

Abstract
Broadly distributed species must cope with diverse and changing environmental conditions, including various forms of stress. Cosmopolitan populations of Drosophila melanogaster are more tolerant to oxidative stress than those from the species’ ancestral range in sub-Saharan Africa, and the degree of tolerance is associated with an insertion/deletion polymorphism in the 3′ untranslated region of the Metallothionein A (MtnA) gene that varies clinally in frequency. We examined oxidative stress tolerance and the transcriptional response to oxidative stress in cosmopolitan and sub-Saharan African populations of D. melanogaster, including paired samples with allelic differences at the MtnA locus. We found that the effect of the MtnA polymorphism on oxidative stress tolerance was dependent on the genomic background, with the deletion allele increasing tolerance only in a northern, temperate population. Genes that were differentially expressed under oxidative stress included MtnA and other metallothioneins, as well as those involved in glutathione metabolism and other genes known to be part of the oxidative stress response or the general stress response. A gene coexpression analysis revealed further genes and pathways that respond to oxidative stress including those involved in additional metabolic processes, autophagy, and apoptosis. There was a significant overlap among the genes induced by oxidative and cold stress, which suggests a shared response pathway to these two stresses. Interestingly, the MtnA deletion was associated with consistent changes in the expression of many genes across all genomic backgrounds, regardless of the expression level of the MtnA gene itself. We hypothesize that this is an indirect effect driven by the loss of microRNA binding sites within the MtnA 3′ untranslated region.
Keywords: adaptation, gene expression, metallothionein, population genetics, transcriptomics
Introduction
Adaptation of a species to changes in environmental conditions caused by seasonal variation, climate change, or range expansion is necessary for survival and proliferation. Understanding the process of such adaptation is an intrinsic goal of evolutionary biologists, however, a major challenge is the identification of adaptive traits and their underlying molecular and genetic bases. Drosophila melanogaster, due to its experimental versatility, well-detailed genetics, and broad geographic distribution, has become a leading model organism for studying the molecular basis of adaptation and environment-based selection. Previous work on this topic typically has involved genomic comparisons between D. melanogaster populations from the ancestral (sub-Saharan Africa) and derived (cosmopolitan) species ranges (Harr et al. 2002; Ometto et al. 2005; Hutter et al. 2008; Glaser-Schmitt and Parsch 2018; Voigt et al. 2019), and may include an environmental gradient such as a latitudinal cline (Sezgin et al. 2004; Bergland et al. 2016; Juneja et al. 2016; Cogni et al. 2017; Erickson et al. 2020).
One example of a clinally varying genetic polymorphism is a 49-bp insertion/deletion (indel) polymorphism in the 3′ untranslated region (UTR) of the Metallothionein A (MtnA) gene (Catalán et al. 2016). The deletion represents the derived allele: it is rare in sub-Saharan African populations and absent in closely related Drosophila species (Lange et al. 1990; Theodore et al. 1991; Stephan et al. 1994). In non-African populations, the deletion is common and increases in frequency with distance from the equator in Europe, North America, and Australia (Catalán et al. 2016; Ramnarine et al. 2019). Functionally, the deletion is associated with increased MtnA expression and greater oxidative stress tolerance (Catalán et al. 2016; Ramnarine et al. 2019). These observations suggest that the MtnA deletion is selectively favored, at least in temperate regions, due to a beneficial effect of providing protection against oxidative stress. Another cosmopolitan polymorphism, Bari-Juvenile hormone epoxy hydrolase (Bari-Jheh), which is a transposable element insertion associated with changes in the regulation of the Juvenile hormone epoxide hydrolase (Jheh) genes and increased oxidative stress tolerance (González et al. 2009; Guio et al. 2014), is also present at high frequency in temperate populations. In Drosophila species, temperate populations have been shown to be more cold tolerant than ancestral, tropical populations (Svetec et al. 2011; von Heckel et al. 2016; Königer and Grath 2018). Given that both the MtnA deletion and Bari-Jheh insertion are more common in regions that experience cold temperatures and large seasonal fluctuations in temperature, it is possible that thermal variability contributes directly to oxidative stress (Joanisse and Storey 1996, 1998; Harrison et al. 2001; Klok et al. 2004; Lalouette et al. 2011; MacMillan et al. 2012), and/or that these two forms of stress induce a common response pathway.
In this study, we examine oxidative stress tolerance and the transcriptional response to oxidative stress in inbred lines derived from cosmopolitan (Germany, Cyprus, and Malaysia) and sub-Saharan African (Zambia) populations of D. melanogaster. For the cosmopolitan populations, we compare paired samples that differ in the presence of the MtnA 3′ UTR deletion, including nearly isogenic lines from Germany and Cyprus in which the remainder of the genome has been homogenized. We find that the MtnA deletion is associated with increased oxidative stress tolerance in the German background, but not the Cypriot or Malaysian background, which highlights the importance of genetic background on the physiological effects of the MtnA deletion. We detected a strong transcriptional response to oxidative stress in all lines, including the up-regulation of MtnA expression, which is consistent with it playing a protective role against oxidative stress. There was significant overlap between the genes up-regulated during oxidative stress and those up-regulated by cold stress, although there were also many genes that responded specifically to one type of stress or the other. Across all backgrounds the presence of the MtnA deletion led to a greater transcriptional response to oxidative stress, which may be caused by the loss of microRNA (miRNA) binding sites within the deleted region of the 3′ UTR, increasing the availability of the miRNAs to regulate other genes.
Materials and methods
Fly culturing and establishment of nearly isogenic lines
Isofemale lines from Munich, Germany (M9 and M12) and Nicosia, Cyprus (C2) were established from wild-caught flies collected in 2014 (Kapun et al. 2020). Each line was derived from a single female and maintained by mass sib–sib mating at each generation (in vials with approximately 50–100 flies transferred per generation). After the initial establishment of the isofemale lines, the MtnA 3′ UTR deletion was found to segregate within these lines, as determined by PCR-genotyping of individual flies (Catalán et al. 2016). These lines were maintained in the same way for an additional 50–60 generations. Subsequently, individual males and females within each isofemale line were mated to generate new inbred lines homozygous for either the deletion or the nondeletion allele. The deletion status of each new inbred line was verified by genotyping both parents, as well as a pool of 10 of their offspring. We additionally examined two isofemale lines from Kuala Lumpur, Malaysia, which had previously been shown to differ in their MtnA 3′ UTR allele (KL1, deletion; KL2, nondeletion) (Catalán et al. 2016), and four isofemale lines from the ancestral range of the species in Siavonga, Zambia (ZI197, ZI254, ZI273, and ZI418) (Pool et al. 2012). All of the Zambian lines had the ancestral, nondeletion MtnA allele (Catalán et al. 2016; Ramnarine et al. 2019). In the following, lines with the MtnA deletion are indicated with a “Δ” at the end of their line name. All stocks were maintained on cornmeal-yeast-molasses medium at 21°C and a 14 h light/10 h dark cycle.
Oxidative stress tolerance assays
Previously, using Drosophila genetic reference panel (DGRP) lines (Mackay et al. 2012) we detected a significant association between the MtnA deletion and increased tolerance to menadione sodium bisulfite (MSB)-induced stress (Ramnarine et al. 2019). Therefore, to determine the effects of genetic background and the MtnA 3′ UTR deletion on oxidative stress tolerance, we assayed the survivorship of adult flies of each line after exposure to MSB (Sigma-Aldrich, Munich, Germany). For each line, males (age 4–6 days) were separated from culture and sorted into replicate groups of 20 flies per vial on standard food (cornmeal-yeast-molasses medium) for 1 day to acclimate. On the following day, 3–5 replicates from each line were transferred to “control” vials containing standard fly food and 5–16 replicates from each line were transferred to “stress” vials containing 75 mM MSB-dosed cornmeal-yeast-molasses medium. Assays were performed in tolerance chambers at 25°C with a 14 h light/10 h dark cycle. Mortality was recorded as the proportion of dead flies in the stress treatment compared to the control after 48 h.
RNA extraction and sequencing
To measure the transcriptional response to oxidative stress, we repeated the MSB stress tolerance assays described above. The German, Malaysian, and Cypriot lines were exposed to stress (or control) condition for 24 h and then total RNA was extracted from two biological replicates per line. Each replicate used 60 whole heads from living male flies. The Zambian flies under both treatments were exposed for a shorter time (18–20 h) to compensate for their high mortality and total RNA was extracted from heads in two replicates per line as described above. This design resulted in a total of 48 samples (12 lines × 2 treatments × 2 biological replicates) that were used for transcriptome profiling. Total RNA was extracted using the RNeasy Mini Kit (Qiagen, Hilden, Germany) following the manufacturer’s protocol. Poly-A mRNA purification, reverse transcription, and high-throughput sequencing were performed by Novogene (Cambridge, UK) using a Novaseq 6000 sequencer (Illumina, San Diego, CA, USA) to generate 150-bp paired-end reads. From the RNA-sequencing we obtained between 29.3 and 67.6 million reads per library, and an average of 97% of these reads were successfully mapped to the D. melanogaster transcriptome (FlyBase, release 6.31) (Thurmond et al. 2019) using NextGenMap (version 0.5.5) (Sedlazeck et al. 2013). The resulting alignments were used to estimate pairwise sequence divergence between strains, considering only sites with a minimum coverage of 20x in both strains.
