

Abstract
Autologous, antigen-specific, tolerogenic dendritic cells (tolDCs) are presently assessed to reverse and possibly cure autoimmune diseases such as type 1 diabetes (T1D). Good Manufacturing Practice production and clinical implementation of such cell therapies critically depend on their stability and reproducible production from healthy donors and, more importantly, patient-derived monocytes. Here the authors demonstrate that tolDCs (modulated using 1,25-dihydroxyvitamin D3 and dexamethasone) displayed similar features, including protein, transcriptome and epigenome profiles, between two international clinical centers and between T1D and healthy donors, validating reproducible production. In addition, neither phenotype nor function of tolDCs was affected by repeated stimulation with inflammatory stimuli, underscoring their stability as semi-mature DCs. Furthermore, tolDCs exhibited differential DNA methylation profiles compared with inflammatory mature DCs (mDCs), and this was already largely established prior to maturation, indicating that tolDCs are locked into an immature state. Finally, approximately 80% of differentially expressed known T1D risk genes displayed a corresponding differential DNA methylome in tolDCs versus mDCs and metabolic and immune pathway genes were also differentially methylated and expressed. In summary, tolDCs are reproducible and stable clinical cell products unaffected by the T1D status of donors. The observed stable, semi-mature phenotype and function of tolDCs are exemplified by epigenetic modifications representative of immature-stage cells. Together, the authors’ data provide a strong basis for the production and clinical implementation of tolDCs in the treatment of autoimmune diseases such as T1D.
Keywords: cell therapy, dendritic cells, DNA methylation, epigenetics, immunotherapy, tolerance induction
Introduction
Dendritic cells (DCs) activate T cells to elicit an inflammatory or anti-inflammatory response, depending on whether the DC is inflammatory or tolerogenic [1]. Tolerogenic DCs (tolDCs) are currently being evaluated as clinical cellular products for therapy in multiple autoimmune disorders, including type 1 diabetes (T1D) [2–8]. T1D is a T-cell-mediated disease in which insulin-producing beta cells are attacked by autoreactive T cells [9]. TolDCs can be generated in vitro from the peripheral blood of T1D patients by isolating and modulating monocytes with 1,25-dihydroxyvitamin D3 (VD3), followed by dexamethasone [10]. TolDCs can subsequently be pulsed with disease-specific peptides to potentiate the capacity to reeducate the immune system in an antigen-specific fashion, which, in the case of T1D, can help to preserve beta cells [11].
Stability of a cellular phenotype or function could be supported by epigenetic regulation of gene expression [12]. The field of epigenetics deals with heritable alterations in gene expression in the absence of changes in the underlying DNA sequence. Epigenetic status is maintained by several mechanisms, including DNA methylation [13–15]. The authors produced tolDCs by treating monocytes with VD3, which acts by binding to the nuclear VD3 receptor. VD3 has long been linked to immunomodulation [16]. Although there are data from various other cell types, the epigenetic effects of VD3 have not yet been explored in human DCs.
The authors found that, in human DCs, VD3 followed by dexamethasone significantly altered the expression of almost half of the transcripts of known T1D risk genes [17,18]. In addition to the effect of VD3 on T1D risk genes, VD3 triggers metabolic changes with upregulation of glycolysis, which is essential for tolerogenic function [19]. TolDCs modulate the immune system by secreting anti-inflammatory cytokines, such as IL-10, and by influencing other immune cells via cell surface markers. TolDCs have low T-cell stimulatory capacities, partly due to low expression of co-stimulatory molecules such as CD86, and are capable of inducing T regulatory cells [20,21]. Moreover, tolDCs express lower levels of HLA-DR compared with inflammatory DCs, resulting in lower T-cell stimulatory capacity in a mixed lymphocyte reaction (MLR) [20]. Because of these properties, tolDCs have been said to have a semi-mature phenotype [22,23]. Immature DCs are not yet inflammatory, and maturation triggers an inflammatory machinery that grants mature DCs (mDCs) the co-stimulatory tools necessary for T-cell priming and activation [24–26]. If arrested in this semi-mature stage, tolDCs would not be affected by further maturation challenges in vivo, securing their anti-inflammatory nature and legacy.
For clinical translation and utility of a cellular product, reproducibility of a stable and effective cell product from different donors is of the utmost importance. Ideally, this reproducibility in manufacturing should be achievable in multiple clinical centers. A safety trial was conducted at Leiden University Medical Center (LUMC), Leiden, the Netherlands, evaluating tolDCs in T1D patients (D-Sense trial) [27], and presently, a phase 1b clinical trial is being set up at the City of Hope Medical Center (COH), Duarte, California, USA, to assess safety and feasibility in C-peptide-positive T1D patients.
In this study, the authors examined the stability of tolDCs by perturbing them with multiple inflammatory stimuli. In addition, the authors studied the reproducibility of tolDCs between two international production centers and between healthy subjects and T1D patients. Finally, the authors explored whether epigenetic modifications induced by VD3 may help to explain the observed stability of tolDCs.
Methods
Donor selection and database generation
Blood samples for tolDC cultures were taken from healthy blood donors and processed at either LUMC or COH. Samples from the D-Sense clinical trial were taken from T1D patients and produced at LUMC [27]. All donors gave informed consent.
DC culture
DCs were cultured as described previously [28]. In short, peripheral blood mononuclear lymphocytes were isolated from buffy coats collected from either healthy or T1D blood donors. CD14+ selection was performed with CD14 microbeads (Miltenyi Biotec, Bergisch Gladbach, Germany), and monocytes were cultured in RPMI 1640 medium supplemented with 8% fetal bovine serum (Greiner Bio-One, Alphen aan den Rijn, the Netherlands), glutamine and penicillin/streptomycin (Life Technologies), recombinant human IL-4 at 500 U/mL (Invitrogen, Breda, the Netherlands) and recombinant human granulocyte-macrophage colony-stimulating factor (GM-CSF) at 800 U/mL (Invitrogen) for 6 days. To induce tolDCs, clinical-grade VD3 at 10−8 M (32222-06-3; Dishman Carbogen Amcis, Veenendaal, the Netherlands) was added on day 0 and day 3. On day 3, dexamethasone at 10−6 M (Sigma-Aldrich) was also added to the tolDC culture. On day 3, culture medium was refreshed by discarding 50% of the medium and adding the same volume and twice concentrated IL-4 and GM-CSF to all cell cultures. On day 6, immature DCs were harvested and matured for 24–48 h by adding a cytokine mix, including GM-CSF, human recombinant IL-1β at 1600 U/mL, human recombinant IL-6 at 500 U/mL and human recombinant tumor necrosis factor alpha (TNF-α) at 335 U/mL (Miltenyi Biotec), and synthetic prostaglandin E2 at 2 μg/mL (Pfizer). Supernatant at day 8 was collected for further analysis of cytokine production. After maturation, DCs were phenotyped by flow cytometry, used for an MLR test, secondly matured or stored in liquid nitrogen. For second maturation with inflammatory stimuli, DCs were rested for 5 days in culture media supplemented with GM-CSF, after which a second round of maturation was performed. DCs were then stimulated with the previously stated cytokine mix, lipopolysaccharide (LPS) (100 ng/mL) or CD40 ligation via co-culture with CD40 ligand (CD40L)-expressing L cells (0.5 × 106 DCs:0.2 × 106 L cells) for 24–48 h. For the second maturation experiments, cells were analyzed immediately after the first and second maturations.
Phenotype analysis
Unless stated otherwise, antibodies for phenotype analysis were purchased from BD Pharmingen (San Diego, CA, USA) and were the following: fluorescein isothiocyanate-conjugated HLA-DR, CD80, IgG2A, CD52 (Bio-Rad, Hercules, CA, USA) and IgG2B (Bio-Rad); phycoerythrin-conjugated CD1a, CD86, IgG1 and CD83 (Beckman Coulter, Brea, CA, USA); phycoerythrin-Cy7- conjugated CD14 (eBioscience, San Diego, CA, USA), ILT-3 (Beckman Coulter) and IgG1 (eBioscience); PercPCy5.5-conjugated CD209 and IgG2B; and allophycocyanin-conjugated IgG1, CD3, CD25, PD-L1 (eBioscience) and CD40 (eBioscience). DCs were incubated with a mix of monoclonal antibodies for 30 min on ice. Cells were washed in fluorescence-activated cell sorting (FACS) buffer containing 1% fetal bovine serum and 0.05% sodium azide (Sigma-Aldrich) and analyzed using FACSCanto or Fortessa (BD Biosciences). Data were analyzed using FACSDiva 8 (BD Biosciences) and FlowJo 10 software (Ashland, Oregon, USA).
Cytokine analysis and MLR
After the culture period, supernatants from mDCs were harvested and analyzed for cytokine analysis with the nine-plex Bio-Plex Pro human cytokine Th1/Th2 assay Luminex kit (Bio-Rad) according to the manufacturer’s protocol. In parallel, the cells were analyzed for T-cell stimulatory capacity in an MLR. The cells were harvested and replated in a flat bottom 96-well plate in different concentrations in triplicate in Iscove’s Modified Dulbecco’s Medium with 10% inactivated human serum (Sanquin, Amsterdam, the Netherlands). Allogeneic CD4+ T cells were obtained from HLA-typed peripheral blood mononuclear lymphocytes using the Dynabeads untouched CD4 T-cell kit (Invitrogen) according to the manufacturer’s protocol. Next, 1 × 104 allogeneic CD4 + T cells were added to the wells, and after 4 days of culture they were pulsed overnight with [3H]-thymidine 0.5 μCi/well. Thymidine incorporation was measured using a liquid scintillation counter (PerkinElmer, Groningen, the Netherlands). The counts per minute of the tolDC condition were divided by the counts per minute of the mDC condition (positive control) and multiplied by 100 to provide the stimulation index (SI). The change in T-cell stimulation from the first maturation for the additional inflammatory stimuli was calculated by the delta SI.
Delta SI (change from first maturation) = SI second maturation − SI first maturation.
Metabolic analysis
The XFe96 extracellular flux analyzer (Seahorse Bioscience, MA, USA) was used to measure the mitochondrial oxygen consumption rate (mpH/min) and extracellular acidification rate (mpH/min). On day 6, immature DCs were harvested and matured with the previously mentioned cytokine mix for 24–48 h in a 96-well Seahorse plate at 4 × 104 cells per well. After maturation, the plate was spun down with slow acceleration, and break off settings and cells were carefully washed. Next, 5 μg/mL human recombinant soluble CD40L (PeproTech, Rocky Hill, NJ, USA) was added for 18 h. After spinning down the plate with slow acceleration and break off settings, DCs were carefully washed three times in either glycolysis stress test assay medium (Dulbecco’s Modified Eagle’s Medium base, 2 mM L-glutamine, pH 7.35) or mitochondrial stress test medium (Dulbecco’s Modified Eagle’s Medium base, 2 mM L-glutamine, 1 mM pyruvate, 25 mM glucose, pH 7.35) and incubated in a non-carbon dioxide incubator at 37°C for 1 h. The following compounds were used for the glycolysis and mitochondrial stress tests: 10 mM glucose, 1.7 μM oligomycin, 50 mM 2-deoxy-D-glucose, 0.5 M carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone, 0.5 μM rotenone and 0.5 μM antimycin A. The plate was analyzed on a XFe96 extracellular flux analyzer (Seahorse Bioscience) using the standard stress test templates. After the assay, the plate was collected and analyzed for cell number using a Celigo cytometer (Nexcelom Bioscience, San Diego, CA, USA). Oxygen consumption rate and extracellular acidification rate values were normalized to cell number.
