

Structured Abstract:
Purpose:
Cohort-based germline variant characterization is the standard approach for pathogenic variant discovery in clinical and research samples. However, the impact of cohort size on the molecular diagnostic yield of joint genotyping is largely unknown.
Methods:
Head-to-head comparison of the molecular diagnostic yield of joint genotyping in two cohorts of 239 cancer patients in the absence and then in the presence of 100 additional germline exomes.
Results:
In 239 testicular cancer patients, four (7.4%, 95%CI:2.1–17.9) of 54 pathogenic variants in the cancer-predisposition and American College of Medical Genetics (ACMG) genes were missed by one or both computational runs of joint genotyping. Similarly, eight (12.1%, 95%CI:5.4–22.5) of 66 pathogenic variants in these genes were undetected by joint genotyping in another independent cohort of 239 breast cancer patients. An exome-wide analysis of putative loss-of-function (pLOF) variants showed that 162 (8.2%, 95%CI:7.1–9.6) pLOF variants were only detected in one analysis run but not the other, while 433 (22.0%, 95%CI:20.2–23.9%) pLOF variants were filtered out by both analyses despite having sufficient sequencing coverage.
Conclusion:
Our analysis of the standard germline variant detection method highlighted a substantial impact of concurrently analyzing additional genomic datasets on the ability to detect clinically relevant germline pathogenic variants.
Keywords: germline genetic analysis, variant calling, Mendelian pathogenic variants, Genome Analysis Toolkit (GATK)
Introduction:
Germline genetic profiling is ubiquitously used to guide molecular-based clinical diagnostic, prognostic, and therapeutic interventions [1]. It was estimated that over one million patients would undergo clinical germline genetic testing in 2019 in the US alone, one-third of which will be for cancer-related indications [2]. Since 2011, clinical and research-based germline variant detection have largely utilized the widely-adopted “Best Practices” of the Genome Analysis Toolkit Joint Genotyping (GATK-JG) [3] which leverages population-wide information from all analyzed samples and high-quality population-based datasets, such as the 1000 Genomes [4] and dbSNP [5], to determine the quality of each identified variant [6–9]. The GATK-JG “Best Practices” strongly recommends performing a cohort-based joint genotyping, with the expectation that the performance of this method is stable for cohorts larger than 30 exomes [10]. However, it is unknown if performing simultaneous germline variant detection of multiple cohorts affects the molecular diagnostic yield of germline variants in any particular sample set.
In this study, we hypothesized that the detection of rare clinically actionable germline alterations in any particular patient sample is sensitive to the genetic data of other germline samples that are being simultaneously analyzed by GATK-JG. To explore this hypothesis, we performed a head-to-head comparison of the germline variant callsets of 239 testicular cancer patients generated by running the standard germline pipeline method on these samples twice, first in the absence and then in the presence of 100 additional germline exome samples. We evaluated the quality score concordance and detection rate of clinically informative pathogenic and putative loss-of-function (pLOF) variants across several clinically relevant gene sets. We then replicated these findings in a similarly sized independent cohort of 239 breast cancer patients whose germline exome data were characterized in the presence and absence of an additional cohort of 100 germline exomes. Identical parameters were used across all analysis runs, and all downstream analyses were limited to germline variants detected in the original cancer cohort (i.e., all germline variants in the additional cohorts of 100 samples, used for joint genotyping, were excluded from all analyses).
Methods
Ethics Statement
Patients’ written informed consent and Institutional Review Board (IRB) approval were obtained by the original studies for all cohorts in this study.
Patient cohorts and genomic data collection
1-. Testicular cancer cohort (Discovery analysis)
Germline whole-exome sequencing (WES) data of 239 patients with testicular germ cell tumors were first used for the performance evaluation of the Genome Analysis Toolkit (GATK), the standard germline variant detection method [7–9,11] (Figure 1). These patients came from three independent cohorts: the Cancer Genome Atlas (TCGA; n=150), the Dana-Farber Cancer Institute (DFCI) TGCT cohort (n=49) [12,13], and the TGCT cohort described by Litchfield et al., 2015 of the United Kingdom (UK) Institute for Cancer Research (ICR) (n=40) [14]. To evaluate the effect of concurrently performing germline analysis on additional samples on the molecular diagnostic yield of GATK Joint Genotyping (GATK-JG), 100 high-quality germline WES samples of cancer-free patients from the Exome Sequencing Project (ESP) of the National Heart, Lung, and Blood Institute (NHLBI) were examined [9]. These samples were only used for the joint genotyping step of GATK. Germline variants detected in these cancer-free samples were entirely removed and were not included in any of the described analyses of this study (Figure 1).
Figure 1: Overview of the study design.
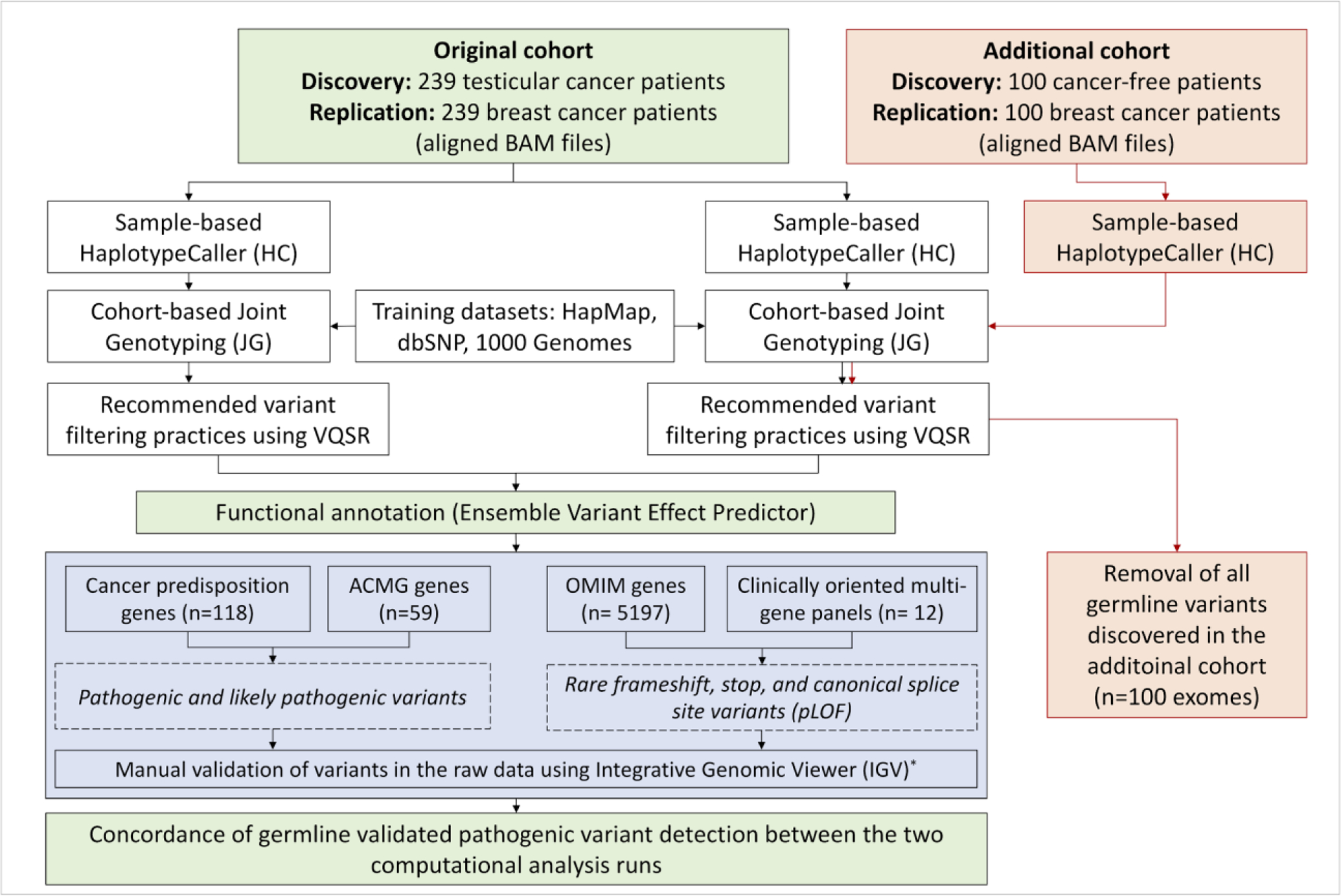
A head-to-head comparison was conducted to evaluate the molecular diagnostic yield of the Genome Analysis Toolkit Joint Genotyping (GATK-JG) based germline variant detection in two independent cohorts of 239 cancer patients in the presence and absence of an additional germline sample set of 100 germline exomes. (BAM: Binary Alignment Map, VQSR: Variant Quality Score Recalibration, ACMG: American College of Medical Genetics and Genomics, OMIM: Online Mendelian Inheritance in Men, pLOF: putative loss-of-function)
2-. Breast cancer cohort (Replication analysis)
