

SUMMARY
A core network of widely expressed proteins within the glutamatergic post-synapse mediates activity-dependent synaptic plasticity throughout the brain, but the specific proteomic composition of synapses differs between brain regions. Here, we address the question, how does proteomic composition affect activity-dependent protein-protein interaction networks (PINs) downstream of synaptic activity? Using quantitative multiplex co-immunoprecipitation, we compare the PIN response of in vivo or ex vivo neurons derived from different brain regions to activation by different agonists or different forms of eyeblink conditioning. We report that PINs discriminate between incoming stimuli using differential kinetics of overlapping and non-overlapping PIN parameters. Further, these “molecular logic rules” differ by brain region. We conclude that although the PIN of the glutamatergic post-synapse is expressed widely throughout the brain, its activity-dependent dynamics show remarkable stimulus-specific and brain-region-specific diversity. This diversity may help explain the challenges in developing molecule-specific drug therapies for neurological disorders.
Graphical Abstract
In brief
Molecular encoding of sensory stimuli occurs through modification of synaptic protein networks, but the underlying rules are poorly defined. Lautz et al. demonstrate that four brain regions respond to eyeblink conditioning by modifying excitatory synapse protein interaction networks using different encoding strategies, despite employing a similar set of proteins.
INTRODUCTION
The glutamatergic synapse is believed to be the fundamental unit of neuronal computation. These computations are conducted by a network of interacting proteins that mediate intracellular signal transduction and synaptic modifications. Proteomic characterization of the glutamatergic postsynapse has identified thousands of “synaptic” proteins in brain-wide preparations (Bayés et al., 2011,2012; Collins et al., 2006; Jordan et al., 2004; Li et al., 2004; Peng et al., 2004), as well as considerable variation between brain regions (Cheng et al., 2006; Lee et al., 2017a; Trinidad et al., 2008). Although core synaptic scaffolds (e.g., DLGs, Homers, and Shanks) or receptors (e.g., AMPAR or NMDAR) are present at most excitatory synapses (albeit at different ratios) (Zhu et al., 2018), hundreds of additional proteins are differentially expressed across brain regions or even across synapse types along a single dendrite (Counts et al., 2006; Esteves and Cardoso, 2020; Roy et al., 2018). These proteins assemble into large multiprotein complexes, sometimes mega-Dalton in size (Frank and Grant, 2017; Frank et al., 2016,2017; Pocklington et al., 2006), the composition of which also shows considerable heterogeneity across brain regions, developmental stages, or downstream of genetic mutations (Lee et al., 2017b; Li et al., 2017). This protein-level diversity is believed to underlie morphological (Zhu et al., 2018) and electrophysiological (Díaz-Quesada et al., 2014; Holderith et al., 2012; Schikorski and Stevens, 1997) diversity among neurons that receive glutamatergic input and to be fundamental to the complexity and information processing capacity of the brain.
Downstream of synaptic activity, modifications to the proteome change the synapse’s response to subsequent activity in a process generally referred to as synaptic plasticity. Measured electrophysiologically, there is great diversity in plasticity, with different cell types responding in different ways to identical stimuli (Gouwens et al., 2019; Jiang et al., 2015; Luo et al., 2017). However, measured biochemically, the level of diversity is less clear. Activation of CamKII (Lisman et al., 2012), mobilization of SynGAP (Araki et al., 2015, 2020; Lautz et al., 2018) phosphorylation of NMDA receptors (NMDARs) (Giannakopoulos et al., 2010; Rosenblum et al., 1996; Trepanier et al., 2012), and release of mGluR5 from Homer scaffolds (Guo et al., 2015; Lautz et al., 2018; Ronesi et al., 2012) have been described for hippocampal or neocortical neurons, and likely occur elsewhere. However, the molecular rules that control plasticity may differ across brain areas; for example, at hippocampal synapses, kinase activation mediates long-term potentiation (LTP), whereas cerebellar LTP is mediated by phosphatase activation (Belmeguenai and Hansel, 2005). At a proteomic level, induction of LTP has been shown to alter the phosphorylation status of 570 sites across 220 postsynaptic proteins in the hippocampus (Coba et al., 2009). Moreover, pharmacological manipulation of ionotropic, metabotropic, and dopamine receptors in the hippocampus activates overlapping phosphorylation networks, such that distinct physiological inputs activate different combinations of biochemical pathways (Coba et al., 2009). How these modifications translate into proteome-wide changes in protein complex composition has not been widely studied.
We previously used a multiplexed co-immunoprecipitation approach to demonstrate that, in neocortical neurons, stimulation of ionotropic versus metabotropic glutamate receptors elicits distinct changes in the composition of, and presumably in the function of, synaptic protein complexes (Lautz et al., 2018). Direct measurement of a network of glutamate synapse proteins joining or dissociating from shared complexes in an activity-dependent manner supported a hypothesis that different combinations of proteins in complex with each other encode units of information. In this model, discrete signaling inputs trigger different combinatorial protein-state-codes, a hypothesis originally proposed by Pawson (1995), allowing neurons to generate situationally appropriate responses to synaptic input. However, given the diversity of synaptic proteomic composition across the brain, it is unclear if the codes we observed in cultured cortical neurons remain consistent, or if biochemical encoding of information varies with proteomic composition across brain regions.
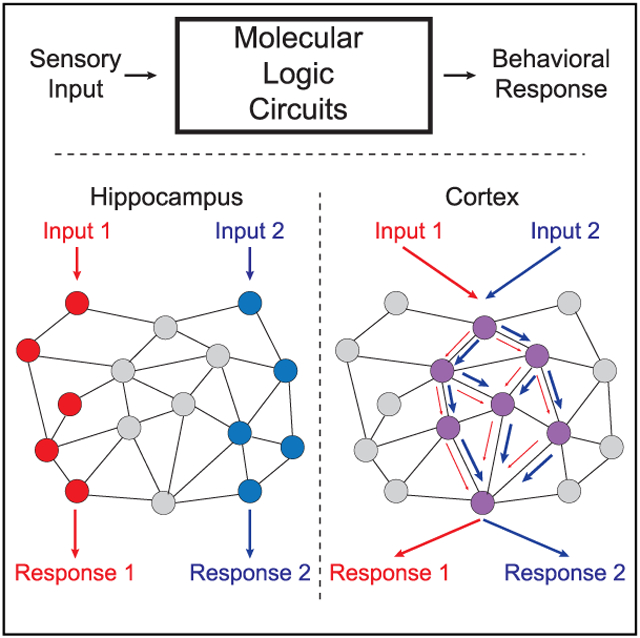
Here, we address the general question of how consistent are experience-dependent modifications in the glutamate synapse protein interaction network (PIN) across brain regions. Are glutamatergic postsynapses from brain regions with distinct proteomic compositions and/or organizations modified by a similar set of “molecular logic” rules? Or alternatively, do differentially expressed proteins fundamentally change the rules of molecular logic circuits, such that identical inputs lead to different responses? We first characterize acute changes in the structure of the glutamatergic post-synapse PIN using cultured neurons in vitro and brain slices ex vivo following ionotropic and metabotropic glutamate receptor activation and the chemical induction of synaptic plasticity. We then induce two different forms of associative learning in awake and behaving mice using classical eye blink conditioning (EBC) and measure the glutamate synapse PIN responses of four brain areas implicated in learning based on electrophysiological and lesion data in the literature (Heiney et al., 2014; Weiss and Disterhoft, 2011,2015; Welsh and Harvey, 1998). We demonstrate that responses to the same stimulus are often quite different across brain areas due to differences in the composition of the protein network. We propose that this diversity of biochemical mechanisms underlies the high information-processing capability of the glutamate post-synapse, and that the expression of this diversity across brain circuits helps define the contribution that each brain area can make to learning.
RESULTS
NMDA, DHPG, and glutamate stimulation produce widespread PIN re-arrangement
PIN networks can be conceptualized as molecular logic circuits, simultaneously translating multiple receptor inputs into intracellular signals, and performing molecular calculations to integrate inputs and select an appropriate response (Figure 1A). In previous studies, we developed a system to investigate the molecular logic coding of a PIN composed of glutamate receptors, scaffolds, and downstream signaling proteins, called quantitative multiplex co-immunoprecipitation (QMI) (Heavner et al., 2020; Lautz et al., 2018, 2019; Smith et al., 2016). QMI uses antibody-coupled flow cytometry beads to quantify changes in targeted proteins in shared complexes with exposed epitopes (PiSCES); we refer to detected interactions as PiSCES instead of protein-protein interactions to highlight that the detected interactions are not necessarily direct, but simply imply shared membership in a larger protein complex. Our QMI panel is composed of 20 protein targets selected based on synaptic expression, known co-association, and linkage to autism, and includes glutamate receptors (NMDA subunits NR1, 2A, 2B, AMPA subunits GluR1 and GluR2, and metabotropic glutamate receptor [mGluR5]), scaffolding proteins (Shank 1 and 3, Homer 1 and 1a, PSD95, SAP97, SAPAP1, and Neuroligin3), and signal transduction effectors (SynGAP, Fyn, CamKII, PI3K, PIKE, and Ube3A) (Brown et al., 2018; Lautz et al., 2018). Importantly, QMI has been validated for antibody specificity (Lautz et al., 2018) and optimized for lysis detergent (Lautz et al., 2019), and specific interactions and protein complexes detected by QMI have been confirmed by immunoprecipitation (IP)-western blotting and size exclusion chromatography (Lautz et al., 2018, 2019).
Figure 1. Glutamate post-synapse PIN kinetics following NMDA and DHPG stimulation of cultured cortical neurons.

(A) Conceptual illustration of a PIN logic circuit linking receptor inputs with cellular outcome. Receptors on the cell surface activate “molecular logic circuits” that link receptor activation to downstream cellular responses.
(B) Experimental design.
(C–F) PCA of 30 s (C), 5 min (D), 15 min (E), and 2 h (F) time points separates aCSF (A, red), glutamate (G, green), NMDA (N, blue), and DHPG (D, purple) treatments. Each data point represents a single biological replicate, N = 48 total.
(G) Average aCSF-normalized time course of PiSCES in the turquoise CNA module for the aCSF, glutamate, NMDA, and DHPG treatments. *, #, and@indicate p < 0.005 for glutamate, NMDA, and DHPG, respectively, by Dunnett’s post hoc test after significant 2-way ANOVA (treatment, F3,48 = 52.31, p < 0.0001; time, F3,48 = 0.13 NS; interaction, F9,48 = 2.92, p < 0.01).
(H) Average aCSF-normalized time course of PiSCES in the yellow CNA module presented as in (G). *, #, and@indicate p < 0.05 for glutamate, NMDA, and DHPG, respectively, by Dunnett’s post hoc tests after significant 2-way ANOVA (treatment, F3,48 = 4.78, p < 0.01; time, F3,48 = 7.94, p < 0.0005; interaction, F9,48 = 1.65, NS).
(I) Heatmap of all PiSCES that were ANC∩CNA significant for the 30 s to 2 h time course expressed as row-normalized MFI. All PiSCES shown are ANC∩CNA significant, meaning they are significant by the ANC statistical test for a comparison between the ACSF condition and at least one time point/treatment and belong to a CNA module that is significantly correlated with NMDA and/or DHPG treatment. CNA modules are illustrated by colored bars on the left, for details on CNA analysis see Figure S2. Each column represents a single biological replicate, N = 48, and each box represents a single PiSCES measurement.
