

Abstract
Acute ozone (O3) exposure is associated with multiple adverse cardiorespiratory outcomes, the severity of which varies across individuals in human populations and inbred mouse strains. However, molecular determinants of response, including susceptibility biomarkers that distinguish who will develop severe injury and inflammation, are not well characterized. We and others have demonstrated that airway macrophages (AMs) are an important resident immune cell type that are functionally and transcriptionally responsive to O3 inhalation. Here, we sought to explore influences of strain, exposure, and strain-by-O3 exposure interactions on AM gene expression and identify transcriptional correlates of O3-induced inflammation and injury across six mouse strains, including five Collaborative Cross (CC) strains. We exposed adult mice of both sexes to filtered air (FA) or 2 ppm O3 for 3 h and measured inflammatory and injury parameters 21 h later. Mice exposed to O3 developed airway neutrophilia and lung injury with strain-dependent severity. In AMs, we identified a common core O3 transcriptional response signature across all strains, as well as a set of genes exhibiting strain-by-O3 exposure interactions. In particular, a prominent gene expression contrast emerged between a low- (CC017/Unc) and high-responding (CC003/Unc) strain, as reflected by cellular inflammation and injury. Further inspection indicated that differences in their baseline gene expression and chromatin accessibility profiles likely contribute to their divergent post-O3 exposure transcriptional responses. Together, these results suggest that aspects of O3-induced respiratory responses are mediated through altered AM transcriptional signatures and further confirm the importance of gene-environment interactions in mediating differential responsiveness to environmental agents.
Keywords: airway macrophages, gene-environment interactions, mouse, ozone, transcriptomics
INTRODUCTION
For decades, ambient ozone (O3) exposure has been recognized as a hazard to human health. Acute O3 exposure causes decrements in pulmonary function, tissue injury, and inflammation in healthy individuals, and these effects are more severe in individuals living with respiratory disease. Moreover, emerging evidence links long-term O3 exposure to development of chronic diseases and disorders including asthma (1–3), allergic rhinitis (4), and potentially chronic obstructive pulmonary disease (COPD) (5). As such, identifying those with enhanced risk and establishing the mechanisms by which O3 exposure promotes and exacerbates disease are important public health objectives.
Controlled exposure studies performed as early as the 1980s have demonstrated that O3-induced respiratory responses are highly reproducible within individuals [including when exposures are separated by long intervals of time (6)] and yet quite variable across a population, even after participant characteristics such as sex, age, and smoking and disease statuses are matched (6–9). Genetic polymorphisms that interact with O3 exposure (“gene-environment interaction” or “GxE”) likely contribute to observed inter-individual variation (10, 11), a notion supported both by studies measuring O3 responses among genetically variable inbred rodent strains (12–17) and through demonstration of the effects of natural genetic variation on responses to other environmental stimuli (18–23). Despite robust evidence of GxE in well-controlled model systems, estimating individual-level causal effects of toxicant exposures remains challenging and few reproducible examples have emerged from human studies. Although this hurdle can be overcome with the use of animal models, most studies examining effects of O3 exposure have primarily used common inbred mouse strains, whose genomes are highly similar (24), limiting power to make broader claims about core drivers of O3-induced responses across genetically diverse individuals or uncover the biology governing divergent outcomes.
To address this research gap, we exposed a panel of Collaborative Cross (CC) recombinant inbred mouse strains that span a broad range of responses to acute O3 exposure and performed genomic analyses of airway macrophages (AMs). While numerous resident and recruited cell types respond readily to O3 exposure, previous studies have established that AMs are a key cell type whose phenotypic and transcriptional plasticity enable nimble responses to O3 (25–30). Thus, studying their activities will likely reveal pathways that are causally upstream of O3-induced pathological and reparative processes.
The CC is a mouse genetic reference population derived from eight founder strains, whose genomes collectively harbor around 40 million single-nucleotide variants (SNVs) (31, 32), thus capturing more than 90% of the genetic variation present in all inbred mouse strains (33). This resource has been used to identify novel candidate genes and variants associated with drug-induced adverse effects (34, 35), responses to pollutants (36), and respiratory innate immunity (37–39). In addition, the CC has been used to study transcriptional correlates of disease features (40–42). Thus, this population is appropriate for understanding genetic contributions to complex disease phenotypes and dissecting causal mechanisms of O3 toxicity.
Here, we exploit O3 response heterogeneity in inbred mouse strains in conjunction with genomic profiling of airway macrophages to: 1) define a core set of transcriptional responses triggered by O3 exposure that are shared, regardless of genetic background; 2) use coexpression analysis along with molecular phenotyping to identify modules of coregulated genes and their correlations with relevant traits; and 3) identify responses that are unique to specific genetic backgrounds. With respect to the last goal, we describe responses that are both qualitatively different (i.e., which genes and pathways) and quantitatively different (i.e., direction and magnitude of effect), focusing on contrasts between a low- and a high-responding strain (CC017/Unc and CC003/Unc, respectively).
MATERIALS AND METHODS
Animals
All experiments described here were approved by the University of North Carolina Institutional Committee on Animal Use and Care (IACUC). Adult (10–12 wk of age) female and male mice were used for the experiments described here. C57BL/6J mice were purchased from the Jackson Laboratory (Bar Harbor, Maine) and acclimated at the University of North Carolina at Chapel Hill for 3 wk before exposure. CC strains CC003/Unc, CC017/Unc, CC025/GeniUnc, CC039/Unc, and CC059/TauUnc were obtained from the UNC Systems Genetics Core Facility as weanlings in June 2018 and aged on investigator’s racks before exposure. In the text, CC strain names are referred to without their corresponding laboratory code (e.g., CC003/Unc is abbreviated as CC003). All animals were housed in groups of 1–5 on ALPHA-Dri bedding (Shepard) with ad libitum food (Envigo 2929) and water, under standard 12-h light/dark cycles.
Ozone Exposure
Animals were exposed to filtered air (FA) or 2 ppm ozone (O3), as described previously (30, 43). In brief, mice were exposed in individual wire-mesh chambers without access to food or water, then returned to their home cages and allowed to recover for 21 h before necropsy. Animals were exposed in sex- and strain-matched pairs, where one mouse was exposed to O3 and the other to FA (n = 3 per sex/treatment/strain except CC0039 where n = 4 females exposed to O3 and 1 male exposed to FA and CC003 where n = 1 female and 2 males exposed to FA). Exposures were performed across three successive days, with one matched pair per sex and strain represented in each batch.
Phenotyping
Lung phenotyping.
Twenty-one hours after cessation of exposure, mice were anesthetized (2 g/kg urethane) and euthanized by inferior vena cava/abdominal aorta exsanguination. Bronchoalveolar lavage (BAL) wash buffer was prepared by diluting 1 mL of 25X cOmplete protease inhibitor cocktail (Roche) stock solution in 24 mL of phosphate-buffered saline. BAL was collected by serially instilling the lungs with 0.5 mL and 1 mL BAL wash buffer. The BAL fluid was centrifuged for 10 min at 400 g, and supernatant from the first fraction was saved and stored at −80°C for biochemical analysis. Pellets from both BAL fractions were pooled, resuspended in red blood cell lysis buffer, and centrifuged once more at 400 g for 10 min. Pellets were resuspended in 500 μL of HBSS. From this suspension, 400 μL was used for isolating airway macrophages, whereas the remainder was used for counting total numbers of viable cells and preparing cytocentrifuge slides for differential cell counts.
Airway macrophage isolation.
The BAL cell suspension was plated with FBS-containing RPMI-1640 in a 24-well nontreated plate and left to rest in a tissue culture incubator for 4 h. Nonadherent cells were removed using a vacuum line. The enriched fraction of airway macrophages (AMs) was removed using Accutase (Gibco) and slow-frozen in two equal aliquots of freezing medium (90% FBS and 10% DMSO). In a separate exposure using only female C57BL/6J mice, we confirmed that this method results in an enriched fraction of AMs devoid of infiltrating monocytes and macrophages (∼80%, Supplemental Fig. S1; all Supplemental figures are available at https://doi.org/10.6084/m9.figshare.14658270.v1).
Protein measurement and cytokine profiling.
Total BAL protein was quantified with the Qubit Total Protein Quantification kit and the Qubit 2.0 fluorometer (Thermo Fisher Scientific). Serum albumin in BAL was measured using the Mouse Serum Albumin ELISA kit (Immunology Consultants Laboratory, Inc.). BAL cytokines were profiled using a premixed MILLIPLEX protein immunoassay (Millipore) that was read on a Bio-Plex 200 multiplex suspension array system (Bio-Rad). A panel of 15 cytokines was measured in total: eotaxin-1/CCL11, G-CSF, GM-CSF, IL-10, IL-12p70, IL-1β, IL-6, IP-10/CXCL10, KC/CXCL1, LIX, MCP-1/CCL2, MIP-1α/CCL3, MIP-1β/CCL4, MIP-2/CXCL2, and TNFα. G-CSF, GM-CSF, IL-12p70, IL-1β, MCP-1, MIP-1α, MIP-2, and TNFα were undetected in > 33% of samples and excluded from further analysis.
Statistical Analysis of Phenotype Data
For inflammatory cell counts, injury measurements, and cytokine profiling presented in Figs. 1, 2, and 3 and Supplemental Fig. S2, raw data (Supplemental Table S1; see https://doi.org/10.6084/m9.figshare.14658288.v1) were transformed where appropriate to resolve heteroscedasticity and ensure normality. Analysis of variance (ANOVA), likelihood-ratio, and t tests were performed in R (version 4.1.1), and results of testing were deemed significant if P values were < 0.05.
RNA Isolation and Sequencing
One aliquot of frozen AMs was gently thawed, spun down at 400 g for 5 min, and washed once with cold PBS to remove freezing medium. AM pellets were lysed directly with 350 μL of buffer RLT, and total RNA was isolated using the Qiagen RNeasy Micro kit. Stranded libraries were prepared using 10 ng of total RNA with the Ovation SoLo RNA-seq Library Preparation kit (NuGEN). Four library pools were created (one matched pair per strain and treatment represented per pool) which were sequenced across four lanes on an Illumina HiSeq 4000 to generate 50-bp, single-end reads.
ATAC-Seq Library Preparation
Frozen AMs were thawed as for RNA isolation, and ATAC-seq libraries were prepared as described previously (44, 45) with minor modifications. AM pellets were gently resuspended in lysis buffer (10 mM Tris-HCl, pH 7.5, 10 mM NaCl, 3 mM MgCl2, 0.1% NP-40 in nuclease-free H2O) and nuclei were pelleted by centrifugation at 500 g for 30 min in a refrigerated swinging bucket centrifuge. The supernatant was gently discarded, nuclei were resuspended in a 25 μL transposition reaction containing 2 μL of Tn5 transposase, and the mixture was incubated at 37°C in a thermomixer set to 1,000 rpm for 1 h. Transposed DNA fragments were purified using a Qiagen MinElute Reaction Cleanup kit. PCR amplification was performed to add indexing primers and increase library yield. Final libraries were purified using Agencourt AMPure XP magnetic beads and quantified via Qubit dsDNA HS Assay kit. Libraries were sequenced on an Illumina HiSeq 4000 to generate 50-bp, paired-end reads.
Gene Expression Analysis
Sequence alignment and transcript quantification.
CC strain-specific pseudogenomes (http://csbio.unc.edu/CCstatus/index.py?run=Pseudo) are only currently only available for GRCm37/mm9. Thus, sequenced reads were aligned to these pseudogenomes or the mm9 C57BL/6J reference genome, where appropriate, using STAR (46) and quantified using RSEM (47).
Differential expression analysis.
To estimate the treatment effects, strain effects, and strain-by-O3 exposure interactive effects on gene expression, a series of models were fit using DESeq2 (48).
The effect of treatment of gene expression was estimated using the following model
| (1) |
where Pair.ID is a factor that indicates sex- and strain-matched pairs, to accommodate the experimental design. Treatment coefficients representing the “treatment effect” were evaluated within the DESeq2 framework using the Wald test, with false-discovery rate (FDR) correction (α = 0.05).
