

Summary
Understanding the molecular determinants that underpin the clinical heterogeneity of non-muscle-invasive bladder cancer (NMIBC) is essential for prognostication and therapy development. Stage T1 disease in particular presents a high risk of progression and requires improved understanding. We present a detailed multi-omics study containing gene expression, copy number, and mutational profiles that show relationships to immune infiltration, disease recurrence, and progression to muscle invasion. We compare expression and genomic subtypes derived from all NMIBCs with those derived from the individual disease stages Ta and T1. We show that sufficient molecular heterogeneity exists within the separate stages to allow subclassification and that this is more clinically meaningful for stage T1 disease than that derived from all NMIBCs. This provides improved biological understanding and identifies subtypes of T1 tumors that may benefit from chemo- or immunotherapy.
Keywords: non-muscle-invasive bladder cancer, NMIBC, stage Ta, stage T1, genomics, transcriptomics, copy number, classification, clinical outcome, PPAR gamma
Graphical abstract
Highlights
-
•
Separate subtyping of stage Ta (non-invasive) and T1 (submucosal invasion) tumors
-
•
Increased biological understanding compared with subtypes derived from all NMIBCs
-
•
Infiltrated subtypes of Ta and T1 with relationship to PPARG activity
-
•
Differential prevalence of cisplatin sensitivity-related mutations in T1 subtypes
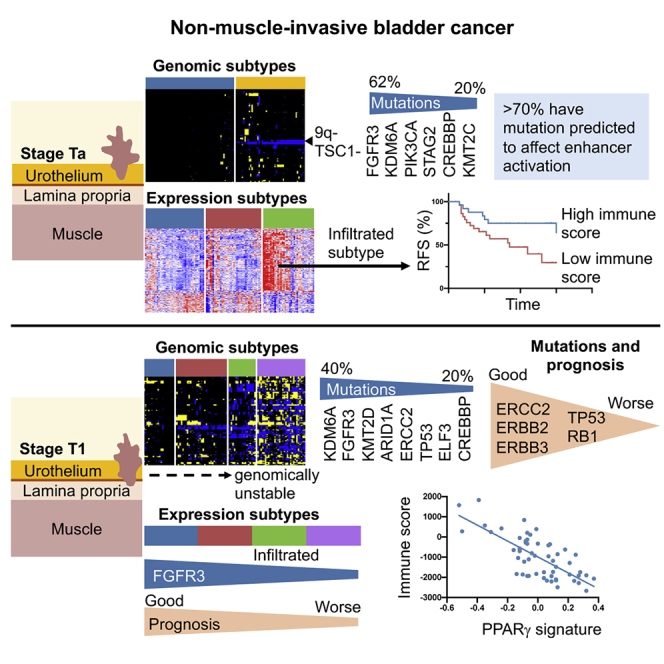
Hurst et al. compare molecular subtyping of all NMIBCs with separate subclassification of stage Ta (non-invasive) and T1 (submucosal invasion) tumors. They show that overall NMIBC subtypes are no more prognostic for T1 tumors than stage alone. However, stage-stratified subclassification identifies clinically relevant features and biological insights and promises improved prognostication.
Introduction
More than 70% of bladder tumors are non-muscle-invasive bladder cancers (NMIBCs), with over 380,000 diagnosed per annum worldwide.1 Affected individuals suffer frequent recurrence, necessitating long-term cystoscopic monitoring, with associated morbidity and high cost. Overall, bladder cancer is more expensive to treat than other cancers because of the cost of managing NMIBC.2 The majority are stage Ta tumors that do not penetrate the epithelial basement membrane, but approximately 20% are stage T1 that invade the submucosa and have a high risk of progression to muscle invasion.3 Knowledge of the molecular landscape of these tumors is incomplete. For T1, prognostic biomarkers are needed, and for all NMIBCs, improved biological understanding should allow development of novel localized therapies to reduce or eliminate risk of recurrence and progression.
NMIBCs are molecularly and clinically heterogeneous. Expression analysis has identified subclasses with relationships to outcome4,5 and expression signatures with prognostic value.6, 7, 8 Analysis of all tumor grades and stages has also identified transcriptional subgroups, one of which, “urothelial-like,” predominates in NMIBC.9 Similarly, DNA copy number and mutation analyses reveal genomic diversity,10, 11, 12, 13 with some genomic signatures showing relationships to outcome.14 However, detailed understanding of the relationships of genomic and expression features and of tumor phenotype is lacking, and the clinical implications of NMIBC subtypes require further clarification.
Longitudinal studies of primary NMIBC and recurrences in the same individual show sequential acquisition of molecular alterations and subclonal evolution.15,16 Instillation of mitomycin C in Ta disease and Bacillus Calmette-Guérin (BCG)or courses of intravesical chemotherapy in T1 disease can cause further genomic alterations, induce transcriptional changes, and impose selective pressure. Thus, for discovery of diagnostic and prognostic molecular information, analysis of primary tumors is important. To improve understanding of the biology of NMIBC and evaluate the potential of molecular subtyping to add information at the time of diagnosis, we focused on primary tumors. We provide the largest whole-exome sequence dataset for T1 tumors and, using copy number, mutation, and transcriptome profiling, explore the values of combining stage Ta and T1 samples and of separating into tumor stage groups for analysis. We show that the latter provides deeper insights into biology and clinical behavior, and suggestions for therapy.
Results
Study design, samples, and analysis platforms
Fresh-frozen tissues from 113 stage Ta and 104 high-grade stage T1 bladder tumors and paired blood samples were analyzed. Apart from 10 T1 samples, all were primary tumors. Follow-up data were available for 107 individuals with Ta disease and 88 with T1 disease (median, 55 months). Recurrence was recorded in 45% of Ta and 47% of T1 cases. A single Ta case and 16 T1 cases progressed to muscle-invasive bladder cancer (MIBC) or metastatic bladder cancer. Clinicopathologic information and analysis platforms are given in Table S1A. All were analyzed for copy number alterations and genome-wide mRNA expression. Whole-exome data were obtained for 58 T1 samples, and all other samples were analyzed using targeted sequencing (Table S2).13
Studies by the UROMOL consortium show that analysis of NMIBC separately from MIBC provides improved biological understanding and prognostic information.4,5 Because Ta and T1 tumors show distinct clinical behavior, we hypothesized that further information could be gained by separate analysis. Thus, we compared analysis of the entire dataset with separate analysis of Ta and T1 samples.
Combined analysis of NMIBC
Unsupervised clustering of copy number (CN) data revealed 4 clusters (CN1–CN4) of increasing genomic complexity, with Ta samples contained largely in CN1 and CN2 and T1 samples in CN3 and CN4 (Figures 1A and 1B). The fraction of genome altered (FGA) increased dramatically from CN1 to CN4 (Figure S1A). High FGA was associated with worse progression-free survival (PFS) (Figure S1B) but not recurrence-free survival (RFS). There was differential distribution of the most common mutations with most TP53 mutations in CN4 and a predominance of FGFR3 mutations in CN1 and CN2 (Figure 1C).
Figure 1.

Combined molecular analysis of stage Ta and T1 NMIBC tumors
(A) CN clusters. Columns, samples; rows, genomic position; yellow, gain; blue, loss. Left: chromosome number. Top: cluster designation and stage.
(B) Relationship of tumor stage to CN subtype.
(C) Distribution of common mutations according to CN subtype.
(D) Relationship of tumor stage to expression subtype.
(E) Relationships of CN and expression subtypes.
(B–E) Chi square test (with Bonferroni correction in B, D, and E).
(F) Top: distribution of CN subtypes, stage, common mutations, and disease progression according to expression subtype. Black, stage T1/progression; red, mutation present. Center: gene expression signatures (standardized Z scores) with differential expression across subtypes (p < 0.0001). Bottom: alignment to UROMOL2021 and LundTax classifications.
(G) Progression-free survival (PFS) stratified according to CN subtype.
(H) PFS according to expression subtype.
(G and H) Log rank analysis.
(I) 12-gene progression risk score7 and weighted CIS score17 in CN subtypes.
(J) 12-gene progression risk score and weighted CIS score in expression subtypes.
(I and J) Kruskal-Wallis test with Dunn’s multiple comparison correction.
Mean, 25th and 75th percentiles, and minimum and maximum values are shown. ∗∗∗∗p < 0.0001, ∗∗∗p < 0.001, ∗∗p < 0.01. See also Figures S1 and S2.
We used two-stagenon-negative matrix factorization (NMF) analysis (k = 2–5) to discover transcriptional subtypes. The two initial groups segregated samples largely according to stage. Independent analysis of these groups generated 4 subtypes (E1–E4) (Figures S1C and 1D). Alignment of transcriptional and CN subtypes showed most alignment between complex CN samples (CN4) and E3. E1 contained largely CN1 and CN2 samples (Figure 1E). TP53 mutations were predominantly in E3, FGFR3 and KDM6A in E1 and E2, and STAG2 in E1 (Figure 1F).
Gene Ontology analysis showed upregulation of categories related to protein synthesis in the first of the two initial groups and cell cycle and immune response in the second, features that dominated when Ta were compared with T1 tumors. In pairwise analysis of E1–E4, E4 had lowest expression of a compiled urothelial differentiation signature.9,18 An FGFR3 mutation-associated signature9 was higher in E1 and E2 and a score for immune cells19 in E4. Cell cycle and DNA repair signatures were also differentially distributed (Figures 1F and S2A–S2D).
RFS was unrelated to CN subtype, but expression subtypes had differential RFS that approached significance, with E3 differing significantly from E2 (Figure S2E). CN and expression subtypes showed differences in PFS (Figures 1G and H) related to expression of progression and carcinoma in situ (CIS) signature scores,7,17,20 with the infiltrated subtype (E4) having a better outcome than E3 (Figures 1F, 1I, and 1J).
These subtypes were clearly associated with tumor stage (Figures 1B and 1D) but did not provide additional prognostic information. Because this is most urgently needed for T1 tumors, we asked whether subtype assignment had prognostic utility when applied to T1 tumors alone. Importantly, we found no differences in PFS between the subtypes (Figure S2F). This suggests that, although diversity within the dataset allowed robust classification, this provided little information beyond the known worse outcome of T1 disease.
The UROMOL study also identified 4 expression classes of NMIBC.5 UROMOL2021 class assignments were derived, and these aligned with good concordance (Figure 1F). Most progression events occurred in UROMOL2021 class 2a/E3, followed by class 2b/E4. E1 and E2 aligned closely with UROMOL2021 classes 1 and 3, which also show higher expression of early cell cycle genes and FGFR3-related signature. UROMOL2021 classes 2a and 2b have higher expression of late cell cycle and DNA repair genes, as in E3 and E4 (Figures 1F and S2B–S2D). We conclude that both cohorts have similar features and that the distinct subclassification methods generated similar results. We also classified the samples according to the LundTax system.21 E1 and E2 samples were almost all classified as UroA. All but one sample classified as genomically unstable (GU) were E3/UROMOL2021 class 2a, and UroB samples were all E4/UROMOL2021 class 2b (Figure 1F). As with the subtypes derived here, neither of these systems showed a significant relationship to PFS in stage T1 samples only (log rank analysis; p = 0.46 and 0.67, respectively).
Independent analysis of stage Ta samples
CN and mutational features
Previously, we defined two groups of Ta tumors, designated genomic subtypes 1 and 2 (GS1 and GS2), demarcated largely by loss of chromosome arm 9q in GS2. Expression data of a subset identified mTORC1 signaling as the most significant difference.13 These subtypes are shown in Figure 2A. In this expanded transcriptional dataset, we confirmed upregulation of late cell cycle, DNA repair, cholesterol synthesis, and unfolded protein response genes in GS2, compatible with loss of the 9q mTORC1 regulator TSC1 (Figure S3A). Despite this difference, these subtypes were not related to grade or RFS, although tumor grade was related to RFS (Figure S3B).
Figure 2.

Independent molecular analysis of stage Ta tumors
(A) CN clusters. Columns, samples; rows, genomic position; yellow, CN gain; blue, CN loss. Left: chromosome number. Top: genomic and expression subtype.
(B) Frequencies of mutations identified by targeted sequencing.
(C) Mutations in chromatin modifier genes that affect enhancer activation status. Blue, mutant; gray, wild type.
(D) TMB as SNVs per megabase according to FGFR3 and KMT2D mutation. Mann-Whitney test.
(E) Recurrence-free survival (RFS) according to PIK3CA mutation status. M, mutant; WT, wild type.
(F) Top: distribution of GS and selected mutations according to expression subtype. Blue, GS1; yellow, GS2; red, mutation present. Lower heatmaps show regulon activity (dES values). Blocks are color coded (left) according to regulon clusters in Figure S4F.
(G) RFS according to expression subtype.
(H) ESTIMATE immune score (left) and expression levels of CD274 (PD-L1) (right) according to expression subtype. Kruskal-Wallis test with Dunn’s multiple comparison correction.
(D and H) Mean, 25th, and 75th percentiles, minimum and maximum values are shown.
(I) RFS according to high (top 25th percentile) and low (lowest 25th percentile) immune score.
(E, G, and I) Log-rank analysis.
∗∗∗∗p < 0.0001, ∗∗∗p < 0.001, ∗∗p < 0.01. See also Figures S3 and S4.
