

Abstract
In contrast to solid cancers, which often require genetic modifications and complex cellular reprogramming for effective metastatic dissemination, leukaemic cells uniquely possess the innate ability for migration and invasion. Dedifferentiated, malignant leukocytes retain the benign leukocytes’ capacity for cell motility and survival in the circulation, while acquiring the potential for rapid and uncontrolled cell division. For these reasons, leukaemias, although not traditionally considered as metastatic diseases, are in fact models of highly efficient metastatic spread. Accordingly, they are often aggressive and challenging diseases to treat. In this Perspective, we discuss the key molecular processes that facilitate metastasis in a variety of leukaemic subtypes, the clinical significance of leukaemic invasion into specific tissues and the current pipeline of treatments targeting leukaemia metastasis.
Metastasis of solid tumours is defined as the emergence of secondary growth away from a primary site. For liquid tumours such as leukaemias that initially present with widespread disease, the precise location of origin in the bone marrow (BM), spleen or lymph node (LN) is, however, often unknown. This ambiguity, as well as the recognition that leukaemic cells ‘inherit’ rather than gradually acquire motility, has resulted in disagreement over whether leukaemia should be considered a metastatic disease. Despite this controversy, disseminated leukaemia cells share many of the properties of metastasizing solid tumour cells. These include derivation from a mutant clone or clones possessing disease-initiating potential1–3, spread via a cascade of molecular events entailing intravasation, extravasation and tissue colonization4–10, specific and reproducible patterns of organ involvement11,12, adaptation to tissue microenvironments that are distinct from that of the cell of origin13,14, and widespread disease that is challenging to treat and can fuel future relapse13,14. The unique selective pressures applied by different organs often lead to heterogeneous clonal evolution, compounding the difficulty in achieving complete treatment response. For these reasons, we consider the process of leukaemia cell dissemination to represent metastasis.
In this Perspective, we argue that leukaemia dissemination is indeed metastatic, and we describe the events in this multi-step process. We review the various sites of leukaemia metastasis and the molecular mechanisms governing this organotropism. Finally, we provide an overview of current and upcoming strategies to target leukaemia metastasis.
Classification and derivation
The term leukaemia is derived from the Greek words ‘leukos’, meaning white, and ‘haima, meaning blood. The name, which is used to reference a broad array of haematopoietic malignancies currently subcategorized according to their morphology, immunophenotype, cytogenetic and molecular abnormalities and clinical features, refers quite literally to the excess of white blood cells seen in the bloodstream of patients with these diseases15. Clinically, these diseases fall within four broad categories, myeloid or lymphoid lineage and acute or chronic, which help to dictate the approach to treatment. The latter categorization refers to the rapidity of clinical onset and disease progression. Although there are a multitude of specific diagnoses, the most commonly seen and studied leukaemias are acute myeloid leukaemia (AML), acute lymphocytic leukaemia/lymphoma (ALL), chronic lymphocytic leukaemia/small lymphocytic lymphoma (CLL/SLL) and chronic myeloid leukaemia (CML). We therefore focus on these entities in this article.
Although circulating malignant white blood cells are a prominent feature of the leukaemias, it should be noted that blood-borne tumour cells can often be detected in epithelial malignancies and other haematopoietic disorders, including lymphomas and multiple myeloma, often in association with more aggressive disease16,17. Furthermore, leukaemia cell involvement is not limited to the bloodstream and haematopoietic organs (BM, spleen, LN and tonsils), and some subtypes show a particular predilection for specific non-haematopoietic tissues (for example, ALL and the central nervous system (CNS)18,19). These observations underscore the premise that wide-ranging, organotropic dissemination is a shared trait among the liquid and solid cancers.
Whereas solid tumour metastasis is a largely inefficient process with a high rate of attrition20, leukaemia cells are motile and can easily traffic throughout the body without the need to acquire a heavy mutational burden and anchorage independence. Leukaemia metastasis is therefore an efficient and cyclical process of tissue homing, colonization and mobilization back into circulation, making leukaemia a deadly and challenging disease to treat, and in the case of the acute leukaemias, an ultimate example of aggressive metastasis.
The haematopoietic stem cell (HSC) is positioned atop the developmental hierarchy of all lymphoid, myeloid and erythroid lineage progenitor and mature haematopoietic cells21. Although the bulk of the HSC population is quiescent and ensconced within regulatory and protective BM niches at any given time, HSCs are in fact motile cells and possess the remarkable ability to swiftly enter circulation and traffic to both haematopoietic and non-haematopoietic organs in response to a variety of inflammatory cues22. Compelling data, recently reviewed3, have shown that AML and CML cells arise from HSCs or myeloid progenitors in which key driver mutations have occurred (for example, the BCR–ABL translocation in CML). Despite the single cell origin of disease, significant heterogeneity arises as mutations accumulate in daughter cells, resulting in what is referred to as subclonal evolution2,23,24. Yet, only a subset of leukaemic cells within a malignant population is believed to harbour the stem cell properties of self-renewal and the ability to initiate disease when engrafted into an immunocompromised host1,3, and these are referred to as leukaemia stem cells (LSCs). LSCs, like other cancer stem cells, are particularly important in the context of metastasis as they are often therapy resistant25–27. Fuelled by the acquisition of new pro-survival and pro-proliferative mutations, they may emerge as a dominant subclone later in the disease course28,29 and serve as a reservoir for disease relapse27. Although the precise cells of origin for CLL and ALL and the relevance of LSCs in these leukaemias remain unclear, the process of clonal evolution, occurring independently at different sites of metastasis, also contributes to the difficulty in curing these diseases30–33.
Reflecting their haematopoietic cells of origin, leukaemia cells co-opt haematopoietic stem and progenitor cell (HSPC) and/or mature leukocyte trafficking mechanisms at almost every step of the metastatic cascade. For example, leukaemic cells respond to environmental cues similar to those that enable HSPCs and leukocytes to exit (‘mobilize’) and enter tissues from peripheral circulation9,34. After extravasating into a new tissue, leukaemia cells, like HSPCs, encounter a complex microenvironment composed of diverse niches capable of fostering either cellular proliferation or dormancy35. Further echoing the cellular origin of leukaemias, much of the microenvironmental crosstalk that regulates leukaemia metastatic growth recapitulates the niche signalling that regulates HSPC repopulation or quiescence22,36.
Patterns of metastasis
One of the hallmarks of metastasis is organ tropism, dictated by the appropriateness of a particular organ’s ‘soil’ as host for the metastatic ‘seed’. Different leukaemia subtypes exhibit unique patterns of metastasis to various tissues including, but not limited to the BM, LN, liver, spleen, CNS, skin and testicles12 (FIG. 1). Although leukaemias are primarily understood to be diseases of the haematopoietic tissues and circulatory system, their metastatic invasion of extramedullary (non-BM) sites holds significant importance for disease progression, severity and outcomes in patients. For example, unique interactions between leukaemic cells and the surrounding microenvironment of different organs may play a role in the heterogeneous responses to treatment observed in some patients. Rituximab, an anti-CD20 monoclonal antibody, has been reported to be more effective in clearing follicular lymphoma from BM than from LNs37, and similarly, its mechanism of cytotoxicity in CLL is believed to vary depending on tumour site38. The metastasis of leukaemia to sanctuary sites such as the CNS and testes, where drugs and the immune system have more restricted penetration, is another important example of the impact of the tissue metastatic profile on disease treatment19,39–41.
Fig. 1 |. Primary metastatic profiles of leukaemia metastasis.
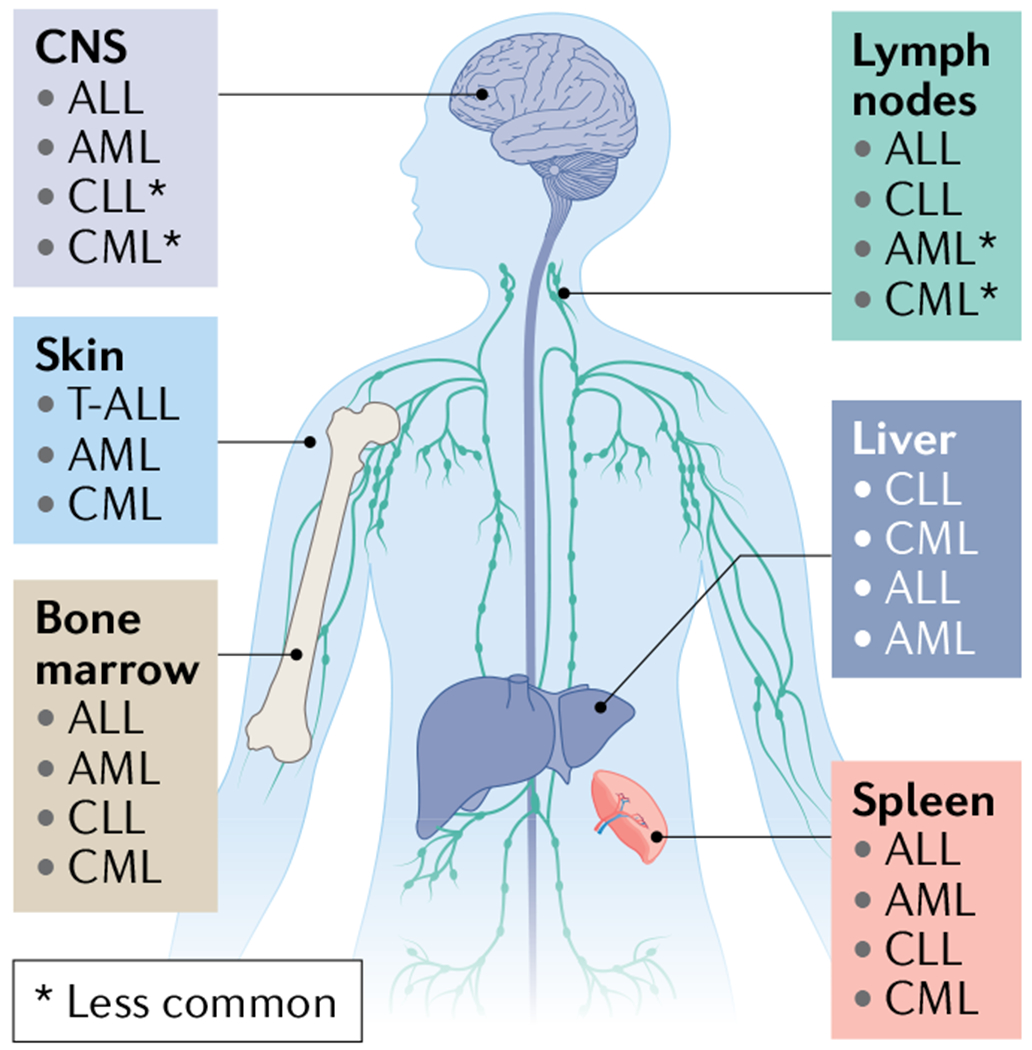
Acute lymphoblastic leukaemia (ALL) cells are typically initially found in the peripheral vascular system and the haematopoietic organs, including the bone marrow (BM)15, lymphatic system and spleen; however, central nervous system (CNS) involvement occurs in 5–10% of adult patients at diagnosis230 and can occur in up to 30–50% of patients in the absence of CNS-directed therapy. WHO classifies ALL and lymphoblastic lymphoma as a spectrum disorder and distinguishes these diseases according to clinical presentation favouring BM (involvement of >20% of blasts) or lymphatic tissue involvement, respectively15. Acute myeloid leukaemia (AML) primarily develops and spreads within the BM and less frequently involves other haematopoietic organs. It may invade non-haematopoietic organs including the gingiva, skin, muscle and CNS, although uncommonly. Chronic lymphocytic leukaemia/small lymphocytic lymphoma (CLL/SLL) is notable for its highly stereotyped clinical evolution: at the earliest stages, bloodstream lymphocytosis is the sole clinical finding, followed over time by the development of lymph node enlargement, then splenomegaly and in advanced stages, progressive BM involvement231. CLL and SLL are considered the same entity, but with differing disease presentation of bloodstream (CLL) or nodal involvement (SLL)232,233. Chronic myeloid leukaemia (CML) primarily involves the BM, peripheral circulation and spleen234. In rare cases including during evolution to blast crisis, CML expansion can result in infiltration into other less common sites, including the lymph nodes, liver, skin or CNS235–237. T-ALL, T cell ALL.
Underpinnings of leukaemia metastasis
The concept of leukaemia as a metastatic disease is bolstered by the recognition that leukaemic cells engage many of the same cell surface adhesion molecules, chemokines, matrix molecules and intracellular motility signalling pathways as solid cancers to invade distant organs. Detailed investigations have revealed considerable molecular conservation between solid and liquid tumours in these processes. Some important molecular components in the leukaemic metastatic cascade are described below.
Selectins.
Selectins (E-, P- and L-selectins) are a family of cell surface glycoproteins expressed on inflamed endothelial cells (E- and P-selectin) and leukocytes (L-selectin), the importance of which in cell trafficking was first established in lymphocytes42 (BOX 1). Later, recognition of selectin ligand expression by leukaemia43 and solid tumour cells44–46 and their association with advanced disease in patients led to investigations defining their important role in tumour metastasis47.
Box 1 |. Leukocyte migration mechanisms.
Tethering and rolling
Leukocyte extravasation begins with tethering (also known as ‘capture’; see figure), the process through which leukocytes first establish contact with the endothelial cells that make up the surface of blood vessels. Leukocytes engage endothelial E- and P-selectin via cell surface carbohydrate ligands such as sialyl Lewis x (SLeX) and P-selectin glycoprotein ligand 1 (PSGL1)244. These adhesions are further promoted by VLA-4, engaging intercellular adhesion molecule 1 (ICAM1) on vascular endothelial cells245.
Slow rolling
As adhesions become stronger, rolling slows down in a phase identified as ‘slow rolling’ wherein leukocyte adhesion to endothelial P- and E-selectin induces an affinity conformation of integrin αLβ2 (also known as lymphocyte function-associated antigen 1 (LFA1)). This allows for the transient binding of ICAM1 on the vascular endothelium and ligand-induced adhesion strengthening246–248.
Firm adhesion
Slow rolling eventually results in leukocytes stopping and firmly adhering to the surface of endothelial cells, a process widely known as ‘arrest’. Arrest is triggered through a chemokine-driven process known as activation, in which endothelial cells are stimulated to express higher levels of adhesive molecules such as ICAM1 and vascular cell adhesion molecule 1 (VCAM1) (REF.249). Thereafter, adhesion molecules are reorganized and clustered to reinforce adhesion250. Before diapedesis, leukocytes crawl along the blood vessel wall via the integrins αMβ2 (also known as MAC-1) and αLβ2 to probe the endothelium and identify sites permissive for crossing251,252.
Transmigration
In most cases, leukocytes extravasate through the endothelial monolayer via a paracellular (diapedesis) route by using membrane protrusions to probe the endothelial surface and identify endothelial cell–cell junctions. This process is mediated by a complex array of integrins, proteases and signalling pathways to ‘squeeze’ through the endothelium253. On rare occasions, leukocytes can extravasate transcellularly (going through endothelial cells) by forming small pockets in the plasma membrane (caveolae) that eventually fuse to form an intracellular channel254.
