

SUMMARY
Cytotoxic T lymphocyte (CTL) responses against tumors are maintained by stem-like memory cells that self-renew, but also give rise to effector-like cells. The latter gradually lose their anti-tumor activity and acquire an epigenetically fixed, hypofunctional state, leading to tumor tolerance. Here, we show that the conversion of stem-like into effector-like CTL involves a major chemotactic reprogramming that includes the upregulation of chemokine receptor CXCR6. This receptor positions effector-like CTL in a discrete perivascular niche of the tumor stroma that is densely occupied by CCR7+ dendritic cells (DC) expressing the CXCR6 ligand CXCL16. CCR7+ DC also express and trans-present the survival cytokine IL-15. CXCR6 expression and IL-15 trans-presentation are critical for the survival and local expansion of effector-like CTL in the tumor microenvironment to maximize their anti-tumor activity before progressing to irreversible dysfunction. These observations reveal a cellular and molecular checkpoint that determines the magnitude and outcome of anti-tumor immune responses.
Keywords: TCF-1, CTL, CCR7+ dendritic cells, CXCR6, CXCL16, IL-15, Tumor Microenvironment, scRNA-seq, TCGA, Multiphoton Intravital Microscopy
eTOC Blurb
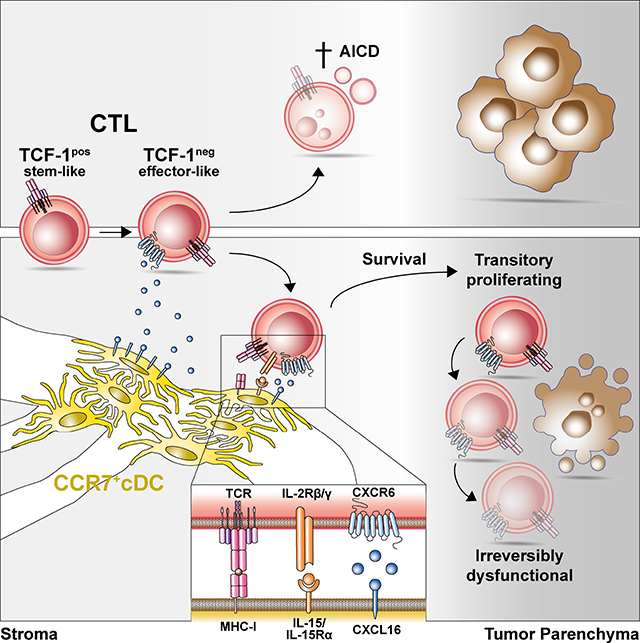
Intravital imaging reveals dense perivascular clusters of IL-12-competent DC3 in the stroma of immunogenic tumors. CXCL16 optimizes their interactions with CXCR6-expressing TCF-1neg effector-like CTL during which they trans-present IL-15 to CTL to protect them from activation-induced cell death, helping to locally sustain the CTL response for effective tumor control. Expression of CXCR6 in the solid tumor microenvironment is the strongest predictor of survival among all chemokine receptors in human cancer patients.
Graphical Abstract
INTRODUCTION
Successful clearance of viral infections by the immune system depends on CD8+ T cells that recognize intracellular pathogen-derived antigens. Clonal expansion of naive cells in lymphoid tissues produces short-lived effector cells that eliminate virally infected cells and produce IFN-γ to amplify the response, as well as precursors for different subsets of memory cells that persist after the infection has been cleared (Kaech and Cui, 2012). In contrast, failure to clear viruses leads to chronic infections and persistent, yet hypofunctional CTL responses characterized by a gradual decline in proliferative capacity, cytokine-secretion, and cytotoxic function of individual cells. This adapted response pattern, often referred to as T cell exhaustion, may serve to avoid immune-pathological damage to host tissues that would result from continued high-level immune activation (Hashimoto et al., 2018; Speiser et al., 2014). However, even exhausted CTL responses continue to limit viral replication (Jin et al., 1999; Schmitz et al., 1999). Many features of this equilibrium state between viruses and the immune system are replicated during immune responses against established tumors. Here, CTL that recognize mutational tumor neoantigens also exert varying levels of immune control but, similar to CTL in chronic viral infection, adopt a hypofunctional state.
Recent studies have revealed the heterogeneity and dynamics of the hypofunctional CTL populations observed in chronic viral infection and cancer (He et al., 2016; Im et al., 2016; Leong et al., 2016; Sade-Feldman et al., 2018; Snell et al., 2018; Utzschneider et al., 2016; Wu et al., 2016). These include stem-like CTL that express the HMG box transcription factor TCF-1 and the SLAM family member Slamf6/Ly108, possess the capacity for self-renewal, and are found primarily in lymphoid tissues but in smaller numbers also at immunological effector sites such as tumors (Miller et al., 2019; Siddiqui et al., 2019). TCF-1pos CTL continually give rise to TCF-1neg effector-like cells that acquire cytotoxic function, but also upregulate inhibitory receptors, such as TIM-3, predicted to attenuate their effector activity. TCF-1neg CTL include cells with a continuum of differentiation states ranging from highly proliferative and functional to irreversibly hypofunctional. Highly proliferative TCF-1neg CTL referred to as transitory CTL express the chemokine receptor CX3CR1 and mediate antiviral control during chronic viral infection. Terminally differentiated TCF-1neg CTL, on the other hand, are characterized by expression of CD101 and stable epigenetic repression of effector genes (Hudson et al., 2019; Li et al., 2019; Philip et al., 2017; Zander et al., 2019).
DC not only initiate anti-tumor responses in tumor-draining lymph nodes (tdLNs), but also support and regulate T cell functions in the tumor microenvironment (TME) (Gerhard et al., 2021; Wculek et al., 2020). Developmental studies have identified two subsets of conventional DC named cDC1 and cDC2 as well as plasmacytoid DC (pDC) as lineages distinct from monocytes, monocyte-derived DC, and macrophages (Murphy et al., 2016). cDC1 are more efficient at cross-presenting tumor cell-derived antigen to CTL (Broz et al., 2014), whereas cDC2 may be more relevant for CD4+ T cell activation (Binnewies et al., 2019).
We recently identified an intratumoral DC state characterized by co-expression of IL12b, Fascin1, and the chemokine receptor gene CCR7, which we initially classified as cDC1 (Garris et al., 2018). Re-analysis of these and additional mouse and human data led us to re-classify these cells as a discrete DC state we named DC3 (Gerhard et al., 2021; Zilionis et al., 2019). Others then reported on similar cell states in mouse and human tumors, to which they referred as LAMP3+ DC (Zhang et al., 2019), mregDC (Maier et al., 2020), or Ccl22+ cDC1 (Zhang et al., 2020). The respective roles of cDC1, cDC2 and DC3 in intratumoral CTL activation and specifically how these cells support the differentiation of stem- to effector-like and to terminally differentiated CTL requires further study.
An unanswered question is how CTL at various stages of differentiation navigate the TME in order to orchestrate their cross-talk with different DC subsets and ultimately to engage with their malignant target cells. Considering their well-established roles in lymphoid tissues, chemokines and their receptors are likely central orchestrators of this process. Inflammatory chemokine receptors such as CXCR3, CCR5, and CCR4 are generally assumed to be important for the recruitment of blood-borne T cells to tumor tissue, although this has only in some cases been directly demonstrated, e.g. for CXCR3 (Mikucki et al., 2015). In addition, CXCR3 guides the local positioning of T cells in both lymphoid and non-lymphoid tissues (Ariotti et al., 2015; Groom et al., 2012). Expression of the CXCR3 ligand CXCL9 specifically by cDC is required for the efficacy of anti-PD-1 cancer immune checkpoint therapy through mechanisms unrelated to T cell trafficking from tdLNs to tumor tissue, hinting at a role for organizing local cDC interactions with tumor-infiltrating CXCR3+ T cells (Chow et al., 2019). However, the full spectrum of chemokine receptors and their ligands expressed in the TME by both immune and non-immune cells, but in particular by CTL subsets, has not been systematically explored. Here we generated a comprehensive account of all chemokine and chemokine receptor genes expressed by all cells of the TME in mouse models of immunogenic cancer in order to provide a road-map for the systematic exploration of their roles in organizing cellular interactions. We identified CXCR6 as the most highly expressed chemokine receptor in tumor-infiltrating CTL, and DC3 as the cell state most highly expressing its ligand CXCL16. Using multiphoton intravital microscopy (MP-IVM) we found that CXCR6 optimizes the positioning of TCF-1neg CTL in perivascular clusters of DC3 in the tumor stroma and uncovered its critical role in rescuing the proliferative transitory CTL subset from activation-induced cell death (AICD) through exposure to trans-presented IL-15 cytokine, which was critical to sustain their population size and anti-tumor function.
RESULTS
CXCR6 is critical for CTL-mediated tumor control
In order to explore chemokine receptors expressed by tumor-infiltrating CTL, we used the immunogenic mouse melanoma model D4M.3A-pOVA (Di Pilato et al., 2019). Tumor single cell suspensions were enriched for immune cells and all single-cell transcriptomes annotated to cell states (see Experimental Details). We detected three main cell clusters containing T and NK cell, myeloid cell, and non-immune cell states, as well as three minor clusters classified as pDC, B cell, and mast cell states (Figures 1A, S1A). Comparisons to published scRNA-seq datasets revealed that T and NK cell states resembled those in MC38 mouse colorectal tumors and CD8 T cell states resembled those in ovalbumin (OVA)-expressing B16.F10 mouse melanoma and in spleens of lymphocytic choriomeningitis virus (LCMV)-infected mice (Figure S1B) (Miller et al., 2019; Zhang et al., 2020). DC states resembled those in KP1.9 mouse lung and in MC38 tumors (Figure S1C), mirroring DC state conservation observed across human solid cancers (Gerhard et al., 2021; Maier et al., 2020; Zhang et al., 2020; Zilionis et al., 2019). Cell state annotation was further validated by marker genes (Figure S1D) and distinct cell state-enriched gene-expression revealing known marker genes (Figure S1E and Table S1A).
Figure 1: CXCR6 is required for CTL-mediated tumor control.
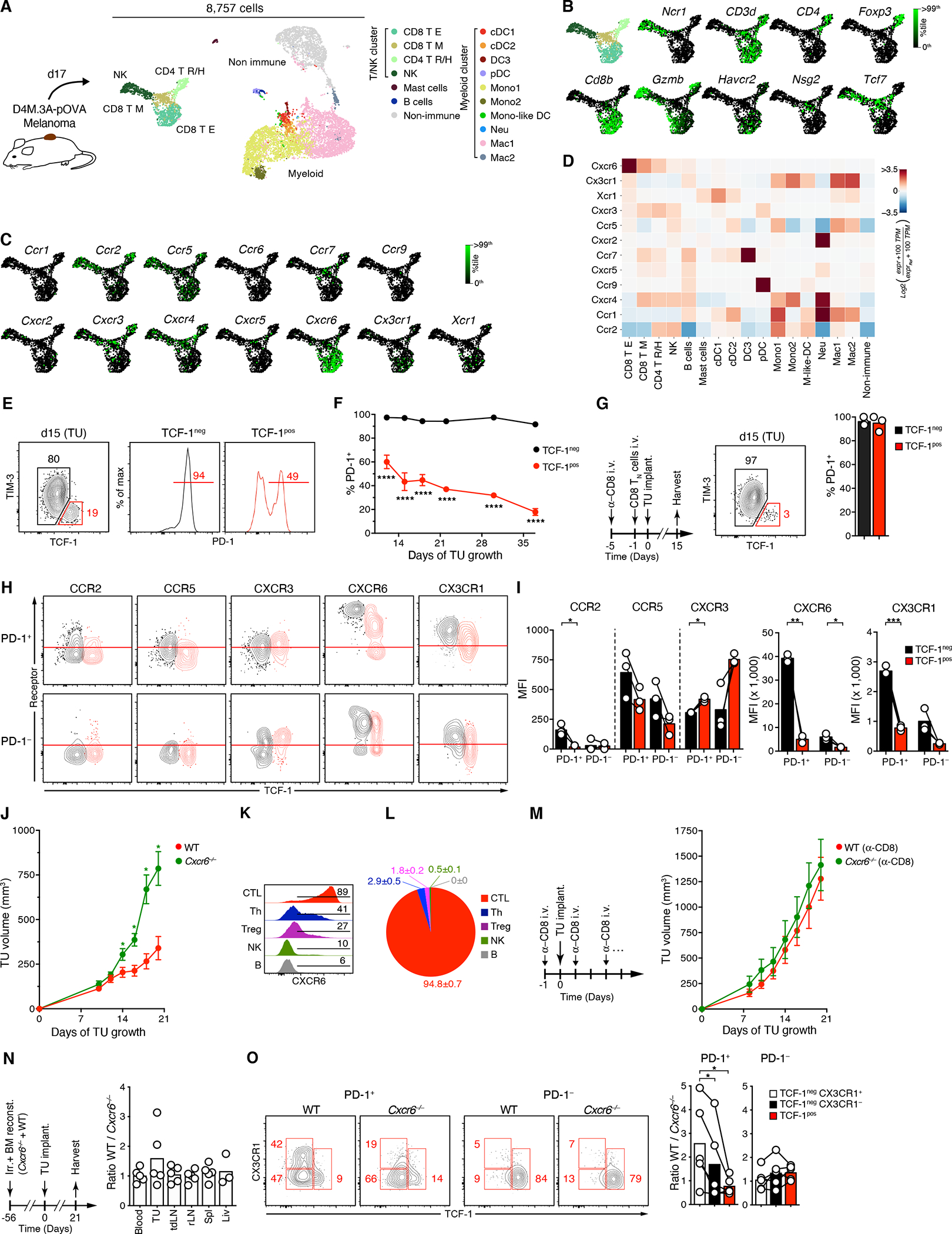
(A) scRNA-seq analysis of D4M.3A-pOVA melanoma expressing the SIINFEKL-peptide fused to histone H2B as a surrogate tumor neoantigen. CD45+ cells were separated from CD45− cells by FACS, recombined at a 9 to 1 ratio, and processed on the InDrops platform.
(B, C) Expression of marker (A) and chemokine receptor genes (B) in the T/NK cluster.
(D) Heatmap of chemokine receptor gene expression in all cell states. 100 TPM = average 100 transcripts per million of all cells. See Table S1B for numerical data underlying the heatmap.
(E, F) PD-1 protein expression by TCF-1pos TIM-3− and TCF-1neg CTL in D4M.3A-pOVA tumors on 15 day (E) and over time (F).
(G) PD-1 protein expression by TCF-1pos TIM-3− and TCF-1neg CTL in 15-day old D4M.3A-pOVA tumors implanted following depletion of endogenous CD8+ cells and transfer of 2.5 × 106 CD45.1 congenic, purified naive CD8+ T cells.
(H, I) Overlaid contour plots of chemokine receptor expression by pre-gated TCF-1pos (red) and TCF-1neg (black) PD-1+ (top) and PD-1− (bottom) CTL subsets from 18 days-old tumors. Red line indicates background fluorescence based on FMO controls (H). Background-corrected MFIs, note varying y-scales (I).
(J) Growth of s.c. D4M.3A-pOVA tumors in the flanks of WT or Cxcr6−/− mice.
(K) CXCR6 expression on tumor-infiltrating CD8+ (CTL), CD4+ Foxp3− (Th), CD4+ Foxp3+ (Treg), NKp46+ CD3− (NK), and B220+ cells (B).
(L) Contribution of each cell type to total CXCR6 expression in the TME based on the product of cell frequency, % CXCR6+ cells, and CXCR6 MFI of CXCR6+ cells.
(M) Growth of s.c. D4M.3A-pOVA tumors in CD8+ T cell-depleted WT or Cxcr6−/− mice.
(N-O) Ratios of CD45.1+ WT to CD45.2+ KO total CD8+ T cells in various tissues (N) and in the TCF-1pos CX3CR1−, TCF-1neg CX3CR1−, and TCF-1neg CX3CR1+ subsets of both PD-1+ and PD-1− tumor-infiltrating CTL (O) when s.c. D4M.3A-pOVA tumors reached a size >150 mm3 in Cxcr6−/− × WT −> WT BMCs.
Data in (E), (G-O) represent at least two independent replicates with similar results. Graphs show means and either individual replicates or ±SEM. */**/***/**** = p<0.05/0.01/0.001/0.0001.
The T/NK cell cluster contained an NK cell state (NK), a CD4+ T cell state containing both regulatory and helper T cells (CD4 T R/H), as well as two CD8+ T cell states annotated as effector-like cells (CD8 T E) and memory cells (CD8 T M). CD8 T E expressed the cytotoxic effector gene Gzmb and Havcr2 (encoding TIM-3), while CD8 T M expressed the memory gene Nsg2 (Best et al., 2013) and Tcf7, which encodes TCF-1 expressed by naive as well as stem-like CTL (Utzschneider et al., 2016) (Figure 1B)
The by far most highly expressed chemokine receptor gene in both CD8 T cell states was Cxcr6, followed by Cx3cr1 in the CD8 T E state, and by Cxcr4, Cxcr3, Ccr7, and lower amounts of Cxcr5 in the CD8 T M state (Figures 1C, 1D, and Table S1B). Cxcr6 was also present, but much less abundant, in some NK and CD4 T R/H cell states. When validating gene expression at the protein level by flow cytometry we used the T cell activation marker PD-1 (Honda et al., 2014) to focus our analysis on tumor-reactive CTL and exclude bystander CTL with other, for instance anti-viral reactivities (Rosato et al., 2019; Scheper et al., 2018; Simoni et al., 2018). While TCF-1neg effector-like cells expressed PD-1 almost uniformly, only a fraction of TIM-3− TCF-1pos stem-like CTL, which gradually declined over time, expressed this receptor (Figures 1E and 1F). When we transferred CD8-depleted mice with congenic, highly purified CD44low CD62Lhi naive CD8+ T cells that require prior activation to enter the TME, all of their tumor-infiltrating progeny, both TCF-1pos and TCF-1neg, expressed PD-1 (Figure 1G), similarly to adoptively transferred clonal populations of TCR transgenic OT-I cells recognizing the tumor cell-expressed SIINFEKL neoepitope (Figure S1F). Hence, PD-1 expression identifies tumor-reactive CTL.
CXCR6 was by far the most highly expressed chemokine receptor on CTL on day 18 of tumor growth, mirroring our transcriptional analysis (Figures 1H and 1I). Three discrete populations with negative/low, intermediate, and high expression were apparent among PD-1+ stem-like CTL, while PD-1+ effector-like CTL uniformly expressed CXCR6 at the highest level. Considering the lineage relationship between TCF-1pos and TCF-1neg cells (Siddiqui et al., 2019; Utzschneider et al., 2016), this pattern suggests that full CXCR6 upregulation immediately precedes or accompanies loss of TCF-1 expression in tumor-reactive PD-1+ CTL. In contrast, PD-1− CTL, both TCF-1pos or TCF-1neg, were either CXCR6-low/negative or -intermediate, but rarely high.
PD-1+ TCF-1neg CTL also upregulated CX3CR1, CCR5, and CCR2, but down-regulated CXCR3. Generally, inflammatory chemokine receptors were more highly expressed by PD-1+ than PD-1− CTL, indicating that they were induced or sustained in the TME through TCR activation (Figures 1H and 1I). Again, CXCR3 formed an exception and was most highly expressed by PD-1− TCF-1pos bystander CTL. Similar expression patterns were observed in a second melanoma model, YUMM1.1, as well as LLC1 lung carcinoma (Figures S2A, B). Loss of TCF-1 expression in stem-like CTL and the emergence of effector-like cells is therefore accompanied by a major chemotactic reprogramming.
To test the role of CXCR6 in tumor immunity, we implanted DM4.3A-pOVA, YUMM1.1, or LLC1 tumors into Cxcr6−/− or WT animals. While at least the melanoma models initially grew at a similar rate, growth of all three tumor types eventually rapidly accelerated in absence of CXCR6, suggesting loss of immune control (Figures 1J and S2C, D). In WT mice, CXCR6 was detected at low levels on CD4+ Th, Foxp3+ Treg, and NK cells (Figures 1K and S2E), but CTL accounted for 95% of total CXCR6 protein in the TME, considering both their frequency and level of expression (Figure 1L), suggesting that CXCR6 deletion affects anti-tumor immunity predominantly through its absence on CTL. Indeed, when we depleted CTL, D4M.3A-pOVA tumors grew similarly in Cxcr6−/− and WT mice (Figure 1M).
