

Summary
In diffuse large B cell lymphoma (DLBCL), tumors belonging to the ABC but not GCB gene expression subgroup rely upon chronic active B cell receptor signaling for viability, a dependency that is targetable by ibrutinib. A phase III trial (“Phoenix”; NCT01855750) showed a survival benefit of ibrutinib addition to R-CHOP chemotherapy in younger patients with non-GCB DLBCL, but the molecular basis for this benefit was unclear. Analysis of biopsies from Phoenix trial patients revealed 3 previously characterized genetic subtypes of DLBCL: MCD, BN2 and N1. The 3-year event-free survival of younger patients (age≤60) treated with ibrutinib plus R-CHOP was 100% in the MCD and N1 subtypes while the survival of patients with these subtypes treated with R-CHOP alone was significantly inferior (42.9% and 50%, respectively). This work provides a mechanistic understanding of the benefit of ibrutinib addition to chemotherapy, supporting its use in younger patients with non-GCB DLBCL.
Keywords: ABC DLBCL, cancer genomics, MYD88, CD79B, NOTCH1, memory B-cell, BTK inhibitor, precision medicine
eTOC Blurb:
Wilson et al show that patients with two genetic subtypes of DLBCL – MCD and N1 – have 100% survival when treated with the BTK inhibitor ibrutinib plus R-CHOP chemotherapy but ≤50% survival when treated with R-CHOP alone. Both subtypes acquire mutations fostering B cell receptor signaling and BTK dependence, accounting for the therapeutic response.
Graphical Abstract
Introduction
The development of targeted therapy in diffuse large B cell lymphoma (DLBCL), which comprises 40% of all lymphomas, is confounded by its extensive molecular heterogeneity. The first molecular classification of DLBCL, based on gene expression profiling, identified three subgroups –ABC, GCB and Unclassified – that explained, in part, the heterogeneous outcome following chemotherapy (Alizadeh et al., 2000; Lenz et al., 2008b; Rosenwald et al., 2002). The classification of DLBCL has recently been transformed by the discovery of genetic subtypes based on patterns of co-occurring genetic alterations (Chapuy et al., 2018; Lacy et al., 2020; Schmitz et al., 2018; Wright et al., 2020). At least 7 genetic subtypes are recognized currently and have been reproducibly identified in 4 large DLBCL cohorts (Runge et al., 2021; Wright et al., 2020). Importantly, the DLBCL genetic subtypes differ with respect to pathogenesis, phenotypic properties, oncogenic survival pathways and responses to therapy (Chapuy et al., 2018; Lacy et al., 2020; Schmitz et al., 2018; Wright et al., 2020). A probabilistic predictor tool termed LymphGen uses the constellation of genetic alterations in an individual DLBCL to assign it to one or more of these genetic subtypes (Wright et al., 2020).
In this new framework, ABC DLBCL is split into 4 genetic subtypes (MCD, BN2, N1, A53) as is GCB DLBCL (EZB-MYC+, EZB-MYC−, ST2, BN2), while Unclassified DLBCL is enriched for the BN2 subtype (Schmitz et al., 2018; Wright et al., 2020). The subtypes of relevance to the present study include MCD, characterized by MYD88L265P and/or CD79B mutations, BN2, characterized by BCL6 translocations and/or NOTCH2 mutations, and N1, characterized by NOTCH1 mutations. Each subtype additionally acquires multiple genetic lesions targeting various oncogenes and tumor suppressors that distinguish tumors of that subtype from other DLBCL tumors.
The viability of ABC DLBCL cells depends on chronic active B-cell receptor (BCR) signaling, which can be blocked by inhibitors of Bruton tyrosine kinase (BTK) (Davis et al., 2010; Young et al., 2019). This insight prompted a phase II study in relapsed/refractory DLBCL of the BTK inhibitor ibrutinib, which induced a 37% response rate in ABC compared to 5% in GCB (Wilson et al., 2015), as predicted pre-clinically (Davis et al., 2010). Current evidence suggests that ABC tumors that belong to the MCD genetic subtype are highly dependent on BCR signaling and downstream activation of the pro-survival NF-κB pathway. More than 80% of MCD tumors have mutations targeting the CD79B subunit of the BCR and/or the signaling adapter MYD88 (Ngo et al., 2011), which cooperatively activate NF-κB via the MYD88-TLR9-BCR (My-T-BCR) multiprotein signaling complex (Phelan et al., 2018). Co-occurrence of CD79B and MYD88L265P mutations, which happens solely in MCD tumors, was associated with an 80% objective response rate to ibrutinib monotherapy (Wilson et al., 2015).
The dependency of ABC DLBCL tumors on chronic active BCR signaling triggered a phase III randomized evaluation of R-CHOP chemotherapy with or without ibrutinib in newly diagnosed patients with non-GCB DLBCL, a group identified by tumor immunohistochemistry and enriched for ABC DLBCL (“Phoenix” Trial; NCT01855750) (Younes et al., 2019). Although the Phoenix study failed to meet its primary survival endpoint, a planned subset analysis in younger patients (age<60) showed that event-free survival (EFS) and overall survival (OS) were, respectively, 10.8% and 12.3% higher in the ibrutinib arm than in the placebo arm, whereas in older patients there was no benefit, apparently due to unanticipated ibrutinib toxicity in this age group (Younes et al., 2019). Despite this clinical evidence, the value of adding ibrutinib to R-CHOP therapy in younger patients remains controversial because the effect of age was a secondary, not primary trial endpoint. To provide a mechanistic basis for the ibrutinib benefit in younger patients, we tested whether the classification of DLBCL into genetic subtypes could identify patients for whom ibrutinib provided a significant survival advantage in the Phoenix clinical trial.
Results
Formalin-fixed and paraffin-embedded (FFPE) DLBCL tumors were obtained from 773 of the 838 patients (92.2%) on the Phoenix trial (Younes et al., 2019), including 189 enrolled in China (“China” cohort) and 584 enrolled elsewhere (“non-China” cohort) (Table S1). Younger patients (age ≤ 60) comprised 44% (n=340) of this cohort. DNA and RNA sequencing of these biopsies was used to identify mutations, translocations, and gene expression phenotypes (see Methods), and the LymphGen algorithm (Wright et al., 2020) was used to assign tumors to genetic subtypes. As a control to gauge the validity of our genetic subtype assignments, we compared the genetic and phenotypic characteristics of the Phoenix subtypes with those of corresponding subtypes in a previously analyzed “NCI cohort” of 574 DLBCL samples, which were obtained from patients receiving standard chemotherapy at multiple academic centers (Schmitz et al., 2018; Wright et al., 2020).
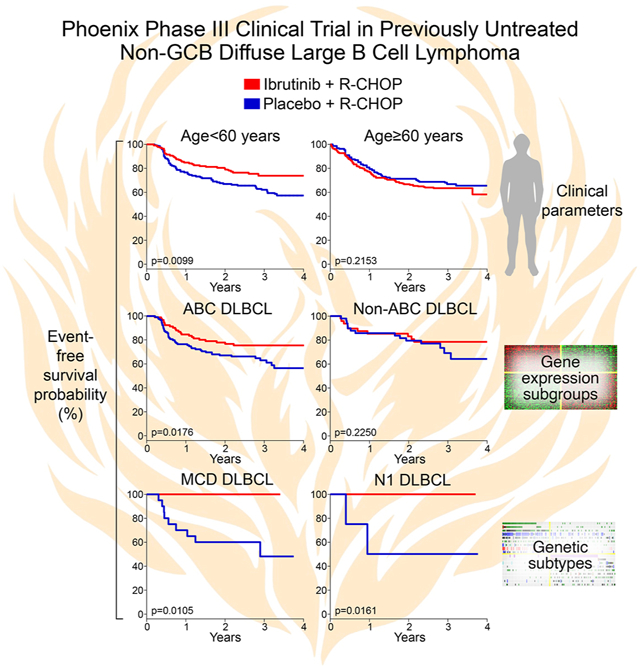
Enrollment on the Phoenix trial was restricted to non-GCB patients as approximated by the “Hans” immunochemical algorithm, which is based on CD10, BCL6 and IRF4 protein expression (Hans et al., 2004). By gene expression profiling, the majority of biopsies from younger Phoenix trial patients were ABC DLBCL (n=239, 70.2%), with the remainder consisting of CD10−negative GCB (n=72, 21.3%) or Unclassified DLBCL (n=29, 8.4%) (Fig. 1A). Ibrutinib provided a survival advantage for patients with ABC DLBCL, as reported (Younes et al., 2019), but this effect was not evident in non-ABC DLBCL, nor in the GCB or Unclassified DLBCL subgroups individually (Fig. 1B, S1A). This provided early evidence that the benefit of ibrutinib depended on molecular attributes of a patient’s tumor.
Figure 1.

Identification of DLBCL genetic subtypes within the Phoenix clinical trial cohort. A. Distribution of gene expression subgroup assignments among tumors from younger (age≤60) Phoenix trial patients. The immunohistochemical method used to enrich for non-GCB DLBCL cases allowed Phoenix trial enrollment of patients with CD10− GCB DLBCL. B. Kaplan-Meier plots of event-free and overall survival in younger (age ≤ 60) patients with ABC or non-ABC DLBCL assigned to the ibrutinib or placebo Phoenix trial arms, as indicated. Shown are log-rank p values for the difference in survival between patients on the ibrutinib and placebo arms. C. Distribution of the genetic subtypes among younger (age ≤ 60), older (age > 60) and all patients in the Phoenix and NCI cohorts. D. Distribution and prevalence of genetic aberrations in DLBCL genetic subtypes in the Phoenix non-China cohort. Missense or inframe deletion/insertion mutations (Mut), protein-truncating mutations (Trunc), and gene rearrangement (Fusion) are shown as indicated. Also shown is the cell-of-origin (COO) gene expression subgroup (NA: Not available), the age category (Younger: age≤60, Older: age>60), and the Phoenix study arm. E. Prevalence and significance of association between genetic aberrations and the DLBCL genetic subtypes. P values compare each subtype to all other cases in the Phoenix cohort. The prevalence of each genetic aberration in non-GCB biopsies from the NCI cohort is shown for comparison.
