

SUMMARY
Alternative lengthening of telomeres (ALT) is a telomere-elongation mechanism observed in ~15% of cancer subtypes. Current models indicate that ALT is mediated by homology-directed repair mechanisms. By disrupting MSH6 gene expression, we show that the deficiency of MutSα (MSH2/MSH6) DNA mismatch repair complex causes striking telomere hyperextension. Mechanistically, we show MutSα is specifically recruited to telomeres in ALT cells by associating with the proliferating-cell nuclear antigen (PCNA) subunit of the ALT telomere replisome. We also provide evidence that MutSα counteracts Bloom (BLM) helicase, which adopts a crucial role in stabilizing hyper-extended telomeres and maintaining the survival of MutSα-deficient ALT cancer cells. Lastly, we propose a model in which MutSα deficiency impairs heteroduplex rejection, leading to premature initiation of telomere DNA synthesis that coincides with an accumulation of telomere variant repeats (TVRs). These findings provide evidence that the MutSα DNA mismatch repair complex acts to restrain unwarranted ALT.
Graphical abstract
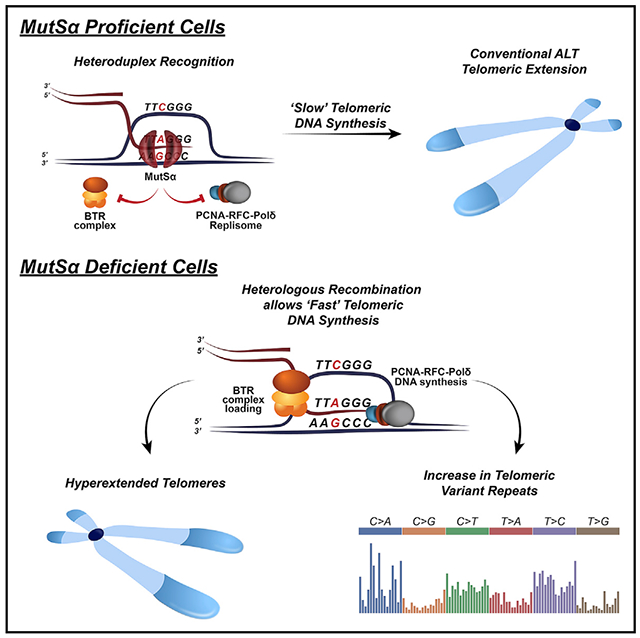
In brief
Barroso-Gonzalez et al. show that the mismatch repair complex MutSα restricts the alternative lengthening of telomeres (ALT) pathway in cancer cells. MutSα has an anti-recombination function and limits recombination between heteroduplex sequences at telomeres, in part by counteracting the Bloom helicase (BLM).
INTRODUCTION
The activation of a telomere elongation mechanism is an essential step in tumorigenesis (Maciejowski and de Lange, 2017). Around 85%–90% of cancer cells reactivate telomerase, a reverse transcriptase dedicated to the synthesis of telomeric sequences (Kim et al., 1994). However, 10%–15% of cancer subtypes do not activate telomerase. These engage a homology-directed repair (HDR) mechanism of telomere length maintenance, termed alternative lengthening of telomeres (ALT) (Bryan et al., 1997). When ALT is activated is unclear, and whether it is stochastic or driven by genetic alterations remains unclear, even though mutations in ATRX (α-thalassemia/mental retardation, X-linked) (Heaphy et al., 2011), DAXX (death-domain associated protein) (Heaphy et al., 2011), histone H3.3 (Schwartzentruber et al., 2012), and other chromatin modifiers have emerged as putative drivers of ALT (Hoang and O’Sullivan, 2020). However, once ALT is initiated, heterogenous telomere lengths are maintained, often at lengths beyond the limits of normal cells. Consequently, the elongated telomeres in ALT-positive cells are intrinsically unstable and prone to replicative stress (Clynes et al., 2015), spontaneous damage (Cesare et al., 2009), and the accumulation of aberrant DNA secondary structures, such as G-quadruplexes (Clynes et al., 2015); DNA:RNA hybrid structures, also known as R-loops (Arora et al., 2014); or even the excess accumulation of TERRA (telomere repeat containing RNA) (Arora et al., 2014; Silva et al., 2021). These destabilizing structures stimulate recurrent DNA repair but, paradoxically, preserve telomere integrity and promote telomere extension to sustain the proliferative advantage of those cancer cells (Arora et al., 2014; Silva et al., 2021).
In contrast with normal unidirectional telomere replication, which starts at sub-telomeric origins in S-phase, ALT initiates at broken DNA that ends during G2-M phases (Dilley et al., 2016). ALT depends on RAD51 and RAD52 DNA recombinases (Cho et al., 2014; Dunham et al., 2000; Min et al., 2017; Verma et al., 2019; Zhang et al., 2019). These facilitate the inter/intra chromosomal pairing of homologous telomere single-stranded DNA (ssDNA) sequences and the formation of homologous recombination intermediates, such as displacement (D)-loops; from which, the synthesis of new telomeric sequences is mediated by a replisome comprising proliferating-cell nuclear antigen (PCNA), RFC1-5, and polymerase δ (Polδ) (Dilley et al., 2016; Zhang et al., 2019). The Bloom (BLM) helicase, acting in concert with TOP3A and RMI1 to form the BTR complex, promotes branch migration and catalyzing of the timely dissolution of telomere homologous recombination (HR) intermediates (Loe et al., 2020; Panier et al., 2019; Sobinoff et al., 2017). Consistent with that important role, the disruption of BTR complex attenuates ALT (Loe et al., 2020). However, the activity of the BTR complex must be counteracted by the SMX complex to prevent chromosome entanglements that could irretrievably destabilize telomeres, causing mitotic catastrophe (Panier et al., 2019; Sobinoff et al., 2017). However, left unrestrained, the SMX complex could prematurely resolve telomere HR intermediates, increasing the rate of telomere sister-chromatid exchanges and telomere deletion (Panier et al., 2019; Sarkar et al., 2015; Sobinoff et al., 2017; Verma et al., 2019; Wilson et al., 2013). Similarly, telomere HR intermediates, formed because of the disruption of other ALT mediators, such as trans-lesion DNA polymerase η (Pol-η) (García-Expósito et al., 2016), the strand-annealing helicase SMARCAL1 (Cox et al., 2016), and the ATPase/translocases FANCM (Lu et al., 2019; Pan et al., 2017; Silva et al., 2019), can be processed by SMX endonucleolytic activity leading to a hyper-recombinogenic ALT phenotype. FANCM and SLX4IP were shown to co-regulate both the BTR and SMX complexes (Lu et al., 2019; Panier et al., 2019), curtailing their unscheduled activity at telomeres and maintaining the balance of pro- and anti-recombinogenic stimuli at telomeres. These studies highlight the complex, coordinated regulation of ALT by multiple DNA repair factors, which is imperative for maintaining telomere length and integrity.
DNA mismatch repair (MMR) has key roles in genome maintenance by correcting DNA replication errors and preventing recombination events between divergent DNA sequences (Jiricny, 2006; Kunkel and Erie, 2015; Li, 2008a). MMR also promotes DNA-damage tolerance to cytotoxic lesions and the expansion of triplet nucleotide repeats (de Wind et al., 1995; Prolla et al., 1998; Strand et al., 1993). Human MMR proteins are MutS and MutL heterodimers. MSH2 heterodimerizes with MSH6 or MSH3 to form MutSα and MutSβ, respectively (Acharya et al., 1996; Iaccarino et al., 1996; Kunkel and Erie, 2015). MutL heterodimers formed between MLH1 and PMS2 (MutLα) and MLH3 (MutLγ), but not PMS1(MutLβ), harbor endonuclease activity (Kunkel and Erie, 2015). PCNA has a prominent role in MMR and interacts with MutS and MutL complexes (Flores-Rozas et al., 2000; Kleczkowska et al., 2001). PCNA’s interaction with MutSα and MutSβ occurs through a conserved motif, termed the PCNA-interaction protein (PIP) box. Both PCNA and the RFC1-5 complex facilitate the initiating steps of MMR, interacting with MutSα and MutSβ at mismatches present in newly replicated DNA strands, which are then excised by the MLH heterodimers acting with EXO1 (exonuclease 1) (Kadyrov et al., 2006; Kawasoe et al., 2016; Umar et al., 1996). Germline mutations in MMR genes and defects in MMR are associated with hereditary non-polyposis colorectal cancer (HNPCC), sporadic colon cancer, and lymphomas (Fishel et al., 1994; Lynch and de la Chapelle, 1999; Parsons et al., 1993).
In addition to MMR, MutSα and MutSβ prevent recombination between similar, but not identical, homeologous DNA sequences, a process termed “heteroduplex rejection” (Jiricny, 2006). Although more infrequent than homologous recombination (HR), heteroduplex-rejection events involving the MutS complexes preserved genome stability by suppressing translocations, deletions, and other genome rearranging events (Putnam et al., 2009). Accordingly, deletion of MSH2 in mice elicits a hyper-recombination phenotype (de Wind et al., 1995). Studies from budding yeast show that heteroduplex rejection requires the RecQ family helicase Sgs1 to dissolve DNA intermediates formed during HR (Myung et al., 2001; Sugawara et al., 2004) . Notably, BLM (Yang et al., 2004), RECQ1 (Doherty et al., 2005), and WRN (Saydam et al., 2007) RecQ helicases interact with MutSα in human cancer cell lines, suggesting their role in recombination is conserved. Interestingly, genetic deletion of msh2 enhanced the survival of yeast strains lacking est2 that encodes the catalytic subunit of telomerase (Rizki and Lundblad, 2001). This was attributed to elimination of the anti-recombinase heteroduplex rejection activity of Msh2, rather than any effect on mutation (Lundblad, 2002; Rizki and Lundblad, 2001). This is significant because break-induced replication (BIR)-mediated telomere maintenance in budding yeast, which resembles ALT (Kramara et al., 2018), allows the mismatch repair system to reject recombination intermediates in certain conditions (Anand et al., 2017). Analogously, MMR deficiency and telomerase inactivation enhances the proliferation of colon carcinoma cells (Bechter et al., 2004). This hints at the activation of an ALT-like telomere-elongation mechanism but was not fully demonstrated. Therefore, even though the MMR pathway clearly influences recombination, including potentially, during telomeres maintenance, whether MMR directly contributes to telomere maintenance in ALT cancer cells remains to be determined.
In this study, we report that the MutSα (MSH2-MSH6) complex occupies telomeres specifically in ALT cancer cells. We show that disrupting MSH6 gene expression, causing MutSα deficiency, provokes a striking hyper-extension of telomeres. Mechanistically, we show MutSα is recruited to telomeres through the ALT replisome factor PCNA. However, MutSα deficiency causes increased telomeric localization of PCNA and POLD3, as well as significant accumulation of BLM helicase at telomeres. Consequently, telomeres are subject to unrestrained telomere DNA synthesis and extension that can be suppressed by depletion of BLM but not WRN, a related RecQ helicase, causing enhanced proliferative defects. We propose that the anti-recombinase activity of MutSα contributes to regulated telomere DNA synthesis by restraining the association of BLM with telomeres. We envision a model in which defective heteroduplex rejection drives premature initiation of telomere DNA synthesis, leading to increased telomere extension. Because of this, and potentially, because of the loss of efficient MMR, telomeres accumulate variant repeat (TVR) sequences that have been associated with the increased genomic instability of ALT cancer cells.
RESULTS
MutSα deficiency leads to telomere hyper-extension in ALT cancer cells
The human MSH6 (MutS homolog 6)-MSH2 (MutS homolog 2) obligate heterodimer, or MutSα complex, was identified as a potential constituent of telomeres in ALT cancer cells in a proteomic analysis of telomere composition (García-Expósito et al., 2016). To investigate a role for MutSα complex in ALT telomere maintenance, MSH6 protein expression was disrupted in ALT-positive U2OS cells using CRISPR/Cas9 (Figure 1A). As predicted because of their obligate and stabilizing interactions, MSH6 disruption reduced MSH2, but not MSH3 protein levels (Figure 1A). The residual MSH2 protein is likely preserved to maintain the MutSβ complex with MSH3. Consistent previous reports (Pani et al., 2007), MSH6 knockout (KO) cells displayed enhanced resistance to cis-platinum (Figure S1A). When compared with control cells, the MSH6 KO U2OS cells exhibited a slightly elevated, but not significant, proliferation in colony-formation assay and a normal cell-cycle profile (Figures S1A and S1B).
Figure 1. ALT telomere hyper-extension because of MutSα deficiency.
