

Abstract
In mammals, histone 3 lysine 4 methylation (H3K4me) is mediated by six different lysine methyltransferases. Among these enzymes, SETD1B (SET domain containing 1b) has been linked to syndromic intellectual disability in human subjects, but its role in the mammalian postnatal brain has not been studied yet. Here, we employ mice deficient for Setd1b in excitatory neurons of the postnatal forebrain, and combine neuron‐specific ChIP‐seq and RNA‐seq approaches to elucidate its role in neuronal gene expression. We observe that Setd1b controls the expression of a set of genes with a broad H3K4me3 peak at their promoters, enriched for neuron‐specific genes linked to learning and memory function. Comparative analyses in mice with conditional deletion of Kmt2a and Kmt2b histone methyltransferases show that SETD1B plays a more pronounced and potent role in regulating such genes. Moreover, postnatal loss of Setd1b leads to severe learning impairment, suggesting that SETD1B‐dependent regulation of H3K4me levels in postnatal neurons is critical for cognitive function.
Keywords: ChIP‐seq, cognitive diseases, histone‐methylation, learning and memory
Subject Categories: Chromatin, Transcription & Genomics; Neuroscience
Comparative analyses of conditional deletion of mammalian H3K4 methyltransferases show a selective role for Setd1b in expression of neuron‐specific genes important for cognitive function.

Introduction
Adult‐onset cognitive diseases, such as for example sporadic Alzheimer’s disease (AD), depend on complex interactions of genetic and environmental risk factors that activate epigenetic processes (Fischer, 2014). In addition, mutations in genes that control epigenetic gene regulation are over‐represented in neurodevelopmental diseases linked to cognitive dysfunction (Kleefstra et al, 2014) (Gabriele et al, 2018). Therefore, targeting the epigenome has emerged as a promising therapeutic avenue to treat neurodevelopmental and adult‐onset cognitive diseases (Fischer, 2014; Nestler et al, 2015) (Larizza & Finelli, 2019). To understand the regulation of epigenetic gene expression in in the context of cognitive function is thus of utmost importance. Increasing evidence hints to an especially important role histone 3 lysine 4 (H3K4me) methylation, a mark linked to active transcription. In mammals, H3K4 methylation is mediated by six different lysine methyltransferases (KMTs), namely KMT2A (Mll1), KMT2B (Mll2), KMT2C (Mll3), KMT2D (Mll4), SETD1A, and SETD1B that catalyze—with some specificity‐mono‐, di‐, and tri‐methylation (Shilatifard, 2012). Importantly, mutations in all of these enzymes are found in neurodevelopmental intellectual disability disorders and a substantial amount of work has focused on the role of these enzymes in cortical development (Gabriele et al, 2018). Neuronal H3K4me is also deregulated in adult‐onset neurodegenerative diseases, such as AD (Gjoneska et al, 2015). Moreover, exposure of mice to a learning task was shown to increase H3K4me levels in postnatal brain (Gupta et al, 2010). However, the role of the different H3K4 KMTs in the postnatal brain is not well understood yet. Thus, it is presently also unclear to what extent postnatal processes may contribute to the cognitive phenotypes observed in intellectual disability disorders that are linked to mutations in the different H3K4 KMTs. Such knowledge is important for the development of therapeutic approaches. Among the six H3K4 KMTs, SETD1B is the least studied enzyme. Virtually nothing is known about the function of SETD1B in postnatal brain, although mutations in the Setd1b gene have been linked to syndromic intellectual disability (Hiraide et al, 2018) (Labonne et al, 2016) (Roston et al, 2020). Therefore, to elucidate the role of Setd1b, we generated mice that lack the gene from excitatory neurons of the postnatal brain and analyzed cognitive function and epigenetic gene expression in young adult mice. Our data reveal that Setd1b is essential for memory formation. Moreover, we provide evidence that Setd1b controls the expression of genes that are characterized by a broad H3K4 trimethylation peak around the transcription start site (TSS) and are intimately linked to neuronal function and learning behavior. A comparative analysis to corresponding data from conditional Kmt2a and Kmt2b knockout mice suggest a distinct role for Setd1b in the regulation of epigenetic gene expression linked to learning processes.
Results
Loss of Setd1b in postnatal forebrain neurons impairs hippocampus‐dependent memory formation
To study the role of Setd1b in the postnatal brain, we crossed mice in which exon 5 of the Setd1b gene is flanked by loxP sites (Bledau et al, 2014) to mice that express CRE‐recombinase under control of the CamKII promoter (Minichiello et al, 1999). This approach ensures deletion of Setd1b from excitatory forebrain neurons of the postnatal brain (cKO mice). Quantitative PCR (qPCR) analysis confirmed decreased expression of Setd1b in the hippocampal Cornu Ammonis (CA) area, the dentate gyrus (DG), and the cortex when compared with corresponding control littermates that carry loxP sites but do not express CRE recombinase (control group). Expression in the cerebellum was not affected, confirming the specificity of the approach (Fig 1A). Residual expression of Setd1b in hippocampal subregions (CA and DG) and cortex is most likely due to the fact that deletion is restricted to excitatory neurons while other cell types are unaffected. In line with the qPCR data, Setd1b protein levels were reduced in the hippocampal CA region of SETD1B cKO mice (Fig 1B). Setd1b cKO mice did not show any gross abnormalities in brain anatomy as evidenced by immunohistological analysis of DAPI staining, staining of marker proteins for neuronal integrity Neuronal N (NeuN), microtubule‐associated protein 2 (MAP2) as well as ionized calcium‐binding adapter molecule 1 (IBA1) as a marker for microglia and glial fibrillary acidic protein (GFAP) as a marker for astrocytes (Fig 1C). Next, we subjected Setd1b cKO and control mice to behavior testing. Notably, it was previously shown that heterozygous mice expressing CRE under control of the CamKII promoter do not differ from wild‐type littermates (Kuczera et al, 2010) (Stilling et al, 2014) and we have confirmed this in the context of this study also for behavior testing (Fig EV1A–G). There was no difference among groups in the open field test, suggesting that explorative behavior and basal anxiety is normal in Setd1b cKO mice (Fig 1D). Short‐term memory was assayed via the Y‐maze and was also similar among groups (Fig 1E). We also tested depressive‐like behavior in the Porsolt‐forced swim test. No difference was observed among groups (Fig 1F). Next, we subjected mice to the Morris Water Maze test to study hippocampus‐dependent spatial memory. While control mice were able to learn the task as indicated by a reduced escape latency throughout the 10 days of training, Setd1b cKO mice were severely impaired (Fig 1G). We also performed a more sensitive analysis using a modified version of the MUST‐C algorithm to measure the different spatial strategies that represent either hippocampus‐dependent or independent abilities (Illouz et al, 2016; Islam et al, 2021). Our results show that Setd1b cKO mice fail to adopt hippocampus‐dependent search strategies such as “direct,” “corrected,” and “short‐chaining” (Fig 1H). Consistently, the cumulative learning score calculated on the basis of these search strategies showed severe learning impairment in Setd1b cKO mice (Fig 1I). Since memory acquisition was severely impaired in Setd1b cKO mice, and we moreover observed a trend for reduced swim speed during a probe test that was performed after the end of the training procedure, memory recall could not be analyzed (Fig EV2A and B). In conclusion, our data show that deletion of Setd1b from excitatory neurons of the postnatal forebrain leads to severe impairment of hippocampus‐dependent learning abilities.
Figure 1. Setd1b is required for hippocampus‐dependent memory.
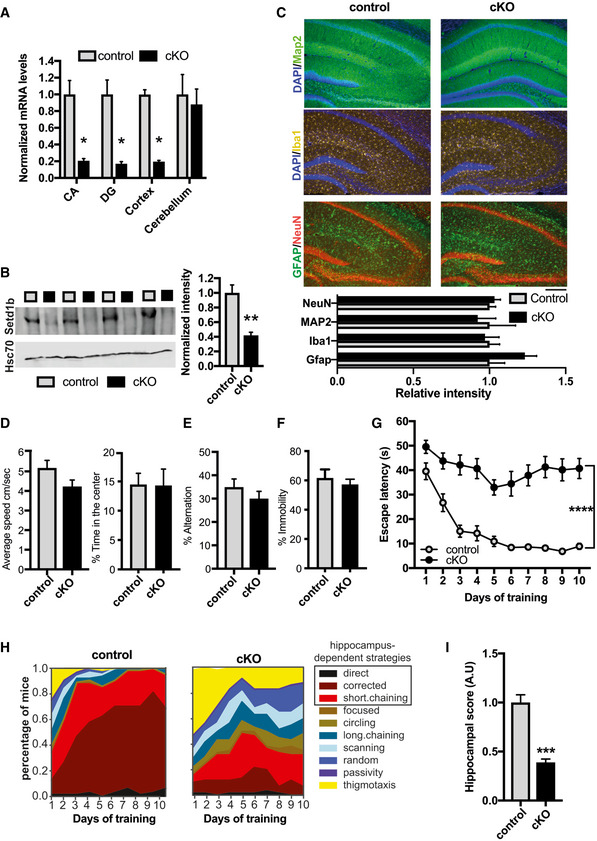
- qPCR analysis shows loss of Setd1b in forebrain regions while levels in the cerebellum are not affected (CA: Control, n = 6; cKO, n = 6. DG: Control, n = 6; cKO, n = 6. Cortex: Control, n = 7; cKO, n = 7. Cerebellum: Control, n = 4; cKO, n = 4). *P‐value < 0.05 (Student's t‐test).
- Immunoblot analysis shows loss of Setd1b protein in the hippocampus of Setd1b cKO mice (Control, n = 4; cKO, n = 4). **P‐value < 0.01 (Student's t‐test).
- Immunohistochemical staining (upper level) for marker proteins of neuronal integrity and quantification (lower panel) show no difference in control and Setd1b cKO mice (NeuN: Control, n = 6; cKO, n = 6; Student's t‐test P‐value = 0.57. MAP2: Control, n = 4; cKO, n = 4; Student's t‐test P‐value = 0.72. Iba1: Control, n = 5; cKO, n = 5; Student's t‐test P‐value = 0.8. Gfap: Control, n = 5; cKO, n = 5; Student's t‐test P‐value = 0.09.). Scale bar: 100 µm.
- Average speed (left panel) and percent time spent in the center (right panel) during exposure to the open field test was similar in control and Setd1b cKO mice (Average speed: Control, n = 15; cKO, n = 15; Student's t‐test P‐value = 0.075. Time spent in center: Control, n = 15; cKO, n = 15; Student's t‐test P‐value = 0.96).
- Short‐term memory was not different between control and Setd1b cKO mice as indicated by similar percent of alternations in the Y‐maze test (Control, n = 15; cKO, n = 15; Student's t‐test P‐value = 0.3).
- Depressive‐like behavior was measured in the Porsolt‐forced swim test. There was no significant difference in the % time spent immobile between groups (Control, n = 8; cKO, n = 8; Student's t‐test P‐value = 0.5).
- Escape latency during water maze training indicated severe learning impairment in Setd1b cKO mice (Control: n = 15, cKO: n = 15. Repeated measures ANOVA, genotype effect: F(1,28) = 82.34, ****P‐value < 0.0001).
- Plots showing the specific search strategies during water maze training. Note the failure of Setd1b cKO mice to adopt hippocampus‐dependent search strategies.
- The cognitive score calculated on the basis of the hippocampal search strategies reflects severe memory impairment in Setd1b cKO mice (Student's t‐test: ***P‐value < 0.001).
Data information: Bar graphs indicate mean, Error bars indicate ± SEM, “n” indicates biological replicates.
Source data are available online for this figure.
Figure EV1. Behavioral analysis of mice expressing CamKII‐driven Cre recombinase.
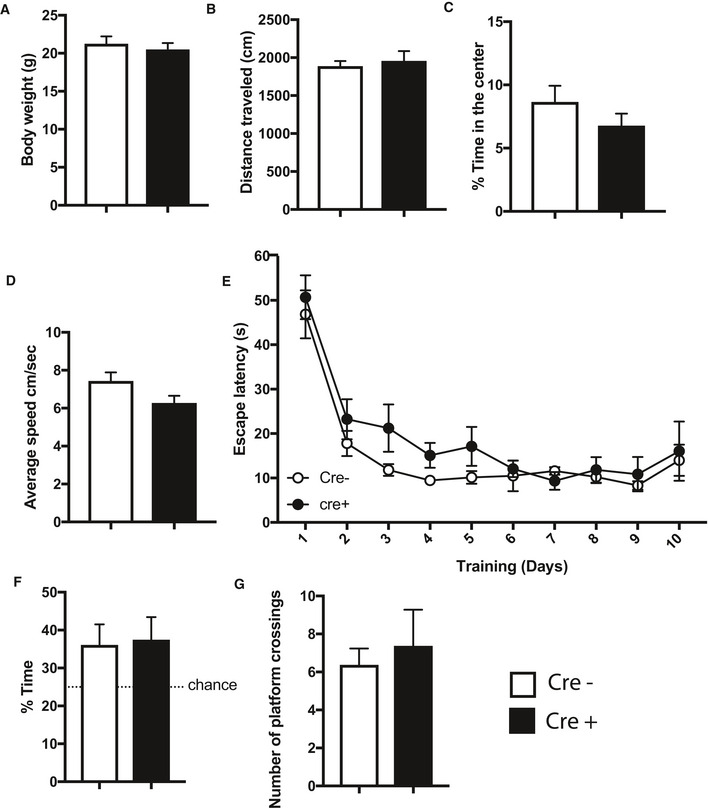
-
ATransgenic mice expressing Cre under control of the CamKII promoter were subjected to behavior testing (n = 8, Cre+) comparing them with wild‐type mice from the same breeding colony that did not express Cre (n = 8; Cre−). No difference was observed in body weight.