Non-GCB DLBCL is comprised primarily of four genetic subtypes that can be discerned using the LymphGen algorithm: MCD, BN2, N1 and A53 (Schmitz et al., 2018; Wright et al., 2020). The LymphGen classifier was established using sequencing data from fresh frozen DLBCL tumor biopsies, unlike the FFPE biopsies from the Phoenix trial that could have adventitial sequence alterations due to fixation. Given this consideration and our wish to identify highly homogenous biological subsets within the Phoenix cohort, we focused on cases with a high probability (≥90%) of DLBCL subtype membership, previously termed “core” subtype cases (Wright et al., 2020). Using this rigorous cutoff, LymphGen classified 203 (26.3%) biopsies, including 110 MCD, 47 BN2, and 28 N1 cases, as well as 18 cases with genetic features of more than one subtype, previously termed “genetically composite” (Wright et al., 2020), and were not considered further. The A53 subtype could not be identified due to the lack of DNA copy number data, which is required for this subtype assignment. The subtype distributions in the Phoenix and NCI cohorts were comparable, after accounting for differences in the cell-of-origin (COO) types of DLBCL tumors included in both studies (see Supplementary Methods; Fig. 1C). The subtype distributions in the Phoenix China and non-China cohorts were also comparable (Figure S1B).
The genetic features that significantly distinguished each subtype from all other DLBCLs (p<0.01; Fig. 1D) occurred with frequencies that mirror those observed among non-GCB DBLCL cases in the NCI cohort (Fig. 1E). Likewise, the subtype-defining genetic lesions were identified comparably in the China and non-China and among younger and older patients in the Phoenix trial, further demonstrating the reproducibility of the genetic subtype assignments (Fig. S1C, S1D).
Phenotypes of DLBCL genetic subtypes
As judged by gene expression signature analysis, DLBCL genetic subtypes have characteristic tumor phenotypes (Schmitz et al., 2018; Wright et al., 2020), which we examined in the Phoenix subtypes. Using the DLBCL cell-of-origin signature (Wright et al., 2003), MCD cases were primarily ABC DLBCL phenotypically, whereas N1 and particularly BN2 were more often GCB and Unclassified, as expected (Schmitz et al., 2018; Wright et al., 2020) (Fig. 2A, Table 1). We next examined the expression of signatures that reflect biological attributes of normal and malignant lymphocytes, many of which are differentially expressed by the DLBCL genetic subtypes (Schmitz et al., 2018; Shaffer et al., 2006; Wright et al., 2020). To determine the relative enrichment of each signature in a given subtype, we compared the average expression of signature genes in subtype samples versus all other samples. We used these relative signature expression values to compare the biological attributes of subtypes defined in the Phoenix and NCI cohorts and observed a high degree of correlation for the MCD (p=0.00039), BN2 (p=0.0086), and N1 (p=0.013) subtypes (Fig. 2B).
Figure 2.

Phenotypic and clinical characteristics of Phoenix genetic subtypes. A. Distribution of DLBCL gene expression subgroups among cases assigned to the MCD, BN2 and N1 genetic subtypes. B. Comparison of gene expression signature averages between genetic subtypes and all other DLBCL samples. Shown are Z scores (see Methods) representing the relative expression of each signature in a given subtype versus all other DLBCLs. Z scores calculated for the Phoenix and NCI cohorts are plotted on the y-axis and x-axis, respectively. The brown dashed lines indicate the Z scores corresponding to a significant difference between the two sample subsets (p=0.01). The yellow shaded areas include those signatures that are more highly expressed in the indicated subtype than in other samples and the blue shaded areas are signatures that are expressed at lower levels in the indicated subtype relative to other samples. The indicated p value is for the correlation of the Z scores from the two cohorts. C. Extranodal involvement in the genetic subtypes, subdivided by anatomic site as indicated. P values are from a 2-way Fisher’s Exact test. ns: non-significant.
Table 1.
Relationship between DLBCL genetic subtypes and clinical parameters
| All cases |
MCD subtype |
BN2 subtype |
N1 subtype |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Total | Young | Old | Total | Young | Old | Total | Young | Old | Total | Young | Old | p-Value | |
|
| |||||||||||||
| Age (average) | 59.8 | 48.1 | 69.0 | 64.1 | 51.7 | 68.9 | 61.3 | 52.1 | 68.7 | 59.6 | 46.5 | 71.0 | 0.13a |
| Gender (% male) | 58.4 | 44.6 | 65.8 | 59.6 | 38.7 | 67.1 | 59.1 | 52.4 | 65.4 | 53.6 | 46.2 | 60.0 | 0.87b |
|
Cell-of-origin | |||||||||||||
| ABC (%) | 76.6 | 70.2 | 81.5 | 97.1 | 90.0 | 100.0 | 73.9 | 71.4 | 76.0 | 84.6 | 90.9 | 80.0 | 0.000053b |
| GCB (%) | 16.8 | 21.4 | 13.3 | 1.0 | 3.3 | 0.0 | 15.2 | 19.0 | 12.0 | 7.7 | 0.0 | 13.3 | 0.00144b |
| UNC (%) | 6.6 | 8.4 | 5.3 | 1.9 | 6.7 | 0.0 | 10.9 | 9.5 | 12.0 | 7.7 | 9.1 | 6.7 | 0.036b |
|
International prognostic index | |||||||||||||
| IPI (average) | 2 | 2 | 3 | 3 | 2 | 3 | 2 | 2 | 3 | 2 | 2 | 3 | 0.83a |
| IPI sans age (average) | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 0.79a |
| IPI 0–1 (%) | 24.3 | 37.6 | 13.9 | 16.4 | 19.4 | 15.2 | 17.0 | 28.6 | 7.7 | 25.0 | 38.5 | 13.3 | 0.53b |
| IPI 2–3 (%) | 62.2 | 58.8 | 64.9 | 67.3 | 67.7 | 67.1 | 70.2 | 71.4 | 69.2 | 53.6 | 46.2 | 60.0 | 0.32b |
| IPI 4–5 (%) | 13.5 | 3.5 | 21.2 | 16.4 | 12.9 | 17.7 | 12.8 | 0.0 | 23.1 | 21.4 | 15.4 | 26.7 | 0.58b |
| age>60 (%) | 56.0 | 0.0 | 100.0 | 71.8 | 0.0 | 100.0 | 55.3 | 0.0 | 100.0 | 53.6 | 0.0 | 100.0 | 0.052b |
| ECOG performance status>1 (%) | 12.0 | 14.1 | 10.4 | 10.0 | 29.0 | 2.5 | 14.9 | 14.3 | 15.4 | 28.6 | 30.8 | 26.7 | 0.051b |
| LDH>1 (%) | 54.9 | 60.9 | 50.1 | 53.6 | 71.0 | 46.8 | 63.8 | 76.2 | 53.8 | 64.3 | 61.5 | 66.7 | 0.39b |
| extranodal sites>1 (%) | 33.8 | 36.5 | 31.6 | 40.9 | 48.4 | 38.0 | 23.4 | 28.6 | 19.2 | 17.9 | 15.4 | 20.0 | 0.020b |
| stage>2 (%) | 76.5 | 77.4 | 75.8 | 74.5 | 80.6 | 72.2 | 83.0 | 81.0 | 84.6 | 82.1 | 84.6 | 80.0 | 0.47b |
UNC, unclassified.
3-way F test comparing total cases in each subtype.
3-way Fisher’s Exact Test comparing total cases in each subtype.
Briefly, MCD was characterized by signatures of oncogenic BCR, NF-κB and PI3 kinase signaling, signatures of subtype-defining transcription factors (IRF4, TBL1XR1, Oct-2), and signatures of cellular proliferation. BN2 expressed a signature of Notch pathway activation as well as signatures of NF-κB, STAT3 and p53 activity. N1 also expressed a Notch activation signature, as well as signatures of cellular quiescence (low proliferation) and the memory B-cell differentiation state.
Clinical attributes of DLBCL genetic subtypes
MCD had a higher proportion of older patients than BN2 and N1 (p=0.019), a distribution mirrored in the NCI cohort (Table 1; Fig. 1C). The Phoenix MCD patients had more frequent extranodal involvement than the other subtypes (p=0.0066; Fig. 2C), as also observed in the NCI cohort (Wright et al., 2020). Previous genetic analysis revealed a striking similarity between MCD and a specific subset of primary extranodal lymphomas, including those that occur in the testis, breast, adrenal, ovary, and uterus (Wright et al., 2020). As expected from previous analysis of the NCI cohort (Wright et al., 2020), secondary involvement of these primary extranodal sites was significantly more frequent in MCD than in BN2 or N1 (p=0.004), which conversely involved other anatomic sites secondarily (p=0.024).
Overall, the genetic subtypes had similar proportions of cases with low (0–1), intermediate (2–3) and high (4–5) values of the International Prognostic Index (IPI) (Table 1). Younger MCD patients had a higher age-adjusted IPI and hence poorer prognosis with chemotherapy than older MCD patients (2.3 vs. 1.6, respectively; p= 0.0018), which was driven by poor ECOG scores and elevated LDH among younger patients.