To explore if the findings from the testicular cancer cohort analysis extend to other cancer datasets that were generated independently for a different cancer type, genomic data of 239 patients with breast cancer (infiltrating duct carcinoma) from The Cancer Genome Atlas (TCGA) were used to further evaluate the performance of GATK-JG. The GATK-JG pipeline was run on germline WES data of these 239 breast cancer patients twice, once in the presence and then in the absence of 100 additional TCGA breast cancer germline exomes. Similarly, the additional samples were only used in the joint genotyping step and were subsequently removed from all analyses.
Sequencing platform, capture kits, and alignment
1-. Testicular cancer cohort analysis
All sequencing data used in the testicular cancer cohort analysis, including the cancer-free cohort, was produced by a variety of Illumina platform machines (HiSeq2500, HiSeq 2000, and Genome AnalyzerIIx). The samples’ Binary Alignment Mapping (BAM) files comprising the four independent cohorts (TCGA, DFCI, ICR, and ESP) were all aligned to the hg19 reference genome using the Burrows-Wheeler Aligner (http://bio-bwa.sourceforge.net/). The exome capture kits utilized in the library preparation of these cohorts were NimbleGen SeqCap EZ Exome Library for the TCGA cohort, SureSelect Human All Exon v.2 Kit for the DFCI cohort, Nextera Rapid Capture Exome kits for the ICR cohort, and Agilent SureSelect Human All Exon 50 Mb for the ESP samples.
2-. Breast cancer cohort analysis
All sequencing data used in the breast cancer cohort analysis was produced using Illumina HiSeq and Illumina Genome Analyzer machines. The samples’ BAM files were aligned to the “hg19” reference genome using the Burrows-Wheeler Aligner. The exome capture kits utilized in the library preparation of these cohorts are the following: Nimblegen EZ Exome v3.0, Nimblegen SeqCap EZ Human Exome Library v2.0, Nimblegen SeqCap EZ Human Exome Library v3.0, and SureSelect Human All Exon 38 Mb v2. All samples included had a primary diagnosis of infiltrating duct carcinoma and were blood-derived germline samples.
Detection of germline variants
Genome Analysis Toolkit (GATK) HaplotypeCaller (HC) pipeline (version 3.7) was used to call germline variants according to the GATK Best Practices [11] (Figure 1). More specifically, we ran GATK HC on each sample individually to call single nucleotide variants (SNVs) and short indels via de-novo assembly of haplotypes of the examined regions. This per sample analysis generates an intermediate file called genomic variant calling format (gVCF) file that has a record for every position of the examined genomic intervals. We then aggregated the generated single-sample gVCFs and performed joint genotyping using GATK “GenotypeGVCFs” as recommended by the current germline variant calling “Best Practices” [11]. At each position of the input gVCFs, GATK “GenotypeGVCFs” module evaluates the genotype likelihood across all the samples and produce one quality score for each unique genomic alteration across the cohort (n=239 germline exomes (original cohort) for the first computational run and n=339 [239 original cohort exomes + 100 additional exomes] for the second computational run), which is then used by the GATK “Variant Quality Score Recalibration (VQSR)” module to perform variant filtering. To filter low-quality calls, VQSR uses highly validated variant callsets (such as dbSNP [5] and the 1000 Genomes [4]) to build a model that can then be applied to calculate the probability of each variant being real. As recommended by the GATK Best Practices, the SNVs VQSR model was trained using HapMap3.3 and 1KG Omni 2.5 SNP sites, and a 99.5% sensitivity threshold was applied to filter variants. In addition, Mills et al. 1KG gold standard and Axiom Exome Plus sites were used for VQSR indel recalibration using a 95% sensitivity threshold [15]. The assignment of quality class (high-quality vs. low-quality variants) was conducted by GATK-VQSR based on the variant’s Tranche and the defined sensitivity levels.GATK “SelectVariants” was used to remove germline variants detected in the additional cohort and keep germline variants only present in the original cohort (n=239). Specific commands and parameters used for the GATK pipeline are summarized in the Supplementary Note.
Selection of Mendelian gene sets
In this study, we analyzed pathogenic variants in 118 established germline cancer-predisposition genes and 59 Mendelian high-penetrance genes deemed clinically actionable by the American College of Medical Genetics (Collectively called the ACMG genes) (Table S1). Given that patients with cancer can also be carriers of disease-causing variants in autosomal recessive and low penetrant genes, we also characterized pLOF variants in 5197 clinically relevant genes in the Online Mendelian Inheritance in Men database (collectively called the OMIM genes) and 12 clinically oriented multi-gene panels (Supplementary Methods) (Tables S1 & S2)
Germline variant pathogenicity evaluation
All detected germline variants in the cancer-predisposition and ACMG gene sets were classified into five categories; benign, likely benign, variants of unknown significance, likely pathogenic and pathogenic using the ACMG guidelines [16]. Only pathogenic and likely pathogenic variants were included in this study (hereafter collectively referred to as pathogenic variants).
Validation of detected germline variants
Validation of the detected pathogenic variants in the cancer predisposition and ACMG gene sets was done in an independent blind fashion by two computational biologists using the “gold-standard” approach of evaluating the variants in the raw genomic data using the integrative Genomics Viewer (IGV) [17,18]. Variants that were called “True Positive” by both examiners were considered real variants. Otherwise, the variant was labeled as an artifactual call (Supplementary Methods).
