

Abstract
The beneficial cardiorenal outcomes of sodium-glucose cotransporter 2 inhibitors (SGLT2i) in patients with type 2 diabetes mellitus (T2DM) have been substantiated by multiple clinical trials, resulting in increased interest in the multifarious pathways by which their mechanisms act. The principal effect of SGLT2i (-flozin drugs) can be appreciated in their ability to block the SGLT2 protein within the kidneys, inhibiting glucose reabsorption, and causing an associated osmotic diuresis. This ameliorates plasma glucose elevations and the negative cardiorenal sequelae associated with the latter. These include aberrant mitochondrial metabolism and oxidative stress burden, endothelial cell dysfunction, pernicious neurohormonal activation, and the development of inimical hemodynamics. Positive outcomes within these domains have been validated with SGLT2i administration. However, by modulating the sodium-glucose cotransporter in the proximal tubule (PT), SGLT2i consequently promotes sodium-phosphate cotransporter activity with phosphate retention. Phosphatemia, even at physiologic levels, poses a risk in cardiovascular disease burden, more so in patients with type 2 diabetes mellitus (T2DM). There also exists an association between phosphatemia and renal impairment, the latter hampering cardiovascular function through an array of physiologic roles, such as fluid regulation, hormonal tone, and neuromodulation. Moreover, increased phosphate flux is associated with an associated increase in fibroblast growth factor 23 levels, also detrimental to homeostatic cardiometabolic function. A contemporary commentary concerning this notion unifying cardiovascular outcome trial data with the translational biology of phosphate is scant within the literature. Given the apparent beneficial outcomes associated with SGLT2i administration notwithstanding negative effects of phosphatemia, we discuss in this review the effects of phosphate on the cardiometabolic status in patients with T2DM and cardiorenal disease, as well as the mechanisms by which SGLT2i counteract or overcome them to achieve their net effects. Content drawn to develop this conversation begins with proceedings in the basic sciences and works towards clinical trial data.
Keywords: Sodium-glucose cotransporter 2, Phosphate, Hyperphosphatemia, Cardiovascular, Canagliflozin, Dapagliflozin, Empagliflozin, Endothelial
Core Tip: Sodium-glucose cotransporter 2 (SGLT2) inhibitors have received increased attention regarding their pleiotropic effects given their markedly impressive performance in cardiovascular outcome trials (CVOT). Preliminary evidence shows that their role as antidiabetic agents is not their sole mechanism in achieving these cardiorenal protective properties. Therefore, investigation in the auxiliary properties that they hold concomitant with glucose control, vindicated by not only CVOTs, but meta-analyses, retrospective studies, and case reports has led to increased interest in delineating their global pharmacodynamic effects across the spectrum of gene expression and molecular modulation to end-organ translational biology. Such a full profile of their effects is not yet understood given the refractory period between clinical evidence supporting their utilization and a proclivity for their implementation in practical clinical environments. In this review, we answer inquiries regarding how via a multifarious avenues, SGLT2 inhibitors, while carrying a negative effect of induced phosphatemia (which is deleterious to the heart), compensate for this phenomenon, retaining their propensity for net cardiac benefit upon pharmacotherapeutic administration under appropriate clinical circumstances.
INTRODUCTION
In addition to serving a role as antidiabetic agents by mediating glycemic control, the cardiovascular benefits associated with sodium-glucose cotransporter 2 inhibitors (SGLT2i) administration is a boon for patients with type 2 diabetes mellitus (T2DM) given that such patients suffer immensely from increased risk of microvascular and macrovascular complications attributable to T2DM, such as diabetic nephropathy and major adverse cardiovascular events (MACE), respectively[1]. It is noteworthy to state these sequelae are not mutually exclusive when one discusses the cardiovascular health of patients with T2DM and the renal considerations of phosphate dynamics with SGLT2i administration on MACE. An example highlighting these interdigitated cardiorenal pathways can be appreciated when considering that cardiovascular demise in patients with T2DM may predispose to acute kidney injury (AKI) through hypoperfusion and consequently, a diminished capacity to efficiently maintain glomerular filtration rate (GFR). Conversely, pathologic nephropathy from T2DM unveils baneful circumstance within the nephron conducive to MACE via mismanagement of fluid and ion flux as well as pathologic neurohormonal activation via the renin-angiotensin-aldosterone system (RAAS)[2-4].
Estimates suggest that T2DM is prevalent in roughly 40% of chronic kidney disease (CKD) patients in the United States[5]. Epidemiological studies further contextualize this association, with one United States Renal Data System (USRDS) report implicating diabetes in 44% of end-stage renal disease (ESRD) cases[6]. Moreover, examinations of 10-year cumulative mortality profiles of participants in the Third National Health and Nutrition Examination Survey (NHANES III) linked with the National Death Index has shown that among individuals with diabetes and kidney disease, standardized mortality was 31.1% (95%CI: 24.7%-37.5%) relative to 7.7% within the reference group, a patient populous defined as that without diabetes or kidney disease (95%CI: 7.0%-8.3%)[7]. This represents a statistically significant absolute risk difference of 23.4% (95%CI: 17.0%-29.9%, P < 0.01). Such emphasis on the baleful aspects of T2DM is important as cardiovascular disease (CVD) burden and T2DM presents with a similarly grim association, as seen in an incidence-based study by Straka et al[8], which followed 29863 patients (5501 with T2DM and 24362 without T2DM). In this study, it was observed that patients with T2DM exhibited a statistically significant relative risk of 1.53 for myocardial infarction (MI), 1.1 for coronary artery disease (CAD), 2.12 for heart failure, and 1.58 for stroke.
With an expanded understanding of the pathophysiologic pathways that stem from T2DM and branch towards its sequelae comes a paradigm shift in which T2DM is no longer focused solely as a disorder of hyperglycemia and aberrant insulin regulation warranting the reduction of hemoglobin A1C (HbA1C) for adequate clinical management[9-12]. Rather, an appraisal of diabetic complications is giving rise to a change in therapeutic approaches that target T2DM sequelae in tandem with glucose and insulin dynamics for expanded, and flexible T2DM treatment strategies. This can be appreciated by the advent of pharmacotherapeutic options with various mechanisms of action and pleiotropic effects, in addition to the Food and Drug Administration’s shift towards expectations that antidiabetic agents being considered for approval undergo scrutiny that is validated by trial data that takes into account the systemic effects of T2DM (with an emphasis on microvascular and macrovascular pathophysiology)[13]. Evidence for this change in doctrine can be seen in the elevation of SGLT2i to more preferential recommended therapeutic options for complicated T2DM by authoritative bodies and medical societies such as the American College of Cardiology (ACC), the American Diabetes Association (ADA), and Kidney Disease Improving Global Outcomes (KDIGO)[14-17].
Affirmation for the beneficial clinical outcomes with SGLT2i pharmacotherapy is validated by favorable results in landmark clinical outcome trials. These aforementioned trials scrutinized the effects of multiple SGLT2i agents (namely, empagliflozin, dapagliflozin, and canagliflozin) in their ability to reduce endpoints defined by cardiovascular mortality, hospitalizations for heart failure, and renal considerations of associated cardiovascular demise such as death from renal failure and end-stage renal disease exacerbation, to name a few[18-21]. These drugs work (as illustrated in Figure 1) by blocking the reabsorption of glucose via the SGLT2 protein, which is responsible for the reuptake of the vast proportion of glucose in the proximal renal tubule. The latter causes glucosuria and decreased serum glucose levels, which inhibits the salient pathophysiologic pathways in T2DM triggered by hyperglycemia. The degree of glucosuria varies depending on drug metabolism, SGLT2 protein expression as well as distribution, and diabetic status. However, narrative reviews backed by quantitative estimates of the nephron’s functional capacity and a general window of SGLT2i efficacy suggest that the glucosuria induced can lead to the excretion of up to 150 g of glucose daily with pharmacotherapy[22].
Figure 1.
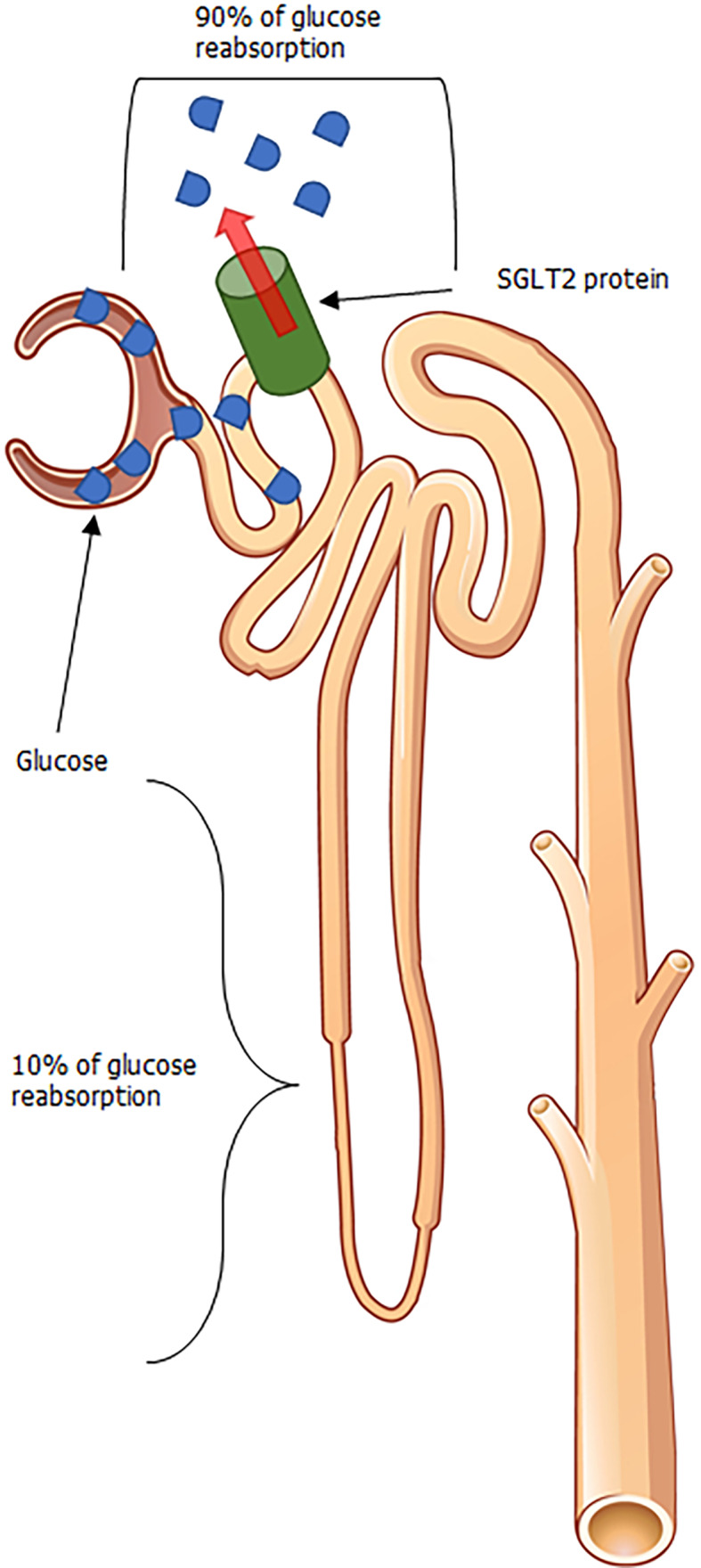
Glycosuria mediated from sodium-glucose cotransporter 2 inhibition. Adapted from OpenStax College, which is licensed under a Creative Commons Attribution 3.0 Unported License. SGLT2: Sodium-glucose cotransporter 2.