Prior data suggest that the glutamate synapse PIN uses a qualitative mechanism to encode distinct signaling inputs: different PiSCES are engaged by 5 min of NMDA versus mGluR stimulation (Lautz et al., 2018). However, in other systems, quantitative (i.e., intensity) (Neier et al., 2019) or kinetic (Toettcher et al., 2013) differences in the engagement of identical signaling intermediates allow cells to distinguish between input types. To further characterize PIN logic-coding in a simple model system, we stimulated days in vitro (DIV) 19–21 cortical neuronal cultures for up to 5 min with glutamate (GLUT), NMDA/glycine, DHPG, or artificial cerebrospinal fluid (aCSF) control and measured the PIN response at 30 s, 5 min, 15 min, and 2 h to establish the qualitative, quantitative, and kinetic differences between different types of synaptic stimulation (Figure 1B). To visualize high-dimensional QMI data, we first applied principal component analysis (PCA). After 30 s, all three treatment conditions separated from aCSF across PC1 (Figure 1C). At 5 min, glutamate and NMDA stimulation continued to separate from aCSF across PC1, whereas DHPG normalized across PC1 but separated across PC2 (Figure 1D), similar to our previous report on only the 5-min time point (Lautz et al., 2018). By 15 min, aCSF and DHPG overlapped, whereas NMDA and glutamate remained separated across PC1 (Figure 1E). At 2 h, the separation between aCSF/DHPG and NMDA/glutamate, while still present, was smaller in magnitude (Figure 1F). Overall, NMDA and glutamate stimulation produced a stronger, more sustained response, whereas DHPG induced a transient response that was initially similar to NMDA/GLUT but diverged at 5 min.
To further explore network-scale temporal dynamics, we performed weighted correlation network analysis (CNA) on all time points (Figure S1). CNA sorted 193 PiSCES measurements above background into 6 modules based on correlated behavior over 48 experiments; two modules significantly correlated with a treatment. One module (“turquoise”) (Figures 1H and 1I) correlated with all treatments, and contained 22 PiSCES that met the strict criteria of module membership (MM) >0.7. All PiSCES in the turquoise module were also significant by a second test developed specifically for QMI data that relies on independent assumptions, adaptive, non-parametric statistical test corrected for multiple comparisons (ANC) (Smith et al., 2016). We refer to these high-confidence PiSCES that changed acutely following treatment as “ANCXCNA significant,” indicating they were identified independently by two statistical approaches. To compare turquoise module behavior across stimulations, we averaged the z-scaled intensity of PiSCES for each treatment/ time point (n = 4 each) and normalized to aCSF (Figure 1H). At 30 s, all three treatment conditions were significantly different from aCSF. In DHPG, the module was no longer significantly different by 5 min, but in NMDA and GLUT, the module activation continued to strengthen, peaking at 5–15 min, and remained significantly different even after 2 h. Thus, although the turquoise module was activated in all treatment conditions, the kinetics of activation distinguished NMDA/GLUT (strong and sustained) from DHPG (weak and transient). A second CNA module, “yellow,” was correlated with only DHPG treatment, and its z-scaled intensity was significantly different from aCSF in the DHPG condition, only at 5 min (Figure 1G), although GLUT stimulation (that targets both NMDA and mGluRs) trended toward significance as well.
We next looked more closely at the specific PiSCES contained within each module by row-normalizing the median fluorescent intensity (MFI) of each ANCXCNA PiSCES in the yellow or turquoise modules and displaying them as a heatmap for aCSF, NMDA, and DHPG treatment (Figure 1I). These data reveal a rapid dissociation of Homer-, Shank-, and SynGAP-containing PiSCES at 30 s, which increased in magnitude at 5 and 15 min in NMDA, but rapidly returned to baseline in DHPG. Even after 2 h, PiSCES remained lower in NMDA and GLUT conditions as compared to aCSF. ANC∩CNA analysis was also performed on each time point independently (Figure S3) and revealed additional time point-specific PiSCES important to synaptic plasticity, such as increased association among PSD95 and AMPA-type glutamate receptors (GluR1/2).
To ensure cells remained healthy and active following glutamate agonist treatment, we performed Ca2+ imaging of cultures for 30 min following a 5-min stimulation. Treatment with glutamate or NMDA/glycine induced a rapid increase in intracellular Ca2+ (Figures S1A and S1B), whereas we were unable to detect any significant changes in intracellular calcium following treatment with DHPG. Upon removal of agonists, intracellular Ca2+ returned to baseline for 30 min, and subsequent depolarization with KCL increased intracellular Ca2+, demonstrating that cells remained alive and receptive to stimuli (Figures S2A and S2B). We also used TUNEL staining to quantify agonist-induced apoptosis at 2 h and did not observe increased rates neuronal apoptosis in any condition (Figures S2C and S2D).
Overall, these data highlight two PiSCES modules, centered on Homer-, Shank-, SynGAP-, and mGluR5-containing interactions that activate following glutamate agonist stimulation. DHPG versus NMDA stimulation is encoded within these modules by quantitative (yellow ON/OFF), qualitative (turquoise weak/strong), and temporal (turquoise fast/slow) parameters.
Chemical LTP activates a different network of PiSCES
Long-term potentiation (LTP) is perhaps the most well studied form of synaptic plasticity and is induced electrophysiologically by pulsed activations of excitatory synaptic inputs. Activating downstream signaling pathways directly with small molecules using “chemical LTP” (cLTP) mimics many of the electrophysiological and biochemical features of LTP in cultured neurons (Kopec et al., 2006; Otmakhov et al., 2004; Zhang et al., 2020). We applied a cLTP protocol (Otmakhov et al., 2004) to DIV 19–21 cultured cortical neurons and performed QMI. PCA revealed separation of control and cLTP neurons (Figure 2A), and ANC∩CNA identified 20 PiSCES that were significantly altered (Figure 2B), consisting of increased Shank1-, Homer1-, and Syn-GAP-containing PiSCES but decreased NL3, PIKE, and Fyn PiSCES. We compared these data to the GLUT 15-min time point because it most closely matched the time of the cLTP treatment (30 min) and involved the activation of the greatest number of PiSCES in the agonist experiments. Nine of 20 PiSCES activated by cLTP were not activated by GLUT, and 23/32 PiSCES activated by GLUT were not activated by cLTP. Of the 11 PiSCES activated by both treatments, the majority (8/11) changed in the opposite direction (e.g., Homer1_SynGAP was increased in cLTP and decreased in GLUT, whereas PIKE_PIKE showed the opposite pattern, Figure 2C). When the log2 fold change of all ANC∩CNA PiSCES was plotted on an x-y axis, cLTP and GLUT application produced strikingly different patterns of PiSCES activation, with the majority of PiSCES showing opposite responses (Figure 2D). These data demonstrate the synaptic PIN under study can adopt multiple input-specific states in a cell culture paradigm and highlight PIN-level differences between cLTP and glutamate receptor stimulation.
Figure 2. Glutamate post-synapse PIN alterations following chemical LTP in cultured cortical neurons.

(A) PCA showing separation of aCSF control (C, red) and cLTP (L, blue).
(B) Heatmap of all PiSCES that were ANC∩CNA significant for the cLTP versus control comparison, meaning they are significant by the ANC statistical test and belong to a CNA module that is significantly correlated with cLTP treatment. Each column represents a single biological replicate, N = 8, and each box represents a single PiSCES measurement.
(C) Graphs comparing the log2 fold change of two example PiSCES, Homer1_SynGAP and PIKE_PIKE, that were ANC∩CNA-significant in both cLTP and 15-min GLUT experiments. Points represent a ratio between the aCSF condition and the respective stimulation, N = 4 per treatment.
(D) x-y plot comparing the log2 fold change (FC) of all PiSCES that were ANC∩CNA significant for either the cLTP experiment (blue), the 15 min glutamate time point (red), or both experiments (purple).
NMDA stimulation of acute brain slices reveals differences across brain areas
We next asked how neurons with known differences in proteomic composition would respond to the same stimulus. We micro-dissected cortical (CTX) or hippocampal (HC) tissue from acute slice preparations stimulated with NMDA/glycine or aCSF for 5 min and ran QMI in parallel to allow direct comparisons (Figure 3A). PCA of QMI data showed separation between CTX and HC, confirming that the proteomic organization of the two tissues is distinct (Figure 3B). Directly comparing CTX versus HC tissue (ignoring NMDA treatment) by ANC∩CNA, we identified 22 PiSCES that were significantly different between brain areas (Figure 3C); notable differences included GluR1_GluR2 and GluR1_GluR1 (higher in HC), AMPA-PSD95 associations (higher in CTX), multiple Homer-containing PiSCES (higher in HC, although Homer detection was higher in CTX), and PI3K/PIKE/NMDAR PiSCES (higher in HC). The large PiSCES differences between tissue types masked the more subtle activity-dependent differences within each tissue. We therefore limited our analysis to comparisons of aCSF versus NMDA within the same brain area using ANC∩CNA and visualized the differences with heatmaps (Figure 3D). In CTX, 14 PiSCES changed significantly with NMDA and 10 changed in HC, with only 5 overlapping (Figure 3F): e.g., GluR1_PSD95 began with higher MFI in CTX, but increased in both CTX and HC; conversely Homer_PSD95 started lower in CTX. For PiSCES that were significant only in CTX (Figure 3E), most were detected at low abundance in aCSF and increased with NMDA, whereas in the HC, these same PiSCES were already at the higher abundance at baseline. For PiSCES that were significant only in HC (Figure 3G), most were detected at a higher abundance in aCSF and decreased with NMDA. The abundance of these PiSCES in CTX started at a lower level and trended toward a decrease with NMDA, but the magnitude of the decrease was not large enough to fulfill strict ANC∩CNA criteria. These data reveal a complex interaction between basal protein expression and activity-dependent PIN encoding of stimuli in CTX and HC.
Figure 3. Glutamate post-synapse PIN dynamics following ex vivo NMDA stimulation of CTX and HC slices.

(A) Experimental design.
(B) PCA of CTX (C, red) and HC (H, blue) following aCSF (A, lighter shade) or NMDA (N, darker shade) treatment. PCA demonstrates separation by brain region but not by NMDA stimulation. Each point represents a biological replicate, N = 16.
(C) Heatmap of all PiSCES that were ANC∩CNA significant for the CTX versus HC comparison, meaning they are significant by the ANC statistical for a comparison between CTX versus HC, and belong to a CNA module that is significantly correlated with brain region. Each column represents a single biological replicate, N = 16, and each box represents a single PiSCES measurement.
(D) Heatmap of all PiSCES that were ANC∩CNA significant for the NMDA versus aCSF comparison, meaning they are significant by the ANC statistical test for a comparison between ACSF versus NMDA in either brain region and belong to a CNA module that is significantly correlated with NMDA treatment. Each column represents a single biological replicate, N = 16, and each box represents a single PiSCES measurement. PiSCES are grouped based on whether they are ANC significant for the ACSF versus NMDA comparison in cortex (top), hippocampus (bottom), or both (middle).
(E–G) Normalized MFIs for PiSCES that were ANC∩CNA significant only in CTX (E), only in HC (G), or both (F). Points represent the average normalized MFI ± SEM for N = 4 biological replicates of a single PiSCES measurement. Lines connect points representing the same PiSCES for visual clarity but do not represent repeated-measures.
In a separate experiment, we repeated QMI after NMDA receptor stimulation in acutely prepared brain stem slices containing the inferior olive (IO), a pre-cerebellar nucleus in the brain stem that undergoes significant NMDA-receptor mediated plasticity of its intrinsic oscillatory activity in order to influence cerebellar function and learning (Welsh and Turecek, 2017). PCA showed considerable overlap between aCSF and NMDA (Figure 4A), but ANC∩CNA identified 7 PiSCES that significantly changed with NMDA (Figure 4B), involving Homer, Shank3, and NMDARs. Batch effects inherent to QMI prevented direct comparisons of protein composition across separate experiments, so we used heatmaps that were row-normalized independently for all three tissue types to observe the behavior of PiSCES that changed significantly with NMDA in any tissue type (Figure 4C). Compared to HC and CTX, the IO was unique (Figure 4D). Zero of 19 PiSCES that were significant with NMDA stimulation in the HC or CTX changed significantly in the IO (by ANC∩CNA), even though the majority were detected. Conversely, 0 of 7 PiSCES that responded to NMDA in the IO responded to NMDA in HC/CTX. Although many PiSCES decreased with NMDA in HC/CTX, all significantly changed PiSCES increased with NMDA in the IO. Collectively, these ex vivo experiments show that chemical stimulation of glutamate receptors or downstream signaling pathways causes the synaptic PIN to undergo stimulus-specific rearrangements that differ by input type and by proteomic composition of the brain area under study.
