

Abstract
Background
Skeletal muscle atrophy is a debilitating complication of many chronic diseases, disuse conditions, and ageing. Genome‐wide gene expression analyses have identified that elevated levels of microRNAs encoded by the H19X locus are among the most significant changes in skeletal muscles in a wide scope of human cachectic conditions. We have previously reported that the H19X locus is important for the establishment of striated muscle fate during embryogenesis. However, the role of H19X‐encoded microRNAs in regulating skeletal mass in adults is unknown.
Methods
We have created a transgenic mouse strain in which ectopic expression of miR‐322/miR‐503 is driven by the skeletal muscle‐specific muscle creatine kinase promoter. We also used an H19X mutant mouse strain in which transcription from the locus is interrupted by a gene trap. Animal phenotypes were analysed by standard histological methods. Underlying mechanisms were explored by using transcriptome profiling and validated in the two animal models and cultured myotubes.
Results
Our results demonstrate that the levels of H19X microRNAs are inversely related to postnatal skeletal muscle growth. Targeted overexpression of miR‐322/miR‐503 impeded skeletal muscle growth. The weight of gastrocnemius muscles of transgenic mice was only 54.5% of the counterparts of wild‐type littermates. By contrast, interruption of transcription from the H19X locus stimulates postnatal muscle growth by 14.4–14.9% and attenuates the loss of skeletal muscle mass in response to starvation by 12.8–21.0%. Impeded muscle growth was not caused by impaired IGF1/AKT/mTOR signalling or a hyperactive ubiquitin–proteasome system, instead accompanied by markedly dropped abundance of translation initiation factors in transgenic mice. miR‐322/miR‐503 directly targets eIF4E, eIF4G1, eIF4B, eIF2B5, and eIF3M.
Conclusions
Our study illustrates a novel pathway wherein H19X microRNAs regulate skeletal muscle growth and atrophy through regulating the abundance of translation initiation factors, thereby protein synthesis. The study highlights how translation initiation factors lie at the crux of multiple signalling pathways that control skeletal muscle mass.
Keywords: Skeletal muscle atrophy, Protein synthesis, H19X, MicroRNAs, miR‐322, miR‐424, miR‐503, miR‐542, AtromiR, Translation initiation
Introduction
Loss of skeletal muscle mass (i.e. muscle atrophy/wasting) is closely associated with poor prognosis in many chronic diseases, including myopathies, muscular dystrophies, cancers, diabetes, sepsis, and heart failure. Muscle loss also contributes to decreased quality of life in ageing. 1 , 2 Muscle atrophy results from a reduced rate of protein synthesis, accelerated protein degradation, or both. The ubiquitin–proteasome system and autophagy are two major proteolytic mechanisms that are activated in skeletal muscle in various atrophying conditions. 3 The activity of ubiquitin–proteasome system and autophagy is regulated through coordinated activation of several signalling pathways, such as nuclear factor‐κB and p38 MAPK. Moreover, the canonical transforming growth factor‐β family members (e.g. myostatin and activin), which function through activin receptors and activation of SMAD2/3 transcription factors, are important drivers of muscle wasting. In contrast, the IGF1/AKT/mTOR pathway increases the rate of protein synthesis leading to skeletal muscle hypertrophy. Activation of this pathway also inhibits muscle protein degradation through distinct mechanisms. 4 , 5 For example, gene expression of two muscle‐specific E3 ubiquitin ligases, MuRF1 and MAFbx/Atrogin‐1, is regulated by transcription factors FoxO1/3, which are removed from the nucleus upon phosphorylation by AKT. 5 , 6 Intriguingly, transforming growth factor‐β inhibits AKT activity through stimulating SMAD signalling. 7 , 8 Accumulating evidence further suggests that non‐coding RNAs play an important role in the regulation of both anabolic and catabolic signalling pathways in skeletal muscle. 9
MicroRNAs (miRNAs) are small non‐coding RNAs that regulate post‐transcriptional gene expression. 10 The abundance of some miRNAs is altered in wasting human muscle biopsies. Eight miRNAs were differentially expressed in rectus abdominis of cachectic and non‐cachectic cancer patients. 11 Thirty‐two differentially expressed miRNAs were identified in the quadriceps of chronic obstructive pulmonary disease patients with a low fat‐free mass index: 26 were down‐regulated, while 6 were up‐regulated. Remarkably, five of the six up‐regulated miRNAs were encoded by the H19X locus located on Xq26.3. 12 , 13 Because H19X‐encoded miRNAs are differentially expressed in skeletal muscle in several cachectic/wasting diseases, 12 , 14 , 15 , 16 we hypothesize that these miRNAs play important roles in the regulation of skeletal muscle mass. Furthermore, it is likely that the increased abundance of H19X miRNAs is specific for muscle atrophy per se but not the underlying diseases.
Our earlier work demonstrated that the H19X‐encoded miR‐322(424)/miR‐503 cluster was highly expressed in bipotent cardiac and skeletal muscle progenitors. During embryonic stem cell differentiation, the cluster plays an essential role in cardiomyocyte specification by targeting and inhibiting neuroectoderm lineage factors. 17 The association of these miRNAs with human muscle atrophy is interesting because it may reflect a common phenomenon—foetal gene reactivation—in which aberrant expression of embryonic genes contributes to pathogenic processes. In this study, using genetic mouse models and molecular approaches, we demonstrate that miR‐322(424)/miR‐503 negatively regulates skeletal muscle mass in adult animals by directly targeting protein translation initiation factors.
Methods
Animal care and use
To generate muscle creatine kinase (MCK)‐miR‐322/miR‐503 transgenic (tg) mice, a ∼300 bp genomic DNA fragment containing the miR‐322/miR‐503 coding sequence was inserted into plasmid pBS MCK promoter (Addgene #12528), originally developed by Bruning et al. 18 Production of the tg mice was performed by Cyagen US. Transgenic offspring were identified by PCR with the following primers: MCKseq‐F: 5′‐GCTGGGATTTCAGGCATGAG‐3′; miR3/5 Internalseq‐R: 5′‐AAGCAGGGTGTGGTGGATAG‐3′.
The H19X mutant (mt) animal harbours a knockout first gene trap allele. It was developed by Park et al. 19 We obtained the strain from Jackson Laboratory (Stock No. 017513, Mirc24tm1Mtm/Mmjax). All animal studies were approved by the Institutional Animal Care and Use Committee at University of Houston.
Exercise training
A 10° uphill treadmill (JOG A DOG) was used to perform the treadmill training. Mice were acclimated to the treadmill for 3 days at 10 min/day and 10 m/min. Starting from the next day, animals run for 30 min at 30 m/min. Exercise training was performed 5 days/week for 4 weeks.
Fasting
Mice were randomly allocated to four groups: one group was fed ad libitum (control), and the other groups were deprived of solid food for 24, 48, or 72 h but had free access to water (starvation).
Grip strength test
Mice were tested at the age of 12 weeks. The device consisted of a scale attached to a triangular pull bar. Grip strength test was measured by allowing the mouse to grasp the bar with forelimb and then pulling the tail with increasing force until it releases its grip. Each mouse was subjected to three consecutive tests with a 15 s interval between tests to obtain the peak value.
EchoMRI
Body composition (% fat and lean mass) was measured in 6 month mice with (without anaesthesia or sedation) an EchoMRI‐100 Body Composition Analyzer (Echo Medical Systems, Houston, TX).
Muscle fibre cross‐sectional area measurement
Transverse sections (10 μm) were cut from OCT‐embedded tibialis anterior (TA) using cryostat (Leica), and sections were stained with Harris' Hematoxylin and Eosin Y (StatLab). TA muscle sections were photographed with a Nikon Ti‐E inverted microscope. Cross‐sectional area (CSA) was assessed at comparable cross sections close to the middle of TA using MyoVision software. We manually defined fibre boundaries and count the whole section, which usually yields 200–500 CSAs excluding regions where fibre boundaries cannot be defined. The histogram contains data from 1500 CSAs.
