

Abstract
Inhibitors of transcriptional protein–protein interactions (PPIs) have high value both as tools and for therapeutic applications. The PPI network mediated by the transcriptional coactivator Med25, for example, regulates stress-response and motility pathways, and dysregulation of the PPI networks contributes to oncogenesis and metastasis. The canonical transcription factor binding sites within Med25 are large (~900 Å2) and have little topology, and thus, they do not present an array of attractive small-molecule binding sites for inhibitor discovery. Here we demonstrate that the depsidone natural product norstictic acid functions through an alternative binding site to block Med25–transcriptional activator PPIs in vitro and in cell culture. Norstictic acid targets a binding site comprising a highly dynamic loop flanking one canonical binding surface, and in doing so, it both orthosterically and allosterically alters Med25-driven transcription in a patient-derived model of triple-negative breast cancer. These results highlight the potential of Med25 as a therapeutic target as well as the inhibitor discovery opportunities presented by structurally dynamic loops within otherwise challenging proteins.
Transcriptional coactivators play an integral role in the regulation of gene expression, serving as hub proteins for transcriptional machinery assembly through interactions with transcriptional activators.1–9 Alterations in the network of coactivator–activator protein–protein interactions (PPIs) contribute to the onset and perpetuation of numerous diseases, leading to significant interest in synthetic probes for mechanistic studies and therapeutic applications.10–16 This is especially true for coactivators whose function is highly context-dependent, required only for a subset of genes or only at particular times or locations in the life cycle of an organism.17 Thus, synthetic modulation of such coactivators would also be context-dependent, providing an advantageous layer of specificity. Recent structural and functional studies of the coactivator Med25 indicate that it falls into this category (Figure 1A); homozygous deletion of Med25, for example, is nonlethal, impacting approximately 900 genes.18 Several lines of evidence indicate that dysregulation of the PPI network of Med25 and the ETV/PEA3 transcriptional activators contributes to oncogenesis as well as metastatic phenotypes in certain breast and prostate cancers, heightening a need for Med25-selective inhibitors.19–22 Here we report the discovery of the natural product norstictic acid as the first such inhibitor.
Figure 1.
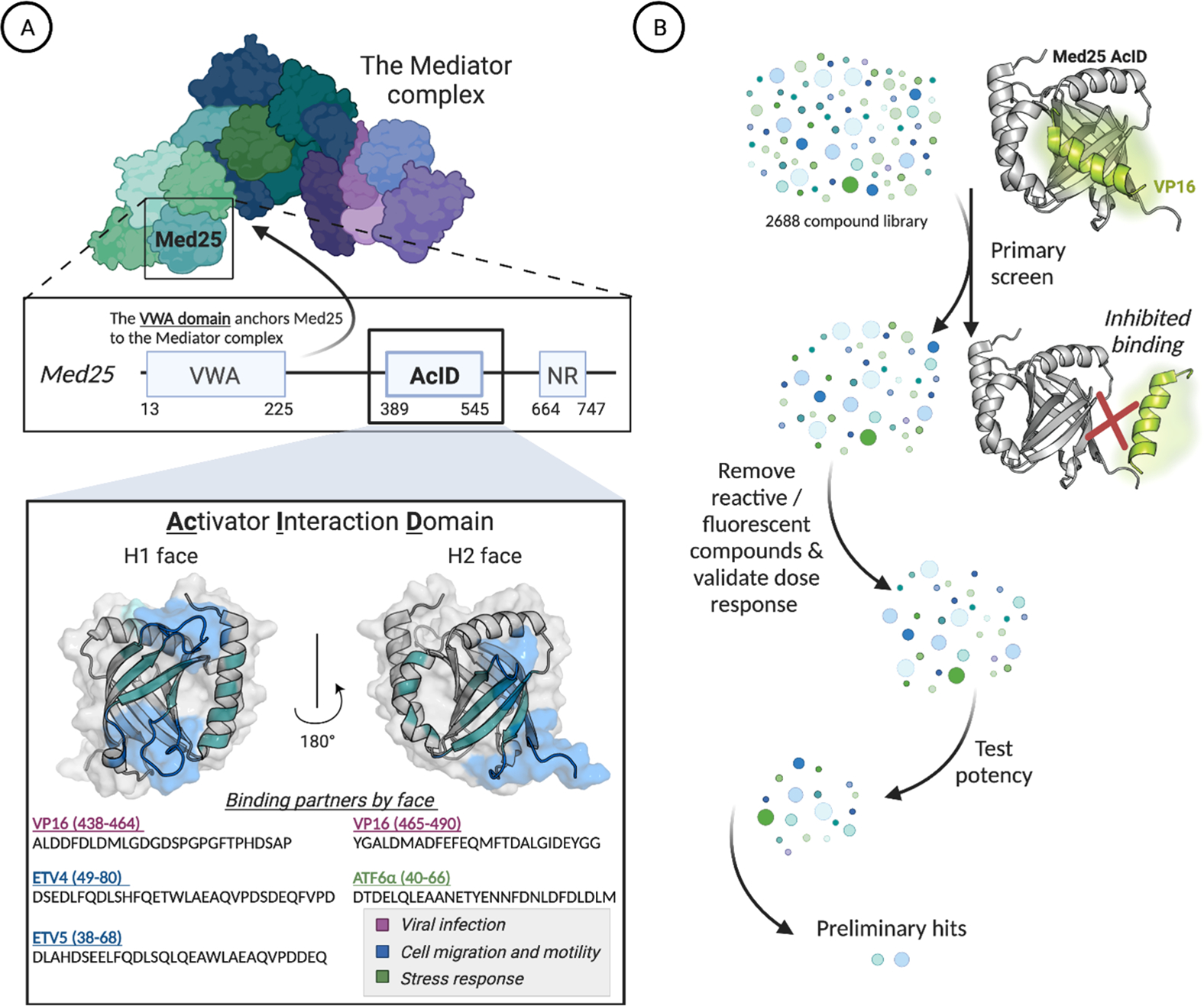
(A) The hub protein Med25 is a subunit of the Mediator coactivator complex. The activator interaction domain (AcID) forms a PPI network with transcriptional activators using two binding surfaces (H1 and H2) and in doing so regulates key cellular processes.19,22–25 PDB entry 2XNF was used to generate figure. (B) Schematic of the high-throughput screen to identify inhibitors of the Med25 AcID PPI network. See the Supporting Information for full screening details.
