

Summary
To meet the needs of synthetic biologists, DNA assembly methods have transformed from simple ‘cut‐and‐paste’ procedures to highly advanced, standardised assembly techniques. Implementing these standardised DNA assembly methods in biotechnological research conducted in non‐model hosts, including Pseudomonas putida and Pseudomonas aeruginosa, could greatly benefit reproducibility and predictability of experimental results. SEVAtile is a Type IIs‐based assembly approach, which enables the rapid and standardised assembly of genetic parts – or tiles – to create genetic circuits in the established SEVA‐vector backbone. Contrary to existing DNA assembly methods, SEVAtile is an easy and straightforward method, which is compatible with any vector, both SEVA‐ and non‐SEVA. To prove the efficiency of the SEVAtile method, a three‐vector system was successfully generated to independently co‐express three different proteins in P. putida and P. aeruginosa. More specifically, one of the vectors, pBGDes, enables genomic integration of assembled circuits in the Tn7 landing site, while self‐replicatory vectors pSTDesX and pSTDesR enable inducible expression from the XylS/Pm and RhaRS/PrhaB expression systems, respectively. Together, we hope these vector systems will support research in both the microbial SynBio and Pseudomonas field.
SEVAtile is a Type IIs‐based assembly approach, which enables the rapid and standardized assembly of genetic parts – or tiles – to create genetic circuits in the established SEVA‐vector backbone. Contrary to existing DNA assembly methods, SEVAtile is an easy and straightforward method, which is compatible with any vector, both SEVA‐ and non‐SEVA. To prove the efficiency of the SEVAtile method, a three‐vector system was successfully generated to independently co‐express three different proteins in Pseudomonas putida and Pseudomonas aeruginosa.

Introduction
Over the past decade, the generation of genetic circuits has become a reproducible and efficient process due to standardised DNA assembly techniques (Decoene et al., 2018). Several of these techniques are based on Type IIs restriction enzymes (RE), including BioBrick, MoClo, GoldenBraid and VersaTile, which enable the rapid construction of transcription units and/or fusion proteins from standard DNA building blocks (Knight, 2003; Sarrion‐Perdigones et al., 2011; Weber et al., 2011; Andreou and Nakayama, 2018; Pollak et al., 2019; Gerstmans et al., 2020). Their standardised design enables the reuse of these building blocks for different constructs, as well as their straightforward exchange between different laboratories, thus saving time and increasing repeatability of results. While standard DNA assembly techniques originated in the field of synthetic biology (SynBio) for model organisms such as Escherichia coli and Saccharomyces cerevisiae, it would be beneficial to extend these approaches to biotechnological research with other, non‐model species including Pseudomonas (Lammens et al., 2020). This genus includes the important human pathogen Pseudomonas aeruginosa, but also contains Pseudomonas putida, an interesting host for recombinant production of toxic and aromatic compounds (Pachori et al., 2019; Calero et al., 2020; Loeschcke and Thies, 2020).
As P. putida is gaining attention as a valuable SynBio host, some SynBio standards and expression systems have already been optimised for this species (Martin‐Pascual et al., 2021). The Standard European Vector Architecture (SEVA) database, for one, offers a wide array of standard, modular vectors for Gram‐negative species, especially P. putida (Silva‐Rocha et al., 2013). Due to their elegant design and the public accessibility, these SEVA vectors are considered the golden standard for the P. putida biotechnology research community. SEVA vectors contain three variable modules (a cargo module, antibiotic resistance marker module and an origin‐of‐replication module), which are interconnected by invariant sequences (Silva‐Rocha et al., 2013). This cargo module usually contains either a standard multiple cloning site (MCS) or an expression device with an insertion site for the user’s gene of interest. Several of these expression devices have been thoroughly characterised in P. putida, in particular the XylS/Pm and RhaRS/PrhaB systems (Martin‐Pascual et al., 2021). Moreover, these two systems have even been shown to function orthogonally to each other in vivo (Calero et al., 2016). Secondly, besides the SEVA standard, standardised DNA assembly techniques have also been introduced in P. putida research. Recently, a new generation of SEVA vectors was developed, compatible with the BioBrick standard and the corresponding A3 assembly method (Damalas et al., 2020). This merger of the SEVA and BioBrick standards could represent a necessary push towards standardisation in the P. putida research community in the future and increase reproducibility and predictability of results.
Besides their use in P. putida‐related research, SEVA‐vectors are also becoming useful tools for P. aeruginosa research (Halfon et al., 2019; Felgner et al., 2020). Together with standardised DNA assembly, they could improve the efficiency of biotechnological research in this host. However, contrary to P. putida, the implementation of standardised DNA assembly techniques in P. aeruginosa research is lagging behind. One could hypothesize that this is due to the inherent complexity of the current standardised DNA assembly methods. Although they allow the construction of practically any genetic circuit with numerous parts, it also makes these techniques complex to set up (Sarrion‐Perdigones et al., 2011; Weber et al., 2011). To introduce both SEVA vectors and DNA assembly standards in the Pseudomonas community in a low‐threshold manner, we here introduce the SEVAtile technique. SEVAtile assembly combines the advantages of the SEVA standard and the VersaTile DNA assembly technique, thus enabling the straightforward and position‐specific assembly of several DNA building blocks in the SEVA vector backbone (Gerstmans et al., 2020). Importantly, SEVAtile assembly is compatible with any vector that lacks BsaI restriction sites, including non‐SEVA vectors, and enables the fast construction of the most common genetic circuits, making this method easier to implement and more accessible than current techniques. To illustrate the advantages of the SEVAtile method, we here generate a three‐vector system for the independent and inducible expression of three different proteins in P. putida and P. aeruginosa, based on the XylS/Pm and RhaRS/PrhaB expression devices as proof‐of‐concept.
Technical implementation
General SEVAtile cloning concept
SEVAtile is a DNA assembly method based on the recently described VersaTile technique, allowing the rapid and semi‐standardised construction of basic genetic circuits using Type IIs REs (Gerstmans et al., 2020) (Fig. 1). With this method, up to six building blocks – called SEVAtiles – can be assembled to form a single construct. Within the construct, each building block is flanked by part‐specific position tags (PTs). These PTs are specific sequences of four nucleotides, which force the tiles to ligate in a specific order by the generation of a dead‐end assembly, e.g. 5′ – PT1 – promoter – PT2 – RBS – PT3 – CDS 1 ‐ PT4 – terminator – PT5 ‐ RBS – PT6 – CDS 2 – PT7 – 3′ (Fig. 1). It is important to note that not all positions are required for use, as long as neighbouring tiles are flanked by the same PT to allow ligation, e.g. 5′ ‐ PT1 ‐ promoter – PT2 – RBS – PT3 – CDS 1 – PT7 ‐ 3′.
Fig. 1.
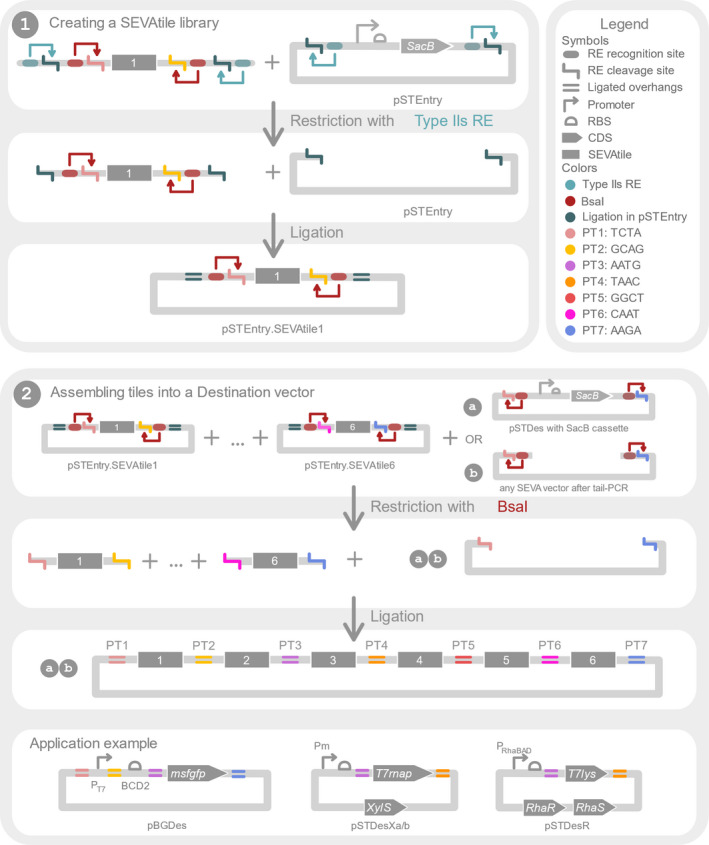
Overview of the general SEVAtile concept. (1) The first panel shows the creation of a SEVAtile library. First, a novel building block is amplified with tail‐PCR to attach two Type IIs recognition and restriction sites at both the 5′ and 3′ end of the building block, further called SEVAtile. The teal sites allow ligation of the SEVAtile into pSTEntry, while the red BsaI sites allow ligation into any destination vector in a specific order. This order is defined by the BsaI cleavage site, which creates a position‐specific overhang of four nucleotides, called position tag (PT). Each of different PTs is indicated with a different colour (see figure legend). Next, both the amplified SEVAtile and the pSTEntry vector are restricted at the teal sites by a Type IIs RE, creating complementary overhangs. After ligation by T4 DNA ligase, the desired product does no longer contain the teal RE recognition sites, thus preventing the desired product from being restricted again by the RE. By repeating this protocol for every wanted SEVAtile, one generates a SEVAtile library of pSTEntry vectors in which the tiles are flanked by the red BsaI recognition sites. (2) Assembly of SEVAtiles into a destination vector is depicted in the second panel. All desired pSTEntry·SEVAtile vectors are mixed with the destination vector of choice. The latter can be both (a) a dedicated SEVAtile destination vector with a SacB cassette for negative selection or (b) a linearised vector flanked by BsaI recognition sites. Restriction with BsaI will cut in the PTs and create four nucleotide overhangs which are specific for the position at which the SEVAtile needs to ligate within the final construct. As such, the SEVAtiles are forced to ligate in the preferred order and orientation, with a minor scar of only four nucleotides in between every tile. The SEVAtile technique allows assembly of up to six SEVAtiles, each linked with a specific PT.
SEVAtile entails three main steps: (1) attaching the correct PTs to each building block using tail‐PCR, (2) the construction of a SEVAtile library in the pSTEntry vector and (3) the rapid rational or (semi‐)combinatorial assembly of SEVAtiles in a SEVAtile‐compatible destination vector. To create a genetic construct with SEVAtile, the construct is first subdivided in functional building blocks, such as the promoter, RBS or CDS (in general called SEVAtile). Then, in the first two steps of the SEVAtile assembly technique, each required building block is individually cloned into the pSTEntry vector to generate a library of SEVAtiles (Fig. 1(1)). Within this pSTEntry vector, the SEVAtile is flanked by the required part‐specific PTs and the recognition site for BsaI, which will cut within the PTs and create position‐specific overhangs. Next, when several pSTEntry·SEVAtile vectors are mixed together and restricted with BsaI, these standard overhangs enable SEVAtiles to assemble in the correct,
predefined order in any SEVAtile destination vector (e.g. promoter‐RBS‐CDS) (Fig. 1(2)). Interestingly, the pSTEntry·SEVAtile vectors can be reused to create different constructs or can be mixed to randomize a position within the assembly, e.g. to test different promoters with the same RBS and reporter.
