

Abstract
Landmark successes in oncoimmunology have led to development of therapeutics boosting the host immune system to eradicate local and distant tumors with impactful tumor reduction in a subset of patients. However, current immunotherapy modalities often demonstrate limited success when involving immunologically cold tumors and solid tumors. Here, we describe the role of various biomaterials to formulate cancer vaccines as a form of cancer immunotherapy, seeking to utilize the host immune system to activate and expand tumor-specific T cells. Biomaterial-based cancer vaccines enhance the cancer-immunity cycle by harnessing cellular recruitment and activation against tumor-specific antigens. In this review, we discuss biomaterial-based vaccine strategies to induce lymphocytic responses necessary to mediate anti-tumor immunity. We focus on strategies that selectively attract dendritic cells via immunostimulatory gradients, activate them against presented tumor-specific antigens, and induce effective cross-presentation to T cells in secondary lymphoid organs, thereby generating immunity. We posit that personalized cancer vaccines are promising targets to generate long-term systemic immunity against patient- and tumor-specific antigens to ensure long-term cancer remission.
Keywords: cancer vaccine, local controlled release, biomaterials, oncoimmunotherapy, in situ delivery
2. Introduction
Clinical successes with oncoimmunotherapeutics have demonstrated the capability of the human immune system to eradicate cancer1–5. Comprehensive treatment modalities modulate host immune signaling pathways via recognition of tumor-specific components to overcome an immunosuppressive tumor microenvironment6–11. Immune checkpoint blockade antibodies (ICB) have revolutionized cancer treatment with remarkable tumor burden reduction in patients with hard to treat cancers 12,13. However, ICB success is limited in solid tumors and only 13% of patients respond across cancer types14–16. Further, majority of responsive patients eventually experience recurrence due to immunotherapy resistance13,16. Consequently, there is a crucial unmet need to increase immunotherapy response rates across all cancer types. To this end, therapeutic cancer vaccines have reemerged to mobilize patient- and tumor-specific antitumor immune response17. By increasing immunogenicity and maintaining specificity, therapeutic cancer vaccines can evade tumor suppressive mechanisms for primary- and metastatic- tumor burden reduction11,18,19.
Cancer vaccine development build on lessons from preventative vaccines for infectious diseases. Current Food and Drug Administration (FDA) approved prophylactic cancer vaccines with viral etiologies include HBV-associated hepatocellular carcinoma (Engerix-B, Recombivas HB, Heplisav-B) and HPV-related mucosal cancers (Gardasil, Cervarix). However, cancers with non-viral etiology are more challenging to develop. Unlike bacteria and viruses, cancerous cells resemble normal cells leading to difficulties in antigenic targeting. Therapeutic cancer vaccines, aiming to elicit in situ targeted responses, must overcome three key challenges. First, an ideal cancer vaccine must identify tumorigenic cells via conserved cell-surface markers without inducing autoimmunity. With the high diversity of tumor cell type and surface markers, there is difficulty in isolating and targeting key immunodominant antigens, potentially leading to tumor escape if unchecked. Such immunodominant antigens are still subject to central and peripheral immune tolerance. Next, to allow generalized antitumor immunity, it must surpass immune equilibrium via targeting key immune activation pathways. Lastly, care must be taken to limit systemic toxicity and off-target effects through vaccine material and design. Currently ex vivo vaccines have a high incidence of generalized constitutive symptoms (ex. fever, muscle aches, nausea, and fatigue) that remit over time20. Thus, improving vaccination safety is paramount for clinical translation.
Failed cancer vaccine trials can be attributed to lack of effective delivery methods21. For example, vaccination using unmodified peptides generated an overall response rate of 3%22, due to difficulty in activating DCs. As such, approaches to target dendritic cells (DC) could potentially improve clinical response. Accordingly, advances in biomaterial-based delivery systems for targeted payload release could enable spatiotemporal presentation to cells and microenvironment, thus enhancing efficacy and reducing potential adverse effects. Given the diversity in vaccination approaches, different biomaterials can be used to overcome delivery challenges specific to each type of vaccine. They range from the nanoscale (e.g. liposomes, nanoparticles) to larger implantable devices or patches as well as injectable scaffolds. Further, biomaterials can be leveraged to deliver immunopharmaceutics via different routes of administration, namely intranasal23, oral24, intramuscular25,26, intravenous (NCT02410733), subcutaneous27–30 and within or adjacent to tumor cavity31,32.
To develop a clinically viable cancer vaccine platform, it is crucial to combine innovations in biomaterials with expanded understanding in cancer immunology (Figure 1). In this review, we will discuss emerging biomaterial-based strategies with a focus on DC-based therapeutic cancer vaccines. We will first elucidate challenges with current therapeutic cancer vaccine strategies, followed by how biomaterial-based platforms can address current obstacles. Next, we will discuss key polymeric and scaffold vaccine approaches. Lastly, we will present perspectives for biomaterial-based translational studies.
Figure 1: Targeting DC mediated cellular immunity utilizing biomaterial-based vaccine strategies.
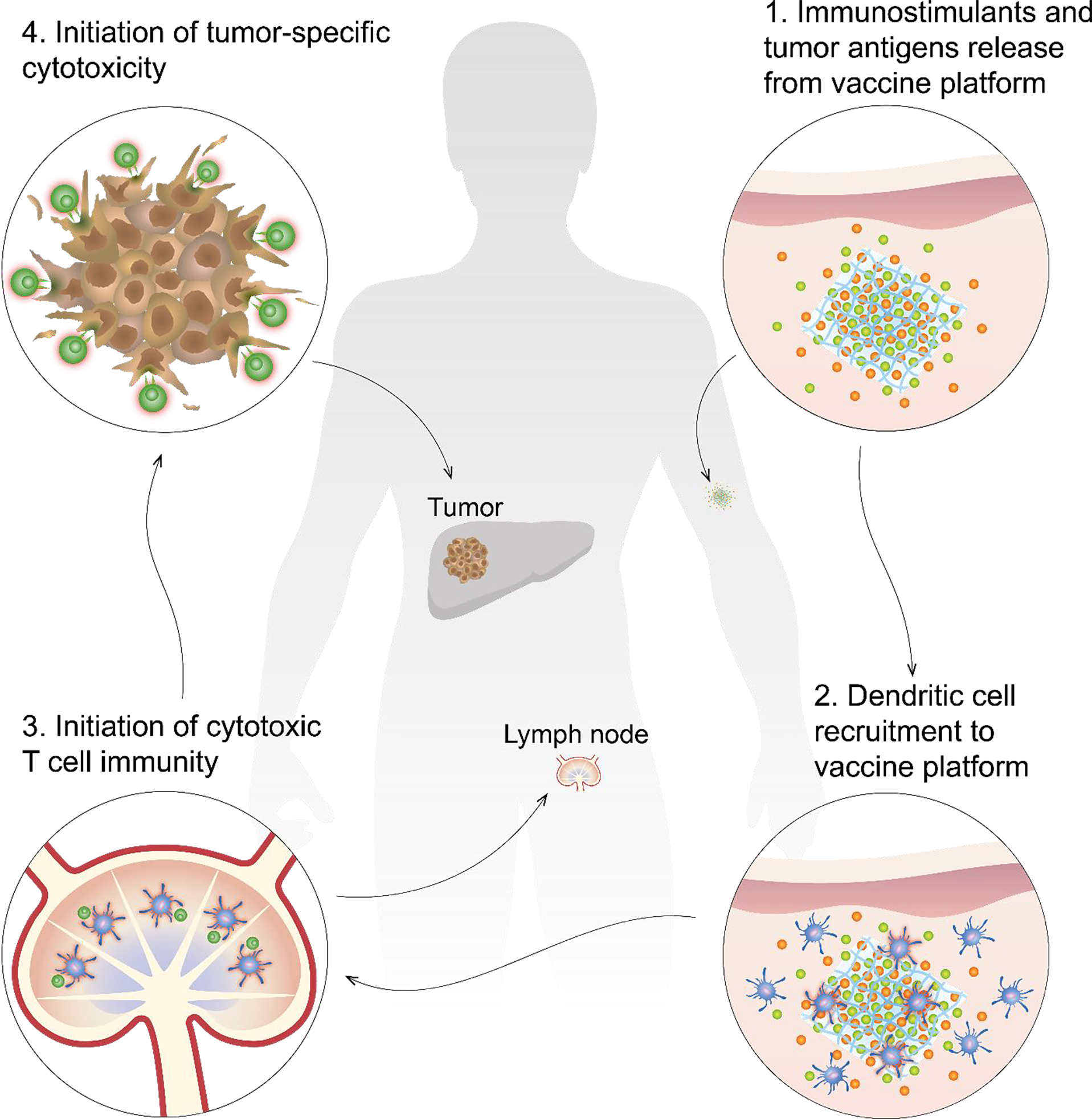
Initiation of cellular immunity via therapeutic cancer vaccines is dependent on a four-step process. 1) Implantation of biomaterial in intended site releases encapsulated immunostimulants (green) and tumor antigens (orange) according to material-specific manner. 2) Immunostimulant release generates a local concentration gradient necessary to recruit dendritic cells (DC, blue) to vaccine platform where they are presented dominant tumor antigens and become activated (red glow). 3) Activated DC (red glow) will traffic to local draining lymph nodes for antigen presentation to CD8+ T cell (green) to initiate cytotoxic immunity. 4) Cytotoxic T cells travel systemically to target cells with featured antigenic target, reducing primary and metastatic burden systemically.
Challenges of current therapeutic vaccine strategies
Numerous therapeutic vaccine strategies and administration routes have been developed with various degrees of clinical and preclinical success (Table 1). Peptide- or protein-based vaccines have high specificity but may limit antigenic responses for widespread cancer eradication33,34. DNA vaccines using unmethylated, repeating cytosine-guanine (CpG) motifs have both an adjuvant and antigen effect by generating a potent and directed antibody response. However, due to their large size and negative charge, they have low cellular uptake and high off-target delivery35,36, requiring improbably high administration doses. mRNA vaccines can encode a variety of antigens with self-adjuvanting effects and pose no infection or mutagenesis risk37,38. Recent success utilizing mRNA vaccines demonstrate high safety, tolerability and degree of protection against SARS-COV-239,40. However, their instability and inefficient delivery, necessitating encapsulation systems linked to severe allergic reactions41,42 may hinder these promising immunogenic effects. Additionally, their paradoxical effects on innate immunity may hinder its effect as an oncotherapeutic43.
Table 1:
Description of various vaccine strategies currently utilized in preclinical studies and clinical trials.
| Vaccine Strategy | Mechanism of Action | Benefit | Limitation |
|---|---|---|---|
| DNA35,36,218,219 | Transfection of plasmids containing immunogenic genes to induce DC antigen uptake stimulating CTL response | • Scalable • High specificity • Long half-life • Immunize against multiple antigens in single plasmid |
• Low efficiency and cellular uptake with high off target delivery • Poor immunogenicity requires high administration doses • Very large size and negative charge • Mutagenesis, autoimmunity and toxicity risk • Difficult to monitor response • Requires high cell manipulation for immunogenicity |
| mRNA38,43 | Delivery of synthetic mRNA that generates specific immunogenic antigen via a carrier molecule | • Scalable • Non-infectious • Non-integrating • Naturally degrading • Stimulate adaptive and innate responses |
• Unstable formulation and inefficient delivery • Requires encapsulation or nanocarrier systems • Delivery systems linked to rare but severe allergic events • Paradoxical suppressive innate immunity |
| Peptide220,221 | Use short antigenic peptide fragments to induce targeted response. Often delivered via liposomal formulations | • Easily produced and delivered • Low toxicity • Easy to monitor immune response • Reduced risk of allergenic or reactogenic responses • Induce immunity against multiple strains by utilization of conserved peptide |
• Require use of carrier molecules for chemical stability • Elicit mainly one type of immune response (either cytotoxic or humoral) • May require booster shots • HLA-restricted • Restricted to certain epitopes • Multivalent formulations are challenging • Require new production and stability assays/protocols for each new antigen |
| DC36,55,58–60,75,222 | Rely on cellular uptake of antigen either via reintroduced ex vivo pulsing or in situ phagocytosis to initiate downstream activation of adaptive immunity Currently most strategies focus on ex vivo activation and re-administration. |
• Scalable • Elicit both humoral and cytotoxic response • Capable of generating memory responses • Not HLA restricted • Variation in antigen type (allogeneic vs autologous, whole-cell vs neoantigen) |
• Ex vivo strategies require expansion, maturation and activation of DCs, necessitating GMP level facilities • Technically challenging • Regulatory challenges due to variability in antigen selection and conferred activity |
Cellular vaccines often target DCs, the most powerful antigen presenting cell44, to initiate cytotoxic immunity. DC-vaccines posit a powerful regulation of key cytotoxic pathways necessary to initiate antitumor immunity45–51. Under investigation for numerous cancer types52, overall clinical efficacy has yet to be achieved52. Further, they may induce mild to moderate toxicity52,53. DC-vaccines are classified by source: autologous (from patient tumors) or allogeneic (lab-generated)54. Autologous vaccines further divert into neoantigen vaccines, utilizing immunogenic specific antigens, or whole-cell vaccines, delivering specific and non-specific antigens. Advances in next-generation sequencing and bioinformatics have transformed our ability to identify and isolate patient-specific neoantigens over weeks to months55–59. While they yield favorable clinical immune responses58,60, their complexity pose significant monetary and labor deterrents57,58. Conversely, whole-cell vaccines simultaneously target multiple immunodominant tumor antigens61–64. This expands their potential to generate favorable robust responses while limiting alloimmune reactivity61,64–67 and tumor escape61. Allogeneic vaccines employ antigens identified from established cancer lines, eliminating challenging manufacturing and commercialization steps. However, lack of personalized antigens manifests poorer clinical efficacy compared to autologous counterparts54. Both sources necessitate numerous interventions requiring a high degree of patient adherence, often difficult to achieve68, even with technology-based outreach efforts69–71.
