

Abstract
Monoclonal antibodies and antibody-derived therapeutics have emerged as a rapidly growing class of biological drugs for the treatment of cancer, autoimmunity, infection, and neurological diseases. To support the development of human antibodies, various display techniques based on antibody gene repertoires have been constructed over the last two decades. In particular, scFv-antibody phage display has been extensively utilized to select lead antibodies against a variety of target antigens. To construct a scFv phage display that enables efficient antibody discovery and optimization, it is desirable to develop a system that allows modular assembly of highly diverse variable heavy chain and light chain (Vκ and Vλ) repertoires. Here we describe modular construction of large non-immune human antibody phage-display libraries built on variable gene cassettes from heavy chain and light chain repertoires (Vκ- and Vλ-light can be made into independent cassettes). We describe utility of such libraries in antibody discovery and optimization through chain shuffling.
Keywords: Antibody gene diversity library, Kappa light chain, Lambda light chain, ScFv phage display, Chain shuffling, Antibody affinity maturation, Antibody optimization, Human monoclonal antibody
1. Introduction
Antibody gene repertoires from non-immune (naïve) human sources have been frequently used to construct antibody-display libraries. To date, several antibody formats, such as single-chain variable fragment (scFv), fragment antigen binding (Fab), or single-domain antibody (sdAb), have been utilized for human antibody-display library generation [1–11]. The scFv form, in which variable heavy chain (VH) and light chain (VL) genes are connected with a flexible linker (typically (Gly4Ser)3 has been widely applied in phage-display [12–20]. Peripheral blood mononuclear cells (PBMC) have been commonly used as a source of human B cells to generate the antibody variable gene libraries. In addition, various primary or secondary lymphoid tissues including bone marrow, lymph node, tonsil, or spleen have also been used as a source of VH and VL (κ/λ) gene repertoires [13, 21–23].
In a commonly used method to create the scFv gene, VH and VL (κ/λ) fragments are separately amplified by two independent polymerase chain reactions (PCR) and then assembled by overlap extension PCR [24, 25]. The heavy and light chain repertoires are thus PCR-amplified at least twice before being spliced into the phage display vector. In addition, library construction using overlapping PCR fragments is often inefficient even with the aid of electroporation. Finally, once assembled and cloned into the display vector, the variable heavy or light chain genes can not be readily removed and replaced due to the lack of appropriate restriction sites. Some research groups have constructed scFv display libraries by the sequential two-step cloning of VL and VH genes into a phagemid vector [26–28]. This has resulted in improved efficiency in library construction. These methods, however, have generally used two-step PCR amplification strategies with the initial set of primers matching the antibody variable genes and the second set of primers containing overhangs for cloning. Ideally, re-amplification steps should be avoided to reduce bias during scFv gene preparation. In addition, previous two-step cloning strategies either do not have restriction sites to allow ready insertion and removal of the variable gene cassettes [17] or less than optimal selection of restriction sites such as the Hind III site [26–28] that is also present in some antibody heavy and light chain genes (http://www2.mrc-lmb.cam.ac.uk/vbase/).
The protocol detailed below describes a modular scheme of library construction where the variable heavy and light chain repertoires are made into cassettes and spliced into the display vector following a single PCR amplification step. VH and VL (κ/λ) genes are amplified by primer sets that encode properly selected flanking restriction enzymes. Separate human Vκ and Vλ gene cassettes are amplified and cloned into a newly designed phagemid vector by the one-step cloning strategy. Likewise, the heavy chain gene cassette is spliced into the phagemid vector by a singe cut and paste action, resulting in an independently constructed heavy chain library. The VH cassette is then restriction-digested and spliced into the light chain display libraries, resulting in independent billion-member VH-Vκ and VH-Vλ scFv phage-display libraries. The antibody gene diversity and library size are assessed. These libraries are utilized to select for scFvs binding to target antigens and to optimize lead antibodies by chain shuffling, which is readily performed due to the modular nature of these display libraries.
2. Materials
2.1. One-step human antibody gene amplification
Eppendorf Mastercycler® pro (Eppendorf, Hauppauge, USA).
OneTaq® PCR master mix (New England BioLabs, Ipswich, USA).
Oligo-nucleotide primers (see Table 1).
Agarose low-EEO (Thermo Fisher Scientific, Waltham, USA).
TAE buffer (40 mM Tris, 20 mM glacial acetic acid, 1 mM EDTA, pH 8.0).
DNA loading buffer [(0.25% Bromophenol blue, 30% Glycerol (v/v)].
QIAquick gel extraction kit (Qiagen, Germantown, USA).
Peripheral Blood Mononuclear Cells (PBMCs) cDNA (Biochain, Newark, USA).
Human Lymph Node QUICK-Clone™ cDNA (Clontech Laboratories, Mountain View, USA).
Table 1.
Oligo-nucleotide primers for one-step PCR amplification of antibody genes and colony PCR.
| Primer | Oligo-nucleotide sequence (5’ – 3’) |
|---|---|
| VH gene PCR amplification | |
