

Abstract
Background aims.
Ex vivo expansion of natural killer (NK) cells is a strategy to produce large numbers of these effector cells for immunotherapy. However, the transfer of bench-top expansion protocols to clinically applicable methods is challenging for NK cell–based therapy because of regulatory aspects and scale-up issues. Therefore, we developed an automated, large-scale NK cell expansion process.
Methods.
Enriched NK cells were expanded with interleukin-2 and irradiated clinical-grade Epstein-Barr virus–transformed lymphoblastoid feeder cells with the use of an automated system in comparison to manual expansion, and the cells were investigated for their functionality, phenotype and gene expression.
Results.
Automated expansion resulted in a mean 850-fold expansion of NK cells by day 14, yielding 1.3 (±0.9) × 109 activated NK cells. Automatically and manually produced NK cells were comparable in target cell lysis, degranulation and production of interferon-γ and tumor necrosis factor-α and had similar high levels of antibody-dependent cellular cytotoxicity against rituximab-treated leukemic cells. NK cells after automated or manual expansion showed similar gene expression and marker profiles. However, expanded NK cells differed significantly from primary NK cells including upregulation of the functional relevant molecules TRAIL and FasL and NK cell–activating receptors NKp30, NKG2D and DNAM-1. Neither automatically nor manually expanded NK cells showed reduced telomere length indicative of a conserved proliferative potential.
Conclusions.
We established an automated method to expand high numbers of clinical-grade NK cells with properties similar to their manually produced counterparts. This automated process represents a highly efficient tool to standardize NK cell processing for therapeutic applications.
Keywords: automation, CliniMACS Prodigy, immunotherapy, natural killer cells, NK cell expansion, NK cell therapy
Introduction
Since the discovery and description of natural killer (NK) cells in 1975, because of their ability to recognize and lyse tumor cells [1], it became more and more clear how NK cells are regulated and recognize their targets and that they play a role in immune surveillance of cancer [2]. Parallel to the proceeding understanding of NK cell biology, they were evaluated in clinical settings [3]. Thus far, the transfer of allogeneic NK cells in addition to standard hematopoietic stem cell transplantation in the treatment of acute myeloid leukemia has proven its feasibility and safety [4]. Furthermore, chemotherapy combined with NK cell infusions has been tested for cancer treatment and seems promising [5–9]. Encouraging results were obtained for acute myeloid leukemia therapy in a study of 10 pediatric patients that reported long-term survival, with all children remaining in remission in a 2- to 4.2-year follow-up [10].
Despite notable progress and development of different strategies to optimize the therapeutic value of NK cells [11], NK cell–based immunotherapy in general had to deal with several challenges, which, until now, limited its efficacy. One major hurdle is to apply sufficient numbers of activated NK cells [12]. An option to increase the number and function of donor-derived NK cells is to expand and activate the cells ex vivo before transfer to the patient. Therefore, NK cell expansion protocols are required that not only efficiently induce NK cell proliferation and activate NK cell function but also fulfill regulatory requirements for safety. Furthermore, compounds used during NK cell expansion must not be harmful to the patient. Different protocols have recently been established that claim to meet these requirements and allow the production of NK cells of clinical-grade quality [13–16].
However, the next challenge is the transfer of these protocols to clinical scale in a manageable, Good Manufacturing Practice (GMP)-compliant way, and cultivation in closed systems such as cell culture bags and G-Rex containers have demonstrated their applicability in this context [17–20]. However, a multitude of necessary hands-on steps complicate the routine use of these scaled-up manual approaches as a standard therapy. In contrast, partial automation of the cell cultivation by use of a bioreactor has shown improved achievability of NK cell production in large scale [16,21,22]. Nevertheless, a complete automation of the activation and expansion procedure of NK cells in production scale remains challenging. Still, fully automated systems are needed to ensure a defined, highly standardized and operator-independent manufacturing procedure that meets clinical requirements at its best. A novel solution to this goal is Miltenyi’s recently introduced technology to perform automated clinical-scale cell processing by use of centrifugation, magnetic cell separation and cell cultivation within a closed system [23]. With this system as a platform, we have developed and optimized an automated NK cell expansion process. The procedure is based on an existing expansion protocol for clinical applications that utilizes an irradiated clinical-grade Epstein-Barr virus–transformed lymphoblastoid cell line (EBV-LCL) as feeder cells to trigger NK cell proliferation and yields pure and highly activated NK cells [13,24]. In this study, we describe the automated expansion procedure as a novel tool for immunotherapy and evaluated the resulting NK cell product on a phenotypic and transcriptomic level as well as for function in comparison to NK cells that have been expanded manually.
Methods
Cells and cell lines
Primary NK cells were obtained from healthy donor buffy coats (Klinikum Dortmund) or leukapheresis products (Hannover Medical School, Hannover, Germany, or Institut für Klinische Trans-fusionsmedizin und Immungenetik Ulm Gemein-nützige GmbH, Ulm, Germany). The EBV-LCL (SMI-EBV-LCL) line was provided by Dr Richard W. Childs (National Heart, Lung and Blood Institute, National Institutes of Health, Bethesda, MD, USA). Human T-cell leukemia cell line 1301 was obtained from Sigma-Aldrich, and K562, Raji and Daudi cell lines were purchased from German Collection of Microorganisms and Cell Cultures (DSMZ, Braunschweig, Germany). All cell lines were maintained in Roswell Park Memorial Institute (RPMI) 1640 (Biowest) supplemented with 10% fetal bovine serum (Biochrom) and 2 mmol/L L-glutamine (PAA).
Antibodies and flow cytometric analysis
Rituximab was from Roche. All other antibodies were obtained from Miltenyi: CD56 (REAfinity antibody [REA]196) was conjugated with allophy-cocyanin (APC)-Vio770. CD3 (BW264/56) was conjugated with VioBlue. CD45 (5B1) was conjugated with VioGreen. Interferon (IFN)-γ (LT27:295), CD57 (TB03), CD159a (REA110), CD244 (REA112), NKp80 (4A4.D10) and CD158e (DX9) were conjugated with fluorescein isothiocyanate (FITC). Tumor necrosis factor (TNF)-α (cA2), CD25 (4E3), CD62L (145/15), CD159c (REA205), CD178 (NOK-1), CD336 (2.29) and CD158b (DX27) were conjugated with phycoerythrin. CD16 (VEP13), CD94 (REA113), CD226 (DX11), CD253 (RIK-2.1), CD314 (BAT221), CX3CR1 (2A9–1), CD158a (REA284) and CD107a (H4A3) were conjugated with APC. NK cells were stained as indicated according to the product manual and analyzed by use of the MACSQuant Analyzer X and MACSQuantify 2.5 software. Dead cells were excluded from the analysis by means of propidium iodide staining. Mouse immunoglobulin (Ig)G1, IgG2a, IgG2b, IgM or REAs conjugated with the respective dyes were used as isotype controls.
NK cell separation
For NK cell isolation from buffy coats, peripheral blood mononuclear cell (PBMC) preparation was performed by means of standard density-gradient centrifugation with the use of Pancoll (PAN-Biotec). NK cells were enriched from PBMCs by means of CD3 depletion with the use of human CD3 MicroBeads (Miltenyi) followed by CD56 enrichment with the use of use of human CD56 MicroBeads (Miltenyi), according to the user manual. For leukapheresis products, the CliniMACS Prodigy instrument and TS310 were used for automated T-cell receptor (TCR)-α/β-CD19 depletion according to the application sheet. Further automated CD56 enrichment with the use of the instrument was achieved by means of Program Enrichment 1, CliniMACS CD56 Reagent and tubing set TS100.