RNA and genomic DNA preparations
A total of 11 donors were used for these studies, of which three DC donors came from COH and three from a previous study conducted at LUMC [17], and the other five donors were from the authors’ D-Sense clinical trial conducted at LUMC. DNA and RNA from these samples were extracted using a Quick-DNA/RNA MiniPrep kit (Zymo Research, Irvine, CA, USA) following the manufacturer’s protocol. Only RNA with an RNA integrity number ≥8 was used in the polyA sequencing library preparation method for RNA sequencing (RNA-seq) (Illumina, San Diego, CA, USA). The genomic DNA was analyzed for DNA methylation levels by the Infinium MethylationEPIC array (Illumina) according to the manufacturer’s instructions. Both assays were performed by the genomics core at COH.
RNA-seq library preparation and sequencing with Illumina HiSeq 2500
RNA-seq libraries were prepared with a messenger RNA HyperPrep kit (KR1352; Kapa Biosystems) according to the manufacturer’s protocol. Briefly, 250 ng of total RNA from each sample was used for polyA RNA enrichment. The enriched messenger RNA underwent fragmentation and first strand complementary DNA (cDNA) synthesis. The combined second cDNA synthesis with 2′-deoxyuridine 5′-triphosphate and A-tailing reaction generated the resulting double-stranded cDNA with deoxyadenosine monophosphate to the 3’ ends. The barcoded adaptors were then ligated to the double-stranded cDNA fragments. A 12-cycle polymerase chain reaction was performed to produce the final sequencing library. The libraries were validated with the Bioanalyzer DNA high sensitivity kit (Agilent) and quantified with Qubit. RNA-seq libraries were sequenced on the Illumina HiSeq 2500 using an SR v4 kit with the single read mode of 51 cycles of read1 and seven cycles of index read. Real-time analysis 2.2.38 software was used to process the image analysis and base calling.
RNA-seq data analysis
Raw RNA-seq reads were trimmed to remove sequencing adapters using Trimmomatic [29] and polyA tails using FASTP [30]. The processed reads were aligned to the human genome (hg19) using STAR 020201 software [31]. HTSeq 0.6.0 software [32] was then applied to generate the count matrix on Reference Sequence (RefSeq) genes with default parameters. The resulting counts were normalized using the trimmed mean of M-values method provided by the edgeR package in R [33] to obtain normalized expression values. For between cell type comparison, general linear models were applied to identify DEGs between two specific cell types using the trimmed mean of M-values normalized expression level as dependent variable and cell type as independent variable, adjusting for disease status and location for each sample. For comparison between different locations (COH versus LUMC) or health status (T1D versus healthy) within one specific cell type, similar models were used with location/health as dependent variable, adjusting for health or location, respectively. Genes with a false discovery rate <0.05 and a fold change (FC) >2 or <0.5 were considered significantly upregulated and downregulated genes, respectively.
Illumina Infinium HD methylation assay
The genomic DNA samples were treated with bisulfite using the EZ DNA methylation kit from Zymo Research with the alternative incubation condition for the Illumina Infinium HD methylation assay according to the manufacturer’s protocol. Briefly, the bisulfite-converted DNA was denatured with 0.1 N sodium hydroxide and amplified for 20–24 h in the 37°C hybridization chamber to produce a sufficient DNA sample. The amplified DNA was enzymatically fragmented at 37°C for 1 h and precipitated for 30 min at 4°C. To hybridize the DNA onto Illumina BeadChips, the precipitated DNA was resuspended using RA1 solution according to the manufacturer’s protocol, and the suspended DNA with an appropriate volume was loaded onto the eight-sample Infinium MethylationEPIC BeadChip (Illumina). The hybridization was performed for over 16 h within a 24-h period. After washing the hybridized BeadChip, the primers hybridized to DNA were extended and incorporated with the labeled nucleotides for the multilayer staining process. The image acquisition was carried out using the Illumina iScan System.
DNA-me profiling and data analysis
After quality controls with Illumina’s GenomeStudio, data were pre-processed using R package minfi. Specifically, background correction was followed by subset quantile within array normalization and quantile normalization. DNA methylation level, or beta value, was then generated for each CpG site and each sample. CpG sites with detection P ≥ 0.01 in at least one sample were excluded. To identify differentially methylated CpGs (DMCs) among groups (mature tolerogenic, immature tolerogenic, mature inflammatory, immature inflammatory), multiple linear regressions were performed using beta values as response variable and group as explanatory variable, adjusting for patients. To identify DMCs among different sites, sites were used as explanatory variable without adjusting for patients. The following criteria were used to select the significant DMCs: (i) P ≤ 0.01, (ii) difference ≥0.15 and (iii) at least one group with mean methylation level ≥0.25. The significance level of difference between two groups genome-wide was visualized using a Manhattan plot generated using R package qqman v.0.1.4. Hierarchical clustering was performed combining all DMCs with Pearson correlation as distance matrix and average linkage using Cluster 3.0. A heatmap was generated using Java TreeView 1.1.6r4.
The significant DMCs were merged into differentially methylated regions (DMRs) if their difference was ≤200 bp. Multiple linear regressions were performed on each of these DMRs using beta value as response variable and group as explanatory variable, adjusting for patient and CpG position, to summarize region-level difference.
CpGs and DMRs were annotated to genomic regions (transcription start site [TSS] 200 [200-bp upstream region of TSS] and TSS1500 [−1500 bp to approximately −200 bp relative to TSS], 5′ untranslated region, coding exon, intron and 3’ untranslated region) relative to RefSeq genes (hg19; University of California Santa Cruz Genome Browser) based on their location. CpGs or DMRs not located in any of the aforementioned regions were considered to locate in intergenic regions. Ingenuity pathway analysis (IPA) (Qiagen) or Database for Annotation, Visualization and Integrated Discovery (DAVID) analysis was applied to the annotated genes of DMCs or DMRs to obtain enriched biological processes or pathways. De novo motif analysis was performed on DMRs between mature tolDCs (mtolDCs) and mDCs using RSAT Metazoa Regulatory Sequence Analysis Tools (http://rsat.sb-roscoff.fr/peak-motifs_form.cgi) to detect enriched motifs using ±250-kb regions of the least differentially 5000 CpGs as background. The top identified significantly enriched motifs were then queried against core non-redundant vertebrates (2018) in JASPAR to identify any known transcription binding sites.
Integration analysis of DNA-me and gene expression
For each DEG identified between mtolDCs and mDCs, DMCs located in eight non-overlapping genomic regions relative to genes were identified. The regions included promoter (2.5 kb upstream of TSS), gene body, approximately 0–5 kb upstream of promoter, approximately 0–5 kb downstream of gene body, approximately 5–50 kb upstream of promoter, approximately 5–50 kb downstream of gene body, approximately 50–500 kb upstream of promoter and approximately 50–500 kb downstream of gene body. The gene lists containing the differentially methylated locus (DML) in each of these regions were also uploaded to IPA for Gene Ontology analysis.
Gene Ontology analysis
Pathway analysis was conducted using DAVID 6.8 [34,35]. GSEAP-reranked analysis was performed using the Gene Set Enrichment Analysis desktop program in Java [36,37] based on a ranked list of whole genes according to their log2 FC and P values.
Analysis on T1D risk genes
T1D risk genes were identified as those genes located on the T1D-associated regions provided by www.t1dbase.org. Specifically, except for one region, all the regions (based on hg38 human genome assembly) were lifted over to hg19 assembly using the University of California Santa Cruz LiftOver tool (http://genome.ucsc.edu/cgi-bin/hgLiftOver) to render them consistent with the genome assembly used by the MethylationEPIC array (Illumina). The 441 RefSeq genes located in these 56 regions were considered potential T1D risk genes, and among these, 62 located in 28 regions showing differential expression between mtolDCs and mDCs were retrieved. DMCs located in these 62 T1D risk DEGs were further identified as described previously. The T1D DEGs and their associated DMLs located within 5-kb flanking regions are shown as circular plots using R package circlize 0.4.3.
Statistical analysis
Data were analyzed with Prism 7 (GraphPad Software). Either an unpaired Student’s t-test or analysis of variance was used to test statistical significance. P < 0.05 was considered significant.
Results
TolDCs retain semi-mature immunophenotype after stimulation with LPS, inflammatory cytokines and CD40L
All human experiments were performed following informed consent and approval from the institutional review boards, in accordance with approved protocols, at both clinical centers. Cells were processed at either LUMC or COH. DCs were derived from purified monocytes from buffy coats of anonymous healthy donors and cultured in six-well plates. Monocytes for the D-Sense clinical trial were obtained by apheresis procedures in T1D patients and cultured in culture bags [27]. All standard operational procedures and reagents in the manufacturing process of DCs were similar, regardless of processing location or clinical status. In short, monocytes were isolated from peripheral blood and treated with GM-CSF and IL-4 to produce inflammatory DCs, and in the case of tolDCs, the culture medium was additionally supplemented with VD3 and dexamethasone. Maturation of inflammatory and tolDCs was achieved by treatment with a cytokine mix (IL-6, TNF, IL-1β and prostaglandin E2).
Upon injection into a patient, mature tolDCs may encounter inflammatory stimuli in vivo that could affect their phenotype and function. To assess whether tolDCs are locked into a definitive semi-mature state, mature tolDCs were restimulated with inflammatory stimuli, and their phenotype and function were subsequently tested. Specifically, after the first cytokine-stimulated maturation and a rest period of 5 days, another inflammatory stimulus with LPS, CD40L or the same cytokine mix followed (Figure 1). The first cytokine-stimulated maturation induced an increase in HLA-DR, CD83 and CD86 expression in inflammatory DCs (mDCs), segregating mature tolDCs from mDCs. After the restimulation, tolDCs largely retained the phenotype acquired in the first step; in particular, they remained HLA-DRlow, CD14+, CD1a−, CD83−, CD86low and PD-L1+ and consistently distinct from inflammatory DCs stimulated in parallel, as examined by flow cytometry. The only exception was CD209 (also called DC-specific ICAM-3-grabbing non-integrin) expression, which decreased upon restimulation in both tolDCs and inflammatory DCs (Figure 1). CD209 is known to recognize ICAM-3 on T cells or ICAM-2 on endothelial cells and thereby has a role in trafficking and T-cell interactions [38,39].
Figure 1.