Most autologous DC-vaccines utilize ex vivo antigen-pulsed activated-DCs53,72. However, due to inefficient homing, less than 5% of administered DCs reach draining lymph nodes73. Thus, ex vivo DC-vaccines require repeated interventions on a prime-boost schedule, in potential combination with other chemotherapeutic or immunostimulatory drugs55,73,74. They are laboriously manufactured under strict regulation within Good Manufacturing Practices (GMP) facilities to ensure patient specificity and consistent production methods68,72. Batch-to-batch variability with complex GMP protocols lead to substantial monetary, time and labor cost with limited clinical benefit52. Therefore, although ex vivo DC-vaccines demonstrate applicability to multiple cancer subtypes, the high development cost and manufacturing challenges may impede widespread clinical use75.
Thus far, Sipuleucel-T, an autologous ex vivo DC-based vaccine, is the only therapeutic cancer vaccine to reach the clinic. It received approval from the US Food and Drug Administration (FDA) in 2010 and European Medical Agencies in 2013 as a last-line therapeutic strategy for metastatic hormone-refractory prostate cancer4. With a tolerable safety profile and no demonstrated dose-limiting toxicity76, immunization induced favorable immune responses correlating with a decline in prostate-specific antigen76. However, in a landmark Phase III trial, progressive disease occurred in 90% of patients77. Although heralded as a paradigm shift, Sipuleucel-T was plagued by notable drawbacks, leading ultimately to its termination of use. First, a single course of treatment cost USD $93,000 for a limited average survival gain of 4 months77, limiting its translatability and use worldwide. Second, due to low manufacturing capacity, only 10% of eligible patients eligible were treated68. Treatment scarcity and accessibility in conjunction with reimbursement problems led to physician endorsement reluctance. Due to complex manufacturing, difficulty in administration and low sales, Dendreon filed for bankruptcy in 2014 and regulatory approval was withdrawn in Europe for commercial reasons68.
In addition to inefficient homing, ex vivo vaccines demonstrate promising efficacy in preclinical studies but cannot be recapitulated clinically52,78. Reasons for clinical failure stem from variable generation, maturation and administration of DCs per dose78. For example, AGS-003, a combination of ex vivo DCs co-electroporated with patient’s amplified tumor RNA and synthetic CD40L RNA and sunitinib, induced moderate immunological activity leading to either partial response (PR) or stable disease (SD)in 62% of patients (NCT00272649)79. Further, expansion of effector memory cytotoxic T-lymphocytes (CTLs) after five doses, correlated to 30 months prolonged progression free survival79. However, subsequent Phase II and III trials were terminated early due to lack of efficacy (NCT01582672)79,80. Likewise, the bioengineered GVAX and FVAX vaccines, composed of irradiated whole tumor cells genetically modified to overexpress cytokines regulating DC homeostasis (granulocyte-macrophage colony stimulating factor [GM-CSF], GVAX)81,82 and development (fms-related tyrosine kinase 3 ligand [Flt3-L], FVAX)83–87, have demonstrated success in Phase I and II clinical trials83,88,89. GVAX led to an overall increase in immunogenicity and mean survival of patients with metastatic prostate cancer, weakly correlating to dose90. Its success led to clinical investigations for other cancers91 including melanoma92, colorectal cancer93,94, acute myeloid leukemia95 and myelodysplastic syndromes96,97. Correspondingly, FVAX vaccination demonstrated a degree of cytotoxic immunity correlated with durability of clinical responses by RECIST criteria83. Additionally, the combination of FVAX with ICB dramatically improved tumor burden and generation of immune responses preclinically98,99, although clinical efficacy remains to be determined.
3. Biomaterials-based cancer vaccine strategies
Need for biomaterial-based DC vaccine strategies
While most DC-vaccines demonstratively induce immunogenicity, many have failed to induce durable and meaningful therapeutic responses in clinical trials52,100 in terms of degree of tumor burden and remission rate101. To address these challenges, innovations in biomaterials, biotechnology and polymer science offer alternative approaches for therapeutic cancer vaccines102. Harnessed since antiquity103, biomaterials are highly versatile synthetic or natural materials used in various medical applications. In cancer, use of biomaterials have advanced drug targeting and delivery104–107 with high clinical efficacy and improvement in patient care108–110. Significant advances in bioengineering and understanding of biological processes have allowed for development of macro- and micro-environments necessary for cellular manipulation111. To reach clinical demand, it is critical to meet four key requirements. First, platforms must be targetable for patient- and tumor-specific antigens. This ensures non-self antigen recognition, ensuring generation of cytotoxic effector T cells opposed to their regulatory counterparts. Second, strategies must be adaptable and versatile against antigenic shift from constant de novo oncogenic mutations. Third, vaccination must generate in situ immunity able to overcome the inherently immunosuppressive tumor microenvironment with efficient immune infiltration. Lastly, immune penetration into target tissue sites must self-propagate durable responses for overall survival and recurrence reduction.
Biomaterial vaccines offer numerous advantages over conventional delivery methods. Conventional systemic delivery of immune adjuvants may have off-target effects causing toxicity. Targeted co-delivery of antigens and adjuvants can limit deleterious effects. By tuning the physical properties of therapeutic cargos, targeting moieties and vehicles (ex. size, morphology, charge, physiochemistry, or porosity), biomaterial vaccines can achieve site-specific delivery with desirable release kinetics. Sustained delivery of antigens and adjuvants via a vehicle or platform could avoid repeated administrations while providing a personalized in situ vaccine platform. These vaccines can generate an immunostimulatory microenvironment to continuously recruit and activate endogenous DCs in situ without further external manipulation or modulation. In this review, we highlight key strategies ranging from the nanoscale, including nano- and microparticles112–121, liposomes122,123 and combinatorial approaches with current front-line therapeutics124, to larger implantable and injectable scaffolding systems29,30,112,125–132 (Figure 2, Table 2).
Figure 2: Biomaterial-based vaccine strategies.
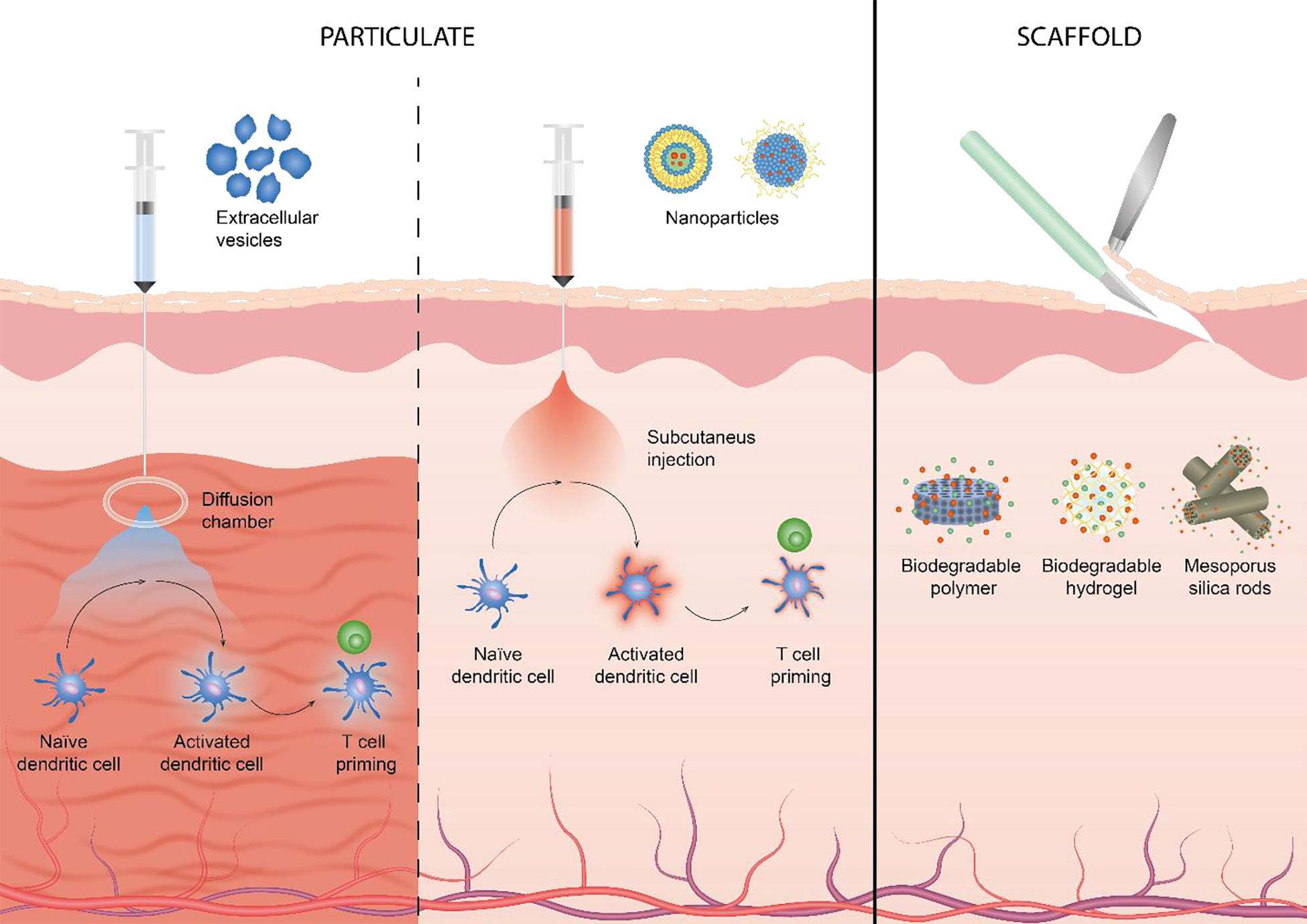
Therapeutic vaccine modalities can be divided into two main categories dependent on primary material modality. Particulate-based vaccines (left) include nanoparticle, including liposomes, and extracellular vesicles such as exosomes. Polymer-based vaccines (right) can either be biodegradable or non-degradable and can be composed of polymers, hydrogels or silica rods.
Table 2:
Brief descriptions of discussed biomaterial-based vaccine scaffold platforms.
| Name | Mechanism of Action | Benefits | Limitations | |
|---|---|---|---|---|
| Nanoparticle | BLP25 (L-BLP25; tecemotide; Stimuvax®)17,20,122,124,133,134,137 NCT02049151 |
• DCs uptake the MUC1 peptide for specificity • Can also be optimized for other highly mutated antigens (ex. HER2Neu) |
• Phase III (NSCLC) with neoadjuvant CR + BLP25 had no impact on OS • Adjuvant CR improved OS • Phase III (Her2+ BC) No impact on OS |
• While promising preclinically, no effect on OS or tumor burden in several Phase III studies led to study cancellation • Most studies done in WEIRD populations |
| Nanodisc118–121 | • Synthetic HDL containing TLR agonists (MPLA, CpG-ODN) with TAAs. • Can also co-deliver chemotherapeutics and ICB |
• When co-delivered with doxorubicin, 88% mice immunized against colorectal carcinoma with 100% rejecting tumor rechallenge. • When co-delivered with ICB, 90% mice eradicated established colorectal carcinoma and melanoma. • Strong humoral response in murine cervical cancer and melanoma model |
• Required neoantigen identification, limiting use due to labor, cost and manufacturing capacity • Small tumor burden studied not representative of clinical burden • Requires multidose therapy with inability to be refilled or rescaled |
|
| Lipid-encapsulated mRNA nanovaccine137 | • Multi-dose every 3 weeks IM injections encoding up to 20 patient-specific neoantigens • Can be co-treated with pembrolizumab |
• Phase I: 85% monotherapy had SD to CR, 66% combination • 2 patients demonstrated CR or PR after previously failing PD-L1 therapy • CTL responses post vaccination against immunized neoantigens with high predicted binding affinity |
• Multidose therapy poses patient adherence problems • Neoantigen identification may be laborious and costly |
|
| NP-CD40141–143 | • PLGA nanoparticles coated with agonistic anti-CD40 with TAAs and adjuvants encapsulated • Tested CpG and polyI:C |
• Murine model reduced tumor burden and prolonged OS • Enhanced DC maturation, CD4+ T proliferation and stimulated IFN-y production in vitro • Selective and efficient immunity in prophylactic and therapeutic challenges |
• Studied tumors were incipient and thus did not recapitulate clinical burden | |
| Tumor cell membrane with particulate core117,145,148 | • Tumor membranes used to coat nanoparticles with immunostimulatory agents • Deliver TAAs in situ • Can be used with ICB |
• 50% melanoma bearing mice had long term survival with ICB combo • Stimulation induced release of IFN-γ, IL-10, IL-12 and IL-4 in human breast cancer patients |
• Required repeated doses • Tumor cell membrane only allows for targeting but can still evade TIME |
|
| Microparticle | Mesoporous silicone vectors12,168 | • Utilizing mesoporous silicone to co-deliver antigenic peptides and dual TLR-agonists | • In prophylactic murine melanoma, suppressed tumor growth with repeated vaccination • Induced antigen-specific T cell expansion and reactivity |
• Required 2 repeated doses • Only tested prophylactically. |
| Calcium carbonate112 | • Encapsulating TLR adjuvants and tumor lysates within calcium carbonate particles | • High loading capacity vs PLGA counterparts • Co-delivery with TLR agonists |
• Only tested in vitro and in silico, limited preclinical data | |
| PLGA113–116 | • Encapsulating autologous tumor lysates with TLR agonists (poly(I:C), MALP-2, CpG motifs)) | • Reduced lung metastasis in 42% mice in 4T1 murine TNBC model | • Repeated doses required | |
| Extracellular Vesicles | Dex156,157 | • Derived from DCs to express co-stimulatory surface markers with TLR adjuvants or mRNA for neoantigens | • Phase 1: safe and well tolerated in advanced NSCLC, melanoma and colorectal cancer. One patient had grade-3 hepatotoxicity • Mixed clinical responses with majority of patients presenting with SD |
• Limited to no antigen-specific T cell reactivity. • Limited evidence of NK cell lytic function weakly correlating with longer PFS |
| Tumor cell derived exosomes (IGV-001)25,26 NCT02507583, NCT01550523 NCT04485949 |
• Express CpG DNA • Autologous tumor cells treated with antisense ODN against IGF-1 |
• Dual surgeries led to increased pain and immobility leading to need for long term enoxaparin for DVT prophylaxis for 3 months with repeated ultrasound evaluations • 6 patients had hematomas, wound complications and DVT • Phase 1 (malignant astrocytomas): 75% had clinical improvement. 37.5% had spontaneous regression at local/metastasis sites • Phase 1 (newly diagnosed glioblastoma with CR): PFS improved by 3.3 months. Improved PFS if de novo mutations in DNA evident in patient |
• Surgically implanted diffusion chambers required multiple surgical events and complicated treatment timeline | |
| Non-hydrogel scaffold | WDVAX131,132 NCT01753089 |
• Encapsulation of tumor lysates, GMCSF and CpG-ODN in PLGA | • Single dose SQ extended survival by 90%. CR of primary and metastatic melanoma in 47% of mice. • Maintained local and systemic T cell population. • Intracranial implantation extended OS in 90% rats with glioma • |
• Four SQ implantations monthly required repeated visits with DVT prophylaxis for pain and immobility. • Complex intracranial implantation PLGA matrix can potentially damage or denature antigen structure • Use of organic solvents may lead to subclinical toxicity • |
| Hydrogel scaffold | Methylacrylated-alginate cryogel29,30,132,192 | • Loaded with tumor lysates, GMCSF and CpG ODN. • Has shape memory properties for needle extrusion in SQ injection • Degradable • |
• Long lasting prophylactic immunity in 80% murine melanoma • In situ immune cell recruitment and trafficking |
• Cannot be refilled necessitating multiple vaccinations • Limited to identified immunogenic tumor models • Rapid burst release of all components within first week |
| Thermosensitive PLGA127,129 | • Chemically modified PLGA with GMCSF and neoantigens | • Longer median survival • Reduction in tumor burden |
• Repeated dosing required | |