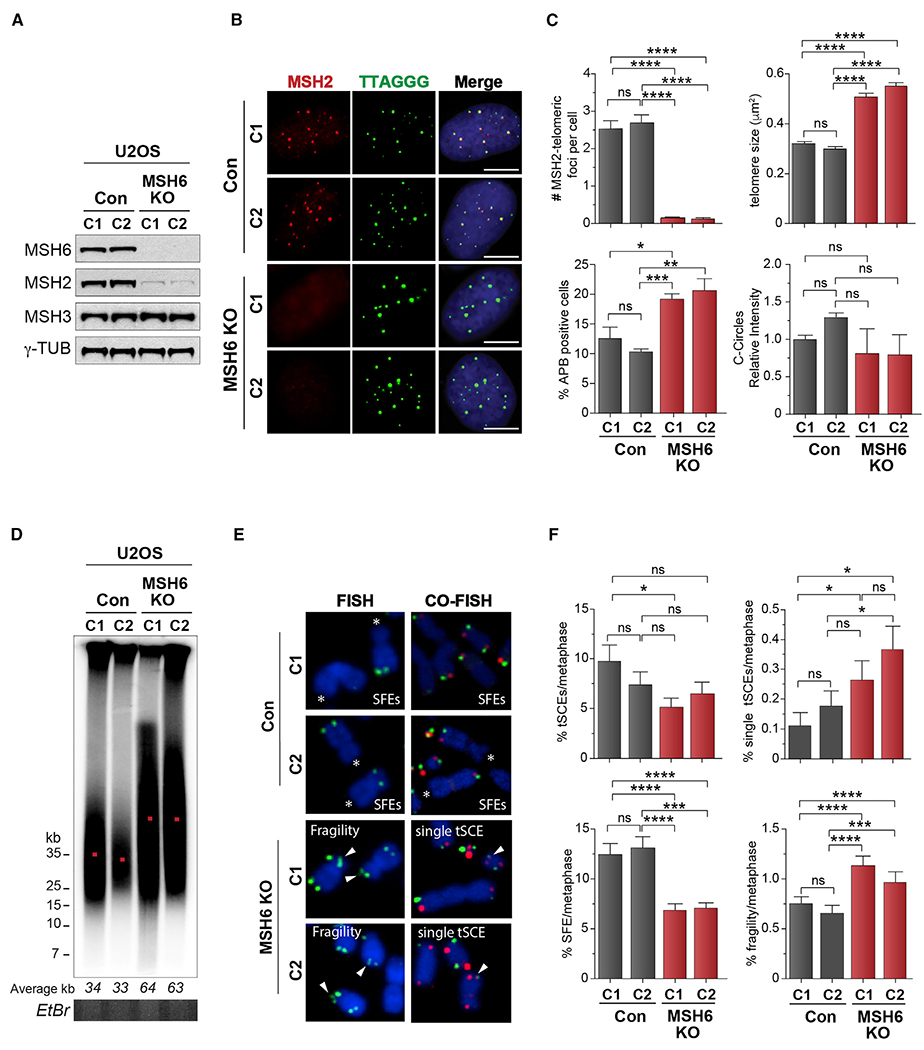
(A) Western blot showing MSH6 disruption in U2OS cells. γ-Tubulin is shown as a loading control.
(B) IF-FISH showing endogenous MSH2 co-localization with telomeres (TTAGGG-FISH) in control but not MSH6 knockout (KO) U2OS cells.
(C) Quantification of MSH2 foci and ALT phenotypes, including the percentage of ALT-associated PML bodies (APBs), telomere focus size, C-circles (CCs) in control, and MSH6 KO clones. For C-circles, levels are presented in relation to those detected in control clone C1.
(D) PFGE telomere length analysis of control (C1 and C2) and MSH6 KO clones (C1 and C2). Telomere size (kb) is shown below, and DNA loading is shown by ethidium bromide (EtBr) gel. Red dot indicates average telomere length
(E) Metaphase spreads prepared by FISH (TelC probe) and chromosome orientation FISH (CO-FISH) (TelC and TelG probes) from control and MSH6 KO clones showing signal free ends (SFEs), telomere fragility, and single inter-telomeric exchanges (single telomere sister chromatid exchanges [t-SCEs]).
(F) Quantifications of the percentage of double t-SCEs, single-telomere SCEs, telomere fragility, and signal-free ends (SFEs) in control and MSH6 KO clones.
All data represent means ± SEM, n = 3 biological replicates. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 (one-way ANOVA). Unless otherwise indicated, all FISH experiments were conducted using TelC FISH probe. All scale bars, 5 μm. See also Figure S1.
The localization of MSH2 to telomeres in ALT U2OS cells, and its loss through disruption of MSH6, were confirmed by immunofluorescence using a specific antibody and telomere fluorescence in situ hybridization (FISH) (Figures 1B and 1C). Notably, intensely bright and large telomere FISH signals that corresponded to ALT-associated promyelocytic leukemia (PML) bodies (APBs) were visible in both MutSα-deficient U2OS clones (Figures 1B, 2C, and S1C). APBs are a hallmark of ALT cancer cells and are important sub-cellular compartments in which telomere DNA synthesis and other processes in ALT take place (Draskovic et al., 2009; Yeager et al., 1999; Zhang et al., 2019). Therefore, their increased size and frequency of cells harboring these structures was suggestive of a potentially strong effect on telomeres. Indeed, increases in APB frequency were also observed after single interfering RNA (siRNA)-mediated depletion of MSH6, but not MSH3, in U2OS cells and in Saos2 and VA13 cancer cell lines that also rely on ALT for telomere lengthening (Figure S1D). Interestingly, we did not detect significant changes in extra-chromosomal telomeric species, such as C-circles, (Figures 1C and S1E), the levels of which have been implicated as representing something akin to a molecular rheostat of ALT activity (Henson et al., 2009), but whose precise origin and role remains unclear. Nonetheless, measurement of telomere length by pulsed-field gel electrophoresis (PFGE) and Southern blot of denatured telomeric DNAs revealed striking hyper-extension of telomeres in MutSα-deficient U2OS clones (c1-c2) and VA13 (c1-c3) (Figures 1D and S1F). Such telomere elongation was not evident in telomerase expressing HeLa LT MutSα KO cells (Figure S1G). Continued expansion in cell culture and telomere PFGE revealed progressive telomere extension in the MutSα-deficient U2OS cells (Figure S1H). Examination of telomeres on metaphase chromosomes showed that MutSα deficiency decreased the overall frequency of telomere sister chromatid exchanges (t-SCEs) and signal-free ends (SFEs) (i.e., no detectable telomere FISH signal) (Figure S1E). Notably, metaphase chromosomes in MutSα-deficient cells exhibited more single-telomere exchanges, involving the lagging strand, and greater evidence of telomere fragility, suggestive of aberrant replication dynamics, which may be consistent with enhanced break-induced replication (BIR) (Roumelioti et al., 2016; Yang et al., 2020)(Figures 1E and 1F). This telomere hyper-extension phenotype and evidence derived from a detailed assessment of ALT-associated telomeric markers suggested that the MutSα complex has a vital role in regulating the ALT telomere elongation mechanism.
Figure 2. MutSα recruitment to telomeres and effect on telomere DNA synthesis.
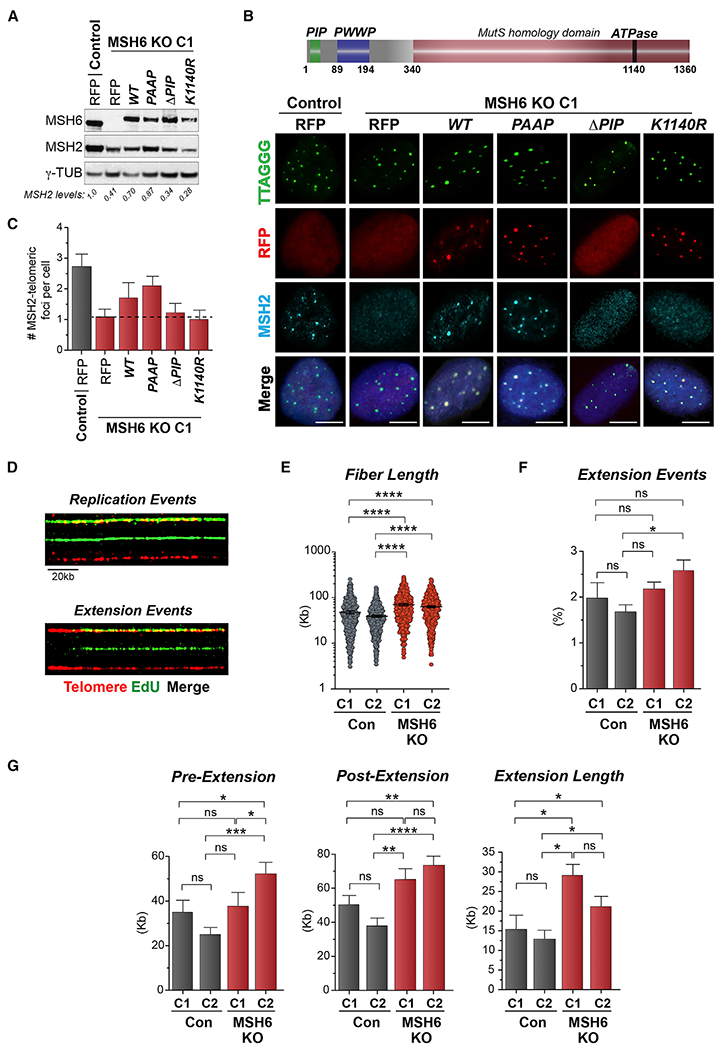
(A) Western blot showing reconstitution of MSH6 with the indicated mutant constructs. γ-Tubulin is shown as a loading control. The level of MSH2 protein in cells expressing the RFP-tagged MSH6 proteins, normalized to γ-tubulin, is indicated beneath the blots.
(B) Top: schematic of functional and homology domains of the MSH6 protein. Bottom: representative immunofluorescence (IF) images of red fluorescent protein (RFP)-tagged MSH6 constructs and MSH2 localizing to TRF1-FokI telomeres.
(C) Quantifications of the number of MSH2 telomeric foci located at WT- or DA-TRF1-FokI telomere DSBs.
(D) Representative example of telomere replication and extension events showing telomere staining (TTAGGG) and synthesis (EdU) by the SMAT assay.
(E) Quantification of telomere length (kb)
(F and G) Percentage of extension events (F) and pre/post and total extension length (kb) (G) of telomere DNA fibers using SMAT in U2OS control and MSH6 KO clones.
All data represent means ± SEM, n = 3 biological replicates. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 (one-way ANOVA). Unless otherwise indicated, all FISH experiments were conducted using TelC FISH probe. All, except (D), scale bars, 5 μm; (D), 20 μm. See also Figure S2.
Localization of MutSα complex at telomeres in ALT cancer cells
To determine how the MutSα complex contributes to ALT, we first examined its recruitment to telomeres. Consistent with prior proteomics studies (García-Expósito et al., 2016), we observed the telomeric localization of RFP-tagged MSH6 and endogenous MSH2 proteins in ALT U2OS cells and not in telomerase expressing HeLa LT cells (Figure S2A). By inducing telomere DNA breaks with transiently expressed FLAG-TRF1-FokI (Cho et al., 2014), followed by an 5-ethynyl-2′-deoxyuridine (EdU) pulse, we observed MSH2 localization to telomeric DSB sites in S-phase cells with pan-nuclear EdU staining and in non-S-phase cells with punctate telomeric EdU foci that indicate sites of active telomere DNA synthesis (Figure S2B). Similar to MSH2 and MSH6, we also observed the localization of the DNA MMR protein MLH1 (human MutL homolog 1) to telomeric DSBs induced by wild-type (WT) TRF1-FokI (Figure S2C). We did not, however, observe the accumulation of MSH2 or MLH1 at AsiSI-induced genomic DNA breaks in U2OS-DiVa cells (Figure S2D). These data again indicated that the MutSα complex resides at telomeres in ALT cells but also accumulates at telomere DSB sites from which ALT can be directly stimulated (Dilley et al., 2016).
Previous studies in budding yeast showed that the MutSα complex localized with the DNA replication machinery independent of mispaired bases. This localization was dependent on interactions with PCNA via an N-terminally located PCNA-inter-acting protein (PIP) box. Curiously, the MutSα pool that localized to replication factories was estimated to account for only a small percentage of mismatch repair (Flores-Rozas et al., 2000; Hombauer et al., 2011a; Kleczkowska et al., 2001). MSH6 also recognizes and binds tri-methylated lysine 36 of histone H3 (H3K36me3) via a PWWP (pro-trp-trp-pro) domain (Li et al., 2013). We generated WT or mutant RFP-tagged MSH6 fusion proteins, including one in which the PIP box was deleted (ΔPIP), a PWWP domain W105/106A mutant (PAAP) (Li et al., 2013), and an ATPase-dead mutant by replacing lysine 1140 with arginine (K1140R) (Iaccarino et al., 1998) (Figures 2A–2C). An RFP-empty protein or those RFP-MSH6 fusion proteins were expressed in MSH6 KO U2OS cells, and RFP+ cells were selected by multiple rounds of cell sorting. By western analysis, we verified that RFP-MSH6 fusion protein expression approximated that of the endogenous MSH6 protein (Figure 2A). Reexpression of the WT and PAAP-MSH6 restored MSH2 protein levels to roughly those of control RFP-expressing cells, whereas neither the ΔPIP nor ATPase-dead MSH6 mutants could (Figure 2A). By immunofluorescence, we observed that the WT localized to telomere breaks and recruited endogenous MSH2, indicative of efficient reconstitution of the MutSα complex (Figures 2B and 2C). The PAAP-MSH6 localized to telomeres and recovered MSH2 like the WT (Figures 2B and 2C). Binding to H3K36me3 is linked with MutSα association at transcribed gene bodies and may be more predominant during G1/S cell cycle phases (Li et al., 2013). Furthermore, whether telomere chromatin is enriched in H3K36me3 is unclear. Interestingly, although the ATPase-dead MSH6 mutant localized to telomeres, MSH2 remained undetectable at telomeres (Figures 2B and 2C). That the catalytic ATPase MSH6 (K1140R) mutant localized to telomeres even in the absence of MSH2 indicated that MSH6 accumulation at telomeres can occur before MutSα complex assembly, once again in the absence of mispairs, as previously suggested (Hombauer et al., 2011a). However, the ΔPIP-MSH6 mutant failed in both localizing to telomeres and in recruiting MSH2, meaning that the interaction between MSH6 and PCNA is required for MutSα complex assembly at telomeres in ALT cells (Figures 2B and 2C). Because PCNA is a central member of the ALT telomere DNA replisome (Dilley et al., 2016), this suggested that MutSα could have a role in telomere DNA synthesis of ALT telomeres.
