

Abstract
CASP is a community experiment to advance methods of computing three-dimensional protein structure from amino acid sequence. Core components are rigorous blind testing of methods and evaluation of the results by independent assessors. In the most recent experiment (CASP14) deep learning methods from one research group consistently delivered computed structures rivalling the corresponding experimental ones in accuracy. In this sense, the results represent a solution to the classical protein folding problem, at least for single proteins. The models have already been shown to be capable of providing solutions for problematic crystal structures, and there are broad implications for the rest of structural biology. Other research groups also substantially improved performance. Here we describe these results and outline some of the many implications. Other related areas of CASP, including modeling of protein complexes, structure refinement, estimation of model accuracy, and prediction of inter-residue contacts and distances, are also described.
Keywords: Protein Structure Prediction, Community Wide Experiment, CASP
INTRODUCTION
CASP (Critical Assessment of Structure Prediction) is an organization whose aim is to advance solutions to the problem of computing protein three-dimensional structure from amino acid sequence information. It’s a community experiment in which those interested in the ‘protein folding problem’ (as it has traditionally been known) are asked to submit computed structures for independent assessment of accuracy. Every two years, CASP identifies a set of modeling targets - proteins for which the experimental structure is about to be solved or is solved but still not public - and provides the corresponding amino acid sequences to the modeling community. Participants are typically required to return computed structures within three weeks. Participating automatic servers are also sent the sequences, and must return models within 72 hours. Submitted structures are analyzed by a team of independent assessors. All models and analyses are made public. Each CASP round culminates with an international conference (held virtually for CASP14) and a special issue of PROTEINS, containing papers by the assessors, selected participants, and an overview of the results. This paper is the overview for the 14th CASP round.
The primary focus of CASP has always been on computing the structures of single proteins and domains. There are two assessments of performance in this area for CASP141,2. Assessing methods for modeling proteins complexes is increasingly important and is done in conjunction with CASPs’ sister organization CAPRI, also providing two assessments3,4. Other assessed categories in this CASP are the prediction of inter-residues contacts and distances5 refinement of initial models6, and estimation of model accuracy7. There is also an analysis of how useful the computed structures are for deducing aspects of function related to molecular recognition8. For the first time, there is a separate assessment of multi-domain assemblies9 with an emphasis on the accuracy of domain interactions.
In CASP14, a total of 97 research groups from 19 countries tested 215 modeling methods and submitted over 67,000 predictions in six prediction categories, maintaining the previous high level of participation in spite of the Covid-19 pandemic. Structures of 52 proteins and protein-protein complexes were received from the experimental community in time for the assessments. 42 were determined using X-ray crystallography, seven using cryo-electron microscopy (cryo-EM), and three by NMR. These were divided into monomeric subunits and, in one case, separate domains (for a large 2180-residue long RNA polymerase, T1044), and released for prediction as 68 tertiary structure modeling targets. For the assessment, the targets were split into domains based on homology and structural integrity, and then re-organized into 96 evaluation units based on the comparison of the performance on individual and combined domains10. Target evaluation units are assigned to one of four classes of modeling difficulty, based on sequence and structure similarity to already experimentally determined structures: ‘TBM-Easy’ (easy template modeling) for straightforward template modeling targets, ‘TBM-Hard’ for more difficult homology modeling targets, ‘FM/TBM’ for those with only remote structural homologies and ‘FM’ (Free modeling) for the most difficult set with no detectable homology to known structures. As discussed later, these divisions are no longer very relevant.
Additionally, ten multidomain targets were assessed for accuracy of domain interactions. Multimolecular assemblies, including eight hetero-complexes, were released for prediction as 22 quaternary structure modeling targets. 12 of those were also selected for the joint CASP/CAPRI experiment. The quaternary structure modeling targets were divided into 29 evaluation units (19 of which were also included in the CASP/ CAPRI experiment). Target details are available at https://predictioncenter.org/casp14/targetlist.cgi and are also discussed in papers in this issue10–12.
Like most aspects of life in 2020, CASP was affected profoundly by the Covid-19 pandemic. The CASP category of data assisted modeling13–15 was not possible because most labs were closed and so not able to generate the necessary data. The CASP14 conference, usually a very intense in-person event, was held virtually. The CASP community also responded to the emergency by working together to compute and evaluate models for 10 of the hardest to model SARS-CoV-2 proteins of unknown structure (see paper in this issue16). This was the most extensive community modeling experiment so far in CASP, and produced interesting results.
Three-dimensional Protein Structure Modeling
CASP14 saw an extraordinary increase in the accuracy of the computed three-dimensional protein structures. One research group, AlphaFold2 from the company DeepMind, submitted models competitive with experimental accuracy for at least 2/3 of the targets (group 427 in the Results tables, available online at https://predictioncenter.org/casp14/results.cgi). Other groups also showed substantial improvement. Figure 1 summarizes performance in terms of backbone accuracy for the best models received in each CASP.
Figure 1:

Trend lines of backbone agreement with experiment for the best models in each of the 14 CASP rounds. Individual target points are shown for the most recent round. The three targets with the lowest agreement with experiment are colored blue (T1027 and T1029, NMR) and red (T0147s1, a subunit of a cryo-EM-derived heteromeric structure with complex inter-subunit interactions). The agreement metric, GDT_TS, is a multi-scale indicator of the closeness of the Cα atoms in a model to those in the corresponding experimental structure. Target difficulty is based on sequence and structure similarity to other proteins with known experimental structures. Performance in CASP14 (top black line) is very impressive, with accuracy approaching and in some cases likely exceeding experimental accuracy for many targets (see later text).
Historically, the most accurate models have been obtained using information about experimentally determined homologous structures (template-based modeling), and the Figure 1 difficulty scale17 (X axis) reflects the degree to which those methods were applicable. As the trend lines for earlier CASPs show, until now, accuracy on the right-hand ‘difficult’ side of the plot was sharply lower. In CASP13 (2018)18, with the introduction of effective deep learning methods, the trend line rose to above 60 on the GDT_TS scale, even for the most difficult targets, a major advance from the previous CASP. Note that the fold of the protein backbone is usually correct at values above 50 on this scale, and so that represented a solution to the problem as classically defined, for most targets.
Astonishingly, the trend curve for CASP14 (the black straight-line) starts at a GDT_TS of about 95, and finishes at about 85 for difficult targets. Because of experimental errors and artifacts, a GDT_TS of 100 is highly unlikely, and previous CASP trend lines intercept the Y axis at about 90, indicating that that is approximately the limit expected. In CASP14, about 2/3 of the 96 targets reached GDT_TS values greater than that, and so are considered competitive with experiment in backbone accuracy.
Although this outstanding performance is dominated by AlphaFold2 (group 427), the dashed black line in Figure 1 shows that other groups also advanced substantially from CASP13. Also of note, performance of servers in CASP14 (dotted black line in Figure 1) is similar to the best performance of all groups in CASP13. This is of particular significance since AlphaFold2 did not submit server models. Thus, other research groups have not only now surpassed AlphaFold’s leading performance in CASP13, they have also made these improved methods available in servers, some of which are publicly accessible. Nevertheless, it is clear that the AlphaFold2 models are generally much more accurate, and the only ones to consistently approach experimental quality. For only four targets did another group obtain a higher GDT_TS.
Figure 2 shows an example of a model with close agreement with experiment. Model and experimental backbone closely overlap almost everywhere. As discussed below, minor differences in loop conformations are often due to crystal packing effects. The helix loop helix motif in the model at the bottom right of the figure corresponds to a disordered region in the experimental structure (for which there is no observed structure). The set of five submitted models contain two different conformations for this region.