Associated with such glucosuria is also the liberation of fluid with SGLT2i therapy among patients with T2DM. In normal, healthy adults, the kidneys have the propensity to filter roughly 180 g of glucose per daily, the vast majority of which is usually reabsorbed at the proximal convoluted tubule (PCT) via sodium-glucose cotransport[23]. Due to insulin resistance exhibited in patients with T2DM, peripheral GLUT4 expression is decreased in T2DM, leading to increased serum glucose, and increased filtered glucose load through the glomerulus once perfusion reaches the nephron. The low affinity-high capacity properties of the SGLT2 protein within the PCT, allows for substantial glucose reabsorption, with reabsorption dynamics reaching up to 90% in certain physiologic scenarios[24,25]. By inhibiting where the majority of glucose reabsorption at the PCT, there remains feeble opportunities for glucose reabsorption to take place along downstream sites within the nephron, resulting in an increase in osmotic pressure to the flow en route to eventual urinary output[26]. Subsequently, an increased osmotic pressure from a rise in SGLT2i mediated increase in luminal glucose prevents the egress of glucose across tubule cells (and by proxy, the interstitial compartments) resulting in increased urinary volume[27-29] and liberation of systemic fluid.
The natriuresis involved in SGLT2i pharmacotherapy has recently been scrutinized and there are mixed views as to the degree of natriuresis involved in SGLT2i therapy, as while there is an osmolarity of sodium that would have been tethered for sodium-glucose cotransport, inhibition does not preclude this sodium from undergoing reabsorption at other sites, with some sites even performing compensatory roles in sodium reabsorption after transient natriuresis has been completed[30]. For example, in the thick ascending loop of Henle the Na-K-Cl cotransporter shunts luminal sodium that would have been excreted in urine. Then sodium is discharged into the blood via ATP-dependent sodium-potassium pump, resulting in retention of sodium. While playing a minor role, sodium reabsorption also takes place in the distal convoluted tubule through a sodium-chloride symporter that harvests urinary sodium that is also discharged via ATP-dependent sodium-potassium pumps. Nevertheless, studies with SGLT2i administration over a 4-week course of empagliflozin resulted in a 30%-60% increase in urinary sodium (which pulls water along for excretion) in patients with T2DM (P ≤ 0.001), and was positively correlated to the degree of glucosuria (P ≤ 0.001)[31]. The cumulative effects of this fluid loss can be appreciated from a cardiovascular perspective as a reduction in blood pressure as vindicated by the landmark trials, implying a reduced afterload. Moreover, studies show that the fluid loss in SGLT2i therapy occur via losses in intravascular compartments, implying a reduction in preload. This dual reprieve in those with heart disease promotes a physiologic status that leads to improved ventricular loading without maladaptive compensatory changes such as remodeling, and may be partially responsible for positive CVOT results, although it shown be mentioned there are many pleiotropic effects of these drugs under investigation[32-34].
SLGT2i also promote cardiovascular benefits in patients with T2DM through auxiliary avenues deeply rooted in diabetic sequelae such as through the blunting of harmful reactive oxygen species (ROS), modulating detrimental neurohormonal activation, improving oxygen flux, and preserving a positive vascular biology profile[35-38]. However, since the capacity for SGLT2i agents hinder the cotransport and reabsorption of sodium and glucose, the sodium gradient is therefore retained for sodium-dependent phosphate cotransporters (SLC34A1 and SLC34A3) as referenced in Figure 2[39-41]. The consequence of this shift in renal cotransporter dynamics is a resultant increase in phosphate reabsorption at the site of the proximal tubule, resulting in hyperphosphatemia. Albeit a recognized a process, ramifications of phosphate flux with SGLT2i administration within the literature have largely been centered upon a controversial dialogue on whether or not these agents contribute to deficits in bone mineral density of significance to subsequently provoke bone fractures[42-44]. Scant in the literature however, is a commentary highlighting the role of serum phosphate changes mediated by SGLT2i on CVD, considering CVOT have vindicated SGLT2i in mitigating CVD burden. Nevertheless, the pharmacology of SGLT2i theoretically induces increased serum phosphate, which is associated with vascular calcification and stiffness, cardiac remodeling, and other pathologic changes conductive to MACE (observed in settings independent of SGLT2i administration), especially in populations with aberrant metabolic derangements such as T2DM and diabetic CKD[45-49]. A discussion of the relative degrees of phosphate induction with SGLT2i therapy in relation to the attenuation of T2DM and its sequelae conferred with therapy (as is supported by multiple clinical trials) is prudent for giving full context on the pharmacology of SGLT2i while affirming that these drugs are still associated with remarkably beneficial cardiovascular benefit.
Figure 2.

Glycosuria mediated from sodium-glucose cotransporter 2 inhibition. Adapted from Servier Medical Art, which is licensed under a Creative Commons Attribution 3.0 Unported License. PT: Proximal tubule; SGLT2i: Sodium-glucose cotransporter 2 inhibitors.
The effects of phosphate on cardiovascular function and also renal physiology which thereby impacts cardiovascular function is well documented. While discussed primarily within the scope of the nephron, hyperphosphatemia as a side effect associated with SGLT2i administration has been vindicated in human studies. In one study by Blau et al[50], among 25 research participants receiving Canagliflozin 300 mg over placebo, a marked increase in sodium excretion with an increase in associated serum phosphate levels were noted, giving validation for the sodium-phosphate cotransporter mechanism of SGLT2i mediated phosphatemia. Moreover, Canagliflozin administration was associated with a 16% increase in serum phosphate levels over placebo (P < 0.001).
In another post-hoc analysis of a double-blind, randomized, crossover trial in 31 patients with T2DM and early-stage diabetic kidney disease (defined as an albumin-to-creatinine ratio between 100 and 3500 m/g, eGFR ≥ 45 mL/min per 1.73 m2, and 11.4% > HbA1c ≥ 7.2%, patients were randomized to dapagliflozin 10 mg daily or placebo. Dapagliflozin administration increased serum phosphate by 9% (95%CI: 4%-15%, P = 0.002)[51]. Interestingly, this increase in phosphate was not correlated with changes in eGFR or 24-h albumin excretion, a known marker of renal and by proxy, cardiovascular impairment[52-54]. Such an increase in phosphate with dapagliflozin administration is noteworthy when considering that a 2016 analysis incorporating over an ethnically diverse cohort of 94989 patients stratified for population-based phosphorus quartiles by ethnicity demonstrated that 0.5 mg/dL serum phosphorus increases were associated with adjusted hazard ratios of increased renal mortality[55]. Examinations of NHANES III with incident ESRD and elevated phosphate (> 4 mg/dL) proved elevated ESRD incidence compared to those with lower phosphate levels (RR = 1.90; 95%CI: 1.03–3.53; P = 0.04)[56]. The latter two studies show a disparity between the dogma of phosphatemia and empiric findings of studies using dapagliflozin.
Associated with hyperphosphatemia is the elevation of fibroblast growth factor 23 (FGF23)[57,58]. This protein has received interest in its clinical applications due to its potential in mediating aberrant metabolic pathways associated with cardiovascular health, as marked by its associated with MACE providing a link between hyperphosphatemia and the heart[59-61]. There is a growing body of evidence that FGF23 is a key player in signaling pathways related or as a biomarker related to distinct pathologic avenues of diminished cardiovascular function- these include left ventricular hypertrophy (LVH), endothelial cell dysfunction, arterial fitness, and atherosclerosis[60,62]. Nevertheless, we can appreciate that SGLT2i, despite promoting hyperphosphatemia (and subsequently FGF23 as marked by the Canagliflozin and Dapagliflozin phosphate studies) mitigate cardiovascular disease buden[50,51]. We speculate that despite being central to pathways that hamper cardiovascular disease outcome, the mechanisms by which phosphatemia and FGF23 expression induce these impairments are also attenuated by SGLT2i in their pleiotropic effects, with even more mechanisms of cardiovascular benefit and thus, leading to a “net” positive outcome. The course of this review will outline these pathways from a phosphate metabolism perspective, and subsequently via commentaries on how SGLT2i overlap with these pathways and go beyond these collective pathways to further support cardiovascular health.
Moreover, the associated osmotic diuresis promotes the loss of sodium and water, which decreases blood pressure and improves oxygen flux and hemodynamic status. For example, in EMPA-REG (Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes), 7020 patients with established coronary artery disease and T2DM were slated to receive 10 mg empagliflozin (n = 2345), 25 mg empagliflozin (n = 2342) or placebo (n = 2333)[63]. The results were reductions in the risk of cardiovascular (CV) death by 38% relative to placebo (3.7% vs 5.9%, HR = 0.62; 95%CI: 0.49-0.77; P < 0.001). An exploratory mediation analysis of EMPA-REG attempted to identify elements influencing CVD death risk reduction with empagliflozin by analyzing post hoc mediators through Cox regression[64]. A significant finding of this audit of trial covariates and their influence in survival identified that hematocrit and hemoglobin mediated roughly 50% of the propensity of empagliflozin to improve CV survival relative to placebo. One possible reason for this rise in red blood cell (RBC) magnitude may be due to more efficient erythropoiesis via the renoprotective properties of SLGTI2[65,66]. Support for this theory can be found in a small-scale study of 66 patients administered empagliflozin over four weeks, with a 31% increase in erythropoietin (P = 0.0078) seen in 64 patients[31]. Similar improvements in erythropoiesis have been observed in other classes of SGLT2i, dapagliflozin and canagliflozin[67,68]. The role for this phenomenon in CV survival is not well studied however and can be attributed from improvements in oxygen flux and global metabolic resource allocation. Alternatively, parsimony would suggest that osmotic diuresis and improved handling of fluid would result in an increase in RBC constituents via dilution-concentration dynamics, and the CV survival observed would be attributable to the mitigation of edema and ventricular stress. Nevertheless, lessons from SGLT2i therapy show us that the improved prospects for patients with T2DM are pleiotropic in nature and extend beyond correction of hyperglycemia. This concept is congruent with the focus in this work that multiple axes promote improved outcomes notwithstanding phosphate metabolism. Such principles will be reinforced throughout this commentary.
PHOSPHATE METABOLISM, DIABETES, HEART DISEASE- LOADING SGLT2 IN THE PICTURE
Within the context of the pleiotropic effects of SLGT2i administration and phosphate metabolism, it is noteworthy to mention that the ramifications of phosphate metabolism occur peripherally and not directly within the heart itself. The SGLT2 protein is not expressed within cardiac tissue, as demonstrated by sequencing studies[69]. Therefore, to make remarks about SGLT2 inhibition within the context of phosphate metabolism, a query of phosphate metabolism beginning within the nephron is warranted.
In normal settings of serum hyperphosphatemia, the parathyroid glands address this homeostatic imbalance through the secretion of parathyroid hormone (PTH). PTH acts on the PT to inhibit sodium/phosphate cotransport, resulting in the excretion of phosphate through urine. There is also a secondary implication of increased sodium flux on the cotransporters hosted on distal portions of the nephron and their relative dynamics Moreover, PTH also has an auxiliary role of promoting phosphate absorption from the small intestine as well as bone, shuttling phosphorus into serum. It also has a role in the activation of vitamin D via secondary hydroxylation, which has cardiorenal implications which will later be discussed. For brevity and completeness, this activation of vitamin D is implicated in the absorption of calcium and phosphate within the intestine, with less reliance on phosphate flux on Vitamin D, resulting in a net decrease of phosphate[70].
