. 2011;286:42545–42554. doi: 10.1074/jbc.M111.306951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kurmi K., Hitosugi S., Wiese E.K., Boakye-Agyeman F., Gonsalves W.I., Lou Z., Karnitz L.M., Goetz M.P., Hitosugi T. Carnitine palmitoyltransferase 1A has a lysine succinyltransferase activity. Cell Rep. 2018;22:1365–1373. doi: 10.1016/j.celrep.2018.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Vella S., Penna I., Longo L., Pioggia G., Garbati P., Florio T., Rossi F., Pagano A. Perhexiline maleate enhances antitumor efficacy of cisplatin in neuroblastoma by inducing over-expression of NDM29 ncRNA. Sci. Rep. 2015;5:18144. doi: 10.1038/srep18144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.He P.J., Ge R.F., Mao W.J., Chung P.S., Ahn J.C., Wu H.T. Oxidative stress induced by carboplatin promotes apoptosis and inhibits migration of HN-3 cells. Oncol. Lett. 2018;16:7131–7138. doi: 10.3892/ol.2018.9563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kleih M., Böpple K., Dong M., Gaißler A., Heine S., Olayioye M.A., Aulitzky W.E., Essmann F. Direct impact of cisplatin on mitochondria induces ROS production that dictates cell fate of ovarian cancer cells. Cell Death Dis. 2019;10:851. doi: 10.1038/s41419-019-2081-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.He F., Ru X., Wen T. NRF2, a transcription factor for stress response and beyond. Int. J. Mol. Sci. 2020;21:E4777. doi: 10.3390/ijms21134777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kant S., Kesarwani P., Guastella A.R., Kumar P., Graham S.F., Buelow K.L., Nakano I., Chinnaiyan P. Perhexiline demonstrates FYN-mediated antitumor activity in glioblastoma. Mol. Cancer Ther. 2020;19:1415–1422. doi: 10.1158/1535-7163.MCT-19-1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Schlaepfer I.R., Rider L., Rodrigues L.U., Gijón M.A., Pac C.T., Romero L., Cimic A., Sirintrapun S.J., Glodé L.M., Eckel R.H., Cramer S.D. Lipid catabolism via CPT1 as a therapeutic target for prostate cancer. Mol. Cancer Ther. 2014;13:2361–2371. doi: 10.1158/1535-7163.MCT-14-0183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Rottenberg S., Disler C., Perego P. The rediscovery of platinum-based cancer therapy. Nat. Rev. Cancer. 2021;21:37–50. doi: 10.1038/s41568-020-00308-y. [DOI] [PubMed] [Google Scholar]
- 114.Ferguson F.M., Gray N.S. Kinase inhibitors: the road ahead. Nat. Rev. Drug Discov. 2018;17:353–377. doi: 10.1038/nrd.2018.21. [DOI] [PubMed] [Google Scholar]
- 115.Florea A.M., Büsselberg D. Anti-cancer drugs interfere with intracellular calcium signaling. Neurotoxicology. 2009;30:803–810. doi: 10.1016/j.neuro.2009.04.014. [DOI] [PubMed] [Google Scholar]
- 116.Shen L., Wen N., Xia M., Zhang Y.U., Liu W., Xu Y.E., Sun L. Calcium efflux from the endoplasmic reticulum regulates cisplatin-induced apoptosis in human cervical cancer HeLa cells. Oncol. Lett. 2016;11:2411–2419. doi: 10.3892/ol.2016.4278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Erickson J.R., Joiner M.L., Guan X., Kutschke W., Yang J., Oddis C.V., Bartlett R.K., Lowe J.S., O’Donnell S.E., Aykin-Burns N., et al. A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell. 2008;133:462–474. doi: 10.1016/j.cell.2008.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Luczak E.D., Anderson M.E. CaMKII oxidative activation and the pathogenesis of cardiac disease. J. Mol. Cell. Cardiol. 2014;73:112–116. doi: 10.1016/j.yjmcc.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Wang Q., Huang L., Yue J. Oxidative stress activates the TRPM2-Ca2+-CaMKII-ROS signaling loop to induce cell death in cancer cells. Biochim. Biophys. Acta Mol. Cell Res. 2017;1864:957–967. doi: 10.1016/j.bbamcr.2016.12.014. [DOI] [PubMed] [Google Scholar]
