

Abstract
In advanced breast cancer, biomarker identification and patient selection using a metastatic tumor biopsy is becoming more necessary. However, the biology of metastasis according to the organ site is largely unknown. Here, we evaluated the expression of 771 genes in 184 metastatic samples across 11 organs, including liver, lung, brain, and bone, and made the following observations. First, all PAM50 molecular intrinsic subtypes were represented across organs and within immunohistochemistry‐based groups. Second, HER2‐low disease was identified across all organ sites, including bone, and HER2 expression significantly correlated with ERBB2 expression. Third, the majority of expression variation was explained by intrinsic subtype and not organ of metastasis. Fourth, subtypes and individual subtype‐related genes/signatures were significantly associated with overall survival. Fifth, we identified 74 genes whose expression was organ‐specific and subtype‐independent. Finally, immune profiles were found more expressed in lung compared to brain or liver metastasis. Our results suggest that relevant tumor biology can be captured in metastatic tissues across a variety of organ sites; however, unique biological features according to organ site were also identified and future studies should explore their implications in diagnostic and therapeutic interventions.
Keywords: breast cancer, gene expression profiling, HER2‐low, metastatic sites, PAM50
We evaluated gene expression in 184 metastatic breast cancer samples across 11 organs. All PAM50 subtypes were represented across metastatic sites and associated with survival. HER2‐low disease was identified across sites. Immune gene expression was higher in lung than brain or liver metastasis. Relevant tumor biology can be captured in metastatic tissue, while each site shows unique biological features.

Abbreviations
- ASCO/CAP
American Society of Clinical Oncologists/College of American Pathologists
- Cor
correlation
- DAVID
database for annotation, visualization and integrated discovery
- ER
estrogen receptor
- FDR
false discovery rate
- FFPE
formalin‐fixed paraffin‐embedded
- GO
gene ontology
- HER2+
HER2‐positive
- HR
hormone receptor
- HR+
hormone receptor‐positive
- IHC
immunohistochemistry
- ISH
in situ hybridization
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- N/A
not available
- OS
overall survival
- P
p‐value
- PCA
principal component analysis
- ROR
risk of recurrence
- SAM
significance analysis of microarrays
- T-DXd
trastuzumab deruxtecan
- TNBC
triple-negative breast cancer
1. Introduction
Advanced or metastatic breast cancer affects multiple organs and is a main cause of cancer death [1]. Common metastatic sites include bone, liver, lung, brain, lymph node, pleura, and skin [1, 2, 3, 4, 5, 6]. Interestingly, the different breast cancer intrinsic subtypes (i.e., luminal A and B, HER2‐enriched, and basal‐like) have distinct preferred metastatic sites [7], and both the tumor cell and the metastatic microenvironment might contribute to this organ specificity [8]. To date, however, the biology of breast cancer metastasis according to organ site remains largely unknown.
In advanced breast cancer, biomarker identification and patient selection using a biopsy from a metastatic lesion is becoming a clinical need. On one hand, it confirms the breast origin of the disease. On the other hand, it allows the identification of predictive biomarkers such as PIK3CA mutations or the expression of PD‐L1, HER2, and hormone receptors (HR). Although tumor tissue of primary disease obtained years before remains of value to identify these biomarkers when available [9, 10], significant biological differences exist between primary and metastatic disease [11]. For instance, loss of estrogen receptor (ER) has been reported in about 20% of cases [12], while 3–10% discordance of HER2 gene amplification exists in primary versus metastatic tissue [13]. Moreover, advanced disease is enriched with new genetic alterations such as ESR1 mutations or the APOBEC genetic signature [14] and with phenotypic changes such as the acquisition of the HER2‐enriched subtype in HR‐positive (HR+)/HER2‐negative disease [11]. Importantly, many of these biological alterations during metastatic disease might lead to resistance and treatment failure [15, 16, 17, 18]. In this direction, clinical trials with novel agents are mandating a metastatic tumor biopsy to select patients based on their tumor’s genomic profile.