To further improve cloning efficiencies of SEVAtile vectors, the original SEVAtile entry and destination vectors carry a SacB cassette. By simply adding sucrose to the plates, transformants carrying the original vector instead of the desired construct will be negatively selected, thus increasing the percentage of correct transformants (Pelicic et al., 1996). It is important to note that SEVAtile cloning is not limited to dedicated SEVAtile destination vectors. Any vector can be made SEVAtile‐compatible by performing a standard tail‐PCR to add the PTs and BsaI recognition sites to the vector, as demonstrated below with the examples of pSTDesR and pBG13, a SEVA sibling vector.
SEVAtile vector design and construction
For this work, four SEVAtile vectors were created by combining modules of SEVA (sibling) vectors and the original VersaTile vectors (Table 1). The vector maps with key characteristics of these SEVAtile vectors are visualised in Fig. 2. The first vector, pSTEntry, was constructed from pSEVA247D and pVTE using Gibson Assembly (Gibson et al., 2009). First, the pRO1600 origin, terminator T1 and terminator T0 were PCR amplified from pSEVA247D with tailed primers (primers pSTEntry_1F/1R/3F/3R, Table S2), whereas the bla gene and VersaTile SacB entry cassette were PCR amplified from pVTE (primers pSTEntry_2F/2R/4F/4R, Table S2). After Gibson Assembly of these four PCR amplicons, the product was used to transform E. coli TOP10 and purified. The full sequence of pSTEntry was verified by Sanger sequencing and is available upon request.
Table 1.
Vectors used in this work.
| Name | Relevant features | Reference |
|---|---|---|
| pSEVA247D | Source for PCR; oriT; oriV(pRO1600/ColE1); DsRed2; KmR | Silva‐Rocha et al. (2013) |
| pVTE | Source for PCR; oriV(ColE1); SacB cassette; AmpR | Gerstmans et al. (2020) |
| pSTEntry | Cloning vector; oriT; oriV(pRO1600/ColE1); SacB cassette; AmpR | This work |
| pSTEntry·PT3‐msfGFP‐PT4 | pSTEntry in which the SacB cassette is substituted with msfGFP, with 5′PT3 AATG and 3′PT4 TAAC | This work |
| pSTEntry·PT3‐msfGFP‐PT7 | pSTEntry in which the SacB cassette is substituted with msfGFP, with 5′PT3 AATG and 3′PT7 TAAGA | This work |
| pSTEntry·PT6‐mCherry‐PT7 | pSTEntry in which the SacB cassette is substituted with mcherry, with 5′PT6 TAATG and 3′PT7 TAAGA | This work |
| pSTEntry·PT1‐PEM7‐PT2 | pSTEntry in which the SacB cassette is substituted with PEM7, with 5′PT1 TCTA and 3′PT2 GCAG | This work |
| pSTEntry·PT2‐BCD2‐PT3 | pSTEntry in which the SacB cassette is substituted with BCD2, with 5′PT2 GCAG and 3′PT3 AATG | This work |
| pSTEntry·PT1‐BCD2‐PT3 | pSTEntry in which the SacB cassette is substituted with BCD2, with 5′PT1 TCTA and 3′PT3 AATG | This work |
| pSTEntry·PT4‐BCD1‐PT6 | pSTEntry in which the SacB cassette is substituted with BCD1, with 5′PT4 TAAC and 3′PT6 TAATG | This work |
| pSTEntry·PT5‐BCD1‐PT6 | pSTEntry in which the SacB cassette is substituted with BCD1, with 5′PT5 GGCT and 3′PT6 TAATG | This work |
| pSTEntry·PT4‐T7terminator(wt)‐PT5 | pSTEntry in which the SacB cassette is substituted with T7terminator(wt),with 5′PT4 TAAC and 3′PT5 GGCT | This work |
| pSTEntry·PT4‐T7terminator(T7)‐PT5 | pSTEntry in which the SacB cassette is substituted with T7terminator(T7), with 5′PT4 TAAC and 3′PT5 GGCT | This work |
| pSTEntry·PT3‐linker‐PT4 | pSTEntry in which the SacB cassette is substituted with a linker, with 5′PT3 AATG and 3′PT4 TAAC | This work |
| pSTEntry·PT3‐T7RNAP‐PT4 | pSTEntry in which the SacB cassette is substituted with T7rnap with 5′PT3 AATG and 3′PT4 TAAC | This work |
| pSTEntry·PT3‐T7lys‐PT4 | pSTEntry in which the SacB cassette is substituted with T7lysozyme, with 5′PT3 AATG and 3′PT4 TAAC | This work |
| pSTEntry·PT1‐PT7‐PT2 | pSTEntry in which the SacB cassette is substituted with PT7, with 5′PT1 TCTA and 3′PT2 GCAG | This work |
| pSEVA248 | Source for PCR; oriT; oriV(pRO1600/ColE1); xylS/Pm→MCS; KmR | Silva‐Rocha et al. (2013) |
| pVTSD2 | Source for PCR; oriV(ColE1); the SacB cassette; LacI; KmR | Gerstmans et al. (2020) |
| pCTX2m | Source for PCR; oriV(pMB1); φCTX int; MCS; TcR | Hoang et al. (2000) |
| pSTDesXa | Destination vector; oriT; oriV(pRO1600/ColE1); xylS/Pm→PT3‐SacB‐PT4; KmR | This work |
| pSTDesXa·empty | pSTDesXa in which the SacB cassette is substituted with a linker | This work |
| pSTDesXa·msfGFP | pSTDesXa in which the SacB cassette is substituted with PT3‐msfGFP‐PT4 | This work |
| pSTDesXa·T7RNAP | pSTDesXa in which the SacB cassette is substituted with PT3‐T7rnap‐PT4 | This work |
| pSTDesXb | Destination vector; oriT; oriV(pRO1600/ColE1); xylS/Pm→ PT3‐SacB‐PT4; Tc | This work R |
| pSTDesXb·empty | pSTDesXb in which the SacB cassette is substituted with a linker | This work |
| pSTDesXb·msfGFP | pSTDesXb in which the SacB cassette is substituted with PT3‐msfGFP‐PT4 | This work |
| pSTDesXb·T7RNAP | pSTDesXb in which the SacB cassette is substituted with PT3‐T7rnap‐PT4 | This work |
| pPS26 | Source for PCR; oriT; oriV(RK2); RhaRS/pRhaBAD USER cassette; KmR | Calero et al. (2016) |
| pPS39 | Source for PCR; oriT; oriV(pRO1600/ColE1); RhaRS/pRhaBAD USER cassette; SmR/SpR | Calero et al. (2016) |
| pSTDesR | Destination vector; oriT; oriV(RK2); RhaRS/pRhaBAD →MCS; SmR/SpR | This work |
| pSTDesR·msfGFP | pSTDesR in which the MCS is substituted with PT3‐msfGFP‐PT4 | This work |
| pSTDesR·T7lys | pSTDesR in which the MCS is substituted with PT3‐T7lysozyme‐PT4 | This work |
| pBG13 | Destination vector; oriT; oriV(R6K); PEM7‐BCD2‐msfGFP fusion; Tn7L and Tn7R extremes; KmR/GmR | Zobel et al. (2015) |
| pBGDes·BCD2‐msfGFP | pBG13 derivative with PT1‐BCD2‐PT3‐msfGFP‐PT4 fusion | This work |
| pBGDes·PEM7‐BCD2‐msfGFP | pBG13 derivative with PT1‐PEM7‐PT2‐BCD2‐PT3‐msfGFP‐PT4 fusion | This work |
| pBGDes·PT7‐BCD2‐msfGFP | pBG13 derivative with PT1‐PT7‐PT2‐BCD2‐PT3‐msfGFP‐PT4 fusion | This work |
| pBGDes·PEM7‐BCD2‐msfGFP‐BCD1‐mCherry | pBG13 derivative with PT1‐PEM7‐PT2‐BCD2‐PT3‐msfGFP‐PT4‐BCD1‐PT6‐mcherry‐PT7 fusion | This work |
| pBGDes·PEM7‐BCD2‐msfGFP‐T7terminator(wt)‐BCD1‐mCherry | pBG13 derivative with PT1‐PEM7‐PT2‐BCD2‐PT3‐msfGFP‐PT4‐T7terminator(wt)‐PT5‐BCD1‐PT6‐mcherry‐PT7 fusion | This work |
| pBGDes·PEM7‐BCD2‐msfGFP‐T7terminator(T7)‐BCD1‐mCherry | pBG13 derivative with PT1‐PEM7‐PT2‐BCD2‐PT3‐msfGFP‐PT4‐T7terminator(T7)‐PT5‐BCD1‐PT6‐mcherry‐PT7 fusion | This work |
| pTNS2 | Helper plasmid; oriV(R6K); tnsABCD; AmpR | Choi et al. (2005) |
According to the journal’s rules, all vector backbones sequences are available upon request, whereas all nucleotide sequences of the inserts are available in Table S1. All pSTEntry‐, pSTDesXa‐, pSTDesXb‐, pSTDesR‐ and pBG13‐derived vectors were created using the SEVAtile technique.
Fig. 2.
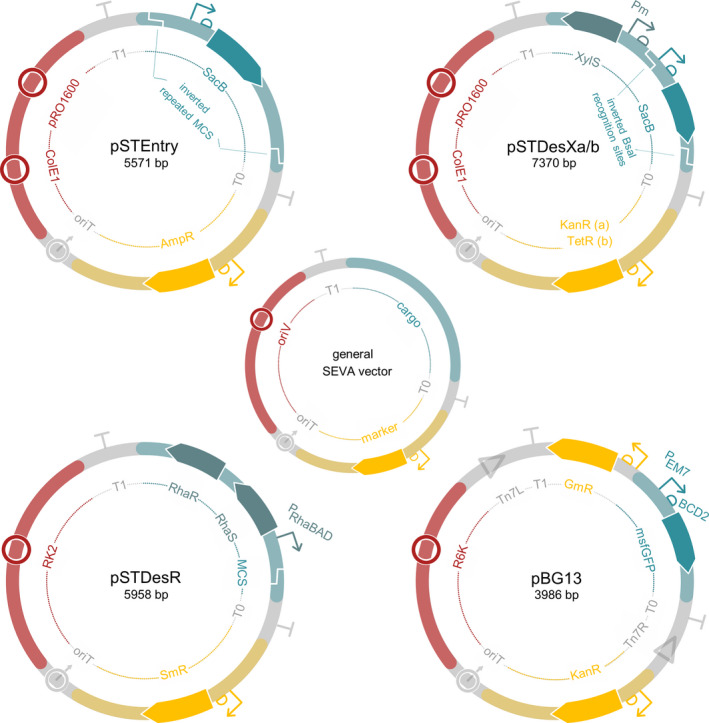
Vectors created in this study and SEVA sibling vector pBG13. All four vectors follow the general SEVA layout, consisting of six modules. Three of these modules are invariant (T0, T1 and oriT), whereas the other three are vector‐dependent (cargo, resistance marker and origin of replication).