Mean non-synonymous mutation frequency from targeted sequencing (Table S2) was 7.9 mutations per megabase (median, 7.55) and C > T and C > G mutations dominated (43% and 25%). The single base substitution (SBS) signatures SBS2 and SBS13 (https://cancer.sanger.ac.uk/signatures/sbs/), which are attributed to activity of the APOBEC family of cytidine deaminases, accounted for 35% of single-nucleotide variants (SNVs), with 29% of tumors showing 50% or more and 61% showing 25% or more such mutations. These mutations were more common in GS2, consistent with increased expression of APOBEC3A, APOBEC3B, and APOBEC3H (Figures S3C and S3D).
As in previous studies by us and others, FGFR3 mutations were most common (62%), followed by KDM6A, PIK3CA, and STAG2. Genes mutated in 10% or more of tumors included KMT2A, KMT2C, KMT2D, CREBBP, EP300, RYR2, CDKN1A, ATM, ZFP36L1, and TSC1 (Figure 2B; Table S1B). FGFR3 and HRAS mutations were mutually exclusive, with one or other in 70%. Compatible with 9q location, TSC1 mutations were more common in GS2 (p = 0.0016). KMT2D mutations were more common in GS1 and KMT2A mutations in GS2 (p = 0.038 and 0.027, respectively).
The histone methyltransferases KMT2C, KMT2D, and KDM6A participate in large multisubunit KMT2C/D COMPASS-like complexes that are recruited to enhancers via interaction with transcription and pioneer factors. KMT2C and KMT2D carry out monomethylation of H3K4, and this, together with H3K27 acetylation by the acetyltransferases CREBBP and EP300, leads to enhancer activation.22 One or more COMPASS-like complex components were mutated in 65% of samples, and 34% had CREBBP or EP300 mutation so that 73% had a mutation predicted to affect enhancer activation (Figure 2C). Of the 7 most common mutations identified, FGFR3 and KMT2D mutations were associated with higher tumor mutational burden (TMB) (Figure 2D). Although FGFR3 mutation is linked to an APOBEC mutational process,23 we found no relationship to APOBEC mutations or expression. Only PIK3CA mutation was related to RFS (Figure 2E), and no relationship of TMB and RFS was found.
Three Ta transcriptional subtypes
NMF analysis identified three expression subtypes (TaE1, TaE2, and TaE3) (Figure S3E). TaE1 and TaE3 contained many GS1 tumors and TaE2 the majority of GS2 (Figures 2A, 2F, and S3F) and high-grade tumors (p = 0.0034). Differences in mutation frequency of STAG2 and KMT2D (p = 0.0028 and 0.0119, respectively) (Figure 2F) and RFS were found (Figure 2G).
We examined differential expression between subtypes (Figure S3G; Table S3). TaE1 was enriched in Gene Ontology categories associated with RNA transcription and protein synthesis (Figure S4A) and many small nucleolar RNAs (snoRNAs) that play a major role in rRNA modification (Figure S4B; Table S4). TaE2 was enriched in features of GS2, including late cell cycle genes, cholesterol homeostasis, fatty acid metabolism, response to hypoxia, and glycolysis. Features related to cell division included “sister chromatid segregation” (Gene Ontology [GO]:819), including STAG2, which had fewest mutations in TaE2 (Figures 2F and S3G). TaE2 also had highest expression of the HIF1α-regulated long non-coding RNA UCA124 (Figure S4C), compatible with activated mTORC1. FGFR3 mutation was directly related to the FGFR3-associated signature (Figure S4D). Expression of transcriptional regulators implicated in urothelial development and differentiation was higher in TaE1 (Figure S4E).
Regulon analysis25, 26, 27 identified 373 with significant activity. These were strongly associated with subtypes, confirming their biologically distinct features (Figure S4F). Two major patterns of activity were revealed, with TaE2 showing differences from TaE1 and TaE3 (Figure 2F), including E2F1, E2F2, and FOXM1 regulons, compatible with enhanced cell cycle activity. SREBF2 activity reflected the preponderance in TaE2 of GS2 tumors with upregulated sterol and lipid synthesis. TaE1 and TaE3 had activity of KLF5 and GRHL2, factors associated with urothelial differentiation.28,29 Both also had higher activity of AR, TP53 and TP63 regulons, and activity of anterior HOXA genes and RXRA was enriched. TaE3 differed from TaE1 in activity of the interferon regulatory factors IRF4 and IRF8 and other factors involved in immune regulation (MSC, EOMES, IKZF1, and SPI1). This analysis also revealed some heterogeneity within the subtypes, suggesting that further subclassification is possible (Figure S4F).
Ta tumors with increased immune infiltration have the lowest recurrence rate
All comparisons with TaE3, which had improved RFS compared with other subtypes, identified “immune response” (GO:6955) (Figure S4G) and terms related to the inflammatory response (Table S3). ESTIMATE immune scores30 and PD-L1 expression were elevated (Figure 2H). Differential RFS was found in individuals with the highest and lowest immune infiltration scores independent of subtype (Figure 2I), but immune scores were not related to mutations or FGA despite more GS1 samples in the infiltrated subtype (Figure S3F).
We interrogated the nature of the immune infiltrate.31 Interferon signaling was highest in TaE3 (p < 0.0001), and significant infiltration by plasma cells (p = 0.002), T cells (p < 0.0001), macrophages (p < 0.0001), monocytes (p = 0.014), and neutrophils (p = 0.001) was detected. Expression profiles for activated CD8+ and effector memory CD8+ T cells, related to cancer immunogenicity,32 and a T regulatory (Treg) cell signature19 were elevated (Figure S4H). Despite expression of immunosuppressive biomarkers, cytolytic activity estimated from granzyme (GZMA) and perforin (PRF1) expression was also higher in TaE3 (Figure S4I). This suggests that longer RFS in TaE3 is related to an upregulated anti-tumor response.
Independent analysis of stage T1 tumors
CN and mutational features
Although stage T1 tumors share features with MIBC, their molecular landscape has not been well studied. We defined four CN subgroups (T1CN1–T1CN4). T1CN1 had few CN alterations, including chromosome 9 deletions, and T1CN3 was dominated by losses rather than gains (Figure 3A). FGA increased from T1CN1 to T1CN4 (Figure S5A). Alterations conformed to our previous findings,10 with frequent losses of 2q (40%), 9p (52%), 9q (52%), 11p (48%), and 17p (40%) and gains of 1q (51%), 5q (27%), 8q (52%), 17q (28%), and 20q (48%). Regions of amplification were identified on 14 chromosome arms. The most common were on 3p25.1 in 7% of samples (NUP210 and IQSEC1), 3p25.2 in 9% (including PPARG and RAF1), 6p22.3 in 8% (including E2F3, SOX4, and CDKAL1), 8q22.2-q22.3 in 6% (including YWHAZ, GRHL2, and KLF10), 11q13.3 in 13% (including CCND1, FGF19, FGF4, and FGF3), and 12q15 in 8% (including MDM2). Homozygous deletions (HD) were infrequent apart from 9p21 (CDKN2A; 12%) (Table S5). The subtypes had similar RFS (p = 0.6) but showed differential PFS and a relationship of FGA to PFS (Figures S5B and S5C). In pairwise comparisons, the major expression differences were upregulation of DNA replication, DNA repair, cell cycle, and cell division gene sets in T1CN4.
Figure 3.

Independent molecular analysis of stage T1 tumors
(A) CN clusters. Columns, samples; rows, genomic position; yellow, CN gain; blue, CN loss. Left: chromosome number. Top: CN subtype and TP53 and FGFR3 mutation status. Black, mutation present.
(B) TMB as SNVs per megabase according to CN subtype. Kruskal-Wallis test. Bars indicate mean and SD.
(C) Expression of APOBEC3A and APOBEC3B in Ta and T1 tumors.
(D) Mutation frequencies in Ta and T1 tumors for genes mutated in 5% or more of samples in either group.
(E) Distribution of FGFR3 and TP53 mutations in CN subtypes.
(F) Distribution of ERCC2 mutations in CN subtypes. Fisher’s exact text.
(G) PFS according to TP53 mutation status. Log rank analysis.
(H) Mutations in DDR genes analyzed by whole-exome sequencing (any of ERCC2, ATM, RB1, ATR, BRCA2, POLE, FANCC, and CHEK2) according to CN subtype.
(E and H) Chi-square test with Bonferroni correction.
(I) TMB as SNVs per megabase according to mutations in DDR genes (any of ERCC2, ATM, RB1, ATR, BRCA2, POLE, FANCC, and CHEK2) in tumors analyzed by whole-exome sequencing.
(C and I) Mann-Whitney test. Mean, 25th and 75th percentiles, minimum and maximum values are shown.
(J) Relationships of DDR gene mutations. Blue, mutant; gray, wild type.
See also Figure S5.
Whole-exome sequencing (mean 87× coverage, 89% of bases > 30×) identified 49,477 somatic SNVs (mean 868 and median 450 per sample). Interestingly, more SNVs were present in the chromosomally stable subtype T1CN1 (Figure 3B), and PFS analysis showed a better outcome for those with medium or high TMB (Figure S5D). C > T transitions (60%) and C > G transversions (17%) dominated. NMF analysis identified two major mutational signatures, one with features of APOBEC-induced mutation, which contributed to more than 50% of mutations in 68% of samples, and a second that contained predominantly C > T and T > C, which likely represents a mixture of common signatures reported previously in bladder cancer (Figure S5E).33 Assessment of SBS signatures34 revealed many dominated by the APOBEC signatures SBS2 and SBS13 (79% of samples) and others containing SBS1 and SBS5; the latter is related to ERCC2 mutation (Figure S5F).35,36 APOBEC3A and APOBEC3B expression was higher than in Ta tumors (Figure 3C).
dNdScv analysis37 identified 23 genes with predicted driver function (Table S6). FAT1, ATM, RHOB, KRAS, and RBM10 fell just below the cutoff for significance, likely because of the small sample size. FGFR3, PIK3CA, and STAG2 mutations were less frequent than in Ta (Figure 3D; Table S1B) but mutations in many genes, including ARID1A, ELF3, ERCC2, and TP53 were more frequent. However, these frequencies did not align closely with those reported in MIBC38 (Figure S5G). Importantly, this intermediate profile was not due to Ta-like or MIBC-like profiles of individual tumors but due to intermediate profiles within tumors (Figure S5H).
Two genes not implicated previously in NMIBC, KAT8 regulatory non-specific lethal (NSL) complex subunit 1 (KANSL1) and G protein subunit alpha 13 (GNA13), were predicted drivers. GNA13 is a subunit of a heterotrimeric G-protein that mediates signaling through specific G-protein-coupled receptors (GPCRs). Four of 5 mutations detected were missense mutations in codon R200. Inactivating mutations in KANSL1, encoding a protein found in chromatin-modifying complexes,39,40 were found in three samples. Mutations in genes involved in the COMPASS-like complex and/or EP300 or CREBBP were frequent (65%). FGFR3 mutations were more common in T1CN1 and T1CN2 and TP53 mutations in T1CN3 and T1CN4 (Figure 3E). ERCC2 mutation was most common in T1CN1 (Figure 3F). Of the 6 most commonly mutated genes, TP53 mutation was associated with worse PFS, as expected41 (Figure 3G), and ERCC2 mutation with favorable PFS (see below).
In MIBC, mutations in ERCC2 and other DNA damage response (DDR) genes predict response to cisplatin-based chemotherapy42, 43, 44, 45 and immune checkpoint inhibitors.46 ERBB2 mutations are also linked to cisplatin response.47 Because individuals with high-risk T1 disease may be considered for such therapies,48 we evaluated the distribution of these mutations. In exome sequence data, 53% of tumors had 1 DDR gene mutation or more, with more in T1CN1 and T1CN4 (Figure 3H), associated with higher TMB (Figure 3I). In the entire series, 50% had ERCC2, RB1, ATM, BRCA2, or ERBB2 mutations (Figure 3J).
ERCC2 mutation was present in 24% of T1 tumors compared with 4% in Ta. Apart from one frameshift, all were missense mutations in the helical motif regions (Figure 4A). Seven of these are functionally inactivating, including N238S and T484M, found in seven and three samples, respectively,49 and 6 new mutations are close to known detrimental mutations (Figure 4B). Compatible with high TMB in T1CN1 (Figure 3B), ERCC2 mutant tumors showed higher TMB (median, 14.84 mutations per megabase; IQR, 10.2–25.04) than wild-type tumors (median, 7.42 mutations per megabase; IQR, 4.64–11.13) (Figure 4C), and mutation was associated with favorable PFS (Figure 4D). Of other DDR genes analyzed separately, only ATM mutations, most not found with ERCC2 mutation, were associated with higher TMB (p = 0.006). No difference in PFS was detected for those with 1 mutation or more in ATM, RB1, or BRCA2.
Figure 4.

ERCC2, ERBB2, and ERBB3 mutations in stage T1 tumors
(A) ERCC2 protein showing positions and frequency of point mutations identified.
(B) Structure of the ERCC2 protein (PDB: 5IVW) showing positions of mutated residues. Red, residues with mutations reported previously to affect function; green, novel mutations.
(C) TMB as SNVs per megabase in ERCC2 mutant and WT samples. Bars indicate mean and SD.
