

ABSTRACT
Macroautophagy/autophagy is elevated to ensure the high demand for nutrients for the growth of cancer cells. Here we demonstrated that MCOLN1/TRPML1 is a pharmaceutical target of oncogenic autophagy in cancers such as pancreatic cancer, breast cancer, gastric cancer, malignant melanoma, and glioma. First, we showed that activating MCOLN1, by increasing expression of the channel or using the MCOLN1 agonists, ML-SA5 or MK6-83, arrests autophagic flux by perturbing fusion between autophagosomes and lysosomes. Second, we demonstrated that MCOLN1 regulates autophagy by mediating the release of zinc from the lysosome to the cytosol. Third, we uncovered that zinc influx through MCOLN1 blocks the interaction between STX17 (syntaxin 17) in the autophagosome and VAMP8 in the lysosome and thereby disrupting the fusion process that is determined by the two SNARE proteins. Furthermore, we demonstrated that zinc influx originating from the extracellular fluid arrests autophagy by the same mechanism as lysosomal zinc, confirming the fundamental function of zinc as a participant in membrane trafficking. Last, we revealed that activating MCOLN1 with the agonists, ML-SA5 or MK6-83, triggers cell death of a number of cancer cells by evoking autophagic arrest and subsequent apoptotic response and cell cycle arrest, with little or no effect observed on normal cells. Consistent with the in vitro results, administration of ML-SA5 in Patu 8988 t xenograft mice profoundly suppresses tumor growth and improves survival. These results establish that a lysosomal cation channel, MCOLN1, finely controls oncogenic autophagy in cancer by mediating zinc influx into the cytosol.
Abbreviation: Abbreviations: 3-MA: 3-methyladenine; AA: amino acid; ATG12: autophagy related 12; Baf-A1: bafilomycin A1; BAPTA-am: 1,2-bis(2-aminophenoxy)ethane-N, N,N’,N’-tetraacetic acid tetrakis-acetoxymethyl ester; co-IP: coimmunoprecipitaion; CQ: chloroquine; DMEM: Dulbecco’s Modified Eagle Medium; FBS: fetal bovine serum; GAPDH: glyceraldehyde- 3-phosphate dehydrogenase; HCQ: hydroxychloroquine; HEK: human embryonic kidney; LAMP1: lysosomal associated membrane protein 1; MAP1LC3/LC3: microtubule associated protein 1 light chain 3; MCOLN1/TRPML1: mucolipin TRP cation channel 1; MTORC1: mechanistic target of rapamycin kinase complex 1; NC: negative control; NRK: normal rat kidney epithelial cells; PBS: phosphate-buffered saline; PtdIns3K: phosphatidylinositol 3-kinase; RPS6KB/S6K: ribosomal protein S6 kinase B; shRNA: short hairpin RNA; siRNA: short interfering RNA; SNARE: soluble N-ethylmaleimide-sensitive factor (NSF) attachment protein receptor; SQSTM1/p62: sequestosome 1; STX17: syntaxin 17; TPEN: N,N,N’,N’-tetrakis(2-pyridylmethyl)ethylenediamine; TTM: tetrathiomolybdate; ULK1: unc-51 like autophagy activating kinase 1; VAMP8: vesicle associated membrane protein 8; Zn2+: zinc.
KEYWORDS: Autophagic arrest, autophagosome-lysosome fusion, cancer, MCOLN1, zinc influx
Introduction
MCOLN1/TRPML1 is a nonselective cation channel specifically localized in the lysosome [1] and plays a critical role in regulating a variety of intracellular processes including endocytosis, exocytosis, lysosomal adaptation and autophagy [2–5]. To date, the established functions of MCOLN1 are attributed to its ability to regulate Ca2+ influx into the cytosol from the lysosome, which is an important second messenger and favors vesicular fusion/fission [6–8].
MCOLN1 has been proposed as a regulator of autophagy. Macroautophagy/autophagy is an evolutionarily conserved degradation process that helps to remove unwanted proteins and damaged organelles and provides the necessary nutrients for cell survival, especially under various stress conditions [9,10]. Fibroblasts from patients with mucolipidosis type IV (MLIV), a disorder caused by mutations in MCOLN1, exhibit impaired autophagic flux showing increased levels of both lipidated MAP1LC3/LC3 (LC3-II) and SQSTM1/p62 (sequestosome 1) [11]. Considering the function of MCOLN1 on regulating Ca2+, the notion that impairment in autophagy in MLIV patients may have resulted from a reduction in lysosomal Ca2+ release due to diminished MCOLN1 activities has been speculated. Supporting this idea, a potential molecular mechanism has been recently suggested for MCOLN1 in the regulation of autophagy, where Ca2+ release from lysosomes through MCOLN1 activates PPP3/calcineurin, which in turn dephosphorylates TFEB (transcription factor EB) [12,13]. TFEB dephosphorylation is thought to promote lysosomal biogenesis and autophagy, thus the activation of MCOLN1 theoretically promotes autophagy by regulating Ca2+ release. This Ca2+-dependent mechanism could, to some extent, explain the impaired autophagic flux with severe autophagosome accumulation exhibited in MLIV fibroblasts.
However, these results connecting MCOLN1 and autophagy are suggestive of an indirect role of MCOLN1 in autophagy. In order to directly investigate the effect of MCOLN1 on autophagy, we boosted MCOLN1’s activity by either using specific MCOLN1 agonists or by increasing its protein expression levels. The agonists and antagonists of MCOLN1 used in the study include ML-SA1 [14], ML-SA5 [15], MK6-83 [16], ML-SI1 [5], and ML-SI3 [5]. Although well characterized, the effects of these agonists and antagonists on modulating MCOLN1 activity were confirmed in our hands by utilizing lysosomal MCOLN1 patch clamp recordings and Ca2+ imaging, respectively (Figure S1A-E [5,17];). Unexpectedly, we found that the activation of MCOLN1 indeed arrests autophagic flux in conjunction with blocked fusion of autophagosomes and lysosomes. Moreover, other than Ca2+, lysosomal zinc influx mediated by MCOLN1 does arrest autophagy by disrupting the interaction between STX17 (syntaxin 17) in the autophagosome and VAMP8 (vesicle associated membrane protein 8) in the lysosome [18,19]. Most importantly, the autophagic arrest induced by stimulating MCOLN1 selectively triggers cell death of a broad range of cancer cells without affecting non-cancer cells, establishing MCOLN1 as a new potential target to combat cancer with minimal side effects on normal cells.
Results
The activation of MCOLN1 arrests autophagic flux by disrupting fusion between autophagosomes and lysosomes
To determine the role of MCOLN1 in autophagy, we systematically monitored changes in autophagy [20] after directly activating MCOLN1 channels in HeLa cells. We firstly confirmed that two synthetic MCOLN1 agonists [15,21,22], ML-SA5 and ML-SA1, not only efficiently stimulated endogenous MCOLN1 channels in HeLa, Patu 8988 t (a human pancreatic cancer cell line) [23] and MCF-7 (a human breast cancer cell line) [24] cells (Figure S1A-C), but also the overexpressed MCOLN1 channels in HEK 293 T cells (Figure. S1D-E), as determined by whole-endolysosome patch-clamp recordings and Ca2+ imaging experiments. Stimulating MCOLN1 with ML-SA5 caused an increase in LC3-II levels at both 1 and 12 h following application (Figure 1A). Notably, the increase in LC3-II levels by 1 μM ML-SA5 for 1 h was comparable to the one by 1 μM bafilomycin A1 (Baf-A1) (Figure 1A), an inhibitor of the vacuolar-type H+ ATPase (V-ATPase) known to block fusion between autophagosomes and lysosomes [25]. The effect of ML-SA5 on LC3-II levels was rapid, showing robust increases within 1 h following application of ML-SA5 (left panel in Figure 1A). The increases in LC3-II levels were also dependent on ML-SA5 concentrations and treatment periods (Figure S1F). Lower doses of ML-SA5 treatments at 0.05 or 0.1 μM also significantly increased LC3-II levels following both 1 h and 4 h of agonist application in HeLa cells (Figure S1G). Furthermore, supporting the involvement of MCOLN1 in autophagy, the ML-SA5-induced increases in LC3-II were completely inhibited by co-application of either ML-SI1 (20 μM) or ML-SI3 (20 μM) (Figure 1B), two MCOLN1 antagonists (Figure S1A, S1B-D) [5]. In sharp contrast, the Baf-A1-evoked increases in LC3-II were unaffected by ML-SI3 (20 μM; Figure 1B), suggesting that ML-SA5 regulates LC3-II levels through stimulating MCOLN1 channel activity.
Figure 1.
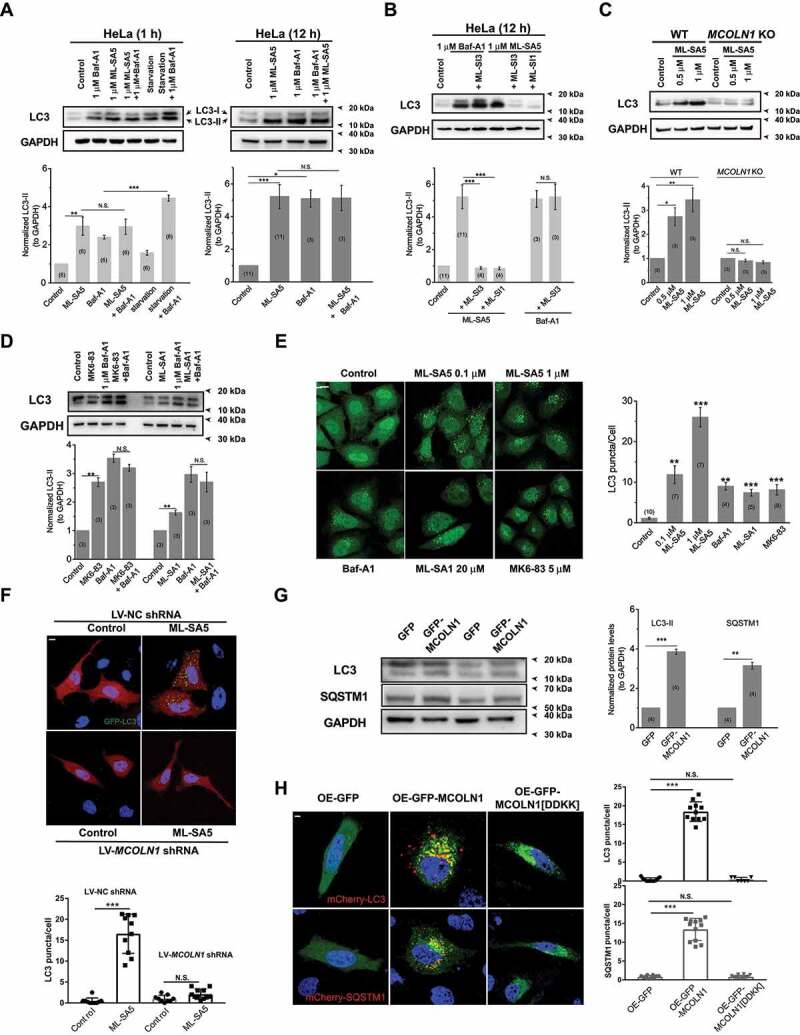
The activation of MCOLN1 specifically regulates autophagy. (A) Both 1 h (left) and 12 h (right) application of ML-SA5 (1 μM) led to an increase in LC3-II levels in HeLa cells, indicated by the normalized LC3-II levels to GAPDH. GAPDH served as a loading control. Starvation (AA-and FBS-free) and Baf-A1 (1 μM) were positive controls for autophagy assessments. Quantification of normalized LC3-II levels under conditions indicated were displayed. (B) The antagonists of MCOLN1, ML-SI1 (20 μM) or ML-SI3 (20 μM), completely inhibited the increases in LC3-II levels triggered by ML-SA5 (1 μM) in HeLa cells, but not ones by Baf-A1 (1 μM). (C) In contrast to WT HAP1 cells, application of ML-SA5 did not lead to an increase in LC3-II levels in MCOLN1 KO HAP1 cells. Deletion of MCOLN1 was confirmed by genotyping (data not shown). (D) Another two MCOLN1 agonists, MK6-83 (5 μM) and ML-SA1 (20 μM), also led to significant increases in LC3-II levels in HeLa cells, respectively. Baf-A1 (1 μM) application did not further facilitate LC3-II increases caused by either MK6-83 or ML-SA1 treatments. All treatments were for 4 h. (E) Four hour treatments of 0.1 μM ML-SA5, 1 μM ML-SA5, 5 μM MK6-83 or 20 μM ML-SA1 caused increased abundance of LC3 puncta in GFP-LC3 stably expressing HEK 293 T (S-GFP-LC3) cell lines, respectively. Baf-A1 (1 μM) was a positive control for inducing LC3 puncta structures. LC3 puncta per cell under the control, ML-SA5 treatments (0.1 μM and 1 μM), MK6-83 (5 μM), ML-SA1 (20 μM) and Baf-A1 (1 μM) were quantified from 4–11 independent experiments (typically 10–40 cells). Scale bar: 10 μm. (F) The LC3 puncta structures induced by the ML-SA5 treatment (1 μM) were profoundly inhibited by reducing MCOLN1 expression with LV-MCOLN1 shRNA in GFP-LC3 transiently expressed HeLa cells. LC3 puncta per cell were quantified from 9–11 cells from at least three independent experiments. Scale bar: 10 μm. (G) GFP-MCOLN1-expressing HeLa cells yielded higher basal LC3-II levels and SQSTM1 levels than GFP-expressing cells, measured by western blot experiments. Transfection efficiency was confirmed by fluorescence microscopy. (H) Overexpressing MCOLN1 significantly induced LC3 or SQSTM1 puncta structures, whereas a channel-dead pore mutant MCOLN1[DDKK] did not induce overt LC3 or SQSTM1 puncta structures in mCherry-LC3 or mCherry-SQSTM1 transiently expressed HeLa cells. LC3 or SQSTM1 puncta per cell were quantified from 7–12 cells from three independent experiments. Scale bar: 10 μm. Means ± SEMs are shown in panels A-H. Significant differences were evaluated using one-way ANOVA followed by Tukey’s test. *P < 0.05; **P < 0.01; ***P < 0.001
ML-SA5 also led to a marked increase in LC3-II levels in wild type (WT) HAP1 cells, but the effects were abolished by the knockout (KO) of MCOLN1 in HAP1 cells (Figure 1C). To further confirm that the ML-SA5 treatment regulates autophagy through mediating MCOLN1 channels, we utilized two less potent MCOLN1 agonists, ML-SA1 [26] and MK6-83 [16]. Similarly, either application of MK6-83 (5 μM) or ML-SA1 (20 μM) led to an increase in LC3-II levels in HeLa cells (Figure 1D). Consistent with our findings using ML-SA5, the increase in LC3-II levels by MK6-83 or ML-SA5 was completely inhibited in MCOLN1 KO HAP1 cells (Figure S1H). Moreover, when reducing expressions of MCOLN2/TRPML2 or MCOLN3/TRPML3 channels that belong to the same family of TRPML with MCOLN1 (Figure S1I), the effects of ML-SA5 on increasing LC3-II levels were not affected (Figure S1J). Collectively, these results demonstrate that ML-SA5 regulates autophagy by specifically stimulating MCOLN1 channels, but not other channels including MCOLN2 and MCOLN3, thus establishing a specific role of MCOLN1 in regulating autophagy.