NK cell expansion
Isolated NK cells were cultivated in TexMACS GMP Medium (Miltenyi) supplemented with 5% human serum type AB (Life Technologies) and 500 U/mL of Proleukin S (Novartis), either together with 100 Gy–irradiated EBV-LCL at a ratio of 1:20 and a starting concentration of 5.25 × 105 total cells/mL or without feeder cells with a seeding density of 5 × 105 cells/mL. NK cell density was checked at days 7, 9, 12 and 14 by means of staining and counting viable CD3−/CD56+ cells with the use of the MACSQuant Analyzer X, and medium was replenished. Manual expansion in T75 flasks was started with 15 mL, and cells were diluted to 5 to 8 × 105 NK cells/mL at the previously mentioned time points. Automated expansion was performed by means of the CliniMACS Prodigy instrument with the use of tubing set TS730, and programs that were generated with a flexible programming suite provided with the instrument. In short, a clinical bag containing the starting cell material was connected to the tubing set through sterile welding, and the cells were transferred automatically to the CentriCult Unit of the instrument. A cultivation program for temperature control and repeated input of CO2 maintained the cultivation conditions comparable to manual cultivation by means of an incubator at 37°C and 5% CO2. The cultivation was initiated with 70 mL, and the volume was increased to 140 mL at day 7 and 280 mL at day 9 by pumping in fresh medium from a reservoir bag. Medium (210 mL) was exchanged at day 12 while the cultivated cells were retained in the CentriCult Unit by centrifugation. Until day 7, the cells were cultivated in a static culture, and, after day 7, short centrifugation intervals of 1 to 2 seconds were used every 30 to 60 seconds to gently mix the cells, allowing high–cell density cell culture. Transfer of small volumes of the cell suspension to a sterile sample pouch was used for sampling during the process.
Cytokine production and degranulation
Cytokine production and degranulation were analyzed by means of flow cytometry. NK cells (2 × 104) per well were seeded in 96-well round-bottom plates alone, as a control, or together with 1 × 104 K562 cells and cultivated for 4 h in RPMI1640 supplemented with Monensin (eBioscience) and CD107a-APC (Miltenyi) according to the user manuals. After labeling with Fixable Aqua Dead Stain (Life Technologies) to exclude dead cells during the analysis, the cells were fixed, permeabilized and stained for IFN-γ, TNF-α and CD56 (Inside Stain Kit, Miltenyi). The latter marker was used to discriminate NK cells from co-cultivated K562 target cells.
Killing assay and antibody-dependent cellular cytotoxicity
Target cell killing of K562, Raji and Daudi cell lines was analyzed by means of a flow cytometry–based assay. Target cells were labeled with CellTrace Violet (Life Technologies) according to the user manual. Labeled target cells (1 × 104) per well were seeded in 96-well round-bottom plates and cultivated alone, as a control, or with NK cells at different NK-to-target ratios as indicated. To analyze antibody-dependent cytotoxicity, 1 μg/mL of rituximab was added directly to the co-culture of NK cells and target cells. After 4 h of incubation, plates were stored at 4°C for 0.5 to 2 h before the viable CellTrace Violet–positive target cells were quantified by use of the MACSQuant Analyzer X. The difference between the number of viable target cells in samples with NK cells and samples without NK cells was defined as killed target cells.
Telomere length analysis
Telomere length was measured by means of flow cytometry with the use of a commercial Telomere PNA Kit/FITC (Dako) according to the user manual. Detection of the samples labeled with fluorescein-conjugated peptide nucleic acid was done with the use of the MACSQuant Analyzer X. As recommended, the cell line 1301 was used as internal control and relative telomere length (RTL) was calculated as follows:
RTL = (mean FL1 sample cells with probe – mean FL1 sample cells without probe) × 2 × 100/(mean FL1 control cells FL1 control cells with probe–mean without probe).
Gene expression
For total RNA isolation, 1 × 106 unexpanded and expanded NK cells per sample were lysed in RA1 buffer (Machery-Nagel) and stored at −20°C. To ensure high purity of the starting material, only CD3-depleted and CD56-enriched NK cells were considered for micro-array analysis. Human total RNA was isolated with the use of the NucleoSpin RNA kit (Macherey-Nagel). RNA quality and integrity were determined with the use of the Agilent RNA 6000 Nano Kit on the Agilent 2100 Bio-analyzer (Agilent Technologies), and RNA integrity numbers were between 8.1 and 10. According to published data, RNA integrity number >6 is of sufficient quality for gene expression profiling experiments [25]. RNA was quantified by measurement of A260 nm on the ND-1000 Spectrophotometer (NanoDrop Technologies).
RNA amplification and labeling
Total RNA from unexpanded and expanded NK cell samples (100 ng each) was used for the amplification and labeling step with the use of the Agilent Low Input Quick Amp Labeling Kit (Agilent Technologies). Yields of complementary RNA measured with the ND-1000 Spectrophotometer (NanoDrop Technologies) were in all cases >5 μg, and dye incorporation rates were in all cases >15fmol/ng.
Hybridization of agilent whole human genome oligo–micro-arrays
Hybridization was performed according to the Agilent 60-mer oligo–micro-array processing protocol with the use of the Agilent Gene Expression Hybridization Kit (Agilent Technologies). Briefly, 600 ng of Cy3-labeled fragmented complementary RNA in hybridization buffer was hybridized overnight (17 h, 65°C) to Agilent SurePrint G3 Human Gene Expression Microarrays 8 × 60K v2 with the use of Agilent’s recommended hybridization chamber and oven. Fluorescence signals of the hybridized Agilent Microarrays were detected with the use of Agilent’s Microarray Scanner System (G2505C, Agilent Technologies). The Agilent Feature Extraction Software (FES 10.7.3.1) was used to read out and process the micro-array image files.
Pre-processing of micro-array data
Raw intensity data were extracted from Feature Extraction output files for Agilent Whole Human Genome Oligo Microarrays 8X60K v2 (Agilent Technologies, Inc) with the use of Rosetta Resolver software (Rosetta, Inpharmatics, LLC). All subsequent calculations were performed with the use of R/Bioconductor and software packages therein [26,27]. Background-corrected intensity values were normalized between arrays by means of quantile normalization [28]. Reliable signal intensities were considered at P ≤ 0.01, according to the Rosetta error model [29]. Log2-transformed normalized intensity values were used for subsequent statistical analysis.
The data set has been uploaded to the NCBI GEO public database: record No. GSE62654.
