

Abstract
The direct position-selective C–4 alkylation of pyridines has been a longstanding challenge in heterocyclic chemistry, particularly from pyridine itself. Historically this has been addressed using pre-functionalized materials to avoid overalkylation and mixtures of regioisomers. This study reports the invention of a simple maleate-derived blocking group for pyridines that enables exquisite control for Minisci-type decarboxylative alkylation at C–4 that allows for inexpensive access to these valuable building blocks. The method is employed on a variety of different pyridines and carboxylic acid alkyl donors, is operationally simple, scalable, and is applied to access known structures in a rapid and inexpensive fashion. Finally, this work points to an interesting strategic departure for the use of Minisci chemistry at the earliest possible stage (native pyridine) rather than current dogma that almost exclusively employs Minisci chemistry as a late-stage functionalization technique.
Graphical Abstract
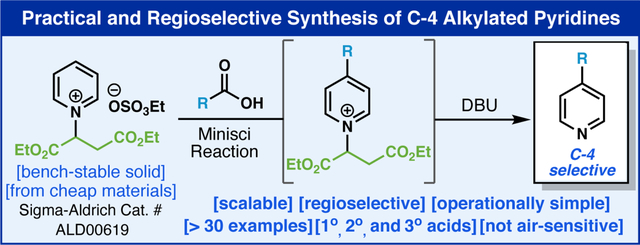
The power of C–H functionalization logic in the context of synthesizing heteroaromatic structures is undeniable.1 Its increasing utility in discovery and medicinal chemistry contexts is a testament to its utility in late-stage derivatization enabling structure activity relationships to be rapidly explored.2 In particular, the venerable Minisci reaction and its many variants have long been recognized as a way to bypass pre-functionalized heterocycles at the carbon.3 Just as the Friedel-Crafts reaction is commonplace for electron rich arene functionalization via electrophilic substitution, free radicals can react with electron deficient heterocycles by capitalizing on innate reactivity.4 In cases where a heterocycle has multiple sites to intercept a free radical, mixtures often result which can be useful in a discovery setting but is problematic when a singular regiochemical outcome is desired (e.g. process scale).5 For example, simple 4-alkylated pyridines (1, Figure 1A) are inaccessible using Minisci chemistry if a single regioisomer is desired. In such cases, C–4 prefunctionalization is necessary and the logical synthon is halopyridine 2. This conundrum has rendered the early-stage application of Minisci chemistry on pyridine and mono-substituted pyridines rare in medicinal chemistry and, to our knowledge, non-existent on process scale. A recent collaborative program6 within the agrochemical industry brought to our attention the need for a simple and inexpensive solution to this unmet challenge in pyridine alkylation for which available methods were not applicable. To be sure, several attempts to solve this problem from starting materials unfunctionalized at the carbon have appeared over the past decade (Figure 1B) mostly based on blocking competitive C–2 sites using transient or covalently linked species at the pyridine nitrogen.7 Nakao’s pioneering studies using bulky Al-based Lewis-acids in an elegant hydroarylation process is limited to olefin donors and must be performed in a glove box.8 The Fier group at Merck invented clever oxime–based pyridinium species that could be employed in three examples of C–C bond formation with carbon-based nucleophiles.9 Finally, the Hong group reported a radical-type addition using N-sulfonamidopyridinium species10 using alkyl bromide donors requiring photochemical initiation and super stoichiometric amounts of an expensive silane [(TMS)3SiH].11 While this is an important precedent it could not be employed easily on process scale as three steps are needed to install the blocking group (1. N-amination using hydroxylamine-O-sulfonic acid, 2. tosylation, and 3. methylation with Meerwein’s salt along with 1 column purification, 1 recrystallization). The Buchwald group reported a Cu-catalyzed, selective C–4 functionalization of pyridine with styrenes without a covalent blocking group, via a novel intramolecular rearrangement mechanism mediated by a pyridine coordinated copper species.7e,7h Herein we disclose a highly practical method featuring on a new blocking group based on a simple fumarate backbone (6a) enabling classic Minisci decarboxylative alkylations to take place with exquisite selectivity at C–4 (Figure 1C) under acid-free conditions. Emblematic of this advance is the preparation of 4-cyclohexylpyridine (8). Subjecting pyridine under four different Minisci-type conditions, the product cannot be accessed in a synthetically useful yield, and a mixture of isomers was observed.
Figure 1.