Figure 4. Effects of NMDA stimulation on the ex vivo IO as compared to CTX and HC.

(A) PCA of aCSF (A, red) and NMDA (N, blue) groups for the IO. Each point represents a biological replicate, N = 8.
(B) Heatmap of all PiSCES that are ANC∩CNA significant for the NMDA versus aCSF comparison for the IO, meaning they are significant by the ANC statistical test for a comparison between ACSF versus NMDA in the IO and belong to a CNA module that is significantly correlated with NMDA treatment.
(C) Heatmaps of all PiSCES that were ANC∩CNA significant for the NMDA versus aCSF comparison in CTX, HC, and IO (CTX and HC data are identical to that in Figure 3). The heatmap is row-normalized separately for each brain region to highlight overall PiSCES behavior while ignoring baseline differences in protein expression. Boxes indicate PiSCES that are ANC∩CNA significant for that brain region: purple boxes indicate significant for both CTX and HC, blue indicates HC, red indicates CTX, and green indicates IO. Although there is overlap in the PiSCES activated by NMDA in HC and CTX, the response in IO is unique.
(D) For each brain area, the averaged normalized MFI value of all PiSCES that were ANC∩CNA significant in that brain area are shown for all other brain areas. For example, the left graph shows PiSCES that significant in the HC also showed similar behavior in CTX, but not in the IO. Conversely, PiSCES that were significant in the IO (right graph) did not change in CTX nor in HC.
Different forms of associative learning elicit region- and learning-specific rearrangements in post-synaptic multiprotein complexes
We next asked how sensory stimuli are processed via PiSCES networks in vivo by inducing two forms of associative learning in awake behaving mice using classical eye blink conditioning (EBC), in which an auditory conditioned stimulus (CS) precedes a periorbital electric shock unconditioned stimulus (US) that elicits a blink reflex (Figure 5A). Over repeated CS-US pairings, mice associate the tone CS with the US and acquire conditioned eye blink responses (CRs) to the tone in anticipation of the US (Welsh and Harvey, 1989; Welsh et al., 2005). In trace EBC, a period of no stimulation (a trace interval) separates CS offset and the US (Figure 5C). In delay EBC, there is no trace interval separating the CS and US (Figure 5D). Trace and delay EBC evoke two forms of associative learning that differ in their essential neural circuitry (Takehara-Nishiuchi, 2018), relative involvement of the forebrain (Cheng et al., 2008; Weiss and Disterhoft, 2011), and dependence upon awareness in humans (Clark and Squire, 1999). An explicitly unpaired control group consisted of mice that received a procedure in which the handling, session duration, number of delivered CSs and USs, and number of reflex eye blinks were identical to the EBC groups, except that the CS and USs were never presented together (Figure 5A). The only parameter that differed between the three groups was the CS-US interval.
Figure 5. Associative learning modifies the glutamate post-synapse PIN in the mPFC.

(A) Experimental design. Eye blinks were detected as muscle activations by intramuscular EMG. CS-US interval for the explicitly unpaired control paradigm was randomized at 30 ± 5 s.
(B and C) Average (±SEM) CRs acquired during trace EBC (B, red) and delay EBC (C, blue) as compared to small reflex muscle activations elicited by the CS during unpaired stimuli (black). Block diagrams underneath EMG traces indicate CS-US timing (n = 8 mice per group).
(D and E) The percentage CRs during 2 daily sessions of trace (D) (red) or delay (E) (blue) EBC in which the percentage CRs exceeded the unpaired control group for both trace EBC (2-way ANOVA: F(1,14) = 35.8, p < 0.0001)and delay EBC (2-way ANOVA: F(1,14) = 10.3, p = 0.006). Asterisks indicate blocks of 10 trials that were significantly different between EBC and unpaired controls by Sidak post hoc testing (*p < 0.05, **p < 0.01).
(F) Peak amplitude of trails in which a CR was recorded for trace (red) and delay (blue) EBC, during the P2 (60–100 ms) or P3 (200–250 ms) time windows. ****p < 0.0001 by ANOVA followed by Dunnett’s post hoc test.
(G) Simplified schematic of brain regions mediating procedural or declarative memories with brain regions that were dissected and lysed for QMI analysis highlighted in colored boxes.
Mice receiving trace (Figure 5D) or delay (Figure 5E) EBC showed robust CR acquisition over 2 EBC sessions, as evidenced by an increase in the percentage CRs that exceeded the CS responding of explicitly unpaired controls. Mice given delay EBC showed a greater increase in the peak amplitude of the CR (Figure 5F), whereas mice given trace conditioning showed a more robust increase in percentage CRs (Figure S4). The use of the explicitly unpaired stimulus control was important to confirm that CR acquisition was specifically due to associative learning and was not due to non-associative influences such as sensitization, an increase in baseline responding, or pseudoconditioning (Welsh and Harvey, 1989). Figures 5B and 5C demonstrate the average topography of CRs acquired to the CS under the trace and delay EBC paradigms, indicating that their temporal kinetics adapted to the CS-US interval (Welsh et al., 2005), and CRs acquired during trace and delay EBC were significantly larger than the small muscle activations elicited reflexively by the tone CS in the unpaired group, both of which are hallmarks of associative learning. Immediately following the second EBC session, the mice were rapidly anesthetized with isoflurane, decapitated, and the medial prefrontal cortex (mPFC), HC, IO, and cerebellum (Cb) were dissected to allow activity-dependent PIN rearrangements to be quantified by QMI (Figure 5G).
In the mPFC, PCA showed separation of trace EBC from unpaired controls across PC1, whereas delay EBC was intermediate (Figure 6B). ANC∩CNA identified 19 PiSCES that were significantly different between trace or delay EBC and controls; 18 for trace, 6 for delay, and 5 that were common to both learning paradigms (Figure 6A). Similar to the NMDA stimulation experiments, dissociations among Shank- and SynGAP-containing PiSCES were prevalent. Interestingly, several mGluR5-containing PiSCES decreased strongly in trace, suggesting widespread dis-engagement of mGluR5 from protein complexes, but these did not change in delay EBC. When we directly compared activity-dependent changes using an x-y plot (Figure 6C), we observed a strong correlation (R2 = 0.71) between the two learning paradigms, but the slope of the regression line (1.32) confirmed that PiSCES activation was stronger in trace EBC. Overall, these data demonstrate that activation of the mPFC during associative learning dissociates specific PiSCES, especially those containing Shank, SynGAP, and mGluR5. The two learning paradigms, which differed only in the timing of the CS-US, activated an overlapping set of PiSCES in the mPFC, but trace EBC produced stronger and more extensive PiSCES activation, perhaps due to the greater necessity of the mPFC for learning during trace EBC.
Figure 6. Effects of associative learning on the glutamate post-synapse PIN in brain regions mediating declarative memory.

(A) Heatmap of row-normalized MFIs for all PiSCES that were ANC∩CNA significant in the delay or trace EBC experiments in medial prefrontal cortical tissue, meaning they are significant by the ANC statistical test in a comparison between trace EBC versus control or delay EBC versus control, and belong to a CNA module that is significantly correlated with EBC. Each column represents a single biological replicate, N = 8 per condition, and each colored box represents a single PiSCES measurement. PiSCES are grouped based on if they are ANC-significant in trace EBC (top), delay EBC (bottom) or both trace and delay EBC (middle).
(B) PCA of cortical QMI data for the unpaired control (C, black), delay EBC (D, blue), and trace EBC (T, red) groups. Each point represents a single mouse, N = 24.
(C) x-y plot comparing the log2 fold change of all PiSCES that were ANC∩CNA significant for only delay EBC (blue), for only trace EBC (red), or for both EBC paradigms (purple) in the cortex.
(D–F) Identical to (A)–(C), except data are from hippocampal tissue.
In the HC, PCA did not clearly distinguish the groups (Figure 6E). However, 4 PiSCES were ANC∩CNA-significant for trace and 22 were significant for delay (Figure 6D). Shank3- and NMDAR-containing PiSCES were most prominently activated for delay EBC, with clear increases in co-associations among NMDARs, FYN, NL3, and the Homer-Shank scaffold, demonstrating strong upregulation of NMDAR-containing complexes. For trace, we observed weak and variable mGluR5_Homer1 and SynGAP_PSD95 dissociation, as well as changes in PI3K and Fyn self-association. When we compared activity-dependent changes using an x-y plot (Figure 6F), the majority of PiSCES that were significant for one form of learning fell below the limit of detection for the other form of learning, although the most strongly activated PiSCES in delay EBC showed a trend toward increasing in trace EBC. Unlike the mPFC, the HC responded differently to the two types of EBC.
We next analyzed the IO, a pre-cerebellar nucleus in the medulla oblongata that is required for both trace and delay EBC (Freeman and Steinmetz, 2011; Welsh and Harvey, 1998; Yang et al., 2015). Similar to the HC, PCA showed no clear separation between control and stimulated conditions (Figure 7B), but ANC∩CNA identified 8 and 10 interactions that were significantly different following delay and trace EBC, respectively, with two PiSCES significant in both conditions (Figure 7A). All significant PiSCES involved increases in the magnitude of interactions, and the proteins involved (e.g., Shank3, NL3, and SAP97) were different from those observed in the mPFC and HC. When we compared activity-dependent PiSCES between trace and delay EBC in the IO (Figure 7C), we found a moderate correlation (R2 = 0.56), suggesting a common set of qualitatively similar changes in PiSCES, with the slope of the regression line (0.71) indicating stronger activation by delay EBC.
Figure 7. Effects of associative learning on the glutamate post-synapse PIN in brain regions mediating procedural memory.

(A) Heatmap of row-normalized MFIs for all PiSCES that were ANC∩CNA-significant in the delay or trace EBC experiments in tissue from the inferior olive, meaning PiSCES are significant by the ANC statistical test in a comparison between trace EBC versus control or delay EBC versus control, and belong to a CNA module that is significantly correlated with EBC. Each column represents a single biological replicate, N = 24, and each colored box represents a single PiSCES measurement. PiSCES were grouped based on if they are ANC-significant in trace EBC (top), delay EBC (bottom), or both trace and delay EBC (middle).
(B) PCA of cortical QMI data for the unpaired control (C, black), delay EBC (D, blue), and trace EBC (T, red) groups. Each point represents a single mouse, N = 24.
(C) x-y plot comparing the log2 fold change of all PiSCES that were ANC∩CNA significant for only delay EBC (blue), for only trace EBC (red), or for both EBC paradigms (purple) in the IO.
(D–F) Identical to (A)–(C) except data are from cerebellar tissue and N = 12.
We next examined the cerebellum, a brain area known to be implicated in EBC (Heiney et al., 2014; Ten Brinke et al., 2017; Weiss and Disterhoft, 2015). PCA showed some separation of control, delay, and trace EBC (Figure 7E). ANC∩CNA identified 12 and 7 interactions that were significantly different following delay and trace EBC, respectively, with three PiSCES significant in both conditions (Figure 7D). A direct comparison of trace and delay EBC (Figure 7F) showed a strong correlation (R2 = 0.81), suggesting a common set of qualitatively similar changes in PiSCES, with the slope of the regression line (0.61) indicating stronger activation by delay EBC.