| Self-assembling tumor peptides31,193 | • Hydrogel with JQ1, ICG and autologous fixed tumor cells to excite and stimulate JQ1 release in situ by NIR irradiation | • Limited tumor recurrence and distant metastasis for 30 days post vaccination • Reduced MDSC population Accelerated DC maturation with increase in CD8T frequency and cytokine release 2.7× |
• Repeated dosing required • Reformulation of tumor cells • Requires NIR irradiation tissue penetration which may be difficult for deep seeded tumors |
|
| Scaffold-particulate | Hydrogel-embedded MSN182 | • Change functional group charge to modulate cell uptake | • Negatively charged MSN have higher cellular uptake | • Lacking in vivo efficacy |
| MSR-embedded MSN183 | • MSR with OVA and CpG-ODN embedded with MSN containing GMCSF | • Dual scaffold generated antigen-presenting DC eliciting potent CD8T response • Synergized with α-CTLA4 antibody further inhibited tumor growth |
• Very high dose of systemic α-CTLA4 antibody required, may lead to toxicity • Regime doesn’t address limitations in adherence |
|
| Mesoporous silica scaffold | High-aspect-ratio MPS scaffolds28 | • 3D microenvironment releasing GMCSF, CpG and antigen fragments | • In prophylactic murine lymphoma model had delayed lymphoma growth compared to bolus vaccine delivery • Enhances systemic serum antibody levels with correlated CD8+T-cell response |
• Rapid burst release of immunostimulants and antigen peptides within 1st day • Resulted in plateau of DC recruitment by day 3 Tested in lymphoma model but vaccines require immunocompetent host to modulate response |
| PEI enhanced MPS | • Tested with GM-CSF, CpG-ODN and antigen • Can be delivered in combination with ICB |
• Eradicated established lung carcinoma in 80% mice • Generated sustained immune memory • With ICB, induced enriched CD8+ T-cell response with enriched in situ cytokine release correlating to degree of lung metastasis eradication |
• Tested on early tumors (5 days growth prior to vaccination) does not resemble clinical tumor burden |
Abbreviations: non-small cell lung carcinoma (NSCLC); receptor tyrosine-protein kinase erbB-2 (Her2Neu); breast cancer (BC); triple negative breast cancer (TNBC); deep vein thrombosis (DVT); overall survival (OS); stable disease (SD); progression free survival (PFS); complete response (CR); near complete response (nCR); western, educated, industrialized, rich and democratic (WEIRD); subcutaneous (SQ); intramuscular (IM); granulocyte macrophage colony stimulating factor (GMCSF); toll-like receptor (TLR); poly(lactic-co-glycolic acid) (PLGA); monophosphoryl lipid A (MPLA); CpG-rich oligonucleotide (CpG-ODN); cytotoxic T-lymphocyte associated protein 4 (CTLA-4); high density lipoproteins (HDL); tumor associated antigens (TAA); immune checkpoint blockade (ICB); chemoradiotherapy (CR); thienotriazolodiazepine (JQ1); near-infrared spectroscopy (NIR); Dendritic cells (DC); myeloid-derived suppressor cell (MDSC); cytotoxic T lymphocyte (CTL); CD8+ T cell (CD8T); mesoporous silica-approaches (MPS); mesoporous silica nanoparticles (MSN); mesoporous silica microrods (MSR)
Particulate-based vaccines
Particulate-based methods have long been used to enhance therapeutic delivery to specific tissues sites without off-target or systemic adverse effects. Here, we will explore key particulate in situ cancer vaccines leveraging liposomes, nanoparticles and cell-fusion particles.
Liposomes and other lipid-based platform strategies
One of the first vaccine carrier systems, liposomes and their derivatives are a favored delivery system due to their potential for rapid clinical translation, as there is a precedent for FDA approved liposomal-based formulations123. Liposomes are comprised of an outer hydrophobic bilayer with an inner hydrophilic core, rendering them suitable for encapsulating both hydrophobic and hydrophilic therapeutic cargo. Due to their inherent versatility and plasticity, liposome formulations carrying adjuvants demonstrate stable formulations with long depot effect at the site of injection123.
Often delivered via intramuscular or subcutaneous injection, liposomes can be easily modulated via addition of stability enhancers such as polyethylene glycol (PEG) and can carry a variety of cargo including novel antigenic fragments and immune adjuvants123. However, once released, lipid-based vaccines cannot be retrieved potentially leading to unequal and uncontrollable responses after administration. Further, significant work is required to increasing loading capacity, improving stability, and minimizing toxicity of liposome-based approaches.
When co-administered with standard-of-care chemotherapy regimens, clinical trials demonstrate favorable Phase I and Phase II results. However, these clinical trials do not lead to clinically relevant impact on overall survival or limiting disease progression133. For example, the BLP25 (L-BLP25; tecemotide; Stimuvax®) vaccine is a liposome-conjugated vaccine containing MUC1 peptide, a commonly overexpressed and aberrantly glycosylated membrane protein in most adenocarcinomas20,122,124,133. In the Phase III START trial, patients with unresectable locally-advanced non-small cell lung cancer received neoadjuvant chemoradiotherapy with BLP25124,133. While treatment did not manifest significant impact in overall survial with neoadjuvant therapy, a small subset of patients who received adjuvant chemotherapy experienced improved overall survival. When repurposed for therapeutic use in human epidermal growth factor receptor 2 (HER2+) breast cancer, no reduction in residual cancer burden was observed in Phase II trials20. An additional follow-up Phase III trial was planned to study vaccine effect patients receiving concurrent chemotherapy (START2, NCT02049151); however, negative results in other subgroups led to trial suspension17. Clear dose-dependent effects in similar lipid-based strategies134 demonstrates the need for a refillable vaccine platform to confer lasting immunity without repeated administration.
While conventional liposomes have mixed clinical efficacy, novel use of lipid-based structures preclinically has led to the development of the nanodisc. Composed of cylindrical synthetic high-density lipoproteins, the nanodisc combines two Toll-like receptor (TLR) agonists, monophosphoryl lipid A (MPLA) and CpG-rich oligonucleotide (CpG-ODN) targeting TLR4 and TLR9 respectively, which can be readily combined with tumor-specific antigenic peptides and proteins118. Nanodisc subcutaneous vaccination demonstrated strong induction of the humoral response leading to antitumor efficacy in cervical cancer 119 and melanoma121 murine models. It can also co-deliver chemotherapeutics, establishing broad induction of antitumor efficacy in 88% of mice immunized against colorectal carcinoma with 100% rejecting tumor rechallenge120. In combination with ICB, such as PD-1 and CTLA-4, the nanodisc can further synergistically improve vaccination efficacy, eradicating 90% of colorectal carcinoma120 and melanoma121.
While these preclinical successes are promising, the nanodisc may pose serious limitations impeding translation. First, it requires neoantigen usage for tumor specificity. As discussed previously, while murine models have predetermined, easily identifiable highly specific and immunogenic neoantigens, finding such antigenic markers in heterogeneous human tumors is laborious. Consequently, the relative simplicity of neoantigen identification in murine models is not immediately translatable to humans and thus limits translation68. Additionally, it has demonstrated efficacy within a small tumor window that is not representative of clinical scenarios with large tumors at diagnosis. Thus, therapeutic efficacy may not be recapitulated in metastatic advanced tumors. Lastly, it requires multi-dose therapy to achieve therapeutic efficacy. This platform is unable to be refilled or rescaled in relation to tumor burden. Clinically, the nanodisc regimen would be no different from multi-dose single-administration of ex vivo vaccines52,89. As such, although the nanodisc is promising, further work is required to improved therapeutic loading with limitation to cancers with known tumor-specific antigens.
Although ICB has garnered widespread FDA approval for a variety of hard-to-treat malignancies, clinical efficacy is delayed and limited to a small subset of patients exhibiting ideal mutational load and host immune profile135,136. One method of improving clinical response hinges on transforming an immunologically cold tumor into a hot immune microenvironment through use of vaccines as a priming agent. Cancer vaccines co-administered with ICBs induce immune infiltration corresponding to increase in efficacy via host-directed therapy. In a Phase I study, a lipid-encapsulated personalized neoantigen mRNA nanovaccine (mRNA-4157, Moderna, NCT03739931) demonstrated high clinical safety, tolerability, immunogenicity and overall response in a variety of malignancy types137. Each vaccine, encoding up to 20 patient-specific neoantigens, was intramuscularly administered up to 9 times every 3 weeks. Patients with metastatic infiltrations were co-treated with pembrolizumab (Keytruda ®) a monoclonal anti-PD-1 antibody, regardless of prior non-response rate to PD-L1 inhibitor therapy. 85% and 66% of patients receiving vaccination monotherapy or combination therapy respectively displayed some effect ranging from stable disease (SD) to complete response (CR) by RECIST criteria. Of these, two patients demonstrated either CR or PR after previously failing PD-L1 inhibitor therapy137. Thus, mRNA-4157 exhibits potential to prime the host immune system to allow further ICB synergistic efficacy.
PLA and PLGA nanoparticles
Nanoparticles display target specificity to tissue of interest, based on size, charge, surface properties and dissemination strategy, and thus have been extensively investigated for oncotherapeutic delivery138. Such nanoparticles can deliver therapeutics to the targeted site with limited off-target accumulation. Poly (lactide-co-glycolide) (PLGA), polylactic acid (PLA) and polyethylene glycol (PEG) are the most commonly used co-polymers to synthesize nanoparticles and their slightly larger counterpart, microparticles, as biodegradable antigenic carriers. Both co-polymers are used in a number of FDA approved therapeutics139. These co-polymers have modifiable degradation rates, ensuring constant delivery of antigens with minimal toxicity139,140.
Variation in coating and encapsulation compounds can help target nanoparticles to potentiate immunogenic responses. Notably, therapeutic application of PLGA nanoparticles coated with agonistic anti-CD40 encapsulating target antigens and adjuvants (NP-CD40) mediated delivery to DCs to induce antigen-specific CTL responses141. NP-CD40 enhanced DC maturation, CD4+ T cell proliferation and subsequently stimulated IFN-γ production in vitro leading to selective and efficient immunity in prophylactic and therapeutic challenges. Vaccination with NP-CD40 in tumor-bearing mice induced statistically smaller tumor burden leading to prolonged overall survival141. In addition to CD40 targeting, other immune adjuvants have been explored as immune potentiating particulate targets. For example, CpG ODN and polyriboinosinic:polyribocitidylic acid (polyI:C) have been extensively investigated as an antigen delivery system targeting DCs142,143. The effect of PLGA particulate systems incorporating TLR ligands on immune response has been reviewed by Silva et al144. Particulate targeting is critical to broadly boost immune responses against specified tumor antigens; however, they do require encapsulation of a known immunogenic antigen and were studied in early stage tumors.
To further enhance nanoparticle targeting and formulation, particulate cores with immunostimulatory agents, can be enveloped using tumor cell membranes to deliver TAAs efficiently to DCs in situ117,145–148. Similar to development of tumor lysates, tumor membranes are isolated via hypotonic lysing and processed via mechanical disruption145. With immune adjuvants, this has shown efficacy in preventing tumor growth in prophylactic149 and therapeutic145 murine melanoma. However, it requires frequent and repeated vaccination to protect mice from tumor growth. Additionally, in combination with ICB, long-term survival was found in half of melanoma-bearing mice145. These preclinical studies demonstrate that although an appropriate antitumor response is elicited, multiple doses with a complicated protocol makes it difficult to develop consistently active personalized vaccines.
Other preclinical strategies hinge on the development of microparticles for antigenic targeting. In murine models, mesoporous silicon vectors can efficiently encapsulate and co-deliver antigenic peptides and dual TLR-agonists, inducing potent antitumor immunity to prolong survival113. For example, calcium carbonate microparticles can encapsulate immune adjuvants and antigen with high loading capacity versus PLGA scaffolds112. Further, PLGA microparticles embedded with tumor lysates demonstrated 42% reduction in breast cancer induced lung via prime-boost vaccination with free TLR adjuvants116. These microparticles can be delivered subcutaneously116, intravenously113 or via oral gavage114,115, inducing effective host immune responses.