However, the metabolism of phosphorus and its associated compounds in the context of contemporary lifestyle and diet with an emphasis on heart disease and T2DM has caused a paradigm shift in how this homeostatic mechanism plays out for such patients. Current diets, especially in the western world, are dense in phosphate and have ramifications in a populous that has a significant burden of T2DM and heart disease[71,72]. Animal studies conducted have shown that both genetically impairing sodium-phosphate cotransporter function as well as hyperphosphatemia-induced via diet in wild-type mammals results in a phenotype akin to metabolic syndrome, which includes loss of lean skeletal muscle mass, increased reactive oxygen species formation, and renal impairment and cardiopulmonary deficits[73]. This dual-approach of the elicitation of increased serum phosphate with similar end-point results confirmed by histologically analyses was cited as giving credence towards the notion that absolute hyperphosphatemia was the end insult responsible for these findings[73]. These findings mirror the components of derangements seen in patients with T2DM and cardiovascular disease.
For example, earlier animal studies show support for hyperinsulinemia promoting reductions in the fractional excretion of phosphate in dogs, caused mainly by abatement in the ratio of tubular fluid to plasma phosphate domineered primarily by proximal tubular phosphate reabsorption (P < 0.02), lending an association between dysfunctional glucose metabolism and phosphate flux with consequences in disease states such as cardiovascular disease[74]. Further evidence for an association between derangements in glucose and phosphate are furthered by one human study, 31P magnetic resonance spectroscopy (MRS) assessing the effects of a hyperglycemic-hyperinsulinemic clamp experiment augmented on study subjects noted an increase in inorganic phosphate and reductions in phosphocreatinine (PCr)[75]. The reductions in PCr have direct implications in heart disease, as one study by Bhella et al[76] recruited healthy patients and those with heart failure with preserved ejection fraction (HFpEF) to perform lower limb exercises with MRI scanning evaluation to assess myocyte function. HFpEF patients were noted to have reduced rates of oxidative phosphorylation rates, with an increase in refractory period to normalization of phosphocreatine when compared to healthy sedentary age-matched controls. Blunted PCr replenishment can cause concern as it functions as a phosphate derivative that skeletal muscle may opt to metabolically activate when ATP reserves are not high enough to sustain a respective workload. Reductions in PCr as seen in hyperphosphatemic states show an increased in the proclivity for cardiomyocytes to undergo apoptosis or irregular phenotypes[77].
Irregularities in phosphate in hyperinsulinemic states as mentioned above have also been directly studied in patients with T2DM, setting the foundation for investigation in diabetic heart disease and phosphate metabolism, with SGLT2i. 31P MRS scanning the vastus lateralis of patients with T2DM compared to healthy, aged-controlled matches showed an absolute decrease in PCr, (PCr 28.6 ± 3.2 vs 24.6 ± 2.4, P < 0.002), which is supported by a negative correlation between PCr and HgbA1C (r = − 0.63, P < 0.01)[78]. These findings support that the diabetic state as well as the cardiovascular state are impacted by sequelae related to hyperphosphatemia, especially in patients with conditions related to insulin resistance where SGLT2i utilization would be warranted given appropriate consideration considering their increased use in patients with diabetes and cardiovascular disease.
Another derangement observed in the Ohnishi study was renal impairment seen in the form of renal arteriole calcification with apoptotic cells upon histology directly associated with heavy phosphate burden, indicative of maladaptive renal calcification. Such observations have been observed in humans, with ramifications of CKD and ESRD[79]. Prevailing theories include phosphate aggravating vascular smooth cells or the buildup of calcium byproducts in the form of nephrocalcinosis[80]. Similar derangements, albeit by different constituents, occur in T2DM. Such processes both however heavily similarly impact the cardiovascular benefits in patients with T2DM. Patients with T2DM experience insulin resistance and as result experience blunted responses to glucose attenuation, precluding euglycemia. The excess of glucose tends to undergo nonenzymatic glycosylation with amine groups of the glomerular basement membrane of the kidney, causing protein leakage which occludes the arterial lumen. This process propensity to impact the efferent arteriole sooner and with more impact than the afferent arteriole, causing an increase in intraluminal pressure and GFR[81].
One consequence of this relative mismatch in luminal caliber is an initial hyperfiltration cascade that ultimately damages the renal mesangium. Subsequent hyalinization of the afferent arteriole decreases GFR, ultimately manifesting as diabetic kidney disease and subsequently, CKD. The decrease in afferent lumen caliber is noted by afferent baroreceptors and elicits renin secretion by the juxtaglomerular cells. Subsequently this promotes the renin-angiotensin-aldosterone system (RAAS) cascade[82]. Ramifications of the RAAS system include an increase in blood pressure as marked by the activity of angiotensin II on the systemic vasculature, causing an increase in afterload as well as sodium retention. Chronically, this can lead to left ventricular hypertrophy and subsequently MACE. We will see that phosphate ultimately impacts the heart in a critical manner similarly, however, the net benefit of SGLT2i through their pleiotropic effects negate the deleterious effects of increased phosphate levels and lead to beneficial cardiorenal outcomes.
Compromised kidneys, such as in the case of phosphate and T2DM flux promotes a physiologic equilibrium that is acclimated to the retention of metabolic toxins which directly serve as insults to the heart and kidney in cardiorenal syndrome as defined by KDIGO[3]. In normal physiologic circumstances, increased congestion caused by edema results in stretch of native mechanosensory nerve fibers scattered throughout the abdominal and pelvic wall[83]. This leads to a phenomenon known as the renorenal reflex, by which activation of this system attenuates efferent renal sympathetic nervous system activity (ERSNA)[84,85]. Decreased ERSNA is associated with a higher threshold for α1-adrenoceptor activation. These receptors play a vital role in the activation of transporters in the proximal tubule that lead to a state of net sodium reabsorption. By decreasing their function, the renorenal reflex induces natriuresis and relief of central congestion. However, chronic kidney disease and ESRD is associated with dysregulation of the renorenal reflex[86,87]. SGLT2i therapy approaches this consequence of renal impairment and promotes homeostatic renal function by offering liberating sodium. Patients with T2DM experience an increase in filtered glucose, which is reabsorbed along with sodium in the proximal tubule with a higher affinity[88]. SGLT2i, mitigates the reabsorption of glucose of up to 90%, with concomitant sodium loss, disinhibiting the juxtaglomerular apparatus from promoting the hyperfiltration loop. Sodium is subsequently diuresed, sparing the kidney from edema and congestion.
In spite of phosphate mediated renal decline, SGLT2i have been shown to improve cardiovascular as well as renal health in patients with T2DM and CKD. CANVAS-Renal (Canagliflozin and cardiovascular and renal events in type 2 diabetes) was one trial that elucidated the effects of canagliflozin on cardiorenal outcomes[89]. Primary outcomes of interest were drawn from composites of cardiovascular death (CVD), nonfatal myocardial infarction (MI), and nonfatal stroke. Death from any cause, CVD related death, albuminuria exacerbation, and heart failure hospitalization. The primary outcome showed improvements in patients receiving canagliflozin compared to placebo (HR = 0.86; 95%CI: 0.75-0.97, P = 0.02)[20]. Cardiovascular secondary outcomes did not demonstrate superiority, (death from any cause, P = 0.24), and thus hypothesis testing was discontinued[20]. However, we will see that other SGLT2i have differential profiles conducive to cardioprotection in spite of phosphate mediated damage, and this example of CANVAS-Renal was included to illustrate that phosphate mediated nephrotoxicity with SGLT2i is not a significant factor as demonstrated by clinical trial data as will be discussed. Interestingly, it is noted that magnesium modifies the cardiovascular mortality profile of patients undergoing hemodialysis in a positive respect[90]. In one meta-analysis, it was shown that SGLT2i improved serum magnesium levels with the potential to attenuate PTH, and was implicated as a potential underlying factor in cardiorenal mortality benefits in one SGLT2i trial[91]. This is just one of the multiple pleiotropic effects of SGLT2i.
As we see the continuous deleterious effects of hyperphosphatemia, a query in its mechanisms naturally arises. Investigations into the mitochondrial physiology has shown that oxidative stress, as seen in patients with T2DM, can exacerbate phosphate mediated toxicity and cause insulin and glucose dysregulation[92]. Mitochondrial deficits are the central theme in reactive oxygen species (ROS) formation belief in which by hyperglycemia mediates ROS formation, which impacts nodes of cardiorenal significance[35].
HYPERPHOSPHATEMIA, SGLT2, AND CARDIORENAL IMPLICATIONS
One prevailing study directly linking increased intracellular phosphate with cardiovascular incidents a la ROS is through endothelial dysfunction, which act by various mechanisms such as nitric oxide mitigation leading to decreased vascular compliance, vascular apoptosis, and the promotion of atherosclerotic plaques[91-93]. It has also been shown to promote arterial stiffness in healthy individuals[94]. The aggregate effect of these processes limits vascular compliance and the ability for the heart to adapt to hemodynamic instability. T2DM overlays this effect by inducing the formation of one member of the ROS family, NF-κB, which increases cardiomyocyte tension through activating processes related to pressure-induced remodeling and fibrosis as well as upregulates pro-coagulant factors such as tissue factor VIII and by proxy, downregulates anticoagulant factors such as plasminogen and urokinase[95,96]. The net effect of both processes would have ramifications in oxygen flux and the propensity for the cardiovascular system to abate vascular injury caused by shear stress, with potential consequences being thrombosis and embolization, and subsequently MACE. An overview of these processes can be seen in Figure 3.
Figure 3.

Examples of deleterious cardiovascular pathways associated with hyperphosphatemia and reactive oxygen species. Adapted from Servier Medical Art, which is licensed under a Creative Commons Attribution 3.0 Unported License. ROS: Reactive oxygen species; NF-κB: Nuclear factor kappa-B.
Maladaptive ramifications of MACE are vast, and may include arrhythmia from hypertrophy and disequilibrium of the electromechanical conduction system of the heart in relation to cardiac geometry, to coronary events or heart failure. Both T2DM and aberrant phosphate toxicity have been implicated in modulating the redox state of mitochondria, causing disruptions in important cardiomyocyte channel regulators such as MAPK and calcium flux via SERCA with the propensity to cause abnormal heart rhythm[97-99].
Yet, evidence with SGLT2i continue to redeem their propensity to bolster the cardiovascular profile of patients with T2DM. The EMPA-REG OUTCOME trial (Empagliflozin Cardiovascular Outcome Event Trial in Type 2 Diabetes Mellitus Patients–Removing Excess Glucose) was one of the initial SLGT2i trials that gave insight into the utilization of SGLT2i for patients with a high cardiovascular risk profile[89]. In the study, three randomized groups were given empagliflozin 10 mg (n = 2345), empagliflozin 25 mg (n = 2342), or placebo (n = 2333). EMPA-REG defined its primary outcomes to note a composite of CVD death, nonfatal MI(s) (not including silent MI), or nonfatality associated with primary endpoint considerations. Unstable angina culminating in hospitalizations were designated as the secondary outcome[90]. Reports from this trial noted relative risk reduction (RRR) of around 13% in the primary outcome group taking both empagliflozin dosages when compared to placebo (HR = 0.86; 95%CI: 0.74-0.99; superiority P = 0.04)[18]. Secondary outcome was not statistically significant (12.8% vs 14.3%, HR = 0.89; 95%CI: 0.78-1.01; superiority P = 0.08).