Finally, we asked if the magnitude of PiSCES changes correlated with individual learning performance in the mice. In the mPFC, two CNA modules that were induced by EBC were also correlated with the percentage CRs, most strongly on day 2 (Figure S5A), but not with CR amplitude. When the individual mice were analyzed without consideration of group, there was a strong correlation between the MFI of a given interaction and the percentage CRs on day 2: for example, Shank1_SynGAP showed a significant negative correlation with MFI (slope = −0.042, F(1,22) = 7.27, p = 0.013, Figure S5B). However, with-group correlations tended to be much weaker, non-significant, and often followed the opposite pattern (trace EBC slope = 0.012, F(1,22) = 0.114, p = 0.75). Several examples are displayed in Figures S5C-S5H. Similar trends existed for Cb, IO, and mPFC (data not shown). Thus, although multiple PiSCES were changed by associative learning, we were unable to correlate the magnitude of the changes to the behavioral performance of individual mice.
Overall, these data show how two distinct forms of associative learning elicit brain-region-specific PIN rearrangements in vivo. Moreover, they reveal that trace and delay EBC elicit qualitatively similar within-region changes in the Cb, IO, and mPFC, but in HC, a different protein-state-dependent encoding paradigm differentiates trace from delay EBC.
DISCUSSION
Molecular logic of stimulus encoding
Far from a static “scaffold,” the post-synaptic density is a dynamic, liquid-liquid-phase-separated (Cai et al., 2021; Feng et al., 2019; Zeng et al., 2016, 2018) structure that rapidly responds to synaptic activity by altering its molecular composition via regulation of protein-protein interactions. Certainly, individual interactions, such as recruitment of AMPARs to PSD95 scaffolds (Buonarati et al., 2019; Ehrlich and Malinow, 2004) or dissociation of mGluR from Homer EVH1 domains (Guo et al., 2015; Ronesi et al., 2012) are well-characterized, but the molecular logic that allows a change in protein interactions to translate into functional cellular “calculations” (Figure 1A) remains understudied, partly due to technical limitations. Here, using QMI, we investigated how networks of interacting proteins are influenced by postsynaptic activation, and how that encoding differs between brain regions.
Cells can use diverse mechanisms to encode signaling inputs. The modular hypothesis, first developed by Pawson and colleagues, suggested that different combinations of proteins recruited to signaling complexes could define different signals (Pawson, 1995). Differences in the temporal dynamics of signals have also been proposed to mediate cellular decisions; notably “weak and fast” versus “strong and sustained” intracellular Ca2+ transients have been proposed to mediate the LTD versus LTP decision (Evans and Blackwell, 2015), in a mechanism that may involve auto-inhibition of CamKII (Hell, 2014; Lisman et al., 2012). In an elegant series of optogenetic experiments, Toettcher et al. (2013) demonstrated that the ERK signal transduction cascade acts as a temporal filter, detecting sustained activation of ERK signaling while rejecting more transient ERK phosphorylation. In addition to qualitative and kinetic logic, some systems simply use the magnitude of signalosome activation to encode different stimuli. In T cell development, the magnitude of activation of a single PIN downstream of the T cell receptor determines whether a cell chooses one of two diametrically opposed outcomes—continued development or apoptotic cell death (Neier et al., 2019). Finally, computational modeling of BMP ligand/receptor interactions revealed a complex and unintuitive combinatorial logic system, in which peak receptor activation is achieved at specific concentrations of ligands determined by receptor sub-type; different combinations of receptors and ligands allow a diversity of cellular logic responses based on concentration gradients (Antebi et al., 2017).
In the brain, the molecular logic rules that allow neurons to distinguish between signaling inputs and synthesize coordinated cellular responses are largely unknown. Here, we first used a simple cell culture model to explore the encoding of two major glutamate receptor classes, metabotropic versus NMDA receptors. We found that qualitative, quantitative, and kinetic mechanisms combine to differentiate signals. As quickly as 30 s, one PIN module was activated uniformly downstream of both receptors. This is congruent with Ca2+-dependent signaling, as intracellular Ca2+ is induced by both NMDA (via direct conduction) and DHPG (via IP3R and release from the ER) (Gladding et al., 2009; Tu et al., 1998). At 5 and 15 min, this same module activated more intensely in NMDA and glutamate conditions, but quickly returned to baseline in DHPG, demonstrating kinetic differentiation of signals consistent with a “weak-and-fast” versus “strong-and-sustained” Ca2+ flux model of LTD versus LTP (Evans and Blackwell, 2015). In stark contrast, cLTP did not resemble either NMDA or DHPG application, and instead produced a distinct pattern of PiSCES changes, which were often in the opposite direction compared to NMDA or glutamate. Although bath application of NMDA has been reported to cause an LTD-like response (Lee et al., 1998; Lüscher and Malenka, 2012), AMPA receptor-scaffold co-associations increased with NMDA/glutamate (implying an increase in synaptic strength) but did not change with cLTP. A limitation of our study is that we did not measure the electrophysiological correlates of our treatments, so we cannot conclude if receptor activation induced an LTP- or LTD-like response. We can conclude that, in a cultured neuron system, the synaptic PIN can obtain multiple independent states (at least three) that correspond to unique input types. These states are defined by differences in composition, intensity, and kinetics of PIN activation.
Proteome-dependent encoding of stimuli
The core proteins targeted by our QMI panel co-exist in multiprotein complexes throughout the brain. However, the proteome of neurons varies by brain area (Cheng et al., 2006; Lee et al., 2017a; Trinidad et al., 2008). Accessory proteins in complex with our QMI targets, or splice variants of the targets themselves, may allow different brain regions to perform unique molecular computations, suited for the particular circuitry of the area. To explore this idea, we directly compared an identical stimulus in two brain regions with known differences in proteome composition—the HC and CTX. When NMDA was applied, the PIN response was broadly similar, involving decreased Homer-Shank scaffold interactions and increased AMPA and PSD95 PiSCES. However, differences in the levels of these PiSCES at baseline between the two areas meant that the dynamic range available for a given PiSCES to change differed. Conversely, the response to NMDA in the IO was unique. PiSCES involving Shank3 and NMDARs were upregulated, as well as a PiSCES containing mGluR5 and GluR2, which may be linked via Shank3 scaffolds (Monteiro and Feng, 2017). In HC/CTX, these PiSCES did not change with NMDA. The different ratios of Shank isoforms expressed in the three brain structures may partially explain these differences, because Shank3 is highly expressed in the IO (Lautz et al., 2021). Overall, our data demonstrate that proteomic differences at baseline allow glutamate receptors and scaffolds to modify their interactions in response to NMDA stimulation in very different ways, allowing for a diversity of computations across brain regions.
PIN activation underlying information encoding in vivo
The EBC paradigms allowed us to present mice with three distinct types of information, while using identical sensory stimuli. Every mouse received the same number of CS and US presentations, but their timing determined which type of conditioning the animal received. Remarkably, the sole difference in timing resulted in different PIN responses within the same brain areas. In the mPFC, the PiSCES activated by EBC were similar to those activated by NMDA/glutamate in neocortical neurons in culture (dissociation of mGluR5, Homer, Shank, and SynGAP), with the intensity of activation and the dis-engagement of mGluR5 distinguishing delay versus trace EBC (note however that age differences between adult mice used in EBC experiments and DIV 19–21 cultures make direct comparisons of results problematic). Consistent with this result, inhibition of NMDA receptors the mPFC has been shown to severely inhibit classical EBC (Takatsuki et al., 2001; Takehara-Nishiuchi et al., 2005). By comparison, in the HC the PiSCES activated in trace versus delay EBC were non-overlapping. Trace EBC weakly activated Fyn, PI3K, mGluR5_Homer1, and SynGAP_PSD95, PiSCES associated with NMDA/glutamate stimulation ex vivo. We speculate that these data are consistent with activation of a sparse population of cells in a background of inactive cells, as might be expected for a hippocampal-dependent task, although other explanations certainly exist. However, delay EBC, which is not dependent on the HC, unexpectedly elicited a strong PIN response. This pattern of activation was not observed in our prior agonist-stimulation experiments, nor have we observed a similar pattern in response to homeostatic scaling, in vitro or in vivo (Heavner et al., 2020). The IO and the Cb, which are essential for the performance of CRs in both trace and delay EBC (Welsh and Harvey, 1998; Zbarska and Bracha, 2012), also activated overlapping sets of PiSCES including Shank3, Fyn, and NL3 PiSCES in response to both EBC types. Overall, the results demonstrate a remarkable diversity of PIN responses to natural stimuli in vivo, which may serve as a basis for the diversity of functions that neuronal subtypes and circuits can perform across the brain.
Why might the HC have responded so strongly to delay EBC, when it is required for trace, but not delay, EBC? Perhaps the hippocampal activation observed here serves an alternative purpose, outside the acquisition of the CR. Rats with hippocampal lesions only exhibit deficits in a conditional EBC paradigm when they must learn to blink only if the CS is preceded by another stimulus (Ross et al., 1984). Humans with hippocampal-temporal lobe anterograde amnesia can acquire a CR, but are unable to describe it (Weiskrantz and Warrington, 1979), and patients with hippocampal-medial temporal lesions acquire a CR, but have impaired conditional discrimination in EBC (Daum et al., 1991, 1992). Although PIN activation in the HC observed here may not reflect acquisition of the CR, it may underlie hippocampal-dependent declarative memory and reflect an awareness of the pairing.
Dysfunction of PIN dynamics in neurological disorders
Synaptic dysfunction is causally associated with numerous neurological disorders including autism spectrum disorder (ASD), schizophrenia, and bipolar disease (Frankle et al., 2003; Heavner and Smith, 2020; Lee et al., 2018; Yin et al., 2012; Zoghbi and Bear, 2012). The synaptic QMI panel itself was designed to target a network of autism-linked gene products (Brown et al., 2018) in order to understand how mutations in any one member of a highly interconnected PIN produces a similar human phenotype in the form of ASD (Heavner et al., 2020). Indeed, we and others have previously demonstrated that mice carrying different ASD-linked mutations show similar disruptions in specific types of plasticity, such as homeostatic scaling (Heavner et al., 2020). As our understanding of ASD biology improves, many (although not all) ASD-linked genes can be placed in a pathway that translates synaptic activity into coordinated changes in neuronal protein networks, over developmental time (Courchesne et al., 2020; Heavner and Smith, 2020). The work presented here further demonstrates that ASD-linked proteins including Homer, Shank, SynGAP, NMDAR2B, and mGluR5 are intimately involved in neuronal computations downstream of both chemical and naturalistic sensory inputs in ethologically valid behavioral paradigms.
It is hoped that as we better understand the mechanisms of pathogenic mutations, we will develop treatments to correct molecular deficits and improve patient outcomes. However, our data highlight a significant challenge in developing a therapeutic: how does one correct signaling deficits that are not consistent across the entire brain? Region specific electrophysiological deficits in numerous models of ASD are well established (Lee et al., 2015). Here, we demonstrate that even the rules governing basic signal transduction networks differ between brain regions when an animal is performing a simple task. That complexity is likely to be greatly amplified when the animal performs two or more different tasks and in the brains of species having a much richer behavioral repertoire than mice. Thus, correcting a signaling deficit in one brain region could have a confounding or even contradictory effect on other regions or other behaviors. While this may complicate therapeutic development, recognizing this diversity may be critical to our integrative understanding of brain function in health and disease.
