

Abstract
Background
Tumour-infiltrating CD3, CD8 lymphocytes and CD68 macrophages are associated with favourable prognosis in localised colorectal cancer, but the effect in metastatic colorectal cancer (mCRC) is not established.
Methods
A Scandinavian population-based cohort of non-resectable mCRC patients was studied. Tissue microarrays (n = 460) were stained with CD3, CD8 and CD68 using fluorescence-based multiplex immunohistochemistry. Associations with clinicopathological variables, overall survival (OS) and progression-free survival were estimated.
Results
Two-thirds of microsatellite instable (MSI) and one-fourth of microsatellite stable (MSS) tumours displayed the highest quartile density of CD8. For CD3 high vs low cases, median OS was 20 vs 16 months (HR: 0.76, 95% CI: 0.59, 0.76, p = 0.025) with 3-year OS of 27 vs 13%. For CD68 high vs low cases, median OS was 23 vs 15 months (HR: 0.69, 95% CI: 0.54, 0.88, p = 0.003) with 3-year OS of 28 vs 12%. MSI, BRAF mutation and CDX2 loss were negative prognostic markers independent of tumour immune infiltration.
Conclusions
In mCRC, high lymphocyte infiltration was found in proportions of MSI and MSS tumours—potential subgroups of immunotherapy response. Tumour-infiltrating CD3 lymphocytes and CD68 macrophages were associated with median and long-term survival. MSI was a significant negative prognostic marker despite high immunogenicity.
Subject terms: Colorectal cancer, Tumour immunology
Background
In recent years, the immune contexture of the tumour microenvironment has gained great attention. This is partly due to the approval of immune checkpoint inhibitors (ICIs) for several malignancies, introducing the era of immunotherapy. By blocking co-inhibitory receptors (e.g. PD-1, CTLA-4) on T-lymphocytes and other immune cells or their ligands (e.g. PD-L1) on tumour cells or immune cells, ICIs enhances the anti-tumour activity of the immune system. It has also been recognised that tumours displaying a dense infiltration of lymphocytes, termed tumour-infiltrating lymphocytes (TILs), are associated with a favourable prognosis in several cancer types [1]. All mature T cells express CD3 antigen, while cytotoxic T cells display both CD3 and CD8 antigens on their surface. The Immunoscore®, being a combined score of CD3- and CD8-positive T-lymphocytes at the tumour centre and invasive margin, is a validated prognostic marker in localised colorectal cancer (CRC) [2, 3]. The prognostic effect of TILs in localised CRC has also been verified in studies using tumour tissue microarrays (TMAs) [4–8]. Still, the impact of immune cell infiltration in the primary tumour of patients with metastatic CRC (mCRC) is not established. Previous prognostic assessments of immune markers in mCRC have mainly been performed on metastases of small and highly selected patient cohorts after secondary metastatic surgery [9–14]. Unless there are signs of obstruction, patients with mCRC generally do not undergo primary tumour surgery and have only small biopsies taken for diagnostic purposes. Therefore, evaluating the prognostic effect of tumour-infiltrating immune cells in TMA is relevant and could be of future clinical importance for mCRC patients.
TILs in CRC are highly associated with tumour microsatellite instability (MSI) status [15]. MSI is a good prognostic marker in localised CRC [2, 16, 17] but is associated with adverse prognosis in mCRC [18, 19]; the reason for this conflict is not understood. MSI is also a predictive marker for immunotherapy with ICIs in mCRC [20], and current NCCN guidelines [21] recommend ICI as first-line treatment for MSI mCRC patients. However, one-third of MSI patients do not benefit from ICI treatment, with immediate disease progression [22, 23]. Assessment of tumour-infiltrating CD8 lymphocytes has been suggested as a potential predictive marker for ICI response [24, 25].
Two major phenotypes of macrophages have been identified with antagonising functions: M1 with pro-inflammatory/anti-tumour effect and M2 with immunosuppressive/cancer progressive effects. CD68 antigen is a transmembrane protein highly expressed by monocytes and macrophages. Tumour-associated macrophages (TAMs) were first believed to express an M2 phenotype, as most studies reveal poor prognosis in different cancers if present [1]. The prognostic effect in CRC has been studied to a limited extent and with conflicting results. However, most studies, including recent meta-analyses [1, 26, 27], suggest that TAM expression is associated with a favourable prognosis. In mCRC, prognostic studies are few, and no studies have been published on population-based cohorts.
Tumour BRAF V600E mutation (BRAFmut) is a well-established poor prognostic marker in mCRC, but the outcome is heterogeneous within this group [28]. Sporadic MSI is associated with BRAFmut [29]. However, previous studies have shown contradictory results on associations between BRAFmut and immune cell infiltration [4–6, 8, 10]. Furthermore, the prognostic effect of BRAFmut in microsatellite stable (MSS) CRC is stratified by the CpG island methylator phenotype [30].
Most studies of prognostic tumour markers are based on clinical trials or referral hospital cohorts. These patients are selected with better performance status, younger age and less comorbidity compared to the general mCRC population. We have previously reported a high incidence of MSI and BRAFmut in a population-based cohort [18, 31], probably due to the poor prognosis in these groups with many patients not fit enough for trial inclusion. The present study included all diagnosed patients with mCRC within three regions during a specific period, reflecting a general population with mCRC. Real-world data on tumour immune cells concerning genetic and molecular tumour alterations could be important for understanding how progress in the use of immunotherapy should proceed. Our study aimed to evaluate the prognostic effect of tumour immune cell infiltration of CD3 and CD8 lymphocytes and CD68 macrophages in an unselected mCRC cohort in relation to tumour molecular alterations.
Methods
Patient cohort
This cohort is a prospectively collected, population-based cohort of all non-resectable mCRC patients referred to the oncology unit of three regional hospitals in Scandinavia: Odense University Hospital (Denmark) (n = 325), Uppsala University Hospital (Sweden) (n = 155), and Haukeland University Hospital (Norway) (n = 316) during 2003–2006 with the last follow-up in 2014. These hospitals cover all oncology treatments in their region. Cases not referred were later identified through the national (Norway and Sweden) and regional (Denmark) cancer registries (n = 49) and were enrolled in the study. The cohort consists of 796 patients.