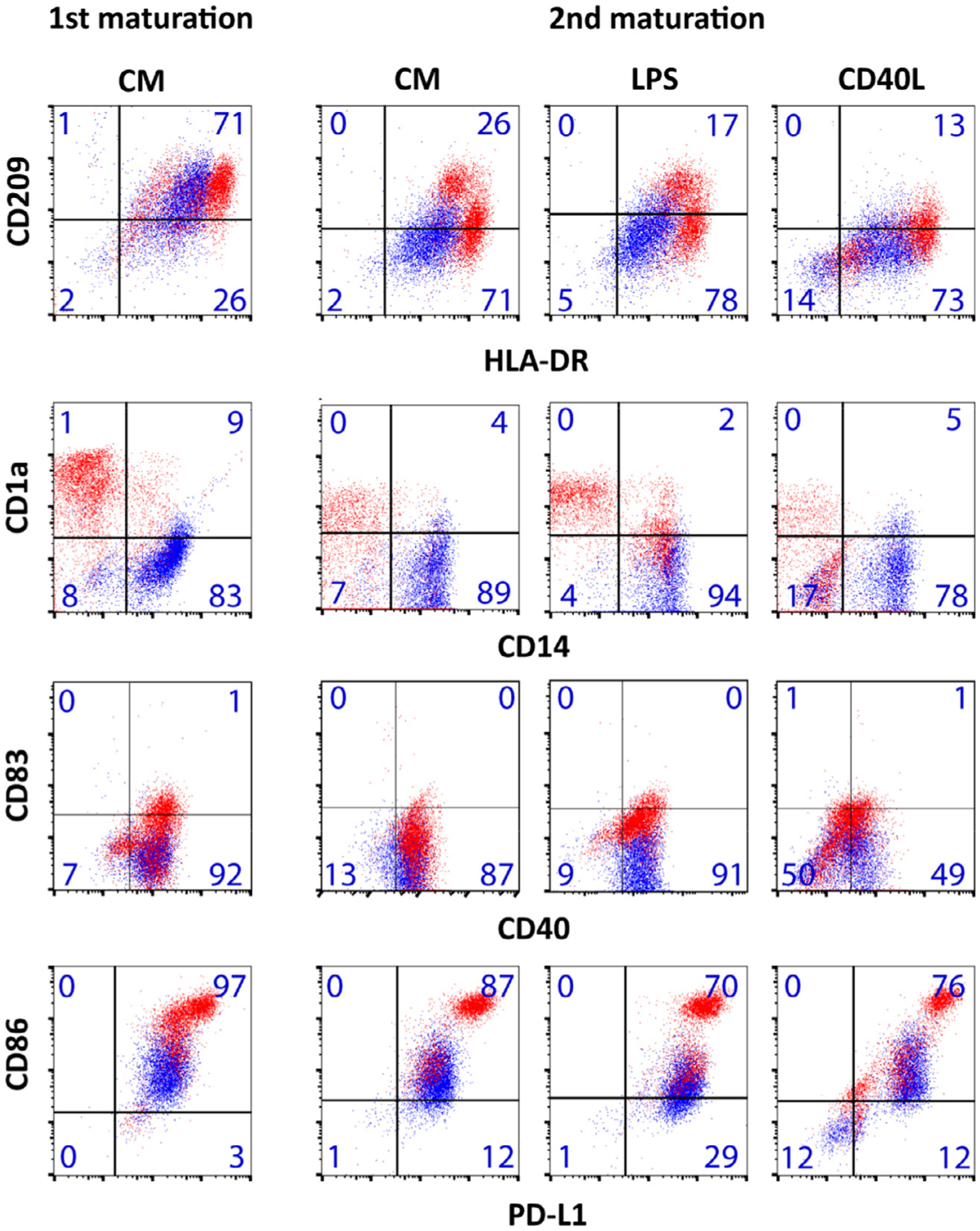
TolDCs have a stable phenotype after second maturation stimuli. Tolerogenic (blue) and inflammatory (red) DCs were first matured with CM and analyzed by flow cytometry. Subsequently, these cells were rested for 5 days and perturbed with another round of maturation stimuli, such as CM, LPS and CD40L, and analyzed. Phenotypic markers were analyzed by flow cytometry, and quadrant gates were set on the corresponding isotype controls. Percentage positivity per gate for tolDCs is noted in each box. Plots are representative of three independent experiments. The phenotype after the 5-day rest period was similar to the first maturation and not shown. With the exception of decreased expression of CD209 upon second maturation stimuli in both tolerogenic and inflammatory DCs, no changes in phenotype were noted upon second maturation. In the case of CD40L-stimulated cells, double-negative events in the lower left quadrant represent CD40L-expressing fibroblasts. ILT-3 was not significantly different between tolerogenic and inflammatory DCs (5.1 ± 12.0% versus 9.1 ± 15.4%, respectively, n = 33 donors, P = 0.18). CM, cytokine mix.
TolDCs retain semi-mature functional aspects and metabolism after additional inflammatory stimuli
In addition to flow cytometry, cytokine and functional analyses were conducted. None of the repeated inflammatory stimuli changed the cytokine profile of tolDCs (Figure 2A). Next, the authors assessed the T-cell stimulatory capacity of DCs in an MLR test in which DCs were co-cultured with allogeneic CD4+ T cells and proliferation of T cells was measured. Mature inflammatory DCs elicited a strong alloreaction, whereas tolDCs only minimally stimulated T cells to proliferate in an MLR (3.5 ± 2.8% of T-cell stimulation by mDCs). Repeated stimulation did not significantly change the low T-cell stimulatory capacity of tolDCs (Figure 2B).
Figure 2.
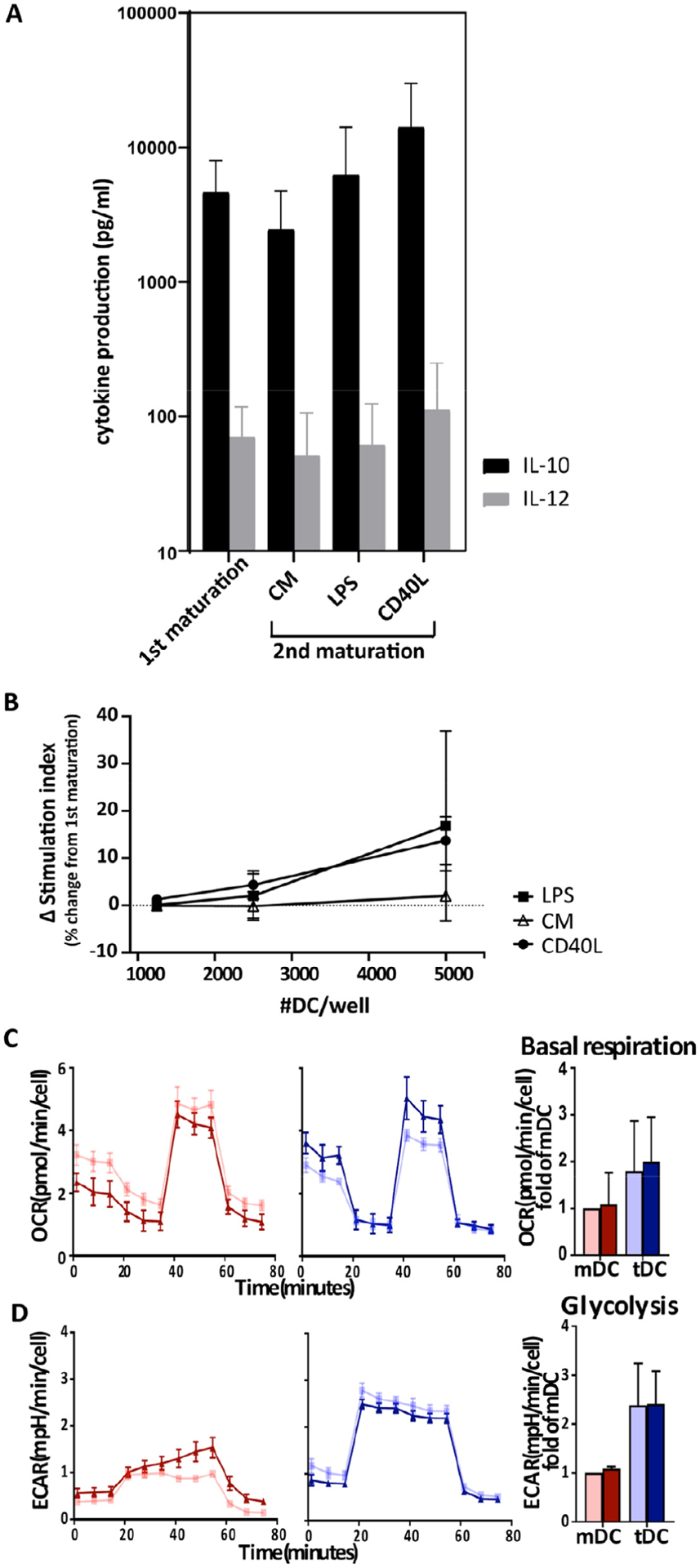
TolDCs have stable function and metabolism after repeated inflammatory stimuli. Tolerogenic and inflammatory DCs were matured with CM. Subsequently, these cells were perturbed with in vivo-simulating stimuli, such as CM, LPS and CD40L. (A) In vivo simulations with CM, LPS or CD40L did not change IL-10 or IL-12 cytokine concentrations in tolDCs (n = 4). Error bars show SD. (B) Relative change in capacity to stimulate T cells (SI) between first maturation and in vivo inflammatory simulations calculated as described earlier. No significant increase in immunogenicity of tolDCs was observed after additional inflammatory stimuli compared with first maturation. Graphs are representative of three independent experiments. (C,D) Seahorse analysis was performed to assess the effect of in vivo-simulating stimulus CD40L (in dark blue and dark red for tolerogenic and inflammatory DCs, respectively) on DC metabolism. DCs that received only the first maturation are in light blue for tolDCs and light red for mDCs. An oxygen consumption (C) and glycolysis stress test (D) was performed (n = 3). OCR and ECAR normalized to cell number are shown with SEM. To the right of the graphs (C,D), bar graphs of summary data of three independent donors show basal respiration and glycolysis calculated from graph data, with error bars showing SD. CD40L did not significantly change basal respiration or glycolysis. CM, cytokine mix; ECAR, extracellular acidification rate; OCR, oxygen consumption rate; SD, standard deviation; SEM, standard error of the mean; SI, stimulation index.
Recently, the authors showed that distinct metabolism is another functional marker for tolDCs [19]. Since CD40 ligation on DCs by T cells directly activates DCs in the context of antigen presentation on HLA class II [1], the authors deemed CD40L the most relevant physiological stimulus for also studying DC metabolism in a Seahorse assay. In concordance with the authors’ previous study [19], TolDCs elicited higher oxygen consumption, glycolysis and glycolytic capacity than mDCs (Figure 2C,D; also see supplementary Figure 1). CD40 ligation did not change oxygen consumption rate, as basal respiration and maximal respiration were unaltered in tolDCs and mDCs. In terms of glycolysis, CD40 ligation sensitized mDCs to oligomycin treatment, which increased extracellular acidification rate, quantified as a significant increase in glycolytic capacity (see supplementary Figure 1). TolDCs, however, remained insensitive to oligomycin treatment and did not exhibit changes in any of the glycolysis parameters upon CD40L treatment.
In summary, tolDCs displayed stable phenotype, function and metabolic activity even after repeated inflammatory stimuli. Therefore, tolDCs appear to be arrested in a semi-mature state.
TolDCs display a differential transcriptome compared with inflammatory DCs that is unaffected by health status or production location
Stability of a cellular product, including reproducible production between different international centers and between healthy subjects and T1D patients, is important for its implementation in the clinic. Therefore, the authors produced tolDCs from healthy subjects and T1D patients that passed validated quality control criteria (low CD86 expression, high CD52 expression [27]) in two international production centers. Subsequently, for more in-depth analysis, a transcriptome study was conducted comparing gene expression by RNA-seq between DCs produced at LUMC and COH. Finally, immature (day 6 of culture) and mature (day 8 of culture) DCs were compared to assess whether tolDCs maintained a stable semi-mature transcriptome.
In agreement with the authors’ previous studies, mature tolDCs showed increased expression of genes involved in glycolysis and oxidative phosphorylation and decreased expression of genes involved in interferon gamma (IFN-γ) signaling, unfolded protein response and antigen processing and presentation (see supplementary Figure 2A) [17]. DAVID pathway analysis confirmed that mature tolDCs displayed decreased cell activation pathways, in particular T-cell activation and response to cytokines, compared with mDCs.