-
B–CThe distance traveled in the open field test (B) and the time spent in the center of the arena (C) were similar amongst groups.
-
DNo difference in the swimming speed was observed among groups when subjected to the water maze test.
-
EEscape latency during water maze training was similar in Cre− and Cre+ mice.
-
F–GDuring the probe test performed after 10 training days, Cre− and Cre+ mice showed similar performance when time spent in the target quadrant (F) and the number of platform crossings (G) were analyzed.
Data information: Bar graphs indicate mean, Error bars indicate ± SEM. “n” indicates biological replicates.
Figure EV2. Analysis of memory recall cannot be interpreted in Setd1b cKO mice.
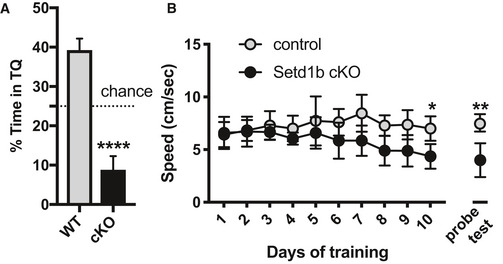
-
A, B(A) Twenty‐four hours after the completion of the 10 day‐training procedure in the water maze paradigm (See Fig 1G) mice were subjected to a probe test. The performance in the probe test, as measured by the time spent in the target quadrant (TQ), is impaired in Setd1b cKO mice (Control: n = 15, cKO: n = 15. ****P < 0.0001; Student's t‐test). These results have to be interpreted with care, however, since faulty interpretation about memory recall is possible in this experiment since the Setd1b cKO mice not even being able to form the memory during the training (See Fig 1G and H). It is moreover important to note that Setd1b cKO mice spent even less time in TQ than would be predicted by chance alone. To address the issue we analyzed the swim speed during the entire experiment (B) and noticed that the swim speed was similar in control and Setd1b cKO mice across the first 5 days of training. There was however a trend for reduced swim speed in Setd1b cKO mice from training day 6 to 10. Although we did not detect significant differences in the swim speed when we analyzed the average values across all training days, a more detailed analysis revealed that swim speed was significantly impaired in Setd1b cKO mice at training day 10 (*P = 0.016; Student's t‐test) and also during the probe test (**P = 0.001; Student's t‐test). These data may help to explain why the Setd1b cKO mice appear to avoid the target quadrant during the one trial 60 s probe test session. The training session consists of a sequence of 4 × 60 s sessions and mice are always placed into the pool from 4 different locations. During the probe test, that consists of a trial of one 60‐s session, animals are always placed in the quadrant opposite to the target quadrant, that is, the quadrant that is the farthest away from the target quadrant. While the reason for the reduced swim speed after 9 days of training is unclear, at present these data do not affect the conclusion that Setd1b cKO mice exhibit impaired learning ability.
Data information: Bar graphs/dots indicate mean, Error bars indicate ± SEM. “n” indicates biological replicates.
Setd1b controls H3K4 methylation peak width in hippocampal excitatory neurons
Although the Morris water maze test is used to specifically measure hippocampus‐dependent learning and memory, we cannot exclude that other brain regions contribute to the observed learning impairment. Nevertheless, our data suggest that the hippocampus would be a suitable brain region to study the molecular function of neuronal Setd1b in the postnatal brain. We reasoned that analyzing the hippocampus would furthermore allow for an optimal comparison to previous studies that investigated epigenetic gene expression from hippocampal neurons and tissue obtained from Kmt2a and Kmt2b cKO mice, that were generated using the same CRE‐driver line as in this study (Kerimoglu et al, 2013, 2017). To this end, we isolated the hippocampal CA region from Setd1b cKO and control mice and prepared nuclei to perform neuron‐specific chromatin immunoprecipitation (ChIP) (Fig 2A). Since Setd1b is a histone 3 lysine 4 (H3K4) methyltransferase, we decided to analyze tri‐methylation (H3K4me3) of histone 3 lysine 4 that is enriched at the TSS of active genes and is associated with euchromatin and active gene expression. H3K4 methylation is believed to be a stepwise process and recent data suggest that the different methylation states (from mono‐ to tri‐methylation) at the TSS of a gene form a gradient reflecting its specific transcriptional state (Soares et al, 2017; Choudhury et al, 2019). Moreover, some specificity for mono‐, di‐, or tri‐methylation has been reported for the different H3K4 KMTs, although biochemical analysis does not always correspond to the in vivo data (Kerimoglu et al, 2017; Bochyńska et al, 2018) (Gabriele et al, 2018). Thus, we also analyzed mono‐methylation of histone 3 at lysine 4 (H3K4me1). In addition, we analyzed histone 3 lysine 9 acetylation (H3K9ac), a euchromatin mark that was shown to partially depend on H3K4 methylation (Kerimoglu et al, 2013). Finally, we also performed ChIP‐Seq for histone 3 lysine 27 acetylation (H3K27ac), another euchromatin mark that is linked to active gene expression and marks promoter elements around the TSS but also enhancer regions and has not been directly linked to H3K4me3 in brain tissue. In all ChIP‐Seq experiments, we observed a clear separation between control and Setd1b cKO samples as evidenced by principal component analysis (Appendix Fig S1A–F).
Figure 2. Setd1b controls H3K4 methylation and H3K4me3 peak width.
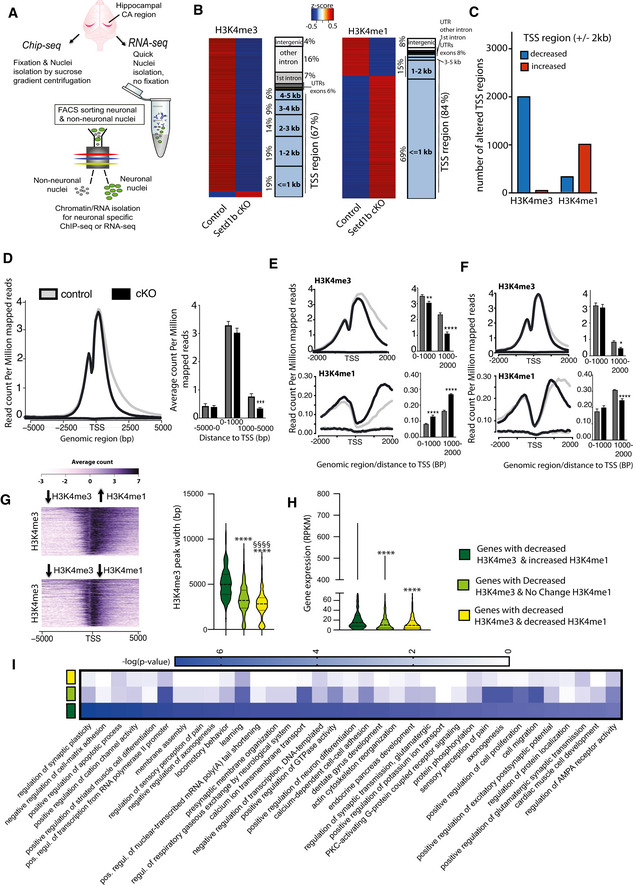
- Experimental scheme showing our approach to perform cell‐type‐specific ChIP‐Seq and RNA‐Seq. For ChIP‐Seq, we employed n = 4 for control and n = 4 for Setd1b cKO.
- Left panel: Heat map showing genomic regions with significantly differing H3K4me3 sites at the close vicinity of TSS (± 2 kb) in Setd1b cKO mice and the overall genomic locations of altered H3K4me3 levels. Right panel shows the same analysis for H3K4me1 (FDR < 0.05 and |fold change| > 1.5). The rectangles left to the corresponding heat maps characterize the genomic distribution of the affected regions as percentage. Blue rectangles represent TSS regions that are further subdivided into bins of 1kB, while grey rectangles represent non‐TSS regions.
- Bar plot showing the number of TSS regions (± 2 kb) with decreased and increased H3K4me3 and H3K4me1 in Setd1b cKO mice (FDR < 0.05 and |fold change| > 1.5).
- Profile plot showing H3K4me3 across all genes with significantly reduced H3K4me3 in Setd1b cKO mice. Left panel shows a bar chart indicating that reduced H3K4me3 in Setd1b cKO mice is mainly occurring downstream of the TSS (*** Student's t‐test P‐value < 0.001).
- Profile plots showing the distribution of H3K4me3 and H3K4me1 at the close vicinity of TSS of genes that show significantly reduced H3K4me3 and increased H3K4me1 in Setd1b cKO mice. Bar graphs on the right show corresponding quantification (Student's t‐test: **P‐value < 0.01, ****P‐value < 0.0001).
- Profile plots showing the distribution of H3K4me3 and H3K4me1 at the TSS of genes that show both reduced H3K4me3 and H3K4me1 in Setd1b cKO mice. Bar graphs on the left show corresponding quantification (Student's t‐test: *P‐value < 0.05, ****P‐value < 0.0001).
- Heatmap (left panel) showing basal state H3K4me3 width around TSS of genes characterized by decreased H3K4me3 in combination with either increased or decreased H3K4me1 in Setd1b cKO mice. Right panel: Quantification of the basal state H3K4me3 width around TSS of genes with decreased H3K4me3 in combination with either increased (472 peaks), decreased (401 peaks) or not altered H3K4me1 (1051 peaks) in Setd1b cKO mice (Kruskal–Wallis test: P‐value < 0.0001. Post‐hoc multiple comparisons, Dunn’s test: increased H3K4me1 vs no change H3K4me1, ****P‐value < 0.0001; increased H3K4me1 vs decreased H3K4me1, ****P‐value < 0.0001; no change H3K4me1 vs decreased H3K4me1, §§§§ P‐value < 0.0001).
- Violin plot showing the basal expression level for the 3 categories of genes that display decreased H3K4me3 in Setd1b cKO mice, now analyzed in wild‐type mice (increased H3K4me1: 456 genes, decreased H3K4me1: 382 genes, not altered H3K4me1: 993 genes). The basal expression level is highest for genes with decreased H3K4me3 in combination with increased H3K4me1 that are characterized by broad H3K4me3 peaks (Kruskal‐Wallis test: P‐value < 0.0001. Post‐hoc multiple comparisons, Dunn’s test: increased H3K4me1 vs no change H3K4me1, ****P‐value < 0.0001; increased H3K4me1 vs decreased H3K4me1, ****P‐value < 0.0001; no change H3K4me1 vs decreased H3K4me1, P‐value > 0.9999). Please note that the number of genes is smaller than the number of peaks in (G) because in some cases there are more than one peaks around TSS of the same gene.
- Heat map showing functional pathways for the 3 categories of genes affected by reduced H3K4me3 in Setd1b cKO mice.
Data information: Error bars indicate SEM. Bar graphs indicate mean, Error bars indicate ±SEM, “n” indicates biological replicates.
Loss of Setd1b leads to a substantial decrease in neuronal H3K4me3 (adjusted P‐value < 0.05 and fold change < −1.5) across the genome while the majority of significant changes are localized to regions in proximity to TSS (Fig 2B and C; Dataset EV1). Similar changes were observed for neuronal H3K9ac and H3K27ac, although fewer regions were affected when compared with H3K4me3 (Fig EV3A and B; Dataset EV2 and EV3). The changes in H3K9ac showed a substantial convergence with altered H3K4me3 (Fig EV3C). Thus, almost all TSS regions (± 2 kb) exhibiting decreased H3K9ac were also marked by decreased H3K4me3, while the TSS regions showing decreased H3K27ac only partially overlapped with those with decreased H3K4me3 (Fig EV3C). These data support previous findings, showing that H3K4me3 is functionally linked to H3K9ac (Kerimoglu et al, 2013) and suggest that the observed changes in H3K27ac might also reflect secondary effects.
Figure EV3. Decreased H3K9ac and H3K27ac in Setd1b cKO mice.
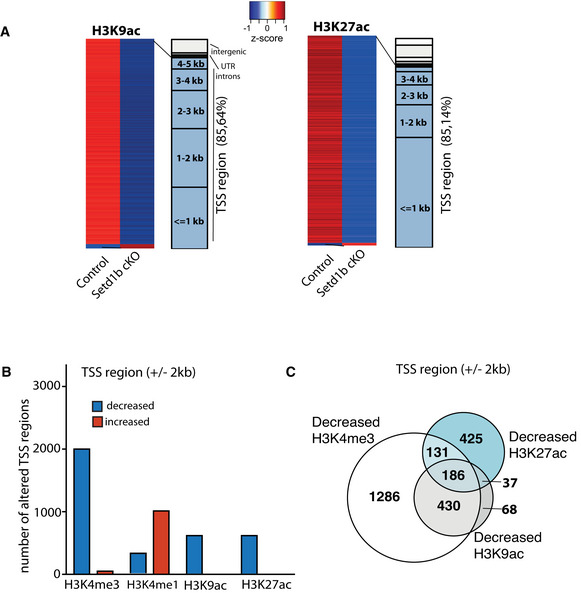
- Left panel: Heat map showing genomic regions with differentially bound H3K9ac sites in the close vicinity of TSS (± 2 kb) in neuronal nuclei from Setd1b cKO mice and the overall genomic locations of altered H3K9ac levels (Control: n = 4; Setd1b cKO: n = 3). Right panel shows the same analysis for H3K27ac (Control: n = 3; Setd1b cKO: n = 4).
- Bar chart showing the number of genes with significantly decreased and increased H3K9ac and H3K27ac marks at the TSS region (± 2 kb). Data for H3K4me3 and H3K4me1 are shown for comparison. As expected, the most affected histone mark is H3K4me3.
- Venn diagram showing that most of the sites exhibiting decreased H3K9ac at the TSS also exhibit reduced H3K4me3, while this is not the case for H3K27ac. TSS, transcription start site.