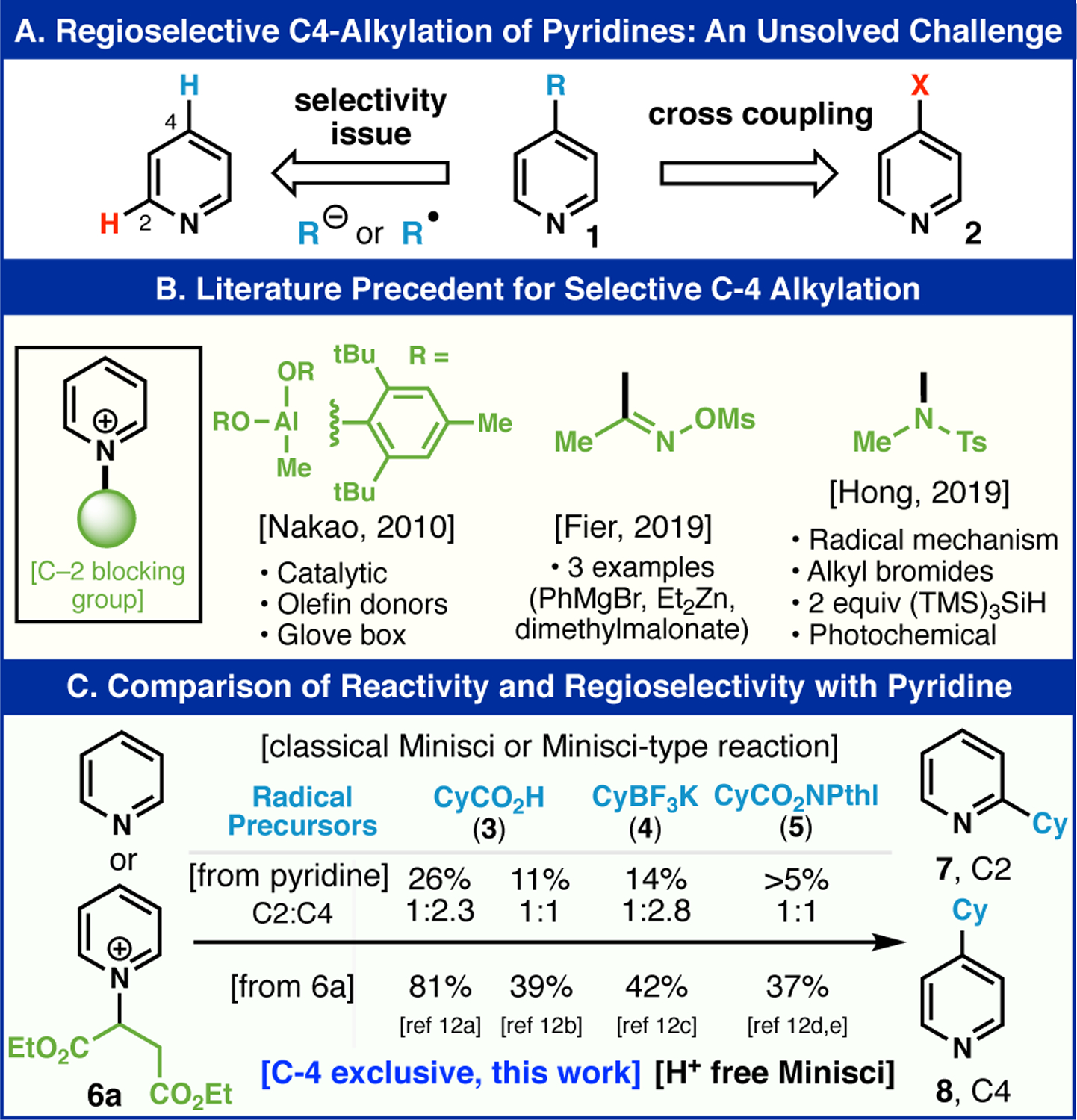
(A) An unsolved challenge in the field of Minisci reaction. (B) Literature precedent. (C) Comparison experiment with pyridine
Tactically, scalable access to valuable structures such as 8 (81% isolated yield from 6a) can now be enabled with a dramatic reduction in cost. From a strategic perspective, this work opens a new dimension of retrosynthetic logic for use of the Minisci transform at an early rather than late stage.
Guided by colleague at Syngenta (E.G.) several criteria needed to be met for a practical blocking group (BG) design, such as: (1) derivation from feedstock materials (ca. $5/mole), (2) simple installation and removal, (3) high stability, ease of handling, and solubility in multiple solvents, and most critically, (4) complete regionchemical control to avoid the need for any chromatography. Towards this end, multiple BGs were explored with most falling into one of two categories (Figure 2A): (1) simple BG installation with either modest or low reactivity under Minisci conditions (BG1, 2, 4, 6) or (2) difficulty in forming a stable BG adduct. After extensive exploration, BG10 emerged as an ideal candidate satisfying all of the criteria laid out above. BG9 was the only other moderately successful one however it exhibited reduced reactivity towards Minisci addition. The preparation of pyridinium 6a with BG10 could be prepared through a simple, chromatography-free, two-step sequence starting from commodity materials (pyridine and maleic acid) followed by esterification. The structure of pyridinium 6a was confirmed by X-ray crystallography and contained the ethyl sulfate as a counter-anion. This crystalline salt represents a straightforward gateway to a variety of C–4 alkylated pyridines (vide infra) and has been commercialized by Sigma-Aldrich (catalog # ALD00619).
