

Abstract
The CBF1 (centromere binding factor 1) gene of Candida glabrata was cloned by functional complementation of the methionine biosynthesis defect of a Saccharomyces cerevisiae cbf1 deletion mutant. The C. glabrata-coded protein, CgCbf1, contains a basic-helix-loop-helix leucine zipper domain and has features similar to those of other budding yeast Cbf1 proteins. CgCbf1p binds in vitro to the centromere DNA element I (CDEI) sequence GTCACATG with high affinity (0.9 × 109 M−1). Bandshift experiments revealed a pattern of protein-DNA complexes on CgCEN DNA different from that known for S. cerevisiae. We examined the effect of altering the CDEI binding site on CEN plasmid segregation, using a newly developed colony-sectoring assay. Internal deletion of the CDEI binding site led only to a fivefold increase in rates of plasmid loss, indicating that direct binding of Cbf1p to the centromere DNA is not required for full function. Additional deletion of sequences to the left of CDEI, however, led to a 70-fold increase in plasmid loss rates. Deletion of the CBF1 gene proved to be lethal in C. glabrata. C. glabrata cells containing the CBF1 gene under the influence of a shutdown promoter (tetO-ScHOP) arrested their growth after 5 h of cultivation in the presence of the reactive drug doxycycline. DAPI (4′,6′-diamidino-2-phenylindole) staining of the arrested cells revealed a significant increase in the number of large-budded cells with single nuclei, 2C DNA content, and short spindles, indicating a defect in the G2/M transition of the cell cycle. Thus, we conclude that Cbf1p is required for chromosome segregation in C. glabrata.
Candida species are major fungal pathogens which can cause both mucosal and systemic infections in humans (for a review, see reference 18). Although Candida albicans is the best known of the pathogenic Candida group, the frequency with which other Candida species are isolated from clinical infections has been steadily increasing during the past few years. A very recent study identified Candida glabrata as the second most commonly isolated Candida species in bloodstream isolates (47). C. glabrata is a common pathogen in immunocompromised persons or those with diabetes mellitus. Depending on the site of infection, C. glabrata is often the second or third most common cause of candidiasis after C. albicans, and C. glabrata infections have been linked to the deaths of compromised, at-risk hospitalized patients (18). The treatment of C. glabrata infections is difficult since the strains are often resistant to antifungal drugs (59).
In contrast to other Candida species, C. glabrata, although asexual, is haploid, which facilitates molecular genetic analysis. C. glabrata is closely related to Saccharomyces cerevisiae, and both strains share high sequence homologies in their genes (30, 37, 62). Recently, a centromere (CEN) was isolated from C. glabrata, and it has been used in the construction of plasmid vectors (31, 32). Centromeres are specific regions of eukaryotic chromosomes that are necessary for chromosome segregation in meiosis and mitosis (for reviews, see references 10 and 21). They provide attachment sites for the spindle microtubules that transport the chromatids during anaphase. A plasmid vector that contains CEN along with an ARS sequence is segregated to daughter cells with high mitotic fidelity and is maintained at low copy numbers. In contrast to the large regional centromeres of humans that span several kilobases on the DNA, the C. glabrata centromere represents a “point centromere” and is only 153 bp in length. It has high homology to the centromeres of S. cerevisiae and those of other budding yeasts (Kluyveromyces lactis, Kluyveromyces marxianus, Saccharomyces uvarum) (12, 23, 24, 26). Two conserved consensus sequences (centromere DNA elements [CDE]), CDEI (8 bp) and CDEIII (26 bp), are separated by a 79-bp nonconserved A+T-rich spacer sequence, CDEII. The species specificities of the CENs of C. glabrata and S. cerevisiae are determined by both CDEII and CDEIII (32). In recent years, several proteins that bind to these elements in S. cerevisiae have been discovered (for a review, see reference 36). A complex that binds to the CDEIII region is the CBF3 complex, which consists of the proteins p110Cbf3a/Ndc10p/Cbf2p, p64Cbf3b/Cep3p, p58Cbf3c/Ctf13p, and p23Cbf3d/Skp1p (35, 36). Other centromere binding proteins are Cse4p, Mif2p (40, 56), and a recently isolated protein complex consisting of Okp1p, Ctf19p, and Mcm21p (25, 46, 48). Loss of these proteins generally results in a lethal phenotype and abnormal mitotic behavior.
Centromere binding factor 1 (Cbf1; also called Cp1 and Cpf1) in S. cerevisiae has been well studied (5, 9, 39). It is an abundant basic-helix-loop-helix leucine zipper protein which binds as a homodimer to the degenerate octanucleotide RTCACRTG (the CDEI region of the centromere, where R is purine) (4, 8, 27). Deletion of the CDEI binding site leads to 10- to 30-fold increases in rates of plasmid loss (16, 43). CBF1 gene disruption leads to a 20-fold increase in chromosome missegregation and a 35% increase in generation time (9, 39). Cbf1 null mutants are methionine auxotrophs, and Cbf1p binding motifs have been found in the promoters of some of the MET genes. Cbf1p interacts directly with other kinetochore proteins, and CBF3 subunits can promote the binding of Cbf1p to the DNA (22). A functional comparison of the centromere binding proteins with transcription factors binding at the MET16 promoter reveals strong analogy between the centromere and the MET16 promoter (22). Cbf1p also binds to the promoters of several other genes (GAL2, TRP1, CYT1), but its potential to activate transcription of these genes is low or undetectable (8, 29, 45). A CBF1 gene from K. lactis has also been cloned (41). In contrast to what occurs in S. cerevisiae, CBF1 deletion is lethal in K. lactis, but it is not clear what exactly causes the lethality. Cbf1 proteins from S. cerevisiae and K. lactis are functionally interchangeable despite low overall homology.
Recently, we have cloned a CBF3d homolog from C. glabrata and showed that it can functionally substitute for its S. cerevisiae counterpart (57). In this study we present the cloning and functional characterization of the CBF1 gene of C. glabrata (CgCBF1). We have developed a color-based colony-sectoring assay for C. glabrata, which enabled us to study the effect of CDEI loss on plasmid stability. Furthermore, we have investigated the effects of CgCbf1p depletion on cell morphology and growth. Our data reveal that, in spite of their high sequence homology, the S. cerevisiae and C. glabrata centromeres are structurally and functionally quite different.
MATERIALS AND METHODS
Yeast and bacterial strains and media.
The yeast strains used in this study and their genotypes are listed in Table 1. Rich medium (yeast-peptone-dextrose [YPD]), synthetic minimal dextrose (SD) medium (0.7% yeast nitrogen base, 2% glucose, 2% agar), and 5-fluoro-orotic acid (5-FOA) medium were prepared as described previously (28). Escherichia coli strain XL-1Blue was used for propagation of plasmids, and E. coli strain BL21(DE3) (Novagen, Madison, Wis.) was used for expression of recombinant proteins. Bacterial media were prepared as described previously (51).
TABLE 1.
List of yeast strains used in this study
| Strain | Genotype | Reference or source |
|---|---|---|
| C. glabrata | ||
| CgHTU2001 | his3 ura3 trp1 | 31 |
| CgTS1 | his3 ura3 trp1 cbf1::TRP1 + p112Cp1 (CBF1 CEN ARS URA3) | This study |
| CgTS3 | his3 ura3 trp1 cbf1::TRP1 TRP1::CBF1-HA | This study |
| CgTS5 | his3 ura3 trp1 cbf1::TRP1 TRP1::CBF1-HA CEN::CEN-TRP1 | This study |
| CgTS7 | his3 ura3 trp1 cbf1::TRP1 TRP1::CBF1-HA CEN::CENΔ1D-TRP1 | This study |
| CgHTUA | his3 ura3 trp1 ade2 | This study |
| ACG22 | his3 ura3 trp1 PScHOP1::tetR::Gal4AD::TRP1 | 42 |
| 97CBF1 | ura3 PScHOP1::tetR::Gal4AD::TRP1 97tCBF1::HIS3 | This study |
| 98CBF1 | ura3 PScHOP1::tetR::Gal4AD::TRP1 98tCBF1::HIS3 | This study |
| 99CBF1 | ura3 PScHOP1::tetR::Gal4AD::TRP1 99tCBF1::HIS3 | This study |
| S. cerevisiae CC718-1A | MATaade2 his3 leu2 trp1 ura3 cbf1::TRP1 | 34 |
Plasmids and generation of yeast strains.
pRSCg1 was constructed by ligating a 2.2-kb SpeI/HindIII fragment from pYep#9.1 (see Fig. 1) into pRS425 (11). p112-Cp1 was created by ligating the blunt-ended SpeI/PstI fragment into the SmaI site of p112-8XM (31). Transformation of yeast with plasmids or with DNA fragments was performed by the lithium acetate method (2).
FIG. 1.

(A) CgCBF1 gene expression complements the methionine biosynthetic defect of an S. cerevisiae cbf1 null mutant strain. Growth of CC718–1A, CC718–1A/pYep#9.1, and CC718–1A/pRSCg1 on SD medium lacking methionine (met) is shown. The plates were incubated for 4 days at 30°C. (B) Genomic map of the cloned DNA insert in plasmid pYep#9.1. Major ORFs were detected and analyzed using BLAST and the Wisconsin Sequence Analysis software package (GCG9). The positions of the ORFs are indicated by arrows. Flanking thick lines represent vector sequences. Restriction sites are shown only if relevant. H, HindIII; S, SpeI.
Plasmids for plasmid loss assays.