MutSα regulates telomere DNA synthesis in ALT cells
To directly assess telomere DNA synthesis in the absence of the MutSα complex, we used the single-molecule analysis of telomeres (SMAT) methodology (Sobinoff et al., 2017). This involves the labeling of cycling cells with the thymidine analogs (e.g., EdU or chloro-deoxyuridine [CldU]), the incorporation of which at telomere is then visualized by stretching DNA fibers on glass slides and performing FISH with telomere-specific peptide nucleic acid (PNA) probes (Sobinoff et al., 2017). Several hundred telomere fibers labeled with EdU on either side or at a single end are scored as normal telomere replication events or telomere extension events, respectively (Figure 2D). Using SMAT, we observed that fibers derived from MutSα-deficient U2OS cells were significantly longer that those from control cells (Figure 2E). This was despite there being no, or only minor, differences in the frequency of detectable extension events (Figure 2F). By measuring the non-labeled (pre-extension) and EdU labeled (post-extension) sections of telomere fibers, we determined the length of telomere extension between control and MutSα-deficient U2OS cells (Figure 2G). This revealed an average net extension of ~12–15 kb in control cells. However, the extension events in the MutSα-deficient U2OS cells were significantly longer, ranging from >20 to 30 kb (Figure 2G). We also considered that the efficiency or rate of DNA synthesis could be accelerated upon MutSα deficiency because of increased residency of factors that mediate ALT telomere DNA synthesis. Accordingly, by immunostaining, we observed increased telomere localization of PCNA and POLD3, two essential subunits of the ALT telomere DNA replisome (Dilley et al., 2016), in the MutSα-deficient U2OS cells (Figures S2E and S2F). It is also possible that, in the absence of heteroduplex rejection BIR initiation can occur more frequently. Within that scenario, the rate of DNA synthesis is unlikely to change, but initiation events that lead to extended DNA synthesis might. Taking the telomere length analysis that was conducted using independent assays in conjunction with data showing enhanced ALT-phenotypes (APBs and single t-SCEs), we envision that the MutSα complex’s anti-recombination function may serve to restrain excessive DNA-synthesis-coupled telomere extension in ALT cancer cells.
BLM helicase supports survival of MutSα-deficient ALT cells
Another key factor in ALT telomere maintenance is BLM helicase (Hoang and O’Sullivan, 2020). Acting in concert with TOP3A and RMI, BLM promotes branch migration and the dissolution of homology-directed repair intermediates at telomeres in cancer cells that rely on ALT (Loe et al., 2020; Min et al., 2019; Panier et al., 2019; Sobinoff et al., 2017). Other studies have shown that BLM depletion prevents the in vitro induction of ALT (O’Sullivan et al., 2014), and complete BLM disruption essentially abolishes ALT-mediated telomere elongation (Loe et al., 2020). BLM, and its homolog in budding yeast SGS1, and the MutSα complex have been shown to physically interact and coordinate heteroduplex rejection (Myung et al., 2001; Pedrazzi et al., 2003; Sugawara et al., 2004; Yang et al., 2004), a process which inhibits recombination between divergent DNA sequences. As with PCNA and POLD3, we detected a marked increase in the accumulation of BLM at telomeres in MutSα-deficient U2OS cells (Figures 3A and 3B). This was evident within individual cells, but also throughout the population of cells. In contrast, MSH2 localization to telomere DSBs could not be detected upon disruption of BLM or RMI1 (Figure S3A). Furthermore, co-depleting BLM prevented the increased formation of APBs that was observed upon MSH6 depletion in U2OS cells (Figure S3B). Similar suppression of APB formation was seen when POLD3 was co-depleted with MSH6 in three distinct ALT cell lines (Figure S3C). These data indicate that, although BLM is required for its recruitment to telomeres, MutSα may act to restrain BLM at telomeres.
Figure 3. MutSα-deficient ALT cells rely on BLM helicase for survival.
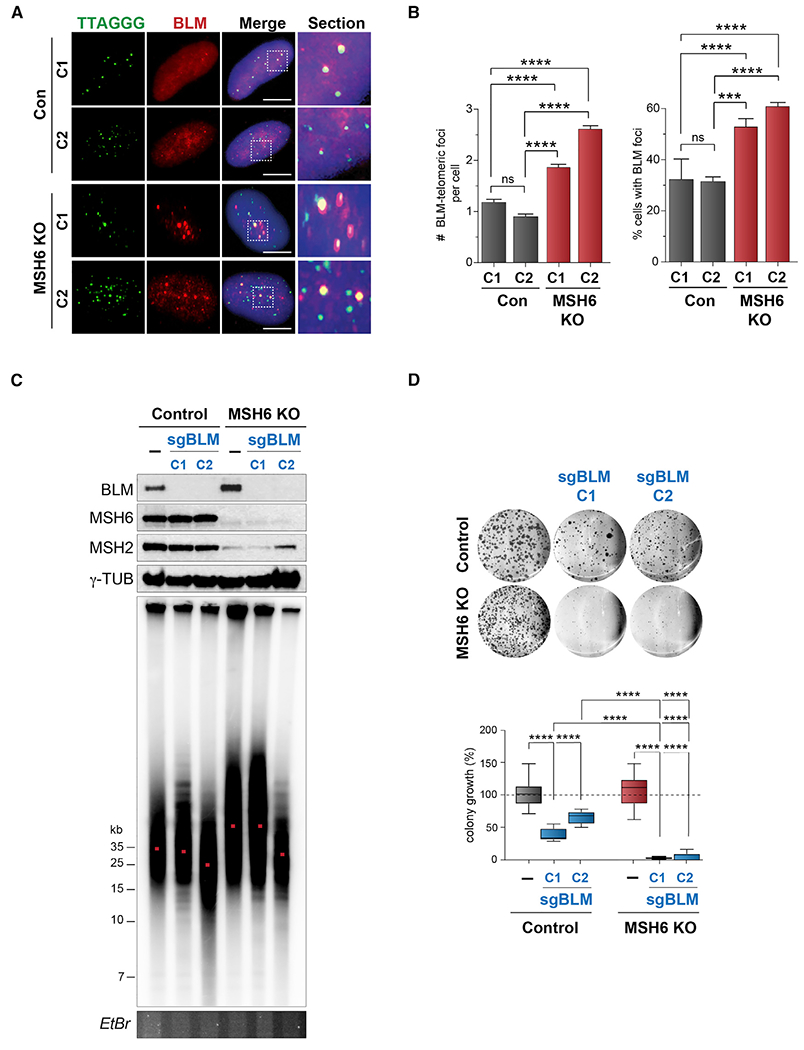
(A) Representative IF images of BLM co-localization with telomeres (TTAGGG-FISH) in U2OS control and MSH6 KO clones.
(B) Quantification of the number of endogenous BLM foci and the percentage of cells presenting telomeric BLM foci in U2OS control and MSH6 KO clones with BLM-telomeric foci.
(C) Top: western blot showing BLM disruption in U2OS control and MSH6 KO clones by CRISPR/Cas9. MSH6 and MSH2 are used to shown MutSα depletion. γ-Tubulin is shown as a loading control. Bottom: telomere-length analysis of BLM KO control and MSH6/BLM double knockout (DKO) clones by PFGE. EtBr gel is used as a DNA loading control.
(D) Images (top) and quantifications (bottom) of colony formation assays with BLM KO U2OS control and MSH6 KO clones. All pairs are quantified relative to their respective control clone.
All data represent means ± SEM, n = 3 biological replicates. ***p < 0.001, ****p < 0.0001 (one-way ANOVA). Unless otherwise indicated, all FISH analyses were conducted using a TelC FISH probe. All scale bars, 5 μm. See also Figure S3.
To characterize the role of BLM in the context of MutSα-deficient ALT telomeres, we disrupted BLM in MSH6 KO U2OS cells, confirming the absence of both BLM and MSH6 proteins by western blot in two independent double KO (DKO) clones (Figure 3C). Control, MSH6 KO, and MSH6/BLM KO clones were expanded in cell culture for ~25 population doublings (PDs), and telomere length was examined by PFGE (Figure 3C). Consistent with reports, we observed the appearance of discrete banding of telomere fragments in BLM KO U2OS cells (Loe et al., 2020). This was proposed to reflect the complete attenuation of ALT-associated HDR (Figure 3C). Indeed, several telomere DNA fragments appeared as lower-molecular-weight bands. Interestingly, the same fragmentation phenomenon was seen in telomere DNA derived from MSH6-BLM DKO U2OS cells (Figure 3C). However, we noted that, in contrast with control/BLM KO U2OS cells, whose ability to form viable colonies was reduced by ~50%, the MSH6-BLM DKO exhibited a complete defect in the ability to form colonies, indicative of catastrophic consequences of combined BLM and MutSα deficiency in ALT U2OS cells (Figure 3D). These results contrasted with observations after the disruption of the related Werner RecQ helicase (WRN), which binds MutSα and has strong links with maintaining telomere integrity (Crabbe et al., 2004; Opresko et al., 2004; Saydam et al., 2007) (Figures S3D and S3E). Although WRN KO cells exhibited substantial growth inhibition and a very modest telomere shortening effect, the growth defect of MSH6-WRN DKO cells was much less pronounced, and telomere length was unaffected (Figures S3D and S3E). Therefore, it seems that the hyper-extension of telomeres caused by MutSα deficiency renders ALT cells more reliant on BLM to maintain telomere integrity and ultimately cellular survival.
The genome-wide and telomeric mutational landscape of MutSα-deficient ALT cells
MutSα disruption and defective MMR can drive genomic mutation patterns in cancer cells. The Catalogue of Somatic Mutations in Cancer (COSMIC) used mathematical modeling of whole cancer genomes derived from 30 tumor types to establish a repertoire of recurrent mutational signatures associated with particular genotoxic stimuli and/or defects in genome maintenance, including MMR (https://cancer.sanger.ac.uk/signatures/) (Alexandrov et al., 2020). These signatures were annotated from the relative use, sequence context, and frequency of single and/or doublet base substitutions, including transitions and/or transversions (i.e., C > A, C > G, C > T, T > A, T > C, and T > G), and small insertions and deletions to delineate the origin and mutational activities driving a particular cancers development.
Whole genomes of control (clones 1 and 2) and MutSα-deficient (clones 1 and 2) U2OS cells were generated by whole-genome sequencing (WGS), aggregated, and parsed into COSMIC to derive common, recurrent mutational signatures. Signatures were extracted as single-base substitutions (SBSs), doublet-base substitutions (DBSs) and small insertion/deletions (indels) and composite signatures generated from the known individual signatures. The signature classified as “unknown,” representing a composite of undefined mutations (Alexandrov et al., 2020), was most abundant in the MutSα-deficient cells (Figures 4A and S4A). SBS signatures (6, 15, 26, and 44) and DBS signatures (10 and 11), which have arisen because of MMR deficiency were next most detected (Figure S4A). However, the overall contribution of SBSs was less than that of DBSs and small insertions/deletions (indels). For the latter, signatures ID1 and ID2 attributed to MMR were the most prominent signatures identified (Figures 4A and S4A). Assessing the mutational patterns, we detected a preponderance of C > A, T > C, and C > T SBSs. C > A alterations were more prominent in CCA and CCT contexts. No bias was seen for T > C transitions (Figure S4B). Most DBSs involved TG > NN changes and occurred predominantly at CA sequences. As with previous observations, most indels involved 1 bp and occurred within runs of 5+ homopolymer runs of T nucleotides (Figure S4B). These data are consistent with MutSα preferentially recognizing and repairing base-base mismatches and small insertion-deletion mispairs of 1–2 nucleotides (Alani, 1996; Drummond et al., 1995; Gradia et al., 1997; Iaccarino et al., 1996; Marsischky et al., 1996) and with those observed in MMR mutant yeast (Strand et al., 1993), nematodes (Meier et al., 2018), and human MMR-deficient cancer cells (Alexandrov et al., 2020).
Figure 4. MutSα deficiency alters the mutation landscape of ALT cancer cells.
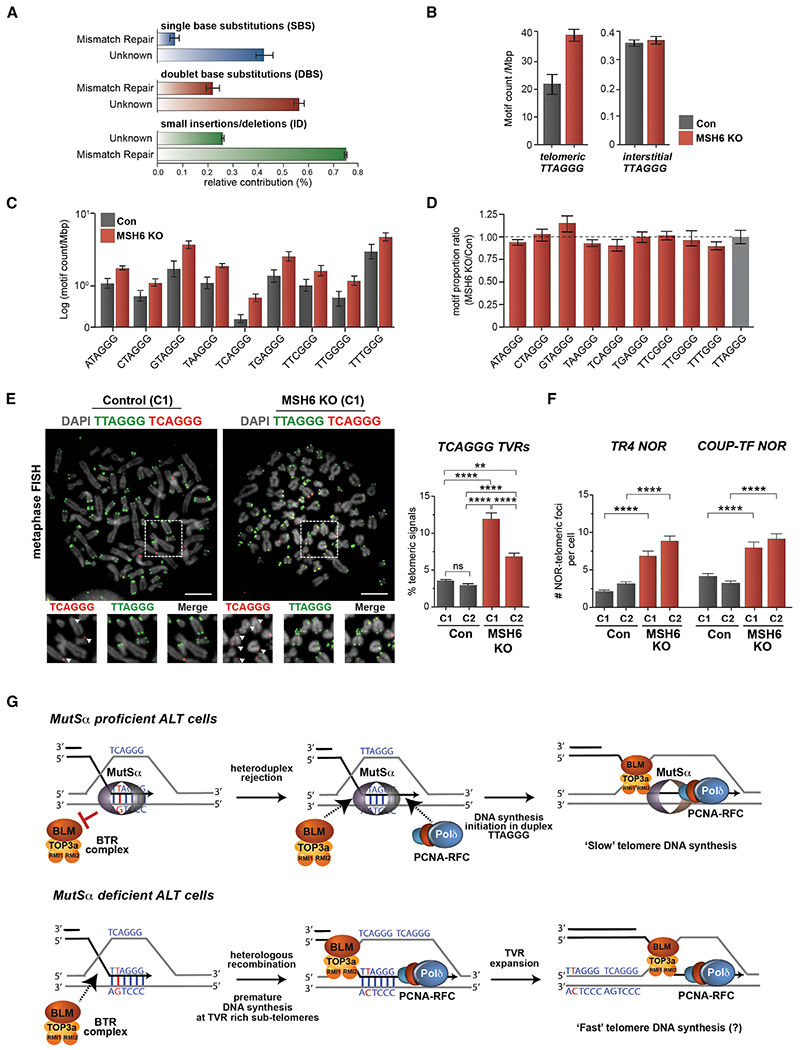
(A) COSMIC single-base substitution (SBS), doublet-base substitutions (DBS), and small insertion/deletion (ID) signatures identified in MSH6 KO clones (C1 and C2), but not in control clones, and their respective etiological contribution.