The domain of Med25 that interacts with activators is the activator interaction domain (AcID) (Figure 1A).19,22–28 This domain contains a seven-stranded β-barrel core flanked with dynamic loops and three α-helices. Like other activator-binding domains (ABDs) within coactivators, AcID does not contain defined binding pockets but rather relies on hydrophobic interfaces, termed H1 and H2, to interact with transcription factors.23,25 These qualities make AcID as well as other coactivators, challenging to target selectively, particularly in an orthosteric mode.17,29 We recently reported that the dynamic substructures within AcID contain allosteric hotspots that regulate cooperativity and selectivity in binding.20,28 Thus, we hypothesized that these substructures, largely loop regions, represent an opportunity for allosteric inhibition. Furthermore, because such substructures are more likely to access conformations with topologically unique binding surfaces, one might anticipate that small-molecule modulators of such sites would exhibit enhanced selectivity compared with purely orthosteric ligands.12,30–34
To identify inhibitors of Med25 AcID, we utilized a high-throughput fluorescence polarization (FP) assay to interrogate a complex of AcID and fluorescein-tagged VP16(465–490) (Figures 1B and S1). As previously reported, this VP16 sequence contains the minimal binding sequence for interaction with AcID (KD = 0.60 ± 0.06 μM) and interacts with the H1 and H2 binding surfaces.20,23 Several commercially available libraries (MS Spectrum 2000, Focused Collections, and BioFocus NCC libraries) with a combined total of 4046 compounds were screened using this format (Z′ = 0.87, 1.6% hit rate; see the Supporting Information for additional details). Compounds with activity >3 SD relative to dimethyl sulfoxide (DMSO) as the negative control were subjected to dose–response assessment with freshly purchased material. Secondary assays against a suite of related PPIs were used to enrich the hit pool for selectivity. From this, the lichen-derived natural products norstictic acid (NA) and psoromic acid (PA) emerged as the best inhibitors, with apparent IC50 values of 2.3 ± 0.1 μM and 3.9 ± 0.3 μM, respectively (Figure 2A,B).
Figure 2.
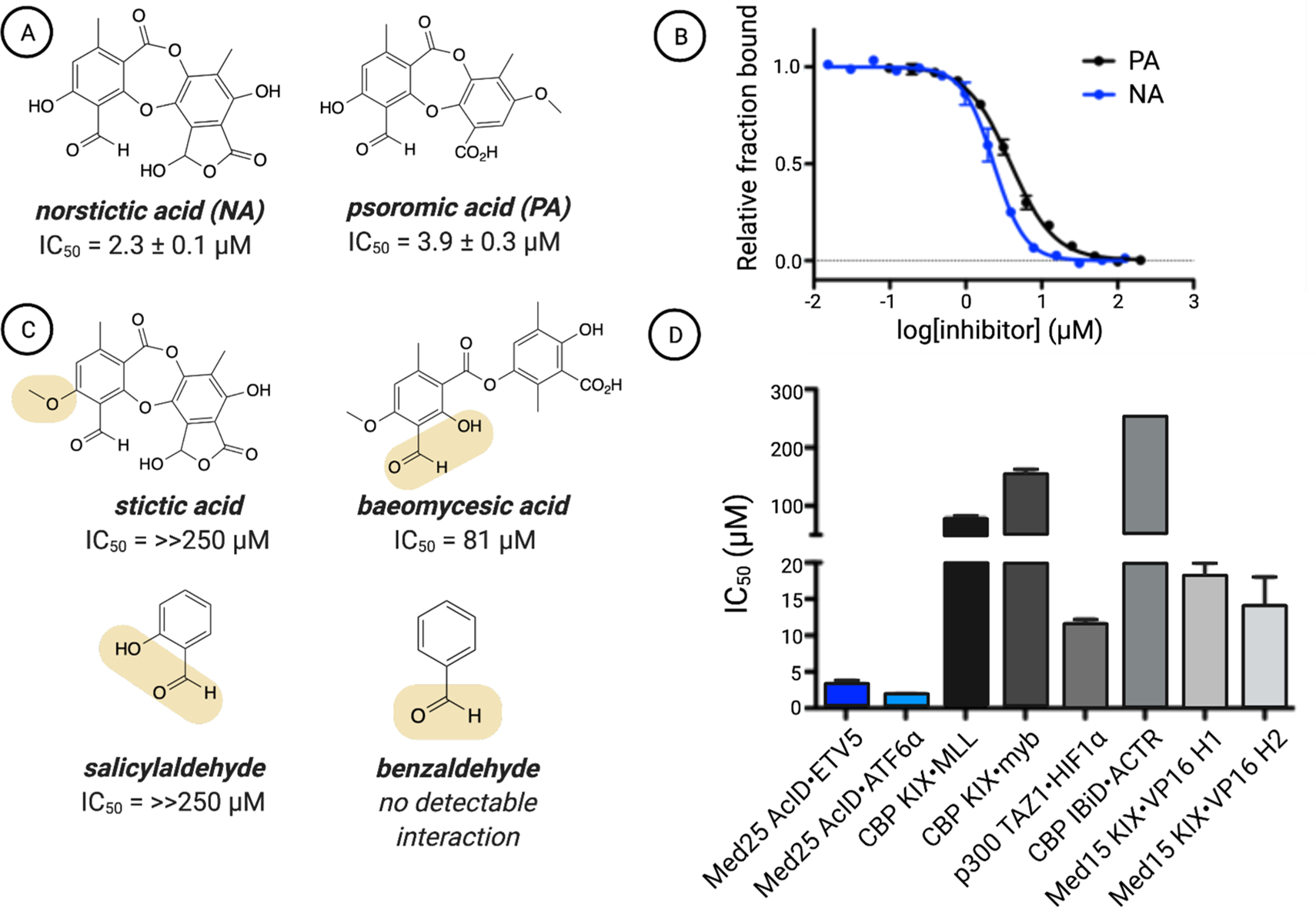
(A) Chemical structures of the top two hits emerging from the screen of Med25 AcID fl-VP16(465–490) along with (B) their apparent IC50 values, which were determined through titrations of either NA or PA against Med25 AcID fl-VP16(465–490) performed in triplicate with the indicated error (SDOM). Full experimental details are reported in the Supporting Information. (C) Assessment of related structures shows that the orthophenoxyaldehyde moiety is important but not sufficient for inhibitory activity. IC50 values were determined via competition fluorescence polarization against Med25 VP16(465–490). Of these molecules, covalent adducts with Med25 were observed only for baeomycesic acid, indicating that the orthophenoxyl group is integral to stable imine formation. (D) Inhibition of related