Figure 2.
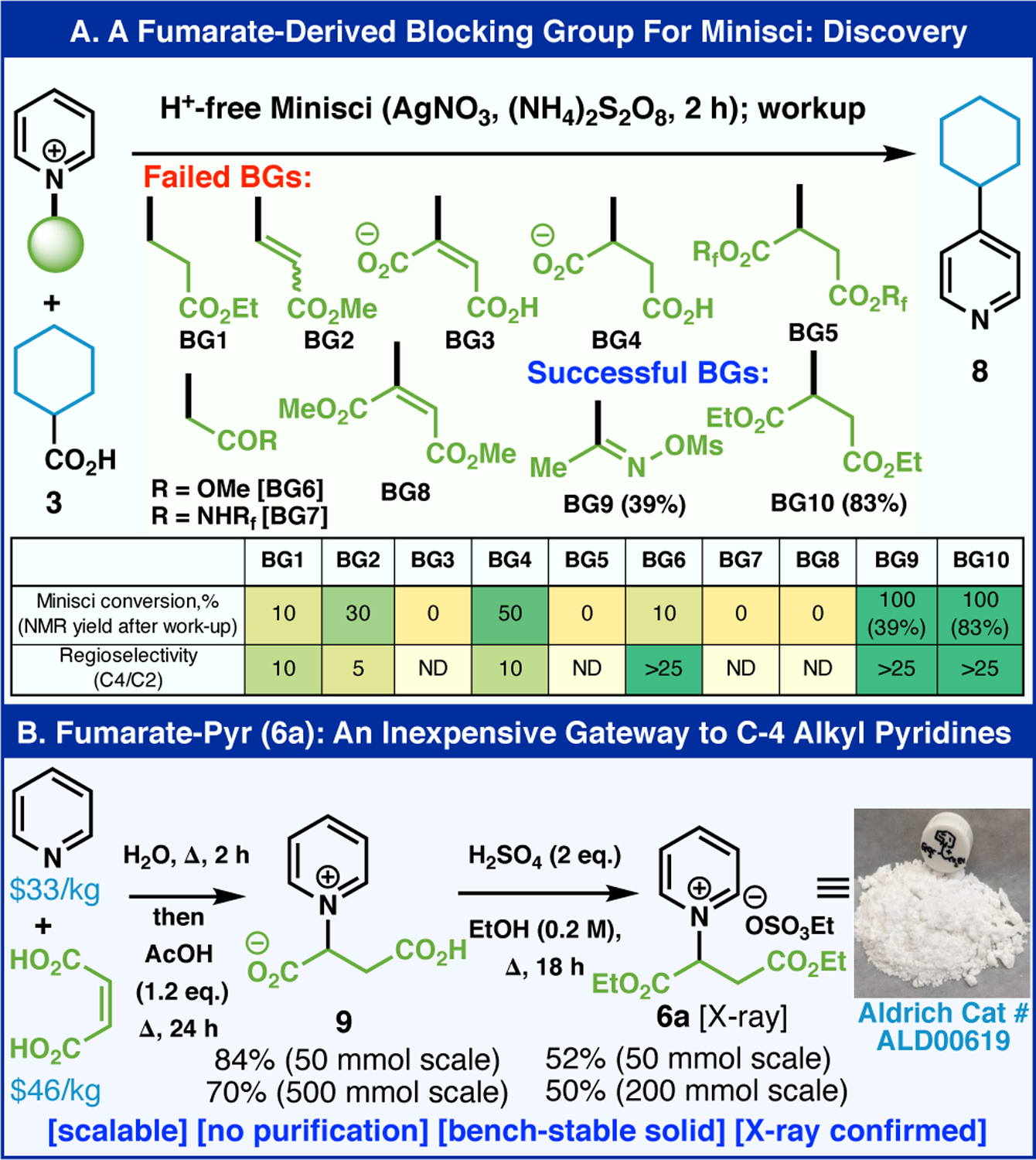
(A) Fumarate-derived blocking group for Minisci reaction in discovery stage. (B) The pyridinium 6a as an inexpensive gateway to C4 alkylated pyridine synthesis.
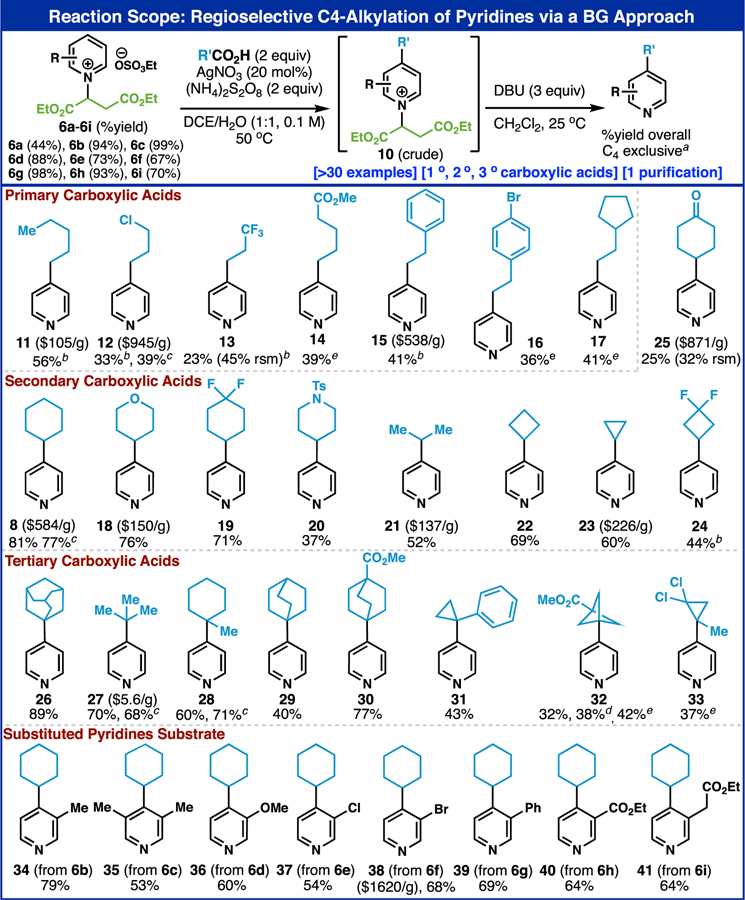
The generalization of this fumarate-based BG approach is illustrated in Table 1 using acid-free Minisci conditions on a range of primary, secondary, and tertiary carboxylic acids. Although these C–4 alkylated pyridines appear simple, it is instructive to comment on the means by which such compounds were previously prepared. In nearly all cases the current method represents a more practical and cost-effective solution. In the case of primary carboxylic acid adducts, pyridine 11 was accessed from C–4 pre-functionalized 4-methylpyridine via lithiation-SN2 with corresponding alkyl bromide (ca. $105/g13b).14 Pyridine 12 was obtained through an analogous sequence using an alkyl bromide containing a protected alcohol requiring subsequent deprotection and chlorination (ca. $945/g13b).15 Pyridine 13, 14 and 17 were previously prepared via photochemical addition on 4-vinyl pyridine.16 Pyridine 15 required a Pd-cross coupling on either 4-vinyl or 4-bromopyridine (Heck17a or Sonogashira,17b respectively) followed by reduction (ca. $530/g13c). Similarly, pyridine 16 can be accessed via reduction of the Heck product of 4-vinylpyridine and an aryl iodide.18
Table 1.