Detection of differentially expressed genes
Differentially expressed genes were called with DESeq2 (version 1.28.1) (Love et al. 2014) as implemented in R (version 4.0.0) (R Core Team 2020). Significant differences between control and stress samples were determined using a Wald test and the P-values subsequently corrected for multiple testing according to Benjamini and Hochberg (1995). Genes with an adjusted P-value less than 0.05 were considered to be differentially expressed. Initially we performed unsupervised clustering (DESeq2 design: “∼population + genotype + treatment”) on the variance-stabilized data and visualized the results with a principal component analysis (PCA). The PCA revealed an extreme outlier, which was one of the Munich (M9) nondeletion stress replicates (Supplementary Figure S1). This sample was excluded from further analyses. To identify genes that shared a common change in expression after MSB exposure across all genetic backgrounds, we focused on the treatment factor and applied a design using only “∼treatment.” Lists of overall up- and downregulated differentially expressed genes associated to treatment (Supplementary Tables S1 and S2) were then generated for subsequent gene ontology (GO) analysis. These lists were also used in comparisons with two external transcriptomic datasets involving cold stress (von Heckel et al. 2016) and oxidative stress (Weber et al. 2012). For this, the log2 fold-change values of the genes matching the list of genes associated to oxidative stress (Supplementary Figures S2 and S3) and cold tolerance (Supplementary Figures S4 and S5) were compared. The significance of the overlap between the oxidative stress response and the cold response genes was determined by a hypergeometric test.
Gene ontology analysis
GO analysis was performed using FlyMine (Lyne et al. 2007) to determine the number of significantly up-regulated and downregulated genes matching a GO term, and REVIGO (Supek et al. 2011) to merge redundant GO terms. A Holm–Bonferroni correction was applied to calculate adjusted P-values (Supplementary Tables S3 and S4). Information from FlyBase (release FB2019_06) (Thurmond et al. 2019) and AMIGO (Carbon et al. 2009) was used to cross-reference genes matching significant GO terms with their annotated function.
Weighted gene coexpression network analysis
To construct gene networks and identify highly relevant (hub) genes and pathways involved in the response to oxidative stress, we applied weighted gene coexpression analysis (WGCNA) (Zhang and Horvath 2005) as implemented in the R package “WGCNA” (version 1.69) using R (version 3.6.3) (Langfelder and Horvath 2008). The WGCNA approach allows for the detection of expression modules or pathways in which many genes show small but consistent changes and, therefore, complements the analysis of individual genes (GO analysis) by means of gene enrichment analysis. In the WGCNA workflow, normalized expression data are used to cluster genes that show similar expression across samples, indicating that the expression of the clustered genes is correlated. Such genes are referred to as being topologically correlated. Coexpression modules are defined by hierarchical clustering of these topological clusters modules. By convention, each module is assigned a color and genes that do not show any coexpression with other genes are placed in the “gray” module. After module identification, the module expression (eigengene) is used to associate the module with categorical information. We performed WGCNA with two design formulas for differential gene expression analysis: (1) considering only the effect of treatment (∼treatment) to build on the identified genes and pathways identified in the GO analysis, and (2) incorporating the effect of the MtnA indel (∼genotype + treatment + genotype: treatment) to identify differentially expressed genes in response to oxidative stress associated to the deletion background relative to the nondeletion background.
Comparison of MtnA deletion and nondeletion lines
To determine how the indel polymorphism in the MtnA 3′ UTR influenced gene expression and the response to oxidative stress, we applied the DESeq2 “∼treatment” design (described above) within each cosmopolitan background separately and then performed the following pairwise comparisons: deletion stress vs deletion control, nondeletion stress vs nondeletion control, deletion stress vs nondeletion stress and deletion control vs nondeletion control (Supplementary Table S6). Adjusted P-values were determined as described above, and genes with an adjusted P < 0.05 were considered as differentially expressed between deletion and nondeletion lines within each treatment. This approach was particularly suited to the comparison of the nearly isogenic lines (Germany and Cyprus) and paired population samples (Malaysia), as it allowed for the comparison of MtnA genotypes without the extensive background variation present in between-strain or between-population comparisons.
Expression analysis of potential miRNA target genes
To determine if some of the expression differences observed between MtnA deletion and nondeletion lines could be caused by the differential binding of miRNAs, we examined the expression of conserved target genes associated with miRNAs predicted to bind within the 49-bp region that is deleted in MtnA 3′ UTR (Catalán et al. 2016). The target genes of these miRNAs were identified using TargetScanFly (version 7.2) (Agarwal et al. 2018). Because miRNAs are typically involved in the negative regulation of gene expression, we focused on the predicted target genes that were downregulated under oxidative stress. We then examined the expression of these genes using a direct contrast in DESeq2 (version 1.28.1) (Love et al. 2014) between deletion and nondeletion lines in the cosmopolitan backgrounds in response to stress (the Zambian lines were excluded because they were fixed for the nondeletion allele). A paired sample Wilcoxon test was applied to test for a directional disparity in the expression of these genes between deletion and nondeletion lines. Finally, the resulting list of genes was tested for functional enrichments using FlyMine (Lyne et al. 2007).
Results
Variation in oxidative stress tolerance among lines and populations
To assay oxidative stress tolerance of the different genotypes and populations, adult males (4–6 days post-eclosion) were placed on food containing 75 mM MSB and their mortality recorded over 48 h. As a control, males of the same age were placed on standard food. Since we observed no mortality on the standard food, we assume that any mortality observed on the MSB food is a consequence of MSB-induced oxidative stress and not any other aspect of the rearing conditions. A comparison of nearly isogenic lines differing specifically at the MtnA locus found that the 3′ UTR deletion was associated with increased oxidative stress tolerance in the German background, but not in the Cypriot background (Figure 1). When comparing the two isofemale lines from Malaysia, the MtnA deletion was associated with a reduction in oxidative stress tolerance. Thus, the effect of the MtnA 3′ UTR deletion on oxidative stress tolerance appears to be highly dependent on the genomic background and is variable among cosmopolitan populations. The Zambian lines, all of which had the ancestral (nondeletion) MtnA allele, were relatively susceptible to oxidative stress. However, there were differences in mortality among the Zambian lines, which suggests that genetic variation for oxidative stress tolerance is also segregating within the ancestral range of the species.
Figure 1.
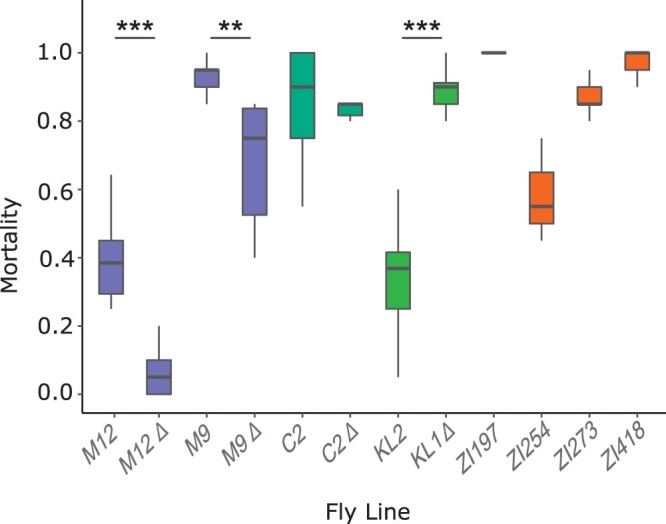
Mortality after 48 h of exposure to MSB-dosed food in lines originating from: Munich, Germany (M9 and M12); Nicosia, Cyprus (C2); Kuala Lumpur, Malaysia (KL), and Siavonga, Zambia (ZI). Differences between MtnA genotypes within the same background were tested with a Wilcoxon test. **P < 0.01, ***P < 0.001.
The transcriptional response to oxidative stress
To investigate the transcriptional response to oxidative stress, we performed high-throughput RNA sequencing (RNA-seq) on the heads of male flies exposed to MSB-dosed and standard food. Across all samples, we found a strong transcriptional response to MSB treatment, with 45% of all expressed genes being differentially expressed in response to oxidative stress (false discovery rate = 5%). In total, 3434 (25%) genes were up-regulated after stress treatment (Supplementary Table S1), while 2839 (20%) were downregulated (Supplementary Table S2). A PCA showed that the majority of the variance (53%) could be explained by the treatment (PC1) (Figure 2), while the second principal component (PC2) mainly separates the samples by population and accounts for only 8% of the variance.
Figure 2.