Tissue retrieval and TMA generation
Formalin-fixed paraffin-embedded tissue blocks were retrieved from the primary tumour in most cases or from a metastatic lesion (6 cases), and corresponding haematoxylin–eosin-stained whole-tissue sections were examined. TMA generation had been performed previously in 460 (58%) cases [32] according to standards used in the Human Protein Atlas [33], with two 1 mm diameter tumour cores extracted per patient. TMA was generated from tissue blocks from surgical resections of the primary tumours in 419 of the 460 (91%) cases. The remaining 41 cases were taken from biopsies (35 cases from the primary tumour and 6 cases from a metastatic lesion). For patients with synchronous disease (n = 249), tissue for TMA generation was collected at diagnosis of metastatic disease, of which 208 were from tissue blocks after surgical resection of primary tumour and 41 from biopsies (35 cases from primary tumour and 6 cases from metastases). For patients with metachronous disease (n = 211), tissue for TMA generation was collected from tissue blocks after prior surgical resection at the time of diagnosis of localised CRC. Lack of inclusion was generally due to small biopsies or necrotic tissue (n = 239), and a proportion was displaced in the archive or had no cancer tissue (n = 97).
Tumour analyses
TMA sections (4 µm) were stained with a multimarker panel including primary antibodies against CD3, CD8 and CD68 using fluorescence-based multiplex immunohistochemistry (Supplementary Methods and Fig. S1). Briefly, a 5-plex stain was designed applying the OpalTM Multiplex IHC method (PerkinElmer/Akoya, USA) with digital image analysis using the Vectra 3.0 Automated Quantitative Pathology Imaging System, 200 (Vectra software version 3, PerkinElmer/Akoya, USA) and the inForm Image Analysis Software (version 2.3, Akoya Biosciences).
Cases with weak staining, poor tissue quality or extensive necrosis were excluded. The cell count of the two tumour cores was assembled for each patient. Cell counts are given as the number of positive cells divided by mm2 (density) in each compartment (tumour epithelium and stroma). Immune cell infiltration was most abundant in the tumour stroma, but concurrent intra-epithelial CD3/CD8-positive immune cell infiltration at lower density was frequently observed. For this reason, we chose to present CD3- and CD8-positive lymphocytes as the density of positive cells of stroma plus epithelium. For CD68-positive macrophages, results are given as density of positive cells in stroma only, as very few positive cells were present in the epithelial compartment.
Tumour molecular alterations
MSI status, BRAF V600E mutation (BRAFmut), KRAS mutation (KRASmut) and CDX2 expression status were available for 591, 595, 485 and 452 patients, respectively, with methods described previously [18, 34]. Results from next-generation DNA tumour sequencing of a customised Ampliseq hotspot targeted panel of 44 cancer-related genes were available for 447 patients [31]. Only the main mutation-driver gene cluster regions for TP53 and APC were covered in the gene panel design and is the most likely explanation for the lower than expected frequency of these mutations in our cohort. In the present study, we examined the most frequently altered genes (>10% of mutated cases) of this panel: BRAF, APC, KRAS, TP53, PIK3CA, and SMAD4. We assembled the previous pyrosequencing and hotspot sequencing panel results for KRASmut and previous immunohistochemistry, pyrosequencing, and hotspot sequencing panel results for BRAF V600E mutation (Supplementary Methods).
Statistics
Non-parametric Mann–Whitney U-test and Spearman’s rank-order correlation were used to analyse associations between the density of each immune cell marker (as continuous variables) and clinical and pathological characteristics. In multivariate analysis, linear regression was used with square root-transformed density of each immune cell marker to resemble a normal distribution. The general assumptions for linear regression about independence, linearity, variance homogeneity and normal distribution were checked by graphical inspection on which an adequate transformation of the response variables was introduced. We also checked that the recommended number of observations per variable was obtained for the linear regression [35]. Results are reported as regression coefficient (B) with 95% confidence intervals (CIs). Overall survival (OS) was calculated from the date of unresectable metastatic disease to death and censored if the patient was alive on 4 February, 2014. Progression-free survival (PFS) was the interval from the date of the first administration of chemotherapy to the date of progression (on computed tomographic scan) or death and censored if the patient was alive without progression on 4 February, 2014. Kaplan–Meier method with log-rank test and Cox multiple regression were used for OS and PFS analyses. Results are reported as hazard ratios (HRs) with 95% CIs. Formal interaction tests were integrated into the Cox models to assess whether effects were different between subgroups, but results must be interpreted carefully due to the low power of such tests. In the fully adjusted (full model) Cox multiple regression analyses, we included variables statistically significant for survival in our cohort and available prognostic variables recommended by Goey et al. [36]. Variables with >25 missing values were excluded from the analyses. We ensured that the recommended number of events per variable for the Cox regression was fulfilled [37]. All analyses were performed using the statistical software program IBM SPSS v25.
Results
Study cohort
Staining of the immune cell markers was successful in 448 of the 460 (97%) cases (Fig. 1). A moderate–strong reproducibility of the average of the two tumour cores obtained from each patient was observed (CD3 intraclass correlation coefficient (ICC): 0,71, p < 0.001; CD8 ICC: 0.74, p < 0.001; CD68 ICC: 0.62, p < 0.001; Fig. S2). Of the 448 patients with successful staining, 8% were MSI, 20% BRAF V600E mutated (BRAFmut), 46% KRAS mutated (KRASmut), 19% CDX2 negative, 54% TP53 mutated (TP53mut), 35% APC mutated (APCmut), 20% PIK3CA mutated (PIK3CAmut), and 12% SMAD4 mutated (SMAD4mut). First-, second- and third-line palliative chemotherapy were initiated in 63, 36 and 16% of patients, respectively. Median OS was 18 months for patients given first-line treatment and 3 months for patients not given chemotherapy. Table 1 summarises the clinical and pathological characteristics and survival of the cohort.
Fig. 1. Fluorescence-based immunohistochemistry staining on tumour tissue microarrays with a multimarker panel of CD3 and CD8 lymphocytes and CD68 macrophages in a Scandinavian population-based cohort of metastatic colorectal cancer.

Representative staining shown in images of well differentiated (a) and poorly differentiated (b) tumours with CD3 in yellow, CD8 in green, CD68 in orange, epithelium (cytokeratin) in red, DAPI in blue in the composite images to the left and white in the higher resolution images to the right.
Table 1.