To further exclude possible off-target effects of ML-SA5, we employed WT normal rat kidney epithelial (NRK) cells and the corresponding atg12 KO NRK cells, in which autophagy is not able to be initiated [27]. In WT NRK cells, ML-SA5 (1 μM) induced robust increase in LC3-II levels, which contrasted sharply to atg12 KO cells where ML-SA5 (1 μM) failed to trigger any detectable LC3-II (Figure S1K). Similarly, the autophagy inhibitor, 3-methyladenine (3-MA) application (10 mM; pretreatment for 2 h), also completely inhibited the effect of ML-SA5 on increasing LC3-II levels in HeLa cells (Figure S1L) and LC3 puncta structures in GFP-LC3 stably expressed HEK 293 T cells (S-GFP-LC3) (Figure S1M). Collectively, these results demonstrate that stimulating MCOLN1 channels generally regulates autophagy in various types of cells, including HeLa, HAP1 and NRK cells.
In consistence with the effects on LC3-II protein levels, following stimulating MCOLN1 channels with ML-SA5, MK6-83, or ML-SA1, GFP-LC3 stably expressed HEK 293 T (S-GFP-LC3) cells displayed overt puncta structure pattern that is indicative of autophagosome accumulation (Figure 1E; representative images from 4 h treatment), which otherwise showed diffused cytosolic distribution under control conditions (Figure 1E). Application of ML-SI3 (20 μM) remarkably rescued LC3 puncta structures induced by ML-SA5 (1 μM; Figure S1M). Likewise, reducing MCOLN1 expressions, by utilizing a lentiviral-MCOLN1 shRNA (LV-MCOLN1 shRNA [28];), profoundly compromised the increases in LC3 puncta structures induced by the treatment of ML-SA5 (1 μM) in GFP-LC3 transiently expressed HeLa cells (Figure 1F). These results again confirmed that the effect of ML-SA5 on LC3 is specifically through modulating MCOLN1 channel activity.
In addition to stimulating MCOLN1 activity with the agonists, increasing MCOLN1 expression levels by transiently overexpressing GFP-MCOLN1 significantly elevated both basal LC3-II and SQSTM1 levels in HeLa cells as compared to control cells transfected with GFP tag alone (Figure 1G) as well as LC3 and SQSTM1 puncta structures in mCherry-LC3 or mCherry-SQSTM1 transiently expressing HeLa cells (Figure 1H). Furthermore, overexpressing a dominant-negative mutant of MCOLN1 [29], MCOLN1[DDKK], did not induce overt LC3 and SQSTM1 puncta structures (Figure 1H), indicating that functional MCOLN1 is required to regulate autophagy. Lastly, transmission electron microscopy (TEM) displayed abundant accumulation of autophagosomes in HeLa cells treated with ML-SA5 (1 μM for 12 h; Figure S1I). Collectively, in agreement with previous studies [11,21], these results provide further evidence indicating that activation of MCOLN1 regulates autophagy.
An increase in LC3-II levels could represent either promoted autophagy induction (autophagy initiation and autophagosome synthesis) or suppressed autophagosome degradation (either suppression of autophagosome-lysosome fusion or impairment of lysosomal degradation, or both). To distinguish between these two possibilities, we combined Baf-A1 with ML-SA5 to block the latter step of autophagy. Baf-A1 did not further facilitate LC3-II levels increased by ML-SA5 alone regardless of the concentration and the duration of the treatments (Figure 1A and S1G). Similarly, Baf-A1 did not further increase LC3-II levels increased by MK6-83 or ML-SA1 (Figure 1D) as well. In contrast, Baf-A1 further facilitated LC3-II levels increased by starvation (amino acid [AA]- and fetal bovine serum [FBS]-free) (Figure 1A). These results demonstrate that MCOLN1 activation mainly exerts an inhibitory effect on autophagosome degradation, which is not further exacerbated by Baf-A1. Consistently, chloroquine (CQ) [30], another commonly used compound that blocks autophagosome degradation, did not further increase LC3-II levels than ML-SA5 alone for both 4 h and 12 h (Figure 2A and S2A), whereas CQ application (10 μM) significantly facilitated the increase in LC3-II levels caused by starvation or rapamycin (50 μM) (Figure S2B). Likewise, ML-SA5 treatments (1 μM) further enhanced LC3-II increases caused by starvation, suggestive of an inhibitory effect of ML-SA5 on autophagy in the same way as CQ (Figure S2C). Furthermore, we examined the levels of SQSTM1, which is degraded efficiently by autophagy per se and therefore a marker of autophagic flux. Consistent with previous reports, both starvation (AA- and FBS-free medium) and treatment with rapamycin resulted in a profound reduction in SQSTM1 levels (Figure 2A). This indicates the active induction of autophagy and complete autophagic flux. In sharp contrast, either ML-SA5 (1 μM) or CQ (10 μM) alone or in combination markedly increased SQSTM1 levels, indicating that MCOLN1 activation arrested autophagic flux. Consistently with our results when utilizing ML-SA5 to stimulate MCOLN1 (Figure 2A), both SQSTM1 protein levels and puncta structures were significantly increased by overexpressing MCOLN1 (Figure 1G,H), suggesting that stimulating MCOLN1 by both the agonists and overexpressing the channel all leads to inhibited autophagy. Moreover, we assessed GFP turnover in cells stably expressing GFP-LC3 (S-GFP-LC3), which is another assay to monitor autophagic flux [31]. We found compromised GFP turnover following application of ML-SA5 (1 μM; Figure S2D) as CQ. Altogether, these results suggest that the activation of MCOLN1 blocks autophagic flux.
Figure 2.
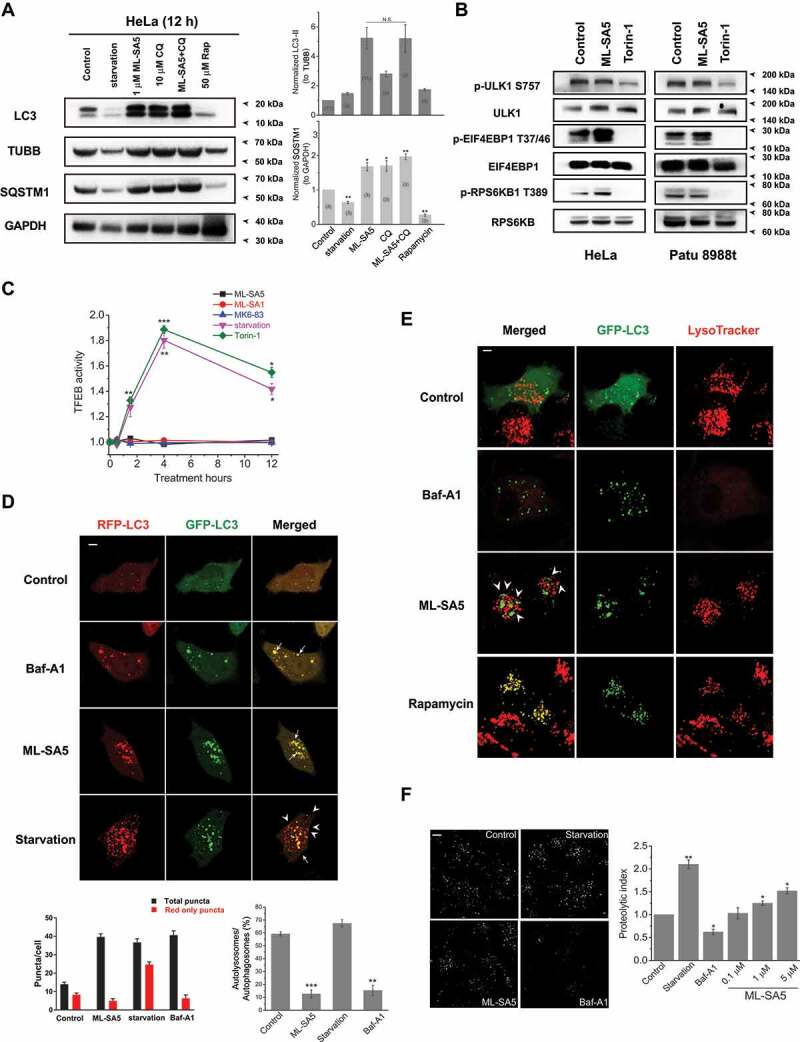
The activation of MCOLN1 arrests autophagic flux by disrupting fusion of autophagosomes and lysosomes. (A) The LC3-II and SQSTM1 levels were measured by western blot under treatments indicated in HeLa cells. Starvation (AA- and FBS-free) and rapamycin (50 μM) treatments served as positive controls for assessing SQSTM1. TUBB/beta-tubulin and GAPDH were loading controls for normalization of LC3-II and SQSTM1, respectively. (B) Phosphorylated (p-) and total ULK1, RPS6KB and EIF4EBP1 were assessed by western blot following ML-SA5 treatments (1 μM) in both HeLa and Patu 8988 t cells, respectively. Torin-1 (1 μM) treatments were positive controls for inhibiting phosphorylation of ULK1, RPS6KB and EIF4EBP1, respectively. Phosphorylated residues at S757 for ULK1, at T37/46 for EIF4EBP1 and at T389 for RPS6KB1 were investigated, respectively. All treatments were for 12 h. (C) TFEB activity was measured along conditions indicated by employing a TFEB-luciferase assay (see methods). The TFEB activities in HeLa cells under ML-SA5 (1 μM), ML-SA1 (20 μM), MK6-83 (5 μM), torin-1 (1 μM) and starvation (AA- and FBS-free) were normalized to control, respectively. n = 4. (D) Abundance of LC3 puncta as reflected by GFP and RFP fluorescence in transiently transfected tandem GFP-RFP-LC3 HeLa cells under different conditions indicated. The arrowheads indicate red puncta only, while the arrows illustrate yellow puncta (merged from red and green puncta). All treatments were for 4 h. Scale bar: 10 μm. The numbers of total (autophagosomes) and red only puncta (autolysosomes) and ratio of autolysosomes to autophagosomes were quantified from three independent experiments (typically n = 4–10 cells). Starvation was used as a positive control for inducing autophagy and Baf-A1 (1 μM) was used as a negative control for inhibiting autophagy. (E) Representative confocal images of co-localizations of autophagosomes (GFP-LC3 puncta) and lysosomes (LysoTracker) in HeLa cells under control, Baf-A1 (1 μM), rapamycin (50 μM) and ML-SA5 treatments (1 μM), respectively. The arrowheads indicate that green puncta and red puncta are adjacent to each other, but not fused. All treatments were for 4 h. Scale bar: 10 μm. (F) Representative confocal images of Cos-1 cells stained by DQ-red-BSA under treatments of control, starvation (AA- and FBS-free), ML-SA5 (1 μM), and Baf-A1 (1 μM), respectively. All treatments were for 4 h. Scale bar: 10 μm. Proteolytic index for different conditions were quantified from five to twenty images. Means ± SEMs are shown in panels A, C, D and F. Significant differences were evaluated using one-way ANOVA followed by Tukey’s test. *P < 0.05; **P < 0.01; ***P < 0.001
To confirm that autophagy initiation is not affected by ML-SA5, we measured levels of autophagy initiation in presence of ML-SA5 for 12 h, by assessing MTOR (mechanistic target of rapamycin kinase) complex 1 (MTORC1) activity. MTOR1 activity was not inhibited, since we observed even stronger phosphorylation on its substrate RPS6KB/S6K (ribosomal protein S6 kinase B) at T389 (p-RPS6KB1) [32] and on EIF4EBP1 (p-EIF4EBP1) at T37/46 residue [33] (Figure 2B). Moreover, phosphorylation of ULK1 (unc-51 like autophagy activating kinase 1) at S757 residue [34] was not altered by ML-SA5 (1 μM for 12 h) treatment, but significantly reduced following the treatment of torin-1 [35] (1 μM; Figure 2B), confirming that autophagosome formation is not affected by the activation of MCOLN1.
Next, we assessed the effects of ML-SA5 on TFEB, another downstream target of MTORC1. TFEB activity was not altered following ML-SA5 (1 μM), or ML-SA1 (20 μM), or MK6-83 (5 μM) treatments at 30 min, 90 min, 4 h and 12 h, as assessed by a TFEB-luciferase assay. On the other hand, starvation (AA- and FBS-free) or torin-1 treatments (1 μM) significantly increased TFEB activity, which validated the TFEB-luciferase assay (Figure 2C). Further, in comparison to nuclear localization triggered by torin-1 (1 μM; representative images from 90 min treatments), TFEB remained mostly in the cytosol under the ML-SA5 treatment, measured by both immunostaining (Figure S2E) and western blot analysis of subcellular fractionated experiments (Figure S2F). Moreover, we assessed changes in transcription levels of several TFEB downstream targets including SQSTM1, LAMP1 and CTSD (cathepsin D) following ML-SA5 treatments. We found that in contrast to starvation, ML-SA5 treatments (1 μM) did not alter the transcription of these TFEB’s common downstream targets (Figure S2G), implying that TFEB activity is not altered by ML-SA5 treatment. Finally, when reducing TFEB expression levels in HeLa cells by utilizing a TFEB siRNA, the effects of ML-SA5 on increasing LC3-II levels were not affected (Figure S2H), confirming that stimulating MCOLN1 inhibits autophagy independent of TFEB activity. In addition to this, levels of the ATG12–ATG5 complex, which could represent autophagy initiation levels, were not affected by ML-SA5 treatment (1 μM; Figure S2I). Taken together, all these results indicate that the activation of MCOLN1 arrests autophagy.
To further explore how the activation of MCOLN1 arrests autophagy, we used tandem RFP-GFP-LC3 transiently expressed HeLa cells to track autophagosomes and autolysosomes. The fluorescent tags GFP and RFP together emit yellow signals in pH-neutral autophagosomes; however, following fusion of autophagosomes and lysosomes, the GFP signal is quenched by the acidic environment of lysosomes, which results in “red signals only.” Therefore, if autophagic flux goes smoothly following ML-SA5 treatments, we expect to see an increase in the abundance of red puncta and less green puncta as acidic lysosomes quench green fluorescence. As expected, we found that starvation (AA- and FBS-free for 4 h; bottom panels) greatly increased red puncta (arrowheads) and less green puncta, which indicates activated autophagic flux (Figure 2D). In contrast to starvation, Baf-A1 (1 μM) or ML-SA5 (1 μM) treatments increased yellow puncta due to increase of both red and green puncta (arrows), which suggests blockage in the late step of autophagy (Figure 2D). This phenomenon could be interpreted as either ML-SA5 blocks fusion of autophagosomes and lysosomes or impairs lysosomal degradation capability. Therefore, we further examined the co-localization of LC3 puncta with lysosome markers by using transiently expressing GFP-LC3 HeLa cells. In comparison with rapamycin treatments, most of LC3 puncta (> 90%; arrowheads) induced by ML-SA5 treatment were LysoTracker (which specifically stains lysosomes)-negative (Figure 2E), thereby indicating that the fusion between autophagosomes and lysosomes is significantly impaired by application of ML-SA5. Similar results were observed in cells transiently overexpressing GFP-LC3 and mCherry-LAMP1 (lysosomal-associated membrane protein 1). Most of increased GFP-LC3 puncta following treatment of ML-SA5 (1 μM) did not co-localize with mCherry-LAMP1 (arrowheads; Figure S2J). These observations indicate that the activation of MCOLN1 significantly disrupts fusion between autophagosomes and lysosomes.