Limitations of the study
Within brain regions, even along the length of a single neuron’s dendrite, synapses display remarkable diversity in size, shape, electrical properties and proteomic composition (Cizeron et al., 2020; Grant and Fransén, 2020; Micheva et al., 2010; Zhu et al., 2018). A limitation of the current study is that we employed bulk preparations of brain areas, and therefore were unable to address this “synapse diversity dilemma” (Grant and Fransén, 2020). Because different brain regions show different proportions of synapse types, as defined by morphological factors and the ratio of PSD95 to SAP97 expression (Zhu et al., 2018), our data presumably reflect the average synaptic composition in each brain area. Despite recent progress toward physically sorting specific synapse types, which may allow high-dimensional biochemical measurements of synapse types within a single brain region (Biesemann et al., 2014), large-scale protein interaction measurements such as QMI are currently limited to bulk preparations defined by regional anatomy. Future work to physically sort synapses from within a single tissue may enable a more nuanced understanding of the biochemical underpinnings of synaptic plasticity.
STAR☆METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Stephen Smith (Seps@uw.edu).
Materials availability
This study did not generate new unique reagents. All reagents used are commercially available.
Data and code availability
QMI data reported here, EBC raw data and raw data from the Ca2+ imaging experiments will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available upon request from the lead contact.
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Animals
CD-1, (Insert EBC strain here), Vglut2-ires-cre (stock: 016963, RRID: IMSR_JAX:016963), and Ai95(RCL-GCaMP6f)-D (stock: 028865, RRID: IMSR_JAX:028865) mice were originally obtained from The Jackson Laboratory (Bar Harbor, Maine) and maintained in an in-house breeding colony. All mice were separated by sex, and housed with littermates, with no more than five mice/cage. Food and water was provided ad libitum. For slice experiments, only p21-30 mice (both male and female) were used. For EBC, p124-314 (both male and female) were used, with treatment group being randomly assigned. The experiments were approved by the Seattle Children’s Research Institute and the University of Washington Animal Care and Use Committees.
Cell Culture
Primary cultures of cortical neurons from were prepared as previously described (Lautz et al., 2019) Briefly, whole cortex from P0-P1, male and female mouse neonates was dissociated using papain (Worthington) and plated at a density of 1.0x106 cells/mL onto 6-well plates treated with poly-D-lysine. Cells were cultured in Neurobasal medium supplemented with 2% B27 and 0.5mM GlutaMax (ThermoFisher) and kept at 37°C, 5% CO2 for 18-21 days. After 3-5 DIV, 5-fluoro-2′-deoxyuridine was added to a final concentration of 5 μM to inhibit glial proliferation.
METHOD DETAILS
Glutamate receptor stimulation
For direct activation of glutamate receptors, neurons were treated with Glutamate (100 μM), NMDA/glycine (100/1 μM), DHPG (100 μM) or control HEPES-aCSF (129 mM NaCl/ 5 mM KCl/ 2 mM CaCl2/ 30 mM Glucose/ 25 mM HEPES; pH 7.4) for up to 5 minutes. For longer time periods, aCSF was removed and replaced with conditioned media. Glutamate (Sigma, G1626; 100 μM or 1 μM) was prepared in HEPES-aCSF. NMDAR agonists NMDA (Sigma, M3262; 100 μM) + glycine (Fisher Scientific, Pittsburgh, PA, USA, BP-381; 10 μM) were dissolved in magnesium-free HEPES-aCSF in the presence of non-NMDA glutamate receptor blockers (CNQX, Tocris, 0190, 40 μM; Nimodipine, Tocris, 0600, 1 μM; LY-367385, Sigma, L4420, 100 μM; MPEP, Tocris, 1212, 10 μM). Type I mGluR agonist DHPG (Tocris, 0805; 100 μM) was dissolved in HEPES-aCSF in the presence of non-mGluR glutamate receptor blockers (CNQX, Tocris, 0190, 40 μM; Nimodipine, Tocris, 0600, 1 μM; D(−)-2-Amino-5 phosphonopentanoic acid (APV), Sigma, A5282, 50 μM). For chemical induction of LTP, neurons were treated as previously described [44]. Briefly, neurons were treated with cLTP solution containing Forskolin (Tocris, 1099, 50 μM), Rolipram (Tocris, 0905, 0.1 μM), and Picrotoxin (Tocris, 1128, 50 μM) dissolved in HEPES-aCSF or control HEPES-aCSF for 30 minutes.
Following treatment, cells were immediately placed on ice and rapidly harvested by scraping in lysis buffer [150 mM NaCl, 50 mM Tris (pH 7.4), 1% NP-40, 10 mM sodium fluoride (Sigma, 201154), 2 mM sodium orthovanadate (Sigma, 450243) + Protease/phosphatase inhibitor cocktails (Sigma, P8340/P5726)], followed by incubation in lysis buffer for 15 min. Lysate was centrifuged for 15 min at high speed to remove nuclei and debris, and protein concentration in the supernatant was determined using a Pierce BCA kit (Pierce, Rockford, IL, USA, 23225).
TUNEL assay and immunocytochemistry
For TUNEL staining and immunocytochemistry, cortical cells were cultured as described above with the following exceptions: cells were plated at 1.0 × 106 cells/mL on glass coverslips treated with poly-D-lysine in 24 well plates. Detection and quantification of apoptosis in cell cultures was determined using a In Situ Cell Death Detection Kit (Roche, 11684795910) according to the manufacturer’s instructions. Briefly, cells were fixed with 4% paraformaldehyde for 1 hour at room temperature, rinsed with PBS, and incubated in permeabilization buffer (0.1% Triton X-100 in 0.1% sodium citrate freshly prepared). Cells were then washed 3x with PBS and stained with TUNEL reaction mixture for 60 min at 37°C in the dark. For negative controls, fixed cells were incubated in Label solution instead of the TUNEL mixture. For positive controls, permeabilized cells were incubated with DNase (3 U/ml in 50 mM Tris-HCL, pH 7.5, 1/ mg/ml BSA: Worthington, LK003170) prior to incubation with TUNEL mixture.
Following TUNEL staining, cells were treated with antibodies toward β3-tubulin (1:500, Biolegend, 801201) and NeuN (1:1000, Abcam, ab177847) overnight at 4°C. The following day, cells were washed 3x with PBS and incubated in Alexa Fluor® 594 AffiniPure Goat Anti-Rabbit IgG (1:10,000, Jackson ImmunoResearch, 111-585-003) and Alexa Fluor® 647 AffiniPure Goat Anti-Mouse IgG (1:10,000, Jackson ImmunoResearch, 115-605-003) for 60 min at RT. Cells were then washed 3x with PBS and mounted in Fluoromount G Mounting Medium, with DAPI (Invitrogen, 00495952). 4 pictures of each coverslip were taken at random, and the NeuN positive cells that were also TUNEL positive was quantified in ImageJ, with a minimum of 500 cells/slip being counted.
2-photon Ca+ imaging
For Ca2+ imaging experiments, primary cortical neurons were cultured on glass coverslips from P0 pups born from crossing Ai95D and Vglut2-ires-cre mice (as described above). We chose vGlut2 because it achieved sparse expression in our cultures to enable imaging of individual neurons, although we acknowledge that this driver will not activate all glutamatergic neurons. The resulting neurons expressed the calcium indicator GCamp6F in Vglut2+ neurons. Two-photon Ca+ imaging was performed using an Olympus FV1000MPE multiphoton laser scanning microscope equipped with a Mai Tai Deepsee Laser. All experiments were performed using a 25x objective immersed in aCSF solution and a 2x digital zoom. For imaging, glass coverslips were placed in a recording chamber, and perfused with oxygenated aCSF at a flow rate of 5-6 ml/min. Acquisition protocols consisted of ~35-min time-lapse sequences measuring changes in the levels of fluorescence using a GFP filter (wavelengths 457-538). To quantify changes in fluorescence intensity, the regions of interest (including both cell body and processes) were selected and mean pixel intensity was measured using the Flowview program. The data was plotted as ΔF/F (baseline defined as average fluorescence in the minute preceding stimulation) relative to time. For stimulation, glass coverslips were perfused with aCSF containing either glutamate (100 μm), NMDA/glycine (100/1 μm), or DHPG (100 μm) for 5 min, after which coverslips were perfused with fresh aCSF lacking agonist. At 30 min, coverslips were perfused with high K+ aCSF (55 mm). Statistical significance was determined using a one-way ANOVA with Dunnet’s post hoc test in Prism Graphpad.
Slice preparation and treatment
Mice were deeply anesthetized with Isofluorane, brains were removed, and coronal cortical and hippocampal slices were sectioned at 400 μm thickness using a vibratome. Slices were immediately hemisected with a sharp razor blade and each half placed in an alternate treatment group, with treatment groups being arbitrarily assigned. For IO slices, brains were removed, hemisected coronally at the midbrain and the cerebellum was removed, and slices sectioned at 400 μm thickness using a vibratome. The IO was then micro-dissected away from surrounding tissue. Slices were initially recovered in NMDG protective recovery solution (93 mM NMDG, 2.5 mM KCl, 1.2 mM NaH2PO4, 30 mM NaHCO3, 20 mM HEPES, 25 mM glucose, 2 mM thiourea, 5 mM Na-ascorbate, 3 mM Na-pyruvate, 0.5 mM CaCl2.4H2O, and 10 mM MgSO4.7H2O; titrated to pH 7.4 with concentrated hydrochloric acid) for 10–15 min at 32–34 °C, then transferred to a modified HEPES holding solution [92 mM NaCl, 2.5 mM KCl, 1.2 mM NaH2PO4, 30 mM NaHCO3, 20 mM HEPES, 25 mM glucose, 2 mM thiourea, 5 mM Na-ascorbate, 3 mM Na-pyruvate, 2 mM CaCl2.4H2O, and 2 mM MgSO4.7H2O; pH 7.4] for an additional 60–90 min recovery at room temperature. Slices were then incubated in HEPES-aCSF x 1h at 32–34 °C to equilibrate, and subsequently stimulated with NMDA/glycine (100/1 μM) or HEPES-aCSF control, similar to cell culture experiments. Following stimulation, slices were homogenized in ice cold 1% NP-40 lysis buffer using 12 strokes of a glass-teflon homogenizer.
Eye blink conditioning
EBC was performed as described in [45]. Adult mice of both sexes (average age in days ± SEM: 198 ± 25 for trace, 198 ± 25 for delay, 199 ± 23 for control; range 124-314) were implanted with stimulation and recording electrodes under Isoflourane anesthesia and were allowed to recover for > 1 week before the EBC sessions. Head-restrained, freely-walking mice (N = 24) were acclimated to the conditioning apparatus for one session, and thereafter conditioned over two daily sessions. The CS was a 2-kHz, 85-dB, 250-ms, free-field tone. The US was an 800-μA, biphasic, 60-Hz, 100-ms shock to the right periorbital region. Eye blinks were recorded by a bipolar intramuscular EMG electrode within the right superior orbicularis oculi muscle (differential amplification relative to a skull-screw reference, 1-kHz sampling, baseline rectified, 200-Hz low-pass filtered). A CR was defined as an EMG signal within the CS-US interval exceeding 6 standard deviations of a stable, 100-ms pre-CS baseline with an onset latency greater than 35 ms and duration exceeding 15 ms. Mice receiving trace or delay EBC experienced 60 CS-US conditioning trials per hour-long session (60 ± 10 s intertrial interval), each trial consisting of a 250 ms CS and a 100 ms US. Mice receiving trace EBC experienced a 500 ms CS-US interval (250 ms trace interval) while mice receiving delay EBC experienced a 250 ms CS-US interval (no trace interval). CSs and USs during the explicitly unpaired paradigm were delivered alone (no more than 3 consecutive trials of the same type) at half the EBC intertrial interval (30 ± 5 s). Immediately following the second training session, the mice were rapidly anesthetized with isoflurane, decapitated, and the mPFC, HC, Cb, and IO were dissected to allow activity-dependent PIN rearrangements to be quantified by QMI. Behavioral comparisons of CR acquisition in trace and delay EBC groups were made to the same unpaired control group using an identical “CS-US” interval as the EBC groups.