- 120.Litchfield D.W. Protein kinase CK2: structure, regulation and role in cellular decisions of life and death. Biochem. J. 2003;369:1–15. doi: 10.1042/BJ20021469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Rabalski A.J., Gyenis L., Litchfield D.W. Molecular pathways: emergence of protein kinase CK2 (CSNK2) as a potential target to inhibit survival and DNA damage response and repair pathways in cancer cells. Clin. Cancer Res. 2016;22:2840–2847. doi: 10.1158/1078-0432.CCR-15-1314. [DOI] [PubMed] [Google Scholar]
- 122.Olsen B.B., Wang S.Y., Svenstrup T.H., Chen B.P., Guerra B. Protein kinase CK2 localizes to sites of DNA double-strand break regulating the cellular response to DNA damage. BMC Mol. Biol. 2012;13:7. doi: 10.1186/1471-2199-13-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Keller D.M., Zeng X., Wang Y., Zhang Q.H., Kapoor M., Shu H., Goodman R., Lozano G., Zhao Y., Lu H. A DNA damage-induced p53 serine 392 kinase complex contains CK2, hSpt16, and SSRP1. Mol. Cell. 2001;7:283–292. doi: 10.1016/s1097-2765(01)00176-9. [DOI] [PubMed] [Google Scholar]
- 124.Siddiqui-Jain A., Bliesath J., Macalino D., Omori M., Huser N., Streiner N., Ho C.B., Anderes K., Proffitt C., O’Brien S.E., et al. CK2 inhibitor CX-4945 suppresses DNA repair response triggered by DNA-targeted anticancer drugs and augments efficacy: mechanistic rationale for drug combination therapy. Mol. Cancer Ther. 2012;11:994–1005. doi: 10.1158/1535-7163.MCT-11-0613. [DOI] [PubMed] [Google Scholar]
- 125.Matsumoto S., Masai H. Regulation of chromosome dynamics by Hsk1/Cdc7 kinase. Biochem. Soc. Trans. 2013;41:1712–1719. doi: 10.1042/BST20130217. [DOI] [PubMed] [Google Scholar]
- 126.Costanzo V., Shechter D., Lupardus P.J., Cimprich K.A., Gottesman M., Gautier J. An ATR- and Cdc7-dependent DNA damage checkpoint that inhibits initiation of DNA replication. Mol. Cell. 2003;11:203–213. doi: 10.1016/s1097-2765(02)00799-2. [DOI] [PubMed] [Google Scholar]
- 127.Zegerman P., Diffley J.F. Checkpoint-dependent inhibition of DNA replication initiation by Sld3 and Dbf4 phosphorylation. Nature. 2010;467:474–478. doi: 10.1038/nature09373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Tenca P., Brotherton D., Montagnoli A., Rainoldi S., Albanese C., Santocanale C. Cdc7 is an active kinase in human cancer cells undergoing replication stress. J. Biol. Chem. 2007;282:208–215. doi: 10.1074/jbc.M604457200. [DOI] [PubMed] [Google Scholar]
- 129.Tsuji T., Lau E., Chiang G.G., Jiang W. The role of Dbf4/Drf1-dependent kinase Cdc7 in DNA-damage checkpoint control. Mol. Cell. 2008;32:862–869. doi: 10.1016/j.molcel.2008.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Rainey M.D., Harhen B., Wang G.N., Murphy P.V., Santocanale C. Cdc7-dependent and -independent phosphorylation of Claspin in the induction of the DNA replication checkpoint. Cell Cycle. 2013;12:1560–1568. doi: 10.4161/cc.24675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Yamada M., Watanabe K., Mistrik M., Vesela E., Protivankova I., Mailand N., Lee M., Masai H., Lukas J., Bartek J. ATR-Chk1-APC/CCdh1-dependent stabilization of Cdc7-ASK (Dbf4) kinase is required for DNA lesion bypass under replication stress. Genes Dev. 2013;27:2459–2472. doi: 10.1101/gad.224568.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Cheng A.N., Lo Y.K., Lin Y.S., Tang T.K., Hsu C.H., Hsu J.T., Lee A.Y. Identification of novel Cdc7 kinase inhibitors as anti-cancer agents that target the interaction with Dbf4 by the fragment complementation and drug repositioning approach. EBioMedicine. 2018;36:241–251. doi: 10.1016/j.ebiom.2018.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Nieman K.M., Kenny H.A., Penicka C.V., Ladanyi A., Buell-Gutbrod R., Zillhardt M.R., Romero I.L., Carey M.S., Mills G.B., Hotamisligil G.S., et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat. Med. 2011;17:1498–1503. doi: 10.1038/nm.2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Wang A.W., Prieto J.M., Cauvi D.M., Bickler S.W., De Maio A. The greater omentum-a vibrant and enigmatic immunologic organ involved in injury and infection resolution. Shock. 