Impaired CTL-mediated tumor control in the absence of CXCR6 may result from reduced recruitment or persistence of tumor-reactive CTL in tumor tissue. To test this, we generated mixed Cxcr6 KO : WT −> WT irradiation bone marrow chimeras (BMCs). Three weeks after implantation of D4M.3A-pOVA tumors, we observed only moderate enrichment of WT over Cxcr6 KO CD8+ T cells in the TME, compared to tdLNs and a range of healthy tissues (Figure 1N). However, focusing the analysis on PD-1+ CTL revealed that WT cells outcompeted their CXCR6-deficient counterparts in the tumor-reactive TCF-1neg effector-like, and in particular in the CX3CR1+ transitory subset (Figure 1O). In contrast, TCF-1pos tumor-reactive, as well as PD-1− bystander cells were not affected by lack of CXCR6. We made analogous observations in non-competitive settings in YUMM1.1 or LLC1 tumors (Figure S2F, G). Hence, CXCR6 enables accumulation of effector-like CTL in tumor tissue, and is critical for their ability to control immunogenic tumors.
Local expansion of transitory effector-like CTL in the TME requires CXCR6
CXCR6 is expressed at low levels on naive CD8+ T cells (Kim et al., 2003; Matloubian et al., 2000), but was already upregulated on PD-1+ TCF-1neg CTL in tdLNs (Figure S2H), and CXCR6-intermediate, mostly TCF-1neg CTL emerged in the blood of tumor-bearing animals (Figure S2I, J). This raised the question whether CXCR6 primarily optimized CTL priming, supported their accumulation locally in the TME, or both. When we adoptively co-transferred naive Cxcr6−/− and WT OT-I cells into hosts with established D4M.3A-pOVA tumors, Cxcr6−/− cells exhibited only a slight delay in their proliferative response and induction of CD69 and CD25 in tdLNs over the first two days (Figures 2A–C and S2K, L), suggesting only a minor role for CXCR6 during CTL priming. Also five days after transfer, when TCF-1neg effector-like OT-I cells had emerged as a minor yet discrete population in tdLNs, but already formed the largest subset in tumor tissue (Figure 2D), WT OT-I cells were still only slightly more numerous than CXCR6-deficient cells at both sites (Figure 2E). However, over the next two days, TCF-1neg CTL dramatically expanded in tumor tissue, but not if they lacked CXCR6 (Figure 2E). During the following seven days, TCF-1neg WT cells again contracted by two thirds, while the already less abundant Cxcr6−/− cells contracted by 90% and almost completely vanished. TCF-1pos CTL followed similar overall trends, but expanded and contracted much less than TCF-1neg cells, irrespective of CXCR6 expression. As a result, TCF-1pos cells remained a minor subset of WT, but formed the majority of Cxcr6−/− cells in the TME at late time-points (Figure 2D). At this time, effector-like WT cells outnumbered their Cxcr6−/− counterparts more than 30 (31.5±11.9)-fold in tumor tissue, while this difference was less pronounced for stem-like cells (7.3±2.7-fold).
Figure 2: Expansion of highly proliferative effector-like CTL in the TME requires CXCR6.
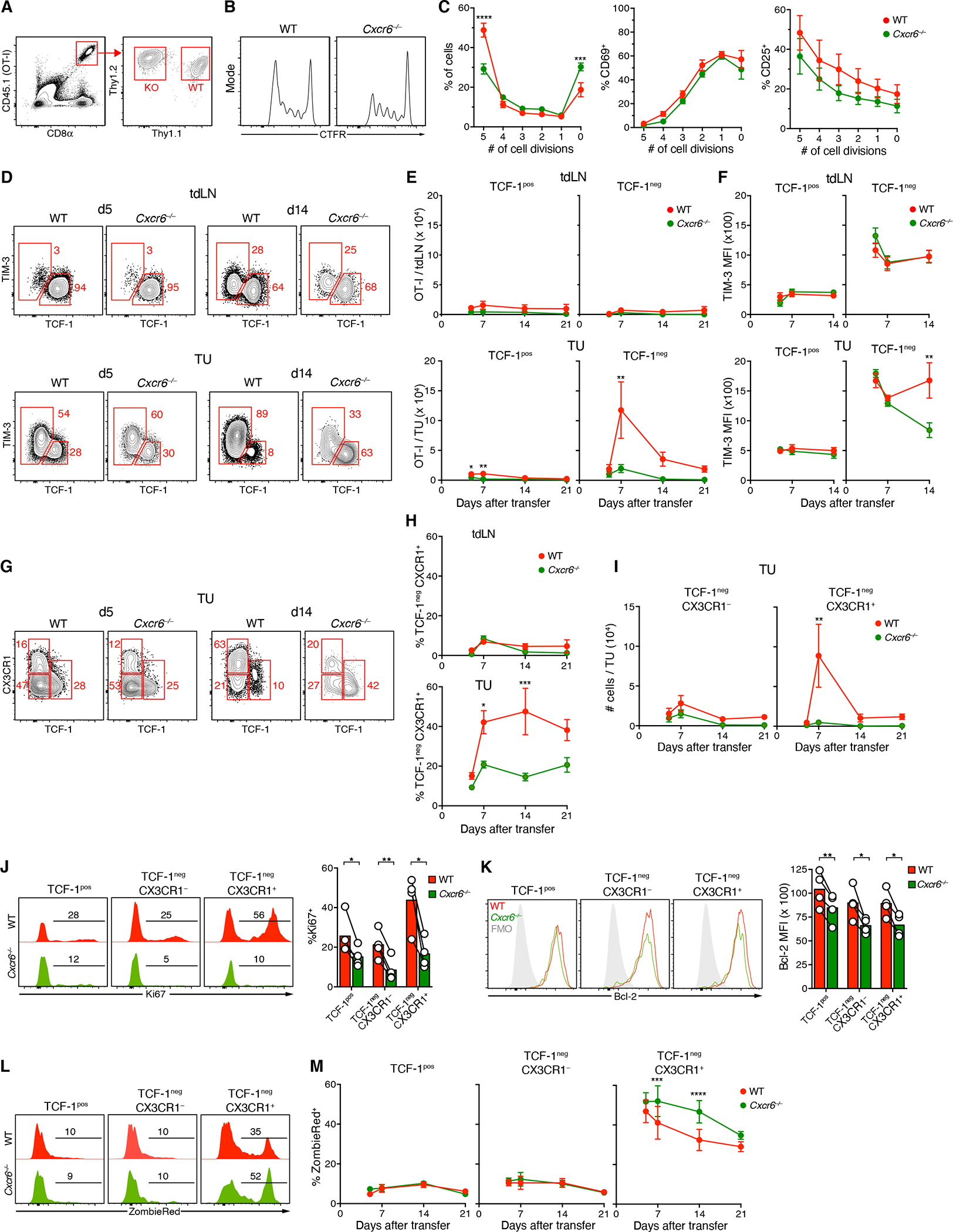
(A-C) Celltrace Far Red (CTFR)-labeled naive Thy1.1+ CD45.1+ WT OT-I and Thy1.2+ CD45.1+ Cxcr6−/− OT-I (2 × 106 cells of each) were i.v. injected into CD45.2 hosts with 14 days-old D4M.3A-pOVA tumors. Proliferation (B, C), CD69 and CD25 expression (C) of OT-I cells in tdLNs after 48 h.
(D-F) Frequencies (D), numbers (E), and TIM-3 expression (F) of TCF-1pos and TCF-1neg OT-I in tdLNs (top) and tumors (bottom) 5 to 21 days following co-injection of 105 naive WT and Cxcr6−/− OT-I cells
(G-I) Frequencies (G, H) and numbers (I) of indicated subsets of WT and Cxcr6−/− OT-I CTL in tdLNs or tumors.
(J, K) Expression of Ki67 (J) and Bcl-2 (K) in subsets defined in (G) on day 21.
(L, M) Ex vivo uptake of viability dye ZombieRed by subsets defined in (G).
Data in (D-M) represent at least two independent replicates with similar results. Graphs show means and either individual replicates or ±SEM. */**/***/**** = p<0.05/0.01/0.001/0.0001.
TIM-3 expression is often characterized as part of a CTL exhaustion program (Jin et al., 2010). Five days after adoptive transfer, WT and Cxcr6−/− TCF-1neg CTL expressed this receptor at similar levels in tumors and dLNs (Figure 2D, F). Subsequently, however, paralleling their near complete lack of intratumoral expansion, Cxcr6−/ − TCF-1neg cells in tumor tissue failed to maintain expression of TIM-3, while expression on their WT counterparts further increased, suggesting an inability of TIM-3+ CTL to persist in the absence of CXCR6.
Despite being considered a marker of CTL exhaustion, TIM-3 is also expressed by the highly functional transitory TCF-1neg CTL subset characterized by maximal expression of T-bet, Granzyme B, as well as CX3CR1 (Hudson et al., 2019; Zander et al., 2019). Similar to PD-1+ polyclonal CTL (Figure 1O), tumor-infiltrating TCF-1pos OT-I CTL were CX3CR1− at all time-points, whereas a comparable fraction of WT and Cxcr6−/− TCF-1neg cells were CX3CR1+ 5 days after transfer (Figure 2G, H). WT, but not Cxcr6−/− OT-I then continued to upregulate CX3CR1, and CX3CR1+ transitory CTL accounted for the vast majority of the intratumoral expansion of WT cells (Figure 2I). Hence, CXCR6 is critical for the rapid accumulation and persistence of CTL with a highly functional effector state, which emerge from TCF-1pos stem-like cells before they adopt an irreversibly hypofunctional state.
T cell population size at effector sites is determined by recruitment, egress, local proliferation, and cell death. To test whether CXCR6 supported proliferation, we examined the cell cycle protein Ki67 in intratumoral CTL subsets. In WT cells, the TCF-1neg CX3CR1+ subset most highly expressed Ki67, confirming prior observations in the context of viral infection (Hudson et al., 2019). In the absence of CXCR6, however, Ki67 expression was reduced in all subsets, but more so in TCF-1neg than in TCF-1pos cells, and most profoundly in the CX3CR1+ subset (Figure 2J). In addition to Ki67, expression of the anti-apoptotic protein Bcl-2 was also reduced in Cxcr6−/− CTL, but this reduction was moderate and comparable between all intratumoral CTL subsets (Figure 2K). Yet, when we assessed the cells’ apoptotic rate based on their ex vivo uptake of the viability dye ZombieRed (a reporter of decreased cell membrane integrity), this dye accumulated mostly in the proliferative CX3CR1+ subset (Figures 2L and 2M). The apoptotic rate of OT-I CTL was similarly low for WT and CXCR6-deficient TCF-1pos and TCF-1neg CX3CR1− cells, but higher for KO than WT cells in the CX3CR1+ state, especially during their intratumoral expansion/contraction phase (Figure 2M). CXCR6 thus regulates the accumulation and persistence specifically of the most highly proliferative, transitory effector cell subset in tumor tissue, at least in large part by supporting their survival. Of note, CX3CR1+ CTL in tdLNs were less affected by lack of CXCR6 (Figure 2H), indicating that this receptor primarily regulates the fate of CTL in the TME.
CXCR6 supports survival of TCF-1neg CTL in the TME to enable their anti-tumor activity
Our observations do not exclude that CXCR6-dependent pre-programming in tdLNs improves subsequent CTL survival in tumor tissue. To examine the role of CXCR6 in TCF-1neg effector-like CTL specifically in the TME, we generated WT and Cxcr6−/− TCF-1neg OT-I cells ex vivo for adoptive transfer studies. IL-12 promotes loss of TCF-1 and conversion of stem-like into effector-like CTL (Danilo et al., 2018). Accordingly, culture of activated OT-I cells in IL-12 and high-dose IL-2 produced TCF-1neg effector-like CTL (“TCF-1neg-like”), while low-dose IL-2 without IL-12 produced Ly108+ TCF-1pos cells (“TCF-1pos-like”) (Figure 3A). Transfer of TCF-1neg-like OT-I into animals with established D4M.3A-pOVA tumors had a pronounced and sustained anti-tumor effect, while the same number of TCF-1pos-like OT-I had a much more moderate effect (Figure 3B). 4 days after transfer, TCF-1neg-injected OT-I cells remained TCF-1neg and uniformly expressed high levels of CXCR6, while TCF-1pos-injected cells remained mostly TCF-1pos and only a fraction were CXCR6-intermediate (Figure 3C). Superior anti-tumor function of TCF-1neg-like OT-I correlated with more efficient recruitment to tumors (Figure 3D) and a greater potential to express IFN-γ, compared to TCF-1pos-like cells, following their recruitment (Figure 3E).
Figure 3: CXCR6 supports survival of TCF-1neg CTL in the TME to enable their anti-tumor activity.
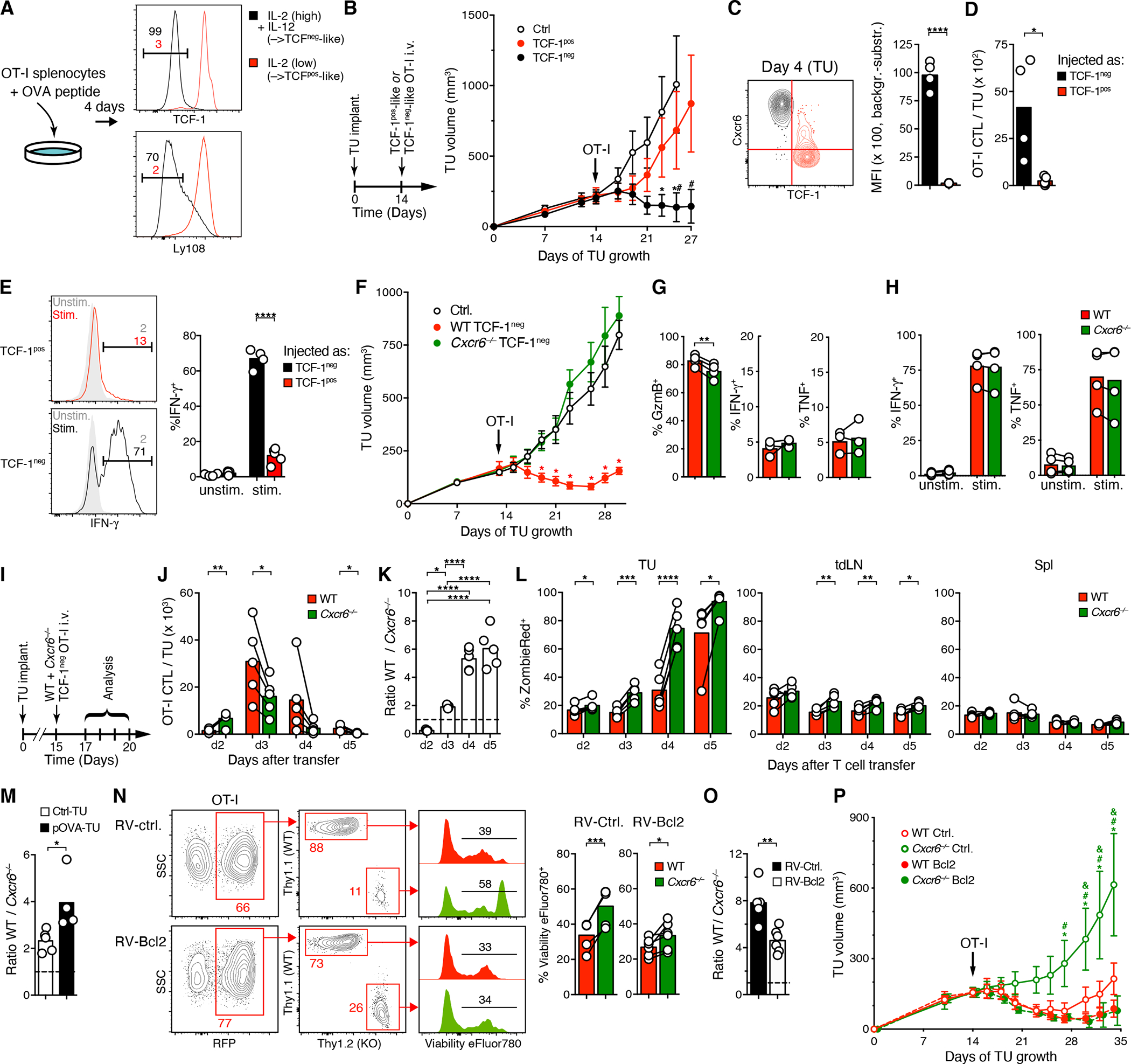
(A) Culture of peptide-activated WT or Cxcr6−/− OT-I splenocytes in low rIL-2 (5 ng/ml) or in high rIL-2 (20 ng/ml) and rIL-12 (10 ng/ml) to generate TCF-1pos-like or TCF-1neg-like OT-I CTL, respectively.
(B) D4M.3A-pOVA tumor growth following i.v. injection of 106 TCF-1pos-like or TCF-1neg-like OT-I CTL.
(C) Overlaid contour plots of CXCR6 expression on tumor-infiltrating OT-I cells 4 days after injection of TCF-1pos-like (red, gated on cells that remained TCF-1pos) or TCF-1neg-like (black) cells into tumor-bearing mice on day 14.
(D, E) Frequency (D) and ex vivo-stimulated IFN-γ expression (E) of the same cells as shown in (C).
(F) Growth of D4M.3A-pOVA tumors following i.v. injection on day 13 of 106 either WT or Cxcr6−/−TCF-1neg-like OT-I as generated in (A) or in non-injected animals (Ctrl.).
(G, H) In situ-expression of Granzyme B, IFN-γ, and TNF (G) and ex vivo-stimulated expression of IFN-γ and TNF (H) by tumor-infiltrating OT-I cells on day 4 following i.v. injection of TCF-1neg-like cells, as described for (F).
(I-L) WT and Cxcr6−/− TCF-1neg-like OT-I cells were co-injected into tumor-bearing mice on day 15 and their respective frequencies and ratios in tumor tissue (J and K) and their ex vivo uptake of the viability dye ZombieRed by cells in tumors, tdLNs, and spleens (L) were assessed at the indicated time-points thereafter.
(M) Ratios of tumor-infiltrating CTL 4 days after injection of WT and Cxcr6−/− TCF-1neg-like OT-I cells (106) into animals implanted with either D4M.3A-pOVA or D4M.3A tumors into their flanks.
(N, O) WT and Cxcr6−/− TCF-1neg-like OT-I cells were retrovirally transduced to express either Bcl-2 and mRFP (RV-Bcl2) or RFP only (RV-ctrl.) and co-injected into tumor-bearing animals on day 14. Four days later, tumor-infiltrating cells were examined for ex vivo uptake of the viability dye Viability eFluor 780 (N) and their input-corrected ratios (O).
(P) Same cells as described for use in (N, O) were injected into separate tumor-bearing animals and tumor growth was monitored. *, #, and & = p<0.05 vs. WT Bcl2, Cxcr6−/− Bcl2, and WT Ctrl., respectively.
Data in A to M represent at least two independent replicates with similar results. Graphs show means and either individual replicates or ±SEM. */**/***/**** = p<0.05/0.01/0.001/0.0001 in all graphs except for (B), (F), and (P).
To test whether CXCR6 enables CTL to control tumors, we injected WT or Cxcr6−/− TCF-1neg-like OT-I into tumor-bearing animals. Lack of CXCR6 did not affect phenotype or function of either TCF-1neg-like or TCF-1pos-like CTL (Figure S3A–B). However, CXCR6-deficient cells were entirely devoid of anti-tumor activity in vivo (Figure 3F), even though tumor-infiltrating Cxcr6−/− CTL continued to express comparable amounts of effector cytokines and only slightly (<10%) less granzyme B in situ (Figure 3G), and produced similar amounts of cytokines upon ex vivo re-stimulation as WT CTL (Figure 3H). To assess the role of CXCR6 in the accumulation of effector-like CTL in tumor tissue, we co-transferred WT and CXCR6-deficient TCF-1neg-like OT-I CTL (Figure 3I).
Unexpectedly, CXCR6-deficient cells were more numerous in tumors than WT cells two days later (Figure 3J, K). This may have resulted from the preferential entrapment of WT cells in the liver (Figure S3E), where the CXCR6 ligand CXCL16 is constitutively expressed in sinusoids (Geissmann et al., 2005), but also indicated that CXCR6 was not essential for extravasation of blood-borne CTL into tumor tissue. The next day, however, when CTL had accumulated in much greater numbers, WT outcompeted KO cells, and this trend intensified when the transferred CTL populations again contracted on days 4 and 5 (Figures 3J, K). The largest increase in the ratio of WT to KO cells coincided with the largest increase in the apoptotic rate of CXCR6-deficient, but not WT CTL between days 3 and 4 (Figures 3K, L), suggesting that premature apoptosis was an important factor in the failure of KO cells to accumulate. The CTL apoptotic rate was much lower in tdLNs, and lowest in spleens, both for WT and KO cells (Figure 3L), indicating that CXCR6-deficient cells were not intrinsically more apoptosis-prone. Apoptotic rates were also low in the liver, even when WT to Cxcr6 KO cell ratios continued to increase at later time-points (Figure S3E, F), suggesting that preferential accumulation of CXCR6-sufficient CTL in this location was independent of superior survival.