The influence of strain on gene expression (“strain effect”) was assessed outside of the paired framework used in Eq. 1, with the following model
| (2) |
where Pair.Label describes the exposure day (i.e., batch). Strain coefficients representing the “strain effect” were tested for significance using the likelihood-ratio test, against a reduced model less the Strain term, subject to FDR correction (α = 0.05).
Finally, “strain-by-O3 exposure effect” coefficients were fit in the paired framework using an extension of Eq. 1
| (3) |
where Strain:Treatment coefficients were tested for significance using the likelihood-ratio test, using the following process. For each gene, a set of six strain-by-O3 exposure coefficients was computed: the first was based on the differential expression for a given strain compared with the mean differential expression across the remaining five strains, and the other five were determined by repeating this procedure individually comparing against each of the other strains. This process was applied for all genes, generating a 13,594 × 6 matrix of strain-by-O3 exposure coefficients. All interactive coefficients were tested for significance using the Wald test, and subject to joint shrinkage using ashr (49) and FDR correction (α = 0.05).
Weighted gene coexpression network analysis.
Network coexpression analysis was performed using WGNCA (50). Expression data for all genes with number of reads greater than total number of samples (n = 46) were included (13,594 genes) and data were subjected to the variance-stabilizing transformation before analysis with WGCNA. One consensus network was generated for samples from both FA and O3-exposed mice using the recommended WGCNA approach for automatic network construction and module detection. We assumed a signed correlation network and used biweight midcorrelations to compute the correlations. We selected a soft threshold of 12 based on a 0.9 threshold for the scale-free topology index. For settings related to dendrogram cutting and module merging, we used the recommendations in the WGCNA tutorial. After module assignment, we examined the correlation of the eigengene (first principal component) for each of these modules with inflammatory, injury, and cytokine metrics measured across the panel of strains. Modules were then prioritized based on the number and significance of these correlations, and gene set enrichment was performed.
We defined hub genes as those with high intramodule connectivity (correlation between a gene and all other genes within a module) and high module membership (correlation between a gene and the module eigengene), which are necessarily related quantities (50).
Gene set enrichment.
Lists of genes were examined for putative biological significance using enrichR (51), with databases used listed in Supplemental Table S4A (see https://doi.org/10.6084/m9.figshare.14658315.v1).
ATAC-Seq Analysis
AM ATAC-seq data from CC003 and CC017 mice were analyzed with the following methods and procedures.
Sequence alignment and preprocessing.
The PEPATAC (52) pipeline with default parameters was used to perform adapter trimming, alignment, and peak calling for ATAC-seq reads, which were aligned to the appropriate pseudogenome. Uniquely mapped reads were converted to mm9 coordinates (excluding those overlapping the mm9 blocklist) to allow for cross-strain comparisons. The union set of peaks were called from each sample and divided into 300-bp overlapping windows which were further subdivided based on their location (proximal: within 5 kb of nearest transcription start site (TSS); distal: beyond 5 kb of nearest TSS).
Differential accessibility analysis.
Similar models were used to perform differential accessibility analysis as described for differential expression analysis, where gene is replaced with number of mapped reads within 300-bp overlapping windows, with a covariate included to account for differences in data quality [transcription start site (TSS) score, as reported by PEPATAC]. Following differential accessibility analysis, significant neighboring windows within 200 bp were merged (FDR < 0.05). Motif analysis was performed using HOMER (53), focusing on enrichment of known motifs.
RESULTS
Cellular and Molecular Markers of O3-Induced Inflammation and Injury Vary by Strain
To investigate the influence of genetic background on O3-induced airway inflammation and injury, we exposed adult, female and male mice from one classical inbred and five CC strains in matched pairs to filtered air (FA) or 2 ppm O3 for 3 h (Fig. 1). Relative to FA-exposed control mice, O3 exposure caused a significant increase in the total number of BAL cells in three of six strains, CC017, CC025, and CC039 (Fig. 2A). Across all strains, neutrophils were significantly increased in mice exposed to O3 relative to their FA counterparts (Fig. 2B), whereas macrophages were only significantly increased in strains where the total BAL cell counts were also statistically significant (CC017, CC025, CC039; Supplemental Fig. S2A). Three-way analysis of variance (ANOVA) with strain, treatment, and sex as factors revealed a significant main effect of strain and treatment. Sex was not a significant predictor for any cellular measurement. We used a likelihood-ratio test (LRT) to compare model fit when including a strain-by-O3 exposure interaction term to the main effects model, which indicated significant interactions for all three cellular measurements (P < 0.0001 for total cells, P < 0.01 for neutrophils, and P < 0.001 for macrophages). Similar effects were seen when considering percentage of neutrophils or macrophages in the BAL (Supplemental Fig. S2, B and C). CC003 and CC039 were the highest responders as judged by the total number and proportion of neutrophils in the BAL, whereas CC017 had the lowest O3-induced inflammation and neutrophilia.
Figure 1.
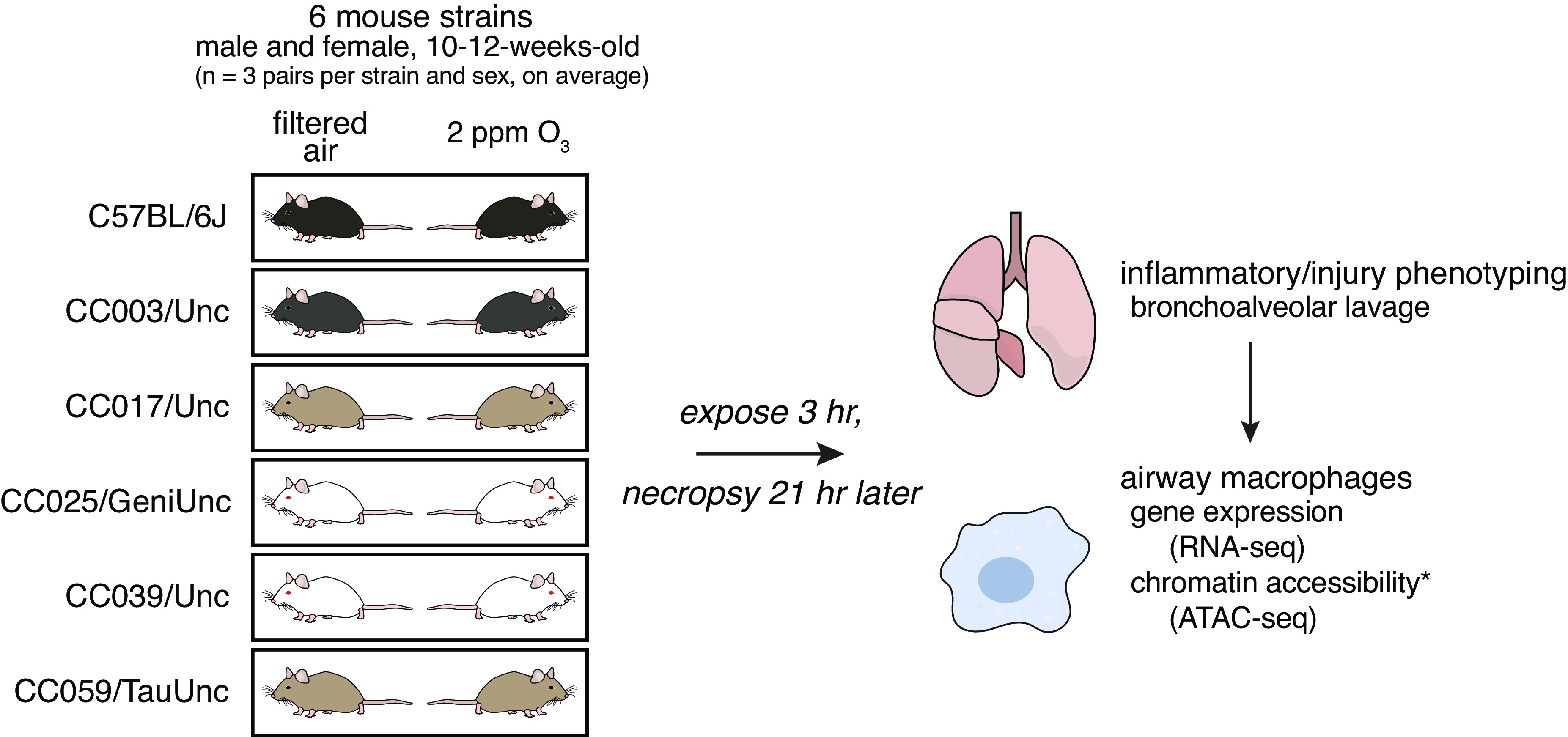
Study design. We exposed adult male and female mice from six strains in sex- and age-matched pairs to filtered air (FA) or 2 ppm ozone (O3) for 3 h and euthanized 21 h later. We performed bronchoalveolar lavage (BAL) to collect cells and protein from the airspace for quantitative inflammatory and injury phenotyping. We also collected airway macrophages by adherence from BAL cells from each mouse and performed gene expression by RNA-seq and, in CC003 and CC017, profiled chromatin accessibility using assay for transposase-accessible chromatin using sequencing (ATAC-seq). (n = 3 mice per sex/treatment/strain except CC0039 where n = 4 females exposed to O3 and 1 male exposed to FA and CC003 where n = 1 female and 2 males exposed to FA).
Figure 2.

O3 exposure causes variable inflammation and injury across six strains of mice. Total cellular inflammation (106; A), neutrophilia (105; B), and total protein concentration (μg/mL; C), a metric of lung injury, were measured in the BAL fluid. Data are presented as box-and-whiskers plots, which display the distribution from the minimum, first quartile, median, third quartile, and maximum. Individual data points are overlaid, with circles representing FA exposed mice and triangles representing O3 exposed mice. Females are closed points and males are open points. All measures had significant strain-by-O3 exposure interaction effects, assessed using a likelihood-ratio test (A: P < 0.0001, B: P < 0.05, C: P < 0.0001). (n = 3 mice per sex/treatment/strain except CC003 where n = 1 female/2 males exposed to FA and CC0039 where n = 4 females/3 males exposed to O3 and 3 females/1 male exposed to FA; *P < 0.05, **P < 0.005, ***P < 0.0005, for within-strain contrasts (t tests) between FA and O3). BAL, bronchoalveolar lavage; FA, filtered air; O3, ozone.
In addition to cellular inflammation, we assessed lung injury induced by acute O3 exposure, as reflected by the concentration of protein in BAL. After FA exposure, all strains had low levels of total protein in the lung lavage fluid (Fig. 2C); however, the increased levels of BAL protein varied by strain after O3 exposure. As with inflammation, we detected a significant main effect of strain and treatment, with no significant effect of sex on total protein concentration, and a highly significant strain-by-O3 exposure interaction (P < 0.0001). Because total protein is a nonspecific marker of serum protein leakage and lung injury, we measured serum albumin by ELISA in a subset of these mice to confirm our findings. Overall, the strain order of the post-O3 exposure effect was similar between total protein and serum albumin measures of lung injury; however, in the FA samples, there was greater variability between strains revealed by the albumin assay (Supplemental Fig. S2D). Interestingly, the rank order of strain effects was not strictly the same when comparing BAL cellular inflammation and total protein, though CC003 and CC039 remained high responders while CC017 had the lowest increase in total protein concentration after O3 exposure of the strains surveyed.