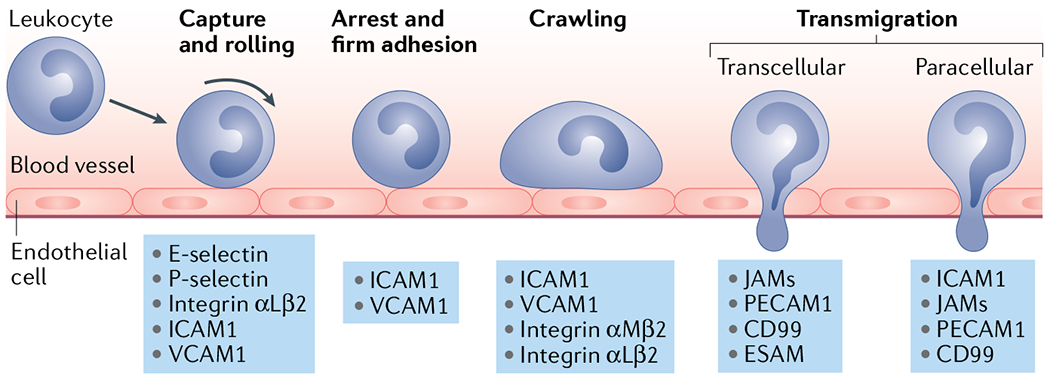
Selectin ligands are known to be expressed by all major categories of leukaemia43, and high levels of E-selectin ligand expression are associated with poor disease outcomes in patients with AML48. Early in vitro studies reported that inhibition of L-selectin and E-selectin binding decreased leukaemia cell adhesion to human umbilical vein endothelial cells, suggesting a functional role in haematogenous metastasis49. In upcoming sections, we highlight examples of selectin-mediated leukaemic metastasis to the BM and LN.
While E- and P-selectin expression is limited to the vasculature, L-selectin can be expressed on the surface of leukaemic cells. In ALL and CLL, L-selectin expression and shedding have been shown to correlate with more highly metastatic disease7,50,51. In fact, patients with CLL who were treated with the PI3Kδ inhibitor idelalisib exhibited decreased L-selectin expression, suggesting an additional pharmacological mechanism by which PI3K inhibition prevents CLL progression7.
Integrins.
Integrins are a family of cell surface molecules that have been shown to have an important role in almost every step of the metastatic cascade from invasion to the activation of pro-survival signalling pathways52. In both solid tumours and leukaemias, integrins enable malignant cells to sense and respond to a variety of cell, extracellular matrix (ECM), cytokine and growth factor binding partners in their tissue microenvironments. During leukaemic cell dissemination, integrins play a key adhesive role, anchoring cells to the vessel lumen during haematogenous metastasis (FIG. 2) and tethering them to the metastatic niche after invasion8,53–55. Integrin activation also stimulates intracellular signalling pathways that augment leukaemia cell motility56.
Fig. 2 |. The leukaemia bone marrow microenvironment.
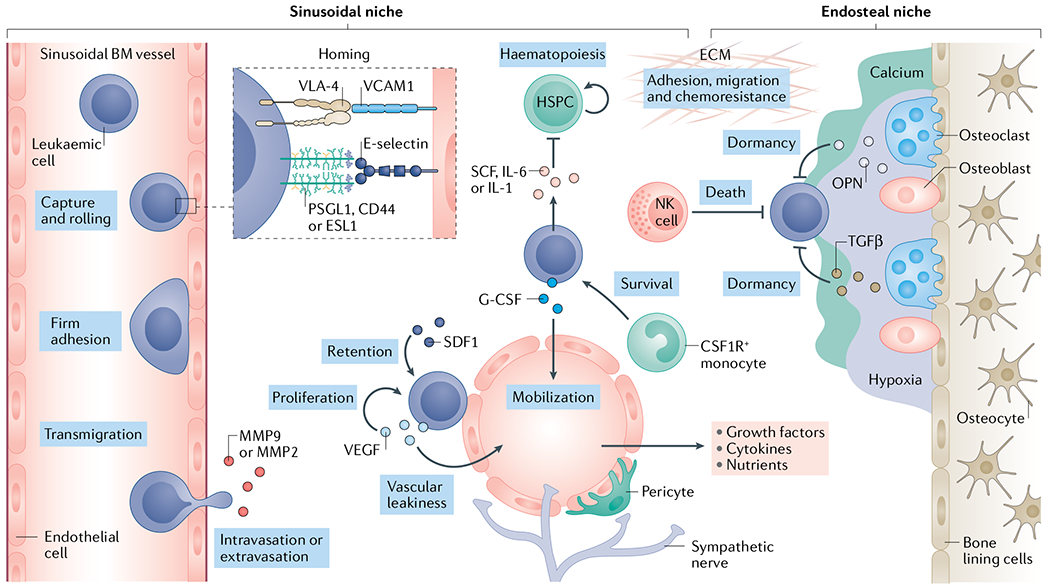
Leukaemia cell homing is mediated by the chemokine stromal cell-derived factor 1 (SDF1)9,99–101 and adhesion molecules such as the integrin VLA-48,53–55 and E-selectin9 expressed by the vascular endothelium of fenestrated, sinusoidal bone marrow153 vessels. In this niche, growth factors, cytokines, extracellular matrix (ECM) components as well as stromal cells such as NG2+ cells, pericytes and colony stimulating factor 1 receptor-positive (CSF1R+) monocytes117 all serve to promote leukaemia cell growth and survival. As leukaemia cells colonize the bone marrow (BM), they deplete resident haematopoietic stem and progenitor cells (HSPCs) through the secretion of stem cell factor (SCF), creating a malignant niche that disrupts normal haematopoiesis123. Leukaemia cells may also migrate to pro-dormancy endosteal niches where osteopontin (OPN)35, transforming growth factor-β (TGFβ)238 and hypoxic conditions support a quiescent state, thus protecting them from chemotherapy. Niche factors such as granulocyte colony-stimulating factor (G-CSF) can mobilize leukaemia cells from the metastatic niche into circulation where they can seed distant sites201–203. Matrix-degrading enzymes such as elastase143, matrix metalloproteinase 2 (MMP2)92 and MMP9 (REFS90,239) can facilitate the extravasation process. Finally, immune cells such as natural killer (NK) cells may limit the survival of leukaemia cells in the BM240,241. PSGL1, P-selectin glycoprotein ligand 1; VCAM1, vascular cell adhesion molecule 1; VEGF, vascular endothelial growth factor.
The integrin VLA-4 (also known as α4β1) is a receptor for vascular cell adhesion molecule 1 (VCAM1) and for ECM fibronectin, and the integrin VLA-5 (also known as α5β1) is a receptor for fibronectin and fibrinogen; both have important roles in tissue homing and retention for the lymphoid leukaemias8,53–55. Clinical investigations of patients with B cell ALL (B-ALL) have also shown a correlation between low-affinity VLA-4 binding and elevated peripheral blood blast numbers, further suggesting that VLA-4 has a crucial role in the retention of lymphoblasts in the BM microenvironment53.
Clinical research has also revealed a role for integrins in leukaemic cell tethering within LNs. In CLL, VLA-4 clustering, which promotes adhesion to VCAM1 expressed on LN endothelium, is induced by B cell receptor activation57. Patients with CLL who were treated with ibrutinib, a small-molecule inhibitor of Bruton tyrosine kinase (BTK) that antagonizes B cell receptor signalling and is approved for treatment of CLL58, displayed LN shrinkage concurrent with transient increases in circulating CLL cells that exhibited decreased adhesion to VCAM1 ex vivo57.
Chemokines, cytokines and growth factors.
Soluble factors such as chemokines, cytokines and growth factors have an integral role in the establishment of metastatic disease. They form a complex intracellular signalling network that guides tumour cell chemotaxis and invasion and colonization in both liquid and solid tumours.
The interaction between stromal cell-derived factor 1 (SDF1; also known as CXCL12) and its receptor, CXCR4, is one of the most studied interactions in leukaemia, as CXCR4 is expressed in most haematological malignancies and is often associated with poor prognosis59,60. SDF1 is synthesized and secreted by a myriad of stromal cells in tissues commonly colonized by leukaemia including the spleen, BM, CNS and skin, where it serves as a potent chemoattractant for leukaemia cells9,61,62 and LSCs10. CXCR4 expression can also be upregulated in response to niche factors such as stem cell factor (SCF; also known as Kit ligand)63 and hypoxia64 and in response to therapy65,66. Other chemokine receptors that have been implicated in leukaemia metastasis67 include CCR7 in the CNS68, spleen69 and LN70, and CCR4 and CCR2 (REFS71,72) in skin.
As in solid tumours, leukaemia metastasis can be enhanced by changes to the microenvironment induced by aberrant cytokine and growth factor secretion. Among the best-studied examples in solid malignancies are the vascular endothelial growth factor (VEGF) family-dependent processes of neoangiogenesis and vascular remodelling, which promote tumour growth and prime pre-metastatic niches73. Although not as intensively studied in the leukaemias, evidence of neoangiogenesis in the BM of patients with ALL74 and AML75 is well documented and will be discussed in upcoming sections.
Extracellular matrix.
Although epithelial cells critically depend on matrix adhesion to prevent anoikis, the ECM also has an important role in leukaemia metastasis as a scaffold to direct adhesion and migration in the metastatic niche. Fibronectin, a major structural component of the basement membrane of BM and LN blood vessels76–78, has been shown to enhance the chemotaxis of AML cells towards SDF1 (REF.79). Similarly, laminin expressed in the basement membrane of CNS vessels serves as a scaffold for B-ALL cell migration towards cerebrospinal fluid (CSL) chemokines in the leptomeninges and also activates leukaemia cell actinomysin contractility pathways through its binding to ALL integrin α6 receptors56. Matrix binding interactions with osteopontin (OPN) and fibronectin have also been shown to anchor leukaemic cells in the nutrient-rich and protective environment of the BM, shielding them from therapeutic killing35,80.
Motility modes.
Similarly to solid tumour cells, leukaemic cells adopt a variety of migratory strategies, including amoeboid-, invadopodia- and lamellipodia-driven movement, that each employ unique and complex molecular signalling pathways81 (BOX 2).
Box 2 |. Leukaemia cell migration modes.
Amoeboid
Leukocytes and leukaemic cells have been shown to migrate using what is known as amoeboid motility. This mode of migration is a highly efficient method employed by leukocytes and cancer cells to squeeze through tissues without having to degrade the extracellular matrix (ECM). Amoeboid migration relies on membrane blebs that are formed when the plasma membrane detaches from the underlying actin cortex allowing cytoplasm to form herniations in the membrane and generate a motile force255. This high contractile force generated through myosin II contractility followed by hydrostatic pressure in the cytoplasm enables the cell to push its way forwards through connective tissues and the ECM256.
Invadopodia
Invadopodia and their close relatives podosomes are F-actin-rich membrane protrusions that play an essential part in degrading the ECM to drive invasion257,258. Invadopodia formation relies on the coordination of CDC42 and tyrosine kinases to form actin bundles that extend into the plasma membrane, aiding cellular invasion. Vesicles carrying matrix-degrading molecules such as matrix metalloproteinases (MMPs) are delivered specifically to invadopodia259, where they can be released into the ECM and break it down260.
Lamellipodia
Lamellipodia are leading edge membrane protrusions that drive directional cell migration in most motile cell types and are occasionally found in migrating leukaemic cells. Lamellipodia consist of thin, flat protrusions that drive the cell forwards by generating a branched actin filament network81. As they migrate, leukocytes activate the RAC–WAVE–ARP2/3 signalling axis that drives actin polymerization, forming a retrograde flow of actin branches that pushes the cell forwards261.
Leukaemic cells, like normal leukocytes, use amoeboid-like migration to efficiently squeeze through tissues without having to degrade the ECM. The BCR–ABL fusion protein, present in CML cells as well as Philadelphia chromosome-positive ALL, can directly phosphorylate and activate cell motility machinery in this pathway. For example, ectopic expression of the p210 variant of the BCR–ABL fusion protein in murine Ba/F3 pro-B cells activates RHOA–ROCK signalling to myosin light chain (MLC) that works in conjunction with the actin-binding protein destrin (also known as actin depolymerizing factor) to promote spontaneous amoeboid motility82,83. The Dbl homology–pleckstrin homology domain in the p210 BCR–ABL chimera was identified to be the specific activator of RHOA, thus providing a further understanding of how BCR–ABL signalling activates cell motility pathways. Amoeboid-like migration may not be exclusive to BCR–ABL+ leukaemia, however. Ongoing work in our group suggests that only some ECM components can induce amoeboid-like migration in BCR–ABL− B-ALL cells, suggesting that the microenvironment also plays an important role in migration mode selection (D.A.S., unpublished observations). Finally, the importance of actomyosin-driven motility in leukaemia progression has been highlighted in recent years by studies showing high expression of the actin-polymerization protein, cortactin, on blasts biopsied from the BM and CSF of patients with relapsed B-ALL84.
Like solid tumour cells85,86, leukaemia cells may transition from one migration mode to another. For example, by suppressing RHOA activity and activating CDC42, BCR–ABL+ leukaemic cells can switch from amoeboid to invadopodia-based migration87. In CML, it has been reported that BCR–ABL constitutively activates the SRC family haematopoietic cell kinase (HCK), which contributes to invadopodia formation88,89.
The process of invadopodia formation is also associated with the release into the extracellular space of matrix-degrading enzymes that facilitate disease spread. For example, in vitro confocal microscopy studies revealed that the fibronectin and VCAM1 degrading enzyme, matrix metalloproteinase 9 (MMP9), co-localizes with invadopodia in CLL cells90. A clinical study found that in patients with CLL, LN infiltrates had high MMP9 content and a substantial reduction in ECM. The authors reported that elevated MMP9 expression correlated with advanced stage and poor prognosis91, suggesting the role of MMP9 in tissue invasion. Additional studies have reported that leukaemic cells can secrete MMP2, which degrades other ECM substrates, including collagen and elastin92,93.
Finally, microenvironmental factors may influence the migration mode adopted by leukaemia cells. In vitro studies have shown that T cell ALL (T-ALL) cells form lamellipodia when plated on a fibroblast monolayer94. Upon contact with fibroblasts, the RAC-specific guanine nucleotide exchange factor, TIAM1, is recruited to the plasma membrane where it interacts with the adhesion molecule CADM1 and activates RAC-dependent actin reorganization and lamellipodia formation. However, leukaemia cell lamellipodia formation may not always depend on contact with an adhesive substrate. For example, soluble extracellular galectins can drive lamellipodia formation in T-ALL cells by binding to integrin β1 and activating downstream PI3K–RAC1–ERK1/2 signalling95.
Navigating bone marrow niches
Most clinicians and scientists are unaccustomed to viewing the BM as a leukaemia metastatic site, yet consideration of the anatomy and pathophysiology of the disease supports the concept that widespread leukaemia BM dissemination is indeed a metastatic process. The collective BM spaces within an individual are largely non-contiguous. For example, no anatomical connection exists between contralateral femurs, yet leukaemia typically presents with involvement of contralateral and distant BM cavities in the femurs, pelvis, sternum, vertebrae and elsewhere96. As leukaemic cells are derived from one mutant clone originating in a single BM space, disseminated BM disease must by necessity arise from leukaemic cells that have exited the primary BM site, travelled through the bloodstream, and recognized and successfully entered a secondary BM location. These processes mirror the metastatic cascade of solid tumour cells. In the solid malignancies, distant spread through a single organ system, for example, contralateral intrapulmonary disease in lung cancer97, is also considered metastasis.
