

Abstract
More than a million Okazaki fragments are synthesized, processed and joined during replication of the human genome. After synthesis of an RNA-DNA oligonucleotide by DNA polymerase α holoenzyme, proliferating cell nuclear antigen (PCNA), a homotrimeric DNA sliding clamp and polymerase processivity factor, is loaded onto the primer-template junction by replication factor C (RFC). Although PCNA interacts with the enzymes DNA polymerase δ (Pol δ), flap endonuclease 1 (FEN1) and DNA ligase I (LigI) that complete Okazaki fragment processing and joining, it is not known how the activities of these enzymes are coordinated. Here we describe a novel interaction between Pol δ and LigI that is critical for Okazaki fragment joining in vitro. Both LigI and FEN1 associate with PCNA-Pol δ during gap-filling synthesis, suggesting that gap-filling synthesis is carried out by a complex of PCNA, Pol δ, FEN1 and LigI. Following ligation, PCNA and LigI remain on the DNA, indicating that Pol δ and FEN1 dissociate during 5’ end processing and that LigI engages PCNA at the DNA nick generated by FEN1 and Pol δ. Thus, dynamic PCNA complexes coordinate Okazaki fragment synthesis and processing with PCNA and LigI forming a terminal structure of two linked protein rings encircling the ligated DNA.
Keywords: DNA replication, DNA synthesis, DNA flap cleavage, DNA ligation, DNA-protein complex, proliferating cell nuclear antigen, sliding clamp, DNA polymerase δ, replication fork, primase
Graphical Abstract
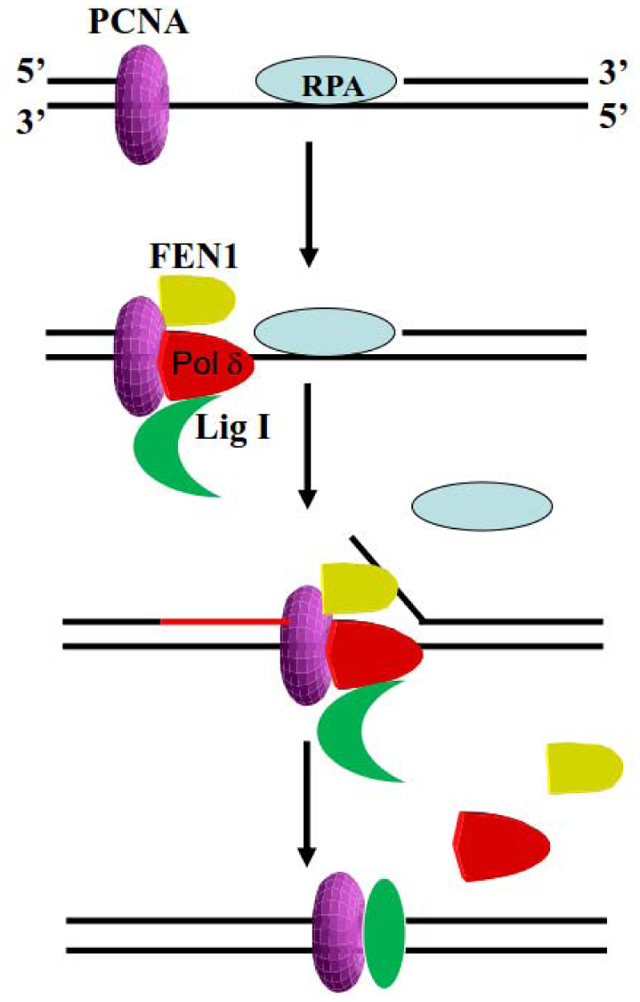
INTRODUCTION
At the replication fork, the lagging strand is synthesized discontinuously as a series of short Okazaki fragments in a cyclical process that occurs more than a million times per human cell cycle [1]. After the initial synthesis of a short RNA-DNA hybrid by the primase and DNA polymerase activities of the Pol α holoenzyme, RFC displaces Pol α from the 3’ terminus of the RNA-DNA hybrid in a reaction that is dependent upon the binding of Replication protein A (RPA) to the single strand DNA template. RFC then loads the homotrimeric DNA sliding clamp PCNA, which in turn recruits and co-ordinates the activities of the enzymes that complete the processing and joining of adjacent Okazaki fragments [1]. PCNA serves as the processivity factor for the Pol δ holoenzyme, which extends the RNA-DNA primer [2]. Three of the four subunits of the human Pol δ holoenzyme, p125, p66 and p12, interact with PCNA via canonical and noncanonical PCNA interacting protein (PIP) motifs [3–6]. When the PCNA-Pol δ complex encounters the 5’ end of the adjacent Okazaki fragment, DNA synthesis slows as the PCNA-Pol δ complex starts to displace the 5’ end of the adjacent RNA-DNA fragment [7]. The resultant 5’ flap is removed by another PCNA-interacting protein, the DNA structure-specific endonuclease FEN1 [8]. This can occur via a dynamic iterative reaction in which Pol δ generates a 1-nucleotide flap that is removed by FEN1 until the ribo- and, presumably, the deoxynucleotides synthesized by Pol α are removed [1, 7]. There is also evidence for an alternative pathway involving a larger 5’ flap that requires processing by the Pif1 DNA helicase, RPA and the DNA2 nuclease prior to flap removal by FEN1 [9]. The nick remaining between Okazaki fragments after flap removal by FEN1 is usually ligated by LigI, which also binds to PCNA [10, 11].
Since eukaryotic PCNA is a homotrimer with three potential binding sites, it has been suggested that it may co-ordinate the synthesis, processing and joining of Okazaki fragments by simultaneously interacting with Pol δ, FEN1 and LigI [12, 13] as has been demonstrated for homotrimeric and heterotrimeric archaeal DNA sliding clamps [14, 15]. While kinetic and enzyme-trapping studies with purified S. cerevisiae proteins provided indirect evidence for a functional complex of the loaded PCNA trimer with both Pol δ and FEN1 [7], yeast PCNA trimers with only one functional binding site can co-ordinate the activities of Pol δ, FEN1 and LigI, albeit less efficiently than PCNA trimers with 2 or 3 functional binding sites, indicating that these factors can interact sequentially with PCNA during Okazaki fragment maturation [16]. Notably, human Pol δ has an additional subunit, p12, that is not present in yeast Pol δ and is less processive than the yeast enzyme due its looser association with PCNA [17–19], suggesting that there are likely to be differences in the mechanisms of Okazaki fragment synthesis, processing and ligation between humans and yeast. Here we describe a novel physical and functional interaction between human Pol δ and LigI that is critical for efficient joining of Okazaki fragments in reconstituted reactions and provide direct evidence for functional DNA-bound PCNA complexes with multiple DNA replication enzymes as well as a stable, PCNA-LigI complex that is retained on ligated DNA.
RESULTS
Association of Pol δ with DNA during Okazaki fragment processing, synthesis and ligation.
To determine whether DNA structure dictates the association and dissociation of DNA replication proteins during Okazaki fragment processing, synthesis and ligation, we constructed a labeled, biotinylated DNA substrate containing a 30-nucleotide gap and a 5-nucleotide 5’ flap (Fig. 1a) that mimics an Okazaki fragment intermediate generated by the Pif1/DNA2 subpathway [9]. Reconstituted reactions containing equimolar amounts of the DNA substrate, RPA, LigI, Pol δ and FEN1, and a 2.5x higher amount of the PCNA trimer in different combinations, enabled us to visualize reaction intermediates and the ligated product (Fig. 1b). Notably, essentially all of the labeled DNA primer was converted into full-length ligated product in the fully reconstituted reaction after incubation at 37°C for 10 min (Fig. 1b, compare lanes 1 and 9). An abnormal ligation product indicated by the asterisk was generated in the fully reconstituted reaction lacking dTTP (Fig. 1b, lane 10) by the mechanism shown in Figure S1. To enable us to monitor association of proteins with the DNA at different stages of the processing reaction, we added protein kinase A phosphorylation sites to PCNA, LigI, Pol δ and FEN1 so that these proteins could be specifically labeled by incubation with protein kinase A. In reactions with labeled PCNA, we observed that the association of labeled PCNA with the DNA substrate was dependent on loading by RFC (Fig. S2, compare lanes 2 with lanes 1 and 3) with loaded PCNA remaining stably associated at levels of about 2.3 monomers or 0.8 PCNA trimers per DNA molecule during the processing and ligation reactions (Fig. S2, lanes 4–9) as expected. In the absence of PCNA and/or RFC, labeled Pol δ was retained on the DNA substrate and synthesized DNA in a non-processive manner (Fig. 1b and c, lanes 1–3). Labeled Pol δ also bound to gapped DNA (Fig. S3, lanes 5–7) and non-ligatable nicked DNA (Fig. S3, lanes 8–10) in the absence of RFC and PCNA, indicating that it has non-specific DNA binding activity. There was, however, a significant increase in the amount of Pol δ retained on PCNA-loaded DNA in the absence of dNTPs, reflecting formation of a specific complex with loaded PCNA (Fig. 1b and c, compare lanes 1 and 5, p≤ 0.0001). In reactions involving gap filling synthesis by PCNA–Pol δ but not ligation (Fig. 1b and c, lanes 4, 6–8), the levels of Pol δ retained on the DNA were higher than non-specific binding (Fig. 1b and c, lanes 1–3, p≤ 0.001) but less than when the assembled PCNA–Pol δ complex was unable to synthesize DNA (Fig. 1b and c, lane 5 p≤ 0.01), suggesting that Pol δ may be less stably bound to PCNA during DNA synthesis, in particular when it encounters a 5’ flap or 5’ end. This is consistent with studies demonstrating that, in comparison to yeast Pol δ, human Pol δ maintains a relatively loose association with PCNA and that stalling of DNA synthesis results in dissociation from PCNA [17]. Finally, the levels of Pol δ retained on the ligated DNA were reduced even further to levels similar to non-specific binding (Fig. 1b and c, compare lane 9 with lanes 1–3).
Fig. 1. Reconstitution of Okazaki fragment synthesis, processing and ligation; association of LigI with reaction intermediates and ligated product.