Moreover, Sato et al[100] examined 46 patients with T2DM given SGLT2i and observed their QTcd (the absolute range of QT-intervals in a 12-lead electrocardiogram, and a surrogate for ventricular depolarization. It was shown that SGLTi pharmacotherapy resulted in a reduction of QTcd by roughly 9%. It is believed that this reduction in QTcd is not related to glycemic control, but blood pressure reduction. These findings are interesting as the show the propensity to regulate ventricular depolarization, and shedding a potential perspective to one pleiotropic effect of mortality reduction in EMPA-REG as a substantial portion of the cohort in of hypertensive EMPA-REG cohort participants were designated as having left ventricular hypertrophy per ECG findings[101,102]. Cardiovascular benefits that deviate from the antidiabetic effects of SGLT2i such as a decrease in blood pressure show that these agents may hold promise in counterbalancing the deleterious effects of ROS and phosphate on multiple fronts.
Another class of SGLT2i, Dapagliflozin, has also been vindicated and shows propensity to leverage the negative vasoconstrictive effects of phosphate and hyperglycemia mediated ROS as demonstrated by Li et al[103]. The ability for SGLT2i to reduce blood pressure was examined via application of dapagliflozin on the aortic rings of male New Zealand white rabbits. Subsequently, vasodilatory events were noted due to the activation of voltage-dependent potassium channels. Moreover, it was shown that SGLT2i had the ability to affect cellular signaling pathways via protein kinase G, which has been speculated to play a role in the opening of calcium-activated potassium channels, with a concomitant influx of positive ions into the vasculature, resulting in cellular hyperpolarization, relaxation, and vasodilation. SGLT2i still had the propensity to induce vasodilation after the aortic rings were removed of their endothelium with nifedipine, a calcium channel blocker, or with administration of nitric oxide inhibitors. Voltage-dependent potassium channels and protein kinase G inhibitors however resulted in amelioration of vasodilation, suggesting that SGLT2i may work via signaling pathways to attenuate extraneous negative players in cardiovascular health.
DECLARE TIMI-58 (Dapagliflozin Effect on CardiovascuLAR Events) is yet another clinical trial redeeming the cardioprotective profile of SGLT2i in spite of phosphate mediated increases with pharmacotherapy. This trial recruited 17160 patients with T2DM[104]. DECLARE TIMI-58 defined their co-primary endpoints as MACE (defined by CVD, MI, or ischemic stroke), with positive results (HR = 0.93; 95%CI: 0.84-1.03). Second co-primary endpoints included HF hospitalization or CVD death composites with results yielding a 18% relative risk reduction (4.9% vs 5.8%; HR = 0.83; 95%CI: 0.73-0.95). Second co-primary endpoints in this study were attributed to a relative risk reduction of 27% regarding heart failure hospitalizations (HR = 0.73; 95%CI: 0.61-0.88).
While the clinical data supports that an increase in phosphate and potential roles it has on vascular calcification, ROS modulation, and exacerbation in the patient with T2DM has no effect and in fact, is shown to improve cardiovascular mortality with SGLT2i, multiple questions arise to how. Glucosuria induced by SGLT2i in patients with T2DM has been shown to reduce the maximal renal glucose transport in addition to the threshold for glucosuria, resulting in a loss of glucose that would otherwise be used to help procure deleterious ROS in the inflamed mitochondria. As demonstrated previously, however, the effects of SGLT2i to taper phosphate effects go beyond antidiabetic properties, and include rhythm control, cellular signaling pathways, hemodynamics, and mineral turnover. More work needs to be done to elucidate the mechanisms of mineral turnover in the nephron given the total variants of SGLT2 proteins in the kidney as well as the pharmacotherapeutic agents, and this work will help understand contextually the full scope of cardiovascular mortality benefits seen with this drug and as validated by clinical trial data.
SGLT2i and other electrolytes: An annotation towards inclusive insight in pharmacodynamics
While this review focused on the effect of phosphate in relation to its changes with SGLT2i therapy correlated with cardiovascular mortality, a brief commentary on the other electrolytes affected by SGLT2i pharmacotherapeutics offers a proclivity to understand the global influence of these drugs. Moreover, these electrolytes have their own cardiorenal effects, and a succinct commentary is warranted given that scope. Therefore, we will include a remark on the current literature using the same standards regarding a focus on evidence-based medicine indexed to translational biology and clinical explanations to share the effects that SGLT2i have on such electrolytes.
A meta-analysis that included a query which successfully harvested random clinical trial (RCT) data of 15309 patients with T2DM taking four SGLTi (canagfliflozin, dapagliflozin, empagliflozin, and iprafliglozin) offers significant insight on the administration of SGLT2i and magnesium levels[89]. Meta-regression analyses for each agent was implemented by the authors of the aforementioned meta-analysis to evaluate the dose-dependent effects for each SGLT2i given the different classes involved. Scrutinization of this study showed that SGLT2i therapy have the propensity to elevate serum magnesium levels 0.08–0.2 mEq/L in T2DM patients compliant with pharmacotherapy. One limitation of this meta-analysis was that patients with CKD are not mentioned in this study. Both magnesium metabolism is impacted in this population group, and there is significant evidence to show a considerable proportion of SGLT2i-eligible patients with CKD are not prescribed in this population despite guideline recommendations for their utilization in this population[19]. Nevertheless, there is some evidence that shows canagliflozin as an agent is beneficial in magnesium retention in patients who have T2DM and CKD (defined as 30 ≤ eGFR ≤ 50 mL/min/1.73 m2) giving reassurance that these agents may be indicated for the regulation of cardiorenal pertinent electrolytes[104,105].
It should be noted in the scope of cardiovascular disease; magnesium levels hold value as supported by another meta-analysis able to aggregate 313,041 patients. Of these patients, 11995 cardiovascular disease risk stratification[106]. It was determined that each increase in 0.2 mmol/L of serum magnesium conferred a 30% decreased risk of CVD acquisition (RR = 0.70; 95%CI: 0.56-0.88 per 0.2 mmol/L step-size in the physiologic range).
There are many interesting insights to gain from the role of other electrolyte influences in cardiovascular mortality, especially when compared to phosphate metabolism and its seemingly deleterious effects that were offset by SGLT2i. Cohort studies using CKD patients as a model for hyperphosphatemia show those with lower magnesium levels exhibited increased cardiovascular mortality[107]. Cardiovascular mortality risk in the setting of hyperphosphatemia has been shown to be blunted in those who have normal to high serum magnesium levels. These same cohort studies show evidence that that magnesium, in-vitro, offsets hyperphosphatemia mediated sequelae conducive to a MACE phenotype, such as the induction of vascular smooth muscle cell calcification that may affect endothelial cell function. Moreover, higher serum magnesium levels hampered the progression of CKD in patients with hyperphosphatemia, highlighting a shared cardiorenal protective nature with SGLT2i.
There are multiple mechanisms by which magnesium may achieve this affect. With respect to the heart, magnesium has been shown to stimulate ATPase, critical for the maintenance of the sodium-potassium pump within the ventricular myocardium[108]. Further investigations have shown that deficiencies in magnesium lead to aberrant potassium and sodium balances, leading to an increased risk of arrhythmia. Moreover, magnesium serves a role as mitochondrial enzyme cofactors, and its dearth leads to a deficient mitochondrial metabolic status, leading to increased reactive oxygen species, a propensity for thrombus formation, and endothelial dysfunction[109]. These antiarrhythmic affects may explain have played a role in the reduction of arrhythmias as marked by stability of QTc observed in one study that attempted to elucidate the affects SGLT2i therapy had on the positive clinical end-outcomes in the EMPA-REG outcome trial, showing that Empagliflozin helped mitigate abnormal heart rhythms, and was attributed to be a major cause of mortality and morbidity reduction[110,111]. Magnesium is also noted to be a GabaA potentiator in the central nervous system, and its binding has been hypothesized to promote a decrease in blood pressure, decreased sympathetic tone, and the mitigation of tachyarrhythmias via parasympathetic and anxiolytic properties[100].
Magnesium wasting is common in patients with T2DM and it is believed that SGLT2i help normalize magnesium levels through an array of mechanisms. For example, T2DM is associated with an increase in transporters that promote magnesium transport across to the luminal side of the nephron lead in diuresis of magnesium, and reduction of hyperglycemia is believed to give a reprieve from this wasting of magnesium[112]. The proposed mechanism are that in-vivo studies have noted increased magnesium increased magnesium reabsorption at the distal convoluted tubule within the nephron through an insulin dependent mechanism that activates increased expression of TRMP6, an ion channel that allows for entry of luminal magnesium ions into the distal convoluted tubule. Within the tubule, a sodium-magnesium that is believed to be putative in nature, represents the proclivity for basolateral transit of magnesium[113]. This process functionally represents the most terminal opportunity for magnesium reabsorption in the nephron beyond the loop of Henle[114]. It should be noted that this mechanism is partially sodium dependent and may involve increased absorption due to the increased tubular retention of sodium that would have otherwise been excreted in the proximal convoluted tubule by the sodium-glucose cotransporter without SGLT2i therapy[114,115]. In insulin resistance, there is a decrease in TRMP6 expression, leading to decreased tubular magnesium ion flow, and loss of magnesium in urine. This is consistent with the association of low magnesium levels in patients with T2DM. Moreover, intra-pancreatic magnesium has been shown to improve insulin sensitivity by serving as a potentiator of depolarization of pancreatic β-cells responsible for insulin release[116]. However, deficits in insulin utilization and magnesium availability may lead to a cycle in which the diabetic status is exacerbated. This represents an opportunity for SGLT2i therapeutics to normalize insulin-magnesium dynamics and could be used to explain some components of positive clinical endpoint response. It should be noted that markedly decreased magnesium levels in lieu of activating parathyroid hormone when mildly depressed, prevent parathyroid hormone secretion. The purported mechanism for this phenomenon is that some basal level of magnesium ions is required for the function of the calcium-sensing receptor (CaSR) expressed in the parathyroid gland and renal tubules, and is responsible for regulating calcium by regulating the release of parathyroid hormone (PTH)[117].
This leads to our second ion of consideration in SGLT2i therapy- calcium. Calcium serves multiple roles as a protein activator or inhibitor, can form pathologic deposits, or mediate neurologic signaling- all important in cardiovascular health. Within electrophysiologic considerations, calcium ions are important during the phase 0 upstroke stage in pacemaker action potentials when voltage-gated calcium channels open, and due to their relatively lower negative resting potential, fast-voltage gated sodium channels expressed remain permanently inactivated. This results in a “lag” effect that the AV node utilizes to prolong potential transmission from the atria to the ventricles for a unified beat. In the myocardium, calcium plays an important role in the maintenance of depolarization. As depolarization begins, intracellular potassium channels open and are released to bring the membrane potential to equilibrium, which would terminate the depolarization sequence. However, voltage-gated calcium channels promote the inward flux of calcium ion that activates further calcium release from the sarcoplasmic reticulum, maintaining a plateau that prolongs myocyte contraction and adequate myocardial tension to initiate the stroke volumes needed to maintain perfusion. It is postulated the calcium channel itself in myocardiocytes is dependent upon calcium for its closing[118]. A hypocalcemic state leads to slower calcium leakage within the myocardial membrane, meaning a longer time to reach a concentration to close the L-Type calcium channels, extending the action potential, and by proxy, initiating QT prolongation[119]. In the previously mentioned study by Blau et al[50], the 25 research participants who received 300 mg Canagliflozin over placebo for 5 d revealed no change in the serum concentration and ionized-calcium concentration among the participants over this period. However, calcium excretion defined as (mmol/d)/(grams of creatinine) as a function of the day on which urine was collected revealed a statistically significant differential decrease in urinary calcium excretion on day 4 (1.50 vs 1.78; P = 0.04)[50]. The subsequent day showed a trend, albeit not statistically significant, towards the same trajectory of urine calcium dynamics (day 5, 1.40 vs 1.66; P = 0.09).