Most patients with leukaemia present at diagnosis with disseminated disease involving the BM, obviating the typical metastatic staging assessments used in solid tumours. As the BM is an important site of metastasis shared across the acute, chronic, myeloid and lymphoid leukaemias, as well as the most readily sampled tissue in patients with leukaemia apart from peripheral blood, much research has focused on the molecular mechanisms of disease spread to this organ. Despite the pervasiveness of disease spread in the BM, this process has been shown to be highly selective, involving specific molecular interactions between leukaemia cells and a subgroup of blood vessels9,98–101.
Similar to circulating normal leukocytes that precisely identify and enter haematopoietic organs or inflamed tissues through vascular interactions102,103, circulating leukaemic cells bind to and exit the vasculature to access new metastatic sites, a process generally known as extravasation and often referred to as diapedesis or transmigration in haematological malignancies4–10. Much of our early understanding of the molecular mechanisms underlying this metastatic process comes from studies of leukocyte trafficking, therefore, it is first helpful to understand the cascade of carefully choreographed events that leukocytes execute to home to secondary lymphoid organs or invade inflamed tissues102,103 (BOX 1). One of the seminal discoveries of leukaemia co-opting normal leukocyte homing mechanisms used intravital microscopy to view the BM microenvironment in mice, in real time9. The authors showed that both circulating pre-B-ALL cells and normal HSPCs home to sinusoidal vessels in the BM that uniquely express SDF1 and E-selectin, molecules with expression typically limited to inflamed vasculature. Mirroring HSC and T lymphocyte interactions with the vessel wall, blood-borne leukaemic cells were observed rolling along, arresting and diapedesing through these BM vascular domains in a manner predominantly dependent on SDF1 and, to a lesser degree, E-selectin9.
Subsequent studies in murine models have demonstrated that diverse leukaemias hijack E-selectin trafficking interactions to disseminate to the BM. Work in a murine CML-like neoplasia model has shown that loss or blockade of selectin–ligand interactions prevents leukaemia cell BM homing5,104. In a positive feedback loop, leukaemic cells that have colonized the BM may encourage additional homing events through secretion of inflammatory cytokines that upregulate endothelial E- and P-selectin expression49. CD44, a cell adhesion molecule and known E-selectin ligand expressed by HSPCs105 and mature white blood cells106, has been shown to play a part in CLL107 and myeloid LSC BM dissemination4,6. Its function as a tissue homing receptor for cancer cells has also been widely documented in solid tumours108,109.
A mounting body of literature has continued to unveil other BM homing mechanisms shared between leukaemia cells and normal leukocytes98–101. The expression of the integrin VLA-4 on leukaemia cells has been correlated with the increased capacity of patient-derived B-ALL cells to home to the BM in mice8. CD98 is another adhesion molecule expressed by HSPCs and leukocytes as well as leukaemic cells, the deletion of which preferentially decreases AML BM repopulation in vivo110.
After diapedesis and entry into a tissue, leukaemic cells must navigate a new microenvironment, a process that involves continued co-option of HSC signalling pathways. It has long been recognized that HSCs occupy unique stem cell regulatory niches in the BM22,36; however, over the past decade it has become increasingly apparent that leukaemic cells also require specialized niches to thrive and that these often overlap with stem cell niches111 (FIG. 2). As described above, the sinusoidal vascular niche serves as the main access point of leukaemic cells and HSPCs migrating in and out of the BM microenvironment9. Once inside the BM, leukaemic cells and stem cells surrounding these large, fenestrated sinusoidal vessels are thought to inhabit a pro-proliferative state supported by key microenvironmental interactions including SDF1–CXCR4 (REF.101) and VLA-4–VCAM1 (REFS80,112), as well as growth factors such as VEGF113–116, and colony stimulating factor 1 receptor-positive (CSF1R+) myeloid cells117.
Leukaemic cells that have entered the BM at the sinusoidal vascular niche also migrate to alternative HSC BM niches, including peri-endosteal, or bony, niches. While the sinusoidal niche tends to be conducive to leukaemia proliferation, long-term, quiescent AML stem cells118 and dormant ALL cells119 encounter pro-dormancy signals in the endosteal niche. In a xenograft mouse model, B-ALL cells were shown to migrate to and persist in the endosteal niche through OPN engagement via the integrin VLA-4 (REF.35); leukaemic cell interaction with the endosteal niche promoted dormancy, resistance to chemotherapy and the persistence of minimal residual disease (MRD) in this model35. This mechanism is conserved from HSCs, which also exhibit OPN-dependent dormancy120. Beyond the endosteal niche, other safe-haven niches have been identified. For example, it was reported that human T-ALL cells transplanted into mice home to adipocyte-rich BM niches where they display decreased metabolic activity and cell-cycle progression, suggesting a dormant phenotype that is also observed in HSPCs occupying ‘fatty’ niches121,122.
Insidiously, leukaemic cell occupation and accumulation in BM niches can induce molecular changes in the host microenvironment that support disease progression by providing a competitive growth advantage over HSPC populations. Our group documented that B-ALL cells disrupt the behaviour of haematopoietic progenitor cells (HPCs) in the BM by secreting SCF, which displaces HPCs from their normal regulatory niches, triggering their loss123. In this way, ALL cells obtain a survival advantage over naive HPCs, co-opting their niches to fuel tumour growth. High levels of leukaemia-derived IL-6 and IL-1 in the BM have also been shown to promote leukaemia cell proliferation while negatively impacting normal haematopoeisis124–126. Leukaemic cells can also disrupt sympathetic nervous system regulation of the HSC niche wherein AML-induced neuropathy impairs the differentiation status of mesenchymal stem/progenitor cells, promoting a pro-leukaemic, as opposed to HSC, niche127.
Niche occupation can also result in vascular remodelling that favours metastatic growth. For example, studies in mouse syngeneic and patient-derived xenograft (PDX) models have demonstrated that AML expansion can cause degradation and remodelling of the BM vascular architecture through pro-inflammatory cytokine release128 or the induction of hypoxia129. This leads to increased vascular permeability, altering levels of supportive growth factors in a manner that favours leukaemic cells over HSCs130. The VEGF family members VEGFA and VEGFC have also been reported to be overexpressed in the BM of patients with AML and to contribute to increased microvessel density116,131. These and other studies suggest that liquid tumours, like solid tumours, can use VEGF paracrine signalling to induce angiogenesis and stimulate endothelial cells to release growth factors that support leukaemia proliferation132. Leukaemic cells may further capitalize on elevated VEGF levels in hypoxic BM metastatic niche conditions through autocrine VEGF signalling133–138.
Dynamic changes in the ECM during disease progression can also promote leukaemia cell invasion. For example, AML cells in the BM can remodel collagen IV, increasing its affinity for the collagen-activated receptor tyrosine kinase, DDR1, on AML cells and promoting their AKT-dependent migration139. B-ALL cells have also been reported to remodel the BM niche by releasing TNF, which stimulates mesenchymal stem cells (MSCs) to produce the ECM-degrading protease MMP9 and thereby enhances leukaemia cell infiltration140.
After taking up residence in the BM, leukaemic cells can later re-enter the bloodstream and populate additional metastatic sites. This process, known generally as intravasation and often referred to as mobilization in haematological malignancies, is essential for disease progression. Disease colonization can promote leukaemia cell mobilization in syngeneic and PDX mouse models by altering the expression of niche molecules such as SDF1 that normally anchor leukaemic cells to the BM9,34. The selectin and integrin interactions that foster leukaemia BM homing have also been shown to have roles in tethering leukaemic cells to the BM niche. E-selectin blockade with the small-molecule inhibitor GMI-1271 was shown to rapidly mobilize leukaemic cells into peripheral circulation in preclinical mouse models of AML141. Similarly, VLA-4 deletion in B-ALL cells impairs both BM homing and retention54. In CML, recent work has shown that conditional deletion of the gene encoding the integrin-binding adaptor protein, kindlin 3 (also known as FERMT3), mobilizes LSCs from the BM in mice142.
Lastly, to promote their own metastatic spread, leukaemic cells can secrete proteases and matrix-degrading enzymes that allow for more efficient mobilization. For example, AML cells can express cell surface elastase that degrades vascular adhesion molecules such as VCAM1 and intercellular adhesion molecule 1 (ICAM1) (REF.143). MMP2 secretion by ALL cells has also been shown to degrade the vascular basement membrane, facilitating intravasation93.
Extramedullary metastasis
All leukaemias possess the ability to metastasize to the BM to varying extents, although some subtypes exhibit a predilection for the extramedullary haematopoietic tissue of the spleen and lymph nodes12 (FIG. 1). As these organs have important roles in haematopoiesis, leukaemia cell infiltration likely reflects the reactivation of haematopoietic programmes, as evidenced by the shared homing mechanisms of normal and malignant leukocytes. However, there are also unique patterns of metastasis to non-haematopoietic organs such as the skin and CNS that do not clearly recapitulate the behaviour of the leukaemia haematopoietic cell of origin. Interesting similarities and differences in the patterns of leukaemic and solid tumour metastasis to these organs exist, and some are highlighted below.
Spleen.
The spleen is an unusual organ for solid tumour metastasis but a frequent site of leukaemia metastasis (FIG. 3). It is unclear why circulating epithelial tumour cells lack the ability to navigate the splenic vasculature or to survive in its microenvironment, although theories based on the spleen’s unique blood flow dynamics and immune composition have been posited144. Multiple studies in the leukaemias, however, have produced insights into specific molecular mechanisms used by leukaemic cells to invade the spleen.
Fig. 3 |. The leukaemia splenic microenvironment.
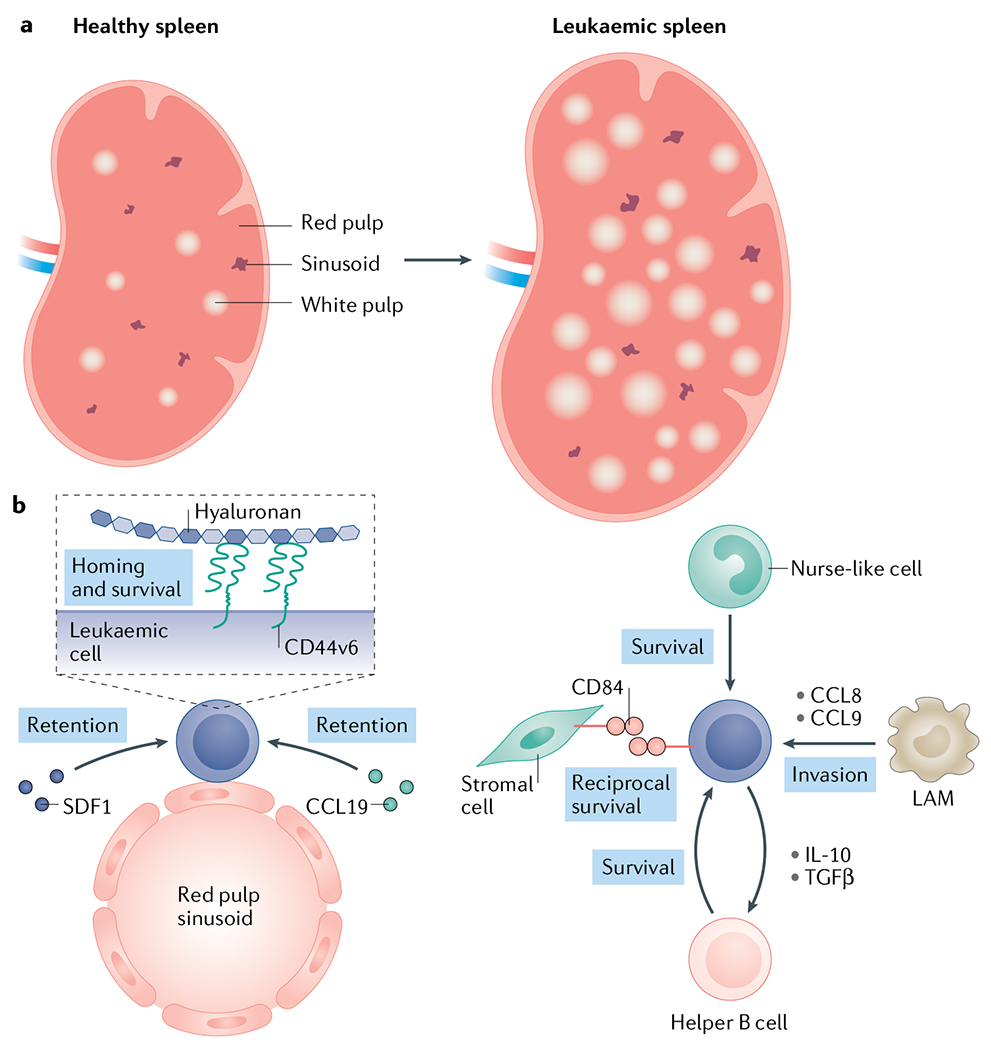
The spleen is a site of leukaemic progression primarily in chronic leukaemias and contains specific microenvironmental niches that can actively promote leukaemia cell homing and support disease expansion. a | As disease progresses, the white pulp expands, leading to a hypertrophic phenotype seen in many patients242,243. b | Leukaemia cells enter the spleen through large, fenestrated sinusoidal vessels in the red pulp. This is mediated by stromal cell-derived factor 1 (SDF1)–CXCR4 (REFS61,145) and CCL19–CCR7 (REF.69) expression in the splenic sinusoidal niche. Ly6C+ leukaemia-associated macrophages (LAMs) may also promote leukaemia cell migration to the spleen through the secretion of the CCL8 and CCL9 chemokines146. Leukaemia proliferation and survival in the spleen is supported by various stromal components such as hyaluronan147, monocyte-derived nurse-like cells167 and helper B cells148. For example, the reciprocal interaction between CD84 expressed by both leukaemia cells and stromal cells can promote the survival of either149. TGFβ; transforming growth factor-β.
As in the BM, leukaemic cells have been found to co-opt SDF1 and other haematopoietic niche chemotactic or adhesive factors to migrate to the spleen. SDF1–CXCR4 interactions have been revealed as key mediators of B-ALL splenic homing, and treatment with CXCR4 antagonist AMD3100 was shown to reduce splenic disease in mouse xenograft models61,145. Molecular mechanisms that specifically regulate spleen metastasis have also been elucidated. For example, a recent study using a syngeneic T-ALL mouse model demonstrated that Ly6C+ leukaemia-associated macrophages that secrete CCL8 and CCL9 chemokines promote leukaemia cell invasion and survival in the spleen146. Inhibiting the respective chemokine receptors, CCR1 and CCR2, decreased splenomegaly but had no effect on BM disease burden146. The chemokine receptor CCR7 and its ligand CCL19 may also have a specific role in T-ALL splenic metastasis. Using a NOTCH1-driven mouse model of T-ALL in which leukaemia cells have a high propensity to metastasize to the spleen compared with BM, investigators showed that leukaemia cells migrate more avidly towards splenocytes than BM cells in vitro, and that migration towards splenocytes, but not BM cells, could be inhibited by CCR7 or CCL19 blockade69. Lastly, in mouse studies using a syngeneic CLL-like cell line, hyaluronan-CD44 variant 6 (REF.147) interactions were shown to regulate splenic but not BM leukaemia homing147.