Statistics and data analysis
Apart from micro-array data analysis, statistical comparisons were performed with the use of unpaired or paired Student’s t-test, as indicated. In the micro-array data sets, significant expression differences were determined per reporter between the following sample groups: freshly isolated NK cells (day 0) and NK cells expanded for 14 days by the automated system CliniMACS Prodigy (P); manually with EBV-LCL feeder cells in T75 flasks (T) and manually without feeder cells only with the use of IL-2–containing media (I). The analysis of variance test with repeated-measurements design was applied by fitting a linear mixed-effects model (random effect: individual donors) on the normalized log2 intensity data. Correction for multiple testing occurred by use of the method of Benjamini & Hochberg (B-H). Further pairwise group comparisons were performed by means of Tukey’s honestly significant differences post hoc test. The following selection criteria were applied: adjusted analysis of variance P values ≤0.05, Tukey P values ≤0.05 and median fold changes ≥2 or ≤−2. Reporters with a detection P value (flag) >0.01 for >3 of 6 samples per sample group were excluded because of insufficient signal reliability. The expression profiles of all reporters with differential gene expression in at least one of the pairwise comparisons was hierarchically clustered (Euclidean distance, complete linkage) and displayed in heat map images centered to the median value per reporter (TM4 suite, MeV_4_8_1) [30]. Calculations were performed with the use of Excel (Microsoft Office Inc) or R/bioconductor [R version 3.1.1 (2014–07–10)]. Because some of the reporters covered on the micro-array platform represent long non-coding RNAs or map to alternative transcripts of the same gene, functional grouping analysis was performed on the gene level with the use of QIAGEN’s Ingenuity Pathway Analysis annotation tools (IPA, QIAGEN Redwood City, www.qiagen.com/ingenuity). Significantly enriched functional groups were identified by use of default settings and a B-H multiple testing P-value cutoff of 0.025.
Results
The production of therapeutic effector cells in a standardized, GMP-compliant and efficient way is challenging for several clinical applications. To minimize the requirement for hands-on technician time and the possibility of contamination, we developed a fully automated cell cultivation process with the use of the CliniMACS Prodigy system and applied it to an NK cell expansion protocol that makes use of IL-2 and EBV-LCL feeder cells to induce NK cell activation and proliferation. The automation covered all cultivation steps that were needed within 14 days, including medium change, gentle cell mixing at high cell densities and sampling for analysis during the expansion process.
Automated or manual NK cell cultivation results in comparable NK cell fold expansion
Purified NK cells from buffy coat samples of 10 donors were either cultivated manually in T-75 flasks or by use of the automated system resulting in comparable increase in NK cell numbers over time (Figure 1A). Automated or manual EBV-LCL feeder cell line–based expansion for 2 weeks led to 850 ± 509-fold or 1344 ± 1135-fold fold increases in NK cell numbers, respectively, with high variability between different donors. To further investigate the effect and necessity to use the feeder cell line, we also cultivated NK cells manually in T-75 flasks without EBV-LCL, representing a long-term expansion with IL-2–containing media alone, which has been used in clinical trials. Without EBV-LCL, only 14 ± 13-fold expansion was achieved, demonstrating the limitation of this approach and the strong proliferation-inducing effect of the LCL cell line on NK cells. On average, starting with only 1.5 × 106 NK cells, a number typically obtained from 20 mL of whole blood, a substantial number (mean, 1.3 × 109) of NK cells could be produced within 14 days by a single run of the automated process. Both the automated and manual expansion resulted in a highly pure NK cell product (>99% CD3−/CD56+), and no T or B cells could be detected.
Figure 1.
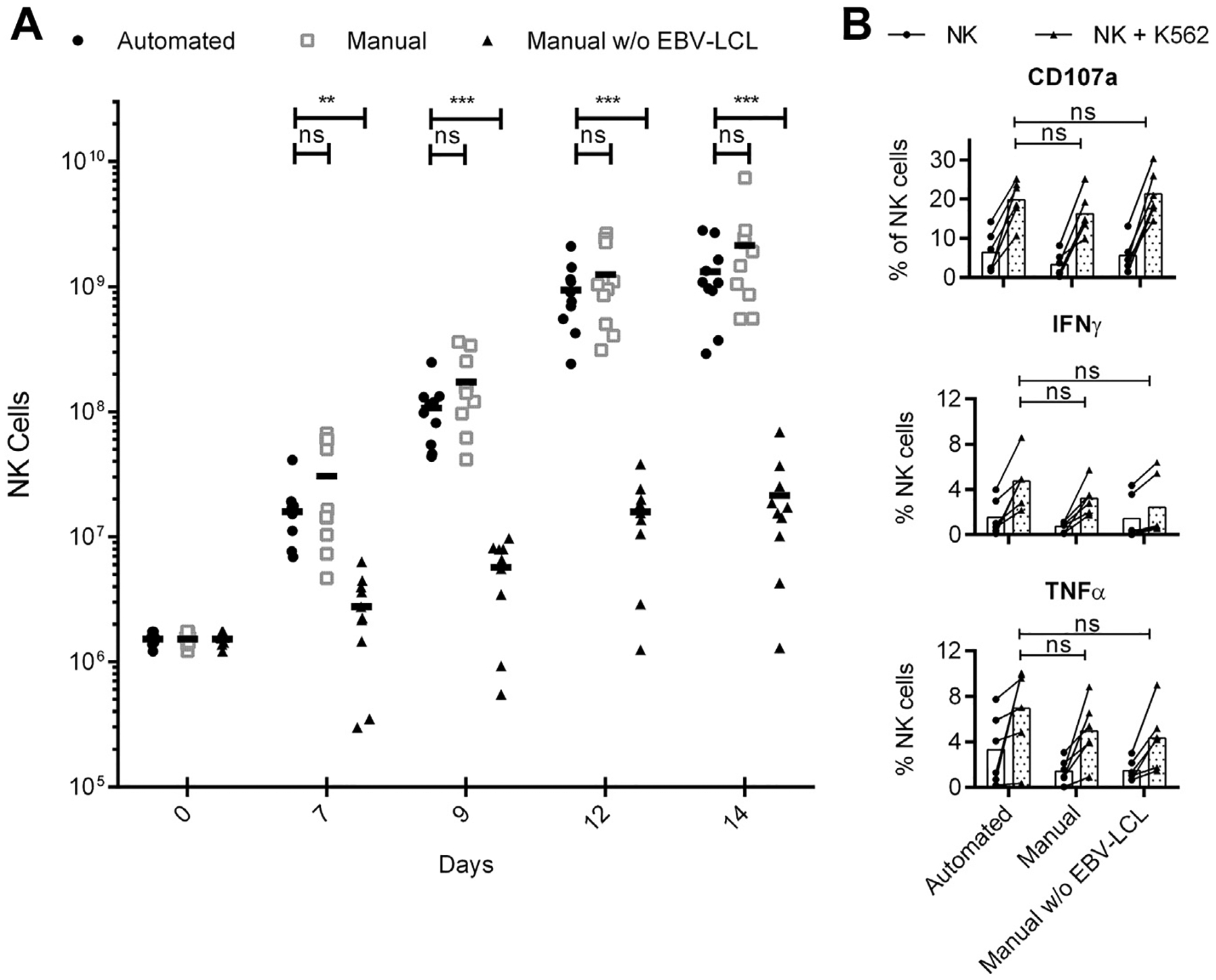
Expansion kinetics and effector functions of differentially expanded NK cells. (A) Automated EBV-LCL–based expansion of NK cells (circles) in comparison to manual NK cell expansion in T flasks with (squares) or without (triangles) EBV-LCL. NK cell numbers displayed for manual expansion are theoretical and were calculated by the NK cell fold expansion obtained in T flasks multiplied by the same starting NK cell number as in the automated approach. (B) Differentially expanded NK cells were tested for their reactivity by staining for CD107a, IFN-γ and TNF-α before (white bars) and after (gray bars) stimulation with K562 target cells. NK cells from 10 (A) or six (B) donors were analyzed; displayed are mean values and P values for paired Student’s t-test, with P < 0.05 considered significant.