One critical question that patients, clinicians, and researchers face is which metastatic lesion is better to biopsy or analyze. In certain cases, the most accessible metastatic lesion is chosen. In other circumstances, different options such as liquid biopsies may be available. A better understanding of the molecular profiles of the different metastatic sites might be of value. For example, PD‐L1 expression in immune cells in triple‐negative breast cancer (TNBC) is not recommended in liver biopsies due to the general lack of immune cells in this organ [19, 20]. Another example is determination of HER2 in bone metastasis, which is generally not recommended for technical reasons due to the decalcification procedures. To improve our understanding of the biology of breast cancer metastasis according to the organ site, we performed a phenotypic and molecular characterization of HER2 and 771 genes in 184 metastatic samples across 11 organs, including liver, lung, brain, and bone.
2. Materials and methods
2.1. Study population
This retrospective and exploratory study included 184 metastatic tumor samples from 176 patients over the age of 18 years with a histologic diagnosis of metastatic breast cancer detected at the time of diagnosis, at first relapse or after disease progression. Tissues were collected from Hospital Clinic of Barcelona (n = 161) and Hospital Universitario 12 de Octubre (n = 23) in Madrid between years 2000 and 2019. To be included, patients were required to have a formalin‐fixed paraffin‐embedded (FFPE) tissue sample from a locoregional or a distant metastatic lesion. Primary tumor biopsies were allowed if the biopsy was obtained in the context of de novo metastatic disease (n = 7). Core biopsies were performed according to the routine clinical practice, and HR and HER2 receptor statuses were determined locally in the metastatic biopsy according to the American Society of Clinical Oncologists (ASCO)/College of American Pathologists (CAP) guidelines [21, 22]. HER2 expression status (positive or negative) assessed by immunohistochemistry (IHC) was available for 163 tumor samples (88.6%), while HER2 detailed expression (HER2‐0, HER2 1+ or HER2 2+, and HER2 3+) was available for 148 tumor samples (80.4%). HER2 in situ hybridization (ISH) was performed in HER2 2+ tumor samples. HER2‐low tumors were defined when HER2 was determined as HER2 1+ or HER2 2+ and ISH‐negative was identified. HR status (positive or negative) assessed by IHC was available for 158 tumor samples (85.9%), while detailed % of ER expression was available for 148 tumor samples (80.4%). Moreover, we included 186 FFPE tumor samples from patients with early‐stage breast cancer from Hospital Clinic of Barcelona representative of all PAM50 subtypes. The hospital institutional ethics committee approved the study in accordance with the principles of Good Clinical Practice, the Declaration of Helsinki, and other applicable local regulations. Written informed consent was obtained from all patients before enrollment. Patient data were obtained from the database of medical records (SAP Logon 730) and Historia Clinica Compartida (HC3). The medical records were retrospectively reviewed to obtain the clinical data analyzed in the study.
Finally, a publicly available dataset of 390 primary tumors with types of metastatic spread and microarray data was interrogated [23].
2.2. Gene expression analysis
RNA was extracted using the High Pure FFPET RNA isolation kit (Roche, Indianapolis, IN, USA) following the manufacturer’s protocol. One to five 10 μm FFPE slides depending on tumor cellularity were used for each tumor sample, and macrodissection was performed, when needed, to avoid normal tissue contamination. A minimum of 100 ng of total RNA was analyzed at the nCounter platform (NanoString Technologies, Seattle, WA, USA) using the Breast Cancer 360 Panel, which measures the expression of 771 breast cancer‐related genes and 5 housekeeping genes (ACTB, MRPL19, PSMC4, RPLP0, and SF3A1) [24]. Expression counts were then normalized using custom scripts in r 3.6.3.
2.3. PAM50 molecular subtypes and gene signatures
All tumors were assigned to an intrinsic molecular subtype of breast cancer (luminal A, luminal B, HER2‐enriched, basal‐like, and normal‐like) using the previously reported PAM50 subtype predictor [25]. For each sample, we calculated scores for 9 signatures including the 5 PAM50 signatures (luminal A, luminal B, HER2‐enriched, basal‐like, and normal‐like) [11], the proliferation signature [26], two risk of recurrence (ROR) signatures at 10 years: ROR score based on subtype (ROR‐S) and based on subtype and proliferation (ROR‐P) as described previously [26], and the previously reported PAM50MET signature, which is based on 17 variables [27]. Gene expression data will be deposited in the Gene Expression Omnibus under the accession number GSE175692.