We also measured a panel of hallmark O3-responsive cytokines in the BAL to further characterize the molecular aspects of airway inflammatory responses (Fig. 3). One strain, CC039, had markedly high concentrations of eotaxin-1 (CCL11) and IL-6 in response to O3 exposure in comparison to the rest of the strains (Fig. 3, A and B). As we observed in a previous study (54), the anti-inflammatory cytokine IL-10 was decreased upon O3 exposure in a strain-dependent manner (Fig. 3C). The chemokine IP-10 (CXCL10) was significantly higher in O3-exposed animals in five of the six strains, with the increase in CC003 not reaching statistical significance, partly due to this chemokine having among the highest average concentration at baseline in this strain (Fig. 3D). The potent neutrophilic chemokine KC (CXCL1) was significantly increased in BAL from O3-exposed mice to varying extents across all strains except C57BL/6J (Fig. 3E). Levels of LIX (CXCL5) tended to be lower in O3-exposed mice compared with FA controls, though this effect was only significant in C57BL/6J (Fig. 3F). Levels of MIP-1β were lower across all strains compared with other analytes and were generally higher in O3-exposed animals (Fig. 3G). Notably, for nearly all cellular and biochemical phenotypes measured, CC strains displayed responses that were both more and less pronounced than the classical inbred strain used (C57BL/6J).
Figure 3.

Respiratory cytokine responses are altered by O3 exposure and vary by strain. Molecular markers of inflammation eotaxin-1 (CCL11; A), IL-6 (B), IL-10 (C), IP-10 (CXCL10; D), KC (CXCL1; E), LIX (CXCL5; F), and MIP-1β (G) were measured using a multiplex cytokine detection kit. Data are presented as box-and-whiskers plots, which display the distribution from the minimum, first quartile, median, third quartile, and maximum. Individual data points are overlaid, with circles representing FA-exposed mice and triangles representing O3-exposed mice. Closed points are female mice, whereas open points are male mice. Points below the assay limit of detection were excluded from analysis. Raw data are available in Supplemental Table S1. Eotaxin-1, KC, and IL-6 also displayed significant strain-by-O3 exposure interactions assessed by a likelihood-ratio test (A: P < 1 × 10−7, B: P < 0.005, C: P < 0.0005). (n = 2 mice per sex/strain/treatment for all strains except CC003 where n = 3 females/1 male exposed to O3 and n = 1 female/2 males exposed to FA and CC0039 where n = 3 females/1 male per treatment; *P < 0.05, **P < 0.005, ***P < 0.0005 for within strain contrasts (t tests) between FA vs. O3). FA, filtered air; O3, ozone.
Finally, we calculated pairwise correlations between these phenotypic traits (Supplemental Fig. S2E). As expected, levels of the anti-inflammatory cytokine IL-10 were negatively correlated with most other inflammatory phenotypes excluding concentrations of LIX, for which there was a positive correlation. Otherwise, nearly all other phenotypes were significantly positively correlated with one another, with correlation coefficients in the range of 0.48–0.85. Some traits were more strongly correlated, including IL-6 and eotaxin-1/CCL11 concentrations, whereas others were less closely linked. This set of correlations suggests that some responses to O3 exposure may be decoupled and subject to different genetic regulation.
A Core Set of O3-Responsive Transcripts Are Shared Across the Panel of Strains, and Group into Coexpression Modules Correlated with Relevant Phenotypes
To comprehensively characterize the gene expression alterations induced by O3 exposure, we performed RNA-seq analysis in airway macrophages (AMs) isolated from a subset of samples from each of the strains (n = 2 mice per sex, treatment, and strain). Mapping statistics are reported in Supplemental Table S2 (see https://doi.org/10.6084/m9.figshare.14658303.v1). Principal components analysis (PCA) of the top 500 most variably expressed genes revealed that samples clearly separate by strain (PC1) and treatment (PC2), and these PCs account for roughly 33% of variance within the data (Fig. 4). Intriguingly, samples only begin to separate by sex at PC9, which accounts for a mere 2.6% of total variance within this subset of genes, indicating that gene expression in this context is only modestly influenced by sex (Supplemental Fig. S3).
Figure 4.

Airway macrophage (AM) gene expression varies by strain and O3 exposure. RNA-seq was performed on a subset of mice exposed to FA or 2 ppm O3. Principal components analysis (PCA) of the top 500 most variably expressed genes reveals separation of samples by strain (PC1) and treatment group (PC2). Circles represent filtered air exposed mice and triangles represent O3-exposed mice. Closed points represent female animals and open points represent male animals. (n = 2 mice per sex/strain/treatment, except CC003 where n = 1 female/2 males per treatment and CC039 where n = 3 females/1 male per treatment). O3, ozone.
We were first interested in identifying genes whose expression is altered by O3 exposure (treatment effect), without explicit consideration of genetic background. To define the set of treatment effect transcripts, we used a paired framework within DESeq2 to regress out the effects of strain and batch and to extract the treatment effect. We identified 2,761 O3-responsive genes with an FDR < 0.05, 773 of which had an absolute log2 fold change > 1. These results are depicted in Fig. 5, and all significantly differentially expressed genes (DEGs), including their full gene names, are reported in Supplemental Table S3A (see https://doi.org/10.6084/m9.figshare.14658309.v1). Four DEGs that we previously identified in a meta-analysis, Thbs1, Lcn2, S100a9, and Cdk1, exhibited sizable changes [log2(fold change): ∼1.6–5.5] in gene expression (30). Other significant DEGs included Scgb1a1, Cyp1b1, and Mmp9. Scgb1a1 is predominantly produced by club cells but was recently implicated in alveolar macrophage immune responses (55) and has been proposed as a biomarker of O3-induced lung injury in humans (56), whereas Cyp1b1 is a well-known detoxification enzyme regulated by activity of the aryl hydrocarbon receptor. Mmp9 is an extensively characterized matrix metalloprotease associated with proinflammatory macrophage activity (57, 58). We also performed pathway analysis with this core set of O3-responsive genes, which identified a wide variety of gene sets previously linked to O3 responses, including neutrophil-mediated immunity, cytokine-mediated processes, regulation of cell proliferation, and responses to oxidative stress (Supplemental Table S4B; see https://doi.org/10.6084/m9.figshare.14658315.v1) (25, 30, 59).
Figure 5.

O3 exposure causes characteristic changes in airway macrophage (AM) gene expression. The union set of the light blue and royal blue dots are all significantly differentially expressed genes (2,761; FDR < 0.05). The royal blue dots alone represent those with absolute log2 fold change > 1 (773). The top 10 most highly differentially expressed genes are labeled (n = 13,594 genes tested). FDR, false-discovery rate.
Next, we were interested in relating our gene expression data to phenotypic data collected on the same mice to identify networks and pathways associated with relevant traits. To achieve this goal, we performed weighted gene coexpression network analysis [WGCNA (50)] on 13,594 genes to jointly cluster data from both FA- and O3-exposed mice into coexpression modules. Using this expression set, we grouped 4,373 genes into 18 modules, each module containing 51 to 1,462 genes (Supplemental Table S3A). The remaining 9,221 transcripts were collected in an unassigned module (“grey60”), largely representing genes that were lowly expressed and/or displayed low variance across both strain and exposure.
Five of the 18 modules displayed strong module-phenotype relationships, with three modules (“red,” “brown,” “blue”) being positively correlated with proinflammatory cytokine secretion, lung injury, and neutrophilia and the remaining two modules (“pink,” “midnightblue”) exhibiting inverse relationships with those phenotypes (Supplemental Fig. S4A). For these five modules, we examined pathway annotations associated with member genes (Supplemental Table S4C). Genes contained in the largest of the five modules (“blue,” 742 genes) were associated with pathways involved in cell cycle progression, DNA replication, and repair. A second module (“brown,” 352 genes) was enriched for pathways generally associated with inflammation such as signaling induced by canonical regulators (e.g., TNFα, IL-6, TGF-β), responses to immune stimuli like lipopolysaccharide (LPS), salmonella, and influenza A, and terms associated with protein modification and processing in the endoplasmic reticulum. The final proinflammatory module (“red,” 188 genes) was enriched for pathways associated with oxidative phosphorylation and other mitochondrial metabolic processes, as well as fatty acid metabolism and thermogenesis. Considering the modules negatively correlated with inflammation, the first (“pink,” 131 genes) only displayed biological associations with one term (“focal adhesion”), though we discovered many genes involved in lipid processing and metabolism after manual inspection of this gene set, including Alox5ap (5-lipoxygenase-activating protein), Fabp1 (fatty acid-binding protein 1), Adipor1 (adiponectin receptor 1), and Abcg1 (ATP-binding cassette subfamily G member 1). Finally, the second anti-inflammatory module (“midnightblue,” 66 genes) was associated with several RNA polymerase II-mediated processes and included early response genes and negative regulators within inflammatory signaling pathways including TNFα signaling via NF-κB.
To uncover putative mechanistic drivers of O3 responses, we identified hub genes (see methods) within the aforementioned modules. For visualization purposes, we highlighted the top 10 hub genes in all modules (except the blue module, from which we selected the top 20), as shown in Supplementary Fig. S4B. To further prioritize hub genes with respect to overall module gene expression and O3 response, we examined their correlation to the module eigengene (also referred to as module membership) and with each phenotype of interest (BAL neutrophil percentage, BAL protein, and levels of IL-10 or KC/CXCL1 in the BAL; Supplemental Fig. S4, C–E). Strikingly, we found a strong negative correlation between the anti-inflammatory pink module hub genes Zfp36, Irs2, Cebpa, and Rara with BAL neutrophilia, protein, and CXCL1. This recapitulates a known relationship between ZFP36 and a variety of cytokines, including CXCL1, where ZFP36 destabilizes their transcripts by binding to the UTRs to target them for degradation (60). In addition, Irs2 is a negative regulator of macrophage activation and pulmonary inflammation in a model of allergic inflammation (61). Cebpa (CEBP-α) is a transcription factor previously profiled in alveolar macrophages after O3 exposure by Fakhrzadeh et al. (62), who found that its levels peaked at 6 h alongside NF-κB signaling; however, in that study, although NF-κB displayed a second peak 48 h after O3 exposure, CEBP-α continued to wane. Finally, Rara is a retinoic acid receptor with no known connection to O3 responses; however, signaling via other related nuclear hormone receptors including the liver X receptor is known to suppress O3-induced inflammation (63).
Unlike the pink module, hub genes for the remaining modules displayed strong correlations with single phenotypes. Within the anti-inflammatory midnightblue module, we observed a negative association between several hub genes including Fosb, Nr4a1, Trib1, Osm, and Pygl, and BAL neutrophilia. Among these hub genes, Fosb and Nr4a1 have documented increases in expression in response to oxidative stress or the O3-derived reaction product 4-hydroxynonenal (4-HNE) (64–66). Trib1 and Pygl have roles in metabolism, and the former gene is known to control tissue macrophage polarization (67). Interestingly, previous studies have indicated that increased Osm (oncostatin M) expression is associated with neutrophilic recruitment via airway epithelium-produced CXCL5 (68), contrary to what we observed here. As opposed to the midnightblue module, the proinflammatory red module hub genes were positively correlated with neutrophilia and comprised a battery of histone transcripts. We noted that hub genes within the proinflammatory brown module, with the exception of Glg1, were modestly correlated with BAL protein levels. Interestingly, though the proinflammatory blue module contained the largest number of genes, we found that its hub genes were poorly correlated with phenotypes except in the case of BAL IL-10. The top blue module hub genes Fignl1, Ncapg2, and Mcm2 were strongly negatively correlated with BAL IL-10, and are all regulators of DNA replication and repair (69–71).
In total, we identified several hub genes with both established and unexplored roles in O3 responses. These hub genes represent candidates for future experimental validation and several may serve as potential novel biomarkers of O3 responsiveness.
Strain Background Modifies Basal and O3-Induced AM Gene Expression
After establishing a set of O3-responsive transcripts that were differentially expressed across all strains, we sought to explore how genetic background influences transcriptomic responses to O3 exposure and examine whether the transcriptome could give insights into biological signatures that distinguish low- and high-responding strains. Specifically, we used a series of modeling approaches to identify genes whose expression is influenced by strain effects (i.e., genetic background) and strain-by-O3 exposure effects.