Figure 2:
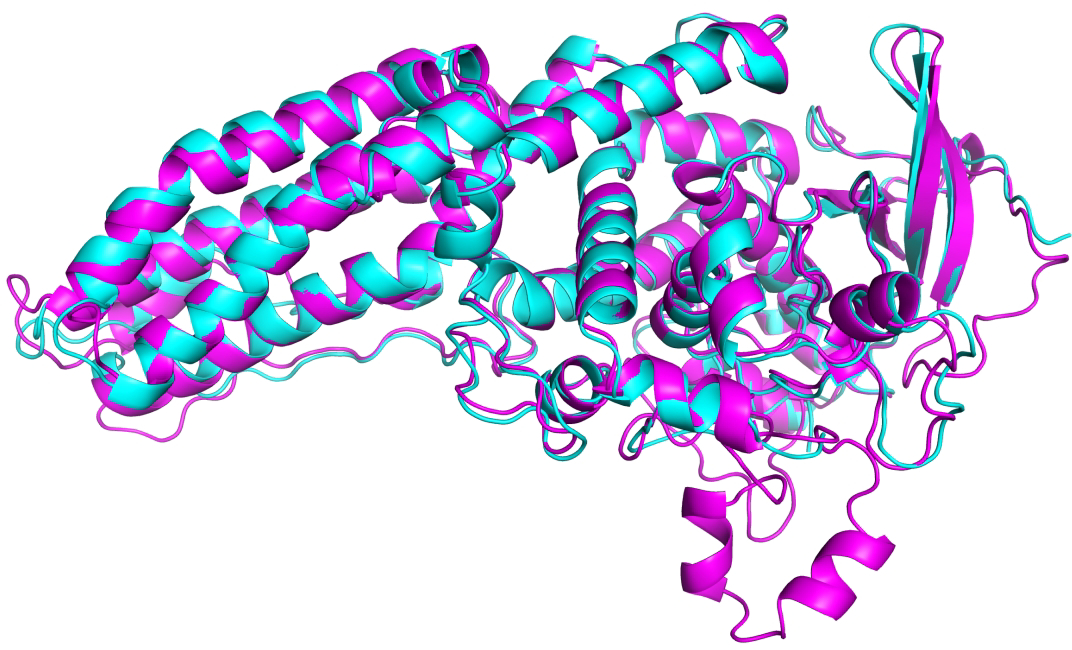
Example of a high accuracy CASP14 model - CASP target T1053, a two-domain bacterial kinase. Model (from AlphaFold2, GDT_TS 93) in magenta, experimental structure (PDB 7m7a, resolution 3.2 Å) in turquoise. Both domains are difficult modeling targets (FM/TBM category).
Figure 3 provides an atomic level view of part of a model of SARS CoV-2 ORF8 (from AlphaFold2) and the corresponding crystal structure (CASP target T1064, FM category, GDT_TS 87) showing impressive atomic level agreement for the main chain as well as side chain atoms.
Figure 3:
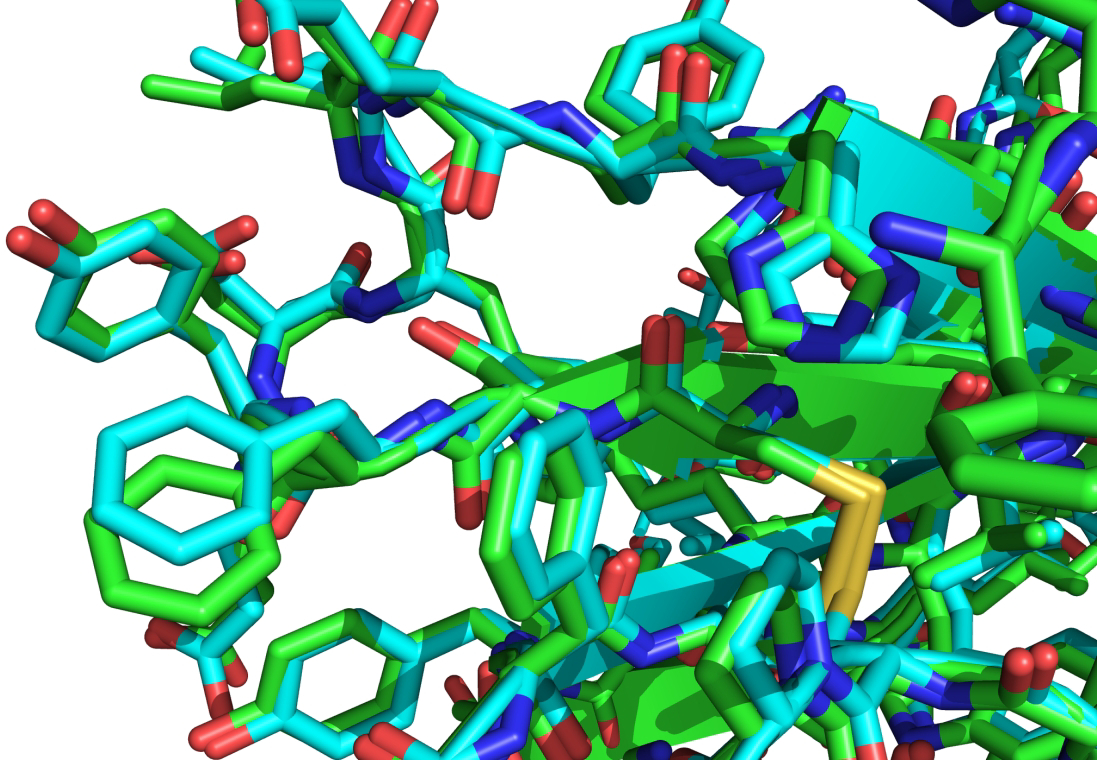
Superposition of a model (from AlphaFold2) of SARS CoV-2 ORF8 (CASP target T1064) and the corresponding experimental structure (PDB 7jtl, resolution 2.0 Å), illustrating the atomic level of agreement with experiment typically found in CASP14.
Remaining sources of disagreement between calculation and experiment
In previous CASPs, with rare exceptions, it was usually safe to assume that differences between models and experiment were dominated by computational error. The high accuracy results in this round required a more careful analysis. Data are limited and some contributing factors are correlated, complicating interpretation. Nevertheless, as outlined below, several distinct influences on agreement with experiment can be identified.
Dependence on Experimental data:
Figure 4A shows the relationship between average best GDT_TS and the quality of experimental data (three ranges of X-ray structure resolution and cryo-EM). The lower agreement with experiment for lower resolution X-ray structures and for cryo-EM structures suggests that experimental structure accuracy may be a factor in limiting the maximum GDT_TS obtained, particularly for values less than 90. There were also three NMR targets in CASP14 (data not included in the figure) two of which are template free (FM) targets with very low GDT_TS values (blue points in Figure 1). Analysis by Gaetano Montelione’s group19, shows that one of these, T1027, is a dynamic structure and the best computed structures may correspond to a member of the ensemble. For the other, the best computed structures agree better with the experimental NOE data than does the experimental structure.