Within the biomarkers assessed in the Canagliflozin study, fibroblast growth factor 23 (FGF23) was tracked. FGF23 is known to be provoked by increased phosphate, and the latter also provokes PTH excretion. Therefore, by proxy, SGLT2i by increasing phosphate, increase both FGF23 and PTH[120,121]. Within this small study, FGF23 levels peaked roughly 12 h after phosphate reached its maximus, consistent with physiologic studies of FGF23 expression[122]. FGF23 acts on the proximal tubule, inhibiting NPT2, a cotransporter of sodium and phosphate (which may further explain why natriuresis is not the predominant mechanism of urinary loss within SGLT2i administration after phosphate levels subsequently reach clinically relevant concentrations)[123]. Interestingly, FGF23 suppresses 1-α-hydroxylation of Vitamin D to activate it , while PTH promotes 1-α-hydroxylation resulting in a mismatch of calcium reabsorption[121]. The dynamics of this small-scale Canagliflozin study that measured FGF23, PTH, and 1,25-dihydroxyvitamin D levels on a daily basis implied that early FGF23 expression resulted in a transient hypocalcemia. The decrease in 1,25-dihydroxyvitamin D theoretically decreased the gastrointestinal harvest calcium ion, further stimulating PTH, which has already been induced by SGLT2i mediated phosphatemia. PTH in this cohort may have resulted in net renal tubular reabsorption of calcium ion by the relative influence of SGLT2i. The effect of hypercalciuria may have been a temporary transient effect of osmotic diuresis, as it resolved by day 5. A study involving higher power may be needed to elucidate the effects of SGLT2i on calcium metabolism, as FGF23 expression has been noted to promote paracrine regulation resulting in left ventricular hypertrophy, contraindicative to net positive CVOT outcomes, while also promoting degenerative vascular changes in the kidney[124].
It seems however based on data related electrolyte flux, calcium may not be as significant player in SGLT2i therapy. The only CVOT trial in which bone fractures (a surrogate for phosphate and calcium metabolism) seemed to exhibit a differential risk for bone fractures was CANVAS, a Canagliflozin based trial. Total fracture incidence for bone fracture was more prominent in the canagliflozin group relative to placebo (15.4 vs 11.9 fractures among participant per 1000 patient-years; HR = 1.26, 95%CI: 1.04–1.52)[20]. However, this trial when compared to the plethora of dapagliflozin and empagliflozin CVOT trials employed a larger proportion of T2DM diagnosis, female gender, and degree of obesity- all factors associated with bone fractures and by proxy, calcium and phosphate flux relative to other SGLT2i. CREDENCE, another canagliflozin trial, did not report a differential fracture risk among patients. Nevertheless, scrutinizing the role of calcium in SGLT2i therapy has important ramifications when discussing the role they pose in cardiac electrophysiology, wasting in those with CKD, their ability to act as signal transduction messengers, and other downstream factors to explain their role in SGLT2i mediated health benefits.
CONCLUSION
Through the use of translational biology, a known side-effect of SGLT2i, phosphatemia, was able to be shown as diminutive relative to the pleiotropic effects of these new class of antidiabetic agents. Commentaries such as these show that despite theoretical contraindications to pharmacotherapy, the full spectrum of drug effects may outweigh what seems to be harmful, resulting in a net positive clinical profile. Yet, more work needs to be done on elucidating the pathways by which SGLT2i act peripherally beyond the nephron. There is a slew of research implicating cytokine modulation, gene expression, and inflammasome activation in avenues not previously discovered that may give new insight into how these agents have propelled their way to shifting from antidiabetic agents to pharmacotherapeutic options with cardiovascular benefits.
Footnotes
Conflict-of-interest statement: The authors do not declare any conflict of interest regarding the publication of this manuscript
Provenance and peer review: Invited article; Externally peer reviewed.
Peer-review model: Single blind
Peer-review started: April 19, 2021
First decision: July 8, 2021
Article in press: November 30, 2021
Specialty type: Cardiac and cardiovascular systems
Country/Territory of origin: United States
Peer-review report’s scientific quality classification
Grade A (Excellent): 0
Grade B (Very good): B
Grade C (Good): 0
Grade D (Fair): 0
Grade E (Poor): 0
P-Reviewer: Higashikawa T S-Editor: Ma YJ L-Editor: A P-Editor: Ma YJ
Contributor Information
Mouhamed Nashawi, Department of Internal Medicine, Baylor Scott and White All Saints Medical Center, Fort Worth, TX 76132, United States. nashawi@livemail.uthscsa.edu.
Mahmoud S Ahmed, Division of Medicine-Cardiology, UT Health San Antonio, San Antonio, TX 78229, United States.
Toka Amin, Division of Medicine-Cardiology, UT Health San Antonio, San Antonio, TX 78229, United States.
Mujahed Abualfoul, Department of Internal Medicine, Faculty of Medicine, Cairo University, Dallas, TX 75203, United States.
Robert Chilton, Department of Internal Medicine, Methodist Dallas Medical Center, Dallas, TX 75203, United States.
References
- 1.Khunti K, Seidu S. Therapeutic Inertia and the Legacy of Dysglycemia on the Microvascular and Macrovascular Complications of Diabetes. Diabetes Care . 2019;42:349–351. doi: 10.2337/dci18-0030. [DOI] [PubMed] [Google Scholar]
- 2.Ruocco G, Palazzuoli A, Ter Maaten JM. The role of the kidney in acute and chronic heart failure. Heart Fail Rev . 2020;25:107–118. doi: 10.1007/s10741-019-09870-6. [DOI] [PubMed] [Google Scholar]
- 3.Rangaswami J, Bhalla V, Blair JEA, Chang TI, Costa S, Lentine KL, Lerma EV, Mezue K, Molitch M, Mullens W, Ronco C, Tang WHW, McCullough PA American Heart Association Council on the Kidney in Cardiovascular Disease and Council on Clinical Cardiology. Cardiorenal Syndrome: Classification, Pathophysiology, Diagnosis, and Treatment Strategies: A Scientific Statement From the American Heart Association. Circulation . 2019;139:e840–e878. doi: 10.1161/CIR.0000000000000664. [DOI] [PubMed] [Google Scholar]
- 4.Zannad F, Rossignol P. Cardiorenal Syndrome Revisited. Circulation . 2018;138:929–944. doi: 10.1161/CIRCULATIONAHA.117.028814. [DOI] [PubMed] [Google Scholar]
- 5.Koro CE, Lee BH, Bowlin SJ. Antidiabetic medication use and prevalence of chronic kidney disease among patients with type 2 diabetes mellitus in the United States. Clin Ther . 2009;31:2608–2617. doi: 10.1016/j.clinthera.2009.10.020. [DOI] [PubMed] [Google Scholar]
- 6.Saran R, Robinson B, Abbott KC, Agodoa LY, Albertus P, Ayanian J, Balkrishnan R, Bragg-Gresham J, Cao J, Chen JL, Cope E, Dharmarajan S, Dietrich X, Eckard A, Eggers PW, Gaber C, Gillen D, Gipson D, Gu H, Hailpern SM, Hall YN, Han Y, He K, Hebert H, Helmuth M, Herman W, Heung M, Hutton D, Jacobsen SJ, Ji N, Jin Y, Kalantar-Zadeh K, Kapke A, Katz R, Kovesdy CP, Kurtz V, Lavalee D, Li Y, Lu Y, McCullough K, Molnar MZ, Montez-Rath M, Morgenstern H, Mu Q, Mukhopadhyay P, Nallamothu B, Nguyen DV, Norris KC, O'Hare AM, Obi Y, Pearson J, Pisoni R, Plattner B, Port FK, Potukuchi P, Rao P, Ratkowiak K, Ravel V, Ray D, Rhee CM, Schaubel DE, Selewski DT, Shaw S, Shi J, Shieu M, Sim JJ, Song P, Soohoo M, Steffick D, Streja E, Tamura MK, Tentori F, Tilea A, Tong L, Turf M, Wang D, Wang M, Woodside K, Wyncott A, Xin X, Zang W, Zepel L, Zhang S, Zho H, Hirth RA, Shahinian V. US Renal Data System 2016 Annual Data Report: Epidemiology of Kidney Disease in the United States. Am J Kidney Dis . 2017;69:A7–A8. doi: 10.1053/j.ajkd.2016.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Afkarian M, Sachs MC, Kestenbaum B, Hirsch IB, Tuttle KR, Himmelfarb J, de Boer IH. Kidney disease and increased mortality risk in type 2 diabetes. J Am Soc Nephrol . 2013;24:302–308. doi: 10.1681/ASN.2012070718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Straka RJ, Liu LZ, Girase PS, DeLorenzo A, Chapman RH. Incremental cardiovascular costs and resource use associated with diabetes: an assessment of 29,863 patients in the US managed-care setting. Cardiovasc Diabetol . 2009;8:53. doi: 10.1186/1475-2840-8-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ismail-Beigi F, Moghissi E, Kosiborod M, Inzucchi SE. Shifting Paradigms in the Medical Management of Type 2 Diabetes: Reflections on Recent Cardiovascular Outcome Trials. J Gen Intern Med . 2017;32:1044–1051. doi: 10.1007/s11606-017-4061-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Davies M, Chatterjee S, Khunti K. The treatment of type 2 diabetes in the presence of renal impairment: what we should know about newer therapies. Clin Pharmacol . 2016;8:61–81. doi: 10.2147/CPAA.S82008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Newman JD, Vani AK, Aleman JO, Weintraub HS, Berger JS, Schwartzbard AZ. The Changing Landscape of Diabetes Therapy for Cardiovascular Risk Reduction: JACC State-of-the-Art Review. J Am Coll Cardiol . 2018;72:1856–1869. doi: 10.1016/j.jacc.2018.07.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Braunwald E. Diabetes, heart failure, and renal dysfunction: The vicious circles. Prog Cardiovasc Dis . 2019;62:298–302. doi: 10.1016/j.pcad.2019.07.003. [DOI] [PubMed] [Google Scholar]
- 13.United States Food and Drug Administration. Type 2 diabetes mellitus: evaluating the safety of new drugs for improving glycemic control guidance for industry. 2020. 2020. Available from: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/type-2-diabetes-mellitus-evaluating-safety-new-drugs-improving-glycemic-control-guidance-industry .