2.4. Statistical analysis
Chi‐square tests were performed to determine the differences in the distribution of variables. Data were subjected to unsupervised hierarchical clustering and principal component analysis (PCA) to identify patterns of expression and to clean the dataset from outliers, 3 out of 184 samples were excluded. Unpaired and multiclass significance analysis of microarrays (SAM) [28], using false discovery rate (FDR), was used to identify differential gene expression across metastatic sites (n = 181). Due to the low number of metastasis from ovary (n = 4), muscle (n = 2), and peritoneum sites (n = 2), these samples were excluded from the multiclass SAM analyses. Logistic regressions were used to identify organ‐specific genes.
Overall survival (OS) was defined as the period of time of first diagnosis of metastatic disease to death or last follow‐up. Censoring was done at 120 months. Estimates of survival were from the Kaplan–Meier curves and tests of differences by the log‐rank test. Univariate and multivariable Cox models were used to test the prognostic significance of each variable. The Bonferroni correction method was used to control the family‐wise error rate in case of multiple comparisons [29]. All differences were considered significant at P‐value < 0.05. All statistical computations were carried out in r 3.6.3 (http://cran.r‐project.org).
2.5. Functional and pathway enrichment analyses
Gene ontology (GO) annotation analysis and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis were analyzed by the Database for Annotation, Visualization and Integrated Discovery (DAVID, http://david.abcc.ncifcrf.gov/) online tool [30]. The list of 771 available genes was used as the background or reference gene list. A P < 0.05 was considered statistically significant.
3. Results
3.1. Patients and samples characteristics
A total of 184 FFPE tumor samples from 176 patients with advanced breast cancer (Fig. 1A) were obtained from bone (18%), brain (12%), breast (13%, including 19 local recurrences and 5 cases of de novo metastatic breast cancer), liver (17%), lung (7%), lymph nodes (9%), muscle (1%), ovary (2%), peritoneum (1%), pleura (5%), and skin (15%). Clinicopathological information for all patients included is summarized in Table 1. RNA expression was analyzed in all samples using the nCounter‐based breast cancer 360™ panel of 771 genes (Fig. 1B). IHC subtypes were available from 171 samples (96.1%), and their distribution was 58.5% HR+/HER2‐negative, 10.5% HER2‐positive (HER2+), and 31% TNBC. IHC subtype was not available (N/A) for 13 samples. Median OS was 63.8 months in patients with HR+/HER2‐negative disease, 35.5 months in patients with HER2+ disease, and 22.1 months in patients with TNBC (Fig. 1C).
Fig. 1.
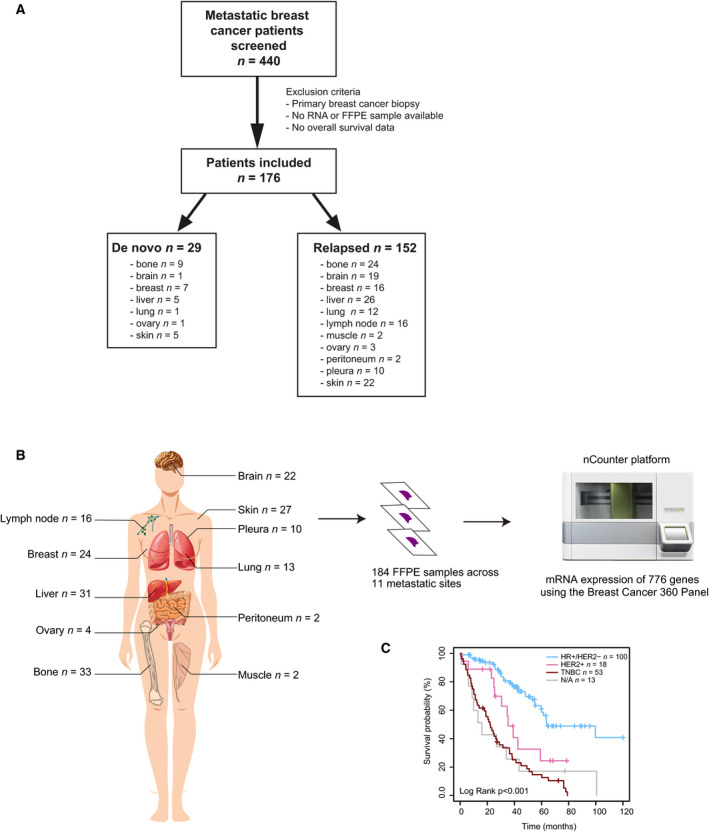
Sample characteristics. (A) Consort diagram reflecting the number of tumor samples evaluated in the study. (B) RNA extracted from 184 FFPE tumor samples obtained from 11 different metastatic sites was analyzed at the nCounter platform using the Breast Cancer 360 Panel. (C) Kaplan–Meier curves of 10‐year OS (log‐rank test) according to IHC subtype.