Reaction scope: regioselective C–4 alkylation of pyridines via a BG approach. Yields of 6b–6l are based on isolated yield from substituted pyiridines (two steps). a) 6a (0.5 mmol), carboxylic acid (1.0 mmol), AgNO3 (20 mol%), (NH4)2S2O8 (1.0 mmol), DCE: H2O=1:1, 0.1 M, 50 °C, 2 h. The regioselectivity was determined by crude NMR after first step and confirmed again after final purification step. b) using carboxylic acid (2.0 mmol, 4 equiv) on the Minisci reaction step and DBU (3.0 mmol, 6 equiv) on the removal step. c) 5.0 mmol scale reaction. d) carboxylic acid was used as a limiting reagent. e) performed in 0.3 M. See Supporting Information for detailed experimental procedures.
|
Numerous secondary carboxylic acids were employed to access such pyridines with high simplicity when placed in context. For example, pyridine 8 has been prepared multiple times leading either to mixtures (e.g., Figure 2B) or requiring C–4 pre-functionalized pyridines (ca. $584/g13d).19 Similarly, pyridine 18 has been accessed from 4-bromopyridine through photochemical and electrochemical reductive couplings or by employing Hong’s BG (Figure 1B) and a Hantzsch ester radical precursor10b (ca. $150/g13e). Pyridine 21 has been accessed either via cross coupling/Hydrogenation20a or C4-selective Grignard addition using TBSOTf to generate a transient BG and reoxidation20b (ca. $100/g13b). Cyclopropyl containing pyridine 23 was accessed either from 4-bromo or 4-Bpin pyridine via Suzuki or Grignard addition/rearomatization (ca. $226/g13a).21 The trivial cyclohexanone pyridine 25 has only been accessed in a controlled fashion using multistep routes with protecting groups and FG manipulations (ca. $871/g13f).22
Many of the quaternary center containing C-4 alkylated pyridines (derived from tertiary carboxylic acids) prepared here are new (29-33) and are likely desirable starting materials for medicinal chemistry programs. Of the known alkylated pyridines in this series, two were prepared as mixtures of regioisomers using radical chemistry (26 and 28)23,24 or via Minisci addition to 4-cyanopyridine.25
The chemistry outlined above is not limited to the parent pyridine 6a but can also be employed on mono (6b, 6d-i) or bis (6c) substituted pyridines. Pyridines 35, 39, and 41 are new compounds and might be challenging to access controllably from the parent pyridines in other ways. Pyridines such as 37, 38 (ca. $1620/g13g), and 40 have previously been synthesized either through Grignard addition/oxidation sequences7c or via Hong’s HAT-based method10d employing BG’s similar to that in Figure 1B.
It is worth noting that pyridines 8, 12, 27 and 28 have been prepared on a gram-scale with no significant reduction in yield. The limitations of this reaction (see SI for full disclosure) stem from the acidic conditions used to install the BG and a lack of tolerance for preexisting C–2 functionality.
Interestingly, when 6a was used in borono-Minisci reaction involving aryl boronic acids as radical precursor,28 lower regioselectivity was observed, leading to a mixture of C-2 and C-4 adducts. This outcome can be rationalized with the compact geometry of the Csp2 radical, which allows the attack on the hindered C-2 position of 6a (see SI for accurate description of the results).
Having facile access to pure C–4 alkylated pyridines open up a new opportunity for early-stage Minisci chemistry to be employed in the synthesis of 2,4-difunctionalized systems. Historically, such heterocycles are prepared by employing Minisci at the end of a sequence in order to obtain more regioselective outcomes. As shown in Figure 3A, a reversal of this traditional choreography is now feasible. Thus, adduct 8 can be submitted to known C–2 selective pyridine functionalizations such as carbamoylation26, cyanation9, and amidation27 to afford pyridines 42, 43, and 44, respectively. This sequence of events is general and can be utilized to obtain 46 (Minisci followed by borono-Minisci28), 47 and 48 (double Minisci), and 49 (Minisci followed by amidation). Conventional retrosynthesis of such compounds would likely involve pre-functionalized handles at the targeted carbon for controlling regiochemistry whereas in the present case innate reactivity and the fumarate-BG overcomes this challenge.
Figure 3.