- 14.American Diabetes Association . 9. Pharmacologic Approaches to Glycemic Treatment: Standards of Medical Care in Diabetes-2020. Diabetes Care . 2020;43:S98–S110. doi: 10.2337/dc20-S009. [DOI] [PubMed] [Google Scholar]
- 15.American Diabetes Association. Addendum. 9. Pharmacologic Approaches to Glycemic Treatment: Standards of Medical Care in Diabetes-2020. Diabetes Care 2020;43(Suppl. 1):S98-S110. Diabetes Care . 2020;43:1979. doi: 10.2337/dc20-ad08a. [DOI] [PubMed] [Google Scholar]
- 16.Das SR, Everett BM, Birtcher KK, Brown JM, Januzzi JL Jr, Kalyani RR, Kosiborod M, Magwire M, Morris PB, Neumiller JJ, Sperling LS. 2020 Expert Consensus Decision Pathway on Novel Therapies for Cardiovascular Risk Reduction in Patients With Type 2 Diabetes: A Report of the American College of Cardiology Solution Set Oversight Committee. J Am Coll Cardiol . 2020;76:1117–1145. doi: 10.1016/j.jacc.2020.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Navaneethan SD, Zoungas S, Caramori ML, Chan JCN, Heerspink HJL, Hurst C, Liew A, Michos ED, Olowu WA, Sadusky T, Tandon N, Tuttle KR, Wanner C, Wilkens KG, Lytvyn L, Craig JC, Tunnicliffe DJ, Howell M, Tonelli M, Cheung M, Earley A, Rossing P, de Boer IH, Khunti K. Diabetes Management in Chronic Kidney Disease: Synopsis of the 2020 KDIGO Clinical Practice Guideline. Ann Intern Med . 2021;174:385–394. doi: 10.7326/M20-5938. [DOI] [PubMed] [Google Scholar]
- 18.Zinman B, Wanner C, Lachin JM, Fitchett D, Bluhmki E, Hantel S, Mattheus M, Devins T, Johansen OE, Woerle HJ, Broedl UC, Inzucchi SE EMPA-REG OUTCOME Investigators. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N Engl J Med . 2015;373:2117–2128. doi: 10.1056/NEJMoa1504720. [DOI] [PubMed] [Google Scholar]
- 19.Wiviott SD, Raz I, Bonaca MP, Mosenzon O, Kato ET, Cahn A, Silverman MG, Zelniker TA, Kuder JF, Murphy SA, Bhatt DL, Leiter LA, McGuire DK, Wilding JPH, Ruff CT, Gause-Nilsson IAM, Fredriksson M, Johansson PA, Langkilde AM, Sabatine MS DECLARE–TIMI 58 Investigators. Dapagliflozin and Cardiovascular Outcomes in Type 2 Diabetes. N Engl J Med . 2019;380:347–357. doi: 10.1056/NEJMoa1812389. [DOI] [PubMed] [Google Scholar]
- 20.Neal B, Perkovic V, Mahaffey KW, de Zeeuw D, Fulcher G, Erondu N, Shaw W, Law G, Desai M, Matthews DR CANVAS Program Collaborative Group. Canagliflozin and Cardiovascular and Renal Events in Type 2 Diabetes. N Engl J Med . 2017;377:644–657. doi: 10.1056/NEJMoa1611925. [DOI] [PubMed] [Google Scholar]
- 21.Perkovic V, Jardine MJ, Neal B, Bompoint S, Heerspink HJL, Charytan DM, Edwards R, Agarwal R, Bakris G, Bull S, Cannon CP, Capuano G, Chu PL, de Zeeuw D, Greene T, Levin A, Pollock C, Wheeler DC, Yavin Y, Zhang H, Zinman B, Meininger G, Brenner BM, Mahaffey KW CREDENCE Trial Investigators. Canagliflozin and Renal Outcomes in Type 2 Diabetes and Nephropathy. N Engl J Med . 2019;380:2295–2306. doi: 10.1056/NEJMoa1811744. [DOI] [PubMed] [Google Scholar]
- 22.Rabizadeh S, Nakhjavani M, Esteghamati A. Cardiovascular and Renal Benefits of SGLT2 Inhibitors: A Narrative Review. Int J Endocrinol Metab . 2019;17:e84353. doi: 10.5812/ijem.84353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shenoy SV, Nagaraju SP, Bhojaraja MV, Prabhu RA, Rangaswamy D, Rao IR. Sodium-glucose cotransporter-2 inhibitors and non-steroidal mineralocorticoid receptor antagonists: Ushering in a new era of nephroprotection beyond renin-angiotensin system blockade. Nephrology (Carlton) . 2021;26:858–871. doi: 10.1111/nep.13917. [DOI] [PubMed] [Google Scholar]
- 24.Bryant NJ, Gould GW. Insulin stimulated GLUT4 translocation - Size is not everything! Curr Opin Cell Biol . 2020;65:28–34. doi: 10.1016/j.ceb.2020.02.006. [DOI] [PubMed] [Google Scholar]
- 25.Vardeny O. The Sweet Spot: Heart Failure Prevention with SGLT2 Inhibitors. Am J Med . 2020;133:182–185. doi: 10.1016/j.amjmed.2019.08.013. [DOI] [PubMed] [Google Scholar]
- 26.Nespoux J, Vallon V. Renal effects of SGLT2 inhibitors: an update. Curr Opin Nephrol Hypertens . 2020;29:190–198. doi: 10.1097/MNH.0000000000000584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weir MR, Januszewicz A, Gilbert RE, Vijapurkar U, Kline I, Fung A, Meininger G. Effect of canagliflozin on blood pressure and adverse events related to osmotic diuresis and reduced intravascular volume in patients with type 2 diabetes mellitus. J Clin Hypertens (Greenwich) . 2014;16:875–882. doi: 10.1111/jch.12425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tanaka H, Takano K, Iijima H, Kubo H, Maruyama N, Hashimoto T, Arakawa K, Togo M, Inagaki N, Kaku K. Factors Affecting Canagliflozin-Induced Transient Urine Volume Increase in Patients with Type 2 Diabetes Mellitus. Adv Ther . 2017;34:436–451. doi: 10.1007/s12325-016-0457-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Patel SM, Hickman MA, Frederich R, Lauring B, Terra S, Johnson SL, Huyck S, Mancuso JP. Evaluation of Osmotic Diuresis and Volume Depletion Events in Patients with Type 2 Diabetes Mellitus (T2DM) Receiving Ertugliflozin. Diabetes . 2018;67 [Google Scholar]
- 30.Gal A, Burton SE, Weidgraaf K, Singh P, Lopez-Villalobos N, Jacob A, Malabu U, Burchell R. The effect of the sodium-glucose cotransporter type-2 inhibitor dapagliflozin on glomerular filtration rate in healthy cats. Domest Anim Endocrinol . 2020;70:106376. doi: 10.1016/j.domaniend.2019.07.004. [DOI] [PubMed] [Google Scholar]
- 31.Ferrannini E, Baldi S, Frascerra S, Astiarraga B, Barsotti E, Clerico A, Muscelli E. Renal Handling of Ketones in Response to Sodium-Glucose Cotransporter 2 Inhibition in Patients With Type 2 Diabetes. Diabetes Care . 2017;40:771–776. doi: 10.2337/dc16-2724. [DOI] [PubMed] [Google Scholar]
- 32.Lu H, Meyer P, Hullin R. Use of SGLT2 inhibitors in cardiovascular diseases: why, when and how? Swiss Med Wkly . 2020;150:w20341. doi: 10.4414/smw.2020.20341. [DOI] [PubMed] [Google Scholar]
- 33.Kanduri SR, Kovvuru K, Hansrivijit P, Thongprayoon C, Vallabhajosyula S, Pivovarova AI, Chewcharat A, Garla V, Medaura J, Cheungpasitporn W. SGLT2 Inhibitors and Kidney Outcomes in Patients with Chronic Kidney Disease. J Clin Med . 2020;9 doi: 10.3390/jcm9092723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.O'Meara E, Verma S. When and How to Use Sodium-Glucose Cotransporter 2 Inhibitors in Patients With Heart Failure With Reduced Ejection Fraction or Chronic Kidney Disease. Can J Cardiol . 2021;37:669–673. doi: 10.1016/j.cjca.2021.01.005. [DOI] [PubMed] [Google Scholar]
- 35.Nashawi M, Sheikh O, Battisha A, Ghali A, Chilton R. Neural tone and cardio-renal outcomes in patients with type 2 diabetes mellitus: a review of the literature with a focus on SGLT2 inhibitors. Heart Fail Rev . 2021;26:643–652. doi: 10.1007/s10741-020-10046-w. [DOI] [PubMed] [Google Scholar]
- 36.Salim HM, Fukuda D, Yagi S, Soeki T, Shimabukuro M, Sata M. Glycemic Control with Ipragliflozin, a Novel Selective SGLT2 Inhibitor, Ameliorated Endothelial Dysfunction in Streptozotocin-Induced Diabetic Mouse. Front Cardiovasc Med . 2016;3:43. doi: 10.3389/fcvm.2016.00043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Abdul-Ghani M, Del Prato S, Chilton R, DeFronzo RA. SGLT2 Inhibitors and Cardiovascular Risk: Lessons Learned From the EMPA-REG OUTCOME Study. Diabetes Care . 2016;39:717–725. doi: 10.2337/dc16-0041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sawada T, Uzu K, Hashimoto N, Onishi T, Takaya T, Shimane A, Taniguchi Y, Yasaka Y, Ohara T, Kawai H. Empagliflozin's Ameliorating Effect on Plasma Triglycerides: Association with Endothelial Function Recovery in Diabetic Patients with Coronary Artery Disease. J Atheroscler Thromb . 2020;27:644–656. doi: 10.5551/jat.50807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thrailkill KM, Clay Bunn R, Nyman JS, Rettiganti MR, Cockrell GE, Wahl EC, Uppuganti S, Lumpkin CK Jr, Fowlkes JL. SGLT2 inhibitor therapy improves blood glucose but does not prevent diabetic bone disease in diabetic DBA/2J male mice. Bone . 2016;82:101–107. doi: 10.1016/j.bone.2015.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Adil M, Khan RA, Kalam A, Venkata SK, Kandhare AD, Ghosh P, Sharma M. Effect of anti-diabetic drugs on bone metabolism: Evidence from preclinical and clinical studies. Pharmacol Rep . 2017;69:1328–1340. doi: 10.1016/j.pharep.2017.05.008. [DOI] [PubMed] [Google Scholar]
- 41.Thrailkill KM, Bunn RC, Uppuganti S, Ray P, Popescu I, Kalaitzoglou E, Fowlkes JL, Nyman JS. Canagliflozin, an SGLT2 inhibitor, corrects glycemic dysregulation in TallyHO model of T2D but only partially prevents bone deficits. Bone . 2020;141:115625. doi: 10.1016/j.bone.2020.115625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bilezikian JP, Watts NB, Usiskin K, Polidori D, Fung A, Sullivan D, Rosenthal N. Evaluation of Bone Mineral Density and Bone Biomarkers in Patients With Type 2 Diabetes Treated With Canagliflozin. J Clin Endocrinol Metab . 2016;101:44–51. doi: 10.1210/jc.2015-1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fralick M, Kim SC, Schneeweiss S, Kim D, Redelmeier DA, Patorno E. Fracture Risk After Initiation of Use of Canagliflozin. Ann Intern Med . 2019;171:80. doi: 10.7326/L19-0320. [DOI] [PubMed] [Google Scholar]
- 44.Zhou Z, Jardine M, Perkovic V, Matthews DR, Mahaffey KW, de Zeeuw D, Fulcher G, Desai M, Oh R, Simpson R, Watts NB, Neal B. Canagliflozin and fracture risk in individuals with type 2 diabetes: results from the CANVAS Program. Diabetologia . 2019;62:1854–1867. doi: 10.1007/s00125-019-4955-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang D, Bi X, Liu Y, Huang Y, Xiong J, Xu X, Xiao T, Yu Y, Jiang W, Zhang J, Zhang B, Zhao J. High Phosphate-Induced Calcification of Vascular Smooth Muscle Cells is Associated with the TLR4/NF-κb Signaling Pathway. Kidney Blood Press Res . 2017;42:1205–1215. doi: 10.1159/000485874. [DOI] [PubMed] [Google Scholar]
- 46.Voelkl J, Egli-Spichtig D, Alesutan I, Wagner CA. Inflammation: a putative link between phosphate metabolism and cardiovascular disease. Clin Sci (Lond) . 2021;135:201–227. doi: 10.1042/CS20190895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tonelli M, Sacks F, Pfeffer M, Gao Z, Curhan G Cholesterol And Recurrent Events Trial Investigators. Relation between serum phosphate level and cardiovascular event rate in people with coronary disease. Circulation . 2005;112:2627–2633. doi: 10.1161/CIRCULATIONAHA.105.553198. [DOI] [PubMed] [Google Scholar]