Statistical Analysis
Two-sided binomial tests were used to calculate the 95% CI of proportions and p values of the likelihood of the filtered variants in both computational runs to be truly absent in a cohort of 239 ancestry matched individuals. P-values <0.05 were considered statistically significant. Bonferroni correction was used to correct for multiple testing when applicable. Statistical analyses were done using “exact2×2” (version 1.5.2), “binom” (version 1.1.1), and “stats” (version 3.5.1) packages on R (version 3.5.1).
Results:
Overall germline variant detection
Two independently sequenced cohorts of patients with testicular and breast cancer were included in this study. The exome-wide median sequencing depth of coverage for the testicular and breast cancer cohorts were 105.9X (IQR=84.8–124.8) and 109.7X (IQR=82.5–125.7) respectively. The mean depth of coverage for the cancer-predisposition, ACMG, and OMIM gene sets were 109.4X (IQR=97.2–124.3), 109.5X (IQR=96.5–124.0), and 106.4X (IQR=92.7–120.0), respectively, for the testicular cancer cohort and 112.5X (IQR=84.5–130.8), 107.8X (IQR=80.8–124.3), and 104.3X (IQR=78.4–121.0) respectively for the breast cancer cohort (Figure S1).
For the testicular cancer analysis, a total of 5,650,748 (99.1% SNVs and 0.9% INDELs) unfiltered rare and common germline variants were evaluated (Supplementary Methods). The variant Quality Tranche, a calibrated score that GATK-JG generates for each variant to represent the likelihood of it being a “true” variant, was concordant between the two analysis runs for only 84.79% (95% CI:84.76–84.82) of all variants while 15.21% (95% CI:15.18–15.24) variants had a different Quality Tranche assignment between the first and second analysis runs. As a result of this Quality Tranche assignment discrepancy, only 92.58% (95% CI:92.56–92.60) of the germline variants in the cancer cohort (n=239) were shared between the final variant callsets of both analysis runs while 134,847 (2.39%; 95%CI:2.37–2.40) variants were only detected in one analysis run (Figure 2A).
Figure 2: Exome-wide analysis of germline variant discovery in the presence and absence of additional genomics datasets.
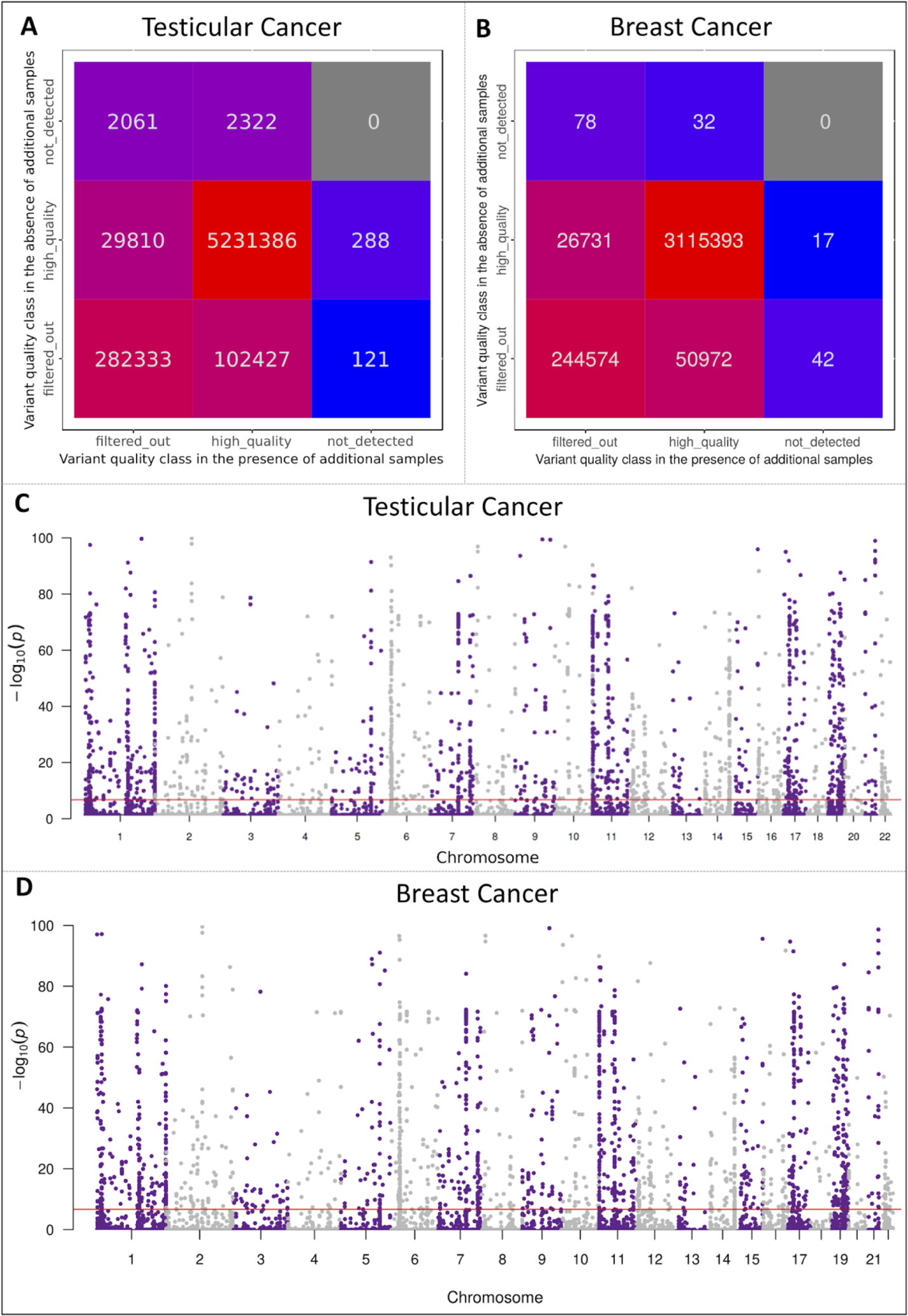
A and B; Confusion matrices of the final quality classification status of the germline variants detected in the testicular and breast cancer cohorts, respectively, between the first and second computational runs. C and D; Manhattan plots of the p-values for the germline variants, filtered by GATK-JG in both computational runs, to be absent by chance in a randomly selected 239 individuals from the European ancestry. A total of 184,827 variants had a p-value <1.76e-07 (depicted in 2C by the horizontal dotted red line) in the testicular cancer cohort and 116,078 variants had a p-value <2.04e-07 (depicted in 2D by the horizontal dotted red line) in the breast cancer cohort, suggesting a non-random underdetection effect of the GATK-JG for common variants across coding regions.
Similarly, a total of 3,437,839 (99.6% SNVs and 0.4% INDELs) unfiltered germline variants were present in the raw germline variant callset of 239 breast cancer patients. However, only 3,115,393 (90.62%; 95% CI:90.59–90.65) of these germline variants were found to be in common between the final variant callsets of both computational runs while 322,446 (9.37%; 95% CI:9.35–9.41) variants were undetected by one or both computational runs (Figure 2B), highlighting a non-trivial cohort size-driven discordance of the detected variant callset in the same patient cohort. The distribution of the population-based minor allele frequency of the detected germline variants can be found in Figure S2.