To investigate the possibility that reduced lysosomal degradation by ML-SA5 may cause the autophagic arrest, we used a DQ-BSA assay to assess the lysosomal degradation capability [5]. We found that in contrast to Baf-A1 treatments (1 μM), a lower dose of ML-SA5 (0.1 μM) did not affect lysosomal degradation capability, while slightly higher doses (1 and 5 μM) increased lysosomal degradation capability (Figure 2F). The results of DQ-BSA experiments excluded the possibility that ML-SA5 blocks autophagy by affecting lysosomal degradation capability. Because lysosomal acidic pH is crucial for lysosomal degradation, we also evaluated lysosomal pH in HeLa cells following ML-SA5 (1 μM for 12 h) treatments. Lysosomal pH appeared not to be altered by ML-SA5 application, assessed by LysoTracker staining (Figure S2K).
Altogether, these findings suggest that activating MCOLN1 unexpectedly arrests autophagy by disrupting fusion between autophagosomes and lysosomes.
Zinc is the downstream effector of MCOLN1 perturbing the fusion between autophagosomes and lysosomes
In the next series of experiments, we sought to identify the downstream effector(s) of MCOLN1 that is responsible for disrupting the fusion between autophagosomes and lysosomes. Because activation of MCOLN1 resulted in autophagosome accumulation within 1 h (Figure 1A), we hypothesized that the activation of MCOLN1 regulates autophagy by mediating the release of divalent cations to the cytosol from the lysosome, where Ca2+, zinc and Fe2+ accumulate [36]. Thus, we aimed to define the cationic effector of MCOLN1 by employing a variety of metal chelators. We found that TPEN, a membrane-permeable zinc chelator [37,38], completely reversed the ML-SA5 (1 μM)-induced increase in LC3-II levels following after 4 h, 12 h and 24 h of treatments (Figure 3A,B and S3A). Notably, in the assay, we used rather low concentration of TPEN (10 μM), which has much higher affinity to zinc over Ca2+ and Mg2+ (Kd Zn = 0.7 fM vs Kd Ca = 67 μM vs Kd Mg = 20 mM) [37]. Furthermore, 1,2-bis(2-aminophenoxy)ethane-N,N,N’,N’-tetraacetic acid tetrakis-acetoxymethyl ester (BAPTA)-AM (10 μM), which is a specific Ca2+ chelator [5,39], had little effect on increased LC3-II levels by ML-SA5, even after pre-treatment with BAPTA-AM (10 μM) (Figure 3A, S3A,B). These results suggest that zinc, but evidently not Ca2+ or Mg2+, might play a role in regulating autophagy by MCOLN1. Moreover, the effect of TPEN on abrogating regulation on autophagy by ML-SA5 is specific, as it was not able to reverse autophagic arrest induced either by Baf-A1 (1 μM) or by CQ (10 μM) (Figure 3A,B and S3C). The lower dose of TPEN at 1 μM also significantly inhibited the increases in LC3-II levels in HeLa cells (Figure S3D). However, it has been known that TPEN has high affinity with other metals as well such as Fe2+ (KdFe2+ = 2.4 fM) and Cu2+ (KdCu2+ = 17 zM). Therefore, to further confirm the role of zinc and to rule out involvement of Fe2+ and Cu2+ in terms of regulating autophagy, we moved on testing other membrane-permeable cation chelators including 1,10 phenanthroline (1,10 Phe), deferoxamine (DFX), N,N’,N’-tris(2-pyridylmethyl)-l,3,5-cis,cis-triaminocyclohexane (tachpyr), and tetrathiomolybdate (TTM). 1,10 Phe has similar affinity to zinc and Cu2+ over other divalent cations at neutral pH within cells [40]. We observed that application of 1,10 Phe (500 μM) significantly reversed the increase of LC3-II by ML-SA5 (1 μM) (Figure 3C-E), thus being able to confirm the role of zinc (or Cu2+) in MCOLN1-induced autophagy inhibition. Moreover, application of TTM (20 μM), a specific Cu2+ chelator [41,42], failed to abolish the effects of ML-SA5 on LC3-II levels (Figure 3C-E), thereby excluding the involvement of Cu2+ in regulating autophagy by MCOLN1. On the other hand, two iron chelators, DFX [43] (20 μM) and tachpyr [44] (20 μM) did not attenuate the effects of ML-SA5 on increasing LC3-II levels (Figure 3C-E). Since tachpyr has much higher affinity to Fe2+ over zinc [45], these results could exclude the possibility that Fe2+ involves in the process of regulating autophagy mediated by MCOLN1.
Figure 3.
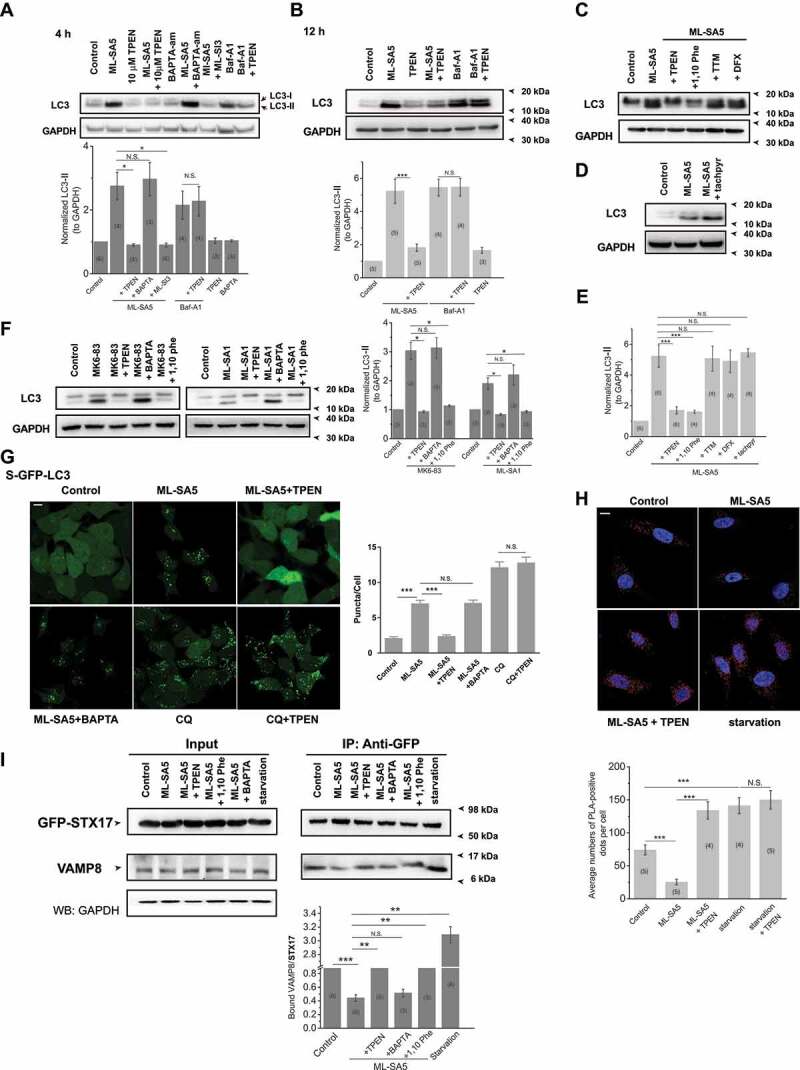
Zinc is the downstream effector of the activation of MCOLN1 that disrupts fusion of autophagosomes and lysosomes. (A, B) Increased LC3-II levels induced by 1 μM ML-SA5 for 4 h (A) or 12 h (B) in HeLa cells were profoundly inhibited by co-application of 1 or 10 μM TPEN, but not by 10 μM BAPTA-am. GAPDH served as a loading control. (C) TPEN (10 μM) and 1,10 phenanthroline (500 μM), but neither TTM (20 μM) nor DFX (20 μM), attenuated the effects of ML-SA5 (1 μM) on increasing LC3-II levels in HeLa cells. All treatments were for 4 h. (D) Application of tachpyr (20 μM) failed to rescue increased LC3-II levels by ML-SA5 (1 μM) in HeLa cells. (E) LC3-II levels were normalized under the control, ML-SA5 (1 μM), ML-SA5+ TPEN (10 μM), ML-SA5 + 1,10 Phe (500 μM), ML-SA5+ TTM (20 μM), ML-SA5+ DFX (20 μM) and ML-SA5+ tachpyr (20 μM). (F) Application of TPEN (10 μM) or 1,10 Phe (500 μM), but not BAPTA-AM (10 μM), completely rescued increased LC3-II levels caused by either MK6-83 (5 μM) or ML-SA1 (20 μM) treatments in HeLa cells. (G) Increased abundance of LC3 puncta by ML-SA5 (1 μM) was completely reduced to control levels by co-application of TPEN (10 μM), whereas increased puncta triggered by CQ (20 μM) were not affected by TPEN application (10 μM), as measured by GFP-LC3 puncta in GFP-LC3 stably expressing HEK 293 T (S-GFP-LC3) cells. All treatments were for 4 h. Statistical analysis of LC3 puncta per cell under different conditions indicated was quantified from typically 50–100 cells from at least three independent experiments. Scale bar: 10 μm. (H) HeLa cells transfected with GFP-STX17 were treated with different chemicals indicated for 8 h before probed with anti-GFP and anti-VAMP8 antibodies and subjected to PLA (red). The effective interaction between STX17 and VAMP8 was illustrated as red dots. Starvation (AA- and FBS-free) significantly increased red dots numbers whereas either ML-SA5 (1 μM), or MK6-83 (5 μM) or ML-SA1 (20 μM) treatments (Figure S3I) remarkably reduced red dots than control. Application of TPEN (10 μM) largely liberated reduced red dots caused by ML-SA5, but not affecting ones increased by starvation. Average numbers of PLA-positive dots were quantified from 69–152 cells from 4–5 independent experiments. Nuclei were indicated by DAPI staining. Scale bar: 20 μm. (I) Co-IP assays showing that interaction between STX17 and VAMP8 was significantly reduced by ML-SA5 (1 μM for 12 h) compared to the control in GFP-STX17-overexpressed HEK 293 T cells, while starvation (AA-and FBS-free for 12 h) increased the interaction. Application of TPEN (10 μM) or 1,10 Phe (500 μM) significantly reversed reduced interaction between Sxt17 and VAMP8 by ML-SA5, but not BAPTA-am (10 μM). GFP-STX17 was immunoprecipitated by GFP antibody, and thus endogenous VAMP8 were co-IPed with STX17. VAMP8 was then normalized to total GFP-STX17 and displayed in the bar graph. Means ± SEMs are shown in panels A, B, E, F, G, H and I. Significant differences were evaluated using one-way ANOVA followed by Tukey’s test. *P < 0.05; **P < 0.01; ***P < 0.001
Likewise, in contrast to BAPTA-AM (10 μM), application of TPEN (1 and 10 μM) or 1,10 Phe (500 μM) profoundly reduced the increases in LC3-II levels induced by ML-SA5 treatments in the cancer cell line, Patu 8988 t cells (Figure S3D), which is consistent with the observations in HeLa cells. Moreover, the increases in LC3-II levels induced by the treatment of MK6-83 (5 μM) and ML-SA1 (20 μM) were also completely inhibited by the application of TPEN (10 μM) or 1,10 Phe (500 μM) (Figure 3F). These results together confirmed that zinc, but not Ca2+, is the effector of MCOLN1-induced autophagy inhibition.
Furthermore, in consistence with changes in LC3-II protein levels, TPEN completely reduced puncta structures triggered by ML-SA5 (1 μM), but not by CQ (10 μM), in GFP-LC3 stably expressing HEK 293 T cells (S-GFP-LC3; Figure 3G). Likewise, the application of TPEN also profoundly reduced the increases in LC3 puncta structures induced by overexpressing MCOLN1 in mCherry-LC3 transiently expressed HeLa cells (Figure S3E). In addition to this, LC3 puncta structures triggered by ML-SA5 in S-GFP-LC3 cells were not affected by BAPTA-AM application (10 μM) (Figure 3G and S3F). Overall, these results indicate that zinc, but not Ca2+ or other cations, is the downstream effector engaged in MCOLN1-induced autophagic arrest.
MCOLN1 is emerging as an important player engaged in cellular zinc homeostasis, while zinc could potentially be a co-factor regulating MCOLN1 gating in the same way as zinc regulates P2X receptor [46]. In particular, previous studies have suggested that MCOLN1 is involved in zinc homeostasis by releasing lysosomal zinc to the cytosol [1,47–49]. We initially confirmed the effects of MCOLN1 on mediating zinc influx from the lysosome by visualizing zinc using a membrane-permeable zinc probe, FluoZin-3 AM (Figure S3G,H). Following ML-SA5 stimulation (1 μM), overloaded zinc (100 μM ZnCl2 loaded for 12 h) moved out of lysosomes and resulted in extremely reduced green fluorescence merged with lysosomes stained by LysoTracker (Figure S3G). Furthermore, cytosolic zinc concentrations were elevated within seconds following ML-SA5 stimulation (1 μM), as verified by zinc images (Figure S3H). These zinc staining and imaging experiments confirmed the involvement of MCOLN1 in mediating lysosomal zinc influx to the cytosol, which is consistent with a recent report suggesting that MCOLN1 mediates lysosomal zinc influx by using a new probe, GZnP3 [49]. Notably, the effect of ML-SA5 on releasing lysosomal zinc is much stronger than ML-SA1 in hippocampal neurons. This is supported by the results that ML-SA5, but not ML-SA1, was able to trigger lysosomal zinc release through endogenous MCOLN1, as detected by the GZnP3 probe. This could explain why the effect of ML-SA5 was greatly stronger than ML-SA1 on increasing LC3-II levels (Figure S1H).
We next tackled the puzzle as to how lysosomal zinc influx disrupts fusion of autophagosomes and lysosomes. Two soluble NSF (N-ethylmaleimide-sensitive factor) attachment protein) receptor (SNARE) proteins, STX17 (syntaxin 17) and VAMP8, have been reported to determine the fusion status of autophagosomes and lysosomes during the autophagy process [18]. Accordingly, we investigated whether zinc influx affects the interaction between these two SNARE proteins following ML-SA5 treatment. Here we used a proximity ligation assay (PLA) to detect the direct interaction between STX17 and VAMP8 under ML-SA5 treatments (see methods). In the PLA assay, we found that starvation (AA- and FBS-free) significantly increased interaction between STX17 and VAMP8, displaying as much more red dots than control, which validated the assay accurately assessing the interaction between STX17 and VAMP8 (Figure 3H). With the validated PLA assay, we found that application of either ML-SA5 (1 μM), ML-SA1 (20 μM), or MK6-83 (5 μM) significantly reduced red dots compared to control, indicating that the interaction between STX17 and VAMP8 is significantly disrupted by stimulating MCOLN1 channels (Figure 3H and S3I). Moreover, application of TPEN significantly reversed the reduction in red dots caused by ML-SA5, but not affecting ones increased by starvation (Figure 3H and S3I). These experimental data demonstrate that MCOLN1-mediated zinc influx disrupts the interaction between STX17 and VAMP8, thus impairing the fusion of autophagosomes and lysosomes and ultimately the autophagy process.