Despite the wide variety of particle-based strategies, only ten have advanced to the clinic in both the United States and Europe150,151. Therapeutic efficacy is limited by delivery efficiency; only 0.7% (median) of a nanoparticle dose is delivered to the intended tumor site138. Therefore, nanoparticles used as cancer vaccines must overcome this fundamental delivery limitation to accelerate translation. Although PLGA nanoparticle coating allows for targeted localization of cargo148,152, the low loading efficiency, specifically for hydrophobic compounds, necessitates large dosing to achieve therapeutic effect139. Due to their size, particle-based therapies are rapidly internalized by phagocytic cells and often require repeated frequent vaccination to achieve intended immunity138. Notably, smaller particles induce the strongest antigen-specific T cell response153. Further, nanoparticles often highly rely on charge-based delivery and thus are limited to a cargo of peptides with defined sequences or naturally charged molecules such as DNA or mRNA154.
Exosomes
Membrane-bound extracellular vesicles, including exosomes, microvesicles and apoptotic bodies, produced from the endosomal compartment of most eukaryotic cells, are used as drug delivery vehicles. Exosomes derived from DCs (Dex) display similar surface expression markers and can induce antigen-specific tumor regression in murine cancer models155. Dex can be engineered to express specific immunostimulatory molecules as well as incorporate TLR ligand adjuvants or mRNAs encoding neoantigens or immune signaling pathways modulators156. Dex is currently being investigated in clinical trials as cancer vaccines for advanced NSCLC and metastatic melanoma156,157. Although disease stabilization was achieved, the limited clinical efficacy emphasizes the need for enhancing Dex activity.
Tumor cell-derived apoptotic bodies engineered to deliver immunostimulatory CpG DNA represents another avenue of research158,159. In two Phase I studies, autologous tumor cells were harvested from patients and treated with antisense oligodeoxynuclotide against insulin-like growth factor receptor-1 (IGV-001) to induce apoptosis and release exosomes containing tumor antigens (NCT02507583, NCT01550523)25,26. Treated tumor cells were encased in a surgically implanted diffusion chamber in the abdomen, generating a slow-release antigen-depot to mediate the host immune system against recurrent glioblastoma and astrocytomas. While IGV-001 is generally well-tolerated, the multistep surgical timeline may contribute to adverse events. Following an invasive debulking craniotomy, loaded chambers are placed between the rectus sheath and rectus abdominis muscle on post-operative day (POD) 1 and removed either on POD 2 or 3, all performed at bedside. Additionally, due to pain and immobility, patients were subjected to long-term thrombosis prophylaxis for 3 months with repeated frequent ultrasound evaluations. Six patients suffered from adverse events including hematomas, wound complication and deep vein thrombosis25,26. While these adverse events were Grade 3 and less, care must be taken to ensure minimal effect on patient quality of life when designing and administering therapeutics. Additionally, protocols manufacturing and administering extracellular vesicles vary wildly leading to concerns and open questions on isolation protocols and related costs, loading techniques, establishing cGMP-grade preparation, therapeutic dosing regimen, and administration methods160. Detailed review of exosomes, representing an innovative strategy for cell-free therapeutic cancer vaccination, are reviewed in detail elsewhere157,161.
In a Phase I trial for malignant astrocytomas, IGV-001 within these commercially available diffusion chambers fitted with 100nm filters resulted in observable clinical improvements in 75% of patients. Further, 37.5% of these patients manifested spontaneous regression at local and metastatic sites25. When applied in newly diagnosed glioblastoma followed by standard chemoradiotherapy, median progression-free survival (PFS) significantly improved by 3.3 months compared to standard of care. Furthermore, PFS was shown to be dose-dependent with an increase of 10.5 months at the highest exposure. Patients with de novo methylation of O6-methylguanine-DNA-methyltransferase promoter (MGMT) had maximum PFS benefit of 38.4 months26. This clinically meaningful improvement correlated with an increase in serum IL-17, likely due to its induction of many immune signaling molecules and cells types26,162. To further investigate the clinical and immunological impact of IGV-001 therapy, enrollment in a Phase II trial is currently open (NCT04485949). Overall, extracellular vesicles represent important facilitators of an antigen-specific cytotoxic response and may be effective in a combinatorial treatment regime. However, significant work is required to overcome manufacturing challenges to induce reproducible mediation of immune responses to reach their full potential.
Injectable mesoporous silica nanoparticles
The aforementioned particulate-based drug delivery systems allow for targeted drug delivery using a specific niche of drugs due to solubility, surface charge and encapsulation efficancy163. However, such delivery systems often demonstrate high variability in delivery efficacy dependent on cellular interactions, especially within the tumor stroma138. Mesoporous silica-based (MPS) approaches aim to address these challenges leveraging their well-established drug carrier properties and high versatility in conjunction with other materials164.
Silica is a well-known biocompatible chemical catalogued as “Generally recognized as safe” by the FDA (ID Code: 14808–60-7). MPS systems are composed of nano-sized spheres or rods of silica in a predetermined non-specific geometric arrangement. This generates a large surface area for cargo loading and cellular infiltration. Addition of functional groups165 and capping treatment166 can further fine-tune idealized chemical and physical properties. MPS platforms can boost dissolution of poorly water soluble drugs167, expanding the potential drug candidates that can be loaded. Prophylactically, MPS nanoparticles with CpG ODN and model antigen demonstrated enhanced DC activation and antigen presentation. It led to induction of antigen-specific cytotoxic responses with significant suppression of tumor growth with immune memory in murine melanoma168, highlighting its feasibility and applicability.
Scaffold-based delivery systems
Scaffold-based vaccines are structures intended to initiate antitumor immunity locally at the implantation or injection site7,169. For in situ cancer vaccines, most deliver stimulatory adjuvants and antigens to induce in situ DC homing and subsequent antigen-specific immune activation. They often target DCs residing in the skin, subcutaneous tissue and circulating in the blood.
In addition to encountering foreign antigens, DCs require presence of a secondary ‘danger signal’ to induce protective T-cell immunity62. Classically, these signals emerge from tissue injury or from cytokine and chemokine secretion from immune activation62,170. While multiple physical adjuvants can induce reaction171–174 and are commonly used in preventative vaccines174,175, biomaterial research demonstrates inorganic materials and man-made technologies can also activate these required danger signals. Thus, in situ cancer vaccines utilizing scaffolds must be designed to address three key criteria. First, they should be macroporous, enabling the infiltration and dispersing of appropriate cells from peripheral tissue without inducing cellular or systemic toxicity176. Ideal pore size can promote cell infiltration, adhesion and activation. However, optimal pore size must be carefully tailored to cell type to ensure homogenous spatial differentiation177,178. Second, bioinert scaffolds should release immune potentiating adjuvants, such as cytokines89 and TLR-agonists179 or their synthetic derivatives, capable of recruiting DCs. Upon antigenic presentation, either via neoantigens or tumor lysates, recruited infiltrated DCs must undergo activation steps necessary for inducing antitumor immunity67. Lastly, an ideal scaffold must be clinically translatable; it should address and obviate any patient adherence issues posed by current non-scaffold based strategies58.
Dual scaffold-particulate combinatorial therapeutics
Scaffold-based strategies can also be used in combination with particulate formulations to enable geometric control of therapeutic cargo release. Spatiotemporal kinetic control is often enabled through use of hydrogel or mesoporous silica micorods (MSR) scaffolds. Such nanocarrier systems have been used in a multitude of delivery modalities180,181 to modulate immunity. For instance, differences in charged functional groups on mesoporous silica nanoparticles (MSN) can direct distribution to certain cell types. Concurrently, 3D printed hydrogels can modulate surface chemistry to augment cellular uptake182. This strategy has been adapted to attune immune responses via loading immunostimulatory therapeutics within MSN. One formulation delivered ovalbumin (OVA) and CpG-ODN via MSN coupled with mesoporous silica microrods (MSR) loaded with GMCSF183. Once activated within the MSR macroporous scaffold, DC were able to generate antigen-specific T cells and inhibit murine melanoma burden locally183. Additionally, synergistic combination with four doses of intraperitoneal α-CTLA4 antibody further inhibited tumor growth. Although this suggests combinatorial vaccines efficacy, the need for repeated systemic α-CTLA4 antibody dosing may cause deleterious immunosuppressive effects. Further, the administration schedule does not address limitations to patient adherence complicating traditional DC vaccines.
Implantable PLGA scaffolds
Vaccine success hinges on the biomaterials used to initiate and sustain the required immune response. Although many scaffolding materials are available, PLGA scaffolds are a cornerstone for implantable scaffold-based vaccines owing to their use in multiple FDA approved pharmaceuticals. In one seminal preclinical study, implantable PLGA scaffolds continuously released GM-CSF and tumor-specific lysates to specifically recruit and activate DCs in situ (WDVAX)132. This subcutaneously implanted scaffold extended survival time in 90% of mice challenged with murine melanoma132. A single vaccination maintained local and systemic immunity, resulting in complete regression of primary and distant melanoma tumors in 47% of mice131. In intracranial glioma, WDVAX extended overall survival in 90% of rats130,184 with a single vaccination. These successes led to an ongoing Phase I clinical trial of WDVAX for Stage IV metastatic melanoma (NCT01753089)185 and licensing by Novartis for commercial use. One scaffold is implanted every 3–4 weeks for total of four implantations per patient. It is important to note clinically, intracranial implantation requires potentially dangerous and lengthy surgical procedures185. Additionally, PLGA matrices may damage or denature antigenic structure. In addition, the need of organic solvents for processing may lead to subclinical toxicity with limited preclinical detection.
Injectable hydrogel-based scaffolds
Injectable hydrogel scaffolds offer an appealing alternative to surgical implantation. Hydrogels can control the rate of adjuvant and antigen delivery while ensuring biocompatibility and biodegradability. These semisolid gels can be loaded with bioactive molecules forming an enriched microenvironment for cellular interaction128,186. In addition to the previously discussed PLGA, two of the most common polymer structures for cellular encapsulation are sodium alginate187 and poloxamer 407 (pluronic F-127, PF127)188,189. Both are commercially available, allowing for straightforward application without the use of harsh organic solvents. Alginate is an anionic biopolymer widely used due to its biocompatibility, hydrophobicity and ease of chemical modifications186. Unless modified, alginate requires ionic crosslinking to undergo the sol-to-gel phase transition required for many biomedical applications128. Thus, vaccine strategies employing unmodified alginate require surgical implantation. Conversely, PF127 is a thermosensitive gel, undergoing gelation at physiological temperature. Thus it is an appealing hydrogel for injectable vaccine delivery188 and sustained release depot of antigens and adjuvants189–191.
Mooney et al. have developed a methyacrylated-alginate cryogel with shape-memory properties for easy injection192. This pre-formed cryogel is loaded with immune adjuvants and tumor lysates similar to WDVAX132. Cryogels generated in situ immune cell recruitment and trafficking and conferred long-lasting prophylactic immunity in 80% of melanoma mice29. However, it has a limited encapsulation efficacy and released 80% of its contents within the first week29. Intended to be degradable, the cryogel scaffold cannot be refilled with additional adjuvants. Thus, multiple vaccinations may be required to release sufficient immune adjuvants and tumor antigens to maintain immunity. This approach is limited to previously identified highly antigenic tumor models and does not extend to different cancer subtypes with both identified and unidentified antigens.
Chemical modification of PLGA can confer thermosensitive properties for in situ gelation. Mice immunized with PLGA hydrogels demonstrate successful DC recruitment and maturation, extending overall median survival time with reduced tumor burden. However, as with other immunization schemes, repeated systemic administrations of tumor antigens and replication-defective transduced DCs were required127. Addressing these limitations, another study immunized mice using PEG-PLGA hydrogel co-encapsulating GM-CSF and ovalbumin nanoparticles. This vaccination strategy released adjuvant and nanoparticles for 13 days, generating cytotoxic immunity prophylactically129. While promising, use of both scaffold- and nanoparticle-based delivery systems increases the drawbacks of associated with both methods.
In parallel, Li et al. have developed an injectable self-assembling hydrogel by chemically modifying key tumor peptides co-loaded with immune modulators31. Preclinically, these hydrogels are co-loaded with thienotriazolodiazepine (JQ1, abromodomain-containing protein 4 inhibitor) and indocyanine green (ICG, photothermal dye capable of excitation in situ by NIR irradiation) along with autologous fixed tumor cells. Use of JQ1 activates DC-mediated antigen uptake and consequent expansion of CD8+T-cell antitumor clones193. Additionally, they promote an activated immune microenvironment by reducing expansion of myeloid-derived suppressive cells193. Light-triggered release of cargo penetrated tissue in the murine 4T1 triple negative breast cancer model. Additionally, it accelerated DC maturation in vivo corresponding to an increase in CD8+T-cell frequency and cytokine release by 2.7-fold31. Vaccination post surgical resection further induced a systemic immune response by blocking recurrence and distant metastasis for up to 30 days. However, this strategy is limited by complex manufacturing and repeated doses with in the surgical bed to ensure immune modulation. Further, efficacy hinges on adequate light penetration into tumor tissues. Tumors seeded deep into the body (ex. colorectal tumors, pancreatic tumors) or in an inaccessible location (ex. lung nodule, prostate tumors) will likely exhibit lower clinical success. Thus, while this strategy is a promising direction, these limitations must be addressed for widespread benefit.
Injectable mesoporous silica-based scaffolds
Previously discussed MPS can be utilized as an implantable 3D scaffold for in situ sustained cytokine release and cellular infiltration27,28,113. Injectable MPS scaffolds utilize high aspect ratio particles for higher cellular uptake compared to spherical particles194,195. Preclinically, mice were injected with spontaneously assembling loaded MPS rods form a macroporous 3D environment prime for cellular trafficking28. Mice prophylactically immunized exhibited significantly delayed lymphoma growth compared to bolus adjuvant delivery28. Although enhanced systemic serum antibody levels was observed, sustained release of immunotherapeutics was not achieved. Burst release of 80% and 60% of CpG and antigen peptide, respectively, occurred within the first day28, resulting in the plateau in DCs recruitment by day 3. Further testing therapeutically with sustained release of adjuvants is warranted.