Characterization of filtered variants in well covered genomic regions
In the testicular cancer cohort, a total of 284,515 (5.03%; 95%CI:5.02–5.05) variants were considered “low quality” or computational artifacts and thus were filtered out by both analysis runs despite having a median sequencing depth of 75 reads (minimum 11 reads, interquartile range: 36–140) and a median variant allelic fraction (VAF) of 49.53%, which is consistent with the expected VAF of true germline variants. Leveraging known minor allele frequency of these variants in the Genome Aggregation Database (gnomAD) [19], we calculated the probability of variants filtered out in both analysis runs to be truly absent from a cohort of 239 randomly selected individuals (Supplementary Methods). Our analysis showed that 166,925 (58.7%; 95% CI:58.5–58.9) filtered variants were common enough in the general population, making it improbable for them to be truly absent in a randomly sampled cohort of this size (adjusted p-value<1.76e-07, Bonferroni correction for 284,515 variants) (Figure 2C).
Performing the same analysis on 239 breast cancer patients showed that of 244,694 germline variants that were filtered out by GATK-GJ in both computational runs, 116,078 (47.4%; 95%CI:47.2–47.6) variants were common enough in the general population, making it unlikely for these variants to be artifactual calls (adjusted p-value<2.04e-07, Bonferroni correction for 244,694 variants) and suggesting a systematic exome-wide variant underdetection of the standard pipeline (Figure 2D).
Impact of concurrently analyzing multiple cohorts on the detection of clinically actionable pathogenic variants
To further explore the impact of the cohort size on variant calling, we systematically characterized all clinically actionable pathogenic germline variants in 118 cancer predisposition genes as well as 59 genes deemed highly actionable by the American College of Medical Genetics (ACMG) (Table S1) in the testicular and breast cancer cohorts (n= 239 patients each). In total, 54 clinically actionable pathogenic variants were identified in the raw variant callset, the unfiltered variant calls from both computational runs, of 239 testicular cancer patients(Supplementary Methods). Of these variants, 50 (92.6%, 95%CI:82.1–97.9) pathogenic variants were detected in both computational runs while two (3.70%, 95%CI:0.5–12.7) pathogenic variants were only detected by GATK-JG when additional samples were used for joint germline variant calling (Figures 3A and 3B). These two variants include a known pathogenic founder frameshift variant in BRCA1 (c.5329dup, p.Gln1777ProfsTer74) (Figure 3C), which is a common high-penetrance cancer-risk variant in the Ashkenazi Jewish population [20], and a frameshift in LDLR gene (c.2397del, p.Val800SerfsTer129) that is associated with familial hypercholesterolemia (Figure 3D). Unexpectedly, our analysis also highlighted two (3.70%, 95%CI:0.5–12.7) known pathogenic cancer risk variants [21,22], a frameshift in BRCA2 (c.9063_9078del, p.Glu3021AspfsTer2) and splice donor site variant in SBDS (c.258+2T>C), that were filtered out by GATK-JG in both analysis runs despite having sufficient sequencing coverage (315 and 75 sequencing reads respectively) and a variant allelic fraction (VAF) supporting a germline heterozygous state (Figure 3E & 3F). In addition to validating these variants in their corresponding raw genomic data, we utilized GATK HaplotypeCaller-generated raw genomic files (BAM) to validate these variants after the tool assembled haplotypes and locally realigned reads (Figures S3A–D).
Figure 3: Detection of rare germline pathogenic in cancer patients using GATK-JG.
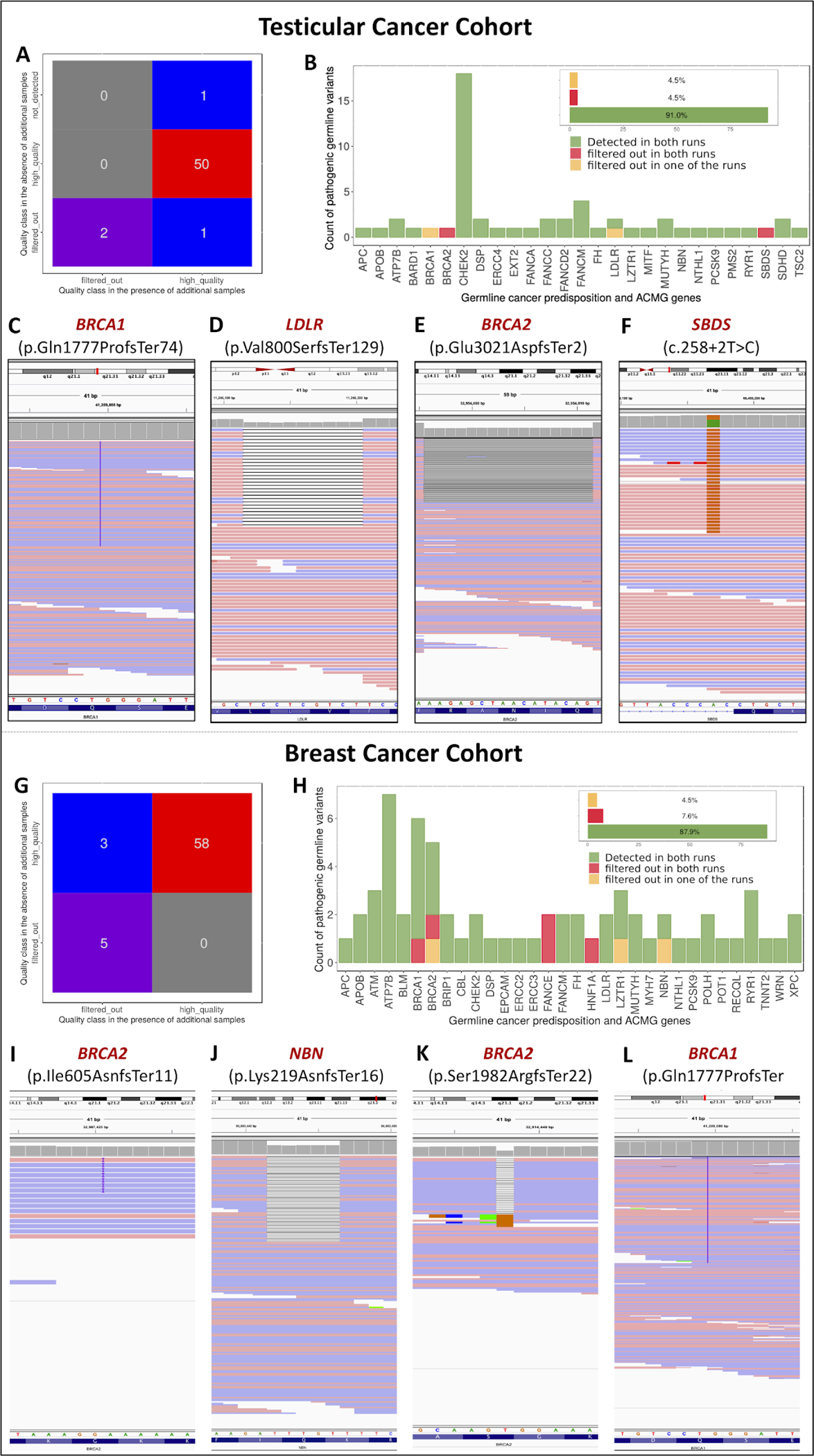
A; A confusion matrix of the quality class assignment of the pathogenic germline variants detected in 239 testicular cancer patients in the cancer-predisposition and ACMG gene sets (n=151) in the presence and absence of the additional cancer-free cohort. B; A total of 50 (92.6%) pathogenic variants were consistently detected by GATK-JG in the testicular cancer cohort (n=239) while 4 (7.4%) clinically actionable pathogenic variants were detected by GATK-JG in only one or none of the computational runs despite being present in the raw genomic data file (C-F), highlighting a substantial limitation of the current standard germline variant detection method. G-H; Conducting similar analyses on an independent cohort of 239 breast cancer patients showed that of 66 pathogenic variants in the raw variant callset, only 58 (87.9%, 95%CI:77.5–94.6) pathogenic variants were considered “high-quality” by GATK-JG while 8 (12.1%, 95%CI:5.4–22.5) variants went undetected by one or both computational runs. I-L; Representative example of pathogenic cancer-risk variants that went undetected by one or both of GATK-JG runs.