Figure 4:
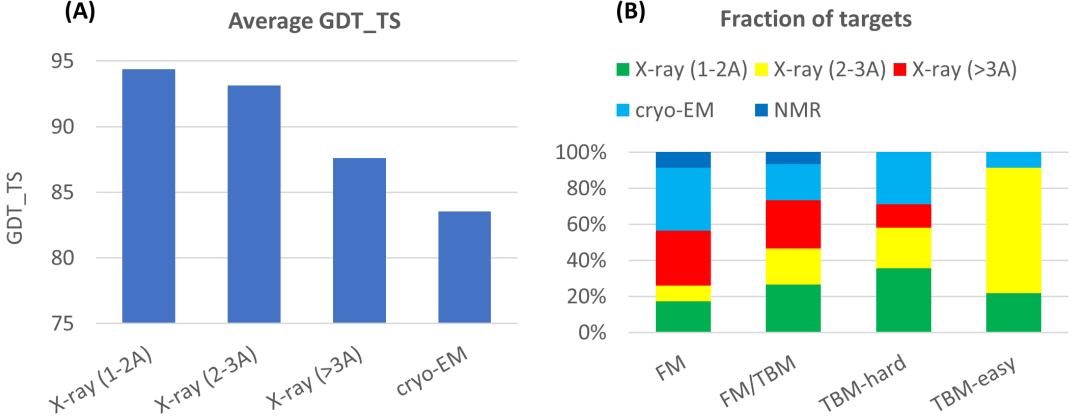
(A) Average agreement of the best CASP14 models with experiment (GDT_TS) for different categories of experimental data. The first three bins show a fall-off as the resolution of X-ray structures decreases, suggesting lower GDT_TS values are partly due to higher experimental error. The effect is most pronounced for Cryo-EM experimental structures (right hand bin, resolution range 2.2 – 3.8 Angstroms). Two of the three NMR targets (not included here) have very low GDT_TS values (see text). (B) Distribution of experimental data type across categories of target difficulty. The large majority of targets in the most difficult category (FM) have low resolution X-ray, Cryo-EM (resolution range 2.2 – 3.8 Angstroms), or NMR data, whereas in the easiest category, 90% of targets are determined from higher resolution X-ray data.
Figure 4B shows that there is a strong relationship between target difficulty categories and the quality of experimental data obtained. That is, proteins belonging to well-studied protein families tend to yield high quality X-ray data. As a consequence, some of the decrease in average agreement with experiment for hardest targets may be due to higher experimental error.
Dependence on modeling difficulty:
Historically, the most accurate models have been obtained by leveraging information on related structures. A surprising feature of the CASP14 results is the small fall-off in agreement with experiment with fall-off in evolutionary information for related structures, especially compared to earlier CASPs. As noted above in Figure 1, the best model trend line starts at 95, but only falls to ~85 for the most difficult targets. Apparently, there is only minor benefit from homologous structure information - models are only marginally more accurate when it is available. Figure 5 shows agreement with experiment (GDT_TS) as a function of the fraction of targets reaching a given level of agreement for different categories of target difficulty. Performance is still strongest for the category of targets with most information from homologous structures available (‘TBM-easy’, green), with the lowest GDT_TS of 90, and many targets with greater than 95. For the most difficult targets (‘FM’, black line), where no assistance from homologous structures is available, performance is slightly lower overall than for the easiest category, but still about 30% of targets achieve a GDT_TS above 90, and 75% have a GDT_TS of 80 or greater. As noted above, two NMR targets with very low GDT_TS (colored blue in Figure 1) pull down the right side of this trend line and a third (red point), discussed below, also contributes. The strong correlations between average GDT_TS, traditional target difficulty, and experimental data quality discussed above (Figure 4) make cause and effect hard to separate conclusively: It is not clear whether the remaining fairly small differences in performance across target categories are because the AlphaFold2 method performs somewhat better when there is information for homologous structures available, or if the performance differences are just due to differences in the average accuracy of the corresponding experimental structures.
Figure 5:
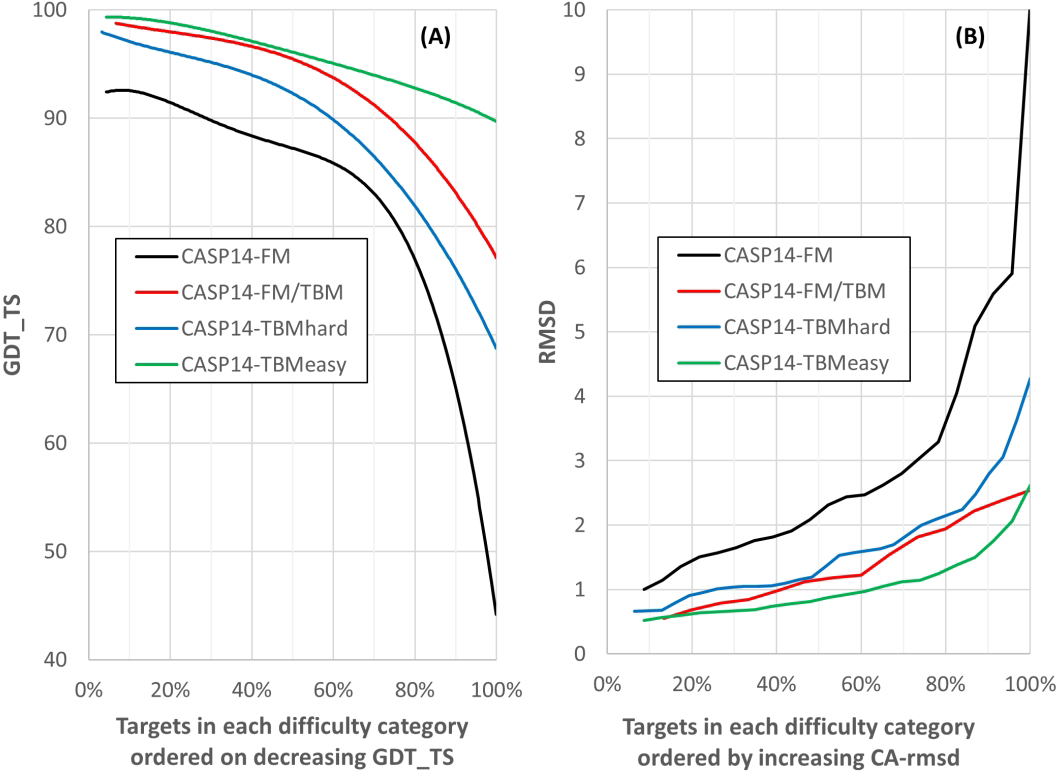
(A) Backbone agreement with experiment (GDT_TS) versus fraction of targets reaching a given level of agreement with experiment in different modeling difficulty categories. Trend lines for targets with the strongest homologous structural information available (‘TBM-easy’) are green, those where homology modeling is more difficult (‘TBM-Hard’) blue, those with only remote structural homologies (‘FM/TBM’) red, and the most difficult set with no detectable homology to known structures (‘FM’) black. Best models for each target. Targets with more information on homologous structures tend to be more accurate, but interpretation of that is complicated (see text). (B) Backbone agreement with experiment (Cα atom Root Mean Square Deviation) for different modeling difficulty categories (best models for each target).
Inter subunit interactions in protein assemblies:
One of the points with low GDT-TS value in Figure 1 (colored red) is a subunit of a 52-mer bacterial flagellum cryo-EM structure (T1047s1, PDB 7bgl20). This is an unusual target in that there are very extensive inter-subunit interactions, including a domain swap21 in which part of the fold of one monomer occupies the corresponding position of a neighboring one. Thus, the monomer conformation is heavily influenced by its neighbors. More moderate conformational changes on forming a multimer are not unusual, and some investigators are developing methods specifically to deal with this issue (see for example22. Participants were not provided with specific multimer assembly information, and so all submitted models are based on an isolated monomer environment. In this sense, this type of difference to experiment is not a computational failure, although it is of course a poorer representation of the in vivo structure.