Succinate dehydrogenase staining
Frozen cross sections of TA muscle were used for succinate dehydrogenase staining. Briefly, slides were incubated in staining buffer containing 0.5 M disodium succinate (Sigma), 20 mM MgCl2, and 0.5 mg/mL of nitro blue tetrazolium (VWR) for 15 min at 37°C. Staining was stopped by immersing the slides in PBS.
Cell culture
C2C12 myoblast culture and differentiation were performed as previously described. 20 C2C12 miR‐322/miR‐503‐tet‐on cells were produced by transfecting cells with pLVX‐miR‐322/miR‐503 17 and pLVX‐tet‐on Advanced, and selected with puromycin (2 μg/mL) and G418 (800 μg/mL).
Luciferase reporter assay
To construct luciferase reporters Luc‐eIF4E‐3′‐UTR, Luc‐eIF4G1‐3′‐UTR, Luc‐eIF4B‐3′‐UTR, Luc‐eIF2B5‐3′‐UTR, and Luc‐eIF3M‐3′‐UTR, we ligated DNA fragments containing the putative targeting sites into the PmeI/XbaI sites of pmirGlo (Promega). Mutant reporters were created by ligating DNA fragments containing tri‐nucleotide mutations within the seed sequence of the targeting sites. The sequences of the inserts are available upon request.
A total of 0.5 μg luciferase reporter and 2 μg miR‐322/miR‐503 plasmid DNA were used to co‐transfect 293T cells. The assay was performed using the Dual‐Luciferase Reporter Assay System (Promega). Luciferase activity was read in a BD Monolight 3010 luminometer.
Western blotting
Western blot assays were carried out with gastrocnemius muscles or C2C12 cells homogenized in RIPA buffer [50 mM Tris–HCl (pH 8.0), 150 mM NaCl, 1% NP‐40, 0.5% sodium deoxycholate, and 0.1% SDS]. Proteins were separated on a 4–12% Bis‐Tris gel (Life Technologies) and transferred onto PVDF membranes. The membranes were incubated with the following primary antibodies at 4°C overnight: anti‐IGF1 receptor (IGF1R) (1:1000; sc713, Santa Cruz), anti‐eIF4E (1:2000; sc9976, Santa Cruz), anti‐phospho‐eIF4E(Ser209) (1:2000; 9741, Cell Signaling), anti‐4EBP1 (1:2000; 9452, Cell Signaling), anti‐phospho‐4EBP1(Ser65) (1:1000; 9451, Cell Signaling), anti‐AKT (1:2000; 9272, Cell Signaling), anti‐phospho‐AKT (Ser473) (1:2000; 4060, Cell Signaling), anti‐puromycin (1:25000; MABE343, Sigma), anti‐eIF3M (1:1000; NBP1‐31045, Novus Biologicals), anti‐eIF4G (1:2000; 2498, Cell Signaling), anti‐eIF4B (1:1000; 3592, Cell Signaling), anti‐eIF2B5 (1:1000; 3595, Cell Signaling), and anti‐phospho‐eIF2α (Ser51) (1:1000; 9721, Cell Signaling). The membranes were next incubated with horseradish peroxidase‐conjugated secondary antibodies to the appropriate IgG (1:2000; Life Technologies) for 90 min. Bound antibodies were visualized with enhanced chemiluminescence reagents (NEL105001EA, PerkinElmer).
Real‐time reverse transcription PCR
Tibialis anterior muscles were used to isolate total RNA using RiboZol RNA extraction reagent (Amresco). Total RNA (100 ng) was subjected to quantitative reverse transcription PCR (RT‐PCR) using TaqMan One‐Step RT‐PCR Master Mix reagent (Eurogentec) and 7900 HT Fast‐Real‐Time PCR System (Applied Biosystems). Copy number for each transcript is calculated relative to that of GAPDH. For miR‐322, miR‐503, and miR‐542, reverse transcription reactions were carried out using TaqMan miRNA Reverse Transcription Kit (Thermo Fisher Scientific) according to the instructions provided. Sequences of primers and probes are available upon request.
Next‐generation sequencing and analysis
RNA‐Seq analysis was performed with wild‐type (wt) and tg mice (n = 3 in each group). In brief, RNA was isolated using RiboZol RNA extraction reagent (Amresco) and DNA‐free DNA Removal kit (Life Technologies). RNA integrity and concentration were determined using Nanodrop 2000c and Agilent 2100 Bioanalyzer. RIN numbers for all samples were greater than 7. mRNA libraries were prepared with NEBNext Ultra II Directional RNA Library Prep Kit for Illumina (New England Biolabs) at University of Houston Seq‐N‐Edit Core. The prepared libraries were pooled and sequenced using NextSeq 500 (Illumina). At least 20M reads were obtained for each sample. Sequence alignment and analysis were performed by Shen et al. with a previously established pipeline. 17 Gene Ontology (GO) analysis was performed on differentially expressed genes (log2FC > 0.75 and FDR‐adjusted P < 0.05) using DAVID (https://david.ncifcrf.gov/). Gene Set Enrichment Analysis was performed using Broad Institute software GSEA.
Statistical analysis
Data were analysed using Microsoft Excel and GraphPad Prism 7.0. All values are expressed as mean ± SEM. Comparisons between two groups were determined by unpaired two‐tailed Student's t‐test. P values less than 0.05 were considered significant.
Results
Expression of miR‐322/miR‐503 is inversely related to postnatal skeletal muscle growth
We have previously demonstrated that the expression of miR‐322(424)/miR‐503 is highly enriched in heart and somites in early embryos. 17 , 19 Here, we first sought to determine whether the tissue distribution and expression level of H19X miRNAs change in postnatal life stages. With the expression specificity in the heart and skeletal muscle maintained (Figure 1A), the level of miR‐322 and miR‐503 decreased progressively (Figure 1B). To understand the potential role of H19X‐encoded miRNAs in regulating muscle mass, we investigated how their levels are regulated in animal models of muscle hypertrophy and atrophy. As a model of muscle hypertrophy, the animals were exercised on a rodent treadmill for 4 weeks, which resulted in a significant increase in soleus, TA, and gastrocnemius muscle mass (Figure 1C). Interestingly, levels of miR‐322, miR‐503, and miR‐542 were markedly lower in TA muscle of exercised mice compared with controls (Figure 1D). As a model of muscle atrophy, the mice were starved for 48 h, which led to a 15.42% reduction in body mass. Muscle mass was reduced for all muscles measured (Figure 1E). The levels of miR‐322 and miR‐542 were significantly increased, whereas the levels of miR‐503 remained comparable in TA muscle of control and starved animals (Figure 1F). Collectively, these results demonstrate an inverse relationship between the levels of H19X miRNAs and muscle mass and suggest that elevated levels of miR‐322, miR‐503, and miR‐542 are incompatible with skeletal muscle growth.
Figure 1.