Besides pSTEntry, three destination vectors were created, called pSTDesXa, pSTDesXb and pSTDesR. Destination vectors pSTDesXa and pSTDesXb contain the XylS/Pm expression system, which is routinely used in P. putida research (Nikel and Lorenzo, 2018; Volke et al., 2019). For the construction of pSTDesXa and pSTDesXb, a strong ribosomal binding site (RBS) and the VersaTile SacB destination cassette were inserted downstream of the Pm promoter in pSEVA248. The SacB cassette was flanked with position tags PT3 and PT4 to allow SEVAtile shuffling of a coding gene into this vector. First, to create pSTDesXa, vector pSEVA248 and the SacB cassette from pVTD2 were amplified with tail‐PCR (primers pSTDesX_1F/1R/2F/2R, Table S2), after which the two PCR products were successfully joined with Gibson Assembly and used to transform E. coli TOP10 (Gibson et al., 2009). This intermediate vector was subsequently subjected to site‐directed mutagenesis by inverse PCR to introduce a potent RBS downstream of Pm according to the manufacturer’s instructions (primers pSTDesX_RBS_F/R, Table S2) (Q5 site‐directed mutagenesis kit; NewEngland BioLabs, Ipswich, MA) (Silva et al., 2017). After transformation of E. coli TOP10 with the ligation product, the vector was purified and sequenced. Although complete Sanger sequencing of pSTDesXa confirmed the desired sequence, the vector still contained an unwanted BsaI recognition site within xylS, which was removed by creating a silent mutation (222G>C) using Q5 site‐directed mutagenesis with primers pSTDesX_BsaIQC_F/R (Table S2) (Silva et al., 2017). This final version of pSTDesXa then served as the basis for pSTDesXb, by substituting the kanamycin marker for the tetracycline resistance gene of pCTX2m using Gibson Assembly (primers pSTDesX_TcR_1F/1R/2F/2R, Table S2) (Gibson et al., 2009). The full sequences of pSTDesXa and pSTDesXb are available upon request.
The third destination vector is called pSTDesR and contains the RhaRS/PrhaB expression system, which enables tightly‐regulated rhamnose‐induced expression of coding sequences in Pseudomonas (Jeske and Altenbuchner, 2010; Calero et al., 2016; Meisner and Goldberg, 2016). The vector was based on pPS26 and pPS39 (Table 1) (Calero et al., 2016) and is compatible with pSTDesXa/b due to different origins of replication and resistance markers. pSTDesR was constructed by substituting the neo gene in pPS26 with the SmR/SpR marker of pPS39 using Gibson Assembly (primers pSTDesR_1F/1R/2F/2R, Table S2) (Gibson et al., 2009). Next, several attempts were made to insert the VersaTile SacB cassette downstream of PrhaB in a similar manner as in pSTDesXa/b, however, none yielded the desired product. Therefore, to make this vector compatible with SEVAtile shuffling, a tail‐PCR on pSTDesR is required to linearize the vector and include an RBS, PTs and BsaI recognition sites (primers pSTDesR_BsaI_F and pSTDesR_RBS_BsaI_R, Table S2). The full sequence of pSTDesR was verified using Sanger sequencing and is available upon request.
SEVAtile cloning
SEVAtile primer design and PCR amplification
Each new SEVAtile is first amplified with tail‐PCR to flank the tile with two Type IIs RE recognition sites, one for cloning into pSTEntry and a second for cloning into a destination vector. The forward and reverse primers for this tail‐PCR have a modular design consisting of three main sections (Fig. 3). The first section contains the Type IIs RE recognition and restriction site for ligation into pSTEntry, which both depend on the choice of a specific RE. One can freely choose between five Type IIs REs (Fig. 3 (2)), as long as their recognition site is absent within the specific SEVAtile sequence. The second region of the primer contains the BsaI recognition site and part‐specific position tag (PT) for cloning of the SEVAtile into a destination vector in a predefined order. For more details on the PTs, the reader is referred to Fig. 3 (3). Importantly, the BsaI recognition site cannot be present within the SEVAtile sequence and should be removed with directed mutagenesis if necessary. Finally, the third section is complementary to the SEVAtile’s 5′ or 3′ end (at least 16 nucleotides) and allows amplification of the tile.
Fig. 3.
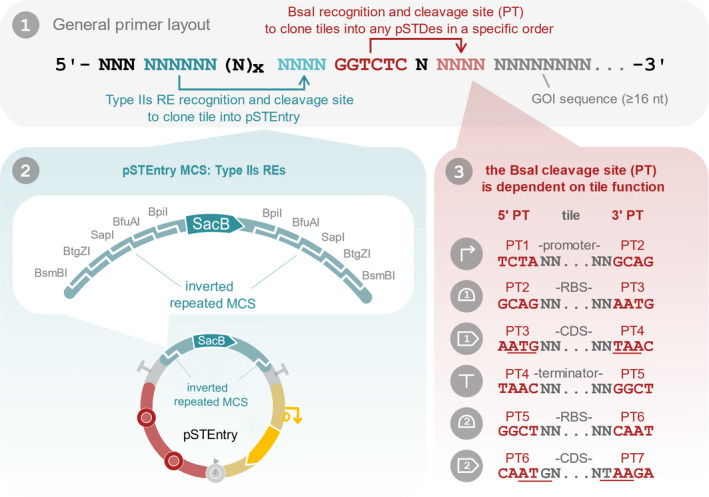
SEVAtile primer design. SEVAtile primers have a modular design, consisting of three main sections indicated in blue, red and grey (panel 1). Nucleotides indicated in black can be freely chosen to optimize the melting temperature of the primer and prevent hairpin formation. The first section, indicated in blue, contains the type IIs RE recognition and cleavage site to clone the SEVAtile in the pSTEntry vector. This vector is equipped with a type IIs RE cloning site, containing the recognition sites for BsmBI, BtgZI, SapI, BfuAI and BpiI (panel 2). The user is free to choose one of these five enzymes for cloning, provided that the recognition site is not present within the sequence of the SEVAtile. As each enzyme will cut the pSTEntry site at a different position, the cleavage site and created overhang is thus enzyme‐dependent. Therefore, the correct overhang for ligation of the SEVAtile in pSTEntry should be included in the primer. The second part of the primer, highlighted in red, enables ligation of the tile in a destination vector (panel 1). BsaI will recognize the invariant BsaI recognition site in this section and create an overhang of four nucleotides as indicated. These four nucleotides depend on the desired position of the SEVAtile in the final construct (panel 3). In total, we have designed seven PTs to connect different parts within a construct as follows: 5′ ‐ PT1 ‐ promoter – PT2 – RBS – PT3 – CDS ‐ PT4 – terminator – PT5 ‐ RBS – PT6 ‐ CDS – PT7 – 3′ vector end. For destination vectors that already contain an expression system, such as pSTDesX and pSTDesR, the promoter and RBS are already present within the vector and a CDS can thus be ligated directly with PT3 and PT4: 5′ ‐ PT3 – CDS ‐ PT4 – 3′. To minimize the impact of the four‐nucleotide scar created by the position tag, start and stopcodons were integrated in PT3, PT4, PT6 and PT7 (underlined in panel 3). The presence of ATG in PT3 and PT6 is especially important for the design of RBS tiles, as the distance between the RBS and ATG should be optimal. The third main section of the primer, indicated in grey, is complementary to the sequence of the desired SEVAtile (panel 1). A minimum of 16 nucleotides is recommended to allow efficient primer binding in the first annealing step of the tail‐PCR reaction.
Next, the primers are used to perform a regular tail‐PCR with Phusion polymerase to amplify the novel SEVAtile, followed by gel electrophoresis to verify the length of the amplicon. The amplicon is purified and ligated into a vector in the next step.
Ligating a SEVAtile into pSTEntry
Amplified SEVAtiles can be ligated into pSTEntry to create a library of tiles. However, this step is optional and can be omitted by ligating the SEVAtile along with other SEVAtiles directly into a destination vector (step 3).
Depending on the Type IIs RE chosen during the primer design, the restriction‐ligation reaction is performed in either a cycling or a non‐cycling manner. The cycling protocol is preferred as it can be performed in a single step and generally generates higher product yield since a dead‐end product is generated that no longer contains the type IIs RE recognition site, contrary to the original vector and SEVAtile PCR product.
Cycling protocol for Type IIs REs SapI, BpiI and BseRI
First, the reaction mixture is prepared by mixing 1× T4 DNA ligase buffer (Thermo Fisher Scientific, Waltham, MA, USA), 1U T4 DNA ligase (Thermo Fisher Scientific), 10U Type IIs RE (SapI, BpiI or BseRI), 100 ng pSTEntry vector DNA and 50 ng of the SEVAtile PCR product in a total volume of 20 µl. This mix is then subjected to 30 restriction‐ligation cycles by alternatingly incubating the mix for 2 min at 37°C and 3 min at 16°C. The reaction ends by inactivating the ligase and REs at 50°C for 5 min and 65°C (or 80°C for BseRI) for 20 min, respectively.
Non‐cycling protocol for Type IIs REs BsmbI, BtgZI and BfuAI
Due to incompatibility of BsmbI, BtgZI and BfuAI’s reaction temperatures with the T4 DNA ligase inactivation temperature, these REs cannot be used in the cycling protocol. Therefore, only two restriction steps and one ligation step is performed as follows: For the first restriction, 1× T4 DNA ligase buffer, 10U Type IIs RE (BsmI, BtgZI or BfuAI), 100 ng pSTEntry vector DNA and 50 ng of the SEVAtile PCR product were mixed in a total volume of 20 µl and incubated for 1 h at the RE’s optimal reaction temperature as provided by the manufacturer. Next, the reaction mix was cooled to 16°C and 1U of T4 DNA ligase was added, after which a ligation step of 90 min at 16°C was performed. Following a second restriction step by incubating the mix once more at the RE’s optimal reaction temperature for 10 min, the RE was inactivated according to the manufacturer’s guidelines.
Chemical transformation and clone analysis
After the restriction‐ligation step, 5 µl of the reaction mix was used for transformation of chemically competent E. coli TOP10 cells (Green and Rogers, 2013). The transformants were plated on selective LB agar supplemented with 100 µg ml−1 ampicillin (LB/Amp100) and 5% (w/v) sucrose (Acros Organics, Geel, Belgium), followed by overnight incubation at 37°C. The next day, colony PCR was performed with the PS1 and PS2 SEVA vector primers (Table S2) to verify the presence of the desired SEVAtile. Five positive clones were inoculated and incubated in LB/Amp100 overnight at 37°C for plasmid purification and Sanger sequencing of the pSTEntry·SEVAtile vector.
Assembling SEVAtiles into a destination vector
Several SEVAtiles can be assembled in a single destination vector to produce the desired expression construct using a cyclic restriction‐ligation protocol, similar to the ligation of SEVAtiles into pSTEntry as described above. Both the destination vector and the SEVAtiles are cleaved with BsaI, creating position‐specific overhangs, thus enabling ligation of the SEVAtiles in the desired order and orientation (Fig. 1).
The reaction mixture consisted of 1× T4 DNA ligation buffer, 1U T4 DNA ligase, 10 U BsaI, 100 ng destination vector DNA and 50 ng of each SEVAtile (SEVAtile PCR product (step 1) or pSTEntry·SEVAtile vector (step 2)) with a total volume of 20 µl. The destination vector can be either a circular vector with SacB cassette, such as pSTDesXa/b, or a linear PCR product of the vector flanked with BsaI recognition sites and PTs, as has been done in this study for pSTDesR and pBG13 with primers pSTDesR_BsaI_RBS_F/R and pBGDes_BsaI_F/R, respectively. Both SacB‐carrying and linearised vectors will generate minimal amounts of false positive colonies upon transformation of E. coli. Next, 50 restriction‐ligation cycles are performed, by repeatedly incubating the reaction mix at 37°C for 2 min, and at 16°C for 3 min. After inactivation of the ligase and BsaI by incubating for 5 min at 50°C and 5 min at 80°C respectively, 5 µl of the reaction product is used for transformation of One Shot® Mach1™ T1 Phage‐Resistant Chemically Competent E. coli cells (Invitrogen, Waltham, MA, USA), unless mentioned otherwise (Green and Rogers, 2013). The transformants are plated on LB agar with the appropriate antibiotics and 5% (w/v) sucrose if a SacB‐carrying destination vector is used. The following day, colony PCR is performed with the appropriate vector primers to verify the presence of the wanted construct (primer PS2 in combination with pSTDesX_F, pSTDesR_F or pBGDes_F, depending on the destination vector, Table S2). Five positive colonies are inoculated and incubated in LB with the appropriate antibiotic overnight at 37°C for plasmid purification and Sanger sequencing of the insert.