Crystal lattice contacts:
A related reason for lower GDT_TS values is the effect of lattice contacts on local conformation in crystal structures. The refinement category assessor, Daniel Rigden, has looked at this for a subset of seven targets, with GDT-TS values ranging from 72 to 936. These are cases where the best models differed from experiment for small regions of the polypeptide chain, and refinement methods were unable to converge to the experimental structure. Of the 105 residues involved, he found 64 to be close to lattice contacts, suggesting the local conformations are determined by the crystal environment (also not provided to the participants). For these regions, the best calculated structures likely provide conformations closer to that found in vivo than those from the crystal structure.
Traditionally, CASP has used the multi-superposition, multiscale GDT-TS measure of agreement between models and experiment as a more robust metric than traditional RMSD when dealing with medium or poor-quality models23,24. In CASP 14, almost all best models are in close enough agreement with experiment for RMSD to be an appropriate metric, and we include it here for those more familiar with its properties than those of GDT_TS. Figure 5B shows the percentage of targets modeled to a given Cα-RMSD for the different target categories, analogous to the GDT_TS cumulative plot in Figure 5A. Supplementary figure 1 shows the mapping between RMSD and GDT_TS for a larger set of CASP14 models. The threshold of 90 GDT_TS corresponds to approximately 1.5 Angstroms RMSD, and 80 corresponds to about 2.5 Angstroms (all residues included, calculated with LGA 24).
Other Modeling Categories
The startling results for 3D structure tend to overshadow the other areas of CASP in this round, where progress was less dramatic. At the same time, several of these areas are likely to benefit directly from type of the deep learning approaches that have been so successful for single proteins.
Multimers and protein complexes:
This CASP saw a very impressive solution to the classical protein folding problem25 - accurate modeling of single protein structures from their amino acid sequences. Since the formulation of that problem, science has moved on - most proteins are not monomers, and most biology involves interactions with other proteins, DNA, RNA, or small molecules. In particular, accurate modeling of protein complexes is now receiving increased attention as the next barrier in computational structural biology likely to be surmounted. In this round, CASP and CAPRI (Critical Assessment of Protein Interactions26) again worked together to assess the accuracy of protein complexes, with two corresponding assessment papers3,4. 39 groups took part in this modeling category, with 22 targets altogether, 12 of which were also considered by CAPRI participants. Most targets are obligate assemblies, and in future CASP needs to include more transient complexes. About a third of the targets were determined by cryo-EM, and the increasing output from that technology is allowing larger and more complicated protein assemblies to be included in CASP.
Three broad types of methods were used for modeling complexes in CASP14. As the PDB becomes more populated with large complexes, opportunities for homology modeling have steadily increased, and targets where that is possible continue to provide the most accurate models3 Where homology modeling is not possible, many groups used classical docking methods in which a search is made for sterically and electrostatically complementary surfaces27. A number of groups have now augmented these with the prediction in interface residue-residue contacts, often employing deep-learning methods, for example22, producing some of the best results, although so far, the gains are fairly modest.
Most methods start with models of individual constituent proteins, and conformational changes accompanying complex formation as well as intertwining of monomers present major challenges. As noted earlier, intertwining was a problem for accurate modeling of bacterial flagellum subunit, and an analysis by the CASP assessor Ezgi Karaca3 showed that at least two of the other CASP14 targets undergo substantial conformational changes on complex formation (targets T1061 and T1070). Three other targets were classified as ‘intertwined’ and two as coiled-coils, and these would also be difficult to predict by starting with monomeric structures.
Although overall progress this round from the previous CASP is small3, there is excitement as to what will happen next time, for several reasons. First, analysis by the function assessors8 shows that simply using more accurate models of the constituent proteins will have a major impact on the effectiveness of classical docking methods. Second, deep learning methods for interface residue contact prediction are still in their infancy. Third, at least one group (Baker) already has a ‘fold and dock’ algorithm intended to model conformational change on binding22. Fourth, that group have recently reported using deep learning methods developed for single proteins to directly predict the structure of multimers28.
Refinement:
The CASP refinement category was introduced in CASP8 (2008) on the basis that informatics methods are ultimately limited in accuracy and so physics or related representations of atomic interactions together with some form of local conformational exploration (such as those provided by conventional molecular dynamics) would be essential to achieving atomic accuracy. The deep learning results in this CASP suggest that assumption was incorrect, so that re-examination of the role of refinement is called for. Further, although earlier CASP rounds have seen substantial progress, in the last two, that has been harder to identify. In CASP14, only four methods on average improve models over the starting structures provided, and no method improves much more than half of the targets6. No refinement for any model approached the accuracy achieved by AlphaFold2 directly.
Why have refinement methods apparently stalled? One reason, as noted after CASP1318, is that modeling methods increasingly incorporate a refinement component, so that further improvement with similar methods has become more difficult. That is, refinement may in fact be improving but most of what the methods can deliver is already being exploited in developing the pre-refinement models. A second more fundamental problem appears to be the rugged nature of the refinement landscape, with local energy barriers preventing convergence to high accuracy, at least using realizable amounts of computer time (identifying the global minimum with current scoring functions does not seem to be an issue29). There are two developments in conformational search methods that suggest future progress may be possible. First, the Baker group have successfully incorporated deep learning prediction of inter-residue distance errors into their refinement procedures22 allowing computational effort to be focused on the parts of the structure most needing it. That strategy led to improved performance on bigger targets in CASP146. Second, a number of groups in the molecular dynamics community are applying new machine learning methods to allow exploration of trajectories in less frustrated latent spaces30.
Estimation of Model Accuracy (EMA):
For any data, it is important to have useful estimates of error. Historically, that has been especially crucial for protein structure models, where accuracy has varied widely from protein to protein and method to method. CASP has a separate category to assess methods for estimation of model accuracy, both globally and locally in a structure. The category has two parts. First, ‘Self Assessment’ - every 3D model that is submitted to CASP is required to have error estimates for each atom in the co-ordinate file, and the accuracy of those data has been considered as part of the overall evaluation metric used by recent assessors. Second, those interested are encouraged to provide accuracy estimates for all the server models submitted to CASP - that is, to develop general methods that can be applied to any model. This is a popular category in CASP, with 70 methods used in CASP14. Methods are divided into two types - those that estimate accuracy based on only the model itself and those that make use of consensus properties across models generated by different modeling methods. Assessment metrics have been stable for some time. For overall accuracy estimation, the gap between the accuracy of the best model and the one ranked highest by an error estimation method (‘top1 loss’) is most useful - how close does an EMA method come to picking the best model available? For local accuracy, an average normalized Cα agreement score is used (‘ASE’, see the assessor’s paper for the full definition7). Negative control baselines are provided by a simple consensus method and an older single model method.