MPS scaffolds can be combined with polyethyeneime (PEI) to increase vaccine efficacy via stimulation of Damage-Associated Molecular Patterns (DAMP) receptors for DC signaling27,125,166. PEI is a potent mucosal adjuvant that forms nanoscale complexes with antigens, inducting DC homing to lymph nodes and cytokine response125. Immune modulation eradicated established lung carcinoma in 80% of mice and generated sustained immune memory27. With ICB, PEI-enhanced scaffold induced a T-cell response with enriched in situ cytokine release correlating to lung metastasis erradication27. PEI greatly enhances this scaffold’s therapeutic value, enabling robust vaccination. Further, it can be loaded with multiple neoantigens simultaneously, inducing immune-mediated responses in synergy with other immunotherapies.
Implantable non-degradable DC homing devices
Our lab has significant expertise in designing implantable medical devices for sustained and constant release of therapeutic cargo for different medical applications106,107,196–200. We have developed an immunostimulatory niche capable of maintaining allogeneic cell encapsulation and transplantation. This 3D-printed device allows for sustained local release of immunosuppressive drugs to ensure local allogeneic graft survival within our scaffold198,199,201. Building on this device for cell transplantation, we developed the NanoLymph, a biomaterial-based cancer vaccine platform capable of generating an immunostimulatory niche to recruit, activate and potentiate DCs in situ202. The NanoLymph is a subcutaneously implantable device with dual reservoir platform for immune adjuvants and hydrogel-encapsulated antigens (Figure 3). Through the sustained and constant elution of immune adjuvants such as GM-CSF and TLR agonists via nanoporous membranes, the NanoLymph generates a cytokine gradient extending into the local tissue. This gradient recruits DCs, allowing them to interface directly with autologous tumor antigens within the cell reservoir. Activated DCs then mobilize to secondary lymphoid organs to trigger antitumor immunity. By providing constant elution of immune adjuvants, continuously recruiting DCs locally, the NanoLymph is designed to address and improve upon previous biomaterial-based vaccine platforms. The ability to be drug-agonistic, personalizable in terms of autologous antigen loading and refillable, offers design features that address unmet needs in implantable platform technologies.
Figure 3: Immune cell recruitment and activation via NanoLymph.
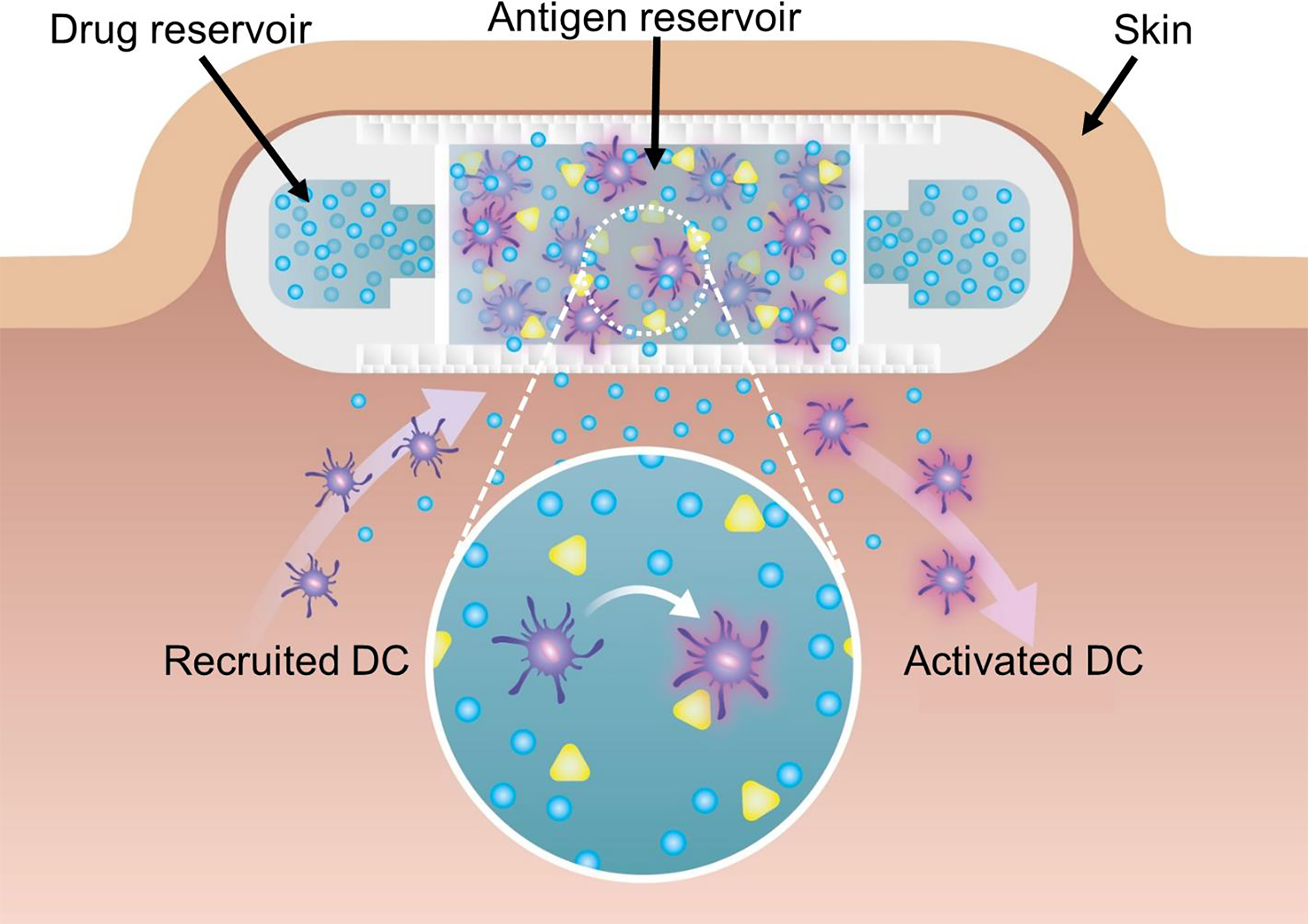
Schematic representation of NanoLymph’s recruitment of DCs through constant and sustained immunostimulant elution into subcutaneous space. Cross-section of NanoLymph demonstrates DCs (purple) are recruited through local release of immune adjuvants (blue) and can interface directly with the implanted NanoLymph. Upon infiltration, DCs encounter presented tumor antigen (green) and subsequently become activated (red glow). These activated DCs are able to exfiltrate the NanoLymph and home to draining lymph nodes to interact with lymphocytes.
4. Considerations for efficient vaccine development and future outlook
Over the past decades, improvements in material science, bioengineering, and cancer immunotherapy have ushered in a new era for cancer vaccines as a promising therapeutic modality. These vaccines can modulate key steps in the cancer immunogenic cycle in two ways. First, vaccine platforms can provide an in situ antigen depot for immunogenic personalized tumor antigens. Second, such engineered strategies aim to provide costimulatory activation to DC cells, potentiating effector cytotoxic T-cell mediated immunity and dampening suppressive regulatory T cell responses203,204. Thus, cancer vaccines aim to induce effector T-cell infiltration and homing to tumors, combating the inherently immunosuppressive tumor microenvironment. Inadequate therapies may target one step of this cycle but fail to spur impactful effects due to failure of other components. Therefore, an optimal immunotherapeutic must be targeted and adapted to immunogenic antigens of any subtype, induce a self-propagated systemic response, penetrate target tissues and establish immune memory to prevent remission and ensure long-term disease-free status. In this review, we discuss how biomaterials can be applied to vaccine strategies to devise clinically-relevant vaccine platforms. We believe that this combination can amplify and enhance clinical response rates of vaccination treatments.
Effective personalized cancer vaccines may serve dual benefit by inducement of the abscopal effect. This elusive effect, traditionally defined through use of radiotherapy, is the regression of a non-treated distant tumor following local treatment205. Often demonstrated in conjunction with radio-immunotherapy, this phenomena supports evidence for local immunogenic cell death (ICD) to catalyze downstream cellular responses mimicking a viral infection206. By generating ICD, targeted local cancer vaccination can induce tumor-specific CTL responses capable of attacking both local and distant tumor sites. Utilization of biomaterials for vaccine formulation ensure constant and effective delivery of antigens to resident APCs, yielding a systemic CTL response similar to the abscopal effect derived from radiotherapy.
Additionally, cancer vaccines used in combination with other oncoimmunotherapies can further potentiate antitumor immune responses. We have already discussed the successes of a nanoparticle formulation utilizing CD40 to target antigenic delivery to DCs141. Use of ICBs, including anti-CTLA-4 or anti PD-1 antibodies, can augment and amplify T cell immunity to generate tumor shrinkage. Preclinically, a PLGA scaffold vaccine co-delivering autologous tumor lysates, polythylenimine (PEI) CpG-ODN in combination with ICB generated a higher infiltration of active cytotoxic T cells and Treg regression consistent with tumor shrinkage in a B16 murine melanoma model207. Moreover, augmentation with CTLA-4 enriched local cytotoxic T cell population by 5- to 10-fold over PD-1 and vaccine alone respectively207. Ongoing clinical trials are characterizing clinical benefit of combinatorial vaccination with ICB modulation. A p53-modified adenovirus-transduced in situ cancer vaccine (INGN-225)208, in combination with nivolumab and ipilimumab, is currently recruiting for small cell lung cancer (NCT03406715). Thus, supplementation of other oncoimmunotherapies may support cancer vaccines in modulating and augmenting tumor-specific immunity.
Clinical translatability of biomaterial vaccine platforms hinge on harnessing an appropriate platform, cancer subtype and animal model for testing. Preclinical experiments often are performed in murine models using the prototypic B16 melanoma or renal carcinoma due to ease of tumor and antigen manipulation. Both are classified as an immunologically “hot” tumors and are highly associated with improved response to immunotherapy via immune blockade209–211. Their high immunogenicity and frequency of mutations lends itself well to generating large amount of neoantigenic peptides available for vaccine targeting212 along with synergistic immunotherapy and vaccine platform designs. While these preclinical studies are critical for vaccine design, focus on non-representative models has led to generation of ineffective vaccines that fail to induce meaningful clinical responses213, especially in other malignancies87. Further, differences between mice and man214 may pose challenges in bringing vaccine to clinical trial. For realistic efficacy assessments, clinically relevant antigens and orthotropic, transgenic patient-derived xenograft models are needed, as well as the use of humanized mice that can more closely recapitulate human immune response. Thus, when designing an appropriate biomaterial-based vaccine platform, suitable testing models must be considered during early phases of research to ensure potential for clinical utility.
Additionally, it is critical to note that a majority of the reviewed vaccination trials were performed in patients from Caucasian backgrounds. Studies that rely on samples drawn entirely from Western, Educated, Industrialized, Rich and Democratic (WEIRD) societies often are the least representative populations for evaluating response to therapy215. WEIRD societies have little demographic variation and often fail to be generalizable across human responses. This risk of selection bias in clinical trials is not limited to the study of cancer vaccines216,217. In order to exclude human variation as an important confounding variable, it is crucial to be inclusive in trial design. These considerations have led to parallel trials for neoadjuvant chemoradiotherapy with tecemotide treatment in East Asian and Japanese patients, to exclude ethnicity as a factor that influences vaccination efficacy122.
While many clinically-viable biomaterial strategies exist, care must be taken to ensure clinical translatability and feasibility when designing preclinical and clinical studies. Clinically-translatable platforms modulate engineered physical properties to target key underlying immunity mechanisms necessary for cancer eradication. Manufacturing and scaling limit a majority of the aforementioned biomaterial vaccine platforms. Unlike vaccines for infectious diseases, cancer vaccines must be tailored for each patient due to high individual heterogeneity in tumor mutation burden. However, platforms utilizing neoantigens require use of complicated next-generation sequencing technologies for adept identification of highly immunogenic personalized neoantigens. Subsequently, this may lead to high batch-to-batch variability in neoantigen generation and loading. Use of tumor lysates provide a pool of tumor-associated antigens to activate a broad spectrum of polyclonal antitumor immune responses. Furthermore, complicated treatment regimens necessitate use of GMP grade materials, further limiting care to tertiary hospital centers. Thus, a collaboration between industry, bioengineers, immunologists and clinicians will further accelerate the generation of an engineered therapeutic cancer vaccine for clinical development and translation.
Funding:
AG receives funding support from the Center for Immunotherapeutic Transport Oncophysics [grant number U54CA210181, pilot award], Houston Methodist Research Institute, through the Department of Defense [grant number W81XWH-20–1-0600], Nancy Owens Breast Cancer Foundation, and Golfers Against Cancer. D.I.V. received joint funding support from Texas A&M University MD/PhD Program and Houston Methodist Research Institute. D.P.H. is supported in part by the National Institutes of Health [grant number R01AI097372] and the W. Bryan Trammell, Jr. Family Presidential Distinguished Chair in Allergy & Immunology.
Footnotes
Conflict of interest: All authors declare no conflict of interest.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Data Availability
As this is a review paper, we did not generate any raw or processed data in preparation of this manuscript.