Table 1.
Clinicopathological characteristics.
| Characteristics | n | |
|---|---|---|
| Median age at diagnosis of metastasis (range) | 54 (24–89) | |
| Menopausal status | Premenopausal | 68 (38.6%) |
| Postmenopausal | 97 (55.1%) | |
| Unknown | 10 (5.7%) | |
| Male | 1 (0.6%) | |
| Type of metastasis | De novo metastasis | 27 (15.3%) |
| Relapsed | 146 (83.0%) | |
| Unknown | 3 (1.7%) | |
| Total number of metastatic sites | < 3 | 76 (43.2%) |
| ≥ 3 | 90 (51.1%) | |
| Unknown | 10 (5.7%) | |
| Site of metastatic biopsy | Locoregional | 50 (27.2%) |
| Distant | 134 (72.8%) | |
| Metastatic spread | Bone‐only | 15 (8.5%) |
| Visceral | 145 (82.4%) | |
| Organ of biopsy | Bone | 33 (17.9%) |
| Brain | 22 (12.0%) | |
| Breast | 24 (13.0%) | |
| Liver | 31 (16.9%) | |
| Lung | 13 (7.1%) | |
| Lymph node | 16 (8.7%) | |
| Muscle | 2 (1.1%) | |
| Ovary | 4 (2.2%) | |
| Peritoneum | 2 (1.1%) | |
| Pleura | 10 (5.4%) | |
| Skin | 27 (14.7%) | |
| IHC group of the metastatic biopsy | HR+/HER2‐ | 100 (54.4%) |
| HER2+ | 18 (9.8%) | |
| TNBC | 53 (28.8%) | |
| Unknown | 13 (7.1%) | |
| PAM50 molecular subtype of the metastatic biopsy | Luminal A | 30 (16.3%) |
| Luminal B | 47 (25.5%) | |
| HER2‐enriched | 42 (22.8%) | |
| Basal‐like | 54 (29.4%) | |
| Normal‐like | 11 (6.0%) | |
| Previous (neo)adjuvant treatment | 129 (73.3%) | |
| Median number of lines of treatment for metastatic disease (range) | 3 (0–13) | |
| Treatments received in the metastatic setting | Endocrine therapy | 111 (63.1%) |
| CDK4/6 inhibitors | 84 (47.7%) | |
| Anti‐HER2 therapies | 28 (15.9%) | |
| Chemotherapy | 125 (71.0%) | |
| Immunotherapy | 10 (5.7%) | |
| Everolimus | 22 (12.5%) | |
| PI3K inhibitors | 18 (10.2%) | |
| AKT inhibitors | 2 (1.1%) | |