PPI networks by NA. Apparent IC50 values were measured via fluorescence polarization against a suite of coactivator domains (CBP KIX, p300 TAZ1, CBP IBiD, Med15 KIX) bound to fluorescein-tagged activators. The values are averages of three independent experiments with the indicated error (SDOM). No error bars are shown for the IC50 against IBiD ACTR because the IC50 was greater than the highest concentration of NA tested (250 μM), and thus, we can accurately report the IC50 only as >250 μM. Full details are reported in the Supporting Information.
Both NA and PA are natural products in the depsidone family containing an orthophenolic aldehyde moiety. The presence of a reactive aldehyde functionality suggested a potential covalent mechanism of action, for example, via imine formation with lysine side chains.35 Consistent with this hypothesis, analysis of NA-treated Med25 AcID using mass spectrometry showed the presence of concentration-dependent covalent adduct(s) (Figure S2). Treatment with the reducing agent NaBH4 led to incorporation of H2 into the adduct, indicating initial formation of a Schiff base followed by reduction (Figure S3). Data from a time-course experiment revealed that significant inhibition was observed at 5 min, with full activity after 30 min (Figure S4). An examination of related structures indicated that the orthophenolic aldehyde is necessary but not sufficient for interaction with Med25 AcID or for inhibitory activity. Stictic acid, in which the phenol is masked as a methyl ether, inhibits Med25 interactions poorly (IC50 > 250 μM) (Figure 2C). Additionally, salicylaldehyde efficiently labels Med25 AcID but does not impact binding of activators (Figure S5). These data suggest that non-covalent interactions play essential roles in the inhibitor function of NA. Consistent with this, NA exhibits remarkable selectivity for Med25 PPIs relative to other coactivators with similar binding surfaces (Figure 2D).36 Notably, NA inhibits Med25 PPIs at both binding surfaces, including those formed with the transcriptional activators ETV5 (H1 binding surface) and ATF6α (H2 binding surface).
Several lines of evidence suggested that the engagement site of NA is a lysine-rich dynamic loop that borders the H2 binding surface (Figure 3A). There are 11 lysine residues within Med25 AcID, six of which are found on dynamic loop regions flanking the two known activator binding surfaces. The binding of the cognate transcriptional activator binding partners was largely unaffected by the replacement of these lysines with arginine either alone or in combination (Figures S6 and S7). Similarly, mutations within the H1 binding surface had minimal impact on both NA binding as determined by mass spectrometric analysis and inhibition in an in vitro binding assay (Figures 3A,B and S8). In contrast, mutation of K519 had a profound effect on NA binding and inhibition. This residue is part of a lysine-rich dynamic loop that flanks the H2 face, and the mutational data indicate that NA can also interact with K520 and K518 within this loop.
Figure 3.