| NcoVH1aF | TCGCAACTGCAATTGCCATGGCCCAGGTKCAGCTGGTGCAG |
| NcoVH1bF | TCGCAACTGCAATTGCCATGGCCCAGGTCCAGCTTGTGCAG |
| NcoVH1cF | TCGCAACTGCAATTGCCATGGCCSAGGTCCAGCTGGTACAG |
| NcoVH1dF | TCGCAACTGCAATTGCCATGGCCCARATGCAGCTGGTGCAG |
| NcoVH2aF | TCGCAACTGCAATTGCCATGGCCCAGATCACCTTGAAGGAG |
| NcoVH2bF | TCGCAACTGCAATTGCCATGGCCCAGGTCACCTTGARGGAG |
| NcoVH3aF | TCGCAACTGCAATTGCCATGGCCGARGTGCAGCTGGTGGAG |
| NcoVH3bF | TCGCAACTGCAATTGCCATGGCCCAGGTGCAGCTGGTGGAG |
| NcoVH3cF | TCGCAACTGCAATTGCCATGGCCGAGGTGCAGCTGTTGGAG |
| NcoVH4aF | TCGCAACTGCAATTGCCATGGCCCAGSTGCAGCTGCAGGAG |
| NcoVH4bF | TCGCAACTGCAATTGCCATGGCCCAGGTGCAGCTACAGCAG |
| NcoVH5aF | TCGCAACTGCAATTGCCATGGCCGARGTGCAGCTGGTGCAG |
| NcoVH6aF | TCGCAACTGCAATTGCCATGGCCCAGGTACAGCTGCAGCAG |
| NcoVH7aF | TCGCAACTGCAATTGCCATGGCCCAGGTSCAGCTGGTGCAA |
| NheJH1–2R | TCTAATTATGGCGCTAGCTGAGGAGACRGTGACCAGGGTGCC |
| NheJH3R | TCTAATTATGGCGCTAGCTGAAGAGACGGTGACCATTGTCCC |
| NheJH4–5R | TCTAATTATGGCGCTAGCTGAGGAGACGGTGACCAGGGTTCC |
| NheJH6R | TCTAATTATGGCGCTAGCTGAGGAGACGGTGACCGTGGTCCC |
| VK gene PCR amplification | |
| XbaVK1aF | TCTGGCGGTGGCTCTAGARACATCCAGATGACCCAG |
| XbaVK1bF | TCTGGCGGTGGCTCTAGAGMCATCCAGTTGACCCAG |
| XbaVK1cF | TCTGGCGGTGGCTCTAGAGCCATCCRGATGACCCAG |
| XbaVK1dF | TCTGGCGGTGGCTCTAGAGTCATCTGGATGACCCAG |
| XbaVK2aF | TCTGGCGGTGGCTCTAGAGATATTGTGATGACCCAG |
| XbaVK2bF | TCTGGCGGTGGCTCTAGAGATRTTGTGATGACTCAG |
| XbaVK3aF | TCTGGCGGTGGCTCTAGAGAAATTGTGTTGACRCAG |
| XbaVK3bF | TCTGGCGGTGGCTCTAGAGAAATAGTGATGACGCAG |
| XbaVK3cF | TCTGGCGGTGGCTCTAGAGAAATTGTAATGACACAG |
| XbaVK4aF | TCTGGCGGTGGCTCTAGAGACATCGTGATGACCCAG |
| XbaVK5aF | TCTGGCGGTGGCTCTAGAGAAACGACACTCACGCAG |
| XbaVK6aF | TCTGGCGGTGGCTCTAGAGAAATTGTGCTGACTCAG |
| XbaVK6bF | TCTGGCGGTGGCTCTAGAGATGTTGTGATGACACAG |
| NotJK1R | AGTCATTCACGACTTGCGGCCGCACGTTTGATTTCCACCTTGGTCCC |
| NotJK2–4R | AGTCATTCACGACTTGCGGCCGCACGTTTGATCTCCASCTTGGTCCC |
| NotJK3R | AGTCATTCACGACTTGCGGCCGCACGTTTGATATCCACTTTGGTCCC |
| NotJK5R | AGTCATTCACGACTTGCGGCCGCACGTTTAATCTCCAGTCGTGTCCC |
| Vλ gene PCR amplification | |
| XbaVλ1aF | TCTGGCGGTGGCTCTAGACAGTCTGTGCTGACTCAG |
| XbaVλ1bF | TCTGGCGGTGGCTCTAGACAGTCTGTGYTGACGCAG |
| XbaVλ1cF | TCTGGCGGTGGCTCTAGACAGTCTGTCGTGACGCAG |
| XbaVλ2F | TCTGGCGGTGGCTCTAGACAGTCTGCCCTGACTCAG |
| XbaVλ3aF | TCTGGCGGTGGCTCTAGATCCTATGWGCTGACTCAG |
| XbaVλ3bF | TCTGGCGGTGGCTCTAGATCCTATGAGCTGACACAG |
| XbaVλ3cF | TCTGGCGGTGGCTCTAGATCTTCTGAGCTGACTCAG |
| XbaVλ3dF | TCTGGCGGTGGCTCTAGATCCTATGAGCTGATGCAG |
| XbaVλ4F | TCTGGCGGTGGCTCTAGACAGCYTGTGCTGACTCAA |
| XbaVλ5dF | TCTGGCGGTGGCTCTAGACAGSCTGTGCTGACTCAG |
| XbaVλ6dF | TCTGGCGGTGGCTCTAGAAATTTTATGCTGACTCAG |
| XbaVλ7dF | TCTGGCGGTGGCTCTAGACAGRCTGTGGTGACTCAG |
| XbaVλ8dF | TCTGGCGGTGGCTCTAGACAGACTGTGGTGACCCAG |
| XbaVλ4–9dF | TCTGGCGGTGGCTCTAGACWGCCTGTGCTGACTCAG |
| XbaVλ10dF | TCTGGCGGTGGCTCTAGACAGGCAGGGCTGACTCAG |
| NotJλ123R | AGTCATTCACGACTTGCGGCCGCACCTAGGACGGTSASCTTGGTCCC |
| NotJλ4–5R | AGTCATTCACGACTTGCGGCCGCACCTAAAACGGTGAGCTGGGTCCC |
| NotJλ7R | AGTCATTCACGACTTGCGGCCGCACCGAGGACGGTCAGCTGGGTGCC |
| Primer for colony PCR of VH and VK/Vλ | |
| ColLMB3F | CAGGAAACAGCTATGAC |
| Colfeseq1R | GAA TTT TCT GTA TGA GGG |
Restriction enzyme sites are underlined.
2.2. Vκ and Vλ library construction
XbaI (New England BioLabs).
NotI-HF (New England BioLabs).
CutSmart® buffer (New England BioLabs).
QIA quick PCR purification kit (Qiagen).
T4 DNA ligase (New England BioLabs).
Microcon Ultracel YM-10 centrifugal filter (Millipore, Billerica, USA).
TG1 electrocompetent cells (Lucigen, Middleton, USA). E.coli TG1 genotype: [F´ traD36 proAB lacIqZ ΔM15] supE thi-1 Δ(lac-proAB) Δ(mcrB-hsdSM)5 (rK- mK-).
Gene Pulser® electroporation cuvettes, 0.1 cm gap (Bio-Rad, Hercules, USA).
Electroporator 2510 (Eppendorf).
2xYT broth (Thermo Fisher Scientific).
50% glucose (Thermo Fisher Scientific), filter-sterilized.
100 mg/ml Ampicillin sodium salt (Sigma, St. Louis, USA), filter-sterilized.
Polystyrene petri-dishes, 150 mm × 15 mm (United Scientific Supplies, Waukegan, USA).
2xYT-AG agar plates (2xYT, 100 μg/ml Ampicillin, 2% glucose, 1.5% agar (w/v)).
Glycerol (Thermo Fisher Scientific), 60% (v/v).
QIAGEN® plasmid mini kit (Qiagen).
Oligo-nucleotide primer set for colony PCR (see Table 1).
14 ml round bottom culture tube (Corning, New York, USA)
2.3. VH cassette cloning and scFv library generation
2.3.1. ScFv library with naïve human VH genes
NcoI-HF (New England BioLabs).
NheI-HF (New England BioLabs).
2.3.2. ScFv-shuffle library with a given human VH gene
Primers: NcoU2scFvF (5’ ATTCCATGGCCCAGGTGCAGCTGCAGGAG 3’) and NheU2scFvR (5’ CAGGCTAGCTGAGGAGACGGTGACCAG 3’).
2.4. scFv-phage packaging and library preparation
M13KO7 helper phage (Thermo Fisher Scientific).
70 mg/ml Kanamycin (Sigma), filter-sterilized.
Polyethylene Glycol (PEG) 8000 (Thermo Fisher Scientific).
PEG/NaCl solution, 5X (20% PEG8000 (w/v), 2.5 M NaCl).
Phosphate buffer saline (PBS): 137 mM NaCl, 2.68 mM KCl, 10 mM Na2HPO4, 2 mM KH2PO4, pH 7.4.
Sodium Azide (NaN3) (Sigma).
2xYT-AK medium (2xYT, 100 μg/ml Ampicillin, 70 μg/ml Kanamycin).
2.5. Selection of phage antibody display library on recombinant antigen
2.5.1. ScFv phage display selection
pFUSE-hIgG1-Fc2 plasmid (InvivoGen, San Diego, USA)
Protein A agarose (Thermo Fisher Scientific)
EZ-Link™ Sulfo-NHS-Biotin (Thermo Fisher Scientific)
Dynabeads® M-280 Streptavidin (Thermo Fisher Scientific)
Dynal® Magnetic rack (Thermo Fisher Scientific)
End-over-end rotator (Barnstead International, Dubuque, USA)
PBSM: PBS, 2% non-fat dry milk (LabScientific, Highlands, USA)
PBSMT: PBS, 2% non-fat dry milk, 0.1% Tween20 (Acros, Geel, Belgium)
Triethylamine (TEA) (Sigma)
2.5.2. Screen by phage ELISA
96 well MaxiSorp™ flat bottom plate (Corning).