Quantitative Multiplex co-immunoprecipitation (QMI)
Generally, our experimental design involved treating identical samples (cell cultures grown in parallel or brain slices from a mouse) with an acute stimulus, then lysing the sample, immunoprecipitating with ~20 unique classes of microspheres each coupled to a different antibody, and probing for co-associating proteins using ~20 fluorophore-coupled probe antibodies. The raw data, collected on a refrigerated flow cytometer, consisted of 105-106 individual bead events, each consisting of ‘gating’ parameters that identify the bead class (i.e., the immunoprecipitating antibody), and the fluorescent intensity of the reporter channel (i.e., the amount of co-associated probe antibody). The data analysis strategy identifies which of ~400 binary PiSCES sampled are significantly different between the ‘unstimulated’ and ‘stimulated’ conditions with an adaptive, non-parametric test corrected for multiple comparisons (ANC), and places such PiSCES into modules of co-varying PiSCES using correlation network analysis (CNA). The analysis strategy, including equations (Smith et al., 2016) and source code (Brown et al., 2019) has been extensively reported and validated in neurons (Heavner et al., 2020; Lautz et al., 2018, 2019).
QMI was performed as described previously (Lautz et al., 2018, 2019; Smith et al., 2016). Briefly, a master mix containing equal numbers of each antibody-coupled Luminex bead was prepared and distributed to lysates containing equal amounts of protein and incubated overnight on a rotator at 4°C (four coupling protocol) (see Brown et al., 2019). The next day, beads from each sample were washed twice in cold Fly-P buffer (50mM tris pH7.4, 100mM NaCl, 1% bovine serum albumin, and 0.02% sodium azide) and distributed into twice as many wells of a 96-well plate as there were probe antibodies (for technical replicates). Biotinylated detection (probe) antibodies were added to the appropriate wells and incubated at 4°C with gentle agitation for 1 hour. The resulting bead-probe complexes were washed 3 times with Fly-P buffer, incubated for 30 minutes with streptavidin PE on ice, washed another 3 times, resuspended in 125μl ice cold Fly-P buffer, and processed for fluorescence using a customized refrigerated Bio-Plex 200 (Smith et al., 2016). The following IP and Probe antibodies were used:
IP probe antibody panels used for QMI
| Target | QMI Component | Clone | Supplier | Cat# | RRID |
|---|---|---|---|---|---|
| Homer1 | IP | AT1F3 | LifeSpan Bioscience | LS-C103482 | AB_2264378 |
| Probe | D3 | Santa Cruz | sc-17842 | AB_627742 | |
| Homer1a | IP | NA | NA | NA | NA |
| Probe | Polym13 | Santa Cruz | sc-8922 | AB_648368 | |
| NL3 | IP | 566209 | ThermoFisher | MA5-24253 | AB_2576897 |
| Probe | G2 | Santa Cruz | sc-271880 | AB_10709173 | |
| panShank | IP | NA | NA | NA | NA |
| Probe | N23B/49 | Neuromab | 75-089 | AB_10672418 | |
| PSD-95 | IP | K28/74 | Biolegend | 810301 | AB_2564749 |
| Probe | K28/43 | Biolegend | 810401 | AB_2564750 | |
| SAPAP | IP | NA | NA | NA | NA |
| Probe | N127/31 | Biolegend | 832601 | AB_2564958 | |
| SAP97 | IP | RPI197.4 | Enzo Life Sciences | ADI-VAM-PS00 | AB_1083921 |
| Probe | polyh60 | Santa Cruz | sc-25661 | AB_2092029 | |
| Shank1 | IP | N22/21 | Neuromab | 75-064 | AB_2270283 |
| Probe | N22/21 | Neuromab | 75-064 | AB_2270283 | |
| Shank3 | IP | N367/62 | Neuromab | 75-344 | AB_2315921 |
| Probe | N69/46 | Neuromab | 75-109 | AB_2187730 |
Scaffold and structural proteins
Receptor proteins
| Target | QMI Component | Clone | Supplier | Cat# | RRID |
|---|---|---|---|---|---|
| GluR1 | IP | N355/1 | Biolegend | 819801 | AB_2564834 |
| Probe | poly1594 | Millipore | AB1504 | AB_2113602 | |
| GluR2 | IP | L21/32 | Biolegend | 810501 | AB_2564751 |
| Probe | polyc20 | Santa Cruz | sc-7610 | AB_2247873 | |
| mGluR5 | IP | 5675 | Millipore | AB5675 | AB_2295173 |
| Probe | N75/3 | Neuromab | 75-115 | AB_10672260 | |
| NMDAR1 | IP | 54.1 | ThermoFisher | 32-0500 | AB_2533060 |
| Probe | polyc20 | Santa Cruz | sc-1467 | AB_670215 | |
| NMDAR2A | IP | N327/95 | Neuromab | 75-288 | AB_2315842 |
| Probe | N3278/38 | Biolegend | 832401 | AB_2564956 | |
| NMDAR2B | IP | N59/20 | Biolegend | 832501 | AB_2564957 |
| Probe | N59/36 | Biolegend | 818701 | AB_2564823 | |
| Cx36 | IP | 51-6200 | ThermoFisher | 51-6200 | AB_2533912 |
| Probe | 1E5H5 | ThermoFisher | 37-4600 | AB_2533320 |
Signaling proteins
| Target | QMI Component | Clone | Supplier | Cat# | RRID |
|---|---|---|---|---|---|
| CaMKII | IP | 6G9 | ThermoFisher | MA1-048 | AB_325403 |
| Probe | polyC6970 | Sigma | C6974 | AB_258984 | |
| Fyn | IP | Fyn15 | Santa Cruz | sc-434 | AB_627642 |
| Probe | Fyn59 | Biolegend | 626502 | RRID:AB_2278824 | |
| PI3K p85 | IP | U5 | ThermoFisher | MA1-74183 | AB_2163452 |
| Probe | AB6 | Millipore | 05-212 | AB_309658 | |
| PIKE | IP | 263A | Bethyl Laboratories | A304-263A | AB_2620459 |
| Probe | DN8 | Rockland | 200-401-DN8 | AB_2612161 | |
| SynGap | IP | D20C7 | Cell Signaling | 5539 | AB_10694401 |
| Probe | PolyR19 | Santa Cruz | sc-8572 | AB_2200750 | |
| Ube3a | IP | H182 | Santa Cruz | sc-25509 | AB_639982 |
| Probe | E4 | Santa Cruz | sc-166689 | AB_2211807 |
QUANTIFICATION AND STATISTICAL ANALYSIS
Data preprocessing and inclusion criteria
For each well from a data acquisition plate, data were processed using the BioPlex200’s built-in software to (i) eliminate doublets on the basis of the doublet discriminator intensity (> 5000 and < 25 000 arbitrary units; Bio-Plex 200), (ii) identify specific bead classes within the bead regions used, and (iii) pair individual bead phycoerythrin fluorescence measurements with their corresponding bead regions. This processing generated a distribution of fluorescence intensity values for each pairwise PiSCES measurement. XML output files were parsed to acquire the raw data for use in MATLAB using the ANC program. No specific analysis was performed on the data to test for outliers.
Adaptive non-parametric analysis with empirical alpha cutoff (ANC)
ANC is used to identify high-confidence, statistically significant differences (corrected for multiple comparisons) in bead distributions on an individual PiSCES basis. ANC analysis was conducted in MATLAB (version 2013a) as described in (Smith et al., 2016). As previously reported, we required that hits must be present in > 70% of experiments (typically three out of four) at an adjusted p < 0.05. The α-cutoff value required per experiment to determine statistical significance was calculated to maintain an overall type I error of 0.05 (adjusted for multiple comparisons with Bonferroni correction), with further empirical adjustments to account for technical variation (see Smith et al., 2016). No assessment of normality was carried out as ANC analysis is a non-parametric test.
Correlation Network Analysis (CNA)
Modules of PiSCES that co-varied with experimental conditions were identified using CNA as described in Brown et al. (2019) and Smith et al. (2016). Briefly, bead distributions used in ANC were collapsed into a single MFI for every PiSCES and averaged across technical replicates for input into the WGCNA package for R (Langfelder and Horvath, 2008). PiSCES with MFI < 100 were removed, and batch effects were corrected using COMBAT (Johnson et al., 2007). For time course analysis, different time points were further batch-corrected using COmbat CO-Normalization Using ConTrols (COCONUT). Power values giving the approximation of scale-free topology were determined using soft thresholding with a power adjacency function. The minimum module size was always set to between 10 and 12, and modules whose eigenvectors significantly correlated with an experimental trait (p < 0.05) were considered “of interest.” PiSCES belonging to module of interest and whose probability of module membership in that module was < 0.05 were considered significantly correlated with that trait.
Principal Component Analysis (PCA)
PCA was performed on Post-COMBAT, log2 transformed MFI values in R studio using the prcomp function.
ANCXCNA
PiSCES that were significant by both ANC and CNA for a given experimental condition were considered significantly altered in that condition.
Data visualization
For heatmap visualization of MFI values of CNA module members, log2 transformed data were input into the Heatmap.2 program in R studio, which normalized the data by row for visualization of multiple analytes spanning a 3-log range. All PiSCES visualized were ANC∩CNA significant for at least one condition, as described in the figure legends. The number of biological replicates for each experiment can be found in figure legends, error bars represent SEM.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| β3-tubulin | Biolegend | Cat #: 801201 |
| NeuN | Abcam | Cat #: ab177847 |
| Alexa Fluor® 594 AffiniPure Goat Anti-Rabbit IgG |
Jackson Immunoresearch | Cat #: 111-585-003 |
| Alexa Fluor® 647 AffiniPure Goat Anti-Mouse IgG |
Jackson Immunoresearch | Cat #: 115-605-003 |
| Chemicals, peptides, and recombinant proteins | ||
| Papain | Worthington | Cat #: LS003119 |
| Neurobasal Medium | GIBCO | Cat #: 21103049 |
| B27 | GIBCO | Cat #: 17504044 |
| GlutaMax | GIBCO | Cat #: 35050061 |
| Poly-D-lysine | Sigma-Aldrich | Cat #: A-003-E |
| 5-Fluoro-2′-deoxyuridine | Sigma-Aldrich | Cat #: F0503 |
| Glutamate | Sigma-Aldrich | Cat #: G1626 |
| NMDA | Sigma-Aldrich | Cat #: M3262 |
| Glycine | Fisher Scientific | Cat #: BP381 |
| CNQX | Tocris | Cat #: 0190 |
| Nimodopine | Tocris | Cat #: 0805 |
| LY-367385 | Sigma-Aldrich | Cat #: L4420 |
| MPEP | Tocris | Cat #: 1212 |
| APV | Sigma-Aldrich | Cat #: A5282 |
| NP-40 | Sigma-Aldrich | Cat #: I8896 |
| Sodium Fluoride | Sigma-Aldrich | Cat #: 201154 |
| Sodium Orthovanadate | Sigma-Aldrich | Cat #: 450243 |
| Protease/Phosphatase Inhibitor Cocktail | Sigma-Aldrich | Cat #: P8340/P5726 |
| NMDG | Sigma-Aldrich | Cat #: M2004 |
| DNase I recombinase | Worthington | Cat #: LK003170 |
| Fluoromount with DAPI | Invitrogen | Cat #: 00495952 |
| Forskolin | Tocris | Cat #: 1099 |
| Rolipram | Tocris | Cat #: 0905 |
| Picrotoxin | Tocris | Cat #: 1128 |
| Critical commercial assays | ||
| Pierce BCA Assay | Pierce | Cat #: 23225 |
| In Situ Cell Death Detection Kit | Roche | Cat #: 11684795910 |
| Experimental models: organisms/strains | ||
| Vglut2-ires-cre | Jackson Laboratories | Cat #: 016963 RRID: IMSR_JAX:016963 |
| Ai95(RCL-GCaMP6f)-D | Jackson Laboratories | Cat #: 028865 RRID: IMSR_JAX:028865 |
| Software and algorithms | ||
| MATLAB | MATLAB | https://www.mathworks.com/products/matlab.html |
| R Studio | R Studio | https://www.rstudio.com/ |
| Flowview | Flowview | N/A |
| GraphPad Prism | Prism | N/A |
Highlights.