To test whether the characteristics of the TME or local TCR stimulation accounted for the death of TCF-1neg-like CTL in tumors, we co-injected WT and Cxcr6−/− OT-I CTL into mice implanted with either D4M.3A-pOVA or D4M.3A control tumors. In the latter, CTL showed higher apoptotic rates than in spleens or tdLNs, but in contrast to OVA peptide-expressing tumors, rates were comparable for WT and Cxcr6−/− cells (Figures S3G, H). The TME is thus generally less supportive of CTL survival than lymphoid tissues, but TCR-driven AICD further promotes CTL apoptosis and this effect is even more pronounced for CXCR6-deficient CTL, contributing to their reduced frequencies (Figure 3M).
Finally, to test whether improving survival can restore the accumulation and anti-tumor function of CXCR6-deficient TCF-1neg OT-I CTL, we used retroviral vectors to express the anti-apoptotic protein Bcl-2 together with mRFP, or mRFP alone, and co-injected WT and Cxcr6−/− cells into tumor-bearing animals. Ectopic Bcl-2 reduced the apoptotic rate of CXCR6-deficient CTL, albeit not fully to the levels in WT cells (Figure 3N). However, when correcting for the input ratios (Figure S3I), Bcl-2 still reduced the ratio of WT to KO cells by half (Figure 3O). Thus, although CXCR6 likely supports the functions of TCF-1neg CTL in multiple ways, its role in preventing AICD contributes to their accumulation. Importantly, ectopic Bcl-2 completely restored the capacity of CXCR6-deficient CTL to control tumor growth (Figure 3P). Promoting survival is therefore a major function of CXCR6 expressed by intratumoral CTL.
The CXCR6 ligand CXCL16 is most highly expressed by the CCR7+ DC3 state
To determine how CXCR6 enhances CTL survival, we examined expression of its ligand, CXCL16, in the TME. Our D4M.3A-pOVA melanoma scRNA-seq data set revealed broad expression primarily in the myeloid cluster. Cxcl16 was however most highly expressed by the DC3 state (Figures 4A, B, and Table S1C). DC3 furthermore expressed the CCR4-ligand CCL22, CCL5, a ligand for CCR5 expressed by some CTL, but especially by NK cells (Böttcher et al., 2018) (Figures 1C, D), as well as the CXCR3 ligands CXCL9 and CXCL10. The latter were however more abundantly expressed by cDC1s, monocytes, and tumor-associated macrophages (TAM). We observed highly similar patterns of chemokine expression in KP1.9 lung tumors (Zilionis et al., 2019) (Figure S4A–C).
Figure 4: The CXCR6 ligand CXCL16 is most highly expressed by the CCR7+ DC3 state.
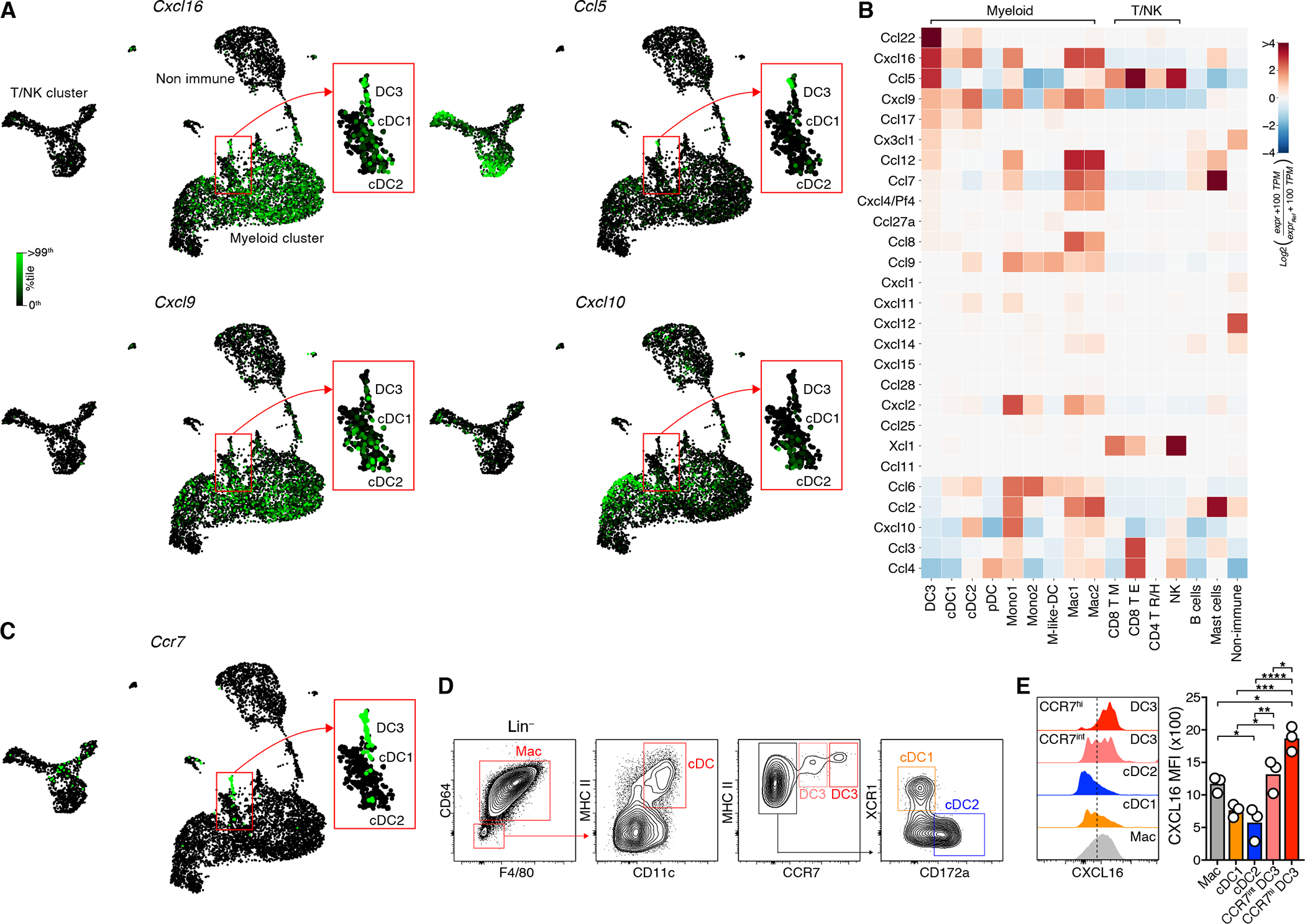
(A-C) Single-cell expression of Cxcl16, Ccl5, Cxcl9, and Cxcl10 (A), of CCR7 (C), and heatmap of chemokine gene expression (B) in D4M.3A-pOVA tumors (Neutrophils not shown). See Table S1C for numerical data underlying the heatmap.
(D, E) Total (intracellular and cell surface) expression of CXCL16 protein in APC types.
(D, E) represent two independent replicates with similar results. Graphs show means and individual replicates. */**/***/**** = p<0.05/0.01/0.001/0.0001.
As previously noted (Maier et al., 2020; Zhang et al., 2020; Zilionis et al., 2019), DC3 expressed large quantities of Ccr7 mRNA (Figures 1D, 4C, and S4D). Flow cytometry further revealed two discrete DC3 subpopulations characterized by intermediate and high expression of CCR7 protein (Figure 4D and S4E). Fractions of both CCR7int and CCR7hi DC3 expressed either the cDC1 markers Xcr1 and CD103, or the cDC2 marker CD172a, suggesting that both can derive from either cDC1 or cDC2 (Figure S4F). Of note, while CCR7int DC3 expressed comparable amounts of CXCL16 protein as F4/80+ TAM, CCR7hi DC3 uniformly expressed even greater quantities, while CCR7− cDC1 and cDC2 expressed little (Figures 4E), corroborating our transcriptomic analysis.
CXCR6 promotes CTL interactions with perivascular clusters of DC3
In light of poor stimulatory activity of TAM for CD8+ T cells (Broz et al., 2014) and low CXCL16 expression by cDC1 and cDC2, we wondered whether CXCR6 organizes cognate DC3 interactions with CTL. To visualize such interactions, we sought to identify a fluorescent reporter system for DC3. Preferential expression of Il12b (encoding for IL-12 p40) in DC3 was previously noted (Maier et al., 2020; Zilionis et al., 2019), and also in D4M.3A-pOVA melanoma as well as KP1.9 lung carcinoma, Il12b mRNA was almost exclusively detected in the DC3 state (Figure 5A and S4G). When we implanted tumors into IL-12 p40-YFP reporter mice, nearly all YFP+ cells in the TME were MHC IIhi, CD11chi, CCR7+ cDC (Figure S4H), and among all cDC only few CCR7int but more than half of CCR7hi DC3 expressed YFP, while CCR7− cDC did not (Figure 5B). Therefore, IL-12 p40-YFP mice allows for selective visualization of the IL-12-competent tumor-infiltrating DC3 subset.
Figure 5: CXCR6 promotes CTL interactions with perivascular clusters of DC3.
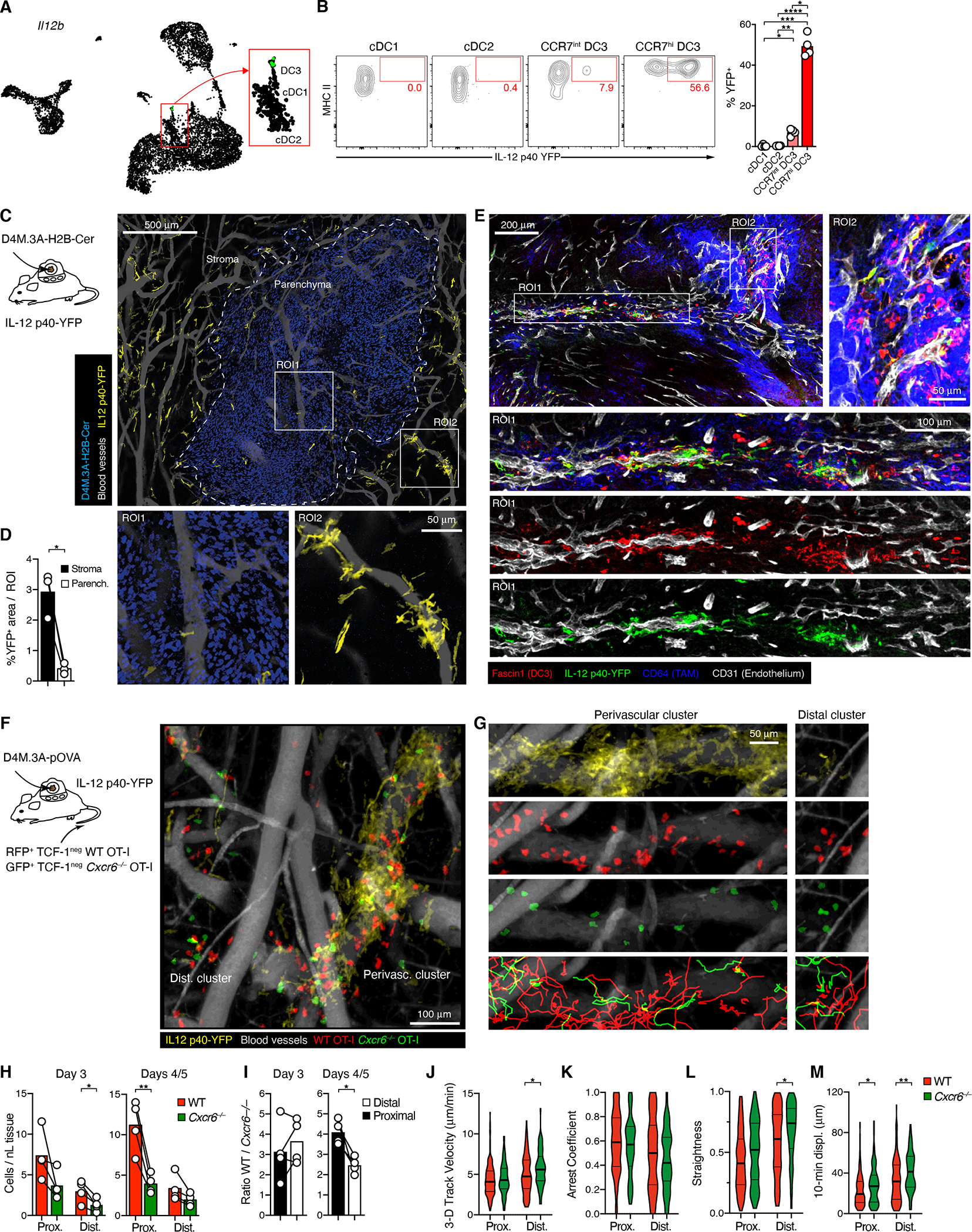
(A) Single-cell expression of Il12b in D4M.3A-pOVA tumors.
(B) Expression of YFP in cDC subsets in 18 days-old tumors in IL-12 p40-YFP reporter mice.
(C) Distribution of YFP+ DC3 in D4M.3A-pOVA-H2B-Cerulean tumors in DSFCs installed on IL-12 YFP reporter mice, as recorded by MP-IVM following injection with QTracker 655 to visualize perfused blood vessels. The image is a collage of 20 individual image stacks. ROIs show representative regions illustrating the characteristic distribution of YFP+ DC3 in tumor parenchyma (ROI1) and stroma (ROI2).
(D) % of area occupied by YFP+ cells in stroma vs. parenchyma
(E) Immuno-stained sections of D4M.3A-pOVA flank tumors 3 days after i.v. injection of CD45.1+ TCF-1neg OT-I into IL-12 p40-YFP reporter mice. Magnified ROIs illustrate overlap of YFP and Fascin1 signal.
(F, G) Migratory behavior of Cxcr6−/− (Green) and WT (Red) TCF-1neg OT-I CTL in the stroma of D4M.3A-pOVA tumors visualized in DSFCs installed on IL-12 p40 YFP mice.
(G) shows DC3 (yellow), WT CTL, KO CTL, and WT and KO CTL migratory tracks (bottom) in ROIs selected for the accumulation of T cells near perivascular DC3 clusters (left) or around smaller DC3 clusters distal to venular vessels (right).
(H, I) Densities (H) and input-corrected ratios (I) of WT and Cxcr6−/− CTL proximal and distal to perivascular DC3 on day 3 (left) or days 4 and 5 (right) after CTL i.v. injection.
(J-M) Median 3D migratory velocities (J), Arrest coefficients (K), Track straightness coefficients (L), and 10-min displacement coefficients (M) of 422 WT and 182 Cxcr6−/− CTL in 4 recordings from two independently performed experiments.
Data in B, D represent two and three independent experiments with similar results. Graphs in B, D, H, and I show means and individual replicates, J-M represent medians and quartiles. */**/**** = p<0.05/0.01/0.0001.
To define the spatial distribution of YFP+ DC3, we implanted H2B-Cerulean-tagged, blue fluorescent D4M.3A melanoma cells into dorsal skinfold chambers (DSFC) installed on IL-12 p40-YFP mice for analysis by MP-IVM. YFP+ DC3 were largely excluded from the tumor parenchyma and instead distributed to the surrounding tumor stroma, where their majority closely aligned with blood vessels, often forming dense perivascular clusters around discrete vessel segments (Figures 5C, D).
To examine whether the spatial pattern of YFP+ DC3 was representative of all DC3, we analyzed histological tumor sections. In line with prior reports (Gerhard et al., 2021; Maier et al., 2020; Zilionis et al., 2019), DC3 Fascin1 transcripts were abundant in DC3 in both D4M.3A-pOVA and KP1.9 tumors (Figure S5A, B), with on average 13.5-fold higher expression than in non-immune cells. Accordingly, Fascin1 protein co-localized with the DC markers MHC II and CD11c (not shown) and showed a similar, preferentially perivascular pattern as observed for YFP in our MP-IVM recordings (Figure 5E). Most cytoplasmic YFP signal co-localized with Fascin1+ cells, although many DC3 were also YFP−. Hence, YFP+ and YFP− DC3 occupied the same perivascular niches. In addition, we occasionally noted Fascin1+ cells aggregated inside the lumina of CD31dim lymphatic vessels (Figure S5C), in line with the role of CCR7 in DC migration to tdLNs (Roberts et al., 2016). Sparse accumulations of CD64dim TAMs often localized to narrow regions directly adjacent to, or partially overlapping with perivascular DC3 clusters, and occasionally these clusters also overlapped with much denser accumulations of CD64bright TAM (Figure 5E).
Transferred TCF-1neg OT-I CTL, as well as all endogenous T cells, accumulated to their highest density around DC3 clusters, irrespective of the density of adjacent TAM accumulations (Figure S5D). T cells were also found in areas dominated by CD64bright TAM, where Fascin1+ cells were sparse and scattered, but did not show the same dense perivascular enrichment as observed around vessels ensheathed by DC3 (Figure S5E, F), indicating that TAM in these areas did not suffice to establish the perivascular niches that attract tumor-reactive TCF-1neg CTL. Thus, both IL-12 p40-positive and -negative DC3 defined perivascular niches of the tumor stroma in which tumor antigen-specific, CXCR6-expressing TCF-1neg CTL as well as other T cells accumulated.
To examine the dynamic behavior of CTL near DC3 clusters and a potential role for CXCR6 in their localization, we co-transferred RFP-expressing WT and GFP-expressing Cxcr6−/− TCF-1neg OT-I into IL-12 p40-YFP animals with D4M.3A-pOVA tumors implanted in DSFC. Accumulation of OT-I CTL was slightly delayed in DSFC- compared to s.c. implanted flank-tumors, and their numbers continued to rise between days three and five after transfer, which was accompanied by an increase in the size of perivascular clusters of YFP+ DC3 (Figures 5F, G, S5G, and Supplemental Movies 1 and 2). Yet, as predicted from our homing studies, WT always outnumbered Cxcr6−/− CTL, both in areas proximal and distal to perivascular DC3 clusters (Figures 5F–H and S5G, H, and Supplemental Movies 3 and 4). At low cell numbers observed on day three, local WT/KO cell ratios were variable, but on days 4 and 5, these ratios were consistently highest in direct vicinity to YFP+ DC3 clusters. Here, WT were four times more abundant than Cxcr6−/− OT-I cells, when correcting for input ratios, while they were only twice as abundant in distal areas (Figure 5I), indicating that CXCR6 promoted CTL accumulation in particular directly adjacent to perivascular DC3.
Although fewer Cxcr6−/− cells accumulated near DC3 clusters, those that did, migrated at similar speeds and arrested at similar rates, yet displaced more effectively relative to their total path lengths traveled than WT cells (Figure 5J–M and Supplemental Movies 2 and 3). This could indicate that CXCR6 subtly optimized CTL interactions with CXCL16+ perivascular APC, resulting in reduced displacement. Independently of CXCR6 expression, CTL migrated more slowly and arrested more frequently than their distal counterparts and rarely departed perivascular areas, suggesting that they were either physically constrained by a perivascular space or retained by antigen-dependent interactions with DC3 and potentially other, non-visualized APCs.
The migratory patterns of WT and Cxcr6−/− CTL were more different from each other in regions distal to perivascular DC3 clusters. Here, CTL occasionally engaged with and arrested around smaller clusters of YFP+ DC3 located distal to blood vessels, but no preferential accumulation of WT compared to Cxcr6−/− CTL was obvious (Figure 5G, right, and Supplemental Movie 4). However, WT cells migrated more slowly, along less straight paths, and displaced less (Figures 5J–M) than Cxcr6−/−cells, suggesting that CXCR6+ CTL were also exposed to CXCL16 in these distal areas. Collectively, these observations suggest that CXCR6 helps position CTL in a perivascular niche densely occupied by DC3, and potentially optimizes their interactions.
DC3 trans-present IL-15, a critical survival signal for effector-like CTL in the TME
Although CXCR6+ TCF-1neg CTL accumulated preferentially around blood vessels surrounded by DC3 clusters, we could not exclude that adjacent or overlapping populations of CD64+ TAM, some of which may also express CXCL16 highly, contributed to their survival. Therefore, to test whether expression of CXCL16 by rare DC3 was functionally relevant or redundant with expression by TAM, we generated Cxcl16 KO : zDCDTR −> B6 mixed BMC, in which cDC (of which only DC3 express CXCL16 highly) can be selectively ablated through diphteria toxin (DT) treatment (Meredith et al., 2012), while leaving macrophages untouched (Figure 6A, B). Even though DT treatment reduced the frequency of cDC in the TME by only two thirds, it raised the apoptotic rate of WT to the level of Cxcr6−/− OT-I, and reduced the ratio of WT to KO cells in tumor tissue by half (Figure 6C, D). Thus, despite their low numbers, CXCL16 expression on DC3 plays a critical role in supporting the survival of intratumoral TCF-1neg CTL.
Figure 6: DC3 trans-present IL-15, a critical survival signal for effector-like CTL in the TME.