First, we identified genes with significant strain effects, accounting for batch, sex, and treatment (Supplemental Table S3A). Overall, there were 3,787 genes (28% of examined transcripts) for which strain was significantly associated with expression (FDR < 0.05), a level that is comparable to a previously published study examining the influence of genetic and environmental effects on peritoneal macrophage gene expression using the Hybrid Mouse Diversity Panel (5,726 out of 12,980 measured; 44.1%) (21). In addition, this figure is slightly greater than the total number of genes influenced by treatment (2,761) defined in the previous section. Next, we identified genes with significant strain-by-O3 exposure effects (Supplemental Table S3A). Rather than testing for interactive effects between strain and treatment jointly across all strains or by examining all 15 possible pairwise strain contrasts, we estimated strain-by-O3 exposure effects for each gene by comparing differential expression (O3 vs. FA) in one strain against the mean differential expression of all other strains. In other words, each coefficient represents the strain-specific relative response for a given gene. In total, 545 genes (4% of examined transcripts) had one or more significant strain-by-O3 exposure effects (FDR < 0.05). Within this list were numerous genes that have been previously implicated in O3 responses (72–77), including cytokines (Ccl2, Ccl6, Ccl7, Ccl9, Cxcl3, Il18, Slpi) and scavenger receptors (Cd36, Colec12, Marco, Msr1) (Fig. 6). Interestingly, there were also 245 genes with both significant strain and strain-by-O3 exposure effects on gene expression, including the genes visualized in Fig. 6.
Figure 6.

Numerous genes display strain-by-O3 exposure effects on expression, including those with putative and known roles in O3 responses. A–C represent immune response genes, whereas D–F are scavenger receptors. Some genes display interactive effects across all strains (e.g., Marco, Msr1), whereas others are selectively observed in only some strains (e.g., Ccl6, Cxcl3, Slpi). Circles represent filtered air exposed mice and triangles represent O3-exposed mice. Closed points represent female animals and open points represent male animals. [n = 2 mice per treatment/sex/strain except CC003 where n = 1 female/2 males per treatment; all genes have one or more significant strain-by-O3 exposure effects on expression (FDR < 0.05)]. FDR, false-discovery rate; O3, ozone.
To identify general patterns of response and guide the choice of specific strains on which to focus downstream analyses, we visualized the matrix of strain-by-O3 exposure coefficients using a PCA biplot, where each gene is an observation (point) and each strain is a loading vector (line) (Fig. 7). Genes clustered along these loading vectors contribute most to the corresponding strain’s overall gene expression response, where positive coefficients indicate that the strain had higher relative change in expression induced by O3 exposure compared with other strains, and vice versa for negative coefficients. Strikingly, in alignment with their divergent inflammatory and injury phenotypes, loadings for CC017, CC003, and CC039 were highest among the six strains. CC017 (a low responder) is nearly orthogonal to both CC003 and CC039 (high responders), whereas the latter two strains exhibit opposing transcriptional responses in line with the divergence observed in their patterns of cytokine secretion (Fig. 3, A, B, E, and G). Within the set of coefficients for CC017, Slc40a1, Plxdc2, and Pla2g7 were among genes with greatest positive contributions to this strain’s loading, whereas Vcan and Cd74 contributed negatively. The former group of genes are known to be involved in macrophage processes including iron homeostasis, IL-10 production via pigment epithelium-derived factor (PEDF), and suppression of proinflammatory platelet factors (54, 78, 79), and the latter set are associated with leukocyte adhesion/migration and neutrophil recruitment (80, 81). Included in CC003’s set of genes with positive coefficients were Card11, Rxra, and Lrch4, though many more genes with negative coefficients contributed to its loading including known anti-inflammatory markers associated with M2 macrophage polarization such as Hif1a, Mmp12, and Clec4a2. Finally, the important immune-related genes Ccl9, Arg1, and Cd86 contributed positively to CC039’s loading, whereas Marco and Osbpl3 were in the group of genes negatively influencing this strain’s loading. Marco is a scavenger receptor whose expression is displayed in Fig. 6E, whereas Osbpl3 is a member of the oxysterol-binding protein family; both may be involved in regulating clearance of O3-generated reaction products (63, 73, 82).
Figure 7.

Strain-by-O3 exposure coefficients for gene expression separate strains by their gene expression responses. PCA was performed with the matrix of strain-by-O3 exposure coefficients. Each point on the graph represents the first two principal components for a single gene (that has six coefficients, one per strain) and the loadings represent the gene expression characteristic for each strain. Symbols for the top 40 genes contributing to strain loadings are labeled, where those mentioned in the text are royal blue. B6, C57B/6J; O3, ozone; PCA, principal components analysis.
Because CC003, CC017, and CC039 displayed highly distinct responses compared with the other strains, we sought to identify strain-by-O3 exposure effects between these pairwise contrasts. Comparing genes whose expression was differentially altered by O3 exposure in CC017 versus CC003, we identified 1,091 significant strain-by-O3 exposure DEGs, whereas in CC039 versus CC017, only 148 genes were significantly DE, and in CC039 versus CC003, 373 genes were significantly DE (FDR < 0.05). Because the first contrast (CC017 vs. CC003) was most pronounced with both the largest number of DEGs identified and the inclusion of a low- and high-responding strain (in terms of BAL neutrophilia and total protein concentration), we chose to perform further analysis with this pair of strains.
CC003 Displays Enhanced Induction of Proinflammatory Pathways Compared with CC017, Which May Be Mediated by Divergent Basal Gene Expression Profiles
The strain-by-O3 exposure effect genes defined in the previous section represent one potential explanation for why CC003 and CC017 face divergent phenotypic consequences after O3 exposure. To explore the biological functions associated with genes DE between CC017 and CC003 and characterize how their responses to O3 differ, we performed pathway analysis with the aforementioned set of 1,091 strain-by-O3 exposure DEGs (partitioned by whether O3-induced expression was enhanced in CC017 vs. CC003; Supplemental Tables S3B and S4D). Within the set of 806 genes with greater O3-induced upregulation in CC017 compared with CC003 (764 with log2 fold change > 1, CC017 vs. CC003), pathways associated with cell cycle-related targets of E2F (Ctcf, Brca1, Brca2, Dck) and the inflammatory response (Il18, Hif1a, Klf6, Ccl2, Ccl7, Il7r) were overrepresented. Conversely, for the 285 genes with greater expression after O3 exposure in CC003 compared with CC017 (256 with log2 fold change < −1, CC017 vs. CC003), pathways involved in adipogenesis (Stat5a, Apoe), IL-2/-5/-7/-9 signaling (Map2k2, Cdk4, Ptpn6, Eif3b, E2f1), and TNFα via NF-κB (Irs2, Junb) were enriched.
An alternative, though not mutually exclusive, explanation is that basal gene expression (i.e., in FA-exposed mice) is a determinant of the contrasting responses between CC003 versus CC017, which we have observed in other studies (83). These baseline differences can result in both additive changes in gene expression (i.e., different intercepts, but the same slope), as well as genotype-environment interactions in expression. We evaluated baseline differences by examining genes with significant strain effects. Between CC017 and CC003, 1,187 genes with significant strain effects were identified, 386 of which were more highly expressed in CC017 and 801 more highly expressed in CC003 (Supplemental Table S3C). Next, we performed pathway analysis to gain insight into the types of biological activities implicated by these 1,187 genes, i.e., pathways that that differ between the strains at baseline (Supplemental Table S4E). Pathways enriched within the set of strain effect genes and that were more highly expressed in CC017 at baseline included those associated with TGF-β signaling (Serpine1, Furin, Klf10), complement and coagulation/clotting cascade (Plau, Vwf, F7, F10, Cfb), and phagocytic pathways (Tlr2, Itgb5, Scarb1). Conversely, pathways associated with genes more highly expressed at baseline in CC003 included glutathione metabolism (Gstm1, Anpep, Mgst2), responses to a variety of pathogens (e.g., influenza A, Bordetella pertussis, Legionella, and Salmonella), and numerous canonical inflammatory pathways (e.g., signaling via IL-2/STAT5, IL-17, IL-6, and Toll-like receptors). In addition, a few pathways were enriched in both sets of genes, including TNFα signaling via NF-κB, xenobiotic metabolism, and oxidative stress response. Taken together, these results suggest that higher expression of canonical inflammatory pathways in CC003 mice prime or accelerate their responses to O3 compared with CC017.
AM Chromatin Accessibility Is Influenced by Strain and Strain-by-O3 Exposure, but Not Treatment Effects, When Comparing CC0017 and CC003
One mechanism through which genetic variation alters gene expression is by modifying the degree of chromatin accessibility, which is associated with changes in gene regulatory activity. Moreover, methods that profile chromatin accessibility such as ATAC-seq enable comprehensive identification of regulatory elements with minimal cell input (44). Hence, we were interested in examining chromatin state differences between CC003 and CC017 using the same analytical frameworks we used for gene expression. To achieve this goal, we generated genome-wide chromatin accessibility profiles using ATAC-seq for a subset of these mice (n = 2 FA- and 3 O3-exposed/strain). We identified 2,030 differentially accessible regions (DARs) with significant strain effects (FDR < 0.05), 963 of which were more accessible in CC017 and 1067 of which more accessible in CC003. Contrary to the strong effects of O3 exposure on transcriptional responses, we did not identify any DARs with significant treatment effects. We note, however, that samples from O3-exposed mice had lower ATAC-seq quality scores; this confounding precluded accurate estimation of treatment effects on chromatin accessibility. We identified 414 regions with significant strain-by-O3 exposure effects on accessibility (FDR < 0.05), 191 of which were more accessible in response to O3 in CC017 and 223 more accessible in response to O3 in CC003. However, because treatment and quality were confounded within this set of data, we focused all downstream analyses on the lists of DARs with significant strain effects.
We examined the set of strain effect DARs for enrichment of transcription factor (TF) motifs using HOMER (53), subset by their position relative to transcription start sites (proximal: < 5 kb; distal: > 5 kb) and their relative accessibility (more accessible in CC017 or CC003). We found that motifs for TFs in the AP-1/ATF family including AP-1, FRA1, FRA2, BATF, and JunB were commonly enriched in distal sites, regardless of whether they were more accessible in CC003 or CC017. To estimate availability of these TFs to bind DARs, we inspected their relative gene expression levels across strains at baseline and after O3 exposure (Supplemental Fig. S5). We discovered that Fos, Junb, and Fosl1 (encoding FRA1) had significant strain effects (Supplemental Fig. S5A–D; adj. P = 1.1 × 10−4; adj. P = 2.4 × 10−2; adj. P = 1.8 × 10−2, respectively). Junb also had significant strain-by-O3 exposure effects (adj. P = 7.7 × 10−3), whereas Jun had suggestive influence of an interactive effect (adj. P = 0.11). In all cases, expression of the given factor was higher in CC003 after O3. These data suggest that a common set of transcription factors differentially regulate gene expression in CC003 versus CC017 by acting on distinct (i.e., strain-specific) regulatory regions; in addition, O3-induced changes in gene expression in CC003 may be more pronounced compared with CC017 because of its increased expression of these transcription factors. Distal CC003-accessible regions also contained a preponderance of motifs for ETS family transcription factors such as ELF3, ELF4, ELF5, and PU.1. We found a near-significant (adj. P = 0.056) strain-by-O3 exposure effect on PU.1 (Spi1) expression, with higher expression in CC003 (Supplemental Fig. S5E). The increased expression of these transcription factors in CC003 may explain why over twice as many genes show increased expression in CC003 compared with the number increased in CC017 (801 vs. 386, respectively).
Like distal CC003-accessible regions, proximal CC003-accessible regions were enriched for ETS factor motifs in the set of strain effect DARs. Proximal regions were differentiated by the presence of motifs for many IRF-family factors including IRF1, IRF3, and IRF8. Expression of Irf8 was significantly influenced by strain effects (adj. P = 4.4 × 10−6), along with a suggestive strain-by-O3 exposure effect (adj. P = 0.073) (Supplemental Fig. S5F), whereas the remaining IRF factors were not influenced by strain and/or treatment effects. In contrast, the list of enriched motifs for proximal CC017-accessible regions was populated by numerous zinc-finger proteins, including some with highly similar binding motifs (e.g., Sp1, Sp2, KLF4, KLF6). Within this set of factors, we determined that Klf6 expression was influenced by a significant strain effect (adj. P = 4.5 × 10−4) and strain-by-O3 exposure effects (adj. P = 4.1 × 10−3; Supplemental Fig. S5G).