2020;53:384–390. doi: 10.1097/SHK.0000000000001428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Mukherjee A., Chiang C.Y., Daifotis H.A., Nieman K.M., Fahrmann J.F., Lastra R.R., Romero I.L., Fiehn O., Lengyel E. Adipocyte-induced FABP4 expression in ovarian cancer cells promotes metastasis and mediates carboplatin resistance. Cancer Res. 2020;80:1748–1761. doi: 10.1158/0008-5472.CAN-19-1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Ladanyi A., Mukherjee A., Kenny H.A., Johnson A., Mitra A.K., Sundaresan S., Nieman K.M., Pascual G., Benitah S.A., Montag A., et al. Adipocyte-induced CD36 expression drives ovarian cancer progression and metastasis. Oncogene. 2018;37:2285–2301. doi: 10.1038/s41388-017-0093-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Sawyer B.T., Qamar L., Yamamoto T.M., McMellen A., Watson Z.L., Richer J.K., Behbakht K., Schlaepfer I.R., Bitler B.G. Targeting fatty acid oxidation to promote anoikis and inhibit ovarian cancer progression. Mol. Cancer Res. 2020;18:1088–1098. doi: 10.1158/1541-7786.MCR-19-1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Gharpure K.M., Pradeep S., Sans M., Rupaimoole R., Ivan C., Wu S.Y., Bayraktar E., Nagaraja A.S., Mangala L.S., Zhang X., et al. FABP4 as a key determinant of metastatic potential of ovarian cancer. Nat. Commun. 2018;9:2923. doi: 10.1038/s41467-018-04987-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Tucker S.L., Gharpure K., Herbrich S.M., Unruh A.K., Nick A.M., Crane E.K., Coleman R.L., Guenthoer J., Dalton H.J., Wu S.Y., et al. Molecular biomarkers of residual disease after surgical debulking of high-grade serous ovarian cancer. Clin. Cancer Res. 2014;20:3280–3288. doi: 10.1158/1078-0432.CCR-14-0445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Nallanthighal S., Rada M., Heiserman J.P., Cha J., Sage J., Zhou B., Yang W., Hu Y., Korgaonkar C., Hanos C.T., et al. Inhibition of collagen XI alpha 1-induced fatty acid oxidation triggers apoptotic cell death in cisplatin-resistant ovarian cancer. Cell Death Dis. 2020;11:258. doi: 10.1038/s41419-020-2442-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Rada M., Nallanthighal S., Cha J., Ryan K., Sage J., Eldred C., Ullo M., Orsulic S., Cheon D.J. Inhibitor of apoptosis proteins (IAPs) mediate collagen type XI alpha 1-driven cisplatin resistance in ovarian cancer. Oncogene. 2018;37:4809–4820. doi: 10.1038/s41388-018-0297-x. [DOI] [PubMed] [Google Scholar]
- 142.Jeon S.M., Chandel N.S., Hay N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature. 2012;485:661–665. doi: 10.1038/nature11066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Qu Q., Zeng F., Liu X., Wang Q.J., Deng F. Fatty acid oxidation and carnitine palmitoyltransferase I: emerging therapeutic targets in cancer. Cell Death Dis. 2016;7:e2226. doi: 10.1038/cddis.2016.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Panieri E., Santoro M.M. ROS homeostasis and metabolism: a dangerous liason in cancer cells. Cell Death Dis. 2016;7:e2253. doi: 10.1038/cddis.2016.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Dianov G.L., Parsons J.L. Co-ordination of DNA single strand break repair. DNA Repair (Amst.) 2007;6:454–460. doi: 10.1016/j.dnarep.2006.10.009. [DOI] [PubMed] [Google Scholar]
- 146.Srinivas U.S., Tan B.W.Q., Vellayappan B.A., Jeyasekharan A.D. ROS and the DNA damage response in cancer. Redox Biol. 2019;25:101084. doi: 10.1016/j.redox.2018.101084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Qin L., Fan M., Candas D., Jiang G., Papadopoulos S., Tian L., Woloschak G., Grdina D.J., Li J.J. CDK1 enhances mitochondrial bioenergetics for radiation-induced DNA repair. Cell Rep. 2015;13:2056–2063. doi: 10.1016/j.celrep.2015.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Weaver A.N., Yang E.S. Beyond DNA repair: additional functions of PARP-1 in cancer. Front. Oncol. 2013;3:290. doi: 10.3389/fonc.2013.00290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Pike L.S., Smift A.L., Croteau N.J., Ferrick D.A., Wu M. Inhibition of fatty acid oxidation by etomoxir impairs NADPH production and increases reactive oxygen species resulting in ATP depletion and cell death in human glioblastoma cells. Biochim. Biophys. Acta. 2011;1807:726–734. doi: 10.1016/j.bbabio.2010.10.022. [DOI] [PubMed] [Google Scholar]