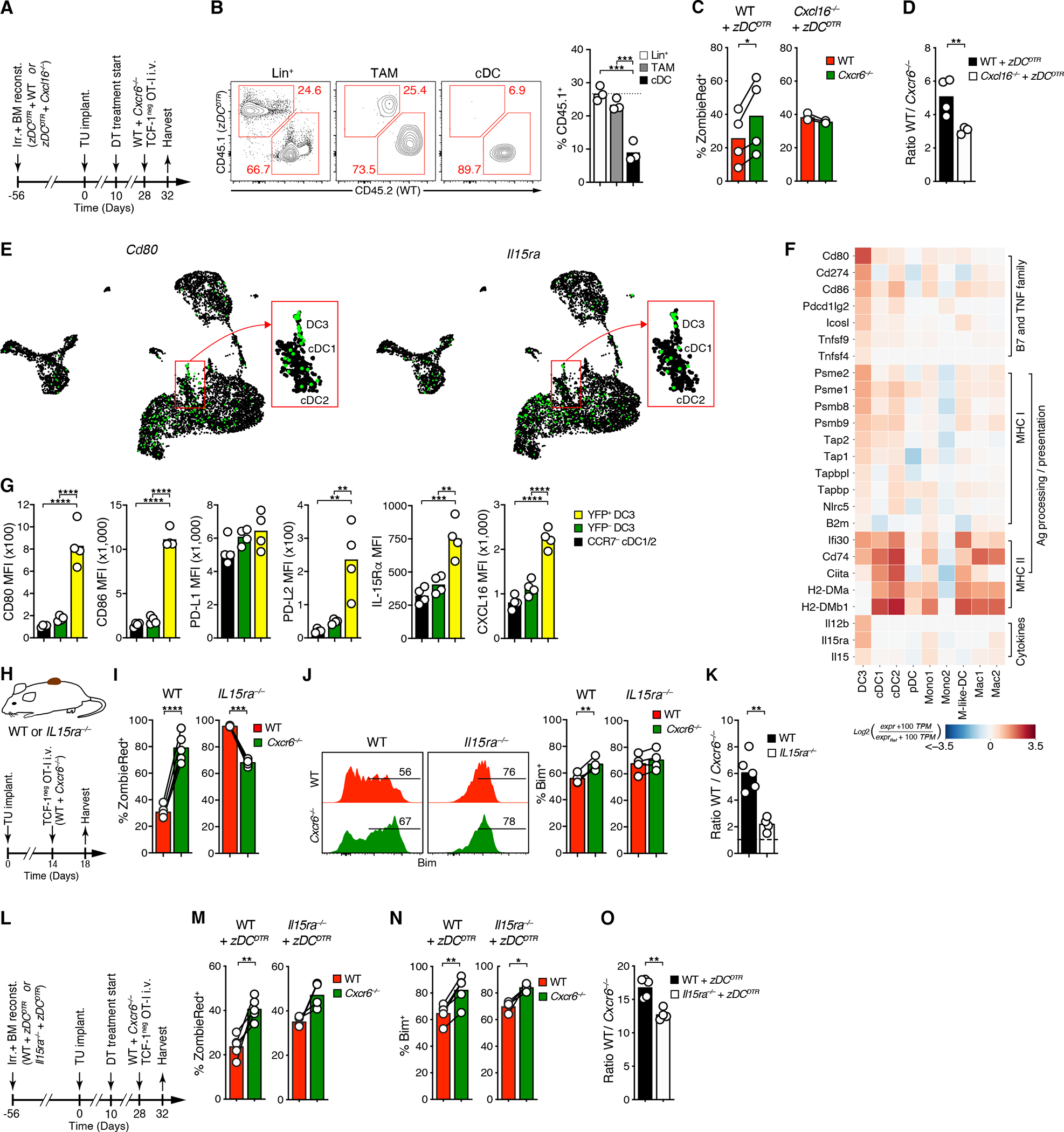
(A) Experimental protocol
(B) Selective depletion of cDC upon DT treatment of WT : zDCDTR −> WT mixed BMCs
(C, D) Ex vivo ZombieRed uptake (C) and ratios (D) of tumor-infiltrating WT and Cxcr6−/− TCF-1neg OT-I CTL
(E, F) Single-cell expression of Cd80 and Il15ra and (E) heatmap analysis of indicated transcripts in the D4M.3A-pOVA TME, ranked within indicated groups (F). See Table S1D for numerical data underlying the heatmap.
(G) Expression of indictated proteins by CCR7− cDC, YFP−, and -YFP+ DC3 in D4M.3A-pOVA tumors in IL-12 p40 YFP mice.
(H-K) Ex vivo uptake of ZombieRed (I), expression of Bim (J), and ratios (K) of WT and Cxcr6−/− TCF-1neg OT-I cells in D4M.3A-pOVA tumors in WT or Il15ra−/− hosts.
(L-O) Same as for (I-K), but following CTL injection into DT-treated Il15ra−/− : zDCDTR −> WT or WT : zDCDTR −> WT mixed BMCs
Graphs in B-D, G, I-K, and M-O show means and individual replicates. */**/***/**** = p<0.01/0.001/0.0001.
In light of this result, we asked which DC3-produced factors contribute to sustaining CTL proliferation and survival. Among all intratumoral APC, DC3 expressed the highest levels of the B7 family co-stimulatory genes Cd80 and Cd86, but also the co-inhibitory genes Cd274 (the PD-L1 gene) and Pdcd1lg2 (the PD-L2 gene), both at the mRNA and the protein level (Figures 6E–G, S6A–C, and Tables S1D, S2B). They also most highly expressed Icosl as well as the TNF superfamily genes Tnfsf4 (OX40 ligand) and Tnfsf9 (4–1BB ligand), known to support CTL anti-tumor function (Schaer et al., 2014). Based on their expression of genes in the MHC I and II antigen processing and presentation pathways, DC3 appeared most specialized in presentation to CD8+ T cells. Among cytokines that support CTL function, the aforementioned IL-12 p40 cytokine chain, but also IL-15 stood out.
In vitro, IL-15 promoted neither the conversion of TCF-1pos stem-like into TCF-1neg effector-like CTL nor their expression of CXCR6, but instead expanded CXCR6hi TCF-1neg cells following their IL-12-driven conversion and CXCR6 up-regulation, possibly by improving the survival of proliferating cells (Figure S6D–F). In vivo, IL-15 signals in T cells require presentation of IL-15 cytokine by IL-15Rα in trans (Dubois et al., 2002; Stonier et al., 2008). DC3 expressed the most Il15 among all DC states, but Mono1 and Mono-like DC states were also abundant sources. However, DC3, and in particular CCR7hi DC3, expressed the highest concentrations of IL-15Rα among all immune cell states (Figures 6E–G and S6A–C), suggesting that they most effectively deliver IL-15 survival signals to CTL. When we co-transferred WT and Cxcr6−/− TCF-1neg OT-I cells into tumor-bearing WT or Il15ra−/− animals (Figure 6H), Cxcr6−/− CTL retrieved from WT hosts exhibited a strongly enhanced apoptotic rate compared to WT OT-I in tumor tissues, but much less so in tdLNs, as shown earlier (Figures 6I, S6G and 3L–O). In contrast, WT CTL were highly apoptotic in Il15ra−/− animals, even more so than Cxcr6−/− CTL, whose apoptotic rate was unchanged (Figure 6I). IL-15 promotes lymphocyte survival in part by repressing gene transcription and promoting proteasomal degradation of the pro-apoptotic factor Bim (Huntington et al., 2007; Uhlin et al., 2005). Accordingly, Bim expression was higher in Cxcr6−/− CTL in WT hosts, but similar to WT cells in Il15ra−/− hosts. (Figure 6J). As a result, the ratio of WT to Cxcr6−/− cells was reduced three-fold in Il15ra−/− hosts in tumor tissue (Figures 6K and S6H).
Some macrophage and monocyte states also expressed IL-15Rα (Figures 6E, F and S6A, B). To test the role of IL-15Rα specifically in cDC, among which DC3 express the highest amounts, we created Il15ra KO : zDCDTR −> B6 mixed BMCs, in which IL-15Rα-sufficient cDC can be selectively ablated (Figure 6L). Despite incomplete ablation of DTR-expressing cDC in tumor tissue in zDCDTR mixed BMCs (Figure 6A, B), apoptotic rate and Bim expression of WT TCF-1neg OT-I CTL increased and approached that of their Cxcr6−/− counterparts, and their preferential accumulation was reduced (Figure 6M–O). Thus, TCF-1neg CTL required CXCR6 to be exposed to IL-15Rα+ cDC, receive IL-15 survival signals, and accumulate in the TME. Among cDC, the DC3 state played a central, non-redundant role, likely because it i) closely co-localized with TCF-1neg CTL, ii) most highly expressed the CXCR6 ligand CXCL16, and iii) also expressed the highest amounts of IL-15Rα.
CXCR6 expression predicts survival in human cancer patients
To assess the role of CXCR6 for human cancer, we examined bulk RNA-Seq data sets from melanoma patients available through the TCGA database. CXCR6 in tumor tissue correlated highly with CD8B expression, on par with CXCR3 and more strongly than CCR5, while the skin homing chemokine receptor CCR10 correlated poorly (Figure 7A). In contrast, CXCR6 correlated less well with expression of CD4 and of a NK cell signature (Figure 7B and S7A) indicating that similar to mice, CXCR6 is also preferentially expressed by CTL in human melanoma. Accordingly, the top third of patients with the highest CXCR6 expression had a greater survival probability than the bottom third, which was similar for CXCR3 and CCR5, but not for CCR10 (Figures 7C and S7B). In contrast, patients with the highest expression of the neutrophil-expressed gene CXCR2 were less likely to survive, as predicted (Engblom et al., 2017). Even though CXCR6 correlated with expression of its ligand CXCL16, with IL12B, and with an NK cell gene signature (Figure S7A, C), it was a better predictor of survival than any of these (Figures 7C and S7D). To avoid the bias of an arbitrary cut-point to operationally divide patients into high and low expressers and at the same time enhance the statistical power, we also compared genes on the basis of their continuous normalized expression levels. In doing so, CXCR6 emerged as the strongest predictor of overall survival in melanoma patients among all chemokine receptor genes, followed by CCR2, CCR5, and CXCR3 (Figure 7D).
Figure 7: CXCR6 expression predicts survival in human cancer patients.
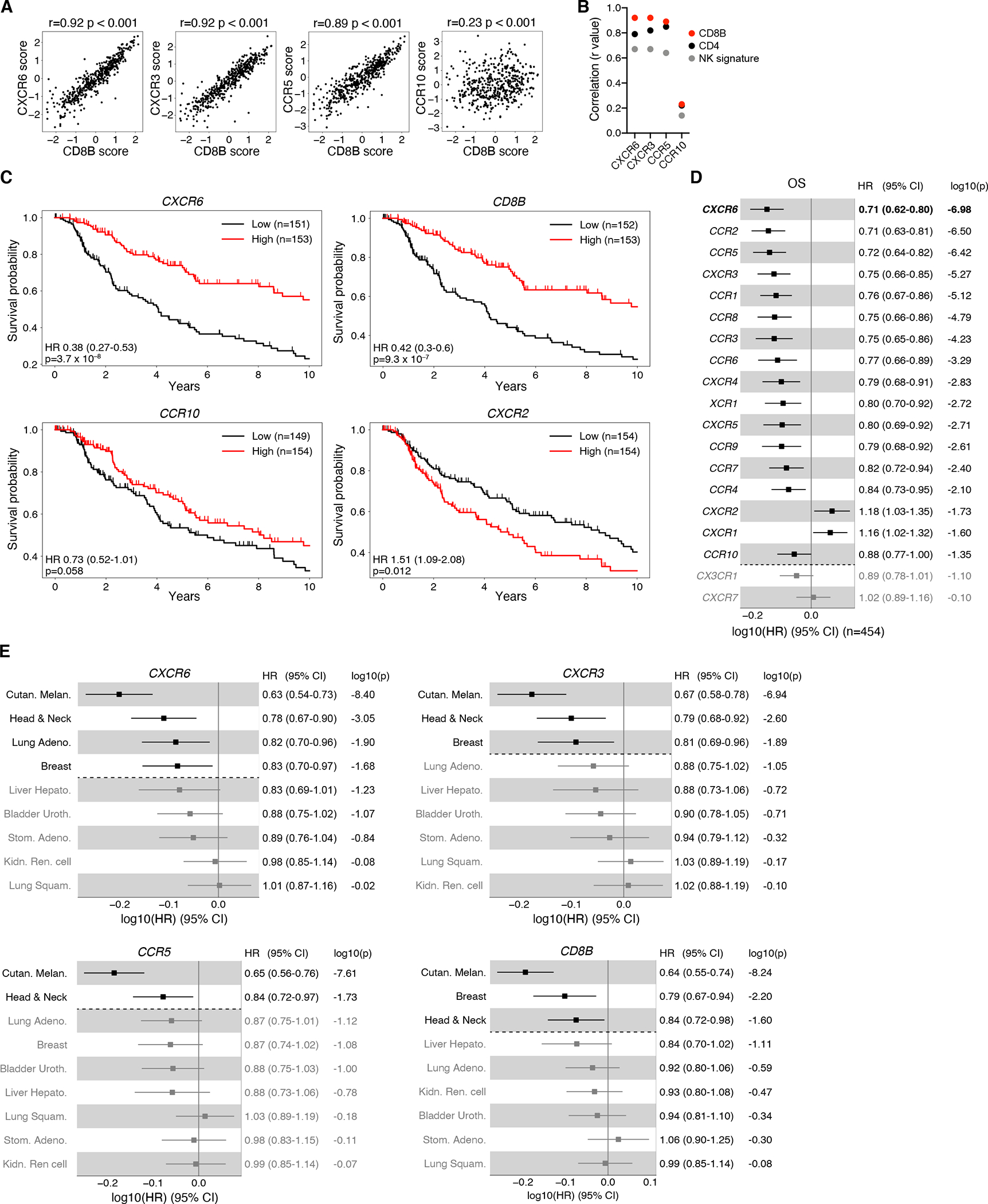
(A) Correlation between chemokine receptor and CD8B expression scores in tumor tissue of 469 TCGA melanoma (SKCM) patients. Spearman’s rank-correlation coefficient r and two-sided P value are shown.
(B) Summary of rank-correlation coefficients between indicated chemokine receptors and CD8B, CD4, or NK signature (NCR1 and SH2D1B) expression scores.
(C) Kaplan-Meier estimates of overall survival comparing the top (“High”) and bottom (“Low”) third of melanoma patients with regard to expression scores of indicated genes. Hazard ratios (HR), 95% confidence intervals (CI), and P values (Wald Chi-Squared test) based on univariate Cox proportional-hazards model (High versus Low). Tick marks indicate censoring.
(D) Association between overall survival and continuous expression score of individual chemokine receptor genes in melanoma patients. HR, 95% CI, and P values (Wald Chi-Squared test) based on univariate Cox proportional-hazards model. Note: a HR of e.g. 0.71 (CXCR6) indicates that at any time during the TCGA melanoma study period patients had a 1–0.71 = 0.29 = 29% reduction in risk of death per one standard deviation increase of normalized log2 transformed CXCR6 expression + pseudo count.
(E) Association between overall survival and continuous expression score of indicated genes in all indicated cancer types, adjusted for Sex (versus Male) and AJCC pathologic tumor stage (versus Stage 0 & I).
Significant (P < 0.05) associations shown in black in (D, E).
To assess the general relevance of CXCR6 for human cancer, we extended this analysis to all TCGA solid tumor types for which at least 186 patients were available, at least 80 events (patient deaths) were recorded, all analyzed genes were detected, and tumor staging was available. When correcting for patient sex and tumor stage, CXCR6 predicted overall survival not only in melanoma, as did CXCR3 and CCR5, but also in head and neck cancer, lung adenocarcinoma, and breast cancer, similar to IL12B and the NK cell signature (Figures 7E and S7E). In contrast, other than in melanoma, CXCR3 was predictive only in head and neck as well as breast, and CCR5 only in head and neck cancer (Figure 7E). Hence, CXCR6 expression most strongly correlated with the presence of CTL in tumor tissue and is the strongest indicator of all chemokine receptor genes for a favorable quality of the immune infiltrate that prolongs patient survival in several immunogenic human cancer types.
DISCUSSION
Our analysis of chemokines and their receptors in the TME revealed the prominent expression of CXCR6 in tumor-infiltrating CTL and its critical role in facilitating their interactions with perivascular CCR7+ DC3 that sustain CD8+ T cell-mediated immune control of tumors. An important aspect of our study was the focus on tumor-reactive CTL, identified by the expression of PD-1. Even though CXCR6 was the most highly expressed receptor on both PD-1+ and PD-1− CTL, chemokine receptor expression in general varied greatly between these populations, and only focusing the analysis on tumor-reactive cells revealed a profound chemotactic reprogramming during the conversion of TCF-1pos into TCF-1neg cells.
In contrast to CXCR6, CXCR3 was preferentially expressed on PD-1− bystander cells, which was unexpected, given the evidence for its role in guiding intratumoral CTL-cDC interactions (Chow et al., 2019). However, in that study CXCR3 unfolded its role in the context of PD-1 targeted immune checkpoint therapy, while the present study examined spontaneous anti-tumor immunity. Furthermore, we did not find CXCR3 to be absent, but only expressed at lower levels on tumor-reactive compared to bystander CTL, and even further down-regulated in the former during their loss of TCF-1. CXCR3 may therefore play a role in supporting interactions of stem-like CTL with intratumoral APCs to promote their accelerated local conversion into effector-like cells, as observed during PD-1 blockade therapy in chronic viral infection or cancer (Miller et al., 2019; Siddiqui et al., 2019; Utzschneider et al., 2016).
The second most highly expressed chemokine receptor on tumor-infiltrating CTL was CX3CR1, which was induced following their TCF-1pos to TCF-1neg conversion, and which identifies the most highly functional CTL subsets in the context of chronic viral infection (Hudson et al., 2019; Zander et al., 2019). CX3CR1 is not required for CTL effector differentiation in lymphoid tissues, but since we observed the highest expression of its sole ligand CX3CL1 on non-immune cells, it may play a role in positioning TCF-1neg CTL in tumor tissue.
CXCR6 has previously received the most attention for its role in lymphocyte homeostasis in the liver, based on the constitutive expression of its ligand CXCL16 in liver sinusoids. There, it supports the maintenance of CXCR6-expressing liver-resident NK, NK T, and CD8+ tissue-resident memory T (Trm) cells (Geissmann et al., 2005; Germanov et al., 2008; Paust et al., 2010; Tse et al., 2014). CXCR6 is also part of a more general tissue-residency gene program (Mackay et al., 2016), and contributes to CD8+ Trm maintenance in skin and lung, where both the membrane-tethered as well as cleaved, soluble form of CXCL16 are expressed by epithelial cells under homeostatic conditions (Lee et al., 2011; Olszak et al., 2012; Scholz et al., 2007; Takamura et al., 2019; Wein et al., 2019; Zaid et al., 2017). It will be of interest to explore whether the mechanisms by which CXCR6 maintains resting memory cells in these settings are related to how it sustains the transitory effector-like CTL pool in the TME, as observed here. In some reports, a role of CXCR6 in lymphocyte survival was demonstrated (Geissmann et al., 2005; Tse et al., 2014), but attributed to its signaling functions that include activation of NF-kB via Akt (Chandrasekar et al., 2004). Here we propose an alternative model, whereby CXCR6 supports lymphocyte survival indirectly by optimizing their interactions with CXCL16-expressing cells that provide survival and other factors. In the TME, CXCL16+ DC3 that express both IL-15 and IL-15Rα play a prominent role. However, since also endothelial and epithelial cells can express CXCL16 and trans-present IL-15 via IL-15Rα (Matloubian et al., 2000; Xie et al., 2020), it is conceivable that CXCR6-mediated cellular interactions are a more general mechanism to facilitate exposure of NK, NK T, and CD8+ memory cells to this important cytokine under steady-state conditions. This model aligns with the central role of IL-15 in the maintenance of both circulating and tissue-resident memory lymphocytes (Waldmann et al., 2020).
Expression of a specific chemokine receptor on tissue-infiltrating immune cells is generally viewed as indicative of its involvement in the recruitment of these cells to that tissue. Based on CXCL16 expression on some endothelia, it is possible that CXCR6 also plays a role in the recruitment of blood-borne CTL to tumor tissue. However, the observed preferential recruitment of CXCR6-deficient compared to WT TCF-1neg CTL to tumors early after i.v. transfer argues against a major role in tissue extravasation. Furthermore, up to a magnitude greater abundance of CXCR6 on tumor-reactive compared to bystander CTL in tumor tissue indicated that it is induced by intratumoral antigen recognition following their recruitment from blood. Accumulation of CXCR6-expressing CTL therefore more likely results from increased exposure to co-stimulatory molecules, most prominently expressed by DC3, which likely enhances their proliferation and function, in addition to the survival benefit through increased exposure to trans-presented IL-15.