Electroporation of P. putida and P. aeruginosa
Electroporation of P. putida KT2440 and P. aeruginosa PAO1 was performed as previously described (Choi et al., 2006). In general, 10 ng of vector DNA was added to transform the host, except for pBGDes vectors, for which 300 ng vector DNA and 500 ng pTNS2 helper plasmid was used to allow genomic integration into the Tn7 landing site. The electroporated cells were plated on selective LB agar and incubated overnight at 30°C or 37° for P. putida KT2440 or P. aeruginosa PAO1, respectively. For P. putida KT2440, media were supplemented with Km50, Sp100 or Gm10 to select for pSTDesXa, pSTDesR or pBGDes, respectively. For P. aeruginosa PAO1, on the other hand, Tc60, Sm200 or Gm30 were added to selective growth media for pSTDesXb, pSTDesR or pBGDes, respectively. For host cells carrying multiple vectors, the electroporations were performed sequentially in the following order: pBGDes, pSTDesX, pSTDesR.
Fluorescent expression assay
To verify the performance of the SEVAtile vectors in both P. putida KT2440 and P. aeruginosa PAO1, fluorescent expression assays were performed. Overnight cultures of two biological replicates of P. putida KT2440 or P. aeruginosa PAO1 carrying the appropriate vectors were prepared in M9 minimal medium containing 1× M9 salts (BD Biosciences), 0.2% citrate (Sigma Aldrich), 2 mM MgSO4 (Sigma Aldrich), 0.1 mM CaCl2 (Sigma Aldrich), 0.5% casein amino acids (LabM; Neogen® Company, Lansing, MI, USA) and the appropriate antibiotics. On the day of the fluorescent assay, each overnight culture was diluted to OD600 0.08–0.12 in fresh M9 medium and incubated until an OD600 of 0.28–0.32 was reached. At this point, two wells of a Corning® 96 Well Black Polystyrene Microplate with Clear Flat Bottom were filled with 50 µl culture of each biological replicate, to which 50 µl fresh M9 medium supplemented with the required antibiotics and inducers was added. The XylS/Pm expression system of pSTDesXa and pSTDesXb was induced with 3‐methyl benzoic acid (3mBz) (Sigma Aldrich) in a range between 0.05 and 10 mM depending on the experiment. On the other hand, the RhaRS/PrhaB system of pSTDesR was induced with a range of 1–100 mM l‐rhamnose (Rha) (Sigma Aldrich) depending on the experiment. Next, msfGFP and OD600 levels were measured every 15 min for 12 h on the CLARIOstar® Plus Microplate Reader (BMG Labtech, Ortenberg, Germany), while incubating at 30°C or 37°C for P. putida KT2440 or P. aeruginosa PAO1, respectively. The fluorescent intensity of msfGFP was measured at 485 nm excitation wavelength and 528 nm emission wavelength with the enhanced dynamic range setting of the apparatus. Similarly, the fluorescence intensity of mCherry was determined at 570 nm excitation wavelength and 620 nm emission wavelength. To convert the relative fluorescence units of msfGFP to absolute units, a calibration curve was added to each experiment (Beal et al., 2018). More specifically, 0, 375, 750 and 1500 nM of 5(6)‐carboxyfluorescein (5(6)‐FAM) (Sigma Aldrich) in phosphate‐buffered saline was added to each plate in duplicate. All relative fluorescent measurements of msfGFP were normalised by dividing the value with the corresponding OD600 value and subsequently converted to the equivalent 5(6)‐FAM concentration. The data were analysed and visualised using JMP 15 Pro (JMP®, Version 15. SAS Institute, Cary, NC, 1989‐2019). Two‐sided P‐values were calculated by performing a pooled t‐test on the appropriate data set.
Application example and discussion
The SEVAtile assembly method is highly efficient to assemble DNA constructs
In this study, three SEVAtile destination vectors were constructed, namely pSTDesXa, pSTDesXb and pSTDesR. Apart from these vectors, the SEVAtile assembly method was also applied to one SEVA sibling vector, pBG13, of which the products were named pBGDes. To demonstrate the efficiency of the SEVAtile method, a total of fourteen destination vectors with different inserts were created, as listed in Table 2. These fourteen vectors contained one up to six building blocks and are used in the following sections for the vector characterisation assay and the T7‐based proof‐of‐concept system. First, all required SEVAtiles were cloned into pSTEntry using SapI according to the SEVAtile protocol (Table 1, primers indicated with ST). Next, the SEVAtiles were cloned into the destination vectors with SEVAtile assembly and used to transform E. coli. The assembly of these fourteen vectors proved to be very efficient, with transformation yields of on average 5.39E+04 colony forming units (CFU) per µg transformed DNA (Table 2). Furthermore, colony PCR analysis showed that for ten out of fourteen constructs, > 97% of the transformants contained the destination vector with the desired insert (Table 2, Fig. S1). These results indicate the efficacy of the SEVAtile assembly method to generate basic genetic circuits. Interestingly, the five vectors with significantly lower transformation efficiency (ranging between 34% and 75%; Table 2) were all variants of pBGDes with a PEM7‐BCD2‐msfGFP expression construct, in which PEM7 is a constitutive promoter and BCD2 a standardised translation initiation element (Mutalik et al., 2013; Zobel et al., 2015). A possible explanation for this observation is that the high msfGFP expression level generated by the strong constitutive PEM7 promoter exerts a negative selection pressure on the E. coli host cell, thus causing the reduced number of CFUs and positive transformants (Lane et al., 2007).
Table 2.
Transformation efficiencies of SEVAtile destination vectors, expressed as CFU per µg transformed DNA and the number of positive transformants after colony analysis.
| Vector | Insert | Host | CFU/µg DNA | Positive transformants |
|---|---|---|---|---|
| pSTDesXa | Empty | E. coli MACH T1 a | 1.20E+03 | 100% (32/32) |
| pSTDesXa | msfGFP | E. coli MACH T1 a | 1.13E+05 | 100% (32/32) |
| pSTDesXa | T7 RNAP | E. coli MACH T1 a | 3.79E+05 | 100% (32/32) |
| pSTDesXb | Empty | E. coli MACH T1 a | 3.84E+03 | 100% (32/32) |
| pSTDesXb | msfGFP | E. coli MACH T1 a | 2.76E+04 | 97% (31/32) |
| pSTDesXb | T7 RNAP | E. coli MACH T1 a | 2.41E+04 | 100% (32/32) |
| pSTDesR | msfGFP | E. coli MACH T1 a | 2.88E+04 | 100% (32/32) |
| pSTDesRT7 | lysozyme | E. coli MACH T1 a | 6.66E+04 | 100% (32/32) |
| pBGDes | BCD2‐msfGFP | E. coli PIR2 b | 4.08E+04 | 100% (32/32) |
| pBGDes | PEM7‐BCD2‐msfGFP | E. coli PIR2 b | 7.20E+02 | 53% (17/32) |
| pBGDes | PT7‐BCD2‐msfGFP | E. coli PIR2 b | 3.00E+04 | 100% (32/32) |
| pBGDes | PEM7‐BCD2‐msfGFP‐BCD1‐mCherry | E. coli PIR2 b | 2.17E+04 | 75% (24/32) |
| pBGDes | PEM7‐BCD2‐msfGFP‐T7terminator(wt)‐BCD1‐mCherry | E. coli PIR2 b | 9.72E+03 | 34% (11/32) |
| pBGDes | PEM7‐BCD2‐msfGFP‐T7terminator(T7)‐BCD1‐mCherry | E. coli PIR2 b | 8.04E+03 | 47% (15/32) |
More detailed vector information is available in Table 1, whereas the full nucleotide sequences of the inserts are listed in Table S1.
The commercially available One Shot® Mach1™ T1 Phage‐Resistant Chemically Competent E. coli cells were used for this transformation.
Chemically competent E. coli PIR2 cells were prepared according to the state‐of‐the‐art Rubidium Chloride method (Green and Rogers, 2013) and used for this transformation, as the R6K origin of pBGDes requires the presence of pir for replication in the transformation host.
pSTDesX and pSTDesR enable inducer‐dependent expression of msfGFP in P. putida KT2440 and P. aeruginosa PAO1
To show the functionality of the SEVAtile destination vectors in P. putida KT2440 and P. aeruginosa PAO1, the msfGFP reporter was cloned into pSTDesXa, pSTDesXb and pSTDesR, which contain the XylS/Pm and RhaRS/PrhaB expression systems, respectively. As the only difference between pSTDesXa and pSTDesXb is their resistance marker, we will further refer to both vectors as pSTDesX. The resulting vectors pSTDesX·msfGFP and pSTDesR·msfGFP were individually electroporated into both Pseudomonas hosts, after which a fluorescence assay was performed to assess the tuneable performance of the XylS/Pm and RhaRS/PrhaB expression systems in response to different inducer concentrations, as described in the Technical Implementation. The results of this assay after 10 h of induction with a 0–10 mM 3mBz concentration range or a 0–100 mM Rha concentration range are depicted in Fig. 4. For P. putida KT2440, the XylS/Pm system on pSTDesX yielded moderate msfGFP expression levels of 15.3 nM 5(6)‐FAM/OD600 for the highest inducer concentration, which was 11‐fold higher than the uninduced sample. These expression levels were lower than expected, as Volke et al. (2019) reported 84‐fold induction levels using a similar construct. We hypothesize that this could be due to the 4nt scar (PT3) between the RBS and msfGFP, which was introduced during the SEVAtile assembly. In contrast to the moderate expression levels of the XylS/Pm system in P. putida, the maximum fluorescence signal of RhaRS/PrhaB after induction with 100 mM Rha was 126 nM 5(6)‐FAM/OD600, 55‐fold higher than the uninduced sample (Fig. 4). In comparison, Calero et al. (2016) reported fold induction levels of around 700‐fold for this system, however, comparison of these results are misleading as a different origin of replication and RBS were used.
Fig. 4.