The physical interaction between STX17 and VAMP8 was further investigated using co-immunoprecipitation (co-IP) assays. Following methods described in details in (Figure S3J), ML-SA5 (1 μM) significantly inhibited the interaction between STX17 and VAMP8, while starvation (AA- and FBS-free) facilitates the interaction [18] (Figure 3I), which validated the co-IP assay. To further investigate whether zinc regulation on the interaction between STX17 and VAMP8 is direct, we added TPEN (10 μM), BAPTA-am (10 μM), and 1,10 Phe (500 μM) after the lysates have been collected, respectively. We found that application of TPEN or 1,10 Phe strongly attenuated eliminated interaction between STX17 and VAMP8 by ML-SA5, but not BAPTA (Figure 3I).
Collectively, these results demonstrate that zinc release from the lysosome mediated by MCOLN1 directly disrupts the interaction between STX17 and VAMP8 and resultant fusion process between autophagosomes and lysosomes.
Zinc influx originating from the extracellular fluid also arrests autophagy
Thus far we uncovered the mechanism by which zinc influx from the lysosome arrests autophagy. To further verify that zinc influx toward the cytosol is the main cause for autophagic arrest induced by MCOLN1 activation, we went on inducing zinc influx to the cytosol by an alternative approach. Here we applied a zinc ionophore, clioquinol [50], to allow zinc to enter the cytosol from the extracellular space (Figure S4A). Due to rapid cell death (~ 4 h) caused by high concentrations of ZnCl2 and clioquinol, we chose 20 μM ZnCl2 (clamped by 10 μM EDTA and 30 μM ZnCl2) (http://web.stanford.edu/~cpatton/webmaxS.htm) + 20 μM clioquinol as our highest test concentration. We found that zinc influx originating from the extracellular fluid specifically regulates autophagy with observation of an increase in LC3-II levels in western blot experiments (20 μM clioquinol + 20 μM ZnCl2; Figure 4A). The regulation on LC3-II levels by extracellular zinc was also dose- and time-dependent (Figure 4B,C). Consistently, LC3 puncta were profoundly induced by application of clioquinol in GFP-LC3 stably expressed HEK 293 T (S-GFP-LC3) cells (Figure 4D). Moreover, increased LC3-II levels and LC3 puncta formation triggered by clioquinol were completely inhibited by co-application of TPEN (10 μM) (Figure 4A-D), confirming that zinc influx originating from the extracellular fluid specifically regulates autophagy. CQ application (10 μM) did not further increase LC3-II levels than clioquinol alone (20 μM clioquinol + 20 μM ZnCl2; Figure 4E), whereas it facilitates LC3-II levels increased by starvation (AA- and FBS-free; Figure 4E), suggesting that zinc influx originating from the extracellular fluid arrests autophagy. Consistent with these observations, SQSTM1 levels were elevated following clioquinol application (20 μM clioquinol + 20 μM ZnCl2; Figure 4F). Furthermore, in contrast to starvation (middle panels in Figure 4G), green puncta and red puncta were equally increased by clioquinol application in GFP-RFP-LC3 transiently expressed HeLa cells (Figure 4G), suggesting that the late step of autophagy is blocked by zinc influx that originates from the extracellular fluid. Moreover, the fusion between autophagosomes and lysosomes was inhibited by clioquinol exposure (Figure S4B). Collectively, these results demonstrate that similar to zinc influx from the lysosome, zinc influx originating from the extracellular fluid also arrests autophagy by perturbing fusion between autophagosomes and lysosomes. Therefore, zinc influx, both from the extracellular fluid and the intracellular zinc stores such as lysosomes, arrests autophagy by disrupting fusion of autophagosomes and lysosomes.
Figure 4.
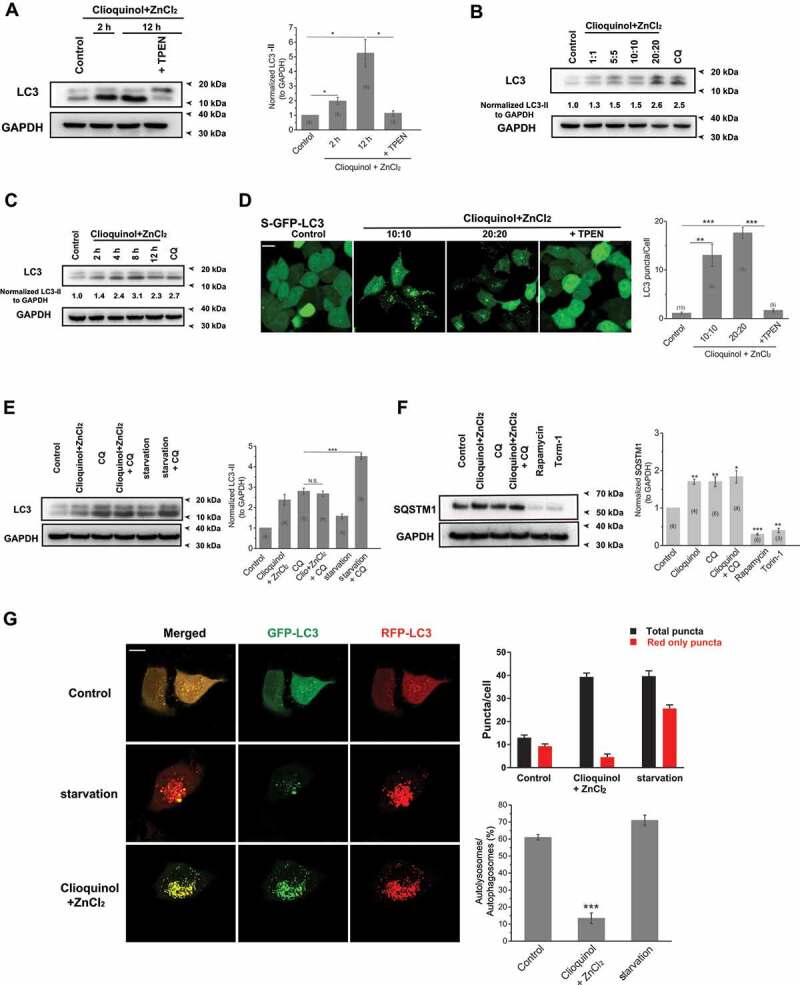
Zinc influx originating from the extracellular fluid arrests autophagy. (A) Zinc influx induced by co-application of ZnCl2 (20 μM) and clioquinol (20 μM) significantly increased LC3-II levels in HeLa cells, confirmed by the western blot experiments. TPEN (10 μM) profoundly reduced increased LC3-II levels induced by ZnCl2 and clioquinol. (B, C) The increases in LC3-II levels caused by co-application of ZnCl2 and clioquinol in HeLa cells were dose- (B; 4 h treatments) and time-dependent (C; 20 μM clioquinol + 20 μM ZnCl2 used here). (D) Co-application of clioquinol and ZnCl2 induced LC3 puncta in GFP-LC3 stably expressing HEK 293 T (S-GFP-LC3) cells and LC3 puncta structures were reduced by co-application of TPEN (10 μM). All treatments were for 4 h. Scale bar: 10 μm. (E) CQ (10 μM) did not further increase LC3-II levels than clioquinol alone (20 μM clioquinol + 20 μM ZnCl2) in HeLa cells, whereas it facilitated LC3-II levels increased by starvation (AA- and FBS-free). (F) SQSTM1 levels were measured under different treatments in HeLa cells including the control, 20 μM clioquinol + 20 μM ZnCl2, CQ (10 μM), CQ+ clioquinol + ZnCl2, rapamycin (50 μM), and torin-1 (1 μM). Rapamycin and torin-1 served as positive controls for inducing autophagy (reducing SQSTM1 levels). (G) In GFP-RFP-LC3 transiently expressed HeLa cells, application of ZnCl2 and clioquinol (20 μM+20 μM) caused both increased green and red puncta in the same pace, while starvation (AA- and FBS-free) induced much more red puncta over green puncta. All treatments were for 4 h. Scale bar: 10 μm. The numbers of total (autophagosomes) and red only puncta (autolysosomes) and ratio of autolysosomes to autophagosomes were quantified from three independent experiments (typically n = 6–13 cells). Error bars indicate Mean ± SEMs in panels A, D, E, F and G. Significant differences were evaluated using one-way ANOVA followed by Tukey’s test. *P < 0.05; **P < 0.01; ***P < 0.001
ML-SA5 is lethal to a variety of cancer cells, but has little effect on normal cells
Cancer cells actively undergo autophagy to conquer nutrients limitation and hypoxia for growth [51]. Therefore, autophagy inhibitors are becoming emerging therapeutics to combat cancer [52,53]. The level of basal autophagy is relatively high in pancreatic ductal adenocarcinoma (PDAC), renal cancer, mammary cancer, and bladder cancer cell lines, suggesting that these cancer cells might highly rely on autophagy for growth [54,55]. In addition, transcriptional levels of MCOLN1 in various human cancer cell lines or human cancerous tissues were significantly lower than the corresponding normal cell lines or human adjacent non-cancerous tissues, respectively (Figure S5A,B). Accordingly, we wondered whether the inhibitory effect on autophagy by activating MCOLN1 is able to influence the growth of PDAC. First, we investigated the effects of ML-SA5 treatment on Patu 8988 t, which is a human PDAC cell line [23]. Intriguingly, we found that cell viability of Patu 8988 t was not affected by application of ML-SA5 (1 μM) in medium containing 10% FBS (Figure S5C). However, when exposing Patu 8988 t cells to ML-SA5 in medium containing 2% FBS (data from 5% and 1% FBS not shown), we observed greater levels of cell death, as verified by Trypan blue staining (arrows indicate dead cells; Figure 5A). Under this particular starvation condition (2% FBS), autophagy in HeLa and Patu 8988 t cells was still arrested following application of ML-SA5 (1 μM), as confirmed by western blot experiments (Figure S5D). These results indicate that ML-SA5-induced cell death is restricted when cells have enough exogenous nutrients.
Figure 5.
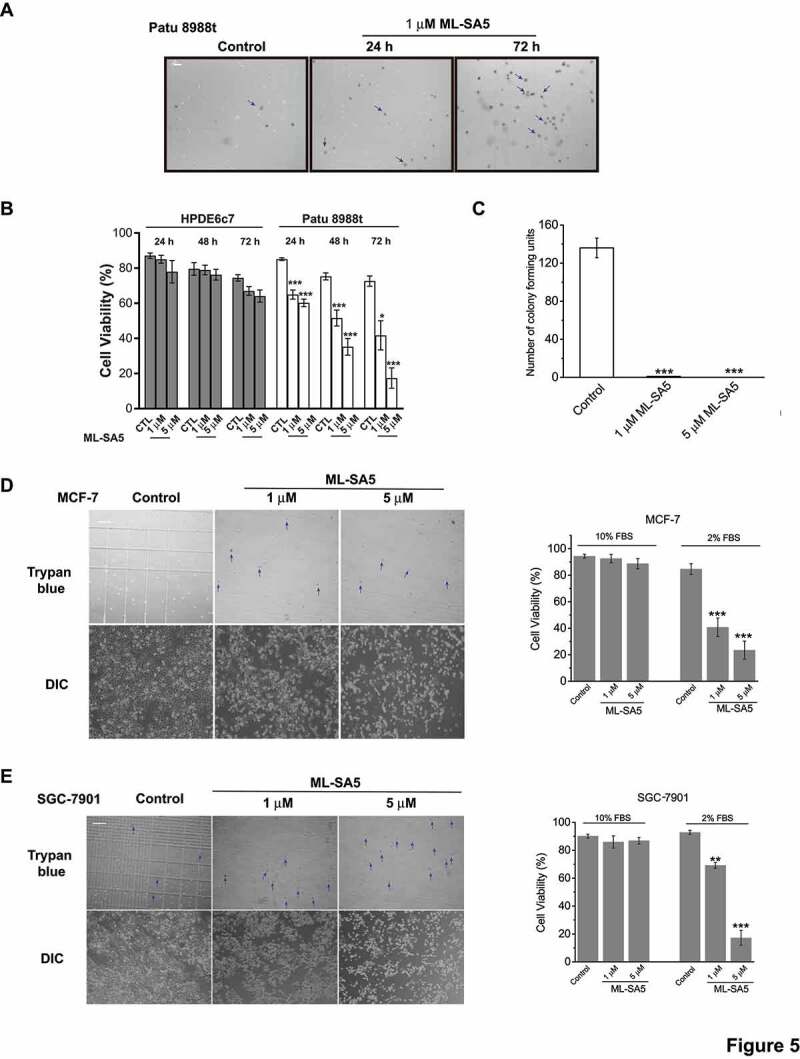
ML-SA5 is lethal to various cancer cells, but has little effect on normal cells. (A) ML-SA5’s application led to significant cell death in Patu 8988 t cells in medium containing 2% FBS, verified by Trypan blue assays. The arrows indicate dead cells stained by Trypan blue solution. Scale bar: 1 cm. (B) Quantifications of HPDE6c7 and Patu 8988 t cell viability under the control, 1 μM ML-SA5 and 5 μM ML-SA5 treatments for 24 h, 48 h, and 72 h, respectively. n = 20–40. (C) Patu 8988 t cell viability represented by colony formation assay were profoundly reduced by both 1 μM and 5 μM ML-SA5 treatments. (D, E) Application of Ml-SA5 led to significant cell death in MCF-7 (D) and SGC-7901 (E) cells, verified by Trypan blue assay. The arrows indicate dead cells stained by Trypan blue solution. Macroscopic DIC images showed that live cell populations in control were much greater than ML-SA5 groups. Scale bar: 1 cm. All treatments were for 48 h. Error bars indicate Mean ± SEMs in panels B, C, D, and E. Significant differences were evaluated using one-way ANOVA followed by Tukey’s test. *P < 0.05; **P < 0.01; ***P < 0.001
In addition to autophagic arrest mediated by ML-SA5 treatments causes cell death of Patu 8988 t, enhanced MTORC1 activities by ML-SA5 are also possible to trigger cell death (Figure 2B). Thus, to rule out the possibility that ML-SA5 causes cell death by enhancing MTORC1 activities, we utilized rapamycin, an MTORC1 inhibitor [56], to dispel enhancement of MTORC1 activities by ML-SA5. Co-treating cells with ML-SA5 (1 μM) and rapamycin (1 μM) significantly reduced cell viability in fed condition (10% FBS) compared to cells treated with either ML-SA5 (1 μM) or rapamycin (1 μM) alone (10% FBS) (Figure S5E). This result indicates that enhanced MTORC1 activity is not the cause of ML-SA5-induced cell death. It is conceivable that autophagic arrest occurs much more severely in the setting of either starvation or co-application of rapamycin, as both of these conditions will increase abundance of autophagosomes due to facilitated formation of autophagosomes (Figure S2I). Therefore, the elimination of enhanced MTORC1 activity may be a prerequisite to cell death triggered by ML-SA5-induced autophagic arrest (also see the discussion). In other words, ML-SA5 triggers cell death only when autophagy is actively undergoing.