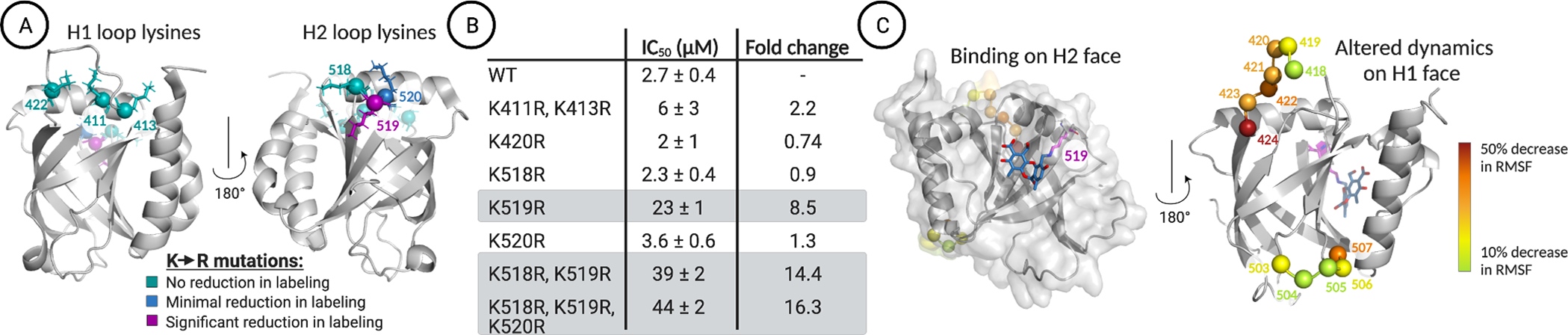
(A) LC–MS analysis of norstictic acid covalent adduct formation with Med25 lysine-to-arginine mutants indicates that K519R leads to the most significant reduction of labeling. No reduction of labeling corresponds to a decrease in abundance of the NA covalent adduct of less than 10%. Minimal reduction in labeling, observed for K520R, corresponds to a 22% reduction in the mass abundance of the NA covalent adduct. Significant reduction in labeling, observed for K519R, corresponds to a 53% reduction in the mass abundance of the +1 covalent adduct. The Supporting Information provides additional quantitative analysis. PDB entry 2XNF was used to generate the figure. (B) Inhibition of the Med25 AcID ETV5 interaction by NA measured using fluorescence polarization. Mutants containing K519R, highlighted in gray, demonstrate the most significant increase in apparent IC50. Values represent averages of three independent experiments with the indicated error (SDOM) (C) (left) Centroid structure of the most populated cluster from molecular dynamics simulations, where NA binds to the H2 face of Med25 and covalently links to K519. (right) The residues that showed the greatest reduction in fluctuations (RMSF) upon activator binding all occur on dynamic substructures on the H1 face.
To develop a structural model of NA binding and function, molecular dynamics simulations of the covalent NA–Med25 AcID complex in which NA is covalently linked to K519 were carried out, and the results were compared with the case of unbound Med25 AcID. As illustrated in Figure 3C, minimal restructuring in the lysine loop adjacent to the H2 binding interface is observed. However, helix α1 shows significant conformational changes, resulting in partial unfolding. More surprising, the only detectable dynamical changes in NA binding occur on the H1 face, with residues in the two loops on that face showing up to 50% reduction in root-mean-square fluctuations (RMSF). Taken together, the data indicate that NA serves as both an orthosteric inhibitor of H2-binding transcription factors (e.g., ATF6α) and an allosteric inhibitor of H1 binding transcriptional activators (e.g., ETV5).
Next, we tested the engagement of full-length Med25 by NA and the resulting impact of PPI formation and function. Thermal shift assays using freshly prepared HeLa nuclear extracts demonstrated that NA stabilizes endogenous Med25 protein, indicating engagement with the AcID motif in the context of full-length protein (Figure 4A).37,38 The ability of NA to block PPIs formed between endogenous Med25 and cognate activators was then assessed. ETV5 is a member of the ETV/PEA3 subfamily of transcriptional activators, comprising ETV5, ETV1, and ETV4. This transcriptional activator trio have nearly identical domains that utilize a PPI with the H1 surface of Med25 for function, and the PPIs are dysregulated in cancer through overexpression of one or both of the binding partners.19,21,39 As shown in Figure 4B, NA treatment of HeLa cells blocks the formation of the Med25 ETV5 complex, consistent with the in vitro binding data in Figure 2D.
Figure 4.
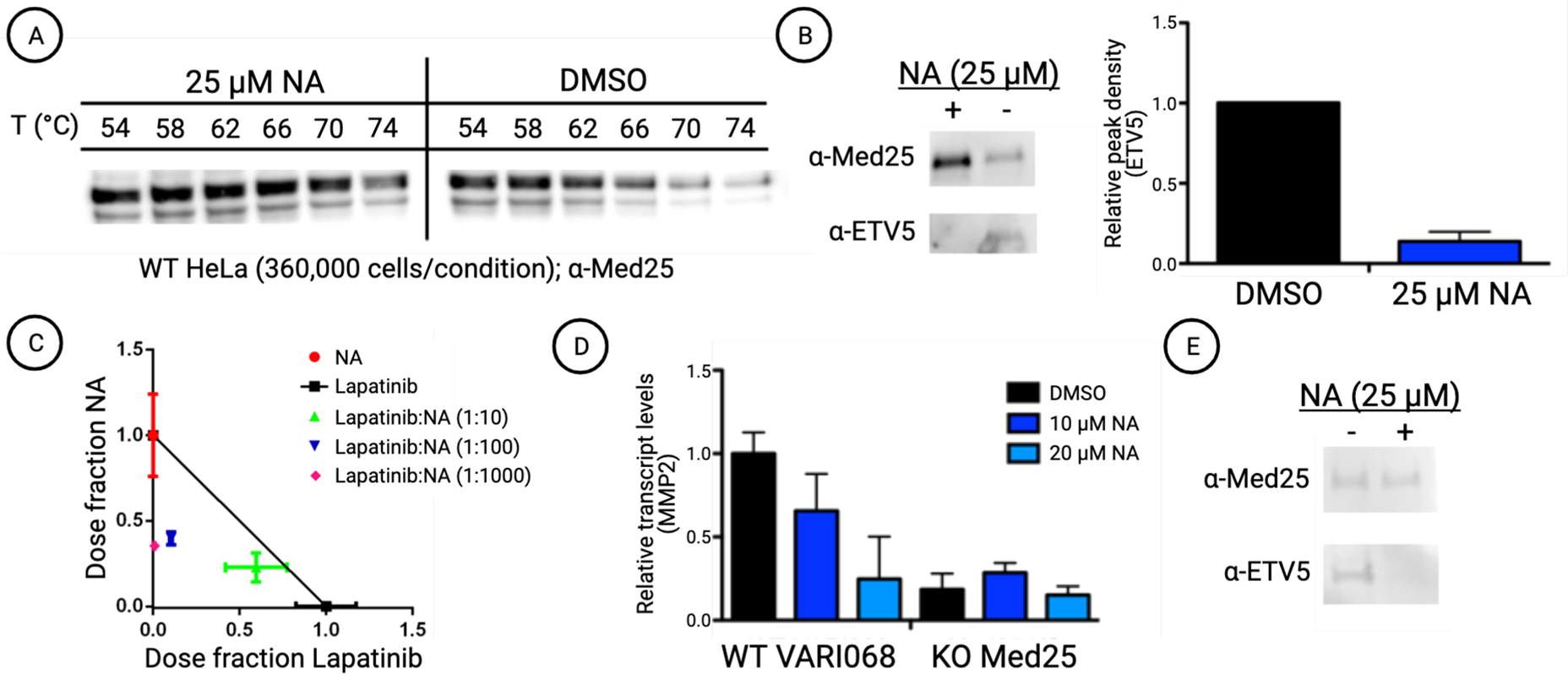