We also observed significantly altered H3K4me1 levels (adjusted P‐value < 0.05 and |fold change| > 1.5) in neurons of Setd1b cKO mice (Fig 2B; Dataset EV4). These changes were also almost exclusively detected in vicinity to the TSS (Fig 2B) but in contrast to the other investigated histone modifications, many of the significantly altered genomic regions exhibited increased H3K4me1 levels in Setd1b cKO mice (Fig 2B and C), suggesting that Setd1b mainly promotes tri‐methylation of H3K4 in neurons. To further interpret these observations, we analyzed the distribution of altered H3K4 methylation across the TSS region including up‐ and downstream regions up to 5 kb. Interestingly, decreased H3K4me3 in cKO manifested itself exclusively downstream of the TSS, indicating that loss of Setd1b may affect peak width (Fig 2D). We decided to further explore this observation and detected two distinct patterns of altered H3K4me. Namely, there was a difference between the genes that exhibit decreased H3K4me3 and at the same time increased H3K4me1 (Fig 2E) and those that show concomitantly decreased H3K4 tri‐ and mono‐methylation around the TSS (Fig 2F). The change in H3K4me3 was most significant in genes with decreased H3K4me3 and increased H3K4me1 and was characterized by a substantially reduced H3K4me3 peak width (Fig 2E), when compared with genes with milder yet significantly decreased H3K4me3 and H3K4me1 (Fig 2F). Findings from other cell types suggest a gradient of H3K4 methylation states in which the proximity of the mark to the TSS is correlated to the level of gene expression. Thus, genes with broader H3K4me3 peaks at the TSS exhibit the highest and most consistent expression levels and represent genes of particular importance for cellular identity and function (Benayoun et al, 2015) (Soares et al, 2017). Our data revealed that the genes which are characterized by decreased H3K4me3 and increased H3K4me1 in Setd1b cKO mice, exhibit significantly broader H3K4me3 peaks under basal conditions, when compared with genes characterized by decreased H3K4me3 but either decreased or unchanged H3K4me1 levels (Fig 2G; Dataset EV5). Interestingly, these genes were also expressed at significantly higher levels under baseline conditions (Fig 2H; Dataset EV6). Gene‐ontology (GO) analysis revealed that the genes with decreased H3K4me3 and increased H3K4me1 and thus having the broadest H3K4me3 peaks under basal conditions, represent pathways intimately linked to the function of excitatory hippocampal neurons (Fig 2I; Dataset EV7). Most importantly, this was not the case for the genes of the other two categories (Fig 2I). Finally, through motif enrichment analysis, we observed that the TSS regions with decreased H3K4me3 and increased H3K4me1 show a significant enrichment for RE1‐Silencing Transcription Factor (REST) consensus binding site (Dataset EV8), which is in line with our GO‐term analysis since the REST complex has previously been shown to repress genes specifically important for synaptic plasticity and specific to neuronal function both during development and aging by recruiting an enzymatic machinery that mediates heterochromatin formation and thus acts as a counterplayer to the H3K4 methyltransferases such as SETD1B (Hwang & Zukin, 2018).
In summary, our data show that loss of Setd1b from hippocampal excitatory neurons leads to distinct changes in neuronal histone methylation and point to a specific role of Setd1b in the H3K4 tri‐methylation and the expression of genes essential for hippocampal excitatory neuronal function.
Setd1b controls the levels of highly expressed hippocampal excitatory neuronal genes characterized by a broad H3K4me3 peak at the TSS
To test the impact of Setd1b on gene expression directly, we analyzed RNA‐sequencing data obtained from neuronal nuclei of the same hippocampi used to generate ChIP‐seq data. This was possible since we had employed a modified fixation protocol that allowed us to perform neuron‐specific ChIP‐seq and RNA‐seq from the same samples (See Figs 2A and EV4A–F). In line with the established role of H3K4me3 in active gene expression, we mainly detected downregulated genes when comparing control to Setd1b cKO mice (adjusted P‐value < 0.1 and |fold change| > 1.2) (Fig 3A, Dataset EV9). The results of the RNA‐seq experiment were confirmed via qPCR for randomly selected up‐ and downregulated genes (Fig. 3B). The comparatively few up‐regulated genes mainly exhibited low expression at baseline (RPKM down‐regulated genes = 18.77 ± 1.45 vs. RPKM up‐regulated genes = 3.08 ± 0.26; Student’s t‐test P‐value < 0.0001). Further analysis revealed that the TSS region of genes down‐regulated in Setd1b cKO mice is characterized by significantly reduced H3K4me3 peak width and increased H3K4me1 (Fig 3C), which was not the case for an equally sized random sample of unaffected genes (Fig 3D). H3K9ac and H3K27ac levels were also reduced at the TSS of the down‐regulated genes (Appendix Fig S2A and B). We also observed that the genes downregulated as a result of Setd1b deletion were characterized by a significantly broader H3K4me3 peak around TSS and higher expression under basal conditions, when compared with unaffected genes (Fig 3E; Dataset EV10 and EV11). GO analysis revealed that the genes down‐regulated in Setd1b cKO mice are linked to synaptic plasticity and learning related processes such as for example ERK1/2 signaling, extracellular matrix organization, or neuropeptide signaling pathways (Sweatt, 2001; Borbély et al, 2013) (Tsilibary et al, 2014) (Fig 3G; Dataset EV12). Specific examples are the Arpp‐21 gene that leads to aberrant dendritic plasticity when down‐regulated (Rehfeld et al, 2018) and has been genetically linked to intellectual disability (Marangi et al, 2013) or the Npy2r gene that leads to memory impairment in corresponding knockout mice (Redrobe et al, 2004) (Fig 3E). Neuropsin (Klk8) is another gene affected in Setd1b cKO mice that plays a complex role in memory function. It regulates the extracellular matrix, leads to spatial reference memory impairment when deleted in mice (Tamura et al, 2006) and has been implicated with mental diseases (Bukowski et al, 2020) (Fig 3E).
Figure EV4. Sorting neuronal and non‐neuronal nuclei for RNA‐Seq analysis.
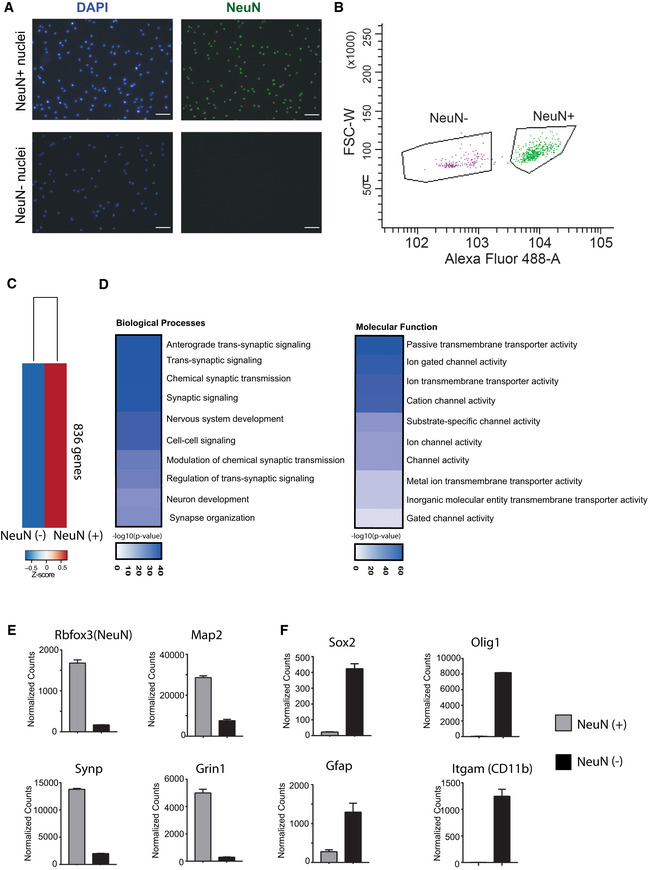
- Representative images showing nuclei that were sorted using the neuronal marker NeuN. Note that no NeuN positive nuclei are detected in the NeuN (−) fraction confirming the purity of the approach. Scale bar: 50 μm.
- Gating strategy for NeuN (+) and NeuN (−) nuclei sorting.
- RNA‐sequencing (n = 2/group) was performed from NeuN (+) and NeuN (−) nuclei and a differential expression analysis was performed. Heat map shows 836 genes specifically enriched in NeuN (+) nuclei when compared to NeuN (−) nuclei. The criteria to select those genes were: adjusted P‐value < 0.01, base mean > 150, fold change > 5.
- GO‐term analysis showing that the top 10 enriched biological processes and molecular functions for the 836 genes enriched in NeuN (+) nuclei all represent specific neuronal processes.
- Normalized expression values obtained from the RNA‐seq experiment showing the expression of selected genes known to be enriched in neurons.
- Normalized expression values of genes that are known to be enriched in non‐neuronal cells including glia cells. Error bars indicate SEM.
Data information: Bar graphs indicate mean, Error bars indicate ± SEM. “n” indicates biological replicates.
Figure 3. Hippocampal Setd1b controls highly expressed learning and memory genes characterized by a broad H3K4me3 peak.
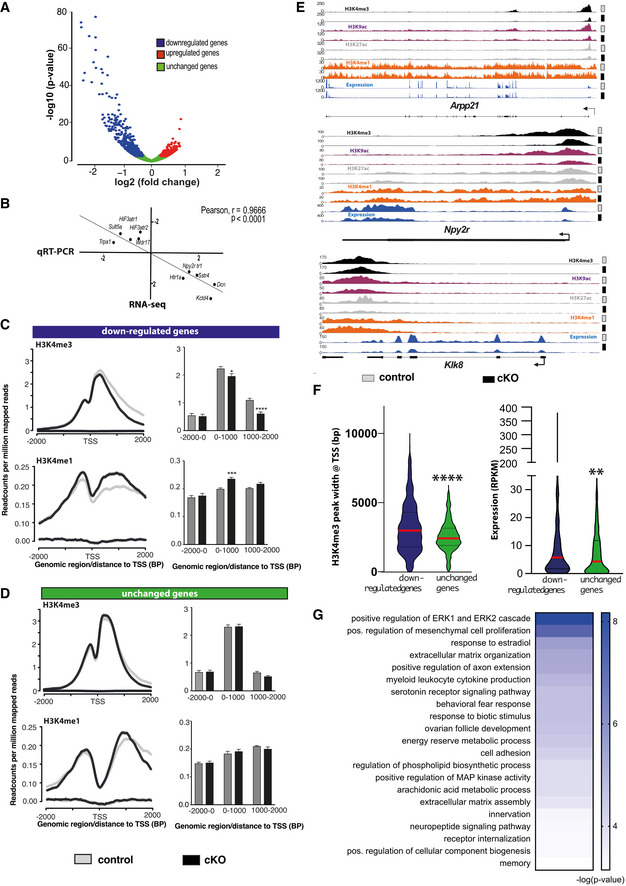
- nucRNA‐seq experiment. Volcano plot showing genes differentially expressed in hippocampal CA neurons of Setd1b cKO mice. n = 3/group.
- Randomly selected genes that were either significantly up‐ (5 genes) or downregulated (5 genes) in the RNA‐seq dataset comparing control vs Setd1b cKO mice were tested via qPCR for validation. The graph shows the corresponding log2 (fold changes) in each experiment. The RNA‐seq and qPCR results show highly significant correlation.
- Profile plots showing H3K4me3 and H3K4me1 at the TSS of genes downregulated in Setd1b cKO mice. Bar plots (right panel) show quantification (Student's t‐test: *P‐value < 0.05, ***P‐value < 0.001, ****P‐value < 0.0001).
- Profile plots showing H3K4me3 and H3K4me1 at the TSS of a random set of genes (equal in number to downregulated genes) that were not altered in Setd1b cKO mice. Bar plot (right panel) shows quantifications.
- Genome browser views showing the 4 analyzed histone modifications and the gene expression pattern for Arpp21, Npy2r, and Klk8 as genes that were previously shown to be involved in synaptic plasticity and learning and memory. Please note that the H3K4me3 peak width is substantially shrinking in Setd1b cKO mice. At the same time there is an obvious increase of H3K4me1 at the TSS of Arpp21 in Setd1b cKO mice.
- Left panel: H3K4me3 peaks are significantly broader in genes that are downregulated in Setd1b cKO mice, when compared with a random set of genes of equal number that were unaffected. Right panel: Genes downregulated in Setd1b cKO mice are characterized by higher baseline expression when compared with a random set of genes of equal number that were unaffected (Mann–Whitney test: ****P‐value < 0.0001, **P‐value < 0.01).
- Heat map showing functional pathways affected by genes downregulated in Setd1b cKO mice.
Data information: Bar graphs indicate mean, Error bars indicate ± SEM. “n” indicates biological replicates. Violin plots: the dashed center line indicates the median, while the upper and lower dotted lines represent the 75th and 25th percentiles, respectively.
In summary, our data further suggest that Setd1b controls a specific set of genes that are characterized by a broad H3K4me3 peak at the TSS, are highly expressed in hippocampal excitatory neurons under basal conditions and play a specific role in synaptic plasticity.