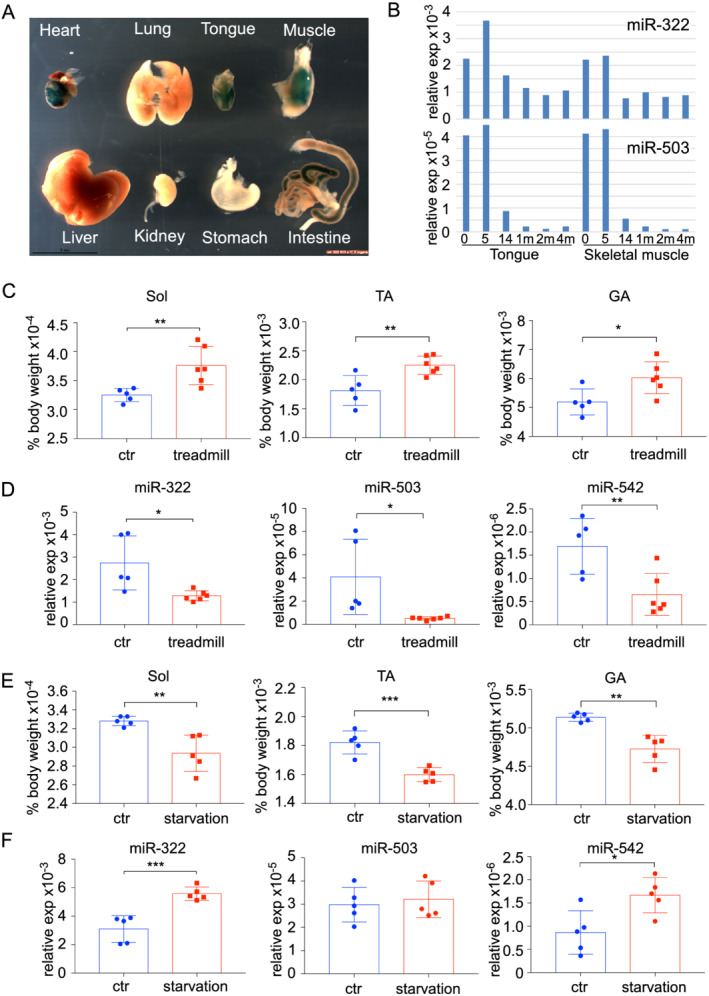
Expression of miR‐322/miR‐503 is inversely related to postnatal skeletal muscle growth. (A) Whole‐mount β‐gal staining of embryonic organs at E17.5. (B) The level of miR‐322/miR‐503 expression is high at birth (0 day) and 5 days in both tongue and hindlimb muscles. It drops at 14 days and sustains a low level afterward. (C) Four weeks' mandatory treadmill training led to hypertrophy of hindlimb muscles (n = 5 vs. 6). (D) Treadmill training resulted in down‐regulation of miR‐322, miR‐503, and miR‐542 in TA muscles (n = 5 vs. 6). (E) Forty‐eight hour starvation caused drops in hindlimb muscle mass (n = 5 vs. 5). (F) Starvation resulted in up‐regulation of miR‐322 and miR‐542 (n = 5 vs. 5). For all panels, * P < 0.05, ** P < 0.01, and *** P < 0.001.
Forced expression of miR‐322/miR‐503 impedes muscle growth
Among the H19X miRNAs, miR‐322, miR‐503, and miR‐542 are most frequently associated with muscle atrophy and cachexia. 12 , 14 , 15 , 16 The miR‐322/miR‐503 cluster has been correlated with a broad range of diseases and has higher expression levels than miR‐542. Therefore, we focused this study to investigating whether miR‐322/miR‐503 plays a role in regulating muscle mass. We generated a tg mouse strain in which the expression of miR‐322/miR‐503 is driven by the MCK promoter (Figure 2A). The MCK promoter was chosen because it is highly restricted to striated muscle, and its activity begins on Embryonic Day 17, which minimizes disturbing the normal function of miR‐322/miR‐503 during early embryogenesis. 18 The expression level of the transgene was ∼2‐fold of the endogenous allele at birth (Figure 2B).
Figure 2.
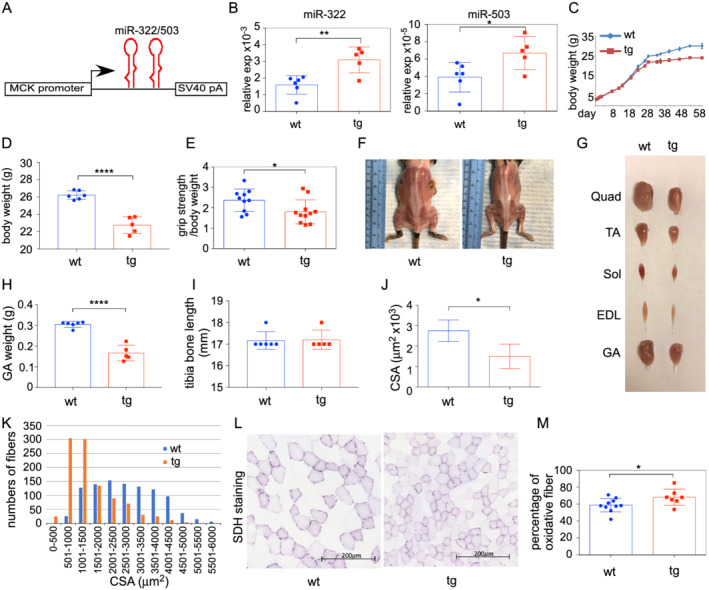
Skeletal muscle‐specific overexpression of miR‐322/miR‐503 impeded muscle growth. (A) Diagram of the transgenic gene expression cassette. The ∼6.5 kb MCK promoter was used to drive miR‐322/miR‐503 expression. (B) Levels of transgene expression in skeletal muscles at birth (n = 6 vs. 5). (C) Transgenic animals had lower body weights than non‐transgenic littermates starting from 28 days of age. (D) The body weights of transgenic animals were significantly lower than non‐transgenic littermates at 4 months of age (n = 6 vs. 5). (E) Transgenic animals had lower grip strength (n = 10 vs. 11). (F) Representative images of transgenic and control animals with back and hindlimb muscles exposed. (G) Representative images of hindlimb muscles from transgenic and control animals. EDL, extensor digitorum longus; GA, gastrocnemius; Quad, quadriceps; Sol, soleus; TA, tibialis anterior. (H) Masses of gastrocnemius muscles were lower in transgenic animals at 4 months of age (n = 6 vs. 5). (I) Tibia bone lengths were comparable between transgenic and control animals (n = 6 vs. 5). (J) Quantification of mean CSA in TA muscles (n = 5 vs. 5). (K) Frequency histograms of CSA in TA muscles (n = 5 vs. 5). (L) Representative images of succinate dehydrogenase activity staining. (M) Quantification of oxidative fibre percentage based on SDH staining (n = 10 vs. 7). For all panels, * P < 0.05, ** P < 0.01, and **** P < 0.0001.
Body masses of tg mice became progressively lower than that of wt since 4 weeks old (Figure 2C). At 4 months, tg mice weighed 13.2% less than their wt littermates (26.22 ± 0.2 vs. 22.75 ± 0.43 g, wt vs. tg) (Figure 2D). Transgenic mice also had lower grip strength (Figure 2E). Autopsy revealed widespread muscle atrophy (Figure 2F and 2G). The weight of gastrocnemius muscles of tg mice was only 54.5% of the weight of wt littermates (0.3048 ± 0.0058 vs. 0.1662 ± 0.0167 g, wt vs. tg) (Figure 2H). There was no significant difference in tibia bone length among tg and wt mice, suggesting that skeletal growth was not affected by the transgene (Figure 2I).
We next performed histomorphometric analysis of skeletal muscles. Results showed that overexpression of miR‐322/miR‐503 caused a decrease in average myofibre CSA (Figure 2J) and a remarkable leftward shift in the myofibre size‐frequency histogram (Figure 2K). The distribution of minimum Feret diameter showed a large shift towards smaller size myofibres in tg mice (Supporting Information, Figure S1B and S1C). Despite a reduction in myofibre size, there were no signs of fibrosis or central nuclei in skeletal muscle of tg mice, indicating that overexpression of miR‐322/miR‐503 does not lead to myopathy or muscle degeneration (Figure S1A). Fast‐to‐slow fibre switching is associated with malnutrition and inflammation, as seen in fasting and glucocorticoid‐induced atrophy. 21 miR‐322/miR‐503 is one of the top miRNAs induced by glucocorticoids in myotubes. 22 Consistently, staining for succinate dehydrogenase activity revealed that tg muscles had an increase in the percentage of oxidative fibres compared with wt littermates (58.7 ± 2.5% vs. 68.1 ± 3.6%, wt vs. tg) (Figure 2L and 2M). Taken together, these results suggest that overexpression of miR‐322/miR‐503 is sufficient to impede skeletal muscle growth in vivo.
miR‐322/miR‐503 inhibits protein synthesis by targeting translation initiation factors
To identify the molecular pathways affected by miR‐322/miR‐503, we performed transcriptome profiling using TA muscles from 4‐month‐old littermate wt and tg mice. This analysis showed that 343 genes were up‐regulated and 300 genes were down‐regulated in the skeletal muscle of tg mice (log2FC > 0.75, P < 0.05). Up‐regulated genes were associated with GO terms insulin receptor signalling pathway, cellular response to hypoxia, multicellular organismal response to stress, and so forth (Figure 3B). GO terms associated with down‐regulated genes were ubiquitin‐dependent protein catabolic process, translational initiation, and so forth (Figure 3C). Overall, an augmented response to stress and hypoxia agrees with the established functions of miR‐424/miR‐503 and is consistent with the atrophy phenotype. However, increased insulin receptor signalling, decreased autophagy, and protein catabolic process are contradictory. To confirm the GO analysis findings, we performed GSEA using the raw expression data (instead of a pre‐ranked gene list). The results showed again that the genes within ‘insulin receptor signalling’ were predominantly up‐regulated (Figure 3D), whereas genes within ‘protein ubiquitination’ were down‐regulated (Figure 3E) in tg mice.