(A) Norstictic acid significantly stabilizes full-length Med25 in HeLa cell extracts. Cellular thermal shift assays were conducted by dosing HeLa cell nuclear extracts with 25 μM NA or equivalent DMSO and subjecting the samples to a range of temperatures. Western blot using a Med25 antibody shows increased band density in NA-dosed samples compared with the control samples, indicating thermal stabilization and target engagement. Quantification and CETSA at additional concentrations are shown in Figure S10. (B) Treatment of HeLa cells with 25 μM NA attenuates the formation of the Med25 ETV5 complex. (left) Representative Western blot showing a reduction in co-immunoprecipitation of ETV5 with Med25. (right) Quantitative assessment of ETV5 co-immunoprecipitation with Med25. The Western blot band density was measured using ImageJ and normalized by comparison to overall Med25 levels. Results are averages of biological triplicates. See Figure S11 for the blot images used. (C) NA shows positive synergy with the on-pathway kinase inhibitor lapatinib in MDA-MB-231 cells. IC50 values of fixed dose ratios of NA and lapatinib were measured after 2 days using a cell viability assay (MTT) and plotted on an isobologram. See the Supporting Information for additional experimental details. (D) Analysis of MMP2 transcript levels by qPCR indicates that NA treatment decreases MMP2 levels to that of a knockout (KO) variant of the cell line. MMP2 transcript levels are normalized to the reference gene RPL19. Results shown are averages of technical triplicate experiments conducted in biological duplicate. (E) Western blot showing that treatment of VARI068 cells with NA attenuates the formation of the Med25 ETV5 complex observed via co-immunoprecipitation.
The Med25 ETV/PEA3 PPIs regulate proliferation, invasion, and migration pathways and in at least a subset of cancers are part of a Her2-driven RAS–RAF–MEK–MAPK circuit.40,41 Thus, if NA blocks Med25 ETV/PEA3 PPIs, positive synergy with a Her2 inhibitor would be anticipated in such systems and would provide additional support for NA inhibition of endogenous Med25. To test this, the combination of NA and lapatinib was tested for synergy by the isobologram method (Figure 4C) in MDA-MB-231 breast cancer cells, an established model.19 As can be seen, strong synergy was observed, consistent with NA engagement of Med25, blocking PPIs formed with ETV/PEA3 activators. This model was further tested in the patient-derived early-passage triple-negative breast cancer cell line VARI068 with robust EGFR expression.42,43 VARI068 cells exhibit approximately 2-fold upregulation of Med25 relative to normal-like nontumorigenic MCF10A cells. CRISPR-Cas9-mediated knockout of Med25 in VARI068 cells led to downregulation of Med25 ETV/PEA3-regulated MMP2 (Figure 4D), and NA treatment led to substantial downregulation of MMP2 relative to vehicle. Furthermore, treatment of VARI068 cells with NA blocks the Med25 ETV5 complex (Figure 4E). Taken together, these data are consistent with a mechanism in which NA engages Med25 in cells and alters its PPI network with downstream effects on tumor phenotype.
The demonstration of NA as a selective orthosteric/allosteric inhibitor of Med25 function validates the importance of dynamic loops in coactivators in molecular recognition and their utility as targets. We also provide evidence that modulation of these protein–protein interactions would provide a useful tool to study cancer at the bench. Our work suggests that Med25 is a potentially viable therapeutic target, thus justifying the search for a drug-like compound for testing the clinical utility of the strategy. We anticipate that NA will be a useful tool for dissecting the Med25–ETV/PEA3 axis in cancers in which Med25 dysregulation is a hallmark. Furthermore, the strategy of targeting dynamic substructures within coactivators should be generalizable beyond Med25.