Comparison of epigenetic neuronal gene expression in Kmt2a, Kmt2b, and Setd1b cKO mice
To further elucidate the role of Setd1b in the regulation of neuronal gene expression, we decided to compare the data from Setd1b cKO mice to those from other mammalian H3K4 KMTs. We have previously generated comparable H3K4me3 and H3K4me1 ChIP‐Seq data from neuronal nuclei obtained from the hippocampal CA region of conditional knockout mice that lack either Kmt2a or Kmt2b from excitatory forebrain neurons using the same CRE‐line as employed in this study (Kerimoglu et al, 2017). Since these experiments were performed at different timepoints, we reanalyzed in parallel the H3K4me3 and H3K4me1 ChIP‐Seq datasets obtained from hippocampal neuronal nuclei of Kmt2a, Kmt2b, and Setd1b cKO mice (cut off adjusted P‐value < 0.1 and |fold change| > 1.2) to ensure reliable comparison. In line with the previous findings, all 3 KMT mutant mice exhibit a substantial number of TSS regions (± 2kb) with decreased H3K4me3 (adjusted P‐value < 0.1 and fold change < −1.2) (Fig 4A; Dataset [Link], [Link], [Link]). Interestingly, we also detected TSS regions with decreased H3K4me1 in all mutant mice, but only in Setd1b cKO mice, a substantial number of TSS regions exhibited increased H3K4me1 (Fig 4B; Dataset [Link], [Link], [Link]). In addition, there was little overlap among the TSS regions with decreased H3K4me3 in Kmt2a, Kmt2b, and Setd1b cKO mice, providing further evidence that Setd1b controls a unique gene expression program in neuronal cells (Fig 4C). To test this hypothesis directly, we decided to compare the corresponding gene expression changes in the hippocampal CA region of the 3 different KMT cKO mice. While the ChIP‐seq data available for Kmt2a and Kmt2b cKO mice had been generated from neuronal nuclei, the corresponding gene expression analysis represents RNA‐seq data obtained from bulk tissue of the hippocampal CA region (Kerimoglu et al, 2017). To ensure optimal comparison of these RNA‐seq data to the gene expression changes in Setd1b cKO mice, we also performed RNA‐seq from the whole tissue of the hippocampal CA region of Setd1b cKO mice and control littermates. We observed 486 genes that were significantly downregulated (adjusted P‐value < 0.1 and fold change < −1.2) when comparing control to Setd1b cKO mice (Fig 4D and E; Dataset EV19). The majority of those genes were also detected via neuronal‐specific RNA‐seq in Setd1b cKO mice that we performed before (see above) (Appendix Fig S3A‐D). While the total number of genes downregulated in the hippocampal CA region of Kmt2a, Kmt2b, and Setd1b cKO mice was comparable (Fig 4D and E), there was little overlap among them (Fig 4E; Dataset [Link], [Link], [Link]). Recently, RNA‐sequencing data was reported for mice that were heterozygous for Setd1a. Although these mutants were heterozygous constitutive knock‐out mice and furthermore cortical tissue was analyzed instead of the hippocampus (Mukai et al, 2019), it is interesting to note that there was virtually no overlap regarding the genes downregulated in Setd1a knock‐out mice, when compared with the data obtained from our Setd1b cKO mice (Appendix Fig S4). Further support for a distinct role of Setd1b in neuronal gene expression was revealed by the finding that the genes downregulated in Setd1b cKO mice exhibited a significant enrichment for neuron‐specific genes, while this was not the case for genes downregulated in Kmt2a cKO or Kmt2b cKO mice (Fig 4F and G).
Figure 4. Comparative analysis of the hippocampal epigenome and transcriptome in Setd1b, Kmt2a, and Kmt2b cKO mice reveals specific regulation of neuron‐enriched genes in Setd1b cKO mice.
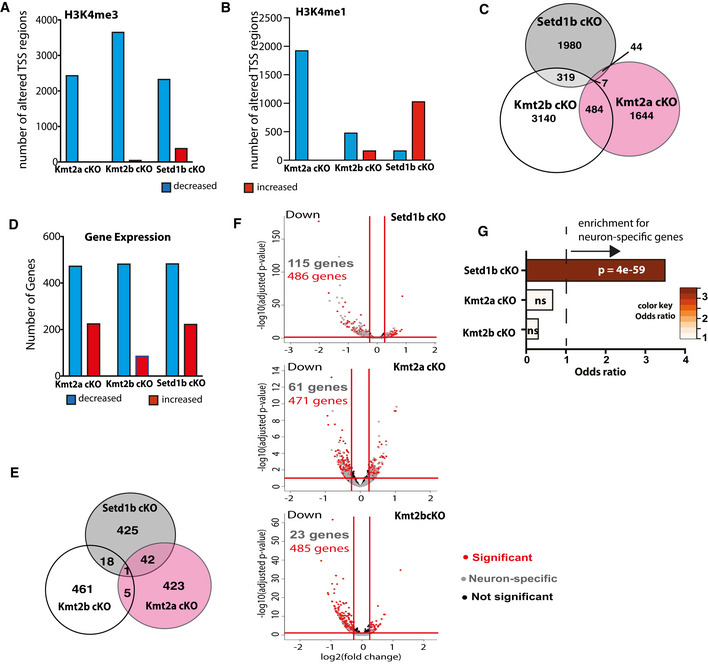
- Bar chart showing the number of TSS regions (± 2 kb) that exhibit significantly altered H3K4me3 (Kmt2a: control, n = 5; cKO, n = 3. Kmt2b: control, n = 6; cKO, n = 5. Setd1b: control, n = 4; cKO, n = 4).
- Bar chart showing the number of TSS regions (± 2 kb) that exhibit significantly altered H3K4me1.
- Venn diagram comparing the TSS regions with significantly decreased H3K4me3 in the 3 respective cKO mice.
- Bar chart showing the number of differentially expressed genes from bulk RNA‐seq in each of the 3 KMT cKO mice. Kmt2a: control, n = 5; cKO, n = 6. Kmt2b: control, n = 8; cKO, n = 11. Setd1b: control, n = 6; cKO, n = 6).
- Venn diagram comparing the significantly downregulated genes amongst the 3 respective cKO mice.
- Volcano plots showing differential gene expression in the three KMT cKO mice (data from bulk RNA‐sequencing results is compared here) together with the overlay of neuron‐specific genes. Although the total number of downregulated genes in each KMT cKO are comparable, more neuron‐specific genes are affected in Setd1b cKO, followed by Kmt2a cKO and Kmt2b cKO.
- Downregulated genes with concomitantly decreased H3K4me3 in each of the 3 KMT cKO mice were tested for the overlap to the 836 neuron‐specific genes we had defined for the hippocampal CA region (See Fig EV4). Only for Setd1b cKO mice a highly significant odds ratio (Fisher’s exact test) was observed, while there was no significant association among neuron‐specific genes and the genes affected in Kmt2a cKO or Kmt2b cKO mice.
Data information: Bar graphs indicate mean, Error bars indicate ± SEM. “n” indicates biological replicates.
While we observed that genes downregulated in Kmt2a or Kmt2b cKO mice also display decreased H3K4me3 at the TSS (Fig 5A), only the genes downregulated in Setd1b cKO mice were characterized by reduced H3K4me3 and also increased H3K4me1 at their TSS (Fig 5A). Consequently, the genes that concomitantly exhibited decreased H3K4me3 levels and were downregulated in Setd1b cKO mice displayed significantly broader H3K4me3 peaks at the TSS (Fig 5B) and were expressed at higher levels under basal conditions when compared with the genes controlled by Kmt2a or Kmt2b (Fig 5C; Dataset EV22 and 23), although it has to be mentioned that the direct comparison of the expression values in Setd1b and Kmt2a cKO mice failed to reach significance (Fig 5C; Dataset EV22 and 23).
Figure 5. Comparative analysis of H3K4me3 peak width in Setd1b, Kmt2a and Kmt2b cKO mice.
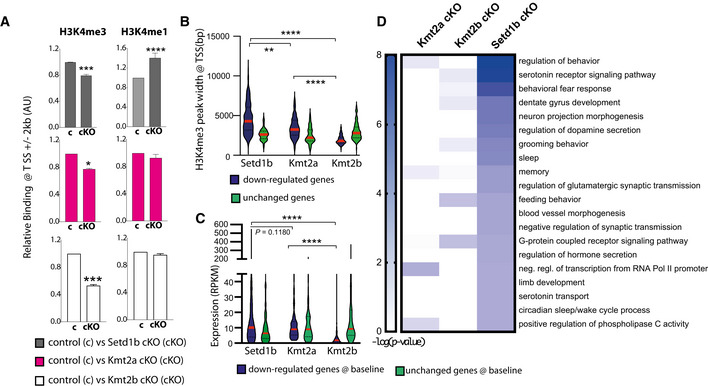
- Left panel: Bar graphs showing H3K4me3 binding around the TSS (± 2 kb) of downregulated genes in either of the 3 KMT cKO mice (Two‐way ANOVA: *P‐value < 0.05, ***P‐value < 0.001). Right panel depicts H3K4me1 binding at the same TSS regions (Two‐way ANOVA: ****P‐value < 0.0001). Note that only in Setd1b cKO mice decreased H3K4me3 is accompanied by significantly increased H3K4me1.
- Genes exhibiting decreased H3K4me3 and reduced expression in Kmt2a, Kmt2b or Setd1b cKO mice were analyzed for H3K4me3 peak width around the TSS under basal conditions. Genes affected in Setd1b cKO mice displayed significantly broader H3K4me3 peak width when compared to genes affected in Kmt2a or Kmt2b cKO mice. H3K4me3 peak widths at random sets of unchanged genes of equal size are shown for comparison. Comparisons between genes with decreased H3K4me3 and expression in the KMT cKOs: Kruskal–Wallis test, P‐value < 0.0001. Post‐hoc multiple comparisons, Dunn’s test: Setd1b cKO vs Kmt2a cKO, **P‐value < 0.01; Setd1b cKO vs Kmt2b cKO, ****P‐value < 0.0001; Kmt2a cKO vs Kmt2b cKO, ****P‐value < 0.0001. Comparisons between “down” and “unchanged” sets in each KMT cKO: Setd1b cKO, Mann–Whitney test P‐value < 0.0001; Kmt2a cKO, Mann–Whitney test P‐value < 0.0001; Kmt2b cKO, Mann–Whitney test P‐value < 0.0001.
- Violin plots showing average basal expression of downregulated genes with decreased H3K4me3 levels at the TSS in Kmt2a, Kmt2b, or Setd1b cKO mice. Genes affected in Setd1b KO mice are expressed at significantly higher levels under basal conditions when compared with genes affected in Kmt2a cKO or Kmt2b cKO mice. Comparisons between genes with decreased H3K4me3 and expression in the KMT cKOs: Kruskal–Wallis test, P‐value < 0.0001. Post‐hoc multiple comparisons, Dunn’s test: Setd1b cKO vs Kmt2a cKO, P‐value = 0.1180; Setd1b cKO vs Kmt2b cKO, ****P‐value < 0.0001; Kmt2a cKO vs Kmt2b cKO, ****P‐value < 0.0001. Comparisons between “down” and “unchanged” sets in each KMT cKO: Setd1b cKO, Mann–Whitney test P‐value < 0.01; Kmt2a cKO, Mann–Whitney test P‐value < 0.05; Kmt2b cKO, t‐test P‐value < 0.0001.
- Heat map showing functional pathways of genes affected in Kmt2a, Kmt2b or Setd1b cKO mice. Note that genes affected by loss of Setd1b specifically represent pathways linked to excitatory neuronal function.
Data information: Bar graphs indicate mean, Error bars indicate ± SEM. “n” indicates biological replicates. Violin plots: the dashed center line indicates the median, while the upper and lower dotted lines represent the 75th and 25th percentiles, respectively
The fact that under basal conditions, genes regulated by Setd1b exhibit a wider H3K4me3 distribution at the TSS compared with those regulated by the other two KMTs suggests that Setd1b spends more time at the TSS and therefore moves further downstream, which is in line with the suggested mode of action for H3K4 KMTs (Soares et al, 2017). This view is further supported by our observation that the number of genomic regions with decreased H3K4me3 was similar in Kmt2a, Kmt2b, and Setd1b cKO mice when we analyzed the first 2 kb downstream of the TSS (Appendix Fig S5A and B). However, significantly more regions were affected in Setd1b cKO mice when we analyzed the region 5 kb downstream the TSS (Appendix Fig S5B). In turn, the genomic regions showing decreased H3K4me3 in Kmt2a cKO and Kmt2b cKO are concentrated in very close vicinity of TSS when compared with the regions affected in Setd1b cKO mice (Appendix Fig S5C–E). These data also support the view that Setd1b promotes efficient tri‐methylation of H3K4 at genes that already underwent mono‐methylation by other KMTs.
In line with our data showing that the genes affected in Kmt2a, Kmt2b, and Setd1b cKO mice substantially differ, functional GO category analysis revealed that genes affected in the 3 KMT cKO mice also represent different functional categories. When compared with the genes affected in Kmt2a or Kmt2b cKO mice, genes that were downregulated and concomitantly exhibited decreased H3K4me3 in Setd1b cKO mice represent pathways intimately linked to learning and memory and the function of hippocampal excitatory neurons such as “regulation of behavior,” or “memory” (Fig 5D; Dataset EV24). Further analysis revealed that the genes affected in Kmt2a cKO mice also partly relate to neuronal plasticity functions (Appendix Fig 6A; Dataset EV24) while the genes affected in Kmt2b cKO mice represent almost exclusively pathways important for basal cellular function such as metabolic processes (Appendix Fig 6B; Dataset EV24). We also compared the genes with significantly reduced H3K4me3 at the TSS region (± 2 kb) among the three KMT cKOs and a study that analyzed H3K4me3 in bulk hippocampus tissue of a mouse model for AD (Gjoneska et al, 2015). Interestingly, the overlap was significantly higher for Setd1b cKO mice (Fig EV5).
Figure EV5. Comparison of H3K4me3 changes in Setd1b cKO, Kmt2a cKO, Kmt2b cKO, and CK‐p25 mice.