Figure 3.
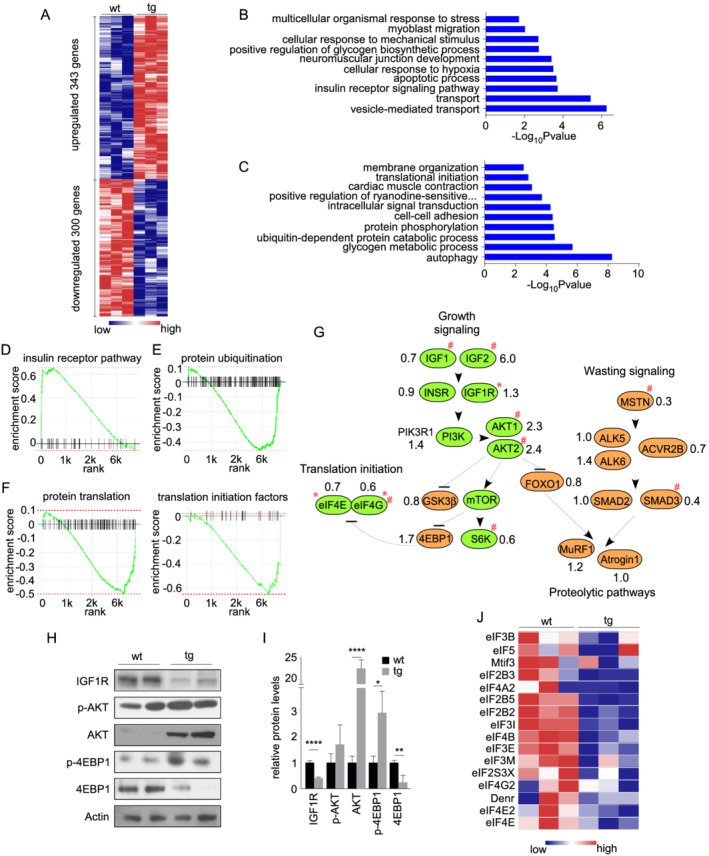
Transcriptome survey identified that the expression of initiation factors was impaired by miR‐322/miR‐503 overexpression. (A) A heat map showing differentially expressed genes between transgenic and control TA muscles (n = 3 vs. 3). GO terms enriched in up‐regulated (C) and down‐regulated (D) genes. GSEA graphs for the top up‐regulated signatures (D, insulin receptor signalling) and top down‐regulated signatures (E, protein ubiquitination; F, translation initiation and initiation factors). (G) Transcript‐level analyses of genes relevant to muscle growth and wasting signalling. Green and brown indicate genes known to play positive and negative roles for muscle growth, respectively. ‘*’ indicates TargetScan‐predicted targets of miR‐322/miR‐503. The numbers indicate fold changes in expression levels (transgenic vs. control). ‘#’ indicates FDR‐adjusted P < 0.05. (H) Western blot assessment of IGF/AKT/mTOR pathway components. (I) Quantification of protein expression of IGF/AKT/mTOR pathway components (n = 4 vs. 4). (J) A heat map of initiation factors down‐regulated by miR‐322/miR‐503 in tg muscles.
Next, we examined the expression of individual genes that have been implicated in muscle atrophy and hypertrophy (Figure 3G). E3 ubiquitin ligases MuRF1 and Atrogin‐1 showed little change; most of their upstream factors were reduced. Real‐time RT‐PCR in additional samples showed that there was no significant difference in levels of MuRF1 and Atrogin‐1 between wt and tg mice (Figure S1D). Further, there was no significant difference in the amount of ubiquitin‐tagged proteins (Figure S1E and S1F), suggesting that the proteolytic pathways are not involved in inducing atrophy in tg animals. In contrast, changes in the level of hypertrophic signalling genes were complex. While the terminal effectors 4EBP1 and p70S6K were up‐regulated and down‐regulated, respectively, upstream factors along the IGF1/AKT/mTOR axis showed mostly increased expression. We next examined protein levels of components of IGF1/AKT/mTOR pathway (Figure 3H and 3I). IGF1R was previously documented as a direct target of miR‐424/miR‐503. 23 Consistently, levels of IGF1R were significantly lower in skeletal muscle of miR‐322/miR‐503 tg animals. However, total AKT and phosphorylated AKT were higher in tg mice. The levels of phosphorylated 4EBP1 protein were higher, despite that the level of total 4EBP1 was lower in tg muscle. Together, these results are consistent with the transcript‐level finding that the IGF1/AKT/mTOR pathway is activated in skeletal muscle after overexpression of miR‐322/miR‐503.
The earlier surprising findings prompted us to look into other pathways involved in anabolic/catabolic processes. We found that the GO term denoting protein translation was identified in down‐regulated genes. GSEA results also demonstrated that genes with terms ‘protein translation’ and ‘translation initiation factors (eIFs)’ were predominantly down‐regulated (Figure 3F and 3J).
We next sought to determine whether miR‐322/miR‐503 directly targets eIFs. A search by TargetScan revealed that eIF4E, eIF4G1, eIF4B, eIF2B5, and eIF3M harbour miR‐424/miR‐503 targeting sites on their 3′‐UTRs (Figure 4A). We appended these 3′‐UTRs individually to the firefly luciferase open reading frame and performed reporter assays by co‐transfecting the reporters with miR‐322/miR‐503 into 293T cells. miR‐322/miR‐503 significantly reduced luciferase activity of the Luc‐eIF4G1‐3′‐UTR, Luc‐eIF4B‐3′‐UTR, Luc‐eIF2B5‐3′‐UTR, and Luc‐eIF3M‐3′‐UTR reporters, whereas point mutations within the putative targeting sites abolished the effect. These results suggest that eIF4G1, eIF4B, eIF2B5, and eIF3M are direct targets of miR‐322/miR‐503 (Figure 4B). The predicted targeting site on eIF4E 3′‐UTR did not mediate a reduction of Luc activity in our assays (Figure 4B). Whether additional targeting sites may mediate the interaction between miR‐322/miR‐503 and eIF4E has not been determined. Further, the transcripts of these eIFs were significantly reduced in tg muscles compared with wt (Figure 4C). The levels of eIF4E, eIF4G1, and eIF4B proteins were markedly lower in tg animals (Figure 4D and 4E), whereas the levels of eIF2B5 and eIF3M proteins were low and variable, and we could not reliably determine their changes. We next confirmed that miR‐322/miR‐503 reduced myofibre size and expression of eIF proteins in established myotubes (Data S1 and Figure S2). Phosphorylation of eIF2α at Ser51 blocks the formation of the pre‐initiation complex and halts global protein translation. 24 However, our results demonstrated that there was no significant difference in the levels of phosphorylated eIF2α in skeletal muscle of wt and tg mice (Figure 4D and 4E). In agreement with the reduced levels of eIFs, the rate of protein synthesis, evaluated by puromycin incorporation assay, was considerably reduced in tg animals (Figures 4F, 4G, S1G, and S1H). Collectively, these results suggest that miR‐322/miR‐503 primarily targets translation initiation factors and attenuates the rate of protein synthesis in skeletal muscle.
Figure 4.