- 48.Wang P, Zhou P, Chen W, Peng D. Combined effects of hyperphosphatemia and hyperglycemia on the calcification of cultured human aortic smooth muscle cells. Exp Ther Med . 2019;17:863–868. doi: 10.3892/etm.2018.7024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brown RB. Diabetes, Diabetic Complications, and Phosphate Toxicity: A Scoping Review. Curr Diabetes Rev . 2020;16:674–689. doi: 10.2174/1573399815666191104113236. [DOI] [PubMed] [Google Scholar]
- 50.Blau JE, Bauman V, Conway EM, Piaggi P, Walter MF, Wright EC, Bernstein S, Courville AB, Collins MT, Rother KI, Taylor SI. Canagliflozin triggers the FGF23/1,25-dihydroxyvitamin D/PTH axis in healthy volunteers in a randomized crossover study. JCI Insight . 2018;3 doi: 10.1172/jci.insight.99123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.de Jong MA, Petrykiv SI, Laverman GD, van Herwaarden AE, de Zeeuw D, Bakker SJL, Heerspink HJL, de Borst MH. Effects of Dapagliflozin on Circulating Markers of Phosphate Homeostasis. Clin J Am Soc Nephrol . 2019;14:66–73. doi: 10.2215/CJN.04530418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Moon H, Chin HJ, Na KY, Joo KW, Kim YS, Kim S, Han SS. Hyperphosphatemia and risks of acute kidney injury, end-stage renal disease, and mortality in hospitalized patients. BMC Nephrol . 2019;20:362. doi: 10.1186/s12882-019-1556-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ortega B, Dey JM, Gardella AR, Proano J, Vaneerde D. Antibody-mediated inhibition of EGFR reduces phosphate excretion and induces hyperphosphatemia and mild hypomagnesemia in mice. Physiol Rep . 2017;5 doi: 10.14814/phy2.13176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Barzilay JI, Davis BR, Ghosh A, Pressel SL, Rahman M, Einhorn PT, Cushman WC, Whelton PK, Wright JT Jr ALLHAT Collaborative Research Group. Rapid eGFR change as a determinant of cardiovascular and renal disease outcomes and of mortality in hypertensive adults with and without type 2 diabetes. J Diabetes Complications . 2018;32:830–832. doi: 10.1016/j.jdiacomp.2018.07.003. [DOI] [PubMed] [Google Scholar]
- 55.Sim JJ, Bhandari SK, Smith N, Chung J, Liu IL, Jacobsen SJ, Kalantar-Zadeh K. Phosphorus and risk of renal failure in subjects with normal renal function. Am J Med . 2013;126:311–318. doi: 10.1016/j.amjmed.2012.08.018. [DOI] [PubMed] [Google Scholar]
- 56.O'Seaghdha CM, Hwang SJ, Muntner P, Melamed ML, Fox CS. Serum phosphorus predicts incident chronic kidney disease and end-stage renal disease. Nephrol Dial Transplant . 2011;26:2885–2890. doi: 10.1093/ndt/gfq808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gupta A, Winer K, Econs MJ, Marx SJ, Collins MT. FGF-23 is elevated by chronic hyperphosphatemia. J Clin Endocrinol Metab . 2004;89:4489–4492. doi: 10.1210/jc.2004-0724. [DOI] [PubMed] [Google Scholar]
- 58.McKenna MJ, Crowley RK, Twomey PJ, Kilbane MT. Renal Phosphate Handling: Independent Effects of Circulating FGF23, PTH, and Calcium. JBMR Plus . 2021;5:e10437. doi: 10.1002/jbm4.10437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ärnlöv J, Carlsson AC, Sundström J, Ingelsson E, Larsson A, Lind L, Larsson TE. Serum FGF23 and risk of cardiovascular events in relation to mineral metabolism and cardiovascular pathology. Clin J Am Soc Nephrol . 2013;8:781–786. doi: 10.2215/CJN.09570912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lu X, Hu MC. Klotho/FGF23 Axis in Chronic Kidney Disease and Cardiovascular Disease. Kidney Dis (Basel) . 2017;3:15–23. doi: 10.1159/000452880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vogt I, Haffner D, Leifheit-Nestler M. FGF23 and Phosphate-Cardiovascular Toxins in CKD. Toxins (Basel) . 2019;11 doi: 10.3390/toxins11110647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Silswal N, Touchberry CD, Daniel DR, McCarthy DL, Zhang S, Andresen J, Stubbs JR, Wacker MJ. FGF23 directly impairs endothelium-dependent vasorelaxation by increasing superoxide levels and reducing nitric oxide bioavailability. Am J Physiol Endocrinol Metab . 2014;307:E426–E436. doi: 10.1152/ajpendo.00264.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zinman B, Inzucchi SE, Lachin JM, Wanner C, Ferrari R, Fitchett D, Bluhmki E, Hantel S, Kempthorne-Rawson J, Newman J, Johansen OE, Woerle HJ, Broedl UC. Rationale, design, and baseline characteristics of a randomized, placebo-controlled cardiovascular outcome trial of empagliflozin (EMPA-REG OUTCOME™) Cardiovasc Diabetol . 2014;13:102. doi: 10.1186/1475-2840-13-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Inzucchi SE, Zinman B, Fitchett D, Wanner C, Ferrannini E, Schumacher M, Schmoor C, Ohneberg K, Johansen OE, George JT, Hantel S, Bluhmki E, Lachin JM. How Does Empagliflozin Reduce Cardiovascular Mortality? Diabetes Care . 2018;41:356–363. doi: 10.2337/dc17-1096. [DOI] [PubMed] [Google Scholar]
- 65.Pirklbauer M, Schupart R, Fuchs L, Staudinger P, Corazza U, Sallaberger S, Leierer J, Mayer G, Schramek H. Unraveling reno-protective effects of SGLT2 inhibition in human proximal tubular cells. Am J Physiol Renal Physiol . 2019;316:F449–F462. doi: 10.1152/ajprenal.00431.2018. [DOI] [PubMed] [Google Scholar]
- 66.Yanai H, Katsuyayama H. A Possible Mechanism for Renoprotective Effect of Sodium-Glucose Cotransporter 2 Inhibitor: Elevation of Erythropoietin Production. J Clin Med Res . 2017;9:178–179. doi: 10.14740/jocmr2857w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Maruyama T, Takashima H, Oguma H, Nakamura Y, Ohno M, Utsunomiya K, Furukawa T, Tei R, Abe M. Canagliflozin Improves Erythropoiesis in Diabetes Patients with Anemia of Chronic Kidney Disease. Diabetes Technol Ther . 2019;21:713–720. doi: 10.1089/dia.2019.0212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lambers Heerspink HJ, de Zeeuw D, Wie L, Leslie B, List J. Dapagliflozin a glucose-regulating drug with diuretic properties in subjects with type 2 diabetes. Diabetes Obes Metab . 2013;15:853–862. doi: 10.1111/dom.12127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chen J, Williams S, Ho S, Loraine H, Hagan D, Whaley JM, Feder JN. Quantitative PCR tissue expression profiling of the human SGLT2 gene and related family members. Diabetes Ther . 2010;1:57–92. doi: 10.1007/s13300-010-0006-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bross R, Shah A, Kopple JD. Nutritional Aspects of Phosphorus Compounds in Foods, in Clinical Aspects of Natural and Added Phosphorus in Foods. Springer 2017: 77-97. [Google Scholar]
- 71.Brown RB, Razzaque MS. Phosphate toxicity and tumorigenesis. Biochim Biophys Acta Rev Cancer . 2018;1869:303–309. doi: 10.1016/j.bbcan.2018.04.007. [DOI] [PubMed] [Google Scholar]
- 72.Erem S, Razzaque MS. Dietary phosphate toxicity: an emerging global health concern. Histochem Cell Biol . 2018;150:711–719. doi: 10.1007/s00418-018-1711-8. [DOI] [PubMed] [Google Scholar]
- 73.Ohnishi M, Razzaque MS. Dietary and genetic evidence for phosphate toxicity accelerating mammalian aging. FASEB J . 2010;24:3562–3571. doi: 10.1096/fj.09-152488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.DeFronzo RA, Goldberg M, Agus ZS. The effects of glucose and insulin on renal electrolyte transport. J Clin Invest . 1976;58:83–90. doi: 10.1172/JCI108463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Petersen KF, Shulman GI. Etiology of insulin resistance. Am J Med . 2006;119:S10–S16. doi: 10.1016/j.amjmed.2006.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bhella PS, Prasad A, Heinicke K, Hastings JL, Arbab-Zadeh A, Adams-Huet B, Pacini EL, Shibata S, Palmer MD, Newcomer BR, Levine BD. Abnormal haemodynamic response to exercise in heart failure with preserved ejection fraction. Eur J Heart Fail . 2011;13:1296–1304. doi: 10.1093/eurjhf/hfr133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Haseler LJ, Hogan MC, Richardson RS. Skeletal muscle phosphocreatine recovery in exercise-trained humans is dependent on O2 availability. J Appl Physiol (1985) . 1999;86:2013–2018. doi: 10.1152/jappl.1999.86.6.2013. [DOI] [PubMed] [Google Scholar]
- 78.Ripley EM, Clarke GD, Hamidi V, Martinez RA, Settles FD, Solis C, Deng S, Abdul-Ghani M, Tripathy D, DeFronzo RA. Reduced skeletal muscle phosphocreatine concentration in type 2 diabetic patients: a quantitative image-based phosphorus-31 MR spectroscopy study. Am J Physiol Endocrinol Metab . 2018;315:E229–E239. doi: 10.1152/ajpendo.00426.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Vervloet MG, van Ballegooijen AJ. Prevention and treatment of hyperphosphatemia in chronic kidney disease. Kidney Int . 2018;93:1060–1072. doi: 10.1016/j.kint.2017.11.036. [DOI] [PubMed] [Google Scholar]
- 80.Moe SM, Chen NX. Mechanisms of vascular calcification in chronic kidney disease. J Am Soc Nephrol . 2008;19:213–216. doi: 10.1681/ASN.2007080854. [DOI] [PubMed] [Google Scholar]
- 81.Kaufman DP, Basit H, Knohl SJ. Physiology, Glomerular Filtration Rate. 2021 Jul 22. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2021 Jan- [PubMed] [Google Scholar]
- 82.Ku E, Lee BJ, Wei J, Weir MR. Hypertension in CKD: Core Curriculum 2019. Am J Kidney Dis . 2019;74:120–131. doi: 10.1053/j.ajkd.2018.12.044. [DOI] [PubMed] [Google Scholar]
- 83.Sata Y, Head GA, Denton K, May CN, Schlaich MP. Role of the Sympathetic Nervous System and Its Modulation in Renal Hypertension. Front Med (Lausanne) . 2018;5:82. doi: 10.3389/fmed.2018.00082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Frame A, Wainford R. The role of the mechanosensitive renal sensory afferent nerve sympathoinhibitory reno-renal reflex in sympathetic outflow, natriuresis, and blood pressure regulation. FASEB J . 2017;31:718–718. [Google Scholar]
- 85.Tanaka S, Okusa MD. Crosstalk between the nervous system and the kidney. Kidney Int . 2020;97:466–476. doi: 10.1016/j.kint.2019.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kopp UC, Smith LA, DiBona GF. Renorenal reflexes: neural components of ipsilateral and contralateral renal responses. Am J Physiol . 1985;249:F507–F517. doi: 10.1152/ajprenal.1985.249.4.F507. [DOI] [PubMed] [Google Scholar]