PBST: PBS, 0.1% Tween20.
Biotin-labeled rabbit anti-fd bacteriophage antibody (Sigma).
Streptavidin-HRP (Horseradish Peroxidase) (Sigma).
TMB substrate solution (Thermo Fisher Scientific).
Hydrochloric acid (Thermo Fisher Scientific).
Plate reader (Synergy HT from Biotek, Winooski, USA).
2.5.3. Flow cytometry analysis of monoclonal phage
Flow cytometry buffer (FCB): PBS, 2% fetal bovine serum (FBS) (Thermo Fisher Scientific)
Streptavidin conjugated with phycoerithrin (PE) (Thermo Fisher Scientific).
HEK293 cell line (ATCC, Manassas, USA).
BD Accuri™ C6 flow cytometer (BD Biosciences, San Jose, USA).
OptiMEM I serum-free medium (Thermo Fisher Scientific).
TransIT 2020 transfection reagent (Mirus Bio, Madison, USA).
3. Methods
The protocols describe modular construction of large naïve human scFv phage-display libraries from independent heavy and light chain gene cassettes. These cassettes are derived from libraries constructed independently from naive human heavy and light chain (Vκ and Vλ) gene repertoires by one-step PCR amplification using primer sets with two restriction enzyme site overhangs. First, the pHEN1 phagemid [29] is modified by inserting stuffer sequences, a (G4S)3 linker flanked by restriction enzyme sites for cloning to generate a new display vector pHEN1-NX (Fig. 1A). Next, primers matched with the N- and C-terminal sequences of VH, Vκ, and Vλ are used for the PCR amplification of each sub-family of antibody variable genes (Fig. 1B). The variable gene fragments are directly digested by two distinct restriction enzymes pairs, NcoI/NheI for VH and XbaI/NotI for VL (κ/λ), and ligated into the modified pHEN1-NX phagemid. Ligation products are desalted and concentrated using a centrifugal filter unit and electro-transformed into electro-competent TG1. In addition to modular construction of the scFv phage-display library, the separately constructed Vκ and Vλ libraries can be used to generate chain shuffled libraries anchored on a previously identified heavy chain for optimization and affinity maturation studies.
Fig. 1. Modified phagemid pHEN1-NX and antibody library generation scheme by one-step PCR and cut-and-paste cloning.
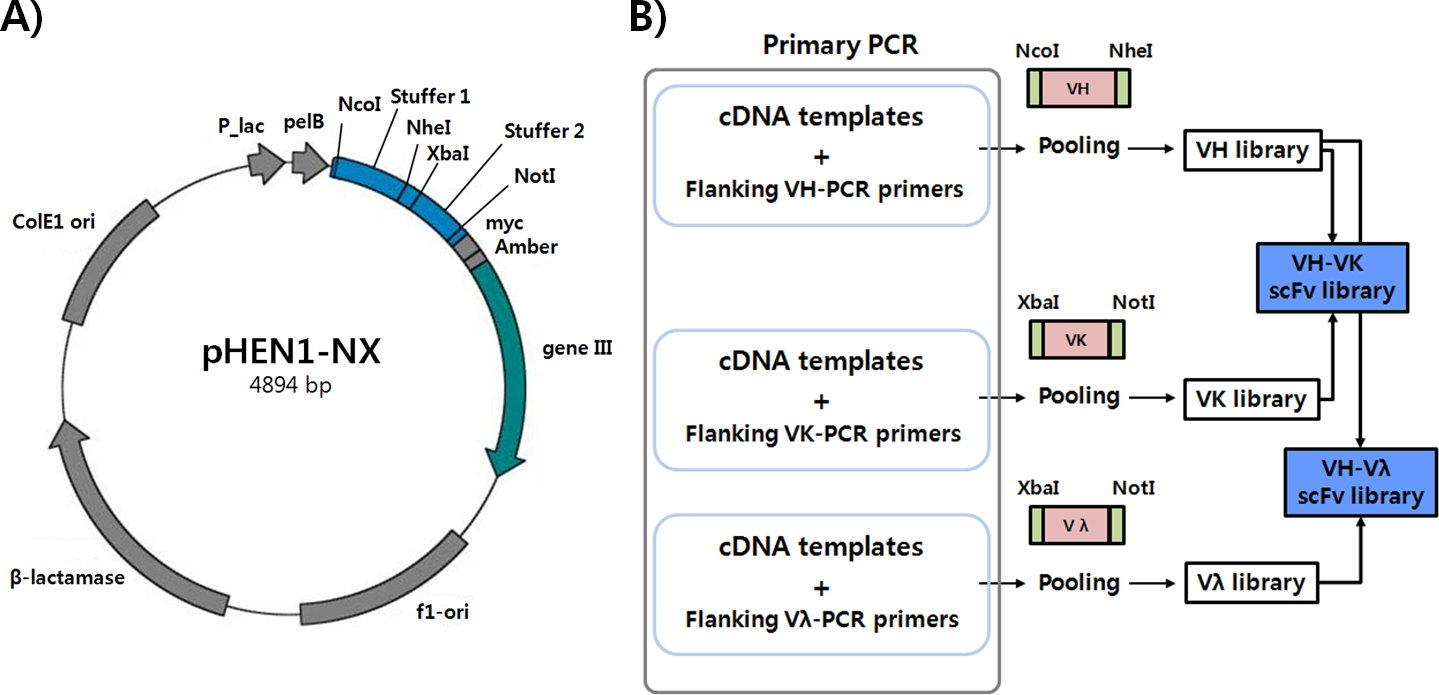
A) The original pHEN1 phagemid was modified by introducing two stuffers with multiple stop codons, a flexible (G4S)3 linker, and two restriction enzyme sites (NheI and XbaI). B) Different primer sets grouped by germline antibody sub-families are used to amplify VH (1, 2–7, 3, 5, 6), Vλ (1, 2, 3, 4–10), and Vκ (1, 2, 3, 4–6). Primer sets include flanking NcoI and NheI sites for VH or XbaI and NotI sites for Vλ and Vκ. Variable fragments amplified by one-step PCR were directly utilized for cloning.
3.1. One-step human antibody gene amplification
Human PBMC and lymph node-derived cDNA samples obtained from bio-sample preparation vendors can be conveniently used as an alternative source for antibody gene fragments (see Note 1).
- Forward primers used for VH, Vκ, and Vλ PCR amplification are described in Table 1. Mix equal molar concentration of each subfamily-based forward primer designated below:
Antibody gene Primer set (final concentration, 10 μM) VH Set#1: NcoVH1aF/NcoVH1bF/NcoVH1cF/NcoVH1dF
Set#2–7: NcoVH2aF/NcoVH2bF/NcoVH4aF/NcoVH4bF/ NcoVH7aF
Set#3: NcoVH3aF/NcoVH3bF/NcoVH3cF
Set#5: NcoVH5aF
Set#6: NcoVH6aFVκ Set#1: XbaVK1aF/XbaVK1bF/XbaVK1cF/XbaVK1dF
Set#2: XbaVK2aF/XbaVK2bF
Set#3: XbaVK3aF/XbaVK3bF/XbaVK3cF
Set#4–6: XbaVK4aF/XbaVK5aF/XbaVK6aF/XbaVK6bFVλ Set#1: XbaVλ1aF/XbaVλ1bF/XbaVλ1cF
Set#2: XbaVλ2F
Set#3: XbaVλ3aF/XbaVλ3bF/XbaVλ3cF/XbaVλ3dF
Set#4–10: XbaVλ4F/XbaVλ5dF/XbaVλ6dF/XbaVλ7dF/XbaVλ8dF/XbaVλ4–9dF/XbaVλ10dF - Make each reverse primer mixture (final concentration, 10 μM) for VH, Vκ, and Vλ by mixing equal molar concentration of each reverse primer (Table 1). Set up mixture for one-step PCR as described below (See Note 2):
Component (concentration) Volume (μl/reaction) ddH2O 22 – 20 OneTaq master mix (2x) 25 Forward primer mixture (10 μM) 1 Reverse primer mixture (10 μM) 1 cDNA template 1 – 3 Add cDNA templates and carry out the PCR for 30 cycles (30 sec at 95°C, 1 min at 55°C, 1 min at 72°C) after pre-incubation for 3 min at 95°C, then complete the PCR by incubating the samples for 7 min at 72°C. Four or six separate PCR amplifications for Vκ/Vλ or VH subfamilies can be performed with reverse primer mixture and each forward primer set.