Effect of ibrutinib within DLBCL genetic subtypes
For patients treated on the placebo arm, the 3-year event-free survival (EFS) estimates were 65% for MCD, 74% for BN2 and 55% for N1, with a median follow-up of 46.2 months (Fig. S2A). These outcomes are better than for R-CHOP-treated patients in the NCI cohort (Fig. S2A) and may reflect the known accrual in phase III lymphoma trials towards patients with better prognosis (Maurer et al., 2018). For patients treated on the ibrutinib arm, we separately analyzed EFS and overall survival (OS) by age because of the increased toxicity observed in patients older than 60 years, an effect not observed on the R-CHOP arm (Younes et al., 2019). As reported, older patients did not benefit from ibrutinib, regardless of genetic subtype (Fig. S2B).
Younger patients with MCD DLBCL (n=31) who received ibrutinib and R-CHOP had a 3-year EFS and OS of 100%, compared to a significantly lower EFS of 48% (p=0.01) and OS of 69.6% (p=0.032) when treated with R-CHOP alone. Although younger non-MCD patients also benefitted from ibrutinib, the effect was smaller than in MCD patients, as indicated by interaction p-values for EFS and OS of 0.0084 and 0.057, respectively (Fig. 3). The survival advantage conferred by ibrutinib was apparent in both the China and non-China cohorts, demonstrating the reproducibility of this effect (Fig. S2C).
Figure 3.

Survival of Phoenix trial patients subdivided by genetic subtype and treatment arm. Shown are Kaplan-Meier plots of event-free and overall survival in younger (age ≤ 60) patients assigned to the MCD, BN2 and N1 genetic subtypes. Also shown are log-rank p values for the difference in survival in the indicated genetic subtype treated with R-CHOP plus ibrutinib or placebo. The interaction p values indicate the significance of the difference in ibrutinib benefit within the indicated genetic subtype compared with all other DLBCLs. “No. at risk” indicates the number of patients remaining without events (for event-free survival) or remaining alive (for overall survival.
Ibrutinib addition was also associated with improved survival in younger patients with N1 DLBCL (n=13). These patients had a 3-year EFS and OS of 100% whereas those treated with R-CHOP alone had a significantly inferior EFS (50%; p=0.0161) and OS (50%; p=0.0134). Ibrutinib had a greater effect on EFS in younger N1 patients than in non-N1 patients (interaction p-value=0.0268). In contrast to MCD and N1, younger patients with BN2 DLBCL (n=21) did not benefit from ibrutinib (Fig. 3).
We suspected that ibrutinib might provide benefit beyond the genetically classified cases because we were deliberately conservative in our assignment of genetic subtypes and also because we could not identify A53 cases with the available data. Indeed, ibrutinib provided a significant survival benefit for younger patients who were not genetically assigned, but not for older patients (Fig. S3A). To explore this further, we extended our genetic classification to include cases with an intermediate LymphGen probability of subtype membership (50–90%), previously called “extended” subtype cases (Wright et al., 2020). We were able to classify 43% (n=332) of Phoenix cases using this relaxed probability cutoff, increasing the number of MCD (n=157) and BN2 (n=129) patients but not N1 patients. For younger MCD patients in this cohort (n=46), those on the ibrutinib treatment arm had 3-year OS and EFS rates of 100% and 89%, respectively, but those on the placebo arm had significantly inferior survival, with OS and EFS rates of 78% (p=0.011) and 58% (p=0.0265), respectively (Fig. S3B). Among younger BN2 patients in this extended cohort (n=60), there was a trend towards ibrutinib benefit as determined by EFS (p=0.06; Fig. S3B). The N1 subtype composition did not change with these relaxed conditions because of the statistically dominance of NOTCH1 in the LymphGen prediction of N1. Patients not genetically classified using this relaxed probability threshold also had significantly improved survival with ibrutinib, albeit to a lesser degree than in MCD and N1 (Fig. S3C).
Genetic and phenotypic attributes of N1 DLBCL
The benefit of ibrutinib in MCD DLBCL is consistent with previous clinical studies showing frequent response of such patients to ibrutinib monotherapy (Lionakis et al., 2017; Wilson et al., 2015) as well as the strong dependence of MCD models on BCR-dependent NF-κB activity (Davis et al., 2010; Phelan et al., 2018). The benefit of ibrutinib in N1 was unanticipated based on previous analysis of its genetic attributes, which was hampered by a small sample size (n=16) (Schmitz et al., 2018). To elucidate the molecular basis for ibrutinib efficacy in N1, we analyzed publicly available tumor sequencing data for NOTCH1 mutations, which are necessary and sufficient for N1 classification. From 4,586 DLBCL cases, we identified 79 (1.7%) NOTCH1-mutant cases (see Supplemental Methods). The cell-of-origin assignment of these cases, when available (n=65), was primarily non-GCB (n=57, 87.7%) (Fig. 4A).
Figure 4.

Genetic, phenotypic, and clinical attributes of N1 DLBCL. A. Distribution of DLBCL gene expression subgroups among NOTCH1-mutant DLBCLs. B. Recurrently mutated genes in NOTCH1-mutant DLBCL. Shown are the type and prevalence of mutations in the indicated genes among NOTCH1-mutant and NOTCH1-wild type DLBCLs. Genes are assigned to functional categories, as indicated. P values are from a Fisher’s Exact Test comparing the prevalence in NOTCH1-mutant and NOTCH1-WT cases. Mut: Non-synonymous mutation; Trunc: Truncating mutation; WT: wild type; ns: non-significant. C. Prevalence of mutations targeting biologic pathways characteristic of N1 DLBCL. Prevalence is based on 1,431 DLBCL NOTCH1-WT and NOTCH1-mutant DLBCL cases for which sequencing data was available for all genes in the indicated pathways. P values are from a Fisher’s Exact Test comparing the prevalence in NOTCH1-mutant and NOTCH1-WT cases. D. Left panel: Average expression of the indicated gene expression signatures in the indicated genetic subtypes in the NCI cohort(Schmitz et al., 2018). Bmem-6: Genes upregulated in memory B and memory B precursors relative to germinal center dark zone and light zone B cell cells (Venturutti et al., 2020); TBL1XR1MutUp-1: Genes up regulated by a TBL1XR1 mutant isoform (Venturutti et al., 2020); BACH2Up-1: Genes upregulated in BACH2+ germinal center light zone cells (Venturutti et al., 2020). Right panel: Average expression of the indicated memory B cell marker genes. ****p<0.0001, ***p<0.001, **p<0.01, *p<0.05. Error bars: SEM. E. Schematic of the BCR-dependent NF-κB pathway showing the prevalence of mutations targeting each pathway component in the N1 (NOTCH1-mutant) and MCD subtypes of DLBCL according to the color scale shown. DAG: diacylglycerol; IP3: inositol triphosphate; Ca++: calcium ion; CBM complex: CARD11-BCL10-MALT1 complex; My-T-BCR: MYD88-TLR9-B cell receptor complex. F. Kaplan-Meier plots of event-free and overall survival in younger (age ≤ 60) and older (age > 60) patients with NOTCH1-mutant DLBCL treated with R-CHOP-like chemotherapy, curated from the published literature.
We next identified genes that were recurrently mutated (2 or more cases) in this larger cohort of N1 cases, 78.0% (39/50) of which were mutated more frequently in N1 than in NOTCH1-wild type (WT) cases (Fig. 4B; Table S2). We grouped these genes into functional categories that revealed new aspects of N1 biology (Fig. 4B). N1 tumors recurrently mutated DTX1, encoding a NOTCH1 transcriptional target and inactivated SPEN, encoding a negative regulator of NOTCH1 signaling. Given the non-GCB phenotype of most N1 tumors, it was somewhat surprising to observe mutations in genes implicated in the pathogenesis of germinal center lymphomas such as GCB DLBCL and Burkitt lymphoma (Schmitz et al., 2014; Schmitz et al., 2018; Victora et al., 2012) (Fig. 4B). Unlike those lymphoma types, however, N1 frequently acquired mutations targeting chromatin proteins that potentially promote memory B-cell differentiation at the expense of plasma cell differentiation, including TBL1XR1, NCOR1, SMRT (NCOR2), BCOR and BACH2 (see Discussion). Compared with MCD and BN2, N1 tumors expressed gene signatures of memory B cells at significantly higher levels, and also had higher expression of genes that are upregulated by mutant TBL1XR1 and by BACH2 (Fig. 2B, 4D). N1 also had the highest expression of T-bet (TBX21) and other markers of a newly defined subset of IgM+ memory B cells (CD27, CXCR3, CD11c (ITGAX); Fig. 4D) (Johnson et al., 2020; Kenderes et al., 2018).
Another recurrent biological them in N1 tumors was genetic inactivation of genes required for tumor immune surveillance, including β2-microglobulin, HLA-A, and transactivators of MHC class I expression (NLRC5, RFX7), thereby hindering antigen presentation, and inactivation of CD58, an NK cell activator. Unexpectedly, N1 tumors also acquired mutations targeting KRAS, NRAS and BRAF in one third of cases, revealing an unanticipated role of Ras/MAP kinase signaling in this lymphoma subtype.
Most notably from the perspective of the Phoenix trial, N1 recurrently acquired mutations targeting components of the BCR-dependent NF-κB pathway, constituting 59.3% (n=47) of cases (Fig. 4B, C). While MCD tumors primarily engage the BCR pathway by acquiring CD79B and MYD88 mutations, N1 mutations preferentially target CD79A, multiple negative regulators of proximal BCR signaling, and downstream mediators of NF-κB activation including phospholipase-Cγ2 (PLCG2), protein kinase Cβ (PRKCB), BCL10, CARD11, IκB kinase (ΙΚΒΚΒ), and A20 (TNFAIP3) (Fig. 4C).