Automatically and manually produced NK cells have similar functionality
The effector functions of the differentially expanded NK cells on stimulation with K562 target cells revealed no differences in the production of the pro-inflammatory cytokines IFN-γ and TNF-α and similar levels of degranulation as an indicator of NK cell cytotoxic function (Figure 1B). We further evaluated NK cell cytotoxicity against the human leukemic cell lines K562, Raji and Daudi (Figure 2A). NK cells obtained by means of the automated or the manual approach showed comparable cytotoxicity against all three target cell lines in a dose-dependent manner, although significant differences in the cytotoxic intensity were observed between different donors. The level of killing tended to be higher compared with NK cells that had been expanded with IL-2 only and without EBV-LCL, but this trend only achieved statistical significance at a 10:1 effector-to-target ratio against K562 cells. Next, we tested whether the killing of target cells could be enhanced by a monoclonal antibody because NK cells are able to induce antibody-dependent cellular cytotoxicity through the Fc receptor CD16. It is known that NK cell activation induces CD16 downregulation [31], and we observed lower levels of CD16 on the surface of the activated and expanded NK cells compared with freshly isolated cells (Figure 2C,D). Still, rituximab significantly increased the NK cell–mediated killing of CD20-positive Raji or Daudi cells for both automatically and manually obtained NK cells, proving that they had the capacity to mediate therapeutically relevant antibody-dependent cellular cytotoxicity (Figure 2B). Of note, rituximab had no effect on the killing of CD20-negative K562 cells (Supplementary Figure 1).
Figure 2.
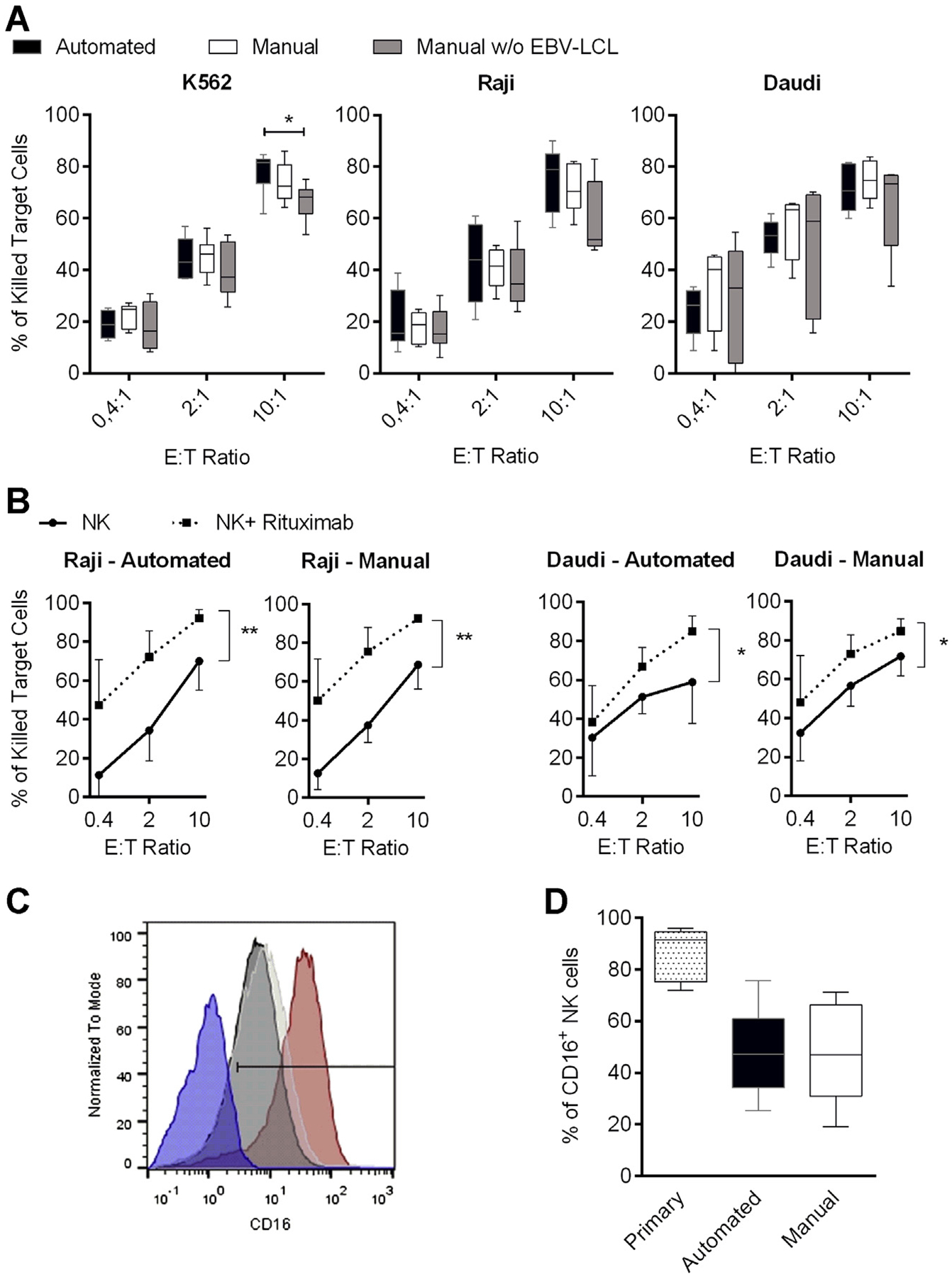
Natural and antibody-dependent cytotoxicity of differentially expanded NK cells. (A) NK cells were expanded for 14 days by means of the automated process (black bars) in comparison to manual NK cell expansion in with (white bars) or without (gray bars) EBV-LCL and analyzed for cytotoxicity against K562, Raji and Daudi cell lines at different effector-to-target (E:T) ratios. (B) Cytotoxicity against CD20-positive Raji or Daudi cells that were untreated (circles) or treated (squares) with 1 μg/mL rituximab for 4 h during the assay. P values indicated are for paired Student’s t-test, with P < 0.05 considered significant. Six or seven donors were analyzed; displayed are mean values, minimum to maximum (whiskers in A) and standard deviation. (C) Representative CD16 expression on NK cells from one donor is shown. Comparison of primary NK cells (red curve) and NK cells after 14 days of automated (black curve) or manual (white curve) expansion in the presence of EBV-LCL is displayed. CD16-negative PBMCs served as a control to determine the positive gate (blue curve). (D) Overview of the CD16 staining of all seven donors used in B is depicted; displayed are mean values, minimum to maximum and standard deviation.
Expanded NK cells do not show a reduction in telomere length
Extensive expansion might reduce the proliferative potential of NK cells for later in vivo applications, which would result in telomere shortening. Therefore, we investigated the telomere length before and after expansion because the proliferative potential of NK cells is crucial for their therapeutic effect after transfer to the patient [6]. After 14 days of expansion, no noticeable difference in the telomere length was detected, independent of the expansion method and expansion intensity, indicating that the proliferative potential of the NK cells was not reduced, even after extensive ex vivo expansion (Supplementary Figure 2).