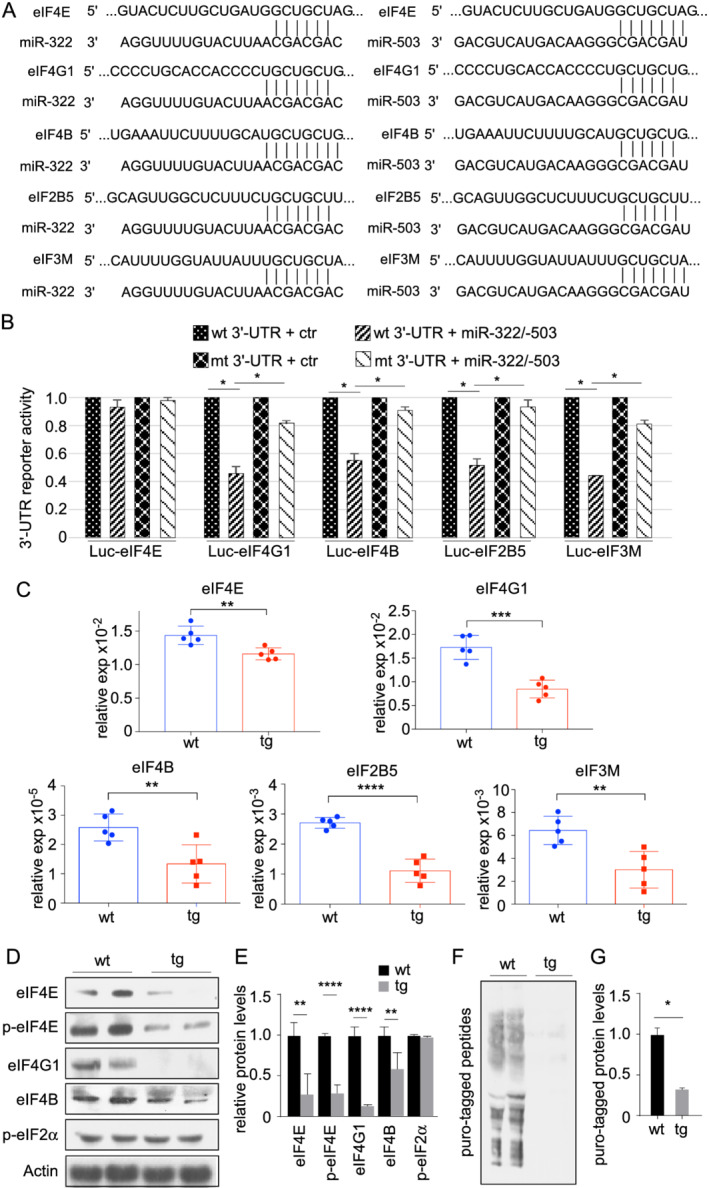
Several initiation factors are directly targeted by miR‐322/miR‐503. (A) Putative miR‐322/miR‐503 recognition sites on the 3′‐UTR of eIF4E, eIF4G1, eIF4B, eIF2B5, and eIF3M, as predicted by TargetScan. (B) miR‐322/miR‐503 inhibited luciferase activity of a firefly luciferase reporter fused with 3′‐UTRs from eIF4G1, eIF4B, eIF2B5, and eIF3M, but not eIF4E. Mutation of the putative recognition sites abolished the inhibition. (C) Confirmation of the down‐regulation of eIF4E, eIF4G1, eIF4B, eIF2B5, and eIF3M in additional transgenic muscles by RT‐PCR (n = 5 vs. 5). (D) Expression of eIF4E, phospho‐eIF4E, eIF4G1, and eIF4B proteins was down‐regulated in transgenic muscles. The expression of phospho‐eIF2α was unchanged. (E) Quantification of protein expression levels of eIFs (n = 4 vs. 4). (F) New protein synthesis was inhibited in transgenic muscles as determined by SUnSET (surface sensing of translation). Mice were labelled with 0.04 μmol/g puromycin for 30 min. (G) Quantification of levels of puromycin‐tagged peptides (n = 4 vs. 4). * P < 0.05, ** P < 0.01, *** P < 0.001, and **** P < 0.0001.
Genetic inactivation of the H19X locus promotes muscle growth and inhibits muscle atrophy
To further establish that H19X miRNAs are negative regulators of skeletal muscle growth, we investigated whether their ablation promotes muscle hypertrophy in vivo. The H19X‐LacZ mouse strain is defective in transcribing H19X miRNAs, because of an upstream poly‐A transcription termination signal (Figure 5A). We confirmed that the expression of miR‐322, miR‐503, and miR‐542 was diminished in skeletal muscles of mt (i.e. H19X‐LacZ) mice (Figure 5B). Mutant mice showed overgrowth starting from 4 weeks of age (not shown). At 6 months, body weight of mt mice was significantly higher compared with age‐matched wt mice (31.88 ± 1.0 vs. 39.5 ± 1.9 g, wt vs. mt) (Figure 5C). Furthermore, wet weight of hindlimb muscle was higher in mt mice than in controls, with the gastrocnemius and quadriceps muscle showing significant differences (gastrocnemius, 0.1608 ± 0.0066 vs. 0.1847 ± 0.0058 g; Quad, 0.2360 ± 0.0114 vs. 0.2639 ± 0.0052 g, wt vs. mt) (Figure 5D). Morphometric analysis depicted a shift towards thicker myofibre in skeletal muscle of mt mice (Figures 5E, S3A, and S3B). There was no significant difference in the percentage of glycolytic and oxidative fibres in skeletal muscle of control and mt mice, as assayed with succinate dehydrogenase staining (Figure 5F and 5G). Altogether, inactivation of the H19X locus causes hypertrophy in both glycolytic and oxidative myofibres.
Figure 5.
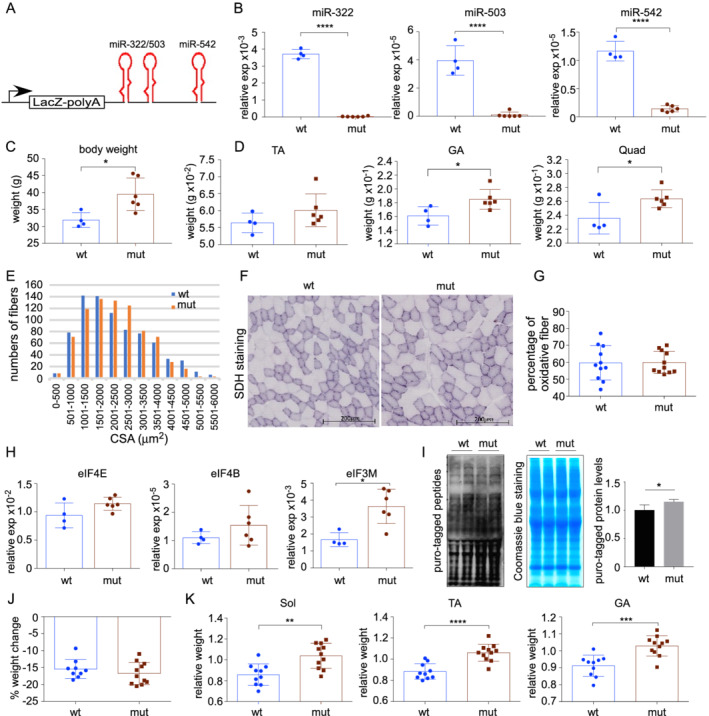
Inactivation of the H19X locus promoted muscle growth and rendered resistance to starvation‐induced atrophy. (A) Diagram of the insertion mutation that blocks downstream transcription in the H19X locus. (B) Quantification of miR‐322, miR‐503, and miR‐542 expression in mutant vs. control muscles (n = 4 vs. 6). (C) Mutant animals had higher body weights than control animals at 6 months old (n = 4 vs. 6). (D) Hindlimb muscle masses were higher in mutant animals than in control animals (n = 4 vs. 6). (E) Frequency histograms of CSA in mutant vs. control muscles (n = 5 vs. 5). (F) Representative images of SDH staining. (G) Percentage of oxidative fibres as determined by SDH staining (n = 11 vs. 11). (H) Quantification of eIF4E, eIF4B, and eIF3M expression in mutant vs. control muscles (n = 4 vs. 6). (I) New protein synthesis was up‐regulated in mutant muscles. (J) Body weight loss was comparable in mutant and control animals after 48 h starvation (n = 9 vs. 11). (K) Interruption of the H19X locus partially rescued starvation‐induced muscle wasting (n = 10 vs. 11). Shown are muscle weight/body weight ratios of starved animals normalized to that of regularly fed control littermates.