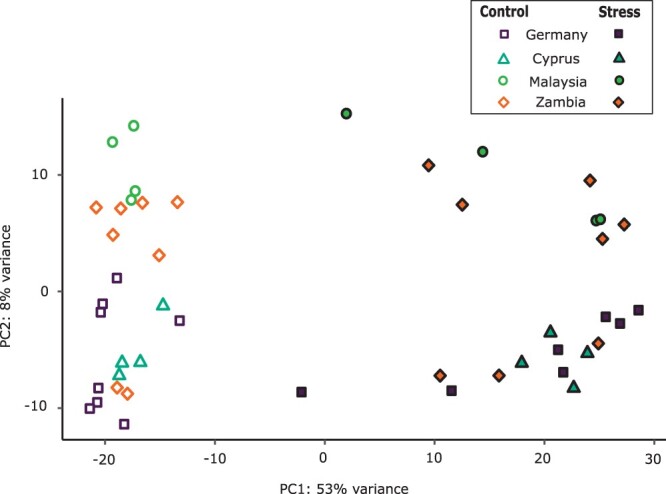
PCA plot showing the amount of variation explained by treatment and population of origin: Munich, Germany (M9 and M12); Nicosia, Cyprus (C2); Kuala Lumpur, Malaysia (KL), and Siavonga, Zambia (ZI). Filled symbols represent the MSB-stress samples, while open symbols represent the control samples.
Functional enrichment of oxidative stress response genes
A GO enrichment analysis of the genes showing a general up- or downregulation of expression in stressed samples relative to the controls revealed various processes broadly related to metabolism and stress response (Supplementary Tables S3 and S4), with the strongest enrichments (ranked by P-value) being for categories related to metabolic processes (Table 1). In addition, we observed a large number of genes in the categories “proteolysis” (up-regulated; P = 2.51E-05) and “animal organ development” (downregulated; P = 3.23E-11).
Table 1.
Top five GO enrichment terms for genes up- or downregulated under oxidative stress
| Regulation | GO term | Description | P-value | Genes |
|---|---|---|---|---|
| Up | 0051186 | Cofactor metabolic process | 1.52E−08 | 98 |
| Up | 0006749 | Glutathione metabolic process | 3.08E−08 | 36 |
| Up | 0006950 | Response to stress | 2.81E−06 | 393 |
| Up | 0006575 | Cellular modified amino acid metabolic process | 4.78E−06 | 48 |
| Up | 0006508 | Proteolysis | 2.51E−05 | 301 |
| Down | 0044281 | Small molecule metabolic process | 3.12E−13 | 264 |
| Down | 0046034 | ATP metabolic process | 2.75E−11 | 77 |
| Down | 0048513 | Animal organ development | 3.23E−11 | 368 |
| Down | 0032543 | Mitochondrial translation | 1.78E−08 | 56 |
| Down | 0006091 | Generation of precursor metabolites and energy | 1.18E−07 | 91 |
A Holm–Bonferroni correction was applied to the P-values.
Expression of candidate genes associated with oxidative stress tolerance
In a previous quantitative trait locus study, seven genes (fog, nACRα-30D, ena, rg, CG9650, Eip75B, and hbn) that were significantly associated with oxidative stress susceptibility were functionally validated by comparing wild-type and mutant alleles (Weber et al. 2012). Of these seven genes, only one was significantly differentially expressed in our dataset (Eip75B), and it shows a relatively small difference in expression between treatments (log2 fold-change = 0.27; Supplementary Figure S2). Thus, there is little evidence that these genes play a role in the oxidative stress response of our samples, at least at the transcriptional level. We further examined the expression of genes associated to epoxide hydrolase, specifically the Jheh genes. The Bari-Jheh transposon, which is present at high frequencies in temperate regions, has been shown to have a cis-regulatory effect on the expression of nearby genes, leading to up-regulation of Jheh1 and Jheh2 and downregulation of Jheh3 under oxidative stress conditions (Guio et al. 2014). In our dataset, we observe the same relationship among these three genes in response to MSB (Supplementary Figure S3), which further supports their functional role in mediating oxidative stress tolerance.
Comparison of genes induced by oxidative stress and cold stress
Previous studies suggested that there may be a common response to both cold and oxidative stress (Lalouette et al. 2011; MacMillan et al. 2016). To investigate this possibility, we compared the genes responding to oxidative stress in our study to those responding to cold stress in a previous study that compared the transcriptional response of European and African strains of D. melanogaster (von Heckel et al. 2016). For this, we focused on a time point of 90 min post-recovery after a 7-h cold shock, which shows the strongest response in terms of gene expression (Colinet et al. 2010; von Heckel et al. 2016). At this time point, 1535 genes were up-regulated and 1979 genes were downregulated in response to cold shock (von Heckel et al. 2016). Of these genes, a significant proportion overlaps with the differentially expressed genes induced by MSB as determined by a hypergeometric test, including 881 up-regulated (P = 1.4E-187) and 832 shared downregulated (P = 1.7E-126) genes. The study of von Heckel et al. (2016) identified 16 genes that respond to cold shock in a population-specific way (Europe vs Africa), indicating a genotype-by-environment interaction. In our data, 11 of these 16 genes showed a significant change in expression under oxidative stress (Supplementary Figure S4). Furthermore, of the 47 cold tolerance candidate genes identified by von Heckel et al. (2016), 34 are significantly differentially expressed under oxidative stress in our dataset (Supplementary Figure S5).
Coexpressed gene modules in response to oxidative stress
Modules of coexpressed genes were identified with weighted gene coexpression network analysis (WGCNA) as implemented in the R package WGCNA by Langfelder and Horvath (2008). WGCNA called 13 modules that contained 7030 genes in total, while an additional 6830 genes were gathered in the gray module, which contains genes that have not been clustered in any module (i.e., unassigned genes; Figure 3). Using an ANOVA, we determined that five modules (turquoise, blue, green, pink, and magenta) out of the original 13 modules showed significant association with treatment (P < 0.001). Two of these modules (turquoise and blue) contained the majority of the associated genes. The turquoise module (3052 genes) corresponds to up-regulation of autophagy categories as well as heat-shock response and programmed cell death, which are components of the generic extra- and/or intra-cellular stress response (Figure 3). Expectedly, there is a strong interconnection of the categories in this module to several heat shock protein (HSP) genes (Supplementary Figure S6A). The blue module (2007 genes) is associated with several metabolic processes that are downregulated in response to stress (Figure 3). Two genes within this module exhibit a particularly large negative fold-change: CG16700 and MFS9 (Major Facilitator Superfamily Transporter 9), both of which are involved in transmembrane activity (Gaudet et al. 2011). An additional three genes within this module also stand out because they are connected to multiple sub-modules, and therefore are likely to affect more pathways: Got1 (Glutamate oxaloacetate transaminase 1), aralar1 (CG2139; a member of the mitochondrial carrier family of genes), and CG18003 (Hao). Got1 and aralar1 are functionally involved in glutamate activity (Gaudet et al. 2011) and connect the categories involving metabolism of carbohydrates, glucose, and amino acids. CG18003 (Hao), which is predicted to have (S)-2-hydroxy-acid oxidase activity, connects fatty acid metabolism and amino acid metabolism (Supplementary Figure S6B).
Figure 3.

Weighted gene coexpression network analysis (WGCNA): (A) Number of genes in the final merged modules, (B) Reactome dotplot of enriched terms for the turquoise module, (C) Reactome dotplot of enriched terms for the blue module. P-values (“p.adjust”) are adjusted according to the method of Benjamini and Hochberg (1995). The number of genes in a pathway is encoded in the dot size (count).
The effect of the MtnA indel polymorphism on gene expression
Using the full dataset, we detected an overall increase in the expression of metallothionein genes in the oxidative stress samples relative to the controls, with highly significant increases for MtnA, MtnD, and MtnE (Figure 4). To examine if the general increase in MtnA could be associated to the indel polymorphism we assessed the expression within each background. We found that the up-regulation of MtnA occurred regardless of whether the deletion or nondeletion variant was present in the 3′ UTR (Figure 5), suggesting that MtnA plays a general and conserved role in the response to oxidative stress. To better determine the effect of the MtnA 3′ UTR indel on expression, we compared the sets of differentially expressed genes within pairs of nearly isogenic lines from Germany (M12 and M9) and Cyprus (C2) that had otherwise homogenized genetic backgrounds, as well as two isofemale lines from Malaysia (KL1 and KL2) that were homozygous for the deletion and nondeletion alleles, respectively. The nearly isogenic lines showed greatly reduced differentiation compared to isofemale lines in both DNA sequence and gene expression (Figure 6), although the extent of this reduction varied among the nearly isogenic pairs. For one of the Munich pairs (M12) genetic differentiation was less than 1% of that between isofemale lines, while for the other pair (M9) it was about 30%. For the Cyprus pair (C2) genetic differentiation was about 4% of that between isofemale lines. In contrast, the isofemale lines from Malaysia (KL1 and KL2), which differ for the MtnA indel, also differed greatly throughout their genomes, showing levels of sequence and expression divergence expected for independent, isofemale lines (Figure 6; Supplementary Table S5). As might be expected, there was considerable overlap among the genes responding to stress in the deletion and nondeletion lines in all backgrounds (Figure 7); however, there were significantly more differentially expressed genes (both up- and downregulated) in the deletion lines relative to the corresponding nondeletion lines in all comparisons (Supplementary Table S6). Thus, the MtnA 3′ UTR deletion appears to have a consistent effect on the transcriptional response to oxidative stress in a way that is independent of MtnA expression (Figure 4) and stress tolerance (Figure 1). A potential explanation for this is that the 3′ UTR deletion may eliminate miRNA binding sites (Catalán et al. 2016), freeing up the interacting miRNAs to regulate the expression of other genes. To test this, we examined the expression of target genes regulated by miRNAs predicted to bind within the 49-bp deletion region in the MtnA 3′ UTR (Catalán et al. 2016). For all of these miRNAs, there was a significant excess of downregulated target genes under oxidative stress (Figure 7), which is consistent with the greater availability of these miRNAs leading to increased negative regulation of their target genes. Furthermore, the downregulated target genes were enriched for those involved in regulatory and transcriptional processes (Supplementary Table S7). Thus, the observed excess of up-regulated genes in the deletion background (Figure 7) could be an indirect effect of miRNA regulation (i.e., the downregulation of miRNA target genes leading to up-regulation of other genes that interact with them).