(D) PFS according to ERCC2 mutation status.
(E) ERBB2 protein, showing positions and frequency of point mutations.
(F) ERBB3 protein, showing positions and frequency of point mutations.
(G) TMB as SNVs per megabase in ERBB2 (left) and ERBB3 (right) mutant and WT T1 samples. Bars indicate mean and SD.
(C and G) Mann-Whitney test.
(H) PFS according to ERBB2 and/or ERBB3 mutation status.
(D and H) Log rank analysis.
ERBB2 and ERBB3 mutations are reported in 12% and 10% of MIBC, respectively.38 Here, mutations were present in 14% and 16% of stage T1 (Figures 4E and 4F; Table S7). Most ERBB2 mutations were potentially activating mutations focused in the extracellular region. 33% had focal gain of the ERBB2 region or all of 17q, and a single tumor had high-level amplification. Mutations were unrelated to CN or expression subtype, but gains were most common (60%) in T1CN4 (p < 0.0001). As in other tumor types,50 8 tumors had mutations in both receptors (p < 0.05). Three of only four potentially inactivating mutations in either gene were in tumors with concomitant point mutations, suggesting selection for mutant-only heterodimers. There was co-occurrence with ERCC2 mutation (Table S7), and, accordingly, ERBB2 and ERBB3 mutations were linked to high TMB (Figure 4G) and longer PFS (Figure 4H). Thus, DNA-based features can subdivide T1 tumors into subtypes with different clinical outcomes and suggest systemic chemotherapy or targeted therapies.
Four T1 transcriptional subtypes
Two-stage NMF analysis revealed four subtypes (T1E1–T1E4) (Figures 5A and S6A). Differences in RFS between T1E1 and T1E4 approached significance (Figure 5B). T1E1 showed best and T1E4 worst PFS. The progression rates at 5 years for subtypes T1E1–T1E4 were in the order 5%, 15%, 17%, and 25%, respectively. This was reflected in progression signature scores (Figures 5C and 5D). Because RFS and PFS curves showed crossing over at early time points after diagnosis (Figures 5B and 5C), we carried out Cox regression analysis to estimate the long-term survival rate under proportional and non-proportional hazard assumptions. This suggested a change in hazard ratios over time (Figure S6B). Relationships to CN subtypes and distribution of common genomic features in these subtypes are shown in Figures 5E and 5F. 6p22.3 (E2F3) amplification and RB1 mutation were only found in T1E3 and T1E4, and RB1 and CDKN2A expression were inversely correlated (r = −0.62). TP53 mutation was higher in T1E2, T1E3, and T1E4, which had more CN alterations (FGA in the order T1E4 > T1E2 > T1E3 > T1E1) (Figure S6C). DDR gene mutations were common in T1E3 and T1E4 (Figure 5G).
Figure 5.

Features of stage T1 expression subtypes
(A) Heatmaps of Z scores for selected GO categories and expression signatures according to expression subtype. Cytosolic ribosome, GO:22626; Translational initiation, GO:6413; DNA replication, GO:6260; Immune response, GO:6955.
(B) RFS according to expression subtype.
(C) PFS according to expression subtype.
(D) 12-gene progression risk score in expression subtypes.
(E) Relationships of CN and expression subtypes.
(F) Selected CN alterations and gene expression according to expression subtype. Black, amplification or mutation; white, normal or WT; yellow, no data.
(G) ERCC2, ATM, and/or RB1 mutations according to expression subtype.
(E and G) Chi-square test with Bonferroni correction.
(H) FGFR3 signature with mutation frequency and differentiation signature in expression subtypes.
(D and H) Kruskal-Wallis test with Dunn’s multiple comparison correction.
(I) Regulon activity profiles according to expression subtypes. Blocks are color coded according to regulon clusters in Figure S6E.
(J) Left: Cox multivariate regression analysis showing the proportional hazards of each regulon, gender, and age, indicating the contribution of each variable to PFS. Right: Kaplan-Meier plot for PFS, stratified by positive versus negative regulon activity status for THAP4 and MYBL1. BH-adjusted p values.
(B, C, and J right) Log rank analysis.
Mean, 25th, and 75th percentiles, minimum and maximum values are shown. ∗∗∗∗p < 0.0001, ∗p < 0.05. See also Figure S6.
Pairwise comparison of the two initial NMF groups revealed higher expression of genes involved in translational initiation, protein targeting, and ribosome biogenesis in the first (T1E1 and T1E2) and of genes related to immune and inflammatory responses in the second (T1E3 and T1E4) (Figure 5A; Table S8). Pairwise comparisons of T1E1–T1E4 showed higher cell cycle, DNA repair, DNA replication, and metabolism-related gene expression in T1E2 and T1E4. Cholesterol and lipid biosynthesis genes and genes involved in the unfolded protein response were highest in T1E4, and categories related to glucose metabolism and canonical glycolysis highest in T1E2. T1E3 was enriched in immune and inflammatory response genes, expressed in the order T1E3 > T1E4 > T1E1 > T1E2 (Figure 5A).
The urothelial differentiation signature was high in T1E1, T1E2, and T1E4 (Figures 5A and 5H). Very low levels of this and of differentiation-associated transcriptional regulators were present in some T1E3 samples (Figure S6D). The FGFR3 signature aligned with mutation frequency (Figure 5H), correlated with early cell cycle gene expression (r = 0.47, p < 0.0001), and was inversely related to late cell cycle gene expression (r = −0.49, p < 0.0001) and CIS signature (r = −0.78, p < 0.0001).
We identified 286 regulons with differential activity (Figures 5I and S6E). E2F1 and FOXM1 activity reflects upregulated cell cycle activity in T1E4 and T1E2. As in Ta, KLF5 and TP63 activity was higher in the more differentiated subtypes. PPARG activity was reduced in T1E3, and T1E4 showed strikingly reduced levels of MYC, nuclear factor κB (NF-κB), and SMAD3 activity. Some highly active regulons in T1E3 (Figure S6E) relate to immune cell function (EOMES, MSC, AIRE, STAT4), but many do not and have no known function or are implicated in neural or testis development or function. We assessed the relationships of regulon differential enrichment scores in relation to RFS and PFS using a multivariate analysis that considered gender and age. HIF1A3 and ZBTB8B activity was associated with reduced RFS (log rank p = 0.027 and 0.04, respectively). Increased PFS was associated with THAP4 and ELF1 activity and reduced PFS with MYBL1 activity (Figure 5J). THAP4 and MYBL1 activities were strongly negatively correlated (r = −0.75, p < 0.0001). Examination of genes correlated with MYBL1 levels revealed negative correlation with TP63 (r = −0.544, p < 0.0001) and the PPARG coactivator PPARGC1B (r = −0.545, p < 0.0001).
A stage T1 subtype with enhanced immune infiltration
ESTIMATE stromal and immune scores were highest in T1E3 (Figure 6A). These were not related to FGA, TMB, or single mutations but had negative relationships with DNA repair gene expression51 (ESTIMATE r = −0.35, p = 0.0002; 60-gene signature19 r = −0.53, p < 0.0001). Deconvolution of expression data provided further insights. Hallmark gene sets for interferon γ (IFNγ) and IFNα responses were upregulated in T1E3 (p < 0.0001 for both). Signatures related to cytotoxic T cells, TBX21, marking Th1 helper cells, IFNG, and cytolytic response markers were present. Markers of Treg cells, interleukin-10 (IL-10) and other immunoregulatory markers (PD-L1 and CTLA4) were upregulated (Figure 6B), suggesting an active but suppressed immune response. A 6-gene T effector signature linked to favorable checkpoint inhibitor response in bladder cancer was upregulated,52 and we noted striking upregulation of a T cell inflamed signature related to response in melanoma53 (Figure S7A). Immune infiltration and PD-L1 expression were higher in samples from females (Figure 6C). However, immune infiltration score or T effector cell signature themselves did not predict outcome.
Figure 6.

Immune infiltration is related to PPARG signaling in NMIBC
(A) ESTIMATE immune signature score in T1 expression subtypes.
(B) Heatmap of Z scores for 60-gene immune cell signature19 in T1 expression subtypes.
(C) 60-gene immune score (left) and PD-L1 expression (right) according to gender in T1 tumors. Mann-Whitney test.
(D) PPARG expression according to T1 expression subtype.
(E) PPARG signature score54 according to T1 expression subtype.
(A, D, and E) Kruskal-Wallis test with Dunn’s multiple comparison correction. ∗∗∗∗p < 0.0001, ∗∗∗p < 0.001, ∗∗p < 0.01.
(A, C, D, and E) Mean, 25th and 75th percentiles, minimum and maximum values are shown.
(F) Correlation of PPARG signature and differentiation score in T1 tumors.
(G) Correlation of PPARG signature and ESTIMATE immune score in T1 tumors.
(H) Correlation of PPARG signature and ESTIMATE immune score in Ta tumors.
(I) Correlation of PPARG signature and DNA repair gene expression in all NMIBCs.
PPARG mutation and expression are related to immune infiltration in NMIBC
The nuclear hormone receptor PPARγ plays a key role in control of urothelial differentiation.55 Conversely, in luminal bladder tumors, amplification, increased expression, and gain of function mutations in it and its binding partner RXRA are implicated in driving disease.56,57 Upregulated PPARG expression and/or RXRA mutation is associated with evasion of immunosurveillance and reduced response to immunotherapy in MIBC.57 Because the situation in NMIBC is not known, we assessed PPARG and RXRA mutations, PPARG gene amplification (3p25.2), PPARG expression, and a PPARG-related transcriptional signature.54
We found PPARG mutations in five and RXRA mutations in three T1 exome-sequenced tumors (14%), including a functionally active mutation (T475M)56 and mutations near or within the ligand-binding domain of PPARG (Q231R, M284I, K291N, M491I). Two RXRA mutations (S427F) also reported in MIBC38 are known to be activating. PPARG gain or amplification was present in 26%, implicating it by mutation or gain in 34% of T1 overall (Table S9). Only one PPARG mutation was identified in 17 Ta exome sequences analyzed and three mutations in RXRA in the entire Ta series (2.6%). No Ta sample had amplification.
PPARG regulon activity, PPARG expression, and the related expression signature were lowest in T1E3 (Figures 5A, 5I, 6D, and 6E). There was strong correlation of differentiation signature and PPARG signature (Figure 6F) and negative correlation of PPARG signature and immune scores (Pearson r = −0.615). We questioned whether correlation of PPARG and differentiation score was due to variations in tumor cell purity. Evaluation of two epithelial cell signatures identified 14 samples with possible lower tumor cell content (Figure S7B). With these removed, a negative relationship of ESTIMATE and PPARG scores remained (Figure 6G). Expression of chemokine chemoattractants for effector T cells that are modulated by PPARG expression in bladder tumor cells57 was high in T1E3 (Figure S7C). Upregulated PPARG signature was associated with 6 of 8 PPARG/RXRA mutations. The two PPARG-mutant samples with no CN gain or upregulation of PPARG or its signature were in T1E3, suggesting that these mutations have no effect (Table S9).
For validation, we evaluated immune and PPARG expression signatures in two independent T1 expression datasets.4,9 In both cases, supervised clustering identified approximately one third of samples with immune signature negatively correlated with PPARG signature (Figures S7D and S7E).
In Ta samples, the lowest PPARG signature was in TaE3, which had the highest immune infiltrate (Figure S7F). Here, correlation with differentiation signature (Figure S7G) and negative correlation with immune and PPARG scores (Figure 6H) were also found.
Finally, we examined other features suggested to relate to immune infiltration and the PPARG signature. In all NMIBCs, there was weak correlation with FGA (r = 0.17, p = 0.01) but a strong relationship with DNA repair gene expression51 (r = 0.41, p < 0.0001) (Figure 6I). This was reflected in association of “DNA repair” (GO:6281) with genes positively associated with PPARG expression (p = 4 × 10−6). Overall, this indicates that, as in MIBC, the strength of PPARG signaling is directly related to immune infiltration and suggests a relationship of PPARG signaling to DNA repair processes.
BCG response
Sixty-three T1 individuals received a full BCG induction course and maintenance. Thirty-eight (60%) suffered no recurrence, and 10 progressed. Most progression cases were in T1E3 and T1E4 (8 of 10), but expression and CN subtype were not related to recurrence. CIS and progression signatures were related to progression (p = 0.015 and 0.03, respectively). No relationships with PPARG signature, ESTIMATE scores, or mutations in DDR genes were found, but only one of the 10 individuals who progressed (at 71 months) had ERCC2 mutation compared with 25% each in the recurrence and no recurrence groups. Median TMB was 10.2 per megabase, 10.2 per megabase, and 5.6 per megabase in those with no recurrence, recurrence, or progression, respectively, but did not reach significance. Transcriptome comparison identified no differential expression between those with and without recurrence. However, there was a relationship of TP53 mutation to post-BCG recurrence (p = 0.02) that was reflected in RFS (p = 0.0074) and PFS (p = 0.0031) (Figures 7A and 7B).
Figure 7.

TP53 mutation and outcome in BCG-treated T1 individuals and alignment of expression subtypes to UROMOL2021 and LundTax classifications
(A) RFS in BCG-treated individuals according to TP53 mutation status.
(B) PFS in BCG-treated individuals according to TP53 mutation status.