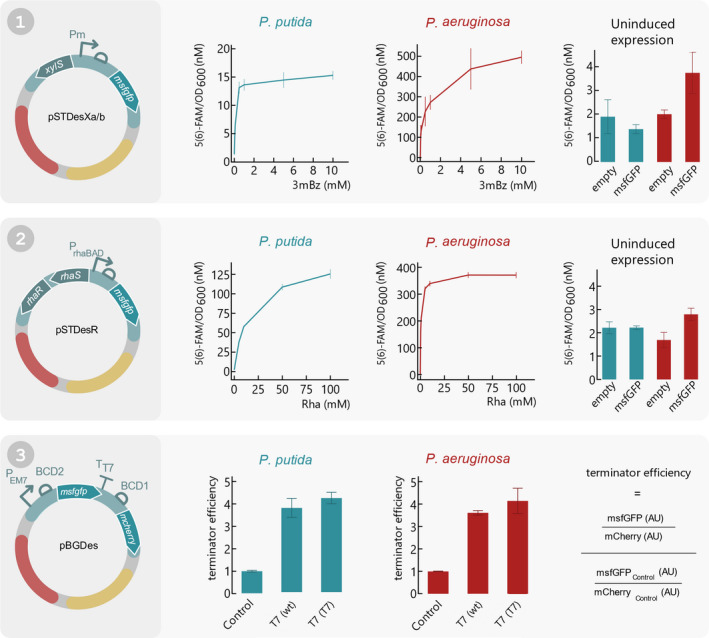
Functionality assay of all SEVAtile destination vectors in P. putida KT2440 and P. aeruginosa PAO1. (1) pSTDesXa⋅msfGFP and pSTDesXb⋅msfGFP were electroporated to P. putida KT2440 and P. aeruginosa PAO1, respectively. A fluorescence assay was performed in which the cells were induced with 0; 0.05; 0.1; 0.5; 1; 5; 10 mM 3mBz and the fluorescence signal and OD600 was monitored for 12 h. The fluorescent signal after 10 h of induction in response to the different inducer concentrations is plotted separately for P. putida KT2440 and P. aeruginosa PA01 (left and middle graph, respectively). Furthermore, to assess the leakiness of XylS/Pm in uninduced conditions, the fluorescence signal of hosts carrying pSTDesXa/b·msfGFP after 10 h of cell growth was compared to an empty control vector (right graph). (2) pSTDesR·msfGFP was electroporated to P. putida KT2440 and P. aeruginosa PAO1, after which the fluorescence intensity and OD600 was monitored for 12 h with 0; 1; 5; 10; 50; 100 mM Rha to assess the performance of RhaRS/PrhaB in both hosts. The left and middle graph display the fluorescence intensity in response to the Rha concentration after 10 h of cell growth for P. putida KT2440 and P. aeruginosa PAO1, respectively. The right graph depicts the fluorescence intensity of an uninduced sample compared to an empty control vector after 10 h of cell growth, for both hosts. (3) To show that the SEVAtile technique allows successful formation of genetic constructs with six building blocks, a terminator trap system was generated (Temme et al., 2012). The terminator is flanked by an msfGFP reporter upstream and mCherry reporter downstream in the pBGDes backbone: pBGDes·PEM7‐BCD2‐msfGFP‐BCD1‐mCherry (control), pBGDes·PEM7‐BCD2‐msfGFP‐T7terminator(wt)‐BCD1‐mCherry and pBGDes·PEM7‐BCD2‐msfGFP‐T7terminator(T7)‐BCD1‐mCherry. These vectors were electroporated to P. putida KT2440 and P. aeruginosa PAO1 with pTNS2 to enable genomic integration of the vector into the host’s Tn7 landing site. The fluorescence intensity levels of msfGFP and mCherry of both hosts carrying the different constructs were monitored for 12 h. The termination efficiency after 10 h of cell growth was calculated as displayed on the right and plotted for both hosts for the control construct and the two different terminators. Data points and bars represent the mean value of four replicates, error bars indicate the standard deviation. Full graphs of fluorescence intensity and OD600 are available in the Supporting Information (Figs S2–S5).
For P. aeruginosa PAO1, on the other hand, XylS/Pm and RhaRS/PrhaB yielded expression levels that were much higher compared to P. putida KT2440. For the highest inducer concentrations, the fluorescence intensities were increased 133‐ and 128‐fold compared to uninduced samples for XylS/Pm and RhaRS/PrhaB respectively, reaching maximal signals of 497 and 371 nM 5(6)‐FAM/OD600 (Fig. 4). For comparison, Meisner et al. observed a 50‐fold induction for the RhaRS/PrhaB system using a β‐galactosidase assay.
Finally, to assess the stringency of both expression systems in pSTDesX·msfGFP and pSTDesR·msfGFP, the fluorescence signal of the uninduced samples were compared to empty control vectors containing a linker instead of the msfGFP reporter. As the results in Fig. 4 indicate, no significant leaky msfGFP expression could be observed for P. putida (P = 0.0651 and P = 0.9554 for pSTDesX and pSTDesR, respectively). This is in contrast to P aeruginosa, in which pSTDesX·msfGFP and pSTDesR·msfGFP did cause significant msfGFP expression in the absence of inducer, compared to the empty control vectors (P = 0.0008 and P = 0.0002 for pSTDesX and pSTDesR, respectively). More specifically, the msfGFP expression levels were almost twice as high for both expression systems compared to the empty vectors (3.73 vs 2.00 nM 5(6)‐FAM/OD600 for pSTDesX and 2.91 vs 1.76 5(6)‐FAM/OD600 for pSTDesR). However, these values are still negligible in comparison to the msfGFP expression levels upon induction and indicate that the vectors have a high level of stringency in both hosts. While the RhaRS/PrhaB system is known to be very stringent in P. putida and P. aeruginosa (Jeske and Altenbuchner, 2010; Calero et al., 2016; Meisner and Goldberg, 2016), the high stringency of the XylS/Pm system was surprising. Although this system is regularly used in P. putida biotechnological research, its high leakiness often remains a well‐known issue (Calero et al., 2016; Volke et al., 2019).
Despite that both hosts belong to the same genus, the expression systems resulted in a very distinct response for P. putida and P. aeruginosa. This was not unexpected considering the differences in the physiology and metabolism of these two species, as P. putida is a soil‐coloniser and P. aeruginosa a human pathogen (Silby et al., 2011). Furthermore, differences in inducer uptake, inducer metabolisation and catabolite repression mechanisms will all directly impact the performance of the expression systems. For example, Jeske and Altenbuchner (2010) demonstrated for the RhaRS/PrhaB system that P. putida KT2440 is unsensitive to catabolite repression and lacks a rhamnose transporter (Jeske and Altenbuchner, 2010). However, further research to elucidate the differences in response of XylS/Pm and RhaRS/PrhaB in P. putida and P. aeruginosa is required.
Genetic circuits in pBGDes are efficiently expressed in P. putida KT2440 and P. aeruginosa PAO1.
First, we confirmed that the pBGDes backbone allows significant gene expression in both P. putida and P. aeruginosa by comparing the msfGFP expression levels of the hosts carrying pBGDes·BCD2‐msfGFP and pBGDes·PEM7‐BCD2‐msfGFP in the host’s Tn7 landing site as a negative and positive control, respectively. In these constructs, PEM7 is a strong constitutive promoter and BCD2 is a standardised translation initiation element (Mutalik et al., 2013; Zobel et al., 2015). As displayed in Fig. 4, significant msfGFP expression levels could be observed for both P. putida and P. aeruginosa carrying the PEM7‐BCD2‐msfGFP construct in contrast to the negative control, namely 16.2 and 45.8 nM 5(6)‐FAM/OD600 after 10 h of cell growth, respectively (P < 0.0001 for both P. putida and P. aeruginosa (Figs S12 and S13)). This proves that the pBGDes backbone allows efficient expression of genetic circuits in these hosts. For P. putida, a highly similar PEM7 ‐BCD2‐msfGFP construct generated high expression levels in previous research, in which an 80‐fold induction level was observed (Zobel et al., 2015). In contrast, in this research only an 8‐fold increase in msfGFP expression was measured. The reason for this difference is unclear, but could be due to the minor differences at the 3′ and 5′ termini of the construct, which could exert context effects (Köbbing et al., 2020).
After proving the functionality of the pBGDes backbone, we next assessed the performance of larger constructs with six building blocks. For this purpose, we recreated the ‘terminator trap’ from Temme et al. (2012), in which a transcriptional terminator is trapped between two fluorescent reporters. Ideally, the terminator does not influence transcription of the upstream reporter, while preventing transcription of the downstream reporter. To this end, both hosts were transformed with pBGDes·PEM7‐BCD2‐msfGFP‐BCD1‐mCherry (control), pBGDes·PEM7‐BCD2‐msfGFP‐T7terminator(wt)‐BCD1‐mCherry and pBGDes·PEM7‐BCD2‐msfGFP‐T7terminator(T7)‐BCD1‐mCherry. In these constructs, PEM7 is a strong constitutive promoter, BCD1 and BCD2 are standardised translation initiation elements and T7terminator(wt) and (T7) are the wildtype and an optimised variant of the T7 terminator from Temme et al. (2012) (Mutalik et al., 2013; Zobel et al., 2015). If the terminator trap functions as expected, the T7 wildtype terminator and the T7 optimised variant should reduce the level of mCherry expression compared to the control. The fluorescence intensity of msfGFP and mCherry was measured for all samples for a 12 h period and the termination efficiency was calculated according to Temme et al. (2012). As illustrated in Fig. 4, the termination efficiency of the wildtype and optimised terminator are 3.44 and 3.84 for P. putida and 3.62 and 4.15 for P. aeruginosa, respectively. These values are consistent with those reported by Temme et al. for E. coli. Based on these results, we conclude that SEVAtile enables the straightforward construction of functional genetic circuits with up to six building blocks.
pSTDesX, pSTDesR and pBGDes form a three‐vector system, enabling co‐expression of three different proteins in P. putida KT2440 and P. aeruginosa PAO1
In this final experiment, we show that pSTDesX, pSTDesR and pBGDes can co‐exist in one host cell and still maintain their functionality. For this purpose, a proof‐of‐concept was designed based on the T7 transcription system and illustrated in Fig. 5 (Studier and Moffatt, 1986). More specifically, the T7 RNA polymerase (RNAP), T7 lysozyme and PT7‐BCD2‐msfGFP were shuffled into pSTDesX, pSTDesR and pBGDes, respectively, and transformed to both hosts as described in the Technical Implementation. The performance of this proof‐of‐concept was assessed by measuring the msfGFP fluorescence intensity in different conditions. Induction of T7 RNAP expression with 0.3 mM 3mBz increased the msfGFP fluorescence signal 17‐fold for P. putida and 13‐fold for P. aeruginosa compared to the uninduced sample (Fig. 5). In the absence of inducers, some leaky expression of the T7 RNAP from pSTDesX is occurring, causing a basal level of msfGFP fluorescence of 46.3 and 107.6 nM 5(6)‐FAM/OD600 for P. putida and P. aeruginosa, respectively after 10 h of growth.
Fig. 5.
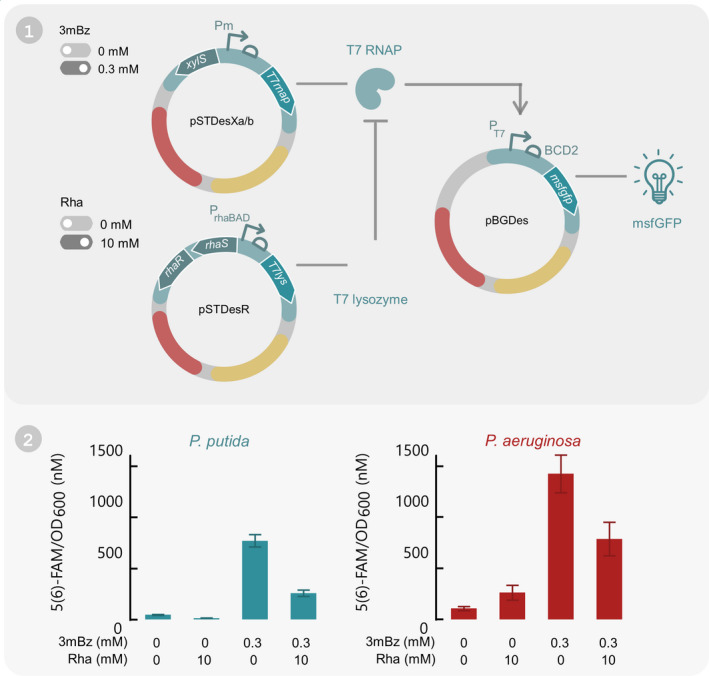
Proof‐of‐concept test to show that all three SEVAtile destination vectors can co‐exist and individually express a gene of interest in P. putida KT2440 and P. aeruginosa PAO1. (1) Graphical overview of the proof‐of‐concept. The T7 transcriptional system was integrated in P. putida KT2440 and P. aeruginosa PAO1 by electroporating pSTDesXa/b·T7RNAP, pSTDesR·T7lysozyme and pBGDes·PT7‐BCD2‐msfGFP together in both hosts. pBGDes is genomically integrated in the host’s Tn7 landing site. In this set‐up, expression of T7 RNAP can be induced with 3mBz, which will cause expression of the msfGFP reporter from the T7 promoter. On the other hand, T7 lysozyme expression is induced with Rha and will inhibit the T7 RNAP. (2) After integration of the T7 transcriptional system in P. putida KT2440 and P. aeruginosa PAO1, both hosts were induced with all four combinations of 0 or 0.3 mM 3mBz and 0 or 10 mM Rha. The fluorescence intensity levels after 10 h of induction are displayed in the graph. Bars represent the mean value of four replicates, error bars indicate the 95%‐confidence interval. Full graphs of fluorescence intensity and OD600 are available in the Supporting Information (Figs S6 and S7).