Further investigations in CLL using murine genetic models have shown that contact with the splenic microenvironment promotes disease growth and survival, perhaps contributing to the preponderance of splenic disease in these patients147–150 (FIG. 3). Interestingly, interactions between CLL cells and splenic stromal cells mediated by the CLL surface receptor, SLAM5 (also known as CD84), can increase the survival of both leukaemic cells and the involved stromal cells, indicating mutually beneficial crosstalk149. CLL cells can also secrete IL-10 and transforming growth factor-β (TGFβ), which promote the differentiation of splenic neutrophils into a helper B cell phenotype148,150. These ‘bystander’ leukocytes then support disease progression by providing pro-growth and anti-apoptotic stimuli148,150.
Lymph nodes.
In dramatic contrast to the spleen, LNs are frequent hosts to solid tumour metastases. Although both are lymphoid tissues, their immune cell, stromal and vascular architecture differ considerably, which may lead to a more permissive immunological environment for solid tumour growth in LNs than in the spleen151. Moreover, solid tumour metastases generally occur in regional LNs draining the primary tumour site, while leukaemic LN metastases are typically widespread throughout the body152,153. This clinical observation suggests that solid tumours depend on lymphatic routes for metastasis, while leukaemias appear capable of distant, haematogenous LN metastasis.
Lessons learnt from the study of leukocyte migration have informed our understanding of the molecular mechanisms of leukaemic LN metastasis. The haematogenous trafficking of normal immune cells in and out of LNs is regulated by specialized post-capillary venules termed high endothelial venules (HEVs)154. HEVs express a myriad of chemokines, adhesion molecules and matrix components that are co-opted by leukaemic cells during the multi-step process of invasion155. As in the BM, intravital imaging of the mouse inguinal LNs has demonstrated that CLL cells roll and crawl on the HEV wall7 prior to extravasation. L-selectin expression by CLL cells has been shown to crucially mediate these adhesive interactions in the HEV lumen7. Furthermore, the chemokines SDF1, CCL19 and CCL21, localized at HEVs, serve as potent chemoattractants for CLL cells entering the LNs70,156,157. CCR7, the receptor for CCL19 and CCL21, is highly expressed by CLL cells and is required for their migration towards these cytokines in vitro158. In mice, CCR7 antibody blockade decreased CLL LN invasion, supporting the importance of the CCR7–ligands axis in CLL LN metastasis159.
Once in the LN tissue, CLL cell surface VLA-4 and CD44 may form a docking complex for MMP9 (REF.160), which may facilitate CLL invasion by degrading the HEV basement membrane and/or LN ECM. Just as leukaemias corrupt BM HSPC niches to promote metastasis, CLL cells have been shown to usurp the germinal centre microenvironment to form a malignant ‘proliferation centre’ where they co-opt stromal cells, chemokines, cytokines and other niche factors to multiply161. CLL cells in the LNs also secrete CCL3 and CCL4, directing the recruitment of T cells and other leukocytes that alter the immune repertoire in this microenvironment to support CLL survival162,163. Uniquely, CLL cells can induce the differentiation of monocytes into large, adherent nurse-like cells that reside in the LNs and support CLL cell growth through the secretion of soluble factors such as SDF1, B cell-activating factor (BAFF) and a proliferation-inducing ligand (APRIL)164–167.
Although LN invasion is also common in ALL, there has not been intensive in vivo investigation into the molecular mechanism involved. However, as ALL cells express many of the molecular components observed in CLL such as VLA-4 (REFS53,54) and CCR7 (REF.69), it is likely that there are similar, shared mechanisms of LN metastasis between these diseases.
The central nervous system.
Although CNS metastasis is rare for most leukaemia subtypes, ALL is a highly neurotropic leukaemia that frequently involves the leptomeninges and CSF surrounding the brain and spinal cord, although rarely the brain parenchyma168. Metastasis to the CNS is found in approximately 5% of patients with ALL at diagnosis and occurs in 30–50% of patients over time, in the absence of prophylactic treatment18,19. Despite routine CNS-directed prophylactic chemotherapy, 5–15% of patients will develop CNS disease, and the prognosis for these patients is poor169,170. Although benign lymphocytes are known to ‘patrol’ the CNS, they are rarely present in the CSF of healthy individuals171,172. By contrast, the pronounced ability of ALL to invade and proliferate within the leptomeninges was shown to be an inherent property of ALL cells173. More recent work using high-throughput sequencing to analyse paired BM and CSF samples from patients with ALL at diagnosis and relapse supports the hypothesis that therapy can induce additional selective pressures that generate CNS-metastatic subclones174.
Three primary mechanisms of CNS invasion have been identified (FIG. 4). First, several studies suggest that leukaemia cells cross the blood–brain barrier to enter the CNS by promoting their own extravasation through VEGF signalling175,176. Various chemokines and chemokine receptors have also been implicated in this process177. In mouse models of oncogenic NOTCH1-driven T-ALL, the CCR7 receptor was shown to play an important role in CNS metastasis by attracting leukaemia cells to CCL19 or CCL21 chemokines expressed by the brain vascular endothelium68. More recent work has demonstrated that the T cell-specific kinase ZAP70 enhances CCR7-dependent migration to the CNS and that ZAP70 expression correlates with CNS metastasis in patients with T-ALL178.
Fig. 4 |. Routes of leukaemia central nervous system invasion.
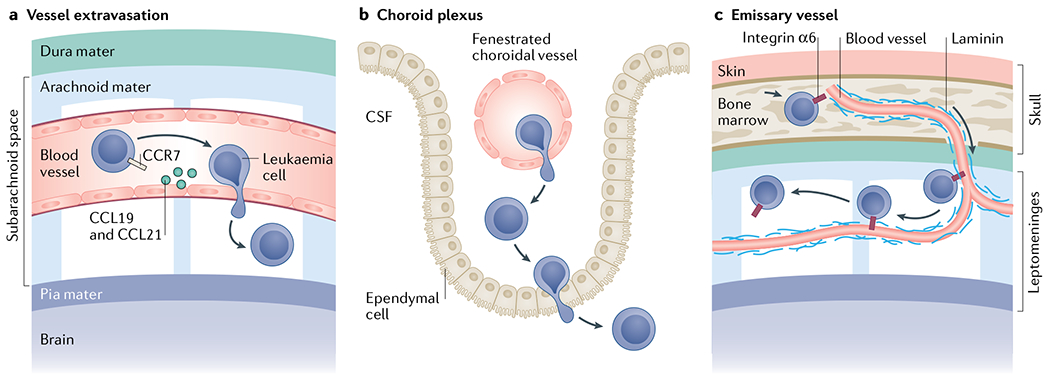
Leukaemia cell invasion of the central nervous system (CNS) mostly relates to acute leukaemias. Leukaemia cells have been shown to invade the CNS by breaching the blood–brain barrier of parenchymal or leptomeningeal vessels68 (part a), crossing the blood–cerebrospinal fluid (CSF) barrier of the choroid plexus179 (part b) or travelling along the abluminal surface of emissary blood vessels that exit the bone marrow through fenestrations in the bone and transition into leptomeningeal vessels (part c)56. The choroid plexus is a secretory tissue in the brain that is responsible for producing CSF. It contains fenestrated vessels and a monolayer of ependymal cells with tight junctions, ion pumps and transporters that filter out many cells, ions and proteins to produce CSF. The meninges comprise three membranous layers–the dura, the arachnoid and the pia mater–that form a continuous physical barrier surrounding the brain parenchyma and spinal cord. The dura mater is the outermost layer that is adjacent to the calvarium (skull) or vertebral bone. The subsequent arachnoid and pia mater are connected and form the leptomeninges. Between these two layers that form the leptomeninges is the subarachnoid space, which is filled with an acellular CSF that physically cushions the brain and spinal cord.
Other work has highlighted the choroid plexus as a specific site of entry for leukaemic cells into the CNS from the peripheral circulation179,180. The choroid plexus is a secretory tissue that produces CSF within the ventricular space in the brain. Recent work using an in vitro model of the blood–CSF barrier demonstrated that paediatric ALL cells are capable of transmigrating across a layer of choroid plexus epithelial cells179, suggesting the choroid plexus as a potential avenue for ALL CNS invasion. However, mouse xenograft studies have failed to find evidence of ALL cell diapedesis through the choroid plexus in vivo, suggesting that the role of the choroid plexus as an access point to the CNS requires further investigation56.
Lastly, our group has reported on a novel avenue for leukaemia cell entry in the CNS that allows ALL to bypass the blood–brain barrier56. We showed that B-ALL cells bind to extracellular matrix laminin surrounding specialized emissary blood vessels in mice. These emissary vessels passage from the skull and vertebral BM through cortical bone fenestrations, emerging as leptomeningeal vasculature in the adjacent CNS, and serve as a direct scaffold for leukaemic cells to migrate from BM to leptomeninges along their surface. Leukaemia cells do not extravasate through these vessels but migrate along their abluminal (outer) surface using integrin α6 to bind to the laminin-positive basement membrane. In a small patient series, leukaemia integrin α6 expression at diagnosis was associated with subsequent CNS relapse56. Although not specifically examining patients with CNS relapse, a study of clinical samples found elevated integrin α5 expression on ALL cells from the CSF at the time of diagnosis compared with BM blasts181, suggesting that additional integrins may have roles in ALL CNS metastasis.
Independently of their route of access, leukaemia cells that enter the CNS reside in a ‘sanctuary site’ of restricted immune cell and chemotherapy access, and we are only beginning to understand the mechanisms that govern this protective environment. In one study it was reported that activated IL-15-producing natural killer (NK) cells controlled peripheral disease in mice but were unable to penetrate the CNS, suggesting a model wherein leukaemic cells enter the CNS to evade NK-mediated cell death182. In another study, ALL cells in the meningeal microenvironment acquired chemoresistance through direct contact with meningeal cells183 and by upregulation of PBX1 (REF.184), a transcription factor known to regulate HSC quiescence.
Skin.
Although there are many forms of cancer that originate in the skin, leukaemias and lymphomas are among the handful of malignancies that infiltrate the skin as a metastatic site. In contrast to solid tumours, which typically demonstrate locoregional skin involvement suggesting local invasion and/or lymphatic spread, leukaemic skin metastases are usually widespread, consistent with blood-borne dissemination185–187. Leukaemic skin metastasis, referred to as leukaemia cutis, is hypothesized to share the mechanisms of benign leukocyte skin infiltration, which is mediated by P-selectin glycoprotein ligand 1 (PSGL1; also known as cutaneous lymphocyte antigen) adhesion to the E-selectin receptor on endothelial cells188. Recently, the molecular pathways responsible for leukaemic cell skin infiltration have been the subject of more intense scrutiny. Foundational work in benign lymphocytes showed that CCR4 deletion impaired CD4+ T cell homing to inflamed skin but not to inflamed peritoneum in a murine transgenic adoptive transfer model, implicating its specific role in cutaneous lymphocyte trafficking189. Although in vivo mechanistic work has not yet been conducted in leukaemia, the observation that CCR4 expression significantly correlates with skin metastasis in patients with T cell leukaemia/lymphoma suggests a skin homing mechanism shared with lymphocytes72.
Data have also shown a role for the chemokine receptor CXCR4 in myelomonocytic AML and lymphoblastic leukaemia invasion into SDF1+ skin niches61,190. Other chemokine receptors have also been identified as important mediators of AML skin infiltration, including CCR5, CXCR7 and CX3CR1 (REF.71). AML blasts isolated from the skin of patients with leukaemia cutis had higher expression of these chemokine receptors than blasts isolated from the BM and peripheral blood, suggesting that local expression of ligands CCL3, SDF1 and CX3CL1 in the skin mediates blast homing and retention.
Targeting metastasis vulnerabilities
The majority of adult leukaemias are not cured by standard therapies. Novel approaches to control disease metastasis could conceivably convert fatal diagnoses into chronic illnesses. In addition, strategies to physically and/or molecularly uncouple leukaemia cells from their protective tissue niches could improve responses to current regimens35,80,112,119,121,191. Below are some examples of the more advanced translational efforts to apply the emerging knowledge of leukaemia metastatic mechanisms to the clinic (TABLE 1).
Table 1 |.
Examples of therapeutics targeting leukaemia engagement with the metastatic niche
| Target | Type | Therapy | Condition | Clinical trial number | Phase | Status | Refs |
|---|---|---|---|---|---|---|---|
| CXCR4 | Small molecule | AMD3100 + MEC | R/R AML | NCT00512252 | I/II | Completed | 198 |
| CXCR4 | Small molecule | AMD3100 + G-CSF (filgastrim) | R/R AML | NCT00906945 | I/II | Completed | 199 |
| CXCR4 | Small molecule | BL-8040 + AraC | R/R AML | NCT01838395 | II | Completed | 227 |
| CXCR4 | Small molecule | Dociparstat + chemotherapy | AML | NCT04571645 | III | Not yet recruiting | 206,208,209 |
| CXCR4 | Small molecule | BL-8040 + nelarabine | R/R T-ALL | NCT02763384 | II | Recruiting | 228 |
| CCR7 | Monoclonal antibody | CAP-100 | R/R CLL | NCT04704323 | I | Not yet recruiting | 159,210 |
| CCR7 | Antibody–drug conjugate | JBH492 | CLL + NHL | NCT04240704 | I | Recruiting | 211 |
| CCR4 | Monoclonal antibody | Mogamulizumab | R/R ATLL | NCT04185220 | I | Active, not recruiting | 213 |
| G-CSF | Recombinant protein | Recombinant G-CSF (filgastrim) + chemotherapy | AML | #NTR230 | III | Completed | 203 |
| E-selectin | Small molecule | GMI-1271 + chemotherapy | R/R AML | NCT03616470 | III | Recruiting | 214 |
| BTK | Small molecule | Ibrutinib | CLL | NCT02048813 | III | Active | 58 |
| PI3Kδ | Small molecule | Idelalisib | CLL | NCT01539512 | III | Completed | 229 |
| PI3Kα/δ/γ | Small molecule | Copanlisib | R/R B-ALL | NCT04803123 | Window of opportunity | Active, not recruiting | 225 |
ALL, acute lymphocytic leukaemia/lymphoma; AML, acute myeloid leukaemia; AraC, cytarabine; ATLL, adult T cell leukaemia/lymphoma; B-ALL, B cell ALL; BTK, Bruton tyrosine kinase; CLL, chronic lymphocytic leukaemia; G-CSF, granulocyte colony-stimulating factor; MEC, mitoxantrone, etoposide and cytarabine; NHL, non-Hodgkin lymphoma; R/R, relapsed/refractory; T-ALL, T cell ALL.
Targeting chemokine receptors.
Molecular interventions to detach leukaemia cells from anti-apoptotic tissue niches, and in the most extreme sense, mobilize them into circulation, could render the disease more susceptible to systemic therapy. The SDF1–CXCR4 axis is the most studied in terms of leukaemia cell mobilization and chemosensitization. In AML in particular, high surface expression of CXCR4 correlates with poor prognosis59. AMD3100 is a specific CXCR4 antagonist that was first approved to mobilize HSPCs into the peripheral blood prior to collection for autologous BM transplant in non-Hodgkin lymphoma and multiple myeloma192,193. Years of work in mouse xenograft models of ALL and AML have demonstrated that blocking the SDF1–CXCR4 axis with AMD3100 mobilizes leukaemic cells into circulation, where they enter active phases of the cell cycle and are susceptible to chemotherapy9,34,194,195. Even in the absence of overt mobilization, inhibiting leukaemia cell interactions with SDF1 has been reported to be pro-apoptotic, further emphasizing the potential of CXCR4 blockade to treat leukaemias196,197.