(A and B) Log-rank analysis.
(C) Alignment of Ta expression subtypes with UROMOL2021 and LundTax classifications.
(D) Alignment of T1 expression subtypes with UROMOL2021 and LundTax classifications.
(E) Heatmap of Z scores for expression of genes associated with LundTax GU subtype in T1 expression subtypes.
(F) Alignment of T1 CN subtypes with UROMOL2021 and LundTax classifications.
Alignment with UROMOL2021 and LundTax classifications
Comparison of stage-specific subtypes with UROMOL2021 and LundTax classifications (Figures 7C, 7D, and 7F) showed virtually all Ta samples classified as Lund “UroA” and many as UROMOL2021 class 1. Many T1E1 cases were UROMOL2021 classes 1, 2b, and 3. All T1E4 and many T1E2 tumors were UROMOL2021 class 2a, compatible with high late-cell-cycle gene expression in these groups (Figure 5A). UROMOL2021 class 2b aligned with T1E3, reflecting higher infiltration. There was strong alignment of T1E1, T1E2, and T1E3 with the Lund Uro subtypes, and only T1E4 samples were classified as GU (p < 0.0001). GU tumors have low FGFR3, CCND1, TP63, and KRT5 expression and common loss of RB1 with commensurate CDKN2A upregulation,21 features of T1E4 (Figure 7E). Lund UroB-classified samples were all T1E3. UroB samples have a lower differentiation signature and features of squamous differentiation, including expression of KRT5 and KRT14,58 consistent with the lowest differentiation signature (Figure 5H), and frequent KRT5 upregulation, and both samples were classified as Ba/Sq in T1E3 (Figure 7D). Alignment of CN subtypes was less clear, but UROMOL2021 class 2a aligned most closely with T1CN3 and T1CN4 (Figure 7F).
Discussion
We show that, when analyzed together, NMIBCs segregate into CN and expression subtypes that align closely with tumor stage and outcome. The transcriptional subtypes were similar to those of the UROMOL consortium.5 In both studies, T1 tumors were predominantly in two subgroups where most progression events occurred, and in each case, the subgroup with the highest immune infiltration had fewer events. Here, these high-risk subtypes also contained LundTax subtypes GU, Ba/Sq, UroB, and UroC, all of which show a worse outcome than UroA when assessed in the TCGA MIBC dataset.21
Although diversity allowed robust classification that was prognostic for the entire NMIBC population, this was not useful in stage T1 subgroup analysis. Importantly, we show that stage-specific subgroups contain sufficient diversity to allow subclassification and that this provides more granular information regarding tumor biology and suggestions for therapy. It also provided prognostic information, although validation in a larger cohort is needed to determine optimal classification.
Our whole-exome analysis of paired T1 samples is the largest to date and provides an improved view of these lesions. Previous studies report that T1 tumors have a mutation spectrum intermediate between Ta tumors and MIBC. We show that this does not indicate the presence of T1 samples with an MIBC-like profile but, rather, that some T1 tumors contain Ta-like and MIBC-like mutations. It is expected that these have distinct phenotypes and that single-individual profiles will be needed for optimal management.
APOBEC enzyme expression and the related mutational profile increased from lowest levels in GS1 Ta tumors to the most genomically altered T1 tumors. In T1, SBS5, which is associated with smoking in bladder cancer,33 and ERCC2 mutation35 was also identified. A relationship of FGA to T1 PFS was strongly related to TP53 and RB1 mutation, identifying high-risk individuals. In contrast, ERCC2 mutant T1CN1 tumors were dominated by point mutations rather than CN events and had favorable outcomes. In addition to known associations with DDR gene mutations,12,59,60 ERBB2 and ERBB3 mutations were associated with high TMB and longer PFS and might suggest bladder preservation in T1 disease.
Two predicted drivers not implicated previously in NMIBC, GNA13 and KANSL1, should be included in future mutation analyses. Mutations of KANSL1, which is implicated as a chromatin modifier,39,40 have been reported in 6% of MIBCs.38 GNA13 is part of a heterotrimeric G-protein complex through which ligand-activated GPCRs signal and has several described activities.61, 62, 63, 64 We identified missense mutations in codon R200, close to the nucleotide binding pocket, two of which (R200G and R200K) are known to cause hyperactivation.65 Twelve of 13 R200 mutations reported to date are in urothelial tumors (COSMIC; accessed January 18, 2021), and codon 200 mutations were found in the urothelium of three of 15 normal individuals.66 Thus, GNA13 may be a key driver and a therapeutic target in a subset of T1 tumors.
The Ta and T1 expression subtypes showed overlap with genomic subtypes. Although the genomic drivers of T1 expression subtypes are unclear, expression features in Ta are strongly influenced by TSC1 (9q) loss and upregulation of mTORC1 activity. In both stages we identified infiltrated subtypes. In Ta but not T1, immune score per se segregated individuals with differential outcomes. In T1, T1E3 and T1E4 were more infiltrated than T1E1 and T1E2, but T1E3, the more highly infiltrated of these subtypes, had improved PFS compared with T1E4. Previously suggested influences on immune infiltration showed no relationships, but our data suggest the importance of DDR gene mutation and downregulation of DNA repair genes. We also implicate PPARG signaling in driving immune evasion and, compatible with reports that PPARG influences DNA repair,67,68 show a relationship of PPARG signaling to DNA repair gene expression. Mutations in PPARG and RXRA are as frequent in T1 as in MIBC,56,57 and gain or amplification of PPARG is common. Modulation of this signaling axis may have therapeutic value alone or in combination with immune checkpoint inhibition.
Regulon analysis confirmed the distinct biology of the expression subtypes and uncovered regulators predicted to determine phenotype. This identified many regulators with still unknown functions, providing a rich resource for future analyses. Importantly, association of MYB proto-oncogene-like 1 (MYBL1) regulon activity with adverse stage T1 outcome and its negative relationship with β-barrel heme protein THAP4 activity, which may play a role in the antioxidant system,69 may identify progression mechanisms.
Therapeutic opportunities for low-risk Ta tumors are limited to intravesical approaches, and targeting FGFR3 and the metabolic vulnerability of GS2 tumors identified here and previously13 are rational opportunities. For individuals with stage T1 disease, immune checkpoint inhibition may be most relevant in infiltrated T1 cases that have signatures associated with a favorable response.52,53,70 Because our data suggest that T1 tumors in females are more infiltrated, retrospective analysis in relation to gender and response in current trials in high-risk NMIBC will be of great interest.
ERCC2 and ERBB2 mutations that are related to chemotherapy response were associated with high TMB and a good outcome. It is unclear whether co-occurrence with ERCC2 mutation reflects an ERCC2-related mutational mechanism or whether there is a functional relationship. ERBB2 and ERBB3 could themselves be suitable targets. Notably, bladder cell lines with two of the mutations found here (ERBB2, S310F; ERBB3, V104L) are sensitive to afatinib,71 and encouraging results have been reported for afatinib in MIBC.72 However, systemic therapy may not be suitable for such cases if association with good outcome is confirmed.
BCG remains the mainstay of treatment for high-risk NMIBC. Because 30%–40% of individuals suffer recurrence, and approximately 10% progress,73,74 predicting response is important. Relationships of response to high TMB,60,75 ERCC2 mutation,60 higher PD-L1 expression,76 and UroVysion fluorescence in situ hybridization (FISH) scores77 have been reported. Here, most post-BCG progression occurred in T1E3 and T1E4. We found a higher but not statistically significant TMB and ERCC2 mutation rate in individuals with a good outcome, no relationship to PD-L1 expression, and no differences in global expression between responders and non-responders. However, TP53 mutation had a striking relationship with outcome, in accordance with some but not all previous studies.12,59,60 The reason for these differences is unclear but indicates the need for more extensive examination of pre-BCG treatment tumor molecular status in relation to outcome and a possible need to consider germline features.
Our data indicate the feasibility of stage-specific classification. We have not generated separate classifiers for Ta and T1 tumors because moderate sample numbers may be suboptimal for generation of single sample classifiers. Because there is the possibility that submucosal invasion might be missed at the time of diagnosis, we envisage that stage Ta and T1 classifiers could be applied to all NMIBCs, and, ideally, because genomic and expression features convey prognostic information, these should be integrated to generate optimum classification.
Limitations of the study
The study is limited by relatively small numbers of samples in each separate stage group. This has not allowed development of single-sample classifiers for these groups or validation of subtype signatures in other datasets. Expanded cohorts of Ta and T1 tumors will be required to confirm the findings and to develop such classifiers. The finding of small numbers of novel mutations in T1 samples (GNA13 and KANSL1) also requires confirmation in a larger cohort. As for T1 tumors, high-grade Ta tumors have a higher risk of progression. Here the majority of Ta samples analyzed were low grade, and only one individual progressed to invasive disease. Thus, it was not possible to identify features associated with high-grade Ta and/or risk of progression.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| Fresh frozen tissue samples | Leeds Multidisciplinary Research Tissue Bank | N/A |
| Venous blood samples from bladder cancer patients | Leeds Multidisciplinary Research Tissue Bank | N/A |
| Critical Commercial Assays | ||
| Affymetrix GeneChip Human Transcriptome Array 2.0 and Genechip WT PLUS Reagent Kit | Affymetrix | Cat# 902310 |
| GeneChip Hybridization, Wash, and Stain Kit | Affymetrix | Cat# 900720 |
| Gentra Puregene Tissue Kit | QIAGEN | Cat# 158667 |
| HiSeq 3000 | Illumina | N/A |
| NEBNext DNA Library Prep Master Mix Set for Illumina | New England BioLabs | Cat# E6040L |
| NEBNext Singleplex Oligos for Illumina | New England BioLabs | Cat# E7350L |
| NEBNext High-Fidelity 2X PCR Master Mix | New England BioLabs | Cat# M0541L |
| QIAamp DNA Mini Kit | QIAGEN | Cat# 51306 |
| RNeasy Plus Micro Kit | QIAGEN | Cat# 74034 |
| SureSelectXT Human All Exon v6 Kit | Agilent Technologies, Inc | Cat# 5190-8864 |
| Deposited Data | ||
| Raw and processed Affymetrix microarray data | This study | GSE:163209 |
| Whole exome sequence data | This study | EGAS00001005765 |
| Targeted sequence data | This study | EGAS00001005766, EGAS00001005767 |
| Software and algorithms | ||
| Affymetrix Expression Console software | Affymetrix | https://www.thermofisher.com/us/en/home/life-science/microarray-analysis.html |
| GenePattern | Broad Institute | https://cloud.genepattern.org |
| GSEA | Subramanian et al.78 | https://www.gsea-msigdb.org/gsea/ |
| Nexus Copy Number | Biodiscovery, Inc | http://www.biodiscovery.com/nexus-copy-number |
| ngCGH | Gartner et al.79 | https://github.com/seandavi/ngCGH |
| R version 3.6.1 | The R Foundation for Statistical Computing | https://www.r-project.org/ |
| R studio for Mac | R-Studio | https://www.rstudio.com/products/rstudio/download/ |
| R2 | Jan Koster, Academic Medical Center, Netherlands | R2: Genomics Analysis and visualization Platform; https://hgserver1.amc.nl:443/ |
| dNdScv | Martincorena et al., 201737 | https://github.com/im3sanger/dndscv |
| MutationalPatterns | Blokzijl et al., 201880 | https://bioconductor.org/packages/release/bioc/html/MutationalPatterns.html |
| ESTIMATE | Yoshihara et al.30 | https://bioinformatics.mdanderson.org/public-software/estimate/ |
| COSMIC mutational signatures v3 | N/A | https://cancer.sanger.ac.uk/signatures/ |
| FASTQC | The Babraham Bioinformatics group | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| Cutadapt | Marcel Martin 2011 | https://github.com/marcelm/cutadapt/ |
| BWA | Li et al.81 | https://github.com/lh3/bwa |
| Picard | Broad Institute | https://broadinstitute.github.io/picard/ |
| GATK3 | Broad Institute | https://accounts.google.com/ServiceLogin?service=cloudconsole&passive=1209600&osid=1&continue=https://console.cloud.google.com/storage/browser/_details/gatk-software/package-archive/gatk/GenomeAnalysisTK-3.8-1-0-gf15c1c3ef.tar.bz2&followup=https://console.cloud.google.com/storage/browser/_details/gatk-software/package-archive/gatk/GenomeAnalysisTK-3.8-1-0-gf15c1c3ef.tar.bz2 |
| ContEst | Broad Institute | http://software.broadinstitute.org/cancer/cga/contest |
| Conpair | Bergmann et al.82 | https://github.com/nygenome/Conpair |
| samtools | Li et al., 200983 | http://samtools.sourceforge.net/ |
| MuTect2 | Benjamin et al.84 | https://accounts.google.com/ServiceLogin?service=cloudconsole&passive=1209600&osid=1&continue=https://console.cloud.google.com/storage/browser/_details/gatk-software/package-archive/gatk/GenomeAnalysisTK-3.8-1-0-gf15c1c3ef.tar.bz2&followup=https://console.cloud.google.com/storage/browser/_details/gatk-software/package-archive/gatk/GenomeAnalysisTK-3.8-1-0-gf15c1c3ef.tar.bz2 |
| MuSE | Fan et al.85 | https://bioinformatics.mdanderson.org/public-software/muse/ |
| VarScan 2 | Koboldt et al.86 | http://massgenomics.org/varscan |
| Strelka2 | Kim et al.87 | https://github.com/Illumina/strelka |
| EBCall | Shiraishi et al.88 | https://github.com/friend1ws/EBCall |
| MeerKat | CBMI, Harvard Medical School | http://compbio.med.harvard.edu/Meerkat/ |
| coxph | Therneau (2021). A Package for Survival Analysis in R. R package version 3.2-13. | https://cran.r-project.org/web/packages/survival/index.html |
| coxphw | Dunkler et al.89 | N/A |
| RTN | Castro et al.25 | http://www.bioconductor.org/packages/release/bioc/html/RTN.html |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Margaret A. Knowles (m.a.knowles@leeds.ac.uk).