Patient and tumour characteristics in a Scandinavian population-based cohort of metastatic colorectal cancer patients with available status on tumour-infiltrating CD3 and CD8 lymphocytes and CD68 macrophages (n = 448).
| Characteristics | Missing, n | |
|---|---|---|
| Age in years, median, range | 70 (24, 96) | |
| Age >75 years, n (%) | 152 (34) | |
| Female, n (%) | 226 (50) | |
| PS ECOG >1, n (%) | 149 (33) | |
| Right-sided, n (%) | 173 (39) | 7 |
| >1 metastatic site, n (%) | 259 (58) | |
| Synchronous metastases, n (%) | 241 (54) | |
| ALP high, n (%) | 218 (55) | 53 |
| Primary tumour resected, n (%) | 411 (92) | |
| Tumour grade 3, n (%) | 93 (22) | 15 |
| Mutations, n (%) | ||
| KRAS | 200 (46) | 15 |
| BRAFV600E | 91 (20) | |
| NRAS | 17 (4) | 50 |
| TP53 | 215 (54) | 50 |
| APC | 139 (35) | 50 |
| PIK3CA | 81 (20) | 50 |
| SMAD4 | 47 (12) | 50 |
| MSI, n (%) | 35 (8) | 1 |
| CDX2 loss, n (%) | 83 (19) | 2 |
| CD3 density, median, mean, 95% CI | 59.58, 123.36, (106.25, 140.47) | |
| CD8 density, median, mean, 95% CI | 40.03, 92.31, (79.08, 105.53) | |
| CD68 density, median, mean, 95% CI | 40.82, 115.21, (97.61, 132.81) | |
| Secondary metastatic surgery, n (%) | 33 (7) | 1 |
| Adjuvant chemotherapy | 67 (15) | |
| First-line chemotherapy, n (%) | 280 (63) | |
| Combination, n (%) | 216 (77) | |
| Monotherapy, n (%) | 64 (23) | |
| OS in months, median, 95% CI | 18 (15.11, 20.89) | |
| PFS in months, median, 95% CI | 8 (7.07, 8.63) | |
| BSC only, n (%) | 167 (37) | |
| OS in months, median, 95% CI | 3 (2.13, 3.87) | |
n number of patients, CI confidence interval, PS ECOG performance status score developed by Eastern Cooperative Oncology Group, Right-sided site of primary colon cancer in ascending colon and transversum, Left-sided site of primary colon cancer in the descending colon, sigmoid and rectum, Synchronous metastases within 6 months after initial diagnoses, ALP alkaline phosphatase, MSI microsatellite instability, MSS microsatellite stable, CD3 density number of tumour-infiltrating CD3 lymphocytes per mm2 tumour tissue microarray, CD8 density number of tumour-infiltrating CD8 lymphocytes per mm2 tumour tissue microarray, CD68 density number of tumour-infiltrating CD68 macrophages per mm2 tumour tissue microarray, OS overall survival, PFS progression-free survival, BSC best supportive care.
Patient and tumour characteristics and treatment
The distribution of CD3, CD8 and CD68 tumour-infiltrating immune cells across the cohort was right-skewed with a median density of 59.6, 40.0 and 40.8 for CD3-, CD8- and CD68-positive cells, respectively (Table 1). Both tumour infiltration of CD3 and CD8 lymphocytes were associated with infiltration of CD68 macrophages (Table S1). Tumour-infiltrating CD3 and CD8 lymphocytes were significantly associated with MSI status, and a trend was observed for CD68 macrophages (Fig. 2 and Table S1). However, tumour immune cell infiltration distribution was heterogeneous with many outliers, especially in MSS cases (Fig. 2). There is no consensus on threshold interpretation of high and low tumour immune cell infiltrations by individual observation in central tumour/TMA. Therefore, we determined the threshold as the median and four percentile groups to describe the distribution. In total, 37 and 60% of MSI cases vs 24 and 22% of MSS cases were in the highest density group (>75 percentile) of tumour-infiltrating CD3 and CD8, respectively (Fig. 2).
Fig. 2. Density of tumour-infiltrating CD3, CD8 lymphocytes and CD68 macrophages according to tumour microsatellite instability (MSI) and microsatellite stable (MSS) status in a Scandinavian population-based cohort of metastatic colorectal cancer.
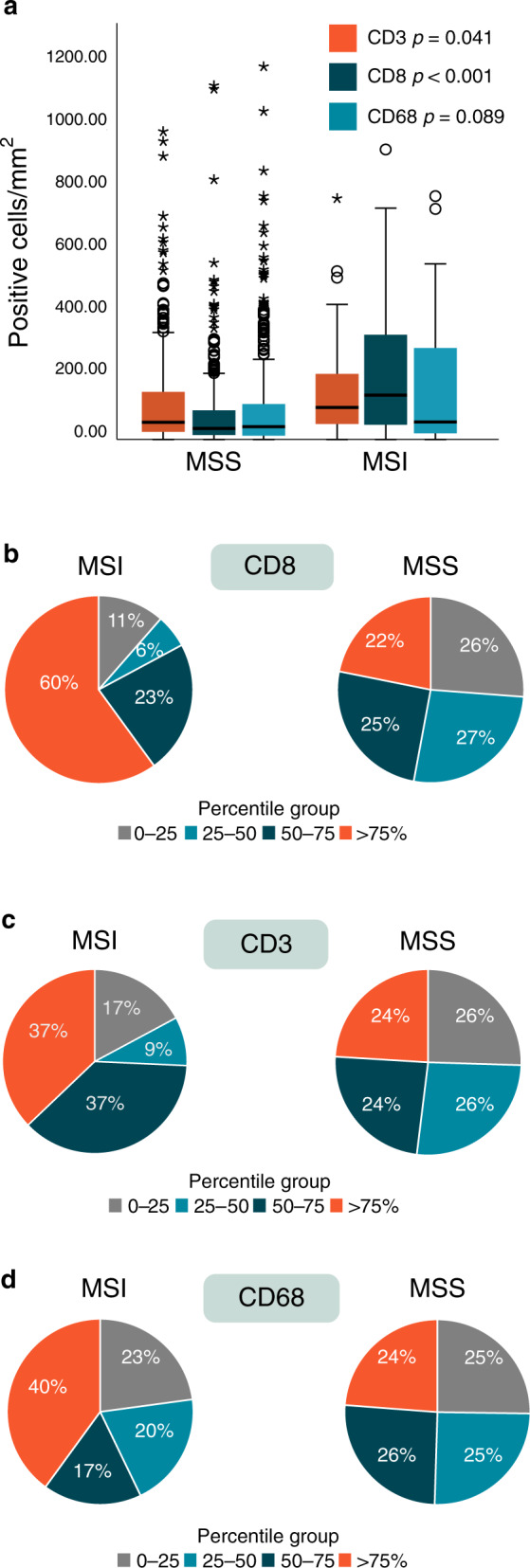
a Density illustrated by boxplot, horizontal line marks the median value, outliers are represented by circles (mild) and stars (extreme); extreme values >1200 are not shown in the figure, p value: Mann–Whitney U-test. b Percentage of cases in the four percentile groups of CD8 density. c Percentage of cases in the four percentile groups of CD3 density. d Percentage of cases in the four percentile groups of CD68 density.