It is thought that the membrane-form of CXCL16 acts as an adhesion molecule, whereas its proteolytically cleaved, soluble form acts as a chemoattractant (Koenen et al., 2017). Based on our transcriptomic analysis, the ADAM10 and 17 proteases that cleave membrane CXCL16 are widely expressed in the melanoma TME by both immune and non-immune cells (data not shown), suggesting that at least fraction of CXCL16 on cell surfaces may be cleaved to support its chemotactic activity. Accumulation of CTL near perivascular DC3 may therefore be driven by their guidance along a gradient of soluble CXCL16 emanating from DC3 and possibly adjacent TAM, or by chemotactic or haptotactic retention of CTL near DC3 after they have already entered the perivascular niche, either from the surrounding tumor tissue or following local extravasation from the adjacent blood vessel. CTL migration among perivascular DC3 clusters was only moderately altered in the absence of CXCR6, indicating that CXCL16 does not support stable adhesive interactions, but it may more subtly optimize CTL-DC3 contacts to promote e.g. IL-15 trans-presentation.
The ontogeny of the CCR7+ DC3 state has not been definitively determined. These cells exhibit transcriptional features of both cDC1 or cDC2, suggesting that they represent a shared activation state of either of these well-defined subsets (Maier et al., 2020). However, a distinct feature of DC3 is their uniform expression of CCR7, which was previously described for a fraction of cells classified as cDC1, but not for cDC2 (Roberts et al., 2016). Beyond the expression of co-stimulatory molecules and cytokines, we found that DC3 overall more strongly expressed genes in the MHC I rather than the MHC II antigen processing and presentation pathway, indicating a specialization in cross-presentation to CTL, a feature generally ascribed to cDC1. On the other hand, we detected mutually exclusive expression of both cDC1 markers CD103 and Xcr1 or the cDC2 marker CD172a on DC3. It therefore seems likely that both cDC1 and cDC2 contribute to the DC3 state and that their relative contribution possible varies with tumor type and immune activation state. Fate-mapping studies should resolve the spectrum of potential DC3 origins. It is also of interest to examine the mechanism by which DC3 are recruited to the perivascular niche, the role of interactions with locally extravasating CTL or NK cells in controlling their activation state, for instance through IFN-γ and IL-12-dependent feedback loops (Garris et al., 2018), and how long CCR7+ DC3 persist in the perivascular niche before potentially entering tumor lymphatics to traffic to draining LNs. Our present findings uncover the central role of the chemokine receptor CXCR6 in positioning tumor-infiltrating CTL in a perivascular niche occupied by an IL-12-competent, activated cDC subset that provides critical survival and proliferation signals to locally sustain the T cell effector response in the TME, and potentially at other immune effector sites.
LIMITATIONS OF THE STUDY
Our description of the perivascular DC3 niche is based on the analysis of a fast-growing, immunogenic mouse melanoma model. Development of this niche and it role for intratumoral CTL differentiation in a wider range of human tumors remains to be further explored.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Thorsten Mempel (tmempel@mgh.harvard.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
scRNA-seq data generated during this study, including includes genes counts pre- and post-normalization, per-cell meta data, as well we the raw FASTQ files, is publicly available on GEO (GSE179111).
The code generated during this study is available at https://github.com/pittetmi/paper-code-data/tree/main/Di_Pilato_et_al_2021
The UMAP visualization of the single-cell transcriptome data is available for interactive exploration at https://kleintools.hms.harvard.edu/tools/springViewer_1_6_dev.html?cgi-bin/client_datasets/dipilato2020/d4m.3a-pova
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
Il15ratm1Ama/J (Lodolce et al., 1998), C57BL/6/J, CD45.1 and Thy1.1 congenic, zDCDTR, (Meredith et al., 2012), and OT-I mice were purchased from Jackson laboratories. Fidel Zavala, Mikael Pittet, and Ulrich von Andrian provided Cxcr6 gfp/gfp knock-in (Cxcr6−/−) mice (Unutmaz et al., 2000), Il12btm1.1Lky/J (IL-12 p40-YFP) mice (Reinhardt et al., 2006) and CAG-mRFP1/J mice (Long et al., 2005), respectively. Animals were maintained in specific pathogen-free facilities at the Massachusetts General Hospital (MGH) and all studies were approved and performed in accordance with guidelines and regulations implemented by the MGH Institutional Animal Care and Use Committee (IACUC).
Tumor cell lines and growth studies
The BRAFV600E × PTENnull melanoma cell lines D4M.3A-H2B-SIINFEKL-Cerulean (D4M.3A-pOVA), D4M.3A-H2B-Cerulean (Di Pilato et al., 2019), Yale University Mouse Melanoma (YUMM) 1.1 and Lewis Lung Carcinoma (LLC) 1 lines were grown in DMEM with 10% FCS and used for experiments when in exponential growth phase. 106 tumor cells were s.c. injected in 100 μl PBS into the flanks of 6–10 weeks-old male mice. Whenever possible, animals were randomized into treatment groups. Tumor volumes were measured on and then every second to third day following the start of treatments and calculated as V = (length × width2)/2.
Generation and use of irradiation bone marrow chimeras
C57BL/6/J were lethally irradiated (950 rad) and i.v. injected with 10 × 106 bone marrow cells from Cxcr6−/− and WT, zDCDTR and Cxcl16−/−, zDCDTR and Il15ra−/−, or zDCDTR and WT mice and allowed at least 6 weeks for hematopoietic reconstitution. 10 days after D4M.3A-pOVA tumor injection, zDCDTR mixed BMCs received a first dose of 20 μg/kg i.p. and subsequently doses of 4 μg/kg of diphtheria toxin every third day until tumor harvest.
T cell cultures and injections
Naive OT-I CD8+ T cells were purified from LNs and spleens of WT and Cxcr6−/− mice by immunomagnetic negative cell selection using the Miltenyi Naive CD8+ T cell isolation kit and 105 cells adoptively transferred into tumor challenged-mice by tail vein injection. In some cases, purified naive CD8+ OT-I cells from WT or Cxcr6−/− mice were labeled with 2 μM CellTrace FarRed in 5 ml of staining buffer (PBS, 1 % FCS) at 37 °C for 20 minutes. After washing, 2 × 106 cells were adoptively transferred i.v. into tumor-challenged mice. In other cases, endogenous CD8+ cells were depleted using 12.5 μg of anti-CD8a mAb, and 5 days later, when no depleting activity of residual mAb was detectable, 2.5 × 106 naive OT-I cells were transferred i.v. and tumors implanted. To generate primed OT-I CTL, splenocytes from WT or Cxcr6−/− OT-1 mice were pulsed with 100 nM SIINFEKL peptide (New England Peptide) at 106 cells/ml in T cell medium (RPMI, 10% FCS, 1% HEPES, 1% sodium pyruvate, 1% GlutaMAX, 1% non-essential amino acids, 55 μM 2-mercaptoethanol) at 37 °C for 24 h. Either 20 ng/ml of murine recombinant IL-2 (mrIL-2) and 10 ng/ml of mrIL-12 (R&D), or 5 ng/ml of mrIL-2 were then added and replenished daily while maintaining cells at 106 cells/ml, to generate TCF-1neg or TCF-1pos OT-1 CTL T cells, respectively. At day 4, 106 TCF-1neg or TCF-1pos were adoptively transferred i.v. into tumor challenged-mice.
METHOD DETAILS
Retroviral vector constructs and transductions
A murine Bcl2 ORF was cloned using Gibson Assembly from the pSFFV-neo Bcl2 plasmid (Addgene plasmid #8776) to replace the ΔNFGR ORF 3’ of the IRES in the MSCV-based, previously described MinW-H2B-mRFP vector (Marangoni et al., 2013), in order to produce MinW-H2B-mRFP-IRES-Bcl2 (referred to as “RV-Bcl2”). Infectious retroviral particles were produced in the supernatants of Platinum E packaging cells stably transduced to express RV-Bcl2 or MinW-H2B-mRFP-IRES-ΔNFGR (“RV-ctrl”). Freshly harvested supernatant was added twice to cultured OT-I T cells (spin-fection at 1000 × g for 90 minutes at 32 °C) on days 2 and 3 after activation. At day 4, 106 TCF-1neg OT-1 CTL (WT or Cxcr6−/−, expressing RV-Bcl2 or RV-ctrl at a purity of >98%) were adoptively transferred i.v. into tumor challenged-mice for tumor growth experiments.
Preparation of single cell suspensions, antibody staining and flow cytometry
LNs and spleens were passed through 40 μm cell strainers. Spleen cell suspensions and blood were lysed with ACK lysis buffer. Tumors were minced into small fragments and treated with 1.5 mg/ml collagenase IV and 50 U/ml DNAse I for 30 minutes at 37 °C under agitation. Livers were also minced into small fragments and treated for 30 minutes at 37°C with 100 μg/ml liberase TM (Sigma Aldrich) and 50 μg/ml DNAse (Roche), passed through a 70 μm cell strainer, and lymphocytes were isolated using Percoll gradient centrifugation.
Cell surface proteins were stained 20 minutes at 4 °C with the following antibodies: α-CD3 (17A2), -CD4 (GK1.5), -CD8α (53–6.7), -CD8β (YTS156.7.7), CD11b (M1/70), -CD11c (N418), - CD25 (PC61.5), -CD45 (30-F11), -CD45.1 (A20), -CD45.2 (104), -CD45R/B220 (RA3–6B2), -CD64 (X54–5/7.1), -CD69 (H1.2F3), -CD90.1 (OX-7), -CD90.2 (30-H12; 53–2.1), - CD172a/SIRPα (P84), -CD197/CCR7) (4B12) -CD279/PD-1 (29F.1A12 or RMP1–30), - CD335/NKp46 (29A1.4), -CX3CR1 (SA011F11), -F4/80 (BM8), -I-A/I-E (M5/114.15.2), -Ly108 (330-AJ), -Ly-6C (HK1.4), -Ly-6G (1A8), -TIM-3 (RMT3–23), -XCR1 (ZET), -CD86 (GL-1), -CD274 (10.F.9G2), - KLRG1 (2F1/KLRG1), -CD25 (PC61), -CD215 (6B4C88), all from BioLegend, -CD103 (M290), -CD273 (TY25), -CD80 (16–10A1), all from BD Biosciences, and α-CD11c (HL3) from eBioscience. Intracellular and nuclear proteins were stained for 60 minutes at room temperature after permeabilization and fixation (Mouse regulatory T cell staining Kit; eBioscience) using antibodies against: α-TCF1/TCF7 (C6329), -BIM (C34C5) from Cell Signaling, α-IFNγ (XMG1.2), -Bcl-2 (BCL/10C4), -T-bet (4B10), -Blimp-1 (5E7), -Granzyme B (GB11), -TNF (MP6-XT22) from BioLegend, -Foxp3 (FJK-16s) from eBioscience, -Fascin1 (55K-2) from Santa Cruz Biotechnology, α-CXCL16 (12–81) and α-Ki67 (B56) from BD Biosciences.
For chemokine receptor analyses only, small minced tumor fragments were mechanically dissociated into single cell suspensions without the use of enzymes using the gentleMACS™ Dissociator (Miltenyi). Cell suspensions were stained for 1h at 37 °C in T cell medium with the following antibodies: α-CD191/CCR1) (S15040E), -CD192/CCR2 (SA203G11), -CD195/CCR5 (HM-CCR5), -CD196/CCR6 (29–2L17), -CD197/CCR7 (4B12), -CD183/CXCR3) (CXCR3–173), -CD185/CXCR5 (L138D7), -CD186/CXCR6 (SA051D1), -CX3CR1 (SA011F11), all from BioLegend.
For all studies, dead cells were stained using the fixable viability violet dyes Zombie Red or Fixable Viability Dye eFluor™ 780 (Invitrogen) for 12 minutes at room temperature, followed by blocking of Fc receptors with TruStain fcX (Biolegend) for 15 minutes at 4 °C. Cells were analyzed on LSR II, LSRFortessa or LSRFortessa X-20 flow cytometers (BD Biosciences) and data were analyzed with FlowJo software version 10.5.3. In order to analyze early and late apoptotic cells, tumors were minced into small fragments, collagenase IV- and DNAse I-digested for 30 minutes at 37 °C. Cell suspensions were treated with TruStain fcX (Biolegend) for 15 minutes at 4°C and stained with fixable viability violet dyes Zombie Red or Fixable Viability Dye eFluor™ 780 and surface proteins binding-antibodies for 20 minutes at 4 °C. Finally, cells were resuspended in Annexin V Binding Buffer (BioLegend) and stained with Annexin V and 7-AAD viability solution (BioLegend) for 15 minutes at 25 °C.
Isolation of CD45+ and CD45− cells from tumor tissue and scRNA-seq
D4M.3A-pOVA injected-mice were sacrificed at day 17 after tumor implantation, tumors were minced into small fragments, which were treated with 1.5 mg/ml collagenase IV and 50 U/ml DNAse I for 30 minutes at 37 °C under agitation. Single cell suspensions were stained with fixable viability violet dye Zombie Red and α-CD45 mAbs (30-F11) (BioLegend) and sorted (BD FACSAria Fusion Cell Sorter). Live CD45+ and CD45− cells were then mixed at a 9:1 ratio and processed using the inDrops V3 scRNA-seq platform (Klein et al., 2015; Zilionis et al., 2017). inDrops Libraries were sequenced on the NextSeq Illumina platform, paired-end mode.
Preparation of mice for MP-IVM studies
C57BL/6/J or IL-12 p40-YFP mice were s.c. injected with 106 D4M.3A-H2B-Cerulean or D4M.3A-H2B-SIINFEKL-Cerulean (pOVA) cells in the right flanks ~1 cm lateral to the midline of the back. 5 to 7 days later, dorsal skin fold chambers (DSFC) were surgically installed on top of the resulting tumors, as described (Marangoni et al., 2013). For analgesia, mice received 5 mg/kg s.c. of carprofen administered before surgery and every 24 h thereafter until termination of the experiment. Intra- and perioperative anesthesia was achieved using isoflurane inhalation. Five days after the surgery, 105 TCF-1neg OT-I CTL T cells were adoptively transferred i.v. and MP-IVM was performed at multiple timepoints thereafter.
MP-IVM recordings
DSFC-bearing mice were anesthetized with isoflurane and the DSFC were mounted on a custom-built stage. In order to visualize blood vessels, mice were retro-orbitally injected with 100 μL of 80 nM QTracker 655 non-targeted quantum dots (Invitrogen) in sterile PBS 10 minutes before image acquisition. The imaging depth varied within the range of 30 – 200 μm below the DSFC cover glass. A DeepSee HP and an Insight 3X Ti:sapphire lasers (Newport/Spectra-Physics) were tuned to 850 and 985 nm, respectively, for balanced multiphoton fluorescence excitation of Cerulean, EGFP, mRFP, QDots. Stacks of 15 to 30 optical sections (512 × 512 pixels) with 3–4 μm z-spacing were acquired every 60 seconds to visualize imaging volumes of 45 to 120 μm in depth. Emitted light and second harmonic signals were detected through 455/50 nm, 525/50 nm, 590/50 nm and 665/65 nm band-pass filter with non-descanned detectors. Data sets were transformed in Imaris 9.5 (Bitplane) to generate maximum intensity projections (MIPs) for export as MP4 movies.
In order to generate static overview images of the TME, overlapping fields of view were acquired as stacks of 4 optical sections (512 × 512 pixels) with 4 μm z-spacing at a single timepoint. Individual images were processed and exported with Imaris. Finally, images were aligned and stitched together in Adobe Illustrator CS6.
Processing and analysis of MP-IVM recordings
MP-IVM recordings were analyzed in Imaris. Individual cell subsets were identified based on the intensity and morphology of the respective 3-dimensional fluorescent objects. Cellular migration was tracked based on automated track generation and manual refinement. To eliminate autofluorescence, corresponding signal intensities outside of the tracked cells were set to 0. Declining signal intensity of intravascular Qdots molecular probe (Invitrogen) was corrected with the “Bleach Correction” tool of Fiji (ImageJ) and smoothened by applying a median filter (3 × 3 × 3) in Imaris. To differentiate CTL cellular behavior proximal and distal to perivascular DC3 clusters, regions of interest (ROI) were created. First, yellow (YFP-) fluorescence was extracted from the “green” and “red” channels using the coloc(alization) tool of Imaris by gating of voxels with overlapping green and red fluorescence. Three-dimensional surfaces were then created based on a manually determined threshold of yellow fluorescence (Figure S5H and Supplemental Movie 5). Continuous surfaces directly adjacent to blood vessel lumina with a diameter 40 μm were selected and defined as proximal ROIs. All other tissue was considered as distal ROI. For CTL motility analyses only, cells in distal ROIs that were visibly interacting with YFP+ DC3 (their fluorescence signal extended into YFP surfaces that had before been manually curated to remove surfaces of yellow signal resulting from overlapping red and green CTL) were excluded. When a track within a ROI was broken into fragments because the tracked cell temporarily left the ROI, the fragments were joined into a single track, irrespective of the temporal gap. Dynamic track parameters (3D track velocity, arrest coefficient, 10-minute displacement) were analyzed in Matlab (Mathworks), using 3-D tracking data. The arrest coefficient was defined as the fraction of time in a trace or segment that a cell was migrating at a velocity below 4 μm/min. The 10-minute displacement parameter describes the average displacement over all observed 10-minute intervals for each cell. Track straightness was extracted from statistical data generated within Imaris and describes the ratio of observed total displacement and total distance traveled by a cell.
Histological analysis of tumor sections
Tumors were harvested, bisected, and fixed using BD cytofix (diluted 1:3 in PBS) for 24 h at 4 °C. Fixed samples were washed twice with PBS and dehydrated with 30% sucrose for 24 h before embedding in OCT. 20 μm sections were prepared using a cryostat and blocked using a buffer containing 1% normal mouse serum, 1% bovine serum albumin, and 0.3% Triton-X 100 for 1 h. Sections were stained with directly conjugated antibodies for 8 h at RT or overnight at 4 °C in a dark humidified chamber, and imaged using a Leica SP8 microscope, as described (Gerner et al., 2012). The following antibodies were used for staining: anti-CD64 (X54–5/7.1, Biolegend), anti-CD11c (N418, BD), anti-CD45.1 (A20, BD Biosciences), anti-MHC-II (M5/114.15.2, Biolegend) conjugated in house with Dy396XL (Dyomics, 396XL-01A), anti-Fascin1 (55K-2, Santa Cruz Biotechnology), anti-CD31 (MEC 13.3, Biolegend) and anti-CD3e (17A2, Biolegend).
Analysis of in situ and ex vivo stimulated cytokine secretion
To detect in situ cytokine secretion, mice were slowly i.v. injected with 500 μg of brefeldin A in 500 μl PBS 5 h before tissue harvest.
To detect cytokine secretion in T cells upon ex vivo re-stimulation, single cell suspensions from tumors were resuspended in T cell medium and added to α-CD3 (clone 145–2C11) and α-CD28 (clone 37.51) mAb-coated (overnight at 10 μg/ml antibody) tissue culture plates for 6 hours at 37 °C in the presence of 1 μg/mL Golgiplug (BD Bioscience) and monensin (Biolegend) and cells processed for intracellular cytokine staining.
In vitro tumor killing assay
2.5 × 105 D4M.3A-pOVA cells were co-cultured with 2.5 × 105 in vitro-generated WT or Cxcr6−/− TCF-1pos or TCF-1neg OT-I CTL for 12 h, and tumor cell death was measured by flow cytometry based on uptake of the viability dye ZombieRed.
CXCR6 in vitro induction
In vitro generated WT TCF-1pos OT-I CTL were cultured for 24 h in mrIL-2 (5 or 20 ng/ml), mrIFNγ (10 ng//ml), mrIL15 (40 ng/ml), or mrIL-12 (10 ng/ml), all from R&D, and stained with α-CD186/CXCR6 (SA051D1) and α-TCF1/TCF7 (C6329).
QUANTIFICATION AND STATISTICAL ANALYSIS
Two-tailed unpaired or paired student’s t-test was used for comparisons between two groups, while two-way ANOVA with either Bonferroni (for tumor growth studies) or SIDAK (for multiple time-point studies) post-tests, or one-way ANOVA with Tukey post-test (for single time-points) were used for comparisons across multiple groups. Kolmogorov-Smirnov test was used for pooled motility analyses. All statistical tests except for TCGA survival analyses were performed with GraphPad Prism software, and p<0.05 was considered statistically significant. No statistical methods were used to predetermine sample size. Investigators were not blinded during experiments and outcome assessment.
Read preprocessing and single cell data filtering
Gene expression counts for individual cells were generated from raw FASTQ files using the bcbio-nextgen RNA-seq pipeline (https://github.com/bcbio/bcbio-nextgen). Reads were aligned to the MM10 mouse genome assembly. Transcriptomes with more than or equal to 350 total counts, less than or equal to 6000 total counts (to filter out doublets), less than 10% of total counts derived from mitochondrial genes, and ratio of number of genes detected to total counts (i.e., the relative number of genes detected) at least higher than 0.8 were retained.