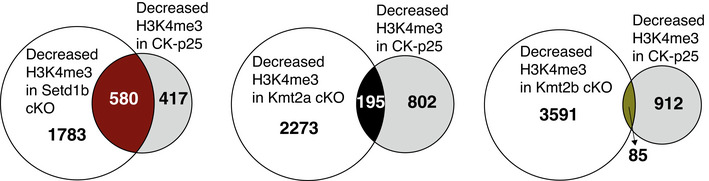
Overlap of TSS regions (± 2 kb) with decreased H3K4me3 in each of the three KMT cKO mice with those decreased in mouse model for Alzheimer’s disease (CK‐p25 mice) (Gjoneska et al, 2015). The pattern of overlapping regions (Setd1b > Kmt2a > Kmt2b) is in agreement with our suggested role of the 3 KMTs in the regulation of neuronal genes important for memory function. Left panel: A highly significant overlap was only seen in case of Setd1b cKO mice (580 out of 997 regions; hypergeometric test: P‐value = 0). Middle panel: The overlap between Kmt2a cKO and CK‐p25 is much smaller (195 TSS regions) but still remains significant according to the hypergeometric test (P‐value = 0.00000292). Right panel: The overlap between Kmt2b cKO and CK‐p25 is in turn negligible (85 TSS regions) and is not significant (hypergeometric test: P‐value = 1). It is important to note that we re‐analyzed our ChIP‐seq data from Kmt2a, Kmt2b, and Setd1b cKO mice together with the data from the CK‐p25 mice and selected only genomic regions which showed H3K4me3 signal in all datasets to allow for a reliable comparison. Thus, the total numbers of H3K4me3 regions that differ between conditions are slightly different to our results reported, for example, in Fig 4A. This is also due to the fact that Gjoneska et al. analyzed bulk hippocampal tissue, whereas our experiments are based on sorted hippocampal neurons from the CA region. Nevertheless, the fact that the overlap is still substantial with Setd1b cKO and lower or negligible in the other two KMT cKOs supports the view that Setd1b is particularly important for regulating genes important for synaptic plasticity and memory function.
In summary, our observations so far support the view that the Setd1b, Kmt2a, and Kmt2b serve distinct functions in excitatory hippocampal neurons of the postnatal brain and that among them Setd1b might be the most relevant for the regulation of learning processes. In line with this, comparison of hippocampus‐dependent learning in Setd1b, Kmt2a, and Kmt2b cKO mice in the Morris water maze task, suggest that learning impairment is more pronounced in Setd1b cKO mice (Fig 6A). To further elucidate the distinct roles of Setd1b, Kmt2a, and Kmt2b, we hypothesized that they might be expressed in different neuronal subtypes. Therefore, we isolated the hippocampus from 3‐month‐old wild‐type mice, sorted the nuclei and performed single nuclear (snuc) RNA‐seq (Fig 6B). We were able to detect all major cell types of the hippocampus and found Kmt2a, Kmt2b, and Setd1b as well as the other three H3K4 KMTs to be expressed in all of these cell types (Fig 6C). We did not observe any particular neuronal cell type with especially high Setd1b expression (Fig 6D). Interestingly, the snucRNA‐seq data revealed higher Kmt2a expression when compared with Setd1b and Kmt2b across cell types including excitatory neurons of the CA region, an effect that was not due to the total amount of reads detected per cell (Fig 6C–E). These data were confirmed via RNAscope. Thus, when we analyzed the pyramidal neurons of the hippocampal CA region, we observed significantly more transcripts/cell for Kmt2a when compared with Kmt2b or Setd1b (Fig 6F). In summary, these data may suggest that highly expressed enzymes such as Kmt2a are essential to ensure neuronal H3K4 methylation sufficient for basal and neuron‐specific cellular processes and that the additional expression of enzymes such as Setd1b is essential to further promote H3K4me3 at genes particularly important for neuronal functions like synaptic plasticity.
Figure 6. Comparative analysis of learning and single nucleus gene expression of Kmt2a, Kmt2b and Setd1b .
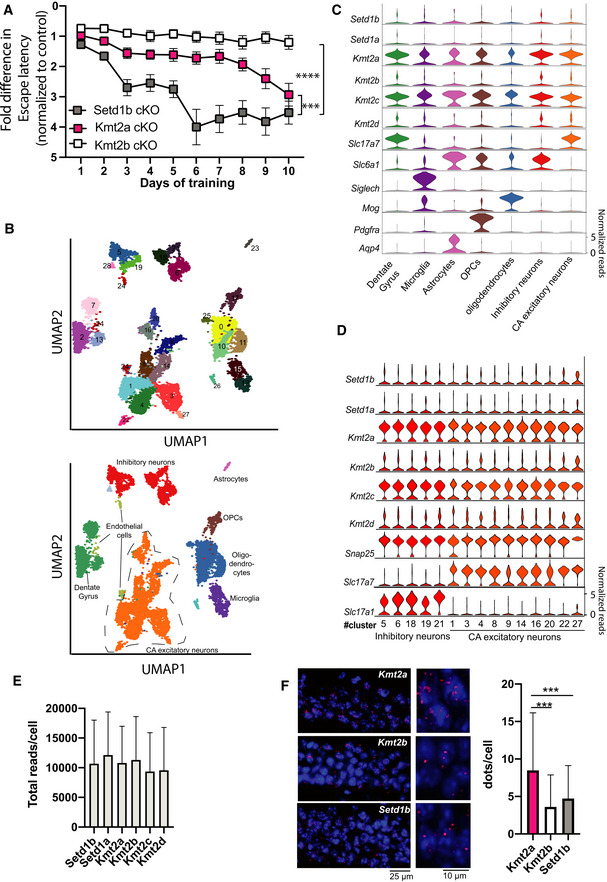
- Comparison of the escape latency in the 3 different KMT cKO mice during 10 days of training. Although we applied the same experimental setting, the experiments were performed at different time points ((Kerimoglu et al, 2013; Kerimoglu et al, 2017), and this study). Thus, for comparison we normalized the data to the corresponding control group. In this plot an increase in the fold change of the normalized escape latency depicts the difference to the corresponding control group. Hence, a higher fold change of the normalized escape latency indicates a greater difference to the corresponding control and thus more severe learning impairment. Setd1b cKO (n = 14) vs Kmt2a cKO (n = 13): Repeated measures ANOVA, genotype effect: F(1,25) = 16.83, ***P‐value < 0.001. Setd1b cKO (n = 14) vs Kmt2b cKO (n = 22): Repeated measures ANOVA, genotype effect: F(1,34) = 70.66, ****P‐value < 0.0001.
- UMAP plot showing the data from 15,661 nuclei. Upper panel shows the different clusters and their corresponding numbers. The lower panel shows the same UMAP plot colored for different groups of cells that are further analyzed in (C) and (D).
- Violin plots showing the expression of the 6 KMTs as well as marker genes for the different hippocampal cell types according the UMAP shown in lower panel B. Note that Kmt2a and Kmt2c are the highest expressed KMTs across cell types, while the other KMTs shown low to moderate expression.
- Violin plots showing the expression of the six KMTs and corresponding marker genes within the different clusters representing inhibitory and excitatory neurons. Note that there is no obvious difference of KMT expression between cell types. In agreement with the data shown in panel C, Kmt2a and Kmt2c exhibit the highest expression levels.
- Bar charts showing the total number of reads/cell that are positive for the corresponding KMT.
- Left panel shows representative images of RNAscope performed for Kmt2a, Kmt2b, and Setd1b. Right panel shows a bar chart quantifying the dots/cell (n = 1500 cells from 2 mice) indicative of the corresponding expression level. Kmt2a expression was significantly higher when compared with Kmt2b or Setd1b (***P‐value < 0.0001, Student's t‐test).
Data information: Bar graphs indicate mean, Error bars indicate ± SEM. “n” indicates biological replicates.
Discussion
We show that postnatal loss of Setd1b from excitatory forebrain neurons impairs learning in mice when measured in the hippocampus‐dependent water maze paradigm. These data are in line with previous findings showing that hippocampal H3K4me3 increases in response to memory training in rodents (Gupta et al, 2010), while its levels are reduced in the hippocampus of a mouse for AD‐like neurodegeneration (Gjoneska et al, 2015) and in postmortem human brain samples of patients suffering from cognitive diseases (Shulha et al, 2012). Our data furthermore support previous genetic studies linking mutations in SETD1B to intellectual disability (Labonne et al, 2016; Hiraide et al, 2018; Roston et al, 2020). Although spatial reference memory measured in the water maze paradigm critically depends on hippocampal function, it is important to note that Setd1b deletion is not specific to the hippocampus and that we cannot exclude that other brain regions and subtle changes in postnatal development may also contribute to the observed phenotype. Impaired hippocampus‐dependent memory has also been observed in mice that lack Kmt2a (Gupta et al, 2010; Kerimoglu et al, 2017) or Kmt2b (Kerimoglu et al, 2013) from excitatory neurons of the postnatal forebrain. Our comparative analysis suggests, however, that the effect is most severe in Setd1b cKO mice. Interestingly, mice heterozygous for Setd1a, the close homologue to Setd1b that is genetically linked to schizophrenia, show no impairment in the water maze task but rather exhibit impaired working memory and schizophrenia‐like phenotypes (Mukai et al, 2019). Distinct roles for Setd1b and Setd1a are also reported for other cellular system (Bledau et al, 2014; Brici et al, 2017; Arndt et al, 2018; Schmidt et al, 2018; Kranz & Anastassiadis, 2020). These data suggest that the different H3K4 KMTs, and at least Kmt2a, Kmt2b, Setd1a, and Setd1b, likely serve distinct functions in the postnatal brain. It will therefore be important to eventually generate corresponding and comparable data for all six H3K4 KMTs.
The molecular characterization of Setd1b cKO further confirms this view. Here, we focused on the analysis of the hippocampus, since this brain region is critical for spatial reference memory, which was severely impaired in Setd1b cKO mice. It will nevertheless be interesting to study other brain regions such as the prefrontal cortex in future experiments. In line with the role of Setd1b in regulating H3K4me3, we observed a substantial decrease of neuronal H3K4me3 levels and the vast majority of these changes were observed close to TSS regions. Many genes with decreased H3K4me3 also exhibited reduced H3K9ac, which is in line with previous data showing that H3K4me3 appears to be a pre‐requisite for H3K9ac, most likely since H3K4 KMTs interact with histone acetyltransferases (Kerimoglu et al, 2013; Mishra et al, 2014; Wang et al, 2009). For example, SETD1B and the histone‐acetyltransferase KAT2A were shown to interact with WDR5, a central component of H3K4 KMT complexes (Lin et al, 2016; Ma et al, 2018), which is interesting since loss of Kat2a from excitatory forebrain neurons also leads to severe impairment of spatial reference memory (Stilling et al, 2014).
Our finding that H3K4me1 levels were increased at a substantial number of TSS regions that exhibited decreased H3K4me3 in Setd1b cKO mice suggests that H3K4 mono‐methylation at the TSS regions of the affected genes is likely mediated by other KMTs in addition to Setd1b. This is in agreement with increasing evidence suggesting that the different H3K4 KMTs exhibit some specificity toward mono‐, di‐, or tri‐methylation. However, specificity likely depends on the cellular context and care has to be taken when interpreting the different in vitro and in vivo data. Nevertheless, our data are in line with studies showing that Setd1b preferentially mediates H3K4‐trimethylation when compared with the other KMTs such as Kmt2a and Kmt2b that are believed to mediate mono‐ and di‐methylation with comparatively low H3K4 tri‐methylation activity (Lee & Skalnik, 2008; Wu et al, 2008; Rao & Dou, 2015; Shinsky et al, 2015; Bochyńska et al, 2018; Kranz & Anastassiadis, 2020).
In agreement with these findings, we show that neuronal H3K4me1 is exclusively reduced in Kmt2a and Kmt2b cKO. Since H3K4me3 is similarly reduced in these cKO mice, our data suggest that neuronal Kmt2a and Kmt2b can also catalyze H3K4 tri‐methylation. In such a scenario, Setd1b might act in concert with other KMTs to further facilitate H3K4me3 at specific genes. This might also explain our finding that genes affected in Setd1b cKO mice exhibit the widest H3K4me3 distribution at their TSS and the highest RNA expression levels under basal conditions. A broad H3K4me3 peak around TSS is indicative of higher transcriptional frequency and RNA expression levels and has thus been linked to the expression of genes that are of particular importance for a given cell type (Benayoun et al, 2015; Park et al, 2020). This is in line with our observation that genes decreased in hippocampal neurons in Setd1b cKO mice are characterized by a broad H3K4me3 peak and represent functional pathways critically involved in synaptic plasticity and learning and memory. In agreement with this, a recent study showed that memory training specifically activates hippocampal genes with broad H3K4me3 peaks at the TSS (Collins et al, 2019). It is therefore interesting to note that the genes affected in Setd1b cKO mice not only showed a broader H3K4me3 peak and higher expression when compared with the genes affected in Kmt2a (it has to be mentioned that this comparison failed to reach statistical significance, P = 0.1) or Kmt2b cKO mice but that also memory performance in the water maze training was more severely affected in Setd1b cKO mice when compared with mice lacking Kmt2a or Kmt2b. These data are also in line with a recent study in mouse embryonic stem cells in which Setd1b was associated with the regulation of highly expressed genes that exhibit a broad H3K4me3 peak, while Kmt2b was linked to the expression of genes with narrow H3K4me3 peaks (Sze et al, 2020). Interestingly, that study suggested a functional redundancy of Setd1b and Setd1a. However, our data suggest that this is not the case in post‐mitotic neurons. Moreover, mutations in either Setd1a or Setd1b lead to distinct neuropsychiatric diseases and unlike Setd1b cKO mice, Setd1a heterozygous mutant mice do not exhibit impairment of long‐term memory consolidation (Mukai et al, 2019).
These findings may suggest that among the three H3K4 KMTs studied here, Kmt2a and Kmt2b are essential to ensure the sufficient expression of genes important for basal cellular processes and in the case of Kmt2a also specific neuronal functions. The additional presence of Setd1b may further facilitate H3K4me3, thereby enabling the preeminent expression of genes specific for hippocampal excitatory neuronal function.