References
- 1.Davila ML, et al. Efficacy and toxicity management of 19–28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med 6, 224ra225 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pardoll DM The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 12, 252–264 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Newick K, O’Brien S, Moon E & Albelda SM CAR T Cell Therapy for Solid Tumors. Annu Rev Med 68, 139–152 (2017). [DOI] [PubMed] [Google Scholar]
- 4.Cheever MA & Higano CS PROVENGE (Sipuleucel-T) in prostate cancer: the first FDA-approved therapeutic cancer vaccine. Clin Cancer Res 17, 3520–3526 (2011). [DOI] [PubMed] [Google Scholar]
- 5.Atezolizumab Combo Approved for PD-L1-positive TNBC. Cancer Discov 9, OF2 (2019). [DOI] [PubMed] [Google Scholar]
- 6.Whiteside TL The tumor microenvironment and its role in promoting tumor growth. Oncogene 27, 5904–5912 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gu L & Mooney DJ Biomaterials and emerging anticancer therapeutics: engineering the microenvironment. Nat Rev Cancer 16, 56–66 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wiig H, Tenstad O, Iversen PO, Kalluri R & Bjerkvig R Interstitial fluid: the overlooked component of the tumor microenvironment? Fibrogenesis Tissue Repair 3, 12 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Anderson KG, Stromnes IM & Greenberg PD Obstacles Posed by the Tumor Microenvironment to T cell Activity: A Case for Synergistic Therapies. Cancer Cell 31, 311–325 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhong S, Jeong JH, Chen Z, Chen Z & Luo JL Targeting Tumor Microenvironment by Small-Molecule Inhibitors. Transl Oncol 13, 57–69 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gajewski TF, Schreiber H & Fu YX Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol 14, 1014–1022 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Popovic A, Jaffee EM & Zaidi N Emerging strategies for combination checkpoint modulators in cancer immunotherapy. J Clin Invest 128, 3209–3218 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wei SC, Duffy CR & Allison JP Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov 8, 1069–1086 (2018). [DOI] [PubMed] [Google Scholar]
- 14.Tahmasebi S, Elahi R & Esmaeilzadeh A Solid Tumors Challenges and New Insights of CAR T Cell Engineering. Stem Cell Rev Rep 15, 619–636 (2019). [DOI] [PubMed] [Google Scholar]
- 15.Haslam A & Prasad V Estimation of the Percentage of US Patients With Cancer Who Are Eligible for and Respond to Checkpoint Inhibitor Immunotherapy Drugs. JAMA Netw Open 2, e192535 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fares CM, Van Allen EM, Drake CG, Allison JP & Hu-Lieskovan S Mechanisms of Resistance to Immune Checkpoint Blockade: Why Does Checkpoint Inhibitor Immunotherapy Not Work for All Patients? Am Soc Clin Oncol Educ Book 39, 147–164 (2019). [DOI] [PubMed] [Google Scholar]
- 17.Vermaelen K Vaccine Strategies to Improve Anti-cancer Cellular Immune Responses. Frontiers in Immunology 10(2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Irvine DJ, Swartz MA & Szeto GL Engineering synthetic vaccines using cues from natural immunity. Nat Mater 12, 978–990 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.De Groot AS, et al. Immune camouflage: relevance to vaccines and human immunology. Hum Vaccin Immunother 10, 3570–3575 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Singer CF, et al. Efficacy and safety of the therapeutic cancer vaccine tecemotide (L-BLP25) in early breast cancer: Results from a prospective, randomised, neoadjuvant phase II study (ABCSG 34). Eur J Cancer 132, 43–52 (2020). [DOI] [PubMed] [Google Scholar]
- 21.Mak IW, Evaniew N & Ghert M Lost in translation: animal models and clinical trials in cancer treatment. Am J Transl Res 6, 114–118 (2014). [PMC free article] [PubMed] [Google Scholar]
- 22.Claesson MH Why current peptide-based cancer vaccines fail: lessons from the three Es. Immunotherapy 1, 513–516 (2009). [DOI] [PubMed] [Google Scholar]
- 23.Phua KK, Staats HF, Leong KW & Nair SK Intranasal mRNA nanoparticle vaccination induces prophylactic and therapeutic anti-tumor immunity. Sci Rep 4, 5128 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hu Q, et al. Engineering nanoparticle-coated bacteria as oral DNA vaccines for cancer immunotherapy. Nano Lett 15, 2732–2739 (2015). [DOI] [PubMed] [Google Scholar]
- 25.Andrews DW, et al. Results of a pilot study involving the use of an antisense oligodeoxynucleotide directed against the insulin-like growth factor type I receptor in malignant astrocytomas. J Clin Oncol 19, 2189–2200 (2001). [DOI] [PubMed] [Google Scholar]
- 26.Andrews DW, et al. Phase Ib Clinical Trial of IGV-001 for Patients with Newly Diagnosed Glioblastoma. Clin Cancer Res (2021). [DOI] [PubMed] [Google Scholar]
- 27.Li AW, et al. A facile approach to enhance antigen response for personalized cancer vaccination. Nat Mater 17, 528–534 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim J, et al. Injectable, spontaneously assembling, inorganic scaffolds modulate immune cells in vivo and increase vaccine efficacy. Nat Biotechnol 33, 64–72 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bencherif SA, et al. Injectable cryogel-based whole-cell cancer vaccines. Nat Commun 6, 7556 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bencherif SA, et al. Injectable preformed scaffolds with shape-memory properties. Proc Natl Acad Sci U S A 109, 19590–19595 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang T, et al. A cancer vaccine-mediated postoperative immunotherapy for recurrent and metastatic tumors. Nat Commun 9, 1532 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fang L, et al. Engineering autologous tumor cell vaccine to locally mobilize antitumor immunity in tumor surgical bed. Science Advances 6, eaba4024 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tornesello AL, Tagliamonte M, Tornesello ML, Buonaguro FM & Buonaguro L Nanoparticles to Improve the Efficacy of Peptide-Based Cancer Vaccines. Cancers (Basel) 12(2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reche P, Flower DR, Fridkis-Hareli M & Hoshino Y Peptide-Based Immunotherapeutics and Vaccines 2017. J Immunol Res 2018, 4568239 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang B, Jeang J, Yang A, Wu TC & Hung CF DNA vaccine for cancer immunotherapy. Hum Vaccin Immunother 10, 3153–3164 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lopes A, Vandermeulen G & Preat V Cancer DNA vaccines: current preclinical and clinical developments and future perspectives. J Exp Clin Cancer Res 38, 146 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Iavarone C, O’Hagan D T, Yu D, Delahaye NF & Ulmer JB Mechanism of action of mRNA-based vaccines. Expert Rev Vaccines 16, 871–881 (2017). [DOI] [PubMed] [Google Scholar]
- 38.Kowalski PS, Rudra A, Miao L & Anderson DG Delivering the Messenger: Advances in Technologies for Therapeutic mRNA Delivery. Mol Ther 27, 710–728 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baden LR, et al. Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. N Engl J Med (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Polack FP, et al. Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine. N Engl J Med 383, 2603–2615 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Caballero ML & Quirce S Excipients as potential agents of anaphylaxis in vaccines: analyzing the formulations of the current authorized COVID-19 vaccines. J Investig Allergol Clin Immunol, 0 (2021). [DOI] [PubMed] [Google Scholar]
- 42.Cabanillas B, Akdis C & Novak N Allergic reactions to the first COVID-19 vaccine: a potential role of Polyethylene glycol? Allergy (2020). [DOI] [PubMed] [Google Scholar]
- 43.Pardi N, Hogan MJ, Porter FW & Weissman D mRNA vaccines - a new era in vaccinology. Nat Rev Drug Discov 17, 261–279 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wallis J, Shenton DP & Carlisle RC Novel approaches for the design, delivery and administration of vaccine technologies. Clin Exp Immunol 196, 189–204 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Steinman RM & Swanson J The endocytic activity of dendritic cells. J Exp Med 182, 283–288 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cella M, Sallusto F & Lanzavecchia A Origin, maturation and antigen presenting function of dendritic cells. Current Opinion in Immunology 9, 10–16 (1997). [DOI] [PubMed] [Google Scholar]
- 47.Banchereau J & Steinman RM Dendritic cells and the control of immunity. Nature 392, 245–252 (1998). [DOI] [PubMed] [Google Scholar]
- 48.Banchereau J, et al. Immunobiology of dendritic cells. Annu Rev Immunol 18, 767–811 (2000). [DOI] [PubMed] [Google Scholar]
- 49.Van Gool SW, Vandenberghe P, de Boer M & Ceuppens JL CD80, CD86 and CD40 provide accessory signals in a multiple-step T-cell activation model. Immunol Rev 153, 47–83 (1996). [DOI] [PubMed] [Google Scholar]
- 50.Ma DY & Clark EA The role of CD40 and CD154/CD40L in dendritic cells. Semin Immunol 21, 265–272 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rotzschke O, et al. Exact prediction of a natural T cell epitope. Eur J Immunol 21, 2891–2894 (1991). [DOI] [PubMed] [Google Scholar]
- 52.Sprooten J, et al. Trial watch: dendritic cell vaccination for cancer immunotherapy. Oncoimmunology 8, e1638212 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Calmeiro J, et al. Biomaterial-based platforms for in situ dendritic cell programming and their use in antitumor immunotherapy. J Immunother Cancer 7, 238 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Srivatsan S, et al. Allogeneic tumor cell vaccines: the promise and limitations in clinical trials. Hum Vaccin Immunother 10, 52–63 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gubin MM, Artyomov MN, Mardis ER & Schreiber RD Tumor neoantigens: building a framework for personalized cancer immunotherapy. J Clin Invest 125, 3413–3421 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schumacher TN & Schreiber RD Neoantigens in cancer immunotherapy. Science 348, 69–74 (2015). [DOI] [PubMed] [Google Scholar]
- 57.Scheetz L, et al. Engineering patient-specific cancer immunotherapies. Nat Biomed Eng 3, 768–782 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hu Z, Ott PA & Wu CJ Towards personalized, tumour-specific, therapeutic vaccines for cancer. Nat Rev Immunol 18, 168–182 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.de Gruijl TD, van den Eertwegh AJ, Pinedo HM & Scheper RJ Whole-cell cancer vaccination: from autologous to allogeneic tumor- and dendritic cell-based vaccines. Cancer Immunol Immunother 57, 1569–1577 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jiang T, et al. Tumor neoantigens: from basic research to clinical applications. J Hematol Oncol 12, 93 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chiang CL, Benencia F & Coukos G Whole tumor antigen vaccines. Semin Immunol 22, 132–143 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gallo PM & Gallucci S The dendritic cell response to classic, emerging, and homeostatic danger signals. Implications for autoimmunity. Front Immunol 4, 138 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Newman JH, et al. Intratumoral injection of the seasonal flu shot converts immunologically cold tumors to hot and serves as an immunotherapy for cancer. Proc Natl Acad Sci U S A (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chiang CL, Coukos G & Kandalaft LE Whole Tumor Antigen Vaccines: Where Are We? Vaccines (Basel) 3, 344–372 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Keenan BP & Jaffee EM Whole cell vaccines--past progress and future strategies. Semin Oncol 39, 276–286 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gonzalez FE, et al. Tumor cell lysates as immunogenic sources for cancer vaccine design. Hum Vaccin Immunother 10, 3261–3269 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Vicari AP, et al. In vivo manipulation of dendritic cell migration and activation to elicit antitumour immunity. Novartis Found Symp 256, 241–254; discussion 254–269 (2004). [DOI] [PubMed] [Google Scholar]
- 68.Jaroslawski S & Toumi M Sipuleucel-T (Provenge((R)))-Autopsy of an Innovative Paradigm Change in Cancer Treatment: Why a Single-Product Biotech Company Failed to Capitalize on its Breakthrough Invention. BioDrugs 29, 301–307 (2015). [DOI] [PubMed] [Google Scholar]
- 69.Kini V & Ho PM Interventions to Improve Medication Adherence: A Review. JAMA 320, 2461–2473 (2018). [DOI] [PubMed] [Google Scholar]
- 70.Lam WY & Fresco P Medication Adherence Measures: An Overview. Biomed Res Int 2015, 217047 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Torres-Robles A, et al. Comparison of Interventions to Improve Long-Term Medication Adherence Across Different Clinical Conditions: A Systematic Review With Network Meta-Analysis. Front Pharmacol 9, 1454 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Figdor CG, de Vries IJ, Lesterhuis WJ & Melief CJ Dendritic cell immunotherapy: mapping the way. Nat Med 10, 475–480 (2004). [DOI] [PubMed] [Google Scholar]
- 73.de Vries IJM, et al. Effective migration of antigen-pulsed dendritic cells to lymph nodes in melanoma patients is determined by their maturation state. Cancer Research 63, 12–17 (2003). [PubMed] [Google Scholar]
- 74.Carreno BM, et al. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science 348, 803–808 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Perez CR & De Palma M Engineering dendritic cell vaccines to improve cancer immunotherapy. Nat Commun 10, 5408 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Small EJ, et al. Immunotherapy of hormone-refractory prostate cancer with antigen-loaded dendritic cells. J Clin Oncol 18, 3894–3903 (2000). [DOI] [PubMed] [Google Scholar]
- 77.Small EJ, et al. Placebo-controlled phase III trial of immunologic therapy with sipuleucel-T (APC8015) in patients with metastatic, asymptomatic hormone refractory prostate cancer. J Clin Oncol 24, 3089–3094 (2006). [DOI] [PubMed] [Google Scholar]
- 78.Schadendorf D, et al. Dacarbazine (DTIC) versus vaccination with autologous peptide-pulsed dendritic cells (DC) in first-line treatment of patients with metastatic melanoma: a randomized phase III trial of the DC study group of the DeCOG. Ann Oncol 17, 563–570 (2006). [DOI] [PubMed] [Google Scholar]