Next, the cells were induced with 10 mM Rha to induce expression of the T7 lysozyme, which should inhibit the T7 RNAP and reduce the basal msfGFP expression level (Moffatt and Studier, 1987; Studier, 1991). This effect can indeed be observed for P. putida, for which the leaky expression was threefold lower compared to the uninduced sample (Fig. 5). For P. aeruginosa, on the other hand, the opposite effect occurred: the basal expression upon induction with 10 mM Rha was doubled (Fig. 5). However, it is important to note that the T7 lysozyme exerted a strong toxic effect on P. aeruginosa cells, reaching an OD600 value of merely 0.24 after 12 h of growth, in contrast to 2.05 for uninduced samples (Fig. S7). Since all fluorescence signals were normalised for the OD600 value, this could cause a distorted image of the results. After induction of both the T7 RNAP and T7 lysozyme, a 6‐ and 7‐fold induction in msfGFP expression levels could be observed for P. putida and P. aeruginosa, respectively, which was significantly lower than the expression levels without lysozyme induction (P < 0.0001 and P = 0.0002 for P. putida and P. aeruginosa, respectively (Figs S16 and S17)).
Overall, these results show that both the T7 RNAP and T7 lysozyme are efficiently and independently produced in the host cells upon induction and that the T7 RNAP is able to express msfGFP from the T7 promoter. Based on this proof‐of‐concept, we conclude that pSTDesX, pSTDesR and pBGDes can be used as an effective three‐vector system in Pseudomonas.
Although these data clearly indicate that the T7 transcription system functions satisfactorily in our three‐vector system in Pseudomonas, further optimisations may be envisioned. The well‐known toxicity of both the T7 RNAP and the T7 lysozyme and the high leakiness of the system have restricted the broad implementation of the T7 transcriptional scheme in Pseudomonas thus far (Szafranski et al., 1997; Shis and Bennett, 2013; Weihmann et al., 2020). Interestingly, recent studies where the T7 lysozyme is replaced with an asRNA or a feedback‐loop to repress the unwanted basal expression of T7 RNAP show promising results in Pseudomonas and would enable the use of this widely used system in Pseudomonas hosts (Kushwaha and Salis, 2015; Liang et al., 2018). A second approach to overcome the toxicity issues is the use of a split version of the T7 RNAP (Shis and Bennett, 2013).
The advantage of our three‐vector system mainly lies in the ‘plug‐and‐play’ aspect of the SEVAtile technique and its general potential beyond one expression system. After generation of a SEVAtile library, the tiles can be quickly ligated in the different destination vectors, enabling the user to screen multiple ‘backbone‐expression system‐GOI’ combinations in a matter of days. As such, the user can easily determine the optimal combination of backbone, expression system and GOI for a given application. Moreover, besides the XylS/Pm and RhaRS/PrhaB expression systems used in this proof‐of‐concept, many other expression systems for Pseudomonas are available in the SEVA format (Silva‐Rocha et al., 2013). The SEVAtile technique could easily be applied to these vectors and thus generate several variations of our three‐vector system. Also non‐SEVA vectors can be used in the SEVAtile technique, such as the recently developed pRGPDuo, which allows independent co‐expression of two GOIs via the LacI/Ptac and TetR/Ptet expression systems (Gauttam et al., 2020). As the backbone of pRGPDuo carries the same origin and resistance marker as pSTDesXa, it should be straightforward to substitute this interesting vector in our current three‐vector system and broaden the possible application range of the SEVAtile technique.
Conclusion
SEVAtile is a highly efficient method to create genetic constructs for Pseudomonas and other potential Gram‐negative bacterial hosts. The Type IIs RE‐approach to assemble DNA parts has previously proven its value in the VersaTile and MoClo standards (Weber et al., 2011; Gerstmans et al., 2020). Although this approach creates scars of (at least) four nucleotides due to the position tags in between parts, the advantages of these position tags are unmistakable. They allow assembly of parts in a specific order and orientation, easy randomisation of parts in a genetic construct and part reuse. Furthermore, the scarring in SEVAtile has been minimised by introducing start and stop codons in the position tags where possible. Hence, the use of position tags minimises cloning effort and primer design. This is in contrast to the well‐known scar‐free Gibson assembly method (Gibson et al., 2009). While this method allows the user total flexibility over the location of ligation and sequence composition, it also requires specific primer design for each construct and has limitations when annealing GC‐rich sequences.
Most advanced DNA assembly methods require the use of specific destination vectors, which represents an additional hurdle for research groups to implement these methods in their current research (Sarrion‐Perdigones et al., 2011; Weber et al., 2011; Gerstmans et al., 2020). To overcome this threshold, SEVAtile enables the user to shuffle parts into any vector of choice, including all currently available SEVA vectors with any expression system or resistance marker, offering great flexibility to the user. In this study, three destination vectors were created and combined successfully in P. putida and P. aeruginosa. Moreover, we showed that pSTDesX and pSTDesR, carrying the XylS/Pm and RhaRS/PrhaB expression systems, enable inducible, tuneable and independent expression of proteins in Pseudomonas. These expression systems have been characterised in Pseudomonas in previous research (Jeske and Altenbuchner, 2010; Calero et al., 2016; Meisner and Goldberg, 2016; Gawin et al., 2017). However, to our knowledge, this study is the first to successfully combine the XylS/Pm and RhaRS/PrhaB in P. aeruginosa and as such extend the engineering toolbox of this important human pathogen. Furthermore, as the SEVAtile technique makes use of the SEVA backbone, the transfer to other interesting Gram‐negative hosts outside the Pseudomonas genus should be feasible and as such broaden the toolset of other non‐model bacteria.
Conflict of interest
D.G., Y.B. and R.L. are inventors on a patent application related to this work filed by Ghent University and the University of Leuven (no. WO/2018/114980, filed on 19 February 2017, published on 28 June 2018). The authors declare that they have no other competing interests.
Supporting information
Table S1. Full nucleotide sequences of all SEVAtiles used in this study. Nucleotides indicated in bold were included in position tags (PT): PT3 AATG, PT4 TAAC, PT6 TAATG and TAATG, PT7 TAAGA.
Table S2. Primers used in this work.
Fig. S1. Colony PCR analysis of 32 transformants of each SEVAtile destination vector made in this study. As a reference, λ‐PstI ladder is used (1 kb GeneRuler for pBGDes·PEM7‐BCD2‐msfGFP‐BCD1‐mCherry, pBGDes·PEM7‐BCD2‐msfGFP‐T7terminator(wt)‐BCD1‐mCherry and pBGDes·PEM7‐BCD2‐msfGFP‐T7terminator(T7)‐BCD1‐mCherry) and the expected amplicon length is indicated on the left of the agarose gel.
Fig. S2. Characterization of pSTDesX. Graphs depicting the OD600, absolute msfGFP fluorescence intensity (AU) and calibrated fluorescence intensity (5(6)‐FAM/OD600) over a 12 hour period in either P. putida KT2440 or P. aeruginosa PAO1 carrying the pSTDesX⋅msfGFP vector. MsfGFP expression was induced with 0; 0.05; 0.1; 0.05; 0.1; 5; 10 mM 3mBz. For clarity, only data of the samples with 0, 1 and 10 mM 3mBz are indicated. Lines represent the mean value of four replicates, error bars indicate the standard deviation.
Fig. S3. Characterization of pSTDesR. Graphs depicting the OD600, absolute msfGFP fluorescence intensity (AU) and calibrated fluorescence intensity (5(6)‐FAM/OD600) over a 12 hour period in either P. putida KT2440 or P. aeruginosa PAO1 carrying the pSTDesR⋅msfGFP vector. MsfGFP expression was induced with 0; 0.5; 1; 5; 10; 50; 100 mM Rha. For clarity, only data of the samples with 0, 10 and 100 mM Rha are shown. Lines represent the mean value of four replicates, error bars indicate the standard deviation.
Fig. S4. Characterization of pBGDes (1). Graphs depicting the OD600, absolute msfGFP fluorescence intensity (AU) and calibrated fluorescence intensity (5(6)‐FAM/OD600) over 12 hours for P. putida KT2440 and P. aeruginosa PAO1 carrying pBGDes⋅PEM7‐BCD2‐msfGFP or pBGDes⋅BCD2‐msfGFP (negative control). Lines represent the mean value of four replicates, error bars indicate the standard deviation.
Fig. S5. Characterization of pBGDes (2). Graphs depicting the OD600, absolute msfGFP fluorescence intensity (AU) and absolute mCherry fluorescence intensity (AU) over 12 hours for P. putida KT2440 and P. aeruginosa PAO1 carrying pBGDes⋅BCD2‐msfGFP (negative control), pBGDes⋅PEM7‐BCD2‐msfGFP‐BCD1‐mCherry (no terminator), pBGDes⋅PEM7‐BCD2‐msfGFP‐T7terminator(wt)‐BCD1‐mCherry (T7terminator (wt)) or pBGDes⋅PEM7‐BCD2‐msfGFP‐ T7terminator(T7)‐BCD1‐mCherry (T7 terminator (T7)). Lines represent the mean value of four replicates, error bars indicate the standard deviation.
Fig. S6. T7‐based proof‐of‐concept in P. putida KT2440. Graphs depicting the OD600, absolute msfGFP fluorescence intensity (AU) and calibrated fluorescence intensity (5(6)‐FAM/OD600) over 12 hours for P. putida KT2440 carrying pBGDes⋅PT7 ‐BCD2‐msfGFP, pSTDesX⋅T7RNAP and pSTDesR⋅T7lysozyme. The cells were induced with all four combinations of 0/0.3 mM 3mBz and 0/10 mM Rha. Lines represent the mean value of four replicates, error bars indicate the standard deviation.
Fig. S7. T7‐based proof‐of‐concept in P. aeruginosa PAO1. Graphs depicting the OD600, absolute msfGFP fluorescence intensity (AU) and calibrated fluorescence intensity (5(6)‐FAM/OD600) over 12 hours for P. aeruginosa PAO1 carrying pBGDes⋅PT7 ‐BCD2‐msfGFP, pSTDesX⋅T7RNAP and pSTDesR⋅T7lysozyme. The cells were induced with all four combinations of 0/0.3 mM 3mBz and 0/10 mM Rha. Lines represent the mean value of four replicates, error bars indicate the standard deviation.
Fig. S8. P‐value determination of pSTDesX leaky expression in P. putida KT2440. Statistical analysis performed with JMP 15 Pro to determine if pSTDesX⋅msfGFP causes significant expression of msfGFP in P. putida KT2440 in absence of inducer, compared to an empty pSTDesX control vector.
Fig. S9. P‐value determination of pSTDesX leaky expression in P. aeruginosa PAO1. Statistical analysis performed with JMP 15 Pro to determine if pSTDesX⋅msfGFP causes significant expression of msfGFP in P. aeruginosa PAO1 in absence of inducer, compared to an empty pSTDesX control vector.
Fig. S10. P‐value determination of pSTDesR leaky expression in P. putida KT2440. Statistical analysis performed with JMP 15 Pro to determine if pSTDesR⋅msfGFP causes significant expression of msfGFP in P. putida KT2440 in absence of inducer, compared to an empty pSTDesR control vector.