Following an initial encouraging phase I/II clinical trial of AMD3100 combined with chemotherapy in patients with relapsed or refractory AML198, a phase I/II clinical trial investigated AMD3100 combined with an additional mobilizing agent, granulocyte colony-stimulating factor (G-CSF) in this population199. Although the precise molecular mechanisms regulating G-CSF-mediated mobilization remain unclear, stromal cell production of G-CSF has been shown to decrease SDF1 levels in the BM niche and induce proteases that cleave VCAM1 (REFS200,201). Some studies have reported a desirable effect of G-CSF priming to enhance chemotherapy in standard-risk patients with AML202,203, while other trials have failed to show a clinical benefit204,205. The dual use of G-CSF and AMD3100 to mobilize leukaemic cells during salvage chemotherapy of patients with relapsed or refractory AML did not, however, improve treatment efficacy, potentially due to the induction of counteractive pro-survival signalling effects by G-CSF199. Despite this initial discouragement, more potent approaches to inhibit leukaemia interactions with SDF1 and thus promote leukaemia cell apoptosis remain in development, including monoclonal antibodies, SDF1 protein mimetics, peptide inhibitors and new small-molecule inhibitors of CXCR4 (REFS10,196,206–208). For example, in a recent phase II clinical trial, the small-molecule CXCR4 inhibitor, dociparstat, was shown to increase event-free survival in combination with chemotherapy in elderly patients with AML208, paving the way for a phase III clinical trial (NCT04571645)209.
In addition to targeting of CXCR4, therapeutic CCR7 and CCR4 chemokine receptor antagonists have advanced to the clinic. Anti-CCR7 monoclonal antibodies were reported to decrease LN invasion in mouse models of CLL as well as inducing CLL antibody-dependent cytotoxicity with minimal effects on normal haematopoietic cells158. This led to the clinical development of the anti-CCR7 monoclonal antibody, CAP-100, which is currently being tested in phase I trials in patients with relapsed or refractory CLL (NCT04704323)210. An antibody–drug conjugate combining anti-CCR7 with the microtubule inhibitor JBH492 is also being tested in patients with CLL in a phase I trial (NCT04240704)211.
As discussed above, CCR4 is often highly expressed by T cell leukaemias and lymphomas and is thought to have a role in skin metastasis72,212. As such, a CCR4 monoclonal antibody is currently being tested in a phase I study in adult T cell leukaemia/lymphoma (ATLL) and cutaneous T cell lymphomas (NCT04185220)213.
Targeting adhesion molecules.
Other promising approaches to target the metastatic niche involve disrupting alternative adhesion molecules, including the selectins and integrins, that contribute to leukaemia blast migration to and retention at metastatic sites.
Two specific E-selectin small-molecule inhibitors that have been studied in leukaemia are GMI-1271 (REFS141,214) and GMI-1359 (REF.215) (a dual E-selectin and CXCR4 inhibitor). In syngeneic murine models, treatment with GMI-1271 mobilized leukaemia cells into circulation and suppressed pro-survival signalling pathways, sensitizing AML blasts to chemotherapy141. The results from a phase I/II clinical trial testing GMI-1271 in combination wth standard chemotherapy in patients with relapsed or refractory AML were recently presented at a conference and reported promising survival outcomes214. This prompted the initiation of an ongoing phase III trial testing GMI-1271 in combination with chemotherapy to evaluate overall survival as a primary end point (NCT03616470)216. In CML, a preclinical study has demonstrated that GMI-1271 administration decreases CML cell contact with the protective BM endothelium and prolongs survival in mice when combined with imatinib, suggesting that investigation of this compound in patients with CML is warranted217.
The dual E-selectin and CXCR4 inhibitor GMI-1359 has also been shown to have potent anti-leukaemia activity in mouse xenograft models of AML215. The ability of GMI-1359 to mobilize cancer cells from the BM and sensitize them to therapy may also be shared across solid tumours, as suggested by preclinical studies in breast cancer218. A phase Ib proof-of-concept clinical trial to test the safety and pharmacodynamics of GMI-1359 in patients with bone-metastatic breast cancer is ongoing219.
Another category of adhesion molecule that has been targeted in leukaemia is the integrin family. In mouse xenograft models of acute leukaemia, administration of the VLA-4 monoclonal antibody natalizumab has been shown to mobilize leukaemic cells from the BM and sensitize them to chemotherapy220. Despite encouraging preclinical studies, clinical trials targeting integrin–ligand interactions have not been as successful221. A more promising approach, however, may be to target integrin receptor expression or downstream effectors indirectly. Ex vivo analysis of peripheral blood samples from patients with CLL showed that treatment with BTK inhibitor ibrutinib abrogates VLA-4-dependent adhesion of CLL cells to fibronectin, suggesting an ability to modulate crosstalk between BTK–integrin signalling pathways222.
A promising target for direct or indirect therapeutic intervention is integrin α6. Integrin α6 is highly expressed in ALL and is associated with CNS metastatic relapse and MRD within the BM56,223. Our lab reported that targeting integrin α6 directly using monoclonal antibodies, or indirectly by decreasing its expression through PI3K inhibition, reduced CNS metastasis and prolonged survival in mouse ALL xenograft models56. Moreover, a recent ALL xenograft study showed that integrin α6 blockade sensitized cells to chemotherapy by blocking SRC family kinase activation of anti-apoptotic signalling224. A window-of-opportunity clinical trial to assess the effect of PI3K inhibition with copanlisib on pharmacodynamic markers of CNS metastasis is currently in development225.
Conclusions
The data reviewed here illustrate the manifold mechanisms of tissue migration employed by leukaemia cells. Careful examination of the anatomy and pathophysiology of leukaemias and of the molecular pathways involved in their spread supports the concept that leukaemia dissemination, as in the solid tumours, is a metastatic process. Indeed, many discoveries of the mechanisms by which tumour cells breach the basement membrane using integrins, selectins and MMPs, then extravasate into the stroma and navigate tissues with amoeboid-like actin dynamics, were first extensively described in normal and malignant leukocytes. Although the multiple basic science discoveries in this space over the past decade have not yet translated to novel, targeted therapies for leukaemias, we are optimistic that, similar to the work culminating in successful cancer immunotherapies, perseverance in this area will reap clinical rewards226. Finally, it is likely that the solid and liquid tumour fields will continue to benefit each other as continued research unveils new mechanisms of metastasis that may be shared between these two disease entities.
Acknowledgements
The authors thank A. Chenn and P. Islam for their discussions and careful review of the manuscript. The authors apologize to all colleagues whose work could not be discussed owing to space limitations.
Competing interests
The authors have received research funding from Bayer Pharmaceutical and Glycomimetics, Inc.
Glossary
- Choroid plexus
A secretory tissue in the brain that produces cerebrospinal fluid.
- Diapedesis
The process of extravasating out of a vessel into the surrounding stroma.
- Endosteal niche
The niche in the bone marrow adjacent to the bone lining (endosteum).
- Germinal centre
Site within the spleen and lymph node where B cells proliferate and differentiate.
- High endothelial venules (HEVs)
Specialized post-capillary venules in the lymph node that allow for the trafficking of immune cells in and out of this lymphoid organ.
- Intravital microscopy
High-resolution imaging of a living organism to study biological events at the cellular level.
- Leptomeninges
The inner two meningeal layers that surround the brain and spinal cord and contain cerebrospinal fluid.
- Minimal residual disease (MRD)
A subclinical amount of disease remaining after therapy that can fuel relapse.
- Nurse-like cells
Monocyte-derived cells that support the survival and growth of chronic lymphocytic leukaemia.
- Sinusoidal vessels
Large vessels found in the bone marrow, spleen, lymph node and liver that contain fenestrations allowing the trafficking of cells across the vascular endothelium.
Footnotes
Peer review information
Nature Reviews Cancer thanks D. Bonnet and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
References
- 1.Lapidot T et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 367, 645–648 (1994). [DOI] [PubMed] [Google Scholar]
- 2.Miles LA et al. Single-cell mutation analysis of clonal evolution in myeloid malignancies. Nature 587, 477–482 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vetrie D, Helgason GV & Copland M The leukaemia stem cell: similarities, differences and clinical prospects in CML and AML. Nat. Rev. Cancer 20, 158–173 (2020). [DOI] [PubMed] [Google Scholar]
- 4.Jin L, Hope KJ, Zhai Q, Smadja-Joffe F & Dick JE Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nat. Med 12, 1167–1174 (2006). [DOI] [PubMed] [Google Scholar]
- 5.Krause DS, Lazarides K, Lewis JB, Von Andrian UH & Van Etten RA Selectins and their ligands are required for homing and engraftment of BCR-ABL1+ leukemic stem cells in the bone marrow niche. Blood 123, 1361–1371 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Krause DS, Lazarides K, von Andrian UH & Van Etten RA Requirement for CD44 in homing and engraftment of BCR-ABL–expressing leukemic stem cells. Nat. Med 12, 1175–1180 (2006). [DOI] [PubMed] [Google Scholar]
- 7.Lafouresse F et al. L-selectin controls trafficking of chronic lymphocytic leukemia cells in lymph node high endothelial venules in vivo. Blood 126, 1336–1345 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Messinger Y, Chelstrom L, Gunther R & Uckun FM Selective homing of human leukemic B-cell precursors to specific lymphohematopoietic microenvironments in SCID mice: a role for the beta 1 integrin family surface adhesion molecules VLA-4 and VLA-5. Leuk. Lymphoma 23, 61–69 (1996). [DOI] [PubMed] [Google Scholar]
- 9.Sipkins DA et al. In vivo imaging of specialized bone marrow endothelial microdomains for tumour engraftment. Nature 435, 969–973 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tavor S et al. CXCR4 regulates migration and development of human acute myelogenous leukemia stem cells in transplanted NOD/SCID Mice. Cancer Res. 64, 2817–2824 (2004). [DOI] [PubMed] [Google Scholar]
- 11.Barcos M et al. An autopsy study of 1206 acute and chronic leukemias (1958 to 1982). Cancer 60, 827–837 (1987). [DOI] [PubMed] [Google Scholar]
- 12.Viadana E, Bross ID & Pickren JW An autopsy study of the metastatic patterns of human leukemias. Oncology 35, 87–96(1978). [DOI] [PubMed] [Google Scholar]
- 13.Döhner H et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 129, 424–447 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Inaba H, Greaves M & Mullighan CG Acute lymphoblastic leukaemia. Lancet 381, 1943–1955 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arber DA et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 127, 2391–2405 (2016). [DOI] [PubMed] [Google Scholar]
- 16.Gharbaran R, Park J, Kim C, Goy A & Suh KS Circulating tumor cells in Hodgkin’s lymphoma — a review of the spread of HL tumor cells or their putative precursors by lymphatic and hematogenous means, and their prognostic significance. Crit. Rev. Oncol. Hematol 89, 404–417 (2014). [DOI] [PubMed] [Google Scholar]
- 17.Granell M et al. Prognostic impact of circulating plasma cells in patients with multiple myeloma: implications for plasma cell leukemia definition. Haematologica 102, 1099–1104 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gokbuget N & Hoelzer D Meningeosis leukaemica in adult acute lymphoblastic leukaemia. J. Neurooncol 38, 167–180 (1998). [DOI] [PubMed] [Google Scholar]
- 19.Cancela CS, Murao M, Viana MB & de Oliveira BM Incidence and risk factors for central nervous system relapse in children and adolescents with acute lymphoblastic leukemia. Rev. Bras. Hematol. Hemoter 34, 436–441 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Valastyan S & Weinberg RA Tumor metastasis: molecular insights and evolving paradigms. Cell 147, 275–292 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang Y, Gao S, Xia J & Liu F Hematopoietic hierarchy – an updated roadmap. Trends Cell Biol. 28, 976–986 (2018). [DOI] [PubMed] [Google Scholar]
- 22.Pinho S & Frenette PS Haematopoietic stem cell activity and interactions with the niche. Nat. Rev. Mol. Cell Biol 20, 303–320 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morita K et al. Clonal evolution of acute myeloid leukemia revealed by high-throughput single-cell genomics. Nat. Commun 11, 5327 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang B et al. Heterogeneity of leukemia-initiating capacity of chronic myelogenous leukemia stem cells. J. Clin. Invest 126, 975–991 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Batlle E & Clevers H Cancer stem cells revisited. Nat. Med 23, 1124–1134 (2017). [DOI] [PubMed] [Google Scholar]
- 26.Visvader JE & Lindeman GJ Cancer stem cells: current status and evolving complexities. Stem Cell 10, 717–728 (2012). [DOI] [PubMed] [Google Scholar]
- 27.Ye H et al. Leukemic stem cells evade chemotherapy by metabolic adaptation to an adipose tissue niche. Cell Stem Cell 19, 23–37 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Holyoake TL & Vetrie D The chronic myeloid leukemia stem cell: stemming the tide of persistence. Blood 129, 1595–1606 (2017). [DOI] [PubMed] [Google Scholar]
- 29.Thomas D & Majeti R Biology and relevance of human acute myeloid leukemia stem cells. Blood 129, 1577–1585 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.de Haas V et al. Quantification of minimal residual disease in children with oligoclonal B-precursor acute lymphoblastic leukemia indicates that the clones that grow out during relapse already have the slowest rate of reduction during induction therapy. Leukemia 15, 134–140 (2001). [DOI] [PubMed] [Google Scholar]
- 31.Ferrando AA & López-Otín C Clonal evolution in leukemia. Nat. Med 23, 1135–1145 (2017). [DOI] [PubMed] [Google Scholar]
- 32.Li A-H, Rosenquist R, Forestier E, Lindh J & Roos G Detailed clonality analysis of relapsing precursor B acute lymphoblastic leukemia: implications for minimal residual disease detection. Leukemia Res. 25, 1033–1045 (2001). [DOI] [PubMed] [Google Scholar]
- 33.Puente XS et al. Non-coding recurrent mutations in chronic lymphocytic leukaemia. Nature 526, 519–524 (2015). [DOI] [PubMed] [Google Scholar]
- 34.Nervi B et al. Chemosensitization of acute myeloid leukemia (AML) following mobilization by the CXCR4 antagonist AMD3100. Blood 113, 6206–6214 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Boyerinas B et al. Adhesion to osteopontin in the bone marrow niche regulates lymphoblastic leukemia cell dormancy. Blood 121, 4821–4831 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Crane GM, Jeffery E & Morrison SJ Adult haematopoietic stem cell niches. Nat. Rev. Immunol 17, 573–590 (2017). [DOI] [PubMed] [Google Scholar]