Finally, we were interested in determining whether strain effects were shared across gene expression and chromatin accessibility, which may suggest corresponding influences on gene regulation and transcription. To this end, we identified which genes possessed significant strain effects on both gene expression and chromatin accessibility located within 100 kb of the corresponding gene’s TSS (± 50 kb of TSS midpoint). We identified 80 genes for which this was the case, an overlap that is highly significant (hypergeometric test P = 1.16 × 10−5; Supplemental Table S3D). This result indicates that differential expression of a subset of genes is associated with baseline alterations in chromatin accessibility and highlights a list of genes whose activity in AMs may contribute to baseline and post-O3 exposure phenotypic differences between CC003 and CC017.
Together, these results indicate that CC003’s enhanced responsiveness to O3 exposure may be mediated by heightened proinflammatory airway macrophage transcriptional activity, which was characterized by increased enrichment of canonical immune signaling pathways both at baseline and after O3 exposure (Table 1). Conversely, CC017 may mount a dampened response to O3 exposure that is coordinated by maintaining anti-inflammatory cytokine secretion and AMs that favor cellular proliferation and repair processes upon O3 exposure with minimal induction of proinflammatory transcriptional activity. Lastly, this analysis suggests that differences in chromatin accessibility enable a small set of transcription factors to have important roles in shaping the baseline expression profiles in CC017 and CC003, and that these variable chromatin profiles may poise each strain for their unique responses to O3 exposure.
Table 1.
Summary of differences between CC017 and CC003 in O3-induced phenotypes and airway macrophage molecular profiles
| CC017 | CC003 | |
|---|---|---|
| Inflammation (total BAL cells, neutrophils) | Low | High |
| Injury (total BAL protein, albumin) | Low | High |
| Cytokines | Muted changes overall; maintained high IL-10 production post-ozone exposure and LIX was unchanged | Significant decrease in IL-10 production, large increase in production of proinflammatory cytokines |
| Baseline gene expression pathways | TGF-β signaling; clotting and complement cascade; phagocytic functions | Glutathione metabolism; pathogen responses; canonical inflammatory signaling cascades |
| Ozone-induced gene expression pathways | Cell cycle progression and cellular proliferation; canonical inflammatory signaling cascades | Adipogenesis; TNF-α signaling via NF-κB; other canonical inflammatory signaling cascades |
| Transcription factors associated with enriched motifs within DARs | Lower expression of most TFs at baseline, and many remain unchanged or only minimally changed by ozone exposure | Higher expression of most TFs at baseline, and higher expression of all TFs after ozone exposure |
DARs, differentially accessible regions; TFs, transcription factors.
DISCUSSION
Although the adverse health consequences of O3 exposure have been appreciated and studied for decades, thorough characterization of the underlying molecular mechanisms and how they develop differentially among individuals is still incomplete. The use of integrative genomics and high-dimensional phenotyping in genetically diverse animal populations such as the CC mouse genetic reference panel provides a means by which to identify universal biomarkers of effect as well as unique biomarkers of susceptibility. In this study, we employed a small panel of CC strains to explore the range of inflammatory, injury, and AM genomic responses elicited by acute O3 exposure. We used statistical approaches to partition the influences of strain, treatment, and strain-by-O3 exposure interactions on variation in gene expression, which enabled 1) selection of broadly defined O3-responsive transcripts; 2) identification of genes whose expression levels are subject to unique genetic and genotype-by-environment interactions; and 3) joint use of these criteria for agnostic selection of CC003 and CC017 as strains of interest for closer scrutiny. We also extended these statistical procedures to examine chromatin accessibility data, an additional, crucial aspect of gene regulation.
This study builds upon previous work examining O3 responses across diverse mouse strains (14, 16, 17) and complements what has been described in classical inbred strains such as C57BL/6J and BALB/cJ. Our results demonstrate that the effects of acute exposure, including inflammatory cell recruitment (namely, neutrophilia), lung injury, and cytokine and chemokine secretion are highly variable across genetic backgrounds, corroborating earlier studies (14, 17, 84). We also demonstrated that O3 exposure causes a characteristic change in AM gene expression shared across multiple genetic backgrounds, and that this signature can be organized into coexpression modules. Genes in these modules are enriched for relevant biological pathways, including canonical immune signaling pathways and cell cycle progression, and have significant correlations with other aspects of response including neutrophilia and cytokine secretion, suggesting that AM gene expression is causally related to these phenotypes. An important future extension of this work will be to incorporate information collected from additional cell types, as AMs are not the lone driver of phenotypic outcomes. For instance, we and others have previously shown that acute O3 exposure causes noticeable changes in the transcriptome of cells lining the conducting airways, in both unique and similar patterns to those elicited in AMs (25, 30); thus, profiling their involvement, perhaps with a combination of genomic modalities that enable precise characterization of individual cells or sorted populations, will enable description of how diverse cellular signals collectively result in a unified O3 response.
Importantly, this study revealed widespread effects of strain and strain-by-O3 exposure interactions on AM gene expression in patterns that mirrored these effects on phenotype. Genes for which we identified a significant effect of strain on expression (termed “strain effect” genes) can effectively be considered expression quantitative trait loci (eQTL), albeit with unknown location, though precedent indicates they are more likely local (and act in “cis”) rather than distal (21, 85). This statement can be extended to the strain-by-O3 exposure effect genes as well; however, previous studies in C. elegans (86), yeast (87), and various cell culture systems (88–91) have shown that context-specific eQTL are often associated with distal signals, in addition to local ones. We found evidence of numerous genes with strain-by-O3 exposure effects, even between strains with similar phenotypic profiles such as CC003/Unc and CC039/Unc, who mounted strong responses to O3 exposure in the form of high airway neutrophilia, lung injury, and elevated cytokine secretion. Although not the focus of the study presented here, this observation suggests that two strains can arrive at convergent phenotypic profiles after traversing a distinct set of transcriptional paths, which motivates the inclusion of molecular parameters alongside higher order phenotypes in studies involving genetically diverse individuals (whether humans or model organisms), as the latter alone might obscure the underlying molecular drivers.
We observed the most striking differences in both baseline and post-O3 exposure gene expression between CC017/Unc and CC003/Unc, which we designated a low- and a high-responding strain, respectively, based on airway neutrophilia and injury. Of note, these strains also exhibit statistically significant differences in BAL neutrophilia and injury after exposure to a lower O3 concentration (1 ppm) (Supplemental Fig. S6). Although these strains have been included in previously published studies examining non-O3 perturbations (92, 93), minimal experimental evidence exists to indicate specific functional differences between the two that might account for their divergent responses to O3 exposure. In a study that screened severity of Zika virus infections across 35 CC strains, CC017/Unc was among the strains with low plasma viral load (2 days postinfection), whereas CC003/Unc had twice as many plasma viral copies (92). Although neither strain went on to develop clinical features of disease and had similar plasma viral load six days postinfection, baseline differences in immune responsiveness may account for early differences in viral load as well as the differences observed in our study. In addition, a study examining variation in liver toxicokinetics of trichloroethylene (TCE) across 50 CC strains demonstrated that CC017/Unc had much lower metabolism of TCE to trichloroacetic acid (TCA, the primary driver of TCE toxicity) than CC003/Unc (94). They concluded that differential metabolism is linked to PPARα activity, in a direct relationship. Although this pathway was not directly implicated in any of the results presented here, it may represent an interesting target for future studies.
Between our two CC strains of interest, we see influences of genetic variation on chromatin architecture, specifically at the level of chromatin accessibility, which provide clues as to the mechanism by which variation affects transcriptional profiles and response. Indeed, these findings have been borne out in integrative studies that jointly profiled chromatin accessibility and gene expression across genetically diverse individuals (85, 95, 96). Though specific variants or even regulatory elements are unlikely to be conserved from mouse to human, general principles governing how they influence gene expression and downstream trait variation are often conserved. Moreover, the number of joint genomic and phenotypic analyses and level of experimental precision that can be achieved using the CC or other genetically diverse mouse populations exceeds that which is currently possible in large-scale human genetic studies, leading to more robust discoveries. Therefore, using systems-level analyses that integrate many layers of regulatory information including chromatin accessibility, gene expression, and protein expression across genetically diverse mouse strains will be highly valuable for identifying specific regulatory mechanisms whose activity is modulated by natural genetic variation and leads to variable O3 responses. Thus, future work that optimizes techniques to measure transcription factor binding and modifications will be essential to draw gene- and pathway-level conclusions about specific mechanisms driving differential expression between these two strains or across larger panels of genetically diverse mice. Results from these studies will illuminate biological pathways that can then be tested in targeted studies in humans.
Finally, we did not observe a significant influence of sex on inflammatory, injury, or cytokine secretion in response to O3 exposure. In addition, the most variably expressed genes were only modestly influenced by sex (separation by sex in principal component 9, which accounted for 2.62% total variance). This contrasts with previous studies demonstrating that O3 responses vary by sex; however, there is not a consensus stating which sex is more sensitive to O3 exposure. Epidemiologic studies have indicated that women have higher rates of hospitalization and mortality following increases in ambient O3 levels (97–99) and experimental studies have indicated that female mice display increased proinflammatory gene expression profiles and secretion of cytokines and chemokines in response to acute O3 exposure (100–102). Otherwise, results regarding sex differences in other indicators of O3 response, including those measured in the study presented here (airway inflammation and injury), have been conflicting (103–105). Future work that examines this question more closely, perhaps in a wider set of CC strains, with additional molecular measurements, or at different time points will be poised to conclusively determine whether sex might interact with genotype to result in divergent outcomes.
In conclusion, we have shown that respiratory responses to O3 exposure are highly variable in a small subset of inbred mouse strains from the CC. A primary future goal will be to identify specific regions of the genome that contribute to interstrain differences in responsiveness (i.e., map QTL) that define an integrative model of response encompassing DNA variation and chromatin architecture through to phenotypic outcomes.
DATA AVAILABILITY
Raw RNA- and ATAC-seq data are deposited in the Sequence Read Archive (BioProject PRJNA728733; https://www.ncbi.nlm.nih.gov/bioproject/PRJNA728733), and corresponding processed data files are available on the Gene Expression Omnibus (SuperSeries GSE174207; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE174207). Source code is available as a GitHub repository: https://github.com/adelaidetovar/ozone-strain-survey.
SUPPLEMENTAL DATA
Supplemental Table S1: https://doi.org/10.6084/m9.figshare.14658288.v1.
Supplemental Table S2: https://doi.org/10.6084/m9.figshare.14658303.v1.
Supplemental Table S3: https://doi.org/10.6084/m9.figshare.14658309.v1.
Supplemental Table S4: https://doi.org/10.6084/m9.figshare.14658315.v1.
Supplemental Figs. S1–S6: https://doi.org/10.6084/m9.figshare.14658270.v1.