Finally, given the limited number of younger N1 patients on the Phoenix placebo arm, we wished to provide further evidence that R-CHOP alone is insufficient to achieve exceptional EFS and OS in this subtype. Among 11 younger patients treated with R-CHOP-like regimens from our literature-curated N1 cohort, 3-year EFS and OS rates were 64% and 73%, respectively, consistent with the survival of comparable Phoenix trial patients who received R-CHOP alone (Fig. 4E).
Discussion
Here we provide a precision medicine framework for the use of BTK inhibitors in previously untreated younger patients with non-GCB DLBCL. We identified two genetic subtypes of DLBCL – MCD and N1 – for which the addition of ibrutinib to R-CHOP chemotherapy improved survival significantly. These two subtypes are highly enriched in genetic lesions that create a dependence on the BCR-dependent NF-κB pathway, providing a mechanistic basis for the observed clinical benefit from ibrutinib. More broadly, ibrutinib was also beneficial in younger patients not classified as MCD or N1, albeit to a lesser degree, suggesting that the utility of BTK inhibitors may extend to other patients with non-GCB DLBCL as well.
Based on pre-clinical and clinical observations, our starting hypothesis was that patients with MCD DLBCL would benefit from the addition of ibrutinib to chemotherapy. The chronic active BCR signaling in MCD renders the tumor cells highly dependent on BTK to activate the pro-survival NF-κB pathway (Davis et al., 2010). This signaling is instigated by binding of the BCR to various self-antigens and by the oncogenic cooperation between the BCR, TLR9, MYD88 and components of the My-T-BCR supercomplex (Phelan et al., 2018; Young et al., 2015). As a result, patients with MCD tumors frequently respond to ibrutinib monotherapy (Wilson et al., 2015), as do patients with primary CNS lymphoma (Lionakis et al., 2017), which bears the MCD genotype (Lionakis et al., 2017; Wright et al., 2020). In addition, the inhibition of NF-κB by ibrutinib is likely to sensitize MCD tumors to R-CHOP given the ability of NF-κB to suppress the apoptotic response to cytotoxic chemotherapy (Mathews Griner et al., 2014; Wang et al., 1998). These considerations together with our analysis of the Phoenix trial firmly establish the rationale for ibrutinib use in the treatment of MCD-like lymphomas.
The sensitivity of N1 DLBCL to ibrutinib plus R-CHOP therapy was unanticipated, largely because the previous analysis of N1 was limited by its relative rarity. The biology of N1 DLBCL came into focus from our genetic analysis of a larger cohort of NOTCH1-mutant tumors gleaned from the literature. N1 DLBCLs engage the BCR-dependent NF-κB pathway by diverse genetic mechanisms in a majority of cases, including CD79A mutations that enhance proximal BCR signaling (Davis et al., 2010; Wilson et al., 2015). In addition, they recurrently acquire mutations targeting multiple negative regulators of proximal BCR signaling – SHP-1 (PTPN6), PRKCD, PTPRO and GRB2 – that might augment the addiction of N1 tumors to BCR signaling. N1 DLBCLs also acquire mutant isoforms of phospholipase-Cγ2 are known to confer partial ibrutinib resistance but nonetheless remain dependent on upstream signaling from the BCR for their enzymatic activity (Wist et al., 2020). N1 tumors also acquire mutations targeting the CARD11-BCL10-MALT1 signaling adapter complex that is critical for chronic active BCR signaling in ABC DLBCL (Lenz et al., 2008a). While some CARD11 mutant isoforms confer strong resistance to ibrutinib, others remain sensitive to upstream signals and therefore may retain ibrutinib sensitivity (Lenz et al., 2008a). The fact that patients with N1 tumors benefitted exceptionally from ibrutinib suggests that the recurrent mutations in PRKCB, BCL10, IKBKB, TNFAIP3 and TNIP1 in these tumors might also potentiate chronic active BCR signaling and reinforce BTK dependency.
While many N1 mutations suggest a germinal center origin, the memory B-cell phenotype that it adopts may be due, in part, to frequent TBL1XR1 mutations that foster memory B-cell differentiation (Venturutti et al., 2020). N1 tumors also frequently acquired truncating mutations targeting the co-repressor BCOR as well as deleterious mutations targeting NCOR1 and SMRT (NCOR2), all of which regulate germinal center biology. BCOR and SMRT/NCOR complexes interact with BCL6 to form functionally distinct co-repressor complexes that enforce the germinal center B cell phenotype (Hatzi et al., 2013). SMRT/NCOR complexes also complex with TBL1XR1 and the transcription factor BACH2, which specifies the memory cell phenotype (Beguelin et al., 2016; Hoshino et al., 2007; Venturutti et al., 2020). Mutant TBL1XR1 isoforms together with SMRT/NCOR complexes preferentially associated with BACH2 at the expense of BCL6, thereby fostering memory B cell differentiation (Venturutti et al., 2020). This mechanism may be pertinent in N1 tumors, which express memory B cell signature genes as well as genes that are upregulated by mutant TBL1XR1 and/or BACH2. N1 tumors particularly resemble a recently defined subset of memory B cells that are IgM+, rely upon the transcription factor T-Bet (TBX21), and express the surface markers CD27, CXCR3 and CD11c (Johnson et al., 2020; Kenderes et al., 2018). This memory B cell subset is capable of long-term self-renewal, which could provide a fertile context for malignant transformation into N1 DLBCL. The memory B cell and TBL1XR1 signatures are less apparent in MCD tumors even though they also acquire TBL1XR1 mutations recurrently. Hypothetically, these TBL1XR1 mutations in MCD may primarily serve to diminish access of BCL6 to SMRT/NCOR co-repressor complexes, thereby derepressing BCL6 target genes. These include genes that are upregulated during BCR-dependent activation of normal B cells and are also characteristically expressed in MCD, such as IRF4, which encodes the master regulatory transcription factor for this genetic subtype (Basso and Dalla-Favera, 2012; Hatzi and Melnick, 2014; Shaffer et al., 2000; Yang et al., 2012).
The present analysis represents the first implementation of the genetic classification system for DLBCL in a clinical trial setting and demonstrates its utility in defining pathogenetically related tumors that respond similarly to therapy. The reproducible classification of tumors using the LymphGen algorithm (Wright et al., 2020) was evidenced by the consistent association of each genetic subtype with a particular tumor phenotype, as judged by gene expression profiling, and clinical attributes, such as the frequent extranodal involvement of MCD tumors. Our success in classifying DLBCL cases using FFPE tumors demonstrates that this method is applicable in clinical trials, which typically collect such biopsies. Given the exceptional response of MCD and N1 tumors to ibrutinib plus R-CHOP, one could consider a precision medicine approach tailored to just these subtypes. Indeed, sequencing of a small number of genes (~20) in DLBCL biopsy specimens could provide this molecular diagnosis. However, the benefit of ibrutinib may not be limited just to these two subtypes. In particular, we were unable identify the A53 subtype in the Phoenix cohort because we lacked the DNA copy number data that is necessary to recognize aneuploidy. Like MCD tumors, A53 tumors acquire frequent mutations targeting the BCR-dependent NF-κB pathway and express BCRs with autoreactive immunoglobulin variable regions (Wright et al., 2020), suggesting that A53 may account for some of the ibrutinib benefit among genetically unassigned patients in our study.
More generally, we believe our study casts the Phoenix trial in a new light by providing a mechanistic basis for the survival benefit of adding ibrutinib to R-CHOP in younger DLBCL patients. The effect of patient age on response to ibrutinib was a secondary, not primary, endpoint of the Phoenix trial. As a result, this highly significant association was considered a subset analysis, implying that the benefit of ibrutinib for younger patients might represent a fortuitous association. Arguing against this view is the fact that the failure of ibrutinib to improve survival in older patients can be explained by the added toxicity of ibrutinib in this age group, resulting in significantly reduced chemotherapy administration. Our observation that the ibrutinib benefit was most pronounced in specific molecular subsets of younger patients is inconsistent with this being a chance finding, but rather indicates that there is a biological basis for the observed survival benefit. The survival benefit in the ibrutinib arm was especially pronounced for the MCD and N1 subtypes, suggesting that the use of ibrutinib with R-CHOP would almost certainly save lives among such patients. However, ibrutinib was also beneficial in younger ABC patients who were not genetically classified in our analysis, albeit to a lesser degree, suggesting that use of a BTK inhibitor with R-CHOP would also be reasonable for these patients. From this line of reasoning, we conclude that it would be rational to consider R-CHOP plus ibrutinib an effective treatment strategy for younger patients with non-GCB DLBCL.
Star methods
Resource availability
Lead Contact
Correspondence and requests regarding this manuscript should be sent to and will be fulfilled by the lead investigator Dr. Louis Staudt (lstaudt@mail.nih.gov). This study did not generate any new unique reagents
Data and code availability
DNA and RNA sequencing data from the non-China cohort are available through the European Genome-phenome archive (EGA) under accession EGAS00001005554. Sequencing data from the China cohort cannot be made available due to Chinese prohibitions on genomic data sharing. Clinical data are available in Table S1. LymphGen code is available at https://doi.org/10.5281/zenodo.3700086.
Method Details
Tumor samples
Formalin-fixed and paraffin-embedded (FFPE) enrolment tumor biopsies were obtained with informed consent from participants in the Phoenix phase III clinical trial (NCT01855750). Full clinical trial results have been reported previously (Younes et al., 2019).