Analyze PCR products on 1.4% TAE agarose gel by electrophoresis. Cut out separately the amplified Vκ, Vλ (~350 bp), and VH (~380 bp) and purify them from agarose gel with a gel extraction kit (see Note 3). For each Vκ, Vλ, and VH subfamily, pool the extraction product together (see Note 4).
Determine the DNA concentration of pooled antibody genes. Store the pooled DNA at 4°C and use them for the following restriction enzyme digestion step.
3.2. Vκ and Vλ library construction
Prepare pHEN1-NX phagemid using a plasmid mini prep kit. Perform a single (XbaI or NotI-HF) and double digestion (XbaI/NotI-HF) to test whether the phagemid can be fully digested.
Digest 5 μg of pHEN1-NX phagemid with XbaI and NotI-HF (5 U/μg of DNA) in 1X buffer provided by the manufacturer for 4 hrs at 37°C. Analyze the digestion product on 0.7% TAE agarose gel and purify the digested vector from agarose gel (see Note 5). Determine the DNA concentration and store the samples at 4°C.
Digest 1 μg of Vκ or Vλ PCR product with XbaI and NotI-HF (20 U/μg of DNA) for 6 hrs at 37°C. Purify the digested Vκ or Vλ fragment by a PCR purification kit (see Note 6) and determine the DNA concentration.
- Perform ligation reaction with the digested pHEN1-NX and Vκ or Vλ as shown below (see Note 7):
Component (concentration) Volume (μl/reaction) ddH2O add up to 100 T4 DNA ligase buffer (10x) 10 pHEN1-NX (40 – 50 ng/μl) 20 – 25 Vκ or Vλ (10 – 20 ng/μl) 15 – 30 T4 DNA ligase (400,000 U/ml) 1 Incubate the ligation mixture at 16°C for 16 hrs and inactivate the ligase at 65°C for 10 min.
Concentrate and desalt the ligation product by using a centrifugal filter with a 50 kDa molecular weight cut-off according to the manufacturer’s instruction (see Note 8).
Thaw one vial of 50 μl electrocompetent TG1 bacteria on ice for 10 min (see Note 9). Distribute 25 μl of TG1 into two pre-chilled 1.5 ml Eppendorf tubes and transfer 5 μl of concentrated and desalted Vκ or Vλ ligation product into each tube. Incubate for 5 min and transfer the mixture into two pre-chilled 0.1 cm electroporation cuvettes. Electroporate at the setting of 1.8 kV/600 ohms/10 μF and add immediately 950 μl recovery medium pre-warmed at 37°C (see Note 10).
Transfer the culture into a 14 ml round bottom tube and incubate at 37°C for 1 hr in a shaker-incubator at 250 rpm.
Spread 1:105 and 1:106 diluents of the culture to determine the titer of transformed bacteria and spread the remaining culture onto three large 2xYT-AG plates (150 mm × 15 mm) per one electroporation reaction. Incubate the plates at 37°C overnight.
Calculate colony titer of each sub-library and scrape transformed bacteria by using 2xYT-AG supplemented with 20% glycerol (~3 ml/large plate). Freeze collected transformants at −80°C as a sub-library stock.
To check Vκ or Vλ gene insertion, randomly pick single colonies from each transformation and perform colony PCR by using a primer set (Table 1). Analyze correct PCR products (~800 bp) on TAE agarose gel and estimate VL-insertion rate based on the electrophoresis result (see Note 11).
Repeat digestion, ligation, and electro-transformation steps to achieve expected size of each Vκ or Vλ library (>~108 cfu/library) (see Note 12). After finalizing transformation reactions, thaw all of collected transformants and mix them together proportionally based on each sub-library size. Aliquot into cryovials (~1 ml) and store each reconstituted Vκ or Vλ library at −80°C.
3.3. VH cassette cloning and scFv library generation
3.3.1. ScFv library with naïve human VH genes
VH cassette library is separately generated using the same methods as described in subheading 3.2 (See Note 13).
Prepare VH- and VL (Vκ or Vλ)-library phagemid using a plasmid mini prep kit. Perform double digestion by using NcoI-HF and NheI-HF to test digestion of the stuffer 1 for VH cloning (see Note 5).
Digest separately 5 μg of VH- and VL-library phagemid with NcoI-HF and NheI-HF (5 U/μg of DNA) in 1X buffer provided by the manufacturer for 4 hrs at 37°C. Analyze the digestion described on 0.7% TAE agarose gel and purify the digested VH gene cassettes (~350 bp) and VL phagemid vector from agarose gel (see Note 5).
Determine the DNA concentration and store samples at 4°C.
- Ligate the digested VL-library phagemid and VH fragments as shown below (see Note 7):
Component (concentration) Volume (μl/reaction) ddH2O Add up to 100 T4 ligase buffer (10x) 10 VL-library phagemid (40 – 50 ng/μl) 20 – 25 VH gene cassette (12 – 16 ng/μl) 15 – 20 T4 ligase (400,000 U/ml) 1 Incubate the ligation reaction at 16°C for 16 hrs and inactivate the ligase at 65°C for 10 min.
Concentrate and desalt the ligation product by using a centrifugal filter with a 50 kDa molecular weight cut-off according to the manufacturer’s instruction (see Note 8).
Thaw one vial of 50 μl electrocompetent TG1 bacteria on ice for 10 min (see Note 9). Distribute 25 μl of bacteria cells into two pre-chilled 1.5 ml Eppendorf tubes and transfer 5 μl of concentrated and desalted VH ligation product into each tube. Incubate for 5 min and transfer the mixture into two pre-chilled 0.1 cm electroporation cuvettes. Electroporate at the setting of 1.8 kV/600 ohms/10 μF and add immediately 950 μl recovery medium pre-warmed at 37°C (see Note 10).
Transfer the culture into a 14 ml round-bottom tube and incubate at 37°C for 1 hr in a shaker-incubator at 250 rpm.
Spread 1:105 and 1:106 diluents of the culture to determine the titer of transformed bacteria and spread the rest onto three large 2xYT-AG plates (150 mm × 15 mm) per one electroporation reaction. Incubate the plates at 37°C overnight.
Calculate colony titer of each VH-inserted sub-library as a scFv sub-library. Scrape transformed bacteria by using 2xYT-AG supplemented with 20% glycerol (~3 ml/large plate). Freeze collected transformants at −80°C as a scFv sub-library stock.