Flow cytometry profiles of automatically and manually expanded NK cells are similar, whereas they differ clearly from primary NK cells
Next, we compared the phenotype of primary and expanded NK cells by means of flow cytometry and stained for 18 selected markers (Figure 3). As previously described [24], the pattern of many relevant NK cell markers changed on ex vivo expansion. Upregulation of TRAIL and FasL as well as the activating NK cell receptors NKp30, NKp44, NKG2D and DNAM-1 indicated an activated state and correlated with the enhanced NK cell function after expansion, which was in line with the results of the preceding functional assays. Besides the strong phenotypic difference between naive and expanded NK cells in general, manually and automatically processed NK cells had a comparable marker profile. However, frequencies of NK cells expressing NKG2C, CX3CR1 and KIR2DL2/DL3 were slightly but still statistically significantly higher after automated expansion compared with manually expanded NK cells that showed slightly higher expression of NKG2A and NKp44 (representative dot plots in Supplementary Figure 3).
Figure 3.
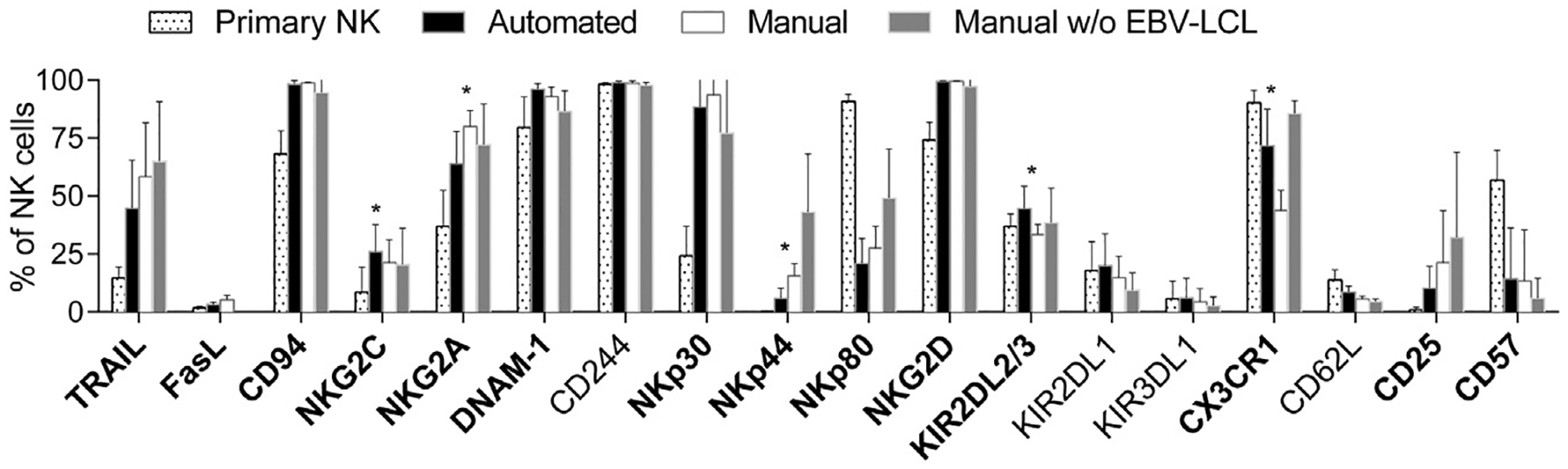
NK cell surface marker expression investigated by means of flow cytometry. NK cells were analyzed by means of flow cytometry for selected surface markers before (primary NK, dotted) or after automated EBV-LCL–based expansion (black bars) in comparison to manual NK cell expansion in T flasks with (white bars) or without (gray bars) EBV-LCL. Five donors were analyzed; mean values and standard deviations are shown. Changes on expansion represented by differences between primary and automatically expanded NK cells were analyzed by means of paired Student’s t-test; markers with P < 0.05 are written in bold letters. In addition, differences between automatically and manually expanded NK cells after co-culture with EBV-LCL were analyzed by means of paired Student’s t-test; markers with P < 0.05 are indicated as significant by a star.
Gene expression analysis reveals only minor differences between NK cells after automated or manual NK cell expansion
We further investigated the NK cells at the gene expression level and performed a whole human genome micro-array with samples from six donors. In total, 13,263 reporters corresponding to differentially expressed transcripts were identified in the comparisons among all sample groups (displayed in a clustered heat map in Figure 4A). Of note, the most prominent expression differences were between freshly isolated and all other expanded NK cell samples.
Figure 4.
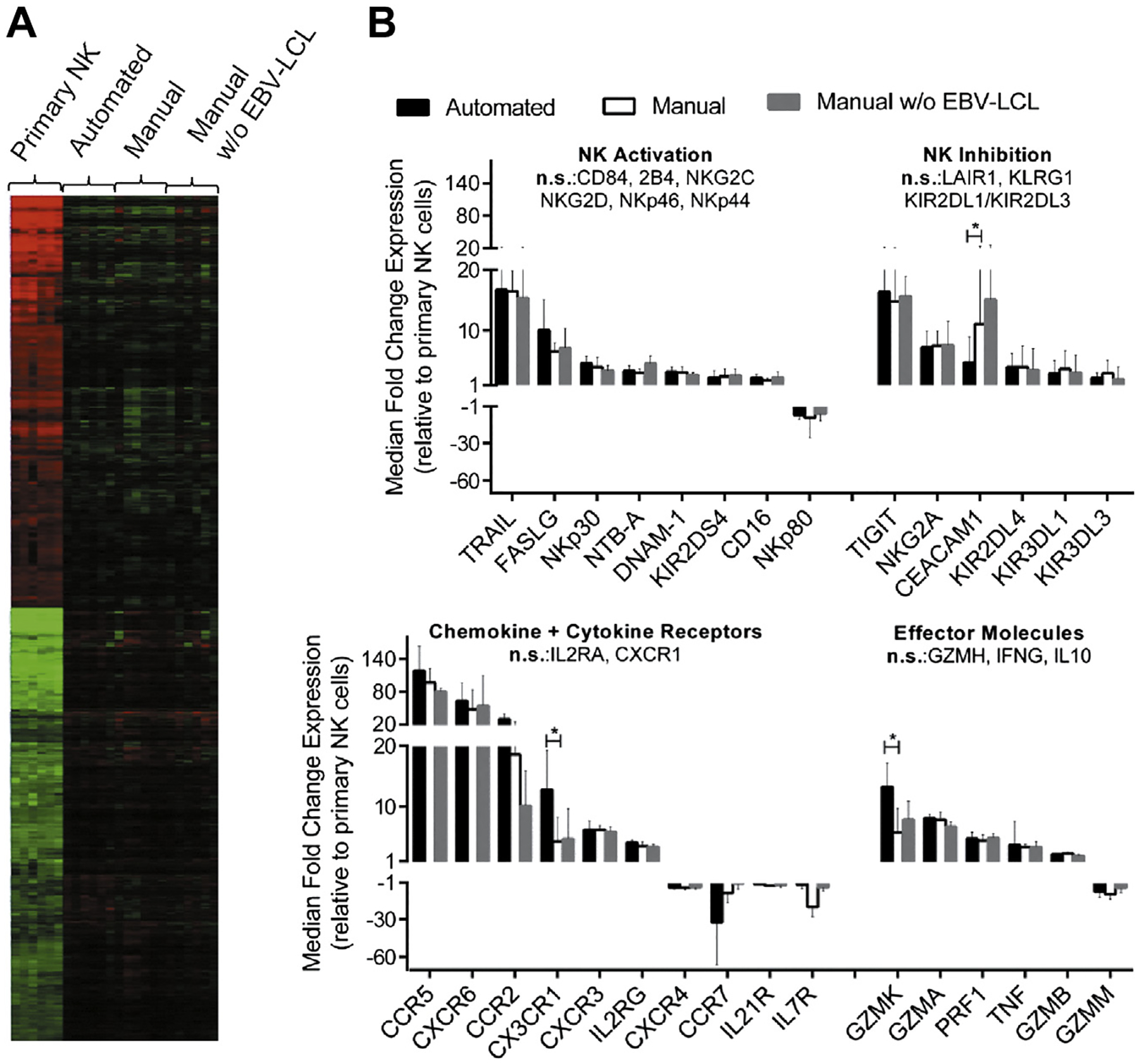
Gene expression profiles of NK cells before and after expansion. Samples from six donors were used for gene expression analysis of primary NK cells and NK cells after automated EBV-LCL–based expansion as well as NK cells after manual expansion in T flasks with or without EBV-LCL. (A) Differentially regulated reporters between the four sample groups were identified by filtering for statistical relevance and reliable signal intensities; median centered values for these reporters are shown in a heat map after hierarchical clustering analysis. Color saturation limits range from log2 intensities of −4 (green) to +4 (red). No changes relative to the reporter-wise median log2 intensity of all samples is displayed in black color. (B) Filtering for NK cell–relevant genes among regulated reporters is displayed for NK cells obtained by the automated process (black bars), manual expansion with EBV-LCL feeders (white bars) or manual expansion without EBV-LCL feeders (gray bars) in relation to primary NK cells. Automated and EBV-LCL–based manual expansion was compared by means of Tukey’s honestly significant differences post hoc test; genes with Tukey P value ≤0.05 and median fold change ≥2 or ≤ −2 are indicated as significant (indicated by stars).