DNA and RNA sequencing
For biopsies from the non-China cohort, targeted sequencing of 99 genes recurrently mutated in lymphoma (Table S3) was performed using the NuGEN Ovation Custom Target Enrichment System to an average sequencing depth of 707x. Whole exome sequencing in the China cohort was performed using a SureSelect Human All Exon V6 capture kit, SureSelectXT Reagent Kit for library preparation kit, and sequenced on a NovoSeq 6000 sequencer to an average sequencing depth of 167x. RNA-seq was performed using TruSeq RNA Access library prep kit and sequenced on a NextSeq 500 instrument, with a target depth of 33 million 100bp paired-end reads per sample.
Bioinformatics and LymphGen classification
Analysis of sequence data and of digital gene expression was performed as previously described (Schmitz et al., 2018). Initial analysis of the RNA-seq data revealed roughly 2.5-fold more sequence variants than were detected by targeted DNA sequencing from the same tumor biopsies, suggesting an increased number of FFPE-generated artifactual variants in the RNA-seq data that necessitated additional filtering. We therefore deemed an RNA-seq variant in a given sample as “verified” if that exact variant was also found in either a) the NuGEN targeted DNA sequencing data for that same sample, or in b) at least five DLBCL biopsy samples within a compilation of 47 whole exome or whole genome sequencing studies that comprised 5,695 DLBCL cases, including nodal, primary extranodal, and transformed DLBCL (2017; Arthur et al., 2018; Bödör et al., 2020; Bohers et al., 2018; Bohers et al., 2015; Braggio et al., 2015; Bruno et al., 2014; Chapuy et al., 2016a; Chapuy et al., 2016b; Chapuy et al., 2018; Chitalia et al., 2019; de Miranda et al., 2014; Dobashi et al., 2018; Dubois et al., 2017; Ducharme et al., 2019; Ennishi et al., 2019b; Fontanilles et al., 2017; Franco et al., 2017; Fukumura et al., 2016; Greenawalt et al., 2017; Hattori et al., 2019; Hattori et al., 2017; Intlekofer et al., 2018; Juskevicius et al., 2017; Juskevicius et al., 2016; Karube et al., 2018; Lacy et al., 2020; Lohr et al., 2012; Ma et al., 2021; Mareschal et al., 2016; Mareschal et al., 2017; Menter et al., 2017; Morin et al., 2016; Morin et al., 2013; Nakamura et al., 2016; Park et al., 2016; Pasqualucci et al., 2011; Reddy et al., 2017; Rossi et al., 2017; Scherer et al., 2016; Schmitz et al., 2018; Sha et al., 2019; Shin et al., 2019; Suehara et al., 2018; Vater et al., 2015; Zhang et al., 2013; Zhou et al., 2018a). For each sample, all verified alterations were retained, as well as those unverified alterations for which the number of mutant reads was above a sample-specific cut-point. This cut-point was chosen so as to maximize the proportion of verified alterations with a mutant count above the cut-point plus the proportion of unverified alterations below the cut-point. Samples were classified into MCD, BN2, and N1 subtypes with the LymphGen algorithm version 2.0 as implemented on the LymphGen website (https://llmpp.nih.gov/lymphgen/index.php).
Analysis of the NCI cohort
To compare the genetic subtype distribution in the Phoenix trial to that of the previously studied NCI cohort (Schmitz et al., 2018; Wright et al., 2020), we needed to account for differences in the type of tumors included in both studies. There is a highly reproducible relationship between the DLBCL genetic subtypes and the cell-of-origin (COO) gene expression subgroups, which can be used to compare studies that includes different percentages of ABC, GCB, and Unclassified subgroups (Wright et al., 2020). For the analysis in Figure 1B, we used this relationship to normalize the prevalence of the MCD, BN2 and N1 subtypes within the NCI cohort by adjusting the COO distribution in the NCI trial to match that of the Phoenix trial.
Analysis of NOTCH1-mutant DLBCL
NOTCH1-mutant DLBCL cases were ascertained from a compilation of whole exome, whole genome, transcriptome and targeted DNA resequencing data from 4,586 biopsies of nodal and primary extranodal DLBCL, curated from 45 studies (2017; Arthur et al., 2018; Bohers et al., 2018; Braggio et al., 2015; Bruno et al., 2014; Chapuy et al., 2016a; Chapuy et al., 2016b; Chapuy et al., 2018; Chitalia et al., 2019; de Miranda et al., 2014; Dobashi et al., 2018; Dubois et al., 2017; Dubois et al., 2016; Ducharme et al., 2019; Ennishi et al., 2019a; Fontanilles et al., 2017; Franco et al., 2017; Greenawalt et al., 2017; Hattori et al., 2019; Intlekofer et al., 2018; Juskevicius et al., 2017; Juskevicius et al., 2016; Karube et al., 2018; Kataoka et al., 2019; Lacy et al., 2020; Lohr et al., 2012; Ma et al., 2021; Mareschal et al., 2016; Mareschal et al., 2017; Menter et al., 2017; Mishina et al., 2021; Morin et al., 2016; Morin et al., 2011; Morin et al., 2013; Park et al., 2016; Pasqualucci et al., 2011; Ramis-Zaldivar et al., 2020; Rossi et al., 2017; Schmitz et al., 2018; Sha et al., 2019; Spina et al., 2018; Vater et al., 2015; Vela et al., 2020; Zhou et al., 2018a; Zhou et al., 2018b).
Quantification and Statistical analysis
P-values for the relationship between subtype and categorical variables (such as mutation incidence or performance status) were calculated with a Fisher’s exact test and were two sided. P-values for the relationship between subtype and numeric variables (such as age or IPI) were calculated with a F-test.
The Z-scores plotted in Figure 2B were calculated as follows: For each signature, we averaged the log signal values for all the genes in that signature, producing a signature average score for each sample. We than calculated a Z-score for the difference between MCD and non-MCD samples according to the following formula:
where μ1, σ1 and n1, respectively represent the mean, standard deviation, and variance of the signature average within the MCD samples, while μ2, σ2, and n2 represent those same quantities within the non-MCD samples. This is effectively the t-statistic for the difference between MCD and non-MCD samples but without the adjustment for sample size, and so it allows us to directly compare the significance of the relationship between subtype and signature average in data sets of different sizes. Z-scores for BN2 and N1 were similarly calculated.
Since many of the signatures included overlapping sets of genes, the signature Z-scores did not represent independent data points, which made the determination of statistical significance challenging. The p-values reported in Figure 2B were estimated from a permutation test as follows: The LymphGen class labels for the NCI and Phoenix cohorts were randomly permuted, severing the connection between class label and gene expression, but retaining the correlation between genes and between signatures within a data set. We then re-calculated the Z-scores for each signature in both data sets and recorded the correlation coefficient between the resulting scores. This was repeated 1,000 times. We estimated the null distribution of the correlation coefficient as a normal distribution with mean and variance equal to the sample mean and variance of the 1,000 permuted correlation values. The reported p-value was the right-tail probability of this distribution at the unpermuted correlation value.
P-values comparing the survival of treatment arms were calculated with a log-rank test and were one-sided in the direction of the hypothesis that patients treated with ibrutinib would have superior survival. P-values for the survival interactions between treatment arm and subtype were calculated with a likelihood ratio test that compared a Cox proportional hazards model that included individual effects for subtype and treatment versus one that included these individual effects and an additional interaction effect. These reported p-values were one-sided in the direction of the hypothesis that the improvement in survival due to ibrutinib treatment would be stronger within patients of the MCD and N1 subtypes.
Supplementary Material
Table S1 - Phoenix trial clinical and molecular features, related to Figure 1
Table S2 - Mutant isoforms identified in NOTCH1-mutant DLBCL samples, related to Figure 4
Table S3 - Targeted DNA resequencing panel, related to STAR Methods
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Critical commercial assays | ||
| Ovation Custom Target Enrichment System | NuGEN | |
| SureSelect Human All Exon V6 capture kit | Illumina | |
| Deposited data | ||
| RNA and DNA sequencing data | This Paper | European Genome-phenome archive EGAS00001005554 |
| NCI RNA and DNA sequence data | Schmitz et al., 2018 | dbGaP: phs001444.v2.p1 |
| Software and algorithms | ||
| Bcl2fastq | https://support.illumina.com/sequencing/sequencing_software/bcl2fastq-conversion-software.html | |
| STAR2 | https://github.com/alexdobin/STAR | |
| SamTools | https://sourceforge.net/projects/samtools/ | |
| Htseq_count | https://htseq.readthedocs.io/en/release_0.11.1/count.html | |
| VarScan2 | https://sourceforge.net/projects/varscan/files/ | |
| BWA | http://bio-bwa.sourceforge.net/ | |
| ANNOVAR | https://annovar.openbioinformatics.org/en/latest/ | |
| SnpEff | http://pcingola.github.io/SnpEff/se_introduction/ | |
| LymphGen | Wright et al. 2020 | https://llmpp.nih.gov/lymphgen/index.php |
Bullet points:
BTK inhibitor ibrutinib plus R-CHOP is effective in younger patients with ABC DLBCL
Genetic subtypes of DLBCL differ in genotype, phenotype, and oncogenic mechanisms
MCD and N1 subtypes acquire mutations that promote chronic active BCR signaling
Patients with the MCD and N1 subtypes have 100% survival with ibrutinib plus R-CHOP
Acknowledgements
Supported by the Intramural Research Program of the National Institutes of Health (NIH), and the Center for Cancer Research, National Cancer Institute (NCI). Next-generation sequencing was supported by Johnson & Johnson.