To check full-length scFv insertion, randomly pick single colonies from each transformation and perform colony PCR by using ColLMB3F (forward) and Colfdseq1R (reverse) primers (Table 1). Analyze correct PCR products (~950 bp) on TAE agarose gel and estimate the percent of scFv-insertion based on the electrophoresis result (see Note 11).
Sequence scFv from step 12 above and analyze VH, Vκ, and Vλ sequences by IgAT tool [30] to evaluate the distribution of antibody subfamilies and the sequences of the third complementarity determining region of the antibody heavy chain (CDR-H3). The subfamily representation of Vκ, and Vλ cassettes (Fig. 2A) or the VH cassette (Fig. 2B) is estimated according to the frequency of each subfamily. CDR-H3 sequences (6 ~ 25 amino acids) from 252 VH-Vκ and VH-Vλ scFvs are analyzed for CDR-H3 length distribution (Fig. 2C).
Repeat digestion, ligation, and electro-transformation steps to achieve expected size (at least ~109 cfu) of the final scFv library. After finalizing transformation reactions, thaw all of collected transformants and mix them together proportionally based on each sub-library size. Aliquot into cryovials (~1 ml) and store at −80°C.
Fig. 2. Sequence analysis of VH-Vκ and VH-Vλ scFv phage libraries.
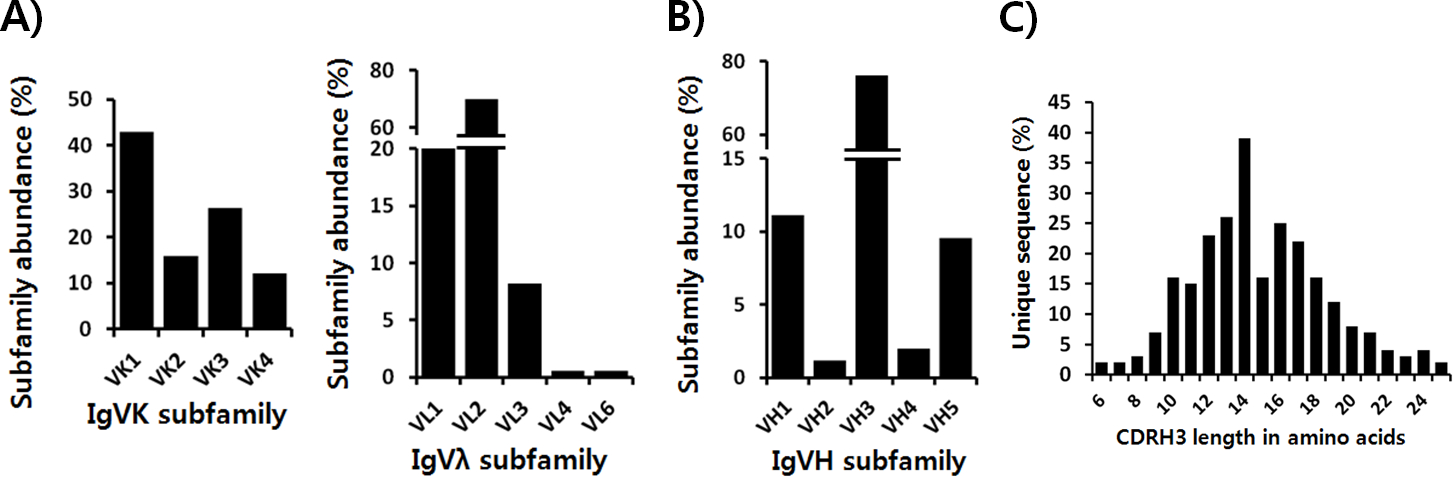
A) Germline subfamily distribution of light and heavy chain sequences in VH-Vκ and VH-Vλ scFv libraries. 182 Vκ (panel A, left), 170 Vλ (panel A, right), or 252 VH (panel B) sequences were obtained from each scFv library and analyzed for frequency of representation of light chain variable region subfamilies. It should be noted that the distribution of subfamilies can be adjusted by adjusting the relative amount of PCR product from each subfamily during VL and VH cassette generation. C) 252 VH sequences derived from constructed scFv libraries were analyzed for length distribution of CDR-H3 regions.
3.3.2. ScFv-shuffle library with a given human VH gene
Perform PCR to amplify the VH fragment of interest with flanking NcoI and NheI sites by using the primer set NcoU2scFvF (5’ ATTCCATGGCCCAGGTGCAGCTGCAGGAG 3’) and NheU2scFvR (5’ CAGGCTAGCTGAGGAGACGGTGACCAG 3’) under the following condition: 30 cycles (30 sec at 95°C, 1 min at 55°C, 1 min at 72°C) after pre-incubation for 3 min at 95°C, with final extension for 7 min at 72°C.
Analyze PCR products (~380 bp) on 1.4% TAE agarose gel by electrophoresis. Gel-purify the amplified VH fragment with a gel extraction kit (see Note 3), and determine the DNA concentration.
Carry out the VH cloning into VL libraries (Vκ or Vλ) to generate a VL-shuffled library as described previously (see Subheading 3.3.1). Repeat digestion, ligation, and electro-transformation steps to achieve desired library size (> ~2 × 108 cfu).
3.4. ScFv-phage packaging and library preparation
Thaw out scFv-phagemid library TG1 stocks and transfer into 500 ml 2xYT-AG medium (OD600 at 0.05 ~ 0.1). Inoculate the culture for 2 hrs in a shaker-incubator at 250 rpm until reaching exponential growth phase (OD600 at ~0.5).
Add M13KO7 helper phage at a MOI (multiplicity of infection) of 20 and incubate the culture at 37°C for 30 min without shaking, followed by 30 min shaking at 120 rpm (see Note 14).
Harvest the bacteria by centrifugation (3,500 × g, 10 min) and remove as much residual glucose as possible.
Resuspend the pellet with 500 ml 2xYT-AK medium in a 2 L shaker flask. Incubate the culture overnight with proper aeration in a shaker-incubator at 250 rpm at 30°C to rescue scFv-phage particles.
Centrifuge the bacteria (4,500 × g, 15 min, 4°C) and transfer 120 ml of the supernatant into four 250 ml centrifuge bottles. Add 30 ml 5X PEG/NaCl solution to each bottle and mix completely by vortexing. Precipitate the phage particles on ice for at least 1 hr.
Centrifuge the precipitated phage suspension (4,500 × g, 15 min, 4°C) and remove as much supernatant as possible. Resuspend the phage pellet from each centrifuge bottle using 2 ml PBS, collect and mix the suspension in a 15 ml Falcon tube.
Centrifuge (4,500 × g, 15 min, 4°C) to remove remaining bacterial debris. Transfer the suspension into a new 15 ml Falcon tube and add 1/5 volume of 5X PEG/NaCl solution, and precipitate the phage on ice for at least 2 hrs.
Centrifuge for 15 min at 4,500 × g at 4°C, and remove supernatant. Resuspend the phage pellet in 3 ml sterile PBS/0.02% NaN3 (w/v). Keep phage stock at 4°C.
To determine phage library titer, dilute rescued phage in 2xYT (1:10 serial dilutions from 1:106 to 1:1010) and infect 180 μl of log phase TG1 with 20 μl of phage diluents. Incubate for 30 min at room temperature without shaking and an additional 30 min in a shaker-incubator at 250 rpm at 37°C, and plate the infected TG1 on 2xYT-AG plate. Incubate the plates at 37°C overnight.
Calculate the phage titer based on the number of colonies on the plate (see Note 15). Add glycerol to 20% into previously resuspended phage stock (see step 8 above) and freeze rescued phage library at −80°C for long-term storage and use.