In contrast, and consistent with the flow cytometry analysis, gene expression after automated or manual expansion with EBV-LCL feeder cells was similar. Less than 2% of all differentially expressed reporters (247 reporters) varied significantly between both sample groups. However, to obtain further insight in associated functions for the small set of differentially expressed genes, functional grouping analysis was performed. The analysis revealed an association of the genes with hematological system development, cellular movement and immune cell trafficking. In particular, genes with known importance in movement of leukocytes were identified (Supplementary Table I), including a group of genes associated with NK cell migration (CMKLR1, CX3CR1, S1PR5, GNLY and CXCR1). The latter set of genes was expressed at slightly higher levels after automated compared with manual expansion.
Nevertheless, the expression profiles of NK cells after automated and manual expansion were strikingly similar and in strong contrast to the many differentially expressed genes between primary and expanded NK cells. Changes between NK cells after expansion with the automated system and naive NK cells before expansion were investigated in more detail; a list of the 100 most upregulated or downregulated genes is shown in Supplementary Tables II and III. As expected for an expansion protocol, functional grouping analysis revealed the most significant functional association of the regulated genes (B-H P values <1.5 × 10 −8) with cell cycle regulation, cell death and survival, DNA replication, DNA recombination and DNA repair, cellular growth and proliferation as well as cellular assembly and organization. Consistent with the results of the flow cytometry analyses, many NK cell markers had a change in their expression levels after expansion (Figure 4B). The most prominent effects were upregulation of TRAIL, FasL, the inhibitory receptor TIGIT and the chemokine receptors CCR2, CCR5 and CXCR6. In addition, granzyme M was downregulated, but other effector molecules that play an important role in tumor killing, such as TNF-α, perforin and granzymes A, B and K, were upregulated.
Automated NK cell expansion can be complemented by a preceding, automated NK cell separation, enabling a fully automated NK cell production process
Finally, we also performed initial NK cell purification steps automated by means of the CliniMACS Prodigy system before the automated cultivation and expansion phase to show that the complete cell processing needed for NK cell expansion, from the starting material to the final cell product inclusive of the NK cell enrichment process, can be achieved with the use of this system. We started with TCR-α/β-CD19 depletion, to ensure efficient removal of potentially harmful contaminating TCR-α/β+ T and B cells, followed by CD56-positive selection in a second step to further enrich for purified NK cells. By use of this strategy, automated NK cell separations from leukapheresis products of three donors were performed and 4,1–4,4 and 2,7–3,1 log depletion of TCR-α/β and B cells was achieved. After subsequent CD56 enrichment, no TCR-α/β and B cells were detected, and NK cells with a purity of 71% to 92% could be obtained while remaining non-NK cells were mainly CD14+ monocytes (12.4% ± 8.8%) and TCR-γδ CD3+CD56+ NK-like T cells (5.3% ± 4.7%). Automated expansion of automatically separated NK cells for 14 days resulted in 390-fold to 1185-fold expansion (Supplementary Figure 4A), within the same range of what was achieved by manually separated NK cells before. The functionality of the fully automatically obtained NK cells was proven by the effective killing of K562, Raji and Daudi cells (Supplementary Figure 4B).
In summary, we expanded substantial numbers of activated NK cells through the use of a completely automated system. These automatically produced NK cells, in comparison to their manually produced counterparts, had similar phenotypic, transcriptional and functional profiles.
Discussion
We successfully developed the automated cell expansion processes by the use of an automated system and report our results on production of activated NK cells for their use in clinical cell therapy applications. For large-scale expansion of clinical-grade cells, NK cell cultures normally are maintained for 14 to 28 days and typically require frequent interventions such as media changes to refresh cytokines and other growth factors as well as to ensure that NK cells are maintained at a concentration that optimizes their growth and viability [32]. We efficiently automated this procedure and generated high NK cell numbers that showed the same in vitro functionality and similar phenotype and gene expression as manually expanded NK cells. The automation within a closed system substantially facilitates the expansion procedure by saving not only time but also minimizing the risk of culture contamination while introducing consistency in the production process. In addition, the automation requires financial investment for the instrument but enables significantly reduced running costs for an actively used clean-room, representing a major expense factor for the cellular product. Therefore, the automated process will be cost-saving in production scale and performance of numerous processes per year.
In line with previous findings, many NK cell–relevant markers and apoptosis-inducing molecules were upregulated on ex vivo expansion and might contribute to an increase of the therapeutic potential of the ex vivo–generated NK cells [24]. In particular, upregulation of TRAIL by expanded NK cells can be utilized to efficiently treat tumors that express TRAIL death receptors and/or are sensitized to TRAIL by drugs such as bortezomib [33].
Although several methods for effective NK cell expansion were developed in the past and have proven their applicability in large-scale by use of manual cultivation systems such as cell culture bags or G-Rex containers [13,14,17–19], there has been virtually no progress in the development of a fully automated and controlled process for clinical-scale NK cell expansion. Sutlu et al. [17] and Lapteva et al. [22], independent of each other, applied an automated Wave Bioreactor system for clinical-grade NK cell expansion from PBMCs, and Spanholtz et al. [21] used the same system to generate clinical-grade NK cells expanded from cord blood hematopoietic progenitor cells. Sutlu et al. [17] concluded that automation of the cultivation is more practical and generated more activated NK cells compared with manual approaches. We confirm the practicability of an automated system, whereas, in our hands, NK cells neither differed significantly in phenotype nor in function, whether they have been cultivated manually or automatically, similar to observations from Lapteva and Spanholtz et al. In comparison, the Wave Bioreactor system requires a high cell number to initiate the culture, which inevitably implicates a manual pre-cultivation until enough cells are generated to start the automated process. In the system presented here, low starting cell numbers were not a limitation because the automation covered the whole cultivation phase including an early static cultivation phase with very low cell numbers, with 106 NK cells being sufficient to initiate the current process.