Footnotes
Declaration of interests
L.M.S., G.W., and D.W.H. are inventors on an NIH patent application covering the LymphGen algorithm. L.M.S., G.W., D.W.H., W.H.W., S.B. and B.H. are inventors on an NIH patent application covering the use of BTK inhibitors in genetic subtypes of DLBCL. B.H., S.B., Y.F., J.V., and M.S. are employees of Johnson & Johnson.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- AACR_Project_GENIE_Consortium (2017). AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov 7, 818–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, Boldrick JC, Sabet H, Tran T, Yu X, et al. (2000). Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 403, 503–511. [DOI] [PubMed] [Google Scholar]
- Arthur SE, Jiang A, Grande BM, Alcaide M, Cojocaru R, Rushton CK, Mottok A, Hilton LK, Lat PK, Zhao EY, et al. (2018). Genome-wide discovery of somatic regulatory variants in diffuse large B-cell lymphoma. Nat Commun 9, 4001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basso K, and Dalla-Favera R (2012). Roles of BCL6 in normal and transformed germinal center B cells. Immunological reviews 247, 172–183. [DOI] [PubMed] [Google Scholar]
- Beguelin W, Teater M, Gearhart MD, Calvo Fernandez MT, Goldstein RL, Cardenas MG, Hatzi K, Rosen M, Shen H, Corcoran CM, et al. (2016). EZH2 and BCL6 Cooperate to Assemble CBX8-BCOR Complex to Repress Bivalent Promoters, Mediate Germinal Center Formation and Lymphomagenesis. Cancer Cell 30, 197–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bödör C, Alpár D, Marosvári D, Galik B, Rajnai H, Bátai B, Nagy Á, Kajtár B, Burján A, Deák B, et al. (2020). Molecular Subtypes and Genomic Profile of Primary Central Nervous System Lymphoma. Journal of neuropathology and experimental neurology 79, 176–183. [DOI] [PubMed] [Google Scholar]
- Bohers E, Viailly PJ, Becker S, Marchand V, Ruminy P, Maingonnat C, Bertrand P, Etancelin P, Picquenot JM, Camus V, et al. (2018). Non-invasive monitoring of diffuse large B-cell lymphoma by cell-free DNA high-throughput targeted sequencing: analysis of a prospective cohort. Blood cancer journal 8, 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohers E, Viailly PJ, Dubois S, Bertrand P, Maingonnat C, Mareschal S, Ruminy P, Picquenot JM, Bastard C, Desmots F, et al. (2015). Somatic mutations of cell-free circulating DNA detected by next-generation sequencing reflect the genetic changes in both germinal center B-cell-like and activated B-cell-like diffuse large B-cell lymphomas at the time of diagnosis. Haematologica 100, e280–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braggio E, Van Wier S, Ojha J, McPhail E, Asmann YW, Egan J, da Silva JA, Schiff D, Lopes MB, Decker PA, et al. (2015). Genome-Wide Analysis Uncovers Novel Recurrent Alterations in Primary Central Nervous System Lymphomas. Clin Cancer Res 21, 3986–3994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruno A, Boisselier B, Labreche K, Marie Y, Polivka M, Jouvet A, Adam C, Figarella-Branger D, Miquel C, Eimer S, et al. (2014). Mutational analysis of primary central nervous system lymphoma. Oncotarget 5, 5065–5075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapuy B, Cheng H, Watahiki A, Ducar MD, Tan Y, Chen L, Roemer MG, Ouyang J, Christie AL, Zhang L, et al. (2016a). Diffuse large B-cell lymphoma patient-derived xenograft models capture the molecular and biological heterogeneity of the disease. Blood 127, 2203–2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapuy B, Roemer MG, Stewart C, Tan Y, Abo RP, Zhang L, Dunford AJ, Meredith DM, Thorner AR, Jordanova ES, et al. (2016b). Targetable genetic features of primary testicular and primary central nervous system lymphomas. Blood 127, 869–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapuy B, Stewart C, Dunford AJ, Kim J, Kamburov A, Redd RA, Lawrence MS, Roemer MGM, Li AJ, Ziepert M, et al. (2018). Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat Med 24, 679–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chitalia A, Swoboda DM, McCutcheon JN, Ozdemirli M, Khan N, and Cheson BD (2019). Descriptive analysis of genetic aberrations and cell of origin in Richter transformation. Leuk Lymphoma 60, 971–979. [DOI] [PubMed] [Google Scholar]
- Davis RE, Ngo VN, Lenz G, Tolar P, Young RM, Romesser PB, Kohlhammer H, Lamy L, Zhao H, Yang Y, et al. (2010). Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature 463, 88–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Miranda NF, Georgiou K, Chen L, Wu C, Gao Z, Zaravinos A, Lisboa S, Enblad G, Teixeira MR, Zeng Y, et al. (2014). Exome sequencing reveals novel mutation targets in diffuse large B-cell lymphomas derived from Chinese patients. Blood 124, 2544–2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobashi A, Togashi Y, Tanaka N, Yokoyama M, Tsuyama N, Baba S, Mori S, Hatake K, Yamaguchi T, Noda T, and Takeuchi K (2018). TP53 and OSBPL10 alterations in diffuse large B-cell lymphoma: prognostic markers identified via exome analysis of cases with extreme prognosis. Oncotarget 9, 19555–19568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubois S, Viailly PJ, Bohers E, Bertrand P, Ruminy P, Marchand V, Maingonnat C, Mareschal S, Picquenot JM, Penther D, et al. (2017). Biological and Clinical Relevance of Associated Genomic Alterations in MYD88 L265P and non-L265P-Mutated Diffuse Large B-Cell Lymphoma: Analysis of 361 Cases. Clin Cancer Res 23, 2232–2244. [DOI] [PubMed] [Google Scholar]
- Dubois S, Viailly PJ, Mareschal S, Bohers E, Bertrand P, Ruminy P, Maingonnat C, Jais JP, Peyrouze P, Figeac M, et al. (2016). Next-Generation Sequencing in Diffuse Large B-Cell Lymphoma Highlights Molecular Divergence and Therapeutic Opportunities: a LYSA Study. Clin Cancer Res 22, 2919–2928. [DOI] [PubMed] [Google Scholar]
- Ducharme O, Beylot-Barry M, Pham-Ledard A, Bohers E, Viailly PJ, Bandres T, Faur N, Frison E, Vergier B, Jardin F, et al. (2019). Mutations of the B-Cell Receptor Pathway Confer Chemoresistance in Primary Cutaneous Diffuse Large B-Cell Lymphoma Leg Type. J Invest Dermatol 139, 2334–2342 e2338. [DOI] [PubMed] [Google Scholar]
- Ennishi D, Jiang A, Boyle M, Collinge B, Grande BM, Ben-Neriah S, Rushton C, Tang J, Thomas N, Slack GW, et al. (2019a). Double-Hit Gene Expression Signature Defines a Distinct Subgroup of Germinal Center B-Cell-Like Diffuse Large B-Cell Lymphoma. J Clin Oncol 37, 190–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ennishi D, Takata K, Beguelin W, Duns G, Mottok A, Farinha P, Bashashati A, Saberi S, Boyle M, Meissner B, et al. (2019b). Molecular and Genetic Characterization of MHC Deficiency Identifies EZH2 as Therapeutic Target for Enhancing Immune Recognition. Cancer Discov 9, 546–563. [DOI] [PubMed] [Google Scholar]
- Fontanilles M, Marguet F, Bohers É, Viailly PJ, Dubois S, Bertrand P, Camus V, Mareschal S, Ruminy P, Maingonnat C, et al. (2017). Non-invasive detection of somatic mutations using next-generation sequencing in primary central nervous system lymphoma. Oncotarget 8, 48157–48168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco F, González-Rincón J, Lavernia J, García JF, Martín P, Bellas C, Piris MA, Pedrosa L, Miramón J, Gómez-Codina J, et al. (2017). Mutational profile of primary breast diffuse large B-cell lymphoma. Oncotarget 8, 102888–102897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukumura K, Kawazu M, Kojima S, Ueno T, Sai E, Soda M, Ueda H, Yasuda T, Yamaguchi H, Lee J, et al. (2016). Genomic characterization of primary central nervous system lymphoma. Acta Neuropathol 131, 865–875. [DOI] [PubMed] [Google Scholar]
- Greenawalt DM, Liang WS, Saif S, Johnson J, Todorov P, Dulak A, Enriquez D, Halperin R, Ahmed A, Saveliev V, et al. (2017). Comparative analysis of primary versus relapse/refractory DLBCL identifies shifts in mutation spectrum. Oncotarget 8, 99237–99244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hans CP, Weisenburger DD, Greiner TC, Gascoyne RD, Delabie J, Ott G, Muller-Hermelink HK, Campo E, Braziel RM, Jaffe ES, et al. (2004). Confirmation of the molecular classification of diffuse large B-cell lymphoma by immunohistochemistry using a tissue microarray. Blood 103, 275–282. [DOI] [PubMed] [Google Scholar]
- Hattori K, Sakata-Yanagimoto M, Kusakabe M, Nanmoku T, Suehara Y, Matsuoka R, Noguchi M, Yokoyama Y, Kato T, Kurita N, et al. (2019). Genetic evidence implies that primary and relapsed tumors arise from common precursor cells in primary central nervous system lymphoma. Cancer Sci 110, 401–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattori K, Sakata-Yanagimoto M, Okoshi Y, Goshima Y, Yanagimoto S, Nakamoto-Matsubara R, Sato T, Noguchi M, Takano S, Ishikawa E, et al. (2017). MYD88 (L265P) mutation is associated with an unfavourable outcome of primary central nervous system lymphoma. Br J Haematol 177, 492–494. [DOI] [PubMed] [Google Scholar]