The vectors used in the sectoring assay were derivatives of pBM2.9 (20). The PstI fragment of pBM2.9 containing the CoxII-ARS fragment was deleted, and the vector was religated, leaving a unique PstI site (pBMΔP). The blunt-ended AatII fragment of pCgACH-3 (31) containing CgHIS3, CgARS, and CgCEN (pBM-HAC-WT) was ligated into the SmaI site of pBMΔP (pBMΔP-HAC). An attempt to exchange the BglII fragment of pBMΔP-HAC with PCR fragments carrying deleted centromeres revealed two more unexpected BglII sites in the plasmid. Therefore, we created a second plasmid in which both BglII sites were deleted, and the SpeI/PstI fragment of pBMΔP-HAC was cloned into this vector (pBMΔPΔB-HAC). Since this plasmid did not complement the red phenotype of CgHTUA, wild-type CEN was replaced with deleted centromeres in pBMΔPΔB-HAC and the SpeI/PstI fragments of the resulting plasmids were cloned back into pBMΔP. Deleted centromeres were generated by PCR using the primer pairs CgΔ292 and CgCEN1R and CgΔ308 and CgCEN1R (Table 2). For site-specific mutagenesis and internal deletion of the CDEI binding site, the wild-type centromere was released as a BglII fragment from pCgACH3 and ligated into the BamHI site of pUC19, creating pUCCgCEN1. This plasmid was subjected to site-specific mutagenesis using a U.S.E. kit (Amersham-Pharmacia, Little Chalfont, Buckinghamshire, United Kingdom) with the primers CgΔIDelete, CgΔIRandom, and CgΔIExchange (Table 2) according to the supplier's instructions, giving pUCCgCENΔ1D, pUCCgCENΔ1R, and pUCCgCENΔ1E, respectively. The mutagenesis was confirmed by sequencing. To create pBM-Δ200 and pBMΔID-Δ200, PBMΔPΔID and pBMΔPAHC were digested with SpeI and NheI, the ends were filled in with T4 polymerase, and the vectors were religated.
TABLE 2.
Oligonucleotides used in this study
| Oligonucleotide | Sequence | Introduced restriction site |
|---|---|---|
| CgΔN270 | TAACGCAGAGCACATCTTTC | |
| CgCEN2 | CGCGTAACTAATGATGCAAT | |
| CgΔN292 | GGGAGATCTTGATGTCTGTCACATG | BglII |
| CgΔN308 | GGGAGATCTATCAAAAACAATTGCA | BglII |
| CgCEN1R | TTGAGATCTTAACTAATGATGCAAT | BglII |
| CgΔIDelete | CAGTGATGTCTGTATCAAAAACAATTGC | |
| CgΔIRandom | CAGTGATGTCTGTGGATCCATCAAAAACAATTGC | |
| CgΔIExchange | CAGTGATGTCTAGCACATCTTCAAAAACAATTGGC | |
| CgCEN6 | CGGGATCCAGCACATCTACCGGATAGAGCGTAAG | BamHI |
| CgCEN6R | AAGGATCCTAACTAATGATGCAAT | BamHI |
| Cg1/1Ec | GGCCGGATCCATGGAGAGAAGCAACC | BamHI |
| Cg1/3Ec | GGTTGGATCCCTATTCCTTAGACTCATC | BamHI |
| Cg1/1A | CCCGGGTACCATGGCCCAACTTTG | KpnI |
| Cg1/1B | TTAACTCGAGCTCCTCCTTCTCATCG | XhoI |
| Cg1/2A | GGGCTCGAGTCTAAGGAATAGATA | XhoI |
| Cg1/2B | AAATCTAGAGCTGAGGTTCATGGA | XbaI |
| Cg1/3 | ACCATTAATGTTGGC | |
| Cg1/4 | AGTATCTTCAGGTAT | |
| Cg1/7 | TTAACTTGGCCATTGATA | MscI |
| Cg1/8 | CCTTGGCATATGTTCCTTAGACTCATCTTC | NdeI |
| Cg1/9 | GAAGATCTATATTGAAAATGATCTGGTAT | BglII |
| Cg1/10 | CCCAAGCTTGTTGGACAGAGAAAAATAG | HindIII |
| Cg1/11 | GGAATTCAGAATGATGGAGAGAAGCAACCGT | EcoRI |
| Cg1/12 | GGGGTACCAGGTTCGTGGCCCTGCTCCTG | KpnI |
| Cg1/13 | GCTCTAGATCTCAATGGACCACGACAACG | XbaI |
| Cg1/14 | GTACTAGTGGTATAAAGCT | SpeI |
| CgCDEI.F | CAGTGATGTCTGTCACATGATCAAA | |
| CgCDEI.R | CTACAGACAGTGTACTAGTTTGTCA | |
| CgACT1 | CGGGGTACCATGGATTCTGGTATGTTCGA | |
| CgACT4 | TATTCACACACGCAGGGACAG | |
| CgHis1 | AGCATGGCGGGTGGTCTC | |
| CgHis2 | CCAGCTCCACCACGGCGA | |
| HA-F | GGAATTCCATATGGTTGTTTACCCATACGAT | NdeI |
| HA-R | CCCAGATCTCTACTAATCTGGAACGTCATATGGATA | BglII |
CgCBF1 gene disruption.
Two DNA fragments (A and B) containing 5′ nontranslated sequences and 5′ coding sequences (fragment A) and 3′ coding sequences and 3′ nontranslated sequences (fragment B) of CBF1 were amplified by PCR using the primer pairs Cg1/1A and -B and Cg1/2A and -B (see Table 2). The two fragments and an XhoI fragment of pCgACT-14 containing CgTRP1 (31) were ligated into the KpnI and XbaI sites of pUC19. The KpnI/XbaI fragment was released from the resulting plasmid and used to transform strain Cg2001HTU/p112-Cp1. Transformants were selected for their ability to grow in the absence of tryptophan. Successful gene replacement was verified by Southern blot analysis. The resulting strain was named CgTS1.
ADE2 disruption
To delete the ADE2 gene in strain Cg2001HTU, an EcoRI/BamHI fragment was released from pAD1A1 (20) and cotransformed with p112-8XM, bearing the S. cerevisiae URA3 (ScURA3) gene. One colony out of 7,400 screened colonies showed a red color. Loss of p112-8XM was selected for by growth on medium containing 5-FOA, and the new strain was named CgHTUA.
Epitope tagging of CgCBF1.
To generate an epitope-tagged version of CgCBF1, three DNA fragments were generated by PCR. Fragment A (primers Cg1/7 and Cg1/8) represented the C-terminal part of the CBF1 coding region, including the last codon before the stop codon. Fragment B (primers HA-F and HA-R) encoded three copies of the 9-amino-acid (aa) hemagglutinin (HA) epitope followed by two TAG stop codons (the template for the PCR was pBFG5, kindly provided by R. Ballester). Fragment C (primers Cg1/9 and Cg1/10) encoded the 3′ nontranslated region of CgCBF1. All three fragments were cut by the corresponding enzymes (Table 2) and ligated simultaneously to pRSCg1 cut with MscI and HindIII. The resulting plasmid was digested with BglII, blunt ended, and ligated with a blunt-ended XhoI fragment coding for CgHIS3 released from pCgACH-3 (31). The SpeI/HindIII fragment was then released from the resulting plasmid (pRSCg1-HA) and transformed into CgTS1. The transformants were screened for their ability to grow on 5-FOA medium and for their inability to grow in the absence of tryptophan, indicating both the functionality of the transcript and integration into the original gene locus. The resulting strain was grown on plates containing 5-FOA to select for loss of the plasmid p112-Cp1, and the selected strain was named CgTS3. The generation of correctly translated protein was confirmed by Western blot analysis (not shown) with a purified monoclonal antibody (MAb), HA.11 16B12 (Berkley Antibody Co.).
Generation of strains for chromatin immunoprecipitation (ChIP) experiments.
CgTRP1 released from pCgACT-14 as an XhoI fragment (31) was blunt ended by filling in protruding ends using T4 DNA polymerase and ligated into pUCCgCEN1 and pUCCgCENΔ1D which had been cut with NheI and blunt ended. Fragments including centromeres plus CgTRP1 (1.5 kb) were released from the plasmids by digestion with EcoRI and SphI and used to transform strain CgTS3. Correct insertion into the centromere locus was confirmed by Southern blot analysis of AseI- and AflIII-digested genomic DNA, using CgCENΔ270 DNA as a probe. To confirm the mutation, the centromeres were amplified from the genomic DNA by PCR and sequenced.
Generation of strains containing CgCBF1 under a controllable promoter.
Region A (nucleotides 749 to 315) and region B (nucleotides 6 to 492) of CgCBF1 were amplified by PCR using the primers Cg1/13 and Cg1/14 and the primers Cg1/11 and 1/12, respectively. Region A was cloned into the SpeI site, and region B was cloned into the EcoRI and KpnI sites of plasmids p97CGH, p98CGH, and p99CGH (42), generating p97CBF1, p98CBF1, and p99CBF1, respectively. The plasmids were linearized with BssHII and used to transform strain ACG22. Homologous recombination was verified by Southern blot analysis of MscI-digested genomic DNA, using radiolabeled region B as a probe (not shown). Three strains, 97CBF1, 98CBF1, and 99CBF1, were obtained, and each contained the CBF1 gene under the influence of a different promoter (42).
DNA sequence analysis.
DNA sequencing of pYep#9.1 (see Fig. 1) was carried out on a shotgun library of the plasmid in an M13 vector. pYep#9.1 DNA (5 μg) was sonicated and size fractionated. The fraction containing DNA fragments between 1 and 1.5 kb in length was ligated into the M13mp18 vector after filling in the protruding ends by T4 polymerase treatment (15). The M13 templates were prepared by the Triton method (38). All templates were sequenced by dye terminator chemistry (Perkin-Elmer), and data were collected using ABI 377 automated sequencers and finally assembled with the computer program Gap 4 (55). Most gaps were closed by performing long runs using dye primer chemistry. Inserts of clones spanning the remaining gaps were amplified using standard forward and reverse primers. PCR bands were purified with a gel extraction kit (Genomed) and sequenced using the reverse universal primer. All other sequence analysis was done by cycle sequencing, and the labeled PCR fragments were analyzed on an ABI 377 automated sequencer.
Expression of recombinant CgCbf1p and ScCbf1p in E. coli.