Single-cell data normalization and dimensionality reduction
Cell counts were normalized with the SCTransform function from the Seurat package (Hafemeister and Satija, 2019). To reduce dimensions, Principal Component Analysis was performed using the function RunPCA from the Seurat package (Butler et al., 2018) set to 15 Principal Components. A nearest neighbor graph and UMAP were created with the functions FindNeighbors and runUMAP from the Seurat package.
Single-cell transcriptome annotation to cell states
Using the Leiden clustering function scanpy.tl.leiden (Wolf et al., 2018), we generated multiple partitions of all single-cell transcriptomes with different numbers of clusters. To define the identity of cells, we used the partition of all single-cell transcriptomes with 436 clusters that each contained on average 20 cells and assigned these clusters to prior annotated transcriptional cell states using a multinomial naïve Bayes classifier (Pedregosa et al., 2011). For annotation, we used whole-transcriptome profiles of FACS-sorted immune cell states from the IMMGEN consortium (Heng and Painter, 2008), immune cell states in healthy and KP1.9 mouse lung tumor tissue (Zilionis et al., 2019), immune cell states in MC38 mouse tumors (Zhang et al., 2020), DC cell states in healthy and KP1.9 mouse lung tumor tissue (Maier et al., 2020), and CD8 T cell states in the spleens of lymphocytic choriomeningitis virus (LCMV) infected mice and in OVA-expressing B16.F10 mouse melanoma tumors (Miller et al., 2019). Side-by-side visualization on the UMAP of our 436 cluster classifications and clusters from all different partitions revealed the clusters or combinations of clusters that corresponded best to similar prior-annotated immune cell states from different studies. Single-cell transcriptome annotations to cell states (Figure 1A) were validated by visualization of marker genes on the UMAP (Figure 1B, 1C, and S1D), cell state enriched genes revealing known marker genes (Figure S1E), and mutual transcriptome correspondence with previously published cell states (Figure S1B and S1C),
Comparison of cell states with previously published cell states
We compared single-cell transcriptomes of T and NK cell states with previously annotated T and NK cell states (Miller et al., 2019; Zhang et al., 2020) and DC states with previously annotated DC states (Maier et al., 2020; Zhang et al., 2020; Zilionis et al., 2019) by calculating a reciprocal similarity score between each T and NK cell or DC state comparison pair, as described (Gerhard et al., 2021). To this end, we asked how confidently a machine-learning classifier model fitted to single-cell transcriptomes of these states from each dataset predicted these states to correspond to states in each other dataset. The reciprocal similarity score is non-vanishing only when two states show mutual correspondence (Gerhard et al., 2021; Zilionis et al., 2019). We used the Linear Support Vector Machines on log2 transformed data machine-learning classifier model implemented in the python package sckikit-learn (v. 0.22.2.post1) (Pedregosa et al., 2011). because we noticed that this classifier performs best for this type of classification problem (Gerhard et al., 2021).
Prior to calculating the reciprocal similarity scores, all previously published single-cell transcriptome datasets were normalized to total-cell counts as described in (Klein et al., 2015). In addition, to filter out outlier genes, each dataset was filtered to only contain the intersecting sets of genes that were detected in at least 5 cells at more than or equal to 150 TPM (average TPM over all cells) within each of the datasets.
Identification of cell state enriched genes
A gene j is an enriched gene cell state ci in heatmap (Figure S1E) if:
Gene j is detected in at least 3 cells at least 100 TPM (average TPM of all cells in C) counts across all cells in C.
Gene j has statistically significantly higher expression (two-tailed Mann- Whitney U test with multiple hypothesis correction, FDR < 5%) in state ci compared to the complement set (all cells C not in cell state ci).
Gene j has maximal average expression in state ci compared to all other states.
Analysis of KP1.9 mouse lung adenocarcinoma scRNA-seq data
Published single cell transcriptome data of FACS-sorted CD45+ cells isolated from two KP1.9 tumor bearing mice (Zilionis et al., 2019) were obtained from GSE127465. The original cell state annotations and 2D visualization of the single cell transcriptome data were used.
TCGA survival analysis
For each cancer type analyzed, TCGA transcriptomics and clinical data were obtained from the Pancancer publication (Liu et al., 2018). Specifically, EBPlusPlusAdjustPANCAN_IlluminaHiSeq_RNASeqV2.geneExp.tsv (RNA-seq expression matrix batch normalized not log2 transformed) and TCGA-CDR-SupplementalTableS1.xlsx were obtained from https://gdc.cancer.gov/about-data/publications/pancanatlas. As we noticed that hazard ratio (HR) 95% confidence intervals started to become large in TCGA solid tumor types for which less than 185 patients and 80 death events were available, all TCGA solid tumor types were included for which at least 185 patients and 80 death events were available. In addition, only TCGA solid tumor types were included for which AJCC pathologic tumor stage and detected expression of all genes analyzed were available (further excluding: COAD, GBM, LAML, LGG, OV, SARC, and UCEC). For each cancer-type, patients included in the transcriptomics data were included in the survival analysis. Patients were excluded if they had missing follow-up time and or death event values. In addition, for each Cox proportional hazards model that was constructed, patients were excluded if they had missing values for additionally included covariates. The TCGA melanoma study included lymph node metastatic site samples because primary tumor sites were often not available (Akbani et al., 2015). Excluding these samples caused loss of statistical power and model stability. Therefore, patients with RNA-Seq performed on lymph node metastatic sites were included for the melanoma survival analysis. Other solid tumor-types did not have a high number of metastatic sites.
To score gene-expression, a pseudo counteracts was added to the gene expression vector of each patient and gene-expression was log2 transformed (Robinson et al., 2010). To allow for a standardized interpretation of the HR (i.e., HR per standard deviation increase), log2 transformed gene-expression + pseudo count was Z-scored. We did not find a difference between the patient ranking of log2 transformed expression + pseudo count and Z-scored log2 transformed expression + pseudo count (tested with the Spearman rank-correlation coefficient).
Kaplan-Meier estimates were constructed using the KaplanMeierFitter function and Cox proportional hazards models were constructed using the CoxPHFitter function from the Lifelines Python package (Davidson-Pilon et al., 2017). The baseline hazard was modelled using the Breslow method (Breslow, 1975). The Wald Chi-Squared test was used to determine whether HRs were significantly different from 0. The Schoenfeld residuals test was used to confirm that all predictor variables satisfied the proportional hazards assumption (P < 0.05) (Schoenfeld, 1982).
In Figure 7E and S7E the Multivariate Cox proportional-hazards model was stratified by AJCC pathologic tumor stage (versus Stage 0 & I) because AJCC pathologic tumor stage III violated the proportional hazards assumption. NCR1 and SH2D1B were used as NK cell signature in Figure S7E because these two genes were expressed separately almost exclusively in NK cells and together only in NK cells in total cell (both CD45+ and CD45−) single-cell transcriptome datasets of human lung cancer, CRC, OV, and BRCA (Qian et al., 2020; Zilionis et al., 2019).
Events (Figure 7C): CXCR6 High=50, Low=91; CD8B High=52, Low=92; CCR10 High=64, Low=77; CXCR2 High=75, Low=79.
Events (Figure 7D): 332
Patients (Figure 7E): Cutan. Melan=408; Head & Neck=444; Lung Adeno=498; Breast=1070; Bladder Uroth.=405; Liver Hepato.=346; Lung Squam.=491; Stom. Adeno=384; Kidn. Ren cell=530.
Events (Figure 7E): Cutan. Melan.=189; Head & Neck=189; Lung Adeno.=181; Breast=140; Bladder Uroth.=177; Liver Hepato.=116; Lung Squam.=210; Stom. Adeno.=147; Kidn. Ren. cell=174.
Supplementary Material
Supplemental Movie 1: Motility of WT and Cxcr6−/− CTL in the TME (Related to Fig. S5G): An MP-IVM recording from a D4M.3A-pOVA tumor implanted into a dorsal skinfold chamber installed on an IL-12 p40-YFP reporter mouse, which was injected 3 days earlier with mRFP-expressing WT (red) and GFP-expressing Cxcr6−/− (green) TCF-1neg OT-I CTL. The animal was also i.v. injected with the QTracker 655 10 min before the recording to monitor perfusion of tumor blood vessels (grey). Each individual frame is a maximum intensity projection of 30 z-stacks spaced 4 μm apart (total thickness of 116 μm). Scale bar = 100 μm. Time is shown in minutes.
Supplemental Movie 2: Motility of WT and Cxcr6−/−CTL in the TME on days 4/5 (Related to Fig. 5F): A similar MP-IVM recording as described for Supplemental Movie 1, but obtained 4 days after injection of TCF-1neg OT-I CTL.
Supplemental Movie 3: Motility of WT and Cxcr6−/− CTL near a large perivascular DC3 cluster (Related to Fig. 5G): A cropped section of Supplemental Movie 2, separately showing YFP+ DC3 (yellow), mRFP+ WT TCF-1neg OT-I (red), and GFP+ Cxcr6−/− TCF-1neg OT-I (green) around a stromal microvessel (grey). Scale bar = 50 μm. Time is shown in minutes.
Supplemental Movie 4: Motility of WT and Cxcr6−/− CTL near a small, distal DC3 cluster (Related to Fig. 5G): A cropped section of Supplemental Movie 2, separately showing mRFP+ WT TCF-1neg OT-I (red) and GFP+ Cxcr6−/− TCF-1neg OT-I (green) around a small cluster of YFP+ DC3 (yellow) distal to larger blood vessels (grey). Scale bar = 50 μm. Time is shown in minutes.
Supplemental Movie 5: Definition of proximal ROIs used for T cell distribution analysis (Related to Fig. S5H): First, all YFP+ objects are rendered as red surfaces. The large red surface representing a continuous network of YFP+ DC3 near a microvessel with ≥40 μm caliber is then subsequently selected as a proximal ROI (see Star Methods).
Figure S1 (Related to Fig. 1). Cell states identified by scRNA-seq analysis of melanoma tissue.
(A) Numbers of single-cell transcriptomes analyzed for each identified cell state in each experimental replicate
(B, C) Heatmaps showing the reciprocal similarity score for cell states identified in the T/NK cluster (C) and the myeloid cluster (D) in this study (Di Pilato), in Miller B. et al, 2019 (Miller and Miller LCMV), Zhang Q. et al. 2019 (Zhang), Maier et al, 2020 (Maier), and Zilionins R. et al, 2019 (Zilionis). The score was calculated using the probability estimates returned by the Linear Support Vector Machine classifier applied to log2-transformed data. The DiPilato-CD8 T E state has internal heterogeneity because it has reciprocal similarity with both the CD8 T E proliferative state and CD8 T E state. Although we identified the subset of the CD8 T E state that is most similar to the CD8 T E proliferative state (marked by Mki67 on UMAP see Figure S1D), we decided not to annotate the CD8 T E proliferative state for the purpose of this paper.
(D) Single-cell expression of the indicated genes.
(E) Expression of cell state-enriched genes (see Table S1A for numerical data underlying the heatmap).
(F) PD-1 expression of OT-I CTL in D4M.3A-pOVA tumors 8 and 22 days following adoptive transfer into tumor-bearing mice.
Figure S2 (Related to Figs. 1 and 2). PD-1 expression on tumor-infiltrating OT-I cells and gating strategies to identify tumor-infiltrating immune cells.
(A, B) Comparison of chemokine receptor expression (background-corrected MFIs) on TCF-1pos and TCF-1neg subsets of tumor-infiltrating PD-1+ CD8+ T cells in s.c. implanted 35 days-old YUMM1.1 (A) or 17 days-old LCC1 (B) tumors. Note varying y-scales on graphs.
(C, D) Tumor growth following s.c. implantation of 106 YUMM1.1 (C) or LCC1 (D) cells into the flanks of C57BL/6 or Cxcr6−/− mice.
(E) Gating strategy to identify CD8+ (CTL) and CD4+ effector (Th) and regulatory T cells (Treg), NKp46+ CD3− NK cells (NK), and B220+ B cells (B) in tumor tissue.
(F, G) Frequencies of TCF-1neg CX3CR1+, TCF-1neg CX3CR1+, and TCF-1pos subsets among PD-1+ and PD-1− tumor-infiltrating CD8+ T cells, respectively, in s.c. implanted 35 days-old YUMM1.1 (F) or 17 days-old LCC1 (G) tumors
(H) Expression of CXCR6 on CD8+ T cells in tdLN 18 days after D4M.3A-pOVA tumor implantation.
(I, J), Expression of CXCR6 on indicated cell types (I), and frequency of CXCR6+ TCF-1neg CD8+ T cells (J) in the peripheral blood of healthy and tumor-bearing (day 21 after implantation) mice.
(K, L) CTFR dilution and either CD69 (K) or CD25 (L) expression by WT and Cxcr6−/− OT-I cells in tdLNs at indicated time-points after co-injection of 2 ×106 naive cells each into mice with established D4M.3A-pOVA tumors.
Data in C, F, and H represent at least two independent replicates with similar results. Graphs show means and individual replicates. */**= p<0.05/0.01 in all graphs except (C) and (D).
Figure S3 (Related to Fig. 3). Role of CXCR6 during priming and expansion of CTL in tdLNs
(A-D) Comparison of in vitro-generated WT and Cxcr6−/− TCF-1pos (top) and TCF-1neg (bottom) OT-CTL with regard to expression of CKRs (A), expression of markers of effector differentiation (B), αCD3/αCD28-stimulated expression of IFN-γ (C), and in vitro cytotoxic killing of D4M.3A-pOVA cells (D).
(E, F) WT and Cxcr6−/− TCF-1neg OT-I cells were co-injected into tumor-bearing mice on day 15 and their ratios (E) and ex vivo uptake of ZombieRed (F) in peripheral blood and liver assessed at the indicated time-points thereafter.
(G, H) AnnexinV-binding (G) and ZombieRed uptake (H) of tumor-infiltrating WT and Cxcr6−/− OT-I CTL 4 days after their injection as in vitro-generated TCF-1neg cells (106) into animals implanted with either D4M.3A-pOVA or D4M.3A flank tumors.
(I) Rates of retroviral transduction (using RV-Bcl2 or RV-ctrl) and injected ratios of transduced WT and Cxcr6−/− TCF-1neg OT-I cells.
Data represent at least two independent replicates with similar results. Graphs show means and either individual replicates or ±SEM. **/***/**** = p<0.01/0.001/0.0001.
Figure S4 (Related to Fig. 4). The CCR7+ DC3 state in KP1.9 lung tumors
(A) UMAP visualization and cell state annotation of previously published scRNA-Seq data from KP1.9 mouse lung tumors (see Experimental Details).
(B, C) Single-cell expression of Cxcl16, Ccl5, Cxcl9, and Cxcl10 (B) and heatmap of chemokine gene expression normalized to median expression value per gene across all cell states in the heatmap (exprRef), detected by scRNA-seq in KP1.9 lung carcinoma. See Table S2A for numerical data underlying the heatmap.100 TPM = average 100 transcripts per million of all cells.
(D) Single-cell expression of Ccr7 in KP1.9 lung tumors.
(E) Numbers of indicated cell types tumor tissue
(F) Expression of indicated cDC subset markers on CCR7neg cDC, CCR7int, and CCR7hi DC3.
(G) Single-cell expression of IL12B in KP1.9 lung tumors.
(H) Frequencies of CD11chi MHC IIhi and CCR7+ MHC IIhi cells among YFP+ and YFP− cells in D4M.3A-pOVA tumors in IL-12 p40 YFP reporter mice.
Insets in (A, B, D, G) show magnified displays of cDC1, cDC2, and DC3 states.
Data in E, F and H represent at least two independent replicates with similar results. Graphs in (E, F) show means and individual replicates. *** = p<0.001
Figure S5 (Related to Fig. 5). T cell distribution relative to DC3 clusters and in CD64+ TEM regions
(A, B) Single-cell expression of Fascin1 in D4M.3A-pOVA (A) and KP1.9 (B) tumors.
(C) Micrograph of a histological D4M3.3A-pOVA tumor section from an IL-12 p40-YFP reporter mouse immune-stained for Fascin1 and CD31 protein. Note aggregate of Fascin1+ cells inside the lumen of a CD31dim lymphatic vessel (LV), surrounded by CD31bright blood vessels (BV).
(D, E) Micrographs of a histological section of a D4M3.3A-pOVA tumor in an IL-12 p40-YFP reporter mouse, immuno-stained for Fascin1, CD64, CD3ε, CD45.1, and CD31 protein. The two images on the left in each row show the same overview, the images on the right show magnified ROIs, as indicated in the overviews.
(F) The positions of all CD64+ TAM, Fascin1+ DC3, and CD45.1+ OT-I cells in a tumor section represented by spots. ROIs indicate locations of overview images in (D) and (E).
(G) DSFCs were installed around D4M.3A-pOVA tumors implanted s.c. into the backs of IL-12 p40 YFP reporter mice, and animals co-injected with mRFP-expressing WT and GFP-expressing Cxcr6−/− TCF-1neg OT-I cells. 3 days later, mice were anesthetized, i.v.-injected with QTracker 655 to visualize perfused tumor vessels and 30 min-long MP-IVM time-lapse recordings were obtained. Perivascular clusters of YFP+ DC3 are indicated.
(H) To generate proximal, 3-dimensional ROIs, surfaces were generated based on thresholded YFP fluorescence signal, and continuous surfaces located directly outside of blood vessels of ≥40 μm diameter were selected.
Figure S6 (Related to Fig. 6). Role of IL-12 and IL-15 in TCF-1pos to TCF-1neg CTL conversion
(A, B) Single-cell expression of Cd80 and Il15ra (A) and heatmap of gene expression of regulators of T cell activation in APCs normalized to median expression value per gene across all cell states in the heatmap (exprRef), detected by scRNA-seq in KP1.9 lung tumors (B). See Table S2B for numerical data underlying the heatmap.100 TPM = average 100 transcripts per million of all cells. Insets in (A) show magnified displays of cDC1, cDC2, and DC3 states
(C) Histograms illustrating expression of indicated regulators of CTL activation in CCR7neg cDC, YFP−, and YFP+ DC3.
(D-F) In vitro-generated TCF-1pos OT-I CTL were treated for 24 h with the indicated combinations of cytokines before analysis for absolute cell numbers (D), as well as TCF-1 and CXCR6 expression (E, F) by the TCF-1pos and TCF-1neg subsets.
(G, H) WT and Cxcr6−/− TCF-1neg OT-I cells were co-injected into D4M.3A-pOVA tumor-bearing WT or Il15ra−/− mice on day 14 of tumor growth and their ex vivo uptake of the viability dye ZombieRed (G) and their ratios (H) in tdLNs of each host assessed 4 days later.
Data in C-F represent at least two independent replicates with similar results. Graphs in (E-H) show means and individual replicates (F-H) or means and SD (E). ** = p<0.01.
Figure S7 (Related to Fig. 7). Extended TCGA analysis
(A) Correlation between chemokine receptor and CD4 (top) or NK cell signature (bottom) expression scores (Z-scored log2 transformed expression) in biopsies from 469 melanoma patients in the SKCM TCGA database. Spearman’s rank-correlation coefficient r and two-sided P value are shown.
(B) Kaplan-Meier estimates of overall survival comparing the survival probabilities of the top (High) and bottom (Low) third of melanoma patients with regard to their expression scores (Z-scored log2 transformed expression) of the indicated genes. Hazard ratios (HR), HR 95% CI, and P values (Wald Chi-Squared Test) from a univariable Cox proportional-hazards model (High versus Low) are shown. Tick marks indicate censoring. Events: CXCR3 High=54, Low=88; CCR5 High=54, Low=86.
(C) Correlation between CXCR6 and CXCL16 or IL12B expression scores (Z-scored log2 transformed expression) in biopsies from 469 melanoma patients in the SKCM TCGA database. Spearman’s rank-correlation coefficient r and two-sided P value are shown.
(D) Kaplan-Meier estimates as described for (B). Events: CXCL16 High=61, Low=80; IL12B High=60, Low=83; NK signature High=58, Low=80
(E) The association between overall survival and the continuous expression score (Z-scored log2 transformed expression) of indicated genes in all indicated cancer types, adjusted for Sex (versus Male) and AJCC pathologic tumor stage (versus Stage 0 & I). Hazard ratios (HR), HR 95% CI, and P values (Wald Chi-Squared Test) from a multivariate Cox proportional-hazards model stratified by AJCC pathologic tumor stage (versus Stage 0 & I) are shown. Multivariate Cox proportional-hazards model was stratified by AJCC pathologic tumor stage (versus Stage 0 & I) because AJCC pathologic tumor stage III violated the proportional hazards assumption. Note that a HR of e.g. 0.75 (CXCL16 Cutan. Melan.) indicates that at any time during the TCGA study period patients had a 1–0.75 = 0.25 = 25% reduction in risk of death per one standard deviation increase of normalized log2 transformed CXCL16 expression + pseudo count.