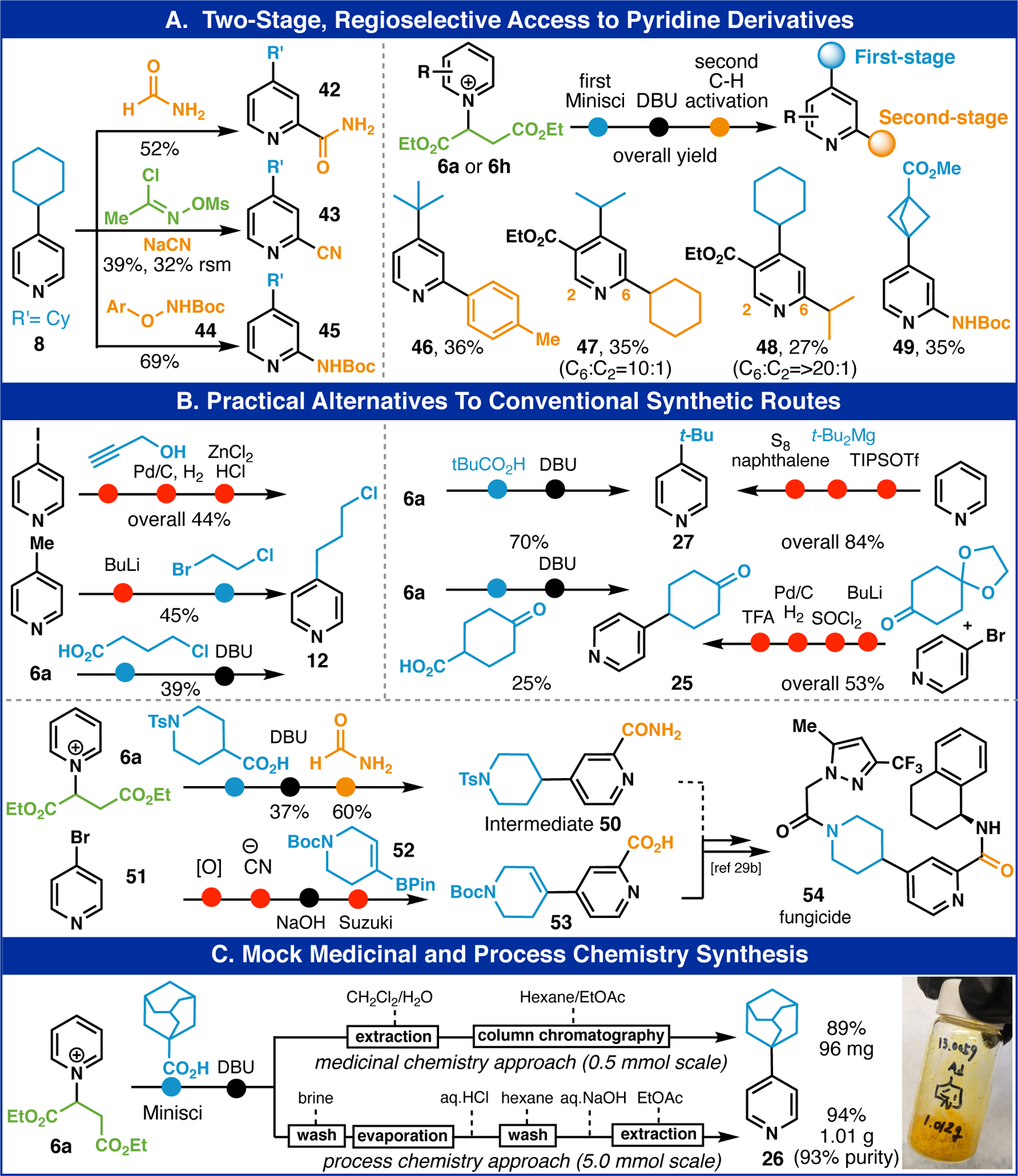
(A) Two-stage derivatization. (B) Practical alternatives to conventional synthetic routes. (C) Mock medicinal and process chemistry synthesis. See Supporting Information for detailed experimental procedures.
As mentioned above (Table 1), many of the pyridines reported herein have been prepared by less direct pathways and this is graphically depicted for pyridines 12, 27, and 25 in Figure 3B. The avoidance of C–4 pre-functionalized pyridines, pyrophoric reagents, and expensive transition metals are highlights of this method. Moreover, a proof of concept is shown for how the fungicide (oomycetes) candidate 54 could conceivably be accessed in a far more practical way from pyridine. Prior studies employed chemistry that was costprohibitive for the agrochemical industry commencing from 51 and employing expensive boronate ester 52, N-oxide chemistry, toxic TMSCN, and a Pd catalyst to access 1,3-disubstituted 53 which required a subsequent hydrogenation to remove the 3,4-unsaturation.29 In contrast, the two-stage Minisci approach from 6a accesses a synthetically equivalent intermediate 50 directly without any of those drawbacks. Finally, as a demonstration of practicality in both medicinal and process scenarios pyridine 26 can be prepared and purified either through column chromatography or through a simple extraction/washing protocol (Figure 3C).
To summarize, a simple solution to the longstanding challenge of practical C–4 alkylation of pyridines has been presented using a simple blocking group derived from inexpensive maleic acid. The resulting pyridinium species is stable and, in many instances, crystalline. The resulting functionalization can be accomplished using classic Minisci conditions without the addition of any acid and proceeds to give a singular adduct at C–4. The scope of this reaction is broad and can be strategically used in concert with other functionalizations or as a stand-alone method to provide high value pyridines that despite their trivial appearance, have posed challenges for direct and inexpensive synthesis in a scalable way.
Supplementary Material
ACKNOWLEDGMENT
Financial support for this work was provided the NIH (GM-118176) and Syngenta Crop Protection. We are thankful to the Vivozon Inc. (Young Scientist Grant and Postdoctoral Fellowship, J.C.) and the George E. Hewitt Foundation (G.L.). We are grateful to Dr. D.-H. Huang and Dr. L. Pasternack (Scripps Research) for NMR spectroscopic assistance, to Dr. J. Chen, B. Sanchez, E. Sturgell (Scripps Research Automated Synthesis Facility), Dr. G. Siuzdak and E. Billings (Scripps Research, The Center of Metabolomics and Mass Spectrometry) for assistance with HRMS, to Dr. M. Gembicky (UCSD) for X-ray crystallographic analysis.
Footnotes
The authors declare no competing financial interest.