Fig. S11. P‐value determination of pSTDesR leaky expression in P. aeruginosa PAO1. Statistical analysis performed with JMP 15 Pro to determine if pSTDesR⋅msfGFP causes significant expression of msfGFP in P. aeruginosa PAO1 in absence of inducer, compared to an empty pSTDesR control vector.
Fig. S12. P‐value determination of pBGDes msfGFP expression in P. putida KT2440. Statistical analysis performed with JMP 15 Pro to determine if pBGDes⋅PEM7 ‐BCD2‐msfGFP causes significant expression of msfGFP in P. putida KT2440, compared to an pBGDes⋅BCD2‐msfGFP control vector.
Fig. S13. P‐value determination of pBGDes msfGFP expression in P. aeruginosa PAO1. Statistical analysis performed with JMP 15 Pro to determine if pBGDes⋅PEM7 ‐BCD2‐msfGFP causes significant expression of msfGFP in P. aeruginosa PAO1, compared to an pBGDes⋅BCD2‐msfGFP control vector.
Fig. S14. P‐value determination of T7 terminator efficiency in P. putida KT2440. Statistical analysis performed with JMP 15 Pro to determine if pBGDes⋅PEM7 ‐BCD2‐msfGFP‐T7terminator(wt)‐BCD1‐mCherry (PEM7 T7 wt) and pBGDes⋅PEM7 ‐BCD2‐msfGFP‐T7terminator(wt)‐BCD1‐mCherry (PEM7 T7 T7) cause significantly less mCherry expression in P. putida KT2440, compared to an pBGDes⋅BCD2‐msfGFP‐BCD1‐mCherry (PEM7 …) control vector.
Fig. S15. P‐value determination of T7 terminator efficiency in P. aeruginosa PAO1. Statistical analysis performed with JMP 15 Pro to determine if pBGDes⋅PEM7 ‐BCD2‐msfGFP‐T7terminator(wt)‐BCD1‐mCherry (PEM7 T7 wt) and pBGDes⋅PEM7 ‐BCD2‐msfGFP‐T7terminator(wt)‐BCD1‐mCherry (PEM7 T7 T7) cause significantly less mCherry expression in P. putida KT2440, compared to an pBGDes⋅BCD2‐msfGFP‐BCD1‐mCherry (PEM7 …) control vector.
Fig. S16. P‐value determination of T7‐based proof‐of‐concept msfGFP expression in P. putida KT2440 with 0.3 mM 3mBz. Statistical analysis performed with JMP 15 Pro to determine if the msfGFP fluorescence intensity of the T7‐based proof‐of‐concept is significantly different with 0 or 10 mM Rha, in the presence of 0.3 mM 3mBz.
Fig. S17. P‐value determination of T7‐based proof‐of‐concept msfGFP expression in P. aeruginosa PAO1 with 0.3 mM 3mBz. Statistical analysis performed with JMP 15 Pro to determine if the msfGFP fluorescence intensity of the T7‐based proof‐of‐concept is significantly different with 0 or 10 mM Rha, in the presence of 0.3 mM 3mBz.
Appendix S1.
Acknowledgements
This project has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (grant agreement No. 819800) and from the Fonds voor Wetenschappelijk Onderzoek Vlaanderen (FWO) as part of the CELL‐PHACTORY project (grant No. G096519N).
Microb. Biotechnol. (2021) 15(1), 370–386
Funding information
This project has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (grant agreement No. 819800) and from the Fonds voor Wetenschappelijk Onderzoek Vlaanderen (FWO) as part of the CELL‐PHACTORY project (grant No. G096519N).
References
- Andreou, A.I. , and Nakayama, N. (2018) Mobius assembly: a versatile golden‐gate framework towards universal DNA assembly. PLoS One 13: 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beal, J. , Haddock‐Angelli, T. , Baldwin, G. , Gershater, M. , Dwijayanti, A. , Storch, M. , et al. (2018) Quantification of bacterial fluorescence using independent calibrants. PLoS One 13: 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calero, P. , Jensen, S.I. , and Nielsen, A.T. (2016) Broad‐host‐range ProUSER vectors enable fast characterization of inducible promoters and optimization of p‐coumaric acid production in Pseudomonas putida KT2440. ACS Synth Biol 5: 741–753. [DOI] [PubMed] [Google Scholar]
- Calero, P. , Volke, D.C. , Lowe, P.T. , Gotfredsen, C.H. , O’Hagan, D. , and Nikel, P.I. (2020) A fluoride‐responsive genetic circuit enables in vivo biofluorination in engineered Pseudomonas putida . Nat Commun 11(5045): 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi, K.H. , Gaynor, J.B. , White, K.G. , Lopez, C. , Bosio, C.M. , Karkhoff‐Schweizer, R.A.R. , and Schweizer, H.P. (2005) A Tn7‐based broad‐range bacterial cloning and expression system. Nat Methods 2: 443–448. [DOI] [PubMed] [Google Scholar]
- Choi, K.H. , Kumar, A. , and Schweizer, H.P. (2006) A 10‐min method for preparation of highly electrocompetent Pseudomonas aeruginosa cells: application for DNA fragment transfer between chromosomes and plasmid transformation. J Microbiol Methods 64: 391–397. [DOI] [PubMed] [Google Scholar]
- Damalas, S.G. , Batianis, C. , Martin‐Pascual, M. , de Lorenzo, V. , and Martins dos Santos, V.A.P. (2020) SEVA 3.1: enabling interoperability of DNA assembly among the SEVA, BioBricks and Type IIS restriction enzyme standards. Microb Biotechnol 13(6): 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decoene, T. , De Paepe, B. , Maertens, J. , Coussement, P. , Peters, G. , De Maeseneire, S.L. , and De Mey, M. (2018) Standardization in synthetic biology: an engineering discipline coming of age. Crit Rev Biotechnol 38: 647–656. [DOI] [PubMed] [Google Scholar]
- Felgner, S. , Preusse, M. , Beutling, U. , Stahnke, S. , Pawar, V. , Rohde, M. , et al. (2020) Host‐induced spermidine production in motile Pseudomonas aeruginosa triggers phagocytic uptake. eLife 9: 1–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauttam, R. , Mukhopadhyay, A. , and Singer, S.W. (2020) Construction of a novel dual‐inducible duet‐expression system for gene (over)expression in Pseudomonas putida . Plasmid 110: 102514. [DOI] [PubMed] [Google Scholar]
- Gawin, A. , Valla, S. , and Brautaset, T. (2017) The XylS/Pm regulator/promoter system and its use in fundamental studies of bacterial gene expression, recombinant protein production and metabolic engineering. Microb Biotechnol 10: 702–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstmans, H. , Grimon, D. , Gutiérrez, D. , Lood, C. , Rodríguez, A. , van Noort, V. , et al. (2020) A VersaTile Driven Platform for rapid hit‐to‐lead development of engineered lysins. Sci Adv 6: eaaz1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson, D.G. , Young, L. , Chuang, R.Y. , Venter, J.C. , Hutchison, C.A. , and Smith, H.O. (2009) Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6: 343–345. [DOI] [PubMed] [Google Scholar]
- Green, R. , and Rogers, E.J. (2013) Methods in enzymology. In Transformation of chemically competent E. coli (Vol. 529, 1st ed.). Amsterdam, Netherlands: Elsevier, pp. 329–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halfon, Y. , Jimenez‐Fernandez, A. , Rosa, R.L. , Portero, R.E. , Johansen, H.K. , Matzov, D. , et al. (2019) Structure of Pseudomonas aeruginosa ribosomes from an aminoglycoside‐resistant clinical isolate. Proc Natl Acad Sci USA 116: 22275–22281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoang, T.T. , Kutchma, A.J. , Becher, A. , and Schweizer, H.P. (2000) Integration‐proficient plasmids for Pseudomonas aeruginosa: Site‐ specific integration and use for engineering of reporter and expression strains. Plasmid 43: 59–72. [DOI] [PubMed] [Google Scholar]
- Jeske, M. , and Altenbuchner, J. (2010) The Escherichia coli rhamnose promoter rhaPBAD is in Pseudomonas putida KT2440 independent of Crp‐cAMP activation. Appl Microbiol Biotechnol 85: 1923–1933. [DOI] [PubMed] [Google Scholar]
- Knight, T. (2003) Idempotent vector design for standard assembly of biobricks. Cambridge, MA, USA: MIT Artificial Intelligence Laboratory; MIT Synthetic Biology Working Group, pp. 1–11. [Google Scholar]
- Köbbing, S. , Blank, L.M. , and Wierckx, N. (2020) Characterization of context‐dependent effects on synthetic promoters. Front Bioeng Biotechnol 8: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushwaha, M. , and Salis, H.M. (2015) A portable expression resource for engineering cross‐species genetic circuits and pathways. Nat Commun 6: 8832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammens, E.‐M. , Nikel, P.I. , and Lavigne, R. (2020) Exploring the synthetic biology potential of bacteriophages for engineering non‐model bacteria. Nat Commun 11(5294): 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane, M.C. , Alteri, C.J. , Smith, S.N. , and Mobley, H.L.T. (2007) Expression of flagella is coincident with uropathogenic Escherichia coli ascension to the upper urinary tract. Proc Natl Acad Sci USA 104: 16669–16674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang, X. , Li, C. , Wang, W. , and Li, Q. (2018) Integrating T7 RNA polymerase and its cognate transcriptional units for a host‐independent and stable expression system in single plasmid. ACS Synth Biol 7: 1424–1435. [DOI] [PubMed] [Google Scholar]
- Loeschcke, A. , and Thies, S. (2020) Engineering of natural product biosynthesis in Pseudomonas putida . Curr Opin Biotechnol 65: 213–224. [DOI] [PubMed] [Google Scholar]
- Martin‐Pascual, M. , Batianis, C. , Bruinsma, L. , Asin‐Garcia, E. , Garcia‐Morales, L. , Weusthuis, R.A. , et al. (2021) A navigation guide of synthetic biology tools for Pseudomonas putida . Biotechnol Adv 49: 107732. [DOI] [PubMed] [Google Scholar]
- Meisner, J. , and Goldberg, J.B. (2016) The Escherichia coli rhaSR‐PrhaBAD inducible promoter system allows tightly controlled gene expression over a wide range in Pseudomonas aeruginosa . Appl Environ Microbiol 82: 6715–6727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffatt, B.A. , and Studier, F.W. (1987) T7 lysozyme inhibits transcription by T7 RNA polymerase. Cell 49: 221–227. [DOI] [PubMed] [Google Scholar]
- Mutalik, V.K. , Guimaraes, J.C. , Cambray, G. , Lam, C. , Christoffersen, M.J. , Mai, Q.‐A. , et al. (2013) Precise and reliable gene expression via standard transcription and translation initiation elements. Nat Methods 10: 354–360. [DOI] [PubMed] [Google Scholar]
- Nikel, P.I. , and de Lorenzo, V. (2018) Pseudomonas putida as a functional chassis for industrial biocatalysis: from native biochemistry to trans‐metabolism. Metab Eng 50: 142–155. [DOI] [PubMed] [Google Scholar]
- Pachori, P. , Gothalwal, R. , and Gandhi, P. (2019) Emergence of antibiotic resistance Pseudomonas aeruginosa in intensive care unit; a critical review. Genes Dis 6: 109–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelicic, V. , Reyrat, J.M. , and Gicquel, B. (1996) Expression of the Bacillus subtilis sacB gene confers sucrose sensitivity on Mycobacteria . J Bacteriol 178: 1197–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollak, B. , Cerda, A. , Delmans, M. , Álamos, S. , Moyano, T. , West, A. , et al. (2019) Loop assembly: a simple and open system for recursive fabrication of DNA circuits. New Phytol 222: 628–640. [DOI] [PubMed] [Google Scholar]