A DNA fragment encoding the entire CgCBF1 open reading frame (ORF) was amplified by PCR with the primer pair Cg1/1Ec and Cg1/3Ec and was cloned into the BamHI site of the expression vector pET28a(+) (Novagen). DNA sequencing revealed a point mutation in codon 355 of the PCR product. The MscI/HindIII fragment of the recombinant plasmid was therefore replaced by the corresponding fragment of pRS-Cg1, resulting in plasmid pETCg1. This plasmid was transformed into E. coli strain BL21(DE3). The histidine-tagged CgCbf1 fusion protein obtained from the E. coli cytosol was purified by affinity chromatography on chelating Sepharose according to a protocol of Novagen. Recombinant ScCbf1p was expressed and purified similarly, using a pETScCbf1 expression plasmid kindly provided by D. Thomas. The protein concentration of the IMAC eluate was determined by a Bradford assay (Bio-Rad, Hercules, Calif.).
Protein extracts.
C. glabrata protein extracts for sodium dodecyl sulfate-gel electrophoresis were made and analysis of proteins on Western blots was done as previously described (28). For analysis of proteins by DNA mobility shift assays, Cg2001HTU cells were grown in 0.5 liter of YPD for 17 h at 30°C. Freshly harvested cell paste (7 g) was washed with water, and the washed pellet was packed into a 10-ml syringe and extruded into a beaker of liquid nitrogen. Cells were mechanically disrupted in liquid nitrogen (53) using a blender at high speed for 10 min. All subsequent steps were performed at 4°C as previously described (35), with the following modifications: the freeze-dried powder was resuspended in 6 ml of buffer A (50 mM KPO4 [pH 7.0], 100 mM β-glycerophosphate, 10 mM NaF, 10 mM EDTA, 10 mM EGTA, 0.5 M dithiothreitol, 1 mM Pefabloc, 2.5 μg of leupeptin, 2.5 μg of pepstatin A per ml, and 2 μg of aprotinin per ml) and stirred gently for 40 min. KCl was added after 10 min to a final concentration of 1 M. After centrifugation (14,000 × g for 30 min), the supernatant (whole-cell extract) was flash frozen in small aliquots and stored at −70°C.
DNA mobility shift assays.
Electrophoretic mobility shift assays were performed as previously described (4, 35) with either poly(dI-dC) (see below) or 6 μg of salmon sperm DNA (Sigma) as the nonspecific competitor. The reaction volume was 20 to 30 μl, and the 4% nondenaturing polyacrylamide gels were electrophoresed at 10 to 15 V/cm at room temperature (RT) either in 0.5× Tris-borate-EDTA (TBE) or in a solution containing 50 or 100 mM glycine and 2 mM EDTA. Specific competitor DNA was prepared by synthesizing complementary single-stranded oligonucleotides carrying the CDEI motif (CgCDEI.F and CgCDEI.R [Table 2]). Oligonucleotides were phosphorylated, annealed, and oligomerized as described previously (60). Concatamerized fragments were radiolabeled by filling in the ends with [α-32P]dATP and [α-32P]dCTP using the Klenow fragment (New England Biolabs).
Determination of a CgCbf1p equilibrium binding constant.
To determine the true equilibrium constant for CgCbf1p binding to CgCEN DNA, we applied a previously described electrophoretic mobility shift method that corrects for nonspecific binding of protein to DNA (3, 4, 7). To determine the apparent binding constant (Kapp), increasing amounts of the 252-bp radiolabeled CgCEN fragment (0.02 to 10 fmol; ∼0.003 to 1.7 ng) were incubated with a constant amount of affinity-purified CgCbf1p (1 μl of a 1:50 dilution; estimated to be 1 to 2 ng by Coomassie blue staining and Bradford analysis) and poly(dI-dC) nonspecific DNA (50 ng). Reaction mixtures (20 μl) were incubated for 15 min on ice in binding buffer containing 15 mM KCl (4), run on 4% polyacrylamide gels in 0.5× TBE, and electrophoresed at 10 V/cm at RT. Gels were preelectrophoresed at 8 V/cm at RT. The concentrations of retarded protein-DNA complexes, [CDs], and of the unbound probe for specific DNA, [Ds], were determined with a molecular phosphorimager (Bio-Rad Laboratories). At equilibrium, the relationship between free and bound specific probe DNAs is given by the following equations:
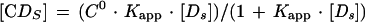 |
1 |
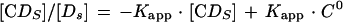 |
2 |
where C0 is the number of CgCbf1p DNA binding sites present. The binding data were plotted according to equation 1 (see Fig. 3A) and equation 2 (see Fig. 3B). Kapp is given by the slope of the line in Fig. 3B, and C0 is given by the x intercept. The equilibrium constant for binding of nonspecific DNA (Kn) was determined from a parallel set of reaction mixtures in which constant amounts of protein (∼1 to 2 ng) and the radiolabeled CgCEN fragment (2.5 fmol; ∼0.4 ng) were incubated together with increasing amounts of nonspecific poly(dI-dC) DNA (480- to 133,000-fold mass excess over the mass of specific DNA). Nonspecific- and specific-DNA equilibrium binding constants (Kn and Ks) were determined using the following equations:
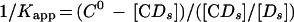 |
3 |
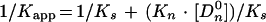 |
4 |
To calculate 1/Kapp from equation 3, the value for C0 determined from Fig. 3B was used. Values for Kn and Ks were determined graphically from equation 4 by plotting 1/Kapp versus the concentration of nonspecific DNA, [Dn0] (see Fig. 3C). Kn/Ks is given by the slope of the line and 1/Ks equals the y intercept. Thus, Kn equals the slope of the line divided by the y intercept. The lines in Fig. 3B and C were fit by least-squares linear regression.
FIG. 3.

Determination of an equilibrium binding constant for CgCbf1p binding to a C. glabrata centromere DNA fragment. (A) Saturation binding curve as determined from experiments using increasing amounts of radiolabeled CgCEN DNA and a constant amount of affinity-purified CgCbf1p in the presence of nonspecific poly(dI-dC) DNA. The concentration of CgCbf1p that bound to CgCEN, [CDs], is plotted versus that of free CgCEN, [Ds]. (B) Scatchard plot of the data in panel A (see Materials and Methods for details). The slope of the line equals Kapp, and the x intercept equals C0, the number of binding sites in the reaction. (C) Determination of the equilibrium constant for nonspecific binding (Kn) to correct Kapp for the contribution made by binding of CgCbf1p to nonspecific DNA. Constant amounts of radiolabeled CgCEN and affinity-purified CgCbf1p were incubated with increasing amounts of poly(dI-dC) DNA. The inverse of Kapp (determined from equation 3, see Materials and Methods) is plotted versus the concentration of nonspecific DNA, [Dn0]. Both Kn and Ks can be determined. The y intercept equals 1/Ks, and the slope of the line is equal to Kn/Ks. Thus, Kn is equal to the slope divided by the y intercept.
In vivo cross-linking and ChIP.
ChIP experiments were carried out as described previously (1, 6). C. glabrata strains were exponentially grown to an optical density (OD) of 1 (∼4 × 107 cells/ml) and fixed with 1% formaldehyde for 15 min at RT. Formaldehyde-induced cross-linking was quenched for 5 min at RT by the addition of glycine to a final concentration of 120 mM. The cells were washed and lysed with glass beads, and the chromatin was sheared by sonication to an average length of 0.8 kb. Purified MAb HA.11 16B12 (Berkeley Antibody Co.) was added to the sheared chromatin at a final concentration of 5 μg/ml, and immunocomplexes were captured with protein A-Sepharose beads for 2 h at 4°C. For DNA analysis, 1 μl of total or 2 μl of immunoprecipitated chromatin was subjected to PCRs (22 cycles) with primers for CgCEN (CgΔN270 and CgCEN2), CgHIS3 (CgHis1 and CgHis2), and CgACT (actin gene) (CgACT1 and CgACT4). The PCR products were electrophoresed on 2% agarose gels, stained with ethidium bromide, and digitally photographed with the AlphaImager 2000 system (Alpha Innotech, San Leandro, Calif.).
Determination of growth rates.
Approximately 1.5 × 105 cells/ml were inoculated into YPD and cultured at 37°C with or without doxycycline (at a final concentration of 10 μg/ml). At the time points indicated in the figures, aliquots were taken and sonicated briefly (∼5 s) to break down aggregates. Growth was monitored by determining the OD at 660 nm and counting the cells in a Neubauer chamber. The number of viable cells was determined by spreading the diluted cultures on YPD plates and counting the number of colonies that had appeared after cultivating the cells for 24 h at 37°C.
Minichromosome stability assays. (i) Sectoring assay
Strain CgHTUA was transformed with pBM-HAC plasmids containing either wild-type or mutated copies of CgCEN, and transformants were grown overnight in selective medium lacking histidine. Dilutions of overnight cultures were plated on SD medium containing limited amounts of adenine (24 μg/ml) plus three supplements (histidine, uracil, and tryptophan; 40 μg each/ml). After growth for 48 h at 30°C, plates were kept at 4°C for 24 h to allow color development. Images were taken at a ×7 magnification under an Olympus stereo microscope and captured with a digital camera.
(ii) Replica-plating assay.
Dilutions of selectively grown overnight cultures of transformants were plated on YPD. After growth for 18 h at 30°C, the colonies were replica plated onto either YPD or selective medium (SD medium with supplements but without histidine). Colonies were counted after 18 to 24 h at 30°C.
Cytological analysis.
For analyzing nuclear morphology, aliquots of ACG22 and 98CBF1 cells were grown for 6 and 9 h, respectively, at 37°C in the presence of doxycycline (10 μg/ml) and were stained with 4′,6′-diamidino-2-phenylindole (DAPI; Roche) as described previously (28). Immunofluorescence was carried out as has been described previously (28) with rat-antitubulin MAb (YOL1/34; Harlan-Sera-Lab, Leicestershire, England) and fluorescein isothiocyanate-conjugated goat anti-rat antibody (Sigma). Cells were examined using an Olympus BX 60 microscope and a 100× objective. Digital images were captured using an Optronics DEI 750 digital color camera and a Micron Mittenia computer. For fluorescence-activated cell sorter (FACS) analysis, the strains were incubated in YPD including 10 μg of doxycycline per ml at 37°C for the time indicated in Fig. 8. Approximately 2 × 107 cells were fixed in 1 ml of 70% ethanol overnight at 4°C. The fixed cells were washed with 1 ml of phosphate-buffered saline and, after treatment with 1 ml of RNase A (1 mg/ml) for 3 h at 37°C, were sonicated for 3 s. Cells were then digested with 0.2 ml of pepsin (5 mg/ml in 55 mM HCl) for 45 min at 37°C and stained overnight at 4°C in 0.5 ml of 10× propidium iodide solution (5 mg of propidium iodide plus 1.42 g of MgCl2 · 6H2O dissolved in 100 ml of 180 mM Tris [pH 7.5]–190 mM NaCl buffer). Approximately 2 × 106 cells were diluted in 2 ml of phosphate-buffered saline in cryotubes and shipped overnight for FACS analysis (Cytometry Research, LLC, San Diego, Calif.).