Both tumour-infiltrating CD3 and CD8 lymphocytes were associated with age when analysed as continuous variables. Increasing density of CD8 lymphocytes was also correlated to right-sided primary tumour and PIK3CAmut (n = 81) and inversely correlated to liver metastasis and APCmut (n = 139) in unadjusted regression analyses (Table S1). In the fully adjusted model, MSI and the negative association to liver metastases remained statistically significant (Table S2). Infiltration of CD68 macrophages was inversely associated with bone metastasis (Tables S1 and S2).
We observed no difference in immune cell infiltration and first-line chemotherapy treatment (Table S3) or secondary metastatic surgery (Table S1). However, increasing tumour infiltration of CD68 macrophages was associated with the initiation of second-line treatment (Table S3).
Overall and progression-free survival
Patients not given chemotherapy, mainly due to poor performance status or old age (n = 168), had a median OS of 3 months. None of the immune cell markers studied had a prognostic impact in this subgroup (Table S4). Patients treated with chemotherapy were used for further survival analyses, making our results more comparable to previous publications.
In patients given first-line palliative chemotherapy (n = 280), increased density of tumour-infiltrating CD3 lymphocytes was associated with improved OS (p = 0.031), and increased density of CD68 was borderline significant (p = 0.063) (Table 2). We further dichotomised the data by the median value, as this has demonstrated associations to survival in previous studies [4, 7, 13, 14]. For CD3 and CD68, a high tumour infiltration was associated with increased OS (HR: 0.76, p = 0.025 and HR: 0.69, p = 0.003 for CD3 and CD68, respectively; Fig. 3). The difference in survival was greater with time, and 3-year OS was 27 vs 13% for CD3 high vs low cases (odds ratio (OR): 2.48) and 28 vs 12% for CD68 high vs low cases (OR: 2.85). Tumour infiltration of CD8 lymphocytes had no significant prognostic impact on OS in our cohort (Table 2 and Fig. 3). None of the tumour-infiltrating immune cell markers had a prognostic impact on PFS after first-line chemotherapy when analysed as a continuous variable (Table S5).
Table 2.
Results from Cox regression of overall survival in a Scandinavian population-based cohort of metastatic colorectal cancer patients treated with first-line chemotherapy.
| Variable | Unadjusted | Fully adjusteda (n = 245, e = 230) | |||||
|---|---|---|---|---|---|---|---|
| n/e | HR | 95% CI | p value | HR | 95% CI | p value | |
| Age, years | 280/265 | 1.01 | (1.00, 1.02) | 0.157 | 1.00 | (0.99, 1.02) | 0.971 |
| Female | 280/265 | 0.97 | (0.76, 1.23) | 0.786 | 0.69 | (0.52, 0.92) | 0.010 |
| PS ECOG | 280/265 | 2.24 | (1.62, 3.09) | <0.001 | 2.15 | (1.49, 3.10) | <0.001 |
| Right-sided | 277/262 | 1.19 | (0.93, 1.54) | 0.171 | 0.92 | (0.67, 1.25) | 0.580 |
| Tumour grade 3 | 274/259 | 1.97 | (1.47, 2.64) | <0.001 | 1.59 | (1.11, 2.28) | 0.012 |
| >1 metastatic site | 280/265 | 1.50 | (1.17, 1.91) | 0.001 | 1.39 | (1.04, 1.87) | 0.026 |
| Synchronous metastasis | 280/265 | 0.93 | (0.73, 1.19) | 0.563 | 0.84 | (0.63, 1.11) | 0.216 |
| Secondary metastasis surgery | 279/264 | 0.26 | (0.17, 0.40) | <0.001 | 0.32 | (0.20, 0.53) | <0.001 |
| ALP > UNL | 265/250 | 1.19 | (1.40, 2.35) | <0.001 | 1.83 | (1.37, 2.46) | <0.001 |
| BRAFV600E mutation | 280/265 | 1.69 | (1.24, 2.30) | 0.001 | 1.70 | (1.09, 2.63) | 0.018 |
| KRAS mutation | 271/256 | 0.97 | (0.76, 1.24) | 0.801 | 1.59 | (1.16, 2.19) | 0.004 |
| CDX2 negative | 278/263 | 2.37 | (1.69, 3.33) | <0.001 | 1.70 | (1.10, 2.62) | 0.017 |
| MSI-H | 279/264 | 3.26 | (1.93, 5.50) | <0.001 | 3.37 | (1.63, 6.97) | 0.001 |
| CD3 density | 280/265 | 0.98 | (0.96, 0.99) | 0.031 | 0.97 | (0.95, 0.99) | 0.027 |
| CD68 density | 280/265 | 0.98 | (0.96, 1.00) | 0.063 | 0.99 | (0.96, 1.01) | 0.224 |
| CD8 density | 280/265 | 0.99 | (0.96, 1.01) | 0.311 | n.i. | ||
n number of patients, e number of events, HR hazard ratio, CI confidence interval, p value from likelihood ratio test, PS ECOG performance status score developed by Eastern Cooperative Oncology Group, Right-sided tumour site of colon cancer in ascending colon and transversum, Synchronous metastases within 6 months after initial diagnoses, ALP > UNL alkaline phosphatase above upper normal limit, MSI-H microsatellite instable high, CD3 density square root transformed number of tumour-infiltrating CD3 lymphocytes per mm2 tumour tissue microarray, CD8 density square root transformed number of tumour-infiltrating CD8 lymphocytes per mm2 tumour tissue microarray, CD68 density square root transformed number of tumour-infiltrating CD68 macrophages per mm2 tumour tissue microarray, n.i. not included. p-values < 0.05 typed in bold.
aLDH high was also statistically significant when included in the multiple regression model but was excluded from the analysis due to missing values.
Fig. 3. Overall survival (OS) according to high (>median) vs low tumour infiltration of CD3 and CD8 lymphocytes and CD68 macrophages in a Scandinavian population-based cohort of metastatic colorectal cancer treated with first-line chemotherapy.

Kaplan–Meier curves were constructed; statistical significance test with the log-rank test for p value and univariate cox regression for hazard ratio (HR) and 95% confidence interval (CI). a OS according to tumour infiltration of CD3. b OS according to tumour infiltration of CD8. c OS according to tumour infiltration of CD68. d OS according to a combined score of tumour-infiltrating CD3 and CD68. e OS according to oxaliplatin-based treatment regimen in cases with low CD3 tumour infiltration only. f OS according to oxaliplatin-based treatment regimen in cases with high CD3 tumour infiltration only. Oxa oxaliplatin, 5FU fluorouracil + folinic acid.