Supplementary Material
ACKNOWLEDGMENTS
The authors acknowledge financial support from the National Institutes of Health (CA242018 to A.K.M.; NIH T32GM008597 to support S.N.D.S.; GM103695 and GM130587 for C.L.B.I), the Breast Cancer Research Foundation (S.D.M., J.A.Y., L.B.), and a Rogel Cancer Center Core Grant (NIH-P30CA046592). Figures were generated using Biorender.com.
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.1c03258.
Experimental procedures for the high-throughput screen and secondary assays; expression and characterization protocols for Med25 AcID mutants used in these studies; protocols for binding assays, Med25 stabilization assays, Med25 co-immunoprecipitation experiments, and cellular assays; and additional data figures (PDF)
Contributor Information
Julie M. Garlick, Life Sciences Institute, University of Michigan, Ann Arbor, Michigan 48109, United States; Department of Chemistry, University of Michigan, Ann Arbor, Michigan 48109, United States.
Steven M. Sturlis, Life Sciences Institute, University of Michigan, Ann Arbor, Michigan 48109, United States; Department of Chemistry, University of Michigan, Ann Arbor, Michigan 48109, United States.
Paul A. Bruno, Life Sciences Institute, University of Michigan, Ann Arbor, Michigan 48109, United States Department of Chemistry, University of Michigan, Ann Arbor, Michigan 48109, United States.
Joel A. Yates, Department of Internal Medicine, Hematology/Oncology, University of Michigan Medical School, Ann Arbor, Michigan 48109, United States
Amanda L. Peiffer, Life Sciences Institute, University of Michigan, Ann Arbor, Michigan 48109, United States Program in Chemical Biology, University of Michigan, Ann Arbor, Michigan 48109, United States.
Yejun Liu, Life Sciences Institute, University of Michigan, Ann Arbor, Michigan 48109, United States; Program in Chemical Biology, University of Michigan, Ann Arbor, Michigan 48109, United States.
Laura Goo, Department of Internal Medicine, Hematology/Oncology, University of Michigan Medical School, Ann Arbor, Michigan 48109, United States.
LiWei Bao, Department of Internal Medicine, Hematology/Oncology, University of Michigan Medical School, Ann Arbor, Michigan 48109, United States.
Samantha N. De Salle, Life Sciences Institute, University of Michigan, Ann Arbor, Michigan 48109, United States Program in Chemical Biology, University of Michigan, Ann Arbor, Michigan 48109, United States.
Giselle Tamayo-Castillo, CIPRONA and School of Chemistry, University of Costa Rica, 11501 San Jose, Costa Rica.
Charles L. Brooks, III, Department of Chemistry, Program in Chemical Biology, and Department of Biophysics, University of Michigan, Ann Arbor, Michigan 48109, United States.
Sofia D. Merajver, Department of Internal Medicine, Hematology/Oncology, University of Michigan Medical School, Ann Arbor, Michigan 48109, United States
Anna K. Mapp, Life Sciences Institute, University of Michigan, Ann Arbor, Michigan 48109, United States; Department of Chemistry and Program in Chemical Biology, University of Michigan, Ann Arbor, Michigan 48109, United States.
REFERENCES
- (1).Krishnamurthy M; Dugan A; Nwokoye A; Fung Y-H; Lancia JK; Majmudar CY; Mapp AK Caught in the Act: Covalent Cross-Linking Captures Activator–Coactivator Interactions in Vivo. ACS Chem. Biol. 2011, 6 (12), 1321–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Sabari BR; Dall’Agnese A; Boija A; Klein IA; Coffey EL; Shrinivas K; Abraham BJ; Hannett NM; Zamudio AV; Manteiga JC; Li CH; Guo YE; Day DS; Schuijers J; Vasile E; Malik S; Hnisz D; Lee TI; Cisse II; Roeder RG; Sharp PA; Chakraborty AK; Young RA Coactivator Condensation at Super-Enhancers Links Phase Separation and Gene Control. Science 2018, 361 (6400), eaar3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Yang F; Vought BW; Satterlee JS; Walker AK; Jim Sun Z-Y; Watts JL; DeBeaumont R; Saito RM; Hyberts SG; Yang S; Macol C; Iyer L; Tjian R; van den Heuvel S; Hart AC; Wagner G; Näär AM An ARC/Mediator Subunit Required for SREBP Control of Cholesterol and Lipid Homeostasis. Nature 2006, 442 (7103), 700–704. [DOI] [PubMed] [Google Scholar]
- (4).Larivière L; Seizl M; Cramer P̀. A Structural Perspective on Mediator Function. Curr. Opin. Cell Biol. 2012, 24 (3), 305–313. [DOI] [PubMed] [Google Scholar]
- (5).Thakur JK; Yadav A; Yadav G Molecular Recognition by the KIX Domain and Its Role in Gene Regulation. Nucleic Acids Res. 2014, 42 (4), 2112–2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Wang H; Dienemann C; Stützer A; Urlaub H; Cheung ACM; Cramer P Structure of the Transcription Coactivator SAGA. Nature 2020, 577 (7792), 717–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Kurokawa R; Kalafus D; Ogliastro M-H; Kioussi C; Xu L; Torchia J; Rosenfeld MG; Glass CK Differential Use of CREB Binding Protein-Coactivator Complexes. Science 1998, 279 (5351), 700–703. [DOI] [PubMed] [Google Scholar]