| Bevacizumab | 15 (8.5%) | |
| PARP inhibitors | 3 (1.7%) | |
| Radiotherapy | 111 (63.1%) |
The intrinsic subtype distribution was 29% basal‐like (n = 54), 26% luminal B (n = 47), 23% HER2‐enriched (n = 42), 16% luminal A (n = 30), and 6% normal‐like (n = 11). Within HR+/HER2‐negative disease (n = 100), subtype distribution was 41% luminal B (n = 41), 27% luminal A (n = 27), 17% HER2‐enriched (n = 17), 11% basal‐like (n = 11), and 4% normal‐like (n = 4). HR+/HER2‐negative metastatic samples were obtained from liver (27%), bone (26%), skin (10%), breast (7%), lymph nodes (7%), pleura (7%), lung (5%), brain (4%), ovary (4%), muscle (2%), and peritoneum (1%) (Fig. 2A). Within HER2+ (n = 18), subtype distribution was 72% HER2‐enriched (n = 13), 11% basal‐like (n = 1), 11% luminal B (n = 2), and 6% normal‐like (n = 2). HER2+ metastatic samples were obtained from bone (28%), brain (17%), breast (17%), lung (17%), lymph nodes (17%), and liver (5%) (Fig. 2B). Within TNBC (n = 53), subtype distribution was 68% basal‐like (n = 36), 15% HER2‐enriched (n = 8), 7% normal‐like (n = 4), 6% luminal B (n = 3), and 4% luminal A (n = 4). TNBC metastatic samples were obtained from skin (30%), breast (23%), brain (15%), lung (7%), lymph nodes (7%), bone (6%), pleura (6%), liver (4%), and peritoneum (2%) (Fig. 2C). Across organs, there were statistically significant differences in subtype distribution (P < 0.001) and IHC groups (P < 0.001) (Tables S1 and S2).
Fig. 2.
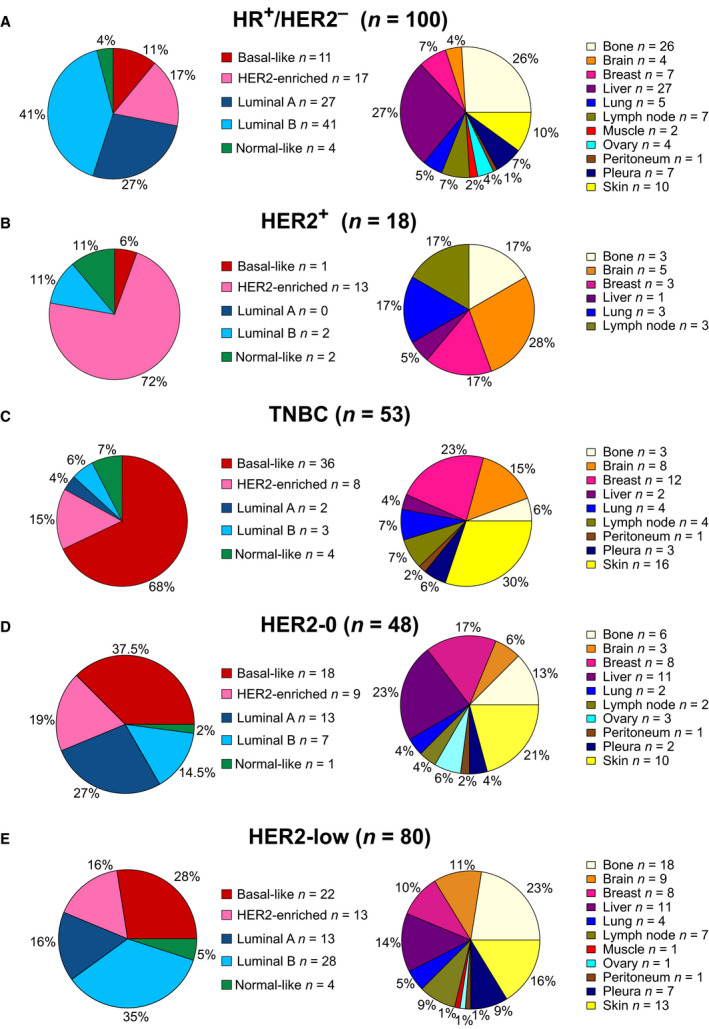
PAM50 subtype and metastatic site distribution in each IHC group. Pie charts depicting the percentage of each PAM50 subtype and the percentage of each metastatic site in (A) HR+/HER2‐negative, (B) HER2+, (C) TNBC, (D) HER2‐0, and (E) HER2‐low tumors.