- Sarrion‐Perdigones, A. , Falconi, E.E. , Zandalinas, S.I. , Juárez, P. , Fernández‐del‐Carmen, A. , Granell, A. , and Orzaez, D. (2011) GoldenBraid: An iterative cloning system for standardized assembly of reusable genetic modules. PLoS One 6: e21622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shis, D.L. , and Bennett, M.R. (2013) Library of synthetic transcriptional AND gates built with split T7 RNA polymerase mutants. Proc Natl Acad Sci USA 110: 5028–5033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silby, M.W. , Winstanley, C. , Godfrey, S.A.C. , Levy, S.B. , and Jackson, R.W. (2011) Pseudomonas genomes: diverse and adaptable. FEMS Microbiol Rev 35: 652–680. [DOI] [PubMed] [Google Scholar]
- Silva, D. , Santos, G. , Barroca, M. , and Collins, T. (2017) Inverse PCR for point mutation introduction. Methods Mol Biol 1620: 87–100. [DOI] [PubMed] [Google Scholar]
- Silva‐Rocha, R. , Martínez‐García, E. , Calles, B. , Chavarría, M. , Arce‐Rodríguez, A. , de las Heras, A. , et al. (2013) The Standard European Vector Architecture (SEVA): a coherent platform for the analysis and deployment of complex prokaryotic phenotypes. Nucleic Acids Res 41: 666–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studier, F.W. (1991) Use of bacteriophage T7 lysozyme to improve an inducible T7 expression system. J Mol Biol 219: 37–44. [DOI] [PubMed] [Google Scholar]
- Studier, F.W. , and Moffatt, B.A. (1986) Use of bacteriophage T7 RNA polymerase to direct selective high‐level expression of cloned genes. J Mol Biol 189: 113–130. [DOI] [PubMed] [Google Scholar]
- Szafranski, P. , Mello, C.M. , Sano, T. , Smith, C.L. , Kaplan, D.L. , and Cantor, C.R. (1997) A new approach for containment of microorganisms: Dual control of streptavidin expression by antisense RNA and the T7 transcription system. Proc Natl Acad Sci USA 94: 1059–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temme, K. , Hill, R. , Segall‐Shapiro, T.H. , Moser, F. , and Voigt, C.A. (2012) Modular control of multiple pathways using engineered orthogonal T7 polymerases. Nucleic Acids Res 40: 8773–8781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volke, D.C. , Turlin, J. , Mol, V. , and Nikel, P.I. (2019) Physical decoupling of XylS/Pm regulatory elements and conditional proteolysis enable precise control of gene expression in Pseudomonas putida . Microb Biotechnol 13: 222–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber, E. , Engler, C. , Gruetzner, R. , Werner, S. , and Marillonnet, S. (2011) A modular cloning system for standardized assembly of multigene constructs. PLoS One 6: e16765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weihmann, R. , Domröse, A. , Drepper, T. , Jaeger, K.‐E. , and Loeschcke, A. (2020) Protocols for yTREX /Tn5‐based gene cluster expression in Pseudomonas putida. Microbial Biotechnology 13(1): 250–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zobel, S. , Benedetti, I. , Eisenbach, L. , De Lorenzo, V. , Wierckx, N. , and Blank, L.M. (2015) Tn7‐based device for calibrated heterologous gene expression in Pseudomonas putida . ACS Synth Biol 4: 1341–1351. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Full nucleotide sequences of all SEVAtiles used in this study. Nucleotides indicated in bold were included in position tags (PT): PT3 AATG, PT4 TAAC, PT6 TAATG and TAATG, PT7 TAAGA.
Table S2. Primers used in this work.
Fig. S1. Colony PCR analysis of 32 transformants of each SEVAtile destination vector made in this study. As a reference, λ‐PstI ladder is used (1 kb GeneRuler for pBGDes·PEM7‐BCD2‐msfGFP‐BCD1‐mCherry, pBGDes·PEM7‐BCD2‐msfGFP‐T7terminator(wt)‐BCD1‐mCherry and pBGDes·PEM7‐BCD2‐msfGFP‐T7terminator(T7)‐BCD1‐mCherry) and the expected amplicon length is indicated on the left of the agarose gel.
Fig. S2. Characterization of pSTDesX. Graphs depicting the OD600, absolute msfGFP fluorescence intensity (AU) and calibrated fluorescence intensity (5(6)‐FAM/OD600) over a 12 hour period in either P. putida KT2440 or P. aeruginosa PAO1 carrying the pSTDesX⋅msfGFP vector. MsfGFP expression was induced with 0; 0.05; 0.1; 0.05; 0.1; 5; 10 mM 3mBz. For clarity, only data of the samples with 0, 1 and 10 mM 3mBz are indicated. Lines represent the mean value of four replicates, error bars indicate the standard deviation.
Fig. S3. Characterization of pSTDesR. Graphs depicting the OD600, absolute msfGFP fluorescence intensity (AU) and calibrated fluorescence intensity (5(6)‐FAM/OD600) over a 12 hour period in either P. putida KT2440 or P. aeruginosa PAO1 carrying the pSTDesR⋅msfGFP vector. MsfGFP expression was induced with 0; 0.5; 1; 5; 10; 50; 100 mM Rha. For clarity, only data of the samples with 0, 10 and 100 mM Rha are shown. Lines represent the mean value of four replicates, error bars indicate the standard deviation.
Fig. S4. Characterization of pBGDes (1). Graphs depicting the OD600, absolute msfGFP fluorescence intensity (AU) and calibrated fluorescence intensity (5(6)‐FAM/OD600) over 12 hours for P. putida KT2440 and P. aeruginosa PAO1 carrying pBGDes⋅PEM7‐BCD2‐msfGFP or pBGDes⋅BCD2‐msfGFP (negative control). Lines represent the mean value of four replicates, error bars indicate the standard deviation.
Fig. S5. Characterization of pBGDes (2). Graphs depicting the OD600, absolute msfGFP fluorescence intensity (AU) and absolute mCherry fluorescence intensity (AU) over 12 hours for P. putida KT2440 and P. aeruginosa PAO1 carrying pBGDes⋅BCD2‐msfGFP (negative control), pBGDes⋅PEM7‐BCD2‐msfGFP‐BCD1‐mCherry (no terminator), pBGDes⋅PEM7‐BCD2‐msfGFP‐T7terminator(wt)‐BCD1‐mCherry (T7terminator (wt)) or pBGDes⋅PEM7‐BCD2‐msfGFP‐ T7terminator(T7)‐BCD1‐mCherry (T7 terminator (T7)). Lines represent the mean value of four replicates, error bars indicate the standard deviation.
Fig. S6. T7‐based proof‐of‐concept in P. putida KT2440. Graphs depicting the OD600, absolute msfGFP fluorescence intensity (AU) and calibrated fluorescence intensity (5(6)‐FAM/OD600) over 12 hours for P. putida KT2440 carrying pBGDes⋅PT7 ‐BCD2‐msfGFP, pSTDesX⋅T7RNAP and pSTDesR⋅T7lysozyme. The cells were induced with all four combinations of 0/0.3 mM 3mBz and 0/10 mM Rha. Lines represent the mean value of four replicates, error bars indicate the standard deviation.
Fig. S7. T7‐based proof‐of‐concept in P. aeruginosa PAO1. Graphs depicting the OD600, absolute msfGFP fluorescence intensity (AU) and calibrated fluorescence intensity (5(6)‐FAM/OD600) over 12 hours for P. aeruginosa PAO1 carrying pBGDes⋅PT7 ‐BCD2‐msfGFP, pSTDesX⋅T7RNAP and pSTDesR⋅T7lysozyme. The cells were induced with all four combinations of 0/0.3 mM 3mBz and 0/10 mM Rha. Lines represent the mean value of four replicates, error bars indicate the standard deviation.
Fig. S8. P‐value determination of pSTDesX leaky expression in P. putida KT2440. Statistical analysis performed with JMP 15 Pro to determine if pSTDesX⋅msfGFP causes significant expression of msfGFP in P. putida KT2440 in absence of inducer, compared to an empty pSTDesX control vector.
Fig. S9. P‐value determination of pSTDesX leaky expression in P. aeruginosa PAO1. Statistical analysis performed with JMP 15 Pro to determine if pSTDesX⋅msfGFP causes significant expression of msfGFP in P. aeruginosa PAO1 in absence of inducer, compared to an empty pSTDesX control vector.
Fig. S10. P‐value determination of pSTDesR leaky expression in P. putida KT2440. Statistical analysis performed with JMP 15 Pro to determine if pSTDesR⋅msfGFP causes significant expression of msfGFP in P. putida KT2440 in absence of inducer, compared to an empty pSTDesR control vector.
Fig. S11. P‐value determination of pSTDesR leaky expression in P. aeruginosa PAO1. Statistical analysis performed with JMP 15 Pro to determine if pSTDesR⋅msfGFP causes significant expression of msfGFP in P. aeruginosa PAO1 in absence of inducer, compared to an empty pSTDesR control vector.
Fig. S12. P‐value determination of pBGDes msfGFP expression in P. putida KT2440. Statistical analysis performed with JMP 15 Pro to determine if pBGDes⋅PEM7 ‐BCD2‐msfGFP causes significant expression of msfGFP in P. putida KT2440, compared to an pBGDes⋅BCD2‐msfGFP control vector.
Fig. S13. P‐value determination of pBGDes msfGFP expression in P. aeruginosa PAO1. Statistical analysis performed with JMP 15 Pro to determine if pBGDes⋅PEM7 ‐BCD2‐msfGFP causes significant expression of msfGFP in P. aeruginosa PAO1, compared to an pBGDes⋅BCD2‐msfGFP control vector.
Fig. S14. P‐value determination of T7 terminator efficiency in P. putida KT2440. Statistical analysis performed with JMP 15 Pro to determine if pBGDes⋅PEM7 ‐BCD2‐msfGFP‐T7terminator(wt)‐BCD1‐mCherry (PEM7 T7 wt) and pBGDes⋅PEM7 ‐BCD2‐msfGFP‐T7terminator(wt)‐BCD1‐mCherry (PEM7 T7 T7) cause significantly less mCherry expression in P. putida KT2440, compared to an pBGDes⋅BCD2‐msfGFP‐BCD1‐mCherry (PEM7 …) control vector.
Fig. S15. P‐value determination of T7 terminator efficiency in P. aeruginosa PAO1. Statistical analysis performed with JMP 15 Pro to determine if pBGDes⋅PEM7 ‐BCD2‐msfGFP‐T7terminator(wt)‐BCD1‐mCherry (PEM7 T7 wt) and pBGDes⋅PEM7 ‐BCD2‐msfGFP‐T7terminator(wt)‐BCD1‐mCherry (PEM7 T7 T7) cause significantly less mCherry expression in P. putida KT2440, compared to an pBGDes⋅BCD2‐msfGFP‐BCD1‐mCherry (PEM7 …) control vector.
Fig. S16. P‐value determination of T7‐based proof‐of‐concept msfGFP expression in P. putida KT2440 with 0.3 mM 3mBz. Statistical analysis performed with JMP 15 Pro to determine if the msfGFP fluorescence intensity of the T7‐based proof‐of‐concept is significantly different with 0 or 10 mM Rha, in the presence of 0.3 mM 3mBz.
Fig. S17. P‐value determination of T7‐based proof‐of‐concept msfGFP expression in P. aeruginosa PAO1 with 0.3 mM 3mBz. Statistical analysis performed with JMP 15 Pro to determine if the msfGFP fluorescence intensity of the T7‐based proof‐of‐concept is significantly different with 0 or 10 mM Rha, in the presence of 0.3 mM 3mBz.
Appendix S1.