Unsupervised hierarchical clustering of 10,854 expressed genes showed discrete cell types in all samples regardless of production site or health status (Figure 3A). When examining the number of differentially expressed genes (DEGs) (Figure 3B; also see supplementary Figures 3, 4), most were found between mature tolerogenic and inflammatory DCs (1663 upregulated and 1333 downregulated genes), with the fewest being found between immature tolerogenic and inflammatory DCs (760 upregulated and 795 downregulated genes). The effect of maturation was more prominent in inflammatory DCs, as they displayed more DEGs between the immature and mature states than tolDCs (1540 upregulated and 1557 downregulated genes versus 1119 upregulated and 1275 downregulated genes, respectively) (see supplementary Figures 5,6), suggesting that mature tolDCs could be more similar to their immature state than inflammatory mDCs are to their immature state.
Figure 3.
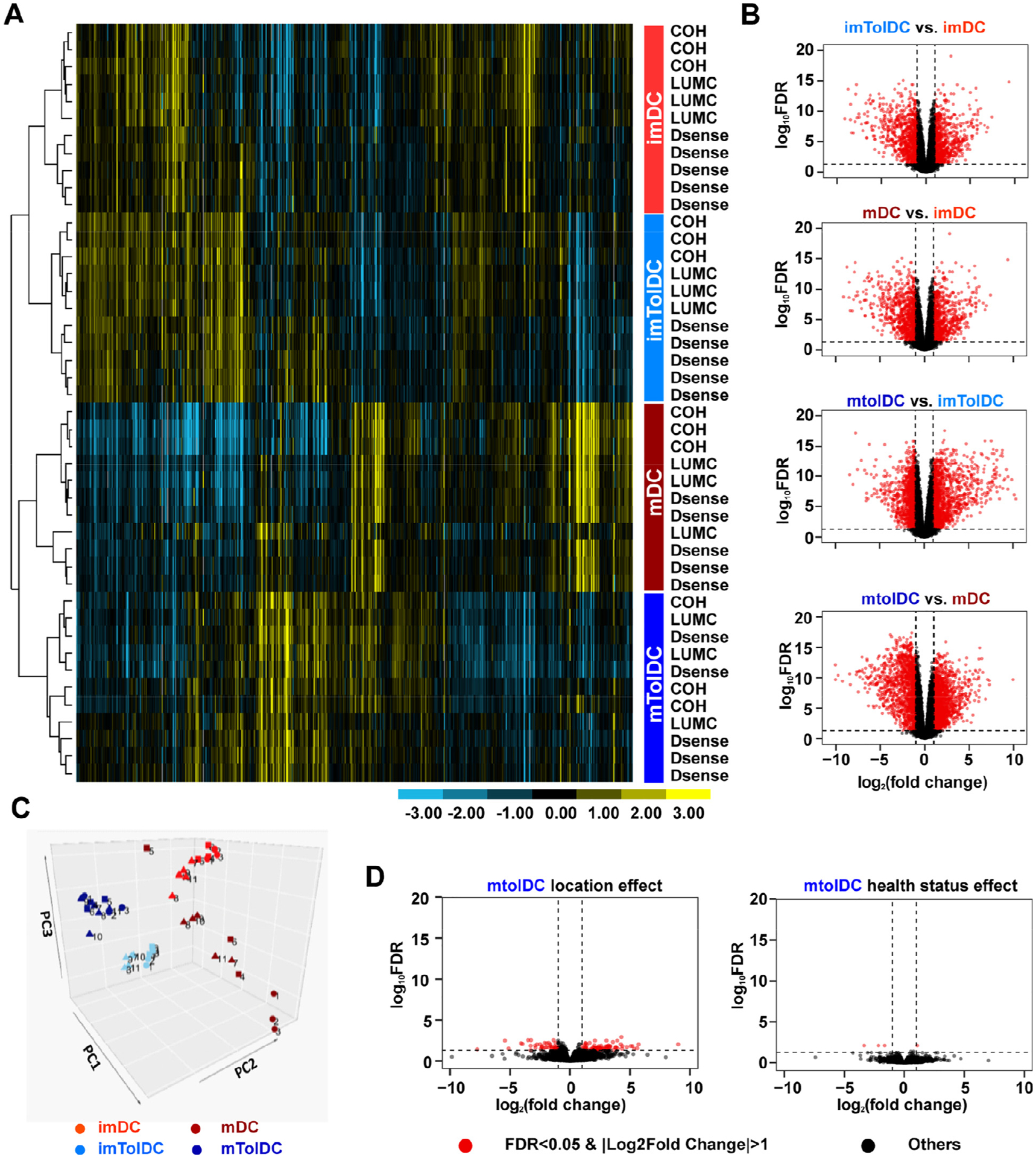
Transcriptomic analysis of tolDCs reveals that they are unaffected by location or T1D status and are more similar to their immature state than inflammatory DCs. RNA was isolated at the immature and mature stages of DC production, and RNA-seq was performed on Illumina HiSeq 2500. Samples clustered based on cell types, which are shown in the colored box to the right of the heatmap. Inflammatory imDCs are shown in light red, mDCs in dark red, imtolDCs in light blue and mtolDCs in dark blue. (A) Unsupervised hierarchical clustering of approximately 10% of 10,854 expressed genes (selected from the 21,121 genes using criterion RPKM > 1 in at least four samples). Each row represents one sample, which is labeled by location to the right of the heatmap, with the color of the label designating the cell type. Data show that samples clustered on cell type rather than location or health status. (B) Volcano plots depict DEGs in different cell type comparisons. For all volcano plots, red dots represent significant genes with an FC > 2 and FDR < 0.05, and black dots represent all other expressed genes. For a zoomed in view of volcano plots and identities of DEGs, see supplementary Figures 3–8. (C) PCA plot of all samples. Squares indicate samples from COH, circles from LUMC and triangles from D-Sense. (D) Volcano plots comparing location and health status in mtolDCs. FDR, false discovery rate; imDC, immature DC; imtolDC, immature tolDC; mtolDC, mature tolDC; PCA, principal component analysis.
In line with this, mature tolDCs showed increased expression of markers associated with an immature phenotype compared with mDCs (CD52, C-C chemokine receptor type 1 [CCR1], CCR5, low affinity immunoglobulin gamma Fc region receptor III-A and mannose receptor) (see supplementary Table 1). In addition, mature tolDCs clustered relatively closer to their immature state than mDCs did in a principal component analysis (Figure 3C). Furthermore, they showed more homogeneity within the immature and mature states than did inflammatory DCs. Besides, clustering was primarily based on cell type, rather than health status or manufacturing center (Figure 3C; also see supplementary Figures 7, 8). Indeed, only a few genes in mature tolDCs were differentially expressed in different centers and between healthy subjects and T1D patients (Figure 3D) compared with the large number of DEGs in the cell type comparisons (Figure 3B).
In summary, the authors’ transcriptomics data corroborated previous findings that mature tolDCs display a reduced capacity to stimulate T cells while having increased metabolic pathways. In addition, the authors’ current study demonstrated that mature tolDCs were more similar to their immature state than their inflammatory counterparts. Overall, tolDCs portrayed some degree of stability in gene expression associated with immature DCs and were largely unaffected by manufacturing location or T1D status.
The differential DNA methylation profile of tolDCs compared with inflammatory DCs is reached at the immature state of the cells and remains unaffected by production site or T1D status
Stability may be explained by epigenetic modifications like DNA methylation [40]. The authors therefore performed DNA methylation profiling with Illumina human MethylationEPIC arrays on DNA samples isolated from the same samples as those used for RNA-seq. With this, the authors identified differentially-methylated CpGs (DMCs) in response to VD3 modulation as described earlier. Data analyses showed that donor health status as well as production site had minimal effect on DNA methylation in mature tolDCs (26 and 13 DMCs, respectively) (see supplementary Table 2). However, a large number of DMCs were noted between immature tolerogenic and inflammatory DCs (2463 DMCs) (Figure 4A,B). After maturation, the number of DMCs between mature tolerogenic and inflammatory DCs increased further to 3457, the majority of which were retained from the immature state (2217 DMCs) (see supplementary Figure 9). This suggests that most of the DNA methylation modifications seen after VD3 treatment were already present at the immature state. Indeed, in the heatmap of DMCs (Figure 4C), the two most distinct clusters are tolerogenic versus inflammatory DCs, rather than immature mDCs versus mDCs. Furthermore, mature tolDCs showed fewer DMCs between their immature states compared with inflammatory DCs (384 and 742 DMCs, respectively) (Figure 4A). Specifically, 475 CpGs were demethylated upon inflammatory DC maturation but remained unchanged upon tolDC maturation (Figure 4C). These were enriched for genes associated with lymphocyte differentiation and leukocyte cell-to-cell adhesion (Q < 0.05) (see supplementary Table 3). Examples of genes in these pathways were CD86, CD25, IL23R, TNF super family member 4, IL7R, CCR6 and nuclear factor κ light chain enhancer of B cells (NFκB) subunit RELB.
Figure 4.
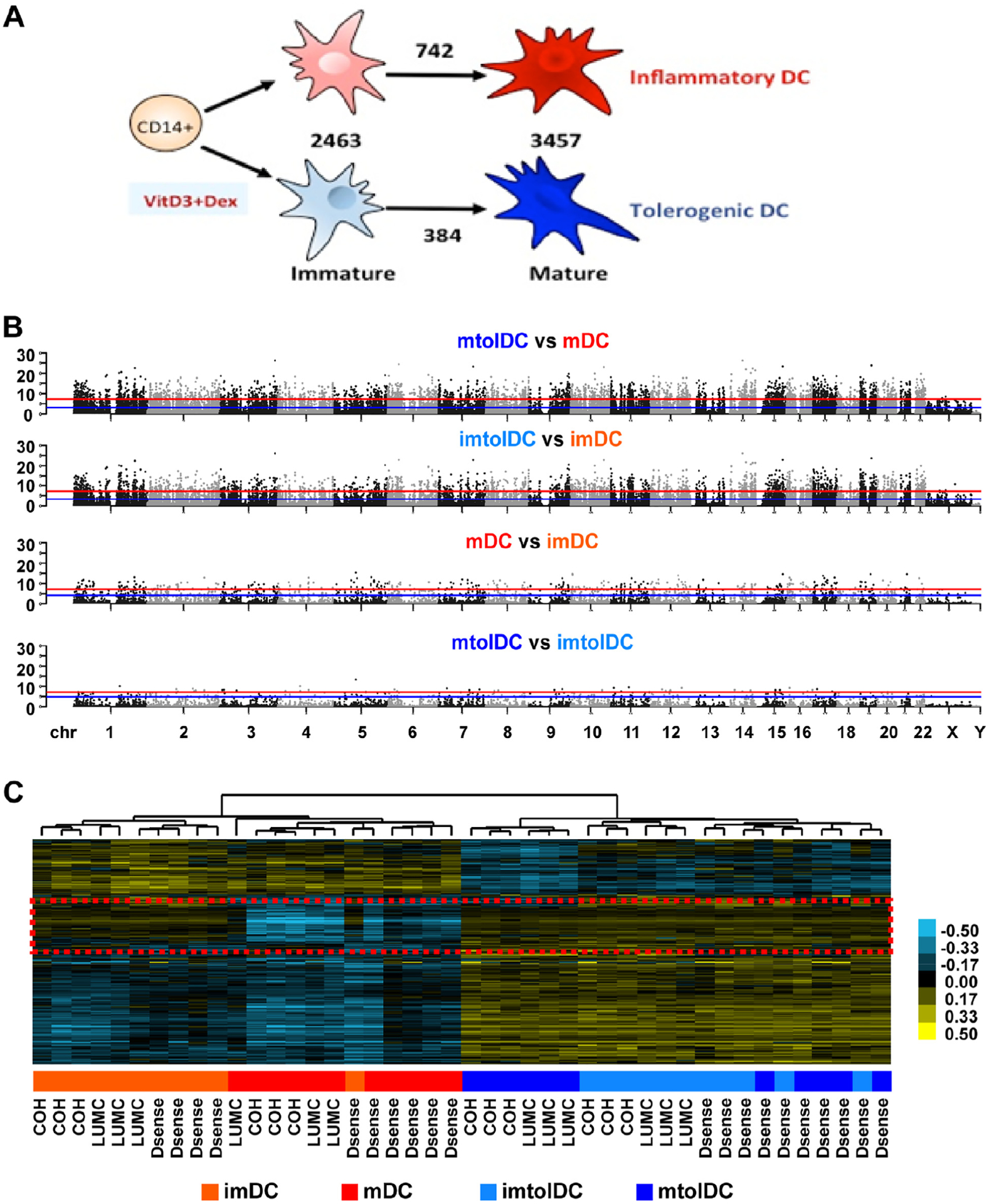
The differential DNA methylation profile of tolerogenic compared with inflammatory DCs is present at the immature state and is unaffected by location or T1D status. Genomic DNA was isolated at the immature and mature stages of DC production and subjected to DNA methylation profiling with Infinium MethylationEPIC arrays. Inflammatory imDCs are shown in light red, mDCs in dark red, imtolDCs in light blue and mtolDCs in dark blue. (A) Schematic representation of mDC versus tolDC culture and numbers of DMCs between different cell type comparisons. (B) Manhattan plots depicting the DNA methylation difference between different cell types, as indicated above the plot, across the human hg19 genome. Each dot represents one CpG, whose genomic location is represented by the x-axis and significance level in logarithm format by the y-axis. The red line represents Bonferroni-adjusted P 0.05, and the blue line represents FDR at 5%. Dots located above the lines are considered DMCs at corresponding confidence level. (C) Heatmap depicting all DMCs identified in at least one comparison shown in B after unsupervised hierarchical clustering analyses. Each row represents one DMC, and each column represents one sample. Blue indicates DNA methylation below the average of all samples, whereas yellow indicates DNA methylation above the average, with the intensity level shown in the color bar. Each sample’s group information is presented using a colored box below the heatmap, with color definitions indicated in the legend at the bottom of the panel and clinic locations where each sample was obtained indicated at the bottom of the heatmap. The red dashed box indicates a region of interest further analyzed in supplementary Table 3. Dex, dexamethasone; FDR, false discovery rate; imDC, immature DC; imtolDC, immature tolDC; mtolDC, mature tolDC.