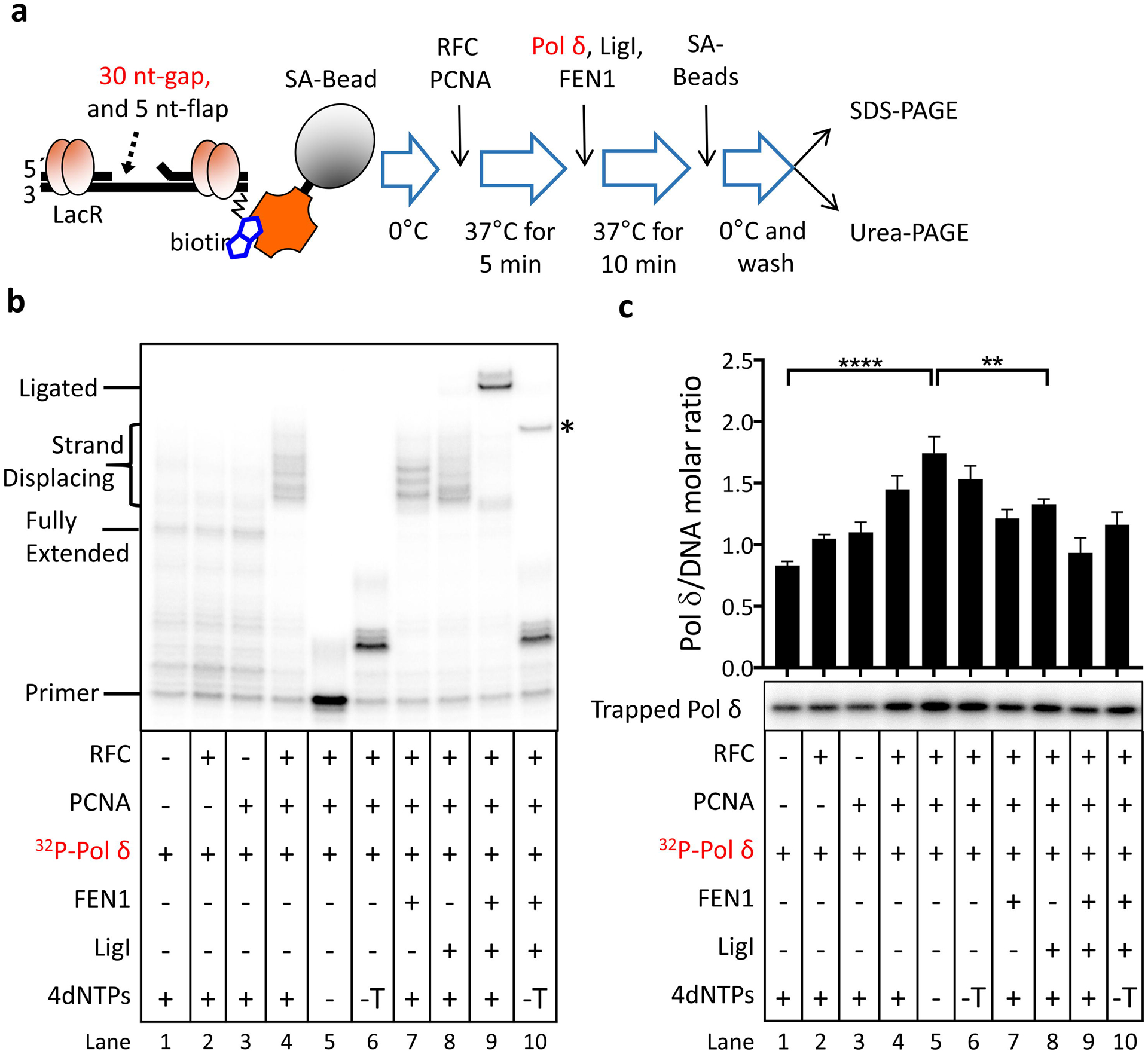
a. Schematic diagram of reconstitution assay. In the fully reconstituted reaction, the labeled DNA substrate (5’P, 1 pmol) with a 30-nucleotide gap and a 5-nucleotide 5’-flap was pre-incubated with 4.5 pmol LacR and 1 pmol RPA prior to incubation with the indicated proteins (1 pmol of each except for 2.5 pmol PCNA trimer) as shown. In reactions with labeled Pol δ, individual proteins (−) and dNTPs (−, no dNTPs; −T, no TTP) were omitted as indicated. b. After separation by denaturing gel electrophoresis, labeled DNA reaction products were detected and quantitated by phosphorimager analysis. The positions of the labeled primer (Primer), the gap filled product (Fully Extended), longer products generated by strand displacement synthesis (Strand Displacing) and the ligated product (Ligated) are indicated on the left. The ligated product appears as a doublet that is most likely due to secondary structure in the ligated oligonucleotide under the gel electrophoresis conditions. An asterisk on the right indicates the position of an aberrant ligation generated as a result of incomplete gap filling synthesis (see Fig. S1). c. Association of Pol δ with the DNA under the different reaction conditions. As shown in Fig. S3, labeled Pol δ was not retained by the streptavidin beads in the absence of the biotinylated DNA.The binding of labeled Pol δ to the DNA is expressed as the ratio of Pol δ retained on the beads to the DNA recovered on the beads. Representative gel of at least three independent experiments with error bars showing standard deviation and with significance calculated and described as indicated in Materials and Methods.
Pol δ and FEN1 compete for binding to DNA bound-PCNA prior to and during gap filling synthesis.
A recent study provided indirect evidence for a PCNA-dependent functional interaction between S. cerevisiae Pol δ and FEN1 that coordinates DNA synthesis and 5’ end processing [7], prompting us to examine the behavior of labeled FEN1 in the reconstituted reactions (Fig. 2). In accord with previous studies showing that the stable association of FEN1 with DNA is dependent upon its interaction with PCNA [20], loading of PCNA onto the DNA substrate resulted in increased retention of FEN1 on the DNA, reaching a level of about 0.5 FEN1 molecules per DNA molecule (Fig. 2, lanes 4 and 9), compared with reactions containing either no or unloaded PCNA (Fig. 2, compare lanes 1, 3 and lane 4). The increased retention of FEN1 on the DNA via binding to loaded PCNA did not markedly enhance flap cleavage (Fig. S4, compare lane 4 with lanes 1–3), indicating that, as expected [21], the loaded PCNA cannot slide to the 5’ flap because of the RPA bound in the gap. The addition of Pol δ under conditions that either prevented initiation of DNA synthesis or supported only partial gap filling significantly reduced the amount of FEN1 retained on PCNA-loaded DNA (Fig. 2, compare lanes 4 and 9 with lanes 7–8) from 0.5 FEN1 molecules per DNA molecule to 0.3 FEN1 per DNA molecule. In contrast, the inclusion of LigI did not reduce the amount of FEN1 retained on the PCNA-loaded DNA (Fig. 2, compare lanes 4 and 9). Thus, it appears that, under these conditions, Pol δ but not LigI competes with FEN1 for binding to loaded PCNA. Efficient flap cleavage was only observed when gap-filling synthesis was completed (Fig. S4, compare lanes 5 and 9 with lanes 6 and 7), suggesting that either FEN1 remains bound to PCNA during gap filling synthesis or it is able to interact with PCNA when the PCNA-Pol δ complex reaches the 5’ flap. Notably, the levels of retained FEN1 were reduced to those observed in reactions with unloaded PCNA either when Pol δ was carrying out strand displacement synthesis (Fig. 2, compare lane 6 with lane 3) or following ligation (Fig. 2, compare lane 10 with lane 3). Thus, it appears that FEN1 is released from DNA-bound PCNA upon encountering the 5’ flap and/or after flap removal.
Fig. 2. Association of FEN1 with Okazaki fragment processing intermediates and ligated product.
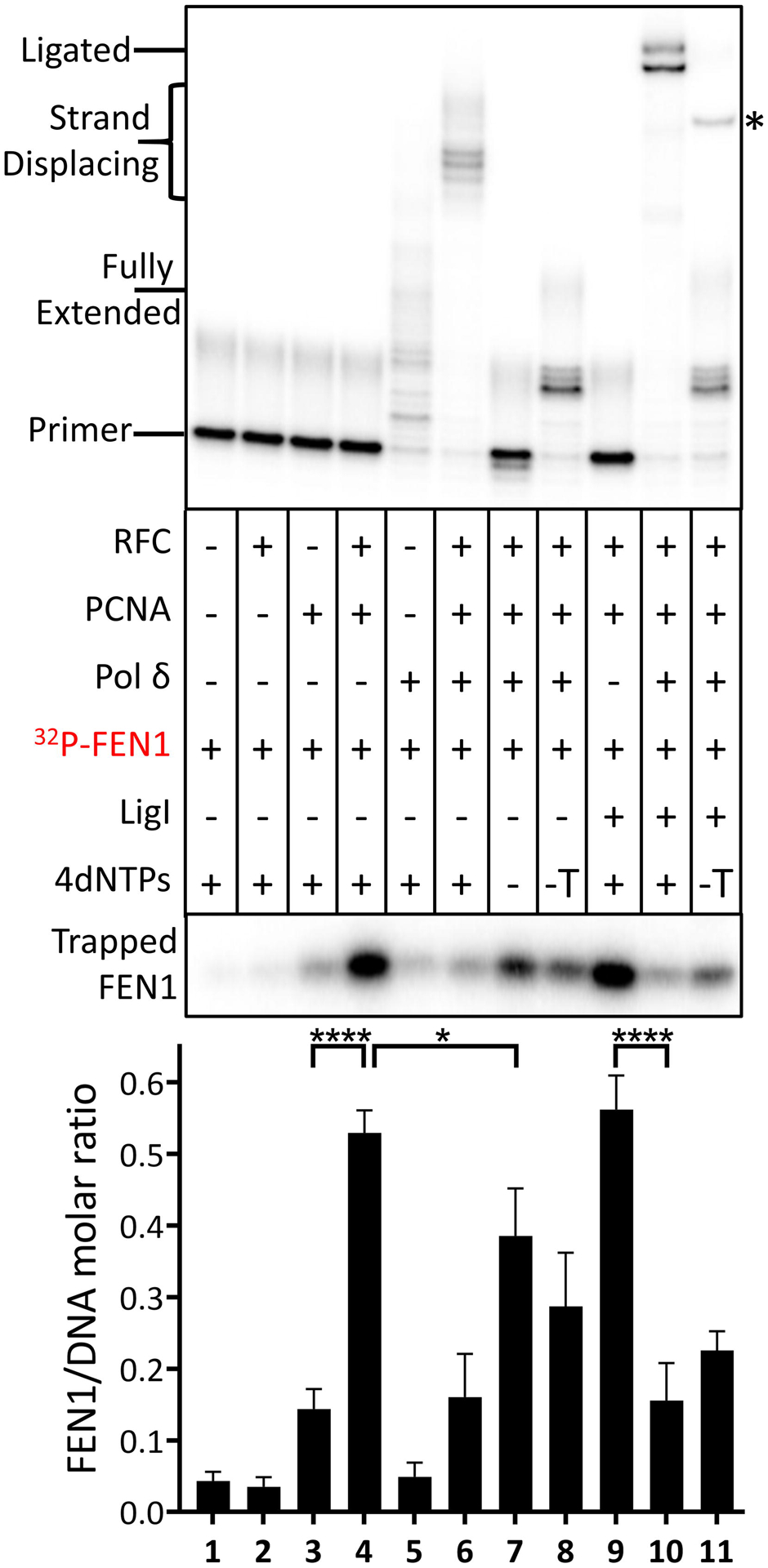
Reconstitution assays were carried out as described in Fig. 1. In reactions with labeled FEN1, individual proteins (−) and dNTPs (−, no dNTPs; −T, no TTP) were omitted as indicated. After separation by denaturing gel electrophoresis, labeled DNA reaction products were detected and quantitated by phosphorimager analysis (Upper panel). The positions of the labeled primer (Primer), the gap filled product (Fully Extended), longer products generated by strand displacement synthesis (Strand Displacing) and the ligated product (Ligated) are indicated on the left. The ligated product appears as a doublet that is most likely due to secondary structure in the ligated oligonucleotide under the gel electrophoresis conditions. An asterisk on the right indicates the position of an aberrant ligation generated as a result of incomplete gap filling synthesis (see Fig. S1). Association of FEN1 with the DNA under the different reaction conditions (Lower Panel). The binding of labeled FEN1 to the DNA is expressed as the ratio of FEN1 retained on the beads to the DNA recovered on the beads. Representative gel of at least three independent experiments with error bars showing standard deviation and with significance calculated and described as indicated in Materials and Methods.
An interaction between LigI and Pol δ mediates the association of Lig1 with DNA.
In accord with a previous study [10], the stable association of LigI with the DNA was dependent upon loaded PCNA (Fig. 3, compare lanes 3 and 4). Surprisingly, similar levels of LigI (approximately 0.2 LigI molecules per DNA molecule) were retained on the the DNA substrate in the presence of Pol δ (Fig. 3, compare lanes 4 and 5). Furthermore, compared to the amount of DNA bound-LigI in the presence of Pol δ alone (Fig. 3, compare lanes 4 and 5), significantly higher levels of LigI were retained on DNA in reactions with an assembled PCNA-Pol δ complex (0.4 to 0.5 LigI molecules per DNA molecule), both prior to initiation of DNA synthesis (Fig. 3, lane 7, p≤ 0.0001) and when DNA synthesis was stalled due either to the absence of dTTP (Fig. 3, lane 8, p≤ 0.0001) or failure to remove the 5’ flap (Fig. 3, lane 6, p≤ 0.01) compared with reactions containing Pol δ alone (Fig. 3, lane 5). This prompted us to ask whether LigI interacts with Pol δ in addition to PCNA [10]. In pull down assays with His-tagged versions of each of the four subunits of human Pol δ holoenzyme [1, 4, 22], a small amount of LigI was specifically retained on the beads liganded by the large catalytic subunit of Pol δ (Fig. 4a) and by the Pol δ holoenzyme (Fig. S5a), suggesting that LigI interacts with the catalytic subunit of Pol δ. In reciprocal experiments, a small amount of purified Pol δ holoenzyme specifically bound to beads liganded by full length LigI but not individual domains [23] (Fig. 4b). Since the weak but specific interaction made further mapping technically challenging, we asked whether the interaction was functionally significant. LigI did not enhance DNA synthesis by Pol δ either in the absence (Fig. S5b, compare lanes 2 and 3) or the presence of RFC and PCNA (Fig. S5b, compare lanes 6 and 7). In contrast, Pol δ enhanced joining by LigI by about 3-fold (Fig. 4c, lanes 2 and 3), a comparable stimulation to that observed in reactions with RFC and PCNA (Fig. 4c, lane 6). As expected, PCNA only enhanced ligation by LigI after it had been loaded onto DNA by RFC (Fig. 4c, compare lane 6 with lanes 2 and 4). Similar levels of ligation were observed in reactions that contained RFC, PCNA and Pol δ (Fig. 4c, compare lanes 7 with lanes 3, 5, and 6), suggesting that the formation of Pol δ-PCNA complexes does not prevent the stimulation of LigI activity by either one or both of these proteins.