(B) Quantifications of the total number of TTAGGG repeats found in either telomeric or interstitial regions of the genome in U2OS control and MSH6 KO clones.
(C) Quantifications of the (log) motif count of telomere variant repeats found in U2OS control and MSH6 KO clones.
(D) Normalized and proportional count of telomere variant repeats relative to TTAGGG repeats. The ratio of TVR to TTAGGG repeats is shown. TVRs whose increase in MSH6 KO cells is proportionally greater than that of the TTAGGG repeat exceed the dotted line.
(E) Representative images of metaphase spread chromosomes from U2OS control and MSH6 KO clones stained with TCAGGG (red) and CCCTAA (green) telomeric probes. Enlarged images are shown below, and arrowheads indicate telomere signals. Quantifications of the percentage of TCAGGG signals localized at telomeres by colocalization with CCCTAA are shown in the graph located to the right of the panels.
(F) Quantification of TR4 and COUP-TF telomeric foci in control and MSH6 U2OS KO clones.
(G) Proposed model for the role of MutSα complex in ALT cells. In MutSα-proficient cells, the MutSα complex binds at the junctions of HR intermediates and recognizes imperfectly paired telomeres formed between canonical and TVR repeats. This leads to heteroduplex rejection and prevents premature helicase/branch migration activity of the BTR complex. Strand invasion continues within duplex telomere DNAs until perfect homologous telomere sequences are identified. In MutSα-deficient cells, defective heteroduplex rejection promotes a BTR complex-mediated premature initiation of telomere DNA synthesis between heterologous telomere sequences, such as within mispaired TVR-rich sub-telomeres, leading to TVR expansion (TCAGGG is shown but model applies to all possible TVRs).
All data, except (A)–(D), represent means ± SEM, n = 3 biological replicates. **p < 0.01, ****p < 0.0001 (one-way ANOVA). For (A) and (B), 95% confidence interval error bars are shown. All scale bars, 5 μm. See also Figure S4.
From the WGS analysis, we extracted and enumerated TTAGGG repeat sequences. As with the telomere length analysis that was performed by PFGE and SMAT, this revealed ~50% more TTAGGG-repeat content that was attributed to bona fide telomeres at chromosomal termini and not interstitial TTAGGG repeats (Figure 4B). Telomeres in ALT cells have an increased content of non-canonical telomere variant repeats (TVR) interspersed between the canonical TTAGGG repeats but are enriched at the distal telomeric regions, closer to sub-telomeric regions (Lee et al., 2014; 2018; Marzec et al., 2015; Sieverling et al., 2020; Varley et al., 2002). These include the more-common TGAGGG (g-type), TCAGGG (c-type), and TTGGGG (j-type) repeats (Sieverling et al., 2020), as well as TTCGGG and TTTGGG TVRs whose abundance might be linked with recurrent truncating mutants in ATRX-DAXX genes that are commonly mutated in ALT cancer (de Nonneville and Reddel, 2021; Sieverling et al., 2020). As with the canonical TTAGGG repeat, the abundance of these TVRs within telomeres increased in the MutSα-deficient cells (Figure 4C) but not at interstitial repeat regions (not shown). Notably, several TVRs exhibited deviating patterns of accumulation with few in fact increasing disproportionally relative to the TTAGGG repeat (Figure 4D). The exception to this being the GTAGGG, and more modestly the CTAGGG, TVRs that were more abundant and the TAAGGG, TCAGGG, and TTTGGG TVRs that were less abundant relative to TTAGGG (Figure 4D).
Despite being one of the least-abundant TVR, TCAGGG has been functionally linked with telomere integrity, particularly in ALT (Conomos et al., 2012; Marzec et al., 2015). The TCAGGG repeat can destabilize TRF2 binding and recognized by nuclear orphan receptors (NORs), TR4, and COUP-TFII (Marzec et al., 2015) that normally reside intra-chromosomally at transcriptionally active regions but also have links with genome destabilization (Lin et al., 2009; Marzec et al., 2015). Therefore, upon further examination, we confirmed an increased abundance of TCAGGG repeats in MutSα-deficient cells by conventional Southern dot-blot using radio-labeled oligonucleotide probes (Figure S4C). Although TCAGGG TVRs were rarely detected at telomeres by sequential FISH on metaphase chromosomes in control cells, they were readily apparent at telomeres on metaphase chromosomes from MutSα-deficient U2OS cells (Figure 4E). Quantification revealed the TCAGGG TVR to be ~2- to 5-fold more abundant than in the control cells (Figure 4E). Similarly, puncta corresponding to TCAGGG repeats could now be visualized on individual telomere DNA fibers from MutSα-deficient U2OS cells by the SMAT assay (Figure S4D). Thus, the absence of MutSα seems to cause the random generation or copying of TVRs in the MutSα-deficient cells, which are largely, but not exclusively, proportional to increases of the canonical TTAGGG telomere DNA repeat (see Discussion and Figure 4G for proposed model).
With respect to whether this caused greater occupancy of NORs at telomeres, indeed, we observed significantly greater recruitment of COUP-TFII and TR4 to telomeres in MutSα-deficient cells than in control U2OS cells (Figures 4F and S4E). Of note, upon the induction of telomere DSBs with TRF1-FokI, we observed that TR4 and COUP-TFII were present at smaller foci but largely excluded from the larger TRF1-FokI DSB sites at which MSH2 accumulated (Figures 4F and S4E). In fact, co-localizations between NORs and MSH2 were rarely observed. This could reflect a pro-active role for the MutSα complex in mitigating TVR formation or suppressing a NOR-FANCD2-SLX4-mediated non-canonical telomere DNA synthesis pathway that was proposed (Xu et al., 2019).
DISCUSSION
In this study, we uncovered that the disruption of the MutSα complex fosters a telomere hyper-recombination phenotype with a striking extension of telomeres to lengths that are substantially greater than typically observed in ALT cancer cells. Additional phenotypes associated with ALT-associated recombination also become increasingly manifest, including more-abundant and larger ALT-associated APBs. However, we did not observe significant alterations in other markers, such as C-circles, and even observed reduced numbers of t-SCEs, despite their associations with pro-recombinogenic activity. Several studies have now shown that t-SCEs in ALT cells most likely arise from non-productive ALT and are inversely correlated with telomere DNA synthesis (Sarkar et al., 2015; Sobinoff et al., 2017). Delving further, we did detect an increase of t-SCEs involving a single chromatid and telomere fragility in MutSα-deficient ALT cells, which is consistent with enhanced conservative DNA synthesis required for efficient ALT (Roumelioti et al., 2016; Yang et al., 2020). It was also notable that MutSα-deficient chromosomes harbored significantly fewer chromatids with undetectable telomeres (signal free ends). This indicated that MutSα deficiency could be leading to the extension and restoration of the shortest telomeres or initiation of synthesis from within TVR-rich sub-telomeric regions (Lee et al., 2018; Varley et al., 2002) rather than duplex TTAGGG repeat DNA sequences. In normal instances, the inter/intra chromosomal pairing of telomeres occurs between homologous telomere sequences (Cho et al., 2014); most are presumably TTAGGG repeats. Pairing and alignment with an imperfect recipient sequence, such as would be the case with TVRs (TCAGGG and GTAGGG, etc.), would likely be rejected through heteroduplex rejection involving the cooperative actions of MutSα complex and RECQ-helicases, such as BLM (Figure 4G). However, in the absence of the MutSα complex, the elimination of this barrier to heterologous recombination would allow premature DNA synthesis initiation within, and copying of, sub-telomeric located TVRs on the extending DNA strands (Figure 4G).
Biochemical studies show that purified MutSα binds to Holliday junctions (HJs) in vitro and multiple RecQ helicases like BLM, RECQ1, and WRN (Doherty et al., 2005; Myung et al., 2001; Saydam et al., 2007; Sugawara et al., 2004; Yang et al., 2004). These studies provided the inference that MutS complexes regulate the recruitment of RecQ-helicases, thereby influencing their activities during HR. Given these precedents, MutSα could fulfill a similar role at telomeres in ALT cells. However, by associating with HJs, MutSα could also serve to counteract the BTR complex, restraining premature helicase and branch migration activity (Figure 4G). That BLM localization and telomere DNA synthesis was significantly enhanced in MutSα-deficient cells would lend support to this aspect of MutSα anti-recombinase function at telomeres. Together with PML, BLM is a primary driver of ALT by forming APBs (Loe et al., 2020; Min et al., 2019). Increased BLM associations in MutSα-deficient cells could cause enhanced sequestration or, perhaps, prolong the residency of telomeres within APBs, thereby facilitating DNA synthesis. However, we found that, even though localization of MutSα was lost in BLM and RMI KO cells, it also required its PIP-mediated interaction with PCNA. Furthermore, the increase in APBs observed in MutSα-deficient cells could be suppressed by depletion of POLD3, which forms the ALT replisome with PCNA. The interaction with PCNA is necessary for correcting errors made during DNA replication and is distinct from its role in heteroduplex rejection (Hombauer et al., 2011b; Kleczkowska et al., 2001). It follows, therefore, that the successful initiation of ALT from homologous paired DNAs and subsequent licensing of BTR-dependent branch migration should involve a change in MutSα function (Figure 4G). It might be attractive to speculate that, once DNA synthesis is licensed, MutSα functional switches from heteroduplex rejection to track with PCNA to catalyze the correction of errors generated during telomere DNA synthesis which, because of its repetitive nature, could be prone to replisome slippage. In addition, if ALT DNA synthesis proceeds slowly by stop-start cycles and is intrinsically error prone, as is the case with BIR, which is the ancestral forerunner of ALT (Deem et al., 2011; Liu et al., 2021), MutSα might act to mitigate excess mutation of telomeric DNA sequences that could affect telomere integrity.
In conclusion, this study uncovers the MutSα complex as an addition to the constellation of DNA repair mechanisms that converge to maintain telomeres by the ALT mechanism (Sobinoff and Pickett, 2017). Components of virtually all DNA repair pathways have been identified as constituents and implicated in ALT (Déjardin and Kingston, 2009; García-Expósito et al., 2016; Grolimund et al., 2013; Hoang et al., 2020; Petti et al., 2019). In addition to the key interactions with BLM (Yang et al., 2004) and PCNA (Hombauer et al., 2011a) that are essential for ALT (Dilley et al., 2016), the MMR machinery has been functionally connected to, among others, the inter-strand crosslink repair protein FANCJ (Peng et al., 2007) and the DNA Pol-η (Peña-Diaz et al., 2012), which similarly, are implicated in ALT (García-Expósito et al., 2016; Zhang et al., 2021). A major future challenge will be to integrate the observations relating to MutSα within the extensive regulatory patchwork that maintains productive ALT. This should provide insights into how those pathways functionally intersect and cooperate (or not), which would be a key advance toward the goals of understanding ALT and toward developing specific therapies.
Limitations of the study
Even though fluctuating patterns of TVRs were observed after MSH6 disruption, few TVRs accumulated disproportionately to the canonical TTAGGG repeat. Furthermore, and rather surprisingly, MutSα-deficient ALT cells proliferated as normal, if not modestly better than control cells, which would imply that telomeres remain functional. This appears to minimize MutSα’s canonical role in MMR as being predominant at telomeres in ALT cells. However, a role for MMR should be not be discounted because MutL heterodimers have been identified by proteomics as constituents of telomeres in ALT cells (García-Expósito et al., 2016), and the accumulation of endogenous MLH1, a subunit of MutLα, at telomeres (this study) have been documented, even though the effect remains unclear. The sustained co-depletion of MutSα in conjunction with MutLα, for instance, could provide important insights. Furthermore, although our assessment was relatively limited, we did not observe strong effects on ALT phenotypes upon depletion of MSH3 (MutSβ). However, this complex acts similarly to MutSα in heteroduplex rejection and might localize to telomeres also. Unlike MutSα, which binds single or indel mispairs, MutSβ preferentially binds to larger loops of mispaired DNAs (Gupta et al., 2011; Warren et al., 2007). Interestingly, MutSβ interacts directly with SLX4 (Young et al., 2020), which, as mentioned, is critical for telomere homeostasis and, so, could act in conjunction with SLX4 at these larger loops formed by mispairing or at MMR-excision intermediates. It will be intriguing to understand whether the differential association of MutSα with BLM and MutSβ with SLX4 is a functional aspect of the antagonism between these complexes to maintain efficient ALT.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Roderick J. O’Sullivan (osullivanr@upmc.edu).
Materials availability
All reagents generated in this study are available upon request to the lead contact and upon signature of the corresponding material transfer agreement, if necessary.
Data and code availability
Whole Genome Sequencing (WGS) Sequence Read Archive data have been deposited at NCBI and are publicly available as of the date of publication. The URL and Accession number are listed in the key resources table.