DNA demethylation can be caused by ten-eleven translocation (TET) enzymes and changes in de novo DNA methylation by DNA methyltransferases [41]. Although the expression of TET1 was undetected by RNA-seq, TET2 was upregulated (FC = 1.50, Q = 0.0050) and TET3 was downregulated (FC = −1.73, Q < 0.0001), whereas DNA methyltransferase 3 α was upregulated in mtolDCs compared with mDCs (FC = 1.73, Q < 0.0001). This is in line with the authors’ finding that tolDCs showed higher DNA methylation levels, as seen in the heatmap, compared with inflammatory DCs. Indeed, 150 hypermethylated and only 37 hypomethylated regions were found in mature tolerogenic compared with inflammatory DCs.
Examining the genomic location of DMCs relative to RefSeq genes, the authors found they are mainly located in introns and intergenic regions, with around 25% of DMCs in coding exons and up to 1500 bp upstream of a TSS (Figure 5A). IPA of genes containing differentially-methylated regions (DMRs) (including 150 hypermethylated regions and 37 hypomethylated regions) in promoter or gene bodies revealed cellular movement, cell death and survival, cell morphology, amino acid metabolism and energy production as the most enriched pathways (P < 0.05) (Figure 5B). In line with this, the top enriched biological processes identified on genes with DMRs were positive regulation of actin filament polymerization and regulation of cell shape (false discovery rate <1%) (see supplementary Table 4). Furthermore, these DMRs were enriched at binding motifs of transcription factors associated with inflammatory genes (JUND, JUN, JUNB, FOS-Like1(FOSL1), FOS, Early growth response gene 3 (EGR3), Kruppel Like Factor 9 (KLF9) which were all upregulated in mature tolDCs compared with inflammatory DCs (Figure 5C).
Figure 5.
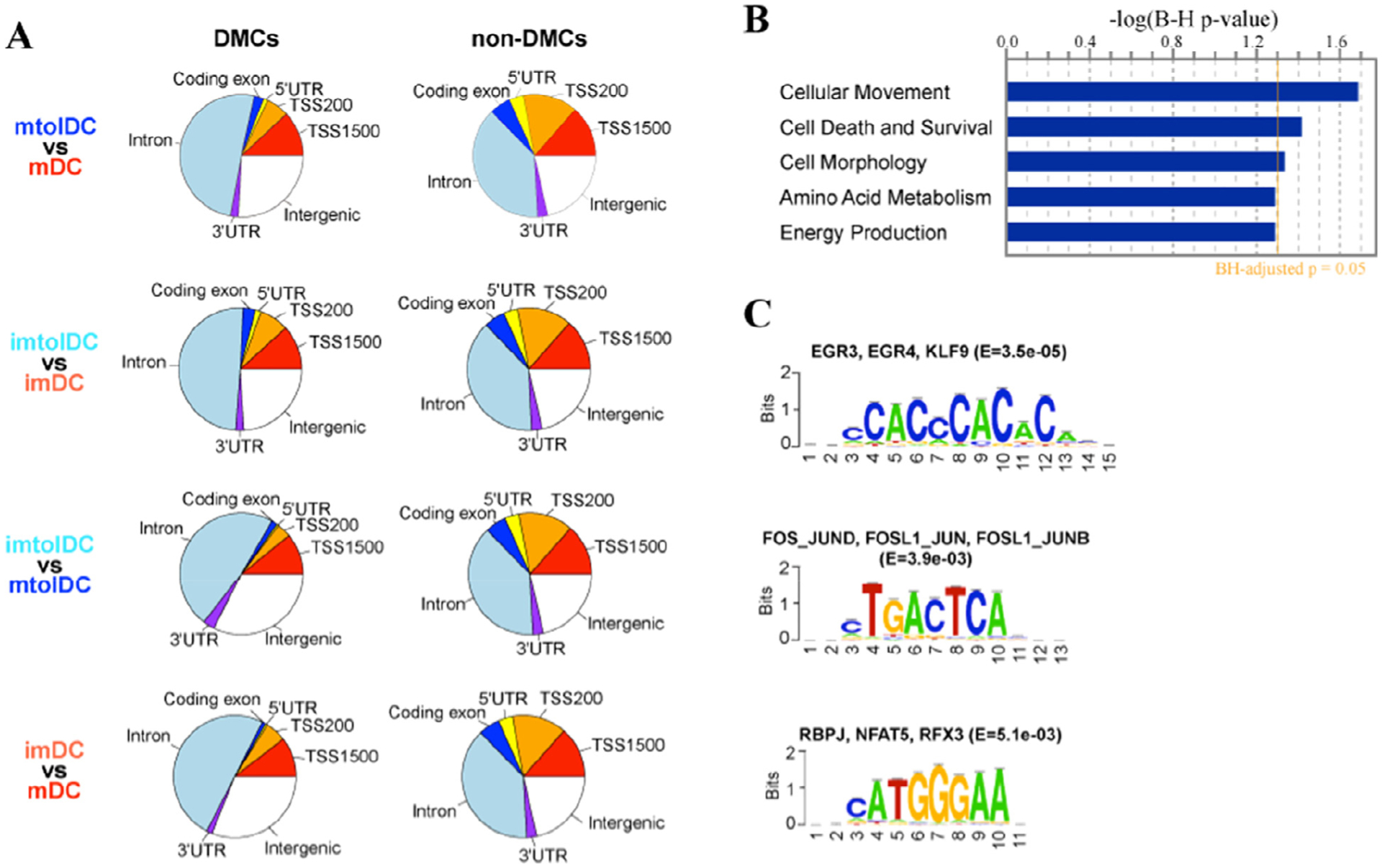
DMC and region analyses between tolerogenic and inflammatory DCs. (A) Pie charts summarizing the genomic location of DMCs relative to RefSeq genes and other non-DMCs covered by MethylationEPIC array. CpGs were annotated to one of the following regions related to RefSeq genes: coding exon, 5′UTR, TSS200 (200 bp upstream of TSS), TSS1500 (1500 bp upstream to 200 bp upstream of TSS), 3′UTR and intron. CpGs not located in any of these regions are considered intergenic. (B) Bar plot of the top enriched biological processes identified on DMRs between mtolDCs and mDCs using IPA. The y-axis represents B-H-adjusted P values in log-transformed format. (C) De novo motif analysis followed by JASPAR vertebrate motif database query using DNA sequences at DMRs. Transcription factors whose binding motifs matched the motifs identified by de novo motif analysis are shown on top of the motif. B-H, Benjamini–Hochberg; imDC, immature DC; imtolDC, immature tolDC; mtolDC, mature tolDC; UTR, untranslated region.
In summary, mature tolDCs have a differential DNA methylation profile compared with mature inflammatory DCs, and this is mostly already present at the immature state. TolDCs retain a hypermethylated state after maturation, whereas inflammatory DCs become demethylated upon maturation. Primarily, genes involved in eliciting an inflammatory immune response are associated with these regions of DNA demethylation in inflammatory DCs. In general, differences in methylation level between mature tolDCs and inflammatory DCS were enriched in pathways involved in cell morphology and movement. Finally, health status and production site had a negligible effect on tolDC DNA methylation.
DEGs associated with DMLs between mature tolerogenic and inflammatory DCs are enriched in immune response and cellular movement pathways
DEGs were aligned with DNA methylation data from both mature tolerogenic and inflammatory DCs. Approximately 80% of DEGs had at least one DMC within 500 kb downstream of their gene body or upstream of their promoter (Figure 6A). Around 20% of DMCs were located directly in the promotor or gene body of the DEG. IPA on genes whose promoters contain DMCs showed that the top canonical pathways were granulocyte adhesion and diapedesis and vitamin D receptor/retinoid X receptor activation, whereas the top upstream regulators were progesterone receptor, jagged canonical notch ligand 2 and NFκB (P < 0.0001). The top upstream regulators, TNF, IL-13 and IFN-γ, were shared between the DEGs containing DMCs in promotors and gene bodies, whereas CD40L was specifically enriched in DMCs of the latter (P < 0.0001). In terms of molecular and cellular functions, both IPAs of DMCs in gene promotors and bodies revealed cellular movement, cell death and survival, cell-to-cell signaling and interaction and cellular development as enriched pathways (P < 0.01).
Figure 6.