Fig. 3. Association of LigI with Okazaki fragment processing intermediates and ligated product.
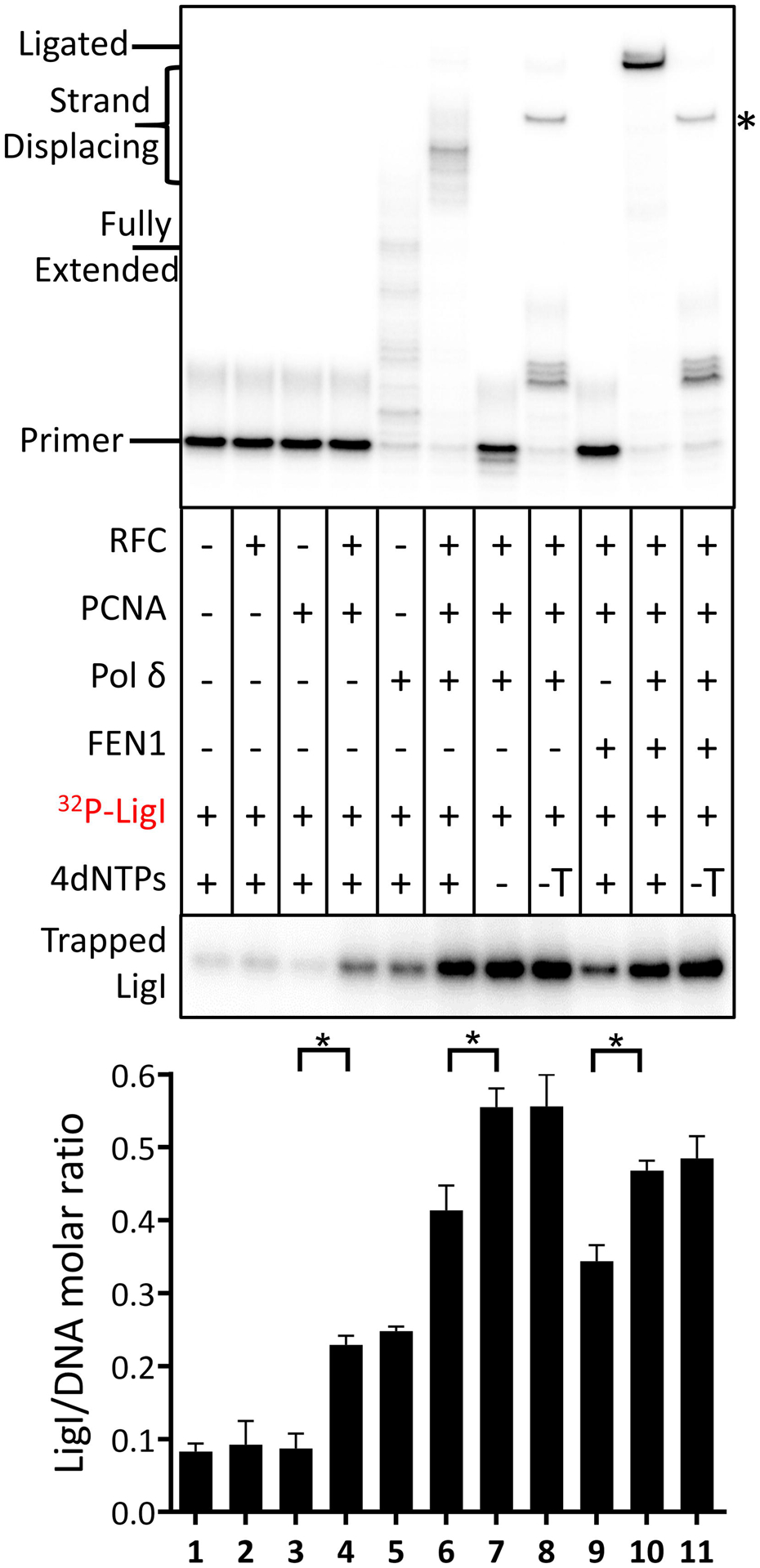
Reconstitution assays were carried out as described in Fig. 1. In reactions with labeled LigI, individual proteins (−) and dNTPs (−, no dNTPs; −T, no TTP) were omitted as indicated. After separation by denaturing gel electrophoresis, labeled DNA reaction products were detected and quantitated by phosphorimager analysis (Upper panel). The positions of the labeled primer (Primer), the gap filled product (Fully Extended), longer products generated by strand displacement synthesis (Strand Displacing) and the ligated product (Ligated) are indicated on the left. The ligated product appears as a doublet that is most likely due to secondary structure in the ligated oligonucleotide under the gel electrophoresis conditions. An asterisk on the right indicates the position of an aberrant ligation generated as a result of incomplete gap filling synthesis (see Fig. S1). Association of LigI with the DNA under the different reaction conditions (Lower panel). The binding of labeled FEN1 to the DNA is expressed as the ratio of LigI retained on the beads to the DNA recovered on the beads. Representative gel of at least three independent experiments with error bars showing standard deviation and with significance calculated and described as indicated in Materials and Methods.
Fig. 4. Interactions between LigI with Pol δ enhances nick ligation by LigI through increased retention on DNA.
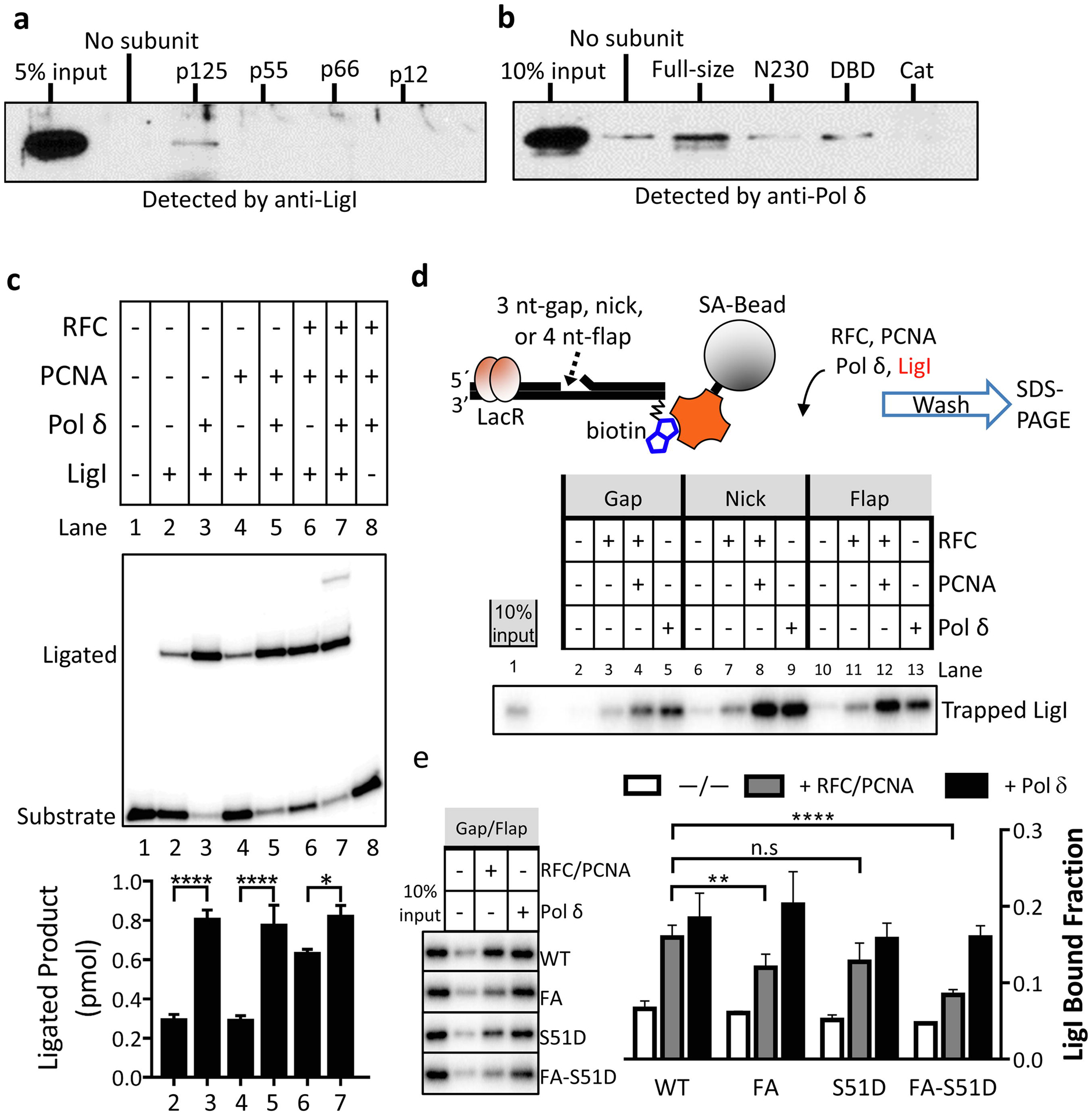
a. Ni-NTA magnetic beads liganded by His-tagged versions (10 pmol of each) of the indicated subunits of Pol δ (p125, p55, p66, p12) or no protein (no subunit) were incubated with 10 pmol LigI as described in Materials and Methods. b. Ni-NTA magnetic beads liganded by His-tagged versions (40 pmol of each) of wild type LigI (Full-size), LigI non-catalytic N terminal domain (N230), LigI DNA binding domain (DBD, residues 233–534) and LigI catalytic domain (Cat resides 533–919) or no protein (no subunit) were incubated with 20 pmol of Pol δ holoenzyme as described in Materials and Methods. Proteins retained on the beads were detected by immunoblotting with the indicated antibody. Inputs for LigI (5% input, panel a) and Pol δ p125 (10% input, panel b) are shown. c. Effect of Pol δ on nick ligation by LigI. The positions of the labeled substrate and ligated product are indicated on the left. Representative gel of at least three independent experiments with error bars showing standard deviation, and with significance calculated as described in Materials and Methods. d. Upper panel: schematic diagram of protein trapping assay. Lac repressor (LacR) and streptavidin-conjugated magnetic bead block both ends of linear DNA duplex, preventing loaded PCNA trimer from sliding off. The nicked DNA substrate is not ligatable due to the absence of a phosphate group at the 5’-terminus of the nick. Lower panel: 32P-Labeled Pol δ (1 pmol) was incubated with either no DNA or the indicated DNA substrate (1 pmol) in the presence (+) or absence (−) of PCNA trimer (2.5 pmol) and RFC (1 pmol) as described in Materials and Methods. Lane 1 represents 10% of input Pol δ. c. Labeled LigI (1 pmol) was incubated with either no DNA or the indicated DNA substrate (1 pmol) in the presence (+) or absence (−) of PCNA trimer (2.5 pmol), RFC (1 pmol) and Pol δ (1 pmol). Lane 1 represents 10% of input LigI. e. Labeled wild type LigI (WT) and LigI mutants (FA, S51D, FA-S51D, 1 pmol of each) were each incubated with either no DNA or a DNA substrate with a 30-nucleotide gap and a 5-nucleotide 5’-flap (1 pmol) in the presence (+) and absence (−) of PCNA trimer (2.5 pmol), RFC (1 pmol) and Pol δ (1 pmol) as indicated. Representative gel of at least three independent experiments with error bars showing standard deviation, statistical significance calculated by a two-way ANOVA with Tukey’s correction for post-hoc unpaired t-tests and asterisks as indicated in Materials and Methods.
Since Pol δ has robust non-specific DNA binding activity (Fig. S3) whereas LigI binds weakly to DNA [23], we examined the effect of Pol δ on the binding of LigI to different linear DNA structures (Fig. 4d). Both Pol δ and loaded PCNA enhanced the retention of labeled LigI on the different DNA substrates to a similar extent (Fig. 4d). In previous studies, we identified mutant versions of LigI that were defective in binding to either PCNA (FA) [11, 24] or RFC (S51D) [25]. While both interactions are critical for LigI functions in cellular DNA replication and repair [24, 25], RFC is not required for the stable interaction of LigI with DNA-loaded PCNA in vitro [10]. As expected [11, 24], binding of the LigI FA mutant to DNA-loaded PCNA was significantly reduced whereas Pol δ-dependent binding of mutant versions of LigI that were defective in interacting with PCNA and/or RFC was not significantly reduced compared with wild type LigI (Fig. 4e). Since pre-loaded PCNA and Pol δ had similar stimulatory effects on DNA binding and DNA joining by LigI (Fig. 4c and d), it appears that both of these proteins enhance ligation by increasing the association of LigI with DNA. While it is possible that the binding of Pol δ to DNA generates a DNA structure that is preferentially bound by LigI, it seems more likely that DNA binding by Pol δ induces a conformational change that enhances LigI binding because the effect is observed with different DNA structures and LigI has only weak non-specific DNA binding activity [23].