3.5. Selection of phage antibody display library on recombinant antigen
3.5.1. ScFv phage display library selection
Clone the cDNA of the target antigen of interest into pFUSE-hIgG1-Fc2 to produce a recombinant Fc-fusion molecule. Purify the fusion protein on a protein A column and biotin-label it using a biotinylation reagent (see Note 16).
Make 15ml 4% PBSM for each round of the selection procedure. Mix an equal volume of phage library and 4% PBSM (total 1 ml, final 2% PBSM) in a 1.5 ml Eppendorf microcentrifuge tube per antigen selection. Rotate the tube end-over-end to mix for 5 min at RT.
Equilibrate 50 μl of streptavidin-coated Dynabeads in PBS and wash once with PBSM. Draw the beads into a pellet with a magnetic rack and remove PBSM from the tube. Add phage antibody library solution (see above Subheading 3.3.1) and resuspend the beads thoroughly. Incubate for 1 hr at RT using an end-over-end rotator.
Draw the beads into a pellet with a magnetic rack and transfer the supernatant containing phage library depleted against the streptavidin beads into a new 1.5 ml Eppendorf tube. Add 0.5~10 nM biotinylated Fc-fusion antigen directly to the counter-selected phage library (see Note 17) and incubate on an end-to-end rotator for 1 hr at RT. While the incubation is in progress, place 50 μl of streptavidin-coated Dynabeads in a new 1.5 ml tube. Equilibrate, wash, and block the beads in 2% PBSM for 1 hr.
Draw blocked beads into a pellet with a magnetic rack and remove the supernatant. Resuspend the beads with 1 ml of the phage library incubated with the antigen. Incubate the phage/biotinylated-antigen/streptavidin-beads mixture for 30 min at RT on an end-to-end rotator.
Draw the phage/antigen/beads complex into a pellet with a magnetic rack for 3 min. Discard carefully the supernatants, wash the beads 5 times with 1 ml PBSMT (see Note 18). After the final washing, resuspend the beads with 1 ml PBS and transfer into a new 1.5 ml Eppendorf tube. Wash twice with PBS.
Elute bound phage from the beads with 0.5 ml of 100 mM TEA by incubation for 10 min on a rotator (see Note 19). Draw the beads and transfer the eluent to a new 1.5 ml tube containing 250 μl of 1 M Tris-HCl (pH 6.8). Immediately neutralize the solution by vortexing.
Add neutralized phage eluent to 8.5 ml of exponential phase TG1 (OD600 at 0.7) and incubate for 1 hr in a shaker-incubator at 120 rpm at 37°C. Plate 1 and 10 μl of the infected TG1 on 2xYT-AG plates (100 mm × 15 mm) to estimate the number of eluted phage.
Centrifuge the remaining TG1 culture at 3,000 × g for 10 min, remove supernatant, resuspend the pellet using 300 μl 2xYT medium, and plate on a large 2xYT-AG plate (150 mm × 15 mm).
Grow overnight at 37°C and collect the TG1 output by scraping using 2.5 ml 2xYT-AG/20% glycerol medium.
Inoculate the TG1 output (OD600 at 0.05~0.1) in 2xYT-AG medium and prepare phage particles for the next round of selection as described in Subheading 3.4 (see Note 20). Culture the bacteria and rescue phage particles as described in Subheading 3.4.
3.5.2. Screen by Phage ELISA
Refer to Subheading 3.5.1 for polyclonal phage preparation from each round output of the selections.
Coat a 96-well microtiter plate with the Fc-fusion antigen (2~10 μg/ml in PBS) by incubating overnight at 4°C. Alternatively, the antigen can be immobilized by incubation for 2 hrs at room temperature.
Discard the antigen solution and block the plate with PBSM (200 μl/well) for 1.5 hrs at room temperature.
Wash the plate 3 times with PBS and add polyclonal phages (109 ~ 1010 pfu/well) resuspended in PBSM for 1 hr.
Discard the solution. Wash the plate 3 times with PBST followed by 2 times with PBS.
Add biotinylated anti-fd bacteriophage antibody (100 μl/well) diluted at 1:1,000 in PBSM and incubate for 1 hr at room temperature.
Discard the primary antibody solution and wash wells 3 times with PBST followed by 2 times with PBS.
Add streptavidin-HRP (100 μl/well) diluted at 1:2,000 in PBSM and incubate for 30 min at room temperature.
Discard the solution and wash wells 3 times with PBST followed by 2 times with PBS.
Add 100 μl of TMB substrate solution into each well and incubate the plate for 1 ~ 10 minutes at room temperature.
Confirm the blue-colored reaction and quench by adding 100 μl/well of 1N HCl solution.
Measure the absorbance at OD450 nm using a plate reader and compare each binding activity of polyclonal phages rescued from unselected library-, 1st-, 2nd-, and 3rd- round outputs. Increasing signals are expected with progressing rounds of selection. Output from the 3rd round of selection is often used for screening of monoclonal phage antibody (described below).
To screen for monoclonal binding phage from polyclonal phage selection output showing positive ELISA signals, individual colonies are separately inoculated into a 96-well plate containing 150 μl/well of 2xYT-AG medium.
After overnight incubation, store the original plate in −80°C after mixing with 50 μl of 60% glycerol. Each bacteria culture (~20 μl) from each well of the original plate is inoculated in a new plate containing 150 μl/well of 2xYT-AG medium and incubated for ~ 2 hrs at 37°C.
Infect the bacteria with helper phages (~2 × 109 pfu/well) for 30 min at 37°C without shaking and an additional 30 min with shaking at 150 rpm.
Pellet the culture by centrifugation at 2,000 × g for 10 min at 4°C. Resuspend the culture with 150 μl/well of 2xYT-AK medium and incubate the plate overnight at 37°C in a shaker-incubator at 150 rpm.
Pellet the bacteria by centrifugation at 2,000 × g for 10 min at 4°C and transfer the supernatant into a new 96-well plate to store at 4°C until analysis.
Use 50 μl of each phage supernatant for ELISA screening using procedures described above in steps 2–12 above in this section. Positive clones are identified and subjected to flow cytometry analysis for binding to the target antigen expressed on the surface of a living cell.
3.5.3. Flow cytometry analysis of monoclonal phage
Refer to steps 13–17 in section 3.5.2 for monoclonal phage production. Use 50 μl of each phage supernatant to analyze binding by flow cytometry.
Prepare GFP- and antigen-expression plasmids for transient transfection into HEK293 cells. Seed HEK293 cells (~1 × 106 cells/well) in a 6-well plate on the day before transfection.
Mix 0.5 μg of GFP-expression plasmid, 1.5 μg of antigen-expression plasmid, and 7.5 μl of transfection reagent thoroughly and let the mixture for 15 minutes at room temperature.
Remove the growth medium from HEK293 culture and transfect the cells with the prepared DNA mixture.
Incubate the cells for 16 ~ 24 hrs, trypsinize and collect the cells by centrifugation at 1,000 × g for 5 min. Resuspend cell pellet with FCB at ~1 × 106 cells/ml.
Transfer 100 μl of cell suspension into a 96 well V-bottom plate. Add 50 μl of monoclonal phage supernatant and incubate for 1 hr at room temperature with shaking.
Centrifuge 96-well plate and wash once with PBS (200 μl/well). Discard PBS and resuspend the cell pellet with 100 μl biotinylated anti-fd bacteriophage antibody (3.5 μg/ml). Incubate the plate for 1 hr at room temperature.