GRANTS
This research was funded by NIH Grants ES024965 and ES024965-S1 to S.N.P.K., a UNC Center for Environmental Health and Susceptibility Pilot Project Award to T.S.F. and S.N.P.K. (P30ES010126), T32 training grants to G.J.S. and W.L.C. (ES007126-35), a Leon and Bertha Golberg Postdoctoral Fellowship from the UNC Curriculum in Toxicology and Environmental Medicine to G.J.S., and UNC Dissertation Completion Fellowships to A.T. and B.P.K.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
A.T., T.S.F., and S.N.P.K. conceived and designed research; A.T., G.J.S., J.M.T., K.M.M., T.P.M., and S.N.P.K. performed experiments; A.T., W.L.C., B.P.K., T.P.M., T.S.F., and S.N.P.K. analyzed data; A.T., W.L.C., G.J.S., B.P.K., T.P.M., T.S.F., and S.N.P.K. interpreted results of experiments; A.T., W.L.C., B.P.K., T.P.M., T.S.F., and S.N.P.K. prepared figures; A.T., W.L.C., T.S.F., and S.N.P.K. drafted manuscript; A.T., W.L.C., G.J.S., K.M.M., T.P.M., T.S.F., and S.N.P.K. edited and revised manuscript; A.T., W.L.C., G.J.S., J.M.T., B.P.K., K.M.M., T.P.M., T.S.F., and S.N.P.K. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors acknowledge the assistance of Daniel Vargas and Jessica Bustamante (technical support), Carlton Anderson (UNC Advanced Analytics Core, Luminex processing), the UNC high-throughput sequencing facility (library preparation, RNA- and ATAC-seq), Courtney Nesline and the UNC Division of Comparative Medicine, and Darla Miller, Ginger Shaw, and Dr. Rachel Lynch of the UNC Systems Genetics Core Facility (Collaborative Cross mice).
Preprint is available at https://doi.org/10.1101/2021.01.29.428733.
REFERENCES
- 1.Akinbami LJ, Lynch CD, Parker JD, Woodruff TJ. The association between childhood asthma prevalence and monitored air pollutants in metropolitan areas, United States, 2001-2004. Environ Res 110: 294–301, 2010. doi: 10.1016/j.envres.2010.01.001. [DOI] [PubMed] [Google Scholar]
- 2.McConnell R, Berhane K, Gilliland F, London SJ, Islam T, Gauderman WJ, Avol E, Margolis HG, Peters JM. Asthma in exercising children exposed to ozone: a cohort study. Lancet 359: 386–391, 2002. doi: 10.1016/S0140-6736(02)07597-9. [DOI] [PubMed] [Google Scholar]
- 3.Thurston GD, Balmes JR, Garcia E, Gilliland FD, Rice MB, Schikowski T, Van Winkle LS, Annesi-Maesano I, Burchard EG, Carlsten C, Harkema JR, Khreis H, Kleeberger SR, Kodavanti UP, London SJ, McConnell R, Peden DB, Pinkerton KE, Reibman J, White CW. Outdoor air pollution and new-onset airway disease. An official American Thoracic Society Workshop report. Ann Am Thorac Soc 17: 387–398, 2020. doi: 10.1513/AnnalsATS.202001-046ST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim BJ, Kwon JW, Seo JH, Kim HB, Lee SY, Park KS, Yu J, Kim HC, Leem JH, Sakong J, Kim SY, Lee CG, Kang DM, Ha M, Hong YC, Kwon HJ, Hong SJ. Association of ozone exposure with asthma, allergic rhinitis, and allergic sensitization. Ann Allergy Asthma Immunol 107: 214–219.e1, 2011. doi: 10.1016/j.anai.2011.05.025. [DOI] [PubMed] [Google Scholar]
- 5.Shin S, Bai L, Burnett RT, Kwong JC, Hystad P, van Donkelaar A, Lavigne E, Weichenthal S, Copes R, Martin RV, Kopp A, Chen H. Air pollution as a risk factor for incident chronic obstructive pulmonary disease and asthma: 15-year population-based cohort study. Am J Respir Crit Care Med 203: 1138–1148, 2021. doi: 10.1164/rccm.201909-1744OC. [DOI] [PubMed] [Google Scholar]
- 6.McDonnell WF 3rd, Horstman DH, Abdul-Salaam S, House DE. Reproducibility of individual responses to ozone exposure. Am Rev Respir Dis 131: 36–40, 1985. doi: 10.1164/arrd.1985.131.S5.S36. [DOI] [PubMed] [Google Scholar]
- 7.Hazucha MJ, Folinsbee LJ, Bromberg PA. Distribution and reproducibility of spirometric response to ozone by gender and age. J Appl Physiol (1985) 95: 1917–1925, 2003. doi: 10.1152/japplphysiol.00490.2003. [DOI] [PubMed] [Google Scholar]
- 8.Holz O, Jörres RA, Timm P, Mücke M, Richter K, Koschyk S, Magnussen H. Ozone-induced airway inflammatory changes differ between individuals and are reproducible. Am J Respir Crit Care Med 159: 776–784, 1999. doi: 10.1164/ajrccm.159.3.9806098. [DOI] [PubMed] [Google Scholar]
- 9.Que LG, Stiles JV, Sundy JS, Foster WM. Pulmonary function, bronchial reactivity, and epithelial permeability are response phenotypes to ozone and develop differentially in healthy humans. J Appl Physiol (1985) 111: 679–687, 2011. doi: 10.1152/japplphysiol.00337.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bauer AK, Kleeberger SR. Genetic mechanisms of susceptibility to ozone-induced lung disease. Ann NY Acad Sci 1203: 113–119, 2010. doi: 10.1111/j.1749-6632.2010.05606.x. [DOI] [PubMed] [Google Scholar]
- 11.Romieu I, Moreno-Macias H, London SJ. Gene by environment interaction and ambient air pollution. Proc Am Thorac Soc 7: 116–122, 2010. doi: 10.1513/pats.200909-097RM. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dye JA, Costa DL, Kodavanti UP. Executive summary: variation in susceptibility to ozone-induced health effects in rodent models of cardiometabolic disease. Inhal Toxicol 27, Suppl 1: 105–115, 2015. doi: 10.3109/08958378.2014.995388. [DOI] [PubMed] [Google Scholar]
- 13.Hatch GE, Crissman K, Schmid J, Richards JE, Ward WO, Schladweiler MC, Ledbetter AD, Kodavanti UP. Strain differences in antioxidants in rat models of cardiovascular disease exposed to ozone. Inhal Toxicol 27, Suppl 1: 54–62, 2015. doi: 10.3109/08958378.2014.954170. [DOI] [PubMed] [Google Scholar]
- 14.Kleeberger SR, Levitt RC, Zhang LY, Longphre M, Harkema J, Jedlicka A, Eleff SM, DiSilvestre D, Holroyd KJ. Linkage analysis of susceptibility to ozone-induced lung inflammation in inbred mice. Nat Genet 17: 475–478, 1997. doi: 10.1038/ng1297-475. [DOI] [PubMed] [Google Scholar]
- 15.Kodavanti UP, Ledbetter AD, Thomas RF, Richards JE, Ward WO, Schladweiler MC, Costa DL. Variability in ozone-induced pulmonary injury and inflammation in healthy and cardiovascular-compromised rat models. Inhal Toxicol 27, Suppl 1: 39–53, 2015. doi: 10.3109/08958378.2014.954169. [DOI] [PubMed] [Google Scholar]
- 16.Prows DR, Shertzer HG, Daly MJ, Sidman CL, Leikauf GD. Genetic analysis of ozone-induced acute lung injury in sensitive and resistant strains of mice. Nat Genet 17: 471–474, 1997. doi: 10.1038/ng1297-471. [DOI] [PubMed] [Google Scholar]
- 17.Savov JD, Whitehead GS, Wang J, Liao G, Usuka J, Peltz G, Foster WM, Schwartz DA. Ozone-induced acute pulmonary injury in inbred mouse strains. Am J Respir Cell Mol Biol 31: 69–77, 2004. doi: 10.1165/rcmb.2003-0001OC. [DOI] [PubMed] [Google Scholar]
- 18.Buscher K, Ehinger E, Gupta P, Pramod AB, Wolf D, Tweet G, Pan C, Mills CD, Lusis AJ, Ley K. Natural variation of macrophage activation as disease-relevant phenotype predictive of inflammation and cancer survival. Nat Commun 8: 16041, 2017. doi: 10.1038/ncomms16041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang W, Carbone MA, Lyman RF, Anholt RRH, Mackay TFC. Genotype by environment interaction for gene expression in Drosophila melanogaster. Nat Commun 11: 5451, 2020. doi: 10.1038/s41467-020-19131-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Idaghdour Y, Czika W, Shianna KV, Lee SH, Visscher PM, Martin HC, Miclaus K, Jadallah SJ, Goldstein DB, Wolfinger RD, Gibson G. Geographical genomics of human leukocyte gene expression variation in southern Morocco. Nat Genet 42: 62–67, 2010. doi: 10.1038/ng.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Orozco LD, Bennett BJ, Farber CR, Ghazalpour A, Pan C, Che N, Wen P, Qi HX, Mutukulu A, Siemers N, Neuhaus I, Yordanova R, Gargalovic P, Pellegrini M, Kirchgessner T, Lusis AJ. Unraveling inflammatory responses using systems genetics and gene-environment interactions in macrophages. Cell 151: 658–670, 2012. doi: 10.1016/j.cell.2012.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reed LK, Williams S, Springston M, Brown J, Freeman K, DesRoches CE, Sokolowski MB, Gibson G. Genotype-by-diet interactions drive metabolic phenotype variation in Drosophila melanogaster. Genetics 185: 1009–1019, 2010. doi: 10.1534/genetics.109.113571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhou S, Luoma SE, St Armour GE, Thakkar E, Mackay TFC, Anholt RRH. A Drosophila model for toxicogenomics: genetic variation in susceptibility to heavy metal exposure. PLoS Genet 13: e1006907, 2017. doi: 10.1371/journal.pgen.1006907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang H, Wang JR, Didion JP, Buus RJ, Bell TA, Welsh CE, Bonhomme F, Yu AH, Nachman MW, Pialek J, Tucker P, Boursot P, McMillan L, Churchill GA, de Villena FP. Subspecific origin and haplotype diversity in the laboratory mouse. Nat Genet 43: 648–655, 2011. doi: 10.1038/ng.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Choudhary I, Vo T, Paudel K, Patial S, Saini Y. Compartment-specific transcriptomics of ozone-exposed murine lungs reveals sex- and cell type-associated perturbations relevant to mucoinflammatory lung diseases. Am J Physiol Lung Cell Mol Physiol 320: L99–L125, 2021. doi: 10.1152/ajplung.00381.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Francis M, Guo G, Kong B, Abramova EV, Cervelli JA, Gow AJ, Laskin JD, Laskin DL. Regulation of lung macrophage activation and oxidative stress following ozone exposure by farnesoid X receptor. Toxicol Sci 177: 441–453, 2020. doi: 10.1093/toxsci/kfaa111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hodge MX, Reece SW, Madenspacher JH, Gowdy KM. In vivo assessment of alveolar macrophage efferocytosis following ozone exposure. J Vis Exp 152: e60109, 2019. doi: 10.3791/60109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mikerov AN, Gan X, Umstead TM, Miller L, Chinchilli VM, Phelps DS, Floros J. Sex differences in the impact of ozone on survival and alveolar macrophage function of mice after Klebsiella pneumoniae infection. Respir Res 9: 24, 2008. doi: 10.1186/1465-9921-9-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sunil VR, Patel-Vayas K, Shen J, Laskin JD, Laskin DL. Classical and alternative macrophage activation in the lung following ozone-induced oxidative stress. Toxicol Appl Pharmacol 263: 195–202, 2012. doi: 10.1016/j.taap.2012.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tovar A, Smith GJ, Thomas JM, Crouse WL, Harkema JR, Kelada SNP. Transcriptional profiling of the murine airway response to acute ozone exposure. Toxicol Sci 173: 114–130, 2020. doi: 10.1093/toxsci/kfz219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Collaborative Cross Consortium. The genome architecture of the Collaborative Cross mouse genetic reference population. Genetics 190: 389–401, 2012. doi: 10.1534/genetics.111.132639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Srivastava A, Morgan AP, Najarian ML, Sarsani VK, Sigmon JS, Shorter JR, Kashfeen A, McMullan RC, Williams LH, Giusti-Rodríguez P, Ferris MT, Sullivan P, Hock P, Miller DR, Bell TA, McMillan L, Churchill GA, de Villena FP. . Genomes of the mouse Collaborative Cross. Genetics 206: 537–556, 2017. doi: 10.1534/genetics.116.198838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Roberts A, Pardo-Manuel de Villena F, Wang W, McMillan L, Threadgill DW. The polymorphism architecture of mouse genetic resources elucidated using genome-wide resequencing data: implications for QTL discovery and systems genetics. Mamm Genome 18: 473–481, 2007. doi: 10.1007/s00335-007-9045-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mosedale M, Cai Y, Eaddy JS, Corty RW, Nautiyal M, Watkins PB, Valdar W. Identification of candidate risk factor genes for human idelalisib toxicity using a Collaborative Cross approach. Toxicol Sci 172: 265–278, 2019. doi: 10.1093/toxsci/kfz199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mosedale M, Kim Y, Brock WJ, Roth SE, Wiltshire T, Eaddy JS, Keele GR, Corty RW, Xie Y, Valdar W, Watkins PB. Editor’s highlight: candidate risk factors and mechanisms for tolvaptan-induced liver injury are identified using a Collaborative Cross approach. Toxicol Sci 156: 438–454, 2017. doi: 10.1093/toxsci/kfw269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lewis L, Borowa-Mazgaj B, de Conti A, Chappell GA, Luo YS, Bodnar W, Konganti K, Wright FA, Threadgill DW, Chiu WA, Pogribny IP, Rusyn I. Population-based analysis of DNA damage and epigenetic effects of 1,3-butadiene in the mouse. Chem Res Toxicol 32: 887–898, 2019. doi: 10.1021/acs.chemrestox.9b00035. [DOI] [PubMed] [Google Scholar]