Original code has been deposited and is publicly available as of the date of publication. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| PCNA | Cell Signaling Technology | Cat# 2586; RRID:AB_2160343 |
| MSH2 | Cell Signaling Technology | Cat# 2850; RRID:AB_2144797 |
| POLD3 | Abnova | Cat# H00010714-M01; RRID:AB_538310 |
| PML | Santa Cruz | Cat# sc-5621; RRID:AB_2166848 |
| FLAG | Cell Signaling Technology | Cat# 14793; RRID:AB_2572291 |
| TR4 | Perseus | Cat# PP-H0107B-00; RRID:AB_2155352 |
| COUP-TFII | Perseus | Cat# PP-H7147-00; RRID:AB_2155627 |
| TRF2 | Novus | Cat# NB110-57130; RRID:AB_844199 |
| BLM | Jan Karlseder | N/A |
| MSH6 | Cell Signaling Technology | Cat# 5424; RRID:AB_10695802 |
| MSH3 | Novus | Cat# NBP1-95114;RRID:AB_11028618 |
| γ-TUB | Sigma-Aldrich | Cat# T6557; RRID:AB_477584 |
| FLAG | Sigma-Aldrich | Cat# F1804; RRID:AB_262044 |
| WRN | Novus-Biological | Cat# NB100-472; RRID:AB_2216076 |
| TRF1 | Jan Karlseder | N/A |
| Chemicals, peptides and recombinant proteins | ||
| Non-targeting control | Dharmacon | D-001810-10-0020 |
| MSH6 | Dharmacon | L-019287 |
| MSH2 | Dharmacon | L-003909 |
| MSH3 | Dharmacon | L-019665 |
| POLD3 | Dharmacon | L-026692 |
| BLM | Dharmacon | L-007287 |
| Benzonase | Merck Millipore | Cat#70746 |
| 4-hydroxytamoxifen | Sigma | Cat#H7904 |
| Doxycycline | Clontech | Cat#631311 |
| Shield Ligand | Clontech | Cat#632189 |
| AluI | New England Biolabs | Cat#R0137 |
| MboI | New England Biolabs | Cat#R0137 |
| f29 Polymerase | New England Biolabs | Cat#M0269 |
| Critical commercial assays | ||
| Click-iT EdU Cell Proliferation Kit for Imaging, Alexa Fluor 555 dye | ThermoFisher | Cat#C10338 |
| Experimental models: Cell lines | ||
| U2OS | this paper | N/A |
| U2OS MSH6 KO C1-C2 | this paper | N/A |
| U2OS Control C1-C2 | this paper | N/A |
| U2OS BLM KO C1-C2 | this paper | N/A |
| U2OS BLM-MSH6 KO C1-C2 | this paper | N/A |
| U2OS WRNKO C1-C2 | this paper | N/A |
| U2OS WRN-MSH6 KO C1-C2 | this paper | N/A |
| U2OS RMI KO | Eros Lazzerini Denchi (Loe et al., 2020) | N/A |
| HeLa LT | this paper | N/A |
| HeLa LT MSH6 KO C1 | this paper | N/A |
| VA13 | Roger Greenberg | N/A |
| VA13 MSH6 KO C1-C3 | Roger Greenberg | N/A |
| VA13 | Roger Greenberg | N/A |
| U2OS TRF1-FokI WT/D450A | Roger Greenberg (Dilley et al., 2016) | N/A |
| Saos2 | this paper | N/A |
| U2OS DiVa | Gaëlle LeGube (Iacovoni et al., 2010) | N/A |
| Hek293T | ATCC | RRID: CVCL_0063 |
| Recombinant DNA | ||
| pSpCas9(BB)-2A-GFP | (Ran et al., 2013) | RRID: Addgene_48138 |
| psPAX2 | Didier Trono | RRID: Addgene_12260 |
| pMD2.G | Didier Trono | RRID: Addgene_12259 |
| FLAG-TRF1-FokI WT/D450A | Roger Greenberg (Dilley et al., 2016) | N/A |
| pTag-RFP-WT-MSH6 | this paper | N/A |
| pTag-RFP-PAAP-MSH6 | this paper | N/A |
| pTag-RFP-K1140-MSH6 | this paper | N/A |
| pTag-RFP-WT-MSH6 | this paper | N/A |
| pTag-RFP | this paper | N/A |
| Oligonucleotides | ||
| MSH6 sgRNA; CACGCGAAGGCGGCCGTGCC | this paper | N/A |
| BLM sgRNA; GGAACGAACTGCTTCAGCAG | this paper | N/A |
| WRN sgRNA 1; GTAAATTGGAAAACCCACGG | (Chan et al., 2019) | N/A |
| WRN sgRNA 2; ATCCTGTGGAACATACCATG | (Chan et al., 2019) | N/A |
| WRN sgRNA 3; GTAGCAGTAAGTGCAACGAT | (Chan et al., 2019) | N/A |
| Control gRNA 1; GTGAACCGCATCGAGCTGAA | this paper | N/A |
| Control gRNA 2; GGAGCGCACCATCTTCTTCA | this paper | N/A |
| Tel C PNA Probe; Alexa 488-(CCCTAA)4 | PNA Bio | Cat#F1004 |
| Tel G PNA Probe; Alexa-594-(TTAGGG)4 | PNA Bio | Cat#F1006 |
| Variant TVR PNA Probe; Cy5-OO-(TGACCC)3 | Panagene | N/A |
| Software and algorithms | ||
| GraphPad Prism | GraphPad | RRID: SCR_002798 |
| Accuri C6 Plus | Becton Dickenson | RRID: SCR_014422 |
| MiniMap2 | https://github.com/lh3/minimap2 | RRID: SCR_018550 |
| Mutect2 | (Van der Auwera et al., 2013) | https://gatk.broadinstitute.org/hc/en-us/articles/360037593851-Mutect2 |
| Genomic sEquence AnalyzeR (GEAR) | https://www.gear-genomics.com/ | N/A |
| Telomere Sequence Mapping | this paper | https://github.com/ChildrensMedicalResearchInstitute/MutSa_Deficiency_ALT |
| Deposited data | ||
| Raw and analyzed data | this paper | https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA776978 SRA: PRJNA776978 |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
Cell lines used in this study are listed in the Key resource table. U2OS, HeLa LT, and Saos2 cell lines were authenticated by STR profiling and confirmed mycoplasma free by ATCC cell line authentication services. Each cell line was cultured in Glutamax-DMEM supplemented with 10% bovine growth serum. Cells were cultured at normal oxygen conditions of 20% O2 and 7.5% CO2. U2OS DiVa cells expressing the restriction enzyme AsiSI fused to the modified estrogen receptor were obtained from Gaëlle Legube (CBI, Toulouse). To trigger nuclear localization of AsiSI and induce AsiSI-dependent breaks, U2OS DiVa cells were treated with 300nM 4-OHT for 4hrs, as described previously (Iacovoni et al., 2010). U2OS cells expressing wild-type (WT) or catalytically inactive (DA) FokI fused to TRF1 were obtained from Roger Greenberg (University of Pennsylvania). TRF1-FokI expression was induced by adding 40ng/mL Doxycycline for ~24hrs followed by 4-OHT (1 μM) and Shieldl ligand (1 μM) for 2hrs, as described (Dilley et al., 2016).
METHOD DETAILS
Direct immunofluorescence
Cells on glass coverslips were washed twice in PBS and fixed with 2% paraformaldehyde (PFA) for 10mins. Cells were permeabilized with 0.1% (w/v) sodium citrate and 0.1% (v/v) Triton X-100 for 5mins and incubated with fresh blocking solution (1 mg/mL BSA, 10% normal goat serum, 0.1% Tween) for 30mins. Primary antibodies were diluted in blocking solution and added to cells for 1hr at RT or overnight in refrigerated conditions. Next, cells were washed 3 times with PBS for 5mins and incubated with Alexa coupled secondary antibodies (488nm, 555nm, 647nm) (Life Technologies) for 1hr at RT. Then, cells were washed 3 times with PBS and mounted on slides with Prolong Gold Anti-fade reagent with DAPI (Life Technologies). Once the Prolong Anti-fade polymerized and cured, cells were visualized by conventional fluorescence with 40X and/or 63X Plan γ objective (1.4 oil) using a Nikon 90I.
IF-FISH
After secondary antibody, cells were washed and then the IF staining was fixed with 2% paraformaldehyde (PFA) for 10mins. PFA was washed off with PBS and coverslips dehydrated with successive washes in 70%, 95% and 100% EtOH for 3mins, allowed to air dry completely. Next, the coverslips were mounted on glass slides with 15 μL per coverslip of hybridization mix (70% deionized Formamide, 1mg/ml of Blocking Reagent [Roche], 10mM Tris-HCl pH 7.4) containing Alexa 488-(CCCTAA)4 (TelC) PNA probe. DNA was denatured by setting the slides on a heating block set to 72°C for 10mins and then incubating overnight at RT in the dark. The coverslips were then washed twice for 15mins with Wash Solution A (70% deionized formamide and 10mM Tris-HCl pH7.2) and three-time with Solution B (0.1M Tris-HCl pH7.2, 0.15M NaCl and 0.08% Tween) for 5mins at RT. EtOH dehydration was repeated as above and finally, the samples were mounted and analyzed as mentioned above.
Western blotting
Cells were harvested with trypsin, quickly washed in PBS, counted with Cellometer AutoT4 (Nexcelom Bioscience) and directly lysed in 4X NuPage LDS sample buffer at 10000 cells per μl. Proteins were gently homogenized using a Nuclease (ThermoFisher), denatured for 10mins at 70°C and resolved by SDS-Page electrophoresis, transferred to nitrocellulose membranes, blocked in 5% milk in TBST for 30mins and probed. For secondary antibodies, HRP-linked anti-rabbit or mouse (Amersham) was used, and the HRP signal was visualized with SuperSignal ECL substrate (Pierce) as per the manufacturer’s instructions.
Generation of KO cells by CRISPR/Cas9
Sequences for guide RNAs used are provided in the Key resource table. To generate knockout cell lines, pSpCas9(BB)-2A-GFP vector (Ran et al., 2013) containing sgRNA was transiently transfected into target cell lines using Lipofectamine 2000. 48hrs after transfection, GFP-positive cells were single-cell sorted in 96 well plates to isolate individual clones. Recovered clones were tested by western blot for the absence of MSH6 protein. Genomic editing was confirmed by whole-genome sequencing. To make MSH6/BLM double KO cells, pSpCas9(BB)-2A-GFP vector containing a BLM sgRNA sequence was transiently transfected into the two different MSH6 KO U2OSas described above. Recovered clones after single-cell sorting were tested by western blot for the absence of BLM protein. To make MSH6/WRN double KO cells, three LentiCRISPR-V2 vectors containing different WRN sgRNA sequences (Chan et al., 2019) were transfected with Lipofectamine 2000 (Life Technologies) into 293FT cells along with psPAX2 and pMD2.G packing plasmids. Media was changed 16hs after transfection. Virus was harvested and filtered 48hrs after media change and used to infect U2OS control and MSH6 KO clones for 2 days. Then, cells were selected with 2 mg/mL puromycin for at least 1 week before performing the experiments. The absence of WRN was tested by western blot.
Telomere restriction fragment analysis by pulsed field gel electrophoresis
Telomere gels were performed using telomere restriction fragment (TRF) analysis. Genomic DNA was digested using AluI and MboI (NEB). 5 μg of DNA was run on a 1% PFGE agarose gel (Bio-Rad) in 0.5 × TBE buffer using the CHEF-DRII system (Bio-Rad) at 6V cm−1; initial switch time 1 s, final switch time 6 s, for 17hrs at 14°C. The gel was then dried for 2hrs at 60°C, denatured in a 0.5N NaOH 1.5M NaCl solution, and neutralized. The gel was hybridized with 32P-labeled (TTAGGG)4 oligonucleotides in Church buffer overnight at 55°C. The next day, the membrane was washed three times in 2 × SSC buffer and once in 2x SSC 0.5% SDS, exposed onto a storage phosphor screen and scanned using Amershan™ Typhoon (GE Healthcare). Telomere length was determined using TeloTool software. To detect TTAGGG and TCAGGG variants from genomic DNA using a Dot Blot system, 2 μg of AluI and MboI (NEB) digested genomic DNA was diluted to 100 μL with 2 × SSC, heated at 95°C for 5mins, cooled on ice and dot-blotted onto a 2 × SSC-soaked nylon membrane. DNA was UV cross-linked onto the membrane and hybridized with P32 end-labeled oligos (CCCTAA)4 or (TCAGGG)4 products. An Alu probe was used as a loading control. Blots were denatured, neutralized, washed, exposed to PhosphoImager screens, scanned using a Typhoon 9400 PhosphoImager (GE Healthcare) and quantified with ImageJ.
Chromosome orientation FISH
U2OS cells were incubated with BrdU and BrdC simultaneously for ~12hrs, and hybridization was performed with Alexa 488-(CCCTAA)4 and Alexa 568-(TTAGGG)4 PNA probes (PNABio). In brief, cell cultures were incubated with 7.5mM BrdU and 2.5mM BrdC for ~12hrs. After removal of nucleotide analogs, Colcemid (GIBCO) was added for ~2hrs, cells were harvested by trypsinization, swelled in 75mM KCl and fixed in 70% Methanol: 30% Acetic Acid. Samples are stored at −20°C for days. Metaphase chromosomes were spread by dropping onto washed slides, then RNase A (0.5 mg/ml) and pepsin treated. Slides were incubated in 2 × SSC containing 0.5 mg/ml Hoechst 33258 for 15mins in the dark and irradiated for 40mins (5.4 × 105 J/m2, energy 5400) at in a UV Stratalinker 2400 (Stratagene). The nicked BrdU/C substituted DNA strands are degraded by Exonuclease III digestion. The slides were then washed in PBS, dehydrated by EtOH washes and allowed to air dry completely. The remaining strands were hybridized with fluorescence-labeled DNA probes of different colors, specific either for the positive telomere strand (TTAGGG)4 (polymerized by lagging strand synthesis) (Alexa 488, green color), or the negative telomere strand (CCCTAA)4, (polymerized by leading strand synthesis) (Alexa-568, red color). Before the hybridization of the first PNA, DNA is denatured by heating at 72°C for 10mins, as in IF-FISH, and then incubated for 2hrs at RT. Slides were washed for 15mins with Wash Solution A (see IF-FISH), dried and then incubated with the second PNA for 2hrs at RT. The slides were then washed again twice for 15mins with Wash Solution A and 3 times with Wash Solution B (see IF-FISH) for 5mins at RT. The second wash contained DAPI (0.5 μg/mL). Finally, cells were dehydrated in EtOH as above and mounted (Vectashield). The resulting chromosomes show dual staining and allow the distinction between leading and lagging strands. Metaphase chromosomes were visualized by a conventional fluorescence microscope with a 63X Plan γ objective (1.4 oil) on a Nikon 90i microscope.