Figure 4.

MSB-induced log2 fold-change in expression (stress/control) of the metallothionein group genes MtnA, MtnB, MtnD, MtnE, and MtnF. The points within each violin plot represent the values of the different inbred lines. Values for MtnC and some data points for MtnB are not included, as expression was below the detection threshold. Asterisks indicate the adjusted P-value from a Wald test as applied in DESeq2 in stress vs control comparisons across all lines. ***P < 0.001.
Figure 5.
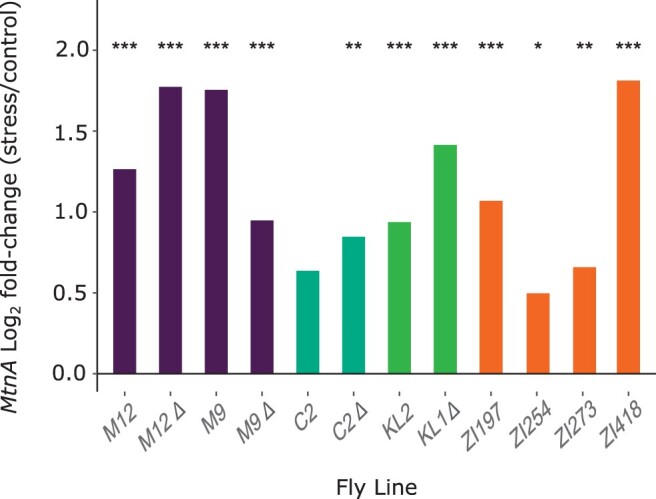
Log2 fold-change in MtnA expression induced by oxidative stress in different backgrounds: Munich, Germany (M9 and M12); Nicosia, Cyprus (C2); Kuala Lumpur, Malaysia (KL), and Siavonga, Zambia (ZI). Lines homozygous for the MtnA 3′ UTR deletion are denoted by “Δ.” Asterisks indicate the adjusted P-value from a Wald test as applied in DESeq2. * P < 0.05, **P < 0.01, ***P < 0.001.
Figure 6.
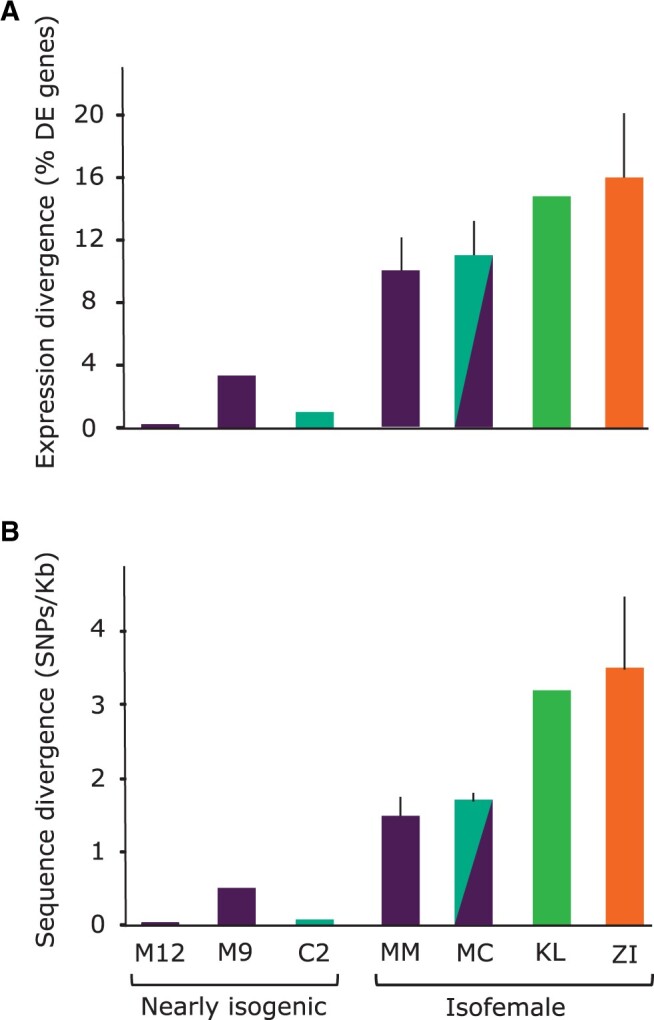
Comparison of variation between nearly isogenic lines from Munich, Germany (M9, M12) and Nicosia, Cyprus (C2), and isofemale lines from Kuala Lumpur, Malaysia (KL), and Siavonga, Zambia (ZI). (A) Gene expression divergence shown as the percentage of differentially expressed (DE) genes at a false discovery rate of 5%. (B) DNA sequence divergence in transcribed regions. “MM” indicates a comparison between the M12 and M9 lines, while “MC” indicates a comparison between both Munich lines and C2. In cases where multiple comparisons were possible, the mean is plotted with error bars indicating the standard deviation.
Figure 7.

Venn diagram showing the number of shared differentially expressed genes under oxidative stress in deletion (“Δ”) and nondeletion lines in the (A) M12, (B) M9, (C) C2, and (D) KL genetic backgrounds.
Coexpression modules that respond differently to oxidative stress between MtnA genotypes
Since we observed that the expression of many genes may be affected by the MtnA 3′ UTR indel polymorphism, we modified our WGCNA design to include genotype (deletion or nondeletion) as a factor within treatment. This design revealed modules of coexpressed genes induced by oxidative stress in the deletion background relative to the nondeletion background. Similar to the treatment only design, the major categories were turquoise (autophagy and general stress response) (Supplementary Figure S7A) and blue (metabolism) (Supplementary Figure S7B). However, we observed a decrease in the number of genes associated to each module and an overall reduction in the range of fold-changes, with many genes showing only small expression differences (log2 fold-change close to 0) (Supplementary Figure 7, A and B). Our analysis also uncovered two genes that showed an amplification of log2 fold-change compared to the treatment only design: Prosα4T2 (Proteasome alpha 4 subunit, Testis-specific 2), which was downregulated (blue module), and CG31792 (ATPase-coupled transmembrane transporter activity), which was up-regulated (turquoise module), in the deletion background under oxidative stress.
Discussion
The effect of the MtnA indel polymorphism on oxidative stress tolerance
The MtnA 3′ UTR deletion polymorphism previously has been associated with an increase in MtnA expression which, in turn, is associated with increased tolerance to oxidative stress (Catalán et al. 2016; Ramnarine et al. 2019). The previous studies, however, compared isofemale lines that differed not only in their MtnA alleles, but also at many loci throughout the genome. This approach makes it impossible to isolate the specific effect of the MtnA indel polymorphism on oxidative stress tolerance, or to detect potential interactions with other variants in the genomic background. The latter may be especially relevant, as oxidative stress tolerance is known to be a highly polygenic trait (Weber et al. 2012; Guio et al. 2014; Harrison et al. 2020). In the current study, we measured oxidative stress tolerance using lines from Germany and Cyprus in which the genetic background, aside from the MtnA locus, was homogenized through inbreeding. While this homogenization was successful in reducing genetic and expression variation (Figure 6), some residual polymorphism remained, especially in one of the German lines. In addition, we measured oxidative stress tolerance in a pair of inbred lines from Malaysia that differed in their MtnA 3′ UTR allele, but did not have a homogenized genomic background, and four independent isofemale lines from Zambia that had the ancestral, nondeletion MtnA allele.