- 150.Han S., Wei R., Zhang X., Jiang N., Fan M., Huang J.H., Xie B., Zhang L., Miao W., Butler A.C., et al. CPT1A/2-mediated FAO enhancement-a metabolic target in radioresistant breast cancer. Front. Oncol. 2019;9:1201. doi: 10.3389/fonc.2019.01201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Pacilli A., Calienni M., Margarucci S., D’Apolito M., Petillo O., Rocchi L., Pasquinelli G., Nicolai R., Koverech A., Calvani M., et al. Carnitine-acyltransferase system inhibition, cancer cell death, and prevention of myc-induced lymphomagenesis. J. Natl. Cancer Inst. 2013;105:489–498. doi: 10.1093/jnci/djt030. [DOI] [PubMed] [Google Scholar]
- 152.Ricciardi M.R., Mirabilii S., Allegretti M., Licchetta R., Calarco A., Torrisi M.R., Foà R., Nicolai R., Peluso G., Tafuri A. Targeting the leukemia cell metabolism by the CPT1a inhibition: functional preclinical effects in leukemias. Blood. 2015;126:1925–1929. doi: 10.1182/blood-2014-12-617498. [DOI] [PubMed] [Google Scholar]
- 153.Samudio I., Konopleva M. Targeting leukemia’s “fatty tooth”. Blood. 2015;126:1874–1875. doi: 10.1182/blood-2015-08-665125. [DOI] [PubMed] [Google Scholar]
- 154.Phan T.T., Shivu G.N., Choudhury A., Abozguia K., Davies C., Naidoo U., Ahmed I., Yousef Z., Horowitz J., Frenneaux M. Multi-centre experience on the use of perhexiline in chronic heart failure and refractory angina: old drug, new hope. Eur. J. Heart Fail. 2009;11:881–886. doi: 10.1093/eurjhf/hfp106. [DOI] [PubMed] [Google Scholar]
- 155.Chong C.R., Sallustio B., Horowitz J.D. Drugs that affect cardiac metabolism: focus on perhexiline. Cardiovasc. Drugs Ther. 2016;30:399–405. doi: 10.1007/s10557-016-6664-3. [DOI] [PubMed] [Google Scholar]
- 156.Vacheron A. [The classic anti-anginal agents and molsidomine] Arch. Mal. Coeur Vaiss. 1983;76:71–75. [PubMed] [Google Scholar]
- 157.Campeau L. The Canadian Cardiovascular Society grading of angina pectoris revisited 30 years later. Can. J. Cardiol. 2002;18:371–379. [PubMed] [Google Scholar]
- 158.Gupta A.K., Winchester D., Pepine C.J. Antagonist molecules in the treatment of angina. Expert Opin. Pharmacother. 2013;14:2323–2342. doi: 10.1517/14656566.2013.834329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Morgan M.Y., Reshef R., Shah R.R., Oates N.S., Smith R.L., Sherlock S. Impaired oxidation of debrisoquine in patients with perhexiline liver injury. Gut. 1984;25:1057–1064. doi: 10.1136/gut.25.10.1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Horowitz J.D., Sia S.T., Macdonald P.S., Goble A.J., Louis W.J. Perhexiline maleate treatment for severe angina pectoris--correlations with pharmacokinetics. Int. J. Cardiol. 1986;13:219–229. doi: 10.1016/0167-5273(86)90146-4. [DOI] [PubMed] [Google Scholar]
- 161.Mondal T., Shivange G.N., Tihagam R.G., Lyerly E., Battista M., Talwar D., Mosavian R., Urbanek K., Rashid N.S., Harrell J.C., et al. Unexpected PD-L1 immune evasion mechanism in TNBC, ovarian, and other solid tumors by DR5 agonist antibodies. EMBO Mol. Med. 2021;13:e12716. doi: 10.15252/emmm.202012716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Krug K., Mertins P., Zhang B., Hornbeck P., Raju R., Ahmad R., Szucs M., Mundt F., Forestier D., Jane-Valbuena J., et al. A curated resource for phosphosite-specific signature analysis. Mol. Cell. Proteomics. 