Materials Availability
This study did not generate new unique reagents.
Experimental model and subject details
Human Tissue Samples and Subject Follow-up Data
Clinical samples and associated clinical data were sourced from the Leeds Multidisciplinary Research Tissue Bank (REC reference: 20/YH/0103). All patients provided written informed consent for the use of their samples for medical research. Cold cup biopsies were collected, snap-frozen and stored in liquid nitrogen. The remainder of the sample was embedded in paraffin for diagnostic assessment. Samples were graded and staged by a consultant urological pathologist (J-A.R) using the 1973 and 2004 WHO and TNM criteria, respectively.90,91 To avoid the risk of under-staging of T1 samples, all samples were re-evaluated by a single urological histopathologist (J-A.R) and when inadequate tissue was present in the surgical specimen, we only included those that were definitively re-evaluated as T1 at re-resection or at cystectomy. Available clinical information including gender and age were collected. Median follow-up time was 55 months (range 3-186 months). This and details of analysis platforms for each sample are given in Table S1A.
Method details
DNA Extraction
Genomic DNA was isolated from frozen tissue sections comprising at least 70% tumor cells using a QIAamp DNA Mini Kit or a Gentra PureGene Tissue Kit. DNA was extracted from venous blood samples using a Nucleon BACC DNA Extraction Kit or by salt precipitation.
Copy Number Analysis
Low pass whole genome sequencing or array-CGH were used to assess copy number alterations in all tumors. Next-generation sequencing libraries were constructed using NEBNext® reagents according to the manufacturer’s instructions. Raw sequencing data was processed as described for whole exome sequencing (below). After BAM file generation, ngCGH was used to compare number of read counts between tumor and matched blood samples using a window size of 1000 reads.79 GC correction and copy number calling using the FASST2 Segmentation Algorithm, a Hidden Markov Model (HMM) based approach, were carried out within the Nexus Copy Number software package. The significance threshold for segmentation was set at 1.0E-5 also requiring a minimum of 3 probes per segment and a maximum probe spacing of 1000 between adjacent probes before breaking a segment. The log ratio thresholds for single copy gain and single copy loss were set at either ± 0.15 or ± 0.2, respectively, with the cut-off employed being determined by examination of individual sample profiles. The log ratio thresholds for two or more copy gain and homozygous loss were set at 1 and −1 respectively. Array-CGH and copy number calling was carried out as described previously.10 To facilitate the combined analysis of copy number data from NGS and array-based platforms, after CN calling all analysis was restricted to those genomic regions associated with BAC clones present on the 1Mb resolution CGH array. To conduct cluster analysis, each individual region associated with a BAC array clone was assigned a copy number class (0 = no copy number alteration; 1 = gain; 2 = high-level amplification; −1 = loss; −2 = high-level loss). One-way unsupervised hierarchical cluster analysis was conducted using Euclidean distance and the Ward method of linkage. For samples analyzed by CGH, fraction of genome altered (FGA) was defined as the percentage of clones reporting significantly altered copy number (gain or loss). For sample analyzed by NGS, FGA was calculated by dividing the sum of the lengths of all regions exhibiting altered copy number by the total length of the genome (hg38). FGA groups were defined as A (< 1%), B (1- < 10%), C (10- < 30%) and D (>30%). Copy number data for the 113 Ta samples and 18 of the 104 T1 samples were reported previously.10,13
RNA Extraction, Gene Expression Profiling and NMF analysis
Total RNA was isolated from frozen tissue sections using a RNeasy Plus Micro Kit and amplified using the Affymetrix GeneChip® WT PLUS Reagent Kit according to manufacturer’s instructions. The resulting cDNA was quantified using OD (NanoDrop). The cDNA was normalized and hybridized onto Affymetrix Human Transcriptome 2.0 microarrays for 16 hours at 45°C. Microarrays were washed and stained using the Affymetrix GeneChip® Hybridization, Wash, and Stain Kit according to manufacturer’s instructions using the Affymetrix GeneChip® Fluidics Station 450. Microarrays were scanned using an Affymetrix GeneChip® 7G microarray scanner. Quality control checks were conducted using Affymetrix® Expression Console Software (RRID:SCR_018718). Affymetrix HTA 2.0 CEL files were normalized as rma_sketch using apt-probeset-summarize from the Affymetrix Power Tools and HTA-2_0.r1.gene.cdf. After normalization, the dataset was loaded into the R2: genomics analysis and visualization platform (http://r2.amc.nl). R2 was used for routine data visualization, data mining and analysis. For NMF analysis, a text file containing the gene-level normalized natural log values for all samples was exported from R2. This datafile was converted to .gct format and preprocessed using the PreprocessDataset module in GenePattern (RRID:SCR_003201) with default settings. The preprocessed datafile was then used as input for the GenePattern NMFConsensus module (v5) with default settings. NMF analysis in Ta samples generated 3 clusters in a single step. For T1 samples two initial NMF clusters were re-analyzed independently, generating four final subtypes.
Whole Exome Sequencing
Libraries were generated using 3 μg of DNA and enriched for exonic regions using the SureSelect Human All Exon V6 Kit (58 tumor:blood pairs) according to the manufacturer’s protocols. Sequencing was performed on an Illumina HiSeq 3000 in paired-end mode with 150 bp read length and eight samples (four tumor:blood pairs) per lane. Base calling and quality control was performed using Illumina’s Real Time Analysis software version 1.6 with standard settings. Sequence files were QC checked using FastQC (v0.10.0) (RRID:SCR_014583) before and after preprocessing. Adaptor contamination and low-quality read ends (< 20) were trimmed using Cutadapt 1.14 (RRID:SCR_011841). Any read in which either pair had a length less than 19 was removed from subsequent analysis.
Alignment was performed using the BWA.1 bwa-mem algorithm81 (RRID:SCR_010910) to the GRCh38 reference genome. To avoid tumor-normal bias, we merged the tumor and its paired normal alignment bam files and performed pre-genotyping processes together. Because we would apply multiple somatic mutation calling tools in addition to MuTect2, we performed local realignment around indels first despite this being an omittable step for MuTect2 pipeline. This step was conducted using the GATK v3.8 RealignerTargetCreator and IndelRealigner (RRID:SCR_001876) in Smith-Waterman mode with reference to 1000 Genomes phase 3 indel sites, and Mills and 1000G gold standard sites.92 Next, we followed the best practice of somatic mutation calling recommended by GATK3 for pre-processing, which includes duplicate reads marking (Picard) and base quality recalibration (GATK3). The tumor-normal pair was then separated and ready for somatic mutation calling. Quality metrics including target coverage [Picard (RRID:SCR_006525) and Samtools83 (RRID:SCR_002105)] and contamination [Contest (RRID:SCR_000595) and Conpair2]82 were evaluated prior to genotyping. In particular for MuTect2 mutation calling, we created the panel-of-normal (PON) based on all the in-house exome sequencing data processed under the same protocol and lab facility (N = 68).
We aimed to develop a tailored somatic mutation calling protocol for our sequencing data. From several tools previously reviewed,82 we evaluated and selected 5 somatic mutation calling tools: MuTect2 (RRID:SCR_000559), MuSE, EBCall (RRID:SCR_006791), VarScan2 (RRID:SCR_006849), and Strelka2 (RRID:SCR_005109),84, 85, 86, 87, 88based on their performance on in-house exome sequencing data benchmarked by different amounts of input DNA (200 ng to 3 μg). We selected mutations called by at least two callers for the downstream analysis. Following this the number of mutations per sample was approximately equal to the median number of mutations generated by the 5 callers. For all callers, post-calling filtration was applied and only variants that passed the build-in filtration by each software were kept. Variant annotation was performed based on Ensembl VEP GRCh38 release 90. Data for 17 Ta samples reported previously13 was re-analyzed using Mutect2. SNV counts were based on Mutect2 calls only.
Targeted Sequencing
Targeted sequencing data from an in-house design for 40 genes was taken from a previous study13 for 18 Ta samples (Table S1A). All other targeted sequencing used a new design for 140 genes. Agilent’s SureDesign tool was used to design a 1.133 Mb SureSelect custom capture for all coding exons of the 140 selected candidate genes (Table S2). The design included a 10 base pair extension to the 3¢ and 5¢ ends of each region. Libraries were generated using 1.2 μg of DNA and enriched for targeted exonic regions according to the manufacturer’s protocols for 112 tumor:blood pairs. Forty eight samples (24 tumor:blood pairs) were run in a single lane on an Illumina HiSeq 3000 in paired-end mode with 150 bp read length. Raw data handling up to FASTQ file generation was as described above.
Tumor mutational burden
Tumor mutational burden (TMB) was calculated as the number of somatic non-synonymous mutations per Mb of targeted DNA.
Mutation Significance Analysis
Putative driver genes were identified from whole exome sequence data from T1 samples using dNdScv37 (RRID:SCR_017093). The dNdScv R package is a suite of maximum-likelihood dN/dS methods designed to quantify selection in cancer and somatic evolution. The package contains functions to quantify dN/dS ratios for missense, nonsense and essential splice site mutations. The background mutation rate of each gene was estimated by combining local information (synonymous mutations in the gene) and global information (variation of the mutation rate across genes, exploiting epigenomic covariates), and controlling for the sequence composition of the gene and mutational signatures. dNdScv uses trinucleotide context-dependent substitution matrices to avoid common mutation biases affecting dN/dS. A pre-computed database for GRCh38 was downloaded for this analysis (https://github.com/im3sanger/dndscv_data/tree/master/data).
Analysis of Mutational Signatures
We used non-negative matrix factorization in the package MutationalPatterns80 to derive signatures and to compare to COSMIC Mutational Signatures (https://cancer.sanger.ac.uk/signatures/) (RRID:SCR_002260) in both whole exome and target capture sequence data.
Gene expression signatures
We used gene lists associated with cell cycle, urothelial differentiation and its regulation (KLF5, PPARG, RXRA, ELF3, FOXA1, GATA3, TP63, GRHL2, GRHL3), FGFR3-related gene expression and DNA repair genes9,18,54,51, and expression signatures described for PPARG signaling,54 bladder CIS,17 progression of NMIBC,7,20 immune infiltration19,31,32 and response to immune checkpoint inhibition.52,53 Signature zscores for these genesets were derived for each sample, weighted where up- and downregulated genes were included. For estimation of immune infiltration we also used ESTIMATE.30 Two epithelial signatures were a compiled list of cytokeratin genes (KRT8, KRT18, KRT19, KRT17, KRT5, KRT6, KRT13, KRT7, KRT20), EPCAM and CDH1, and a list of low-variance epithelial-expressed genes correlated with E-cadherin and EPCAM in urothelial tumor datasets (SPINT2, TACSTD2, EPCAM, KRT19, RAB25, GPR56, SDC1, SPINT1, CDH1, DDR1, CDS1, RAB5B, LAD1, PRPRF).
Classification of samples using UROMOL2021 and LundTax classifiers
Samples were classified according to the UROMOL2021 classes.5 A Pearson correlation was computed between each sample’s gene expression profile and each of the four centroids (the mean gene expression profile for each class) corresponding to the four NMIBC classes. Samples were then assigned to the class with the highest sample-centroid correlation. The UROMOL2021 NMIBC classifier is available as a web application at http://www.nmibc-class.dk.
The R package multiclassPairs93 was used to generate a single sample version of the Lund classification system (LundTax).21 Tumors were classified according to LundTax into Urothelial-like (Uro), Genomically Unstable (GU), Basal/Squamous-like (Ba/Sq), Mesenchymal-like (Mes-like), and small cell/neuroendocrine like (Sc/Ne-like) using gene expression data. Uro samples were classified to Uro subclasses, UroA, UroB, and UroC.
Regulon analysis
To identify regulators of molecular subtypes identified, we analyzed regulatory networks (regulons) for a comprehensive set of 1547 transcription factors94 using RTN.25, 26, 27 Gene level normalization (using SST-RMA) and signal summarization was conducted using Affymetrix® Expression Console Software. We inferred the regulons using the R package RTN (version 2.13.2), which is described elsewhere.25,95 Briefly, gene expression matrices for a set of samples were used to estimate the associations between a transcription factor and all of its potential targets. We used Mutual Information (MI) to identify potential regulator-target associations, and Spearman’s correlation to assign the direction of an inferred association. Associations with MI below a minimum threshold were eliminated by permutation analysis (BH-adjusted p value < 1x10−5), and unstable interactions were removed by bootstrapping, to create a regulatory network. Regulons were additionally processed by the ARACNe algorithm to enrich the regulons with direct TF-target interactions.96 We estimated regulon activity by a two-tailed gene set enrichment analysis (GSEA-2T), which is described elsewhere.25,27 The GSEA-2T was performed in R97 using the RTN package.25 We fitted a Cox proportional hazards regression to further assess regulon activity and survival, using the RTNsurvival package.27 For the Kaplan-Meier curve, we stratified the cohort into 2 groups – positive and negative dES – and evaluated differences between the groups for 200-month PFS and RFS, using a Log-rank test. This package reports BH-adjusted P values98.