- 37.Ghielmini M et al. Prolonged treatment with rituximab in patients with follicular lymphoma significantly increases event-free survival and response duration compared with the standard weekly x 4 schedule. Blood 103, 4416–4423 (2004). [DOI] [PubMed] [Google Scholar]
- 38.Maloney DG, Smith B & Rose A Rituximab: mechanism of action and resistance. Semin. Oncol 29, 2–9 (2002). [DOI] [PubMed] [Google Scholar]
- 39.Gökbuget N et al. Adult patients with acute lymphoblastic leukemia and molecular failure display a poor prognosis and are candidates for stem cell transplantation and targeted therapies. Blood 120, 1868–1876 (2012). [DOI] [PubMed] [Google Scholar]
- 40.Kamps WA et al. Intensive treatment of children with acute lymphoblastic leukemia according to ALL-BFM-86 without cranial radiotherapy: results of Dutch Childhood Leukemia Study Group Protocol ALL-7 (1988–1991). Blood 94, 1226–1236 (1999). [PubMed] [Google Scholar]
- 41.Miniero R et al. Relapse after first cessation of therapy in childhood acute lymphoblastic leukemia: a 10-year follow-up study. Italian Association of Pediatric Hematology-Oncology (AIEOP). Med. Pediatr. Oncol 24, 71–76 (1995). [DOI] [PubMed] [Google Scholar]
- 42.McEver RP Selectins: initiators of leucocyte adhesion and signalling at the vascular wall. Cardiovasc. Res 107, 331–339 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Spertini C et al. Acute myeloid and lymphoblastic leukemia cell interactions with endothelial selectins: critical role of PSGL-1, CD44 and CD43. Cancers (Basel) 11, 1253 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Myung JH et al. Direct measurements on CD24-mediated rolling of human breast cancer MCF-7 cells on E-selectin. Anal. Chem 83, 1078–1083 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Renkonen J, Paavonen T & Renkonen R Endothelial and epithelial expression of sialyl Lewisx and sialyl Lewisa in lesions of breast carcinoma. Int. J. Cancer 74, 296–300 (1997). [DOI] [PubMed] [Google Scholar]
- 46.Shirure VS, Henson KA, Schnaar RL, Nimrichter L & Burdick MM Gangliosides expressed on breast cancer cells are E-selectin ligands. Biochem. Biophys. Res. Commun 406, 423–429 (2011). [DOI] [PubMed] [Google Scholar]
- 47.Natoni A, Macauley MS & O’Dwyer ME Targeting selectins and their ligands in cancer. Front. Oncol 6, 1–12 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chien SS et al. E-selectin ligand expression by leukemic blasts is associated with prognosis in patients with AML. Blood 132, 1513 (2018). [Google Scholar]
- 49.Stucki A et al. Endothelial cell activation by myeloblasts: molecular mechanisms of leukostasis and leukemic cell dissemination. Blood 97, 2121–2129 (2001). [DOI] [PubMed] [Google Scholar]
- 50.Dagdemir A, Ertem U, Duru F & Kirazli S Soluble L-selectin increases in the cerebrospinal fluid prior to meningeal involvement in children with acute lymphoblastic leukemia. Leuk. Lymphoma 28, 391–398 (1998). [DOI] [PubMed] [Google Scholar]
- 51.Spertini O et al. High levels of the shed form of L-selectin are present in patients with acute leukemia and inhibit blast cell adhesion to activated endothelium. Blood 84, 1249–1256 (1994). [PubMed] [Google Scholar]
- 52.Hamidi H & Ivaska J Every step of the way: integrins in cancer progression and metastasis. Nat. Rev. Cancer 18, 533–548 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Blenc AM, Chiagev A, Sklar L & Larson RS VLA-4 affinity correlates with peripheral blood white cell count and DNA content in patients with precursor B-ALL. Leukemia 17, 641–643 (2003). [DOI] [PubMed] [Google Scholar]
- 54.Filshie R, Gottlieb D & Bradstock K VLA-4 is involved in the engraftment of the human pre-B acute lymphoblastic leukaemia cell line NALM-6 in SCID mice. Br. J. Haematol 102, 1292–1300 (1998). [DOI] [PubMed] [Google Scholar]
- 55.Hu Z & Slayton WB Integrin VLA-5 and FAK are good targets to improve treatment response in the philadelphia chromosome positive acute lymphoblastic leukemia. Front. Oncol 4, 1–10 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yao H et al. Leukaemia hijacks a neural mechanism to invade the central nervous system. Nature 560, 55–60 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tissino E et al. Functional and clinical relevance of VLA-4 (CD49d/CD29) in ibrutinib-treated chronic lymphocytic leukemia. J. Exp. Med 215, 681–697 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shanafelt TD et al. Ibrutinib–rituximab or chemoimmunotherapy for chronic lymphocytic leukemia. N. Engl. J. Med 381, 432–443 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Konoplev S et al. Overexpression of CXCR4 predicts adverse overall and event-free survival in patients with unmutatedFLT3 acute myeloid leukemia with normal karyotype. Cancer 109, 1152–1156 (2007). [DOI] [PubMed] [Google Scholar]
- 60.van den Berk LCJ et al. Disturbed CXCR4/CXCL12 axis in paediatric precursor B-cell acute lymphoblastic leukaemia. Br. J. Haematol 166, 240–249 (2014). [DOI] [PubMed] [Google Scholar]
- 61.Crazzolara R et al. High expression of the chemokine receptor CXCR4 predicts extramedullary organ infiltration in childhood acute lymphoblastic leukaemia. Br. J. Haematol 115, 545–553 (2001). [DOI] [PubMed] [Google Scholar]
- 62.De Lourdes Perim A, Amarante MK, Guembarovski RL, De Oliveira CEC & Watanabe MAE CXCL12/CXCR4 axis in the pathogenesis of acute lymphoblastic leukemia (ALL): a possible therapeutic target. Cell Mol. Life Sci 72, 1715–1723 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jin L et al. CXCR4 up-regulation by imatinib induces chronic myelogenous leukemia (CML) cell migration to bone marrow stroma and promotes survival of quiescent CML cells. Mol. Cancer Therapeutics 7, 48–58 (2008). [DOI] [PubMed] [Google Scholar]
- 64.Fiegl M et al. CXCR4 expression and biologic activity in acute myeloid leukemia are dependent on oxygen partial pressure. Blood 113, 1504–1512 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fei F et al. Development of resistance to dasatinib in Bcr/Abl-positive acute lymphoblastic leukemia. Leukemia 24, 813–820 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sison EAR, McIntyre E, Magoon D & Brown P Dynamic chemotherapy-induced upregulation of CXCR4 expression: a mechanism of therapeutic resistance in pediatric AML. Mol. Cancer Res 11, 1004–1016 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Burger JA Chemokines and chemokine receptors in chronic lymphocytic leukemia (CLL): from understanding the basics towards therapeutic targeting. Semin. Cancer Biol 20, 424–430 (2010). [DOI] [PubMed] [Google Scholar]
- 68.Buonamici S et al. CCR7 signalling as an essential regulator of CNS infiltration in T-cell leukaemia. Nature 459, 1000–1004 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ma S et al. Notch1-induced T cell leukemia can be potentiated by microenvironmental cues in the spleen. J. Hematol. Oncol. 7, 71 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Till KJ, Lin K, Zuzel M & Cawley JC The chemokine receptor CCR7 and alpha4 integrin are important for migration of chronic lymphocytic leukemia cells into lymph nodes. Blood 99, 2977–2984 (2002). [DOI] [PubMed] [Google Scholar]
- 71.Faaij CMJM et al. Chemokine/chemokine receptor interactions in extramedullary leukaemia of the skin in childhood AML: differential roles for CCR2, CCR5, CXCR4 and CXCR7. Pediatric Blood Cancer 55, 344–348 (2010). [DOI] [PubMed] [Google Scholar]
- 72.Ishida T et al. Clinical significance of CCR4 expression in adult T-cell leukemia/lymphoma: its close association with skin involvement and unfavorable outcome. Clin. Cancer Res 9, 3625–3634 (2003). [PubMed] [Google Scholar]
- 73.Apte RS, Chen DS & Ferrara N VEGF in signaling and disease: beyond discovery and development. Cell 176, 1248–1264 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Perez-Atayde AR et al. Spectrum of tumor angiogenesis in the bone marrow of children with acute lymphoblastic leukemia. Am. J. Pathol 150, 815–821 (1997). [PMC free article] [PubMed] [Google Scholar]
- 75.Hussong JW, Rodgers GM & Shami PJ Evidence of increased angiogenesis in patients with acute myeloid leukemia. Blood 95, 309–313 (2000). [PubMed] [Google Scholar]
- 76.Nombela-Arrieta C & Manz MG Quantification and three-dimensional microanatomical organization of the bone marrow. Blood Adv. 1, 407–416 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Van Der Velde-Zimmermann D et al. Fibronectin distribution in human bone marrow stroma: matrix assembly and tumor cell adhesion via α5β1 integrin. Exp. Cell Res 230, 111–120 (1997). [DOI] [PubMed] [Google Scholar]
- 78.Reilly J, Nash J, Mackie M & McVerry B Distribution of laminin and fibronectin in normal and pathological lymphoid tissue. J. Clin. Pathol 38, 849–854 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Voermans C, van Heese WPM, de Jong I, Gerritsen WR & van der Schoot CE Migratory behavior of leukemic cells from acute myeloid leukemia patients. Leukemia 16, 650–657 (2002). [DOI] [PubMed] [Google Scholar]
- 80.Matsunaga T et al. Interaction between leukemic-cell VLA-4 and stromal fibronectin is a decisive factor for minimal residual disease of acute myelogenous leukemia. Nat. Med 9, 1158–1165 (2003). [DOI] [PubMed] [Google Scholar]
- 81.Ridley AJ Life at the leading edge. Cell 145, 1012–1022 (2011). [DOI] [PubMed] [Google Scholar]
- 82.Daubon T et al. Differential motility of p190bcr-abl- and p210 bcr-abl-expressing cells: respective roles of Vav and Bcr-Abl GEFs. Oncogene 27, 2673–2685 (2008). [DOI] [PubMed] [Google Scholar]
- 83.Rochelle T et al. p210-bcr-abl induces amoeboid motility by recruiting ADF/destrin through RhoA/ROCK1. FASEB J. 27, 123–134 (2013). [DOI] [PubMed] [Google Scholar]
- 84.Velázquez-Avila M et al. High cortactin expression in B-cell acute lymphoblastic leukemia is associated with increased transendothelial migration and bone marrow relapse. Leukemia 33, 1337–1348 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Friedl P & Wolf K Plasticity of cell migration: a multiscale tuning model. J. Cell Biol 188, 11–19 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pankova K, Rosel D, Novotny M & Brabek J The molecular mechanisms of transition between mesenchymal and amoeboid invasiveness in tumor cells. Cell Mol. Life Sci 67, 63–71 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Daubon T, Rochelle T, Bourmeyster N & Génot E Invadopodia and rolling-type motility are specific features of highly invasive p190bcr-abl leukemic cells. Eur. J. Cell Biol 91, 978–987 (2012). [DOI] [PubMed] [Google Scholar]
- 88.Cougoule C et al. Three-dimensional migration of macrophages requires Hck for podosome organization and extracellular matrix proteolysis. Blood 115, 1444–1452 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Poincloux R, Cougoule C, Daubon T, Maridonneau-Parini I & Le Cabec V Tyrosine-phosphorylated STAT5 accumulates on podosomes in Hck-transformed fibroblasts and chronic myeloid leukemia cells. J. Cell. Physiol 213, 212–220 (2007). [DOI] [PubMed] [Google Scholar]
- 90.Redondo-Muñoz J et al. MMP-9 in B-cell chronic lymphocytic leukemia is up-regulated by α4β1 integrin or CXCR4 engagement via distinct signaling pathways, localizes to podosomes, and is involved in cell invasion and migration. Blood 108, 3143–3151 (2006). [DOI] [PubMed] [Google Scholar]
- 91.Kamiguti AS et al. The role of matrix metalloproteinase 9 in the pathogenesis of chronic lymphocytic leukaemia. Br. J. Haematol 125, 128–140 (2004). [DOI] [PubMed] [Google Scholar]
- 92.Sawicki G, Matsuzaki A & Janowska-Wieczorek A Expression of the active form of MMP-2 on the surface of leukemic cells accounts for their in vitro invasion. J. Cancer Res. Clin. Oncol 124, 245–252 (1998). [DOI] [PubMed] [Google Scholar]
- 93.Spiegel A et al. Unique SDF-1 – induced activation of human precursor-B ALL cells as a result of altered CXCR4 expression and signaling. Blood 103, 2900–2907 (2004). [DOI] [PubMed] [Google Scholar]
- 94.Masuda M et al. CADM1 interacts with Tiam1 and promotes invasive phenotype of human T-cell leukemia virus type I-transformed cells and adult T-cell leukemia cells. J. Biol. Chem 285, 15511–15522 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cárcamo C et al. Galectin-8 binds specific β1 integrins and induces polarized spreading highlighted by asymmetric lamellipodia in Jurkat T cells. Exp. Cell Res 312, 374–386 (2006). [DOI] [PubMed] [Google Scholar]
- 96.Navarro SM et al. Musculoskeletal imaging findings of hematologic malignancies. RadioGraphics 37, 881–900 (2017). [DOI] [PubMed] [Google Scholar]
- 97.Detterbeck FC et al. The IASLC lung cancer staging project: background data and proposed criteria to distinguish separate primary lung cancers from metastatic foci in patients with two lung tumors in the forthcoming eighth edition of the TNM classification for lung cancer. J. Thorac. Oncol 5, 651–665 (2016). [DOI] [PubMed] [Google Scholar]
- 98.Lapidot T & Kollet O The essential roles of the chemokine SDF-1 and its receptor CXCR4 in human stem cell homing and repopulation of transplanted immune-deficient NOD/SCID and NOD/SCID/B2m(null) mice. Leukemia 16, 1992–2003 (2002). [DOI] [PubMed] [Google Scholar]
- 99.Menu E et al. The involvement of stromal derived factor 1alpha in homing and progression of multiple myeloma in the 5TMM model. Haematologica 91, 605–612 (2006). [PubMed] [Google Scholar]
- 100.Passaro D et al. CXCR4 is required for leukemia-initiating cell activity in T cell acute lymphoblastic leukemia. Cancer Cell 27, 769–779 (2015). [DOI] [PubMed] [Google Scholar]
- 101.Pitt LA et al. CXCL12-producing vascular endothelial niches control acute T cell leukemia maintenance. Cancer Cell 27, 755–768 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Nourshargh S & Alon R Leukocyte migration into inflamed tissues. Immunity 41, 694–707 (2014). [DOI] [PubMed] [Google Scholar]
- 103.Vestweber D How leukocytes cross the vascular endothelium. Nat. Rev. Immunol 15, 692–704 (2015). [DOI] [PubMed] [Google Scholar]
- 104.Wicklein D et al. E- and P-selectins are essential for repopulation of chronic myelogenous and chronic eosinophilic leukemias in a scid mouse xenograft model. PLoS ONE 8, e70139 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sackstein R et al. Ex vivo glycan engineering of CD44 programs human multipotent mesenchymal stromal cell trafficking to bone. Nat. Med 14, 181–187 (2008). [DOI] [PubMed] [Google Scholar]