2019;18:576–593. doi: 10.1074/mcp.TIR118.000943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Hornbeck P.V., Kornhauser J.M., Latham V., Murray B., Nandhikonda V., Nord A., Skrzypek E., Wheeler T., Zhang B., Gnad F. 15 years of PhosphoSitePlus®: integrating post-translationally modified sites, disease variants and isoforms. Nucleic Acids Res. 2019;47(D1):D433–D441. doi: 10.1093/nar/gky1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Keshava Prasad T.S., Goel R., Kandasamy K., Keerthikumar S., Kumar S., Mathivanan S., Telikicherla D., Raju R., Shafreen B., Venugopal A., et al. Human Protein Reference Database--2009 update. Nucleic Acids Res. 2009;37:D767–D772. doi: 10.1093/nar/gkn892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Liberzon A., Subramanian A., Pinchback R., Thorvaldsdóttir H., Tamayo P., Mesirov J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics. 2011;27:1739–1740. doi: 10.1093/bioinformatics/btr260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Johnson W.E., Li C., Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8:118–127. doi: 10.1093/biostatistics/kxj037. [DOI] [PubMed] [Google Scholar]
- 167.Savage S.R., Shi Z., Liao Y., Zhang B. Graph algorithms for condensing and consolidating gene set analysis results. Mol. Cell. Proteomics. 2019;18(8, suppl 1):S141–S152. doi: 10.1074/mcp.TIR118.001263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Liao Y., Wang J., Jaehnig E.J., Shi Z., Zhang B. WebGestalt 2019: gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 2019;47(W1):W199–W205. doi: 10.1093/nar/gkz401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Schneider C.A., Rasband W.S., Eliceiri K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Gu Z., Eils R., Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. 2016;32:2847–2849. doi: 10.1093/bioinformatics/btw313. [DOI] [PubMed] [Google Scholar]
- 171.Huttlin E.L., Jedrychowski M.P., Elias J.E., Goswami T., Rad R., Beausoleil S.A., Villén J., Haas W., Sowa M.E., Gygi S.P. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell. 2010;143:1174–1189. doi: 10.1016/j.cell.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Rose C.M., Isasa M., Ordureau A., Prado M.A., Beausoleil S.A., Jedrychowski M.P., Finley D.J., Harper J.W., Gygi S.P. Highly multiplexed quantitative mass spectrometry analysis of ubiquitylomes. Cell Syst. 2016;3:395–403.e4. doi: 10.1016/j.cels.2016.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Erickson B.K., Jedrychowski M.P., McAlister G.C., Everley R.A., Kunz R., Gygi S.P. Evaluating multiplexed quantitative phosphopeptide analysis on a hybrid quadrupole mass filter/linear ion trap/orbitrap mass spectrometer. Anal. Chem. 2015;87:1241–1249. doi: 10.1021/ac503934f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Xi R., Hadjipanayis A.G., Luquette L.J., Kim T.M., Lee E., Zhang J., Johnson M.D., Muzny D.M., Wheeler D.A., Gibbs R.A., et al. Copy number variation detection in whole-genome sequencing data using the Bayesian information criterion. Proc. Natl. Acad. Sci. USA. 2011;108:E1128–E1136. doi: 10.1073/pnas.1110574108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Mermel C.H., Schumacher S.E., Hill B., Meyerson M.L., Beroukhim R., Getz G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011;12:R41. doi: 10.1186/gb-2011-12-4-r41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Bauer D.F. Constructing confidene sets using rank statistics. J. Am. Stat. Assoc. 1972;67:687–690. [Google Scholar]
- 177.Laird N.M., Ware J.H. Random-effects models for longitudinal data. Biometrics. 1982;38:963–974. [PubMed] [Google Scholar]