In multiple Cox regression analysis of patients given first-line chemotherapy, including other important prognostic factors in mCRC, tumour infiltration of CD3 lymphocytes was an independent good prognostic marker for OS (HR: 0.97, p = 0.027; Table 2). MSI, BRAFmut and CDX2-negative status were significant negative prognostic markers for OS. Dichotomised by median values, high tumour density of CD3 lymphocytes and CD68 macrophages were significant prognostic markers (HR: 0.73, p = 0.029 and HR: 0.61, p = 0.001, respectively; Table S6).
Survival in subgroups of tumour molecular markers
In the subgroup analyses according to MSI status, the favourable prognostic impact of high (>median) tumour infiltration of CD3 and CD68 cells was only evident in the MSS subgroup (median OS 21 vs 16 months, p = 0.008 and 24 vs 15 months, p = 0.001, respectively; Fig. S3). However, we had very few patients in the MSI subgroup (n = 16), with four patients presenting tumour MSI status and low infiltration of CD3 lymphocytes (Fig. S3). The interaction tests were not significant (p = 0.945 and p = 0.775).
In subgroup analyses according to BRAF status, the prognostic effect of high tumour infiltration of CD3 and CD68 cells was only evident in the BRAF wild type (BRAFwt) subgroup (median OS 21 vs 17 months, p = 0.016 and median OS 26 vs 15 months, p = 0.002, respectively; Fig. S3), with no prognostic effect in BRAFmut (n = 52) cases. Again, there were few cases in these subgroup analyses, with only 24 patients with BRAFmut and low tumour density of CD3 lymphocytes, and the interaction tests were not significant (p = 0.528 and p = 0.313).
The prognostic effect of tumour-infiltrating CD3 and CD68 cells was only evident in CDX2-positive cases, as none of the studied immune cells had a prognostic impact on CDX2-negative cases (Fig. S3). However, there were few patients in this subgroup analysis, and the interaction tests were not significant (p = 0.981 and p = 0.706).
MSI, BRAFmut and CDX2 loss were negative prognostic markers regardless of tumour immune cell infiltration (Table S7).
No survival benefit of oxaliplatin-containing combination chemotherapy in patients with low tumour infiltration of CD3 lymphocytes
Dichotomised by the median value, patients with high tumour density of CD3 lymphocytes had better survival after first-line fluorouracil–oxaliplatin-based chemotherapy compared to fluorouracil monotherapy (median OS 23 vs 9 months, p < 0.001). No survival advantage of oxaliplatin-based chemotherapy was observed in patients with low tumour density of CD3 lymphocytes (17 vs 16 months, p = 0.321; Fig. 3).
Higher prognostic impact with a combined score of tumour-infiltrating CD3 lymphocytes and CD68 macrophages
Since a high density of CD3 and CD68 tumour-infiltrating cells were prognostic, we calculated a combined score of these immune cell markers, dichotomised by the median value. Patients with a high tumour density of CD3 and CD68 cells had a median OS of 25 months compared to 15 months in patients with low infiltration of CD3 and CD68 cells (p = 0.002; Fig. 3). When selecting MSS/BRAFwt cases, median OS was 29 months if combined high density of CD3 and CD68 cells and 15 months if combined low density of CD3 and CD68 cells (p < 0.001).
Discussion
Our study is, as far as we know, the first study reporting the prognostic effect of tumour-infiltrating CD3 and CD8 lymphocytes and CD68 macrophages in a non-selected mCRC patient population. Exploring population-based cohorts is important since trial and referral hospital cohorts are highly selected with few elderly patients, less comorbidity, and better performance status. Our previous studies have revealed that molecular alterations and other characteristics markedly differ between trial/referral centre patients and mCRC patients in the general population [18, 31]. In our series of chemotherapy-treated patients, tumour-infiltrating CD3 lymphocytes was an independent good prognostic marker for OS, with apparently greater influence on long-term OS than median survival. This finding is supported by a previous study of mCRC patients who underwent metastatic surgery [10]. However, tumour-infiltrating CD8 lymphocytes had no prognostic impact, in line with a recent report of 109 mCRC patients [38]. Craig et al. recently published results on mCRC patients included in a randomised study of first-line chemotherapy and found that a combined score of tumour-infiltrating CD3 and CD8 lymphocytes was significantly associated with improved OS when adjusted for age, sex, MSI status and treatment [8]. In line with our results, a small study of 68 mCRC patients who underwent first-line treatment found no association with PFS and TILs [39]. Other studies, including all stages of CRC, with a limited number of mCRC patients, reported good prognostic effects of TILs in multivariate analyses corrected for tumour stage [6, 7].
MSI is recognised as a predictive marker for ICI in mCRC. One reason is believed to be due to the observed higher tumour immune cell infiltration in these tumours. However, not all patients with MSI tumours respond clinically [22], and 29% of the patients had immediate progression on pembrolizumab in the randomised first-line study by André et al. [23]. Assessment of TILs has been suggested as a potential predictive marker for ICI response [24, 25], although studies are lacking. In a publication of ICI treatment in 85 MSI mCRC patients, cases with high TILs (≥2 TILs) showed a better response rate, PFS and OS than those with low TILs [40]. Interestingly, a small study of patients with localised CRC given neoadjuvant ICI demonstrated a significantly higher tumour density of CD8+PD-1+ lymphocytes in MSS cases that responded to treatment [41]. Our study observed that, even though there is a highly significant correlation between MSI status and immune cell infiltration, the distribution is heterogeneous in the two groups with many outliers, especially in the MSS subgroup. When dividing the density of TILs into four percentile groups, we found that 60% of our MSI cases and 22% of MSS had the highest (>75 percentile) tumour CD8 lymphocyte infiltration, subgroups that potentially could indicate ICI benefit. Future studies are needed to confirm the predictive effect of TILs on ICI treatment with consensus on threshold and methods to determine high and low TILs subgroups.
In patients treated with chemotherapy, tumour infiltration of CD3 TILs revealed a good prognosis, also after correcting for the associated MSI status. In contrast to the prognostic dependence of TILs in studies of MSI in localised CRC [2], we found that MSI is an independent marker of poor prognosis, regardless of tumour immune cell infiltration. Different immune-escape mechanisms have been reported in MSI CRC studies [42, 43]. Future studies are needed to explore whether immune-escape mechanisms, despite high immunogenicity, are the reason for the heterogeneous prognosis of MSI in mCRC compared to early stages. In line with previous knowledge, MSI, BRAFmut and CDX2 loss were significant negative prognostic markers. We could now add that this was seen regardless of immune cell infiltration. Within the major groups of patients, i.e. with MSS, BRAFwt and CDX2 expression, immune infiltration was prognostic. The limited number of patients within the poor prognosis groups (MSI, BRAFmut, CDX2 loss) prevented firm conclusions; however, we could not detect any prognostic importance of intratumoural immune cells in these groups. Thus, tumour immune cell infiltration could not explain the heterogeneous prognosis within the group with BRAFmut tumours.