Quantification and statistical analysis
Statistical analysis of transcriptome data was done in R2. The statistical test LIMMA with FDR 0.01 was applied to identify genes differentially expressed between copy number and expression subtypes. FDR adjusted p values (q values) are given in Tables S3 and S8. Other statistical tests Fisher’s exact, chi square, Mann-Whitney, Kruskal-Wallis tests and correlation analyses, survival curve generation using the Kaplan-Meier method and curve comparison using the Log-rank (Mantel-Cox) test were carried out using GraphPad Prism 8.2 for Mac. Comparison between groups used the Mann-Whitney or Kruskal-Wallis tests for continuous variables and Fisher’s exact test and Chi square test for categorical variables. A post hoc Dunn’s test was used for multiple comparison correction following Kruskal-Wallis analyses. Bonferroni adjustment of chi square p values was used. A significance level of 0.05 was used in all tests.
We used the ‘cox.zph’ R function (Therneau, 2021; R package version 3.2-13, 2021; https://cran.r-project.org/web/packages/survival/index.html) to assess the proportional hazards assumption of a Cox Regression analysis, assessing the association between the ‘group’ covariate and the outcomes RFS and PFS, and displayed the Schoenfeld residuals. The Schoenfeld plot gives an estimate of the time-dependent coefficient beta. If the proportional hazards assumption holds then the beta function is a horizontal line.99 Systematic departures from the horizontal line indicate non-proportional hazards. This was not observed in the RFS and PFS Schoenfeld plots and the ‘cox.zph’ test was not statistically significant. However, we saw that survival curves crossed over for both RFS and PFS data at early time points, which suggested the presence of non-proportional hazards. As the testing power for the corresponding regression parameter can be reduced in the presence of non-proportional hazards,89 we carried out Cox regression analysis to estimate long-term survival rate under both proportional and non-proportional hazards assumptions. The Cox regression model under proportional hazards assumption was carried out using the ‘coxph’ function from the survival R package (Therneau, 2021; R package version 3.2-13, 2021. https://cran.r-project.org/web/packages/survival/index.html) and under non-proportional assumption was carried out using the ‘coxphw’ function from the coxphw R Package.89,100
Gene Ontology, KEGG Pathway and Gene Set Enrichment Analysis
Significantly upregulated genes (LIMMA test, false discovery rate p value = 0.01) in each subtype comparison was used as input for Gene Ontology (GO) biological process analysis in R2. A cut off of nominal p value < 0.05 was implemented. Gene Set Enrichment Analysis v3.078 (RRID:SCR_003199) was carried out using all genes; genomic and expression subtypes were assigned as phenotypes and permuted 1000 times, the test dataset was collapsed to gene symbols and run against gene sets in the Hallmarks database (v7.1).
Acknowledgments
We thank Dmitry Gordenin for helpful discussions of mutational signatures and Matthieu Rederstorff for advice on snoRNA expression. We are indebted to all individuals who gave consent for their samples to be used. This work was funded by Yorkshire Cancer Research (L376PA) and Cancer Research UK (C57387/A21777).
Author contributions
Conceptualization, C.D.H. and M.A.K.; methodology, C.D.H., G.C., F.M.P., M.A.A.C., N.-a.-d.S.M., P.E., A.R.J.L., S.V.L., J.K., A.G.R., I.M., and M.A.K.; software, M.A.A.C., N.-a.-d.S.M., P.E., A.R.J.L., S.V.L., A.G.R., and I.M.; formal analysis, C.D.H., G.C., F.M.P., M.A.A.C., N.-a.-d.S.M., P.E., O.A., A.R.J.L., S.V.L., and M.A.K.; investigation, C.D.H., G.C., F.M.P., E.V.I.B., O.A., and J.E.B.; resources, S.J., J.-A.R., J.C.B., J.K., I.M., L.D., and M.H.; data curation, C.D.H., G.C., F.M.P., and J.C.B.; writing – original draft, C.D.H., G.C., M.A.A.C., N.-a.-d.S.M., S.V.L., and M.A.K.; writing – review & editing, all authors; supervision, M.A.K.; project administration, M.A.K.; funding acquisition, M.A.K., S.J., and J.-A.R.
Declaration of interests
M.A.K. has an advisory/consulting role at Janssen, LOXO Oncology, QED, and Rainier Therapeutics. L.D. has sponsored research agreements with C2i-genomics, Natera, AstraZeneca, and Ferring and an advisory/consulting role at Ferring. L.D. has received a speaker honorarium from Roche.
Published: December 21, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2021.100472.
Supplemental information
Data and code availability
-
•
Microarray data are available at the Gene Expression Omnibus under accession number GSE163209. Raw sequencing data are available at The European Genome-phenome Archive (EGA) under accession numbers EGAS00001005765, EGAS00001005766 and EGAS00001005767.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Ferlay J., Colombet M., Soerjomataram I., Mathers C., Parkin D.M., Piñeros M., Znaor A., Bray F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer. 2019;144:1941–1953. doi: 10.1002/ijc.31937. [DOI] [PubMed] [Google Scholar]
- 2.Svatek R.S., Hollenbeck B.K., Holmäng S., Lee R., Kim S.P., Stenzl A., Lotan Y. The economics of bladder cancer: costs and considerations of caring for this disease. Eur. Urol. 2014;66:253–262. doi: 10.1016/j.eururo.2014.01.006. [DOI] [PubMed] [Google Scholar]
- 3.Jordan B., Meeks J.J. T1 bladder cancer: current considerations for diagnosis and management. Nat. Rev. Urol. 2019;16:23–34. doi: 10.1038/s41585-018-0105-y. [DOI] [PubMed] [Google Scholar]
- 4.Hedegaard J., Lamy P., Nordentoft I., Algaba F., Høyer S., Ulhøi B.P., Vang S., Reinert T., Hermann G.G., Mogensen K., et al. Comprehensive Transcriptional Analysis of Early-Stage Urothelial Carcinoma. Cancer Cell. 2016;30:27–42. doi: 10.1016/j.ccell.2016.05.004. [DOI] [PubMed] [Google Scholar]
- 5.Lindskrog S.V., Prip F., Lamy P., Taber A., Groeneveld C.S., Birkenkamp-Demtröder K., Jensen J.B., Strandgaard T., Nordentoft I., Christensen E., et al. An integrated multi-omics analysis identifies prognostic molecular subtypes of non-muscle-invasive bladder cancer. Nat. Commun. 2021;12:2301. doi: 10.1038/s41467-021-22465-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang R., Morris D.S., Tomlins S.A., Lonigro R.J., Tsodikov A., Mehra R., Giordano T.J., Kunju L.P., Lee C.T., Weizer A.Z., Chinnaiyan A.M. Development of a multiplex quantitative PCR signature to predict progression in non-muscle-invasive bladder cancer. Cancer Res. 2009;69:3810–3818. doi: 10.1158/0008-5472.CAN-08-4405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dyrskjøt L., Reinert T., Algaba F., Christensen E., Nieboer D., Hermann G.G., Mogensen K., Beukers W., Marquez M., Segersten U., et al. Prognostic Impact of a 12-gene Progression Score in Non-muscle-invasive Bladder Cancer: A Prospective Multicentre Validation Study. Eur. Urol. 2017;72:461–469. doi: 10.1016/j.eururo.2017.05.040. [DOI] [PubMed] [Google Scholar]
- 8.Patschan O., Sjödahl G., Chebil G., Lövgren K., Lauss M., Gudjonsson S., Kollberg P., Eriksson P., Aine M., Månsson W., et al. A Molecular Pathologic Framework for Risk Stratification of Stage T1 Urothelial Carcinoma. Eur. Urol. 2015;68:824–832. doi: 10.1016/j.eururo.2015.02.021. discussion 835–836. [DOI] [PubMed] [Google Scholar]
- 9.Sjödahl G., Lauss M., Lövgren K., Chebil G., Gudjonsson S., Veerla S., Patschan O., Aine M., Fernö M., Ringnér M., et al. A molecular taxonomy for urothelial carcinoma. Clin. Cancer Res. 2012;18:3377–3386. doi: 10.1158/1078-0432.CCR-12-0077-T. [DOI] [PubMed] [Google Scholar]
- 10.Hurst C.D., Platt F.M., Taylor C.F., Knowles M.A. Novel tumor subgroups of urothelial carcinoma of the bladder defined by integrated genomic analysis. Clin. Cancer Res. 2012;18:5865–5877. doi: 10.1158/1078-0432.CCR-12-1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nordentoft I., Lamy P., Birkenkamp-Demtröder K., Shumansky K., Vang S., Hornshøj H., Juul M., Villesen P., Hedegaard J., Roth A., et al. Mutational context and diverse clonal development in early and late bladder cancer. Cell Rep. 2014;7:1649–1663. doi: 10.1016/j.celrep.2014.04.038. [DOI] [PubMed] [Google Scholar]
- 12.Pietzak E.J., Bagrodia A., Cha E.K., Drill E.N., Iyer G., Isharwal S., Ostrovnaya I., Baez P., Li Q., Berger M.F., et al. Next-generation Sequencing of Nonmuscle Invasive Bladder Cancer Reveals Potential Biomarkers and Rational Therapeutic Targets. Eur. Urol. 2017;72:952–959. doi: 10.1016/j.eururo.2017.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hurst C.D., Alder O., Platt F.M., Droop A., Stead L.F., Burns J.E., Burghel G.J., Jain S., Klimczak L.J., Lindsay H., et al. Genomic Subtypes of Non-invasive Bladder Cancer with Distinct Metabolic Profile and Female Gender Bias in KDM6A Mutation Frequency. Cancer Cell. 2017;32:701–715.e7. doi: 10.1016/j.ccell.2017.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van Kessel K.E.M., van der Keur K.A., Dyrskjøt L., Algaba F., Welvaart N.Y.C., Beukers W., Segersten U., Keck B., Maurer T., Simic T., et al. Molecular Markers Increase Precision of the European Association of Urology Non-Muscle-Invasive Bladder Cancer Progression Risk Groups. Clin. Cancer Res. 2018;24:1586–1593. doi: 10.1158/1078-0432.CCR-17-2719. [DOI] [PubMed] [Google Scholar]
- 15.Lamy P., Nordentoft I., Birkenkamp-Demtröder K., Thomsen M.B., Villesen P., Vang S., Hedegaard J., Borre M., Jensen J.B., Høyer S., et al. Paired exome analysis reveals clonal evolution and potential therapeutic targets in urothelial carcinoma. Cancer Res. 2016;76:5894–5906. doi: 10.1158/0008-5472.CAN-16-0436. [DOI] [PubMed] [Google Scholar]
- 16.van Tilborg A.A., de Vries A., de Bont M., Groenfeld L.E., van der Kwast T.H., Zwarthoff E.C. Molecular evolution of multiple recurrent cancers of the bladder. Hum. Mol. Genet. 2000;9:2973–2980. doi: 10.1093/hmg/9.20.2973. [DOI] [PubMed] [Google Scholar]
- 17.Dyrskjøt L., Kruhøffer M., Thykjaer T., Marcussen N., Jensen J.L., Møller K., Ørntoft T.F. Gene expression in the urinary bladder: a common carcinoma in situ gene expression signature exists disregarding histopathological classification. Cancer Res. 2004;64:4040–4048. doi: 10.1158/0008-5472.CAN-03-3620. [DOI] [PubMed] [Google Scholar]
- 18.Eriksson P., Aine M., Veerla S., Liedberg F., Sjödahl G., Höglund M. Molecular subtypes of urothelial carcinoma are defined by specific gene regulatory systems. BMC Med. Genomics. 2015;8:25. doi: 10.1186/s12920-015-0101-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Danaher P., Warren S., Dennis L., D’Amico L., White A., Disis M.L., Geller M.A., Odunsi K., Beechem J., Fling S.P. Gene expression markers of Tumor Infiltrating Leukocytes. J. Immunother. Cancer. 2017;5:18. doi: 10.1186/s40425-017-0215-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dyrskjøt L., Zieger K., Kruhøffer M., Thykjaer T., Jensen J.L., Primdahl H., Aziz N., Marcussen N., Møller K., Orntoft T.F. A molecular signature in superficial bladder carcinoma predicts clinical outcome. Clin. Cancer Res. 2005;11:4029–4036. doi: 10.1158/1078-0432.CCR-04-2095. [DOI] [PubMed] [Google Scholar]
- 21.Marzouka N.A., Eriksson P., Rovira C., Liedberg F., Sjödahl G., Höglund M. A validation and extended description of the Lund taxonomy for urothelial carcinoma using the TCGA cohort. Sci. Rep. 2018;8:3737. doi: 10.1038/s41598-018-22126-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fagan R.J., Dingwall A.K. COMPASS Ascending: Emerging clues regarding the roles of MLL3/KMT2C and MLL2/KMT2D proteins in cancer. Cancer Lett. 2019;458:56–65. doi: 10.1016/j.canlet.2019.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shi M.J., Meng X.Y., Lamy P., Banday A.R., Yang J., Moreno-Vega A., Chen C.L., Dyrskjøt L., Bernard-Pierrot I., Prokunina-Olsson L., Radvanyi F. APOBEC-mediated Mutagenesis as a Likely Cause of FGFR3 S249C Mutation Over-representation in Bladder Cancer. Eur. Urol. 2019;76:9–13. doi: 10.1016/j.eururo.2019.03.032. [DOI] [PubMed] [Google Scholar]