Similarly, our analysis of the germline WES data of 239 breast cancer patients identified 66 pathogenic variants in cancer predisposition and ACMG gene sets that were present in the unfiltered variant callset. However, only 58 (87.9%, 95%CI:77.5–94.6) of these pathogenic variants were considered “high-quality” by GATK-JG while 8 (12.1%, 95%CI:5.4–22.5) variants went undetected by one or both computational runs (Figures 3G & 3H). Germline variants that were only detected by one computational run included a well established pathogenic frameshift in BRCA2 (p.Ile605AsnfsTer11) (Figure 3I) and a known pathogenic variant in NBN (p.Lys219AsnfsTer16) that leads to premature termination and nonsense mediated decay of the protein transcript (Figure 3J). In addition, several pathogenic cancer-predisposition variants went undetected by both GATK-JG runs including a truncating pathogenic variant in BRCA2 (p.Ser1982ArgfsTer22) (Figure 3K) and a pathogenic founder frameshift variant in BRCA1 (p.Gln1777ProfsTer74) that is prevalent in Ashkenazi Jewish population [20] (Figure 3L), which was also seen in the testicular cancer cohort (Figure 3C).
Notably, germline pathogenic variants in the cancer-predisposition and ACMG gene sets that were missed by one or both computational runs in the testicular cancer cohort included one SNV and 3 indel variants while those pathogenic variants missed by one or both computational analyses in the breast cancer cohort included 3 SNVs and 5 indels.
Detection of pLOF variants in 5197 clinically relevant Mendelian genes
Next, we sought to assess the impact of concurrent genotyping of multiple cohorts on identifying autosomal recessive and low penetrant autosomal dominant putative loss-of-function (pLOF) variants across 5197 clinically relevant genes in our cancer cohorts (Supplementary Methods) (Table S1). Of 1964 rare pLOF variants in the raw variant callset in the testicular cancer cohort (n=239), only 69.7% (n=1369, 95%CI:67.7–71.7) variants were detected by both analysis runs while 8.2% (n=162, 95%CI:7.1–9.6) pLOF variants were only detected in one analysis run but not the other one (Figure 4A), demonstrating instability in GATK-JG performance for identifying rare truncating variants that are of potential clinical interest. Furthermore, 433 (22.0%, 95%CI:20.2–23.9) pLOF variants were considered low-quality variants or artifacts and were thus filtered out in both analyses despite having sufficient sequencing coverage (median:49 reads, interquartile range:18–78) and a VAF consistent with the germline heterozygous state (median:43%, interquartile range:35–57). To explore if germline variants that were filtered out in both analysis runs represent high-quality calls that were erroneously filtered out by GATK-JG, we randomly selected 100 variants for manual evaluation using the Integrative Genomic Viewer (IGV) (Supplementary Methods) [18]. Of these variants, 39% (95% CI:29.4–49.3) were validated in raw genomic data files including germline pLOF variants in MPO (p.Met519ProfsTer21) and LIPT1 (p.Lys123AsnfsTer8) (Figures 4B and 4C), suggesting a non-trivial false-negative rate (8.6%; 95% CI:7.4–9.9) of GATK-JG for rare germline pLOF variants that should be prioritized for further evaluation of pathogenicity and disease-association. In addition, we also went on to confirm these variants in the raw genomic files (BAM files) generated by GATK HaplotypeCaller (Figures S3E and S3F).
Figure 4: Detection of rare germline pLOF variants in cancer patients using GATK-GJ.
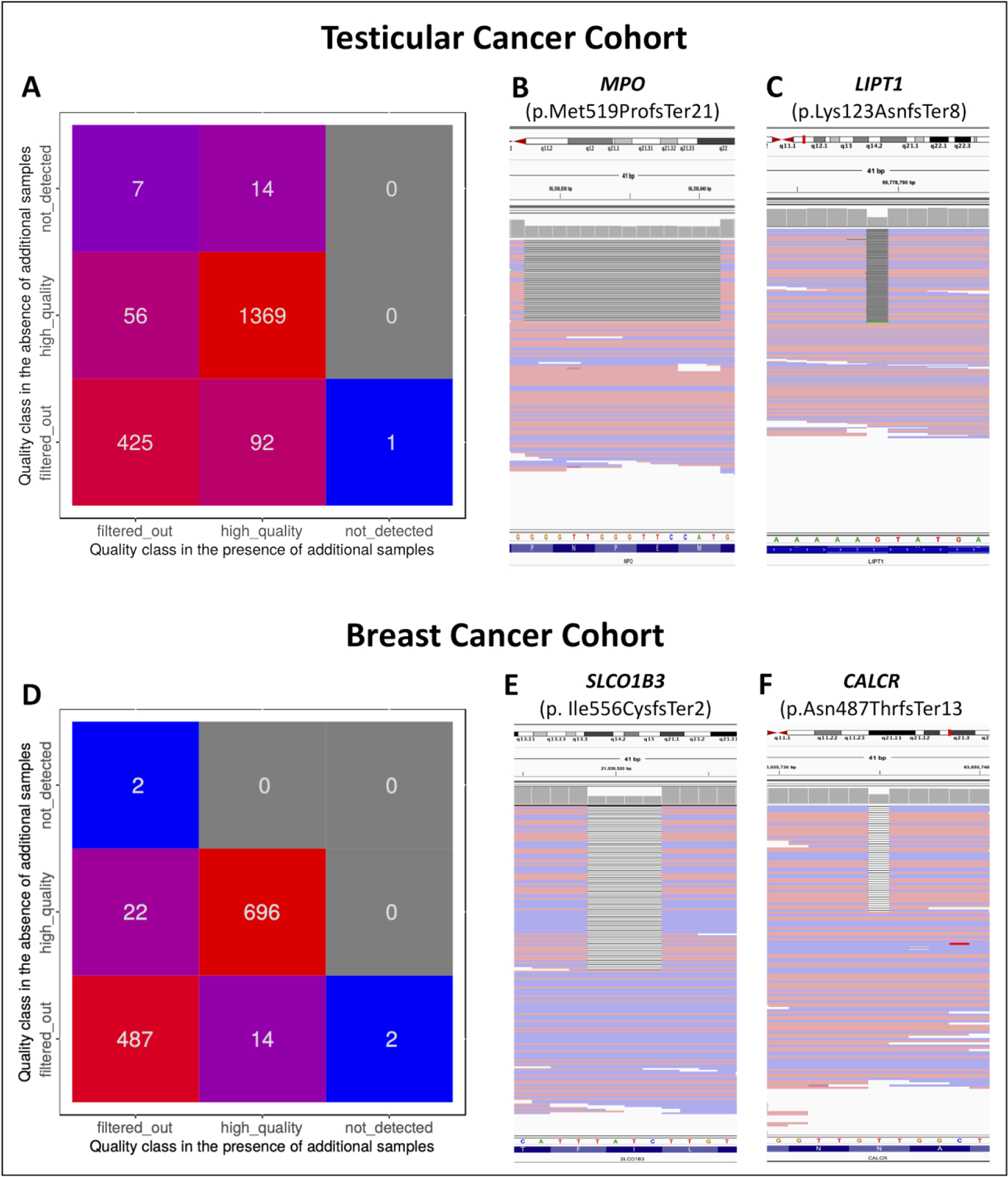