Dashed lines separate cancer types for which the prediction reaches statistical significance (P < 0.05) from those for which it does not, shown in grey.
Patients (n): Cutan. Melan=408; Head & Neck=444; Lung Adeno=498; Breast=1070; Bladder Uroth.=405; Liver Hepato.=346; Lung Squam.=491; Stom. Adeno=384; Kidn. Ren cell=530.
Events: Cutan. Melan.=189; Head & Neck=189; Lung Adeno.=181; Breast=140; Bladder Uroth.=177; Liver Hepato.=116; Lung Squam.=210; Stom. Adeno.=184; Kidn. Ren. cell=174.
Supplemental Table S1: Numerical data underlying heatmaps (Related to Figs. S1E, 1D, 4B, and 6F)
Highlights.
CXCR6 is critical for sustained tumor control mediated by CD8+ cytotoxic T cells (CTL)
CXCR6 optimizes CTL interactions with the CCR7+ DC3 state of conventional DC
DC3 trans-present IL-15 to TCF-1neg effector-like CTL to sustain their survival in the TME
DC3 densely cluster in T cell-rich perivascular niches of the tumor stroma
ACKNOWLEDGEMENTS
We thank the Harvard Single Cell Core, the Harvard Chan Bioinformatics Core, and Allon Klein for assistance in the scRNA-seq as well as TCGA survival analyses. Funding was through National Institutes of Health (NIH) grants R01 AI123349 (to T.R.M.), P01-CA240239 (to T.R.M. and M.J.P.), R01 AI084880, R01 CA218579, R01 AI123349, R01 CA206890 (to M.J.P), R01 CA204028 (to A.D.L.), R01 AI134713, R21AI142667 (to M.Y.G.), the BMBF project Oncoattract, the MSC network for Optimizing Adoptive T Cell Therapy of Cancer funded by the H2020 Program of the European Union (grant 955575), the Bavarian Ministry for Economic Affairs (CARMOUFLAGE), ERC Grant 756017, i-Target: Immunotargeting of Cancer funded by the Elite Network of Bavaria (to S.K). T.R.M. was also supported by a Melanoma Research Alliance Established Investigator Award (Award: #827872), and by a research grant from the Melanoma Research Foundation, M.J.P. in part by the ISREC Foundation, M.D.P. by an EMBO fellowship (ALTF534-2015) and a Marie Curie Global Fellowship (750973), J.N.P. by a DFG research fellowship (PR 1652/1-1), R.K.R. by the German Academic Scholarship Foundation and by a LMU Prosa Scholarship, A.J.O. by the Swiss National Science Foundation Early Postdoctoral Mobility Award (P3BEP3_165406) and the American Association Cancer Research Basic Science Fellowship Program (18-40-01-OZGA), and D.R.P and F.Z. by the Bloomberg Philanthropies.
Footnotes
DECLARATION OF INTERESTS
S.K. has filed a patent application (PCT/EP2016/074644) related to the use of CXCR6-transduced T cells in tumor therapy. All other authors declare no competing interests.
REFERENCES
- Akbani R, Akdemir KC, Aksoy BA, Albert M, Ally A, Amin SB, Arachchi H, Arora A, Auman JT, Ayala B, et al. (2015). Genomic Classification of Cutaneous Melanoma. Cell 161, 1681–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apetoh L, Smyth MJ, Drake CG, Abastado J-P, Apte RN, Ayyoub M, Blay J-Y, Bonneville M, Butterfield LH, Caignard A, et al. (2015). Consensus nomenclature for CD8(+) T cell phenotypes in cancer. Oncoimmunology 4, e998538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ariotti S, Beltman JB, Borsje R, Hoekstra ME, Halford WP, Haanen JBAG, de Boer RJ, and Schumacher TNM (2015). Subtle CXCR3-Dependent Chemotaxis of CTLs within Infected Tissue Allows Efficient Target Localization. J. Immunol. 195, 5285–5295. [DOI] [PubMed] [Google Scholar]
- Best JA, Blair DA, Knell J, Yang E, Mayya V, Doedens A, Dustin ML, Goldrath AW, Monach P, Shinton SA, et al. (2013). Transcriptional insights into the CD8 + T cell response to infection and memory T cell formation. Nat. Immunol. 14, 404–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binnewies M, Mujal AM, Pollack JL, Combes AJ, Hardison EA, Barry KC, Tsui J, Ruhland MK, Kersten K, Abushawish MA, et al. (2019). Unleashing Type-2 Dendritic Cells to Drive Protective Antitumor CD4 + T Cell Immunity. Cell 177, 556–571.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Böttcher JP, Bonavita E, Chakravarty P, Blees H, Cabeza-Cabrerizo M, Sammicheli S, Rogers NC, Sahai E, Zelenay S, and Reis e Sousa C. (2018). NK Cells Stimulate Recruitment of cDC1 into the Tumor Microenvironment Promoting Cancer Immune Control. Cell 172, 1022–1037.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breslow NE (1975). Analysis of Survival Data under the Proportional Hazards Model. Int. Stat. Rev. / Rev. Int. Stat. 43, 45–57. [Google Scholar]
- Broz ML, Binnewies M, Boldajipour B, Nelson AE, Pollack JL, Erle DJ, Barczak A, Rosenblum MD, Daud A, Barber DL, et al. (2014). Dissecting the Tumor Myeloid Compartment Reveals Rare Activating Antigen-Presenting Cells Critical for T Cell Immunity. Cancer Cell 26, 638–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandrasekar B, Bysani S, and Mummidi S (2004). CXCL16 signals via Gi, phosphatidylinositol 3-kinase, Akt, I kappa B kinase, and nuclear factor-kappa B and induces cell-cell adhesion and aortic smooth muscle cell proliferation. J. Biol. Chem. 279, 3188–3196. [DOI] [PubMed] [Google Scholar]
- Chow MT, Ozga AJ, Servis RL, Frederick DT, Lo JA, Fisher DE, Freeman GJ, Boland GM, and Luster AD (2019). Intratumoral Activity of the CXCR3 Chemokine System Is Required for the Efficacy of Anti-PD-1 Therapy. Immunity 50, 1498–1512.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danilo M, Chennupati V, Silva JG, Siegert S, and Held W (2018). Suppression of Tcf1 by Inflammatory Cytokines Facilitates Effector CD8 T Cell Differentiation. Cell Rep. 22, 2107–2117. [DOI] [PubMed] [Google Scholar]
- Davidson-Pilon C, Kalderstam J, Kuhn B, Fiore-Gartland P, Alex A, Stark K, Anton S, Besson L, Jona Gadgil, H., et al. (2017). CamDavidsonPilon/lifelines: 0.10.1. Zenodo June 11. [Google Scholar]
- Dubois S, Mariner J, Waldmann TA, and Tagaya Y (2002). IL-15Rα recycles and presents IL-15 in trans to neighboring cells. Immunity 17, 537–547. [DOI] [PubMed] [Google Scholar]
- Engblom C, Pfirschke C, Zilionis R, Da Silva Martins J, Bos SA, Courties G, Rickelt S, Severe N, Baryawno N, Faget J, et al. (2017). Osteoblasts remotely supply lung tumors with cancer-promoting SiglecFhigh neutrophils. Science 358, eaal5081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garris CS, Arlauckas SP, Kohler RH, Trefny MP, Garren S, Piot C, Engblom C, Pfirschke C, Siwicki M, Gungabeesoon J, et al. (2018). Successful Anti-PD-1 Cancer Immunotherapy Requires T Cell-Dendritic Cell Crosstalk Involving the Cytokines IFN-γ and IL-12. Immunity 49, 1148–1161.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geissmann F, Cameron TO, Sidobre S, Manlongat N, Kronenberg M, Briskin MJ, Dustin ML, and Littman DR (2005). Intravascular immune surveillance by CXCR6+ NKT cells patrolling liver sinusoids. PLoS Biol. 3, e113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhard GM, Bill R, Messemaker M, Klein AM, and Pittet MJ (2021). Tumor-infiltrating dendritic cell states are conserved across solid human cancers. J. Exp. Med. 218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germanov E, Veinotte L, Cullen R, Chamberlain E, Butcher EC, and Johnston B (2008). Critical role for the chemokine receptor CXCR6 in homeostasis and activation of CD1d-restricted NKT cells. J. Immunol. 181, 81–91. [DOI] [PubMed] [Google Scholar]
- Gerner MY, Kastenmuller W, Ifrim I, Kabat J, and Germain RN (2012). Histo-cytometry: A method for highly multiplex quantitative tissue imaging analysis applied to dendritic cell subset microanatomy in lymph nodes. Immunity 37, 364–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groom JR, Richmond J, Murooka TT, Sorensen EW, Sung JH, Bankert K, von Andrian UH, Moon JJ, Mempel TR, and Luster AD (2012). CXCR3 chemokine receptor-ligand interactions in the lymph node optimize CD4+ T helper 1 cell differentiation. Immunity 37, 1091–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafemeister C, and Satija R (2019). Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 20, 296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto M, Kamphorst AO, Im SJ, Kissick HT, Pillai RN, Ramalingam SS, Araki K, and Ahmed R (2018). CD8 T Cell Exhaustion in Chronic Infection and Cancer: Opportunities for Interventions. Annu. Rev. Med. 69, 301–318. [DOI] [PubMed] [Google Scholar]
- He R, Hou S, Liu C, Zhang A, Bai Q, Han M, Yang Y, Wei G, Shen T, Yang X, et al. (2016). Follicular CXCR5-expressing CD8+ T cells curtail chronic viral infection. Nature 537, 412–416. [DOI] [PubMed] [Google Scholar]
- Heng TSP, and Painter MW (2008). The Immunological Genome Project: networks of gene expression in immune cells. Nat. Immunol. 9, 1091–1094. [DOI] [PubMed] [Google Scholar]
- Honda T, Egen JG, Lämmermann T, Kastenmüller W, Torabi-Parizi P, and Germain RN (2014). Tuning of Antigen Sensitivity by T Cell Receptor-Dependent Negative Feedback Controls T Cell Effector Function in Inflamed Tissues. Immunity 40, 235–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson WH, Gensheimer J, Hashimoto M, Wieland A, Valanparambil RM, Li P, Lin J-X, Konieczny BT, Im SJ, Freeman GJ, et al. (2019). Proliferating Transitory T Cells with an Effector-like Transcriptional Signature Emerge from PD-1(+) Stem-like CD8(+) T Cells during Chronic Infection. Immunity 51, 1043–1058.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntington ND, Puthalakath H, Gunn P, Naik E, Michalak EM, Smyth MJ, Tabarias H, Degli-Esposti MA, Dewson G, Willis SN, et al. (2007). Interleukin 15-mediated survival of natural killer cells is determined by interactions among Bim, Noxa and Mcl-1. Nat. Immunol. 8, 856–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Im SJ, Hashimoto M, Gerner MY, Lee J, Kissick HT, Burger MC, Shan Q, Hale JS, Lee J, Nasti TH, et al. (2016). Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 537, 417–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin H-T, Anderson AC, Tan WG, West EE, Ha S-J, Araki K, Freeman GJ, Kuchroo VK, and Ahmed R (2010). Cooperation of Tim-3 and PD-1 in CD8 T-cell exhaustion during chronic viral infection. Proc. Natl. Acad. Sci. U. S. A. 107, 14733–14738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin X, Bauer DE, Tuttleton SE, Lewin S, Gettie A, Blanchard J, Irwin CE, Safrit JT, Mittler J, Weinberger L, et al. (1999). Dramatic rise in plasma viremia after CD8(+) T cell depletion in simian immunodeficiency virus-infected macaques. J. Exp. Med. 189, 991–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaech SM, and Cui W (2012). Transcriptional control of effector and memory CD8+ T cell differentiation. Nat. Rev. Immunol. 12, 749–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim CH, Nagata K, and Butcher EC (2003). Dendritic Cells Support Sequential Reprogramming of Chemoattractant Receptor Profiles During Naive to Effector T Cell Differentiation. J. Immunol. 171, 152–158. [DOI] [PubMed] [Google Scholar]
- Klein AM, Mazutis L, Akartuna I, Tallapragada N, Veres A, Li V, Peshkin L, Weitz DA, and Kirschner MW (2015). Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell 161, 1187–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenen A, Babendreyer A, Schumacher J, Pasqualon T, Schwarz N, Seifert A, Deupi X, Ludwig A, and Dreymueller D (2017). The DRF motif of CXCR6 as chemokine receptor adaptation to adhesion. PLoS One 12, e0173486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee LN, Ronan EO, de Lara C, Franken KLMC, Ottenhoff THM, Tchilian EZ, and Beverley PCL (2011). CXCR6 is a marker for protective antigen-specific cells in the lungs after intranasal immunization against Mycobacterium tuberculosis. Infect. Immun. 79, 3328–3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leong YA, Chen Y, Ong HS, Wu D, Man K, Deleage C, Minnich M, Meckiff BJ, Wei Y, Hou Z, et al. (2016). CXCR5(+) follicular cytotoxic T cells control viral infection in B cell follicles. Nat. Immunol. 17, 1187–1196. [DOI] [PubMed] [Google Scholar]
- van der Leun AM, Thommen DS, and Schumacher TN (2020). CD8+ T cell states in human cancer: insights from single-cell analysis. Nat. Rev. Cancer 20, 218–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, van der Leun AM, Yofe I, Lubling Y, Gelbard-Solodkin D, van Akkooi A.C.J., van den Braber M, Rozeman EA, Haanen JBAG, Blank CU, et al. (2019). Dysfunctional CD8 T Cells Form a Proliferative, Dynamically Regulated Compartment within Human Melanoma. Cell 176, 775–789.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Lichtenberg T, Hoadley KA, Poisson LM, Lazar AJ, Cherniack AD, Kovatich AJ, Benz CC, Levine DA, Lee AV, et al. (2018). An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 173, 400–416.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodolce JP, Boone DL, Chai S, Swain RE, Dassopoulos T, Trettin S, and Ma A (1998). IL-15 receptor maintains lymphoid homeostasis by supporting lymphocyte homing and proliferation. Immunity 9, 669–676. [DOI] [PubMed] [Google Scholar]
- Long JZ, Lackan CS, and Hadjantonakis A-K (2005). Genetic and spectrally distinct in vivo imaging: embryonic stem cells and mice with widespread expression of a monomeric red fluorescent protein. BMC Biotechnol. 5, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay LK, Minnich M, Kragten NAM, Liao Y, Nota B, Seillet C, Zaid A, Man K, Preston S, Freestone D, et al. (2016). Hobit and Blimp1 instruct a universal transcriptional program of tissue residency in lymphocytes. Science 352, 459–463. [DOI] [PubMed] [Google Scholar]
- Maier B, Leader AM, Chen ST, Tung N, Chang C, LeBerichel J, Chudnovskiy A, Maskey S, Walker L, Finnigan JP, et al. (2020). A conserved dendritic-cell regulatory program limits antitumour immunity. Nature 580, 257–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marangoni F, Murooka TT, Manzo T, Kim EY, Carrizosa E, Elpek NM, and Mempel TR (2013). The Transcription Factor NFAT Exhibits Signal Memory during Serial T Cell Interactions with Antigen-Presenting Cells. Immunity 38, 237–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matloubian M, David A, Engel S, Ryan JE, and Cyster JG (2000). A transmembrane CXC chemokine is a ligand for HIV-coreceptor Bonzo. Nat. Immunol. 1, 298–304. [DOI] [PubMed] [Google Scholar]
- Meredith MM, Liu K, Darrasse-Jeze G, Kamphorst AO, Schreiber HA, Guermonprez P, Idoyaga J, Cheong C, Yao K-H, Niec RE, et al. (2012). Expression of the zinc finger transcription factor zDC (Zbtb46, Btbd4) defines the classical dendritic cell lineage. J. Exp. Med. 209, 1153–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikucki ME, Fisher DT, Matsuzaki J, Skitzki JJ, Gaulin NB, Muhitch JB, Ku AW, Frelinger JG, Odunsi K, Gajewski TF, et al. (2015). Non-redundant requirement for CXCR3 signalling during tumoricidal T-cell trafficking across tumour vascular checkpoints. Nat. Commun. 6, 7458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller BC, Sen DR, Al Abosy R, Bi K, Virkud YV, LaFleur MW, Yates KB, Lako A, Felt K, Naik GS, et al. (2019). Subsets of exhausted CD8 + T cells differentially mediate tumor control and respond to checkpoint blockade. Nat. Immunol. 20, 326–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy TL, Grajales-Reyes GE, Wu X, Tussiwand R, Briseño CG, Iwata A, Kretzer NM, Durai V, and Murphy KM (2016). Transcriptional Control of Dendritic Cell Development. Annu. Rev. Immunol. 34, 93–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olszak T, An D, Zeissig S, Vera MP, Richter J, Franke A, Glickman JN, Siebert R, Baron RM, Kasper DL, et al. (2012). Microbial exposure during early life has persistent effects on natural killer T cell function. Science 336, 489–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paust S, Gill HS, Wang B-Z, Flynn MP, Moseman EA, Senman B, Szczepanik M, Telenti A, Askenase PW, Compans RW, et al. (2010). Critical role for the chemokine receptor CXCR6 in NK cell-mediated antigen-specific memory of haptens and viruses. Nat. Immunol. 11, 1127–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, Blondel M, Prettenhofer P, Weiss R, Dubourg V, et al. (2011). Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 12, 2825–2830. [Google Scholar]
- Philip M, Fairchild L, Sun L, Horste EL, Camara S, Shakiba M, Scott AC, Viale A, Lauer P, Merghoub T, et al. (2017). Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature 545, 452–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Pilato M, Kim EY, Cadilha BL, Prüßmann JN, Nasrallah MN, Seruggia D, Usmani SM, Misale S, Zappulli V, Carrizosa E, et al. (2019). Targeting the CBM complex causes Treg cells to prime tumours for immune checkpoint therapy. Nature 570, 112–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian J, Olbrecht S, Boeckx B, Vos H, Laoui D, Etlioglu E, Wauters E, Pomella V, Verbandt S, Busschaert P, et al. (2020). A pan-cancer blueprint of the heterogeneous tumor microenvironment revealed by single-cell profiling. Cell Res. 30, 745–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhardt RL, Hong S, Kang S-J, Wang Z, and Locksley RM (2006). Visualization of IL-12/23p40 in vivo reveals immunostimulatory dendritic cell migrants that promote Th1 differentiation. J. Immunol. 177, 1618–1627. [DOI] [PubMed] [Google Scholar]
- Roberts EW, Broz ML, Binnewies M, Headley MB, Nelson AE, Wolf DM, Kaisho T, Bogunovic D, Bhardwaj N, and Krummel MF (2016). Critical Role for CD103(+)/CD141(+) Dendritic Cells Bearing CCR7 for Tumor Antigen Trafficking and Priming of T Cell Immunity in Melanoma. Cancer Cell 30, 324–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosato PC, Wijeyesinghe S, Stolley JM, Nelson CE, Davis RL, Manlove LS, Pennell CA, Blazar BR, Chen CC, Geller MA, et al. (2019). Virus-specific memory T cells populate tumors and can be repurposed for tumor immunotherapy. Nat. Commun. 10, 567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sade-Feldman M, Yizhak K, Bjorgaard SL, Ray JP, de Boer CG, Jenkins RW, Lieb DJ, Chen JH, Frederick DT, Barzily-Rokni M, et al. (2018). Defining T Cell States Associated with Response to Checkpoint Immunotherapy in Melanoma. Cell 175, 998–1013.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaer DA, Hirschhorn-Cymerman D, and Wolchok JD (2014). Targeting tumor-necrosis factor receptor pathways for tumor immunotherapy. J. Immunother. Cancer 2, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheper W, Kelderman S, Fanchi LF, Linnemann C, Bendle G, de Rooij MAJ, Hirt C, Mezzadra R, Slagter M, Dijkstra K, et al. (2018). Low and variable tumor reactivity of the intratumoral TCR repertoire in human cancers. Nat. Med. 359, 1350. [DOI] [PubMed] [Google Scholar]
- Schmitz JE, Kuroda MJ, Santra S, Sasseville VG, Simon MA, Lifton MA, Racz P, Tenner-Racz K, Dalesandro M, Scallon BJ, et al. (1999). Control of viremia in simian immunodeficiency virus infection by CD8+ lymphocytes. Science 283, 857–860. [DOI] [PubMed] [Google Scholar]
- Schoenfeld D (1982). Partial Residuals for The Proportional Hazards Regression Model. Biometrika 69, 239–241. [Google Scholar]
- Scholz F, Schulte A, Adamski F, Hundhausen C, Mittag J, Schwarz A, Kruse M-L, Proksch E, and Ludwig A (2007). Constitutive expression and regulated release of the transmembrane chemokine CXCL16 in human and murine skin. J. Invest. Dermatol. 127, 1444–1455. [DOI] [PubMed] [Google Scholar]