Perform centrifugation and washing steps as above. Resuspend the cell pellet with 100 μl PE-conjugated streptavidin (2 μg/ml). Incubate the plate for 30 min at room temperature.
After washing the cells, resuspend the pellet thoroughly with 200 μl PBS. Determine cell-binding by flow cytometer by gating the GFP positive population and analyze mean fluorescence intensity in appropriate channel for PE. The non-transfected parental HEK293 is used as the negative control.
Acknowledgment
Work in our laboratory is supported by grants from the National Institutes of Health/National Cancer Institute (R01 CA171315, R01 CA118919, and R01 CA129491). NKL received fellowship support from Basic Science Research Program of the National Research Foundation of Korea (NRF) that is funded by the Ministry of Education, Science and Technology (2013R1A6A3A03060495).
Footnotes
Pooled cDNA from over 400 healthy donors was used in our library construction although there is no linear correlation between the pool size and the diversity of the eventual antibody library.
The amount of cDNA template required for optimal antibody gene PCR amplification should be determined by direct testing.
Because the purity of the PCR products is critical for downstream procedures including restriction enzyme digestion and ligation, we routinely incorporate additional column washing steps into the gel extraction protocol. After spinning the solubilized gel pieces through the nucleic acid purification column, we perform a column wash step with gel solubilization solution to remove any remaining gel fragments. We then wash the column twice with wash buffer, taking care to ensure that all residual wash buffer is removed from the column prior to elution.
Consider the desired final library size and prepare enough PCR product for subsequent steps. Approximately 0.5 μg of digested PCR product is needed for each ligation reaction and three ligation reactions are sufficient for 2–4 transformations. Although commercially available electro-competent TG1 is rated at 4×1010 – 1011 cfu/μg (presumably tested using purified plasmids), in our hands, the transformation efficiency of ligation products generally falls in the range of 0.5–2.0 × 108 cfu/transformation.
The pHEN1-NX phagemid has two stuffer sequences (stuffer 1 and 2) for VH and VL cloning. The stuffer 1 and 2 are sized at 198 bp and 150 bp, respectively. Digestion with NcoI-HF/NheI-HF or XbaI/NotI-HF releases the stuffer 1 or 2 from the vector. Cut out carefully the larger vector band from TAE agarose gel to avoid contamination of the stuffer fragments. Both stuffer fragments contain multiple stop codons. If either stuffer remains in the phagemid, no gene III fusion product will be made.
Use ddH2O pre-warmed at 70°C to efficiently elute the digested antibody gene fragments from the DNA purification column. Incubate the DNA column for 5 min at room temperature and spin for DNA elution.
High transformation efficiency by electroporation is facilitated by high DNA concentration in the concentrated and desalted ligation product (100–300 ng/μl). To generate enough ligation products for subsequent concentration and desalting steps, it is preferable to set up multiple small volume ligations (100 μl) rather than one large volume ligation. The ligation reactions can then be pooled and concentrated together.
Desalting and concentrating the ligation products are crucial for efficient electro-transformation. The purification of ligation products using this column-based method can improve the electro-transformation efficacy by desalting and concentrating at the same time. About 10–20 μl of concentrated ligation reaction should be isolated from this step. Check the DNA concentration to confirm that it is in the desired range (100–300 ng/μl).
Pre-made TG1 electro-competent cells have high electro-transformation efficiency (~4 × 1010 cfu/μg as claimed by the manufacturer, approximately 0.5 ~ 2.0 × 108 cfu/transformation in our hands, see Note 4). 50 μl of TG1 electrocompetent cells are suitable for two separate electroporations according to the manufacturer’s instruction.
The recovery medium provided with the electro-competent TG1 cells should be used for this step according to the manufacturer’s instruction.
To exclude sub-libraries with poor VL- or VH-insertion rate, only include potential sub-libraries qualified by colony PCR (more than 80% of full-length VL or VH inserts). To validate the scFv-phage library, analyze DNA sequences of single colonies randomly picked from each sub-library by using the ColLMB3F (forward) and Colfdseq1R (reverse) primers. Estimate the final complexity of the scFv-phage library by aligning all sequenced clones with germline sub-family sequences in antibody gene databases (e.g., VBASE2).
Theoretical diversity in variable heavy or light chains is determined by combinatorial rearrangements, junctional flexibility, addition of P-(palindromic) and N-(non-templated) nucleotides, and somatic hypermutation. Experimental data showed the estimated diversity of VL segments may be about 106 - 107 in a scFv library [31]. Thus Vκ or Vλ libraries that possess more than 108 diverse clones would be sufficient for scFv-library generation and affinity engineering by light chain-shuffling.
All materials used for VH gene cassette library generation are the same as listed in Subheading 2.2, except NcoI-HF and NheI-HF restriction enzymes. Because the quality of antibody library is mainly determined by the diversity of VH genes rather than that of VL genes, sufficient VH library size should be achieved for the high diversity of final libraries. Thus, allowing for further utilization of VH library (e.g., heavy chain shuffling, common light chain library generation..), the size of VH cassette library should reach at least 1~2 × 109 diversity.
An OD600 at 0.5 is equivalent to about 4.0 × 108 cells/ml. So the number of M13KO7 helper phage that should be added into the bacteria culture can be estimated as follows; 4.0 × 108 (cells/ml) × 500 (ml) × 20 (MOI) = 4.0 × 1012 pfu. The M13KO7 from the manufacture (Thermo Fisher Scientific) has a titer of 1 × 1011 pfu/ml. We routinely prepare a higher titer M13KO7 stock from infected TG1 to make the concentration at 1012 pfu/ml.
The phage titer rescued from 500 ml culture is generally expected at about 1012 cfu/ml or higher.
pFUSE-hIgG1-Fc2 plasmid encoding cDNA of target antigen can be transiently transfected into cell lines suitable for protein production, such as HEK-293A or CHO cell lines. Transient transfection is commonly performed using polyethylenimine (PEI), a stable cationic polymer. Transfected cells are maintained in serum-free media, and the media can be collected twice over a period of 1 ~ 2 weeks. Produced Fc-fusions are purified using a gravity column packed with Protein A beads and buffer-exchanged to PBS. EZ-Link™ Sulfo-NHS-Biotin was used to biotin-label purified Fc-fusion followed by neutralization with 1 M Tris-HCl and re-purification by gel filtration.
To control stringency during the selection rounds, the concentration of biotinylated Fc-fusion target antigen can be decreased as the selection proceeds (e.g., 5 nM for first round, 1 nM for second round, etc.).
To remove weak binders, washing steps with PBSMT can be increased in later selection rounds (e.g., 5 washes in the first round, 10 washes in the second round, etc.).
Exposure to the high pH TEA solution during elution can negatively affect the infectivity of eluted phage. Do not exceed 10 min during the elution step.
Total output of TG1 infected with phage eluent is generally between 105 ~ 107 cfu. It is generally recommended to perform a next round of selection with several hundred copies of each phage clone. Therefore, using a smaller volume (~ 50 ml) to inoculate the output bacteria from previous round is sufficient to amplify and prepare phages for the next round.