A; Evaluating rare germline truncating variants in clinically relevant genes (n=5197), detected by GATK-JG in the testicular cancer cohort (n=239) in the presence and absence of the 100 additional germline WES samples, showed a substantial discrepancy of the final germline callsets between the two computational runs. B & C; Two representative examples of pLOF variants that were filtered out by GATK-JG in both analysis runs (due to low GATK-generated Quality Tranches) but existed in the raw genomic data (Binary Alignment Map [BAM] file) of testicular cancer patients. The observed 14bp deletion in MPO (c.1555_1568del) is a known pathogenic variant that has been reported previously by clinical laboratories in several patients with myeloperoxidase deficiency (OMIM: 254600), an autosomal recessive condition associated with a higher risk of disseminated candidiasis. Similarly, LIPT1:c.369del is a known likely pathogenic variant that has been seen in patients with Lipoyltransferase 1 deficiency, another autosomal recessive condition associated with delayed psychomotor development, cerebellar atrophy, bradycardia, and liver dysfunction. D; Performing an exome-wide analysis of germline pLOF variants in an independently sequenced 239 breast cancer patients showed similarly substantial cohort size-driven variability in the ability to detect these potentially relevant germline alterations. E & F; Two representative examples of pLOF variants that were filtered out by GATK-JG in both computational runs but existed in the raw germline genomic data of breast cancer patients.
Using the same analysis approach, we systematically surveyed the pLOF variants in 5197 clinically relevant genes in the independently sequenced 239 germline exomes of breast cancer patients. Of 1223 pLOF variants that were discovered in this cohort, only 696 (56.9%; 95% CI:54.1–59.7) pLOF variants were detected in both computational runs while 36 (2.9%; 95% CI:2.1–4.1) pLOF variants were only detected in one of the analysis runs (Figure 4D). Similarly, a large fraction of the pLOF (n=491; 40.1%; 95% CI:37.4–43.0) variants in the breast cancer cohort were filtered out by both computational runs despite having a VAF suggestive of a germline heterozygous state (median:38%, interquartile range:33–46) and sufficient sequencing coverage (median:57 reads, interquartile range:26–127) (Figure 4E and 4F). To explore if some of these variants exist in the raw genomic data of the breast cancer cohort, we randomly selected 100 pLOF that were filtered out in both computational runs for manual evaluation. Again, our analysis showed that 50% (95% CI:39.8–60.2) of the manually evaluated pLOF variants were present in the raw genomic data of these patients, suggesting a missing rate of 24.1% (95% CI:16.1–33.7) for rare germline pLOF variants.
Similar to pathogenic variants in the cancer predisposition and ACMG gene sets, germline pLOF variants in the OMIM genes that were missed by one or both computational runs in the testicular cancer cohort included 132 (22.2%, 95%CI: 18.9–25.7) SNVs and 463 (77.8%, 95%CI: 74.3–81.1) indels while those pathogenic variants missed by one or both computational analyses in the breast cancer cohort included 315 (59.8%, 95%CI: 55.4–64.0) SNVs and 212 (40.2%, 95%CI: 36.0–44.6) indels.
Detection of pLOF variants in 12 commonly used clinical multi-gene panels
Lastly, we evaluated the effect of concurrently analyzing additional genomic datasets on the molecular diagnostic yield of 12 commonly used phenotype-specific multi-gene panels (MGPs) (Supplementary Methods) (Table S2). Overall, more rare pLOF variants were identified in the testicular cancer cohort when GATK-JG concurrently analyzed an additional set of 100 exomes compared with when GATK-JG was run on the original testicular cancer cohort (n=239) alone (9 MGPs, 75%, 95%CI:42.8–94.5 vs. 2 MGPs, 16.7%, 95%CI:2.1–48.4 respectively, with similar performance in one MGP, 8.3%, 95%CI:0.2–38.5) (Figure 5A). Notably, of the evaluated 1911 pLOF variants, 150 (7.8, 95%CI:6.7–9.1) pLOF variants were only identified in one of the analysis runs (median: 5 pLOF per gene panel, interquartile range:3–20) while 365 (19.1, 95%CI:17.4–20.9) pLOF variants were filtered out in both analysis runs (median: 15 pLOF per gene panel, interquartile range:10–28) (Figure 5A).
Figure 5: Performance of GATK-JG in detecting pathogenic and pLOF variants in 12 clinically oriented phenotype-specific multi-gene panels.