We then systematically studied cell death caused by ML-SA5 in various cell types in medium containing 2% FBS. First, we monitored cell viability of HPDE6c7 cell, a normal human pancreatic duct epithelial cell line [57], in which the basal autophagy level is relatively lower [54] than Patu 8988 t cells (Figure S5F). Consistent with the observation in pancreatic cancer cell line, the basal autophagy in human pancreatic cancer tissues was much higher than the adjacent non-cancerous tissues (Figure S5F). The similar pattern was also observed in human gastric cancer (Figure S5F). ML-SA5 did not cause overt cell death in HPDE6c7 (Figure 5B). In either case of HPDE6c7 or Patu 8988 t cells, ML-SA5 (1 μM) or CQ (10 μM) further increased LC3-II levels (Figure S5G), indicating that ML-SA5 arrests autophagy in both of HPDE6c7 and Patu 8988 t cells, but to different extents according to their distinct basal autophagy levels.
On the other hand, cell death was triggered in Patu 8988 t cells following ML-SA5 treatment in a time- and concentration-dependent fashion (Figure 5A,B). To further verify cell death caused by treatments of ML-SA5, we performed a colony formation assay (Figure 5C and S5H) and a cell counting kit 8 (CCK 8) assay (Figure S5I), respectively. In both assays, ML-SA5 resulted in significant cell death in Patu 8988 t cells. Furthermore, ML-SA5 (1 μM for 48 h) was also lethal to another PDAC cell line, Panc-1 (Figure S5J). In addition to ML-SA5, we evaluated the regulatory effects of MK6-83 on cell viability of Patu 8988 t cells. We found that MK6-83 (5 μM) has comparable effects on cell lethality in Patu 8988 t cells compared to ML-SA5 (Figure S5K). These results establish a role of autophagic arrest induced by the activation of MCOLN1 in growth of pancreatic cancer.
Additionally, we observed that ML-SA5 had lethal effects on other cancer cell lines including MCF-7, a human mammary cell line (Figure 5D and S5L), SGC-7901, a human gastric cancer cell line (Figure 5E), A-375, a human malignant melanoma cell line, and U-87 MG, a human malignant glioma cell line (see the related manuscript). Notably, ML-SA5 was nontoxic when exposing to MCF 10A and GES-1 cells, a normal human mammary epithelial cell line and a normal gastric epithelial cell line, respectively (Figure S6A-D). Overall, similar to other autophagy inhibitors such as CQ and Baf-A1 (Figure S5M), ML-SA5 possesses potent anti-neoplastic activities against multiple cancers without affecting viability of normal cells. Interestingly, ML-SA5 had little effect on several lung cancer cell lines including A549 and NCI-H1299 cells, which might be due to their low reliance on autophagy for growth (Figure S6E,F).
We next explored the possible causes for the observed reduction of viability in cancer cell lines following ML-SA5 treatment. Firstly, to determine whether cell death triggered by ML-SA5 is mediated through autophagic arrest, we used 3-MA (10 mM; pretreatment for 2 h) to block initiation of autophagy prior to adding ML-SA5. We observed that Patu 8988 t cell death triggered by ML-SA5 was completely abrogated by pre-treatment with 3-MA (Figure 6A), in which autophagic arrest by ML-SA5 was already rescued (Figure S1L,M). Another PtdIns3K inhibitor, wortmannin (100 nM; pretreatment for 30 min), also significantly rescued ML-SA5-induced cell death in both Patu 8988 t and SGC-7901 cells (Figure S7A). Likewise, when disrupting autophagy initiation by knocking down of ATG5 in Patu 8988 t cells and MCF-7 cells (Figure S7B), cell death triggered by ML-SA5 was significantly reduced in the cells (Figure 6B and S7C). These results suggest that autophagic arrest by ML-SA5 is most likely the primary cause for cell death in the cancer cell lines studied. Secondly, in order to confirm the role of zinc influx mediated by MCOLN1 in cell death triggered by ML-SA5, we used lower dose of TPEN (1 μM) that was nontoxic to cancer cells and was able to reverse autophagic arrest by ML-SA5 (Figure S3D, S3E and S7D). We found that application of TPEN (1 μM) completely rescued cancer cell death caused by ML-SA5 (1 μM) not only in Patu 8988 t (Figure 6A), but also in SGC-7901 cells (Figure S7E), strongly supporting the notion that autophagic arrest induced by the zinc influx through MCOLN1 triggers cancer cells death. Thirdly, we confirmed the specific role of MCOLN1 in cell death induced by ML-SA5 in Patu 8988 t, MCF-7 and SGC-7901 cells by utilizing both an antagonist of MCOLN1, ML-SI3 (20 μM), and an MCOLN1 siRNA [58] (Figure S7F-H). Co-application of ML-SI3 (20 μM) or MCOLN1 siRNA remarkably rescued cell death triggered by ML-SA5 (1 μM) in Patu 8988 t (Figure 6A-C), MCF-7 (Figure S7I) or SGC-7901 (Figure S7J) cells. Taken together, these results indicate that the activation of MCOLN1 leads to cancer cell death by triggering zinc influx-mediated autophagic arrest.
Figure 6.
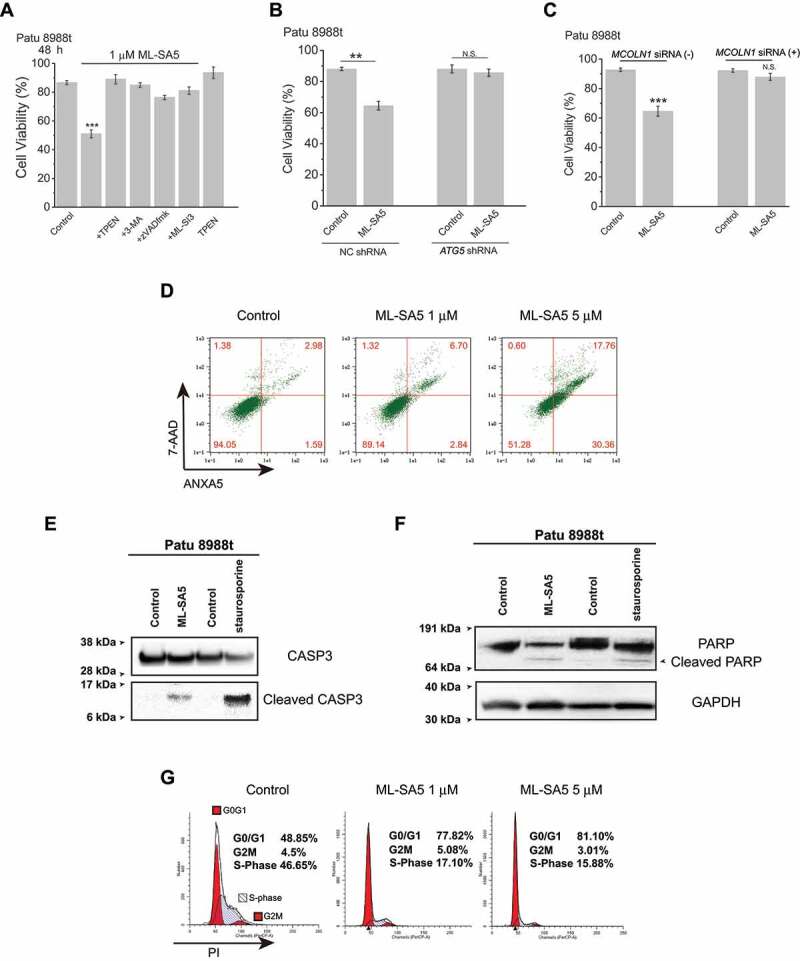
Following autophagic arrest mediated by ML-SA5, induction of apoptosis and cell cycle arrest triggers cell death in the cancer cells. (A) Patu 8988 t cell death triggered by 1 μM ML-SA5 (48 h) were strongly rescued by co-application of either 3-MA (10 mM, pretreatment for 2 h), or TPEN (1 μM), or zVADfmk (20 μM), or ML-SI3 (20 μM), respectively. n = 10–40. (B) Knockdown of ATG5 genes significantly attenuated cell death triggered by ML-SA5 treatments (1 μM for 48 h) in Patu 8988 t cells. n = 3. (C) ML-SA5 was not lethal to Patu 8988 t cells when MCOLN1 was efficiently knocked down by the MCOLN1 siRNA, evaluated by Trypan blue assay. n = 3–5. (D-F) Induction of apoptosis following ML-SA5 treatments (1 μM) in Patu 8988 t cells was confirmed by detection of ANXA5/annexin V-7-AAD (D), cleaved CASP3 (caspase 3) (E) and cleaved PARP (F). All treatments were for 48 h. (G) Flow cytometry images displaying that G0/G1 phase arrest was triggered upon application of ML-SA5 for 48 h at both 1 μM and 5 μM in Patu 8988 t cells in medium containing 2% FBS, stained by PI. Error bars indicate Mean ± SEMs in panels A, B and C. Significant differences were evaluated using one-way ANOVA followed by Tukey’s test. **P < 0.01; ***P < 0.001
On the other hand, as a subsequent event, apoptosis usually occurs [59], and can therefore contribute to cell death following autophagic arrest. In our hands, we observed that apoptosis was strongly induced after 48 h ML-SA5 treatments in Patu 8988 t cells in medium containing 2% FBS. This was confirmed by both flow cytometry and activities of CASP3 (caspase-3) and PARP (Figure 6D-F). Co-application of zVADfmk [60] (20 μM) that is a caspase inhibitor with ML-SA5 significantly reduced cell death induced by ML-SA5 in Patu 8988 t cells (Figure 6A), confirming that apoptosis following autophagic arrest is one of the causes of cell death triggered by ML-SA5. Notably, in fed condition, ML-SA5 did not induce apoptosis (Figure S7K), indicating that certain/threshold levels of autophagic arrest are required to induce apoptosis. In addition to assessing cell apoptosis, cell cycles were monitored in Patu 8988 t cells by flow cytometry. We found that ML-SA5-induced autophagic arrest resulted in G0/G1 phase arrest when autophagy was highly active (Figure 6G and S7L). These results confirmed that following autophagic arrest induced by the activation of MCOLN1, the set of induced apoptosis and arrested cell cycle participates in impairing viability of the cancer cells.
Our cell culture experiments indicate that ML-SA5 possesses potent anti-tumor cell activities without affecting normal cells. We then sought to test anti-neoplastic potentials of ML-SA5 in vivo by using Patu 8988 t xenografts mice model. Consistent with the in vitro results of experiments on Patu 8988 t cell viability, administration of ML-SA5 (5 μM) dramatically improved mouse survival and suppressed tumor growth in xenografted mice (Figure 7A,B). After 14 d of ML-SA5 intratumoral injection (5 μM), the final tumor volume of ML-SA5 group was reduced to half the size of tumors in the control group (Figure 7C and S7M). And administration of ML-SA5 remarkably suppressed tumor growth likely through inhibiting autophagy through MCOLN1, as autophagy inhibition is strongly triggered in the tumor tissues from ML-SA5 group, reflected by the increased LC3 and SQSTM1 puncta structures in the tumor tissues (Figure 7D-E and S7N). Furthermore, growth inhibition and apoptosis were demonstrated by a reduction in the proliferation marker MKI67/ki67 (Figure 7D-E) and an increase in TUNEL staining (Figure 7D-E) in tumor tissues from ML-SA5 group, respectively. Due to toxicity of 3-MA and TPEN, only co-administration of zVADfmk (20 μM) or ML-SI3 (20 μM) with ML-SA5 (5 μM) was performed. Both zVADfmk and ML-SI3 strongly reversed delayed tumor growth caused by ML-SA5 administration (Figure 7F and S7O). In addition, in comparison with the profoundly reduced tumor sizes by the ML-SA5 administration in mice injected with a negative control (NC) shRNA, the sizes of tumors injected with a lentiviral-MCOLN1 shRNA (LV-MCOLN1 shRNA) were not significantly affected by the ML-SA5 administration (Figure 7G). These results confirmed the specific role of MCOLN1 in tumor growth.
Figure 7.
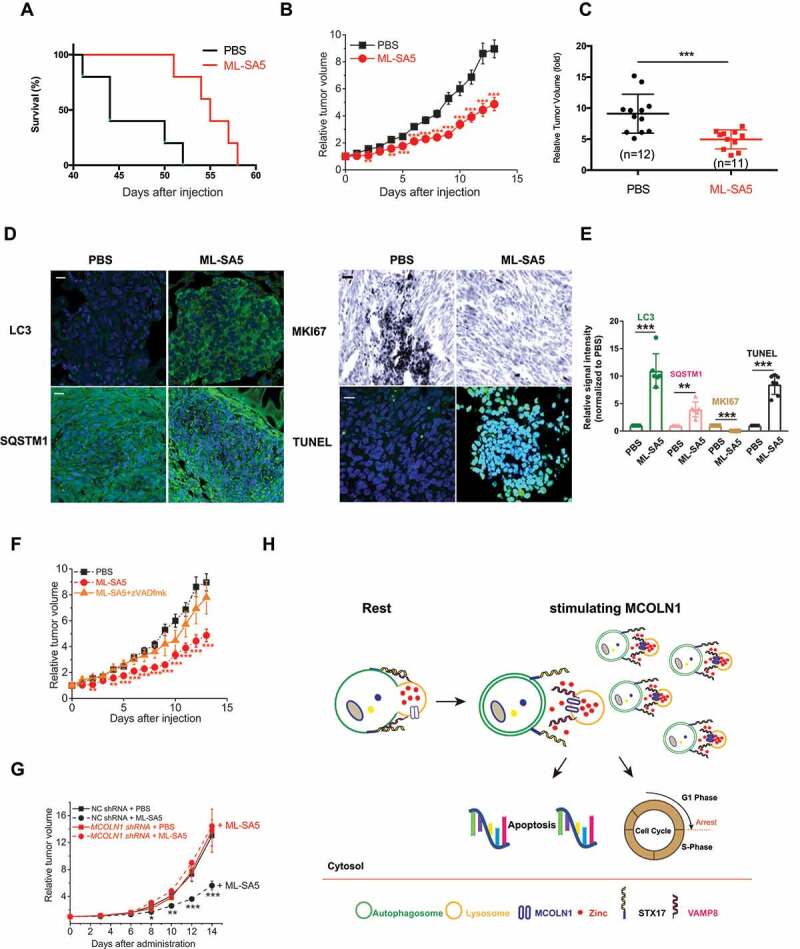
Administration of ML-SA5 profoundly promotes survival and suppresses tumor growth in Patu 8988 t xenograft mice. (A) ML-SA5 administration (5 μM) improved survival of mice affected by Patu 8988 t. Two-tailed logrank, **P = 0.0066. (B) The tumor xenograft growth in PBS injection group and ML-SA5 (5 μM) injection group were monitored over the course of 14 d, respectively. n = 11–12. (C) Final relative tumor volumes were compared between PBS injection group (n = 12) and ML-SA5 (5 μM; n = 11) injection group (intratumoral injection) and shown as Mean ± SEMs. Average values were indicated by solid lines among scattered solid circles. ***P < 0.001. (D) Representative sections of Patu 8988 t tumors from PBS injection group and ML-SA5 injection group stained for LC3, SQSTM1, TUNEL, and MKI67, respectively. Scale bar: 1 μm. (E) Intensity of LC3, SQSTM1, TUNEL, and MKI67 in tumor tissues from ML-SA5 group were normalized to PBS group, respectively. (F) The suppressed tumor growth by ML-SA5 (5 μM) injection was dramatically reversed by co-injection of zVADfmk (20 μM), while injection of zVADfmk alone did not affect tumor growth (see Fig. S7O). PBS injection and ML-SA5 injection groups were replotted from (B) and displayed as dashed lines. n = 5–12. (G) ML-SA5 administration (5 μM) for 14 d significantly reduced the sizes of the tumors in mice injected with a LV-NC shRNA, while it did not significantly affect the sizes of the tumors in mice injected with a LV-MCOLN1 shRNA. n = 7–8. (H) The working model illustrates molecular mechanism by which the activation of MCOLN1 arrests autophagy and triggers cancer cell death by evoking subsequent apoptosis and cell cycle arrest. Error bars indicate Mean ± SEMs. Significant differences were evaluated using one-way ANOVA followed by Turkey’s test. **P < 0.01; ***P < 0.001
Discussion
In this study, we uncovered the molecular mechanism by which stimulating MCOLN1 arrests autophagy (Figure 7H). Activation of MCOLN1 induces lysosomal zinc influx to the cytosol. The zinc influx in turn perturbs fusion of autophagosomes and lysosomes by eliminating the interaction between STX17 and VAMP8, which ultimately arrests autophagy. Subsequently, arrested autophagy leads to induction of apoptosis and G0/G1 cell cycle arrest, all of which confer ML-SA5 potent anti-neoplastic activities against multiple cancer cells.