- Hatzi K, Jiang Y, Huang C, Garrett-Bakelman F, Gearhart MD, Giannopoulou EG, Zumbo P, Kirouac K, Bhaskara S, Polo JM, et al. (2013). A hybrid mechanism of action for BCL6 in B cells defined by formation of functionally distinct complexes at enhancers and promoters. Cell reports 4, 578–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatzi K, and Melnick A (2014). Breaking bad in the germinal center: how deregulation of BCL6 contributes to lymphomagenesis. Trends Mol Med 20, 343–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshino H, Nishino TG, Tashiro S, Miyazaki M, Ohmiya Y, Igarashi K, Horinouchi S, and Yoshida M (2007). Co-repressor SMRT and class II histone deacetylases promote Bach2 nuclear retention and formation of nuclear foci that are responsible for local transcriptional repression. J Biochem 141, 719–727. [DOI] [PubMed] [Google Scholar]
- Intlekofer AM, Joffe E, Batlevi CL, Hilden P, He J, Seshan VE, Zelenetz AD, Palomba ML, Moskowitz CH, Portlock C, et al. (2018). Integrated DNA/RNA targeted genomic profiling of diffuse large B-cell lymphoma using a clinical assay. Blood cancer journal 8, 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JL, Rosenthal RL, Knox JJ, Myles A, Naradikian MS, Madej J, Kostiv M, Rosenfeld AM, Meng W, Christensen SR, et al. (2020). The Transcription Factor T-bet Resolves Memory B Cell Subsets with Distinct Tissue Distributions and Antibody Specificities in Mice and Humans. Immunity 52, 842–855 e846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juskevicius D, Jucker D, Klingbiel D, Mamot C, Dirnhofer S, and Tzankov A (2017). Mutations of CREBBP and SOCS1 are independent prognostic factors in diffuse large B cell lymphoma: mutational analysis of the SAKK 38/07 prospective clinical trial cohort. J Hematol Oncol 10, 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juskevicius D, Lorber T, Gsponer J, Perrina V, Ruiz C, Stenner-Liewen F, Dirnhofer S, and Tzankov A (2016). Distinct genetic evolution patterns of relapsing diffuse large B-cell lymphoma revealed by genome-wide copy number aberration and targeted sequencing analysis. Leukemia 30, 2385–2395. [DOI] [PubMed] [Google Scholar]
- Karube K, Enjuanes A, Dlouhy I, Jares P, Martin-Garcia D, Nadeu F, Ordóñez GR, Rovira J, Clot G, Royo C, et al. (2018). Integrating genomic alterations in diffuse large B-cell lymphoma identifies new relevant pathways and potential therapeutic targets. Leukemia 32, 675–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kataoka K, Miyoshi H, Sakata S, Dobashi A, Couronné L, Kogure Y, Sato Y, Nishida K, Gion Y, Shiraishi Y, et al. (2019). Frequent structural variations involving programmed death ligands in Epstein-Barr virus-associated lymphomas. Leukemia 33, 1687–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenderes KJ, Levack RC, Papillion AM, Cabrera-Martinez B, Dishaw LM, and Winslow GM (2018). T-Bet(+) IgM Memory Cells Generate Multi-lineage Effector B Cells. Cell reports 24, 824–837 e823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacy SE, Barrans SL, Beer PA, Painter D, Smith AG, Roman E, Cooke SL, Ruiz C, Glover P, Van Hoppe SJL, et al. (2020). Targeted sequencing in DLBCL, molecular subtypes, and outcomes: a Haematological Malignancy Research Network report. Blood 135, 1759–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenz G, Davis RE, Ngo VN, Lam L, George TC, Wright GW, Dave SS, Zhao H, Xu W, Rosenwald A, et al. (2008a). Oncogenic CARD11 mutations in human diffuse large B cell lymphoma. Science 319, 1676–1679. [DOI] [PubMed] [Google Scholar]
- Lenz G, Wright G, Dave SS, Xiao W, Powell J, Zhao H, Xu W, Tan B, Goldschmidt N, Iqbal J, et al. (2008b). Stromal gene signatures in large-B-cell lymphomas. N Engl J Med 359, 2313–2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lionakis MS, Dunleavy K, Roschewski M, Widemann BC, Butman JA, Schmitz R, Yang Y, Cole DE, Melani C, Higham CS, et al. (2017). Inhibition of B Cell Receptor Signaling by Ibrutinib in Primary CNS Lymphoma. Cancer Cell 31, 833–843 e835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohr JG, Stojanov P, Lawrence MS, Auclair D, Chapuy B, Sougnez C, Cruz-Gordillo P, Knoechel B, Asmann YW, Slager SL, et al. (2012). Discovery and prioritization of somatic mutations in diffuse large B-cell lymphoma (DLBCL) by whole-exome sequencing. Proc Natl Acad Sci U S A 109, 3879–3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma MCJ, Tadros S, Bouska A, Heavican T, Yang H, Deng Q, Moore D, Akhter A, Hartert K, Jain N, et al. (2021). Subtype-specific and co-occurring genetic alterations in B-cell non-Hodgkin lymphoma. Haematologica. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mareschal S, Dubois S, Viailly PJ, Bertrand P, Bohers E, Maingonnat C, Jaïs JP, Tesson B, Ruminy P, Peyrouze P, et al. (2016). Whole exome sequencing of relapsed/refractory patients expands the repertoire of somatic mutations in diffuse large B-cell lymphoma. Genes Chromosomes Cancer 55, 251–267. [DOI] [PubMed] [Google Scholar]
- Mareschal S, Pham-Ledard A, Viailly PJ, Dubois S, Bertrand P, Maingonnat C, Fontanilles M, Bohers E, Ruminy P, Tournier I, et al. (2017). Identification of Somatic Mutations in Primary Cutaneous Diffuse Large B-Cell Lymphoma, Leg Type by Massive Parallel Sequencing. J Invest Dermatol 137, 1984–1994. [DOI] [PubMed] [Google Scholar]
- Mathews Griner LA, Guha R, Shinn P, Young RM, Keller JM, Liu D, Goldlust IS, Yasgar A, McKnight C, Boxer MB, et al. (2014). High-throughput combinatorial screening identifies drugs that cooperate with ibrutinib to kill activated B-cell-like diffuse large B-cell lymphoma cells. Proc Natl Acad Sci U S A 111, 2349–2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurer MJ, Ghesquieres H, Link BK, Jais JP, Habermann TM, Thompson CA, Haioun C, Allmer C, Johnston PB, Delarue R, et al. (2018). Diagnosis-to-Treatment Interval Is an Important Clinical Factor in Newly Diagnosed Diffuse Large B-Cell Lymphoma and Has Implication for Bias in Clinical Trials. J Clin Oncol 36, 1603–1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menter T, Juskevicius D, Alikian M, Steiger J, Dirnhofer S, Tzankov A, and Naresh KN (2017). Mutational landscape of B-cell post-transplant lymphoproliferative disorders. Br J Haematol 178, 48–56. [DOI] [PubMed] [Google Scholar]
- Mishina T, Oshima-Hasegawa N, Tsukamoto S, Fukuyo M, Kageyama H, Muto T, Mimura N, Rahmutulla B, Nagai Y, Kayamori K, et al. (2021). Genetic subtype classification using a simplified algorithm and mutational characteristics of diffuse large B-cell lymphoma in a Japanese cohort. Br J Haematol. [DOI] [PubMed] [Google Scholar]
- Morin RD, Assouline S, Alcaide M, Mohajeri A, Johnston RL, Chong L, Grewal J, Yu S, Fornika D, Bushell K, et al. (2016). Genetic Landscapes of Relapsed and Refractory Diffuse Large B-Cell Lymphomas. Clin Cancer Res 22, 2290–2300. [DOI] [PubMed] [Google Scholar]
- Morin RD, Mendez-Lago M, Mungall AJ, Goya R, Mungall KL, Corbett RD, Johnson NA, Severson TM, Chiu R, Field M, et al. (2011). Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature 476, 298–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin RD, Mungall K, Pleasance E, Mungall AJ, Goya R, Huff RD, Scott DW, Ding J, Roth A, Chiu R, et al. (2013). Mutational and structural analysis of diffuse large B-cell lymphoma using whole-genome sequencing. Blood 122, 1256–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Tateishi K, Niwa T, Matsushita Y, Tamura K, Kinoshita M, Tanaka K, Fukushima S, Takami H, Arita H, et al. (2016). Recurrent mutations of CD79B and MYD88 are the hallmark of primary central nervous system lymphomas. Neuropathology and applied neurobiology 42, 279–290. [DOI] [PubMed] [Google Scholar]
- Ngo VN, Young RM, Schmitz R, Jhavar S, Xiao W, Lim KH, Kohlhammer H, Xu W, Yang Y, Zhao H, et al. (2011). Oncogenically active MYD88 mutations in human lymphoma. Nature 470, 115–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HY, Lee SB, Yoo HY, Kim SJ, Kim WS, Kim JI, and Ko YH (2016). Whole-exome and transcriptome sequencing of refractory diffuse large B-cell lymphoma. Oncotarget 7, 86433–86445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasqualucci L, Trifonov V, Fabbri G, Ma J, Rossi D, Chiarenza A, Wells VA, Grunn A, Messina M, Elliot O, et al. (2011). Analysis of the coding genome of diffuse large B-cell lymphoma. Nat Genet 43, 830–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelan JD, Young RM, Webster DE, Roulland S, Wright GW, Kasbekar M, Shaffer AL 3rd, Ceribelli M, Wang JQ, Schmitz R, et al. (2018). A multiprotein supercomplex controlling oncogenic signalling in lymphoma. Nature 560, 387–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramis-Zaldivar JE, Gonzalez-Farre B, Balague O, Celis V, Nadeu F, Salmeron-Villalobos J, Andres M, Martin-Guerrero I, Garrido-Pontnou M, Gaafar A, et al. (2020). Distinct molecular profile of IRF4-rearranged large B-cell lymphoma. Blood 135, 274–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy A, Zhang J, Davis NS, Moffitt AB, Love CL, Waldrop A, Leppa S, Pasanen A, Meriranta L, Karjalainen-Lindsberg ML, et al. (2017). Genetic and Functional Drivers of Diffuse Large B Cell Lymphoma. Cell 171, 481–494.e415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenwald A, Wright G, Chan WC, Connors JM, Campo E, Fisher RI, Gascoyne RD, Muller-Hermelink HK, Smeland EB, Giltnane JM, et al. (2002). The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N Engl J Med 346, 1937–1947. [DOI] [PubMed] [Google Scholar]