Our results indicate that the effect of the MtnA deletion on oxidative stress tolerance is highly dependent of the genomic background. In the two pairs of nearly isogenic lines from Germany, the deletion lines showed a significant reduction in mortality under oxidative stress compared to the corresponding nondeletion lines (Figure 1). Although there was a relatively high amount of residual polymorphism between the paired lines in one of the German backgrounds (M9), the concordant results observed in the two backgrounds suggest a consistent effect of the MtnA 3′ UTR indel polymorphism on oxidative stress tolerance. However, we cannot rule out a contribution from other polymorphic loci in the genome, especially in the M9 background. In contrast to the German background, the deletion had no detectable effect on oxidative stress tolerance in the Cypriot background, and was associated with a significant increase in mortality under stress in the Malaysian background (Figure 1). Thus, we find evidence for sign epistasis, in which the deletion has opposite effects depending on the genetic background. Note, however, that because the Malaysian background is not homogenized and there is considerable genetic and expression variation between the Malaysian lines (Figure 6), we cannot attribute the observed difference in stress tolerance solely to the MtnA indel polymorphism. Interestingly, the benefit of the deletion allele appears to be correlated with its frequency in the tested populations, with the greatest benefit in Germany (frequency = 0.91), no benefit in Cyprus (frequency = 0.65), and a significant cost in Malaysia (frequency = 0.45) (Catalán et al. 2016; Ramnarine et al. 2019). Thus, it may be that the beneficial effect of the MtnA deletion is dependent on an interaction with alleles at other loci that are in high frequency in temperate populations, which could contribute to its clinal distribution (Catalán et al. 2016), but confirmation of this trend would require additional experiments with a larger number of populations. The deletion is absent from the ancestral species range in sub-Saharan Africa, aside from rare alleles that appear to be caused by recent admixture with non-African populations (Ramnarine et al. 2019), suggesting that it conveys no selective advantage in sub-Saharan populations.
Transcriptomic data implicate several metabolic processes in oxidative stress response
The genes that show a consistent response to MSB exposure across all populations and genotypes can be considered part of the general oxidative stress response. In the following, we discuss the genes, functional categories, and expression modules that are part of this general response, as revealed by our transcriptomic analyses.
Metallothioneins have been proposed to function as free radical scavengers and as antioxidants against reactive oxygen and nitrogen species (Thornalley and Vasák 1985; Abel and de Ruiter 1989; Cai et al. 2000). These genes are typically thought to have distinctive but overlapping functions involving metal ion homeostasis and detoxification (Egli et al. 2006; Atanesyan et al. 2011; Gaudet et al. 2011; Luo et al. 2020). In D. melanogaster, expression changes in MtnA have been demonstrated to be associated with oxidative stress tolerance (Catalán et al. 2016; Ramnarine et al. 2019). A new finding of the current study is that the expression of MtnA and two other metallothionein genes (MtnD and MtnE) is induced by oxidative stress. Furthermore, MtnD and MtnE show an even higher induction of expression under stress than MtnA, while MtnB, MtnC, and MtnF show no sign of induction (Figure 4). These results suggest that the members of the metallothionein family have diversified in both function and regulation.
GO enrichment analysis revealed a strong connection between oxidative stress and metabolism, specifically redox reactions involving glutathione (GSH), and particularly glutathione-S-transferase genes (GSTs). Redox status is often measured as the ratio of GSH (reduced state) to glutathione disulfide (GSSG; oxidized state) and used as a marker for cellular toxicity due to oxidative stress (Bonilla et al., 2006). Therefore, GSTs may prove to be of further interest because of their functional association to cellular protection against oxidative stress-inducing agents (Mockett et al. 1999; Whitworth et al. 2005; Hayes and McLellan 2009). In this context, the mechanism by which MSB affects biological systems may be particularly relevant, as it involves reactive oxygen species generation through quinone redox cycling and/or electrophilic attack involving cellular nucleophiles, both of which are modulated by oxidation or conjugation of glutathione (Akman et al. 1985; Baker et al. 1996; Wallace et al. 2005; Elgawish et al. 2013). Treatment with menadione has therefore been associated to oxidative damage due to lipid peroxidation, depletion of cellular glutathione, and variation in expression of glutathione metabolic genes (Chang et al. 1992; Kavitha and Chandra 2014; Thomas et al. 2016). In addition, changes in the expression of GSTs may be linked to cold tolerance. For example, Glutathione S-transferase E8 (GstE8), which was identified in this study, was also identified as a cold tolerance candidate and as a genotype-environment interaction gene by von Heckel et al. (2016). In fact, many of the differentially expressed genes identified by von Heckel et al. (2016) and by our study overlap, which provides further evidence for there being shared pathways between cold tolerance and oxidative stress tolerance. It has been previously proposed that at the metabolic level, cold tolerance adaptations may involve changes in neighboring pathways such as those related to redox balance (MacMillan et al. 2016). In previous studies, MtnA expression showed a variable response to cold stress (MacMillan et al. 2016; von Heckel et al. 2016; Königer and Grath 2018; Teets and Hahn 2018), which contrasts with the uniform induction of MtnA under oxidative stress. This suggests that at least some of the metallothioneins respond specifically to oxidative stress and are not induced by stress in general. Nonetheless, the up-regulated GO term “response to stress” supports the idea that oxidative stress is linked to other stress responses. Within this category, we examined the top 20 differentially expressed genes with the largest log2 fold-change. Of these, 10 belong to the HSP gene family. The other genes include: Ets21C, Fst, Gadd45, mthl7, MtnE, TotB, Cht9, GstE1, Arc1, and CG31659. With the exception of GstE1, these genes are not known for their association to oxidative stress specifically but are involved in varied pathway responses to diverse stress stimuli. Notably, among these genes is one of the few genes implicated specifically in cold tolerance, Fst (Frost) (Colinet et al. 2010), which further suggests a mechanistic relationship between cold stress and oxidative stress.
Among the GO terms with the lowest P-values and the greatest downregulation under oxidative stress are a small number of genes related to (1) regulatory processes: Thymidylate synthase (Ts) and cardinal (cd); (2) oxidation-reduction and catabolic processes: beta-hydroxy acid dehydrogenase (Had1), Stearoyl-CoA 9-desaturase (CG9747), Alcohol dehydrogenase (Adh), and Hexokinase-C (Hex-C); and (3) tracheal system development: Rebuf (reb), filzig (flz), and breathless (btl). Notably, previous work has indicated that several genes involved in tracheal system and/or cuticular development were up-regulated with cold acclimation (MacMillan et al. 2016). Thus, our observation that genes associated with tracheal system development are downregulated under oxidative stress may imply that the physiological response mechanisms differ between these two stresses. In addition, we observed an up-regulation of genes that were associated to the GO term “proteolysis.” Since several of the top differentially expressed genes in this category are functionally associated to cleavage of peptide bonds and have endopeptidase (e.g., Jon25Bii) or protease (e.g., snk) activity (Gaudet et al. 2011), we hypothesize that the up-regulation of these genes is in response to damage or modification of cellular proteins (Finkel and Holbrook 2000; Das et al. 2001; Pickering et al. 2013).
A potential limitation of performing GO enrichment analysis with lists of significantly differentially expressed genes is that one primarily observes how an individual gene relates to a functional category. This may overlook correlated expression patterns among genes or genes that have a significant impact based on associations with other genes. Therefore, we re-analyzed our data using WGCNA to identify further candidate genes (Langfelder and Horvath 2008). This approach can assist in the identification of clusters of correlated genes or putative pathways, represented as modules that may be involved in oxidative stress tolerance based on gene expression profiles. Using WGCNA, we can isolate key nodes that have multiple edges, because the malfunction of such genes would affect all connected genes and therefore represent possible interesting candidates for further investigation. In both WGCNA designs (“∼treatment” and “∼genotype: treatment”), the two main modules can be categorized as (1) autophagy and apoptosis (turquoise) and (2) metabolic processes (blue) (Figure 3). In the turquoise module, we observed the up-regulation of genes involved in the general stress response, including a group of highly connected HSP genes and genes related to ubiquitin protein ligase binding (Ubi-p5E, Ubi-p63E, and CG11700), which also connect multiple sub-modules. This may indicate a connection between oxidative stress response genes and those involved in the ubiquitin proteasome pathway (Gaudet et al. 2011; Shang and Taylor 2011). On the other hand, in the blue module we observed the gene glutamate oxaloacetic transaminase (Got1), which is involved in glutamate biosynthesis (Gaudet et al. 2011), and aralar1, which is a mitochondrial and calcium-binding carrier that catalyzes the calcium-dependent exchange of cytoplasmic glutamate with mitochondrial aspartate across the mitochondrial inner membrane (Gaudet et al. 2011). Thus, altered expression of Got1 and aralar1 may reflect either a direct effect of the ROS burden and/or a systematic attempt to attenuate oxidative stress conditions through metabolic management of glutamate (Schinder et al. 1996; Rival et al. 2004; Peng et al. 2019). When the MtnA genotype is included as a factor, Prosα4T2 (blue module) and CG31792 (turquoise module) were revealed as potentially relevant to oxidative stress tolerance in the deletion background relative to the nondeletion background. Prosα4T2, involved in proteasomal ubiquitin-independent protein catabolic process, has also been suggested to have important functional roles in spermatogenesis, which may indicate a trade-off between oxidative stress tolerance and male reproduction (Belote et al. 1998; Tsakiri et al. 2013).