Human LigI and PCNA remain associated with ligated DNA.
Since Okazaki fragment synthesis, processing and ligation is a cyclical process, the protein factors involved must be released from the DNA following ligation for subsequent cycles. In accord with studies showing that RFC does not efficiently unload PCNA from DNA [26, 27], loaded PCNA remained stably associated with ligated DNA (Fig. S2, lane 8). Notably, the amount of LigI associated with the fully ligated product was not significantly different than the levels detected during gap filling synthesis by an assembled PCNA-Pol δ complex (Fig. 3, compare lane 10 with lane 6) and was significantly higher than the levels bound to loaded PCNA in the absence of Pol δ (Fig. 3, compare lane 10 with lanes 4 and 9, p≤ 0.05). In contrast, the amounts of FEN1 (Fig. 2, compare lane 10 with lane 3) and Pol δ (Fig. 1c, compare lane 9 with lanes 1–3) retained by the fully ligated product were reduced to the low levels retained in the absence of DNA-bound-PCNA, corresponding to non-specific binding in the absence of loaded PCNA. Thus, it appears that Pol δ and FEN1 are released from loaded PCNA following the completion of gap filling synthesis, flap removal and/or ligation whereas LigI remains stably associated with ligated DNA along with PCNA.
To examine the contribution of LigI interactions with PCNA and RFC on ligation in the reconstituted reaction, we carried out reactions with the FA, S51D and FA/S51D mutant versions of LigI (Fig. 5a). As expected, there were no significant differences in gap-filling and strand displacement DNA synthesis in reconstituted reactions with the mutant versions of LigI compared with wild type LigI (Fig. 5a). Surprisingly, there was also no reduction in either the initial rate (Fig. S6) or total amount of DNA joining (Fig. 5a) in reactions with mutant versions of LigI that were defective in binding to PCNA and/or RFC. Inactivation of the LigI PIP box did, however, result in lower amounts of the mutant LigI retained on DNA (Fig. 5b) during strand displacement synthesis and following ligation, indicating that, at least under these conditions, reduced stable association of LigI with DNA does not reduce the overall efficiency of either DNA synthesis or ligation. In the absence of a version of LigI that is selectively defective in interacting with Pol δ, we examined the activity of a catalytic fragment of LigIII [28, 29] that has similar nick joining activity to LigI under these reaction conditions (Fig. S7) in reconstituted Okazaki fragment processing and joining reactions. Notably, in contrast to reactions containing equivalent amounts of LigI protein and nick ligation activity, no significant ligation occurred in fully reconstituted reactions with the LigIII catalytic fragment despite efficient gap filling DNA synthesis (Fig. 5c). Together, our results strongly argue that the interaction of LigI with Pol δ underlies the specific function of this enzyme in joining DNA nicks generated as a consequence of DNA synthesis and flap removal by Pol δ and FEN1, respectively.
Fig. 5. LigI mutants defective in interacting with PCNA and/or RFC but not LigIII catalytic fragment substitute for wild type LigI in Okazaki fragment synthesis, processing and ligation.
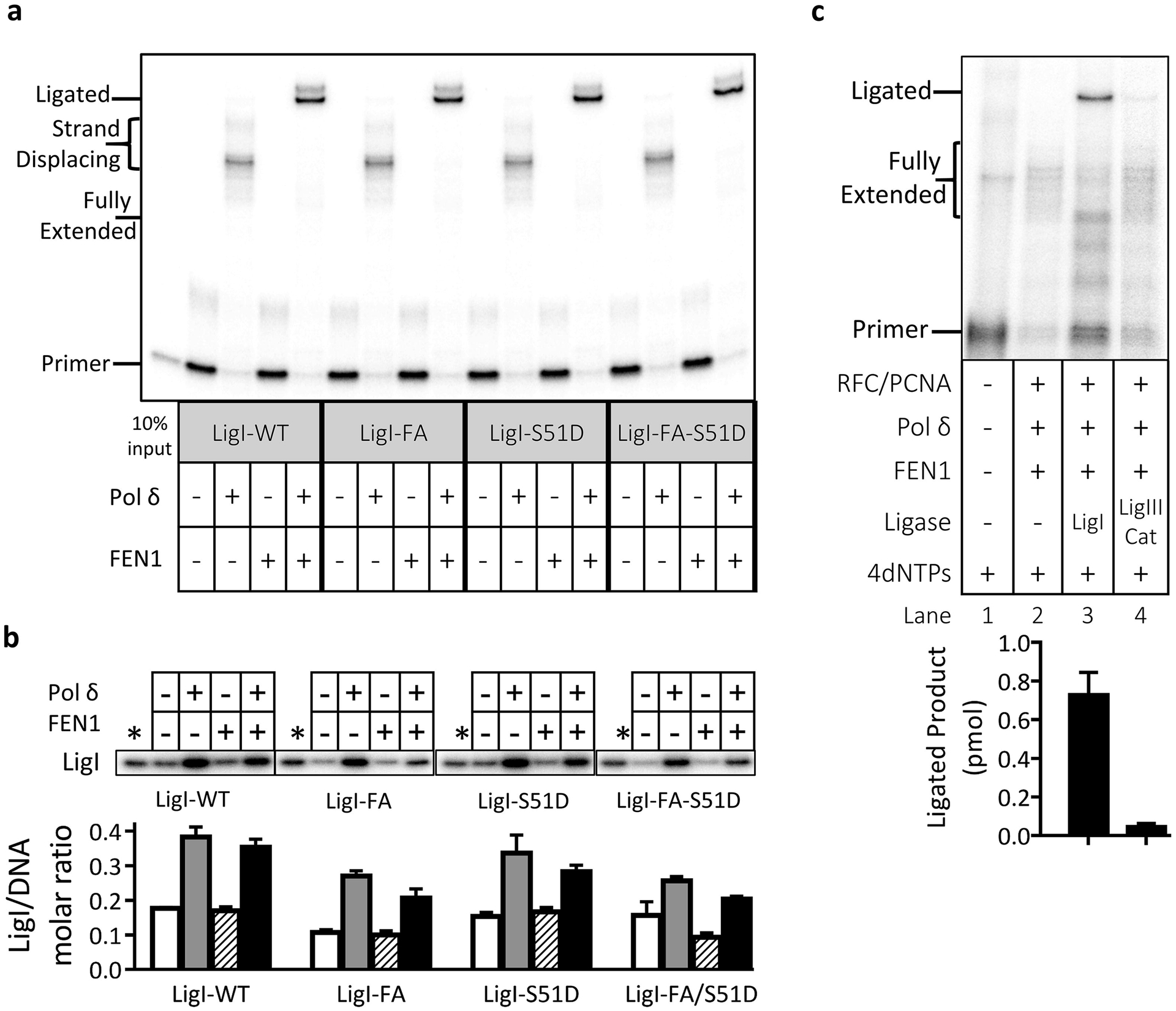
Reconstitution assays were carried out with wild type LigI (LigI-WT) and mutant versions of LigI defective in interacting with PCNA (LigI-FA), with RFC (LigI-S51D) or with both PCNA and RFC (LigI-FA-S51D) as described in Fig. 1. All reactions contained labeled LigI, RPA, PCNA, RFC and the four dNTPS. Pol δ and FEN1 were present (+) or absent (−) as indicated. a. After separation by denaturing gel electrophoresis, labeled DNA reaction products were detected and quantitated by phosphorimager analysis. The positions of the labeled primer (Primer), the gap filled product (Fully Extended), longer products generated by strand displacement synthesis (Strand Displacing) and the ligated product (Ligated) are indicated on the left. The ligated product appears as a doublet that is most likely due to secondary structure in the ligated oligonucleotide under the gel electrophoresis conditions. b. Association of wild type and mutant versions of LigI with the DNA under the different reaction conditions. The binding of wild type and mutant versions of labeled LigI to the DNA is expressed as the ratio of LigI retained on the beads to the DNA recovered on the beads. Representative gel of two independent experiments with error bars showing standard deviation. c. Reconstituted assays with either LigI or the catalytic subunit of LigIII as indicated. Representative gel of at least three independent experiments with error bars showing standard deviation.
Stacked ring structure of complex formed by LigI and PCNA on DNA.
To gain insights into the structure of the complex formed by LigI with DNA-bound PCNA, we pre-incubated the purified proteins with linear duplex DNA containing a single non-ligatable nick and then purified DNA-protein complexes by size exclusion chromatography (SEC). In reactions containing equimolar amounts of LigI, PCNA trimer and DNA, there was a single early-eluting peak that contained PCNA, LigI and DNA whereas with a two-fold excess of LigI, there were two major peaks with the second peak corresponding to LigI alone (Fig. 6a). Compared with the elution positions of globular protein standards, the estimated molecular masses of PCNA, LigI-DNA and LigI-PCNA-DNA were ~160, 290, and 450 kDa, respectively. However, the elution volume of LigI-containing samples cannot be used to accurately determine molar mass or stoichiometry because of the non-globular, unstructured N-terminal region of LigI [10, 23]. Using multi-angle light scattering (MALS) to determine the molar masses of proteins eluting from the SEC column (Fig. 6b) resulted in values of 103 kDa for PCNA, 124 kDa for LigI-DNA and 240 kDa for LigI-PCNA-DNA that were close to the theoretical values for a PCNA trimer (98.1 kDa), a LigI monomer complexed with the linear duplex DNA(123 kDa) and a LigI monomer and PCNA trimer complexed with the linear duplex DNA (221.1 kDa, Fig. 6c). This is in accord with the estimated stoichiometry of the LigI-PCNA complexes formed by adenylated LigI with PCNA loaded onto circular DNA molecules [10].
Fig. 6. DNA-bound PCNA binds one molecule of LigI.
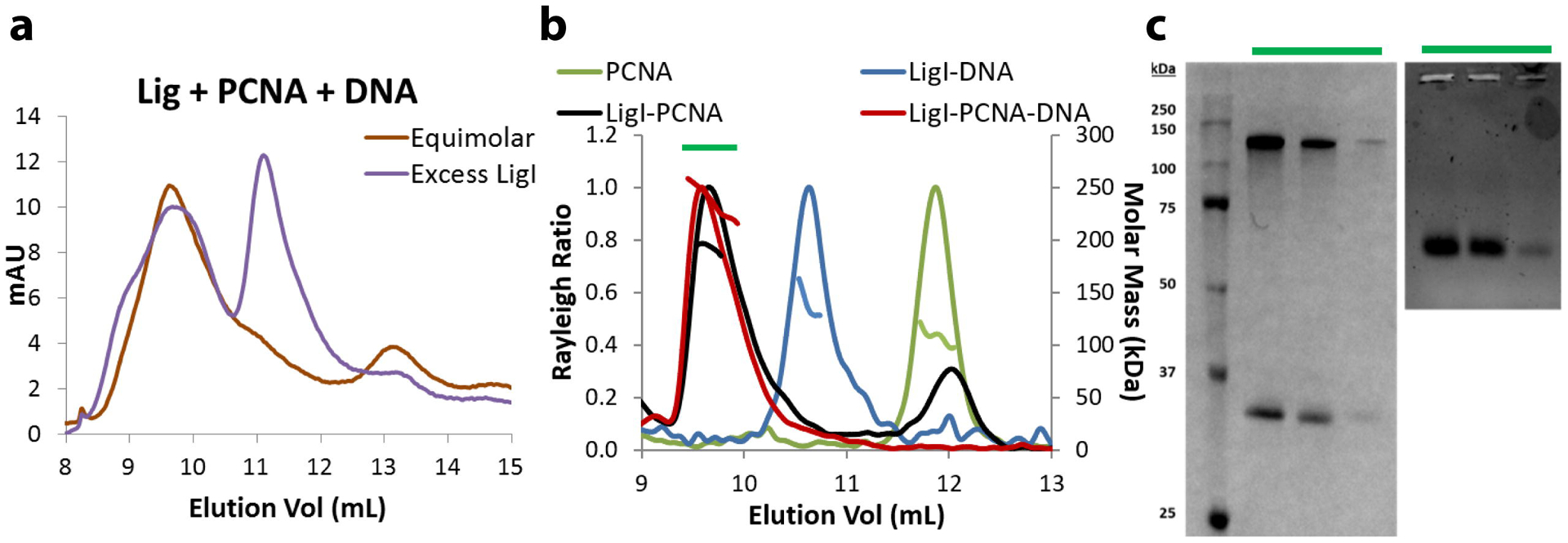
Gel filtration to assess LigI-PCNA-DNA complex monodispersity and stoichiometry a. An equimolar mixture of LigI, PCNA, and non-ligatable nicked DNA eluted as a single peak (brown trace). A mixture containing excess LigI produced a second, smaller peak (purple trace) that corresponds to LigI alone. b. SEC-MALS determined correct molar masses of PCNA (green trace), LigI-DNA (blue trace), LigI-PCNA (black trace) and LigI-PCNA-DNA (red trace), confirming equimolar stoichiometry. c. SDS-PAGE (left) and agarose (right) gels identifying LigI, PCNA and DNA within the peak gel filtration fractions.