Metaphase FISH
U2OS cells treated with Colcemid (GIBCO) were for ~2hrs were harvested by trypsinization, swelled in 75mM KCl and fixed in 70% Methanol: 30% Acetic Acid. Samples were stored at −20°C for at least one day before dropping chromosomes. Metaphase chromosomes on microscope slides were rehydrated in PBS for 5mins, then fixed with PBS containing 3.7% formaldehyde for 5mins at RT. Washed 3 times in PBS, and then RNase A (0.5 mg/ml) and pepsin treated. Slides were fixed again in PBS containing 3.7% formaldehyde for 5mins at RT. Washed three times in PBS and dehydrated by EtOH washes and allowed to air dry completely. Metaphases were hybridized at 72°C for 10mins using an Alexa Fluor 488 conjugated-C strand telomere PNA probe. Hybridize overnight in the dark at room temperature in a humidified chamber. The next day, wash solutions A and B were performed as described above for CO-FISH. Finally, cells were dehydrated in EtOH as above and mounted (Vectashield). For TCAGGG/TTAGGG staining on metaphase spreads, a Cy5 conjugated-TCAGGG was hybridized at 80°C for 5mins and incubated overnight in the dark at room temperature before. The next day, cells were washed in solution A, solution B and finally ethanol dehydrated as before. Air-dried slides were then hybridized at 72°C for 10mins using a FITC conjugated-C strand telomere PNA probe and incubated for 3hrs at room temperature. Slides were sequentially washed in solution A, solution B, PBS, and dH2O. Air-dried slides were finally mounted (Vectashield). Metaphase chromosomes were visualized by a conventional fluorescence microscope with a 63X Plan γ objective (1.4 oil) on a Nikon 90i microscope.
TRF1-FokI mediated induction of telomeric DSBs
U2OS cells were transfected with FLAG-tagged (WT)- or (D450A)-TRF1-FokI. 24hrs later, cells were processed for immunofluorescence using either MSH2 or MLH1 antibodies. To detect MSH2 at damage telomeres in S- and Non-S phase cells, cells were transfected with WT-TRF1-FokI for 24hrs. Then, cells were pulsed the with 10 μM EdU (Sigma) for 2hrs before fixation with 4% PFA for 10mins. After permeabilization (0.5% Triton X-100 for 5mins), cells were incubated with MSH2 antibody. Following secondary antibody, Click-it chemistry was applied to detect incorporated EdU.
siRNA Transfections
For siRNA knockdown, siRNA Smartpools from Dharmacon (GE) were used (See Key resource table). Briefly, 200,000 and 700,000 cells were seeded per well of a 6-well plate and 10cm dish containing growth medium without antibiotics, respectively. ~6hrs later cells were transfected. siRNAs and Dharmafect were diluted in OptiMEM (Life Technologies). A working siRNA concentration of 50nM was used. We used 2.5 μL and 10 μL Dharmafect transfection reagent per well and 10cm plate, respectively. Transfection medium was replaced with complete culture media 24hrs later or cells were split for the desired application and harvested at 72hrs post-transfection.
C-circle assay
CC assay was performed as described previously. Genomic DNA was purified, digested with AluI and MboI and cleaned up by phenol-chloroform extraction and precipitation. DNA was diluted in ultraclean water and concentrations were exhaustively measured to the indicated quantity (30, 15, 7.5ng) using a Nanodrop (ThermoFisher). Samples (10 μ.l) were combined with 10 μL BSA (NEB; 0.2 mg/ml), 0.1% Tween, 0.2mM each dATP, dGTP, dTTP and 1 × Φ29 Buffer (NEB) in the presence or absence of 7.5U ΦDNA polymerase (NEB). Samples were incubated at 30°C for 8hrs and then at 65°C for 20mins. Reaction products were diluted to 100 μL with 2 × SSC and dot-blotted onto a 2 × SSC-soaked nylon membrane. DNA was UV cross-linked onto the membrane and hybridized with a P32 end-labeled TelC (CCCTAA)4 oligo probe to detect C-circle amplification products. All blots were washed, exposed to PhosphoImager screens, scanned using a Typhoon 9400 PhosphoImager (GE Healthcare) and quantified with ImageJ. In all reactions Φ29 was omitted as a negative control DNA was used.
Colony formation assays
1000 cells were seeded in 6 well plates in triplicate and cultured for 7 days before fixation and staining in a 1% crystal violet solution. For cisplatin experiments, 1000 cells were plates, 24hrs later treated or not with 2.5 μM cisplatin for another 24hrs, washed and finally cultured for 7 days. Plates were images and analyzed with the Protein Simple FluorChem system, which was used to count positively stained colonies and calculate total cell coverage per well.
Flow cytometry
Cells in each condition were collected and washed twice in cold 1X PBS. Cell pellets were resuspended in 100 μL cold PBS. Cells were fixed by slowly adding 1mL cold ethanol 70%. Incubate cells overnight at −20°C. The next day, wash cells three times with an excess of PBS. After the last wash, remove all PBS and resuspend cells in 500 μL PI staining solution containing 2mgr RNase and 200 μg propidium iodide in PBS. Stain cells overnight at 4°C. The next day, fluorescence was analyzed by flow cytometry (Accuri C6, BD).
Single-molecule analysis of telomeres (SMAT)
Cells were labeled with 15 μM EdU for 5 h before harvesting by trypsinization. Cells were embedded in low-melting agarose plugs then subjected to proteinase K digestion overnight. Plugs were dissolved with agarose (Thermo Scientific) according to the manufacturer’s instructions. Molecular combing was performed using the Molecular Combing System (Genomic Vision S.A.) with a constant stretch factor of 2 kb/μm using vinyl silane coverslips (20 × 20 mm; Genomic Vision S.A.), according to the manufacturer’s instructions. After combing, coverslips were dried for 4hrs at 60°C. The quality and integrity of combed DNA fibers were checked using the YoYo-1 counterstain (Molecular Probes). Coverslips were denatured for 10mins in the alkali-denaturing buffer (0.5M NaOH + 1MNaCl), followed by a neutralization step (3X 5mins PBS washes). Telomeric DNA was visualized by sequential hybridization first with a TAMRA-OO-KKK(TTAGGG)3 PNA probe (Panagene). Coverslips were washed for 5mins at 42°C (50% Formamide and 1X SSC final concentration), 3x 5mins wash no agitation at 60°C (2x SSC) and 1x 5mins wash medium shaking at RT (2x SSC and 0.1% Tween). Coverslips were then overlaid with Cy5-OO-(TGACCC)3 and hybridized again overnight at RT. Following the 2nd PNA probe hybridization, samples were fixed for 10mins (4% formaldehyde) and Click-it chemistry for EdU was applied. Telomere fibers were detected on a Zeiss Axio Imager microscope with the ApoTome module and analyzed with Zen software (Zeiss)12.
Genomic DNA preparation for whole-genome sequencing
Genomic DNA was extracted using QIAGEN® Genomic DNA Preparation. Briefly, 4x106 cells were collected by trypsinization, washed twice in cold PBS and finally resuspended in 0.5mL ice-cold PBS1X. Cytoplasmic fraction was first extracted twice with buffer C1. Purified nuclear fraction was completely resuspended in buffer G2.25 μL Proteinase K was added, and samples were incubated at 50°C for 1hr. DNA was then loaded on pre-equilibrated Genomic-tip 20/G with buffer QBT. The column was then washed twice with buffer QC, and DNA was finally eluted using 2mL of buffer QF. Genomic DNA was precipitated using 0.7 volumes isopropanol and finally resuspended in 200 μL 10mM Tris-HCl pH 8.
DNA sequencing and variant calling
Samples were sequenced using paired-ended Illumina NovaSeq sequencing. Reads were mapped to the hg38 reference genome using Minimap2 (Li, 2018b). Mutations were called using Mutect2 (Van der Auwera et al., 2013). The control samples were used as a background to call for mutations in MSH6 KO genome samples. Additionally, a panel of normals from the 1000 genome project mapped to hg38 was used to remove common somatic mutations. Figure S5 shows the workflow.
Mutational signatures
Single base substitutions (sbs), doublet base substitutions (dbs) and small insertions and deletions (indels) were extracted from the vcf. data using in-house Genomic sEquence AnalyzeR (GEAR) software. Signature spectra were calculated using non-negative matrix factorization using COSMIC signatures v3.1 (Alexandrov et al., 2020; Tate et al., 2019).
Telomere length estimation
Telomeric sequences were filtered by extracting reads with five or more consecutive combinations of telomeric motifs including TTAGGG and 18 other variants (VTAGGG, TVAGGG, TTBGGG, TTAHGG, TTAGHG, and TTAGGH), as well as reverse complement motifs. The resulting reads were mapped to the hg38 reference genome. From the resulting bam file, reads mapped to the reference genome at the interstitial region with map quality greater than 20 were excluded. The rest of the reads were extracted from the bam file and saved into a fastq file. From here on, these sequences are referred to as telomeric reads. The telomeric reads were mapped to a 200 bp telomeric reference template with pure TTAGGG motifs using GEAR (URLs provided in Key resource table). For each read, the matching region, SBS, DBS and indels were considered. Telomere length was estimated by counting the number of motifs in the matching region of each read and normalizing by the total number of reads in each sample. Figure S6 shows the workflow.
Telomere variants
The frequency of telomere variants was compared between the control samples and the MSH6 KO samples to quantify differences in the 18 extracted variants relative to TTAGGG. Sbs with base quality larger than or equal to 20 and with the mean base quality of the two 5′ and 3′ adjacent bases larger than or equal to 20 was considered. Similarly, for indels, only those with mean base quality larger than or equal to 20 were considered. The number of motifs for the each of the 18 telomeric variants were calculated in the samples. Then, for each of the motifs, the ratio between the count in the MSH6 KO samples and the control samples was calculated.
QUANTIFICATION AND STATISTICAL ANALYSIS
All data in this study was analyzed in GraphPad Prism and Microsoft Excel. Statistical tests used are indicated in the figure legend accompanying each figure. Typically, unless otherwise stated n refers to the number of independent experiments and SEM refers to standard error of means. Sample size was not pre-determined.
Supplementary Material
Highlights.
Loss of MutSα mismatch repair complex causes telomere hyper-extension by ALT pathway
MutSα counteracts BLM and limits premature initiation of telomere extension
Simultaneous loss of MutSα and BLM leads to cell death
ACKNOWLEDGMENTS
We are grateful to members of the O’Sullivan laboratory for comments on the manuscript. We thank Eros Lazzerini Denchi, Jan Karlseder, Roger Greenberg, and Gaëlle Legube for sharing reagents. We thank Patricia Opresko, Kara Bernstein, and Jacob Stewart-Ornstein for providing access to instrumentation. M.L.L. is recipient of John S. Lazo Cancer Pharmacology Fellowship from the Department of Pharmacology & Chemical Biology, University of Pittsburgh School of Medicine. Facilities at the UPMC Hillman Cancer Center were supported by Comprehensive Cancer Center support grant NCI P30CA047904. Research funding was provided to investigators from the following agencies: R.J.O., NCI 5R01CA207209 and R37CA263622 and American Cancer Society RSG-18-038-01-DMC; S.C.W, NIH 1S10OD019973. The work was also supported by the National Health and Medical Research Council of Australia(1162886) to H.A.P. P.G. acknowledges the Luminesce Alliance, Innovation for Children’s Health, for its contribution and support. Luminesce Alliance is affiliated with the University of Sydney and the University of New South Wales, Sydney.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.110088.
DECLARATION OF INTERESTS
The authors have no conflicts of interest to declare.