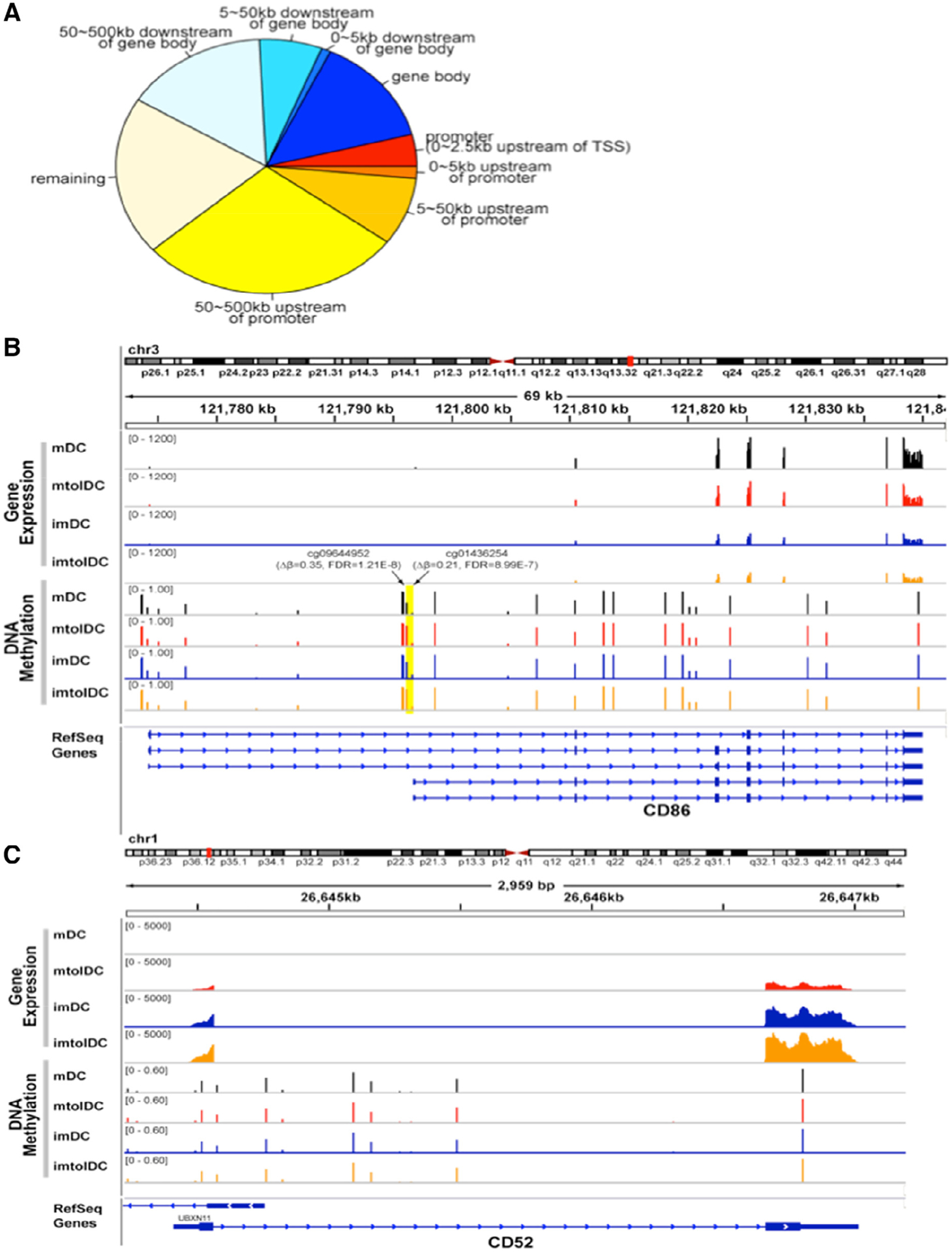
The majority of DEGs are associated with DMRs between mature tolerogenic and inflammatory DCs. (A) Pie chart representing all DEGs, categorized depending on the genomic location of DMCs associated with that DEG. The promotor region was identified as being 0–2.5 kb upstream of the TSS. When there was no associated DMC with that DEG, it was categorized as “remaining.” (B) Methylation of the CD86 gene. Average gene expression of CD86 was obtained using the scaled coverage of the 11 samples in each group. Average DNA methylation level at each CpG on each group was calculated based on normalized DNA methylation level. Two DMLs (cg09644952 and cg01436254) were noted in the proximal promotor of CD86 (yellow highlighted area), and expression was lower in tolDCs compared with inflammatory DCs (log2FC = −0.49, Q = 0.009). (C) Similar methods were used for gene expression and DNA methylation of CD52. Although a significant difference in gene expression of CD52 was noted between tolerogenic and inflammatory DCs (log2FC = 4.48, Q < 0.0001), no DMLs near the gene body or in the promotor were observed. imDC, immature DC; imtolDC, immature tolDC; mtolDC, mature tolDC.
The authors also identified 121 DEGs containing multiple DMCs in their promoters (approximately 0–2.5 kb upstream of TSS). Among them were multiple DEGs involved in free fatty acid metabolism. Acyl-CoA thioesterase 7, for instance, had five DMCs in its promotor region, whereas acyl-CoA synthetase long chain family member 1 had three and solute carrier family 27 member 3 had one, and their expression was upregulated in tolDCs compared with mDCs. The chemokines, chemokine ligand 24 (CCL24) and CCL13, had two DMCs in their promotor as well and were upregulated and downregulated, respectively. Another immunological mediator, interleukin 1 receptor antagonist (IL1RN), which is a decoy protein for the IL-1 receptor, was strongly upregulated in mature tolDCs (log2FC = 2.51, Q = 0.0003) and had four DMCs in its promotor region compared with mature inflammatory DCs. In terms of the release criteria for the D-Sense clinical trial, CD86 had significantly decreased expression (log2FC = −0.49, Q = 0.009), whereas CD52 was highly upregulated (log2FC = 4.48, Q < 0.0001) in tolDCs versus mDCs. Interestingly, two highly significantly hypermethylated loci (cg01436254 and cg09644952) were identified in the proximal promoters of two shorter isoforms of CD86 (NM_176892 and NM_006889) in mature tolDCs versus inflammatory DCs, whereas CD52 had one DMC in the 500-kb flanking region of its TSS (Figure 6B,C). Moreover, the DNA methylation levels at cg01436254 in tolDCs were very similar to the levels seen in immature tolDCs and immature mDCs. In summary, the majority of genes differentially expressed between tolerogenic and inflammatory DCs were associated with DMCs, among which many are important for tolDC function.
The majority of T1D risk genes differentially expressed between tolerogenic and inflammatory DCs are associated with DMLs
The authors previously reported that VD3 alters the expression of approximately 30% of T1D risk genes in DCs [17]. Here the authors found that 62 out of 198 (31%) expressed genes located in T1D-associated regions (T1D risk genes) were differentially expressed (including 35 upregulated and 27 downregulated genes) in tolerogenic versus inflammatory DCs (Figure 7). This was corroborated by the observation that the T1D Kyoto Encyclopedia of Genes and Genomes gene set that consists of 43 genes associated with T1D was significantly downregulated when comparing immature tolerogenic with inflammatory DCs as well as mature tolerogenic with inflammatory DCs (see supplementary Figure 2A,B). Out of the 62 identified T1D risk DEGs, 52 genes (84%) had DMLs within the 500-kb flanking region of the TSS. Of these, 12 genes (19%) contained DMCs at their gene bodies and/or in the 5-kb flanking region. These included eight downregulated genes (Cytotoxic T-Lymphocyte Associated Protein 4 (CTLA4), C-Type Lectin Domaine Family 2 Member D (CLEC2D), CLEC16A, IL2RA, Cyclin Dependent Kinase 2 (CDK2), RAB5B, Class II Major Histocompatibility Transactivator (CIITA), IKAROS Family Zinc Finger 1 (IKZF1)) and four upregulated genes (CCR1, CLN3, Apolipoprotein B Receptor (APOBR), T cell Activation RhoGTPase Activating protein (TAGAP)) in mature tolDCs versus mDCs (Figure 7). The most significant DMCs for each of these genes were hypermethylated in mtolDCs versus mDCs (Figure 7). In summary, VD3 alters the expression of T1D risk genes in DCs, and the altered expression of at least a subset of these genes might be regulated by DNA methylation, revealing a role of epigenetic modifications in this process.
Figure 7.
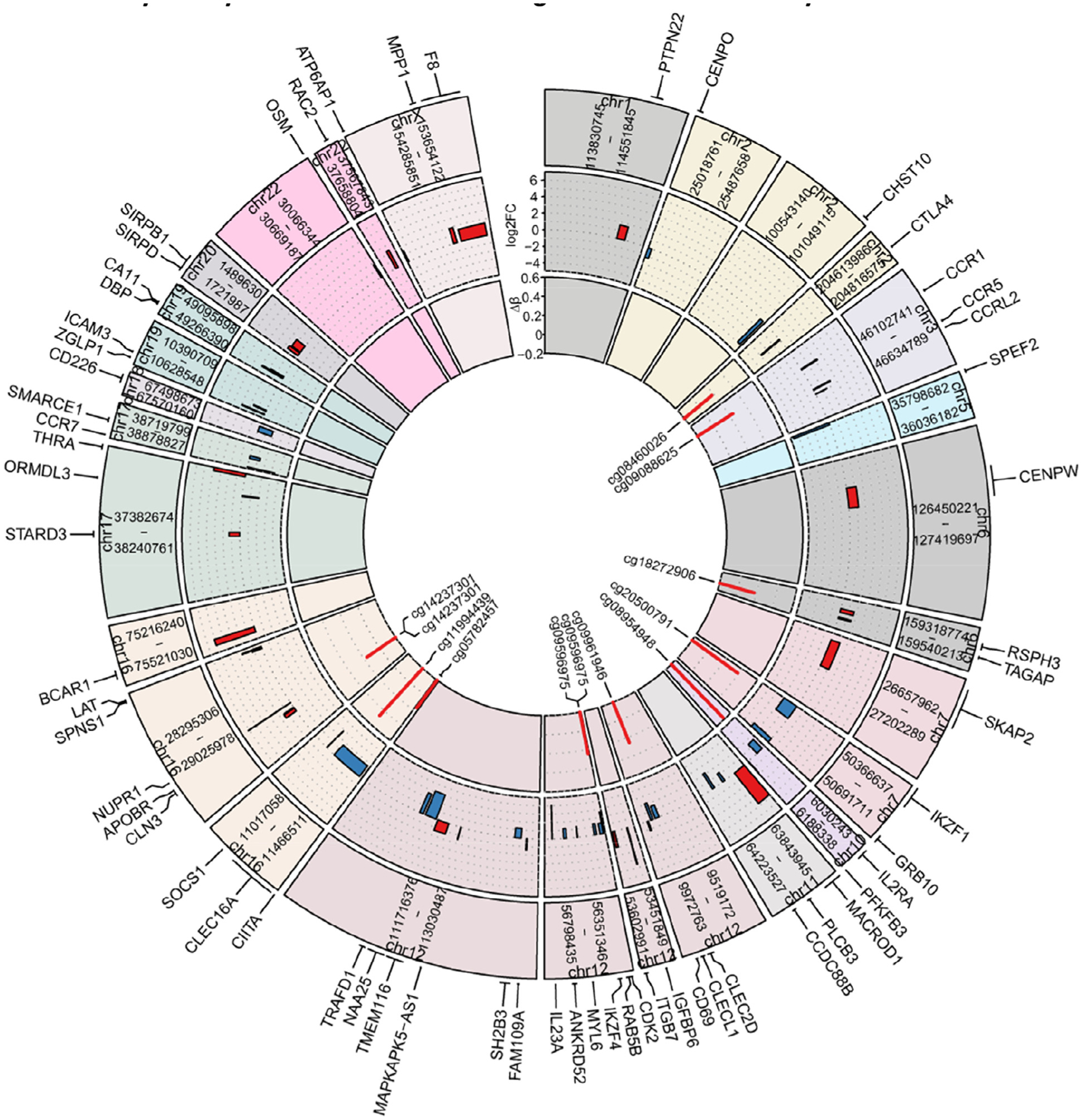
Multiple T1D risk genes were differentially expressed and associated with DMLs between tolerogenic and inflammatory DCs. Outer layer of graph shows the genomic locations of 62 differentially expressed T1D risk genes between mtolDCs (n = 11) and inflammatory mDCs (n = 11) by name in a circos plot. Middle layer of graph shows the gene expression difference in log2FC between tolDCs and mDCs visualized by box plots, where the x-axis represents the genomic locations of the genes (from TSS to end site) in the corresponding T1D-suspectible regions and the y-axis represents log2FC. Upregulated expression in tolDCs versus mDCs is depicted in red and downregulated expression in blue. Inner layer of graph shows that among the 62 genes, 12 contained DMCs at their gene bodies and/or 5-kb flanking regions (5 kb upstream of TSS [promoter] and 5 kb downstream of gene bodies). The methylation level difference (Δβ, mtolDCs versus mDCs) of the most significant DMCs (IDs shown inside the plot) for each of these 12 genes is plotted in the inner layer of the graph, with red representing hypermethylation and blue representing hypomethylation. IDs, identifiers; mtolDCs, mature tolDCs.