In our study, patients with low infiltration of CD3 TILs had no survival benefit of oxaliplatin-based chemotherapy compared to fluorouracil monotherapy, in contrast to patients with high CD3 tumour TILs. These results must be evaluated with caution due to the limited number of patients. The IDEA study of stage III CRC recently reported that patients with a low Immunoscore® have no disease-free survival benefit of 6 months compared to 3 months adjuvant FOLFOX [44]. They hypothesise that patients with a low number of TILs have no effect of the immunogenic cell death otherwise induced by oxaliplatin due to weak immunogenicity or immunosuppressive environment in these tumours. In another recent stage III CRC publication, patients with high Immunoscore® had the most benefit from adjuvant chemotherapy in terms of recurrence-free survival [45]. These findings support our observation that only patients with a high density of CD3 TILs have a survival benefit from a more intensified oxaliplatin-based chemotherapy regimen. Patients with high TILs seem to have a generally better prognosis with improved benefit of both oxaliplatin-based chemotherapy and ICI treatment.
Although the extent of tumour-infiltrating CD3 lymphocytes is prognostic for patients treated with chemotherapy in our mCRC cohort, the prognostic impact seems more important in studies of early-stage CRC [2, 8]. The prognostic effect did not reach statistical significance in patients not given chemotherapy, but this could be due to their short survival. However, MSI, BRAFmut and CDX2 loss were all significant markers of poor prognosis, indicating that those traits influence the spontaneous behaviour, whereas the immune infiltration recorded here does not. Based on previous observation of different immune-escape mechanisms with CRC progression [46–48], we hypothesise that certain immune-escape mechanisms could explain the inferior prognostic importance of immune infiltration in mCRC compared to localised CRC.
The prognostic effect of tumour-infiltrating macrophages in CRC have been studied to a lesser extent, and results are conflicting. However, most studies, including a recent meta-analysis [1, 22, 23], reports a favourable prognosis with higher infiltration at the invasive tumour margin. Nevertheless, this did not reach significance in studies of tumour centre infiltration. In our study, high infiltration of CD68 macrophages was significantly associated with a favourable prognosis. Still, the prognostic effect was no longer significant when interpreting the results as a continuous variable and might indicate that the prognostic impact of CD68 is less certain. A spectrum of different TAMs has been identified with diverse functions, and our study could not differentiate between these phenotypes.
In a population-based cohort of mCRC patients, tumour infiltration of CD3 lymphocytes and CD68 macrophages are independent markers for prolonged OS in chemotherapy-treated patients, especially for long-term survival. Two-thirds of MSI and one-fourth of MSS cases displayed the highest quartile tumour CD8 lymphocyte infiltration, potential predictive subgroups of ICI effect. Although immune cell infiltration is highly associated with MSI status, MSI is still an independent negative prognostic marker in these patients, regardless of immune infiltration.
Limitations
Our cohort is population based and includes more elderly patients and patients with worse performance status than studies reporting cohorts from referral hospitals and clinical trials. Patients without sufficient tumour material for inclusion in TMA construction were not included in the analyses. This was generally due to small biopsies where primary tumour surgery was not performed or due to necrotic/fibrotic tissue after neoadjuvant radiotherapy for rectal cancer. According to our previous publication, patients without sufficient tumour material for TMA generation had poor performance status, received less treatment and had a worse prognosis [32]. However, this selection bias is present for most studies including tumour tissue sampling. Lack of tumour tissue from metastases prevented evaluation of tumour-infiltrating immune cells at secondary metastatic sites. However, as this is a prospectively collected population-based cohort, there was no clinical practice to obtain biopsies from metastases unless uncertainties of origin. In population-based cohorts, treatments are more heterogeneous compared to trial cohorts. This cohort was collected >10 years ago. Even though palliative chemotherapy treatment options have not changed much during the past decade, intensive regimens and metastatic surgery are more often used today. We had few patients within subgroups of tumour molecular alterations that make conclusions uncertain. In observational studies, there is a risk of missed effects due to multi-collinearity, which is why we have also published unadjusted estimates. Moreover, multiple testing deflates the p values and presented p values should be interpreted with caution.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Supplementary information
Acknowledgements
We would like to thank Randi Eikeland (Haukeland University Hospital, Bergen, Norway) for data management.
Author contributions
HS, PP and BG designed the study and was responsible for patient collection. CHB, JB and RAL were responsible for multiplex IHC analyses and digital image analyses. KA collected and assembled all patient data. LN was responsible for the targeted gene panel DNA sequencing analyses. KA and GEE performed the statistical analyses. KA drafted the manuscript with HS, who supervised the project together with BG, OD and RAL. All authors contributed to manuscript revision and approved the submitted version.
Funding information
This study has received grants from Lions Cancer Foundation (Sweden), Selanders Foundation (Sweden), Uppsala University Hospital (Sweden), the Swedish Cancer Society, the Norwegian Cancer Society and the South-Eastern Norway Regional Health Authorities.
Data availability
Data can be made available upon reasonable request.
Ethics approval and consent to participate
This study was performed in accordance with the Declaration of Helsinki. Written informed consent was obtained from all patients seen at the clinics. The study was approved by the regional ethical committees in Norway (Regional Committee for Medical and Health Research Ethics - REC West), Sweden (Regional Ethical Committee Uppsala) and Denmark (The Regional Scientific Ethical Committees for Southern Denmark).
Consent to publish
This manuscript does not contain any individual person data in any form.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s41416-021-01586-5.