REFERENCES
- 1).(a) Yu J-Q; Shi Z, CH activation Springer: 2010; Vol. 292. [Google Scholar]; (b) Brückl T; Baxter RD; Ishihara Y; Baran PS, Innate and Guided C–H Functionalization Logic. Acc. Chem. Res 2012, 45, 826–839. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Murakami K; Yamada S; Kaneda T; Itami K, C–H Functionalization of Azines. Chem. Rev 2017, 117, 9302–9332. [DOI] [PubMed] [Google Scholar]
- 2).(a) Brow DG; Bostrom J, Analysis of Past and Present Synthetic Methodologies on Medicinal Chemistry: Where Have All the New Reactions Gone? J. Med. Chem 2016, 59, 4443–4458. [DOI] [PubMed] [Google Scholar]; (b) Cernak T; Dykstra KD; Tyagarajan S; Vachal P; Krska SW, The Medicinal Chemist’s Toolbox for Late-stage Functionalization of Drug-like Molecules. Chem. Soc. Rev 2016, 45, 546–576. [DOI] [PubMed] [Google Scholar]; (c) Bostrom J; Brown DG; Young RJ; Keseru GM, Expanding the Medicinal Chemistry Synthetic Toolbox. Nat. Rev. Drug. Discov 2018, 17, 709–727. [DOI] [PubMed] [Google Scholar]
- 3).(a) For reviews of Minisci reaction see;Minisci F; Galli R; Cecere M; Malatesta V; Caronna T, Nucleophilic Character of Alkyl Radicals: New Syntheses by Alkyl Radicals Generated in Redox Processes. Tetrahedron Lett 1968, 9, 5609–5612. [Google Scholar]; (b) Duncton MAJ, Minisci reactions: Versatile CH-functionalizations for Medicinal Chemists. Med. Chem. Comm 2011, 2, 1135–1161. [Google Scholar]; (c) Proctor RSJ; Phipps RJ, Recent Advances in Minisci-Type Reactions. Angew. Chem. Int. Ed 2019, 58, 13666–13699. [DOI] [PubMed] [Google Scholar]; (d) Wang WG; Wang SF, Recent Advances in Minisci-type Reactions and Applications in Organic Synthesis. Cur. Org. Chem 2021, 25, 894–934. [Google Scholar]
- 4).Ishihara Y; Montero A; Baran PS The Portable Chemist’s Consultant: A Survival Guide for Discovery, Process, and Radiolabeling Macintosh Publishing, 2013. (electronic book) https://books.apple.com/us/book/the-portable-chemists-consultant/id618463142 (accessed May 3, 2021). [Google Scholar]
- 5).O’Hara F; Blackmond DG; Baran PS, Radical-Based Regioselective C–H Functionalization of Electron-Deficient Heteroarenes: Scope, Tunability, and Predictability. J. Am. Chem. Soc 2013, 135, 12122–12134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6).Michaudel Q; Ishihara Y; Baran PS, Academia–Industry Symbiosis in Organic Chemistry. Acc. Chem. Res 2015, 48, 712–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7).(a) Corey EJ; Tian Y, Selective 4-Arylation of Pyridines by a Nonmetalloorganic Process. Org. Lett 2005, 7, 5535–5537. [DOI] [PubMed] [Google Scholar]; (b) Tsai CC; Shih WC; Fang CH; Li CY; Ong TG; Yap GPA, Bimetallic Nickel Aluminun Mediated Para-Selective Alkenylation of Pyridine: Direct Observation of η2, η1-Pyridine Ni(0)-Al(III) Intermediates Prior to C-H Bond Activation. J. Am. Chem. Soc 2010, 132, 11887–11889. [DOI] [PubMed] [Google Scholar]; (c) Chen Q; du Jourdin XM; Knochel P, Transition-Metal-Free BF3-Mediated Regioselective Direct Alkylation and Arylation of Functionalized Pyridines Using Grignard or Organozinc Reagents. J. Am. Chem. Soc 2013, 135, 4958–4961. [DOI] [PubMed] [Google Scholar]; (d) Ma X; Dang H; Rose JA; Rablen P; Herzon SB, Hydroheteroarylation of Unactivated Alkenes Using N-Methoxyheteroarenium Salts. J. Am. Chem. Soc 2017, 139, 5998–6007. [DOI] [PubMed] [Google Scholar]; (e) Gribble MW; Guo S; Buchwald SL, Asymmetric Cu-Catalyzed 1,4-Dearomatization of Pyridines and Pyridazines without Preactivation of the Heterocycle or Nucleophile. J. Am. Chem. Soc 2018, 140, 5057–5060. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Zhang W-B; Yang X-T; Ma J-B; Su Z-M; Shi S-L, Regioand Enantioselective C–H Cyclization of Pyridines with Alkenes Enabled by a Nickel/N-Heterocyclic Carbene Catalysis. J. Am. Chem. Soc 2019, 141, 5628–5634. [DOI] [PubMed] [Google Scholar]; (g) Wang Y; Li R; Guan W; Li Y; Li X; Yin J; Zhang G; Zhang Q; Xiong T; Zhang Q, Organoborohydride-catalyzed Chichibabin-type C4-position alkylation of pyridines with alkenes assisted by organoboranes. Chem. Sci 2020, 11, 11554–11561. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Gribble MW; Liu RY; Buchwald SL, Evidence for Simultaneous Dearomatization of Two Aromatic Rings under Mild Conditions in Cu(I)-Catalyzed Direct Asymmetric Dearomatization of Pyridine. J. Am. Chem. Soc 2020, 142, 11252–11269. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Obradors C; List B, Azine Activation via Silylium Catalysis. J. Am. Chem. Soc 2021, 143, 6817–6822. For representative review, see: [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Bull JA; Mousseau JJ; Pelletier G; Charette AB, Synthesis of Pyridine and Dihydropyridine Derivatives by Regio- and Stereoselective Addition to N-Activated Pyridines. Chem. Rev 2012, 112, 2642–2713. [DOI] [PubMed] [Google Scholar]; (k) Rossler SL; Jelier BJ; Magnier E; Dagousset G; Carreira EM; Togni A, Pyridinium Salts as Redox-Active Functional Group Transfer Reagents. Angew. Chem. Int. Ed 2020, 59, 9264–9280. [DOI] [PubMed] [Google Scholar]; (l) Zhou F-Y; Jiao L, Recent Developments in Transition-Metal-Free Functionalization and Derivatization Reactions of Pyridines. Synlett 2021, 32, 159–178. [Google Scholar]