Discussion
Ensuring reproducible and stable cellular products is crucial for the implementation of a cellular therapy, as the protocol should deliver stable and similar cell products regardless of processing site or health status. Other cellular therapies, such as mesenchymal stromal cells, have been under scrutiny lately, as reproducibility between centers and even donors within the same center has been poor [42]. In the authors’ study, it was found that tolDCs were reproducible between centers and between healthy subjects and T1D patients. In addition, tolDCs seemed more homogeneous in phenotype and function than inflammatory DCs. The variability among inflammatory DCs supports the authors’ experience that using these as a reference for the release of tolDCs for clinical use can be troublesome. In an effort to standardize the production of tolDCs and improve reproducibility, minimum information regarding tolerogenic antigen-presenting cells was introduced in recent years [43]. The next step would be to standardize tolDC production on the basis of clinical therapeutic efficacy, which is currently still lacking. In this respect, the authors are presently developing independent and stable release criteria for tolDCs, which might include CD86 and CD52 [27], which the current study has now shown to be epigenetically regulated in tolDCs.
The effect of T1D on the production of tolDCs was also studied at the transcriptome and methylome levels. At transcription level, only minimal differences between tolDCs from T1D patients and healthy controls were noted. Overall, only four DEGs were found in the mature tolDCs of T1D donors compared with healthy controls, and none were related to immunological function. Recently, a report warned about the negative impact of hyperglycemia in T1D patients on tolDC function [44,45]. It should be noted, however, that all D-Sense T1D patients had tight blood glucose control since hemoglobin A1c > 64 mmol/mol (8%) was an exclusion criterion of this trial [27]. Several other groups have reported that monocytes and DCs of T1D patients are different from healthy subjects, but these studies used protocols different from that used by the authors [46–49]. Although monocyte frequencies were similar between T1D patients and healthy subjects, monocytes from T1D patients had alterations in their endoplasmic reticulum and oxidative stress pathways at the RNA level [49,50]. Furthermore, the authors have reported previously that monoyctes of T1D patients have 155 hypomethylated and 247 hypermethylated regions at the DNA methylation level compared with healthy subjects [49,51,52]. The authors’ current Figure 1 data, however, match experiences from investigators studying tolDCs for rheumatoid arthritis or multiple sclerosis, reporting no effect of health status on tolDC production [53,54].
Modulation by VD3 overrides clinical phenotypes, which is especially relevant in the context of T1D, where VD3 supplementation in early childhood reduces the risk of developing T1D later in life [55,56]. The protective effect of VD3 on T1D development could be due to the binding of the vitamin D receptor to autoimmune risk genes [57]. Indeed, VD3 changed the expression of half of the risk genes associated with multiple sclerosis in mouse T cells [58]. In accordance with the authors’ previous study [17], one third of candidate T1D risk genes were differentially expressed, implying that VD3 supplementation may override genetic risk predisposition for T1D. Furthermore, the authors found in this study that up to 80% of these genes contain DMCs between tolDCs and mDCs in the promoter, gene body or nearby region. This is important, as epigenetics have been reported to influence the expression of T1D risk genes in the monocytes of T1D patients compared with healthy controls [59,60]. VD3 may reduce this disparity in T1D patients, and the present results support the notion that VD3 supplementation early in life has a longstanding protective effect, as DNA methylation is thought to be a stable marker [12,55].
The stability of tolDCs was validated in two stages. First, mature tolDCs resisted perturbation with inflammatory stimuli, in line with what has been observed for tolDCs in rheumatoid arthritis [53]. Out of all phenotypic, functional and metabolic markers tested, only CD209 decreased in both tolerogenic and inflammatory DCs after additional inflammatory stimuli. With CD209 also downregulated upon anti-inflammatory treatment (i.e., dexamethasone) [61], low CD209 could be associated with an anti-inflammatory phenotype. These results are reassuring, as concerns have been raised about the possibility of an in vivo conversion of tolDCs to a pro-inflammatory phenotype [62].
Second, epigenetic studies revealed that several thousand DMLs found between tolerogenic and inflammatory DCs were mostly already present at the immature state. IL1RN is one of the few genes consistently upregulated in tolDCs across several previous studies as well as the authors’ present study [63,64]. In addition, the authors showed that IL1RN contained several DMCs in its promotor region, which may explain the consistent expression of IL1RN across studies. IPA of genes associated with both differential expression and methylation revealed enrichment of NFκB, TNF, IFN-γ, CD40L and IL-13, suggesting a stable, epigenetically controlled regulation of these important inflammatory pathways. TNF signaling proved to be crucial in inducing regulatory T cells from naive CD4 T cells by tolDCs, and CCL24 attracts naive CD4 T cells [21]. CCL24 was upregulated, whereas CCL13, which is associated with chronic inflammatory diseases, was downregulated in tolDCs versus inflammatory DCs [65,66]. Both chemokines were differentially methylated in their promotor regions, pointing to the induction of a stable anti-inflammatory environment by tolDCs.
The authors’ data also support the observation that maturation of immune-activating DCs results in widespread DNA demethylation [67]. Instead, matured tolDCs mostly retained the DNA methylation status of their immature phase, which is in accordance with the hypothesis that tolDCs are “locked” in an immature state. The transcription factors KLF9 and JUNB were similarly upregulated in immature DCs and mature tolDCs, as opposed to mature inflammatory DCs [63]. Furthermore, the enrichment of KLF9 and JUNB binding sites at DMRs between tolDCs and inflammatory DCs suggests potential important roles of these transcription factors in gene expression regulation of tolDCs via DNA methylation.
The region that was differentially demethylated in inflammatory DCs compared with tolDCs was associated with lymphocyte differentiation and activation. This finding corroborates the higher T-cell stimulatory capacity of inflammatory DCs compared with tolDCs that is consistently found at the RNA, protein and functional level. Furthermore, the DNA methylation levels at these same CpGs are comparable in immature inflammatory DCs and tolDCs (both before and after maturation), pointing toward a common functional asset of reduced T-cell activation by these cell types. Other characteristics are diverging, however. The authors show, namely, that tolDCs do not merely retain their immature state but have an independent resetting of their phenotype, as has been recently proposed [63]. For instance, morphology is an easily detectable and distinguishable feature separating tolDCs from inflammatory DCs. Within 3 days of culture, tolDCs can be separated from inflammatory DCs by visual means on the basis of their spindle shape and plate adherence, which persists throughout the 8 days of culture [20]. Indeed, DNA methylation could play an important role in cell morphology, as the most significant enriched biological function in genes containing DMCs in mature tolDCs versus inflammatory DCs proved related to cell morphology. By contrast, differences in immunological pathways dominate at the transcriptome level [17]. Interestingly, these pathways were enriched in genes containing DMCs upon maturation of inflammatory DCs, suggesting the involvement of DNA methylation in these pathways as well. This points to the divergence of tolDCs from inflammatory DCs in terms of cell shape and cellular movement, whereas they retain the hypoimmunogenic features of immature DCs. Some caution is warranted when interpreting the role of DNA methylation in gene expression, however, as gene expression can precede DNA methylation in response to activation of monocyte-derived DCs [68].
Conclusions
VD3 plus dexamethasone induced epigenetic modifications at key loci in tolDCs, which may contribute to the observed stability of tolDCs in phenotype and function. Furthermore, reproducibility was shown with regard to phenotypic, transcriptomic and methylomic tolDC profiles regardless of manufacturing center or health status. Together, this reinforces the feasibility of and attraction for the implementation of tolDCs as a stable, reproducible immunomodulatory therapeutic strategy in T1D and other autoimmune disorders.
Supplementary Material
Acknowledgments
The authors are grateful to Drs Xiwei Wu and Min-Hsuan Chen of the Integrative Genomics Core for assistance with data analyses.
Funding
This study was funded by the Wanek Family Project for Type 1 Diabetes (BOR and RN) and the National Institutes of Health (RN; DP3 DK106917). TN and JZ are financially supported by the Dutch Diabetes Research Foundation/Stichting DON and Dutch Arthritis Society. Research reported in this publication includes work performed in the City of Hope Integrative Genomics Core supported by the National Cancer Institute of the National Institutes of Health (P30CA33572).
Footnotes
Declaration of Competing Interest
The authors have no commercial, proprietary or financial interest in the products or companies described in this article.
Supplementary materials
Supplementary material associated with this article can be found in the online version at doi:10.1016/j.jcyt.2020.12.003.