REFERENCES
- Acharya S, Wilson T, Gradia S, Kane MF, Guerrette S, Marsischky GT, Kolodner R, and Fishel R (1996). hMSH2 forms specific mispair-binding complexes with hMSH3 and hMSH6. Proc. Natl. Acad. Sci. USA 93, 13629–13634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alani E (1996). The Saccharomyces cerevisiae Msh2 and Msh6 proteins form a complex that specifically binds to duplex oligonucleotides containing mismatched DNA base pairs. Mol. Cell. Biol 16, 5604–5615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, Tian Ng AW, Wu Y, Boot A, Covington KR, Gordenin DA, Bergstrom EN, et al. PCAWG Mutational Signatures Working Group; PCAWG Consortium (2020). The repertoire of mutational signatures in human cancer. Nature 578, 94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand R, Beach A, Li K, and Haber J (2017). Rad51-mediated double-strand break repair and mismatch correction of divergent substrates. Nature 544, 377–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arora R, Lee Y, Wischnewski H, Brun CM, Schwarz T, and Azzalin CM (2014). RNaseH1 regulates TERRA-telomeric DNA hybrids and telomere maintenance in ALT tumour cells. Nat. Commun 5, 5220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bechter OE, Zou Y, Walker W, Wright WE, and Shay JW (2004). Telomeric recombination in mismatch repair deficient human colon cancer cells after telomerase inhibition. Cancer Res. 64, 3444–3451. [DOI] [PubMed] [Google Scholar]
- Bryan TM, Englezou A, Dalla-Pozza L, Dunham MA, and Reddel RR (1997). Evidence for an alternative mechanism for maintaining telomere length in human tumors and tumor-derived cell lines. Nat. Med 3, 1271–1274. [DOI] [PubMed] [Google Scholar]
- Cesare AJ, Kaul Z, Cohen SB, Napier CE, Pickett HA, Neumann AA, and Reddel RR (2009). Spontaneous occurrence of telomeric DNA damage response in the absence of chromosome fusions. Nat. Struct. Mol. Biol 16, 1244–1251. [DOI] [PubMed] [Google Scholar]
- Chan EM, Shibue T, McFarland JM, Gaeta B, Ghandi M, Dumont N, Gonzalez A, McPartlan JS, Li T, Zhang Y, et al. (2019). WRN helicase is a synthetic lethal target in microsatellite unstable cancers. Nature 568, 551–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho NW, Dilley RL, Lampson MA, and Greenberg RA (2014). Interchromosomal homology searches drive directional ALT telomere movement and synapsis. Cell 159, 108–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clynes D, Jelinska C, Xella B, Ayyub H, Scott C, Mitson M, Taylor S, Higgs DR, and Gibbons RJ (2015). Suppression of the alternative lengthening of telomere pathway by the chromatin remodelling factor ATRX. Nat. Commun 6, 7538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conomos D, Stutz MD, Hills M, Neumann AA, Bryan TM, Reddel RR, and Pickett HA (2012). Variant repeats are interspersed throughout the telomeres and recruit nuclear receptors in ALT cells. J. Cell Biol 199, 893–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox KE, Maréchal A, and Flynn RL (2016). SMARCAL1 resolves replication stress at ALT telomeres. Cell Rep. 14, 1032–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabbe L, Verdun RE, Haggblom CI, and Karlseder J (2004). Defective telomere lagging strand synthesis in cells lacking WRN helicase activity. Science 306, 1951–1953. [DOI] [PubMed] [Google Scholar]
- de Nonneville A, and Reddel RR (2021). Alternative lengthening of telomeres is not synonymous with mutations in ATRX/DAXX. Nat. Commun 12, 1552–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wind N, Dekker M, Berns A, Radman M, and te Riele H (1995). Inactivation of the mouse Msh2 gene results in mismatch repair deficiency, methylation tolerance, hyperrecombination, and predisposition to cancer. Cell 82, 321–330. [DOI] [PubMed] [Google Scholar]
- Deem A, Keszthelyi A, Blackgrove T, Vayl A, Coffey B, Mathur R, Chabes A, and Malkova A (2011). Break-induced replication is highly inaccurate. PLoS Biol. 9, e1000594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Déjardin J, and Kingston RE (2009). Purification of proteins associated with specific genomic Loci. Cell 136, 175–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dilley RL, Verma P, Cho NW, Winters HD, Wondisford AR, and Greenberg RA (2016). Break-induced telomere synthesis underlies alternative telomere maintenance. Nature 539, 54–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doherty KM, Sharma S, Uzdilla LA, Wilson TM, Cui S, Vindigni A, and Brosh RM Jr. (2005). RECQ1 helicase interacts with human mismatch repair factors that regulate genetic recombination. J. Biol. Chem 280, 28085–28094. [DOI] [PubMed] [Google Scholar]
- Draskovic I, Arnoult N, Steiner V, Bacchetti S, Lomonte P, and Londoño-Vallejo A (2009). Probing PML body function in ALT cells reveals spatiotemporal requirements for telomere recombination. Proc. Natl. Acad. Sci. USA 106, 15726–15731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond JT, Li GM, Longley MJ, and Modrich P (1995). Isolation of an hMSH2-p160 heterodimer that restores DNA mismatch repair to tumor cells. Science 268, 1909–1912. [DOI] [PubMed] [Google Scholar]
- Dunham MA, Neumann AA, Fasching CL, and Reddel RR (2000). Telomere maintenance by recombination in human cells. Nat. Genet 26, 447–450. [DOI] [PubMed] [Google Scholar]
- Fishel R, Lescoe MK, Rao MR, Copeland NG, Jenkins NA, Garber J, Kane M, and Kolodner R (1994). The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer. Cell 77, 167. [PubMed] [Google Scholar]
- Flores-Rozas H, Clark D, and Kolodner RD (2000). Proliferating cell nuclear antigen and Msh2p-Msh6p interact to form an active mispair recognition complex. Nat. Genet 26, 375–378. [DOI] [PubMed] [Google Scholar]
- García-Expósito L, Bournique E, Bergoglio V, Bose A, Barroso-González J, Zhang S, Roncaioli JL, Lee M, Wallace CT, Watkins SC, et al. (2016). Proteomic profiling reveals a specific role for translesion DNA polymerase η in the alternative lengthening of telomeres. Cell Rep. 17, 1858–1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gradia S, Acharya S, and Fishel R (1997). The human mismatch recognition complex hMSH2-hMSH6 functions as a novel molecular switch. Cell 91, 995–1005. [DOI] [PubMed] [Google Scholar]
- Grolimund L, Aeby E, Hamelin R, Armand F, Chiappe D, Moniatte M, and Lingner J (2013). A quantitative telomeric chromatin isolation protocol identifies different telomeric states. Nat. Commun 4, 2848. [DOI] [PubMed] [Google Scholar]
- Gupta S, Gellert M, and Yang W (2011). Mechanism of mismatch recognition revealed by human MutSβ bound to unpaired DNA loops. Nat. Struct. Mol. Biol 19, 72–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heaphy CM, de Wilde RF, Jiao Y, Klein AP, Edil BH, Shi C, Bettegowda C, Rodriguez FJ, Eberhart CG, Hebbar S, et al. (2011). Altered telomeres in tumors with ATRX and DAXX mutations. Science 333, 425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henson JD, Cao Y, Huschtscha LI, Chang AC, Au AYM, Pickett HA, and Reddel RR (2009). DNA C-circles are specific and quantifiable markers of alternative-lengthening-of-telomeres activity. Nat. Biotechnol 27, 1181–1185. [DOI] [PubMed] [Google Scholar]
- Hoang SM, and O’Sullivan RJ (2020). Alternative lengthening of telomeres: building bridges to connect chromosome ends. Trends Cancer 6, 247–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoang SM, Kaminski N, Bhargava R, Barroso-González J, Lynskey ML, García-Expósito L, Roncaioli JL, Wondisford AR, Wallace CT, Watkins SC, et al. (2020). Regulation of ALT-associated homology-directed repair by polyADP-ribosylation. Nat. Struct. Mol. Biol 27, 1152–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hombauer H, Campbell CS, Smith CE, Desai A, and Kolodner RD (2011a). Visualization of eukaryotic DNA mismatch repair reveals distinct recognition and repair intermediates. Cell 147, 1040–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hombauer H, Srivatsan A, Putnam CD, and Kolodner RD (2011b). Mismatch repair, but not heteroduplex rejection, is temporally coupled to DNA replication. Science 334, 1713–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iaccarino I, Palombo F, Drummond J, Totty NF, Hsuan JJ, Modrich P, and Jiricny J (1996). MSH6, a Saccharomyces cerevisiae protein that binds to mismatches as a heterodimer with MSH2. Curr. Biol 6, 484–486. [DOI] [PubMed] [Google Scholar]
- Iaccarino I, Marra G, Palombo F, and Jiricny J (1998). hMSH2 and hMSH6 play distinct roles in mismatch binding and contribute differently to the ATPase activity of hMutSα. EMBO J. 17, 2677–2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iacovoni JS, Caron P, Lassadi I, Nicolas E, Massip L, Trouche D, and Legube G (2010). High-resolution profiling of gammaH2AX around DNA double strand breaks in the mammalian genome. EMBO J 29, 1446–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiricny J (2006). The multifaceted mismatch-repair system. Nat. Rev. Mol. Cell Biol 7, 335–346. [DOI] [PubMed] [Google Scholar]
- Kadyrov FA, Dzantiev L, Constantin N, and Modrich P (2006). Endonucleolytic function of MutLα in human mismatch repair. Cell 126, 297–308. [DOI] [PubMed] [Google Scholar]
- Kawasoe Y, Tsurimoto T, Nakagawa T, Masukata H, and Takahashi TS (2016). MutSα maintains the mismatch repair capability by inhibiting PCNA unloading. eLife 5, e15155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim NW, Piatyszek MA, Prowse KR, Harley CB, West MD, Ho PL, Coviello GM, Wright WE, Weinrich SL, and Shay JW (1994). Specific association of human telomerase activity with immortal cells and cancer. Science 266, 2011–2015. [DOI] [PubMed] [Google Scholar]
- Kleczkowska HE, Marra G, Lettieri T, and Jiricny J (2001). hMSH3 and hMSH6 interact with PCNA and colocalize with it to replication foci. Genes Dev. 15, 724–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramara J, Osia B, and Malkova A (2018). Break-induced replication: The where, the why, and the how. Trends Genet. 34, 518–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkel TA, and Erie DA (2015). Eukaryotic mismatch repair in relation to DNA replication. Annu. Rev. Genet 49, 291–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M, Hills M, Conomos D, Stutz MD, Dagg RA, Lau LMS, Reddel RR, and Pickett HA (2014). Telomere extension by telomerase and ALT generates variant repeats by mechanistically distinct processes. Nucleic Acids Res. 42, 1733–1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M, Teber ET, Holmes O, Nones K, Patch A-M, Dagg RA, Lau LMS, Lee JH, Napier CE, Arthur JW, et al. (2018). Telomere sequence content can be used to determine ALT activity in tumours. Nucleic Acids Res. 46, 4903–4918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G-M (2008a). Mechanisms and functions of DNA mismatch repair. Cell Res. 18, 85–98. [DOI] [PubMed] [Google Scholar]
- Li H (2018b). Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Mao G, Tong D, Huang J, Gu L, Yang W, and Li G-M (2013). The histone mark H3K36me3 regulates human DNA mismatch repair through its interaction with MutSα. Cell 153, 590–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C, Yang L, Tanasa B, Hutt K, Ju B-G, Ohgi K, Zhang J, Rose DW, Fu X-D, Glass CK, and Rosenfeld MG (2009). Nuclear receptor-induced chromosomal proximity and DNA breaks underlie specific translocations in cancer. Cell 139, 1069–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Yan Z, Osia BA, Twarowski J, Sun L, Kramara J, Lee RS, Kumar S, Elango R, Li H, et al. (2021). Tracking break-induced replication shows that it stalls at roadblocks. Nature 590, 655–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loe TK, Li JSZ, Zhang Y, Azeroglu B, Boddy MN, and Denchi EL (2020). Telomere length heterogeneity in ALT cells is maintained by PML-dependent localization of the BTR complex to telomeres. Genes Dev. 34, 650–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu R, O’Rourke JJ, Sobinoff AP, Allen JAM, Nelson CB, Tomlinson CG, Lee M, Reddel RR, Deans AJ, and Pickett HA (2019). The FANCM-BLM-TOP3A-RMI complex suppresses alternative lengthening of telomeres (ALT). Nat. Commun 10, 2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundblad V (2002). Telomere maintenance without telomerase. Oncogene 21, 522–531. [DOI] [PubMed] [Google Scholar]
- Lynch HT, and de la Chapelle A (1999). Genetic susceptibility to non-polyposis colorectal cancer. J. Med. Genet 36, 801–818. [PMC free article] [PubMed] [Google Scholar]
- Maciejowski J, and de Lange T (2017). Telomeres in cancer: Tumour suppression and genome instability. Nat. Rev. Mol. Cell Biol 18, 175–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsischky GT, Filosi N, Kane MF, and Kolodner R (1996). Redundancy of Saccharomyces cerevisiae MSH3 and MSH6 in MSH2-dependent mismatch repair. Genes Dev. 10, 407–420. [DOI] [PubMed] [Google Scholar]
- Marzec P, Armenise C, Pérot G, Roumelioti F-M, Basyuk E, Gagos S, Chibon F, and Déjardin J (2015). Nuclear-receptor-mediated telomere insertion leads to genome instability in ALT cancers. Cell 160, 913–927. [DOI] [PubMed] [Google Scholar]
- Meier B, Volkova NV, Hong Y, Schofield P, Campbell PJ, Gerstung M, and Gartner A (2018). Mutational signatures of DNA mismatch repair deficiency in C. elegans and human cancers. Genome Res. 28, 666–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min J, Wright WE, and Shay JW (2017). Alternative lengthening of telomeres mediated by mitotic DNA synthesis engages break-induced replication processes. Mol. Cell. Biol 37, 405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min J, Wright WE, and Shay JW (2019). Clustered telomeres in phase-separated nuclear condensates engage mitotic DNA synthesis through BLM and RAD52. Genes Dev. 33, 814–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myung K, Datta A, Chen C, and Kolodner RD (2001). SGS1, the Saccharomyces cerevisiae homologue of BLM and WRN, suppresses genome instability and homeologous recombination. Nat. Genet 27, 113–116. [DOI] [PubMed] [Google Scholar]