- 106.McDonald B & Kubes P Interactions between CD44 and hyaluronan in leukocyte trafficking. Front. Immunol 6, 68 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Gutjahr JC, Greil R & Hartmann TN The role of CD44 in the pathophysiology of chronic lymphocytic leukemia. Front. Immunol 6, 177 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Chen C, Zhao S, Karnad A & Freeman JW The biology and role of CD44 in cancer progression: therapeutic implications. J. Hematol. Oncol 11, 64 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Senbanjo LT & Chellaiah MA CD44: a multifunctional cell surface adhesion receptor is a regulator of progression and metastasis of cancer cells. Front. Cell Dev. Biol 5, 18 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Bajaj J et al. CD98-mediated adhesive signaling enables the establishment and propagation of acute myelogenous leukemia. Cancer Cell 30, 792–805 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Méndez-Ferrer S et al. Bone marrow niches in haematological malignancies. Nat. Rev. Cancer 20, 285–298 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Jacamo R et al. Reciprocal leukemia-stroma VCAM-1/VLA-4-dependent activation of NF-κB mediates chemoresistance. Blood 123, 2691–2702 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Dias S et al. Autocrine stimulation of VEGFR-2 activates human leukemic cell growth and migration. J. Clin. Invest 106, 511–521 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Fiedler W et al. Vascular endothelial growth factor, a possible paracrine growth factor in human acute myeloid leukemia. Blood 89, 1870–1875 (1997). [PubMed] [Google Scholar]
- 115.Liersch R et al. Expression of VEGF-C and its receptor VEGFR-3 in the bone marrow of patients with acute myeloid leukaemia. Leuk. Res 32, 954–961 (2008). [DOI] [PubMed] [Google Scholar]
- 116.Padró T et al. Overexpression of vascular endothelial growth factor (VEGF) and its cellular receptor KDR (VEGFR-2) in the bone marrow of patients with acute myeloid leukemia. Leukemia 16, 1302–1310 (2002). [DOI] [PubMed] [Google Scholar]
- 117.Witkowski MT et al. Extensive remodeling of the immune microenvironment in B cell acute lymphoblastic leukemia. Cancer Cell 37, 867–882.e812 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ishikawa F et al. Chemotherapy-resistant human AML stem cells home to and engraft within the bone-marrow endosteal region. Nat. Biotechnol 25, 1315–1321 (2007). [DOI] [PubMed] [Google Scholar]
- 119.Ebinger S et al. Characterization of rare, dormant, and therapy-resistant cells in acute lymphoblastic leukemia. Cancer Cell 30, 849–862 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Stier S et al. Osteopontin is a hematopoietic stem cell niche component that negatively regulates stem cell pool size. J. Exp. Med 201, 1781–1791 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Cahu X et al. Bone marrow sites differently imprint dormancy and chemoresistance to T-cell acute lymphoblastic leukemia. Blood Adv. 1, 1760–1772 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Naveiras O et al. Bone-marrow adipocytes as negative regulators of the haematopoietic microenvironment. Nature 460, 259–263 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Colmone A et al. Leukemic cells create bone marrow niches that disrupt the behavior of normal hematopoietic progenitor cells. Science 322, 1861–1865 (2008). [DOI] [PubMed] [Google Scholar]
- 124.Carey A et al. Identification of interleukin-1 by functional screening as a key mediator of cellular expansion and disease progression in acute myeloid leukemia. Cell Rep. 18, 3204–3218 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Welner RS et al. Treatment of chronic myelogenous leukemia by blocking cytokine alterations found in normal stem and progenitor cells. Cancer Cell 27, 671–681 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Zhang B et al. Altered microenvironmental regulation of leukemic and normal stem cells in chronic myelogenous leukemia. Cancer Cell 21, 577–592 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Hanoun M et al. Acute myelogenous leukemia-induced sympathetic neuropathy promotes malignancy in an altered hematopoietic stem cell niche. Cell Stem Cell 15, 365–375 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Duarte D, Hawkins ED, Akinduro O, Purton LE & Carlin LM Inhibition of endosteal vascular niche remodeling article inhibition of endosteal vascular niche remodeling rescues hematopoietic stem cell loss in AML. Stem Cell 22, 64–77.e66 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Passaro D et al. Increased vascular permeability in the bone marrow microenvironment contributes to disease progression and drug response in acute myeloid. Cancer Cell 32, 324–341 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kebelmann-Betzing C et al. Characterization of cytokine, growth factor receptor, costimulatory and adhesion molecule expression patterns of bone marrow blasts in relapsed childhood B cell precursor all. Cytokine 13, 39–50 (2001). [DOI] [PubMed] [Google Scholar]
- 131.Hou HA et al. Expression of angiopoietins and vascular endothelial growth factors and their clinical significance in acute myeloid leukemia. Leuk. Res 32, 904–912 (2008). [DOI] [PubMed] [Google Scholar]
- 132.Schmidt T & Carmeliet P Angiogenesis: a target in solid tumors, also in leukemia? Hematol. Am. Soc. Hematol Educ. Program 2011, 1–8 (2011). [DOI] [PubMed] [Google Scholar]
- 133.Norén-Nyström U et al. Vascular density in childhood acute lymphoblastic leukaemia correlates to biological factors and outcome. Br. J. Haematol 146, 521–530 (2009). [DOI] [PubMed] [Google Scholar]
- 134.Aguayo A et al. Plasma vascular endothelial growth factor levels have prognostic significance in patients with acute myeloid leukemia but not in patients with myelodysplastic syndromes. Cancer 95, 1923–1930 (2002). [DOI] [PubMed] [Google Scholar]
- 135.De Bont ESJM, Rosati S, Jacobs S, Kamps WA & Vellenga E Increased bone marrow vascularization in patients with acute myeloid leukaemia: a possible role for vascular endothelial growth factor. Br. J. Haematol 113, 296–304 (2001). [DOI] [PubMed] [Google Scholar]
- 136.Hiramatsu A et al. Disease-specific expression of VEGF and its receptors in AML cells: possible autocrine pathway of VEGF/type1 receptor of VEGF in t(15;17) AML and VEGF/type2 receptor of VEGF in t(8;21) AML. Leuk. Lymphoma 47, 89–95 (2006). [DOI] [PubMed] [Google Scholar]
- 137.Padro T et al. Increased angiogenesis in the bone marrow of patients with acute myeloid leukemia. Blood 8, 2637–2644 (2000). [PubMed] [Google Scholar]
- 138.Santos SCR & Dias S Internal and external autocrine VEGF/KDR loops regulate survival of subsets of acute leukemia through distinct signaling pathways. Blood 103, 3883–3889 (2004). [DOI] [PubMed] [Google Scholar]
- 139.Favreau AJ, Vary CPH, Brooks PC & Sathyanarayana P Cryptic collagen IV promotes cell migration and adhesion in myeloid leukemia. Cancer Med. 3, 265–272 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Verma D et al. Bone marrow niche-derived extracellular matrix-degrading enzymes influence the progression of B-cell acute lymphoblastic leukemia. Leukemia 34, 1540–1552 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Barbier V et al. Endothelial E-selectin inhibition improves acute myeloid leukaemia therapy by disrupting vascular niche-mediated chemoresistance. Nat. Commun 11, 2042 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Krenn PW, Koschmieder S & Fässler R Kindlin-3 loss curbs chronic myeloid leukemia in mice by mobilizing leukemic stem cells from protective bone marrow niches. Proc. Natl Acad. Sci. USA 117, 24326 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Tavor S et al. Motility, proliferation, and egress to the circulation of human AML cells are elastase dependent in NOD / SCID chimeric mice. Blood 106, 2120–2127 (2005). [DOI] [PubMed] [Google Scholar]
- 144.Lam KY & Tang V Metastatic tumors to the spleen: a 25-year clinicopathologic study. Arch. Pathol. Lab. Med 124, 526–530 (2000). [DOI] [PubMed] [Google Scholar]
- 145.Juarez J et al. CXCR4 antagonists mobilize childhood acute lymphoblastic leukemia cells into the peripheral blood and inhibit engraftment. Leukemia 21, 1249–1257 (2007). [DOI] [PubMed] [Google Scholar]
- 146.Yang F et al. Monocyte-derived leukemia-associated macrophages facilitate extramedullary distribution of T-cell acute lymphoblastic leukemia cells. Cancer Res. 80, 3677–3691 (2020). [DOI] [PubMed] [Google Scholar]
- 147.Gutjahr JC et al. Microenvironment-induced CD44v6 promotes early disease progression in chronic lymphocytic leukemia. Blood 131, 1337–1349 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Gatjen M et al. Splenic marginal zone granulocytes acquire an accentuated neutrophil B-cell helper phenotype in chronic lymphocytic leukemia. Cancer Res. 76, 5253–5265 (2016). [DOI] [PubMed] [Google Scholar]
- 149.Marom A et al. CD84 mediates CLL-microenvironment interactions. Oncogene 36, 628–638 (2017). [DOI] [PubMed] [Google Scholar]
- 150.Saulep-Easton D et al. The BAFF receptor TACI controls IL-10 production by regulatory B cells and CLL B cells. Leukemia 30, 163–172 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Pereira ER, Jones D, Jung K & Padera TP The lymph node microenvironment and its role in the progression of metastatic cancer. Semin. Cell Dev. Biol 38, 98–105 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Habermann TM & Steensma DP Lymphadenopathy. Mayo Clin. Proc 75, 723–732 (2000). [DOI] [PubMed] [Google Scholar]
- 153.Libman H Generalized lymphadenopathy. J. Gen. Intern. Med 2, 48–58 (1987). [DOI] [PubMed] [Google Scholar]
- 154.Ruddle NH High endothelial venules and lymphatic vessels in tertiary lymphoid organs: characteristics, functions, and regulation. Front. Immunol 7, 491 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Fecteau JF & Kipps TJ Structure and function of the hematopoietic cancer niche: focus on chronic lymphocytic leukemia. Front. Biosci 4 S, 61–73 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Calpe E et al. ZAP-70 enhances migration of malignant B lymphocytes toward CCL21 by inducing CCR7 expression via IgM-ERK1/2 activation. Blood 118, 4401–4410 (2011). [DOI] [PubMed] [Google Scholar]
- 157.Gunn MD et al. A chemokine expressed in lymphoid high endothelial venules promotes the adhesion and chemotaxis of naive T lymphocytes. Proc. Natl Acad. Sci. USA 95, 258–263 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Alfonso-Pérez M et al. Anti-CCR7 monoclonal antibodies as a novel tool for the treatment of chronic lymphocyte leukemia. J. Leukoc. Biol 79, 1157–1165 (2006). [DOI] [PubMed] [Google Scholar]
- 159.Cuesta C, Munoz-Callega C, Loscertales J, Terron F & Mol W CAP-100: first-in-class antibody for CCR7+ hematological malignancies. J. Clin. Oncol 37, e19008 (2019). [Google Scholar]
- 160.Redondo-Muñoz J et al. a4B1 integrin and 190-kDa CD44v constitute a cell surface docking complex for gelatinase B/MMP-9 in chronic leukemic but not in normal B cells. Blood 112, 169–178 (2008). [DOI] [PubMed] [Google Scholar]
- 161.Kipps TJ et al. Chronic lymphocytic leukaemia. Nat. Rev. Dis. Prim 3, 16096 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Burger JA et al. High-level expression of the T-cell chemokines CCL3 and CCL4 by chronic lymphocytic leukemia B cells in nurselike cell cocultures and after BCR stimulation. Blood 113, 3050–3058 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Hartmann EM, Rudelius M, Burger JA & Rosenwald A CCL3 chemokine expression by chronic lymphocytic leukemia cells orchestrates the composition of the microenvironment in lymph node infiltrates. Leuk. Lymphoma 57, 563–571 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Burger JA et al. Blood-derived nurse-like cells protect chronic lymphocytic leukemia B cells from spontaneous apoptosis through stromal cell-derived factor-1. Blood 96, 2655–2663 (2000). [PubMed] [Google Scholar]
- 165.Burger M et al. Small peptide inhibitors of the CXCR4 chemokine receptor (CD184) antagonize the activation, migration, and antiapoptotic responses of CXCL12 in chronic lymphocytic leukemia B cells. Blood 106, 1824–1830 (2005). [DOI] [PubMed] [Google Scholar]
- 166.Nishio M et al. Nurselike cells express BAFF and APRIL, which can promote survival of chronic lymphocytic leukemia cells via a paracrine pathway distinct from that of SDF-1 alpha. Blood 106, 1012–1020 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Tsukada N, Burger JA, Zvaifler NJ & Kipps TJ Distinctive features of “nurselike” cells that differentiate in the context of chronic lymphocytic leukemia. Blood 99, 1030–1037 (2002). [DOI] [PubMed] [Google Scholar]
- 168.Price RA & Johnson WW The central nervous system in childhood leukemia: I. The arachnoid. Cancer 31, 520–533 (1973). [DOI] [PubMed] [Google Scholar]
- 169.Brower JV, Saha S, Rosenberg SA, Hullett CR & Ian Robins H Management of leptomeningeal metastases: Prognostic factors and associated outcomes. J. Clin. Neurosci 27, 130–137 (2016). [DOI] [PubMed] [Google Scholar]
- 170.Nayar G et al. Leptomeningeal disease: current diagnostic and therapeutic strategies. Oncotarget 8, 73312–73328 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Mastorakos P & McGavern D The anatomy and immunology of vasculature in the central nervous system. Sci. Immunol 4, eaav0492 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Ransohoff RM & Engelhardt B The anatomical and cellular basis of immune surveillance in the central nervous system. Nat. Rev. Immunol 12, 623–635 (2012). [DOI] [PubMed] [Google Scholar]
- 173.Williams MT et al. The ability to cross the blood-cerebrospinal fluid barrier is a generic property of acute lymphoblastic leukemia blasts. Blood 127, 1998–2006 (2016). [DOI] [PubMed] [Google Scholar]
- 174.Bartram J et al. High throughput sequencing in acute lymphoblastic leukemia reveals clonal architecture of central nervous system and bone marrow compartments. Haematologica 103, 110–114 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Munch V et al. Central nervous system involvement in acute lymphoblastic leukemia is mediated by vascular endothelial growth factor. Blood 130, 643–654 (2017). [DOI] [PubMed] [Google Scholar]
- 176.Kinjyo I, Bragin D, Grattan R, Winter SS & Wilson BS Leukemia-derived exosomes and cytokines pave the way for entry into the brain. J. Leukoc. Biol 105, 741–753 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Ramirez M et al. Chemokines in leukemic infiltration of the central nervous system in childhood acute lymphoblastic leukemia. Blood 114, 651 (2009).19443658 [Google Scholar]
- 178.Alsadeq A et al. The role of ZAP70 kinase in acute lymphoblastic leukemia infiltration into the central nervous system. Haematologica 102, 346–355 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Marz M et al. Pediatric acute lymphoblastic leukemia — conquering the CNS across the choroid plexus. Leuk. Res 71, 47–54 (2018). [DOI] [PubMed] [Google Scholar]
- 180.Naumann JA & Gordon PM In vitro model of leukemia cell migration across the blood–cerebrospinal fluid barrier. Leuk. Lymphoma 58, 1747–1749 (2017). [DOI] [PubMed] [Google Scholar]
- 181.Scharff BFSS et al. A comprehensive clinical study of integrins in acute lymphoblastic leukemia indicates a role of alpha 6/CD49f in persistent minimal residual disease and alpha 5 in the colonization of cerebrospinal fluid. Leuk. Lymphoma 7, 1714–1718 (2020). [DOI] [PubMed] [Google Scholar]