Evaluation of the expression levels of eIFs showed that eIF3M was significantly higher in mt mice, whereas increases in eIF4E, eIF4G1, eIF4B, and eIF2B5 were not significant (Figure 5H and data not shown). Perhaps there are parallel or compensatory mechanisms that also contribute to the regulation of eIF abundance; hence, the effect of loss of H19X miRNAs was partially masked. There was a significant increase in protein synthesis in mt muscles (Figure 5I). In contrast, the amount of ubiquitin‐tagged proteins was not changed (Figure S3C and S3D), suggesting H19X miRNAs control muscle protein synthesis but not degradation.
We demonstrated earlier that starvation stimulates the expression of miR‐322 and miR‐542, two miRNAs that are associated with muscle atrophy and cachexia in human chronic diseases. We tested whether inactivating the H19X locus can rescue starvation‐induced skeletal muscle atrophy. In H19X mt and control animals, 48 h starvation resulted in comparable loss of body weight (15.4 ± 0.9% in wt and 16.7 ± 0.9% in mt) (Figure 5J). Importantly, mt animals maintained more hindlimb muscle mass, as shown by 12.8–21.0% increases in muscle weight to body weight ratio (Figure 5K). Body composition analyses showed that mt animals have more fat (Figure S4A–S4C). After starvation, more fat than muscle was lost in mt animals (Figure S4D and S4E). These results suggest that inactivation of the H19X locus exerted a system‐wide anabolic effect that changes body composition and preserves muscle while consuming fat during starvation.
Discussion
H19X‐encoded miR‐322/miR‐503 has lineage‐specifying roles for the cardiac and skeletal muscles during development. 17 Here, we showed that skeletal muscle expression of miR‐322/miR‐503 diminishes after birth. While exercise suppresses the expression of miR‐322/miR‐503 even further, starvation up‐regulates it. With mouse genetic models, we showed that ectopic miR‐322/miR‐503 is sufficient to impede muscle growth, whereas inactivation of the H19X locus affecting miR‐322/miR‐503 and other miRNAs promotes muscle growth and reduces muscle wasting upon starvation. These results are consistent with the notion that miR‐322/miR‐503 is a novel regulator of skeletal muscle mass in distinct conditions.
miR‐322/miR‐503 is among the most up‐regulated miRNAs in serum‐starved fibroblasts. 25 In actively proliferating cells, overexpression of miR‐322/miR‐503 promotes cell cycle exit by targeting cyclin D1, cyclin D3, cyclin E1, and CDC25A. 26 , 27 It appears that miR‐322/miR‐503 plays distinct roles in skeletal muscle and cultured myotubes, both of which are terminally differentiated non‐cycling cells. Our results demonstrate that starvation induces the expression of miR‐322/miR‐503, which in turn inhibits protein translation by targeting translation initiation factors in muscle cells. It is widely established that cells shut down protein translation during prolonged periods of nutrient starvation as an adaptive means for survival. 28 , 29 , 30 Starvation attenuates the IGF1/AKT/mTOR pathway, leading to dephosphorylation of 4EBP1 and preventing formation of initiation complex eIF4F. The present study demonstrates that miR‐322/miR‐503 represents a new dimension in the suppression of translation initiation during nutrient starvation. Further, regulation over global protein translation is a conserved cellular response to many stresses, such as oxygen shock (hypoxia and oxidative stress), temperature shock (heat shock and cold shock), and DNA damage. 31 miR‐424 is among the most up‐regulated miRNAs during hypoxic and ischaemic stresses 32 , 33 , 34 ; therefore, it will be interesting to determine if miR‐322/miR‐503 is a common effector of stress‐induced inhibition of translation initiation.
Post‐translational modification of eIFs and associated proteins is an important node of regulation over protein translation. In this study, eIF4E, eIF4G1, eIF4B, eIF2B5, and eIF3M were significantly down‐regulated by miR‐322/miR‐503, with eIF4G1, eIF4B, eIF2B5, and eIF3M being directly targeted. eIF4E, eIF4G1, and eIF4B are components of the eIF4F complex. Therefore, it is possible that lower eIF4E, eIF4G1, and eIF4B levels reduce the formation of eIF4F complexes. eIF2B5 is a subunit of eIF2B, which is the guanine nucleotide exchange factor for eIF2 and is responsible for converting inactive eIF2‐GDP to the active eIF2‐GTP. Mutations in eIF2B subunits decrease guanine nucleotide exchange factor activity and underlie encephalopathy in eIF2B‐related diseases. 35 eIF3M is a subunit of eIF3, which acts as a scaffold for the 40S ribosome and other eIFs to coordinate responses to stimuli that call for protein translation. Knockout of eIF3M results in embryonic growth retardation and perinatal lethality. 36 Given the large number of eIFs that were down‐regulated by ectopic miR‐322/miR‐503, we did not test if the abundance of some eIFs was more important than others in affecting muscle growth. Our work supports the notion that the abundance of eIFs is a node of regulation in protein translation and that miR‐322/miR‐503 is an effector of such regulation (Figure 6).
Figure 6.
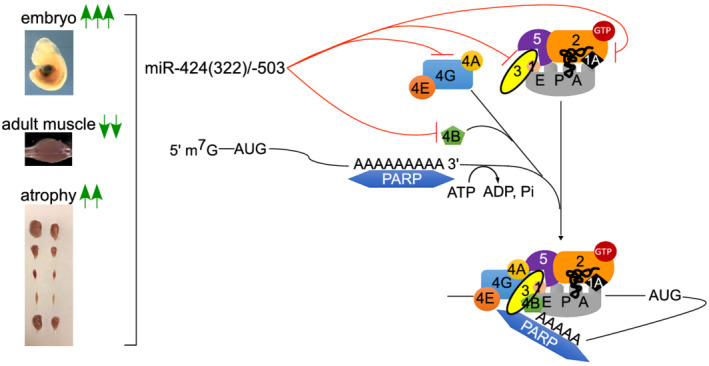
A working model of miR‐322(424)/miR‐503 in regulating multiple eIFs during translation initiation.
A recent study demonstrated that miR‐424‐5p targets genes essential for ribosomal RNA transcription: PolR1A and upstream binding transcription factor (UBTF) during muscle wasting. 14 We did not find changes in the abundance of PolR1A, UBTF, or rRNAs in either miR‐322/miR‐503 tg or H19X mt animals (not shown). It is possible that the effect of miR‐424‐5p on rRNA synthesis is compensated by other mechanisms in vivo. Nonetheless, these studies agree that miR‐322(424)/miR‐503 regulates fundamental processes converging on protein translation. Sarkar et al. reported that miR‐322/miR‐503 was induced during myotube formation in C2C12 cells, promoting cell cycle quiescence by directly targeting Cdc25A. 37 Although cell cycle exit is important for muscle differentiation, loss of miR‐322/miR‐503 seems tolerable, because H19X mt animals are not defective in muscle differentiation.
As miR‐322/miR‐503 was most highly expressed in embryonic muscle progenitors, we propose that the reactivation of miR‐322/miR‐503 in stress situations befits the theory of ‘foetal gene reactivation’. Stress signals drive miR‐322/miR‐503 expression as an adaptation mechanism to shutting down translation to allow other vital processes; however, persistent high levels of miR‐322/miR‐503 block muscle growth and in turn causing muscle atrophy. The present study for the first time links H19X‐encoded miRNAs to the regulation of muscle growth. Neutralizing the activities of these miRNAs may provide a new means for alleviating muscular atrophy.
Conflict of interest
None declared.
Funding
This study was supported by research funds from University of Houston (to Y.L. and R.J.S.), Texas Heart Institute (to R.J.S.), American Heart Association grants (11SDG5260033 and 16GRNT27760164 to Y.L.), Congressionally Directed Medical Research Programs (PR162075 to Y.L.), and National Institutes of Health grants (AR059810 and AR068313 to A.K.).