- O’Sullivan RJ, Arnoult N, Lackner DH, Oganesian L, Haggblom C, Corpet A, Almouzni G, and Karlseder J (2014). Rapid induction of alternative lengthening of telomeres by depletion of the histone chaperone ASF1. Nat. Struct. Mol. Biol 21, 167–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opresko PL, Otterlei M, Graakjaer J, Bruheim P, Dawut L, Kølvraa S, May A, Seidman MM, and Bohr VA (2004). The Werner syndrome helicase and exonuclease cooperate to resolve telomeric D loops in a manner regulated by TRF1 and TRF2. Mol. Cell 14, 763–774. [DOI] [PubMed] [Google Scholar]
- Pan X, Drosopoulos WC, Sethi L, Madireddy A, Schildkraut CL, and Zhang D (2017). FANCM, BRCA1, and BLM cooperatively resolve the replication stress at the ALT telomeres. Proc. Natl. Acad. Sci. USA 114, E5940–E5949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pani E, Stojic L, El-Shemerly M, Jiricny J, and Ferrari S (2007). Mismatch repair status and the response of human cells to cisplatin. Cell Cycle 6, 1796–1802. [DOI] [PubMed] [Google Scholar]
- Panier S, Maric M, Hewitt G, Mason-Osann E, Gali H, Dai A, Labadorf A, Guervilly J-H, Ruis P, Segura-Bayona S, et al. (2019). SLX4IP antagonizes promiscuous BLM activity during ALT maintenance. Mol. Cell 76, 27–43.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons R, Li GM, Longley MJ, Fang WH, Papadopoulos N, Jen J, de la Chapelle A, Kinzler KW, Vogelstein B, and Modrich P (1993). Hypermutability and mismatch repair deficiency in RER+ tumor cells. Cell 75, 1227–1236. [DOI] [PubMed] [Google Scholar]
- Pedrazzi G, Bachrati CZ, Selak N, Studer I, Petkovic M, Hickson ID, Jiricny J, and Stagljar I (2003). The Bloom’s syndrome helicase interacts directly with the human DNA mismatch repair protein hMSH6. Biol. Chem 384, 1155–1164. [DOI] [PubMed] [Google Scholar]
- Peña-Diaz J, Bregenhorn S, Ghodgaonkar M, Follonier C, Artola-Borán M, Castor D, Lopes M, Sartori AA, and Jiricny J (2012). Noncanonical mismatch repair as a source of genomic instability in human cells. Mol. Cell 47, 669–680. [DOI] [PubMed] [Google Scholar]
- Peng M, Litman R, Xie J, Sharma S, Brosh RM Jr., and Cantor SB (2007). The FANCJ/MutLα interaction is required for correction of the cross-link response in FA-J cells. EMBO J. 26, 3238–3249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petti E, Buemi V, Zappone A, Schillaci O, Broccia PV, Dinami R, Matteoni S, Benetti R, and Schoeftner S (2019). SFPQ and NONO suppress RNA:DNA-hybrid-related telomere instability. Nat. Commun 10, 1001–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prolla TA, Baker SM, Harris AC, Tsao JL, Yao X, Bronner CE, Zheng B, Gordon M, Reneker J, Arnheim N, et al. (1998). Tumour susceptibility and spontaneous mutation in mice deficient in Mlh1, Pms1 and Pms2 DNA mismatch repair. Nat. Genet 18, 276–279. [DOI] [PubMed] [Google Scholar]
- Putnam CD, Hayes TK, and Kolodner RD (2009). Specific pathways prevent duplication-mediated genome rearrangements. Nature 460, 984–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F (2013). Genome engineering using the CRISPR-Cas9 system. Nat. Protoc 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizki A, and Lundblad V (2001). Defects in mismatch repair promote telomerase-independent proliferation. Nature 411, 713–716. [DOI] [PubMed] [Google Scholar]
- Roumelioti F-M, Sotiriou SK, Katsini V, Chiourea M, Halazonetis TD, and Gagos S (2016). Alternative lengthening of human telomeres is a conservative DNA replication process with features of break-induced replication. EMBO Rep. 17, 1731–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar J, Wan B, Yin J, Vallabhaneni H, Horvath K, Kulikowicz T, Bohr VA, Zhang Y, Lei M, and Liu Y (2015). SLX4 contributes to telomere preservation and regulated processing of telomeric joint molecule intermediates. Nucleic Acids Res. 43, 5912–5923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saydam N, Kanagaraj R, Dietschy T, Garcia PL, Peña-Diaz J, Shevelev I, Stagljar I, and Janscak P (2007). Physical and functional interactions between Werner syndrome helicase and mismatch-repair initiation factors. Nucleic Acids Res. 35, 5706–5716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartzentruber J, Korshunov A, Liu X-Y, Jones DTW, Pfaff E, Jacob K, Sturm D, Fontebasso AM, Quang D-AK, Tönjes M, et al. (2012). Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 482, 226–231. [DOI] [PubMed] [Google Scholar]
- Sieverling L, Hong C, Koser SD, Ginsbach P, Kleinheinz K, Hutter B, Braun DM, Cortés-Ciriano I, Xi R, Kabbe R, et al. PCAWG-Structural Variation Working Group; PCAWG Consortium (2020). Genomic footprints of activated telomere maintenance mechanisms in cancer. Nat. Commun 11, 733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva B, Pentz R, Figueira AM, Arora R, Lee YW, Hodson C, Wischnewski H, Deans AJ, and Azzalin CM (2019). FANCM limits ALT activity by restricting telomeric replication stress induced by deregulated BLM and R-loops. Nat. Commun 10, 2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva B, Arora R, Bione S, and Azzalin CM (2021). TERRA transcription destabilizes telomere integrity to initiate break-induced replication in human ALT cells. Nat. Commun 12, 3760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobinoff AP, and Pickett HA (2017). Alternative lengthening of telomeres: DNA repair pathways converge. Trends Genet. 33, 921–932. [DOI] [PubMed] [Google Scholar]
- Sobinoff AP, Allen JA, Neumann AA, Yang SF, Walsh ME, Henson JD, Reddel RR, and Pickett HA (2017). BLM and SLX4 play opposing roles in recombination-dependent replication at human telomeres. EMBO J. 36, 2907–2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strand M, Prolla TA, Liskay RM, and Petes TD (1993). Destabilization of tracts of simple repetitive DNA in yeast by mutations affecting DNA mismatch repair. Nature 365, 274–276. [DOI] [PubMed] [Google Scholar]
- Sugawara N, Goldfarb T, Studamire B, Alani E, and Haber JE (2004). Heteroduplex rejection during single-strand annealing requires Sgs1 helicase and mismatch repair proteins Msh2 and Msh6 but not Pms1. Proc. Natl. Acad. Sci. USA 101, 9315–9320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM, Bindal N, Boutselakis H, Cole CG, Creatore C, Dawson E, et al. (2019). COSMIC: The catalogue of somatic mutations in cancer. Nucleic Acids Res. 47 (D1), D941–D947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umar A, Buermeyer AB, Simon JA, Thomas DC, Clark AB, Liskay RM, and Kunkel TA (1996). Requirement for PCNA in DNA mismatch repair at a step preceding DNA resynthesis. Cell 87, 65–73. [DOI] [PubMed] [Google Scholar]
- Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J, et al. (2013). From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinformatics 43, 11.10.1–11.10.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varley H, Pickett HA, Foxon JL, Reddel RR, and Royle NJ (2002). Molecular characterization of inter-telomere and intra-telomere mutations in human ALT cells. Nat. Genet 30, 301–305. [DOI] [PubMed] [Google Scholar]
- Verma P, Dilley RL, Zhang T, Gyparaki MT, Li Y, and Greenberg RA (2019). RAD52 and SLX4 act nonepistatically to ensure telomere stability during alternative telomere lengthening. Genes Dev. 33, 221–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren JJ, Pohlhaus TJ, Changela A, Iyer RR, Modrich PL, and Beese LS (2007). Structure of the human MutSα DNA lesion recognition complex. Mol. Cell 26, 579–592. [DOI] [PubMed] [Google Scholar]
- Wilson JSJ, Tejera AM, Castor D, Toth R, Blasco MA, and Rouse J (2013). Localization-dependent and -independent roles of SLX4 in regulating telomeres. Cell Rep. 4, 853–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Qin J, Wang L, Lee H-J, Kao C-Y, Liu D, Songyang Z, Chen J, Tsai M-J, and Tsai SY (2019). Nuclear receptors regulate alternative lengthening of telomeres through a novel noncanonical FANCD2 pathway. Sci. Adv 5, eaax6366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Q, Zhang R, Wang XW, Linke SP, Sengupta S, Hickson ID, Pedrazzi G, Perrera C, Stagljar I, Littman SJ, et al. (2004). The mismatch DNA repair heterodimer, hMSH2/6, regulates BLM helicase. Oncogene 23, 3749–3756. [DOI] [PubMed] [Google Scholar]
- Yang Z, Takai KK, Lovejoy CA, and de Lange T (2020). Break-induced replication promotes fragile telomere formation. Genes Dev. 34, 1392–1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeager TR, Neumann AA, Englezou A, Huschtscha LI, Noble JR, and Reddel RR (1999). Telomerase-negative immortalized human cells contain a novel type of promyelocytic leukemia (PML) body. Cancer Res. 59, 4175–4179. [PubMed] [Google Scholar]
- Young SJ, Sebald M, Shah Punatar R, Larin M, Masino L, Rodrigo-Brenni MC, Liang C-C, and West SC (2020). MutSβ stimulates holliday junction resolution by the SMX complex. Cell Rep. 33, 108289. [DOI] [PubMed] [Google Scholar]
- Zhang J-M, Yadav T, Ouyang J, Lan L, and Zou L (2019). Alternative lengthening of telomeres through two distinct break-induced replication pathways. Cell Rep. 26, 955–968.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J-M, Genois M-M, Ouyang J, Lan L, and Zou L (2021). Alternative lengthening of telomeres is a self-perpetuating process in ALT-associated PML bodies. Mol. Cell 81, 1027–1042.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Whole Genome Sequencing (WGS) Sequence Read Archive data have been deposited at NCBI and are publicly available as of the date of publication. The URL and Accession number are listed in the key resources table.
Original code has been deposited and is publicly available as of the date of publication. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| PCNA | Cell Signaling Technology | Cat# 2586; RRID:AB_2160343 |
| MSH2 | Cell Signaling Technology | Cat# 2850; RRID:AB_2144797 |
| POLD3 | Abnova | Cat# H00010714-M01; RRID:AB_538310 |
| PML | Santa Cruz | Cat# sc-5621; RRID:AB_2166848 |
| FLAG | Cell Signaling Technology | Cat# 14793; RRID:AB_2572291 |
| TR4 | Perseus | Cat# PP-H0107B-00; RRID:AB_2155352 |
| COUP-TFII | Perseus | Cat# PP-H7147-00; RRID:AB_2155627 |
| TRF2 | Novus | Cat# NB110-57130; RRID:AB_844199 |
| BLM | Jan Karlseder | N/A |
| MSH6 | Cell Signaling Technology | Cat# 5424; RRID:AB_10695802 |
| MSH3 | Novus | Cat# NBP1-95114;RRID:AB_11028618 |
| γ-TUB | Sigma-Aldrich | Cat# T6557; RRID:AB_477584 |
| FLAG | Sigma-Aldrich | Cat# F1804; RRID:AB_262044 |
| WRN | Novus-Biological | Cat# NB100-472; RRID:AB_2216076 |
| TRF1 | Jan Karlseder | N/A |
| Chemicals, peptides and recombinant proteins | ||
| Non-targeting control | Dharmacon | D-001810-10-0020 |
| MSH6 | Dharmacon | L-019287 |
| MSH2 | Dharmacon | L-003909 |
| MSH3 | Dharmacon | L-019665 |
| POLD3 | Dharmacon | L-026692 |
| BLM | Dharmacon | L-007287 |
| Benzonase | Merck Millipore | Cat#70746 |
| 4-hydroxytamoxifen | Sigma | Cat#H7904 |
| Doxycycline | Clontech | Cat#631311 |
| Shield Ligand | Clontech | Cat#632189 |
| AluI | New England Biolabs | Cat#R0137 |
| MboI | New England Biolabs | Cat#R0137 |
| f29 Polymerase | New England Biolabs | Cat#M0269 |
| Critical commercial assays | ||
| Click-iT EdU Cell Proliferation Kit for Imaging, Alexa Fluor 555 dye | ThermoFisher | Cat#C10338 |
| Experimental models: Cell lines | ||
| U2OS | this paper | N/A |
| U2OS MSH6 KO C1-C2 | this paper | N/A |
| U2OS Control C1-C2 | this paper | N/A |
| U2OS BLM KO C1-C2 | this paper | N/A |
| U2OS BLM-MSH6 KO C1-C2 | this paper | N/A |
| U2OS WRNKO C1-C2 | this paper | N/A |
| U2OS WRN-MSH6 KO C1-C2 | this paper | N/A |
| U2OS RMI KO | Eros Lazzerini Denchi (Loe et al., 2020) | N/A |
| HeLa LT | this paper | N/A |
| HeLa LT MSH6 KO C1 | this paper | N/A |
| VA13 | Roger Greenberg | N/A |
| VA13 MSH6 KO C1-C3 | Roger Greenberg | N/A |
| VA13 | Roger Greenberg | N/A |
| U2OS TRF1-FokI WT/D450A | Roger Greenberg (Dilley et al., 2016) | N/A |
| Saos2 | this paper | N/A |
| U2OS DiVa | Gaëlle LeGube (Iacovoni et al., 2010) | N/A |
| Hek293T | ATCC | RRID: CVCL_0063 |
| Recombinant DNA | ||
| pSpCas9(BB)-2A-GFP | (Ran et al., 2013) | RRID: Addgene_48138 |
| psPAX2 | Didier Trono | RRID: Addgene_12260 |
| pMD2.G | Didier Trono | RRID: Addgene_12259 |
| FLAG-TRF1-FokI WT/D450A | Roger Greenberg (Dilley et al., 2016) | N/A |
| pTag-RFP-WT-MSH6 | this paper | N/A |
| pTag-RFP-PAAP-MSH6 | this paper | N/A |
| pTag-RFP-K1140-MSH6 | this paper | N/A |
| pTag-RFP-WT-MSH6 | this paper | N/A |
| pTag-RFP | this paper | N/A |
| Oligonucleotides | ||
| MSH6 sgRNA; CACGCGAAGGCGGCCGTGCC | this paper | N/A |
| BLM sgRNA; GGAACGAACTGCTTCAGCAG | this paper | N/A |
| WRN sgRNA 1; GTAAATTGGAAAACCCACGG | (Chan et al., 2019) | N/A |
| WRN sgRNA 2; ATCCTGTGGAACATACCATG | (Chan et al., 2019) | N/A |
| WRN sgRNA 3; GTAGCAGTAAGTGCAACGAT | (Chan et al., 2019) | N/A |
| Control gRNA 1; GTGAACCGCATCGAGCTGAA | this paper | N/A |
| Control gRNA 2; GGAGCGCACCATCTTCTTCA | this paper | N/A |
| Tel C PNA Probe; Alexa 488-(CCCTAA)4 | PNA Bio | Cat#F1004 |
| Tel G PNA Probe; Alexa-594-(TTAGGG)4 | PNA Bio | Cat#F1006 |
| Variant TVR PNA Probe; Cy5-OO-(TGACCC)3 | Panagene | N/A |
| Software and algorithms | ||
| GraphPad Prism | GraphPad | RRID: SCR_002798 |
| Accuri C6 Plus | Becton Dickenson | RRID: SCR_014422 |
| MiniMap2 | https://github.com/lh3/minimap2 | RRID: SCR_018550 |
| Mutect2 | (Van der Auwera et al., 2013) | https://gatk.broadinstitute.org/hc/en-us/articles/360037593851-Mutect2 |
| Genomic sEquence AnalyzeR (GEAR) | https://www.gear-genomics.com/ | N/A |
| Telomere Sequence Mapping | this paper | https://github.com/ChildrensMedicalResearchInstitute/MutSa_Deficiency_ALT |
| Deposited data | ||
| Raw and analyzed data | this paper | https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA776978 SRA: PRJNA776978 |