- 24.Xue M., Li X., Li Z., Chen W. Urothelial carcinoma associated 1 is a hypoxia-inducible factor-1α-targeted long noncoding RNA that enhances hypoxic bladder cancer cell proliferation, migration, and invasion. Tumour Biol. 2014;35:6901–6912. doi: 10.1007/s13277-014-1925-x. [DOI] [PubMed] [Google Scholar]
- 25.Castro M.A., de Santiago I., Campbell T.M., Vaughn C., Hickey T.E., Ross E., Tilley W.D., Markowetz F., Ponder B.A., Meyer K.B. Regulators of genetic risk of breast cancer identified by integrative network analysis. Nat. Genet. 2016;48:12–21. doi: 10.1038/ng.3458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chagas V.S., Groeneveld C.S., Oliveira K.G., Trefflich S., de Almeida R.C., Ponder B.A.J., Meyer K.B., Jones S.J.M., Robertson A.G., Castro M.A.A. RTNduals: an R/Bioconductor package for analysis of co-regulation and inference of dual regulons. Bioinformatics. 2019;35:5357–5358. doi: 10.1093/bioinformatics/btz534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Groeneveld C.S., Chagas V.S., Jones S.J.M., Robertson A.G., Ponder B.A.J., Meyer K.B., Castro M.A.A. RTNsurvival: an R/Bioconductor package for regulatory network survival analysis. Bioinformatics. 2019;35:4488–4489. doi: 10.1093/bioinformatics/btz229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bell S.M., Zhang L., Mendell A., Xu Y., Haitchi H.M., Lessard J.L., Whitsett J.A. Kruppel-like factor 5 is required for formation and differentiation of the bladder urothelium. Dev. Biol. 2011;358:79–90. doi: 10.1016/j.ydbio.2011.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fishwick C., Higgins J., Percival-Alwyn L., Hustler A., Pearson J., Bastkowski S., Moxon S., Swarbreck D., Greenman C.D., Southgate J. Heterarchy of transcription factors driving basal and luminal cell phenotypes in human urothelium. Cell Death Differ. 2017;24:809–818. doi: 10.1038/cdd.2017.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yoshihara K., Shahmoradgoli M., Martínez E., Vegesna R., Kim H., Torres-Garcia W., Treviño V., Shen H., Laird P.W., Levine D.A., et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013;4:2612. doi: 10.1038/ncomms3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nirmal A.J., Regan T., Shih B.B., Hume D.A., Sims A.H., Freeman T.C. Immune Cell Gene Signatures for Profiling the Microenvironment of Solid Tumors. Cancer Immunol. Res. 2018;6:1388–1400. doi: 10.1158/2326-6066.CIR-18-0342. [DOI] [PubMed] [Google Scholar]
- 32.Charoentong P., Finotello F., Angelova M., Mayer C., Efremova M., Rieder D., Hackl H., Trajanoski Z. Pan-cancer Immunogenomic Analyses Reveal Genotype-Immunophenotype Relationships and Predictors of Response to Checkpoint Blockade. Cell Rep. 2017;18:248–262. doi: 10.1016/j.celrep.2016.12.019. [DOI] [PubMed] [Google Scholar]
- 33.Alexandrov L.B., Ju Y.S., Haase K., Van Loo P., Martincorena I., Nik-Zainal S., Totoki Y., Fujimoto A., Nakagawa H., Shibata T., et al. Mutational signatures associated with tobacco smoking in human cancer. Science. 2016;354:618–622. doi: 10.1126/science.aag0299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alexandrov L.B., Kim J., Haradhvala N.J., Huang M.N., Tian Ng A.W., Wu Y., Boot A., Covington K.R., Gordenin D.A., Bergstrom E.N., et al. PCAWG Mutational Signatures Working Group. PCAWG Consortium The repertoire of mutational signatures in human cancer. Nature. 2020;578:94–101. doi: 10.1038/s41586-020-1943-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim J., Mouw K.W., Polak P., Braunstein L.Z., Kamburov A., Kwiatkowski D.J., Rosenberg J.E., Van Allen E.M., D’Andrea A., Getz G. Somatic ERCC2 mutations are associated with a distinct genomic signature in urothelial tumors. Nat. Genet. 2016;48:600–606. doi: 10.1038/ng.3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Taber A., Christensen E., Lamy P., Nordentoft I., Prip F., Lindskrog S.V., Birkenkamp-Demtröder K., Okholm T.L.H., Knudsen M., Pedersen J.S., et al. Molecular correlates of cisplatin-based chemotherapy response in muscle invasive bladder cancer by integrated multi-omics analysis. Nat. Commun. 2020;11:4858. doi: 10.1038/s41467-020-18640-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Martincorena I., Raine K.M., Gerstung M., Dawson K.J., Haase K., Van Loo P., Davies H., Stratton M.R., Campbell P.J. Universal Patterns of Selection in Cancer and Somatic Tissues. Cell. 2017;171:1029–1041.e21. doi: 10.1016/j.cell.2017.09.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Robertson A.G., Kim J., Al-Ahmadie H., Bellmunt J., Guo G., Cherniack A.D., Hinoue T., Laird P.W., Hoadley K.A., Akbani R., et al. TCGA Research Network Comprehensive Molecular Characterization of Muscle-Invasive Bladder Cancer. Cell. 2017;171:540–556.e25. doi: 10.1016/j.cell.2017.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Smith E.R., Cayrou C., Huang R., Lane W.S., Côté J., Lucchesi J.C. A human protein complex homologous to the Drosophila MSL complex is responsible for the majority of histone H4 acetylation at lysine 16. Mol. Cell. Biol. 2005;25:9175–9188. doi: 10.1128/MCB.25.21.9175-9188.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dou Y., Milne T.A., Tackett A.J., Smith E.R., Fukuda A., Wysocka J., Allis C.D., Chait B.T., Hess J.L., Roeder R.G. Physical association and coordinate function of the H3 K4 methyltransferase MLL1 and the H4 K16 acetyltransferase MOF. Cell. 2005;121:873–885. doi: 10.1016/j.cell.2005.04.031. [DOI] [PubMed] [Google Scholar]
- 41.Sarkis A.S., Dalbagni G., Cordon-Cardo C., Zhang Z.-F., Sheinfeld J., Fair W.R., Herr H.W., Reuter V.E. Nuclear overexpression of p53 protein in transitional cell bladder carcinoma: a marker for disease progression. J. Natl. Cancer Inst. 1993;85:53–59. doi: 10.1093/jnci/85.1.53. [DOI] [PubMed] [Google Scholar]
- 42.Van Allen E.M., Mouw K.W., Kim P., Iyer G., Wagle N., Al-Ahmadie H., Zhu C., Ostrovnaya I., Kryukov G.V., O’Connor K.W., et al. Somatic ERCC2 mutations correlate with cisplatin sensitivity in muscle-invasive urothelial carcinoma. Cancer Discov. 2014;4:1140–1153. doi: 10.1158/2159-8290.CD-14-0623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu D., Plimack E.R., Hoffman-Censits J., Garraway L.A., Bellmunt J., Van Allen E., Rosenberg J.E. Clinical Validation of Chemotherapy Response Biomarker ERCC2 in Muscle-Invasive Urothelial Bladder Carcinoma. JAMA Oncol. 2016;2:1094–1096. doi: 10.1001/jamaoncol.2016.1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Plimack E.R., Dunbrack R.L., Brennan T.A., Andrake M.D., Zhou Y., Serebriiskii I.G., Slifker M., Alpaugh K., Dulaimi E., Palma N., et al. Defects in DNA Repair Genes Predict Response to Neoadjuvant Cisplatin-based Chemotherapy in Muscle-invasive Bladder Cancer. Eur. Urol. 2015;68:959–967. doi: 10.1016/j.eururo.2015.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Teo M.Y., Bambury R.M., Zabor E.C., Jordan E., Al-Ahmadie H., Boyd M.E., Bouvier N., Mullane S.A., Cha E.K., Roper N., et al. DNA Damage Response and Repair Gene Alterations Are Associated with Improved Survival in Patients with Platinum-Treated Advanced Urothelial Carcinoma. Clin. Cancer Res. 2017;23:3610–3618. doi: 10.1158/1078-0432.CCR-16-2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Teo M.Y., Seier K., Ostrovnaya I., Regazzi A.M., Kania B.E., Moran M.M., Cipolla C.K., Bluth M.J., Chaim J., Al-Ahmadie H., et al. Alterations in DNA Damage Response and Repair Genes as Potential Marker of Clinical Benefit From PD-1/PD-L1 Blockade in Advanced Urothelial Cancers. J. Clin. Oncol. 2018;36:1685–1694. doi: 10.1200/JCO.2017.75.7740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Groenendijk F.H., de Jong J., Fransen van de Putte E.E., Michaut M., Schlicker A., Peters D., Velds A., Nieuwland M., van den Heuvel M.M., Kerkhoven R.M., et al. ERBB2 Mutations Characterize a Subgroup of Muscle-invasive Bladder Cancers with Excellent Response to Neoadjuvant Chemotherapy. Eur. Urol. 2016;69:384–388. doi: 10.1016/j.eururo.2015.01.014. [DOI] [PubMed] [Google Scholar]
- 48.Hahn N.M., Necchi A., Loriot Y., Powles T., Plimack E.R., Sonpavde G., Roupret M., Kamat A.M. Role of Checkpoint Inhibition in Localized Bladder Cancer. Eur. Urol. Oncol. 2018;1:190–198. doi: 10.1016/j.euo.2018.05.002. [DOI] [PubMed] [Google Scholar]
- 49.Li Q., Damish A.W., Frazier Z., Liu D., Reznichenko E., Kamburov A., Bell A., Zhao H., Jordan E.J., Gao S.P., et al. ERCC2 Helicase Domain Mutations Confer Nucleotide Excision Repair Deficiency and Drive Cisplatin Sensitivity in Muscle-Invasive Bladder Cancer. Clin. Cancer Res. 2019;25:977–988. doi: 10.1158/1078-0432.CCR-18-1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Milewska M., Cremona M., Morgan C., O’Shea J., Carr A., Vellanki S.H., Hopkins A.M., Toomey S., Madden S.F., Hennessy B.T., Eustace A.J. Development of a personalized therapeutic strategy for ERBB-gene-mutated cancers. Ther. Adv. Med. Oncol. 2018;10 doi: 10.1177/1758834017746040. 1758834017746040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wood R.D., Mitchell M., Lindahl T. Human DNA repair genes, 2005. Mutat. Res. 2005;577:275–283. doi: 10.1016/j.mrfmmm.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 52.Rosenberg J.E., Hoffman-Censits J., Powles T., van der Heijden M.S., Balar A.V., Necchi A., Dawson N., O’Donnell P.H., Balmanoukian A., Loriot Y., et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet. 2016;387:1909–1920. doi: 10.1016/S0140-6736(16)00561-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ayers M., Lunceford J., Nebozhyn M., Murphy E., Loboda A., Kaufman D.R., Albright A., Cheng J.D., Kang S.P., Shankaran V., et al. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Invest. 2017;127:2930–2940. doi: 10.1172/JCI91190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Biton A., Bernard-Pierrot I., Lou Y., Krucker C., Chapeaublanc E., Rubio-Pérez C., López-Bigas N., Kamoun A., Neuzillet Y., Gestraud P., et al. Independent component analysis uncovers the landscape of the bladder tumor transcriptome and reveals insights into luminal and basal subtypes. Cell Rep. 2014;9:1235–1245. doi: 10.1016/j.celrep.2014.10.035. [DOI] [PubMed] [Google Scholar]
- 55.Varley C.L., Stahlschmidt J., Lee W.C., Holder J., Diggle C., Selby P.J., Trejdosiewicz L.K., Southgate J. Role of PPARgamma and EGFR signalling in the urothelial terminal differentiation programme. J. Cell Sci. 2004;117:2029–2036. doi: 10.1242/jcs.01042. [DOI] [PubMed] [Google Scholar]