- 79.Amin A, et al. Survival with AGS-003, an autologous dendritic cell-based immunotherapy, in combination with sunitinib in unfavorable risk patients with advanced renal cell carcinoma (RCC): Phase 2 study results. J Immunother Cancer 3, 14 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mougel A, Terme M & Tanchot C Therapeutic Cancer Vaccine and Combinations With Antiangiogenic Therapies and Immune Checkpoint Blockade. Front Immunol 10, 467 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Shi Y, et al. Granulocyte-macrophage colony-stimulating factor (GM-CSF) and T-cell responses: what we do and don’t know. Cell Res 16, 126–133 (2006). [DOI] [PubMed] [Google Scholar]
- 82.van de Laar L, Coffer PJ & Woltman AM Regulation of dendritic cell development by GM-CSF: molecular control and implications for immune homeostasis and therapy. Blood 119, 3383–3393 (2012). [DOI] [PubMed] [Google Scholar]
- 83.Fong L, et al. Altered peptide ligand vaccination with Flt3 ligand expanded dendritic cells for tumor immunotherapy. Proc Natl Acad Sci U S A 98, 8809–8814 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Manz MG, Weissman IL, Cozzio A, Merad M & Karsunky H Flt3 Ligand Regulates Dendritic Cell Development from Flt3+ Lymphoid and Myeloid-committed Progenitors to Flt3+ Dendritic Cells In Vivo. Journal of Experimental Medicine 198, 305–313 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.O’Keeffe M, et al. Effects of administration of progenipoietin 1, Flt-3 ligand, granulocyte colony-stimulating factor, and pegylated granulocyte-macrophage colony-stimulating factor on dendritic cell subsets in mice. Blood 99, 2122–2130 (2002). [DOI] [PubMed] [Google Scholar]
- 86.Ali OA, Tayalia P, Shvartsman D, Lewin S & Mooney DJ Inflammatory cytokines presented from polymer matrices differentially generate and activate DCs in situ. Adv Funct Mater 23, 4621–4628 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Pyzer AR, Avigan DE & Rosenblatt J Clinical trials of dendritic cell-based cancer vaccines in hematologic malignancies. Hum Vaccin Immunother 10, 3125–3131 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Dranoff G, et al. Vaccination with irradiated tumor cells engineered to secrete murine granulocyte-macrophage colony-stimulating factor stimulates potent, specific, and long-lasting anti-tumor immunity. Proc Natl Acad Sci U S A 90, 3539–3543 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Dranoff G GM-CSF-based cancer vaccines. Immunol Rev 188, 147–154 (2002). [DOI] [PubMed] [Google Scholar]
- 90.Higano CS, et al. Phase 1/2 dose-escalation study of a GM-CSF-secreting, allogeneic, cellular immunotherapy for metastatic hormone-refractory prostate cancer. Cancer 113, 975–984 (2008). [DOI] [PubMed] [Google Scholar]
- 91.Yan WL, Shen KY, Tien CY, Chen YA & Liu SJ Recent progress in GM-CSF-based cancer immunotherapy. Immunotherapy 9, 347–360 (2017). [DOI] [PubMed] [Google Scholar]
- 92.Lipson EJ, et al. Safety and immunologic correlates of Melanoma GVAX, a GM-CSF secreting allogeneic melanoma cell vaccine administered in the adjuvant setting. J Transl Med 13, 214 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kim VM, et al. Neoantigen-based EpiGVAX vaccine initiates antitumor immunity in colorectal cancer. JCI Insight 5(2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yarchoan M, et al. A phase 2 study of GVAX colon vaccine with cyclophosphamide and pembrolizumab in patients with mismatch repair proficient advanced colorectal cancer. Cancer Med 9, 1485–1494 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Borrello IM, et al. Granulocyte-macrophage colony-stimulating factor (GM-CSF)-secreting cellular immunotherapy in combination with autologous stem cell transplantation (ASCT) as postremission therapy for acute myeloid leukemia (AML). Blood 114, 1736–1745 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Robinson TM, et al. Pilot trial of K562/GM-CSF whole-cell vaccination in MDS patients. Leuk Lymphoma 59, 2801–2811 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zhang R, Billingsley MM & Mitchell MJ Biomaterials for vaccine-based cancer immunotherapy. J Control Release 292, 256–276 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Curran MA, Montalvo W, Yagita H & Allison JP PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci U S A 107, 4275–4280 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Duraiswamy J, Freeman GJ & Coukos G Therapeutic PD-1 pathway blockade augments with other modalities of immunotherapy T-cell function to prevent immune decline in ovarian cancer. Cancer Res 73, 6900–6912 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sabado RL, Balan S & Bhardwaj N Dendritic cell-based immunotherapy. Cell Res 27, 74–95 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Pitt JM, et al. Resistance Mechanisms to Immune-Checkpoint Blockade in Cancer: Tumor-Intrinsic and -Extrinsic Factors. Immunity 44, 1255–1269 (2016). [DOI] [PubMed] [Google Scholar]
- 102.Pons-Faudoa FP, Ballerini A, Sakamoto J & Grattoni A Advanced implantable drug delivery technologies: transforming the clinical landscape of therapeutics for chronic diseases. Biomed Microdevices 21, 47 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Batra J, et al. Comparison of Skin Staples and Standard Sutures for Closing Incisions After Head and Neck Cancer Surgery: A Double-Blind, Randomized and Prospective Study. J Maxillofac Oral Surg 15, 243–250 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Chew SA & Danti S Biomaterial-Based Implantable Devices for Cancer Therapy. Adv Healthc Mater 6(2017). [DOI] [PubMed] [Google Scholar]
- 105.Leach DG, Young S & Hartgerink JD Advances in immunotherapy delivery from implantable and injectable biomaterials. Acta Biomater 88, 15–31 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Chua CYX, et al. Nanofluidic drug-eluting seed for sustained intratumoral immunotherapy in triple negative breast cancer. J Control Release 285, 23–34 (2018). [DOI] [PubMed] [Google Scholar]
- 107.Liu HC, et al. Potentiating anti-tumor efficacy through radiation and sustained intratumoral delivery of anti-CD40 and anti-PDL1. Int J Radiat Oncol Biol Phys (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Coccia M Deep learning technology for improving cancer care in society: New directions in cancer imaging driven by artificial intelligence. Technology in Society 60(2020). [Google Scholar]
- 109.Hartshorn CM, et al. Nanotechnology Strategies To Advance Outcomes in Clinical Cancer Care. ACS Nano 12, 24–43 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Sharma M, et al. Cancer Nanotechnology-An Excursion on Drug Delivery Systems. Anticancer Agents Med Chem 18, 2078–2092 (2018). [DOI] [PubMed] [Google Scholar]
- 111.Bhat S & Kumar A Biomaterials and bioengineering tomorrow’s healthcare. Biomatter 3(2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lybaert L, et al. Cancer Cell Lysate Entrapment in CaCO3 Engineered with Polymeric TLR-Agonists: Immune-Modulating Microparticles in View of Personalized Antitumor Vaccination. Chemistry of Materials 29, 4209–4217 (2017). [Google Scholar]
- 113.Zhu M, et al. Co-delivery of tumor antigen and dual toll-like receptor ligands into dendritic cell by silicon microparticle enables efficient immunotherapy against melanoma. J Control Release 272, 72–82 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Mattila JP, Mirandola L & Chiriva-Internati M Development of a M cell-targeted microparticulate platform, BSK02, for oral immunization against the ovarian cancer antigen, sperm protein 17. J Biomed Mater Res B Appl Biomater 107, 29–36 (2019). [DOI] [PubMed] [Google Scholar]
- 115.Parenky AC, Akalkotkar A, Mulla NS & D’Souza MJ Harnessing T-cell activity against prostate cancer: A therapeutic microparticulate oral cancer vaccine. Vaccine 37, 6085–6092 (2019). [DOI] [PubMed] [Google Scholar]
- 116.Gross BP, Wongrakpanich A, Francis MB, Salem AK & Norian LA A therapeutic microparticle-based tumor lysate vaccine reduces spontaneous metastases in murine breast cancer. AAPS J 16, 1194–1203 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Dionisi M, et al. Tumor-Derived Microvesicles Enhance Cross-Processing Ability of Clinical Grade Dendritic Cells. Front Immunol 9, 2481 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kuai R, Ochyl LJ, Bahjat KS, Schwendeman A & Moon JJ Designer vaccine nanodiscs for personalized cancer immunotherapy. Nat Mater 16, 489–496 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kuai R, et al. Dual TLR agonist nanodiscs as a strong adjuvant system for vaccines and immunotherapy. J Control Release 282, 131–139 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kuai R, et al. Elimination of established tumors with nanodisc-based combination chemoimmunotherapy. Sci Adv 4, eaao1736 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kuai R, et al. Subcutaneous Nanodisc Vaccination with Neoantigens for Combination Cancer Immunotherapy. Bioconjug Chem 29, 771–775 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wu YL, et al. INSPIRE: A phase III study of the BLP25 liposome vaccine (L-BLP25) in Asian patients with unresectable stage III non-small cell lung cancer. BMC Cancer 11, 430 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Schwendener RA Liposomes as vaccine delivery systems: a review of the recent advances. Ther Adv Vaccines 2, 159–182 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Butts C, et al. Tecemotide (L-BLP25) versus placebo after chemoradiotherapy for stage III non-small-cell lung cancer (START): a randomised, double-blind, phase 3 trial. The Lancet Oncology 15, 59–68 (2014). [DOI] [PubMed] [Google Scholar]
- 125.Wegmann F, et al. Polyethyleneimine is a potent mucosal adjuvant for viral glycoprotein antigens. Nat Biotechnol 30, 883–888 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Song H, et al. Injectable polypeptide hydrogel-based co-delivery of vaccine and immune checkpoint inhibitors improves tumor immunotherapy. Theranostics 9, 2299–2314 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Liu Y, et al. In situ modulation of dendritic cells by injectable thermosensitive hydrogels for cancer vaccines in mice. Biomacromolecules 15, 3836–3845 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Singh NK & Lee DS In situ gelling pH- and temperature-sensitive biodegradable block copolymer hydrogels for drug delivery. J Control Release 193, 214–227 (2014). [DOI] [PubMed] [Google Scholar]
- 129.Sun Z, et al. Injectable Hydrogels Coencapsulating Granulocyte-Macrophage Colony-Stimulating Factor and Ovalbumin Nanoparticles to Enhance Antigen Uptake Efficiency. ACS Appl Mater Interfaces 10, 20315–20325 (2018). [DOI] [PubMed] [Google Scholar]
- 130.Ali OA, et al. Biomaterial-based vaccine induces regression of established intracranial glioma in rats. Pharm Res 28, 1074–1080 (2011). [DOI] [PubMed] [Google Scholar]
- 131.Ali OA, Emerich D, Dranoff G & Mooney DJ In situ regulation of DC subsets and T cells mediates tumor regression in mice. Sci Transl Med 1, 8ra19 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Ali OA, Huebsch N, Cao L, Dranoff G & Mooney DJ Infection-mimicking materials to program dendritic cells in situ. Nat Mater 8, 151–158 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Wurz GT, Kao CJ, Wolf M & DeGregorio MW Tecemotide: an antigen-specific cancer immunotherapy. Hum Vaccin Immunother 10, 3383–3393 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.North S & Butts C Vaccination with BLP25 liposome vaccine to treat non-small cell lung and prostate cancers. Expert Rev Vaccines 4, 249–257 (2005). [DOI] [PubMed] [Google Scholar]
- 135.Chan TA, Wolchok JD & Snyder A Genetic Basis for Clinical Response to CTLA-4 Blockade in Melanoma. N Engl J Med 373, 1984 (2015). [DOI] [PubMed] [Google Scholar]
- 136.McGranahan N, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 351, 1463–1469 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Burris HA, et al. A phase I multicenter study to assess the safety, tolerability, and immunogenicity of mRNA-4157 alone in patients with resected solid tumors and in combination with pembrolizumab in patients with unresectable solid tumors. Journal of Clinical Oncology 37, 2523–2523 (2019). [Google Scholar]
- 138.Wilhelm S, et al. Analysis of nanoparticle delivery to tumours. Nature Reviews Materials 1(2016). [Google Scholar]
- 139.Essa D, Kondiah PPD, Choonara YE & Pillay V The Design of Poly(lactide-co-glycolide) Nanocarriers for Medical Applications. Frontiers in Bioengineering and Biotechnology 8(2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Peres C, et al. Poly(lactic acid)-based particulate systems are promising tools for immune modulation. Acta Biomater 48, 41–57 (2017). [DOI] [PubMed] [Google Scholar]
- 141.Rosalia RA, et al. CD40-targeted dendritic cell delivery of PLGA-nanoparticle vaccines induce potent anti-tumor responses. Biomaterials 40, 88–97 (2015). [DOI] [PubMed] [Google Scholar]
- 142.Mueller M, Schlosser E, Gander B & Groettrup M Tumor eradication by immunotherapy with biodegradable PLGA microspheres--an alternative to incomplete Freund’s adjuvant. Int J Cancer 129, 407–416 (2011). [DOI] [PubMed] [Google Scholar]
- 143.Schliehe C, et al. CD8- dendritic cells and macrophages cross-present poly(D,L-lactate-co-glycolate) acid microsphere-encapsulated antigen in vivo. J Immunol 187, 2112–2121 (2011). [DOI] [PubMed] [Google Scholar]
- 144.Silva JM, Videira M, Gaspar R, Préat V & Florindo HF Immune system targeting by biodegradable nanoparticles for cancer vaccines. J Control Release 168, 179–199 (2013). [DOI] [PubMed] [Google Scholar]
- 145.Fang RH, et al. Cancer Cell Membrane-Coated Nanoparticles for Anticancer Vaccination and Drug Delivery. Nano Letters 14, 2181–2188 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Shi GN, et al. Enhanced antitumor immunity by targeting dendritic cells with tumor cell lysate-loaded chitosan nanoparticles vaccine. Biomaterials 113, 191–202 (2017). [DOI] [PubMed] [Google Scholar]
- 147.Zhang R, Leeper CN, Wang X, White TA & Ulery BD Immunomodulatory vasoactive intestinal peptide amphiphile micelles. Biomater Sci 6, 1717–1722 (2018). [DOI] [PubMed] [Google Scholar]
- 148.Iranpour S, Nejati V, Delirezh N, Biparva P & Shirian S Enhanced stimulation of anti-breast cancer T cells responses by dendritic cells loaded with poly lactic-co-glycolic acid (PLGA) nanoparticle encapsulated tumor antigens. J Exp Clin Cancer Res 35, 168 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Kroll AV, et al. Nanoparticulate Delivery of Cancer Cell Membrane Elicits Multiantigenic Antitumor Immunity. Adv Mater 29(2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.de Lázaro I & Mooney DJ A nanoparticle’s pathway into tumours. Nature Materials 19, 486–487 (2020). [DOI] [PubMed] [Google Scholar]