MCOLN1 is a nonselective cationic channel and executes functions attributed to its mediation on releases of lysosomal Fe2+ [1] and Ca2+ [15,61,62]. By mediating lysosomal zinc release, our study proposes a new function of MCOLN1 on autophagy. However, it is conceivable that various cationic fluxes through MCOLN1 underlie their corresponding functions, respectively, upon MCOLN1’s opening. Therefore, when studying a particular function related to MCOLN1, we have to be extremely cautious to distinguish where it comes from by using specific chelators. For instance, we used relatively low concentrations of TPEN (1–10 μM) and 1,10 phenanthroline as zinc-specific chelators, BAPTA as Ca2+-specific chelator, deferoxamine and tachpyr as iron chelators, and tetrathiomolybdate as Cu2+ chelator to dissect possible involvements of these cations in the autophagy process mediated by MCOLN1. With the results from these chelators, we are therefore able to draw the conclusion that MCOLN1-induced autophagy inhibition is regulated by zinc influx.
On the one hand, our conclusion, to some extent, contradicts with the hypothesis that lysosomal Ca2+ exit through MCOLN1 promotes autophagy by activating PPP3/calcineurin and subsequent de-phosphorylation of TFEB [13,63]. Thus, we carefully investigated TFEB activity under ML-SA5 treatments. ML-SA5 application did not change TFEB activity for different periods (Figure 2C), while starvation and torin-1 treatments significantly triggered TFEB activity. Moreover, we investigated TFEB subcellular localization under ML-SA5 treatments for 30 min, 90 min and 4 h as well by immunostaining experiments. We observed that TFEB mostly remained in the cytosol (> 90%; images for 90 min were shown), which is sharply contrast to nuclear localization of TFEB triggered by the torin-1 treatments (Figure S2E). In addition, we investigated TFEB subcellular localization by western blot analysis of sub-cellular fractionated experiments (Figure S2F). Further, changes in transcriptions of several TFEB downstream targets including SQSTM1, LAMP1 and CTSD upon ML-SA5 treatment were assessed. We found that in contrast to starvation, ML-SA5 treatments did not alter either of transcription of these TFEB’s common downstream targets (Figure S2G), implying that TFEB activity is not altered by ML-SA5 treatments. Collectively, with all these assays, we conclude that ML-SA5 treatments regulate autophagy through MCOLN1 channels independent of TFEB activity. These observations were supported independently by the results that stimulating MCOLN1 channels by overexpressing the channels does not significantly trigger TFEB nuclear translocation reported by Zhang at al [64].
In agreement with the statement of Scotto Rosato et al., MCOLN1’s regulation in autophagy is independent of TFEB activity [63]. Our findings have suggested that the activation of MCOLN1 disrupts the fusion between autophagosome and lysosome, however, Scotto Rosato et al., proposed an opposite effect of MCOLN1 on promoting autophagy initiation. There are also a few reports suggesting that overexpressing MCOLN1 blocks autophagosome-lysosome fusion. This was determined by the RFP-GFP-LC3 tandem plasmid and displayed as the disrupted autophagic flux [65]. Those results highly agree with our observations.
As autophagy is a dynamic process, either promoted initiation or blocked autophagic flux leads to increased autophagosome accumulation, represented as increased LC3-II levels or LC3 puncta structures. Therefore, together with evaluating LC3 levels, approaches such as SQSTM1 measurements and tracking autophagic flux by monitoring the tandem GFP-RFP-LC3 are required to illustrate how exactly autophagy is regulated. In addition, monitoring autophagy parameters at different timing points might not be able to catch all of the dynamic changes of the autophagic flux. As such, the prolonged fusion disruption, displayed as autophagosome accumulation, might be read as the changes in autophagy initiation. Also, the different observations on the role of MCOLN1 in autophagy might be explained by differences in the experimental conditions used and levels of MCOLN1 in various cells used by the different groups.
On the other hand, presumably, lysosomal Ca2+ should be beneficial for fusion of autophagosomes and lysosomes, as cytosolic application of Ca2+ does promote interaction between STX17 and VAMP8 (data not shown), even though we do not know the origin of the Ca2+ involved in the process. However, following stimulation of MCOLN1, the ultimate outcome is the arrested autophagy due to the overwhelming regulation of zinc over Ca2+ on fusion of autophagosomes and lysosomes, as regulation on autophagy by ML-SA5 is not abrogated by application of BAPTA-am at all, but completely reduced by TPEN or 1,10 phenanthroline. Our study suggests that zinc can oppose the effects of Ca2+ on the interaction of STX17 and VAMP8 when zinc and Ca2+ are simultaneously extruded from the lysosome following MCOLN1 activation. However, it is unlikely to illustrate and dissect individual effects of zinc or Ca2+ on regulating autophagy separately in physiological context. The different environmental cues may mobilize MCOLN1 to release the corresponding cations for regulating various cellular events, which need to be elucidated experimentally. Nevertheless, we do establish a new fundamental role of zinc in membrane trafficking. This conclusion is strongly supported by the results on the effect of zinc influx from the extracellular fluid that arrests autophagy by the same mechanism as lysosomal zinc. These results together suggest that zinc does regulate fusion process between autophagosomes and lysosomes.
Our results have demonstrated that activation of MCOLN1 by ML-SA5 enhances MTORC1 activities as confirmed by increased levels of RPS6KB phosphorylation (Figure 2B), thereby implying that autophagosome formation is, in part, suppressed by the activation of MCOLN1. In other words, activation of MCOLN1 suppresses autophagy initiation on one hand and prevents fusion of autophagosomes and lysosomes on the other hand. Upon activation of MCOLN1, only when inhibited formation of autophagosomes is relieved by the specified conditions such as starvation and rapamycin treatments, then available autophagosomes accumulate and ultimately lead to induction of apoptosis and cell cycle arrest. These dual-effects by ML-SA5 nicely set up a “hurdle” for over-regulation of autophagy via MCOLN1 activation. This mechanism could explain why cell death did not occur in non-cancer cells in which basal autophagy is rather low. Thus, these findings suggest that ML-SA5 administration will be highly effective only in nutrient- and oxygen-deprived environments such as the environment of tumor, thus giving ML-SA5 great clinical utility potential.
Another important finding of our work is that lysosomal zinc engages in fusion of autophagosomes and lysosomes by eliminating the interaction between STX17 and VAMP8. To our knowledge, this is the first report that identifies a role for zinc in organelle trafficking. This regulation applies to zinc influx originating from the extracellular fluid, and presumably also to other intracellular sources of zinc such as the unidentified intracellular zinc stores reported [66]. Zinc plays a critical role in a vast array of cellular functions including gene expression, enzymatic activities, and stability of proteins. Here we provide the evidence that zinc prevents the interaction between STX17 and VAMP8 and resultant fusion process between autophagosomes and lysosomes [67]. However, the mechanism by which zinc prevents the interaction between STX17 and VAMP8 remains unknown. We postulate that zinc may participate in regulating the conformation of one or both of STX17 and VAMP8, as it does in another SNARE protein, TMEM163/SV31 [68]. However, this theory needs to be proved experimentally in the near future. HOPS, a well-known tethering complex, is essential for autophagosome and lysosome fusion events [69]. And it has been reported that the HOPS complex regulates autophagosome-lysosome fusion through interaction with STX17 [70,71]. It is possible that the HOPS complex, which contains a C-terminal zinc finger, is recruited by the zinc flux mediated by MCOLN1 to regulate the interaction between STX17 and VAMP8. Given that the complexity of the molecular mechanism or protein conformation changes on how the zinc regulates the fusion process between autophagosomes and lysosomes, this needs to be evaluated on a more granular level. Nevertheless, it does not change our finding that zinc flux through MCOLN1, as a mediator, prevents the interaction between two SNARE proteins. On the other hand, owing to the feature of arresting autophagy, elevating cytosolic zinc levels appears to be another efficient way to combat cancer. Thus, taking advantage of zinc on triggering cell death without affecting normal cells through specifically arresting autophagy in cancer cells can be a novel and new therapeutic approach to prevent tumor growth. However, an overdose of zinc is rather toxic as it leads to mitochondrial dysfunction and increases ROS levels [72]. Therefore, it is essential to perform additional studies using zinc ionophores such as clioquinol and zinc pyrithione for therapeutic purposes.
This study also sheds a new light on developing specific MCOLN1 agonists as a new type of autophagy inhibitor that can be used to combat cancer. Over the past decade, our depth of knowledge and understanding of therapeutic potential of autophagy inhibition for treating cancer has vastly been improved. To this end, more pharmacological molecules that are designed to suppress autophagy have been developed such as 3-MA, wortmannin, LY294002, CQ, and hydroxychloroquine (HCQ) [53]. For example, class III of phosphoinositide 3 kinases (PtdIns3Ks) inhibitors including 3-MA, wortmannin and LY294002 prevent autophagosome formation and in turn arrest autophagy. However, those inhibitors are not specific to inhibit autophagy and paradoxically activate autophagy on higher doses. In particular, CQ and its derivative HCQ that are FDA-approved anti-malaria drugs, have been extensively studied and tested in clinical trials. Although CQ and HCQ showed moderate anti-neoplastic effects, their effects largely come from the modulation of pathways other than autophagy inhibition. Moreover, the mechanism by which CQ and HCQ inhibit autophagy is still not understood. Therefore, developing molecules that specifically regulate autophagy will surely broaden clinical utility in combating cancer. Here we have reported that the MCOLN1 agonists, ML-SA1, ML-SA5, and MK6-83, specifically arrest autophagy by perturbing fusion of autophagosomes and lysosomes. Moreover, we have demonstrated that these agonists possess potent anti-neoplastic effects without affecting normal cells, therefore giving MCOLN1 great potential for clinical therapeutic use.
In summary, we have identified a regulatory network that was activating MCOLN1 channels arrests autophagy to trigger cancer cell death. Importantly, this network provides two clear targets, MCOLN1 and cytosolic zinc levels, that can be exploited as novel approaches to treat cancer.
Materials and methods
Mammalian cell culture and transfection
HeLa cells, Cos-1 cells, HEK 293 T cells, Patu 8988 t cells, and HPDE6c7 cells were purchased from Shanghai Gefan Biotechnology. Co., Ltd. (GE042, GE098, GE102, GE322 and GE324) MCF-7, MCF-10A, SGC-7901, and GES-1 were obtained from KeyGEN BioTECH (KG031, KG218, KG520 and KG552). GFP-LC3 stable HEK 293 T cells were a gift from Dr. Ling Chen at Guangzhou Institutes of Biomedicine and Health, Chinese Academy Sciences and atg12 KO NRK cells were a gift from Dr. Li Yu at the School of Life Science in the Tsinghua University. MCOLN1 KO HAP1 cells were purchased from Horizon discovery (C361). All cells were grown at 37°C in a 1:1 mixture of Dulbecco’s modified Eagle’s medium (DMEM; Gibco, 11,965,175) supplemented with 10% fetal bovine serum (FBS; Gibco, 10,091,155) in a humidified 5% CO2 incubator. Unless indicated otherwise, experiments of cell viability measurement were performed with the following timeline: cells (~20 x 104/ml) were plated in medium with 10% FBS. After 24 h, cells were changed to 2% FBS with or without drugs. Treatments were applied for 24 to 72 h periods as indicated. For transfection, cells were transfected with Lipofectamine 2000 (Invitrogen, 11,668,027). Culture media were refreshed 18–24 h post-transfection, and cells were imaged 48 h post-transfection to allow sufficient recovery from transfection stress.
Plasmids and siRNAs
GFP-LC3 (21,073), mCherry-LAMP1 (45,147), GFP-MCOLN1 (62,960), TFEB promoter-luciferase reporter (66,801), and GFP-RFP-LC3 (84,573) were obtained from Addgene (deposited by Drs. Tamotsu Yoshimori, Amy Palmer, Paul Luzio, Albert La Spada and Noburu Mizushima, respectively). mCherry-VAMP8 and GFP-STX17 were kindly provided by Drs. Hong Zhang (National Institute of Biological Sciences, Beijing, China), Edward A Fon (Department of Neurology and Neurosurgery, McGill University), Amy Kiger (Department of Cell & Developmental Biology, University of California, San Diego), and Qiming Sun (School of Basic Medical Sciences, Zhejiang University). Negative control and MCOLN1 siRNAs [28,58] were synthesized from GenePharma company. The ATG5-specific target sequence used was 5-AAGCAACTCTGGATGGGATTGC-3, corresponding to nucleotides 744–765 of the human ATG5 mRNA. The human TFEB siRNA#1 (5′-AGACGAAGGUUCAACAUCA-3′) and human TFEB siRNA #2 (5′-CUACAUCAAUCCUGAAAUG-3′) were tested to knock down TFEB expression levels. The MCOLN1 shRNA was inserted into a lentiviral vector for in vivo injection.