Visualization of the purified LigI-PCNA-DNA complexes by negative stain electron microscopy revealed particles approximately 11 nm in diameter (Fig. S8a). Single particles were selected and extracted for 2D analysis (Fig. S8b). Clustering of the particles revealed fairly homogeneous classes with a thin line traversing two densities (Fig. 7a). The dumbbell-shaped density (purple bracket) corresponds to the PCNA ring, the second density is LigI (red bracket) and the thin line (arrow) is nicked DNA. In contrast to the PCNA ring, the appearance of LigI was more variable. While some classes showed LigI pivoting away from PCNA (see yellow square in Fig. 7a), most classes have LigI stacked on PCNA (see blue square in Fig. 7a). 3D classification was used to separate the different conformations (Fig. S9). Particles belonging to the two conformations were grouped and each conformer was refined to 20 and 22 Å resolution (Fig. S8c). In about one-third of the particles, the LigI density is separate from the PCNA ring, except for a thin connection between the proteins (Fig. S10) whereas the majority of particles appeared as stacked rings (Fig. 7b). The crystal structures of PCNA (PDB 1AXC) and the LigI-DNA complex (PDB 1X9N) that lacks the N-terminal 262 residues of LigI were docked into the lower and upper rings, respectively. Although readily observed in 2D averages, there is only partial 3D density for modelling DNA in the space between the rings. Therefore, the crystal structure of the 19-nucleotide linear nicked DNA complexed with LigI [23] was extended to reflect the longer 32-nucleotide DNA used in this experiment, with this longer duplex spanning the length of the complex.
Fig. 7. Structure of DNA-bound PCNA-LigI complex.
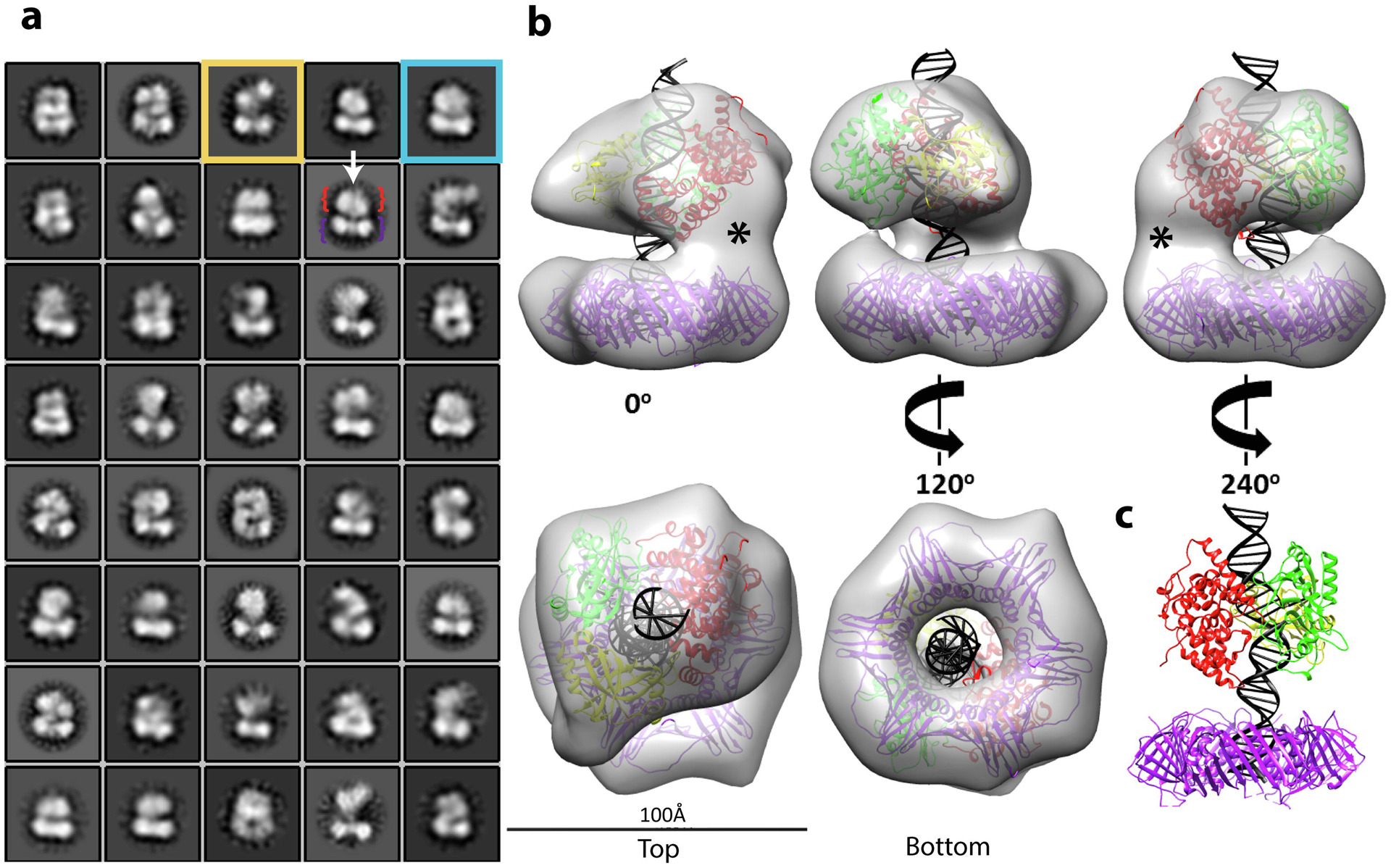
Single particle EM analysis of complexes formed by LigI and PCNA were co-purified with non-ligatable nicked DNA. a. 2D classification reveals DNA (white arrow) traversing two separate densities: PCNA (purple bracket) and LigI (red bracket). The boxsize is 210 Å in length and width, the circular mask is 150 Å. Classes show open (yellow box) and stacked rings (blue box) conformations. b. The stacked rings 3D map (~20 Å resolution) of the complex is shown at the indicated rotations. Crystal structures of PCNA (PDB 1AXC) and LigI-DNA (PDB 1X9N) were docked into the lower and upper rings, respectively, with extended nicked DNA. LigI was positioned with its DBD (red) adjacent to the density linking to PCNA. The empty density (*) is attributable to the unstructured N-terminal region of LigI. The LigI AdD (green) mediates a second point of contact with PCNA and the OBD (yellow) completely encircles the nicked DNA. c. A 240° view of the modeled crystal structures.
Although the dimensions of the LigI-PCNA stacked rings are similar to those determined for the Pyrococcus furiosus DNA ligase (Lig)-PCNA-DNA complex [30], including two points of contact between DNA ligase and PCNA in both structures, the distribution of protein volume between the two structures is clearly different (Fig. S10). Specifically, there is more substantial contact between the rings in the human complex that is not accounted for by the docked atomic coordinates (Fig. 7b, asterisk). While the archaeal Lig contacts PCNA through two domains, DNA binding (DBD) and adenylation, involved in catalysis [30], the primary PCNA binding site in LigI is an N-terminal PIP box that is separated from the second binding site within the LigI DBD by an unstructured region that is not required for enzymatic activity [11, 24, 31, 32]. Furthermore, the OB-fold domain of the Pfu Lig partially engages the DNA without encircling it [30]. The LigI-PCNA stacked ring structures are presumably formed as a consequence of the human LigI catalytic fragment containing the OB-fold domain completely encircling the DNA as observed in the crystal structure of the LigI-nicked DNA complex [23] (Fig. 7c). Together, our results indicate that joining of Okazaki fragments occurs within a DNA-bound stacked ring structure formed by PCNA and LigI and that these proteins remain linked to the ligated DNA.
DISCUSSION
Through interactions with Pol δ, RFC, FEN1 and LigI, the PCNA DNA sliding clamp co-ordinates the gap filling, 5’end processing and ligation reactions that complete the synthesis and joining of Okazaki fragments on the lagging strand at the replication fork [1–11]. Although it is possible that up to three different replication proteins may be able to simultaneously interact with homotrimeric PCNA that is topologically linked to duplex DNA, direct experimental evidence for such complexes in eukaryotes has been lacking. To provide insights into the proteins associated with homotrimeric PCNA during different stages of Okazaki fragment synthesis, processing and ligation, we established a reconstituted reaction with purified human RFC, RPA, Pol δ, LigI, and PCNA, in which a gapped duplex DNA substrate with a 5’ flap is efficiently converted into an intact duplex product (Fig. 1). Analysis of DNA-protein complex formation and dissociation during Okazaki fragment processing and ligation provides direct evidence that gap filling synthesis is carried out by a complex of PCNA, Pol δ, FEN1, LigI and possibly FEN1, in which LigI interacts with Pol δ. Furthermore, after the completion of gap filling synthesis and flap removal, a DNA-bound complex of PCNA and LigI remains stably associated with the ligated DNA. It should be noted that, with the exception of RFC, the proteins used in the reconstitution assays were purified after expression in E. coli and so lacked the post-translational modifications that occur in human cells [33–35]. Thus, the model shown in Figure 8 does not address the contribution of post-translational modifications in co-ordinating Okazaki fragment synthesis, processing and ligation.
Fig. 8. Model for PCNA-dependent Okazaki fragment processing and joining by Pol δ, FEN1 and LigI.