The effect of the MtnA indel polymorphism on gene expression
In pairwise comparisons of cosmopolitan lines that differed in their MtnA indel genotype, we observed that a large portion of the differentially expressed genes were shared between lines, but that there was a greater number of differentially expressed genes under stress in the deletion background than in the nondeletion background (Figure 7). This differential expression did not always correspond to the expression level of the MtnA gene (Figure 5), suggesting that the deletion influences the expression of other genes through a mechanism that is independent of its effect on MtnA. One possibility is that the polymorphic region of the MtnA 3′ UTR contains binding sites for miRNAs, and when this region is deleted the miRNAs are free to regulate the expression of other genes. Consistent with this interpretation, several miRNAs have been predicted to have target sites that overlap with the 49-bp deletion region of the MtnA 3′ UTR (Catalán et al. 2016). Furthermore, the predicted target genes of these miRNAs show greater downregulation under stress in the deletion background compared to the nondeletion background (Figure 8). This would be expected if there is direct regulation of the target genes by these miRNAs, as miRNAs typically function in the post-transcriptional regulation of gene expression by inducing mRNA degradation (Chen and Rajewsky 2007). However, there was also a significant excess of up-regulated genes under stress in the deletion background. This up-regulation is not likely to be mediated directly by miRNAs, which are predominantly involved in the negative regulation of gene expression. Instead, the up-regulation may be an indirect effect of miRNA regulation. That is, the downregulation of one gene by a miRNA leads to the up-regulation of a second gene through the action of one or more intermediate genes involved in a regulatory network. This interpretation is supported by the finding that genes in the functional categories: “regulation of gene expression” (59 genes), and particularly “negative regulation of gene expression” (35 genes), were overrepresented among the genes that were differentially expressed between deletion and nondeletion lines under oxidative stress (Supplementary Table S7). In addition, many of these genes fall into the general categories with the lowest P-values, “regulation of biological processes” (124 genes) and “regulation of cellular processes” (118 genes). One of the more common mechanisms associated to these terms is regulation of transcriptional activity via RNA Polymerase II (Supplementary Table S7), which is a key component in regulating gene expression. Interestingly, there were just three miRNA target genes associated to the ontology term “response to oxidative stress” but with potentially significant impact due to their diverse functionality and effect on transcriptional activity. One of these target genes is Adar (Adenosine deaminase acting on RNA) an RNA-editing enzyme that has been proposed to facilitate temperature-dependent adaptive evolution through an epigenetic mechanism and downregulation of this gene has been shown to be associated to an increase in expression of genes encoding known ROS scavengers (Chen et al. 2004; Gaudet et al. 2011; Rieder et al. 2015; Duan et al. 2017). The other two targets are: alph (alphabet) a Serine/Threonine specific protein phosphatase that is involved in the mitogen-activated protein kinase (MAPK) cascade and per (period), a gene essential for biological clock functions (Gaudet et al. 2011). Alph may represent a potential candidate to investigate our hypothesis concerning the adaptive benefits and indirect regulatory capabilities of the MtnA indel-associated miRNAs. Alph is involved in the negative regulation of Jun N-terminal kinase (JNK) cascade, therefore downregulation of this gene augments JNK signaling, which induces protective genes that confer increased oxidative stress tolerance (Wang et al. 2003; Baril et al. 2009). Another example here is the downregulation of per which is phenotypically associated to pathological effects on circadian rhythms and aging but may also have metabolic benefits which control longevity (Krishnan et al. 2009; Ulgherait et al. 2020).
Figure 8.

Log2 fold-changes (stress/control) of downregulated target genes of miRNAs with predicted binding sites within the MtnA 3′ UTR deletion region (miR-284-3p, miR-956-3p, miR-9c-5p, and bantam-3p). Two target genes that fall below the limits of the y-axis (log2 fold-change <−1.5) are not shown in the plot, but were included in the statistical analysis. Differences between the deletion and nondeletion background were tested using a paired sample Wilcoxon test. **P < 0.01, ***P < 0.001.
In summary, we have demonstrated that the molecular and genetic mechanisms involved in oxidative stress tolerance are strongly associated to metabolism and DNA/protein damage and repair. Moreover, our transcriptomic data suggest that this response overlaps with genes responding to cold tolerance and that metallothionein genes (MtnA, MtnD, and MtnE) may operate collaboratively in response to oxidative stress. To further investigate the functional elements associated to these two stress responses and their roles in clinal or local adaptation, future studies may utilize combined or parallel oxidative stress and cold stress treatments on lines with precisely controlled genomic backgrounds. In addition, a candidate gene approach involving oxidative and cold stress with single gene knockout/knockdown lines could be employed. With regards to the MtnA indel polymorphism and its effect on oxidative stress tolerance, our data imply that the associated phenotype is dependent on alleles at other loci in the genetic background. Therefore, a logical progression of this study may involve experimental work including expression, tolerance, and transcriptomic analyses of a larger number of natural population backgrounds that have been subjected to direct genetic manipulation of the MtnA indel region using gene editing techniques. Finally, we provide preliminary evidence for the involvement of miRNAs in the response to oxidative stress, which may be part of the mechanism by which the MtnA 3′ UTR deletion affects the expression of MtnA and other genes. In the future, reporter genes, small-RNA sequencing, and/or transgenic lines (e.g., UAS-miRNA) could be used to confirm specific miRNA-mRNA interactions and to functionally evaluate the regulatory capabilities of miRNAs associated with the MtnA 3′ UTR.
Data availability
Gene expression data are available at GEO under the accession number GSE174813. All other data are provided in the online supplementary materials are available at figshare: https://doi.org/10.25387/g3.14778540.
Acknowledgments
The authors thank Hilde Lainer, Aleksei Belyi, Amanda Glaser-Schmitt, and Eliza Argyridou for help with fly maintenance and sample preparation. The authors extend our appreciation to all current and previous members of the Division of Evolutionary Biology, as well as the Graduate School Life Science Munich (LSM) at Ludwig-Maximilians-Universität (LMU). The manuscript was improved thanks to comments from two anonymous reviewers.
Funding
This work was supported by the Deutsche Forschungsgemeinschaft research unit “Seasonal temperature acclimation in Drosophila” (FOR2682) project 347368302 (J.P.) and Deutsche Forschungsgemeinschaft project 271330745 (S.G.).