Synaptic protein interaction networks differentiate NMDA versus DHPG stimulation
NMDA stimulation produces network responses that differ by brain region
Eyeblink conditioning induces region- and paradigm-specific network changes
Proteomic composition and sensory input determine protein network response
ACKNOWLEDGMENTS
The authors would like to thank Dr. Adam Schrum and members of the Smith lab, especially Dr. Whitney Heavner and Devin Wehle, for helpful discussions concerning the data and manuscript. The authors also thank Liza Severs and the Ramirez lab for their gift of Ai95D and Vglut2-ires-cre mice. This work was supported by the NIH (R01-MH113545 to S.E.P.S. and R01-NS31224 to J.P.W.).
Footnotes
CONFLICT OF INTEREST
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.110076.
REFERENCES
- Antebi YE, Linton JM, Klumpe H, Bintu B, Gong M, Su C, McCardell R, and Elowitz MB (2017). Combinatorial Signal Perception in the BMP Pathway. Cell 170, 1184–1196.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki Y, Zeng M, Zhang M, and Huganir RL (2015). Rapid dispersion of SynGAP from synaptic spines triggers AMPA receptor insertion and spine enlargement during LTP. Neuron 85, 173–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki Y, Hong I, Gamache TR, Ju S, Collado-Torres L, Shin JH, and Huganir RL (2020). SynGAP isoforms differentially regulate synaptic plasticity and dendritic development. eLife 9, e56273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayés A, van de Lagemaat LN, Collins MO, Croning MD, Whittle IR, Choudhary JS, and Grant SG (2011). Characterization of the proteome, diseases and evolution of the human postsynaptic density. Nat. Neurosci 14, 19–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayés A, Collins MO, Croning MD, van de Lagemaat LN, Choudhary JS, and Grant SG (2012). Comparative study of human and mouse postsynaptic proteomes finds high compositional conservation and abundance differences for key synaptic proteins. PLoS ONE 7, e46683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belmeguenai A, and Hansel C (2005). A role for protein phosphatases 1, 2A, and 2B in cerebellar long-term potentiation. J. Neurosci 25, 10768–10772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biesemann C, Grønborg M, Luquet E, Wichert SP, Bernard V, Bungers SR, Cooper B, Varoqueaux F, Li L, Byrne JA, et al. (2014). Proteomic screening of glutamatergic mouse brain synaptosomes isolated by fluorescence activated sorting. EMBO J. 88, 157–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown EA, Lautz JD, Davis TR, Gniffke EP, VanSchoiack AAW, Neier SC, Tashbook N, Nicolini C, Fahnestock M, Schrum AG, and Smith SEP (2018). Clustering the autisms using glutamate synapse protein interaction networks from cortical and hippocampal tissue of seven mouse models. Mol. Autism 9, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown EA, Neier SC, Neuhauser C, Schrum AG, and Smith SEP (2019). Quantification of Protein Interaction Network Dynamics using Multiplexed Co-Immunoprecipitation. J. Vis. Exp (150) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buonarati OR, Hammes EA, Watson JF, Greger IH, and Hell JW (2019). Mechanisms of postsynaptic localization of AMPA-type glutamate receptors and their regulation during long-term potentiation. Sci. Signal 12, eaar6889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Q, Zeng M, Wu X, Wu H, Zhan Y, Tian R, and Zhang M (2021). CaMKIIα-driven, phosphatase-checked postsynaptic plasticity via phase separation. Cell Res. 31, 37–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng D, Hoogenraad CC, Rush J, Ramm E, Schlager MA, Duong DM, Xu P, Wijayawardana SR, Hanfelt J, Nakagawa T, et al. (2006). Relative and absolute quantification of postsynaptic density proteome isolated from rat forebrain and cerebellum. Mol. Cell. Proteomics 5, 1158–1170. [DOI] [PubMed] [Google Scholar]
- Cheng DT, Disterhoft JF, Power JM, Ellis DA, and Desmond JE (2008). Neural substrates underlying human delay and trace eyeblink conditioning. Proc. Natl. Acad. Sci. USA 105, 8108–8113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cizeron M, Qiu Z, Koniaris B, Gokhale R, Komiyama NH, Fransén E, and Grant SGN (2020). A brainwide atlas of synapses across the mouse life span. Science 369, 270–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark RE, and Squire LR (1999). Human Eyeblink Classical Conditioning: Effects of Manipulating Awareness of the Stimulus Contingencies. Psychol. Sci 10, 14–18. [Google Scholar]
- Coba MP, Pocklington AJ, Collins MO, Kopanitsa MV, Uren RT, Swamy S, Croning MD, Choudhary JS, and Grant SG (2009). Neurotransmitters drive combinatorial multistate postsynaptic density networks. Sci. Signal 2, ra19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins MO, Husi H, Yu L, Brandon JM, Anderson CN, Blackstock WP, Choudhary JS, and Grant SG (2006). Molecular characterization and comparison of the components and multiprotein complexes in the postsynaptic proteome. J. Neurochem 97 (Suppl 1), 16–23. [DOI] [PubMed] [Google Scholar]
- Counts SE, Nadeem M, Lad SP, Wuu J, and Mufson EJ (2006). Differential expression of synaptic proteins in the frontal and temporal cortex of elderly subjects with mild cognitive impairment. J. Neuropathol. Exp. Neurol 65, 592–601. [DOI] [PubMed] [Google Scholar]
- Courchesne E, Gazestani VH, and Lewis NE (2020). Prenatal Origins of ASD: The When, What, and How of ASD Development. Trends Neurosci. 43, 326–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daum I, Channon S, Polkey CE, and Gray JA (1991). Classical conditioning after temporal lobe lesions in man: impairment in conditional discrimination. Behav. Neurosci 105, 396–408. [DOI] [PubMed] [Google Scholar]
- Daum I, Channon S, and Gray JA (1992). Classical conditioning after temporal lobe lesions in man: sparing of simple discrimination and extinction. Behav. Brain Res 52, 159–165. [DOI] [PubMed] [Google Scholar]
- Díaz-Quesada M, Martini FJ, Ferrati G, Bureau I, and Maravall M (2014). Diverse thalamocortical short-term plasticity elicited by ongoing stimulation. J. Neurosci 34, 515–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich I, and Malinow R (2004). Postsynaptic density 95 controls AMPA receptor incorporation during long-term potentiation and experience-driven synaptic plasticity. J. Neurosci 24, 916–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteves AR, and Cardoso SM (2020). Differential protein expression in diverse brain areas of Parkinson’s and Alzheimer’s disease patients. Sci. Rep 10, 13149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans RC, and Blackwell KT (2015). Calcium: amplitude, duration, or location? Biol. Bull 228, 75–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Z, Chen X, Zeng M, and Zhang M (2019). Phase separation as a mechanism for assembling dynamic postsynaptic density signalling complexes. Curr. Opin. Neurobiol 57, 1–8. [DOI] [PubMed] [Google Scholar]
- Frank RA, and Grant SG (2017). Supramolecular organization of NMDA receptors and the postsynaptic density. Curr. Opin. Neurobiol 45, 139–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank RA, Komiyama NH, Ryan TJ, Zhu F, O’Dell TJ, and Grant SG (2016). NMDA receptors are selectively partitioned into complexes and supercomplexes during synapse maturation. Nat. Commun 7, 11264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank RAW, Zhu F, Komiyama NH, and Grant SGN (2017). Hierarchical organization and genetically separable subfamilies of PSD95 postsynaptic supercomplexes. J. Neurochem 142, 504–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankle WG, Lerma J, and Laruelle M (2003). The synaptic hypothesis of schizophrenia. Neuron 39, 205–216. [DOI] [PubMed] [Google Scholar]
- Freeman JH, and Steinmetz AB (2011). Neural circuitry and plasticity mechanisms underlying delay eyeblink conditioning. Learn. Mem 18, 666–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannakopoulos M, Kouvelas ED, and Mitsacos A (2010). Experience-dependent regulation of NMDA receptor subunit composition and phosphorylation in the retina and visual cortex. Invest. Ophthalmol. Vis. Sci 51, 1817–1822. [DOI] [PubMed] [Google Scholar]
- Gladding CM, Fitzjohn SM, and Molnár E (2009). Metabotropic glutamate receptor-mediated long-term depression: molecular mechanisms. Pharmacol. Rev 61, 395–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouwens NW, Sorensen SA, Berg J, Lee C, Jarsky T, Ting J, Sunkin SM, Feng D, Anastassiou CA, Barkan E, et al. (2019). Classification of electrophysiological and morphological neuron types in the mouse visual cortex. Nat. Neurosci 22, 1182–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant SGN, and Fransén E (2020). The Synapse Diversity Dilemma: Molecular Heterogeneity Confounds Studies of Synapse Function. Front. Synaptic Neurosci 12, 590403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Ceolin L, Collins KA, Perroy J, and Huber KM (2015). Elevated CaMKIIα and Hyperphosphorylation of Homer Mediate Circuit Dysfunction in a Fragile X Syndrome Mouse Model. Cell Rep. 13, 2297–2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heavner WE, and Smith SEP (2020). Resolving the Synaptic versus Developmental Dichotomy of Autism Risk Genes. Trends Neurosci. 43, 227–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heavner WE, Speed H, Lautz JD, Gniffke EP, Immendorf KB, Welsh JP, and Smith SEP (2020). Homeostatic Plasticity Requires Remodeling of the Homer-Shank Interactome. bioRxiv, 2020.2003.2026.010314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heiney SA, Wohl MP, Chettih SN, Ruffolo LI, and Medina JF (2014). Cerebellar-dependent expression of motor learning during eyeblink conditioning in head-fixed mice. J. Neurosci 34, 14845–14853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hell JW (2014). CaMKII: claiming center stage in postsynaptic function and organization. Neuron 81, 249–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holderith N, Lorincz A, Katona G, Rózsa B, Kulik A, Watanabe M, and Nusser Z (2012). Release probability of hippocampal glutamatergic terminals scales with the size of the active zone. Nat. Neurosci 15, 988–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Shen S, Cadwell CR, Berens P, Sinz F, Ecker AS, Patel S, and Tolias AS (2015). Principles of connectivity among morphologically defined cell types in adult neocortex. Science 350, aac9462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson WE, Li C, and Rabinovic A (2007). Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 8, 118–127. [DOI] [PubMed] [Google Scholar]
- Jordan BA, Fernholz BD, Boussac M, Xu C, Grigorean G, Ziff EB, and Neubert TA (2004). Identification and verification of novel rodent postsynaptic density proteins. Mol. Cell. Proteomics 3, 857–871. [DOI] [PubMed] [Google Scholar]
- Kopec CD, Li B, Wei W, Boehm J, and Malinow R (2006). Glutamate receptor exocytosis and spine enlargement during chemically induced long-term potentiation. J. Neurosci 26, 2000–2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P, and Horvath S (2008). WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9, 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lautz JD, Brown EA, Williams VanSchoiack AA, and Smith SEP (2018). Synaptic activity induces input-specific rearrangements in a targeted synaptic protein interaction network. J. Neurochem 146, 540–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lautz JD, Gniffke EP, Brown EA, Immendorf KB, Mendel RD, and Smith SEP (2019). Activity-dependent changes in synaptic protein complex composition are consistent in different detergents despite differential solubility. Sci. Rep 9, 10890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lautz JD, Zhu Z, Speed HE, Smith SEP, and Welsh JP (2021). Shank3 mutations impair electrical synapse scaffolding and transmission in mouse brain. bioRxiv. 10.1101/2021.03.25.437056. [DOI] [Google Scholar]