- 87.Okusa MD, Rosin DL, Tracey KJ. Targeting neural reflex circuits in immunity to treat kidney disease. Nat Rev Nephrol . 2017;13:669–680. doi: 10.1038/nrneph.2017.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lovshin JA, Gilbert RE. Are SGLT2 inhibitors reasonable antihypertensive drugs and renoprotective? Curr Hypertens Rep . 2015;17:551. doi: 10.1007/s11906-015-0551-3. [DOI] [PubMed] [Google Scholar]
- 89.Carbone S, Dixon DL. The CANVAS Program: implications of canagliflozin on reducing cardiovascular risk in patients with type 2 diabetes mellitus. Cardiovasc Diabetol . 2019;18:64. doi: 10.1186/s12933-019-0869-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sakaguchi Y, Fujii N, Shoji T, Hayashi T, Rakugi H, Iseki K, Tsubakihara Y, Isaka Y Committee of Renal Data Registry of the Japanese Society for Dialysis Therapy. Magnesium modifies the cardiovascular mortality risk associated with hyperphosphatemia in patients undergoing hemodialysis: a cohort study. PLoS One . 2014;9:e116273. doi: 10.1371/journal.pone.0116273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tang H, Zhang X, Zhang J, Li Y, Del Gobbo LC, Zhai S, Song Y. Elevated serum magnesium associated with SGLT2 inhibitor use in type 2 diabetes patients: a meta-analysis of randomised controlled trials. Diabetologia . 2016;59:2546–2551. doi: 10.1007/s00125-016-4101-6. [DOI] [PubMed] [Google Scholar]
- 92.Nguyen TT, Quan X, Hwang KH, Xu S, Das R, Choi SK, Wiederkehr A, Wollheim CB, Cha SK, Park KS. Mitochondrial oxidative stress mediates high-phosphate-induced secretory defects and apoptosis in insulin-secreting cells. Am J Physiol Endocrinol Metab . 2015;308:E933–E941. doi: 10.1152/ajpendo.00009.2015. [DOI] [PubMed] [Google Scholar]
- 93.Ruan L, Chen W, Srinivasan SR, Xu J, Toprak A, Berenson GS. Relation of serum phosphorus levels to carotid intima-media thickness in asymptomatic young adults (from the Bogalusa Heart Study) Am J Cardiol . 2010;106:793–797. doi: 10.1016/j.amjcard.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Di Marco GS, Hausberg M, Hillebrand U, Rustemeyer P, Wittkowski W, Lang D, Pavenstädt H. Increased inorganic phosphate induces human endothelial cell apoptosis in vitro. Am J Physiol Renal Physiol . 2008;294:F1381–F1387. doi: 10.1152/ajprenal.00003.2008. [DOI] [PubMed] [Google Scholar]
- 95.Di Marco GS, König M, Stock C, Wiesinger A, Hillebrand U, Reiermann S, Reuter S, Amler S, Köhler G, Buck F, Fobker M, Kümpers P, Oberleithner H, Hausberg M, Lang D, Pavenstädt H, Brand M. High phosphate directly affects endothelial function by downregulating annexin II. Kidney Int . 2013;83:213–222. doi: 10.1038/ki.2012.300. [DOI] [PubMed] [Google Scholar]
- 96.Ix JH, De Boer IH, Peralta CA, Adeney KL, Duprez DA, Jenny NS, Siscovick DS, Kestenbaum BR. Serum phosphorus concentrations and arterial stiffness among individuals with normal kidney function to moderate kidney disease in MESA. Clin J Am Soc Nephrol . 2009;4:609–615. doi: 10.2215/CJN.04100808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zuo L, Chuang CC, Hemmelgarn BT, Best TM. Heart failure with preserved ejection fraction: Defining the function of ROS and NO. J Appl Physiol (1985) . 2015;119:944–951. doi: 10.1152/japplphysiol.01149.2014. [DOI] [PubMed] [Google Scholar]
- 98.Vazzana N, Ranalli P, Cuccurullo C, Davì G. Diabetes mellitus and thrombosis. Thromb Res . 2012;129:371–377. doi: 10.1016/j.thromres.2011.11.052. [DOI] [PubMed] [Google Scholar]
- 99.Tse G, Yan BP, Chan YW, Tian XY, Huang Y. Reactive Oxygen Species, Endoplasmic Reticulum Stress and Mitochondrial Dysfunction: The Link with Cardiac Arrhythmogenesis. Front Physiol . 2016;7:313. doi: 10.3389/fphys.2016.00313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sato T, Miki T, Ohnishi H, Yamashita T, Takada A, Yano T, Tanno M, Tsuchida A, Miura T. Effect of sodium-glucose co-transporter-2 inhibitors on impaired ventricular repolarization in people with Type 2 diabetes. Diabet Med . 2017;34:1367–1371. doi: 10.1111/dme.13424. [DOI] [PubMed] [Google Scholar]
- 101.Jeong EM, Liu M, Sturdy M, Gao G, Varghese ST, Sovari AA, Dudley SC Jr. Metabolic stress, reactive oxygen species, and arrhythmia. J Mol Cell Cardiol . 2012;52:454–463. doi: 10.1016/j.yjmcc.2011.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Li H, Shin SE, Seo MS, An JR, Choi IW, Jung WK, Firth AL, Lee DS, Yim MJ, Choi G, Lee JM, Na SH, Park WS. The anti-diabetic drug dapagliflozin induces vasodilation via activation of PKG and Kv channels. Life Sci . 2018;197:46–55. doi: 10.1016/j.lfs.2018.01.032. [DOI] [PubMed] [Google Scholar]
- 103.He P, Mann-Collura O, Fling J, Edara N, Razzaque MS. Elevated phosphate mediates extensive cellular toxicity: from abnormal proliferation to excessive cell death. bioRxiv . 2020 [Google Scholar]
- 104.Malik M, Batchvarov VN. Measurement, interpretation and clinical potential of QT dispersion. J Am Coll Cardiol . 2000;36:1749–1766. doi: 10.1016/s0735-1097(00)00962-1. [DOI] [PubMed] [Google Scholar]
- 105.Yale JF, Bakris G, Cariou B, Yue D, David-Neto E, Xi L, Figueroa K, Wajs E, Usiskin K, Meininger G. Efficacy and safety of canagliflozin in subjects with type 2 diabetes and chronic kidney disease. Diabetes Obes Metab . 2013;15:463–473. doi: 10.1111/dom.12090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Yale JF, Bakris G, Cariou B, Nieto J, David-Neto E, Yue D, Wajs E, Figueroa K, Jiang J, Law G, Usiskin K, Meininger G DIA3004 Study Group. Efficacy and safety of canagliflozin over 52 weeks in patients with type 2 diabetes mellitus and chronic kidney disease. Diabetes Obes Metab . 2014;16:1016–1027. [Google Scholar]
- 107.Del Gobbo LC, Imamura F, Wu JH, de Oliveira Otto MC, Chiuve SE, Mozaffarian D. Circulating and dietary magnesium and risk of cardiovascular disease: a systematic review and meta-analysis of prospective studies. Am J Clin Nutr . 2013;98:160–173. doi: 10.3945/ajcn.112.053132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Sakaguchi Y, Hamano T, Isaka Y. Effects of Magnesium on the Phosphate Toxicity in Chronic Kidney Disease: Time for Intervention Studies. Nutrients . 2017;9 doi: 10.3390/nu9020112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Fischer PW, Giroux A. Effects of dietary magnesium on sodium-potassium pump action in the heart of rats. J Nutr . 1987;117:2091–2095. doi: 10.1093/jn/117.12.2091. [DOI] [PubMed] [Google Scholar]
- 110.DiNicolantonio JJ, O'Keefe JH, Wilson W. Subclinical magnesium deficiency: a principal driver of cardiovascular disease and a public health crisis. Open Heart . 2018;5:e000668. doi: 10.1136/openhrt-2017-000668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sato T, Miki T, Furukawa S, Matsuura B, Hiasa Y, Ohnishi H, Tanno M, Miura T. Longitudinal impact of dapagliflozin treatment on ventricular repolarization heterogeneity in patients with type 2 diabetes. J Diabetes Investig . 2019;10:1593–1594. doi: 10.1111/jdi.13063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Raghav A, Ahmad J. Glycated albumin in chronic kidney disease: Pathophysiologic connections. Diabetes Metab Syndr . 2018;12:463–468. doi: 10.1016/j.dsx.2018.01.002. [DOI] [PubMed] [Google Scholar]
- 113.Piuri G, Zocchi M, Della Porta M, Ficara V, Manoni M, Zuccotti GV, Pinotti L, Maier JA, Cazzola R. Magnesium in Obesity, Metabolic Syndrome, and Type 2 Diabetes. Nutrients . 2021;13 doi: 10.3390/nu13020320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Curry JN, Yu ASL. Magnesium Handling in the Kidney. Adv Chronic Kidney Dis . 2018;25:236–243. doi: 10.1053/j.ackd.2018.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Franken GAC, Adella A, Bindels RJM, de Baaij JHF. Mechanisms coupling sodium and magnesium reabsorption in the distal convoluted tubule of the kidney. Acta Physiol (Oxf) . 2021;231:e13528. doi: 10.1111/apha.13528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kostov K. Effects of Magnesium Deficiency on Mechanisms of Insulin Resistance in Type 2 Diabetes: Focusing on the Processes of Insulin Secretion and Signaling. Int J Mol Sci . 2019;20 doi: 10.3390/ijms20061351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Tinawi M. Disorders of Calcium Metabolism: Hypocalcemia and Hypercalcemia. Cureus . 2021;13:e12420. doi: 10.7759/cureus.12420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Grandi E, Pasqualini FS, Pes C, Corsi C, Zaza A, Severi S. Theoretical investigation of action potential duration dependence on extracellular Ca2+ in human cardiomyocytes. J Mol Cell Cardiol . 2009;46:332–342. doi: 10.1016/j.yjmcc.2008.12.002. [DOI] [PubMed] [Google Scholar]
- 119.Zühlke RD, Pitt GS, Deisseroth K, Tsien RW, Reuter H. Calmodulin supports both inactivation and facilitation of L-type calcium channels. Nature . 1999;399:159–162. doi: 10.1038/20200. [DOI] [PubMed] [Google Scholar]
- 120.Ye Y, Zhao C, Liang J, Yang Y, Yu M, Qu X. Effect of Sodium-Glucose Co-transporter 2 Inhibitors on Bone Metabolism and Fracture Risk. Front Pharmacol . 2018;9:1517. doi: 10.3389/fphar.2018.01517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Erythropoulou-Kaltsidou A, Polychronopoulos G, Tziomalos K. Sodium-Glucose Co-Transporter 2 Inhibitors and Fracture Risk. Diabetes Ther . 2020;11:7–14. doi: 10.1007/s13300-019-00724-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Burnett SM, Gunawardene SC, Bringhurst FR, Jüppner H, Lee H, Finkelstein JS. Regulation of C-terminal and intact FGF-23 by dietary phosphate in men and women. J Bone Miner Res . 2006;21:1187–1196. doi: 10.1359/jbmr.060507. [DOI] [PubMed] [Google Scholar]
- 123.Weber TJ, Quarles LD. Molecular Control of Phosphorus Homeostasis and Precision Treatment of Hypophosphatemic Disorders. Curr Mol Biol Rep . 2019;5:75–85. doi: 10.1007/s40610-019-0118-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Leifheit-Nestler M, Haffner D. Paracrine Effects of FGF23 on the Heart. Front Endocrinol (Lausanne) . 2018;9:278. doi: 10.3389/fendo.2018.00278. [DOI] [PMC free article] [PubMed] [Google Scholar]