Conflicts of interest
The authors declare that there is no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Literature cited
- Abel J, de Ruiter N.. 1989. Inhibition of hydroxyl-radical-generated DNA degradation by metallothionein. Toxicol Lett. 47:191–196. doi:10.1016/0378–4274(89)90075-1. [DOI] [PubMed] [Google Scholar]
- Agarwal V, Subtelny AO, Thiru P, Ulitsky I, Bartel DP.. 2018. Predicting microRNA targeting efficacy in Drosophila. Genome Biol. 19:152.doi:10.1186/s13059-018–1504-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akman SA, Dietrich M, Chlebowski R, Limberg P, Block JB.. 1985. Modulation of cytotoxicity of menadione sodium bisulfite versus leukemia L1210 by the acid-soluble thiol pool. Cancer Res. 45:5257–5262. [PubMed] [Google Scholar]
- Atanesyan L, Günther V, Celniker SE, Georgiev O, Schaffner W.. 2011. Characterization of MtnE, the fifth metallothionein member in Drosophila. J Biol Inorg Chem. 16:1047–1056. doi:10.1007/s00775-011–0825-4. [DOI] [PubMed] [Google Scholar]
- Baker H, DeAngelis B, Frank O, Khalil M, Hutner SH, et al. 1996. Antioxidant survey to assess antagonism to redox stress using a prokaryotic and an eukaryotic system. Experientia. 52:597–599. doi:10.1007/BF01969736. [DOI] [PubMed] [Google Scholar]
- Baril C, Sahmi M, Ashton-Beaucage D, Stronach B, Therrien M.. 2009. The PP2C alphabet is a negative regulator of stress-activated protein kinase signaling in Drosophila. Genetics. 181:567–579. doi:10.1534/genetics.108.096461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belote JM, Miller M, Smyth KA.. 1998. Evolutionary conservation of a testes-specific proteasome subunit gene in Drosophila. Gene. 215:93–100. doi:10.1016/s0378-1119(98)00256-x. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y.. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B Methodol. 57:289–300. doi:10.1111/j.2517–6161.1995.tb02031.x. [Google Scholar]
- Bergland AO, Tobler R, González J, Schmidt P, Petrov D.. 2016. Secondary contact and local adaptation contribute to genome-wide patterns of clinal variation in Drosophila melanogaster. Mol Ecol. 25:1157–1174. doi:10.1111/mec.13455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonilla E, , Medina-LeendertzS, , VillalobosV, , MoleroL, , Bohórquez A.. 2006. Paraquat-induced oxidative stress in drosophila melanogaster: effects of melatonin, glutathione, serotonin, minocycline, lipoic acid and ascorbic acid. Neurochem Res. 31:1425–1432. doi: 10.1007/s11064-006-9194-8. [DOI] [PubMed] [Google Scholar]
- Cai L, Klein JB, Kang YJ.. 2000. Metallothionein inhibits peroxynitrite-induced DNA and lipoprotein damage. J Biol Chem. 275:38957–38960. doi:10.1074/jbc.C000593200. [DOI] [PubMed] [Google Scholar]
- Carbon S, Ireland A, Mungall CJ, Shu S, Marshall B, et al. ; the AmiGO Hub, the Web Presence Working Group. 2009. AmiGO: online access to ontology and annotation data. Bioinformatics. 25:288–289. doi:10.1093/bioinformatics/btn615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catalán A, Glaser-Schmitt A, Argyridou E, Duchen P, Parsch J.. 2016. An indel polymorphism in the MtnA 3′ untranslated region is associated with gene expression variation and local adaptation in Drosophila melanogaster. PLoS Genet. 12:e1005987.doi:10.1371/journal.pgen.1005987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang M, Shi M, Forman HJ.. 1992. Exogenous glutathione protects endothelial cells from menadione toxicity. Am J Physiol. 262:L637–L643. doi:10.1152/ajplung.1992.262.5.L637. [DOI] [PubMed] [Google Scholar]
- Chen K, Rajewsky N.. 2007. The evolution of gene regulation by transcription factors and microRNAs. Nat Rev Genet. 8:93–103. doi:10.1038/nrg1990. [DOI] [PubMed] [Google Scholar]
- Chen L, Rio DC, Haddad GG, Ma E.. 2004. Regulatory role of dADAR in ROS metabolism in Drosophila CNS. Brain Res Mol Brain Res. 131:93–100. doi:10.1016/j.molbrainres.2004.08.013. [DOI] [PubMed] [Google Scholar]
- Cogni R, Kuczynski K, Koury S, Lavington E, Behrman EL, et al. 2017. On the long-term stability of clines in some metabolic genes in Drosophila melanogaster. Sci Rep. 7:42766.doi:10.1038/srep42766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colinet H, Fai Lee S, Hoffmann A.. 2010. Functional characterization of the Frost gene in Drosophila melanogaster: importance for recovery from Chill Coma. PLoS One. 5:e10925.doi:10.1371/journal.pone.0010925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das N, Levine RL, Orr WC, Sohal RS.. 2001. Selectivity of protein oxidative damage during aging in Drosophila melanogaster. Biochem J. 360:209–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan Y, Dou S, Luo S, Zhang H, Lu J.. 2017. Adaptation of A-to-I RNA editing in Drosophila. PLoS Genet. 13:e1006648.doi:10.1371/journal.pgen.1006648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egli D, Domènech J, Selvaraj A, Balamurugan K, Hua H, et al. 2006. The four members of the Drosophila metallothionein family exhibit distinct yet overlapping roles in heavy metal homeostasis and detoxification. Genes Cells. 11:647–658. doi:10.1111/j.1365–2443.2006.00971.x. [DOI] [PubMed] [Google Scholar]
- Elgawish MS, Shimomai C, Kishikawa N, Ohyama K, Wada M, et al. 2013. Development and validation of the first assay method coupling liquid chromatography with chemiluminescence for the simultaneous determination of menadione and its thioether conjugates in rat plasma. Chem Res Toxicol. 26:1409–1417. doi:10.1021/tx400253k. [DOI] [PubMed] [Google Scholar]
- Erickson PA, Weller CA, Song DY, Bangerter AS, Schmidt P, Bergland AO.. 2020. Unique genetic signatures of local adaptation over space and time for diapause, an ecologically relevant complex trait, in Drosophila melanogaster. PLoS Genet. 16:e1009110.doi:10.1371/journal.pgen.1009110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkel T, Holbrook NJ.. 2000. Oxidants, oxidative stress and the biology of ageing. Nature. 408:239–247. doi:10.1038/35041687. [DOI] [PubMed] [Google Scholar]
- Gaudet P, Livstone MS, Lewis SE, Thomas PD.. 2011. Phylogenetic-based propagation of functional annotations within the gene ontology consortium. Brief Bioinform. 12:449–462. doi:10.1093/bib/bbr042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaser-Schmitt A, Parsch J.. 2018. Functional characterization of adaptive variation within a cis-regulatory element influencing Drosophila melanogaster growth. PLoS Biol. 16:e2004538.doi:10.1371/journal.pbio.2004538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González J, Macpherson JM, Petrov DA.. 2009. A recent adaptive transposable element insertion near highly conserved developmental loci in Drosophila melanogaster. Mol Biol Evol. 26:1949–1961. doi:10.1093/molbev/msp107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guio L, Barrón MG, González J.. 2014. The transposable element Bari-Jheh mediates oxidative stress response in Drosophila. Mol Ecol. 23:2020–2030. doi:10.1111/mec.12711. [DOI] [PubMed] [Google Scholar]
- Harr B, Kauer M, Schlotterer C.. 2002. Hitchhiking mapping: a population-based fine-mapping strategy for adaptive mutations in Drosophila melanogaster. Proc Natl Acad Sci USA. 99:12949–12954. doi:10.1073/pnas.202336899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison BR, Wang L, Gajda E, Hoffman EV, Chung BY, et al. 2020. The metabolome as a link in the genotype-phenotype map for peroxide resistance in the fruit fly, Drosophila melanogaster. BMC Genomics. 21:341.doi:10.1186/s12864-020–6739-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison JF, Camazine S, Marden JH, Kirkton SD, Rozo A, et al. 2001. Mite not make it home: tracheal mites reduce the safety margin for oxygen delivery of flying honeybees. J Exp Biol. 204:805–814. [DOI] [PubMed] [Google Scholar]
- Hayes JD, McLellan LI.. 2009. Glutathione and glutathione-dependent enzymes represent a co-ordinately regulated defence against oxidative stress. Free Radic Res. 31:273–300 doi:10.1080/10715769900300851. [DOI] [PubMed] [Google Scholar]
- Hutter S, Saminadin-Peter SS, Stephan W, Parsch J.. 2008. Gene expression variation in African and European populations of Drosophila melanogaster. Genome Biol. 9: r12.doi:10.1186/gb-2008-9-1-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joanisse D, Storey K.. 1996. Oxidative stress and antioxidants in overwintering larvae of cold-hardy goldenrod gall insects. J Exp Biol. 199:1483–1491. [DOI] [PubMed] [Google Scholar]
- Joanisse DR, Storey KB.. 1998. Oxidative stress and antioxidants in stress and recovery of cold-hardy insects. Insect Biochem Mol Biol. 28:23–30. doi:10.1016/S0965-1748(97)00070-2. [Google Scholar]
- Juneja P, Quinn A, Jiggins FM.. 2016. Latitudinal clines in gene expression and cis-regulatory element variation in Drosophila melanogaster. BMC Genomics. 17:981.doi:10.1186/s12864-016–3333-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapun M, Barrón MG, Staubach F, Obbard DJ, Wiberg RAW, et al. 2020. Genomic analysis of European Drosophila melanogaster populations reveals longitudinal structure, continent-wide selection, and previously unknown DNA viruses. Mol Biol Evol. 37:2661–2678. doi:10.1093/molbev/msaa120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavitha S, Chandra TS.. 2014. Oxidative stress protection and glutathione metabolism in response to hydrogen peroxide and menadione in riboflavinogenic fungus Ashbya gossypii. Appl Biochem Biotechnol. 174:2307–2325. doi:10.1007/s12010-014–1188-4. [DOI] [PubMed] [Google Scholar]
- Klok CJ, Sinclair BJ, Chown SL.. 2004. Upper thermal tolerance and oxygen limitation in terrestrial arthropods. J Exp Biol. 207:2361–2370. doi:10.1242/jeb.01023. [DOI] [PubMed] [Google Scholar]