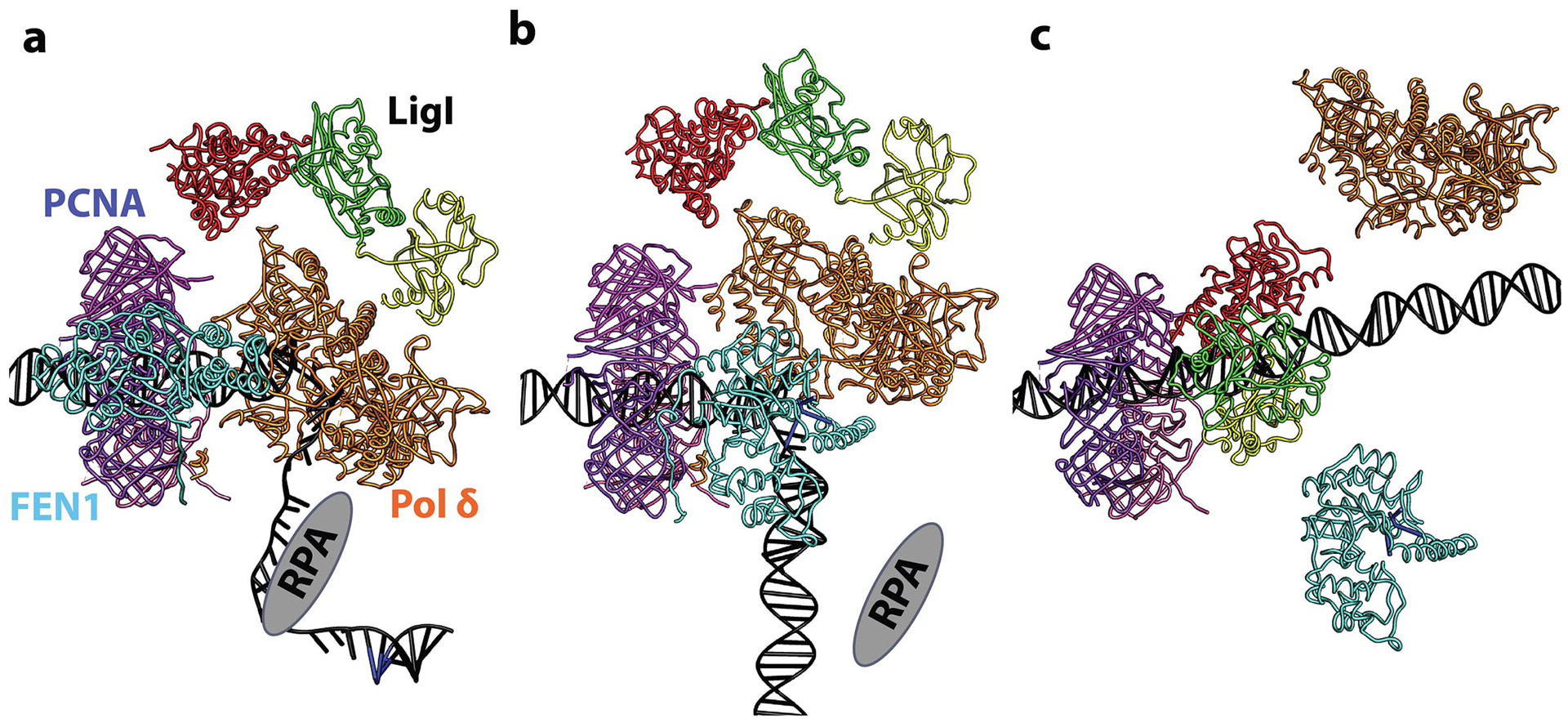
After synthesis of a RNA-DNA primer by Pol α holoenzyme, RFC loads PCNA onto the RPA-coated DNA at the primer template junction. a. RFC-loaded PCNA (purple, pink, magenta; PDB 1U76) in complex with Pol δ (brown; PDB 3IAY) on a primer-template junction generated by Pol α in a RPA (grey)-dependent manner. FEN1 (cyan; PDB 1UL1 chain X) also binds PCNA whereas LigI associates with Pol δ, most likely in the extended conformation. b. Gap filling synthesis by Pol δ displaces RPA and initiates strand displacement synthesis, generating a 5’ flap (blue nucleotides). With Pol δ disengaged from DNA, but possibly still tethered to PCNA, FEN1 (PDB 5K97 positioned as chain Y from PDB 1UL1) carries out flap cleavage, thereby generating a ligatable nick. c. Dissociation of Pol δ and FEN1 from PCNA enables LigI to enage both the DNA nick and PCNA trimer forming a stable stacked ring structure that remains linked to the DNA after ligation.
The interaction with DNA-bound PCNA markedly enhances FEN1 nuclease activity [8, 20, 36]. There are, however, contradictory reports as to the effects of the LigI interaction with PCNA on nick ligation activity that appear to reflect differences in the structure of the nicked DNA substrate[10, 37]. Interestingly, larger amounts of FEN1 than LigI were retained by DNA bound-PCNA (Figs. 2 and 3, compare lanes 4). Although this could be due to differences in the affinity and stability of binding to PCNA, it may also reflect differences in the stoichiometry of the complexes formed by LigI and FEN1 with PCNA. PCNA-LigI complexes formed on nicked circular [10] and linear DNA (Fig. 6) contained one LigI per PCNA trimer. In contrast, complexes with three FEN1 molecules bound to a PCNA trimer have been observed, albeit in the absence of DNA[38]. It is possible that the number of FEN1 molecules bound by a PCNA trimer will be different when PCNA is topologically linked to duplex DNA and when the PCNA-FEN1 complex encounters a 5’ flap [39–41]. In our studies, the FEN1-PCNA complex formed prior to DNA synthesis is restricted to the primer-template region and unable to engage the 5’ flap because of the presence of RPA in the single strand gap [21].
While there is evidence that a three subunit version of human Pol δ, which is equivalent to the yeast holoenzyme, functions in response to DNA damage and replicative stress [42, 43], our studies were performed with the four subunit exonuclease-proficient version of human Pol δ [17, 18]. The formation of PCNA-Pol δ complexes that were either poised to initiate DNA synthesis or had partially completed gap-filling synthesis had opposite effects on the association of FEN1 and LigI with PCNA-loaded DNA. Retention of FEN1 was reduced but not eliminated (Fig. 2, compare lanes 7–8 with lanes 4 and 9). Since at least three subunits of the Pol δ holoenzyme, p125, p68 and p12, have PCNA binding sites [3–6], we suggest that, after loading of PCNA by RFC onto the gapped DNA coated by RPA (Fig. 8a), the Pol δ holoenzyme engages binding sites on at least two of the subunits of the PCNA homotrimer, thereby limiting the number of FEN1 molecules bound per PCNA trimer. This model is consistent with the S. cerevisiae PCNA-dependent functional interactions between Pol δ and FEN1 [7] and the recently published structure of a human Pol δ-PCNA-FEN1 complex determined by cryo-electron microscopy (EM), in which the Pol δ holoenzyme contacts two of the PCNA monomers and FEN1 the remaining one [44].
In contrast to FEN1, the addition of Pol δ increased the retention of LigI on PCNA-loaded DNA (Fig. 3). This may reflect either the additive nature of LigI interactions with Pol δ (Fig. 4) and PCNA [10] or, more likely, increased binding of LigI to Pol δ because of changes in the conformation of Pol δ when it is complexed with PCNA. Irrespective of the precise mechanism of assembly, our studies demonstrate that LigI forms a stable complex with PCNA-Pol δ during gap-filling DNA synthesis (Figs. 3 and 8a). Since previous studies have shown that expression of mutant versions of LigI that were defective in binding to either PCNA or the clamp loader RFC in LigI-deficient cells did not correct abnormalities in DNA replication and DNA damage repair [25, 45, 46], it was surprising that there was no reduction in ligation efficiency in reconstituted reactions with mutant versions of LigI that were defective in either one or both of these protein-protein interactions (Fig. 5a). Moreover, the LigIII catalytic fragment was unable to substitute for LigI in the reconstituted reaction (Fig. 5c), strongly arguing that the interaction between Pol δ and LigI is critical for efficient ligation under these conditions. Thus, our studies, together with published results with FEN1 [7] and recently reported cryo-EM structure of a human Pol δ-PCNA-FEN1 complex [44], suggest that PCNA, FEN1, Pol δ and LigI form a functional complex during gap-filling DNA synthesis (Fig. 8).
When DNA synthesis by PCNA-Pol δ extended beyond the gap due to the absence of FEN1 and/or LigI, the levels of DNA-bound PCNA remained constant (Fig. S2) whereas Pol δ levels were reduced Fig.1c (compare lanes 5 with 7 and 8). This presumably reflects the relative instability of the human PCNA-Pol δ complex and the tendency of human Pol δ to dissociate from PCNA when DNA synthesis is slowed or stalled after it reaches the 5’ end of the gap [17]. Furthermore, the levels of Pol δ and FEN1 retained on ligated DNA generated in the fully reconstituted reaction were reduced to near the levels of DNA binding observed in the absence of DNA-bound PCNA. This suggests that, when a complex of PCNA, Pol δ, FEN1 and LigI reaches the 5’ flap, this encounter and the subsequent removal of the preformed flap to generate a DNA nick reduces the binding affinity of Pol δ and FEN1 for DNA-bound PCNA. During DNA replication, the PCNA-Pol δ complex will encounter the 5’ end of the RNA-DNA oligonucleotide synthesized by Pol α-primase. While the relative contribution of the 5’ end processing subpathways (7, 9) and the regulation of 5’ end resection is not known, there is intriguing evidence that, in S. cerevisiae, the site of Okazaki fragment joining is dictated by nucleosome positioning [47].
The predicted role for DNA structure in dictating protein-protein interactions with PCNA is consistent with studies showing that a yeast PCNA homotrimer with one functional PIP box binding site is capable of co-ordinating gap filling synthesis, end processing and ligation by sequential switching of Pol δ, FEN1 and LigI [16]. Furthermore, our evidence for a PCNA complex, in which FEN1 engages one PCNA subunit and Pol δ engages either one or two PCNA subunits with LigI primarily interacting with Pol δ provides an explanation as to why PCNA homotrimers with two or more functional PIP box binding sites were more effective at supporting gap filling synthesis, end processing and ligation compared with the PCNA homotrimer with one functional PIP box binding site [16]. In contrast to Pol δ and FEN1, the levels of PCNA and LigI retained on ligated DNA were similar to the high levels observed during gap filling DNA synthesis (Figs. 3 and S2). While this was expected for PCNA because RFC does not have robust PCNA unloading activity [26], it was not for LigI. Interestingly, while disruption of the LigI-PCNA interaction did not reduce the efficiency of ligation, less of the mutant LigI was retained on the ligated DNA (Figs. 5a and b), indicating that, while LigI is delivered to the DNA nick via its interaction with PCNA-bound Pol δ, it is retained there by an interaction with DNA-bound PCNA [10]. We suggest that removal of the 5’ flap and generation of a ligatable nick induces the dissociation of Pol δ and FEN1 from DNA-bound PCNA (Figs. 8b and c). This enables LigI to not only encircle and productively engage the DNA nick [23] but to also bind to the DNA-bound PCNA trimer [10, 32].
Although we do not have structural information about the LigI-PCNA complex remaining on the ligated DNA, we have visualized LigI-PCNA complexes formed on linear duplex DNA with a non-ligatable nick by negative stain EM (Fig. 7). The observation of particles resembling stacked rings linked by a string is consistent with the ring structure of PCNA and the compact clamp-structure of LigI around nicked DNA [23, 48]. While this stacked ring structure is similar to that of the DNA-bound complex of P. furiosus Lig-PCNA [30], the interaction interface between the rings is larger for the human complex. Unlike human LigI, the catalytic fragment of the P. furiosus Lig does not completely encircle the nicked DNA [30]. Furthermore, the Pfu Lig DBD is mainly responsible for PCNA binding with the adjacent AdD providing another point of contact [30]. In contrast, the principal link of human LigI to PCNA is via an N-terminal PIP box that is separated from the LigI DBD by a long flexible linker region [10, 32]. It is likely that it is this relatively unstructured linker region that contributes to the larger interface between the rings in the human PCNA-LigI complex with the LigI AdD possibly engaging another PCNA subunit. While the stacked rings structure of the human PCNA-LigI complex provides an explanation as to why only one LigI molecule binds per PCNA, our EM analysis also revealed a conformation in which LigI appeared to be mostly separated from the the PCNA ring while remaining tethered, presumably via the N-terminal PIP-box (Fig. S10). The earlier than expected elution of LigI-DNA complexes either with or without PCNA during SEC (Fig. 6b) indicates that LigI is not stably engaged with the nicked DNA in the compact clamp structure but instead is sampling different conformations as it binds to both PCNA and DNA [23] with the catalytic LigI catalytic domain possibly in an extended conformation as observed for the Sulfolobus solfataricus DNA ligase complexed with PCNA in the absence of DNA [49]. Thus, we envisage that the conformation of the human LigI-PCNA complex left on the DNA following ligation is likely to be dynamic with the catalytic fragment of LigI shuttling between between closed clamp and extended conformations.
Although loss of PCNA binding did not reduce ligation efficiency in the reconstituted reaction (Fig. 5a), the inability of a PCNA binding mutant version of LigI to correct the replication and repair defect in extracts from LigI-deficient cells provides compelling evidence that the LigI-PCNA interaction is biologically significant (11). While PCNA binding may primarily be for the subnuclear targeting of LigI [24], the reduced ability of a PCNA binding mutant version of LigI to correct replication defect in extracts from LigI-deficient cells is indicative of a direct role for this interaction at the replication fork [11]. It is possible that the PCNA-LigI complex left on the ligated DNA serves as the signal for PCNA unloading and release of LigI from the ligated DNA. Elegant studies in S. cerevisiae have shown that another member of the RFC clamp loading/unloading family, Elg1-RFC, catalyzes the unloading of PCNA from replicated DNA in a reaction that is dependent upon ligation of Okazaki fragments [50]. Thus, Elg1-RFC may interact with the PCNA-LigI complex, releasing both of these proteins from the ligated DNA for subsequent cycles of Okazaki fragment synthesis, processing and ligation.
MATERIALS AND METHODS
Recombinant proteins.