3.2. HER2‐low disease according to organ site
The HER2‐low category using IHC (i.e., HER2 1+ or HER2 2+/ISH‐negative) is becoming an important biomarker for predicting benefit from antibody–drug conjugates such as trastuzumab deruxtecan (T‐DXd) [29, 31]. However, the identification of HER2‐low disease according to organ site, with a special emphasis in bone metastasis, is unknown. To address it, we explored data of HER2 expression and ERBB2 amplification in 146 samples (i.e., HER2‐0 n = 48, HER2‐low n = 80, and HER2+ n = 18). In HER2‐0 tumors (n = 48), the PAM50 distribution was 37.5% basal‐like (n = 18), 27% luminal A (n = 13), 19% HER2‐enriched (n = 9), 14.5% luminal B (n = 7), and 2% normal‐like (n = 1); HER2‐0 samples were obtained from liver (23%), skin (21%), breast (17%), bone (13%), brain (6%), ovary (6%), lung (4%), lymph nodes (4%), pleura (4%), and peritoneum (2%) (Fig. 2D). In HER2‐low metastatic tumors (n = 80), the PAM50 distribution was 35% luminal B (n = 28), 28% basal‐like (n = 22), 16% luminal A (n = 13), 16% HER2‐enriched (n = 13), and 5% normal‐like (n = 4). No significant difference in subtype distribution was identified between HER2‐0 and HER2‐low (P = 0.091), while a significant difference in subtype distribution was identified between HER2‐low and HER2+ (P < 0.001). Importantly, HER2‐low disease was identified in all metastatic sites: bone (23%), liver (14%), skin (16%), brain (11%), breast (10%), lymph nodes (9%), pleura (9%), lung (5%), muscle (1%), ovary (1%), and peritoneum (1%) (Fig. 2E). No significant difference in organ distribution was identified between HER2‐0 and HER2‐low (P = 0.414) nor between HER2‐low and HER2+ (P = 0.207).
Technical aspects might affect IHC staining of metastatic lesions, including bone metastasis. To address this potential issue, we correlated the expression of HER2 protein levels with ERBB2 mRNA and the expression of ER protein levels with ESR1 mRNA. Overall, ERBB2 mRNA was found significantly correlated with HER2 protein levels (HER2‐0, HER2‐low, or HER2+) (Spearman Cor = 0.531, P < 0.001). Compared to HER2‐0 disease, ERBB2 mRNA levels were found increased 1.20‐fold, 4.44‐fold, and 14.04‐fold in HER2 1+, HER2 2+/ISH‐negative, and HER2+ disease, respectively (Fig. 3A). In bone metastases, which are usually not accepted for inclusion in clinical trials, ERBB2 mRNA levels were also significantly correlated with HER2 protein levels (Spearman Cor = 0.604, P < 0.001) (Fig. 3B). We also found significant correlation between ERBB2 mRNA and HER2 protein levels in brain metastasis (Spearman Cor = 0.697, P = 0.002), breast metastasis (Spearman Cor = 0.754, P = 0.002), lung metastasis (Cor = 0.882, P = 0.003), and lymph node metastasis (Spearman Cor = 0.410, P = 0.05). Importantly, a strong correlation between ER protein expression determined by IHC and ESR1 mRNA was observed across all metastatic samples (Pearson Cor = 0.82, P < 0.001) and across bone metastatic samples (Pearson Cor = 0.85, P < 0.001) (Fig. S1), suggesting that good quality gene expression data can be obtained from bone samples.
Fig. 3.