- Königer A, Grath S.. 2018. Transcriptome analysis reveals candidate genes for cold tolerance in Drosophila ananassae. Genes. 9:624.doi:10.3390/genes9120624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan N, Kretzschmar D, Rakshit K, Chow E, Giebultowicz JM.. 2009. The circadian clock gene period extends healthspan in aging Drosophila melanogaster. Aging (Albany NY). 1:937–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalouette L, Williams CM, Hervant F, Sinclair BJ, Renault D.. 2011. Metabolic rate and oxidative stress in insects exposed to low temperature thermal fluctuations. Comp Biochem Physiol A Mol Integr Physiol. 158:229–234. doi:10.1016/j.cbpa.2010.11.007. [DOI] [PubMed] [Google Scholar]
- Lange BW, Langley CH, Stephan W.. 1990. Molecular evolution of Drosophila metallothionein genes. Genetics. 126:921–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P, Horvath S.. 2008. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 9:559.doi:10.1186/1471–2105-9–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S.. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15:550.doi:10.1186/s13059-014–0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo M, Finet C, Cong H, Wei H, Chung H.. 2020. The evolution of insect metallothioneins. Proc Biol Sci. 287:20202189.doi:10.1098/rspb.2020.2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyne R, Smith R, Rutherford K, Wakeling M, Varley A, et al. 2007. FlyMine: an integrated database for Drosophila and Anopheles genomics. Genome Biol. 8: r129.doi:10.1186/gb-2007-8-7-r129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay TFC, Richards S, Stone EA, Barbadilla A, Ayroles JF, et al. 2012. The Drosophila melanogaster genetic reference panel. Nature. 482:173–178. doi:10.1038/nature10811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacMillan HA, Knee JM, Dennis AB, Udaka H, Marshall KE, et al. 2016. Cold acclimation wholly reorganizes the Drosophila melanogaster transcriptome and metabolome. Sci Rep. 6:28999.doi:10.1038/srep28999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacMillan HA, Williams CM, Staples JF, Sinclair BJ.. 2012. Metabolism and energy supply below the critical thermal minimum of a chill-susceptible insect. J Exp Biol. 215:1366–1372. doi:10.1242/jeb.066381. [DOI] [PubMed] [Google Scholar]
- Mockett RJ, Sohal RS, Orr WC.. 1999. Overexpression of glutathione reductase extends survival in transgenic Drosophila melanogaster under hyperoxia but not normoxia. FASEB J. 13:1733–1742. doi:10.1096/fasebj.13.13.1733. [DOI] [PubMed] [Google Scholar]
- Ometto L, Glinka S, De Lorenzo D, Stephan W.. 2005. Inferring the effects of demography and selection on Drosophila melanogaster populations from a chromosome-wide scan of DNA variation. Mol Biol Evol. 22:2119–2130. doi:10.1093/molbev/msi207. [DOI] [PubMed] [Google Scholar]
- Peng J-J, Lin S-H, Liu Y-T, Lin H-C, Li T-N, et al. 2019. A circuit-dependent ROS feedback loop mediates glutamate excitotoxicity to sculpt the Drosophila motor system. eLife. 8:e47372.doi:10.7554/eLife.47372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickering AM, Vojtovich L, Tower J, Davies KJA.. 2013. Oxidative stress adaptation with acute, chronic and repeated stress. Free Radic Biol Med. 55:109–118. doi:10.1016/j.freeradbiomed.2012.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pool JE, Corbett-Detig RB, Sugino RP, Stevens KA, Cardeno CM, et al. 2012. Population genomics of sub-Saharan Drosophila melanogaster: African diversity and non-African admixture. PLoS Genet. 8:e1003080.doi:10.1371/journal.pgen.1003080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramnarine TJS, Glaser-Schmitt A, Catalán A, Parsch J.. 2019. Population genetic and functional analysis of a cis-regulatory polymorphism in the Drosophila melanogaster Metallothionein A gene. Genes. 10:147.doi:10.3390/genes10020147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. 2020. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing. (Accessed: 2020 November 11). https://www.R-project.org/.
- Rieder LE, Savva YA, Reyna MA, Chang Y-J, Dorsky JS, et al. 2015. Dynamic response of RNA editing to temperature in Drosophila. BMC Biol. 13:1.doi:10.1186/s12915-014–0111-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rival T, Soustelle L, Strambi C, Besson M-T, Iché M, et al. 2004. Decreasing glutamate buffering capacity triggers oxidative stress and neuropil degeneration in the Drosophila Brain. Curr Biol. 14:599–605. doi:10.1016/j.cub.2004.03.039. [DOI] [PubMed] [Google Scholar]
- Schinder AF, Olson EC, Spitzer NC, Montal M.. 1996. Mitochondrial dysfunction is a primary event in glutamate neurotoxicity. J Neurosci. 16:6125–6133. doi:10.1523/JNEUROSCI.16–19-06125.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedlazeck FJ, Rescheneder P, von Haeseler A.. 2013. NextGenMap: fast and accurate read mapping in highly polymorphic genomes. Bioinformatics. 29:2790–2791. doi:10.1093/bioinformatics/btt468. [DOI] [PubMed] [Google Scholar]
- Sezgin E, Duvernell DD, Matzkin LM, Duan Y, Zhu C-T, et al. 2004. Single-locus latitudinal clines and their relationship to temperate adaptation in metabolic genes and derived alleles in Drosophila melanogaster. Genetics. 168:923–931. doi:10.1534/genetics.104.027649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang F, Taylor A.. 2011. Ubiquitin–proteasome pathway and cellular responses to oxidative stress. Free Radic Biol Med. 51:5–16. doi:10.1016/j.freeradbiomed.2011.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephan W, Rodriguez VS, Zhou B, Parsch J.. 1994. Molecular evolution of the Metallothionein gene Mtn in the melanogaster species group: results from Drosophila ananassae. Genetics. 138:135–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supek F, Bošnjak M, Škunca N, Šmuc T.. 2011. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS One. 6:e21800.doi:10.1371/journal.pone.0021800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svetec N, Werzner A, Wilches R, Pavlidis P, Alvarez-Castro JM, et al. 2011. Identification of X-linked quantitative trait loci affecting cold tolerance in Drosophila melanogaster and fine mapping by selective sweep analysis. Mol Ecol. 20:530–544. doi:10.1111/j.1365-294X.2010.04951.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teets NM, Hahn DA.. 2018. Genetic variation in the shape of cold-survival curves in a single fly population suggests potential for selection from climate variability. J Evol Biol. 31:543–555. doi: 10.1111/jeb.13244. [DOI] [PubMed] [Google Scholar]
- Theodore L, Ho A-S, Maroni G.. 1991. Recent evolutionary history of the metallothionein gene Mtn in Drosophila. Genet Res. 58:203–210. doi:10.1017/S0016672300029955. [DOI] [PubMed] [Google Scholar]
- Thomas NO, Shay KP, Kelley AR, Butler JA, Hagen TM.. 2016. Glutathione maintenance mitigates age-related susceptibility to redox cycling agents. Redox Biol. 10:45–52. doi:10.1016/j.redox.2016.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornalley PJ, Vasák M.. 1985. Possible role for metallothionein in protection against radiation-induced oxidative stress. Kinetics and mechanism of its reaction with superoxide and hydroxyl radicals. Biochim Biophys Acta. 827:36–44. doi:10.1016/0167-4838(85)90098-6. [DOI] [PubMed] [Google Scholar]
- Thurmond J, Goodman JL, Strelets VB, Attrill H, Gramates LS, Marygold SJ, et al. ; FlyBase Consortium. 2019. FlyBase 2.0: the next generation. Nucleic Acids Res. 47:D759–D765. doi:10.1093/nar/gky1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsakiri EN, Sykiotis GP, Papassideri IS, Gorgoulis VG, Bohmann D, et al. 2013. Differential regulation of proteasome functionality in reproductive vs somatic tissues of Drosophila during aging or oxidative stress. FASEB J. 27:2407–2420. doi: 10.1096/fj.12–221408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulgherait M, Chen A, McAllister SF, Kim HX, Delventhal R, et al. 2020. Circadian regulation of mitochondrial uncoupling and lifespan. Nat Commun. 11:1927.doi:10.1038/s41467-020–15617-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voigt S, Erpf AC, Stephan W.. 2019. Decreased temperature sensitivity of vestigial gene expression in temperate populations of Drosophila melanogaster. Genes. 10:498.doi:10.3390/genes10070498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Heckel K, Stephan W, Hutter S.. 2016. Canalization of gene expression is a major signature of regulatory cold adaptation in temperate Drosophila melanogaster. BMC Genomics. 17:574.doi:10.1186/s12864-016–2866-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace MA, Bailey S, Fukuto JM, Valentine JS, Gralla EB.. 2005. Induction of phenotypes resembling CuZn-superoxide dismutase deletion in wild-type yeast cells: an in vivo assay for the role of superoxide in the toxicity of redox-cycling compounds. Chem Res Toxicol. 18:1279–1286. doi:10.1021/tx050050n. [DOI] [PubMed] [Google Scholar]
- Wang MC, Bohmann D, Jasper H.. 2003. JNK signaling confers tolerance to oxidative stress and extends lifespan in Drosophila. Dev Cell. 5:811–816. doi:10.1016/s1534-5807(03)00323-x. [DOI] [PubMed] [Google Scholar]
- Weber AL, Khan GF, Magwire MM, Tabor CL, Mackay TFC, et al. 2012. Genome-wide association analysis of oxidative stress resistance in Drosophila melanogaster. PLoS One. 7:e34745.doi:10.1371/journal.pone.0034745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitworth AJ, Theodore DA, Greene JC, Benes H, Wes PD, et al. 2005. Increased glutathione S-transferase activity rescues dopaminergic neuron loss in a Drosophila model of Parkinson’s disease. Proc Natl Acad Sci USA. 102:8024–8029. doi:10.1073/pnas.0501078102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Horvath S.. 2005. A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol. 4:Article17. doi:10.2202/1544-6115.1128. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Gene expression data are available at GEO under the accession number GSE174813. All other data are provided in the online supplementary materials are available at figshare: https://doi.org/10.25387/g3.14778540.