Untagged wild type LigI was purified as described [51]. Wild-type (WT) and mutant versions, FA (F8A/F9A), S51D and FA-S51D [11, 25], of LigI with a poly(histidine (His)) and a C-terminal target site for phosphorylation by cAMP-dependent protein kinase (PKA) were expressed in Rosetta2(DE3)pLacI cells and purified by serial chromatography on HisTrap and HiTrap Q columns (GE Healthcare). His-tagged fragments of LigI corresponding to the non-catalytic N-terminal domain (N230, residues 2–230), the DBD (residues 233–534) and the catalytic domain (Cat, residues 533–919) were expressed in Rosetta2(DE3)pLacI cells and purified by affinity chromatography on a HisTrap column (GE Healthcare). For SEC-MALS and EM studies, His-tagged LigI was purified by HisTrap and HiTrap Heparin column chromatography. His-tagged LigIII catalytic fragment [28, 29] was expressed in BL21(DE3) cells and affinity chromatography on a HisTrap column (GE Healthcare).
Wild type human Pol δ holoenzyme and E. coli LacR were expressed and purified as previously described [52]. His-tagged human PCNA with an N-terminal PKA site was expressed in Rosetta2(DE3)pLacI cells and purified by serial chromatography on HisTrap and HiTrap Q (GE Healthcare). His-tagged human FEN1 with an N-terminal PKA site was expressed in Rosetta2(DE3)pLacI cells and purified by serial chromatography on HisTrap and HiTrap SP (GE Healthcare). His-tagged RFC was purified from High Five insect cells that had been co-infected with a baculovirus encoding RFC1 with a C-terminal His-tag and the four small RFC subunits by serial chromatography on HisTrap and HiTrap SP (GE Healthcare). Human RPA was expressed and purified as described [53].
Oligonucleotides.
The oligonucleotides used in this study, which are shown in Supplementary Information (Table S1), were synthesized by Integrated DNA Technologies and purified by denaturing polyacrylamide gel electrophoresis.
Okazaki fragment joining assay.
The DNA substrate was prepared by annealing of 32P-labeled Oligo-top-7, 5-nucleotide flapped Oligo-top-8 and Oligo-bottom-2. Individual proteins were labeled with 32P at their attached PKA site immediately before the assay. The reaction was conducted in a final 40 μL mixture containing 20 mM HEPES-NaOH pH7.5, 130 mM NaCl, 10 mM MgCl2, 0.25 mg/mL BSA, 0.01% Triton X-100, 2 mM ATP, 1 pmol of the DNA substrate, 25 μM of indicated dNTPs and 4.5 pmol LacR, initiated by addition of 1 pmol RPA on ice, followed by addition of 1 pmol RFC and 7.5 pmol PCNA as indicated and incubation at 37°C for 5 min. Subsequently 1 pmol Pol δ, 1 pmol FEN1 and 1 pmol LigI were added as indicated. After incubation at 37°C for another 10 min, the reaction mixture was transferred on ice, and the casein-blocked streptavidin-conjugated magnet beads were added. Following incubation on ice for 10 min, the beads were washed with 100 μL Washing buffer. Proteins retained by the beads were released by incubation in SDS-containing buffer at 37°C for 10 min. After separation by SDS-PAGE, labeled proteins were detected and quantitated by phosphorimager analysis. DNA retained on the beads was released by incubation in 20 mM EDTA-containing formamide at 95°C for 5 min. After electrophoresis through a denaturing 12% polyacrylamide gel, labeled DNA was detected and quantitated by phosphorimager analysis. As shown previously [54], the absence of RPA had no significant effect on DNA synthesis by Pol δ but it did result in increased non-specific binding of LigI to the DNA substrate so RPA was included in all reconstituted reactions.
Bead pull down assays.
His-tagged Pol δ subunits (10 pmol of each) were individually pre-incubated with Ni-NTA Magnetic Agarose Beads (Qiagen) for 1 h at 4°C prior to the addition of WT, untagged LigI (10 pmol) in binding buffer (50 mM HEPES-NaOH pH7.5, 100 mM NaCl, 30 mM imidazole, 0.01% Triton X-100 and proteinase inhibitors) and continued incubation at 4°C for 1 h. After washing with the binding buffer twice, the beads were heated in SDS-sample buffer to release bound proteins, which were then separated by SDS-polyacrylamide gel electrophoresis. LigI was detected by immunoblotting with a polyclonal anti-LigI antibody [10].
For pull down assays with His-tagged domains of LigI, the Pol δ holoenzyme was digested with PreScission protease to remove the His16 tag from the p12 subunit, generating a His-tag-free version of the Pol δ holoenzyme. Ni-NTA Magnetic Agarose Beads were pre-incubated with 40 pmol of each of the DNA ligase domains prior to incubation with 20 pmol of His-tag-free Pol δ in binding buffer at 4°C for 1 h. After washing twice with binding buffer, samples were subjected to electrophoresis in an SDS-containing polyacrylamide gel and immunoblotting with polyclonal anti-Pol δ antibody (GTX70151, GeneTex).
Ligation assay.
32P-labeled Oligo-top-1 and 5’-phosphorylated Oligo-top-2, were annealed with Oligo-bottom-1 to form a ligatable substrate. The reaction was conducted at 37°C for 15 min in a 40 μL mixture containing 20 mM HEPES-NaOH pH7.5, 100 mM NaCl, 10 mM MgCl2, 0.1 mg/mL BSA, 0.01% Triton X-100, 2 mM ATP, 40 μM four dNTPs, 100 fmol of the DNA substrate and where indicated: 100 fmol RFC, 250 fmol PCNA trimer, 100 fmol Pol δ and 100 fmol LigI. After the reaction was terminated by the addition of SDS to a final concentration of 0.5%, labeled DNA was recovered with streptavidin-conjugated magnet beads (Dynabeads M-280 Streptavidin, Dynal), electrophoresed through a denaturing 15% polyacrylamide gel and detected and quantitated by phosphorimager analysis.
DNA binding assay.
A DNA substrate carrying a 3-nucleotide gap was prepared by annealing of Oligo-top-3, Oligo-top-5 and Oligo-bottom-1. A DNA substrate with an unligatable nick was prepared by annealing of Oligo-top-4, Oligo-top-5 and Oligo-bottom-1. A DNA substrate with a 4-nucleotide flap was prepared by annealing of Oligo-top-4, Oligo-top-6 and Oligo-bottom-1. These DNA substrates were retained by streptavidin-conjugated magnet beads and blocked with Blocker Casein (Thermo Fisher). Where indicated, the following proteins: 1 pmol RFC, 2.5 pmol PCNA trimer, 1 pmol Pol δ, 1 pmol LigI and 7 pmol LacR were incubated in a 40 μL mixture containing 20 mM HEPES-NaOH pH7.5, 130 mM NaCl, 10 mM MgCl2, 0.25 mg/mL BSA, 0.01% Triton X-100, 2 mM ATP with 1 pmol of the DNA substrate attached to the beads. For detection of DNA binding by Pol δ, the PKA site attached to the p66 subunit was labeled with 32P by PKA (New England Biolabs). For detection of DNA binding by LigI, the PKA site attached its C-terminus was labeled with 32P as above. After incubation at 37°C for 10 min, the beads were washed with 100 μL Washing buffer (20 mM HEPES-NaOH pH7.5, 130 mM NaCl, 10 mM MgCl2, 0.01% Triton X-100). Proteins retained on the beads were separated by SDS-PAGE followed by detection and quantitation of labeled proteins by phosphorimager analysis.
SEC-MALS.
Combinations of LigI, PCNA and non-ligatable nicked DNA were assembled at 10 μM in low-salt gel filtration buffer (GFB; 20 mM HEPES pH7.5, 50 mM NaCl, 10 mM MgCl2) with 1 mM ATP and incubated at room temperature for 15 minutes, followed by injection onto a Superdex 200 Increase 10/300 GL gel filtration column at room temperature equilibrated in GFB using an AKTAmicro system (GE Healthcare) combined with Dawn HELEOS II MALS and OptiLab T-rEX online refractive index detectors (Wyatt Technology). Data were processed using ASTRA Version 6.1.6.5 (Wyatt Technology) following calibration with BSA.
Negative stain electron microscopy and image analysis.
Carbon-coated copper grids (Electron Microscopy Sciences) were glow-discharged (Agar Scientific) immediately before adsorbing 5 μL of fractions containg the LigI-PCNA-DNA complex from the Superdex 200 Increase 10/300 GL gel filtration column for 1 minute, followed by removal of excess volume and addition of 5 μL freshly prepared 1.5% uranyl formate (Electron Microscopy Sciences) for 1 minute. Samples were imaged at room temperature using a FEI Tecnai T12 transmission electron microscope equipped with a LaB6 filament operated at 120 keV. Images were acquired using a FEI Eagle 4k × 4k CCD camera at 67,000× magnification, corresponding to a pixel size of 1.64 Å. The SerialEM program [55] was used to acquire 419 micrographs at defocus values between 0.5–2 μm.
All image analysis was performed using RELION [56]. None of the steps used contrast transfer function correction nor imposed symmetry. An automated reference-free particle picking algorithm identified ~228,000 coordinates [57]. Particles were two-fold downsampled and extracted in 64 × 64 pixel boxes (210 Å box). A final dataset of ~49,500 particles was generated through successive reference-free 2D classifications using a circular 150 Å mask and default parameters [56]. The P. furiosus Lig-PCNA-DNA negative stain EM map (EMD 1485) was low-pass filtered to 60 Å and 3D classification (150 Å mask, six classes) using all particles. Particles belonging to similar 3D classes were grouped for separate refinements (31,910 and 17,594 particles into the stacked rings and open conformations, respectively). 3D auto-refinement was carried out using a 150 Å circular mask. The resolution was computed using the gold-standard Fourier shell correlation at the 0.5 criterion [58]. Crystal structures were docked into the EM map using UCSF Chimera [59]. Ideal B-form duplex DNA was added to the termini of the 19-nucleotide DNA co-crystallized with the LigI catalytic fragment (PDB 1X9N) to reflect the 32-nucleotide DNA used in the negative stain EM experiment. The LigI coordinates with the extended DNA was docked within the upper ring density and the resulting extended DNA traversed the center of the lower ring.
Data Analysis.
Densitometric analysis was performed using NIH software ImageJ. Molar ratio, percent ligation, and total product calculations were performed in Microsoft Excel and shown graphically using GraphPad Prism 6.0. Statistical significance in each data set, unless otherwise noted in figure legends, is calculated by a one-way ANOVA with Tukey’s corrections for post-hoc unpaired t-tests and indicated as * (p≤ 0.05), ** (p≤ 0.01), *** (p≤ 0.001), **** (p≤ 0.0001), or n.s. (p> 0.05).
Supplementary Material
Highlights.
How does PCNA co-ordinate Okazaki fragment synthesis, processing and ligation?
Physical and functional interaction between DNA polymerase d and DNA ligase I
Both DNA ligase I and FEN1 associate with DNA polymerase d-PCNA during gap filling DNA synthesis
A double-dumbbell complex of DNA ligase I and PCNA remains on the ligated DNA
Dynamic PCNA complexes co-ordinate Okazaki fragment synthesis, processing and ligation
Acknowledgements:
We thank Dr. Annahita Sallmyr for assistance with preparing the figures. This work was supported by the University of New Mexico Comprehensive Cancer Center and National Institute of Health Grants R01 GM57479 (to A.E.T.), P01 CA92584 and P30 CA118100, and a Discovery grant from the National Science and Engineering Research Council of Canada (RGPIN-2015-05776 to J.M.P.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Accession numbers: EMBD accession no. ID EMD-21958
Conflict of Interest:
The authors declare that they have no conflicts of interest with the contents of this article.