In recent CASPs there has not been substantial progress in methods performance. On the other hand, particularly for global estimates, the methods appear to be usefully accurate for selecting close to the best model available, with an average loss of about 10 GDT_TS units for the most effective methods, both single model and consensus based. However, a recent more real-life test suggests there is something misleading about the CASP evaluation framework. As noted earlier and reported in a paper in this issue16, the CASP community worked together to generate models for 10 of the SARS2 proteins that had no experimental structure and where homology modeling was not effective. The result was a large set of models for each of these ten targets. To be useful to the broader scientific community, it was necessary to somehow select one model for each target and to provide global and local accuracy estimates. A large set of accuracy estimates were also collected for the models. It turned out there was very little agreement as to which were the most accurate structures. The Venclovas group devised a consensus accuracy estimate method to address this problem in the short term. Subsequently, two of these structures have been solved experimentally so making it possible to check how well model selection worked. The SARS2 paper16 shows these data. By far the most accurate models are from the AlphaFold group, consistent with the later CASP results. But only one EMA method selected an AlphaFold model as the best, for only one of the targets, and generally the AlphaFold models were not highly ranked.
This failure may reflect unusual properties of the AlphaFold models. For single model methods this problem has been recognized before31 - it is difficult to devise a general method. The large gap in accuracy between the AlphaFold models and others also likely defeated the consensus methods - the best models were far from any consensus measure. Whatever the cause, it is clear that CASP should carefully reconsider how assessment is done for this category.
Results based on estimates of error provided by model builders themselves are much more encouraging. The AlphaFold2 method outputs estimated Cα errors directly from the structure modeling deep learning network. The average normalized accuracy in estimated Cα error (ASE) for their CASP14 models is 0.91 (out a maximum of possible 1.0), suggesting this approach to error estimation can be very effective. Some other groups are well placed to provide this type of estimate in future. Interestingly, some of the less accurate AlphaFold2 error estimates are for targets where there is doubt about the quality of the experimental structures, such as the NMR targets discussed earlier. That is, a low ASE for a model may turn out to be a useful indicator of low experimental structure reliability.
Inter-residue Contacts and Distances:
It was first proposed that evolutionary sequence information could be used to predict which pairs of amino acids are in three-dimensional contact in 1994, and in 1996 CASP2 introduced a category to encourage development of such methods. After initial progress, for about 14 years, from 2000 to 2014 (CASP11), the methods showed no significant improvement, in spite the huge quantities of relevant sequence data that became available in that period. The CASP accuracy metric stuck at around 20% on the most confidently predicted set of long-range contacts32. But in 2016, the accuracy from the best groups almost doubled to just under 50%33. Apparently, this was a consequence of the introduction of improved classical statistical methods34. That level of accuracy was still too low to have a big impact on the three-dimensional structure accuracy though. In 2018 (CASP13), accuracy improved again, to around 70%, this time as the result of the use of deep learning convolutional network methods35. A number of CASP participants also began using these methods to predict a continuous probability function for inter-residue distances rather than just a binary yes/no for contacts36. Together, these developments did result in the CASP13 major jump in 3D accuracy seen in Figure 1.
For CASP14, we maintained the category on binary contact prediction and extended it to include prediction of inter-residue probability distributions5. There was no further improvement in the contact accuracy. That may reflect the fact that most creative energy went into the development of distance probability methods. This first assessment of probability accuracy showed a strong signal using newly developed metrics and provides a baseline against which to measure progress in future CASPs. The currently most successful deep learning methods depend on predicting these distributions.
DISCUSSION
Objective testing in CASP14 has shown that the problem of computing atomic accuracy protein structures from amino acid sequence is solved, at least for single, ordered, proteins. The improvements in model accuracy by AlphaFold2 and the other leading groups almost all arise from more advanced use of deep learning methods, discussed in 37. At the CASP14 conference, AlphaFold2 outlined four significant changes from their CASP13 methodologies and a detailed methodology paper describing these and many specifics has recently been published38. The changes are: (a) An additional stage of the neural network architecture which produces three-dimensional coordinates rather than ending with inter-residue probability distributions as was done in CASP13. (b) Replacement of convolutional operations with attention learning39. Convolutions do not appear ideal for distogram or contact map feature extraction, and in fact it is surprising they work as well as they do. Attention learning is a rapidly advancing branch of deep learning40 that in principle allows identification of the most important information flows in a network. (c) Some protein specific features, such as covalent geometry, were introduced into the network structure, partly tailoring the network to the specifics of the problem. (d) The network directly outputs confidence estimates for the position of each residue in the structure, and as noted earlier, these are impressively accurate.
Most (but not all) previous participants in CASP have been academic research groups. AlphaFold2 are from a company, and the CASP organizers recognize they operate under different restraints. For conference presentations, CASP expects that methods descriptions equivalent to that normally found in a published paper will be available, a level that was not reached by AlphaFold2 at that time. However, as noted earlier, full methodology has now been published38, and there is a shorter CASP special issue paper discussing details of the CASP results41. As a practical matter, CASP is unable to insist on full code and data availability though it is of course encouraged. In any case, extensive AlphaFold2 code has in fact now been released. Nevertheless, the delay has been a source of controversy. Previous experience suggests it may not have been critical. AlphaFold were also the most successful participant in the previous CASP, and although more method information was provided at the conference, full details were only published many months later, and there was no code release. In spite of this, as Figure 1 shows, other research groups had substantially surpassed AlphaFold’s performance by CASP14, and most importantly, publicly available servers were also performing at a level similar to AlphaFold’s CASP13 level. There is intense activity in the modeling community now, exploring the new techniques, and the Baker group has already reported modeling accuracy similar to that of AlphaFold2 using deep learning methods28.
The success of a company in this field has lessons for the academic community. In CASP13, DeepMind’s methods were very clearly and directly built on ideas and methods pioneered in the CASP community. In CASP14 they very effectively implemented their own new insights. Partly, this reflects the fact that they have unparalleled expertise in deep learning. It has also been suggested that the computing resources used are beyond what is available to a normal academic group, though this is unclear. The human resources used are also larger than a typical academic group. On the other hand, the total human resources and computer power deployed by other CASP participants likely far exceeds DeepMind’s – but it was fragmented over multiple competitive groups. In the US in particular, the funding system encourages this kind of small-scale approach. What happened here should be a reason to carefully examine and adjust funding models.
The AlphaFold2 methodology consistently produces models competitive in accuracy with the best experimental results, and subatomic scale differences from experiment are the norm. In this sense, it is an almost complete solution to the problem of computing three-dimensional structure from amino acid sequence. But there have been some objections raised to calling it a solution to the classical ‘protein folding’ problem25. To some that requires two further conditions to be fulfilled: there is no dependence on evolutionary information (a folding protein does not know the sequence of its relatives) and there is some explicit inclusion of the folding process. On the first of these, recent CASPs have seen a dramatic reduction in the dependence of model accuracy on sequence alignment depth, a key ingredient in the classical contact prediction methods that preceded deep learning33 and also a usual input into deep learning networks. Figure 6 shows this dependency over recent CASPs for the subset of the hardest (FM) targets.