- 37.Ferris MT, Aylor DL, Bottomly D, Whitmore AC, Aicher LD, Bell TA, Bradel-Tretheway B, Bryan JT, Buus RJ, Gralinski LE, Haagmans BL, McMillan L, Miller DR, Rosenzweig E, Valdar W, Wang J, Churchill GA, Threadgill DW, McWeeney SK, Katze MG, Pardo-Manuel de Villena F, Baric RS, Heise MT. Modeling host genetic regulation of influenza pathogenesis in the Collaborative Cross. PLoS Pathog 9: e1003196, 2013. doi: 10.1371/journal.ppat.1003196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gralinski LE, Ferris MT, Aylor DL, Whitmore AC, Green R, Frieman MB, Deming D, Menachery VD, Miller DR, Buus RJ, Bell TA, Churchill GA, Threadgill DW, Katze MG, McMillan L, Valdar W, Heise MT, Pardo-Manuel de Villena F, Baric RS. Genome wide identification of SARS-CoV susceptibility loci using the Collaborative Cross. PLoS Genet 11: e1005504, 2015. doi: 10.1371/journal.pgen.1005504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kelada SN, Carpenter DE, Aylor DL, Chines P, Rutledge H, Chesler EJ, Churchill GA, Pardo-Manuel de Villena F, Schwartz DA, Collins FS. Integrative genetic analysis of allergic inflammation in the murine lung. Am J Respir Cell Mol Biol 51: 436–445, 2014. doi: 10.1165/rcmb.2013-0501OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bubier JA, Philip VM, Quince C, Campbell J, Zhou Y, Vishnivetskaya T, Duvvuru S, Blair RH, Ndukum J, Donohue KD, Foster CM, Mellert DJ, Weinstock G, Culiat CT, O’Hara BF, Palumbo AV, Podar M, Chesler EA. Microbe associated with sleep revealed by a novel systems genetic analysis of the microbiome in Collaborative Cross mice. Genetics 214: 719–733, 2020. doi: 10.1534/genetics.119.303013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Price A, Okumura A, Haddock E, Feldmann F, Meade-White K, Sharma P, Artami M, Lipkin WI, Threadgill DW, Feldmann H, Rasmussen AL. Transcriptional correlates of tolerance and lethality in mice predict Ebola virus disease patient outcomes. Cell Rep 30: 1702–1713.e6, 2020. doi: 10.1016/j.celrep.2020.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Salimova E, Nowak KJ, Estrada AC, Furtado MB, McNamara E, Nguyen Q, Balmer L, Preuss C, Holmes JW, Ramialison M, Morahan G, Rosenthal NA. Variable outcomes of human heart attack recapitulated in genetically diverse mice. NPJ Regen Med 4: 5, 2019. doi: 10.1038/s41536-019-0067-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Smith GJ, Walsh L, Higuchi M, Kelada SNP. Development of a large-scale computer-controlled ozone inhalation exposure system for rodents. Inhal Toxicol 31: 61–72, 2019. doi: 10.1080/08958378.2019.1597222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10: 1213–1218, 2013. doi: 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lavin Y, Winter D, Blecher-Gonen R, David E, Keren-Shaul H, Merad M, Jung S, Amit I. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell 159: 1312–1326, 2014. doi: 10.1016/j.cell.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29: 15–21, 2013. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12: 323, 2011. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15: 550, 2014. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stephens M. False discovery rates: a new deal. Biostatistics 18: 275–294, 2017. doi: 10.1093/biostatistics/kxw041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9: 559, 2008. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, McDermott MG, Monteiro CD, Gundersen GW, Ma’ayan A. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 44: W90–W97, 2016. doi: 10.1093/nar/gkw377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Smith JP, Ryan Corces M, Xu J, Reuter VP, Chang HY, Sheffield NC. PEPATAC: An optimized ATAC-seq pipeline with serial alignments (Preprint). bioRxiv 2020. doi: 10.1101/2020.10.21.347054. [DOI] [PMC free article] [PubMed]
- 53.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38: 576–589, 2010. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Du Y, Yang M, Wei W, Huynh HD, Herz J, Saghatelian A, Wan Y. Macrophage VLDL receptor promotes PAFAH secretion in mother’s milk and suppresses systemic inflammation in nursing neonates. Nat Commun 3: 1008, 2012. doi: 10.1038/ncomms2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xu M, Yang W, Wang X, Nayak DK. Lung secretoglobin Scgb1a1 influences alveolar macrophage-mediated inflammation and immunity. Front Immunol 11: 584310, 2020. doi: 10.3389/fimmu.2020.584310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Blomberg A, Mudway I, Svensson M, Hagenbjörk-Gustafsson A, Thomasson L, Helleday R, Dumont X, Forsberg B, Nordberg G, Bernard A. Clara cell protein as a biomarker for ozone-induced lung injury in humans. Eur Respir J 22: 883–888, 2003. doi: 10.1183/09031936.03.00048203. [DOI] [PubMed] [Google Scholar]
- 57.Gong Y, Hart E, Shchurin A, Hoover-Plow J. Inflammatory macrophage migration requires MMP-9 activation by plasminogen in mice. J Clin Invest 118: 3012–3024, 2008. doi: 10.1172/JCI32750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Min D, Moore AG, Bain MA, Breit SN, Lyons JG. Activation of macrophage promatrix metalloproteinase-9 by lipopolysaccharide-associated proteinases. J Immunol 168: 2449–2455, 2002. doi: 10.4049/jimmunol.168.5.2449. [DOI] [PubMed] [Google Scholar]
- 59.Bauer AK, Rondini EA, Hummel KA, Degraff LM, Walker C, Jedlicka AE, Kleeberger SR. Identification of candidate genes downstream of TLR4 signaling after ozone exposure in mice: a role for heat-shock protein 70. Environ Health Perspect 119: 1091–1097, 2011. doi: 10.1289/ehp.1003326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Carballo E, Lai WS, Blackshear PJ. Feedback inhibition of macrophage tumor necrosis factor-α production by tristetraprolin. Science 281: 1001–1005, 1998. doi: 10.1126/science.281.5379.1001. [DOI] [PubMed] [Google Scholar]
- 61.Dasgupta P, Dorsey NJ, Li J, Qi X, Smith EP, Yamaji-Kegan K, Keegan AD. The adaptor protein insulin receptor substrate 2 inhibits alternative macrophage activation and allergic lung inflammation. Sci Signal 9: ra63, 2016. doi: 10.1126/scisignal.aad6724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fakhrzadeh L, Laskin JD, Laskin DL. Ozone-induced production of nitric oxide and TNF-α and tissue injury are dependent on NF-κB p50. Am J Physiol Lung Cell Mol Physiol 287: L279–L285, 2004. doi: 10.1152/ajplung.00348.2003. [DOI] [PubMed] [Google Scholar]
- 63.Speen AM, Kim HH, Bauer RN, Meyer M, Gowdy KM, Fessler MB, Duncan KE, Liu W, Porter NA, Jaspers I. Ozone-derived oxysterols affect liver X receptor (LXR) signaling: a potential role for lipid-protein adducts. J Biol Chem 291: 25192–25206, 2016. doi: 10.1074/jbc.M116.732362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Francis M, Groves AM, Sun R, Cervelli JA, Choi H, Laskin JD, Laskin DL. Editor’s highlight: CCR2 regulates inflammatory cell accumulation in the lung and tissue injury following ozone exposure. Toxicol Sci 155: 474–484, 2017. doi: 10.1093/toxsci/kfw226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Murphy SR, Oslund KL, Hyde DM, Miller LA, Van Winkle LS, Schelegle ES. Ozone-induced airway epithelial cell death, the neurokinin-1 receptor pathway, and the postnatal developing lung. Am J Physiol Lung Cell Mol Physiol 307: L471–L481, 2014. doi: 10.1152/ajplung.00324.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang H, Liu H, Iles KE, Liu R-M, Postlethwait EM, Laperche Y, Forman HJ. 4-Hydroxynonenal induces rat gamma-glutamyl transpeptidase through mitogen-activated protein kinase-mediated electrophile response element/nuclear factor erythroid 2-related factor 2 signaling. Am J Respir Cell Mol Biol 34: 174–181, 2006. doi: 10.1165/rcmb.2005-0280OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Satoh T, Kidoya H, Naito H, Yamamoto M, Takemura N, Nakagawa K, Yoshioka Y, Morii E, Takakura N, Takeuchi O, Akira S. Critical role of Trib1 in differentiation of tissue-resident M2-like macrophages. Nature 495: 524–528, 2013. doi: 10.1038/nature11930. [DOI] [PubMed] [Google Scholar]
- 68.Traber KE, Hilliard KL, Allen E, Wasserman GA, Yamamoto K, Jones MR, Mizgerd JP, Quinton LJ. Induction of STAT3-dependent CXCL5 expression and neutrophil recruitment by oncostatin-M during pneumonia. Am J Respir Cell Mol Biol 53: 479–488, 2015. doi: 10.1165/rcmb.2014-0342OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Evrin C, Clarke P, Zech J, Lurz R, Sun J, Uhle S, Li H, Stillman B, Speck C. A double-hexameric MCM2-7 complex is loaded onto origin DNA during licensing of eukaryotic DNA replication. Proc Natl Acad Sci USA 106: 20240–20245, 2009. doi: 10.1073/pnas.0911500106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kim JH, Shim J, Ji MJ, Jung Y, Bong SM, Jang YJ, Yoon EK, Lee SJ, Kim KG, Kim YH, Lee C, Lee BI, Kim KT. The condensin component NCAPG2 regulates microtubule-kinetochore attachment through recruitment of polo-like kinase 1 to kinetochores. Nat Commun 5: 4588, 2014. doi: 10.1038/ncomms5588. [DOI] [PubMed] [Google Scholar]