At present, we cannot conclusively answer the question how Setd1b affects the expression of a specific set of hippocampal genes. Our snucRNA‐seq analysis showed that Setd1b is expressed at similar levels in all types of excitatory neurons of the hippocampal CA region suggesting that its specific function of facilitating learning behavior is not driven by its expression in a specific neuronal subtype. The fact that loss of either Kmt2a, Kmt2b, or Setd1b leads to distinct changes in neuronal gene expression might also be due to the specific subunit compositions of the KMT complexes. Previous data suggest that H3K4 KMTs associate with different co‐activators (Yokoyama et al, 2004; Dreijerink et al, 2006; Lee et al, 2006; Scacheri et al, 2006); Shilatifard, 2012; Bochyńska et al, 2018; Kranz & Anastassiadis, 2020). While all H3K4 KMTs are believed to interact with a number of core subunits (Kranz & Anastassiadis, 2020), data from other cell types than neurons show that KMT2A and KMT2B can bind the transcriptional regulator multiple endocrine neoplasia type 1 (Menin) (Hughes et al, 2004). In contrast, SETD1B was shown to interact with CXXC finger protein 1 (CFP1) and WDR82 which binds to RNA‐polymerase II. This is in line with data suggesting that SETD1A and SETD1B are the major KMTs found at TSS regions (Bi et al, 2011; Deng et al, 2013). Future studies will need to further explore this possibility.
It is however, interesting to note that the genes with decreased H3K4me3 in the hippocampus of Kmt2a or Kmt2b cKO mice were enriched for binding sites of ETS and E2F transcription factors, respectively (Kerimoglu et al, 2017). In case of Setd1b cKO mice, we observed an enrichment for REST, a key transcription factor inhibiting the expression of neuron‐specific genes (Hwang & Zukin, 2018). This observation may help to understand why loss of Setd1b mainly affected the expression of neuron‐specific genes important for memory function. While future research should aim to further elucidate the specific role of SETD1B, it is interesting to note that the H3K4 Demethylase KMD5C was found to regulate REST target genes (Tahiliani et al, 2007).
Taking into account that decreased neuronal H3K4me3 levels have been observed in cognitive and neurodegenerative diseases, therapeutic strategies that reinstate specifically the expression of neuronal‐enriched genes controlled by SETD1B might be particularly helpful. We suggest that the epigenetic drugs currently tested in pre‐clinical and clinical settings for cognitive diseases should also be analyzed for their potential to reinstate the H3K4me3 peak width at the TSS of genes linked to neuronal function and learning behavior.
The aim of this study was to analyze the role of Setd1b on learning behavior when deleted postnatally and to explore its impact on epigenetic gene expression and compare these data with findings obtained from postnatal deletion of Kmt2a and Kmt2b. While we observe that Setd1b is required for memory function and controls the expression of genes linked to neuronal plasticity, it will be important to study the functional and structural consequences in the corresponding neuronal networks. At present such data is rare. A recent study employed heterozygous Kmt2a constitutive knockout mice and found decreased dendritic spine density in the ventral hippocampal CA1 region (Vallianatos et al, 2020). To compare the role of the different H3K4 KMTs in synaptic morphology and network activity during development and in the postnatal brain is thus an important task for future research.
In summary, we identify H3K4 methyltransferase Setd1b as a crucial factor regulating genes important for hippocampal excitatory neuronal function, which are involved in synaptic plasticity and learning.
Materials and Methods
Animals
All animals used in this study were of the same genetic background (C57BL/6J) mice and of 3–6 months of age. The experimental groups were age and sex matched. Mice were kept in standard home cages with food and water provided ad libitum. All experiments were performed according to the animal protection law of the state of Lower Saxony. The floxed Setd1b mice were first described by Bledau et al (2014). The CRE mice were first described by Minichiello et al (1999) and were used in previous studies related to Kmt2a and Kmt2b (Kerimoglu et al, 2013; Kerimoglu et al, 2017). Deletion of the target gene is restricted to the forebrain excitatory neurons and is initiated between second and third postnatal weeks.
Behavior experiments
Water maze
The behavioral experiments were performed as described previously (Kerimoglu et al, 2017). For in depth feature analysis from water maze data, a modified version of MUST‐C algorithm was used (Illouz et al, 2016; Islam et al, 2021). In brief, different strategies were defined based on trajectories mice employed in each trial. Cumulative score for hippocampal‐dependent strategy score was calculated as follow:
n, total trial number; Sdfi, Frequency of direct strategy in ith trial; Sdc, given strategy score for direct search; 10; mc, total number of mice; Sscfi, Frequency of short chaining in ith trial; Sscc, given strategy score for short chaining search; 9.5; Slcfi, Frequency of short chaining in ith trial; Slcc, given strategy score for long chaining search; 7.
Y‐maze and open field
The open field tests were performed as described previously (Islam et al, 2021). Working memory was accessed with a Y‐shaped plastic runway (10 × 40 cm arms, walls 40 cm high). The mouse was placed into the Y‐maze in the triangle‐shaped central platform and left to freely explore the maze for 5 min. Percentage of spontaneous alterations (choice of “novel” arm: when animal goes into different arm then before) was recorded and scored via a videosystem (TSE).
Tissue isolation and processing
ChIP‐Seq and bulk RNA‐Seq experiments from NeuN (+) nuclei were performed from the hippocampal cornu ammonis (CA) region, which consists of the hippocampus without the dentate gyrus and was isolated as described before (Kerimoglu et al, 2013; Kerimoglu et al, 2017). For single nuclear RNA‐sequencing whole hippocampal tissue was isolated. The tissues were dissected and flash frozen in liquid nitrogen and stored at −80°C until further processing.
Cell‐type specific nuclear RNA isolation and sequencing
Frozen CA tissues from the left and right hemisphere of two mice were pooled together and processed on ice to maintain high RNA integrity. Tissue was homogenized using a plastic pestle in a 1.5‐ml Eppendorf tube containing 500‐µl EZ prep lysis buffer (Sigma, NUC101‐1KT) with 30 strokes. The homogenate was transferred into 2‐ml microfuge tubes, lysis buffer was added up to 2 ml and incubated on ice for 7 min. After centrifuging for 5 min at 500 g, the supernatant was removed and the nuclear pellet was resuspended into 2 ml of lysis buffer and incubated again on ice (7 min). After centrifuging for 5 min at 500 g, the supernatant was removed and the nuclei pellet was resuspended into 500‐μl of nuclei storage buffer (NSB: 1 × PBS; Invitrogen, 0.5% RNase‐free BSA; Serva, 1:200 RNaseIN plus inhibitor; Promega, 1 × EDTA‐free protease inhibitor; Roche) and filtered through 40 µm filter (BD falcon) with additional 100 µl of NSB to collect residual nuclei from the filter. Nuclei were stained with anti‐NeuN‐Alexa488‐conjugated antibody (1:1,000) for 45 min and washed once with NSB. Stained nuclei were then FACS‐sorted with FACSaria III using 85 µm nozzle. Nuclei were gated by their size, excluding doublets and neuronal nuclei were separated from non‐neuronal nuclei by their NeuN‐Alexa488 fluorescence signal. Sorted nuclei were collected into a 15‐ml falcon tube precoated with NSB, spun down and RNA was isolated using Trizol LS. After addition of chloroform according to the Trizol LS protocol, aqueous phase was collected and RNA was isolated by using Zymo RNA clean and concentrator‐5 kit with DNAse treatment. Resulting RNA concentration were measured in Qubit and RNA‐seq was performed using 100 ng of neuronal RNA with illumina TruSeq RNA Library Prep Kit. Since glial nuclei are smaller and contains very little amount of RNA, neuronal nuclear RNA was scaled down and 1 ng from both neuronal and glial nuclear RNA was used to make RNA‐seq libraries using Takara SMART‐Seq v4 Ultra Low Input RNA Kit. Libraries were sequenced using single‐end 75 bp in Nextseq 550 or single‐end 50 bp in HiSeq 2000, respectively.
Cell‐type specific chromatin isolation and ChIP sequencing
Frozen tissues were homogenized, formaldehyde (1%)‐fixed for 10 min and quenched with 125 mM of glycine for 5 min. Debris was removed by sucrose gradient centrifugation. The resulting nuclear pellet was stained with anti‐NeuN‐Alexa488‐conjugated antibody (1:1,000) for 25 min and washed 3 times with PBS. Stained nuclei were then FACS‐sorted with FACSaria III using 85 µm nozzle. Nuclei were gated similarly as described previously (Halder et al, 2016). Sorted nuclei were collected into a 15‐ml falcon tube and transferred into 1.5‐ml tubes. The nuclear pellet was flash‐frozen in liquid nitrogen and saved at −80°C for further processing. For chromatin shearing, the pellet was resuspended into 100‐μl RIPA buffer (containing 1% SDS) and sonicated for 25 cycles in Diagenode bioruptor plus with high power and 30 cycles on/30 cycles off. Chromatin shearing was checked by taking a small aliquot and decrosslinking the DNA by 30 min RNAse and 2 h of proteinase K treatment. DNA was isolated using SureClean Plus protocol. Sheared chromatin size was determined using Bioanalyzer 2100 (DNA high sensitivity kit) and the concentration was measured using Qubit 2.0 fluorometer (DNA high sensitivity kit). 0.3 µg of chromatin was used along with 1 µg of antibody to do ChIP for H3K4me3 (Abcam ab8580), H3K4me1 (Abcam ab8895), H3K27ac (Abcam ab4729) and H3K9ac (Millipore 07‐352). ChIP was performed as previously described (Halder et al, 2016). The resulting ChIP DNA was subjected to library preparation using NEBNext Ultra II DNA library preparation kit and sequenced for single end 50 bp at illumina HiSeq 2000.
ChIP‐Seq analysis
Pre‐Processing, Profile Plots, Peak Calling, Differential Binding Analysis, Annotation, Motif Enrichment Analysis, and Visualization.
Base calling and fastq conversion were performed using Illumina pipeline. Quality control was performed using fastqc (www.bioinformatics.babraham.ac.uk/projects/fastqc). Reads were mapped to mm10 mouse reference genome with STAR aligner v2.3.0.w. PCR duplicates were removed by rmdup‐s function of samtools (Li et al, 2009). BAM files with unique reads belonging to the same group were merged into a single BAM file with the merge function of samtools. Profile plots were created from these merged BAM files with NGSPlot (Shen et al, 2014).
Peak calling was performed using MACS2 (Feng et al, 2012) against the input corresponding to the particular group (i.e., control or cKO) using q < 0.1. Consensus peaksets were generated for each histone modification individually using the Diffbind package of Bioconductor (Ross‐Innes et al, 2012) with the command dba.count and the parameter minOverlap = 1. Using the latter parameter ensured that peaks passing the significance threshold even only in one sample were included in the consensus peakset for further analysis, thereby minimizing the possibility of overlooking genomic regions that may fall behind the threshold albeit having biological importance. Then, these consensus peaksets were intersected with each other using the intersect function of bedtools with default parameters, providing one common peakset representing all four histone modifications. The differential binding analysis for each histone mark between control and Setd1b cKO was then performed using Diffbind (Ross‐Innes et al, 2012) with this common peakset as input.
For the comparison of H3K4me3 changes in Kmt2a cKO, Kmt2b cKO, and Setd1b cKO common peaksets for each individual histone mark from three separate ChIP‐Seq experiments were extracted in the same way as above. In this case, first, consensus peaksets for a histone mark from each individual ChIP‐Seq experiment (i.e., “Control vs Kmt2a cKO,” “Control vs Kmt2b cKO,” and “Control vs Setd1b cKO”) were determined using Diffbind as above. Then, these consensus peaksets were intersected with each other as above, and as a result we obtained a set of common regions detected in all three independent experiments that possess H3K4me3 mark. For the purpose of comparing the effects of the three KMT knockdowns on H3K4me3, the differential binding analysis for each individual ChIP‐Seq experiment was performed using this common peakset as input.
We also wanted to compare the changes in H3K4me1 in these three KMT cKO mice. But additionally, based on our prior observations from Setd1b cKO mice, we wanted to also investigate the interplay between H3K4me3 and H3K4me1, and whether it differs between the three KMT cKO mice. We therefore first came up with the common peakset for H3K4me1 from all three ChIP‐Seq experiments as described above. Then, we intersected this common H3K4me1 peakset with the common H3K4me3 peakset and came up with a peakset representing regions containing both H3K4me3 and H3K4me1 marks in all three separate experiments (“me3_me1_3_kmt_ckos”). The differential binding analyses and comparisons relating to H3K4me1 in the three KMT cKOs were performed using “me3_me1_3_kmt_ckos” as input. Diffbind package was used for differential binding analysis with in‐built DESEQ2 option for differential analysis (Ross‐Innes et al, 2012). For the initial analysis of Setd1b cKO alone, we used the cut‐off “adjusted P‐value < 0.05 and |fold change| > 1.5” to determine significance. For comparing differential binding in Kmt2a cKO, Kmt2b cKO, and Setd1b cKO mice, we used a more lenient cut‐off—“adjusted P‐value < 0.1 and |fold change| > 1.2” in order to avoid missing out on overlaps and/or common trends that might be biologically relevant. The annotation of the genomic regions and transcription factor motif enrichment analysis were performed with HOMER (Heinz et al, 2010). ChIPSeeker package of Bioconductor (Yu et al, 2015) was used to calculate the proportion of the types of genomic regions in a given set and to create corresponding pie charts. Graph Pad Prism was used to generate violin plots. Outliers were removed using the Rout methods (Q = 1%) in GraphPad Prism. Integrated Genome Browser (IGB) was used to make visualizations representing the distribution of histone marks and RNA expression over selected genes (Nicol et al, 2009).