- 151.Challenging paradigms in tumour drug delivery. Nat Mater 19, 477 (2020). [DOI] [PubMed] [Google Scholar]
- 152.Dhas NL, Kudarha RR, Acharya NS & Acharya SR Polymeric Immunonanoparticles Mediated Cancer Therapy: Versatile Nanocarriers for Cell-Specific Cargo Delivery. Crit Rev Ther Drug Carrier Syst 35, 1–64 (2018). [DOI] [PubMed] [Google Scholar]
- 153.Joshi VB, Geary SM & Salem AK Biodegradable particles as vaccine delivery systems: size matters. Aaps j 15, 85–94 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.de Lazaro I & Mooney DJ A nanoparticle’s pathway into tumours. Nat Mater 19, 486–487 (2020). [DOI] [PubMed] [Google Scholar]
- 155.Wahlund CJE, et al. Exosomes from antigen-pulsed dendritic cells induce stronger antigen-specific immune responses than microvesicles in vivo. Scientific Reports 7, 17095 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Pitt JM, et al. Dendritic cell–derived exosomes for cancer therapy. The Journal of Clinical Investigation 126, 1224–1232 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Xu Z, Zeng S, Gong Z & Yan Y Exosome-based immunotherapy: a promising approach for cancer treatment. Molecular Cancer 19, 160 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Morishita M, Takahashi Y, Matsumoto A, Nishikawa M & Takakura Y Exosome-based tumor antigens-adjuvant co-delivery utilizing genetically engineered tumor cell-derived exosomes with immunostimulatory CpG DNA. Biomaterials 111, 55–65 (2016). [DOI] [PubMed] [Google Scholar]
- 159.Morishita M, Takahashi Y, Nishikawa M, Ariizumi R & Takakura Y Enhanced Class I Tumor Antigen Presentation via Cytosolic Delivery of Exosomal Cargos by Tumor-Cell-Derived Exosomes Displaying a pH-Sensitive Fusogenic Peptide. Mol Pharm 14, 4079–4086 (2017). [DOI] [PubMed] [Google Scholar]
- 160.Markov O, Oshchepkova A & Mironova N Immunotherapy Based on Dendritic Cell-Targeted/-Derived Extracellular Vesicles-A Novel Strategy for Enhancement of the Anti-tumor Immune Response. Frontiers in pharmacology 10, 1152–1152 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Naseri M, Bozorgmehr M, Zöller M, Ranaei Pirmardan E & Madjd Z Tumor-derived exosomes: the next generation of promising cell-free vaccines in cancer immunotherapy. OncoImmunology 9, 1779991 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Zenobia C & Hajishengallis G Basic biology and role of interleukin-17 in immunity and inflammation. Periodontol 2000 69, 142–159 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Bamrungsap S, et al. Nanotechnology in therapeutics: a focus on nanoparticles as a drug delivery system. Nanomedicine (Lond) 7, 1253–1271 (2012). [DOI] [PubMed] [Google Scholar]
- 164.Castillo RR & Vallet-Regi M Functional Mesoporous Silica Nanocomposites: Biomedical applications and Biosafety. Int J Mol Sci 20(2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Castillo RR, Lozano D & Vallet-Regi M Mesoporous Silica Nanoparticles as Carriers for Therapeutic Biomolecules. Pharmaceutics 12(2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Wang G, et al. Robust vaccine formulation produced by assembling a hybrid coating of polyethyleneimine-silica. Chem Sci 7, 1753–1759 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Heikkila T, et al. Mesoporous silica material TUD-1 as a drug delivery system. Int J Pharm 331, 133–138 (2007). [DOI] [PubMed] [Google Scholar]
- 168.Cha BG, Jeong JH & Kim J Extra-Large Pore Mesoporous Silica Nanoparticles Enabling Co-Delivery of High Amounts of Protein Antigen and Toll-like Receptor 9 Agonist for Enhanced Cancer Vaccine Efficacy. ACS Cent Sci 4, 484–492 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Koshy ST & Mooney DJ Biomaterials for enhancing anti-cancer immunity. Curr Opin Biotechnol 40, 1–8 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Sozzani S, Del Prete A & Bosisio D Dendritic cell recruitment and activation in autoimmunity. J Autoimmun 85, 126–140 (2017). [DOI] [PubMed] [Google Scholar]
- 171.Hailemichael Y, et al. Persistent antigen at vaccination sites induces tumor-specific CD8(+) T cell sequestration, dysfunction and deletion. Nat Med 19, 465–472 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Martinon S, et al. Chemical and Immunological Characteristics of Aluminum-Based, Oil-Water Emulsion, and Bacterial-Origin Adjuvants. J Immunol Res 2019, 3974127 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Temizoz B, Kuroda E & Ishii KJ Vaccine adjuvants as potential cancer immunotherapeutics. Int Immunol 28, 329–338 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Batista-Duharte A, Lindblad EB & Oviedo-Orta E Progress in understanding adjuvant immunotoxicity mechanisms. Toxicol Lett 203, 97–105 (2011). [DOI] [PubMed] [Google Scholar]
- 175.Eisenbarth SC, Colegio OR, O’Connor W, Sutterwala FS & Flavell RA Crucial role for the Nalp3 inflammasome in the immunostimulatory properties of aluminium adjuvants. Nature 453, 1122–1126 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Wang X, et al. The effect of fiber size and pore size on cell proliferation and infiltration in PLLA scaffolds on bone tissue engineering. J Biomater Appl 30, 1545–1551 (2016). [DOI] [PubMed] [Google Scholar]
- 177.Murphy CM, Duffy GP, Schindeler A & O’Brien F J Effect of collagen-glycosaminoglycan scaffold pore size on matrix mineralization and cellular behavior in different cell types. J Biomed Mater Res A 104, 291–304 (2016). [DOI] [PubMed] [Google Scholar]
- 178.Tytgat L, et al. Evaluation of 3D Printed Gelatin-Based Scaffolds with Varying Pore Size for MSC-Based Adipose Tissue Engineering. Macromol Biosci 20, e1900364 (2020). [DOI] [PubMed] [Google Scholar]
- 179.Kaczanowska S, Joseph AM & Davila E TLR agonists: our best frenemy in cancer immunotherapy. J Leukoc Biol 93, 847–863 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Batista P, Castro PM, Madureira AR, Sarmento B & Pintado M Recent insights in the use of nanocarriers for the oral delivery of bioactive proteins and peptides. Peptides 101, 112–123 (2018). [DOI] [PubMed] [Google Scholar]
- 181.Calori IR, Braga G, de Jesus P.d.C.C., Bi H & Tedesco ACJEPJ Polymer scaffolds as drug delivery systems. 129, 109621 (2020). [Google Scholar]
- 182.Baumann B, et al. Control of Nanoparticle Release Kinetics from 3D Printed Hydrogel Scaffolds. Angew Chem Int Ed Engl 56, 4623–4628 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Nguyen TL, Cha BG, Choi Y, Im J & Kim J Injectable dual-scale mesoporous silica cancer vaccine enabling efficient delivery of antigen/adjuvant-loaded nanoparticles to dendritic cells recruited in local macroporous scaffold. Biomaterials 239, 119859 (2020). [DOI] [PubMed] [Google Scholar]
- 184.Ali OA, et al. The efficacy of intracranial PLG-based vaccines is dependent on direct implantation into brain tissue. J Control Release 154, 249–257 (2011). [DOI] [PubMed] [Google Scholar]
- 185.Dolgin E Cancer vaccines: Material breach. Nature 504, S16–17 (2013). [DOI] [PubMed] [Google Scholar]
- 186.Cirillo G, Spizzirri UG, Curcio M, Nicoletta FP & Iemma F Injectable Hydrogels for Cancer Therapy over the Last Decade. Pharmaceutics 11(2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Espona-Noguera A, et al. Tunable injectable alginate-based hydrogel for cell therapy in Type 1 Diabetes Mellitus. Int J Biol Macromol 107, 1261–1269 (2018). [DOI] [PubMed] [Google Scholar]
- 188.Escobar-Chavez JJ, et al. Applications of thermo-reversible pluronic F-127 gels in pharmaceutical formulations. J Pharm Pharm Sci 9, 339–358 (2006). [PubMed] [Google Scholar]
- 189.Diniz IM, et al. Pluronic F-127 hydrogel as a promising scaffold for encapsulation of dental-derived mesenchymal stem cells. J Mater Sci Mater Med 26, 153 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Rafael D, et al. Sterilization Procedure for Temperature-Sensitive Hydrogels Loaded with Silver Nanoparticles for Clinical Applications. Nanomaterials (Basel) 9(2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Hyun H, et al. In vitro and in vivo release of albumin using a biodegradable MPEG-PCL diblock copolymer as an in situ gel-forming carrier. Biomacromolecules 8, 1093–1100 (2007). [DOI] [PubMed] [Google Scholar]
- 192.Shih TY, et al. Injectable, Tough Alginate Cryogels as Cancer Vaccines. Adv Healthc Mater 7, e1701469 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Riganti C, et al. Bromodomain inhibition exerts its therapeutic potential in malignant pleural mesothelioma by promoting immunogenic cell death and changing the tumor immune-environment. Oncoimmunology 7, e1398874 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Meng H, et al. Aspect ratio determines the quantity of mesoporous silica nanoparticle uptake by a small GTPase-dependent macropinocytosis mechanism. ACS Nano 5, 4434–4447 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Cong VT, Gaus K, Tilley RD & Gooding JJ Rod-shaped mesoporous silica nanoparticles for nanomedicine: recent progress and perspectives. Expert Opin Drug Deliv 15, 881–892 (2018). [DOI] [PubMed] [Google Scholar]
- 196.Ferrati S, et al. Leveraging nanochannels for universal, zero-order drug delivery in vivo. J Control Release 172, 1011–1019 (2013). [DOI] [PubMed] [Google Scholar]
- 197.Di Trani N, et al. Nanofluidic microsystem for sustained intraocular delivery of therapeutics. Nanomedicine 16, 1–9 (2019). [DOI] [PubMed] [Google Scholar]
- 198.Paez-Mayorga J, et al. Enhanced In Vivo Vascularization of 3D-Printed Cell Encapsulation Device Using Platelet-Rich Plasma and Mesenchymal Stem Cells. Adv Healthc Mater 9, e2000670 (2020). [DOI] [PubMed] [Google Scholar]
- 199.Paez-Mayorga J, et al. Neovascularized implantable cell homing encapsulation platform with tunable local immunosuppressant delivery for allogeneic cell transplantation. Biomaterials 257, 120232 (2020). [DOI] [PubMed] [Google Scholar]
- 200.Pons-Faudoa FP, et al. Viral load Reduction in SHIV-Positive Nonhuman Primates via Long-Acting Subcutaneous Tenofovir Alafenamide Fumarate Release from a Nanofluidic Implant. Pharmaceutics 12(2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Farina M, et al. Transcutaneously refillable, 3D-printed biopolymeric encapsulation system for the transplantation of endocrine cells. Biomaterials 177, 125–138 (2018). [DOI] [PubMed] [Google Scholar]
- 202.Viswanath DI, et al. Engineered implantable vaccine platform for continuous antigen-specific immunomodulation Biomaterials (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Chen DS & Mellman I Oncology meets immunology: the cancer-immunity cycle. Immunity 39, 1–10 (2013). [DOI] [PubMed] [Google Scholar]
- 204.Chen DS & Mellman I Elements of cancer immunity and the cancer-immune set point. Nature 541, 321–330 (2017). [DOI] [PubMed] [Google Scholar]
- 205.Demaria S & Formenti SC The abscopal effect 67 years later: from a side story to center stage. Br J Radiol 93, 20200042 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Zhang X & Niedermann G Abscopal Effects With Hypofractionated Schedules Extending Into the Effector Phase of the Tumor-Specific T-Cell Response. Int J Radiat Oncol Biol Phys 101, 63–73 (2018). [DOI] [PubMed] [Google Scholar]
- 207.Ali OA, Lewin SA, Dranoff G & Mooney DJ Vaccines Combined with Immune Checkpoint Antibodies Promote Cytotoxic T-cell Activity and Tumor Eradication. Cancer Immunol Res 4, 95–100 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Chiappori AA, Soliman H, Janssen WE, Antonia SJ & Gabrilovich DI INGN-225: a dendritic cell-based p53 vaccine (Ad.p53-DC) in small cell lung cancer: observed association between immune response and enhanced chemotherapy effect. Expert Opin Biol Ther 10, 983–991 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Maleki Vareki S High and low mutational burden tumors versus immunologically hot and cold tumors and response to immune checkpoint inhibitors. J Immunother Cancer 6, 157 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Kawashima A, et al. Immunological classification of renal cell carcinoma patients based on phenotypic analysis of immune check-point molecules. Cancer Immunol Immunother 67, 113–125 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Yakirevich E & Patel NR Tumor mutational burden and immune signatures interplay in renal cell carcinoma. Ann Transl Med 8, 269 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Turajlic S, et al. Insertion-and-deletion-derived tumour-specific neoantigens and the immunogenic phenotype: a pan-cancer analysis. Lancet Oncol 18, 1009–1021 (2017). [DOI] [PubMed] [Google Scholar]
- 213.Klebanoff CA, Acquavella N, Yu Z & Restifo NP Therapeutic cancer vaccines: are we there yet? Immunol Rev 239, 27–44 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Mestas J & Hughes CC Of mice and not men: differences between mouse and human immunology. J Immunol 172, 2731–2738 (2004). [DOI] [PubMed] [Google Scholar]
- 215.Henrich J, Heine SJ & Norenzayan A The weirdest people in the world? Behav Brain Sci 33, 61–83; discussion 83–135 (2010). [DOI] [PubMed] [Google Scholar]
- 216.Schooling CM Selection bias in population-representative studies? A commentary on Deaton and Cartwright. Soc Sci Med 210, 70 (2018). [DOI] [PubMed] [Google Scholar]
- 217.Uschner D, Hilgers RD & Heussen N The impact of selection bias in randomized multi-arm parallel group clinical trials. PLoS One 13, e0192065 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Oh J, et al. Phase II study of Vigil(R) DNA engineered immunotherapy as maintenance in advanced stage ovarian cancer. Gynecol Oncol 143, 504–510 (2016). [DOI] [PubMed] [Google Scholar]
- 219.Tondini E, et al. A poly-neoantigen DNA vaccine synergizes with PD-1 blockade to induce T cell-mediated tumor control. Oncoimmunology 8, 1652539 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Dey S, et al. Peptide vaccination directed against IDO1-expressing immune cells elicits CD8(+) and CD4(+) T-cell-mediated antitumor immunity and enhanced anti-PD1 responses. J Immunother Cancer 8(2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Li W, Joshi MD, Singhania S, Ramsey KH & Murthy AK Peptide Vaccine: Progress and Challenges. Vaccines (Basel) 2, 515–536 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Copier J & Dalgleish A Whole-cell vaccines: A failure or a success waiting to happen? Curr Opin Mol Ther 12, 14–20 (2010). [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
As this is a review paper, we did not generate any raw or processed data in preparation of this manuscript.