- 71.Yuan J, Chen J. FIGNL1-containing protein complex is required for efficient homologous recombination repair. Proc Natl Acad Sci USA 110: 10640–10645, 2013. doi: 10.1073/pnas.1220662110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Che L, Jin Y, Zhang C, Lai T, Zhou H, Xia L, Tian B, Zhao Y, Liu J, Wu Y, Wu Y, Du J, Li W, Ying S, Chen Z, Shen H. Ozone-induced IL-17A and neutrophilic airway inflammation is orchestrated by the caspase-1-IL-1 cascade. Sci Rep 6: 18680, 2016. doi: 10.1038/srep18680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dahl M, Bauer AK, Arredouani M, Soininen R, Tryggvason K, Kleeberger SR, Kobzik L. Protection against inhaled oxidants through scavenging of oxidized lipids by macrophage receptors MARCO and SR-AI/II. J Clin Invest 117: 757–764, 2007. doi: 10.1172/JCI29968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kesic MJ, Meyer M, Bauer R, Jaspers I. Exposure to ozone modulates human airway protease/antiprotease balance contributing to increased influenza a infection. PLoS One 7: e35108, 2012. doi: 10.1371/journal.pone.0035108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Michalec L, Choudhury BK, Postlethwait E, Wild JS, Alam R, Lett-Brown M, Sur S. CCL7 and CXCL10 orchestrate oxidative stress-induced neutrophilic lung inflammation. J Immunol 168: 846–852, 2002. doi: 10.4049/jimmunol.168.2.846. [DOI] [PubMed] [Google Scholar]
- 76.Robertson S, Colombo ES, Lucas SN, Hall PR, Febbraio M, Paffett ML, Campen MJ. CD36 mediates endothelial dysfunction downstream of circulating factors induced by O3 exposure. Toxicol Sci 134: 304–311, 2013. doi: 10.1093/toxsci/kft107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhao Q, Simpson LG, Driscoll KE, Leikauf GD. Chemokine regulation of ozone-induced neutrophil and monocyte inflammation. Am J Physiol Lung Cell Mol Physiol 274: L39–L46, 1998. doi: 10.1152/ajplung.1998.274.1.L39. [DOI] [PubMed] [Google Scholar]
- 78.Cheng G, Zhong M, Kawaguchi R, Kassai M, Al-Ubaidi M, Deng J, Ter-Stepanian M, Sun H. Identification of PLXDC1 and PLXDC2 as the transmembrane receptors for the multifunctional factor PEDF. Elife 3: e05401, 2014. doi: 10.7554/eLife.05401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Donovan A, Lima CA, Pinkus JL, Pinkus GS, Zon LI, Robine S, Andrews NC. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab 1: 191–200, 2005. doi: 10.1016/j.cmet.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 80.Chang MY, Kang I, Gale M Jr, Manicone AM, Kinsella MG, Braun KR, Wigmosta T, Parks WC, Altemeier WA, Wight TN, Frevert CW. Versican is produced by Trif- and type I interferon-dependent signaling in macrophages and contributes to fine control of innate immunity in lungs. Am J Physiol Lung Cell Mol Physiol 313: L1069–L1086, 2017. doi: 10.1152/ajplung.00353.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Takahashi K, Koga K, Linge HM, Zhang Y, Lin X, Metz CN, Al-Abed Y, Ojamaa K, Miller EJ. Macrophage CD74 contributes to MIF-induced pulmonary inflammation. Respir Res 10: 33, 2009. doi: 10.1186/1465-9921-10-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kosmider B, Loader JE, Murphy RC, Mason RJ. Apoptosis induced by ozone and oxysterols in human alveolar epithelial cells. Free Radic Biol Med 48: 1513–1524, 2010. doi: 10.1016/j.freeradbiomed.2010.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chappell GA, Israel JW, Simon JM, Pott S, Safi A, Eklund K, Sexton KG, Bodnar W, Lieb JD, Crawford GE, Rusyn I, Furey TS. Variation in DNA-damage responses to an inhalational carcinogen (1,3-butadiene) in relation to strain-specific differences in chromatin accessibility and gene transcription profiles in C57BL/6J and CAST/EiJ mice. Environ Health Perspect 125: 107006, 2017. doi: 10.1289/EHP1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kleeberger SR, Bassett DJ, Jakab GJ, Levitt RC. A genetic model for evaluation of susceptibility to ozone-induced inflammation. Am J Physiol Lung Cell Mol Physiol 258: L313–L320, 1990. doi: 10.1152/ajplung.1990.258.6.L313. [DOI] [PubMed] [Google Scholar]
- 85.Keele GR, Quach BC, Israel JW, Chappell GA, Lewis L, Safi A, Simon JM, Cotney P, Crawford GE, Valdar W, Rusyn I, Furey TS. Integrative QTL analysis of gene expression and chromatin accessibility identifies multi-tissue patterns of genetic regulation. PLoS Genet 16: e1008537, 2020. doi: 10.1371/journal.pgen.1008537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Li Y, Alvarez OA, Gutteling EW, Tijsterman M, Fu J, Riksen JAG, Hazendonk E, Prins P, Plasterk RHA, Jansen RC, Breitling R, Kammenga JE. Mapping determinants of gene expression plasticity by genetical genomics in C. elegans. PLoS Genet 2: e222, 2006. doi: 10.1371/journal.pgen.0020222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Smith EN, Kruglyak L. Gene-environment interaction in yeast gene expression. PLoS Biol 6: e83, 2008. doi: 10.1371/journal.pbio.0060083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Fairfax BP, Humburg P, Makino S, Naranbhai V, Wong D, Lau E, Jostins L, Plant K, Andrews R, McGee C, Knight JC. Innate immune activity conditions the effect of regulatory variants upon monocyte gene expression. Science 343: 1246949, 2014. doi: 10.1126/science.1246949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kim-Hellmuth S, Bechheim M, Pütz B, Mohammadi P, Nédélec Y, Giangreco N, Becker J, Kaiser V, Fricker N, Beier E, Boor P, Castel SE, Nöthen MM, Barreiro LB, Pickrell JK, Müller-Myhsok B, Lappalainen T, Schumacher J, Hornung V. Genetic regulatory effects modified by immune activation contribute to autoimmune disease associations. Nat Commun 8: 266, 2017. doi: 10.1038/s41467-017-00366-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lee MN, Ye C, Villani AC, Raj T, Li W, Eisenhaure TM, Imboywa SH, Chipendo PI, Ran FA, Slowikowski K, Ward LD, Raddassi K, McCabe C, Lee MH, Frohlich IY, Hafler DA, Kellis M, Raychaudhuri S, Zhang F, Stranger BE, Benoist CO, De Jager PL, Regev A, Hacohen N. Common genetic variants modulate pathogen-sensing responses in human dendritic cells. Science 343: 1246980, 2014. doi: 10.1126/science.1246980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Smirnov DA, Morley M, Shin E, Spielman RS, Cheung VG. Genetic analysis of radiation-induced changes in human gene expression. Nature 459: 587–591, 2009. doi: 10.1038/nature07940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Manet C, Simon-Lorière E, Jouvion G, Hardy D, Prot M, Conquet L, Flamand M, Panthier JJ, Sakuntabhai A, Montagutelli X. Genetic diversity of Collaborative Cross mice controls viral replication, clinical severity, and brain pathology induced by Zika virus infection, independently of Oas1b. J Virol 94: e01034-19, 2020. doi: 10.1128/JVI.01034-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Xue J, Hutchins EK, Elnagheeb M, Li Y, Valdar W, McRitchie S, Sumner S, Ideraabdullah FY. Maternal liver metabolic response to chronic vitamin D deficiency is determined by mouse strain genetic background. Curr Dev Nutr 4: nzaa106, 2020. doi: 10.1093/cdn/nzaa106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Venkatratnam A, Furuya S, Kosyk O, Gold A, Bodnar W, Konganti K, Threadgill DW, Gillespie KM, Aylor DL, Wright FA, Chiu WA, Rusyn I. Editor’s highlight: Collaborative Cross mouse population enables refinements to characterization of the variability in toxicokinetics of trichloroethylene and provides genetic evidence for the role of PPAR pathway in its oxidative metabolism. Toxicol Sci 158: 48–62, 2017. doi: 10.1093/toxsci/kfx065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Alasoo K, Rodrigues J, Mukhopadhyay S, Knights AJ, Mann AL, Kundu K, Hale C, Dougan G, Gaffney DJ; HIPSCI Consortium. Shared genetic effects on chromatin and gene expression indicate a role for enhancer priming in immune response. Nat Genet 50: 424–431, 2018. doi: 10.1038/s41588-018-0046-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Degner JF, Pai AA, Pique-Regi R, Veyrieras JB, Gaffney DJ, Pickrell JK, De Leon S, Michelini K, Lewellen N, Crawford GE, Stephens M, Gilad Y, Pritchard JK. DNase I sensitivity QTLs are a major determinant of human expression variation. Nature 482: 390–394, 2012. doi: 10.1038/nature10808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Liang L, Cai Y, Barratt B, Lyu B, Chan Q, Hansell AL, Xie W, Zhang D, Kelly FJ, Tong Z. Associations between daily air quality and hospitalisations for acute exacerbation of chronic obstructive pulmonary disease in Beijing, 2013-17: an ecological analysis. Lancet Planet Health 3: e270–e279, 2019. doi: 10.1016/S2542-5196(19)30085-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Medina-Ramón M, Schwartz J. Who is more vulnerable to die from ozone air pollution? Epidemiology 19: 672–679, 2008. doi: 10.1097/EDE.0b013e3181773476. [DOI] [PubMed] [Google Scholar]
- 99.Stafoggia M, Forastiere F, Faustini A, Biggeri A, Bisanti L, Cadum E, Cernigliaro A, Mallone S, Pandolfi P, Serinelli M, Tessari R, Vigotti MA, Perucci CA; EpiAir Group. Susceptibility factors to ozone-related mortality: a population-based case-crossover analysis. Am J Respir Crit Care Med 182: 376–384, 2010. doi: 10.1164/rccm.200908-1269OC. [DOI] [PubMed] [Google Scholar]
- 100.Cabello N, Mishra V, Sinha U, DiAngelo SL, Chroneos ZC, Ekpa NA, Cooper TK, Caruso CR, Silveyra P. Sex differences in the expression of lung inflammatory mediators in response to ozone. Am J Physiol Lung Cell Mol Physiol 309: L1150–L1163, 2015. doi: 10.1152/ajplung.00018.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Fuentes N, Roy A, Mishra V, Cabello N, Silveyra P. Sex-specific microRNA expression networks in an acute mouse model of ozone-induced lung inflammation. Biol Sex Differ 9: 18, 2018. doi: 10.1186/s13293-018-0177-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mishra V, DiAngelo SL, Silveyra P. Sex-specific IL-6-associated signaling activation in ozone-induced lung inflammation. Biol Sex Differ 7: 16, 2016. doi: 10.1186/s13293-016-0069-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Birukova A, Cyphert-Daly J, Cumming RI, Yu YR, Gowdy KM, Que LG, Tighe RM. Sex Modifies acute ozone-mediated airway physiologic responses. Toxicol Sci 169: 499–510, 2019. doi: 10.1093/toxsci/kfz056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Cho Y, Abu-Ali G, Tashiro H, Brown TA, Osgood RS, Kasahara DI, Huttenhower C, Shore SA. Sex differences in pulmonary responses to ozone in mice. Role of the microbiome. Am J Respir Cell Mol Biol 60: 198–208, 2019. doi: 10.1165/rcmb.2018-0099OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kasahara DI, Wilkinson JE, Cho Y, Cardoso AP, Huttenhower C, Shore SA. The interleukin-33 receptor contributes to pulmonary responses to ozone in male mice: role of the microbiome. Respir Res 20: 197, 2019. doi: 10.1186/s12931-019-1168-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table S1: https://doi.org/10.6084/m9.figshare.14658288.v1.
Supplemental Table S2: https://doi.org/10.6084/m9.figshare.14658303.v1.
Supplemental Table S3: https://doi.org/10.6084/m9.figshare.14658309.v1.
Supplemental Table S4: https://doi.org/10.6084/m9.figshare.14658315.v1.
Supplemental Figs. S1–S6: https://doi.org/10.6084/m9.figshare.14658270.v1.
Data Availability Statement
Raw RNA- and ATAC-seq data are deposited in the Sequence Read Archive (BioProject PRJNA728733; https://www.ncbi.nlm.nih.gov/bioproject/PRJNA728733), and corresponding processed data files are available on the Gene Expression Omnibus (SuperSeries GSE174207; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE174207). Source code is available as a GitHub repository: https://github.com/adelaidetovar/ozone-strain-survey.