FIG. 8.

Phenotype of a CBF1 gene shutdown mutant. (A) Generation of strain 98CBF1 by replacing the CgCBF1 promoter with the controllable promoter tetO::ScHOP1 (43). (B) Effect of doxycycline on the growth of cells. ACG22 (wild-type) (□ and ⋄) and 98CBF1 (mutant) (▵ and ●) cells were cultured without (□ and ▵) or with (⋄ and ●) 10 μg of doxycycline per ml. The number of viable cells was determined by spreading diluted aliquots of the cultures at the indicated time points on YPD and counting the number of colonies that had appeared after incubating the plates for 24 h at 37°C. (C) DAPI-stained mutant and wild-type cells grown for 6 h with 10 μg of doxycycline (dox) per ml. (D) Immunofluorescence of an arrested mutant cell (after 6 h with doxycycline). Spindle microtubules were detected with an antitubulin antibody followed by fluorescein isothiocyanate-conjugated goat anti-rat antibody. (E and F) Flow cytometry profiles of mutant and wild-type cells. (E) Mutant cells show a partial arrest in G2. The cells were grown in the presence of doxycycline for the indicated time points and processed for the analysis of their DNA contents by staining with propidium iodide (see Materials and Methods for details). The x axis is a measure of the fluorescence caused by the propidium iodide, whereas the y axis represents the relative numbers of cells. As a control, wild-type cells (7 h) were treated with 30 μg of nocodazole per ml for 3 h prior to processing. (F) The mutant cells are larger than the wild-type cells. A forward scatter analysis of the flow cytometry data reveals the relative size distributions of mutant and wild-type cells after 6 h of growth in the presence of doxycycline. The y axis represents the relative number of cells.
Nucleotide sequence accession number.
The sequences are available in the GenBank database under the accession number AF233343.
RESULTS
The CgCBF1 gene functionally complements methionine auxotrophy of an S. cerevisiae cbf1 deletion mutant.
In S. cerevisiae, CBF1 gene deletion results in plasmid instability, slow growth, and methionine auxotrophy (39). Therefore, we decided to attempt cloning CBF1 from C. glabrata by functional complementation of the methionine biosynthesis defect of an S. cerevisiae cbf1 null mutant. A C. glabrata genomic library (52) was transformed into the S. cerevisiae cbf1 null mutant strain CC718-1A (34). The cells were first grown under conditions that selected for ScURA3 on the library vector pYep24 and were then replica plated onto minimal medium lacking methionine. After several days at 30°C, one clone of about 2,000 transformants tested was found to be growing without methionine. The plasmid DNA was isolated by shuttling to E. coli and subsequently was retransformed into CC718-1A. The plasmid could correct the methionine auxotrophy of CC718-1A (Fig. 1A). To avoid time-consuming subcloning of the 13-kb insert, the entire plasmid was sequenced by an automated sequencing approach (see Materials and Methods for details). Sequencing revealed that the insert contained five major ORFs whose translation products could be successfully aligned to proteins from the database (Fig. 1B). One of them was identified as CgCBF1, and a vector containing this ORF was proved able to complement the CBF1 gene disruption in CC718-1A (Fig. 1A).
Cbf1 proteins from budding yeasts share common features.
The sequence of CgCBF1 predicts a protein of 441 aa with a calculated molecular mass of 51.4 kDa. CgCbf1p shares general features of Cbf1ps from other budding yeasts, including S. cerevisiae and K. lactis (39, 41). It is a basic helix-loop-helix leucine zipper protein with an acidic isoelectric point of 4.6 and has a negative net charge of −55.9. Interestingly, the negative net charge of CgCbf1p is much higher than that of ScCbf1p and K. lactis Cbf1p (KlCbf1p) (−19.5 and −34, respectively). The N terminus contains patches of acidic amino acids that are characteristic for transcription factors. Amino acid sequence alignment of CgCbf1p with the sequences of other budding yeast Cbf1ps revealed high homologies in the C-terminal regions but low overall homology (Fig. 2). The homologies (identities) of the C termini are 66% (46%) between CgCbf1p (aa 263 to 386) and ScCbf1p (aa 205 to 329) and 65% (46%) between CgCbf1p and KlCbf1p (aa 231 to 355). The overall homology (identity) between CgCbf1p and both of the other budding yeast Cbf1ps is 34% (25%). Interestingly, both the homology (identity) of the C termini and the overall homology (identity) between ScCbf1p and KlCbf1p are higher, namely, 82% (68%) and 44% (35%).
FIG. 2.

Cbf1 proteins of budding yeasts are homologous in their C-terminal regions but show low overall homology. Shown is a CLUSTAL V protein sequence alignment of CgCbf1, KlCbf1, and ScCbf1. Conserved amino acids are highlighted. The basic helix-loop-helix leucine zipper domain is boxed.
A soluble recombinant histidine-tagged version of CgCbf1p (rCgCbf1p) expressed in E. coli cells was shown to alter the migration of a C. glabrata centromere (CgCEN) DNA fragment in gel mobility shift assays (Fig. 3). A prominent slowly migrating complex and a minor, slightly faster-migrating complex were observed. Both complexes could be specifically competed by an annealed and concatemerized oligomer containing the CDEI binding site (data not shown). Binding of Cbf1p to the centromere is not species specific since CgCEN could be shifted by ScCbf1p and CEN3 from S. cerevisiae was shifted by CgCbf1p (data not shown). To determine the equilibrium constant for rCgCbf1p binding to CgCEN, we chose a method that has been used to determine the equilibrium binding constant of ScCbf1p (4). A constant amount of rCbf1p is titrated with increasing amounts of CgCEN, and the binding reactions are analyzed by a shift assay (Fig. 3). A typical titration curve is shown in Fig. 3A, and the Scatchard plot of the data is shown in Fig. 3B. The apparent binding constant calculated from these data is 2.6 × 1010 M−1. The concentration of active rCgCbf1 in the reaction mixture could also be determined from the data and was calculated to be 1.6 × 1010 M−1, or 16.5 μg/ml. Given a protein concentration of 100 μg/ml as determined by a protein assay, the rCgCbf1p preparation contains 16.5% active protein. The equilibrium binding constant for nonspecific binding of Cbf1p to poly(dI-dC) was determined from a parallel set of experiments where constant amounts of rCgCbf1 and CgCEN were titrated with increasing amounts of poly(dI-dC) (Fig. 3C). The binding constant for nonspecific binding (Kapp) was determined to be 5.3 × 105 M−1. The true equilibrium binding constant (Ks) for rCgCbf1 binding to CgCEN was calculated from these data to be 0.9 × 109 M−1. The true equilibrium constant determined for ScCbf1p binding to CEN3 of S. cerevisiae is 3 × 108 M−1 (4, 61). Thus, CgCbf1 binds with a threefold higher affinity to CgCEN than ScCbf1p does to ScCEN3.
Bandshift experiments reveal multiple CgCEN-bound proteins.
We next examined C. glabrata crude extracts and CEN DNA in mobility shift assays. Fragment mobility shift assays have been used in the past to identify centromere binding proteins in S. cerevisiae (4, 35, 44, 61). In contrast to the migration patterns obtained in bandshift experiments with purified rCgCbf1 (Fig. 3), the use of crude extracts led to the specific formation of four different protein-DNA complexes with CgCEN in which the first 270 bp were deleted (CgCENΔ270) (Fig. 4A). The three largest complexes but not complex I could be specifically competed with a CDEI oligomer and thus contain CgCbf1p (Fig. 4A). When the CgCEN DNA was incubated with increasing amounts of crude extract, we first observed the formation of complex I, followed by the formation of complexes III and IV and then complex II (Fig. 4B). Direct comparison of the migration patterns of complexes obtained with purified rCgCbf1 and crude extracts led to the conclusion that complexes II and III correspond to the major and minor complexes observed with rCgCbf1, taking into consideration that the molecular weight of histidine-tagged rCgCbf1 is ∼10% greater than that of the wild-type protein (Fig. 4B). Surprisingly, the migration pattern of a bandshift with a CEN DNA fragment lacking sequences to the left of CDEI (CgCENΔ292 DNA) did not show complexes IV and I, whereas as expected the migration behavior of the rCgCbf1-DNA complex was unaltered (Fig. 4B). Complex I formation was also abolished when the sequences to the right of CDEIII were less than 27 bp (not shown).
FIG. 4.

Gel mobility shift assays with C. glabrata crude extracts and a labeled CgCEN DNA fragment. Shown are autoradiograms of protein-DNA complexes that were formed after incubation of CgCEN DNA with C. glabrata crude extracts and the results of fractionation on nondenaturing gels as described previously (35). (A) Specific competition of complex formation with a CDEI oligomer. Radioactively labeled CEN DNA (5 pmol) was incubated with 2 μl of C. glabrata crude extract in the presence or absence of increasing amounts of an unlabeled, concatemerized CDEI binding site. The numbers on the right indicate the four different complexes that were observed with wild-type CEN DNA. (B) Concentration dependence of protein-DNA complex formation. The same amount of radiolabeled CEN DNA used to obtain the results in panel A was incubated with increasing amounts of crude extract. (C) Centromere mutants that were tested in bandshift experiments and in vivo plasmid loss assays (Fig. 5). WT, wild type. (D) Protein-DNA complex formation on mutated centromere DNAs. Centromere DNA fragments were generated by PCR and radiolabeled as described in Materials and Methods. Centromere DNAs (5 pmol) were incubated with either 2 μl of crude extract or with a 1:20 dilution of purified rCbf1p. (E) Protein-DNA complex formation of CgCbf1p-depleted cells. A crude extract made from C. glabrata 98CBF1 cells (Fig. 8) grown for 7 h in the presence of 10 g of doxycycline per ml was incubated with CEN DNA.