- (8).York B; O’Malley BW Steroid Receptor Coactivator (SRC) Family: Masters of Systems Biology*. J. Biol. Chem. 2010, 285 (50), 38743–38750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Näär AM; Lemon BD; Tjian R Transcriptional Coactivator Complexes. Annu. Rev. Biochem. 2001, 70 (1), 475–501. [DOI] [PubMed] [Google Scholar]
- (10).Bushweller JH Targeting Transcription Factors in Cancer — from Undruggable to Reality. Nat. Rev. Cancer 2019, 19 (11), 611–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Mapp AK; Pricer R; Sturlis S Targeting Transcription Is No Longer a Quixotic Quest. Nat. Chem. Biol. 2015, 11 (12), 891–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Garlick JM; Mapp AK Selective Modulation of Dynamic Protein Complexes. Cell Chem. Biol. 2020, 27 (8), 986–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Breen ME; Mapp AK Modulating the Masters: Chemical Tools to Dissect CBP and P300 Function. Curr. Opin. Chem. Biol. 2018, 45, 195–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Tien JC-Y; Xu J Steroid Receptor Coactivator-3 as a Potential Molecular Target for Cancer Therapy. Expert Opin. Ther. Targets 2012, 16 (11), 1085–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Nierenberg AA; Ghaznavi SA; Sande Mathias I; Ellard KK; Janos JA; Sylvia LG Peroxisome Proliferator-Activated Receptor Gamma Coactivator-1 Alpha as a Novel Target for Bipolar Disorder and Other Neuropsychiatric Disorders. Biol. Psychiatry 2018, 83 (9), 761–769. [DOI] [PubMed] [Google Scholar]
- (16).Yu Y; Su X; Qin Q; Hou Y; Zhang X; Zhang H; Jia M; Chen Y Yes-Associated Protein and Transcriptional Coactivator with PDZ-Binding Motif as New Targets in Cardiovascular Diseases. Pharmacol. Res. 2020, 159, 105009. [DOI] [PubMed] [Google Scholar]
- (17).Pricer R; Gestwicki JE; Mapp AK From Fuzzy to Function: The New Frontier of Protein-Protein Interactions. Acc. Chem. Res. 2017, 50 (3), 584–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).El Khattabi L; Zhao H; Kalchschmidt J; Young N; Jung S; Van Blerkom P; Kieffer-Kwon P; Kieffer-Kwon K-R; Park S; Wang X; Krebs J; Tripathi S; Sakabe N; Sobreira DR; Huang S-C; Rao SSP; Pruett N; Chauss D; Sadler E; Lopez A; Nóbrega MA; Aiden EL; Asturias FJ; Casellas R A Pliable Mediator Acts as a Functional Rather Than an Architectural Bridge between Promoters and Enhancers. Cell 2019, 178 (5), 1145–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Verger A; Baert J-L; Verreman K; Dewitte F; Ferreira E; Lens Z; de Launoit Y; Villeret V; Monté D The Mediator Complex Subunit MED25 Is Targeted by the N-Terminal Transactivation Domain of the PEA3 Group Members. Nucleic Acids Res. 2013, 41 (9), 4847–4859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Henderson AR; Henley MJ; Foster NJ; Peiffer AL; Beyersdorf MS; Stanford KD; Sturlis SM; Linhares BM; Hill ZB; Wells JA; Cierpicki T; Brooks CL; Fierke CA; Mapp AK Conservation of Coactivator Engagement Mechanism Enables Small-Molecule Allosteric Modulators. Proc. Natl. Acad. Sci. U. S. A. 2018, 115 (36), 8960–8965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Pellecchia A; Pescucci C; De Lorenzo E; Luceri C; Passaro N; Sica M; Notaro R; De Angioletti M Overexpression of ETV4 Is Oncogenic in Prostate Cells through Promotion of Both Cell Proliferation and Epithelial to Mesenchymal Transition. Oncogenesis 2012, 1 (7), No. e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Landrieu I; Verger A; Baert J-L; Rucktooa P; Cantrelle F-X; Dewitte F; Ferreira E; Lens Z; Villeret V; Monté D Characterization of ERM Transactivation Domain Binding to the ACID/PTOV Domain of the Mediator Subunit MED25. Nucleic Acids Res. 2015, 43 (14), 7110–7121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Vojnic E; Mourão A; Seizl M; Simon B; Wenzeck L; Larivière L; Baumli S; Baumgart K; Meisterernst M; Sattler M; Cramer P Structure and VP16 Binding of the Mediator Med25 Activator Interaction Domain. Nat. Struct. Mol. Biol. 2011, 18 (4), 404–409. [DOI] [PubMed] [Google Scholar]
- (24).Sela D; Conkright JJ; Chen L; Gilmore J; Washburn MP; Florens L; Conaway RC; Conaway JW Role for Human Mediator Subunit MED25 in Recruitment of Mediator to Promoters by Endoplasmic Reticulum Stress-Responsive Transcription Factor ATF6α. J. Biol. Chem. 2013, 288 (36), 26179–26187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Bontems F; Verger A; Dewitte F; Lens Z; Baert J-L; Ferreira E; Launoit Y. de; Sizun C; Guittet E; Villeret V; Monté D NMR Structure of the Human Mediator MED25 ACID Domain. J. Struct. Biol. 2011, 174 (1), 245–251. [DOI] [PubMed] [Google Scholar]