- Siddiqui I, Schaeuble K, Chennupati V, Fuertes Marraco S.A., Calderon-Copete S, Pais Ferreira D., Carmona SJ, Scarpellino L, Gfeller D, Pradervand S, et al. (2019). Intratumoral Tcf1 + PD-1 + CD8 + T Cells with Stem-like Properties Promote Tumor Control in Response to Vaccination and Checkpoint Blockade Immunotherapy. Immunity 50, 195–211.e10. [DOI] [PubMed] [Google Scholar]
- Simoni Y, Becht E, Fehlings M, Loh CY, Koo S-L, Teng KWW, Yeong JPS, Nahar R, Zhang T, Kared H, et al. (2018). Bystander CD8(+) T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature 557, 575–579. [DOI] [PubMed] [Google Scholar]
- Snell LM, MacLeod BL, Law JC, Osokine I, Elsaesser HJ, Hezaveh K, Dickson RJ, Gavin MA, Guidos CJ, McGaha TL, et al. (2018). CD8(+) T Cell Priming in Established Chronic Viral Infection Preferentially Directs Differentiation of Memory-like Cells for Sustained Immunity. Immunity 49, 678–694.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speiser DE, Utzschneider DT, Oberle SG, Münz C, Romero P, and Zehn D (2014). T cell differentiation in chronic infection and cancer: functional adaptation or exhaustion? Nat. Rev. Immunol. 14, 768–774. [DOI] [PubMed] [Google Scholar]
- Stonier SW, Ma LJ, Castillo EF, and Schluns KS (2008). Dendritic cells drive memory CD8 T-cell homeostasis via IL-15 transpresentation. Blood 112, 4546–4554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takamura S, Kato S, Motozono C, Shimaoka T, Ueha S, Matsuo K, Miyauchi K, Masumoto T, Katsushima A, Nakayama T, et al. (2019). Interstitial-resident memory CD8+ T cells sustain frontline epithelial memory in the lung. J. Exp. Med. 216, 2736–2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tse S-W, Radtke AJ, Espinosa DA, Cockburn IA, and Zavala F (2014). The chemokine receptor CXCR6 is required for the maintenance of liver memory CD8(+) T cells specific for infectious pathogens. J. Infect. Dis. 210, 1508–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlin M, Sandalova E, Masucci MG, and Levitsky V (2005). Help signals provided by lymphokines modulate the activation and apoptotic programs induced by partially agonistic peptides in specific cytotoxic T lymphocytes. Eur. J. Immunol. 35, 2929–2939. [DOI] [PubMed] [Google Scholar]
- Unutmaz D, Xiang W, Sunshine MJ, Campbell J, Butcher E, and Littman DR (2000). The primate lentiviral receptor Bonzo/STRL33 is coordinately regulated with CCR5 and its expression pattern is conserved between human and mouse. J. Immunol. 165, 3284–3292. [DOI] [PubMed] [Google Scholar]
- Utzschneider DT, Charmoy M, Chennupati V, Pousse L, Ferreira DP, Calderon-Copete S, Danilo M, Alfei F, Hofmann M, Wieland D, et al. (2016). T Cell Factor 1-Expressing Memory-like CD8+ T Cells Sustain the Immune Response to Chronic Viral Infections. Immunity 45, 415–427. [DOI] [PubMed] [Google Scholar]
- Waldmann TA, Miljkovic MD, and Conlon KC (2020). Interleukin-15 (dys)regulation of lymphoid homeostasis: Implications for therapy of autoimmunity and cancer. J. Exp. Med. 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wculek SK, Cueto FJ, Mujal AM, Melero I, Krummel MF, and Sancho D (2020). Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol. 20, 7–24. [DOI] [PubMed] [Google Scholar]
- Wein AN, McMaster SR, Takamura S, Dunbar PR, Cartwright EK, Hayward SL, McManus DT, Shimaoka T, Ueha S, Tsukui T, et al. (2019). CXCR6 regulates localization of tissue-resident memory CD8 T cells to the airways. J. Exp. Med. 216, 2748–2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf FA, Angerer P, and Theis FJ (2018). SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 19, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu T, Ji Y, Ashley Moseman E, Xu HC, Manglani M, Kirby M, Anderson SM, Handon R, Kenyon E, Elkahloun A, et al. (2016). The TCF1-Bcl6 axis counteracts type I interferon to repress exhaustion and maintain T cell stemness. Sci. Immunol. 1, eaai8593–eaai8593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie CB, Jiang B, Qin L, Tellides G, Kirkiles-Smith NC, Jane-wit D, and Pober JS (2020). Complement-activated interferon-γ–primed human endothelium transpresents interleukin-15 to CD8+ T cells. J. Clin. Invest. 130, 135060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaid A, Hor JL, Christo SN, Groom JR, Heath WR, Mackay LK, and Mueller SN (2017). Chemokine Receptor–Dependent Control of Skin Tissue–Resident Memory T Cell Formation. J. Immunol. 199, 2451–2459. [DOI] [PubMed] [Google Scholar]
- Zander R, Schauder D, Xin G, Nguyen C, Wu X, Zajac A, and Cui W (2019). CD4(+) T Cell Help Is Required for the Formation of a Cytolytic CD8(+) T Cell Subset that Protects against Chronic Infection and Cancer. Immunity 51, 1028–1042.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Li Z, Skrzypczynska KM, Fang Q, Zhang W, O’Brien SA, He Y, Wang L, Zhang Q, Kim A, et al. (2020). Single-Cell Analyses Inform Mechanisms of Myeloid-Targeted Therapies in Colon Cancer. Cell 181, 442–459.e29. [DOI] [PubMed] [Google Scholar]
- Zhang Q, He Y, Luo N, Patel SJ, Han Y, Gao R, Modak M, Carotta S, Haslinger C, Kind D, et al. (2019). Landscape and Dynamics of Single Immune Cells in Hepatocellular Carcinoma. Cell 179, 829–845.e20. [DOI] [PubMed] [Google Scholar]
- Zilionis R, Nainys J, Veres A, Savova V, Zemmour D, Klein AM, and Mazutis L (2017). Single-cell barcoding and sequencing using droplet microfluidics. Nat. Protoc. 12, 44–73. [DOI] [PubMed] [Google Scholar]
- Zilionis R, Engblom C, Pfirschke C, Savova V, Zemmour D, Saatcioglu HD, Krishnan I, Maroni G, Meyerovitz CV, Kerwin CM, et al. (2019). Single-Cell Transcriptomics of Human and Mouse Lung Cancers Reveals Conserved Myeloid Populations across Individuals and Species. Immunity 50, 1317–1334.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Movie 1: Motility of WT and Cxcr6−/− CTL in the TME (Related to Fig. S5G): An MP-IVM recording from a D4M.3A-pOVA tumor implanted into a dorsal skinfold chamber installed on an IL-12 p40-YFP reporter mouse, which was injected 3 days earlier with mRFP-expressing WT (red) and GFP-expressing Cxcr6−/− (green) TCF-1neg OT-I CTL. The animal was also i.v. injected with the QTracker 655 10 min before the recording to monitor perfusion of tumor blood vessels (grey). Each individual frame is a maximum intensity projection of 30 z-stacks spaced 4 μm apart (total thickness of 116 μm). Scale bar = 100 μm. Time is shown in minutes.
Supplemental Movie 2: Motility of WT and Cxcr6−/−CTL in the TME on days 4/5 (Related to Fig. 5F): A similar MP-IVM recording as described for Supplemental Movie 1, but obtained 4 days after injection of TCF-1neg OT-I CTL.
Supplemental Movie 3: Motility of WT and Cxcr6−/− CTL near a large perivascular DC3 cluster (Related to Fig. 5G): A cropped section of Supplemental Movie 2, separately showing YFP+ DC3 (yellow), mRFP+ WT TCF-1neg OT-I (red), and GFP+ Cxcr6−/− TCF-1neg OT-I (green) around a stromal microvessel (grey). Scale bar = 50 μm. Time is shown in minutes.
Supplemental Movie 4: Motility of WT and Cxcr6−/− CTL near a small, distal DC3 cluster (Related to Fig. 5G): A cropped section of Supplemental Movie 2, separately showing mRFP+ WT TCF-1neg OT-I (red) and GFP+ Cxcr6−/− TCF-1neg OT-I (green) around a small cluster of YFP+ DC3 (yellow) distal to larger blood vessels (grey). Scale bar = 50 μm. Time is shown in minutes.
Supplemental Movie 5: Definition of proximal ROIs used for T cell distribution analysis (Related to Fig. S5H): First, all YFP+ objects are rendered as red surfaces. The large red surface representing a continuous network of YFP+ DC3 near a microvessel with ≥40 μm caliber is then subsequently selected as a proximal ROI (see Star Methods).
Figure S1 (Related to Fig. 1). Cell states identified by scRNA-seq analysis of melanoma tissue.
(A) Numbers of single-cell transcriptomes analyzed for each identified cell state in each experimental replicate
(B, C) Heatmaps showing the reciprocal similarity score for cell states identified in the T/NK cluster (C) and the myeloid cluster (D) in this study (Di Pilato), in Miller B. et al, 2019 (Miller and Miller LCMV), Zhang Q. et al. 2019 (Zhang), Maier et al, 2020 (Maier), and Zilionins R. et al, 2019 (Zilionis). The score was calculated using the probability estimates returned by the Linear Support Vector Machine classifier applied to log2-transformed data. The DiPilato-CD8 T E state has internal heterogeneity because it has reciprocal similarity with both the CD8 T E proliferative state and CD8 T E state. Although we identified the subset of the CD8 T E state that is most similar to the CD8 T E proliferative state (marked by Mki67 on UMAP see Figure S1D), we decided not to annotate the CD8 T E proliferative state for the purpose of this paper.
(D) Single-cell expression of the indicated genes.
(E) Expression of cell state-enriched genes (see Table S1A for numerical data underlying the heatmap).
(F) PD-1 expression of OT-I CTL in D4M.3A-pOVA tumors 8 and 22 days following adoptive transfer into tumor-bearing mice.
Figure S2 (Related to Figs. 1 and 2). PD-1 expression on tumor-infiltrating OT-I cells and gating strategies to identify tumor-infiltrating immune cells.
(A, B) Comparison of chemokine receptor expression (background-corrected MFIs) on TCF-1pos and TCF-1neg subsets of tumor-infiltrating PD-1+ CD8+ T cells in s.c. implanted 35 days-old YUMM1.1 (A) or 17 days-old LCC1 (B) tumors. Note varying y-scales on graphs.
(C, D) Tumor growth following s.c. implantation of 106 YUMM1.1 (C) or LCC1 (D) cells into the flanks of C57BL/6 or Cxcr6−/− mice.
(E) Gating strategy to identify CD8+ (CTL) and CD4+ effector (Th) and regulatory T cells (Treg), NKp46+ CD3− NK cells (NK), and B220+ B cells (B) in tumor tissue.
(F, G) Frequencies of TCF-1neg CX3CR1+, TCF-1neg CX3CR1+, and TCF-1pos subsets among PD-1+ and PD-1− tumor-infiltrating CD8+ T cells, respectively, in s.c. implanted 35 days-old YUMM1.1 (F) or 17 days-old LCC1 (G) tumors
(H) Expression of CXCR6 on CD8+ T cells in tdLN 18 days after D4M.3A-pOVA tumor implantation.
(I, J), Expression of CXCR6 on indicated cell types (I), and frequency of CXCR6+ TCF-1neg CD8+ T cells (J) in the peripheral blood of healthy and tumor-bearing (day 21 after implantation) mice.
(K, L) CTFR dilution and either CD69 (K) or CD25 (L) expression by WT and Cxcr6−/− OT-I cells in tdLNs at indicated time-points after co-injection of 2 ×106 naive cells each into mice with established D4M.3A-pOVA tumors.
Data in C, F, and H represent at least two independent replicates with similar results. Graphs show means and individual replicates. */**= p<0.05/0.01 in all graphs except (C) and (D).
Figure S3 (Related to Fig. 3). Role of CXCR6 during priming and expansion of CTL in tdLNs
(A-D) Comparison of in vitro-generated WT and Cxcr6−/− TCF-1pos (top) and TCF-1neg (bottom) OT-CTL with regard to expression of CKRs (A), expression of markers of effector differentiation (B), αCD3/αCD28-stimulated expression of IFN-γ (C), and in vitro cytotoxic killing of D4M.3A-pOVA cells (D).
(E, F) WT and Cxcr6−/− TCF-1neg OT-I cells were co-injected into tumor-bearing mice on day 15 and their ratios (E) and ex vivo uptake of ZombieRed (F) in peripheral blood and liver assessed at the indicated time-points thereafter.
(G, H) AnnexinV-binding (G) and ZombieRed uptake (H) of tumor-infiltrating WT and Cxcr6−/− OT-I CTL 4 days after their injection as in vitro-generated TCF-1neg cells (106) into animals implanted with either D4M.3A-pOVA or D4M.3A flank tumors.
(I) Rates of retroviral transduction (using RV-Bcl2 or RV-ctrl) and injected ratios of transduced WT and Cxcr6−/− TCF-1neg OT-I cells.
Data represent at least two independent replicates with similar results. Graphs show means and either individual replicates or ±SEM. **/***/**** = p<0.01/0.001/0.0001.
Figure S4 (Related to Fig. 4). The CCR7+ DC3 state in KP1.9 lung tumors
(A) UMAP visualization and cell state annotation of previously published scRNA-Seq data from KP1.9 mouse lung tumors (see Experimental Details).
(B, C) Single-cell expression of Cxcl16, Ccl5, Cxcl9, and Cxcl10 (B) and heatmap of chemokine gene expression normalized to median expression value per gene across all cell states in the heatmap (exprRef), detected by scRNA-seq in KP1.9 lung carcinoma. See Table S2A for numerical data underlying the heatmap.100 TPM = average 100 transcripts per million of all cells.
(D) Single-cell expression of Ccr7 in KP1.9 lung tumors.
(E) Numbers of indicated cell types tumor tissue
(F) Expression of indicated cDC subset markers on CCR7neg cDC, CCR7int, and CCR7hi DC3.
(G) Single-cell expression of IL12B in KP1.9 lung tumors.
(H) Frequencies of CD11chi MHC IIhi and CCR7+ MHC IIhi cells among YFP+ and YFP− cells in D4M.3A-pOVA tumors in IL-12 p40 YFP reporter mice.
Insets in (A, B, D, G) show magnified displays of cDC1, cDC2, and DC3 states.
Data in E, F and H represent at least two independent replicates with similar results. Graphs in (E, F) show means and individual replicates. *** = p<0.001
Figure S5 (Related to Fig. 5). T cell distribution relative to DC3 clusters and in CD64+ TEM regions
(A, B) Single-cell expression of Fascin1 in D4M.3A-pOVA (A) and KP1.9 (B) tumors.
(C) Micrograph of a histological D4M3.3A-pOVA tumor section from an IL-12 p40-YFP reporter mouse immune-stained for Fascin1 and CD31 protein. Note aggregate of Fascin1+ cells inside the lumen of a CD31dim lymphatic vessel (LV), surrounded by CD31bright blood vessels (BV).
(D, E) Micrographs of a histological section of a D4M3.3A-pOVA tumor in an IL-12 p40-YFP reporter mouse, immuno-stained for Fascin1, CD64, CD3ε, CD45.1, and CD31 protein. The two images on the left in each row show the same overview, the images on the right show magnified ROIs, as indicated in the overviews.
(F) The positions of all CD64+ TAM, Fascin1+ DC3, and CD45.1+ OT-I cells in a tumor section represented by spots. ROIs indicate locations of overview images in (D) and (E).
(G) DSFCs were installed around D4M.3A-pOVA tumors implanted s.c. into the backs of IL-12 p40 YFP reporter mice, and animals co-injected with mRFP-expressing WT and GFP-expressing Cxcr6−/− TCF-1neg OT-I cells. 3 days later, mice were anesthetized, i.v.-injected with QTracker 655 to visualize perfused tumor vessels and 30 min-long MP-IVM time-lapse recordings were obtained. Perivascular clusters of YFP+ DC3 are indicated.
(H) To generate proximal, 3-dimensional ROIs, surfaces were generated based on thresholded YFP fluorescence signal, and continuous surfaces located directly outside of blood vessels of ≥40 μm diameter were selected.
Figure S6 (Related to Fig. 6). Role of IL-12 and IL-15 in TCF-1pos to TCF-1neg CTL conversion
(A, B) Single-cell expression of Cd80 and Il15ra (A) and heatmap of gene expression of regulators of T cell activation in APCs normalized to median expression value per gene across all cell states in the heatmap (exprRef), detected by scRNA-seq in KP1.9 lung tumors (B). See Table S2B for numerical data underlying the heatmap.100 TPM = average 100 transcripts per million of all cells. Insets in (A) show magnified displays of cDC1, cDC2, and DC3 states
(C) Histograms illustrating expression of indicated regulators of CTL activation in CCR7neg cDC, YFP−, and YFP+ DC3.
(D-F) In vitro-generated TCF-1pos OT-I CTL were treated for 24 h with the indicated combinations of cytokines before analysis for absolute cell numbers (D), as well as TCF-1 and CXCR6 expression (E, F) by the TCF-1pos and TCF-1neg subsets.
(G, H) WT and Cxcr6−/− TCF-1neg OT-I cells were co-injected into D4M.3A-pOVA tumor-bearing WT or Il15ra−/− mice on day 14 of tumor growth and their ex vivo uptake of the viability dye ZombieRed (G) and their ratios (H) in tdLNs of each host assessed 4 days later.
Data in C-F represent at least two independent replicates with similar results. Graphs in (E-H) show means and individual replicates (F-H) or means and SD (E). ** = p<0.01.
Figure S7 (Related to Fig. 7). Extended TCGA analysis
(A) Correlation between chemokine receptor and CD4 (top) or NK cell signature (bottom) expression scores (Z-scored log2 transformed expression) in biopsies from 469 melanoma patients in the SKCM TCGA database. Spearman’s rank-correlation coefficient r and two-sided P value are shown.
(B) Kaplan-Meier estimates of overall survival comparing the survival probabilities of the top (High) and bottom (Low) third of melanoma patients with regard to their expression scores (Z-scored log2 transformed expression) of the indicated genes. Hazard ratios (HR), HR 95% CI, and P values (Wald Chi-Squared Test) from a univariable Cox proportional-hazards model (High versus Low) are shown. Tick marks indicate censoring. Events: CXCR3 High=54, Low=88; CCR5 High=54, Low=86.
(C) Correlation between CXCR6 and CXCL16 or IL12B expression scores (Z-scored log2 transformed expression) in biopsies from 469 melanoma patients in the SKCM TCGA database. Spearman’s rank-correlation coefficient r and two-sided P value are shown.
(D) Kaplan-Meier estimates as described for (B). Events: CXCL16 High=61, Low=80; IL12B High=60, Low=83; NK signature High=58, Low=80
(E) The association between overall survival and the continuous expression score (Z-scored log2 transformed expression) of indicated genes in all indicated cancer types, adjusted for Sex (versus Male) and AJCC pathologic tumor stage (versus Stage 0 & I). Hazard ratios (HR), HR 95% CI, and P values (Wald Chi-Squared Test) from a multivariate Cox proportional-hazards model stratified by AJCC pathologic tumor stage (versus Stage 0 & I) are shown. Multivariate Cox proportional-hazards model was stratified by AJCC pathologic tumor stage (versus Stage 0 & I) because AJCC pathologic tumor stage III violated the proportional hazards assumption. Note that a HR of e.g. 0.75 (CXCL16 Cutan. Melan.) indicates that at any time during the TCGA study period patients had a 1–0.75 = 0.25 = 25% reduction in risk of death per one standard deviation increase of normalized log2 transformed CXCL16 expression + pseudo count.
Dashed lines separate cancer types for which the prediction reaches statistical significance (P < 0.05) from those for which it does not, shown in grey.
Patients (n): Cutan. Melan=408; Head & Neck=444; Lung Adeno=498; Breast=1070; Bladder Uroth.=405; Liver Hepato.=346; Lung Squam.=491; Stom. Adeno=384; Kidn. Ren cell=530.
Events: Cutan. Melan.=189; Head & Neck=189; Lung Adeno.=181; Breast=140; Bladder Uroth.=177; Liver Hepato.=116; Lung Squam.=210; Stom. Adeno.=184; Kidn. Ren. cell=174.
Supplemental Table S1: Numerical data underlying heatmaps (Related to Figs. S1E, 1D, 4B, and 6F)
Data Availability Statement
scRNA-seq data generated during this study, including includes genes counts pre- and post-normalization, per-cell meta data, as well we the raw FASTQ files, is publicly available on GEO (GSE179111).
The code generated during this study is available at https://github.com/pittetmi/paper-code-data/tree/main/Di_Pilato_et_al_2021
The UMAP visualization of the single-cell transcriptome data is available for interactive exploration at https://kleintools.hms.harvard.edu/tools/springViewer_1_6_dev.html?cgi-bin/client_datasets/dipilato2020/d4m.3a-pova