- 8).Nakao Y; Yamada Y; Kashihara N; Hiyama T, Selective C-4 Alkylation of Pyridine by Nickel/Lewis Acid Catalysis. J. Am. Chem. Soc 2010, 132, 13666–13668. [DOI] [PubMed] [Google Scholar]
- 9).Fier PS, A Bifunctional Reagent Designed for the Mild, Nucleophilic Functionalization of Pyridines. J. Am. Chem. Soc 2017, 139, 9499–9502. [DOI] [PubMed] [Google Scholar]
- 10).(a) Moon Y; Park B; Kim I; Kang G; Shin S; Kang D; Baik MH; Hong S, Visible Light Induced Alkene Aminopyridylation using N-aminopyridinium Salts as Bifunctional Reagents. Nat. Commun 2019, 10, 4117. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kim I; Park S; Hong S, Functionalization of Pyridinium Derivatives with 1,4-Dihydropyridines Enabled by Photoinduced Charge Transfer. Org. Lett 2020, 22, 8730–8734. [DOI] [PubMed] [Google Scholar]; (c) Shin S; Lee S; Choi W; Kim N; Hong S, Visible-Light-Induced 1,3-Aminopyridylation of [1.1.1] Propellane with N-Aminopyridinium Salts. Angew. Chem. Int. Ed 2021, 60, 7873–7879. [DOI] [PubMed] [Google Scholar]; (d) Lee W; Jung S; Kim M; Hong S, Site-Selective Direct C-H Pyridylation of Unactivated Alkanes by Triplet Excited Anthraquinone. J. Am. Chem. Soc 2021, 143, 3003–3012. [DOI] [PubMed] [Google Scholar]; (e) Kim M; You E; Park S; Hong S, Divergent Reactivity of Sulfinates with Pyridinium Salts Based on One- versus Two-electron Pathways. Chem. Sci, 2021, 12, 6629–6637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11).Jung S; Shin S; Park S; Hong S, Visible-Light-Driven C4-Selective Alkylation of Pyridinium Derivatives with Alkyl Bromides. J. Am. Chem. Soc 2020, 142, 11370–11375. [DOI] [PubMed] [Google Scholar]
- 12).(a) Minisci F; Bernardi R; Bertini F; Galli R; Perchinummo M, Nucleophilic character of alkyl radicals—VI: A New Convenient Selective Alkylation of Heteroaromatic Bases. Tetrahedron 1971, 27, 3575–3579. [Google Scholar]; (b) Galloway JD; Mai DN; Baxter RD, Silver-Catalyzed Minisci Reactions Using Selectfluor as a Mild Oxidant. Org. Lett 2017, 19, 5772–5775. [DOI] [PubMed] [Google Scholar]; (c) Presset M; Fleury-Brégeot N; Oehlrich D; Rombouts F; Molander GA, Synthesis and Minisci Reactions of Organotrifluoroborato Building Blocks. J. Org. Chem 2013, 78, 4615–4619. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Liu Y; Xue L; Shi B; Bu F; Wang D; Lu L; Shi R; Lei A, Catalyst-free electrochemical decarboxylative cross-coupling of N-hydroxyphthalimide esters and N-heteroarenes towards C(sp3)–C(sp2) bond formation. Chem. Commun 2019, 55, 14922–14925. [DOI] [PubMed] [Google Scholar]; (e) Niu K; Song L; Hao Y; Liu Y; Wang Q, Electrochemical decarboxylative C3 alkylation of quinoxalin-2(1H)-ones with N-hydroxyphthalimide esters. Chem. Commun 2020, 56, 11673–11676. [DOI] [PubMed] [Google Scholar]
- 13).(a) Reagent price based on. Sigma-Aldrich.; (b) Combi-Blocks.; (c) BLD Pharm.; (d) Enamine.; (e) Oakwood Chemical.; (f) AstaTech.; (g) Aurum Pharmtech (accessed April 8th, 2021)
- 14).Howell JM; Feng K; Clark JR; Trzepkowski LJ; White MC, Remote Oxidation of Aliphatic C–H Bonds in Nitrogen-Containing Molecules. J. Am. Chem. Soc 2015, 137, 14590–14593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15).(a) Kassiou M; Read RW; Shi X-Q, Synthesis and Evaluation of Halogenated Dibenzodiazepines as Muscarinic Receptor Ligands. Bioorg. Med. Chem. Lett 1997, 7, 799–804. [Google Scholar]; (b) Hoang V-H; Tran P-T; Cui M; Ngo VTH; Ann J; Park J; Lee J; Choi K; Cho H; Kim H; Ha H-J; Hong H-S; Choi S; Kim Y-H; Lee J, Discovery of Potent Human Glutaminyl Cyclase Inhibitors as Anti-Alzheimer’s Agents Based on Rational Design. J. Med. Chem 2017, 60, 2573–2590. [DOI] [PubMed] [Google Scholar]
- 16).(a) Straathof NJW; Cramer SE; Hessel V; Noel T, Practical Photocatalytic Trifluoromethylation and Hydrotrifluoromethylation of Styrenes in Batch and Flow. Angew. Chem. Int. Ed 2016, 55, 15549–15553. [DOI] [PubMed] [Google Scholar]; (b) Wang YW; Deng LF; Zhang X; Mou ZD; Niu DW, A Radical Approach to Making Unnatural Amino Acids: Conversion of C-S Bonds in Cysteine Derivatives into C-C bonds. Angew. Chem. Int. Ed 2021, 60, 2155–2159. [DOI] [PubMed] [Google Scholar]; (c) Quan Y; Song Y; Shi W; Xu Z; Chen JS; Jiang X; Wang C; Lin W, Metal–Organic Framework with Dual Active Sites in Engineered Mesopores for Bioinspired Synergistic Catalysis. J. Am. Chem. Soc 2020, 142, 8602–8607. [DOI] [PubMed] [Google Scholar]