- Lee HK, Kameyama K, Huganir RL, and Bear MF (1998). NMDA induces long-term synaptic depression and dephosphorylation of the GluR1 subunit of AMPA receptors in hippocampus. Neuron 21, 1151–1162. [DOI] [PubMed] [Google Scholar]
- Lee J, Chung C, Ha S, Lee D, Kim DY, Kim H, and Kim E (2015). Shank3-mutant mice lacking exon 9 show altered excitation/inhibition balance, enhanced rearing, and spatial memory deficit. Front. Cell. Neurosci 9, 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Kang H, Lee B, Zhang Y, Kim Y, Kim S, Kim WK, and Han K (2017a). Integrative Analysis of Brain Region-specific Shank3 Interactomes for Understanding the Heterogeneity of Neuronal Pathophysiology Related to SHANK3 Mutations. Front. Mol. Neurosci 10, 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Kim SG, Lee B, Zhang Y, Kim Y, Kim S, Kim E, Kang H, and Han K (2017b). Striatal Transcriptome and Interactome Analysis of Shank3-overexpressing Mice Reveals the Connectivity between Shank3 and mTORC1 Signaling. Front. Mol. Neurosci 10, 201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Zhang Y, Kim S, and Han K (2018). Excitatory and inhibitory synaptic dysfunction in mania: an emerging hypothesis from animal model studies. Exp. Mol. Med 50, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li KW, Hornshaw MP, Van Der Schors RC, Watson R, Tate S, Casetta B, Jimenez CR, Gouwenberg Y, Gundelfinger ED, Smalla KH, and Smit AB (2004). Proteomics analysis of rat brain postsynaptic density. Implications of the diverse protein functional groups for the integration of synaptic physiology. J. Biol. Chem 279, 987–1002. [DOI] [PubMed] [Google Scholar]
- Li J, Zhang W, Yang H, Howrigan DP, Wilkinson B, Souaiaia T, Evgrafov OV, Genovese G, Clementel VA, Tudor JC, et al. (2017). Spatiotemporal profile of postsynaptic interactomes integrates components of complex brain disorders. Nat. Neurosci 20, 1150–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman J, Yasuda R, and Raghavachari S (2012). Mechanisms of CaMKII action in long-term potentiation. Nat. Rev. Neurosci 13, 169–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo H, Hasegawa K, Liu M, and Song WJ (2017). Comparison of the Upper Marginal Neurons of Cortical Layer 2 with Layer 2/3 Pyramidal Neurons in Mouse Temporal Cortex. Front. Neuroanat 11, 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüscher C, and Malenka RC (2012). NMDA receptor-dependent long-term potentiation and long-term depression (LTP/LTD). Cold Spring Harb. Perspect. Biol 4, a005710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micheva KD, Busse B, Weiler NC, O’Rourke N, and Smith SJ (2010). Single-synapse analysis of a diverse synapse population: proteomic imaging methods and markers. Neuron 68, 639–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteiro P, and Feng G (2017). SHANK proteins: roles at the synapse and in autism spectrum disorder. Nat. Rev. Neurosci 18, 147–157. [DOI] [PubMed] [Google Scholar]
- Neier SC, Ferrer A, Wilton KM, Smith SEP, Kelcher AMH, Pavelko KD, Canfield JM, Davis TR, Stiles RJ, Chen Z, et al. (2019). The early proximal αβ TCR signalosome specifies thymic selection outcome through a quantitative protein interaction network. Sci. Immunol 4, eaal2201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otmakhov N, Khibnik L, Otmakhova N, Carpenter S, Riahi S, Asrican B, and Lisman J(2004). Forskolin-induced LTP in the CA1 hippocampal region is NMDA receptor dependent. J. Neurophysiol 91, 1955–1962. [DOI] [PubMed] [Google Scholar]
- Pawson T (1995). Protein modules and signalling networks. Nature 373, 573–580. [DOI] [PubMed] [Google Scholar]
- Peng J, Kim MJ, Cheng D, Duong DM, Gygi SP, and Sheng M (2004). Semiquantitative proteomic analysis of rat forebrain postsynaptic density fractions by mass spectrometry. J. Biol. Chem 279, 21003–21011. [DOI] [PubMed] [Google Scholar]
- Pocklington AJ, Cumiskey M, Armstrong JD, and Grant SG (2006). The proteomes of neurotransmitter receptor complexes form modular networks with distributed functionality underlying plasticity and behaviour. Mol. Syst. Biol 2, 2006.0023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronesi JA, Collins KA, Hays SA, Tsai NP, Guo W, Birnbaum SG, Hu JH, Worley PF, Gibson JR, and Huber KM (2012). Disrupted Homer scaffolds mediate abnormal mGluR5 function in a mouse model of fragile X syndrome. Nat. Neurosci 15, 431–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenblum K, Dudai Y, and Richter-Levin G (1996). Long-term potentiation increases tyrosine phosphorylation of the N-methyl-D-aspartate receptor subunit 2B in rat dentate gyrus in vivo. Proc. Natl. Acad. Sci. USA 93, 10457–10460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross RT, Orr WB, Holland PC, and Berger TW (1984). Hippocampectomy disrupts acquisition and retention of learned conditional responding. Behav. Neurosci 98, 211–225. [DOI] [PubMed] [Google Scholar]
- Roy M, Sorokina O, McLean C, Tapia-González S, DeFelipe J, Armstrong JD, and Grant SGN (2018). Regional Diversity in the Postsynaptic Proteome of the Mouse Brain. Proteomes 6, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schikorski T, and Stevens CF (1997). Quantitative ultrastructural analysis of hippocampal excitatory synapses. J. Neurosci 17, 5858–5867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SE, Neier SC, Reed BK, Davis TR, Sinnwell JP, Eckel-Passow JE, Sciallis GF, Wieland CN, Torgerson RR, Gil D, et al. (2016). Multiplex matrix network analysis of protein complexes in the human TCR signalosome. Sci. Signal 9, rs7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takatsuki K, Kawahara S, Takehara K, Kishimoto Y, and Kirino Y (2001). Effects of the noncompetitive NMDA receptor antagonist MK-801 on classical eyeblink conditioning in mice. Neuropharmacology 41, 618–628. [DOI] [PubMed] [Google Scholar]
- Takehara-Nishiuchi K (2018). The Anatomy and Physiology of Eyeblink Classical Conditioning. Curr. Top. Behav. Neurosci 37, 297–323. [DOI] [PubMed] [Google Scholar]
- Takehara-Nishiuchi K, Kawahara S, and Kirino Y (2005). NMDA receptor-dependent processes in the medial prefrontal cortex are important for acquisition and the early stage of consolidation during trace, but not delay eyeblink conditioning. Learn. Mem 12, 606–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ten Brinke MM, Heiney SA, Wang X, Proietti-Onori M, Boele HJ, Bakermans J, Medina JF, Gao Z, and De Zeeuw CI (2017). Dynamic modulation of activity in cerebellar nuclei neurons during pavlovian eyeblink conditioning in mice. eLife 6, e28132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toettcher JE, Weiner OD, and Lim WA (2013). Using optogenetics to interrogate the dynamic control of signal transmission by the Ras/Erk module. Cell 155, 1422–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trepanier CH, Jackson MF, and MacDonald JF (2012). Regulation of NMDA receptors by the tyrosine kinase Fyn. FEBS J. 279, 12–19. [DOI] [PubMed] [Google Scholar]
- Trinidad JC, Thalhammer A, Specht CG, Lynn AJ, Baker PR, Schoepfer R, and Burlingame AL (2008). Quantitative analysis of synaptic phosphorylation and protein expression. Mol. Cell. Proteomics 7, 684–696. [DOI] [PubMed] [Google Scholar]
- Tu JC, Xiao B, Yuan JP, Lanahan AA, Leoffert K, Li M, Linden DJ, and Worley PF (1998). Homer binds a novel proline-rich motif and links group 1 metabotropic glutamate receptors with IP3 receptors. Neuron 21, 717–726. [DOI] [PubMed] [Google Scholar]
- Weiskrantz L, and Warrington EK (1979). Conditioning in amnesic patients. Neuropsychologia 17, 187–194. [DOI] [PubMed] [Google Scholar]
- Weiss C, and Disterhoft JF (2011). Exploring prefrontal cortical memory mechanisms with eyeblink conditioning. Behav. Neurosci 125, 318–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss C, and Disterhoft JF (2015). The impact of hippocampal lesions on trace-eyeblink conditioning and forebrain-cerebellar interactions. Behav. Neurosci 129, 512–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh JP, and Harvey JA (1989). Cerebellar lesions and the nictitating membrane reflex: performance deficits of the conditioned and unconditioned response. J. Neurosci 9, 299–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh JP, and Harvey JA (1998). Acute inactivation of the inferior olive blocks associative learning. Eur. J. Neurosci 10, 3321–3332. [DOI] [PubMed] [Google Scholar]
- Welsh JP, and Turecek J (2017). Implications of electrical synapse plasticity in the inferior olive. In Network Functions and Plasticity, Jing J, ed. (Academic Press; ), pp. 265–299. [Google Scholar]
- Welsh JP, Yamaguchi H, Zeng XH, Kojo M, Nakada Y, Takagi A, Sugimori M, and Llinás RR (2005). Normal motor learning during pharmacological prevention of Purkinje cell long-term depression. Proc. Natl. Acad. Sci. USA 102, 17166–17171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Lei C, Feng H, and Sui JF (2015). The neural circuitry and molecular mechanisms underlying delay and trace eyeblink conditioning in mice. Behav. Brain Res 278, 307–314. [DOI] [PubMed] [Google Scholar]
- Yin DM, Chen YJ, Sathyamurthy A, Xiong WC, and Mei L (2012). Synaptic dysfunction in schizophrenia. Adv. Exp. Med. Biol 970, 493–516. [DOI] [PubMed] [Google Scholar]
- Zbarska S, and Bracha V (2012). Assessing the role of inferior olivary sensory signaling in the expression of conditioned eyeblinks using a combined glutamate/GABAA receptor antagonist protocol. J. Neurophysiol 107, 273–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng M, Shang Y, Araki Y, Guo T, Huganir RL, and Zhang M (2016). Phase Transition in Postsynaptic Densities Underlies Formation of Synaptic Complexes and Synaptic Plasticity. Cell 166, 1163–1175.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng M, Chen X, Guan D, Xu J, Wu H, Tong P, and Zhang M (2018). Reconstituted Postsynaptic Density as a Molecular Platform for Understanding Synapse Formation and Plasticity. Cell 174, 1172–1187.e16. [DOI] [PubMed] [Google Scholar]
- Zhang B, Fang W, Ma W, Xue F, Ai H, and Lu W (2020). Differential Roles of GluN2B in Two Types of Chemical-induced Long Term Potentiation-mediated Phosphorylation Regulation of GluA1 at Serine 845 in Hippocampal Slices. Neuroscience 433, 144–155. [DOI] [PubMed] [Google Scholar]
- Zhu F, Cizeron M, Qiu Z, Benavides-Piccione R, Kopanitsa MV, Skene NG, Koniaris B, DeFelipe J, Fransén E, Komiyama NH, and Grant SGN (2018). Architecture of the Mouse Brain Synaptome. Neuron 99, 781–799.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoghbi HY, and Bear MF (2012). Synaptic dysfunction in neurodevelopmental disorders associated with autism and intellectual disabilities. Cold Spring Harb. Perspect. Biol 4, a009886. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
QMI data reported here, EBC raw data and raw data from the Ca2+ imaging experiments will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available upon request from the lead contact.