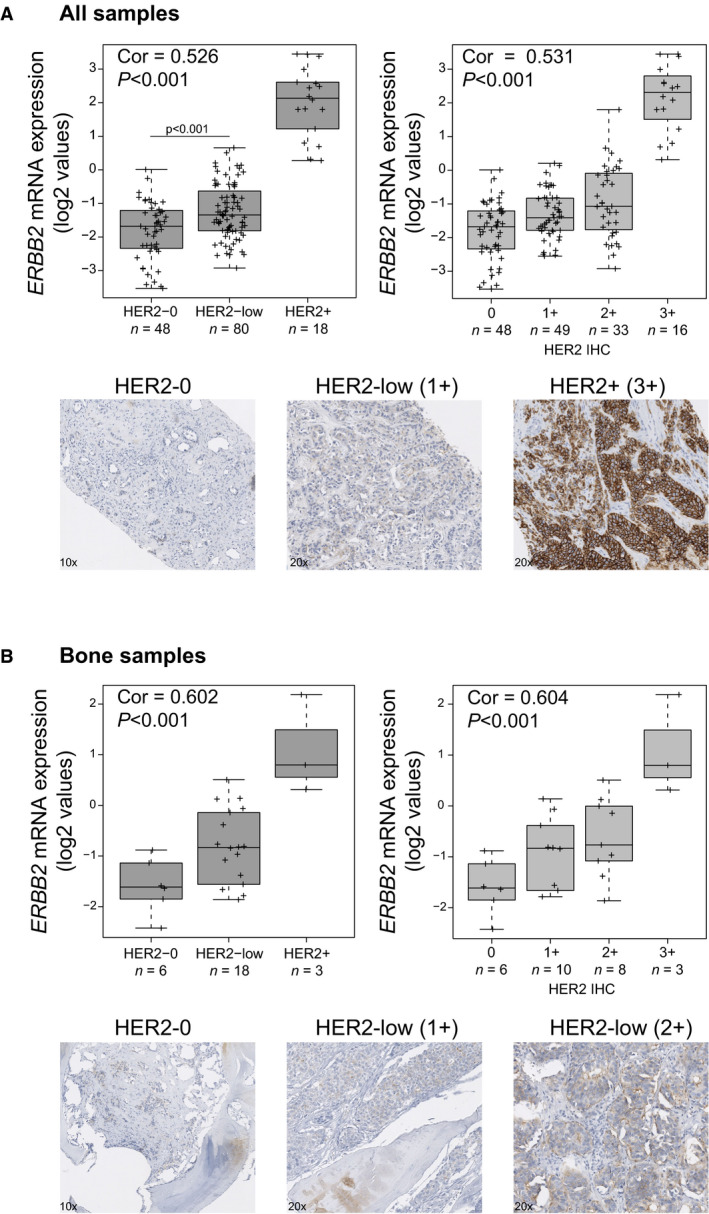
ERBB2 mRNA correlates with HER2 protein expression. ERBB2 mRNA expression (log2 values) across HER2 IHC categories (i.e., HER2‐0, HER2‐low, and HER2+ or HER2 0+, 1+, 2+, and 3+) in (A) all metastatic sites and (B) bone metastasis. Spearman correlation was determined between ERBB2 mRNA and HER2 protein expression. Examples of HER2 staining are represented at 10× and 20×.
3.3. Effect of organ site in gene expression profiling
The influence of organ site in gene expression profiling of metastatic breast cancer has not been formally addressed. To start approaching it, we first assessed the expression of 771 breast cancer‐related genes across the 184 metastatic tumor samples and 11 organ sites. Three out of 184 samples were identified as outliers and were excluded from the gene expression analysis (Fig S2). Unsupervised hierarchical clustering (Fig. S3) and principal component analysis (Fig. 4) revealed that the intrinsic subtypes explain a greater amount of gene expression variability than organ of metastasis. Secondly, we combined gene expression data from 186 patients with early‐stage breast cancer representative of all subtypes in the 181 metastatic dataset. The combined dataset (n = 367) of tumors obtained from early‐stage and metastatic breast cancer revealed that the 2 main principal components (i.e., PC1 and PC2) are also explained by intrinsic subtype (Fig. S4).
Fig. 4.