Figure 6:
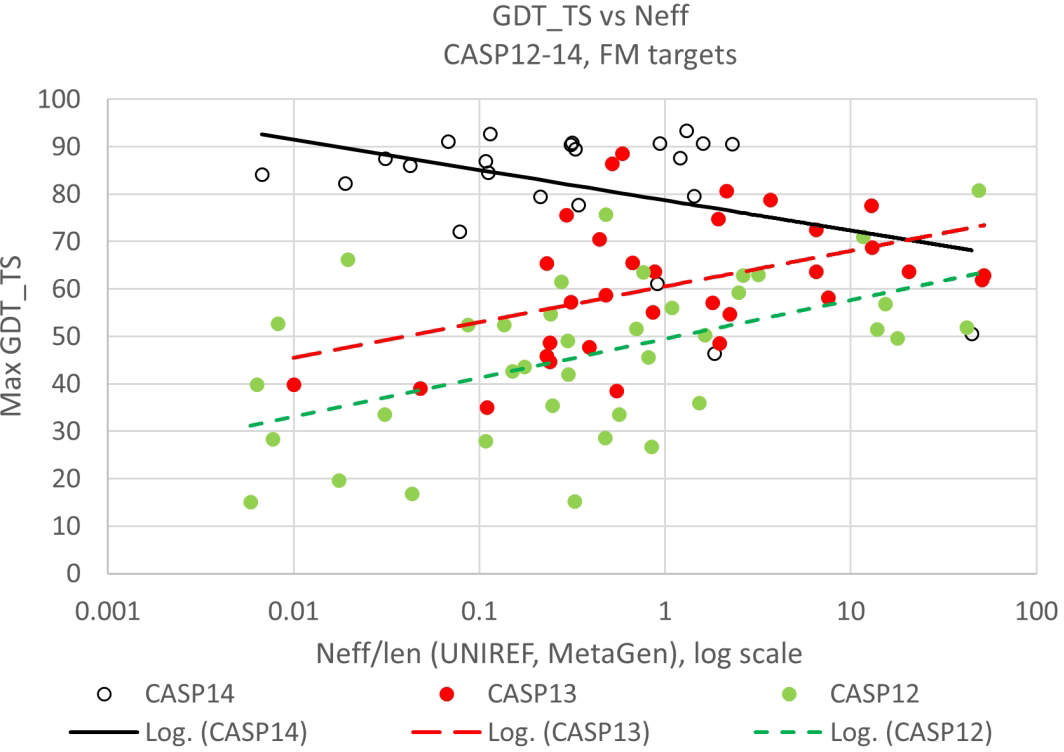
Best model backbone agreement with experiment (GDT_TS) as a function of log normalized sequence alignment depth (Neff/len) for targets with no detectable homology to known structures (‘Free Modeling’ (‘FM’)). Data for the most recent three CASPs. For this subset of the hardest ‘FM’ targets, dependence on alignment depth seen in earlier CASPs is not seen in CASP14.
In CASP12 (2016), where best performance was dominated by methods dependent on predicting three dimensional contacts between residue pairs using classical statistical methods, there is a pronounced fall-off in accuracy for shallower alignments. In CASP13, where convolutional neural networks had become the most effective methods, there is a similar dependency, though with an overall increase in accuracy. Strikingly, in CASP14, where more sophisticated deep learning methods were most successful, there is no accuracy fall-off with decreasing alignment depth. In their analysis of a much larger benchmarking sample38 the AlphaFold2 group did find that for shallow alignments (less than 30 sequences) there is a remaining dependence on depth. However, the accuracy spread in that region is large, and there are still highly accurate models, some with only a single sequence available. Very few models are really low accuracy. So, especially if the less demanding criterion of fold rather than atomic accuracy is used, the method is effective for single sequences.
The second objection - inclusion of the folding process - is more nuanced. There are two factors involved. One is a belief that a protein sequence that can successfully fold must not only have a well-defined global free energy minimum but must also incorporate specific features dictating a preferred folding pathway. This concept arises from a 1968 paper42 which argues that the conformational space of a protein is so large that there must be a very specific pathway by which the conformation progresses from the unfolded to folded state. That motivated many experimental and computational investigations of possible pathways in the subsequent decades. In fact, as has often been pointed out (for example43) this is a fallacious argument, since the free energy falls progressively as the protein folds, providing sufficient guidance44. Local conformational restraints also greatly reduce the size of the space45. More concrete evidence for this conclusion is provided by the large number of designed proteins that have now been made, with no explicit design of a pathway46.
The further concern is an unease that we do not know what the machine is doing, and therefore still do not understand key aspects of the physics of the process. This is probably the first solution of a serious scientific problem by artificial intelligence, and we will face more of these issues in future. The AlphaFold2 method is clearly not just using pattern recognition - at this level of accuracy there are astronomically more atomic configurations than are present in the PDB - in some serious sense the machine generalizes from the training data to an extent analogous to the way in which a physics force field is a generalization that is applicable to all atomic configurations. Does that mean that the network learns the force field? Not in the way we understand the term. For example, there are two free modeling targets with zinc binding sites and another target with two bound hemes. The parts of these structures interacting with the ligands are accurately modeled, even though the ligands are absent in the calculation.
A frequently asked question is if any of the new methods contribute to modeling of disorder and dynamics. There are limited data from CASP to fully address this, and the terms mean different things to different people. Some CASP targets do have local disorder and flexibility and it appears that AlphaFold2 sometimes produces a variety of structures for these regions. The difficulty here is one that affects the whole disorder field - a lack of experimental data with which to assess performance. There is also at least one example in CASP14 of flexibility between domains being reproduced (T1024)47. As discussed earlier, conformational flexibility associated with docking to other molecules is already being addressed by members of the CASP community22. As already noted, the new report from the Baker group uses the deep learning system developed for monomers to directly built multimers, in some circumstances obviating the problem48. Given adequate ligand docking methods (see below), allosteric conformational changes should be addressable. Short time scale classical dynamics do not appear to be within the scope of the methods, so there is still a role there for molecular dynamics.
The level of modeling accuracy seen in this CASP has many implications for structural biology. We have already seen that the structures are accurate enough to help solve structures, both X-ray and cryo-EM12. Three difficult target structures were solved with molecular replacement using crystallographic data and CASP14 models. An additional atomic structure was derived based on a lower resolution cryo-EM density map. A post-CASP study of the extent to which other targets could have been solved using molecular replacement by Randy Read and colleagues found that only two of those tested did not have models good enough for molecular replacement49. So it is clear that going forward, the solution of crystal structures in particular will very frequently be done using these models, often greatly speeding the process. As well as providing a powerful aid for solving structures the methods will create more general synergy between computation and experiment. For example, a sequence alignment error in one of the target experimental structures was corrected with the aid of a model12. The correction hinged on accurately identifying which of two proline residues is in the cis conformation rather than trans, a very finely balanced energy difference with about only one in 15 prolines in protein structures adopting the cis form50. All this will be aided by the availability of servers such as that already released by the Baker group48, and by databases of computed structures, such as the one launched by DeepMind and the EMBL.
As discussed earlier, there is reason to believe that the new methods will also be extended to protein complexes. As with protein docking, the CASP function assessors have shown that improved accuracy of structure models will improve the performance of current ligand docking methods8, with implications for screening ligand specificity across all proteins. Deep learning methods for small ligand docking have been developing in parallel to the protein structure work51 and a new community experiment (CACHE) similar to CASP is about to be launched to evaluate these. There are obvious implications for drug design and repurposing if the methods are as effective as claimed.
Supplementary Material
ACKNOWLEDGEMENTS
CASP is only possible through the generosity and support of three groups of people: the data providers, the assessors, and the participants. A total of 39 experimental groups from 15 countries provided targets for CASP14.