- 178.Stordal B., Timms K., Farrelly A., Gallagher D., Busschots S., Renaud M., Thery J., Williams D., Potter J., Tran T., et al. BRCA1/2 mutation analysis in 41 ovarian cell lines reveals only one functionally deleterious BRCA1 mutation. Mol. Oncol. 2013;7:567–579. doi: 10.1016/j.molonc.2012.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Butler K.A., Hou X., Becker M.A., Zanfagnin V., Enderica-Gonzalez S., Visscher D., Kalli K.R., Tienchaianada P., Haluska P., Weroha S.J. Prevention of human lymphoproliferative tumor formation in ovarian cancer patient-derived xenografts. Neoplasia. 2017;19:628–636. doi: 10.1016/j.neo.2017.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Navarrete-Perea J., Yu Q., Gygi S.P., Paulo J.A. Streamlined tandem mass tag (SL-TMT) protocol: an efficient strategy for quantitative (phospho)proteome profiling using tandem mass tag-synchronous precursor selection-MS3. J. Proteome Res. 2018;17:2226–2236. doi: 10.1021/acs.jproteome.8b00217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Rappsilber J., Ishihama Y., Mann M. Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal. Chem. 2003;75:663–670. doi: 10.1021/ac026117i. [DOI] [PubMed] [Google Scholar]
- 182.Udeshi N.D., Mertins P., Svinkina T., Carr S.A. Large-scale identification of ubiquitination sites by mass spectrometry. Nat. Protoc. 2013;8:1950–1960. doi: 10.1038/nprot.2013.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Ting L., Rad R., Gygi S.P., Haas W. MS3 eliminates ratio distortion in isobaric multiplexed quantitative proteomics. Nat. Methods. 2011;8:937–940. doi: 10.1038/nmeth.1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.McAlister G.C., Nusinow D.P., Jedrychowski M.P., Wühr M., Huttlin E.L., Erickson B.K., Rad R., Haas W., Gygi S.P. MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal. Chem. 2014;86:7150–7158. doi: 10.1021/ac502040v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Schroeder M.J., Shabanowitz J., Schwartz J.C., Hunt D.F., Coon J.J. A neutral loss activation method for improved phosphopeptide sequence analysis by quadrupole ion trap mass spectrometry. Anal. Chem. 2004;76:3590–3598. doi: 10.1021/ac0497104. [DOI] [PubMed] [Google Scholar]
- 186.Erickson B.K., Mintseris J., Schweppe D.K., Navarrete-Perea J., Erickson A.R., Nusinow D.P., Paulo J.A., Gygi S.P. Active instrument engagement combined with a real-time database search for improved performance of sample multiplexing workflows. J. Proteome Res. 2019;18:1299–1306. doi: 10.1021/acs.jproteome.8b00899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Elias J.E., Gygi S.P. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods. 2007;4:207–214. doi: 10.1038/nmeth1019. [DOI] [PubMed] [Google Scholar]
- 188.Wang P., Tang H., Zhang H., Whiteaker J., Paulovich A.G., Mcintosh M. Normalization regarding non-random missing values in high-throughput mass spectrometry data. Pac. Symp. Biocomput. 2006:315–326. [PubMed] [Google Scholar]
- 189.Paster E.V., Villines K.A., Hickman D.L. Endpoints for mouse abdominal tumor models: refinement of current criteria. Comp. Med. 2009;59:234–241. [PMC free article] [PubMed] [Google Scholar]
- 190.Subramanian, et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. PNAS. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Liberzon, et al. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Systems. 2015;1:417–425. doi: 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Poux S., Arighi C.N., Magrane M., Bateman A., Wei C.-H., Lu Z., Boutet E., Bye-A-Jee H., Famiglietti M.L., Roechert B., The UniProt Consortium On expert curation and scalability: UniProtKB/Swiss-Prot as a case study. Bioinformatics. 2017;33:3454–3460. doi: 10.1093/bioinformatics/btx439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Chen L.S., Paul D., Prentice R.L., Wang P. A regularized Hotelling’s T2 test for pathway analysis in proteomic studies. J. Am. Stat. Assoc. 