References
- [1].Steinman RM. Decisions about dendritic cells: past, present, and future. Annual review of immunology 2012;30:1–22. [DOI] [PubMed] [Google Scholar]
- [2].Bell G, et al. Autologous tolerogenic dendritic cells for rheumatoid and inflammatory arthritis. Annals of the rheumatic diseases 2017;76:227–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Benham H, et al. Citrullinated peptide dendritic cell immunotherapy in HLA risk genotype-positive rheumatoid arthritis patients. Science translational medicine 2015;7:290ra287. [DOI] [PubMed] [Google Scholar]
- [4].Giannoukakis N, Phillips B, Finegold D, Harnaha J, Trucco M. Phase I (safety) study of autologous tolerogenic dendritic cells in type 1 diabetic patients. Diabetes care 2011;34:2026–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Jauregui-Amezaga A, et al. Intraperitoneal administration of autologous tolerogenic dendritic cells for refractory Crohn’s disease: a phase I study. Journal of Crohn’s and Colitis 2015;9:1071–8. [DOI] [PubMed] [Google Scholar]
- [6].Roep BO, Wheeler DCS, Peakman M. Antigen-based immune modulation therapy for type 1 diabetes: the era of precision medicine. Lancet Diabetes Endocrinol 2019;7:65–74. [DOI] [PubMed] [Google Scholar]
- [7].Willekens B, et al. Tolerogenic dendritic cell-based treatment for multiple sclerosis (MS): a harmonised study protocol for two phase I clinical trials comparing intradermal and intranodal cell administration. BMJ open 2019;9:e030309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Mansilla MJ, et al. Cryopreserved vitamin D3-tolerogenic dendritic cells pulsed with autoantigens as a potential therapy for multiple sclerosis patients. Journal of neuroinflammation 2016;13:113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Atkinson MA, Eisenbarth GS, Michels AW. Type 1 diabetes. The Lancet 2014;383:69–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Unger WW, Laban S, Kleijwegt FS, van der Slik AR, Roep BO. Induction of Treg by monocyte-derived DC modulated by vitamin D3 or dexamethasone: differential role for PD-L1. European journal of immunology 2009;39:3147–59. [DOI] [PubMed] [Google Scholar]
- [11].Suwandi J, Toes R, Nikolic T, Roep B. Inducing tissue specific tolerance in autoimmune disease with tolerogenic dendritic cells. Clin Exp Rheumatol 2015;33: 97–103. [PubMed] [Google Scholar]
- [12].Mohn F, Schübeler D. Genetics and epigenetics: stability and plasticity during cellular differentiation. Trends in Genetics 2009;25:129–36. [DOI] [PubMed] [Google Scholar]
- [13].Jerram ST, Dang MN, Leslie RD. The role of epigenetics in type 1 diabetes. Current diabetes reports 2017;17:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kato M, Natarajan R. Epigenetics and epigenomics in diabetic kidney disease and metabolic memory. Nat Rev Nephrol 2019;15:327–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Chen ZX, Riggs AD. DNA methylation and demethylation in mammals. J Biol Chem 2011;286:18347–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Vanherwegen A-S, Gysemans C, Mathieu C. Vitamin D endocrinology on the cross-road between immunity and metabolism. Molecular and cellular endocrinology 2017;453:52–67. [DOI] [PubMed] [Google Scholar]
- [17].Nikolic T, et al. Differential transcriptome of tolerogenic versus inflammatory dendritic cells points to modulated T1D genetic risk and enriched immune regulation. Genes and immunity 2017;18:176. [DOI] [PubMed] [Google Scholar]
- [18].Robertson CC, Rich SS. Genetics of type 1 diabetes. Curr Opin Genet Dev 2018;50:7–16. [DOI] [PubMed] [Google Scholar]
- [19].Ferreira GB, et al. Vitamin D3 induces tolerance in human dendritic cells by activation of intracellular metabolic pathways. Cell reports 2015;10:711–25. [DOI] [PubMed] [Google Scholar]
- [20].van Halteren AG, et al. Redirection of human autoreactive T-cells upon interaction with dendritic cells modulated by TX527, an analog of 1,25 dihydroxyvitamin D3. Diabetes 2002;51:2119–25. [DOI] [PubMed] [Google Scholar]
- [21].Kleijwegt FS, et al. Critical role for TNF in the induction of human antigen-specific regulatory T cells by tolerogenic dendritic cells. The Journal of Immunology 2010;185:1412–8. [DOI] [PubMed] [Google Scholar]
- [22].Mahnke K, Qian Y, Knop J, Enk AH. Induction of CD4+/CD25+ regulatory T cells by targeting of antigens to immature dendritic cells. Blood 2003;101:4862–9. [DOI] [PubMed] [Google Scholar]
- [23].Schinnerling K, Garcia-Gonzalez P, Aguillon JC. Gene Expression Profiling of Human Monocyte-derived Dendritic Cells—Searching for Molecular Regulators of Tolerogenicity. Front Immunol 2015;6:528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Jonuleit H, Schmitt E, Schuler G, Knop J, Enk AH. Induction of interleukin 10–producing, nonproliferating CD4+ T cells with regulatory properties by repetitive stimulation with allogeneic immature human dendritic cells. Journal of Experimental Medicine 2000;192:1213–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Roncarolo M-G, Levings MK, Traversari C. Differentiation of T regulatory cells by immature dendritic cells. Journal of Experimental Medicine 2001;193:F5–F10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Mahnke K, Qian Y, Knop J, Enk AH. Induction of CD4+/CD25+regulatory T cells by targeting of antigens to immature dendritic cells. Blood 2003;101:4862–9. [DOI] [PubMed] [Google Scholar]
- [27].Nikolic T, et al. Safety and feasibility of intradermal injection with tolerogenic dendritic cells pulsed with proinsulin peptide—for type 1 diabetes. Lancet Diabetes Endocrinol 2020;8(6):470–2. [DOI] [PubMed] [Google Scholar]
- [28].Ferreira GB, et al. Differential protein pathways in 1,25-dihydroxyvitamin d(3) and dexamethasone modulated tolerogenic human dendritic cells. Journal of proteome research 2012;11:941–71. [DOI] [PubMed] [Google Scholar]
- [29].Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 2014;30:2114–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Chen S, Zhou Y, Chen Y, Gu J. Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018;34:i884–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Dobin A, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2012;29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Anders S, Huber W. Differential expression analysis for sequence count data. Genome biology 2010;11:R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Robinson MD, McCarthy DJ, Smyth GK. EdgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010;26:139–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature Protocols 2009;4:44–57. [DOI] [PubMed] [Google Scholar]
- [35].Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 2009;37:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Mootha VK, et al. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nature genetics 2003;34:267. [DOI] [PubMed] [Google Scholar]
- [37].Subramanian A, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences 2005;102:15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Geijtenbeek TB, et al. Identification of DC-SIGN, a novel dendritic cell–specific ICAM-3 receptor that supports primary immune responses. Cell 2000;100:575–85. [DOI] [PubMed] [Google Scholar]
- [39].Geijtenbeek TB, et al. DC-SIGN–ICAM-2 interaction mediates dendritic cell trafficking. Nature immunology 2000;1:353–7. [DOI] [PubMed] [Google Scholar]
- [40].Mohn F, Schubeler D. Genetics and epigenetics: stability and plasticity during cellular differentiation. Trends Genet 2009;25:129–36. [DOI] [PubMed] [Google Scholar]
- [41].Lyko F The DNA methyltransferase family: a versatile toolkit for epigenetic regulation. Nature Reviews Genetics 2017;19:81. [DOI] [PubMed] [Google Scholar]
- [42].Wilson A, Hodgson-Garms M, Frith JE, Genever P. Multiplicity of Mesenchymal Stromal Cells: Finding the Right Route to Therapy. Frontiers in Immunology 2019;10(16):1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Lord P, et al. Minimum information about tolerogenic antigen-presenting cells (MITAP): a first step towards reproducibility and standardisation of cellular therapies. PeerJ 2016;4:e2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Palová Jelínková L, Grohová A, Dáňová K, Spisek R. Cell based therapy for type 1 diabetes: should we take hyperglycemia into account? Frontiers in immunology 2019;10:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Dáňová K, et al. Tolerogenic Dendritic Cells from Poorly Compensated Type 1 Diabetes Patients Have Decreased Ability To Induce Stable Antigen-Specific T Cell Hyporesponsiveness and Generation of Suppressive Regulatory T Cells. The Journal of Immunology 2017;198:729–40. [DOI] [PubMed] [Google Scholar]
- [46].Mollah ZU, et al. Abnormal NF-κB function characterizes human type 1 diabetes dendritic cells and monocytes. The Journal of Immunology 2008;180:3166–75. [DOI] [PubMed] [Google Scholar]
- [47].Jansen A, van Hagen M, Drexhage HA. Defective maturation and function of antigen-presenting cells in type 1 diabetes. The Lancet 1995;345:491–2. [DOI] [PubMed] [Google Scholar]
- [48].Allen JS, et al. Plasmacytoid dendritic cells are proportionally expanded at diagnosis of type 1 diabetes and enhance islet autoantigen presentation to T-cells through immune complex capture. Diabetes 2009;58:138–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Irvine KM, et al. Peripheral blood monocyte gene expression profile clinically stratifies patients with recent-onset type 1 diabetes. Diabetes 2012;61:1281–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Oras A, Peet A, Giese T, Tillmann V, Uibo R. A study of 51 subtypes of peripheral blood immune cells in newly diagnosed young type 1 diabetes patients. Clin Exp Immunol 2019;198:57–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Oras A, Peet A, Giese T, Tillmann V, Uibo R. A study of 51 subtypes of peripheral blood immune cells in newly diagnosed young type 1 diabetes patients. Clinical & Experimental Immunology 2019;198(1):57–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Chen Z, et al. Epigenomic profiling reveals an association between persistence of DNA methylation and metabolic memory in the DCCT/EDIC type 1 diabetes cohort. Proceedings of the National Academy of Sciences 2016;113:E3002–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Harry RA, Anderson AE, Isaacs JD, Hilkens CM. Generation and characterisation of therapeutic tolerogenic dendritic cells for rheumatoid arthritis. Annals of the rheumatic diseases 2010;69:2042–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Raϊch-Regué D, et al. Stable antigen-specific T-cell hyporesponsiveness induced by tolerogenic dendritic cells from multiple sclerosis patients. European Journal of Immunology 2012;42:771–82. [DOI] [PubMed] [Google Scholar]
- [55].Hyppönen E, Läärä, Reunanen A, Järvelin M-R, Virtanen SM. Intake of vitamin D and risk of type 1 diabetes: a birth-cohort study. The Lancet 2001;358:1500–3. [DOI] [PubMed] [Google Scholar]
- [56].Pozzilli P, et al. Low levels of 25-hydroxyvitamin D3 and 1, 25-dihydroxyvitamin D3 in patients with newly diagnosed type 1 diabetes. Hormone and Metabolic Research 2005;37:680–3. [DOI] [PubMed] [Google Scholar]
- [57].Ramagopalan SV, et al. A ChIP-seq defined genome-wide map of vitamin D receptor binding: associations with disease and evolution. Genome research 2010;20:1352–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Zeitelhofer M, et al. Functional genomics analysis of vitamin D effects on CD4+ T cells in vivo in experimental autoimmune encephalomyelitis. Proceedings of the National Academy of Sciences 2017;114:E1678–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Miao F, et al. Profiles of epigenetic histone post-translational modifications at type 1 diabetes susceptible genes. Journal of Biological Chemistry 2012;287: 16335–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Elboudwarej E, et al. Hypomethylation within gene promoter regions and type 1 diabetes in discordant monozygotic twins. J Autoimmun 2016;68:23–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Relloso M, et al. DC-SIGN (CD209) expression is IL-4 dependent and is negatively regulated by IFN, TGF-β, and anti-inflammatory agents. The Journal of Immunology 2002;168:2634–43. [DOI] [PubMed] [Google Scholar]
- [62].Lim DS, Kang MS, Jeong JA, Bae YS. Semi-mature DC are immunogenic and not tolerogenic when inoculated at a high dose in collagen-induced arthritis mice. European journal of immunology 2009;39:1334–43. [DOI] [PubMed] [Google Scholar]
- [63].Schinnerling K, García-González P, Aguillón JC. Gene Expression Profiling of Human Monocyte-derived Dendritic Cells—Searching for Molecular Regulators of Tolerogenicity. Frontiers in Immunology 2015;6:528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Ferreira GB, et al. Differential protein pathways in 1,25-dihydroxyvitamin D3 and dexamethasone modulated tolerogenic human dendritic cells. Journal of proteome research 2011;11:941–71. [DOI] [PubMed] [Google Scholar]
- [65].Mendez-Enriquez E, Garcia-Zepeda E. The multiple faces of CCL13 in immunity and inflammation. Inflammopharmacology 2013;21:397–406. [DOI] [PubMed] [Google Scholar]
- [66].Koga Y, Matsuzaki A, Suminoe A, Hattori H, Hara T. Expression of cytokine-associated genes in dendritic cells (DCs): comparison between adult peripheral blood- and umbilical cord blood-derived DCs by cDNA microarray. Immunology Letters 2008;116:55–63. [DOI] [PubMed] [Google Scholar]
- [67].Zhang X, et al. DNA methylation dynamics during ex vivo differentiation and maturation of human dendritic cells. Epigenetics & chromatin 2014;7:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Pacis A, et al. Gene activation precedes DNA demethylation in response to infection in human dendritic cells. bioRxiv 2018;116(14):6938–43. 358531. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.