We once again thank the assessment teams for their thorough and insightful analyses: Lisa Kinch for target analysis; Nick Grishin, Lisa Kinch, and Dustin Schaeffer for topology and domain assessment; Andrei Lupas, Joana Pereira, and Marcus Hartmann for high accuracy analysis; Dan Rigden for refinement; Alfonso Valencia and Rosalba Lepore for inter-residue distances and contacts; Chaok Seok for accuracy estimation; Ezgi Karaca for protein assemblies; Marc Lensink and Shoshana Wodak for CAPRI assessment; and Sandor Vajda and Dima Kozakov for function analysis. Thanks also to Gaetano Montelione for analysis of performance on NMR targets and Randy Read for molecular replacement tests. As always, for participants, it takes courage to expose their methods to such intense and public scrutiny. We greatly appreciate the 97 research groups who submitted their work to this CASP. We again thank PROTEINS for providing a mechanism for peer reviewed publication of the outcome of the experiment.
The CASP Prediction Center at UC Davis is supported by a grant from the US National Institute of General Medical Sciences (NIGMS/NIH), R01GM100482 to KF.
Footnotes
The authors declare they have no conflicts of interest.
REFERENCES
- 1.Pereira J, Simpkin AJ, Hartmann MD, Rigden DJ, Keegan RM, Lupas AN. High-accuracy protein structure prediction in CASP14. Proteins 2021. doi: 10.1002/prot.26171. Online ahead of print. [DOI] [PubMed] [Google Scholar]
- 2.Kinch LN, Pei J, Kryshtafovych A, Schaeffer RD, Grishin NV. Topology evaluation of models for difficult targets in the 14th round of the critical assessment of protein structure prediction. Proteins 2021. doi: 10.1002/prot.26172. Online ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ozden B, Kryshtafovych A, Karaca E. Assessment of the CASP14 Assembly Predictions. Proteins 2021. doi: 10.1002/prot.26199. Online ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lensink MF, Brysbaert G, Mauri T, Nadzirin N, Velankar S, Chaleil RAG, Clarence T, Bates PA, Kong R, Liu B, Yang G, Liu M, Shi H, Lu X, Chang S, Roy RS, Quadir F, Liu J, Cheng J, Antoniak A, Czaplewski C, Gieldo NA, Kogut M, Lipska AG, Liwo A, Lubecka EA, Maszota-Zieleniak M, Sieradzan AK, Slusarz R, Wesolowski PA, ZiEba K, Del Carpio Munoz CA, Ichiishi E, Harmalkar A, Gray JJ, Bonvin A, Ambrosetti F, Honorato RV, Jandova Z, Jimenez-Garcia B, Koukos PI, Van Keulen S, Van Noort CW, Reau M, Roel-Touris J, Kotelnikov S, Padhorny D, Porter KA, Alekseenko A, Ignatov M, Desta I, Ashizawa R, Sun Z, Ghani U, Hashemi N, Vajda S, Kozakov D, Rosell M, Rodriguez-Lumbreras LA, Fernandez-Recio J, Karczynska A, Grudinin S, Yan Y, Li H, Lin P, Huang SY, Christoffer C, Terashi G, Verburgt J, Sarkar D, Aderinwale T, Wang X, Kihara D, Nakamura T, Hanazono Y, Gowthaman R, Guest JD, Yin R, Taherzadeh G, Pierce BG, Barradas-Bautista D, Cao Z, Cavallo L, Oliva R, Sun Y, Zhu S, Shen Y, Park T, Woo H, Yang J, Kwon S, Won J, Seok C, Kiyota Y, Kobayashi S, Harada Y, Takeda-Shitaka M, Kundrotas PJ, Singh A, Vakser IA, DapkUnas J, Olechnovic K, Venclovas C, Duan R, Qiu L, Zhang S, Zou X, Wodak SJ. Prediction of protein assemblies, the next frontier: The CASP14-CAPRI experiment. Proteins 2021. doi: 10.1002/prot.26222. Online ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ruiz-Serra V et al. Assessing the accuracy of contact and distance predictions in CASP14. Proteins 2021. [CASP14 special issue]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Simpkin AJ, Sanchez Rodriguez F, Mesdaghi S, Kryshtafovych A, Rigden DJ. Evaluation of model refinement in CASP14. Proteins 2021. doi: 10.1002/prot.26185. Online ahead of print. doi: 10.1002/prot.26209. Online ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kwon S, Won J, Kryshtafovych A, Seok C. Assessment of protein model structure accuracy estimation in CASP14: Old and new challenges. Proteins 2021. doi: 10.1002/prot.26192. Online ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Egbert M, Ghani U, Ashizawa R, Kotelnikov S, Nguyen T, Desta I, Hashemi N, Padhorny D, Kozakov D, Vajda S. Assessing the binding properties of CASP14 targets and models. Proteins 2021. doi: 10.1002/prot.26209. Online ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schaeffer RD, Kinch L, Kryshtafovych A, Grishin NV. Assessment of domain interactions in CASP14. Proteins 2021. doi: 10.1002/prot.26225. Online ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kinch LN, Schaeffer RD, Kryshtafovych A, Grishin NV. Target classification in the 14th round of the critical assessment of protein structure prediction (CASP14). Proteins 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alexander L et al. Target highlights in CASP14: analysis of models by structure providers. Proteins 2021. [CASP14 special issue]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kryshtafovych A, Moult J, Albrecht R, Chang GA, Chao K, Fraser A, Greenfield J, Hartmann MD, Herzberg O, Josts I, Leiman PG, Linden SB, Lupas AN, Nelson DC, Rees SD, Shang X, Sokolova ML, Tidow H, Team AF. Computational models in the service of X-ray and cryo-EM structure determination. Proteins 2021. doi: 10.1002/prot.26223. Online ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hura GL, Hodge CD, Rosenberg D, Guzenko D, Duarte JM, Monastyrskyy B, Grudinin S, Kryshtafovych A, Tainer JA, Fidelis K, Tsutakawa SE. Small angle X-ray scattering-assisted protein structure prediction in CASP13 and emergence of solution structure differences. Proteins 2019;87(12):1298–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fajardo JE, Shrestha R, Gil N, Belsom A, Crivelli SN, Czaplewski C, Fidelis K, Grudinin S, Karasikov M, Karczynska AS, Kryshtafovych A, Leitner A, Liwo A, Lubecka EA, Monastyrskyy B, Pages G, Rappsilber J, Sieradzan AK, Sikorska C, Trabjerg E, Fiser A. Assessment of chemical-crosslink-assisted protein structure modeling in CASP13. Proteins 2019;87(12):1283–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sala D, Huang YJ, Cole CA, Snyder DA, Liu G, Ishida Y, Swapna GVT, Brock KP, Sander C, Fidelis K, Kryshtafovych A, Inouye M, Tejero R, Valafar H, Rosato A, Montelione GT. Protein structure prediction assisted with sparse NMR data in CASP13. Proteins 2019;87(12):1315–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kryshtafovych A, Moult J, Billings WM, Della Corte D, Fidelis K, Kwon S, Olechnovic K, Seok C, Venclovas C, Won J. Modeling SARS-CoV2 proteins in the CASP-commons experiment. Proteins 2021. doi: 10.1002/prot.26231. Online ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kryshtafovych A, Fidelis K, Moult J. CASP10 results compared to those of previous CASP experiments. Proteins 2014;82 Suppl 2:164–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kryshtafovych A, Schwede T, Topf M, Fidelis K, Moult J. Critical assessment of methods of protein structure prediction (CASP)-Round XIII. Proteins 2019;87(12):1011–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang YP. et al. Assessment of Prediction Methods for Protein Structures Determined by NMR in CASP14: Impact of AlphaFold2. Proteins 2021. [CASP14 special issue]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johnson S, Furlong EJ, Deme JC, Nord AL, Caesar JJE, Chevance FFV, Berry RM, Hughes KT, Lea SM. Molecular structure of the intact bacterial flagellar basal body. Nat Microbiol 2021;6(6):712–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu Y, Eisenberg D. 3D domain swapping: as domains continue to swap. Protein Sci 2002;11(6):1285–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baek M, Anishchenko I, Park H, Humphreys IR, Baker D. Protein oligomer modeling guided by predicted interchain contacts in CASP14. Proteins 2021. doi: 10.1002/prot.26197. Online ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zemla A, Venclovas Č, Moult J, Fidelis K. Processing and evaluation of predictions in CASP4. Proteins: Structure, Function, and Bioinformatics 2001;45(S5):13–21. [DOI] [PubMed] [Google Scholar]