A series of mutant centromeres was generated (Fig. 4C), and the in vitro binding of Cbf1p to these centromeres was tested in bandshift experiments (Fig. 4D). In addition to mutants in which the CEN DNA was truncated from the 5′ end, we created three mutants in which the CDEI site is deleted internally. In the CEN mutant ΔID, the CDEI binding site is partially deleted; in ΔIR, the CDEI binding site is replaced by a BamHI site; and in ΔIE, the CDEI site is exchanged for a motif found in position 280 which closely resembles a CDEI binding site (Fig. 4C). With CEN DNA fragments in which the first 308 bp, including the CDEI binding site, were deleted (Δ308), no complexes could be detected (Fig. 4D). Centromere DNA fragments containing an internally deleted CDEI binding site led to formation of complex I only (Fig. 4D). Taken together, these results indicate that complex I forms independently of the presence of CDEI and that complex IV contains both CgCbf1p and a protein(s) bound in complex I. The exact constitution of complex I will require purification of the bound protein(s) by DNA affinity column chromatography.
Influence of the CDEI binding site on mitotic fidelity of CEN plasmid segregation.
In order to determine the effect of mutations of the CDEI binding site upon CEN function, we tested the effects of deletion of the CDEI binding site in a colony-sectoring assay for C. glabrata, which enabled us to score for the loss frequencies of CEN-bearing plasmids (Fig. 5). To establish this assay, an ade2 null mutant strain was created by deleting ADE2 from strain CgHTU2001. The new strain, CgHTUA, has a red color due to the accumulation of intermediates of purine biosynthesis (Fig. 5N) (50). The various C. glabrata centromeres to be tested for CEN function were then cloned into an ARS plasmid (pBMΔP) which carried CgADE2 and CgHIS3 as selectable markers (see Materials and Methods for details). Strain CgHTUA transformed with pBM-WT gave white colonies, since the ade2 deletion was fully complemented by the wild-type copy of the CgADE2 gene on the plasmid. To monitor the loss of plasmids, transformed colonies were scored for the appearance of red colony color (Fig. 5B to D and F to M). “Half-sectored” colonies have lost or misseggregated the plasmid during the first cell division. Plasmid loss rate is thus equal to the number of half-red colonies (or greater than the number of half-sectored colonies) divided by the total number of colonies after the number of colonies that were entirely red was subtracted. Although this assay does not distinguish between nonsegregation (2:0), plasmid loss (1:0), and other aberrant transmission events like the sophisticated systems developed for S. cerevisiae (33, 54), it is much more sensitive and accurate than the replica-plating assays previously used (31). The plasmid loss rate per generation for a plasmid containing wild-type CEN (450 bp, depicted in Fig. 5A) was determined to be 1.6 × 10−2 (1.7%), which is comparable to plasmid stabilities determined for CEN-based plasmids in S. cerevisiae (Table 3). Deletion of sequences to the left of CDEI (Δ292) resulted in a twofold increase in plasmid loss per generation compared to the plasmid loss of the wild type. Further deletion of the CDEI domain, however, led to such a dramatic increase in plasmid loss per generation that it could not be monitored by the sectoring assay (Δ308). A replica-plating assay (Table 4) showed that the plasmid loss frequency of pBM-Δ308 is increased 70-fold relative to that of the wild type. A similar result, namely, a drop in plasmid stability from 61 to 17% after deletion of CDEI, had been reported previously (31). Plasmids completely lacking the centromere have a 107-fold-increased relative rate of plasmid loss. Surprisingly, the mutants with internally deleted CDEI binding sites showed only a four- to fivefold increase in rates of minichromosome loss (Table 3). When 200 bp of the CDEI-flanking region was deleted in addition to the CDEI deletion (ΔID-Δ200), the loss rate was increased by 15-fold, which is 3-fold higher than that of a CEN construct lacking only CDEI (ΔID) (Table 3). We thus concluded that direct Cbf1p binding to CDEI is not essential for full centromere function, as long as centromere sequences of sufficient length are present to the left of CDEI. However, in the absence of those sequences (i.e., with vector sequences substituting), CDEI is crucial for centromere function and its deletion leads to a dramatic loss of CEN function.
FIG. 5.

Color-based colony-sectoring assay for CEN plasmid missegregation in C. glabrata. A visual assay was established to accurately measure the rates of plasmid loss per generation (see Materials and Methods). Strain CgHTUA was transformed with derivatives of the vector pBMΔP containing the various centromeres depicted in Fig. 4C (for details, see Materials and Methods). (A) CgHTUA/pBM-WT; (B) CgHTUA/pBM-Δ292; (C) CgHTUA/pBM-ΔID-Δ200; (D) CgHTUA/pBM-Δ308; (E) CgHTUA/pBM-ΔCEN; (F to N) colonies in various stages of sectoring; (F) CgHTUA-pBM-WT; (N) CgHTUA.
TABLE 3.
Rates of plasmid loss observed with CDEI mutant centrometers (sectoring assay)
| Plasmid | Total no. of coloniesa | Mean frequency of loss (± SD)/ cell divisionb | Relative frequency of loss |
|---|---|---|---|
| pBM-WT | 2,927 (2) | 1.6 (±1.2) × 10−2 | 1 |
| pBM-Δ200 | 7,663 (3) | 2.8 (±0.4) × 10−2 | 1.75 |
| pBM-Δ292 | 8,791 (4) | 3.3 (±0.7) × 10−2 | 2 |
| pBM-ΔID | 7,504 (3) | 7.8 (±1.0) × 10−2 | 4.9 |
| pBM-ΔIR | 8,926 (4) | 8.3 (±1.6) × 10−2 | 5.2 |
| pBM-ΔIE | 6,469 (3) | 6.7 (±0.4) × 10−2 | 4.2 |
| pBM-ΔID-Δ200 | 3,429 (3) | 2.5 (±1.1) × 10−1 | 15.6 |
| pBM-Δ308 | 693 | ++ | NDc |
| pBM-ΔCEN | 763 | ++ | ND |
Numbers in parentheses are numbers of independent experiments whose results were summarized.
Loss rate/cell division = number of half-sectored colonies/(total number of colonies − number of red colonies). ++, too high to quantitate by the sectoring assay.
ND, not determined.
TABLE 4.
Rates of plasmid loss observed with CDEI mutant centromeres determined by replica-plating assay
| Plasmid | No. of colonies on YPDa | No. of colonies on selective medium | Plasmid loss frequency (± SD)b | Relative loss frequencyc |
|---|---|---|---|---|
| pBM-WT | 428 (1) | 425 | 7 × 10−3 | 1 |
| PBM-Δ278-6/6R | 3,872 (3) | 1,790 | 5.4 (±0.6) × 10−1 | 77 |
| pBM-Δ308 | 1,858 (2) | 940 | 4.9 (±0.1) × 10−1 | 70 |
| pBM-ΔCEN | 2,608 (2) | 647 | 7.5 (±0.3) × 10−1 | 107 |
Numbers in parenthesese indicate the numbers of independent experiments whose results were summarized.
Number of colonies on YPD − number of colonies on selective medium/number of colonies on YPD.
Normalized to the loss frequency of the wild type.
CgCBF1 deletion is lethal in C. glabrata.
Since C. glabrata is a haploid organism without a known sexual cycle, it does not have a second allele that might cover a lethal gene disruption. Therefore, before inactivating genomic CBF1, a plasmid containing a copy of CgCBF1 along with ScURA3 as a selection marker (p112-Cp1) was introduced into the C. glabrata strain Cg2001HTU. Plasmid stability was maintained by the presence of CgARS and CgCEN sequences on the plasmid (31). The new strain, CgHTU/p112-Cp1, was transformed with a DNA fragment containing CgTRP1 flanked on each side by 400 bp of 5′ and 3′ coding and noncoding sequences of CgCBF1 (Fig. 6A). Transformants were selected on medium lacking tryptophan, and stable transformants were replica plated onto medium containing 5-FOA to select for loss of p112-Cp1. Out of the 25 colonies that were replica plated, 7 were not able to grow on 5-FOA medium. Southern analysis of genomic DNAs from the transformants revealed that, in those colonies that could not grow on 5-FOA medium, CgCBF1 had been successfully deleted. In contrast, colonies that were able to grow on 5-FOA medium still contained the wild-type gene (Fig. 6B), indicating that CgCBF1 is necessary for the viability of the strain. In two-thirds of the transformants, the CgCBF1 gene had not been replaced but the CgTRP1 gene was integrated nevertheless. Nonhomologous-recombination events have been reported to occur with high frequency in C. glabrata and might be the cause for high numbers of random integrants (14). We found similarly high numbers of nonhomologous recombination events in all gene replacements we have attempted with C. glabrata. Furthermore, the Southern analysis and genetic data suggest that CBF1 is a single-copy gene in C. glabrata.
FIG. 6.

CBF1 gene deletion is lethal in C. glabrata. (A) Gene replacement of CgCBF1. The 2.2-kb SpeI/HindIII fragment containing the CgCBF1 gene (Fig. 1) was subcloned and is shown as part of pRS-Cg1. For the gene knockout, the CBF1 ORF was replaced by the CgTRP1 gene in a homologous recombination event. Hatched bars (Regions A and B) represent ∼400 bp of homologous sequences on each side of the CgTRP1 gene generated by PCR. Flanking thick lines represent vector sequences. Restriction sites are shown only if relevant. H, HindIII; S, SpeI. (B) Southern blot of genomic DNAs from C. glabrata transformants. Genomic DNAs were extracted from strain CgHTU2001 (lane 1), strain CgHTU2001/random (a transformant that was able to grow after being replated on 5-FOA medium) (lane 2), and the cbf1 deletion strain CgTS1/p112-Cp1 (lane 3) and digested with EcoRI. CgCBF1 was probed with a radioactively labeled PCR fragment generated with primers Cg1/3 and Cg1/4 (Table 2). The labeled band in lane 3 (CgTS1/p112-Cp1) corresponds to the CgCBF1 copy on plasmid p112-Cp1.