- 17).(a) Efange SMN; Michelson RH; Remmel RP; Boudreau RJ; Dutta AK; Freshler A, Flexible N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine Analog: Synthesis and Monoamine Oxidase Catalyzed Bioactivation. J. Med. Chem 1990, 33, 3133–3138. [DOI] [PubMed] [Google Scholar]; (b) Takhi M; Hosahalli S; Panigrahi SK; Mahadari MK; Kottam SR; Abd Rahman N; Yusof R Substituted Pyridine Derivatives as FABI Inhibitors, PCT WO 2013 80222 A1 June 6, 2013. [Google Scholar]
- 18).Kalashnikov VV; Tomilova LG, Catalytic Reduction of an α,β-Disubstituted Alkene with Sodium Borohydride in the Presence of Tetra-tert-butylphthalocyanine Complexes. Mendeleev Commun 2007, 17, 343–344. [Google Scholar]
- 19).(a) Minisci F; Fontana F, Mechanism of the Gif-Barton Type Alkane Functionalization by Halide and Pseudohalide Ions. Tetrahedron Lett 1994, 35, 1427–1430. [Google Scholar]; (b) For selected examples from pyridines prefunctionalized at the carbon see: Molander GA; Argintaru OA; Aron I; Dreher SD, Nickel-Catalyzed Cross-Coupling of Potassium Aryl- and Heteroaryltrifluoroborates with Unactivated Alkyl Halides. Org. Lett 2010, 12, 5783–5785. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Basch CH; Liao J; Xu J; Piane JJ; Watson MP, Harnessing Alkyl Amines as Electrophiles for Nickel-Catalyzed Cross Couplings via C–N Bond Activation. J. Am. Chem. Soc 2017, 139, 5313–5316. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Perry IB; Brewer TF; Sarver PJ; Schultz DM; DiRocco DA; MacMillan DWC, Direct Arylation of Strong Aliphatic C–H bonds. Nature 2018, 560, 70–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20).(a) Friedfeld MR; Margulieux GW; Schaefer BA; Chirik PJ, Bis(phosphine)cobalt Dialkyl Complexes for Directed Catalytic Alkene Hydrogenation. J. Am. Chem. Soc 2014, 136, 13178–13181. [DOI] [PubMed] [Google Scholar]; (b) Akiba K; Iseki Y; Wada M, A Convenient Method for the Regioselective Synthesis of 4-Alkyl(aryl)pyridines using Pyridinium Salts. Bull. Chem. Soc. Jpn 1984, 57, 1994–1999. [Google Scholar]
- 21).(a) Lewis RT; Jones P; Petrocchi A; Reyna N; Hamilton M; Cross J; Tremblay M; Leonard PG Compounds, PCT WO 2018 136887 July 26, 2018. [Google Scholar]; (b) Panda S; Coffin A; Nguyen QN; Tantillo DJ; Ready JM, Synthesis and Utility of Dihydropyridine Boronic Es ters. Angew. Chem. Int. Ed 2016, 55, 2205–2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22).Zhou J; Jiang Q; Fu P; Liu S; Zhang S; Xu S; Zhang Q, Syntheses of 4-(Heteroaryl)cyclohexanones via Palladium-Catalyzed Ester α-Arylation and Decarboxylation. J. Org. Chem 2017, 82, 9851–9858. and reference therein. [DOI] [PubMed] [Google Scholar]
- 23).(a) Barton DHR; Bévière SD; Chavasiri W, The Functionalization of Saturated Hydrocarbons. Part 25. Ionic Substitution Reactions in GoAggIV Chemistry: the Formation of Carbon- halogen Bonds. Tetrahedron 1994, 50, 31–46. [Google Scholar]; (b) Barniol-Xicota M; Gazzarrini S; Torres E; Hu Y; Wang J; Naesens L; Moroni A; Vázquez S, Slow but Steady Wins the Race: Dissimilarities among New Dual Inhibitors of the Wild-Type and the V27A Mutant M2 Channels of Influenza A Virus. J. Med. Chem 2017, 60, 3727–3738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24).Pitre SP; Muuronen M; Fishman DA; Overman LE, Tertiary Alcohols as Radical Precursors for the Introduction of Tertiary Substituents into Heteroarenes. ACS Catal 2019, 9, 3413–3418. [Google Scholar]
- 25).Gao L; Wang G; Cao J; Chen H; Gu Y; Liu X; Cheng X; Ma J; Li S, Lewis Acid-Catalyzed Selective Reductive Decarboxylative Pyridylation of N-Hydroxyphthalimide Esters: Synthesis of Congested Pyridine-Substituted Quaternary Carbons. ACS Catal 2019, 9, 10142–10151. [Google Scholar]
- 26).Han W; Jin F; Zhao Q; Du H; Yao L, Acid-Free Silver-Catalyzed Cross-Dehydrogenative Carbamoylation of Pyridines with Formamides. Synlett 2016, 27, 1854–1859. [Google Scholar]
- 27).Fier PS; Kim S; Cohen RD, A Multifunctional Reagent Designed for the Site-Selective Amination of Pyridines. J. Am. Chem. Soc 2020, 142, 8614–8618. [DOI] [PubMed] [Google Scholar]
- 28).Seiple IB; Su S; Rodriguez RA; Gianatassio R; Fujiwara Y; Sobel AL; Baran PS, Direct C−H Arylation of Electron-Deficient Heterocycles with Arylboronic Acids. J. Am. Chem. Soc 2010, 132, 13194–13196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29).(a) Sulzer-Mosse S; Cederbaum F; Lamberth C; Berthon G; Umarye J; Grasso V; Schlereth A; Blum M; Waldmeier R, Synthesis and fungicidal activity of N-thiazol-4-yl-salicylamides, a new family of anti-oomycete compounds. Bioorg. Med. Chem 2015, 23, 2129–2138. [DOI] [PubMed] [Google Scholar]; (b) Lamberth C, Episodes from the Continuous Search for Solutions against Downy Mildew Diseases. Chimia 2019, 73, 571–580. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.