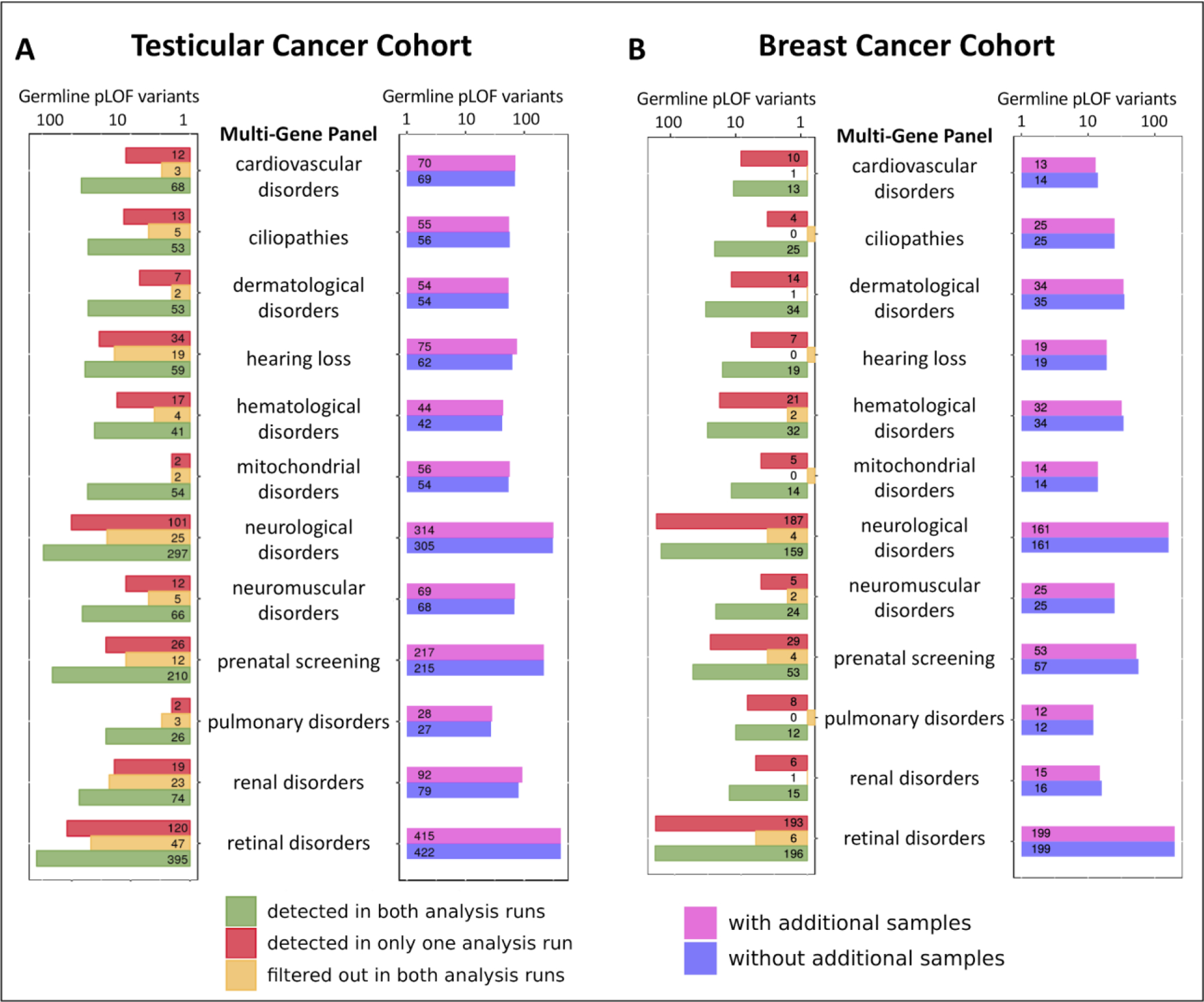
In the testicular cancer cohort (n=239), more pLOF variants were considered “high quality” in the presence of additional samples for GATK-JG (A). However, GATK-JG detected more pLOF variants in the analyzed MGPs in the breast cancer cohort (n=239) when the germline exomes of this cohort were analyzed in the absence of any other genomic dataset (B). Overall, these findings demonstrated significant variability of GATK-JG ability to detect pLOF variants in clinically relevant genes.
However, performing the same analysis on germline data of the breast cancer cohort (n=239) showed a clear tendency to detect more pLOF in the MGPs when this dataset is analyzed by GATK-JG in the absence of the additional 100 exome dataset (7 MGPs, 58.3%, 95%CI:27.7–84.8 with similar performance in 5 MGP, 41.7%, 95%CI:15.2–72.3) (Figure 5B), suggesting a stochastic nature of GATK-JG performance when additional genomic datasets are included. Lastly, similar to the testicular cancer analysis, 21 (1.9%; 95%CI:1.2–2.9) and 489 (44.2%; 95%CI:41.3–47.2) of 1106 pLOF variants present in the raw germline callset went undetected by one and both computational runs, respectively (Figure 5B).
Detection of germline genetic variants using 50 vs. 100 additional germline exomes
To investigate whether the observed higher detection rate of GATK-JG when concurrently analyzing additional samples has an additive effect, we compared the number of high quality heterozygous germline variants detected in the breast cancer cohort (n=239) when no additional samples, 50 additional germline samples, and 100 additional germline samples were used for joint genotyping (Supplementary Methods). Our analysis showed that although 67,326 additional heterozygous germline variants were detected in this cohort when concurrently analyzed with 100 additional germline exomes compared with when no additional cohort is used (3,873,154 vs 3,805,828 respectively), analyzing germline data of 239 breast cancer patients with 50 additional germline exomes unexpectedly detected 107,058 fewer high quality heterozygous variants than when no additional samples were concurrently characterized (3,698,770 vs 3,805,828 respectively) (Figure S4A). Importantly, this variability of the number of idnetified germline variants in the breast cancer cohort was seen across all autosomal and sex chromosomes (Figure S4B & S4C), highlighting a systematic exome-wide stochastic effect that does not seem to be limited to particular genes or genomic regions.
Discussion:
Collectively, our analysis of GATK-JG, the standard germline variant detection method commonly used for clinical and research studies, highlighted a substantial impact of concurrently analyzing additional genomic datasets on the detection of rare and common germline variants in any particular sample. In the testicular cancer cohort, additional rare pathogenic and pLOF germline variants were detected in the analyzed 239 germline exomes of these patients when additional genomic datasets were included in the “Joint Genotyping” step. However, analyzing an independent cohort of 239 patients with breast cancer showed that GATK-JG detected more pathogenic and pLOF variants when this patient cohort was genotyped without any additional genomic dataset, suggesting a stochastic nature of GATK-JG sensitivity when additional datasets are concurrently analyzed. This stochastic nature of GATK-JG performance was also seen when exploring the effect of performing germline variant detection in the presence of an additional cohort of different sizes, where while using 100 additional exomes resulted in detecting more high-quality variants than baseline (i.e., when no additional samples are used), using 50 additional exomes resulted in detecting fewer high-quality germline variants, resulting in a lower detection rate than baseline.
Collectively, our analysis of two independent cohorts of cancer patients suggests that GATK-JG’s ability to detect rare pathogenic and pLOF variants in any particular germline sample is significantly influenced by the number of samples that are being concurrently analyzed, resulting in substantially variable sensitivity and detection rate for these clinically informative variants. Such variable performance can result in missing clinically-actionable pathogenic variants in a non-trivial fraction of patients who undergo clinical germline genetic testing. Indeed, our analysis of the cancer predisposition and ACMG gene sets showed that four of 239 (1.67%, 95%CI:0.46–4.23) testicular cancer patients and eight of 239 (3.35%, 95%CI:1.46–6.49) breast cancer patients had clinically actionable pathogenic variants that went undetected in one or both computational analyses. Furthermore, this variable performance, along with the arbitrary user-defined filter cutoffs that GATK-JG uses, can greatly limit the ability to reproduce large germline analyses even when the raw genomic data are accessible. Such issues can be potentially mitigated by adopting a sample-based analysis approach that leverages deep learning and other related algorithms which have shown promising results for superior variant detection performance in “The Genome in a Bottle” ground-truth set [23,24]. However, until sample-based deep learning approaches are fully adopted, detection of rare clinically relevant germline variants using GATK should utilize internal or publically available genomic datasets that may improve the molecular diagnostic yield of joint genotyping-based variant detection.