Obtaining H3K4me3 width at Promoters
For promoters with decreased H3K4me3 and different H3K4me1 trends at TSS proximal regions in Setd1b cKO
First, we performed differential binding analysis in “Control vs Setd1b cKO” for H3K4me3 and H3K4me1 using the “me3/me1/k9ac/k27ac common peakset.” Then we extracted the TSS proximal regions (± 2,000 bp from TSS) with significantly decreased H3K4me3 in Setd1b cKO (adjusted P‐value < 0.05 and fold change < −1.5). These TSS proximal regions were then separated into three groups depending on the concomitant change of H3K4me1; (i) increased H3K4me1 (fold change > 1.5), (ii) decreased H3K4me1 (fold change < −1.5), and (iii) not changed H3K4me1 (the rest). Then each of these TSS proximal regions was intersected to the consensus H3K4me3 peakset with bedtools using intersect function with the option “‐u.” The latter option ensures that the full original feature in the first input (in this case always the H3K4me3 peakset), not just the overlapping portion, is reported once the overlap is found. In this way the full extent of H3K4me3 distribution around the target genomic region can be appreciated. Finally, the width was calculated by subtracting chromosome start coordinates from chromosome end coordinates and adding 1 (chromosome end − chromosome start + 1).
For promoters with decreased H3K4me3 at TSS proximal regions in different KMT cKOs
For the purpose of comparison between the three KMT cKO mice, we chose a more lenient cut‐off (adjusted P‐value < 0.1 and fold change < −1.2) in order to avoid as much as possible excluding genomic regions showing the same trend but not making it past the threshold in some of the cases. The TSS proximal regions (± 2,000 bp) with significantly decreased H3K4me3 in each KMT cKO were overlapped to the consensus H3K4me3 peakset using intersect u. Again, in this way we were able to capture the full extent of H3K4me3 around the selected target regions. The width at the resulting region was then calculated by chromosome end − chromosome start + 1. A random list of the same size was also generated from the TSS proximal regions with unchanged H3K4me3 for each cKO using the same procedure. The criteria for unchanged H3K4me3 were adjusted P‐value > 0.5 and |log2(fold change)| > 0.1.
For downregulated genes
Again, to obtain a more comprehensive view of downregulation of gene expression and its concordance with peak width, we chose a more lenient cut‐off; adjusted P‐value < 0.1 and fold change < −1.2.
From the consensus H3K4me3 peakset, the promoter regions were selected and annotated for their gene names using HOMER. To capture the whole extent of H3K4me3 along the promoters as much as possible, we used the default standard for promoter identification implemented by ChIPSeeker (± 5,000 bp; see above). From the resulting annotated file, the promoter regions belonging to significantly downregulated genes were extracted and the widths were calculated as above. In each case, a random list of the same size from genes with unchanged expression was also generated using the same procedure. The criteria for unchanged expression were adjusted P‐value > 0.5 and |log2(fold change)| < 0.1.
For the genes with both decreased TSS proximal H3K4me3 and downregulated expression in different KMT cKOs
The bed files generated in Section 2 by the overlap of the consensus H3K4me3 peakset with TSS proximal regions having significantly decreased H3K4me3 were annotated for gene names using HOMER. From the resulting annotated file, the genomic coordinates belonging to the downregulated genes were extracted and the widths were calculated. Again, in each case a random list of the same size containing genes with unchanged expression and H3K4me3 was selected using the same procedure and the same criteria as above.
RNA‐Seq analysis
Base calling, fastq conversion, quality control, mapping of reads were performed as described before (Kerimoglu et al, 2017). Bulk RNA‐Seq was mapped with STAR on the whole genome and reads were counted with featureCounts thus considering spliced as well as unspliced transcripts. Differential expression was analyzed with DESeq2 package of Bioconductor (Love et al, 2014). RPKM values were calculated using edgeR package of Bioconductor. We used the cut‐off “adjusted P‐value < 0.1 and |fold change| > 1.2” to determine significance.
Functional GO enrichment analysis
Enrichment for functional GO categories was performed with the topGO package of Bioconductor (https://bioconductor.org/packages/release/bioc/html/topGO.html), using the weighted analysis option as described in the manual with default settings. As a result, a weighted P‐value for each GO category was calculated (described in the manual). In each separate analysis, only the GO categories (biological processes) with a weighted P‐value < 0.005 were selected and shown in the figures. All genes in the genome were used as the reference set (i.e., gene universe). The whole list of the GO categories, including the ones highlighted in the figures, can be found in supplemental figures 7, 12, and 24.
Single‐nucleus RNA‐Seq
Unfixed NeuN+ neuronal nuclei were isolated as mentioned above (section: Cell‐type‐specific nuclear RNA isolation and sequencing). Sorted neuronal nuclei were counted in a Neubauer chamber with 10% trypan blue (in PBS) and nuclei concentration were adjusted to 1,000 nuclei/µl. The nuclei were further diluted to capture and barcode either 2,000 or 4,000 nuclei according to Chromium single cell 3ʹ reagent kit v3 (10X genomics). Single nuclei barcoding, GEM formation, reverse transcription, cDNA synthesis, and library preparation were performed according to 10X genomics guidelines. Finally, the library was sequenced in Illumina NextSeq 550 according to manufacturer’s protocol. Gene counts were obtained by aligning reads to the mm10 genome (GRCm38.p4) (NCBI:GCA_000001635.6) using CellRanger software (v.4.0.0) (10X Genomics). The CellRanger count pipeline was used to generate a gene‐count matrix by mapping reads to the pre‐mRNA as reference to account for unspliced nuclear transcripts. The CellRanger aggr pipeline was used to generate an aggregated normalized gene‐count matrix from seven datasets. The final count matrix contained 15,661 cells with a mean of 56.150 total read counts over protein‐coding genes.
The SCANPY package was used for pre‐filtering, normalization, and clustering (Wolf et al, 2018). Initially, nuclei that reflected low‐quality cells (either too many or too few reads, nuclei isolated almost exclusively, nuclei expressing <10% of house‐keeping genes (Eisenberg & Levanon, 2013)) were excluded remaining in 15,656 nuclei. Next, counts were scaled by the total library size multiplied by 10,000, and transformed to log space. A total of 3,771 highly variable genes were identified based on dispersion and mean, the technical influence of the total number of counts was regressed out, and the values were rescaled. Principal component analysis (PCA) was performed on the variable genes, and UMAP was run on the top 50 principal components (PCs) (Becht et al, 2018). The top 50 PCs were used to build a k‐nearest‐neighbors cell–cell graph with k = 100 neighbors. Subsequently, spectral decomposition over the graph was performed with 50 components, and the Leiden clustering algorithm was applied to identify nuclei clusters. We confirmed that the number of PCs captures almost all the variance of the data. For each cluster, we assigned a cell‐type label using manual evaluation of gene expression for sets of known marker genes. Violin plots for marker genes were created using the stacked_violin function as implemented in SCANPY.
RNAscope
For the detection of Setd1b, Kmt2a, and Kmt2b, the RNAscope® Fluorescent Multiplex Assay (ACD Biosystems) was used according to the manufacturer’s instructions for fresh‐frozen tissue. Briefly, mice (n = 2) were sacrificed by cervical dislocation and the brains were removed quickly, flash‐frozen using liquid nitrogen and embedded in OCT. Then, 20 µm coronal sections were cut and stored at −80°C until further use. For the pre‐treatment, the sections were fixed with cold 10% neutral‐buffered formalin at 4°C for 15 min and afterward dehydrated using 50, 70, and 100% ethanol. Then, the samples were incubated with Protease IV for 30 min at RT. The probes were hybridized to the tissue for 2 h at 40°C using the HybEZ™ Humidifying System (ACD Biosystems). Probes were applied to detect Setd1b, Kmt2a, or Kmt2b. Positive and negative control probes were also used to control for signal sensitivity and specificity. The probes were then amplified following the manufacturer’s protocol. Kmt2a and Kmt2b were labeled with Atto647 and Setd1b with Atto550. Images were obtained on a Leica DMi8 microscope using a 40x air objective and the software CellProfiler (Carpenter et al, 2006) was used for downstream analysis.
Author contributions
AM initiated the project as part of her PhD thesis, performed behavior experiments, performed and analyzed immunohistochemistry, and analyzed mutant mice; MSS generated cell‐type specific ChIP/RNA‐seq and single nucleus RNA‐seq data and contributed to the analysis of cell‐type‐specific RNA‐seq, CK analyzed and interpreted ChIP‐seq and RNA‐seq data and supervised all bioinformatic data analysis, DMF analyzed single nucleus RNA‐seq data, MRI and JZ contributed to the behavioral analysis, LK, XX, and EMZ helped with the generation of single nucleus RNA‐seq; TPC helped with statistical analysis of data, SS, PDJ, and GE performed and supervised RNAscope experiments, RP and JC help with immunoblot analysis, AK and AFS provided material and analyzed data, AM, MSS, CK, and AF designed experiments and generated figures. CK and AF wrote the paper.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Dataset EV5
Dataset EV6
Dataset EV7
Dataset EV8
Dataset EV9
Dataset EV10
Dataset EV11
Dataset EV12
Dataset EV13
Dataset EV14
Dataset EV15
Dataset EV16
Dataset EV17
Dataset EV18
Dataset EV19
Dataset EV20
Dataset EV21
Dataset EV22
Dataset EV23
Dataset EV24
Source Data for Figure 1
Acknowledgments
This work was supported by the following third party funds to AF: The ERC consolidator grant DEPICODE (648898), the BMBF projects ENERGI (01GQ1421A) and Intergrament (01ZX1314D), the DFG under Germany’s Excellence Strategy ‐ EXC 2067/1 390729940 and SFB1286 as well as funds from the German Center for Neurodegenerative Diseases (DZNE) and the ERA‐NET Neuron. AM and SS are students of the International Max Planck Research School (IMPRS) for Genome Science. MSS and RI are students of the IMPRS for Neuroscience. Open access funding enabled and organized by Projekt DEAL.
The EMBO Journal (2022) 41: e106459
Contributor Information
Cemil Kerimoglu, Email: cemil.kerimoglu@dzne.de.
André Fischer, Email: andre.fischer@dzne.de.
Data availability
GEO accession GSE180326 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE180326).