CgCbf1p is present at the centromere in vivo.
One way to explain that internal deletion of the CDEI binding site results in only a mild loss of CEN function would be that Cbf1p is still at the centromere and can carry out its function without binding to CDEI. In order to test this hypothesis, we investigated the in vivo interaction of CgCbf1p with the CEN DNA in the presence or absence of CDEI, employing a ChIP assay (1). A new strain, CgTS3, that expressed an HA-tagged version of CgCbf1 (CgCbf1-HA) was created (Fig. 7A). CgCEN in this strain was then replaced with CEN in which the CDEI site was deleted and which carried the CgTRP1 gene inserted 100 bp upstream of CDEI in the centromere to allow for selection (CgTS7). As mentioned above, the loss rate of plasmids carrying this centromere mutation was increased 15-fold over that of the wild type. As a control, CgCEN was also replaced with a wild-type CEN bearing a CgTRP1 insertion at the same position (CgTS5). Both strains grew normally in rich medium (YPD). After the growth of both strains to an OD of 1, the cells were treated with formaldehyde to cross-link cellular structures and then lysed and sonicated to shear the chromatin. Subsequently, HA-specific antiserum was used to immunoprecipitate chromatin containing tagged Cbf1p. After reversing the cross-links, coimmunoprecipitation of CEN DNA was probed by PCR using the primers CgΔ270 and CgCEN2. The CEN DNA was found to be present in the CgCbf1-HA immunoprecipitate but not in the mock-treated control of chromatin from strain CgTS5 (Fig. 7B). To test for the specificity of the CEN DNA coprecipitation, two additional PCRs were carried out with primers against the C. glabrata actin gene (CgACT) and CgHIS3. Neither of these sequences was found in the CgCbf1-HA immunoprecipitate. In chromatin from strain CgTS7 carrying a centromere with a deleted CDEI site, HA-Cbf1p was not detected (Fig. 7B). These results demonstrate that CgCbf1p specifically associates with the centromere in vivo as long as an intact CDEI binding site is present. However, because of the relative insensitivity of ChIP analysis, the failure to detect CEN chromatin-bound CgCbf1p in the absence of a CDEI binding site does not conclusively demonstrate that CgCbf1p is absent in the ΔID mutant CEN chromatin.
FIG. 7.

CgCbf1p binds in vivo at the centromere. (A) An HA-tagged version of CgCBF1 was created by employing a PCR-based strategy (see Materials and Methods for details). (B) Coimmunoprecipitation (ChIP) of CEN DNA and CgCbf1-HA. Formaldehyde-cross-linked chromatin (15 min of fixation) prepared from CgCbf1p-HA epitope-tagged strains CgTS5 (wild type) and CgTS7 (ΔI) was immunoprecipitated with anti-HA antibody or mock treated (no antibody [AB]). Experimental reaction mixtures were prepared in duplicate. Total input material (1 μl of chromatin solution) and coimmunoprecipitated DNA (2 μl of chromatin solution) were analyzed by PCR using primers specific to CgCEN (180 bp) and two noncentromeric loci, CgACT1 (308 bp) and CgHIS3 (224 bp) (see Table 2 for primers). Aliquots of the PCRs were loaded on 2% agarose gels and analyzed. α, anti.
CgCbf1p-depleted cells show defects in chromosome segregation.
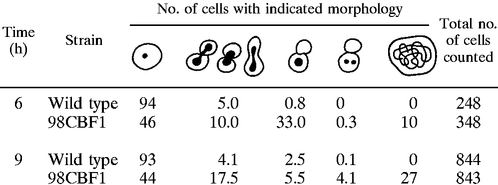
We observed that after overnight incubation on 5-FOA plates, the cbf1 null mutant cells had formed a thin layer, indicating that they were able to perform a few cell divisions before they were depleted of CgCbf1p and ceased to grow. DAPI staining of these cells revealed a significant percentage of large-budded cells with a single mass of DNA bridging the neck (data not shown). This cell morphology is highly suggestive of a defect in chromosome segregation. Since the toxicity of 5-FOA also led to morphologic changes in wild-type cells, we decided to bring the gene under the influence of a controllable promoter to study the effects of Cbf1p depletion in a more controlled way. We employed a system described by Nakayama et al. (42) to create a doxycycline-controllable cassette including the tetracycline operator chimeric promoter (tetO-ScHOP1), which was cloned in front of the CgCBF1 ORF by homologous recombination (Fig. 8A). Strain 98CBF1 showed normal growth compared to that of the wild type (ACG22), indicating that the introduced tetO-ScHOP promoter is sufficiently active to drive expression of CgCBF1 (Fig. 8B). In contrast to wild-type cells, however, 98CBF1 cells arrested growth after cultivation for 5 h in the presence of 10 μg of doxycycline per ml (Fig. 8B). A gel mobility shift assay with crude extract derived from arrested cells and CEN DNA showed the elimination of complexes II, III, and IV, indicating that CgCbf1p is no longer present in the cells (Fig. 4E). A cell count of DAPI-stained cells that had been incubated for 6 h in the presence of doxycycline showed a substantial fraction (33%; n = 348) of large-budded cells in which the DNA had stayed in the mother cell (Table 5; Fig. 8C). After 9 h, a large number of 98CBF1 cells (27%; n = 843) had become enlarged with diffuse or undetectable nuclei (Table 5). A closer examination of the large-budded cells by immunostaining with an antitubulin antibody showed that they had abnormally short spindles (Fig. 8D). The relative DNA contents of the cells was determined by flow cytometry (FACS analysis) (Fig. 8E). Logarithmically growing 98CBF1 cells showed an accumulation of cells with 2C DNA content after 6 h of growth in the presence of doxycycline. The wild-type cells were predominantly (>95%) in G1 phase throughout the experiment. Flow cytometry was also used to compare the sizes of mutant and wild-type cells by measuring their forward scatter, which is a measure of cell size (Fig. 8F). The mean sizes of the mutant cells were significantly increased over that of wild-type cells after 6 h of growth in the presence of doxycycline. Taken together, this phenotype indicates a defect in chromosome transmission at the G2/M transition in the cell cycle. A similar G2 delay is seen in kinetochore structural mutants at their nonpermissive temperatures in S. cerevisiae, for example, in strains with mutations at ndc10-1 (19), cep3-1 or -2 (58), ctf13-30 (17), skp1-4 (13), okp1-5 (46), ctf19Δ1 (25), and mcm21 (48). It is thus likely that Cbf1p carries out its essential function in some aspect of the chromosome segregation process in C. glabrata, although the results do not conclusively demonstrate a centromere defect.
TABLE 5.
Phenotypes of cells after shutdown of the CgCBF1 gene
 |
DISCUSSION
CgCbf1 can functionally complement a cbf1 deletion in S. cerevisiae.
We have cloned the CBF1 gene from C. glabrata by functional complementation of the methionine defect of an S. cerevisiae cbf1 null mutant. The overall homology between C. glabrata and S. cerevisiae Cbf1p is only 34%, and it is thus remarkable that CgCbf1p can substitute for its S. cerevisiae counterpart. It has been shown previously that the first 209 aa of the protein are not required for transcription of the MET genes in S. cerevisiae (39). The C-terminal region containing the basic helix-loop-helix leucine zipper domains are highly conserved between the two yeasts and seem to be sufficient to carry out the function(s) of Cbf1p at the MET promoters of S. cerevisiae. A similar result has been reported for KlCbf1p, which may also complement the methionine deficiency of the S. cerevisiae cbf1 null mutant. While KlCbf1p and ScCbf1p are functionally interchangeable, i.e., ScCbf1p can complement the lethality of the Klcbf1 gene disruption (41), attempts to grow the Cgcbf1 null mutant on 5-FOA medium after introducing a plasmid bearing a copy of the ScCBF1 gene under its own promoter were unsuccessful (data not shown). Although other promoters of S. cerevisiae have been shown to be functional in C. glabrata (e.g., the ScURA3 gene [31]), it is possible that a promoter defect inhibits the functional expression of ScCbf1p in C. glabrata. The overall growth of the Sccbf1 null mutant carrying the CgCBF1 gene is very slow and indicates that CgCbf1p cannot fully substitute for all the functions of ScCbf1p.
Structure-function analysis of the CDEI region in the C. glabrata centromere.
CgCbf1p binds to CDEI-like sequences, as has been found for Cbf1ps from other budding yeasts, but CgCbf1p has a threefold-higher binding affinity for CgCEN than the average binding affinity of ScCbf1p to the 16 centromeres of S. cerevisiae (61). In CEN DNA fragment mobility shift assays with crude extracts we observed a specific DNA binding activity that required sequences to the left of CDEI to be able to bind to the DNA (complex I). In the absence of CDEI, this binding activity could still be detected at the centromere. Bandshifts with crude extracts from CgCbf1p-depleted cells showed the elimination of complexes II, III, and IV but not complex I, suggesting that complex I does not contain intact or fragmented CgCbf1p. In vivo, deletion of flanking DNA alone or of CDEI alone resulted in relatively minor effects on CEN function, whereas deletion of both the flanking region and CDEI completely inactivated the centromere. This result strongly suggests that in addition to binding to CDEI, CgCbf1p or another protein or proteins bind to the DNA region to the left of CDEI. The ChIP experiments and gel shift assays using CgCbf1p-depleted extracts suggest that the protein that binds to the CDEI-flanking region is a new unidentified protein or protein complex. The combined results indicate that centromere function requires either binding of CgCbf1p to the CDEI binding site or the presence of this new protein or protein complex. The exact nature of this protein will need to be determined via DNA affinity column chromatography. Nothing similar has been reported to exist in S. cerevisiae; thus, in this respect the C. glabrata centromere is different than CEN in S. cerevisiae, although the CEN DNA sequences are quite homologous between the two yeasts.
As yet, no evidence for a CDEIII binding activity in C. glabrata extracts was found, although definitive experiments are still to be carried out. It is possible that the uncharacterized band (complex I) seen in fragment mobility shift assays is due to binding of a CBF3-like subunit protein, since deletion of CDEIII sequences also prevents the formation of this complex.