Supplementary Material
Acknowledgments
We thank all individuals who participated in this study. Drs. AlDubayan and Van Allen had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. This work was supported by American Society of Clinical Oncology (ASCO) Conquer Cancer Foundation Career Development Award (CCF CDA) -CDA#13167 (S.H.A.), the Prostate Cancer Foundation Young Investigator Award -YIA#18YOUN02 (S.H.A), the PCF-V Foundation Challenge Award (E.M.V.), the National Institutes of Health R37CA222574 (E.M.V.), R01 CA227388 (E.M.V.), and King Abdulaziz City for Science and Technology grant #12-MED2226-46 (M.A.). The funding organizations were not responsible for the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication. The results published here are in part based upon data generated by The Cancer Genome Atlas managed by the NCI and NHGRI. Information about TCGA can be found at http://cancergenome.nih.gov.
Competing interests
Dr. Van Allen has the following disclosures; advisory and/or consulting for Tango Therapeutics, Genome Medical, Invitae, Illumina, and Ervaxx; research support from Novartis and BMS; equity in Tango Therapeutics, Genome Medical, Syapse, Ervaxx, and Microsoft; travel reimbursement from Roche and Genentech; and institutional patents (ERCC2 mutations and chemotherapy response, chromatin mutations and immunotherapy response, and methods for clinical interpretation). Other authors reported no conflicts of interest.
Footnotes
Ethics approval and consent to participate
All individuals in this study consented to institutional review board-approved protocols that allowed for comprehensive genetic analysis of germline samples (methods). This study conforms to the Declaration of Helsinki.
Data and software availability
The raw sequence data for all cohorts utilized in this study can be obtained through dbGaP (https://www.ncbi.nlm.nih.gov/gap) or as described in their original papers (See methods). All software tools used in this study are publicly available.
References:
- 1.AlDubayan SH. Leveraging Clinical Tumor-Profiling Programs to Achieve Comprehensive Germline-Inclusive Precision Cancer Medicine. JCO Precision Oncology. 2019; 1–3. [DOI] [PubMed] [Google Scholar]
- 2.Bergin J DNA Sequencing Market: Size, Trends, Share & Research Report 2023. [cited 25 Nov 2019]. Available: https://www.bccresearch.com/market-research/biotechnology/dna-sequencing-emerging-tech-applications-report.html
- 3.Best Practices for Variant Calling GATK. Github; Available: https://github.com/broadinstitute/gatk-docs [Google Scholar]
- 4.1000 Genomes Project Consortium, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, et al. A global reference for human genetic variation. Nature. 2015;526: 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sherry ST, Ward M, Sirotkin K. dbSNP—database for single nucleotide polymorphisms and other classes of minor genetic variation. Genome Res. 1999. Available: http://genome.cshlp.org/content/9/8/677.short [PubMed] [Google Scholar]
- 6.DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43: 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bohannan ZS, Mitrofanova A. Calling Variants in the Clinic: Informed Variant Calling Decisions Based on Biological, Clinical, and Laboratory Variables. Comput Struct Biotechnol J. 2019;17: 561–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536: 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tennessen JA, Bigham AW, O’Connor TD, Fu W, Kenny EE, Gravel S, et al. Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science. 2012;337: 64–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Geraldine_VdAuwera. Calling variants on cohorts of samples using the HaplotypeCaller in GVCF mode. In: GATK-Forum; [Internet]. 6 March 2014. [cited 30 Mar 2020]. Available: https://gatkforums.broadinstitute.org/gatk/discussion/3893/calling-variants-on-cohorts-of-samples-using-the-haplotypecaller-in-gvcf-mode [Google Scholar]
- 11.der Auwera GAV, Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, del Angel G, et al. From FastQ Data to High-Confidence Variant Calls: The Genome Analysis Toolkit Best Practices Pipeline. Current Protocols in Bioinformatics. 2013. pp. 11.10.1–11.10.33. doi: 10.1002/0471250953.bi1110s43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Taylor-Weiner A, Zack T, O’Donnell E, Guerriero JL, Bernard B, Reddy A, et al. Genomic evolution and chemoresistance in germ-cell tumours. Nature. 2016;540: 114–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.AlDubayan SH, Pyle LC, Gamulin M, Kulis T, Moore ND, Taylor-Weiner A, et al. Association of Inherited Pathogenic Variants in Checkpoint Kinase 2 (CHEK2) With Susceptibility to Testicular Germ Cell Tumors. JAMA Oncol. 2019;5: 514–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Litchfield K, Summersgill B, Yost S, Sultana R, Labreche K, Dudakia D, et al. Whole-exome sequencing reveals the mutational spectrum of testicular germ cell tumours. Nat Commun. 2015;6: 5973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mills RE, Pittard WS, Mullaney JM, Farooq U, Creasy TH, Mahurkar AA, et al. Natural genetic variation caused by small insertions and deletions in the human genome. Genome Research. 2011. pp. 830–839. doi: 10.1101/gr.115907.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Richards S, ; on behalf of the ACMG Laboratory Quality Assurance Committee, Aziz N, Bale S, Bick D, Das S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine. 2015. pp. 405–423. doi: 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Robinson JT, Thorvaldsdóttir H, Wenger AM, Zehir A, Mesirov JP. Variant Review with the Integrative Genomics Viewer. Cancer Res. 2017;77: e31–e34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, et al. Integrative genomics viewer. Nat Biotechnol. 2011;29: 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581: 434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Abeliovich D, Kaduri L, Lerer I, Weinberg N, Amir G, Sagi M, et al. The founder mutations 185delAG and 5382insC in BRCA1 and 6174delT in BRCA2 appear in 60% of ovarian cancer and 30% of early-onset breast cancer patients among Ashkenazi women. Am J Hum Genet. 1997;60: 505–514. [PMC free article] [PubMed] [Google Scholar]
- 21.VCV000052738.1 - ClinVar - NCBI [cited 8 Nov 2019]. Available: https://www.ncbi.nlm.nih.gov/clinvar/variation/52738/
- 22.VCV000003196.6 - ClinVar - NCBI [cited 8 Nov 2019]. Available: https://www.ncbi.nlm.nih.gov/clinvar/variation/3196/
- 23.Poplin R, Chang P-C, Alexander D, Schwartz S, Colthurst T, Ku A, et al. A universal SNP and small-indel variant caller using deep neural networks. Nat Biotechnol. 2018;36: 983–987. [DOI] [PubMed] [Google Scholar]
- 24.Train a CNN model for filtering variants. In: GATK-CNNVariantTrain; [Internet]. [cited 8 Oct 2019]. Available: https://software.broadinstitute.org/gatk/documentation/tooldocs/4.0.4.0/org_broadinstitute_hellbender_tools_walkers_vqsr_CNNVariantTrain.php [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw sequence data for all cohorts utilized in this study can be obtained through dbGaP (https://www.ncbi.nlm.nih.gov/gap) or as described in their original papers (See methods). All software tools used in this study are publicly available.