REFERENCES
- [1].Burgers PMJ, Kunkel TA. Eukaryotic DNA Replication Fork. Annual review of biochemistry. 2017;86:417–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Chilkova O, Stenlund P, Isoz I, Stith CM, Grabowski P, Lundstrom EB, et al. The eukaryotic leading and lagging strand DNA polymerases are loaded onto primer-ends via separate mechanisms but have comparable processivity in the presence of PCNA. Nucleic acids research. 2007;35:6588–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Shikata K, Ohta S, Yamada K, Obuse C, Yoshikawa H, Tsurimoto T. The human homologue of fission Yeast cdc27, p66, is a component of active human DNA polymerase delta. J Biochem. 2001;129:699–708. [DOI] [PubMed] [Google Scholar]
- [4].Xie B, Mazloum N, Liu L, Rahmeh A, Li H, Lee MY. Reconstitution and characterization of the human DNA polymerase delta four-subunit holoenzyme. Biochemistry. 2002;41:13133–42. [DOI] [PubMed] [Google Scholar]
- [5].Zhang P, Mo JY, Perez A, Leon A, Liu L, Mazloum N, et al. Direct interaction of proliferating cell nuclear antigen with the p125 catalytic subunit of mammalian DNA polymerase delta. The Journal of biological chemistry. 1999;274:26647–53. [DOI] [PubMed] [Google Scholar]
- [6].Li H, Xie B, Zhou Y, Rahmeh A, Trusa S, Zhang S, et al. Functional roles of p12, the fourth subunit of human DNA polymerase delta. The Journal of biological chemistry. 2006;281:14748–55. [DOI] [PubMed] [Google Scholar]
- [7].Stodola JL, Burgers PM. Resolving individual steps of Okazaki-fragment maturation at a millisecond timescale. Nat Struct Mol Biol. 2016;23:402–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Wu X, Li J, Li X, Hsieh CL, Burgers PM, Lieber MR. Processing of branched DNA intermediates by a complex of human FEN-1 and PCNA. Nucleic Acids Res. 1996;24:2036–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Pike JE, Henry RA, Burgers PM, Campbell JL, Bambara RA. An alternative pathway for Okazaki fragment processing: resolution of fold-back flaps by Pif1 helicase. The Journal of biological chemistry. 2010;285:41712–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Levin DS, Bai W, Yao N, O’Donnell M, Tomkinson AE. An interaction between DNA ligase I and proliferating cell nuclear antigen: implications for Okazaki fragment synthesis and joining. Proc Natl Acad Sci U S A. 1997;94:12863–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Levin DS, McKenna AE, Motycka TA, Matsumoto Y, Tomkinson AE. Interaction between PCNA and DNA ligase I is critical for joining of Okazaki fragments and long-patch base-excision repair. Curr Biol. 2000;10:919–22. [DOI] [PubMed] [Google Scholar]
- [12].Warbrick E PCNA binding through a conserved motif. Bioessays. 1998;20:195–9. [DOI] [PubMed] [Google Scholar]
- [13].Maga G, Hubscher U. Proliferating cell nuclear antigen (PCNA): a dancer with many partners. J Cell Sci. 2003;116:3051–60. [DOI] [PubMed] [Google Scholar]
- [14].Dionne I, Nookala RK, Jackson SP, Doherty AJ, Bell SD. A heterotrimeric PCNA in the hyperthermophilic archaeon Sulfolobus solfataricus. Mol Cell. 2003;11:275–82. [DOI] [PubMed] [Google Scholar]
- [15].Mayanagi K, Ishino S, Shirai T, Oyama T, Kiyonari S, Kohda D, et al. Direct visualization of DNA baton pass between replication factors bound to PCNA. Sci Rep. 2018;8:16209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Dovrat D, Stodola JL, Burgers PM, Aharoni A. Sequential switching of binding partners on PCNA during in vitro Okazaki fragment maturation. Proc Natl Acad Sci U S A. 2014;111:14118–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Hedglin M, Pandey B, Benkovic SJ. Stability of the human polymerase delta holoenzyme and its implications in lagging strand DNA synthesis. Proc Natl Acad Sci U S A. 2016;113:E1777–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Liu L, Mo J, Rodriguez-Belmonte EM, Lee MY. Identification of a fourth subunit of mammalian DNA polymerase delta. J Biol Chem. 2000;275:18739–44. [DOI] [PubMed] [Google Scholar]
- [19].Gerik KJ, Li X, Pautz A, Burgers PM. Characterization of the two small subunits of Saccharomyces cerevisiae DNA polymerase delta. J Biol Chem. 1998;273:19747–55. [DOI] [PubMed] [Google Scholar]
- [20].Gomes XV, Burgers PM. Two modes of FEN1 binding to PCNA regulated by DNA. Embo J. 2000;19:3811–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Hedglin M, Benkovic SJ. Replication Protein A Prohibits Diffusion of the PCNA Sliding Clamp along Single-Stranded DNA. Biochemistry. 2017;56:1824–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Zhou Y, Meng X, Zhang S, Lee EY, Lee MY. Characterization of human DNA polymerase delta and its subassemblies reconstituted by expression in the MultiBac system. PloS one. 2012;7:e39156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Pascal JM, O’Brien PJ, Tomkinson AE, Ellenberger T. Human DNA ligase I completely encircles and partially unwinds nicked DNA. Nature. 2004;432:473–8. [DOI] [PubMed] [Google Scholar]
- [24].Montecucco A, Rossi R, Levin DS, Gary R, Park MS, Motycka TA, et al. DNA ligase I is recruited to sites of DNA replication by an interaction with proliferating cell nuclear antigen: identification of a common targeting mechanism for the assembly of replication factories. Embo J. 1998;17:3786–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Peng Z, Liao Z, Dziegielewska B, Matsumoto Y, Thomas S, Wan Y, et al. Phosphorylation of serine 51 regulates the interaction of human DNA ligase I with replication factor C and its participation in DNA replication and repair. J Biol Chem. 2012;287:36711–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Bylund GO, Burgers PM. Replication protein A-directed unloading of PCNA by the Ctf18 cohesion establishment complex. Mol Cell Biol. 2005;25:5445–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Yao NY, Johnson A, Bowman GD, Kuriyan J, O’Donnell M. Mechanism of proliferating cell nuclear antigen clamp opening by replication factor C. The Journal of biological chemistry. 2006;281:17528–39. [DOI] [PubMed] [Google Scholar]
- [28].Cotner-Gohara E, Kim IK, Tomkinson AE, Ellenberger T. Two DNA-binding and Nick Recognition Modules in Human DNA Ligase III. J Biol Chem. 2008;283:10764–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Mackey ZB, Niedergang C, Murcia JM, Leppard J, Au K, Chen J, et al. DNA ligase III is recruited to DNA strand breaks by a zinc finger motif homologous to that of poly(ADP-ribose) polymerase. Identification of two functionally distinct DNA binding regions within DNA ligase III. J Biol Chem. 1999;274:21679–87. [DOI] [PubMed] [Google Scholar]
- [30].Mayanagi K, Kiyonari S, Saito M, Shirai T, Ishino Y, Morikawa K. Mechanism of replication machinery assembly as revealed by the DNA ligase-PCNA-DNA complex architecture. Proc Natl Acad Sci U S A. 2009;106:4647–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Tomkinson AE, Lasko DD, Daly G, Lindahl T. Mammalian DNA ligases. Catalytic domain and size of DNA ligase I. J Biol Chem. 1990;265:12611–7. [PubMed] [Google Scholar]
- [32].Song W, Pascal J, Ellenberger T, Tomkinson AE. The DNA binding domain of human DNA ligase I interacts with both nicked DNA and the DNA sliding clamps, PCNA and hRad9-hRad1-hHus1. DNA Repair (Amst). 2009;8:912–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Guo Z, Kanjanapangka J, Liu N, Liu S, Liu C, Wu Z, et al. Sequential posttranslational modifications program FEN1 degradation during cell-cycle progression. Mol Cell. 2012;47:444–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Ferrari G, Rossi R, Arosio D, Vindigni A, Biamonti G, Montecucco A. Cell cycle-dependent phosphorylation of human DNA ligase I at the cyclin-dependent kinase sites. J Biol Chem. 2003;278:37761–7. [DOI] [PubMed] [Google Scholar]
- [35].Slade D Maneuvers on PCNA Rings during DNA Replication and Repair. Genes (Basel). 2018;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Li X, Li J, Harrington J, Lieber MR, Burgers PM. Lagging strand DNA synthesis at the eukaryotic replication fork involves binding and stimulation of FEN-1 by proliferating cell nuclear antigen. J Biol Chem. 1995;270:22109–12. [DOI] [PubMed] [Google Scholar]
- [37].Tom S, Henricksen LA, Park MS, Bambara RA. DNA ligase I and proliferating cell nuclear antigen form a functional complex. J Biol Chem. 2001;276:24817–25. [DOI] [PubMed] [Google Scholar]
- [38].Sakurai S, Kitano K, Yamaguchi H, Hamada K, Okada K, Fukuda K, et al. Structural basis for recruitment of human flap endonuclease 1 to PCNA. Embo J. 2005;24:683–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Rashid F, Harris PD, Zaher MS, Sobhy MA, Joudeh LI, Yan C, et al. Single-molecule FRET unveils induced-fit mechanism for substrate selectivity in flap endonuclease 1. Elife. 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Tsutakawa SE, Thompson MJ, Arvai AS, Neil AJ, Shaw SJ, Algasaier SI, et al. Phosphate steering by Flap Endonuclease 1 promotes 5’-flap specificity and incision to prevent genome instability. Nat Commun. 2017;8:15855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Xu H, Shi R, Han W, Cheng J, Xu X, Cheng K, et al. Structural basis of 5’ flap recognition and protein-protein interactions of human flap endonuclease 1. Nucleic Acids Res. 2018;46:11315–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Lin SH, Wang X, Zhang S, Zhang Z, Lee EY, Lee MY. Dynamics of enzymatic interactions during short flap human Okazaki fragment processing by two forms of human DNA polymerase delta. DNA Repair (Amst). 2013;12:922–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Zhang S, Zhou Y, Trusa S, Meng X, Lee EY, Lee MY. A novel DNA damage response: rapid degradation of the p12 subunit of dna polymerase delta. J Biol Chem. 2007;282:15330–40. [DOI] [PubMed] [Google Scholar]
- [44].Lancey C, Tehseen M, Raducanu VS, Rashid F, Merino N, Ragan TJ, et al. Structure of the processive human Pol delta holoenzyme. Nat Commun. 2020;11:1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Levin DS, Vijayakumar S, Liu X, Bermudez VP, Hurwitz J, Tomkinson AE. A conserved interaction between the replicative clamp loader and DNA ligase in eukaryotes: implications for Okazaki fragment joining. J Biol Chem. 2004;279:55196–201. [DOI] [PubMed] [Google Scholar]
- [46].Vijayakumar S, Dziegielewska B, Levin DS, Song W, Yin J, Yang A, et al. Phosphorylation of human DNA ligase I regulates its interaction with replication factor C and its participation in DNA replication and DNA repair. Mol Cell Biol. 2009;29:2042–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Smith DJ, Whitehouse I. Intrinsic coupling of lagging-strand synthesis to chromatin assembly. Nature. 2012;483:434–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Krishna TS, Kong XP, Gary S, Burgers PM, Kuriyan J. Crystal structure of the eukaryotic DNA polymerase processivity factor PCNA. Cell. 1994;79:1233–43. [DOI] [PubMed] [Google Scholar]
- [49].Pascal JM, Tsodikov OV, Hura GL, Song W, Cotner EA, Classen S, et al. A Flexible Interface between DNA Ligase and PCNA Supports Conformational Switching and Efficient Ligation of DNA. Mol Cell. 2006;24:279–91. [DOI] [PubMed] [Google Scholar]
- [50].Kubota T, Nishimura K, Kanemaki MT, Donaldson AD. The Elg1 replication factor C-like complex functions in PCNA unloading during DNA replication. Mol Cell. 2013;50:273–80. [DOI] [PubMed] [Google Scholar]
- [51].Chen X, Pascal J, Vijayakumar S, Wilson GM, Ellenberger T, Tomkinson AE. Human DNA ligases I, III, and IV-purification and new specific assays for these enzymes. Methods Enzymol. 2006;409:39–52. [DOI] [PubMed] [Google Scholar]
- [52].Fazlieva R, Spittle CS, Morrissey D, Hayashi H, Yan H, Matsumoto Y. Proofreading exonuclease activity of human DNA polymerase delta and its effects on lesion-bypass DNA synthesis. Nucleic Acids Res. 2009;37:2854–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Henricksen LA, Umbricht CB, Wold MS. Recombinant replication protein A: expression, complex formation, and functional characterization. J Biol Chem. 1994;269:11121–32. [PubMed] [Google Scholar]
- [54].Podust LM, Podust VN, Floth C, Hubscher U. Assembly of DNA polymerase delta and epsilon holoenzymes depends on the geometry of the DNA template. Nucleic Acids Res. 1994;22:2970–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Mastronarde DN. Automated electron microscope tomography using robust prediction of specimen movements. J Struct Biol. 2005;152:36–51. [DOI] [PubMed] [Google Scholar]
- [56].Scheres SH. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J Struct Biol. 2012;180:519–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Kimanius D, Forsberg BO, Scheres SH, Lindahl E. Accelerated cryo-EM structure determination with parallelisation using GPUs in RELION-2. Elife. 2016;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Scheres SH, Chen S. Prevention of overfitting in cryo-EM structure determination. Nat Methods. 2012;9:853–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, et al. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–12. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.