Ethics statement
The authors certify that they comply with the ethical guidelines for authorship and publishing in the Journal of Cachexia, Sarcopenia and Muscle. 38 Animal studies have been approved by University of Houston Institutional Animal Care and Use Committee and have therefore been performed in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments. The manuscript does not contain clinical studies or patient data.
Supporting information
Figure S1. Histological and molecular analyses of MCK‐miR‐322/‐503 muscles. (A) H&E staining of TA muscle sections. Scale bar, 100 μm. (B) Tg muscles had smaller mean min. Ferret diameters (n = 5 vs. 5). *, p < 0.05. (C) Frequency histogram of min. Ferret diameters (n = 5 vs. 5). (D) Expression levels of E3 ubiquitin ligases Atrogin‐1 and MuRF1 in tg vs. control muscles (n = 7 vs. 6). ns, not significant. (E) Measurement of levels of Ub‐tagged peptides by western blot. Coomassie blue staining serves as loading control. (F) Quantification of levels of ubiquitin‐tagged peptides (n = 4 vs. 4). ns, not significant. (G) Measurement of levels of puromycin‐tagged peptides by western blot. Coomassie blue staining serves as loading control. (H) Quantification of levels of puro‐tagged peptides (n = 4 vs. 4). *, p < 0.05.
Figure S2. miR‐322/‐503 overexpression induces myotube atrophy. (A) Dox induced ∼2 folds of miR‐322 and miR‐503 expression in a tet‐on temporal expression system in C2C12 cells. (B) Micrographs of C2C12‐derived myotubes with induced expression of miR‐322 and miR‐503 at day 1–3 and day 4–6 of differentiation. (C) Quantification of myotube areas in miR‐322/‐503‐overexpressing cells. (D) Analyses of eIF protein expression in miR‐322/‐503‐overexpressing myotubes. (E) Quantification of protein levels of eIFs in miR‐322/‐503‐overexpressing myotubes (n = 3 vs. 3).
Figure S3. Histological and molecular analyses of muscles from H19X mutant mice. (A) H&E staining of H19X mutant muscle sections. Scale bar, 100 μm. (B) Quantification of mean CSA in TA muscles (n = 5 vs. 5). (C) Frequency histogram and mean of min. Ferret diameters (n = 5 vs. 5). (D) Measurement of levels of ubiquitin‐tagged peptides by western blot. Coomassie blue staining serves as loading control. (E) Quantification of levels of ubiquitin‐tagged peptides (n = 4 vs. 4). ns, not significant.
Figure S4. Body composition analyses of H19X mutant mice before and after starvation. (A) Body mass of 6‐month wild‐type and mutant mice at baseline. (B) Lean mass content is less in mutant mice. *, p < 0.05. (C) Fat mass is greater in mutant mice. *, p < 0.05. (D) The loss of lean mass is less in mutant mice after 48 h starvation. *, p < 0.05. (E) The loss of fat mass is greater in mutant mice after 48 h starvation. N = 3 in each group. wt, wild‐type; mt, mutant.
Data S1. Supporting Information.
Acknowledgements
We thank Michael McManus at University of California and Didier Trono at École Polytechnique Fédérale de Lausanne for providing the reagents.
Liang R., Shen X., Wang F., Wang X., DesJarlais A., Syed A., Saba R., Tan Z., Yu F., Ji X., Shrestha S., Ren Y., Yang J., Park Y., Schwartz R. J., Soibam B., McConnell B. K., Stewart M. D., Kumar A., and Liu Y. (2021) H19X‐encoded miR‐322(424)/miR‐503 regulates muscle mass by targeting translation initiation factors, Journal of Cachexia, Sarcopenia and Muscle, 12, 2174–2186, 10.1002/jcsm.12827
References
- 1. Bonaldo P, Sandri M. Cellular and molecular mechanisms of muscle atrophy. Dis Model Mech 2013;6:25–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jackman RW, Kandarian SC. The molecular basis of skeletal muscle atrophy. Am J Physiol Cell Physiol 2004;287:C834–C843. [DOI] [PubMed] [Google Scholar]
- 3. Cohen S, Nathan JA, Goldberg AL. Muscle wasting in disease: molecular mechanisms and promising therapies. Nat Rev Drug Discov 2015;14:58–74. [DOI] [PubMed] [Google Scholar]
- 4. Latres E, Amini AR, Amini AA, Griffiths J, Martin FJ, Wei Y, et al. Insulin‐like growth factor‐1 (IGF‐1) inversely regulates atrophy‐induced genes via the phosphatidylinositol 3‐kinase/Akt/mammalian target of rapamycin (PI3K/Akt/mTOR) pathway. J Biol Chem 2005;280:2737–2744. [DOI] [PubMed] [Google Scholar]
- 5. Sandri M, Sandri C, Gilbert A, Skurk C, Calabria E, Picard A, et al. Foxo transcription factors induce the atrophy‐related ubiquitin ligase atrogin‐1 and cause skeletal muscle atrophy. Cell 2004;117:399–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Stitt TN, Drujan D, Clarke BA, Panaro F, Timofeyva Y, Kline WO, et al. The IGF‐1/PI3K/Akt pathway prevents expression of muscle atrophy‐induced ubiquitin ligases by inhibiting FOXO transcription factors. Mol Cell 2004;14:395–403. [DOI] [PubMed] [Google Scholar]
- 7. McFarlane C, Plummer E, Thomas M, Hennebry A, Ashby M, Ling N, et al. Myostatin induces cachexia by activating the ubiquitin proteolytic system through an NF‐κB‐independent, FoxO1‐dependent mechanism. J Cell Physiol 2006;209:501–514. [DOI] [PubMed] [Google Scholar]
- 8. Morissette MR, Cook SA, Foo S, McKoy G, Ashida N, Novikov M, et al. Myostatin regulates cardiomyocyte growth through modulation of Akt signaling. Circ Res 2006;99:15–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Simionescu‐Bankston A, Kumar A. Noncoding RNAs in the regulation of skeletal muscle biology in health and disease. J Mol Med (Berl) 2016;94:853–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ambros V. The functions of animal microRNAs. Nature 2004;431:350–355. [DOI] [PubMed] [Google Scholar]
- 11. Narasimhan A, Ghosh S, Stretch C, Greiner R, Bathe OF, Baracos V, et al. Small RNAome profiling from human skeletal muscle: novel miRNAs and their targets associated with cancer cachexia. J Cachexia Sarcopenia Muscle 2017;8:405–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Garros RF, Paul R, Connolly M, Lewis A, Garfield BE, Natanek SA, et al. MicroRNA‐542 promotes mitochondrial dysfunction and SMAD activity and is elevated in intensive care unit‐acquired weakness. Am J Respir Crit Care Med 2017;196:1422–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Necsulea A, Soumillon M, Warnefors M, Liechti A, Daish T, Zeller U, et al. The evolution of lncRNA repertoires and expression patterns in tetrapods. Nature 2014;505:635–640. [DOI] [PubMed] [Google Scholar]
- 14. Connolly M, Paul R, Farre‐Garros R, Natanek SA, Bloch S, Lee J, et al. miR‐424‐5p reduces ribosomal RNA and protein synthesis in muscle wasting. J Cachexia Sarcopenia Muscle 2018;9:400–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. de Andrade HM, de Albuquerque M, Avansini SH, de S Rocha C, Dogini DB, Nucci A, et al. MicroRNAs‐424 and 206 are potential prognostic markers in spinal onset amyotrophic lateral sclerosis. J Neurol Sci 2016;368:19–24. [DOI] [PubMed] [Google Scholar]
- 16. Kovanda A, Leonardis L, Zidar J, Koritnik B, Dolenc‐Groselj L, Ristic Kovacic S, et al. Differential expression of microRNAs and other small RNAs in muscle tissue of patients with ALS and healthy age‐matched controls. Sci Rep 2018;8:5609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shen X, Soibam B, Benham A, Xu X, Chopra M, Peng X, et al. miR‐322/‐503 cluster is expressed in the earliest cardiac progenitor cells and drives cardiomyocyte specification. Proc Natl Acad Sci U S A 2016;113:9551–9556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bruning JC, Michael MD, Winnay JN, Hayashi T, Horsch D, Accili D, et al. A muscle‐specific insulin receptor knockout exhibits features of the metabolic syndrome of NIDDM without altering glucose tolerance. Mol Cell 1998;2:559–569. [DOI] [PubMed] [Google Scholar]