Function of CgCbf1p at the C. glabrata centromere.
Deletion of the CgCBF1 gene is lethal in C. glabrata, and repression of CgCbf1p synthesis results in severe defects in chromosome segregation. The effects of CgCbf1p depletion on C. glabrata cells, namely, the accumulation of large-budded cells in which the DNA remains with the mother cells, cell enlargement, a 2C DNA content, and short spindles, are indicative of defects in the G2/M transition in the cell cycle. Taken together with the results of the sectoring assays, which show a 70-fold drop in plasmid stability upon deletion of CDEI and flanking sequences, this finding strongly suggests that CgCbf1p is important for centromere function in C. glabrata.
While CDEI is essentially required for CEN function when flanking sequences are missing, deletion of CDEI, leaving sequences upstream of CDEI intact, does not have a dramatic effect on plasmid stability and is not lethal when it is introduced into the genome. Furthermore, we did not find any evidence by ChIP analysis for the presence of CgCbf1p at the centromere in vivo when the CDEI binding site was deleted. These results raise the question of whether direct binding of CgCbf1p to CDEI is necessary for proper chromosome segregation. It is possible that a modified form of CgCbf1p is part of a protein complex that binds to the CEN region at a site different from that of CDEI, perhaps in the flanking region to the left of CDEI, and supplies an essential centromere or kinetochore function. In a similar way, a mammalian basic helix-loop-helix transcription factor, the leukemia oncoprotein SCL, has been reported to carry out its function in hematopoietic development without direct binding to the DNA (49). Whereas direct DNA binding of SCL was dispensable for some of its functions, it was essentially required for others. One assumes that, in the absence of direct binding to the DNA, CgCbf1p would be held in place by protein-protein interactions. These might be too weak to remain stable during the relatively harsh washing conditions of the ChIP assays. Thus, it is difficult to draw a definite conclusion from the results from the ChIP experiments.
In addition to its role in CEN function, CgCbf1p may also act as a transcription factor for a protein involved in the chromosome segregation process. The regulated protein may be a kinetochore protein or another protein(s) involved in processes of mitosis. An auxotrophic growth defect caused by the lack of CgCbf1p at other promoters (e.g., MET) is most likely not the reason for the lethality of the CBF1 gene deletion in C. glabrata since the cbf1 null strain does not grow on rich media. To gain further insight into the exact mechanism of CgCbf1p function at the C. glabrata centromere, identification and characterization of other kinetochore proteins will be necessary. These remain the aims of future studies.
Cbf1p as a target for chemotherapy against C. glabrata infections.
Classic antifungal drugs are known for their toxic side effects. A number of new antifungal drugs which are currently under investigation in clinical trials have been developed in recent years. More research is needed to find suitable antifungal drug targets that would lead to the discovery of drugs that are less harmful to the human body. The budding yeast centromere proteins are suitable selective targets for antifungal drug screens since centromeres of this class are structurally different than the large “regional” centromeres found on human chromosomes. Thus, drugs directed toward the inactivation of budding yeast centromere proteins should be quite selective and relatively nontoxic to animal cells. Since the lack of active CgCbf1p in C. glabrata is fatal and causes severe chromosome segregation defects, this protein may be an excellent selective target for chemotherapy against candidiasis.
ACKNOWLEDGMENTS
This work was supported by the Thueringen and German Ministries of Science (TMWFK and BMBF) and by NIH grant CA-11034 to J. Carbon from the National Cancer Institute.
We thank D. Sanglard for providing the C. glabrata library; R. Ballester, P. Joyce, K. Kitada, N. Nakayama, and D. Thomas for providing strains and plasmids; J. Hegemann for the polyclonal antibody to ScCbf1; B. Matsumoto for help with microscopy and colony photography; and D. McLaren for artwork. We also thank P. Hemmerich, G. Wieland, and the members of the Clarke and Carbon labs for helpful discussions; R. Eck, K. Sanyal, and M. Baum for help with experiments; and S. Ohndorf for technical assistance with the cloning of CBF1.
REFERENCES
- 1.Aparicio O M, Weinstein D M, Bell S P. Components and dynamics of DNA replication complexes in S. cerevisiae: redistribution of MCM proteins and Cdc45 during S-phase. Cell. 1997;91:59–69. doi: 10.1016/s0092-8674(01)80009-x. [DOI] [PubMed] [Google Scholar]
- 2.Aususbel F M, Brent R, Kingston R E, Moore D D, Seidmann J G, Smith J A, Struhl K, editors. Current protocols in molecular biology. New York, N.Y: Wiley; 1995. [Google Scholar]
- 3.Baker R E, Gabrielsen O, Hall B D. Effects of tRNATyr point mutations on the binding of yeast RNA polymerase III transcription factor C. J Biol Chem. 1986;261:5275–5282. [PubMed] [Google Scholar]
- 4.Baker R E, Fitzgerald-Hayes M, O'Brien T C. Purification of the yeast centromere binding protein CP1 and a mutational analysis of its binding site. J Biol Chem. 1989;264:10843–10850. [PubMed] [Google Scholar]
- 5.Baker R E, Masison D C. Isolation of the gene encoding the Saccharomyces cerevisiae centromere-binding protein CP1. Mol Cell Biol. 1990;10:2458–2467. doi: 10.1128/mcb.10.6.2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baum M, Clarke L. Fission yeast homologs of human CENP-B have redundant functions affecting cell growth and chromosome segregation. Mol Cell Biol. 2000;20:2852–2864. doi: 10.1128/mcb.20.8.2852-2864.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boulanger P, Yoshinaga S K, Berk A J. DNA-binding properties and characterization of human transcription factor TFIIIC2. J Biol Chem. 1987;262:15098–15105. [PubMed] [Google Scholar]
- 8.Bram R J, Kornberg R D. Isolation of a Saccharomyces cerevisiae centromere DNA-binding protein, its human homolog, and its possible role as a transcription factor. Mol Cell Biol. 1987;7:403–409. doi: 10.1128/mcb.7.1.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cai M, Davis R. Yeast centromere binding protein Cbf1, of the helix-loop-helix protein family, is required for chromosome stability and methionine prototrophy. Cell. 1990;61:437–446. doi: 10.1016/0092-8674(90)90525-j. [DOI] [PubMed] [Google Scholar]
- 10.Choo K H A, editor. The centromere. Oxford, United Kingdom: Oxford University Press; 1997. [Google Scholar]
- 11.Christianson T W, Sikorski R S, Dante M, Shero J H, Hieter P. Multifunctional yeast high-copy-number shuttle vectors. Gene. 1992;110:119–122. doi: 10.1016/0378-1119(92)90454-w. [DOI] [PubMed] [Google Scholar]
- 12.Clarke L, Carbon J. Isolation of a yeast centromere and construction of functional small circular chromosomes. Nature. 1980;287:504–509. doi: 10.1038/287504a0. [DOI] [PubMed] [Google Scholar]
- 13.Connelly C, Hieter P. Budding yeast SKP1 encodes an evolutionarily conserved kinetochore protein required for cell cycle progression. Cell. 1996;86:275–285. doi: 10.1016/S0092-8674(00)80099-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cormack B, Falkow S. Efficient homologous and illegitimate recombination in the opportunistic yeast pathogen Candida glabrata. Genetics. 1999;151:979–987. doi: 10.1093/genetics/151.3.979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Craxton M. Cosmid sequencing. Methods Mol Biol. 1993;23:149–67. doi: 10.1385/0-89603-248-5:149. [DOI] [PubMed] [Google Scholar]
- 16.Cumberledge S, Carbon J. Mutational analysis of meiotic and mitotic centromere function in Saccharomyces cerevisiae. Genetics. 1987;117:203–212. doi: 10.1093/genetics/117.2.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Doheny K F, Sorger P K, Hyman A A, Tugendreich S, Spencer F, Hieter P. Identification of essential components of the S. cerevisiae kinetochore. Cell. 1993;73:761–774. doi: 10.1016/0092-8674(93)90255-O. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fidel P, Vazquez J A, Sobel J D. Candida glabrata: review of epidemiology, pathogenesis, and clinical disease with comparison to C. albicans. Clin Microbiol Rev. 1999;12:80–96. doi: 10.1128/cmr.12.1.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goh P-Y, Kilmartin J V. NDC10: a gene involved in chromosome segregation in Saccharomyces cerevisiae. J Cell Biol. 1993;121:503–512. doi: 10.1083/jcb.121.3.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hanic-Joyce P J, Joyce P B M. A high-copy-number ADE2-bearing plasmid for transformation of Candida glabrata. Gene. 1998;211:395–400. doi: 10.1016/s0378-1119(98)00157-7. [DOI] [PubMed] [Google Scholar]
- 21.Hegemann J H, Fleig U. The budding yeast centromere. Bioessays. 1993;15:451–460. doi: 10.1002/bies.950150704. [DOI] [PubMed] [Google Scholar]
- 22.Hemmerich P, Stoyan T, Wieland G, Koch M, Lechner J, Diekmann S. Interaction of yeast kinetochore proteins with centromere-protein/transcription factor Cbf1. Proc Natl Acad Sci USA. 2000;97:12583–12588. doi: 10.1073/pnas.97.23.12583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heus J J, Zonneveld B J M, Steensma H Y, Van den Berg J A. Centromeric DNA of Kluyveromyces lactis. Curr Genet. 1990;18:517–522. doi: 10.1007/BF00327022. [DOI] [PubMed] [Google Scholar]