Western blotting
Standard western blotting procedures were used. Antibodies used for western blots include anti-LC3 (Sigma-Aldrich, L8918), anti-TUBB/beta-tubulin (Cell Signaling Technology, 2146), anti-SQSTM1 (Cell Signaling Technology, 5114), anti-VAMP8 (Cell Signaling Technology, 13,060), anti-CASP3/caspase 3 (Cell Signaling Technology, 9662), anti-cleaved CASP3 (Cell Signaling Technology, 9661), anti-PARP (Cell signaling Technology, 9542), anti-CTSD/cathepsin D (Cell Signaling Technology, 2284), anti-GAPDH (Cell Signaling Technology, 2118), anti-ATG5 (Cell Signaling Technology, 2630), anti-Histone (Cell Signaling Technology, 9715), and anti-GFP (Abcam, ab290).
qPCR analysis
Total RNA from various cancer cells and corresponding normal cells was isolated using RNAiso Trizol reagent (Sangon Biotech, China), and reverse-transcribed with PrimeScript™ RT Master Mix (Takara, Japan) according to the manufacturer’s instructions. Then a TB GreenTM Premix EX TaqTM II (Takara, Japan) in a LightCycler 480 qPCR instrument (Roche) was used for qRT-PCR. qRT-PCR was performed in triplicate and the results were normalized against GAPDH. Relative fold expressions were calculated with the comparative threshold cycle (2-ΔΔCt) method. The sequences of all primers for qRT-PCR are listed in below:
mMCOLN1 forward: 5′AAACACCCCAGTGTCTCCAG-3′
mMCOLN1 reverse: 5′GAATGACACCGACCCAGACT-3′
rMCOLN1-F:TCCATGTGACAAGTTCCGGG
rMCOLN1-R:AGAGGTGTCGGAAGGCAATG
hMCOLN1-F:TCTTCCAGCACGGAGACAAC
hMCOLN1-R:GCCACATGAACCCCACAAAC
Co-immunoprecipitation
Transfected cells from 10 cm dishes were incubated with 1 ml of NP40 lysis buffer (1% NP-40 [Sigma-Aldrich, 9,016,459], 137.5 mM NaCl [Sigma-Aldrich, S9988], and 10% glycerol [Sigma-Aldrich, G5516] in 20 mM Tris-HCl [Sigma-Aldrich, 1,185,531], adjusted to pH 7.4) for 30 min at 4°C. Lysates were centrifuged at 16,000 g for 10 min, and supernatants were incubated with 1 μg of primary antibodies at 4°C for 12 h. After addition of 20 μl of protein A/G plus-agarose (Santa Cruz Biotechnology, sc-2003), lysates were incubated at 4°C for 1 h with gentle shaking. Agarose beads were then collected through centrifugation at 500 g for 5 min. Beads were washed three times thoroughly in lysis buffer, and then heated to 95°C for 5 min in NuPAGE loading buffer (ThermoFisher Scietific, NP0007). Proteins were blotted with anti-GFP (Cell Signaling Technology, 2555) and anti-VAMP8 (Cell Signaling Technology, 13,060; 1:500).
TFEB-luciferase assay
The luciferase reporter gene assay was measured as described previously [73]. Briefly, HEK 293 T cells were seeded in a 35-mm dish to reach 80–90% confluence for overnight. TFEB promoter-luciferase reporter plasmid was transfected into the cells with the aid of Lipofectamine 2000. Cells were then additionally co-transfected with pGL4.75 (Renilla-luc) for normalization. After 6 h post-transfection, cells were seeded in a 12-well plate and grown for another 16 h with DMEM containing 10% FBS. Then the cells were treated with different drugs as indicated. Luciferase and Renilla activities were determined using a Dual-luciferase Reporter Assay kit (Promega, E1910). The results were averaged from at least three independent experiments.
Trypan blue assay
Freshly detached cells were diluted with same volume of trypan blue solution. Ten μL of sample was loaded into an hemacytometer and examined immediately under a microscope at low magnification. The number of blue staining cells and the number of total cells were counted. Cell viability was determined as [1.00 – (Number of blue cells/Number of total cells)].
CCK 8 assay
Cells were plated in a 96-well plate (100 μl/well). Ten μl of the CCK-8 solution (Dojindo, CK04) was added to each well of the plate. Plates were incubated for 1–4 h in the incubator and the absorbance was measured at 450 nm using a microplate reader.
Colony formation
An appropriate number of Patu 8988 t cells per well were seeded on a 6-well plate. Cells were incubated for 12 h in a CO2 incubator at 37°C to allow cells to attach to the wells. Cells were then treated with drugs as indicated for approximately 2 weeks until cells in the control well had formed colonies. Medium was removed, cells were washed with PBS (ThermoFisher Scientific, 10,010,049), and fixed with 4% PFA (ThermoFisher Scientific, R37814). Finally, cells were stained with 0.3% crystal violet for 1 h.
Flow cytometry
For flow cytometry analyses, Patu 8988 t cells were treated with different drugs as indicated. Cells were collected, washed with PBS (ThermoFisher Scientific, 10,010,049) twice, and stained using ANXA5/annexin V-7-AAD (Sigma-Aldrich, HR8285) for apoptosis assay, H2DCFDA (ThermoFisher Scientific, D399) for ROS measurement, and propidium iodide (PI; ThermoFisher Scientific, P1304) for cell cycle assay, according to the manufacturer’s instructions. Stained cells were analyzed with a BD FACSAria III flow cytometer and data was processed using FlowJo software.
Mouse xenografts
Experiments on mice were performed in accordance with Xuzhou Medical University Animal Facility and Use Committee-approved protocols. 1–5 × 106 Patu 8988 t cells were re-suspended in 50% Matrigel (BD Biosciences, 356,234) to a final volume of 100 μL and then injected slowly into the subcutaneous tissue of ~6-week-old BALB/c nude mice. When diameter of tumors reached 10 mm, PBS or ML-SA5 was injected daily into the solid tumors. Tumor volumes were measured daily and calculated by using the equation (length X width2)/2 for 14 d. Mice were sacrificed and tumors were collected for immunohistochemistry and immunoblotting.
Immunofluorescence and immunohistology staining
The tumor tissues of mice were fixed with 4% paraformaldehyde, embedded with paraffin, and cut at 6 μm. Histological sections were dewaxed, antigen retrieved with citric acid buffer, blocked with 3% hydroperoxide for 10 min, and then incubated with antibody against LC3 (Sigma-Aldrich, L8918; 1:200) or SQSTM1 (Progen, GP62-C; 1:300) at 4°C overnight and incubated with secondary antibody (Alexa Fluor 488; Jackson ImmunoResearch, 611,545,215) for 1.5 h at room temperature. The sections were counterstained with hematoxylin for 5 min, dehydrated, cleared and mounted in neutral gum. TUNEL staining was performed according to the instructions (Roche, 11,684,795,910). The tissue slices were counterstained with 4ʹ,6-diamidino-2-phenylindole (DAPI; ThermoFisher Scientific, D9542) for 3 min. Diaminobenzidine (Sigma-Aldrich, D8001) was used as a chromogen to visualize the antibody/antigen complex. For Immunohistochemistry experiments, tumor tissues were fixed and cryosectioned. Processed tissue sections were first incubated with MKI67/ki67 antibody (Santa Cruz Biotechnology, sc23900). Biotinylated secondary antibody was added, followed by the addition of preformed ABC reagent (Santa Cruz Biotechnology, sc2018). The target protein was visualized by incubation with peroxidase substrate. All sections were observed using the confocal laser scanning microscope.
Proximity ligation assay (PLA)
For detecting the interaction between STX17 and VAMP8, HeLa cells transfected with GFP-STX17 treated with different chemicals for 8 h before probed with anti-GFP and anti-VAMP8 antibodies and subjected to PLA. In detail, PLA was conducted using a PLA kit (Sigma-Aldrich, DUO9008, DUO92002 and DUO92004) according to the manufacturer’s instructions. Treated cells were fixed, permeabilized and blocked for 1 h with Duolink® Blocking Solution (Sigma-Aldrich, DUO82047). Then the cells were incubated with both mouse anti-GFP (Proteintech, 66,002-1-Ig) and rabbit anti-VAMP8 (Cell Signaling Technology, 13,060) at 4°C overnight. Images were obtained with a QImaging Rolera XR digital camera on an Olympus X81 microscope. The effective interaction between STX17 and VAMP8 was illustrated as red dots. Determination of the number of PLA dots was performed using the ImageJ software (NIH).
Ca2+ and zinc imaging
Ca2+ and zinc imaging were carried out within 2–3 h after plating while overexpressing-MCOLN1-cells exhibited a round morphology. Cells were loaded with 5 μM Fura-2 AM (ThermoFisher Scientific, F1201) or 2.5 μM FluoZin-3 AM (ThermoFisher Scientific, F24195) in the culture medium at 37°C for 1 h. Fura-2 ratios (F340/F380) or FluoZin-3 AM fluorescence (Ex494/Em516) were used to monitor changes in intracellular [Ca2+] or [Zn2+] upon stimulation, respectively. Lysosomal Ca2+ release was measured under a “zero” Ca2+ external solution. To maximally visualize zinc influx upon stimulation, cells used in zinc imaging were pretreated with 100 μM ZnCl2 overnight.
Confocal imaging and Transmission Electron Microscope (TEM)
Cells were grown on glass coverslips and fixed with 4% PFA. After three washes with PBS, the coverslips were mounted with Fluoromount-G (Southern Biotech, 010001) and processed for imaging using an Olympus confocal microscope. For TEM experiments, HeLa cells were treated with DMSO (Sigma-Aldrich, D2650) or ML-SA5 for 12 h prior to collecting. The collected samples were then centrifuged and processed in the Transmission Electron Microscopy Core at Jinan Weiya Electron Microscopy Company.
Whole-endolysosome electrophysiology
Endolysosomal electrophysiology was performed on isolated endolysosomes using a modified patch-clamp method [1]. Briefly, cells were treated with 1 μM vacuolin-1 (MCE, HY-118,630), a lipid-soluble polycyclic triazine that can increase the size of endosomes and lysosomes selectively [74] for at least 1 h and up to 12 h. Large vacuoles (up to 5 μm in diameter; capacitance = 1.9 ± 0.1 pF, n = 35 vacuoles) were observed in most vacuolin-treated cells. To isolate vacuoles, a patch pipette (electrode) was pressed against individual cells and then was pulled away quickly to sever the cell membrane. Whole-endolysosome recordings were then performed on enlarged vacuoles from cells that were released into the dish.
Unless otherwise stated, the bath (internal/cytoplasmic) solution contained 140 mM K+-gluconate (Sigma-Aldrich, P1847), 4 mM NaCl (Sigma-Aldrich, S9988), 2 mM MgCl2 (Sigma-Aldrich, 208,337), and 10 mM HEPES (Sigma-Aldrich, H3375), pH adjusted with KOH to 7.2. Solutions with 0.1–10 μM free Ca2+ were prepared by combining various concentrations of EGTA (Sigma-Aldrich, E3478) and CaCl2 (Sigma-Aldrich, C4901), as calculated with the MaxcHeLator program (http://maxcHeLator.stanford.edu/CaEGTA-TS.htm). The pipette (lumenal) solution contained 145 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 20 mM HEPES, and 10 mM glucose (Sigma-Aldrich, G8270), pH adjusted to 4.6 with NaOH. Data were acquired with an Axopatch 2A patch-clamp amplifier and a Digidata® 1440 digitizer and recorded with pClamp 10.0 software (Axon Instruments). Whole-endolysosome currents were digitized at 10 kHz and filtered at 2 kHz. All experiments were conducted at room temperature (21–23°C), and all recordings were analyzed in pCLAMP10 (Axon Instruments) and Origin 8.0 (OriginLab) software.
DQ-BSA proteolytic assay
DQ-red-BSA was used as an artificial substrate to evaluate lysosomal proteolytic degradation [22,75]. Briefly, cells were treated with DQ-red-BSA (ThermoFisher Scientific, D12051; 10 μg/ml) for 2 h at 37°C. After removal of extracellular DQ-red-BSA, cells were starved (amino acid-free and serum withdrawal) to trigger autophagic degradation. Upon cleavage, DQ-red-BSA exhibited red fluorescence, as detected by confocal imaging.
Zinc staining
HEK293T cells were seeded on glass coverslips and loaded with 100 μM ZnCl2 (Sigma-Aldrich, 208,086) for 12 h together with or without ML-SA5. Cells were washed 3 times with PBS, and then loaded with 2.5 μM FluoZin-3 AM (ThermoFisher Scientific, F24195) and 100 nM LysoTracker Red DND-99 (ThermoFisher Scientific, L7528) for no less than 30 min. Confocal microscopy was performed using Olympus confocal microscopes.
Reagents and chemicals
All reagents were dissolved and stored in DMSO (Sigma-Aldrich, D2650) or water. Baf-A1 (HY-100,558), rapamycin (HY-10,219), zVADfmk (HY-16658B) was obtained from Med Chem Express. Torin 1 (S2827) was purchased from Selleck. DQ-BSA-red (D12051), FluoZin-3 AM (F24195), Fura-2 AM (F1201), BAPTA AM (B6769), LysoTracker Red DND-99 (L7528), PI (P1304), and H2DCFDA (D399) were from ThermoFisher Scientific. ANXA5/annexin V-7-AAD (HR8285), chloroquine (C6628), 3-MA (M9281), TPEN (P4413), 1,10 phenanthroline (P9375), deferoxamine (D9533), MK6-83 (SML1509), and tetrathiomolybdate (323,446) were purchased from Sigma-Aldrich. Trypan blue (K940) was from VWR. CCK 8 kit (CK04) was purchased from Dojindo. Tachpyr was synthesized in Wenxi ChemBio Co., Ltd. ML-SA1, ML-SA5, ML-SI1, and ML-SI3 were kindly provided by Dr. Haoxing Xu.
Statistical Analysis
All experiments are repeated at least 3 times. Data are presented as the means ± standard errors of the mean (SEMs). Statistical significance of differences was evaluated using ANOVA followed by Tukey’s test. P values < 0.05 were considered statistically significant.
Supplementary Material
Acknowledgments
This work was supported by National Natural Science Foundation of China (NSFC) grants (81772559 to W. W; 81971212 to F. G; 81600967 to C. L), NSF grants of the Jiangsu Province (BK20170262 to W. W), Jiangsu Specially-Appointed Professor award to W.W, Jiangsu Province Innovative and Entrepreneurial Talent program to W.W and Jiangsu Province Innovative and Entrepreneurial Team program to W.W. We are grateful to Drs. Hong Zhang (National Institute of Biological Sciences, Beijing, China), Li Yu (School of Life Sciences, Tsinghua University), Edward A Fon (Department of Neurology and Neurosurgery, McGill University), Amy Kiger (Department of Cell & Developmental Biology, University of California, San Diego), Ling Chen (Guangzhou institutes of biomedicine and health, Chinese academy of sciences), and Qiming Sun (School of Basic Medical Sciences, Zhejiang University) for sharing the plasmids and cell lines. We are grateful to Dr. Hailey Jansen (Libin Cardiovascular Institute, University of Calgary, Canada) for critical reading through the manuscript and appreciate the encouragement and helpful comments from other members of the Wang laboratory.
Funding Statement
This work was supported by the National Natural Science Foundation of China [81772559]; National Natural Science Foundation of China [82071225]; National Natural Science Foundation of China [81971212].
Disclosure statement
The authors disclose no competing interests.