- 56.Rochel N., Krucker C., Coutos-Thévenot L., Osz J., Zhang R., Guyon E., Zita W., Vanthong S., Hernandez O.A., Bourguet M., et al. Recurrent activating mutations of PPARγ associated with luminal bladder tumors. Nat. Commun. 2019;10:253. doi: 10.1038/s41467-018-08157-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Korpal M., Puyang X., Jeremy Wu Z., Seiler R., Furman C., Oo H.Z., Seiler M., Irwin S., Subramanian V., Julie Joshi J., et al. Evasion of immunosurveillance by genomic alterations of PPARγ/RXRα in bladder cancer. Nat. Commun. 2017;8:103. doi: 10.1038/s41467-017-00147-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bernardo C., Eriksson P., Marzouka N.A., Liedberg F., Sjödahl G., Höglund M. Molecular pathology of the luminal class of urothelial tumors. J. Pathol. 2019;249:308–318. doi: 10.1002/path.5318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Meeks J.J., Carneiro B.A., Pai S.G., Oberlin D.T., Rademaker A., Fedorchak K., Balasubramanian S., Elvin J., Beaubier N., Giles F.J. Genomic characterization of high-risk non-muscle invasive bladder cancer. Oncotarget. 2016;7:75176–75184. doi: 10.18632/oncotarget.12661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bellmunt J., Kim J., Reardon B., Perera-Bel J., Orsola A., Rodriguez-Vida A., Wankowicz S.A., Bowden M., Barletta J.A., Morote J., et al. Genomic Predictors of Good Outcome, Recurrence, or Progression in High-Grade T1 Non-Muscle-Invasive Bladder Cancer. Cancer Res. 2020;80:4476–4486. doi: 10.1158/0008-5472.CAN-20-0977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rasheed S.A.K., Leong H.S., Lakshmanan M., Raju A., Dadlani D., Chong F.T., Shannon N.B., Rajarethinam R., Skanthakumar T., Tan E.Y., et al. GNA13 expression promotes drug resistance and tumor-initiating phenotypes in squamous cell cancers. Oncogene. 2018;37:1340–1353. doi: 10.1038/s41388-017-0038-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Siehler S. Regulation of RhoGEF proteins by G12/13-coupled receptors. Br. J. Pharmacol. 2009;158:41–49. doi: 10.1111/j.1476-5381.2009.00121.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yagi H., Asanoma K., Ohgami T., Ichinoe A., Sonoda K., Kato K. GEP oncogene promotes cell proliferation through YAP activation in ovarian cancer. Oncogene. 2016;35:4471–4480. doi: 10.1038/onc.2015.505. [DOI] [PubMed] [Google Scholar]
- 64.Zhang Z., Tan X., Luo J., Cui B., Lei S., Si Z., Shen L., Yao H. GNA13 promotes tumor growth and angiogenesis by upregulating CXC chemokines via the NF-κB signaling pathway in colorectal cancer cells. Cancer Med. 2018;7:5611–5620. doi: 10.1002/cam4.1783. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 65.Maziarz M., Park J.C., Leyme A., Marivin A., Garcia-Lopez A., Patel P.P., Garcia-Marcos M. Revealing the Activity of Trimeric G-proteins in Live Cells with a Versatile Biosensor Design. Cell. 2020;182:770–785.e16. doi: 10.1016/j.cell.2020.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lawson A.R.J., Abascal F., Coorens T.H.H., Hooks Y., O’Neill L., Latimer C., Raine K., Sanders M.A., Warren A.Y., Mahbubani K.T.A., et al. Extensive heterogeneity in somatic mutation and selection in the human bladder. Science. 2020;370:75–82. doi: 10.1126/science.aba8347. [DOI] [PubMed] [Google Scholar]
- 67.Li C.G., Mahon C., Sweeney N.M., Verschueren E., Kantamani V., Li D., Hennigs J.K., Marciano D.P., Diebold I., Abu-Halawa O., et al. PPARγ Interaction with UBR5/ATMIN Promotes DNA Repair to Maintain Endothelial Homeostasis. Cell Rep. 2019;26:1333–1343.e7. doi: 10.1016/j.celrep.2019.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Khandekar M.J., Banks A.S., Laznik-Bogoslavski D., White J.P., Choi J.H., Kazak L., Lo J.C., Cohen P., Wong K.K., Kamenecka T.M., et al. Noncanonical agonist PPARγ ligands modulate the response to DNA damage and sensitize cancer cells to cytotoxic chemotherapy. Proc. Natl. Acad. Sci. USA. 2018;115:561–566. doi: 10.1073/pnas.1717776115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.De Simone G., di Masi A., Vita G.M., Polticelli F., Pesce A., Nardini M., Bolognesi M., Ciaccio C., Coletta M., Turilli E.S., et al. Mycobacterial and Human Nitrobindins: Structure and Function. Antioxid. Redox Signal. 2020;33:229–246. doi: 10.1089/ars.2019.7874. [DOI] [PubMed] [Google Scholar]
- 70.Necchi A., Raggi D., Gallina A., Ross J.S., Farè E., Giannatempo P., Marandino L., Colecchia M., Lucianò R., Bianchi M., et al. Impact of Molecular Subtyping and Immune Infiltration on Pathological Response and Outcome Following Neoadjuvant Pembrolizumab in Muscle-invasive Bladder Cancer. Eur. Urol. 2020;77:701–710. doi: 10.1016/j.eururo.2020.02.028. [DOI] [PubMed] [Google Scholar]
- 71.Tamura S., Wang Y., Veeneman B., Hovelson D., Bankhead A., 3rd, Broses L.J., Lorenzatti Hiles G., Liebert M., Rubin J.R., Day K.C., et al. Molecular Correlates of In Vitro Responses to Dacomitinib and Afatinib in Bladder Cancer. Bladder Cancer. 2018;4:77–90. doi: 10.3233/BLC-170144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Choudhury N.J., Campanile A., Antic T., Yap K.L., Fitzpatrick C.A., Wade J.L., 3rd, Karrison T., Stadler W.M., Nakamura Y., O’Donnell P.H. Afatinib Activity in Platinum-Refractory Metastatic Urothelial Carcinoma in Patients With ERBB Alterations. J. Clin. Oncol. 2016;34:2165–2171. doi: 10.1200/JCO.2015.66.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cambier S., Sylvester R.J., Collette L., Gontero P., Brausi M.A., van Andel G., Kirkels W.J., Silva F.C., Oosterlinck W., Prescott S., et al. EORTC Nomograms and Risk Groups for Predicting Recurrence, Progression, and Disease-specific and Overall Survival in Non-Muscle-invasive Stage Ta-T1 Urothelial Bladder Cancer Patients Treated with 1-3 Years of Maintenance Bacillus Calmette-Guérin. Eur. Urol. 2016;69:60–69. doi: 10.1016/j.eururo.2015.06.045. [DOI] [PubMed] [Google Scholar]
- 74.Fernandez-Gomez J., Solsona E., Unda M., Martinez-Piñeiro L., Gonzalez M., Hernandez R., Madero R., Ojea A., Pertusa C., Rodriguez-Molina J., et al. Club Urológico Español de Tratamiento Oncológico (CUETO) Prognostic factors in patients with non-muscle-invasive bladder cancer treated with bacillus Calmette-Guérin: multivariate analysis of data from four randomized CUETO trials. Eur. Urol. 2008;53:992–1001. doi: 10.1016/j.eururo.2007.10.006. [DOI] [PubMed] [Google Scholar]
- 75.Bastos D.A., Mattedi R.L., Barreiro R., dos Santos F.F., Buzatto V., Masotti C., Souza J.M., de Lima M.Z.T., Friguglietti G.W., Dzik C., et al. Genomic Biomarkers and Underlying Mechanism of Benefit from BCG Immunotherapy in Non-Muscle Invasive Bladder Cancer. Bladder Cancer. 2020;6:171–186. [Google Scholar]
- 76.Kates M., Matoso A., Choi W., Baras A.S., Daniels M.J., Lombardo K., Brant A., Mikkilineni N., McConkey D.J., Kamat A.M., et al. Adaptive Immune Resistance to Intravesical BCG in Non-Muscle Invasive Bladder Cancer: Implications for Prospective BCG-Unresponsive Trials. Clin. Cancer Res. 2020;26:882–891. doi: 10.1158/1078-0432.CCR-19-1920. [DOI] [PubMed] [Google Scholar]
- 77.Kamat A.M., Dickstein R.J., Messetti F., Anderson R., Pretzsch S.M., Gonzalez G.N., Katz R.L., Khanna A., Zaidi T., Wu X., et al. Use of fluorescence in situ hybridization to predict response to bacillus Calmette-Guérin therapy for bladder cancer: results of a prospective trial. J. Urol. 2012;187:862–867. doi: 10.1016/j.juro.2011.10.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gartner J.J., Davis S., Wei X., Lin J.C., Trivedi N.S., Teer J.K., Meltzer P.S., Rosenberg S.A., Samuels Y., NISC Comparative Sequencing Program Comparative exome sequencing of metastatic lesions provides insights into the mutational progression of melanoma. BMC Genomics. 2012;13:505. doi: 10.1186/1471-2164-13-505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Blokzijl F., Janssen R., van Boxtel R., Cuppen E. MutationalPatterns: comprehensive genome-wide analysis of mutational processes. Genome Med. 2018;10:33. doi: 10.1186/s13073-018-0539-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bergmann E.A., Chen B.J., Arora K., Vacic V., Zody M.C. Conpair: concordance and contamination estimator for matched tumor-normal pairs. Bioinformatics. 2016;32:3196–3198. doi: 10.1093/bioinformatics/btw389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Benjamin D., Sato T., Cibulskis K., Getz G., Stewart C., Lichtenstein L. Calling Somatic SNVs and Indels with Mutect2. bioRxiv. 2019 doi: 10.1101/861054. [DOI] [Google Scholar]
- 85.Fan Y., Xi L., Hughes D.S., Zhang J., Zhang J., Futreal P.A., Wheeler D.A., Wang W. MuSE: accounting for tumor heterogeneity using a sample-specific error model improves sensitivity and specificity in mutation calling from sequencing data. Genome Biol. 2016;17:178. doi: 10.1186/s13059-016-1029-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Koboldt D.C., Zhang Q., Larson D.E., Shen D., McLellan M.D., Lin L., Miller C.A., Mardis E.R., Ding L., Wilson R.K. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22:568–576. doi: 10.1101/gr.129684.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kim S., Scheffler K., Halpern A.L., Bekritsky M.A., Noh E., Källberg M., Chen X., Kim Y., Beyter D., Krusche P., Saunders C.T. Strelka2: fast and accurate calling of germline and somatic variants. Nat. Methods. 2018;15:591–594. doi: 10.1038/s41592-018-0051-x. [DOI] [PubMed] [Google Scholar]
- 88.Shiraishi Y., Sato Y., Chiba K., Okuno Y., Nagata Y., Yoshida K., Shiba N., Hayashi Y., Kume H., Homma Y., et al. An empirical Bayesian framework for somatic mutation detection from cancer genome sequencing data. Nucleic Acids Res. 2013;41:e89. doi: 10.1093/nar/gkt126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Dunkler D., Ploner M., Schemper M., Heinze G. Weighted Cox Regression Using the R Package coxphw. J. Stat. Softw. 2018;84 [Google Scholar]
- 90.Eble J.N., Sauter G., Epstein J.I., Sesterhenn I.A. IARC Press Lyon; 2004. World Health Organization Classification of Tumours. Pathology and Genetics of Tumours of the Urinary System and Male Genital Organs. [Google Scholar]
- 91.Mostofi F.K., Davis C.J., Jr., Sesterhenn I.A., Sobin L.H. Springer; 1973. Histological typing of urinary bladder tumours. [Google Scholar]
- 92.Mills R.E., Luttig C.T., Larkins C.E., Beauchamp A., Tsui C., Pittard W.S., Devine S.E. An initial map of insertion and deletion (INDEL) variation in the human genome. Genome Res. 2006;16:1182–1190. doi: 10.1101/gr.4565806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Marzouka N.A., Eriksson P. multiclassPairs: An R package to train multiclass pairbased classifier. Bioinformatics. 2021:btab088. doi: 10.1093/bioinformatics/btab088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lambert S.A., Jolma A., Campitelli L.F., Das P.K., Yin Y., Albu M., Chen X., Taipale J., Hughes T.R., Weirauch M.T. The Human Transcription Factors. Cell. 2018;172:650–665. doi: 10.1016/j.cell.2018.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Fletcher M.N., Castro M.A., Wang X., de Santiago I., O’Reilly M., Chin S.F., Rueda O.M., Caldas C., Ponder B.A., Markowetz F., Meyer K.B. Master regulators of FGFR2 signalling and breast cancer risk. Nat. Commun. 2013;4:2464. doi: 10.1038/ncomms3464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Margolin A.A., Wang K., Lim W.K., Kustagi M., Nemenman I., Califano A. Reverse engineering cellular networks. Nat. Protoc. 2006;1:662–671. doi: 10.1038/nprot.2006.106. [DOI] [PubMed] [Google Scholar]
- 97.R Core Team . R Foundation for Statistical Computing; 2012. A language and environment for statistical computing. [Google Scholar]
- 98.Benjamini Y., Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. B. 1995;57:289–300. [Google Scholar]
- 99.Grambsch P., Therneau T. Proportional hazards tests and diangnostics based on weighted residuals. Biometrika. 1994;81:515–526. [Google Scholar]
- 100.Schemper M., Wakounig S., Heinze G. The estimation of average hazard ratios by weighted Cox regression. Stat. Med. 2009;28:2473–2489. doi: 10.1002/sim.3623. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Microarray data are available at the Gene Expression Omnibus under accession number GSE163209. Raw sequencing data are available at The European Genome-phenome Archive (EGA) under accession numbers EGAS00001005765, EGAS00001005766 and EGAS00001005767.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.