2011;106:1345–1360. doi: 10.1198/jasa.2011.ap10599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Oberg A.L., Heinzen E.P., Hou X., Al Hilli M.M., Hurley R.M., Wahner Hendrickson A.E., Goergen K.M., Larson M.C., Becker M.A., Eckel-Passow J.E., et al. Statistical analysis of comparative tumor growth repeated measures experiments in the ovarian cancer patient derived xenograft (PDX) setting. Sci. Rep. 2021;11:8076. doi: 10.1038/s41598-021-87470-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Differential proteogenomic features were identified that associated with 3 hypotheses, as follows. Hypothesis 1: Platinum induces changes in both sensitive and resistant cell lines (i.e., a combined analysis of all cell lines). Hypothesis 2: Sensitive and Resistant cell lines differ at baseline (mock). Hypothesis 3: Platinum-induced changes differ between Sensitive and Resistant cell lines. On the “Assn test Summary” worksheet, the number of significant proteins, PTMs or RNAs under each hypothesis are shown in each cell, with hyperlinks to the results in other worksheets. In the result tabs, for Hypothesis 1, “FC_time_8 ” is 8Hr / 0Hr fold change for all cell lines (both sensitive and resistant). “FC_time_24 ” is 24Hr / 0Hr fold change. (FC > 1 indicates upregulation after treatment; FC < 1 indicates downregulation.) For Hypothesis 3, “Dir_int_8” or “Dir_int_24” are directions whether the fold changes over baseline (FC = 8hr/0hr or 24hr/0hr) is larger or smaller in sensitive cell lines as compared to resistant cell lines. “p-value” and “FDR” are the Wilcoxon statistic values. A single sample overview of proteins and posttranslational modifications that were both identified and quantified is provided in the Data Summary worksheet.
Number of significant pathways in each of the 3 hypotheses (described in the Table S1 legend) are shown in each cell, with hyperlink to results from the “Pathway test summary” worksheet. In the result worksheets, “size” is the number of genes observed in the data from the pathway. “p-value” and “FDR” are the Wilcoxon statistic values. “dir” is directions whether the expression of the pathways is up or down (8 or 24 hour compared to 0 hour for Hypothesis 1; sensitive compared to resistant for Hypothesis 2 and 3).
related to Figure 4 A) Gene level DNA copy number data derived from segmented CNV profiles based on WGS (See STAR Methods). For a given cell line and a given gene, its CNV number is set to be the log ratio measure of the CNV-segment the gene belongs to. For the column names: “Start” is the starting location on the chromosome for the CNV segment the gene belongs to; “End” is the end location; “PEA1.S” through “PEO23.R” are the CNV (log ratio measure) of genes for each of the 6 cell lines. B) Integrated multiomic association studies conducted on all three cell line pairs to identify differences between sensitive and resistant cell lines. For the column names: “Start” is the starting location on the chromosome for the CNV segment the gene belongs to; “End” is the end location. For a given gene, “CNV_FC” is the average CNV (log) fold change (FC) across the three pairs of cell lines, where the log FC of each pair of cell line is calculated as the differences between its log ratio CNV in the sensitive cell line and that in the paired resistant cell line; “CNV_p,” “CNV_FDR” are the p value and FDR of the paired test. Similarly, “prot_FC” or “rna_FC” are the mean of log2(sensitive/resistant) of the protein abundance or RNA in the paired tests between sensitive and resistant cell lines; “prot_p” and “rna_p” are p values of the corresponding paired tests; “prot_RNA_cnv_p” is the combined p value based on protein, RNA and CNV; “prot_RNA_cnv_FDR” is the combined FDR based on protein, RNA and CNV.
Data Availability Statement
All LC-MS/MS proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository1 with the dataset identifier PRIDE:PXD020764 (http://proteomecentral.proteomexchange.org/cgi/GetDataset?ID=PXD020764). All RNA sequencing data have been deposited to the National Center for Biotechnology Information Gene Expression Omnibus (GEO)2 with GEO Series accession GEO:GSE163152 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE163152). All whole genome sequencing data have been deposited to the National Center for Biotechnology Information Sequencing Read Archive (SRA)3 with the BioProject accession # SRA:PRJNA684350 (https://www.ncbi.nlm.nih.gov/sra/?term=PRJNA684350).