References
- Arndt K, Kranz A, Fohgrub J, Jolly A, Bledau AS, Di Virgilio M, Lesche M, Dahl A, Höfer T, Stewart AF et al (2018) SETD1A protects HSCs from activation‐induced functional decline in vivo . Blood 131: 1311–1324 [DOI] [PubMed] [Google Scholar]
- Becht E, McInnes L, Healy J, Dutertre CA, Kwok I, Ng LG, Ginhoux F, Newell EW (2018) Dimensionality reduction for visualizing single‐cell data using UMAP. Nat Biotechnol 37: 38–44 [DOI] [PubMed] [Google Scholar]
- Benayoun BA, Pollina EA, Ucar D, Mahmoudi S, Karra K, Wong ED, Devarajan K, Daugherty AC, Kundaje AB, Mancini E et al (2015) H3K4me3 breadth is linked to cell identity and transcriptional consistency. Cell 165: 1281–1286 [DOI] [PubMed] [Google Scholar]
- Bi Y, Lv Z, Wang Y, Hai T, Huo R, Zhou Z, Zhou Q, Sha J (2011) WDR82, a key epigenetics‐related factor, plays a crucial role in normal early embryonic development in mice. Biol Reprod 84: 756–764 [DOI] [PubMed] [Google Scholar]
- Bledau AS, Schmidt K, Neumann K, Hill U, Ciotta G, Gupta A, Torres DC, Fu J, Kranz A, Stewart AF et al (2014) H3K4 methyltransferase Setd1a is first required at the epiblast stage, whereas Setd1b becomes essential after gastrulation. Development 141: 1022–1035 [DOI] [PubMed] [Google Scholar]
- Bochyńska A, Lüscher‐Firzlaff J, Lüscher B (2018) Modes of interaction of KMT2 Histone H3 Lysine 4 methyltransferase/COMPASS complexes with chromatin. Cells 7: 17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borbély E, Scheich B, Helyes Z (2013) Neuropeptides in learning and memory. Neuropeptides 47: 439–450 [DOI] [PubMed] [Google Scholar]
- Brici D, Zhang Q, Reinhardt S, Dahl A, Hartmann H, Schmidt K, Goveas N, Huang J, Gahurova L, Kelsey G et al (2017) The histone 3 lysine 4 methyltransferase Setd1b is a maternal effect gene required for the oogenic gene expression program. Development 144: 2606–2617 [DOI] [PubMed] [Google Scholar]
- Bukowski L, Chernomorchenko AMF, Starnawska A, Mors O, Staunstrup NH, Børglum AD, Qvist P (2020) Neuropsin in mental healt. J Physiol Sci 70: 26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, Guertin DA, Chang JH, Lindquist RA, Moffat J et al (2006) Cell Profiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol 7: R100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhury R, Singh S, Arumugam S, Roguev A, Stewart AF (2019) The Set1 complex is dimeric and acts with Jhd2 demethylation to convey symmetrical H3K4 trimethylation. Genes Dev 33: 550–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins BE, Sweatt JD, Greer CB (2019) Broad domains of histone 3 lysine 4 trimethylation are associated with transcriptional activation in CA1 neurons of the hippocampus during memory formation. Neurobiol Learn Mem 5: 149–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng C, Li Y, Liang S, Cui K, Salz T, Yang H, Tang Z, Gallagher PG, Qiu YI, Roeder R et al (2013) USF1 and hSET1A mediated epigenetic modifications regulate lineage differentiation and HoxB4 transcription. PLoS Genet 9: e1003524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreijerink KM, Mulder KW, Winkler GS, Hoppener JW, Lips CJ, Timmers HT (2006) Menin links estrogen receptor activation to histone H3K4 trimethylation. Can Res 66: 4929–4935 [DOI] [PubMed] [Google Scholar]
- Eisenberg E, Levanon EY (2013) Human housekeeping genes, revisited. Trends Genet 29: 569–574 [DOI] [PubMed] [Google Scholar]
- Feng J, Liu T, Qin B, Zhang Y, Liu XS (2012) Identifying ChIP‐seq enrichment using MACS. Nat Protoc 7: 1728–1740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer A (2014) Targeting histone‐modifications in Alzheimer's disease. What is the evidence that this is a promising therapeutic avenue? Neuropsychopharmacology 80: 95–012 [DOI] [PubMed] [Google Scholar]
- Gabriele M, Lopez Tobon A, D'Agostino G, Testa G (2018) The chromatin basis of neurodevelopmental disorders: Rethinking dysfunction along the molecular and temporal axes. Prog Neuropsychopharmacol Biol Psychiatry 84: 306–327 [DOI] [PubMed] [Google Scholar]
- Gjoneska E, Pfenning AR, Mathys H, Quon G, Kundaje A, Tsai LH, Kellis M (2015) Conserved epigenomic signals in mice and humans reveal immune basis of Alzheimer's disease. Nature 518: 365–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, Kim SY, Artis S, Molfese DL, Schumacher A, Sweatt JD, Paylor RE, Lubin FD (2010) Histone methylation regulates memory formation. J Neurosci 30: 3589–3599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halder R, Hennion M, Vidal RO, Shomroni O, Rahman R‐U, Rajput A, Centeno TP, van Bebber F, Capece V, Vizcaino JCG et al (2016) DNA methylation changes in plasticity genes accompany the formation and maintenance of memory. Nat Neurosci 19: 102–110 [DOI] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK (2010) Simple combinations of lineage‐determining transcription factors prime cis‐regulatory elements required for macrophage and B cell identities. Mol Cell 38: 576–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiraide T, Nakashima M, Yamoto K, Fukuda T, Kato M, Ikeda H, Sugie Y, Aoto K, Kaname T, Nakabayashi K et al (2018) De novo variants in SETD1B are associated with intellectual disability, epilepsy and autism. Hum Genet 137: 95–104 [DOI] [PubMed] [Google Scholar]
- Hughes CM, Rozenblatt‐Rosen O, Milne TA, Copeland TD, Levine SS, Lee JC, Hayes DN, Shanmugam KS, Bhattacharjee A, Biondi CA et al (2004) Menin associates with a trithorax family histone methyltransferase complex and with the hoxc8 locus. Mol Cell 4: 587–597 [DOI] [PubMed] [Google Scholar]
- Hwang J‐Y, Zukin RS (2018) REST, a master transcriptional regulator in neurodegenerative disease. Curr Opin Neurobiol 48: 193–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Illouz T, Madar R, Louzon Y, Griffioen KJ, Okun E (2016) Unraveling cognitive traits using the Morris water maze unbiased strategy classification (MUST‐C) algorithm. Brain Behav Immun 52: 132–144 [DOI] [PubMed] [Google Scholar]
- Islam MR, Lbik D, Sakib MS, Hofmann M, Berulava T, Jiménez Mausbach M, Cha J, Goldberg M, Vakhtang E, Schiffmann C et al (2021) Epigenetic gene expression links heart failure to memory impairment. Embo Mol Med 13: e11900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerimoglu C, Agis‐Balboa RC, Kranz A, Stilling R, Bahari‐Javan S, Benito‐Garagorri E, Halder R, Burkhardt S, Stewart AF, Fischer A (2013) Histone‐methyltransferase mll2 (kmt2b) is required for memory formation in mice. J Neurosci 33: 3452–3464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerimoglu C, Sakib MS, Jain G, Benito E, Burkhardt S, Capece V, Kaurani L, Halder R, Agís‐Balboa RC, Stilling R et al (2017) KMT2A and KMT2B mediate memory function by affecting distinct genomic regions. Cell Rep 20: 538–548 [DOI] [PubMed] [Google Scholar]
- Kleefstra T, Schenck A, Kramer JM, van Bokhoven H (2014) The genetics of cognitive epigenetics. Neuropharmacology 80: 83–94 [DOI] [PubMed] [Google Scholar]
- Kranz A, Anastassiadis KT (2020) The role of SETD1A and SETD1B in development and disease. Biochim Biophys Acta Gene Regul Mech 1863: 194578 [DOI] [PubMed] [Google Scholar]
- Kuczera T, Stilling RM, Hsia HE, Bahari‐Javan S, Irniger S, Nasmyth K, Sananbenesi F, Fischer A (2010) The anaphase promoting complex is required for memory function in mice. Learn Mem 18: 49–57 [DOI] [PubMed] [Google Scholar]
- Labonne JD, Lee KH, Iwase S, Kong IK, Diamond MP, Layman LC, Kim CH, Kim HG (2016) An atypical 12q24.31 microdeletion implicates six genes including a histone demethylase KDM2B and a histone methyltransferase SETD1B in syndromic intellectual disability. Hum Genet 135: 757–771 [DOI] [PubMed] [Google Scholar]
- Larizza L, Finelli P (2019) Developmental disorders with intellectual disability driven by chromatin dysregulation: Clinical overlaps and molecular mechanisms. Clin Genet 95: 231–240 [DOI] [PubMed] [Google Scholar]
- Lee JH, Skalnik DG (2008) Wdr82 is a C‐terminal domain‐binding protein that recruits the Setd1A Histone H3‐Lys4 methyltransferase complex to transcription start sites of transcribed human genes. Mol Cell Biol 28: 609–618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Lee DK, Dou Y, Lee J, Lee B, Kwak E, Kong YY, Lee SK, Roeder RG, Lee JW (2006) Coactivator as a target gene specificity determinant for histone H3 lysine 4 methyltransferases. Proc Natl Acad Sci U S A 103: 15392–15397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R (2009) The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H, Min Z, Tao Q (2016) The MLL/Setd1b methyltransferase is required for the Spemann's organizer gene activation in Xenopus. Mech Dev 142: 1–9 [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol 15: 550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma M, Zhang Y, Weng M, Hu Y, Xuan Y, Hu Y, Lv K (2018) lncRNA GCAWKR promotes gastric cancer development by scaffolding the chromatin modification factors WDR5 and KAT2A. Mol Ther 26: 2658–2668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marangi G, Orteschi D, Milano V, Mancano G, Zollino M (2013) Interstitial deletion of 3p22.3p22.2 encompassing ARPP21 and CLASP2 is a potential pathogenic factor for a syndromic form of intellectual disability: a co‐morbidity model with additional copy number variations in a large family. Am J Med Genet A 161: 2890–2893 [DOI] [PubMed] [Google Scholar]
- Minichiello L, Korte M, Wolfer D, Kühn R, Unsicker K, Cestari V, Rossi‐Arnaud C, Lipp HP, Bonhoeffer T, Klein R (1999) Essential role for TrkB receptors in hippocampus‐mediated learning. Neuron 24: 401–414 [DOI] [PubMed] [Google Scholar]
- Mishra BP, Zaffuto KM, Artinger EL, Org T, Mikkola HK, Cheng C, Djabali M, Ernst P (2014) The histone methyltransferase activity of MLL1 is dispensable for hematopoiesis and leukemogenesis. Cell Rep 7: 1239–1247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukai J, Cannavò E, Crabtree GW, Sun Z, Diamantopoulou A, Thakur P, Chang C‐Y, Cai Y, Lomvardas S, Takata A et al (2019) Recapitulation and reversal of schizophrenia‐related phenotypes in Setd1a‐deficient mice. Neuron 104: 471–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestler EJ, Peña CJ, Kundakovic M, Mitchell A, Akbarian S (2015) Epigenetic basis of mental illness. Neuroscientist 8: 447–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicol JW, Helt GA, Blanchard SG Jr, Raja A, Loraine AE (2009) The Integrated Genome Browser: free software for distribution and exploration of genome‐scale datasets. Bioinformatics 25: 2730–2731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S, Kim GW, Kwon SH, Lee JS (2020) Broad domains of histone H3 lysine 4 trimethylation in transcriptional regulation and disease. FEBS J 287: 2891–2902 [DOI] [PubMed] [Google Scholar]
- Rao RC, Dou Y (2015) Hijacked in cancer: the KMT2 (MLL) family of methyltransferases. Nat Rev Cancer 15: 334–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redrobe JP, Dumont Y, Herzog H, Quirion R (2004) Characterization of neuropeptide Y, Y(2) receptor knockout mice in two animal models of learning and memory processing. J Mol Neurosci 22: 159–166 [DOI] [PubMed] [Google Scholar]
- Rehfeld F, Maticzka D, Grosser S, Knauff P, Eravci M, Vida I, Backofen R, Wulczyn FG (2018) The RNA‐binding protein ARPP21 controls dendritic branching by functionally opposing the miRNA it hosts. Nat Commun 9: 1235–1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross‐Innes CS, Stark R, Teschendorff AE, Holmes KA, Ali HR, Dunning MJ, Brown GD, Gojis O, Ellis IO, Green AR et al (2012) Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 481: 389–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roston A, Evans D, Gill H, McKinnon M, Isidor B, Cogné B, Mwenifumbo J, van Karnebeek C, An J, Jones SJM et al (2020) SETD1B‐associated neurodevelopmental disorder. J Med Genet 58: 196–204 [DOI] [PubMed] [Google Scholar]
- Scacheri PC, Davis S, Odom DT, Crawford GE, Perkins S, Halawi MJ, Agarwal SK, Marx SJ, Spiegel AM, Meltzer PS et al (2006) Genome‐wide analysis of menin binding provides insights into MEN1 tumorigenesis. PLoS Genet 2: e51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt K, Zhang Q, Tasdogan A, Petzold A, Dahl A, Arneth BM, Slany R, Fehling HJ, Kranz A, Stewart AF et al (2018) The histone 3 lysine 4 methyltransferase Setd1b is essential for hematopoietic stem and progenitor cell homeostasis in mice. eLife 7: e27157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L, Shao N, Liu X, Nestler E (2014) ngs.plot: Quick mining and visualization of next‐generation sequencing data by integrating genomic databases. BMC Genom 15: 284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shilatifard A (2012) The COMPASS family of histone H3K4 methylases: mechanisms of regulation in development and disease pathogenesis. Annu Rev Biochem 81: 65–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinsky SA, Monteith KE, Viggiano S, Cosgrove MS (2015) Biochemical reconstitution and phylogenetic comparison of human SET1 family core complexes involved in histone methylation. J Biol Chem 290: 6361–6375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulha HP, Cheung I, Whittle C, Wang J, Virgil D, Lin CL, Guo Y, Lessard A, Akbarian S, Weng Z (2012) Epigenetic signatures of autism: trimethylated H3K4 landscapes in prefrontal neurons. Arch Gen Psychiatry 69: 314–324 [DOI] [PubMed] [Google Scholar]
- Soares LM, He PC, Chun Y, Suh H, Kim T, Buratowski S (2017) Determinants of histone H3K4 methylation patterns. Mol Cell 68: 773–785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stilling RM, Rönicke R, Benito E, Urbanke H, Capece V, Burkhardt S, Bahari‐Javan S, Barth J, Sananbenesi F, Schütz AL et al (2014) K‐Lysine acetyltransferase 2A regulates a hippocampal gene‐expression network linked to memory formation. EMBO J 33: 1912–1927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweatt JD (2001) The neuronal MAP kinase cascade: a biochemical signal integration system subserving synaptic plasticity and memory. J Neurochem 76: 1–10 [DOI] [PubMed] [Google Scholar]
- Sze CC, Ozark PA, Cao K, Ugarenko M, Das S, Wang LU, Marshall SA, Rendleman EJ, Ryan CA, Zha D et al (2020) Coordinated regulation of cellular identity–associated H3K4me3 breadth by the COMPASS family. Sci Adv 6: eaaz4764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahiliani M, Mei P, Fang R, Leonor T, Rutenberg M, Shimizu F, Li J, Rao A, Shi Y (2007) The histone H3K4 demethylase SMCX links REST target genes to X‐linked mental retardation. Nature 447: 601–605 [DOI] [PubMed] [Google Scholar]
- Tamura H, Ishikawa Y, Hino N, Maeda M, Yoshida S, Kaku S, Shiosaka S (2006) Neuropsin is essential for early processes of memory acquisition and Schaffer collateral long‐term potentiation in adult mouse hippocampus in vivo . J Physiol 570: 541–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsilibary E, Tzinia A, Radenovic L, Stamenkovic V, Lebitko T, Mucha M, Pawlak R, Frischknecht R, Kaczmarek L (2014) Neural ECM proteases in learning and synaptic plasticity. Prog Brain Res 214: 135–157 [DOI] [PubMed] [Google Scholar]
- Vallianatos CN, Raines B, Porter RS, Bonefas KM, Wu MC, Garay PM, Collette KM, Seo YA, Dou Y, Keegan CE et al (2020) Mutually suppressive roles of KMT2A and KDM5C in behaviour, neuronal structure, and histone H3K4 methylation. Commun Biol 3: 278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Zang C, Cui K, Schones DE, Barski A, Peng W, Zhao K (2009) Genome‐wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell 138: 1019–1031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf FA, Angerer P, Theis FJ (2018) SCANPY: large‐scale single‐cell gene expression data analysis. Genome Biol 19: 15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M, Wang PF, Lee JS, Martin‐Brown S, Florens L, Washburn M, Shilatifard A (2008) Molecular regulation of H3K4 trimethylation by Wdr82, a component of human Set1/COMPASS. Mol Cell Biol 28: 7337–7344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama A, Wang Z, Wysocka J, Sanyal M, Aufiero DJ, Kitabayashi I, Herr W, Cleary ML (2004) Leukemia proto‐oncoprotein MLL forms a SET1‐like histone methyltransferase complex with menin to regulate Hox gene expression. Mol Cell Biol 24: 5639–5649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Wang LG, He QY (2015) ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 31: 2382–2383 [DOI] [PubMed] [Google Scholar]