In addition and in contrast to other expansion approaches, we show that the entire cell cultivation phase, including the preceding cell separation steps, can be fully automated by a single instrument, enabling complete cell processing from the starting material to the final “ready-to-use” cell product. To start the NK cell expansion with enriched NK cells is beneficial because it results in high-purity expanded NK cells without contaminating T cells in the final cell product [13,34].
On average, we could generate 1.3 × 109 activated NK cells with one instrument within 2 weeks, enough to treat a typical 70- to 100-kg patient with 1 to 2 × 107 NK cells/kg. This would fall within the range of NK cells typically used in investigational trials of adoptive NK cell immunotherapy [3]. Nevertheless, the optimal dose for NK cell injections has not yet been determined, and, because no dose dependent side effects have been observed, NK cell injections of 108 to 109/kg are imaginable in future [32]. Whereas bioreactor systems provide a volume of up to 3 L for cultivation and can yield 2 × 109 NK cells derived from umbilical cord blood hematopoietic stem cells [16] or 9.8 × 109 NK cells from expansion of PBMCs [21,22], our current system is equipped with a medium scale culture volume of only 300 mL, which allowed a maximum NK cell number of 2.7 × 109. Thus, if mega-doses of NK cells are needed, several process runs or multiple devices might be necessary for one application. Integration of a bioreactor in the system may be another option to enable fully automated processes that can include cell separation followed by an initial expansion and a late cultivation phase at higher culture volumes. With the current instrumentation, it is already possible to perform a continuously running process that maintains the NK cells in the expansion phase with repeated harvesting of cells whenever the maximum cell density is reached.
On the other hand, the number of NK cells that is required for the transfer to the a patient may be lower, if better in vivo persistence and expansion of the transferred cells could be achieved through post-infusion maneuvers such as cytokine administration. Clinical trials showed that it is possible to induce in vivo NK cell expansion in humans by means of IL-2 injection or endogenous production of IL-15 that can be stimulated by distinct immune suppressive regimen [7,35]. Importantly, NK cells produced with the use of the automated method showed no noticeable telomere length shortening after expansion, indicating that the cells do not become senescent, and the regular proliferative potential is conserved potentially, allowing ex vivo–expanded NK cells to further expand in vivo. However, this must be proven in a suitable animal model or clinical application.
Another aspect to consider is that NK cells are heterogeneous in phenotype and function, with only a fraction of NK cell subsets driving their major cytotoxic effects [36,37]. As a consequence, effective NK cell–based immunotherapy may not necessarily require the transfer of a high number of bulk NK cells but the transfer of sufficient cells of therapeutically relevant NK cell subsets. Detailed characterization of these subsets is pending, but, because the automated expansion system inimitably combines the feature of cell separation and the possibility to use a very low starting NK cell number for expansion, it would be possible to isolate only a rare, therapeutically relevant NK cell subset and expand these cells to clinically needed numbers within a single process, representing a promising future strategy.
In the present study, we automated our system through the use of TCR-α/β-CD19 depletion followed by CD56 enrichment as a novel strategy to enrich for bulk NK cells in clinical scale from leukapheresis products because TCR-α/β depletion has been shown to be superior over conventional CD3 depletion as the result of a more efficient removal of TCR-α/β T cells, which are responsible for graft-versus-host disease [38]. We achieved a highly efficient depletion of TCR-α/β cells and obtained NK cells that had the same potential to proliferate and showed the same functionality after expansion as NK cells, which were obtained by CD3 depletion and CD56 enrichment. The latter method has been used as a standard strategy for NK cell separation in clinical scale in investigational therapeutic settings [20,39,40].
In view of the need to develop standardized methods to expand NK cells that are being investigated for their therapeutic potential in the clinic, this automated process enables easy up-scaling for centralized manufacturing of the therapeutic cell product. Furthermore, the closed system allows scale-out strategies and decentralized cell processing directly at the location of use, avoiding the need for cell shipping, which often represents a logistic challenge, and, if done incorrectly, can compromise the product quality [22].
In conclusion, the automation of the entire NK cell expansion process presented in the present report represents a novel procedure with the use of a single instrument that allows for the efficient production of clinical-grade NK effector cells. Because all processing steps are done automated in a closed system, this provides the highest standards for GMP conformity. Most importantly, this automated process represents a highly efficient and much-needed tool to standardize NK cell processing for therapeutic applications.
Supplementary Material
Acknowledgments
We would like to thank all staff of Cell Biology K at Miltenyi and in particular Katharina Drechsel, Juliane Stuth, Mareke Brüning, Elmar Fahrendorff, Florian Siegismund, Stefan Tomiuk, Silvia Rüberg, Michail Knauel, Jürgen Schmitz and Stefan Miltenyi for support or/and technical assistance in enabling this work.
Footnotes
Disclosure of interests: Markus Granzin, Stephanie Soltenborn, Sabine Mueller, Jutta Kollet and Volker Huppert are employees of Miltenyi Biotec. The authors have no commercial, proprietary, or financial interest in the products or companies described in this article.
Supplementary data
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.jcyt.2015.03.611.
References
- [1].Kiessling R, Klein E, Pross H, Wigzell H. “Natural” killer cells in the mouse. II. Cytotoxic cells with specificity for mouse Moloney leukemia cells. Characteristics of the killer cell. Eur J Immunol 1975;5:117–21. [DOI] [PubMed] [Google Scholar]
- [2].Vivier E, Raulet DH, Moretta A, Caligiuri MA, Zitvogel L, Lanier LL, et al. Innate or adaptive immunity? The example of natural killer cells. Science 2011;331:44–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Cheng M, Chen Y, Xiao W, Sun R, Tian Z. NK cell-based immunotherapy for malignant diseases. Cell Mol Immunol 2013;10:230–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Stern M, Passweg JR, Meyer-Monard S, Esser R, Tonn T, Soerensen J, et al. Pre-emptive immunotherapy with purified natural killer cells after haploidentical SCT: a prospective phase II study in two centers. Bone Marrow Transplant 2013;48:433–8. [DOI] [PubMed] [Google Scholar]
- [5].Curti A, Ruggeri L, D’Addio A, Bontadini A, Dan E, Motta MR, et al. Successful transfer of alloreactive haploidentical KIR ligand-mismatched natural killer cells after infusion in elderly high risk acute myeloid leukemia patients. Blood 2011;118:3273–9. [DOI] [PubMed] [Google Scholar]
- [6].Miller JS, Soignier Y, Panoskaltsis-Mortari A, McNearney SA, Yun GH, Fautsch SK, et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood 2005;105: 3051–7. [DOI] [PubMed] [Google Scholar]