- 19. Park CY, Jeker LT, Carver‐Moore K, Oh A, Liu HJ, Cameron R, et al. A resource for the conditional ablation of microRNAs in the mouse. Cell Rep 2012;1:385–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Peng X, Shen X, Chen X, Liang R, Azares AR, Liu Y. Celf1 regulates cell cycle and is partially responsible for defective myoblast differentiation in myotonic dystrophy RNA toxicity. Biochim Biophys Acta 2015;1852:1490–1497. [DOI] [PubMed] [Google Scholar]
- 21. Ciciliot S, Rossi AC, Dyar KA, Blaauw B, Schiaffino S. Muscle type and fiber type specificity in muscle wasting. Int J Biochem Cell Biol 2013;45:2191–2199. [DOI] [PubMed] [Google Scholar]
- 22. Shen H, Liu T, Fu L, Zhao S, Fan B, Cao J, et al. Identification of microRNAs involved in dexamethasone‐induced muscle atrophy. Mol Cell Biochem 2013;381:105–113. [DOI] [PubMed] [Google Scholar]
- 23. Llobet‐Navas D, Rodriguez‐Barrueco R, Castro V, Ugalde AP, Sumazin P, Jacob‐Sendler D, et al. The miR‐424(322)/503 cluster orchestrates remodeling of the epithelium in the involuting mammary gland. Genes Dev 2014;28:765–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science 2011;334:1081–1086. [DOI] [PubMed] [Google Scholar]
- 25. Rissland OS, Hong SJ, Bartel DP. MicroRNA destabilization enables dynamic regulation of the miR‐16 family in response to cell‐cycle changes. Mol Cell 2011;43:993–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Xu YY, Wu HJ, Ma HD, Xu LP, Huo Y, Yin LR. MicroRNA‐503 suppresses proliferation and cell‐cycle progression of endometrioid endometrial cancer by negatively regulating cyclin D1. FEBS J 2013;280:3768–3779. [DOI] [PubMed] [Google Scholar]
- 27. Llobet‐Navas D, Rodriguez‐Barrueco R, de la Iglesia‐Vicente J, Olivan M, Castro V, Saucedo‐Cuevas L, et al. The microRNA 424/503 cluster reduces CDC25A expression during cell cycle arrest imposed by transforming growth factor β in mammary epithelial cells. Mol Cell Biol 2014;34:4216–4231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Samarel AM, Parmacek MS, Magid NM, Decker RS, Lesch M. Protein synthesis and degradation during starvation‐induced cardiac atrophy in rabbits. Circ Res 1987;60:933–941. [DOI] [PubMed] [Google Scholar]
- 29. Magnusson K, Wahren J, Ekman L. Protein synthesis in skeletal muscle during starvation and refeeding: comparison of data from intact muscle and muscle biopsy material. Metabolism 1990;39:1113–1117. [DOI] [PubMed] [Google Scholar]
- 30. Essen P, McNurlan MA, Wernerman J, Milne E, Vinnars E, Garlick PJ. Short‐term starvation decreases skeletal muscle protein synthesis rate in man. Clin Physiol 1992;12:287–299. [DOI] [PubMed] [Google Scholar]
- 31. Holcik M, Sonenberg N. Translational control in stress and apoptosis. Nat Rev Mol Cell Biol 2005;6:318–327. [DOI] [PubMed] [Google Scholar]
- 32. Ghosh G, Subramanian IV, Adhikari N, Zhang X, Joshi HP, Basi D, et al. Hypoxia‐induced microRNA‐424 expression in human endothelial cells regulates HIF‐α isoforms and promotes angiogenesis. J Clin Invest 2010;120:4141–4154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhang D, Shi Z, Li M, Mi J. Hypoxia‐induced miR‐424 decreases tumor sensitivity to chemotherapy by inhibiting apoptosis. Cell Death Dis 2014;5:e1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liu P, Zhao H, Wang R, Wang P, Tao Z, Gao L, et al. MicroRNA‐424 protects against focal cerebral ischemia and reperfusion injury in mice by suppressing oxidative stress. Stroke 2015;46:513–519. [DOI] [PubMed] [Google Scholar]
- 35. Dietrich J, Lacagnina M, Gass D, Richfield E, Mayer‐Proschel M, Noble M, et al. EIF2B5 mutations compromise GFAP+ astrocyte generation in vanishing white matter leukodystrophy. Nat Med 2005;11:277–283. [DOI] [PubMed] [Google Scholar]
- 36. Zeng L, Wan Y, Li D, Wu J, Shao M, Chen J, et al. The m subunit of murine translation initiation factor eIF3 maintains the integrity of the eIF3 complex and is required for embryonic development, homeostasis, and organ size control. J Biol Chem 2013;288:30087–30093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sarkar S, Dey BK, Dutta A. MiR‐322/424 and ‐503 are induced during muscle differentiation and promote cell cycle quiescence and differentiation by down‐regulation of Cdc25A. Mol Biol Cell 2010;21:2138–2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2019. J Cachexia Sarcopenia Muscle 2019;10:1143–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Histological and molecular analyses of MCK‐miR‐322/‐503 muscles. (A) H&E staining of TA muscle sections. Scale bar, 100 μm. (B) Tg muscles had smaller mean min. Ferret diameters (n = 5 vs. 5). *, p < 0.05. (C) Frequency histogram of min. Ferret diameters (n = 5 vs. 5). (D) Expression levels of E3 ubiquitin ligases Atrogin‐1 and MuRF1 in tg vs. control muscles (n = 7 vs. 6). ns, not significant. (E) Measurement of levels of Ub‐tagged peptides by western blot. Coomassie blue staining serves as loading control. (F) Quantification of levels of ubiquitin‐tagged peptides (n = 4 vs. 4). ns, not significant. (G) Measurement of levels of puromycin‐tagged peptides by western blot. Coomassie blue staining serves as loading control. (H) Quantification of levels of puro‐tagged peptides (n = 4 vs. 4). *, p < 0.05.
Figure S2. miR‐322/‐503 overexpression induces myotube atrophy. (A) Dox induced ∼2 folds of miR‐322 and miR‐503 expression in a tet‐on temporal expression system in C2C12 cells. (B) Micrographs of C2C12‐derived myotubes with induced expression of miR‐322 and miR‐503 at day 1–3 and day 4–6 of differentiation. (C) Quantification of myotube areas in miR‐322/‐503‐overexpressing cells. (D) Analyses of eIF protein expression in miR‐322/‐503‐overexpressing myotubes. (E) Quantification of protein levels of eIFs in miR‐322/‐503‐overexpressing myotubes (n = 3 vs. 3).
Figure S3. Histological and molecular analyses of muscles from H19X mutant mice. (A) H&E staining of H19X mutant muscle sections. Scale bar, 100 μm. (B) Quantification of mean CSA in TA muscles (n = 5 vs. 5). (C) Frequency histogram and mean of min. Ferret diameters (n = 5 vs. 5). (D) Measurement of levels of ubiquitin‐tagged peptides by western blot. Coomassie blue staining serves as loading control. (E) Quantification of levels of ubiquitin‐tagged peptides (n = 4 vs. 4). ns, not significant.
Figure S4. Body composition analyses of H19X mutant mice before and after starvation. (A) Body mass of 6‐month wild‐type and mutant mice at baseline. (B) Lean mass content is less in mutant mice. *, p < 0.05. (C) Fat mass is greater in mutant mice. *, p < 0.05. (D) The loss of lean mass is less in mutant mice after 48 h starvation. *, p < 0.05. (E) The loss of fat mass is greater in mutant mice after 48 h starvation. N = 3 in each group. wt, wild‐type; mt, mutant.
Data S1. Supporting Information.