- (26).Mittler G; Stühler T; Santolin L; Uhlmann T; Kremmer E; Lottspeich F; Berti L; Meisterernst M A Novel Docking Site on Mediator Is Critical for Activation by VP16 in Mammalian Cells. EMBO J. 2003, 22 (24), 6494–6504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Yang F; DeBeaumont R; Zhou S; Näär AM The Activator-Recruited Cofactor/Mediator Coactivator Subunit ARC92 Is a Functionally Important Target of the VP16 Transcriptional Activator. Proc. Natl. Acad. Sci. U. S. A. 2004, 101 (8), 2339–2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Henley MJ; Linhares BM; Morgan BS; Cierpicki T; Fierke CA; Mapp AK Unexpected Specificity within Dynamic Transcriptional Protein–Protein Complexes. Proc. Natl. Acad. Sci. U. S. A. 2020, 117 (44), 27346–27353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Ran X; Gestwicki JE Inhibitors of Protein–Protein Interactions (PPIs): An Analysis of Scaffold Choices and Buried Surface Area. Curr. Opin. Chem. Biol. 2018, 44, 75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Chakraborty S; Inukai T; Fang L; Golkowski M; Maly DJ Targeting Dynamic ATP-Binding Site Features Allows Discrimination between Highly Homologous Protein Kinases. ACS Chem. Biol. 2019, 14 (6), 1249–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Lake EW; Muretta JM; Thompson AR; Rasmussen DM; Majumdar A; Faber EB; Ruff EF; Thomas DD; Levinson NM Quantitative Conformational Profiling of Kinase Inhibitors Reveals Origins of Selectivity for Aurora Kinase Activation States. Proc. Natl. Acad. Sci. U. S. A. 2018, 115 (51), E11894–E11903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Sandhu M; Touma AM; Dysthe M; Sadler F; Sivaramakrishnan S; Vaidehi N Conformational Plasticity of the Intracellular Cavity of GPCR–G-Protein Complexes Leads to G-Protein Promiscuity and Selectivity. Proc. Natl. Acad. Sci. U. S. A. 2019, 116 (24), 11956–11965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Hollingsworth SA; Kelly B; Valant C; Michaelis JA; Mastromihalis O; Thompson G; Venkatakrishnan AJ; Hertig S; Scammells PJ; Sexton PM; Felder CC; Christopoulos A; Dror RO Cryptic Pocket Formation Underlies Allosteric Modulator Selectivity at Muscarinic GPCRs. Nat. Commun. 2019, 10 (1), 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Fang Z; Grütter C; Rauh D Strategies for the Selective Regulation of Kinases with Allosteric Modulators: Exploiting Exclusive Structural Features. ACS Chem. Biol. 2013, 8 (1), 58–70. [DOI] [PubMed] [Google Scholar]
- (35).Bandyopadhyay A; Gao J Targeting Biomolecules with Reversible Covalent Chemistry. Curr. Opin. Chem. Biol. 2016, 34, 110–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Majmudar CY; Højfeldt JW; Arevang CJ; Pomerantz WC; Gagnon JK; Schultz PJ; Cesa LC; Doss CH; Rowe SP; Vásquez V; Tamayo-Castillo G; Cierpicki T; Brooks CL; Sherman DH; Mapp AK Sekikaic Acid and Lobaric Acid Target a Dynamic Interface of the Coactivator CBP/P300. Angew. Chem., Int. Ed. 2012, 51 (45), 11258–11262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Molina DM; Jafari R; Ignatushchenko M; Seki T; Larsson EA; Dan C; Sreekumar L; Cao Y; Nordlund P Monitoring Drug Target Engagement in Cells and Tissues Using the Cellular Thermal Shift Assay. Science 2013, 341 (6141), 84–87. [DOI] [PubMed] [Google Scholar]
- (38).Jafari R; Almqvist H; Axelsson H; Ignatushchenko M; Lundbäck T; Nordlund P; Molina DM The Cellular Thermal Shift Assay for Evaluating Drug Target Interactions in Cells. Nat. Protoc. 2014, 9 (9), 2100–2122. [DOI] [PubMed] [Google Scholar]
- (39).Aytes A; Mitrofanova A; Kinkade CW; Lefebvre C; Lei M; Phelan V; LeKaye HC; Koutcher JA; Cardiff RD; Califano A; Shen MM; Abate-Shen C ETV4 Promotes Metastasis in Response to Activation of PI3-Kinase and Ras Signaling in a Mouse Model of Advanced Prostate Cancer. Proc. Natl. Acad. Sci. U. S. A. 2013, 110 (37), E3506–E3515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Kar A; Gutierrez-Hartmann A Molecular Mechanisms of ETS Transcription Factor-Mediated Tumorigenesis. Crit. Rev. Biochem. Mol. Biol. 2013, 48 (6), 522–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Shepherd TG; Kockeritz L; Szrajber MR; Muller WJ; Hassell JA The Pea3 Subfamily Ets Genes Are Required for HER2/Neu-Mediated Mammary Oncogenesis. Curr. Biol. 2001, 11 (22), 1739–1748. [DOI] [PubMed] [Google Scholar]
- (42).Aw Yong KM; Ulintz PJ; Caceres S; Cheng X; Bao L; Wu Z; Jiagge EM; Merajver SD Heterogeneity at the Invasion Front of Triple Negative Breast Cancer Cells. Sci. Rep. 2020, 10 (1), 5781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Liu M; Liu Y; Deng L; Wang D; He X; Zhou L; Wicha MS; Bai F; Liu S Transcriptional Profiles of Different States of Cancer Stem Cells in Triple-Negative Breast Cancer. Mol. Cancer 2018, 17 (1), 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.