Supplementary material
Supplemental data for this article can be accessed here
References
- [1].Dong XP, Cheng X, Mills E, et al. The type IV mucolipidosis-associated protein TRPML1 is an endolysosomal iron release channel. Nature. 2008;455(7215):992–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Fares H, Greenwald I.. Regulation of endocytosis by CUP-5, the Caenorhabditis elegans mucolipin-1 homolog. Nat Genet. 2001;28(1):64–68. [DOI] [PubMed] [Google Scholar]
- [3].Medina DL, Fraldi A, Bouche V, et al. Transcriptional activation of lysosomal exocytosis promotes cellular clearance. Dev Cell. 2011;21(3):421–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].LaPlante JM, Sun M, Falardeau J, et al. Lysosomal exocytosis is impaired in mucolipidosis type IV. Mol Genet Metab. 2006;89(4):339–348. [DOI] [PubMed] [Google Scholar]
- [5].Wang W, Gao Q, Yang M, et al. Up-regulation of lysosomal TRPML1 channels is essential for lysosomal adaptation to nutrient starvation. Proc Natl Acad Sci U S A. 2015;112(11):E1373–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Pryor PR, Mullock BM, Bright NA, et al. The role of intraorganellar Ca(2+) in late endosome-lysosome heterotypic fusion and in the reformation of lysosomes from hybrid organelles. J Cell Biol. 2000;149(5):1053–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Luzio JP, Rous BA, Bright NA, et al. Lysosome-endosome fusion and lysosome biogenesis. J Cell Sci. 2000;113(Pt 9):1515–1524. [DOI] [PubMed] [Google Scholar]
- [8].LaPlante JM, Ye CP, Quinn SJ, et al. Functional links between mucolipin-1 and Ca2+-dependent membrane trafficking in mucolipidosis IV. Biochem Biophys Res Commun. 2004;322(4):1384–1391. [DOI] [PubMed] [Google Scholar]
- [9].Klionsky DJ. Autophagy revisited: a conversation with Christian de Duve. Autophagy. 2008;4(6):740–743. [DOI] [PubMed] [Google Scholar]
- [10].Klionsky DJ. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol. 2007;8(11):931–937. [DOI] [PubMed] [Google Scholar]
- [11].Vergarajauregui S, Connelly PS, Daniels MP, et al. Autophagic dysfunction in mucolipidosis type IV patients. Hum Mol Genet. 2008;17(17):2723–2737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Sardiello M, Palmieri M, di Ronza A, et al. A gene network regulating lysosomal biogenesis and function. Science. 2009;325(5939):473–477. [DOI] [PubMed] [Google Scholar]
- [13].Medina DL, Di Paola S, Peluso I, et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat Cell Biol. 2015;17(3):288–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Schmiege P, Fine M, Blobel G, et al. Human TRPML1 channel structures in open and closed conformations. Nature. 2017;550(7676):366–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Sahoo N, Gu M, Zhang X, et al. Gastric acid secretion from parietal cells is mediated by a Ca(2+) efflux channel in the tubulovesicle. Dev Cell. 2017;41(3):262–73 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Chen CC, Keller M, Hess M, et al. A small molecule restores function to TRPML1 mutant isoforms responsible for mucolipidosis type IV. Nat Commun. 2014;5(1):4681. [DOI] [PubMed] [Google Scholar]
- [17].Garrity AG, Wang W, Collier CM, et al. The endoplasmic reticulum, not the pH gradient, drives calcium refilling of lysosomes. Elife. 2016;5. DOI: 10.7554/eLife.15887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Itakura E, Kishi-Itakura C, Mizushima N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell. 2012;151(6):1256–1269. [DOI] [PubMed] [Google Scholar]
- [19].McLelland GL, Lee SA, McBride HM, et al. Syntaxin-17 delivers PINK1/parkin-dependent mitochondrial vesicles to the endolysosomal system. J Cell Biol. 2016;214(3):275–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Klionsky DJ, Abdel-Aziz AK, Abdelfatah S, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition). Autophagy. 2021;17(1):1–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Zhang X, Cheng X, Yu L, et al. MCOLN1 is a ROS sensor in lysosomes that regulates autophagy. Nat Commun. 2016;7(1):12109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Wang W, Zhang X, Gao Q, et al. A voltage-dependent K+ channel in the lysosome is required for refilling lysosomal Ca2+ stores. J Cell Biol. 2017;216(6):1715–1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Elsasser HP, Lehr U, Agricola B, et al. Establishment and characterisation of two cell lines with different grade of differentiation derived from one primary human pancreatic adenocarcinoma. Virchows Arch B Cell Pathol Incl Mol Pathol. 1992;61(1):295–306. [DOI] [PubMed] [Google Scholar]
- [24].Sugarman BJ, Aggarwal BB, Hass PE, et al. Recombinant human tumor necrosis factor-alpha: effects on proliferation of normal and transformed cells in vitro. Science. 1985;230(4728):943–945. [DOI] [PubMed] [Google Scholar]
- [25].Yamamoto A, Tagawa Y, Yoshimori T, et al. Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct Funct. 1998;23(1):33–42. [DOI] [PubMed] [Google Scholar]
- [26].Shen D, Wang X, Xu H. Pairing phosphoinositides with calcium ions in endolysosomal dynamics: phosphoinositides control the direction and specificity of membrane trafficking by regulating the activity of calcium channels in the endolysosomes. Bioessays. 2011;33(6):448–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Malhotra R, Warne JP, Salas E, et al. Loss of Atg12, but not Atg5, in pro-opiomelanocortin neurons exacerbates diet-induced obesity. Autophagy. 2015;11(1):145–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Kiselyov K, Chen J, Rbaibi Y, et al. TRP-ML1 is a lysosomal monovalent cation channel that undergoes proteolytic cleavage. J Biol Chem. 2005;280(52):43218–43223. [DOI] [PubMed] [Google Scholar]
- [29].Dong XP, Shen D, Wang X, et al. PI(3,5)P(2) controls membrane trafficking by direct activation of mucolipin Ca(2+) release channels in the endolysosome. Nat Commun. 2010;1(1):38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Yoon YH, Cho KS, Hwang JJ, et al. Induction of lysosomal dilatation, arrested autophagy, and cell death by chloroquine in cultured ARPE-19 cells. Invest Ophthalmol Vis Sci. 2010;51(11):6030–6037. [DOI] [PubMed] [Google Scholar]
- [31].Ni HM, Bockus A, Wozniak AL, et al. Dissecting the dynamic turnover of GFP-LC3 in the autolysosome. Autophagy. 2011;7(2):188–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Lee SB, Kim S, Lee J, et al. ATG1, an autophagy regulator, inhibits cell growth by negatively regulating S6 kinase. EMBO Rep. 2007;8(4):360–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Nyfeler B, Bergman P, Triantafellow E, et al. Relieving autophagy and 4EBP1 from rapamycin resistance. Mol Cell Biol. 2011;31(14):2867–2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Bach M, Larance M, James DE, et al. The serine/threonine kinase ULK1 is a target of multiple phosphorylation events. Biochem J. 2011;440(2):283–291. [DOI] [PubMed] [Google Scholar]
- [35].Kang SA, Pacold ME, Cervantes CL, et al. mTORC1 phosphorylation sites encode their sensitivity to starvation and rapamycin. Science. 2013;341(6144):1236566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Xu H, Ren D. Lysosomal physiology. Annu Rev Physiol. 2015;77(1):57–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Radford RJ, Lippard SJ. Chelators for investigating zinc metalloneurochemistry. Curr Opin Chem Biol. 2013;17(2):129–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Schatten H, Eisenstark A. Salmonella: methods and protocols. Methods Mol Biol. 2007;394:v–vi. [DOI] [PubMed] [Google Scholar]
- [39].Tsien RY. New calcium indicators and buffers with high selectivity against magnesium and protons: design, synthesis, and properties of prototype structures. Biochemistry. 1980;19(11):2396–2404. [DOI] [PubMed] [Google Scholar]
- [40].Chouyyok W, Yantasee W, Shin Y, et al. Transition metal ion capture using functional mesoporous carbon made with 1,10-phenanthroline. Inorg Chem Commun. 2009;12(11):1099–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Smirnova J, Kabin E, Jarving I, et al. Copper(I)-binding properties of de-coppering drugs for the treatment of Wilson disease. alpha-Lipoic acid as a potential anti-copper agent. Sci Rep. 2018;8(1):1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Khan G, Merajver S. Copper chelation in cancer therapy using tetrathiomolybdate: an evolving paradigm. Expert Opin Investig Drugs. 2009;18(4):541–548. [DOI] [PubMed] [Google Scholar]
- [43].Kontoghiorghe CN, Kontoghiorghes GJ. Efficacy and safety of iron-chelation therapy with deferoxamine, deferiprone, and deferasirox for the treatment of iron-loaded patients with non-transfusion-dependent thalassemia syndromes. Drug Des Devel Ther. 2016;10:465–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Heath JL, Weiss JM, Lavau CP, et al. Iron deprivation in cancer–potential therapeutic implications. Nutrients. 2013;5(8):2836–2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Planalp RP, Przyborowska AM, Park G, et al. Novel cytotoxic chelators that bind iron(II) selectively over zinc(II) under aqueous aerobic conditions. Biochem Soc Trans. 2002;30(4):758–762. [DOI] [PubMed] [Google Scholar]
- [46].Schwiebert EM, Liang L, Cheng NL, et al. Extracellular zinc and ATP-gated P2X receptor calcium entry channels: new zinc receptors as physiological sensors and therapeutic targets. Purinergic Signal. 2005;1(4):299–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Eichelsdoerfer JL, Evans JA, Slaugenhaupt SA, et al. Zinc dyshomeostasis is linked with the loss of mucolipidosis IV-associated TRPML1 ion channel. J Biol Chem. 2010;285(45):34304–34308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Kukic I, Lee JK, Coblentz J, et al. Zinc-dependent lysosomal enlargement in TRPML1-deficient cells involves MTF-1 transcription factor and ZnT4 (Slc30a4) transporter. Biochem J. 2013;451(2):155–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Minckley TF, Zhang C, Fudge DH, et al. Sub-nanomolar sensitive GZnP3 reveals TRPML1-mediated neuronal Zn(2+) signals. Nat Commun. 2019;10(1):4806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Oyama TM, Ishida S, Okano Y, et al. Clioquinol-induced increase and decrease in the intracellular Zn2+ level in rat thymocytes. Life Sci. 2012;91(23–24):1216–1220. [DOI] [PubMed] [Google Scholar]
- [51].Mathew R, Karantza-Wadsworth V, White E. Role of autophagy in cancer. Nat Rev Cancer. 2007;7(12):961–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Lotze MT, Maranchie J, Appleman L. Inhibiting autophagy: a novel approach for the treatment of renal cell carcinoma. Cancer J. 2013;19(4):341–347. [DOI] [PubMed] [Google Scholar]
- [53].Chude CI, Amaravadi RK. Targeting autophagy in cancer: update on clinical trials and novel inhibitors. Int J Mol Sci. 2017;18(6). DOI: 10.3390/ijms18061279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Yang S, Wang X, Contino G, et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011;25(7):717–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Garbar C, Mascaux C, Giustiniani J, et al. Chemotherapy treatment induces an increase of autophagy in the luminal breast cancer cell MCF7, but not in the triple-negative MDA-MB231. Sci Rep. 2017;7(1):7201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Cota D, Proulx K, Smith KA, et al. Hypothalamic mTOR signaling regulates food intake. Science. 2006;312(5775):927–930. [DOI] [PubMed] [Google Scholar]
- [57].Ouyang H, Mou L, Luk C, et al. Immortal human pancreatic duct epithelial cell lines with near normal genotype and phenotype. Am J Pathol. 2000;157(5):1623–1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Miedel MT, Rbaibi Y, Guerriero CJ, et al. Membrane traffic and turnover in TRP-ML1-deficient cells: a revised model for mucolipidosis type IV pathogenesis. J Exp Med. 2008;205(6):1477–1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Maiuri MC, Zalckvar E, Kimchi A, et al. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007;8(9):741–752. [DOI] [PubMed] [Google Scholar]
- [60].Slee EA, Zhu H, Chow SC, et al. Benzyloxycarbonyl-Val-Ala-Asp (OMe) fluoromethylketone (Z-VAD.FMK) inhibits apoptosis by blocking the processing of CPP32. Biochem J. 1996;315(1):21–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Samie M, Wang X, Zhang X, et al. A TRP channel in the lysosome regulates large particle phagocytosis via focal exocytosis. Dev Cell. 2013;26(5):511–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Cheng X, Zhang X, Gao Q, et al. The intracellular Ca(2)(+) channel MCOLN1 is required for sarcolemma repair to prevent muscular dystrophy. Nat Med. 2014;20(10):1187–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Scotto Rosato A, Montefusco S, Soldati C, et al. TRPML1 links lysosomal calcium to autophagosome biogenesis through the activation of the CaMKKbeta/VPS34 pathway. Nat Commun. 2019;10:5630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Zhang X, Chen W, Gao Q, et al. Rapamycin directly activates lysosomal mucolipin TRP channels independent of mTOR. PLoS Biol. 2019;17(5):e3000252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Sun X, Yang Y, Zhong XZ, et al. A negative feedback regulation of MTORC1 activity by the lysosomal Ca2+ channel MCOLN1 (mucolipin 1) using a CALM (calmodulin)-dependent mechanism. Autophagy. 2018;14(1):38–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Abiria SA, Krapivinsky G, Sah R, et al. TRPM7 senses oxidative stress to release Zn2+ from unique intracellular vesicles. Proc Natl Acad Sci U S A. 2017;114(30):E6079–E88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Kochanczyk T, Drozd A, Krezel A. Relationship between the architecture of zinc coordination and zinc binding affinity in proteins–insights into zinc regulation. Metallomics. 2015;7(2):244–257. [DOI] [PubMed] [Google Scholar]
- [68].Barth J, Zimmermann H, Volknandt W. SV31 is a Zn2+-binding synaptic vesicle protein. J Neurochem. 2011;118(4):558–570. [DOI] [PubMed] [Google Scholar]
- [69].Lorincz P, Juhasz G. Autophagosome-Lysosome Fusion. J Mol Biol. 2020;432(8):2462–2482. [DOI] [PubMed] [Google Scholar]
- [70].Jiang P, Nishimura T, Sakamaki Y, et al. The HOPS complex mediates autophagosome-lysosome fusion through interaction with syntaxin 17. Mol Biol Cell. 2014;25(8):1327–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Hunter MR, Scourfield EJ, Emmott E, et al. VPS18 recruits VPS41 to the human HOPS complex via a RING-RING interaction. Biochem J. 2017;474(21):3615–3626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Chimienti F, Aouffen M, Favier A, et al. Zinc homeostasis-regulating proteins: new drug targets for triggering cell fate. Curr Drug Targets. 2003;4(4):323–338. [DOI] [PubMed] [Google Scholar]
- [73].Dar AA, Belkhiri A, Ecsedy J, et al. Aurora kinase A inhibition leads to p73-dependent apoptosis in p53-deficient cancer cells. Cancer Res. 2008;68(21):8998–9004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Cerny J, Feng Y, Yu A, et al. The small chemical vacuolin-1 inhibits Ca2+-dependent lysosomal exocytosis but not cell resealing. EMBO Rep. 2004;5(9):883–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Yue W, Hamai A, Tonelli G, et al. Inhibition of the autophagic flux by salinomycin in breast cancer stem-like/progenitor cells interferes with their maintenance. Autophagy. 2013;9(5):714–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.