- 24.Zemla A LGA: A method for finding 3D similarities in protein structures. Nucleic Acids Res 2003;31(13):3370–3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Anfinsen CB. Principles that govern the folding of protein chains. Science 1973;181(4096):223–230. [DOI] [PubMed] [Google Scholar]
- 26.Lensink MF, Nadzirin N, Velankar S, Wodak SJ. Modeling protein-protein, protein-peptide, and protein-oligosaccharide complexes: CAPRI 7th edition. Proteins 2020;88(8):916–938. [DOI] [PubMed] [Google Scholar]
- 27.Vakser IA. Protein-protein docking: from interaction to interactome. Biophys J 2014;107(8):1785–1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baek M, DiMaio F, Anishchenko I, Dauparas J, Ovchinnikov S, Lee GR, Wang J, Cong Q, Kinch LN, Schaeffer RD, Millan C, Park H, Adams C, Glassman CR, DeGiovanni A, Pereira JH, Rodrigues AV, van Dijk AA, Ebrecht AC, Opperman DJ, Sagmeister T, Buhlheller C, Pavkov-Keller T, Rathinaswamy MK, Dalwadi U, Yip CK, Burke JE, Garcia KC, Grishin NV, Adams PD, Read RJ, Baker D. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021;373(6557):871–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tsai J, Bonneau R, Morozov AV, Kuhlman B, Rohl CA, Baker D. An improved protein decoy set for testing energy functions for protein structure prediction. Proteins 2003;53(1):76–87. [DOI] [PubMed] [Google Scholar]
- 30.Wang Y, Lamim Ribeiro JM, Tiwary P. Machine learning approaches for analyzing and enhancing molecular dynamics simulations. Curr Opin Struct Biol 2020;61:139–145. [DOI] [PubMed] [Google Scholar]
- 31.Won J, Baek M, Monastyrskyy B, Kryshtafovych A, Seok C. Assessment of protein model structure accuracy estimation in CASP13: Challenges in the era of deep learning. Proteins 2019;87(12):1351–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Monastyrskyy B, D’Andrea D, Fidelis K, Tramontano A, Kryshtafovych A. New encouraging developments in contact prediction: Assessment of the CASP11 results. Proteins 2016;84 Suppl 1:131–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schaarschmidt J, Monastyrskyy B, Kryshtafovych A, Bonvin A. Assessment of contact predictions in CASP12: Co-evolution and deep learning coming of age. Proteins 2018;86 Suppl 1:51–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.de Juan D, Pazos F, Valencia A. Emerging methods in protein co-evolution. Nat Rev Genet 2013;14(4):249–261. [DOI] [PubMed] [Google Scholar]
- 35.Kandathil SM, Greener JG, Jones DT. Prediction of interresidue contacts with DeepMetaPSICOV in CASP13. Proteins 2019; 87(12):1092–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Senior AW, Evans R, Jumper J, Kirkpatrick J, Sifre L, Green T, Qin C, Zidek A, Nelson AWR, Bridgland A, Penedones H, Petersen S, Simonyan K, Crossan S, Kohli P, Jones DT, Silver D, Kavukcuoglu K, Hassabis D. Protein structure prediction using multiple deep neural networks in CASP13. Proteins 2019; 87(12):1141–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Laine E et al. Protein sequence-to-structure learning: Is this the end(-to- end revolution)? Proteins 2021. [CASP14 special issue]. [DOI] [PubMed] [Google Scholar]
- 38.Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Zidek A, Potapenko A, Bridgland A, Meyer C, Kohl SAA, Ballard AJ, Cowie A, Romera-Paredes B, Nikolov S, Jain R, Adler J, Back T, Petersen S, Reiman D, Clancy E, Zielinski M, Steinegger M, Pacholska M, Berghammer T, Bodenstein S, Silver D, Vinyals O, Senior AW, Kavukcuoglu K, Kohli P, Hassabis D. Highly accurate protein structure prediction with AlphaFold. Nature 2021; 596(7873):583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bates RJ, Evans RA, Figurnov M, Gren TFG, Jumper J, Pritzel A, Senior A; Protein Structure Prediction from Amino Acid Sequences Using Self-Attention Neural Networks. US patent US2021166779A1, DeepMind technologies Limited, 2021
- 40.Niu Z, Zhong G, Yu H. A Review on the attention mechanism of deep learning. Neurocomputing 2021;452:48–62. [Google Scholar]
- 41.Jumper J, et al. AlphaFold at CASP14. Proteins 2021. [CASP14 special issue]. [Google Scholar]
- 42.Levinthal C Are there Pathways for Protein Folding? JChimPhys 1968;65:44–45. [Google Scholar]
- 43.Thomas DJ. Concepts in protein folding. FEBS Lett 1992;307(1):10–13. [DOI] [PubMed] [Google Scholar]
- 44.Dill KA, MacCallum JL. The protein-folding problem, 50 years on. Science 2012;338(6110):1042–1046. [DOI] [PubMed] [Google Scholar]
- 45.Fidelis K, Stern PS, Bacon D, Moult J. Comparison of systematic search and database methods for constructing segments of protein structure. Protein Eng 1994;7(8):953–960. [DOI] [PubMed] [Google Scholar]
- 46.Baker D What has de novo protein design taught us about protein folding and biophysics? Protein Sci 2019;28(4):678–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Del Alamo D, Govaerts C, McHaourab HS. AlphaFold2 predicts the inward-facing conformation of the multidrug transporter LmrP. Proteins 2021;89(9):1226–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Baek M, DiMaio F, Anishchenko I, Dauparas J, Ovchinnikov S, Lee GR, Wang J, Cong Q, Kinch LN, Schaeffer RD, Millan C, Park H, Adams C, Glassman CR, DeGiovanni A, Pereira JH, Rodrigues AV, van Dijk AA, Ebrecht AC, Opperman DJ, Sagmeister T, Buhlheller C, Pavkov-Keller T, Rathinaswamy MK, Dalwadi U, Yip CK, Burke JE, Garcia KC, Grishin NV, Adams PD, Read RJ, Baker D. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021. doi: 10.1126/science.abj8754 Online ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Millan C, Keegan RM, Pereira J, Sammito MD, Simpkin AJ, McCoy AJ, Lupas AN, Hartmann MD, Rigden DJ, Read RJ. Assessing the utility of CASP14 models for molecular replacement. Proteins 2021. doi: 10.1002/prot.26214. Online ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.MacArthur MW, Thornton JM. Influence of proline residues on protein conformation. J Mol Biol 1991;218(2):397–412. [DOI] [PubMed] [Google Scholar]
- 51.Brown N, Ertl P, Lewis R, Luksch T, Reker D, Schneider N. Artificial intelligence in chemistry and drug design. J Comput Aided Mol Des 2020;34(7):709–715. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.