- Rossi D, Diop F, Spaccarotella E, Monti S, Zanni M, Rasi S, Deambrogi C, Spina V, Bruscaggin A, Favini C, et al. (2017). Diffuse large B-cell lymphoma genotyping on the liquid biopsy. Blood 129, 1947–1957. [DOI] [PubMed] [Google Scholar]
- Runge HFP, Lacy S, Barrans S, Beer PA, Painter D, Smith A, Roman E, Burton C, Crouch S, Tooze R, and Hodson DJ (2021). Application of the LymphGen classification tool to 928 clinically and genetically-characterised cases of diffuse large B cell lymphoma (DLBCL). Br J Haematol 192, 216–220. [DOI] [PubMed] [Google Scholar]
- Scherer F, Kurtz DM, Newman AM, Stehr H, Craig AF, Esfahani MS, Lovejoy AF, Chabon JJ, Klass DM, Liu CL, et al. (2016). Distinct biological subtypes and patterns of genome evolution in lymphoma revealed by circulating tumor DNA. Science translational medicine 8, 364ra155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz R, Ceribelli M, Pittaluga S, Wright G, and Staudt LM (2014). Oncogenic mechanisms in Burkitt lymphoma. Cold Spring Harbor perspectives in medicine 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz R, Wright GW, Huang DW, Johnson CA, Phelan JD, Wang JQ, Roulland S, Kasbekar M, Young RM, Shaffer AL, et al. (2018). Genetics and Pathogenesis of Diffuse Large B-Cell Lymphoma. N Engl J Med 378, 1396–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sha C, Barrans S, Cucco F, Bentley MA, Care MA, Cummin T, Kennedy H, Thompson JS, Uddin R, Worrillow L, et al. (2019). Molecular High-Grade B-Cell Lymphoma: Defining a Poor-Risk Group That Requires Different Approaches to Therapy. J Clin Oncol 37, 202–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaffer AL, Wright G, Yang L, Powell J, Ngo V, Lamy L, Lam LT, Davis RE, and Staudt LM (2006). A library of gene expression signatures to illuminate normal and pathological lymphoid biology. Immunological reviews 210, 67–85. [DOI] [PubMed] [Google Scholar]
- Shaffer AL, Yu X, He Y, Boldrick J, Chan EP, and Staudt LM (2000). BCL-6 represses genes that function in lymphocyte differentiation, inflammation, and cell cycle control. Immunity 13, 199–212. [DOI] [PubMed] [Google Scholar]
- Shin SH, Kim YJ, Lee D, Cho D, Ko YH, Cho J, Park WY, Park D, Kim SJ, and Kim WS (2019). Analysis of circulating tumor DNA by targeted ultra-deep sequencing across various non-Hodgkin lymphoma subtypes. Leuk Lymphoma 60, 2237–2246. [DOI] [PubMed] [Google Scholar]
- Spina V, Bruscaggin A, Cuccaro A, Martini M, Di Trani M, Forestieri G, Manzoni M, Condoluci A, Arribas A, Terzi-Di-Bergamo L, et al. (2018). Circulating tumor DNA reveals genetics, clonal evolution, and residual disease in classical Hodgkin lymphoma. Blood 131, 2413–2425. [DOI] [PubMed] [Google Scholar]
- Suehara Y, Sakata-Yanagimoto M, Hattori K, Nanmoku T, Itoh T, Kaji D, Yamamoto G, Abe Y, Narita K, Takeuchi M, et al. (2018). Liquid biopsy for the identification of intravascular large B-cell lymphoma. Haematologica 103, e241–e244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vater I, Montesinos-Rongen M, Schlesner M, Haake A, Purschke F, Sprute R, Mettenmeyer N, Nazzal I, Nagel I, Gutwein J, et al. (2015). The mutational pattern of primary lymphoma of the central nervous system determined by whole-exome sequencing. Leukemia 29, 677–685. [DOI] [PubMed] [Google Scholar]
- Vela V, Juskevicius D, Gerlach MM, Meyer P, Graber A, Cathomas G, Dirnhofer S, and Tzankov A (2020). High throughput sequencing reveals high specificity of TNFAIP3 mutations in ocular adnexal marginal zone B-cell lymphomas. Hematol Oncol 38, 284–292. [DOI] [PubMed] [Google Scholar]
- Venturutti L, Teater M, Zhai A, Chadburn A, Babiker L, Kim D, Beguelin W, Lee TC, Kim Y, Chin CR, et al. (2020). TBL1XR1 Mutations Drive Extranodal Lymphoma by Inducing a Pro-tumorigenic Memory Fate. Cell 182, 297–316 e227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Victora GD, Dominguez-Sola D, Holmes AB, Deroubaix S, Dalla-Favera R, and Nussenzweig MC (2012). Identification of human germinal center light and dark zone cells and their relationship to human B-cell lymphomas. Blood 120, 2240–2248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CY, Mayo MW, Korneluk RG, Goeddel DV, and Baldwin AS Jr. (1998). NF-kappaB antiapoptosis: induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science 281, 1680–1683. [DOI] [PubMed] [Google Scholar]
- Wilson WH, Young RM, Schmitz R, Yang Y, Pittaluga S, Wright G, Lih CJ, Williams PM, Shaffer AL, Gerecitano J, et al. (2015). Targeting B cell receptor signaling with ibrutinib in diffuse large B cell lymphoma. Nat Med 21, 922–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wist M, Meier L, Gutman O, Haas J, Endres S, Zhou Y, Rosler R, Wiese S, Stilgenbauer S, Hobeika E, et al. (2020). Noncatalytic Bruton’s tyrosine kinase activates PLCgamma2 variants mediating ibrutinib resistance in human chronic lymphocytic leukemia cells. J Biol Chem 295, 5717–5736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright G, Tan B, Rosenwald A, Hurt EH, Wiestner A, and Staudt LM (2003). A gene expression-based method to diagnose clinically distinct subgroups of diffuse large B cell lymphoma. Proc Natl Acad Sci U S A 100, 9991–9996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright GW, Huang DW, Phelan JD, Coulibaly ZA, Roulland S, Young RM, Wang JQ, Schmitz R, Morin RD, Tang J, et al. (2020). A Probabilistic Classification Tool for Genetic Subtypes of Diffuse Large B Cell Lymphoma with Therapeutic Implications. Cancer Cell 37, 551–568 e514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Shaffer AL 3rd, Emre NC, Ceribelli M, Zhang M, Wright G, Xiao W, Powell J, Platig J, Kohlhammer H, et al. (2012). Exploiting synthetic lethality for the therapy of ABC diffuse large B cell lymphoma. Cancer Cell 21, 723–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younes A, Sehn LH, Johnson P, Zinzani PL, Hong X, Zhu J, Patti C, Belada D, Samoilova O, Suh C, et al. (2019). Randomized Phase III Trial of Ibrutinib and Rituximab Plus Cyclophosphamide, Doxorubicin, Vincristine, and Prednisone in Non-Germinal Center B-Cell Diffuse Large B-Cell Lymphoma. J Clin Oncol 37, 1285–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young RM, Phelan JD, Wilson WH, and Staudt LM (2019). Pathogenic B-cell receptor signaling in lymphoid malignancies: New insights to improve treatment. Immunological reviews 291, 190–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young RM, Wu T, Schmitz R, Dawood M, Xiao W, Phelan JD, Xu W, Menard L, Meffre E, Chan WC, et al. (2015). Survival of human lymphoma cells requires B-cell receptor engagement by self-antigens. Proc Natl Acad Sci U S A 112, 13447–13454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Grubor V, Love CL, Banerjee A, Richards KL, Mieczkowski PA, Dunphy C, Choi W, Au WY, Srivastava G, et al. (2013). Genetic heterogeneity of diffuse large B-cell lymphoma. Proc Natl Acad Sci U S A 110, 1398–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou XA, Louissaint A Jr., Wenzel A, Yang J, Martinez-Escala ME, Moy AP, Morgan EA, Paxton CN, Hong B, Andersen EF, et al. (2018a). Genomic Analyses Identify Recurrent Alterations in Immune Evasion Genes in Diffuse Large B-Cell Lymphoma, Leg Type. J Invest Dermatol 138, 2365–2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Liu W, Xu Z, Zhu H, Xiao D, Su W, Zeng R, Feng Y, Duan Y, Zhou J, and Zhong M (2018b). Analysis of Genomic Alteration in Primary Central Nervous System Lymphoma and the Expression of Some Related Genes. Neoplasia 20, 1059–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 - Phoenix trial clinical and molecular features, related to Figure 1
Table S2 - Mutant isoforms identified in NOTCH1-mutant DLBCL samples, related to Figure 4
Table S3 - Targeted DNA resequencing panel, related to STAR Methods
Data Availability Statement
DNA and RNA sequencing data from the non-China cohort are available through the European Genome-phenome archive (EGA) under accession EGAS00001005554. Sequencing data from the China cohort cannot be made available due to Chinese prohibitions on genomic data sharing. Clinical data are available in Table S1. LymphGen code is available at https://doi.org/10.5281/zenodo.3700086.