- 182.Frishman-Levy L et al. Central nervous system acute lymphoblastic leukemia: role of natural killer cells. Blood 125, 3420–3431 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Jonart LM et al. Disrupting the leukemia niche in the central nervous system attenuates leukemia chemoresistance. Haematologica 105, 2130–2140 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Gaynes JS et al. The central nervous system microenvironment influences the leukemia transcriptome and enhances leukemia chemoresistance. Haematologica 102, 136–139 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Barnhill RL et al. Atypical spitzoid melanocytic neoplasms with angiotropism: a potential mechanism of locoregional involvement. Am. J. Dermatopathol 33, 236–243 (2011). [DOI] [PubMed] [Google Scholar]
- 186.Wong C, Helm M, Kalb R, Helm T & Zeitouni N The presentation, pathology, and current management strategies of cutaneous metastasis. North. Am. J. Med. Sci 5, 499–504 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Miyashiro D & Sanches JA Cutaneous manifestations of adult T-cell leukemia/lymphoma. Semin. Diagnostic Pathol 37, 81–91 (2020). [DOI] [PubMed] [Google Scholar]
- 188.Berg EL et al. The cutaneous lymphocyte antigen is a skin lymphocyte homing receptor for the vascular lectin endothelial cell-leukocyte adhesion molecule-1. J. Exp. Med 174, 1461–1466 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Campbell JJ, O’Connell DJ & Wurbel M-A Cutting edge: chemokine receptor CCR4 is necessary for antigen-driven cutaneous accumulation of CD4 T cells under physiological conditions. J. Immunol 178, 3358 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Mohle R et al. Functional response of leukaemic blasts to stromal cell-derived factor-1 correlates with preferential expression of the chemokine receptor CXCR4 in acute myelomonocytic and lymphoblastic leukaemia. Br. J. Haematol 110, 563–572 (2000). [DOI] [PubMed] [Google Scholar]
- 191.Schlesinger M & Bendas G Contribution of very late antigen-4 (VLA-4) integrin to cancer progression and metastasis. Cancer Metastasis Rev 34, 575–591 (2015). [DOI] [PubMed] [Google Scholar]
- 192.DiPersio JF et al. Phase III prospective randomized double-blind placebo-controlled trial of plerixafor plus granulocyte colony-stimulating factor compared with placebo plus granulocyte colony-stimulating factor for autologous stem-cell mobilization and transplantation for patients with non-Hodgkin’s lymphoma. J. Clin. Oncol 27, 4767–4773 (2009). [DOI] [PubMed] [Google Scholar]
- 193.DiPersio JF et al. Plerixafor and G-CSF versus placebo and G-CSF to mobilize hematopoietic stem cells for autologous stem cell transplantation in patients with multiple myeloma. Blood 113, 5720–5726 (2009). [DOI] [PubMed] [Google Scholar]
- 194.Zeng Z et al. Targeting the leukemia microenvironment by CXCR4 inhibition overcomes resistance to kinase inhibitors and chemotherapy in AML. Blood 113, 6215–6224 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Welschinger R et al. Plerixafor (AMD3100) induces prolonged mobilization of acute lymphoblastic leukemia cells and increases the proportion of cycling cells in the blood in mice. Exp. Hematol 41, 293–302.e291 (2013). [DOI] [PubMed] [Google Scholar]
- 196.Liesveld JL et al. Effects of AMD3100 on transmigration and survival of acute myelogenous leukemia cells. Leuk. Res 31, 1553–1563 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Tavor S et al. The CXCR4 antagonist AMD3100 impairs survival of human AML cells and induces their differentiation. Leukemia 22, 2151–5158 (2008). [DOI] [PubMed] [Google Scholar]
- 198.Uy GL et al. A phase 1/2 study of chemosensitization with the CXCR4 antagonist plerixafor in relapsed or refractory acute myeloid leukemia. Blood 119, 3917–3924 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Uy GL et al. A phase 1/2 study of chemosensitization with plerixafor plus G-CSF in relapsed or refractory acute myeloid leukemia. Blood Cancer J. 7, 2–5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Greenbaum AM & Link DC Mechanisms of G-CSF-mediated hematopoietic stem and progenitor mobilization. Leukemia 25, 211–217 (2011). [DOI] [PubMed] [Google Scholar]
- 201.Petit I, Ponomaryov T, Zipori D & Tsvee L G-CSF induces stem cell mobilization by decreasing bone marrow SDF-1 and up-regulating CXCR4. Nat. Immunol 3, 687–694 (2002). [DOI] [PubMed] [Google Scholar]
- 202.Löwenberg B et al. Effect of priming with granulocyte colony-stimulating factor on the outcome of chemotherapy for acute myeloid leukemia. N. Engl. J. Med 349, 743–752 (2003). [DOI] [PubMed] [Google Scholar]
- 203.Pabst T et al. Favorable effect of priming with granulocyte colony-stimulating factor in remission induction of acute myeloid leukemia restricted to dose escalation of cytarabine. Blood 119, 5367–5373 (2012). [DOI] [PubMed] [Google Scholar]
- 204.Dombret H et al. A controlled study of recombinant human granulocyte colony-stimulating factor in elderly patients after treatment for acute myelogenous leukemia. N. Engl. J. Med 332, 1678–1683 (1995). [DOI] [PubMed] [Google Scholar]
- 205.G H et al. A randomized, double-blind, placebo-controlled, phase III study of filgrastim in remission induction and consolidation therapy for adults with de novo acute myeloid leukemia. Blood 90, 4710–4718 (1997). [PubMed] [Google Scholar]
- 206.Huselton E et al. Updated study results of CX-01, an inhibitor of CXCL12/CXCR4, and azacitidine for the treatment of hypomethylating agent refractory AML and MDS. Blood 134, 3915 (2019). [Google Scholar]
- 207.Pillozzi S et al. Peptides and small molecules blocking the CXCR4/CXCL12 axis overcome bone marrow-induced chemoresistance in acute leukemias. Oncol. Rep 41, 312–324 (2019). [DOI] [PubMed] [Google Scholar]
- 208.Kovacsovics T et al. A randomized phase II trial of CX-01 with standard therapy in elderly patients with acute myeloid leukemia (AML). J. Clin. Oncol 37, 7001 (2019). [Google Scholar]
- 209.US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT04571645 (2020). [DOI] [PubMed]
- 210.US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT04704323 (2021). [DOI] [PubMed]
- 211.US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT04240704 (2021). [DOI] [PubMed]
- 212.Yoshie O & Matsushima K CCR4 and its ligands: from bench to bedside. Int. Immunol 27, 11–20 (2015). [DOI] [PubMed] [Google Scholar]
- 213.US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT04185220 (2019). [DOI] [PubMed]
- 214.DeAngelo DJ et al. Uproleselan (GMI-1271), an E-selectin antagonist, improves the efficacy and safety of chemotherapy in relapsed/refractory (R/R) and newly diagnosed older patients with acute myeloid leukemia: final, correlative, and subgroup analyses. Blood 132, 331 (2018). [Google Scholar]
- 215.Zhang W et al. Dual E-selectin/CXCR4 antagonist GMI-1359 exerts efficient anti-leukemia effects in a FLT3 ITD mutated acute myeloid leukemia patient-derived xenograft murine model. Blood 128, 3519 (2016). [Google Scholar]
- 216.US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT03616470 (2009). [DOI] [PubMed]
- 217.Godavarthy PS et al. The vascular bone marrow niche influences outcome in chronic myeloid leukemia via the E-selectin - SCF/TAL1 - CD44 axis. Haematologica 105, 136–147 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Price TT et al. Dormant breast cancer micrometastases reside in specific bone marrow niches that regulate their transit to and from bone. Sci. Transl Med 8, 340ra373 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT04197999 (2019). [DOI] [PubMed]
- 220.Hsieh YT et al. Integrin alpha4 blockade sensitizes drug resistant pre-B acute lymphoblastic leukemia to chemotherapy. Blood 121, 1814–1818 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Hamidi H, Pietilä M & Ivaska J The complexity of integrins in cancer and new scopes for therapeutic targeting. Br. J. Cancer 115, 1017–1023 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Herman SEM et al. Treatment with ibrutinib inhibits BTK- and VLA-4-dependent adhesion of chronic lymphocytic leukemia cells in vivo. Clin. Cancer Res 21, 4642–4651 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.DiGiuseppe JA, Fuller SGSG & Borowitz MJ Overexpression of CD49f in precursor B-cell acute lymphoblastic leukemia: Potential usefulness in minimal residual disease detection. Cytometry B Clin. Cytometry 76B, 150–155 (2009). [DOI] [PubMed] [Google Scholar]
- 224.Gang EJ et al. Integrin α6 mediates drug resistance of acute lymphoblastic B-cell leukemia. Blood 136, 210–223 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT04803123 (2021). [DOI] [PubMed]
- 226.Dobosz P & Dzieciątkowski T The intriguing history of cancer immunotherapy. Front. Immunol 10, 2965 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT01838395 (2013). [DOI] [PubMed]
- 228.US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT02763384 (2016). [DOI] [PubMed]
- 229.Furman RR et al. Idelalisib and rituximab in relapsed chronic lymphocytic leukemia. N. Engl. J. Med 370, 997–1007 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Jabbour E et al. Central nervous system prophylaxis in adults with acute lymphoblastic leukemia: current and emerging therapies. Cancer 116, 2290–2300 (2010). [DOI] [PubMed] [Google Scholar]
- 231.Ten Hacken E & Burger JA Microenvironment interactions and B-cell receptor signaling in chronic lymphocytic leukemia: implications for disease pathogenesis and treatment. Biochim. Biophys. Acta 1863, 401–413 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Santos FP & O’Brien S Small lymphocytic lymphoma and chronic lymphocytic leukemia: are they the same disease? Cancer J. 18, 396–403 (2012). [DOI] [PubMed] [Google Scholar]
- 233.Tees MT & Flinn IW Chronic lymphocytic leukemia and small lymphocytic lymphoma: two faces of the same disease. Expert Rev Hematol. 10, 137–146 (2017). [DOI] [PubMed] [Google Scholar]
- 234.McKenna MK et al. Splenic microenvironment is important in the survival and growth of chronic lymphocytic leukemia in mice. J. Immunol 198 (Suppl. 1), 130–20 (2017). [Google Scholar]
- 235.Lad D et al. CLL: common leukemia; uncommon presentations. Indian J. Hematol. Blood Transfus 32, 268–275 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Altintas A, Cil T, Kilinc I, Kaplan MA & Ayyildiz O Central nervous system blastic crisis in chronic myeloid leukemia on imatinib mesylate therapy: a case report. J. Neurooncol 84, 103–105 (2007). [DOI] [PubMed] [Google Scholar]
- 237.Rajappa S, Uppin SG, Raghunadharao D, Rao IS & Surath A Isolated central nervous system blast crisis in chronic myeloid leukemia. Hematol. Oncol 22, 179–181 (2004). [DOI] [PubMed] [Google Scholar]
- 238.Dong M & Blobe GC Role of transforming growth factor-β in hematologic malignancies. Blood 107, 4589–4596 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Bauvois B, Dumont J, Mathiot C & Kolb JP Production of matrix metalloproteinase-9 in early stage B-CLL: suppression by interferons. Leukemia 16, 791–798 (2002). [DOI] [PubMed] [Google Scholar]
- 240.Romee R et al. Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci. Transl Med 8, 357ra123 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Yan Y et al. Antileukemia activity of a natural killer cell line against human leukemias. Clin. Cancer Res 4, 2859–2868 (1998). [PubMed] [Google Scholar]
- 242.Swerdlow S et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues (Medicine) 4th Edn, Vol. 2 (World Health Organization; 2017). [Google Scholar]
- 243.Lampert IA, Hegde U & Van Noorden S The splenic white pulp in chronic lymphocytic leukaemia: a microenvironment associated with CR2 (CD21) expression, cell transformation and proliferation. Leuk. Lymphoma 1, 319–326 (1990). [DOI] [PubMed] [Google Scholar]
- 244.Somers WS, Tang J, Shaw GD & Camphausen RT Insights into the molecular basis of leukocyte tethering and rolling revealed by structures of P- and E-selectin bound to SLe X and PSGL-1. Cell 103, 467–479 (2000). [DOI] [PubMed] [Google Scholar]
- 245.Alon R et al. α4β1-dependent adhesion strengthening under mechanical strain is regulated by paxillin association with the α4-cytoplasmic domain. J. Cell Biol 171, 1073–1084 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Kuwano Y, Spelten O, Zhang H, Ley K & Zarbock A Rolling on E- or P-selectin induces the extended but not high-affinity conformation of LFA-1 in neutrophils. Blood 116, 617–624 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Shamri R et al. Lymphocyte arrest requires instantaneous induction of an extended LFA-1 conformation mediated by endothelium-bound chemokines. Nat. Immunol 6, 497–506 (2005). [DOI] [PubMed] [Google Scholar]
- 248.Zarbock A, Ley K & McEver RP Leukocyte ligands for endothelial selectins: specialized glycoconjugates that mediate rolling and signaling under flow. Blood 118, 6743–6751 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Alon R & Shulman Z Chemokine triggered integrin activation and actin remodeling events guiding lymphocyte migration across vascular barriers. Exp. Cell Res 317, 632–641 (2010). [DOI] [PubMed] [Google Scholar]
- 250.Barreiro O et al. Dynamic interaction of VCAM-1 and ICAM-1 with moesin and ezrin in a novel endothelial docking structure for adherent leukocytes. J. Cell Biol 157, 1233–1245 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Phillipson M et al. Intraluminal crawling of neutrophils to emigration sites: a molecularly distinct process from adhesion in the recruitment cascade. J. Exp. Med 203, 2569–2575 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Shulman Z et al. Lymphocyte crawling and transendothelial migration require chemokine triggering of high-affinity LFA-1 integrin. Immunity 30, 384–396 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Nourshargh S, Hordijk PL & Sixt M Breaching multiple barriers: leukocyte motility through venular walls and the interstitium. Nat. Rev. Mol. Cell Biol 11, 366–378 (2010). [DOI] [PubMed] [Google Scholar]
- 254.Carman CV & Springer TA A transmigratory cup in leukocyte diapedesis both through individual vascular endothelial cells and between them. J. Cell Biol 167, 377–388 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Charras G & Sahai E Physical influences of the extracellular environment on cell migration. Nat. Rev. Mol. Cell Biol. 15, 813–824 (2014). [DOI] [PubMed] [Google Scholar]
- 256.Bovellan M, Fritzsche M, Stevens C & Charras G Death-associated protein kinase (DAPK) and signal transduction: blebbing in programmed cell death. FEBS J. 277, 58–65 (2010). [DOI] [PubMed] [Google Scholar]
- 257.Buccione R, Caldieri G & Ayala I Invadopodia: specialized tumor cell structures for the focal degradation of the extracellular matrix. Cancer Metastasis Rev. 28, 137–149 (2009). [DOI] [PubMed] [Google Scholar]
- 258.Schoumacher M, Louvard D & Vignjevic D Cytoskeleton networks in basement membrane transmigration. Eur. J. Cell Biol 90, 93–99 (2011). [DOI] [PubMed] [Google Scholar]
- 259.Poincioux R, Lizárraga F & Chavrier P Matrix invasion by tumour cells: a focus on MT1-MMP trafficking to invadopodia. J. Cell Sci 122, 3015–3024 (2009). [DOI] [PubMed] [Google Scholar]
- 260.Linder S The matrix corroded: podosomes and invadopodia in extracellular matrix degradation. Trends Cell Biol. 17, 107–117 (2007). [DOI] [PubMed] [Google Scholar]
- 261.Krause M & Gautreau A Steering cell migration: lamellipodium dynamics and the regulation of directional persistence. Nat. Rev. Mol. Cell Biol 15, 577–590 (2014). [DOI] [PubMed] [Google Scholar]