- 24.Huberman J, Pridmore R, Jager D, Zonneveld B, Phillipsen P. Centromeric DNA from Saccharomyces uvarum is functional in Saccharomyces cerevisiae. Chromosoma. 1986;94:162–168. [Google Scholar]
- 25.Hyland K, Kingsbury J, Koshland D, Hieter P. Ctf19p: a novel kinetochore protein in Saccharomyces cerevisiae and a potential link between the kinetochore and mitotic spindle. J Cell Biol. 1999;145:15–28. doi: 10.1083/jcb.145.1.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Iborra F, Ball M M. Kluyveromyces marxianus small DNA fragments contain both autonomous replicative and centromeric elements that also function in Kluyveromyces lactis. Yeast. 1994;10:1621–1629. doi: 10.1002/yea.320101211. [DOI] [PubMed] [Google Scholar]
- 27.Jiang W, Phillipsen P. Purification of a protein binding to the CDEI subregion of Saccharomyces cerevisiae centromere DNA. Mol Cell Biol. 1989;9:5585–5593. doi: 10.1128/mcb.9.12.5585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kaiser C, Michaelis S, Mitchell A. Methods in yeast genetics. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1994. [Google Scholar]
- 29.Kim S Y, Mellor J, Kingsman A J, Kingsman S M. An AT rich region of dyad symmetry is a promoter element in the yeast TRP1 gene. Mol Gen Genet. 1988;211:472–476. doi: 10.1007/BF00425703. [DOI] [PubMed] [Google Scholar]
- 30.Kitada K, Yamaguchi E, Arisawa M. Cloning of the Candida glabrata TRP1 and HIS3 genes, and construction of their disruptant strains by sequential integrative transformation. Gene. 1995;165:203–206. doi: 10.1016/0378-1119(95)00552-h. [DOI] [PubMed] [Google Scholar]
- 31.Kitada K, Yamaguchi E, Arisawa M. Isolation of a Candida glabrata centromere and its use in construction of plasmid vectors. Gene. 1996;175:106–108. doi: 10.1016/0378-1119(96)00132-1. [DOI] [PubMed] [Google Scholar]
- 32.Kitada K, Yamaguchi E, Hamada K, Arisawa M. Structural analysis of a Candida glabrata centromere and its functional homology to the Saccharomyces cerevisiae centromere. Curr Genet. 1997;31:122–127. doi: 10.1007/s002940050185. [DOI] [PubMed] [Google Scholar]
- 33.Koshland D, Hieter P. Visual assay for chromosome ploidy. Methods Enzymol. 1987;155:351–372. doi: 10.1016/0076-6879(87)55024-8. [DOI] [PubMed] [Google Scholar]
- 34.Kuras L, Thomas D. Identification of the yeast methionine biosynthetic genes that require the centromere binding factor 1 for their transcriptional activation. FEBS Lett. 1995;367:15–18. doi: 10.1016/0014-5793(95)00528-h. [DOI] [PubMed] [Google Scholar]
- 35.Lechner J, Carbon J. A 240 kd multisubunit protein complex, CBF3, is a major component of the budding yeast centromere. Cell. 1991;64:717–725. doi: 10.1016/0092-8674(91)90501-o. [DOI] [PubMed] [Google Scholar]
- 36.Lechner J, Ortiz J. The Saccharomyces cerevisiae kinetochore. FEBS Lett. 1996;389:70–74. doi: 10.1016/0014-5793(96)00563-7. . (Minireview.) [DOI] [PubMed] [Google Scholar]
- 37.Mannarelli, B. M., and C. P. Kurtzman. 1998. Rapid identification of Candida albicans and other human pathogenic yeasts by using short oligonucleotides in a PCR. 36:1634–1641. [DOI] [PMC free article] [PubMed]
- 38.Mardis E R. High-throughput detergent extraction of M13 subclones for fluorescent DNA sequencing. Nucleic Acids Res. 1994;22:2173–2175. doi: 10.1093/nar/22.11.2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mellor J, Jiang W, Funk M, Rathjen J, Barnes C A, Hinz J H, Phillippsen P. CPF1, a yeast protein which functions in centromeres and promoters. EMBO J. 1990;9:4017–4026. doi: 10.1002/j.1460-2075.1990.tb07623.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Meluh P B, Koshland D. Evidence that the MIF2 gene of Saccharomyces cerevisiae encodes a centromere protein with homology to the mammalian centromere protein CENP-C. Mol Biol Cell. 1995;6:793–807. doi: 10.1091/mbc.6.7.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mulder W, Winkler A A, Scholten I H J M, Zonneveld B J M, de Winde J, de Steensma H J, Grivell L A. Centromere promoter factors (CPF1) of the yeasts Saccharomyces cerevisiae and Kluyveromyces lactis are functionally exchangeable, despite low overall homology. Curr Genet. 1994;26:198–207. doi: 10.1007/BF00309548. [DOI] [PubMed] [Google Scholar]
- 42.Nakayama H, Izuta M, Nagahashi S, Sihta E Y, Sato Y, Yamazaki T, Arisawa M, Kitada K. A controllable gene-expression system for the pathogenic fungus Candida glabrata. Microbiology. 1998;144:2407–2415. doi: 10.1099/00221287-144-9-2407. [DOI] [PubMed] [Google Scholar]
- 43.Niedenthal R, Stoll R, Hegemann J H. In vivo characterization of the Saccharomyces cerevisiae centromere DNA element I, a binding site for the helix-loop-helix protein CPF1. Mol Cell Biol. 1991;11:3545–3553. doi: 10.1128/mcb.11.7.3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ng R, Carbon J. Mutational and in vitro protein-binding studies on centromere DNA from Saccharomyces cerevisiae. Mol Cell Biol. 1987;7:4522–4534. doi: 10.1128/mcb.7.12.4522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Oechsner U, Bandlow B. Interaction of the yeast centromere and promoter factor, Cbf1p, with the cytochrome c1 upstream region and functional implications on regulated gene expression. Nucleic Acids Res. 1996;24:2395–2403. doi: 10.1093/nar/24.12.2395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ortiz J, Stemmann O, Rank S, Lechner J. A putative protein complex consisting of Ctf19, Mcm21, and Okp1 represents a missing link in the budding yeast kinetochore. Genes Dev. 1999;13:1140–1155. doi: 10.1101/gad.13.9.1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pfaller M A, Messer S A, Hollis R J, Jones R N, Doern G V, Brandt M E, Hajjeh R A. Trends in species distribution and susceptibility to fluconazole among blood stream isolates of Candida species in the United States. Diagn Microbiol Infect Dis. 1999;33:217–222. doi: 10.1016/s0732-8893(98)00160-6. [DOI] [PubMed] [Google Scholar]
- 48.Poddar A, Roy N, Sinha P. MCM21 and MCM22, two novel genes of the yeast Saccharomyces cerevisiae are required for chromosome transmission. Mol Microbiol. 1999;31:349–360. doi: 10.1046/j.1365-2958.1999.01179.x. [DOI] [PubMed] [Google Scholar]
- 49.Porcher C, Lia E C, Fujiwara Y, Zon L I, Orkin S H. Specification of hematopoietic and vascular development by the bHLH transcription factor SCL without direct DNA binding. Development. 1999;126:4603–4615. doi: 10.1242/dev.126.20.4603. [DOI] [PubMed] [Google Scholar]
- 50.Roman H. Studies of gene mutation in Saccharomyces cerevisiae. Cold Spring Harbor Symp Quant Biol. 1956;21:175–185. doi: 10.1101/sqb.1956.021.01.015. [DOI] [PubMed] [Google Scholar]
- 51.Sambrook J, Fritsch E F, Maniatis T. Molecular cloning: a laboratory manual. 2nd ed. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 52.Sanglard D, Ischer F, Calabrese D, Majcherczyk P A, Bille J. The ATP binding cassette transporter gene CgCDR1 from Candida glabrata is involved in the resistance of clinical isolates to azole antifungal agents. Antimicrob Agents Chemother. 1999;43:2753–2765. doi: 10.1128/aac.43.11.2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sorger P K, Ammerer G, Shore D. Identification and purification of sequence-specific DNA-binding proteins. In: Creighton T E, editor. Protein function, a practical approach. Oxford, United Kingdom: IRL Press at Oxford University Press; 1989. pp. 199–223. [Google Scholar]
- 54.Spencer F, Gerring S L, Connelly C, Hieter P. Mitotic chromosome transmission fidelity mutants in Saccharomyces cerevisiae. Genetics. 1990;124:237–249. doi: 10.1093/genetics/124.2.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Staden R. The Staden sequence analysis package. Mol Biotechnol. 1996;5:233–241. doi: 10.1007/BF02900361. [DOI] [PubMed] [Google Scholar]
- 56.Stoler S, Keith K C, Curnick K E, Fitzgerald-Hayes M. A mutation in CSE4, an essential gene encoding a novel chromatin-associated protein in yeast, causes chromosome nondisjunction and cell cycle arrest at mitosis. Genes Dev. 1995;9:573–586. doi: 10.1101/gad.9.5.573. [DOI] [PubMed] [Google Scholar]
- 57.Stoyan T, Eck R, Lechner J, Hemmerich P, Kuenkel W, Diekmann S. Cloning of a centromere binding factor 3D (CBF3D) gene from Candida glabrata. Yeast. 1999;15:793–798. doi: 10.1002/(SICI)1097-0061(19990630)15:9<793::AID-YEA415>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 58.Strunnikov A V, Kingsbury J, Koshland D. CEP3 encodes a centromere protein of Saccharomyces cerevisiae. J Cell Biol. 1995;128:749–760. doi: 10.1083/jcb.128.5.749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Van Den Bossche H, Dromer F, Improvisi I, Lozano-Chiu M, Rex J H, Sanglar D. Antifungal drug resistance in pathogenic fungi. Med Mycol. 1998;36(Suppl. I):119–128. [PubMed] [Google Scholar]
- 60.Vinson C R, LaMarco K L, Johnson P F, Landschulz W H, McKnight S L. In situ detection of sequence-specific DNA binding activity specified by a recombinant bacteriophage. Genes Dev. 1988;2:801–806. doi: 10.1101/gad.2.7.801. [DOI] [PubMed] [Google Scholar]
- 61.Wieland G, Hemmerich P, Koch M, Stoyan T, Hegemann J, Diekmann S. Determination of the binding constants of the centromere protein Cbf1 to all 16 centromere DNAs of Saccharomyces cerevisiae. Nucleic Acids Res. 2001;29:1054–1060. doi: 10.1093/nar/29.5.1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhou P, Szczypka M S, Young R, Thiele D J. A system for gene cloning and manipulation in the yeast Candida glabrata. Gene. 1994;142:135–140. doi: 10.1016/0378-1119(94)90368-9. [DOI] [PubMed] [Google Scholar]