References
- 1.Fridman WH, Zitvogel L, Sautes-Fridman C, Kroemer G. The immune contexture in cancer prognosis and treatment. Nat Rev Clin Oncol. 2017;14:717–34. doi: 10.1038/nrclinonc.2017.101. [DOI] [PubMed] [Google Scholar]
- 2.Pages F, Mlecnik B, Marliot F, Bindea G, Ou FS, Bifulco C, et al. International validation of the consensus Immunoscore for the classification of colon cancer: a prognostic and accuracy study. Lancet. 2018;391:2128–39. doi: 10.1016/S0140-6736(18)30789-X. [DOI] [PubMed] [Google Scholar]
- 3.Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313:1960–4. doi: 10.1126/science.1129139. [DOI] [PubMed] [Google Scholar]
- 4.Glaire MA, Domingo E, Sveen A, Bruun J, Nesbakken A, Nicholson G, et al. Tumour-infiltrating CD8(+) lymphocytes and colorectal cancer recurrence by tumour and nodal stage. Br J Cancer. 2019;121:474–82. doi: 10.1038/s41416-019-0540-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nosho K, Baba Y, Tanaka N, Shima K, Hayashi M, Meyerhardt JA, et al. Tumour-infiltrating T-cell subsets, molecular changes in colorectal cancer, and prognosis: cohort study and literature review. J Pathol. 2010;222:350–66. doi: 10.1002/path.2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berntsson J, Svensson MC, Leandersson K, Nodin B, Micke P, Larsson AH, et al. The clinical impact of tumour-infiltrating lymphocytes in colorectal cancer differs by anatomical subsite: a cohort study. Int J Cancer. 2017;141:1654–66. doi: 10.1002/ijc.30869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Edin S, Kaprio T, Hagstrom J, Larsson P, Mustonen H, Bockelman C, et al. The prognostic importance of CD20(+) B lymphocytes in colorectal cancer and the relation to other immune cell subsets. Sci Rep. 2019;9:19997. doi: 10.1038/s41598-019-56441-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Craig SG, Humphries MP, Alderdice M, Bingham V, Richman SD, Loughrey MB, et al. Immune status is prognostic for poor survival in colorectal cancer patients and is associated with tumour hypoxia. Br J Cancer. 2020;123:1280–88. [DOI] [PMC free article] [PubMed]
- 9.Mlecnik B, Van den Eynde M, Bindea G, Church SE, Vasaturo A, Fredriksen T, et al. Comprehensive intrametastatic immune quantification and major impact of immunoscore on survival. J Natl Cancer Inst. 2018. 10.1093/jnci/djx123. [DOI] [PubMed]
- 10.Kwak Y, Koh J, Kim DW, Kang SB, Kim WH, Lee HS. Immunoscore encompassing CD3+ and CD8+ T cell densities in distant metastasis is a robust prognostic marker for advanced colorectal cancer. Oncotarget. 2016;77:81778–90. [DOI] [PMC free article] [PubMed]
- 11.Van den Eynde M, Mlecnik B, Bindea G, Fredriksen T, Church SE, Lafontaine L, et al. The link between the multiverse of immune microenvironments in metastases and the survival of colorectal cancer patients. Cancer Cell. 2018;34:1012.e3–26.e3. doi: 10.1016/j.ccell.2018.11.003. [DOI] [PubMed] [Google Scholar]
- 12.Wang Y, Lin HC, Huang MY, Shao Q, Wang ZQ, Wang FH, et al. The Immunoscore system predicts prognosis after liver metastasectomy in colorectal cancer liver metastases. Cancer Immunol Immunother. 2018;67:435–44. doi: 10.1007/s00262-017-2094-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brunner SM, Kesselring R, Rubner C, Martin M, Jeiter T, Boerner T, et al. Prognosis according to histochemical analysis of liver metastases removed at liver resection. Br J Surg. 2014;101:1681–91. doi: 10.1002/bjs.9627. [DOI] [PubMed] [Google Scholar]
- 14.Tanis E, Julie C, Emile JF, Mauer M, Nordlinger B, Aust D, et al. Prognostic impact of immune response in resectable colorectal liver metastases treated by surgery alone or surgery with perioperative FOLFOX in the randomised EORTC study 40983. Eur J Cancer. 2015;51:2708–17. doi: 10.1016/j.ejca.2015.08.014. [DOI] [PubMed] [Google Scholar]
- 15.Bruni D, Angell HK, Galon J. The immune contexture and Immunoscore in cancer prognosis and therapeutic efficacy. Nat Rev Cancer. 2020;20:662–80. [DOI] [PubMed]
- 16.Merok MA, Ahlquist T, Royrvik EC, Tufteland KF, Hektoen M, Sjo OH, et al. Microsatellite instability has a positive prognostic impact on stage II colorectal cancer after complete resection: results from a large, consecutive Norwegian series. Ann Oncol. 2013;24:1274–82. doi: 10.1093/annonc/mds614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dienstmann R, Mason MJ, Sinicrope FA, Phipps AI, Tejpar S, Nesbakken A, et al. Prediction of overall survival in stage II and III colon cancer beyond TNM system: a retrospective, pooled biomarker study. Ann Oncol. 2017;28:1023–31. doi: 10.1093/annonc/mdx052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aasebo KO, Dragomir A, Sundstrom M, Mezheyeuski A, Edqvist PH, Eide GE, et al. Consequences of a high incidence of microsatellite instability and BRAF-mutated tumors: a population-based cohort of metastatic colorectal cancer patients. Cancer Med. 2019;8:3623–35. doi: 10.1002/cam4.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wensink GE, Elferink MAG, May AM, Mol L, Hamers PAH, Bakker SD, et al. Survival of patients with deficient mismatch repair metastatic colorectal cancer in the pre-immunotherapy era. Br J Cancer. 2020;124:399–406. [DOI] [PMC free article] [PubMed]
- 20.Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372:2509–20. [DOI] [PMC free article] [PubMed]
- 21.NCCN. Colon cancer (Version 2.2021). Access date february 18 2021. Available from https://www.nccn.org/professionals/physician_gls/pdf/colon.pdf.