Reference:
- 1.Nelson AL (2010) Antibody fragments: hope and hype. MAbs. 2, 77–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chan CEZ, Lim APC, MacAry PA et al. (2014) The role of phage display in therapeutic antibody discovery. International Immunology. 26, 649–657. doi: 10.1093/intimm/dxu082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marks JD, Hoogenboom HR, Bonnert TP et al. (1991) By-passing immunization. Human antibodies from V-gene libraries displayed on phage. J Mol Biol. 222, 581–97. [DOI] [PubMed] [Google Scholar]
- 4.Clackson T, Hoogenboom HR, Griffiths AD et al. (1991) Making antibody fragments using phage display libraries. Nature. 352, 624–8. doi: 10.1038/352624a0 [DOI] [PubMed] [Google Scholar]
- 5.de Haard H.J. vNN, Reurs A, Hufton SE, Roovers RC, Henderikx P, de Bruine AP, Arends JW, Hoogenboom HR (1999) A large non-immunized human Fab fragment phage library that permits rapid isolation and kinetic analysis of high affinity antibodies. J. Biol. Chem. 274, 18218–18230. [DOI] [PubMed] [Google Scholar]
- 6.Barbas CF 3rd, Kang AS, Lerner RA et al. (1991) Assembly of combinatorial antibody libraries on phage surfaces: the gene III site. Proc Natl Acad Sci U S A. 88, 7978–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hawkins RE, Russell SJ, Winter G (1992) Selection of phage antibodies by binding affinity. Mimicking affinity maturation. J Mol Biol. 226, 889–96. [DOI] [PubMed] [Google Scholar]
- 8.Reiter Y, Schuck P, Boyd LF et al. (1999) An antibody single-domain phage display library of a native heavy chain variable region: isolation of functional single-domain VH molecules with a unique interface. J Mol Biol. 290, 685–98. doi: 10.1006/jmbi.1999.2923 [DOI] [PubMed] [Google Scholar]
- 9.Lorimer IA, Keppler-Hafkemeyer A, Beers RA et al. (1996) Recombinant immunotoxins specific for a mutant epidermal growth factor receptor: targeting with a single chain antibody variable domain isolated by phage display. Proc Natl Acad Sci U S A. 93, 14815–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dooley H, Flajnik MF, Porter AJ (2003) Selection and characterization of naturally occurring single-domain (IgNAR) antibody fragments from immunized sharks by phage display. Mol Immunol. 40, 25–33. [DOI] [PubMed] [Google Scholar]
- 11.Hairul Bahara NH, Chin ST, Choong YS et al. (2016) Construction of a Semisynthetic Human VH Single-Domain Antibody Library and Selection of Domain Antibodies against alpha-Crystalline of Mycobacterium tuberculosis. J Biomol Screen. 21, 35–43. doi: 10.1177/1087057115609144 [DOI] [PubMed] [Google Scholar]
- 12.Sanchez-Martin D, Sorensen MD, Lykkemark S et al. (2015) Selection strategies for anticancer antibody discovery: searching off the beaten path. Trends in Biotechnology. 33, 292–301. doi: 10.1016/j.tibtech.2015.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schwimmer LJ, Huang B, Giang H et al. (2013) Discovery of diverse and functional antibodies from large human repertoire antibody libraries. J Immunol Methods. 391, 60–71. doi: 10.1016/j.jim.2013.02.010 [DOI] [PubMed] [Google Scholar]
- 14.Liu B and Marks JD (2000) Applying phage antibodies to proteomics: selecting single chain Fv antibodies to antigens blotted on nitrocellulose. Anal Biochem. 286, 119–28. doi: 10.1006/abio.2000.4788 [DOI] [PubMed] [Google Scholar]
- 15.Liu B, Huang L, Sihlbom C et al. (2002) Towards proteome-wide production of monoclonal antibody by phage display. J Mol Biol. 315, 1063–73. doi: 10.1006/jmbi.2001.5276 [DOI] [PubMed] [Google Scholar]
- 16.Liu B, Conrad F, Cooperberg MR et al. (2004) Mapping tumor epitope space by direct selection of single-chain Fv antibody libraries on prostate cancer cells. Cancer Res. 64, 704–10. [DOI] [PubMed] [Google Scholar]
- 17.Sheets MD, Amersdorfer P, Finnern R et al. (1998) Efficient construction of a large nonimmune phage antibody library: the production of high-affinity human single-chain antibodies to protein antigens. Proc Natl Acad Sci U S A. 95, 6157–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ruan W, Sassoon A, An F et al. (2006) Identification of clinically significant tumor antigens by selecting phage antibody library on tumor cells in situ using laser capture microdissection. Mol Cell Proteomics. 5, 2364–73. doi: 10.1074/mcp.M600246-MCP200 [DOI] [PubMed] [Google Scholar]
- 19.An F, Drummond DC, Wilson S et al. (2008) Targeted drug delivery to mesothelioma cells using functionally selected internalizing human single-chain antibodies. Mol Cancer Ther. 7, 569–78. doi: 10.1158/1535-7163.MCT-07-2132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhu X, Bidlingmaier S, Hashizume R et al. (2010) Identification of internalizing human single-chain antibodies targeting brain tumor sphere cells. Mol Cancer Ther. 9, 2131–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Venet S, Kosco-Vilbois M, Fischer N (2013) Comparing CDRH3 diversity captured from secondary lymphoid organs for the generation of recombinant human antibodies. Mabs. 5, 690–698. doi: 10.4161/mabs.25592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yip YL, Hawkins NJ, Clark MA et al. (1997) Evaluation of different lymphoid tissue sources for the construction of human immunoglobulin gene libraries. Immunotechnology. 3, 195–203. [DOI] [PubMed] [Google Scholar]
- 23.Vaughan TJ, Williams AJ, Pritchard K et al. (1996) Human antibodies with sub-nanomolar affinities isolated from a large non-immunized phage display library. Nat Biotechnol. 14, 309–14. doi: 10.1038/nbt0396-309 [DOI] [PubMed] [Google Scholar]
- 24.Lennard S (2002) Standard protocols for the construction of scFv libraries. Methods Mol Biol. 178, 59–71. [DOI] [PubMed] [Google Scholar]
- 25.Hust M and Dubel S (2004) Mating antibody phage display with proteomics. Trends Biotechnol. 22, 8–14. doi: 10.1016/j.tibtech.2003.10.011 [DOI] [PubMed] [Google Scholar]
- 26.Welschof M, Terness P, Kipriyanov SM et al. (1997) The antigen-binding domain of a human IgG-anti-F(ab’)2 autoantibody. Proc Natl Acad Sci U S A. 94, 1902–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hust M, Frenzel A, Meyer T et al. (2012) Construction of human naive antibody gene libraries. Methods Mol Biol. 907, 85–107. doi: 10.1007/978-1-61779-974-7_5 [DOI] [PubMed] [Google Scholar]
- 28.Kugler J, Wilke S, Meier D et al. (2015) Generation and analysis of the improved human HAL9/10 antibody phage display libraries. BMC Biotechnol. 15, 10. doi: 10.1186/s12896-015-0125-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hoogenboom HR, Griffiths AD, Johnson KS et al. (1991) Multi-subunit proteins on the surface of filamentous phage: methodologies for displaying antibody (Fab) heavy and light chains. Nucleic Acids Res. 19, 4133–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rogosch T, Kerzel S, Hoi KH et al. (2012) Immunoglobulin analysis tool: a novel tool for the analysis of human and mouse heavy and light chain transcripts. Front Immunol. 3, 176. doi: 10.3389/fimmu.2012.00176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Glanville J, Zhai W, Berka J et al. (2009) Precise determination of the diversity of a combinatorial antibody library gives insight into the human immunoglobulin repertoire. Proc Natl Acad Sci U S A. 106, 20216–21. doi: 10.1073/pnas.0909775106 [DOI] [PMC free article] [PubMed] [Google Scholar]