- [7].Geller MA, Cooley S, Judson PL, Ghebre R, Carson LF, Argenta PA, et al. A phase II study of allogeneic natural killer cell therapy to treat patients with recurrent ovarian and breast cancer. Cytotherapy 2011;13:98–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Bachanova V, Burns LJ, Mckenna DH, Curtsinger J, Panoskaltsis-Mortari A, Lindgren BR, et al. Allogeneic natural killer cells for refractory lymphoma. Cytotherapy 2010; 59:1739–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Iliopoulou EG, Kountourakis P, Karamouzis MV, Doufexis D, Ardavanis A, Baxevanis CN, et al. A phase I trial of adoptive transfer of allogeneic natural killer cells in patients with advanced non-small cell lung cancer. Cancer Immunol Immunother 2010;59:1781–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Rubnitz JE, Inaba H, Ribeiro RC, Pounds S, Rooney B, Bell T, et al. NKAML: a pilot study to determine the safety and feasibility of haploidentical natural killer cell transplantation in childhood acute myeloid leukemia. J Clin Oncol 2010;28:955–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Leung W. Infusions of allogeneic natural killer cells as cancer therapy. Clin Cancer Res 2014;20:3390–400. [DOI] [PubMed] [Google Scholar]
- [12].Klingemann HG. Cellular therapy of cancer with natural killer cells—where do we stand? Cytotherapy 2013;15: 1185–94. [DOI] [PubMed] [Google Scholar]
- [13].Berg M, Lundqvist A, McCoy P Jr, Samsel L, Fan Y, Tawab A, et al. Clinical-grade ex vivo-expanded human natural killer cells up-regulate activating receptors and death receptor ligands and have enhanced cytolytic activity against tumor cells. Cytotherapy 2009;11:341–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Barkholt L, Alici E, Conrad R, Sutlu T, Gilljam M, Stellan B, et al. Safety analysis of ex vivo-expanded NK and NK-like T cells administered to cancer patients: a phase I clinical study. Immunotherapy 2009;1:753–64. [DOI] [PubMed] [Google Scholar]
- [15].Fujisaki H, Kakuda H, Shimasaki N, Imai C, Ma J, Lockey T, et al. Expansion of highly cytotoxic human natural killer cells for cancer cell therapy. Cancer Res 2009;69: 4010–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Spanholtz J, Preijers F, Tordoir M, Trilsbeek C, Paardekooper J, de Witte T, et al. Clinical-grade generation of active NK cells from cord blood hematopoietic progenitor cells for immunotherapy using a closed-system culture process. PLoS One 2011;6:e20740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Lapteva N, Durett AG, Sun J, Rollins LA, Huye LL, Fang J, et al. Large-scale ex vivo expansion and characterization of natural killer cells for clinical applications. Cytotherapy 2012; 14:1131–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Siegler U, Meyer-Monard S, Jorger S, Stern M, Tichelli A, Gratwohl A, et al. Good manufacturing practice-compliant cell sorting and large-scale expansion of single KIR-positive alloreactive human natural killer cells for multiple infusions to leukemia patients. Cytotherapy 2010;12:750–63. [DOI] [PubMed] [Google Scholar]
- [19].Luhm J, Brand JM, Koritke P, Hoppner M, Kirchner H, Frohn C. Large-scale generation of natural killer lymphocytes for clinical application. J Hematother Stem Cell Res 2002;11:651–7. [DOI] [PubMed] [Google Scholar]
- [20].Koehl U, Brehm C, Huenecke S, Zimmermann S-Y, Kloess S, Bremm M, et al. Clinical grade purification and expansion of NK cell products for an optimized manufacturing protocol. Front Oncol 2013;3:118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Sutlu T, Stellan B, Gilljam M, Quezada HC, Nahi H, Gahrton G, et al. Clinical-grade, large-scale, feeder-free expansion of highly active human natural killer cells for adoptive immunotherapy using an automated bioreactor. Cytotherapy 2010;12:1044–55. [DOI] [PubMed] [Google Scholar]
- [22].Lapteva N, Szmania SM, van Rhee F, Rooney CM. Clinical grade purification and expansion of natural killer cells. Crit Rev Oncog 2014;19:121–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Apel M, Brüning M, Granzin M, Essl M, Stuth J, Blaschke J, et al. Integrated Clinical Scale Manufacturing System for Cellular Products Derived by Magnetic Cell Separation, Centrifugation and Cell Culture. Chemie Ing Tech 2013;85: 103–10. [Google Scholar]
- [24].Park KU, Jin P, Sabatino M, Feng J, Civini S, Khuu H, et al. Gene expression analysis of ex vivo expanded and freshly isolated NK cells from cancer patients. J Immunother Hagerst Md 1997 2010;33:945–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Fleige S, Pfaffl MW. RNA integrity and the effect on the real-time qRT-PCR performance. Mol Aspects Med 2006;27: 126–39. [DOI] [PubMed] [Google Scholar]
- [26].Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol 2004;5:R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].R Core Team (2013). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. ISBN 3-900051-07-0, URL http://www.R-project.org/. [Google Scholar]
- [28].Speed TP, Bolstad BM, Irizarry RA, Astrand M. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 2003;19:185–93. [DOI] [PubMed] [Google Scholar]
- [29].Weng L, Dai H, Zhan Y, He Y, Stepaniants SB, Bassett DE. Rosetta error model for gene expression analysis. Bioinformatics 2006;22:1111–21. [DOI] [PubMed] [Google Scholar]
- [30].Saeed AI, Bhagabati NK, Braisted JC, Liang W, Sharov V, Howe EA, et al. TM4 microarray software suite. Methods Enzymol 2006;411:134–93. [DOI] [PubMed] [Google Scholar]
- [31].Romee R, Foley B, Lenvik T, Wang Y, Zhang B, Ankarlo D, et al. NK cell CD16 surface expression and function is regulated by a disintegrin and metalloprotease-17 (ADAM17). Blood 2013;121:3599–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Childs RW, Berg M. Bringing natural killer cells to the clinic: ex vivo manipulation. Hematology Am Soc Hematol Educ Program 2013;2013:234–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Lundqvist A, Abrams SI, Schrump DS, Alvarez G, Suffredini D, Berg M, et al. Bortezomib and depsipeptide sensitize tumors to tumor necrosis factor-related apoptosis-inducing ligand: a novel method to potentiate natural killer cell tumor cytotoxicity. Cancer Res 2006;66:7317–25. [DOI] [PubMed] [Google Scholar]
- [34].Berg M, Childs R. Ex-vivo expansion of NK cells: what is the priority–high yield or high purity? Cytotherapy 2010;12: 969–70. [DOI] [PubMed] [Google Scholar]
- [35].Parkhurst MR, Riley JP, Dudley ME, Rosenberg SA. Adoptive transfer of autologous natural killer cells leads to high levels of circulating natural killer cells but does not mediate tumor regression. Clin Cancer Res 2011;17:6287–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Vanherberghen B, Olofsson PE, Forslund E, Sternberg-Simon M, Khorshidi MA, Pacouret S, et al. Classification of human natural killer cells based on migration behavior and cytotoxic response. Blood 2013;121:1326–34. [DOI] [PubMed] [Google Scholar]
- [37].Horowitz A, Strauss-Albee DM, Leipold M, Kubo J, Nemat-Gorgani N, Dogan OC, et al. Genetic and environmental determinants of human NK cell diversity revealed by mass cytometry. Sci Transl Med 2013;5:208ra145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Handgretinger R. Negative depletion of CD3(+) and TcRαβ(+) T cells. Curr Opin Hematol 2012;19:434–9. [DOI] [PubMed] [Google Scholar]
- [39].Iyengar R, Handgretinger R, Babarin-Dorner A, Leimig T, Otto M, Geiger TL, et al. Purification of human natural killer cells using a clinical-scale immunomagnetic method. Cytotherapy 2003;5:479–84. [DOI] [PubMed] [Google Scholar]
- [40].Passweg JR, Tichelli A, Meyer-Monard S, Heim D, Stern M, Kuhne T, et al. Purified donor NK-lymphocyte infusion to consolidate engraftment after haploidentical stem cell transplantation. Leukemia 2004;18:1835–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.