- 22.Overman MJ, Lonardi S, Wong KYM, Lenz HJ, Gelsomino F, Aglietta M, et al. Durable clinical benefit with nivolumab plus ipilimumab in DNA mismatch repair-deficient/microsatellite instability-high metastatic colorectal cancer. J Clin Oncol. 2018:36:773–9. [DOI] [PubMed]
- 23.André T, Shiu K-K, Kim TW, Jensen BV, Jensen LH, Punt C, et al. Pembrolizumab in microsatellite-instability–high advanced colorectal cancer. N Engl J Med. 2020;383:2207–18. doi: 10.1056/NEJMoa2017699. [DOI] [PubMed] [Google Scholar]
- 24.Havel JJ, Chowell D, Chan TA. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat Rev Cancer. 2019;19:133–50. doi: 10.1038/s41568-019-0116-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Galon J, Mlecnik B, Hermitte F, Marliot F, Bifulco C, Lugli A, et al. MSI status plus immunoscore to select metastatic colorectal cancer patients for immunotherapies. Ann Oncol. 2018;29:x4. doi: 10.1093/annonc/mdy493.011. [DOI] [Google Scholar]
- 26.Alexander PG, McMillan DC, Park JH. The local inflammatory response in colorectal cancer - type, location or density? A systematic review and meta-analysis. Cancer Treat Rev. 2020;83:101949. doi: 10.1016/j.ctrv.2019.101949. [DOI] [PubMed] [Google Scholar]
- 27.Yang Z, Zhang M, Peng R, Liu J, Wang F, Li Y, et al. The prognostic and clinicopathological value of tumor-associated macrophages in patients with colorectal cancer: a systematic review and meta-analysis. Int J Colorectal Dis. 2020;35:1651–61. doi: 10.1007/s00384-020-03686-9. [DOI] [PubMed] [Google Scholar]
- 28.Loupakis F, Intini R, Cremolini C, Orlandi A, Sartore-Bianchi A, Pietrantonio F, et al. A validated prognostic classifier for V600EBRAF-mutated metastatic colorectal cancer: the ‘BRAF BeCool’ study. Eur J Cancer. 2019;118:121–30. doi: 10.1016/j.ejca.2019.06.008. [DOI] [PubMed] [Google Scholar]
- 29.Carethers JM, Jung BH. Genetics and genetic biomarkers in sporadic colorectal cancer. Gastroenterology. 2015;149:1177.e3–90.e3. doi: 10.1053/j.gastro.2015.06.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vedeld HM, Merok M, Jeanmougin M, Danielsen SA, Honne H, Presthus GK, et al. CpG island methylator phenotype identifies high risk patients among microsatellite stable BRAF mutated colorectal cancers. Int J Cancer. 2017;141:967–76. doi: 10.1002/ijc.30796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nunes L, Aasebo K, Mathot L, Ljungstrom V, Edqvist PH, Sundstrom M, et al. Molecular characterization of a large unselected cohort of metastatic colorectal cancers in relation to primary tumor location, rare metastatic sites and prognosis. Acta Oncol. 2020;59:417–26. doi: 10.1080/0284186X.2019.1711169. [DOI] [PubMed] [Google Scholar]
- 32.Sorbye H, Dragomir A, Sundstrom M, Pfeiffer P, Thunberg U, Bergfors M, et al. High BRAF mutation frequency and marked survival differences in subgroups according to KRAS/BRAF mutation status and tumor tissue availability in a prospective population-based metastatic colorectal cancer cohort. PLoS ONE. 2015;10:e0131046. doi: 10.1371/journal.pone.0131046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, et al. Proteomics. Tissue-based map of the human proteome. Science. 2015;347:1260419. doi: 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- 34.Aasebo K, Dragomir A, Sundstrom M, Mezheyeuski A, Edqvist PH, Eide GE, et al. CDX2: a prognostic marker in metastatic colorectal cancer defining a better BRAF mutated and a worse KRAS mutated subgroup. Front Oncol. 2020;10:8. doi: 10.3389/fonc.2020.00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Austin PC, Steyerberg EW. The number of subjects per variable required in linear regression analyses. J Clin Epidemiol. 2015;68:627–36. doi: 10.1016/j.jclinepi.2014.12.014. [DOI] [PubMed] [Google Scholar]
- 36.Goey KKH, Sorbye H, Glimelius B, Adams RA, Andre T, Arnold D, et al. Consensus statement on essential patient characteristics in systemic treatment trials for metastatic colorectal cancer: supported by the ARCAD Group. Eur J Cancer. 2018;100:35–45. doi: 10.1016/j.ejca.2018.05.010. [DOI] [PubMed] [Google Scholar]
- 37.Vittinghoff E, McCulloch CE. Relaxing the rule of ten events per variable in logistic and Cox regression. Am J Epidemiol. 2007;165:710–8. doi: 10.1093/aje/kwk052. [DOI] [PubMed] [Google Scholar]
- 38.Millen R, Hendry S, Narasimhan V, Abbott R, Croxford M, Gibbs P, et al. CD8(+) tumor-infiltrating lymphocytes within the primary tumor of patients with synchronous de novo metastatic colorectal carcinoma do not track with survival. Clin Transl Immunol. 2020;9:e1155. doi: 10.1002/cti2.1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marechal R, De Schutter J, Nagy N, Demetter P, Lemmers A, Deviere J, et al. Putative contribution of CD56 positive cells in cetuximab treatment efficacy in first-line metastatic colorectal cancer patients. BMC Cancer. 2010;10:340. doi: 10.1186/1471-2407-10-340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Loupakis F, Depetris I, Biason P, Intini R, Prete AA, Leone F, et al. Prediction of benefit from checkpoint inhibitors in mismatch repair deficient metastatic colorectal cancer: role of tumor infiltrating lymphocytes. Oncologist. 2020;25:481–7. [DOI] [PMC free article] [PubMed]
- 41.Chalabi M, Fanchi LF, Dijkstra KK, Van den Berg JG, Aalbers AG, Sikorska K, et al. Neoadjuvant immunotherapy leads to pathological responses in MMR-proficient and MMR-deficient early-stage colon cancers. Nat Med. 2020;26:566–76. doi: 10.1038/s41591-020-0805-8. [DOI] [PubMed] [Google Scholar]
- 42.Grasso CS, Giannakis M, Wells DK, Hamada T, Mu XJ, Quist M, et al. Genetic mechanisms of immune evasion in colorectal cancer. Cancer Discov. 2018;8:730–49. doi: 10.1158/2159-8290.CD-17-1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Llosa NJ, Cruise M, Tam A, Wicks EC, Hechenbleikner EM, Taube JM, et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov. 2015;5:43–51. doi: 10.1158/2159-8290.CD-14-0863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pages F, Andre T, Taieb J, Vernerey D, Henriques J, Borg C, et al. Prognostic and predictive value of the Immunoscore in stage III colon cancer patients treated with oxaliplatin in the prospective IDEA France PRODIGE-GERCOR cohort study. Ann Oncol. 2020;31:921–9. [DOI] [PubMed]
- 45.Mlecnik B, Bifulco C, Bindea G, Marliot F, Lugli A, Lee JJ, et al. Multicenter International Society for Immunotherapy of Cancer Study of the Consensus Immunoscore for the Prediction of Survival and Response to Chemotherapy in Stage III Colon Cancer. J Clin Oncol. 2020;38:3638–51. [DOI] [PMC free article] [PubMed]
- 46.Angelova M, Mlecnik B, Vasaturo A, Bindea G, Fredriksen T, Lafontaine L, et al. Evolution of metastases in space and time under immune selection. Cell. 2018;175:751.e16–65.e16. doi: 10.1016/j.cell.2018.09.018. [DOI] [PubMed] [Google Scholar]
- 47.Pancione M, Giordano G, Remo A, Febbraro A, Sabatino L, Manfrin E, et al. Immune escape mechanisms in colorectal cancer pathogenesis and liver metastasis. J Immunol Res. 2014;2014:686879. doi: 10.1155/2014/686879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Galon J, Bruni D. Tumor immunology and tumor evolution: intertwined histories. Immunity. 2020;52:55–81. doi: 10.1016/j.immuni.2019.12.018. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data can be made available upon reasonable request.