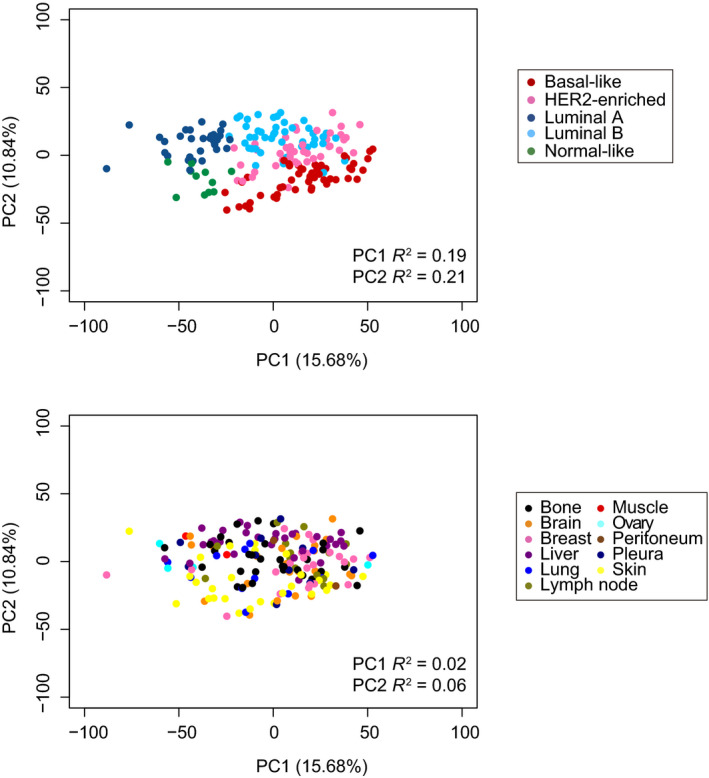
Principal component analysis. Unsupervised PCA of 181 metastatic samples with coloring of PAM50 molecular subtype and metastatic site. % of gene expression variability explained by each PC, and PC1 and PC2 R2 values obtained from simple linear regression models are show.
3.4. Genes and biological processes associated with organ site
To explore differences in gene expression across metastatic sites, we performed a multiclass SAM analysis. Using a FDR < 5%, we identified a total of 631 differentially expressed genes (81.1%) across organs (Table S3). Some examples were IBSP, which was highly expressed in bone metastasis; FGF1, which was highly expressed in brain metastasis; PCK1, which was highly expressed in liver metastasis; CAV1, which was highly expressed in lung metastasis; or KRT14, which was highly expressed in skin metastasis. Next, we performed a two‐class unpaired SAM analysis between each organ versus the rest of samples to identify genes whose expression is associated with each organ of metastasis. Using a FDR < 5%, we identified a total of 518 upregulated genes (67.2%) across organs (i.e., 204 in bone, 201 in skin, 109 in brain, 91 in liver, 29 in lung, and 7 in breast) (Table S4 and Fig. S5A). We did not identify any upregulated gene in lymph node or pleural metastases, nor any common gene upregulated in all metastatic sites (Fig. S5B). We also compared the bone samples of patients with bone‐only metastasis vs patients with metastasis in bone and other sites, and we could not find any significant differential expressed gene (Table S4).
We then carried out functional enrichment GO analyses using the upregulated gene lists, and although these analyses were limited by a minority of genes in each gene list, they revealed biological processes and pathways significantly enriched (P < 0.05) in each metastatic site (Table S5). In bone metastasis, ossification, the bone morphogenetic protein (BMP), the TGF‐beta, and the Hippo signaling pathways were enriched. In brain metastases, enriched GO and pathways included regulation of transcription and GTPase activity and cell migration and also brain‐related processes such as nervous system development, chemical synaptic transmission, adult behavior, dopamine synapse, or amphetamine addiction. In liver metastases, we identified enrichment in processes such as oxidation–reduction, glucose metabolism, chromatin remodeling, cholesterol esterification or vasodilation, and the AMPK and calcium signaling pathway. In lung metastasis, enriched GO and pathways included regulation of transcription, immune response, regulation of nitric oxide regulation, regulation of IL6 production, or regulation of vasoconstriction. Finally, enriched GO and pathways in skin metastasis included cell adhesion, angiogenesis, extracellular matrix organization, proteolysis, wound healing, epidermis development, collagen‐related processes and ERK and Notch pathways and lipid metabolism (Fig. S6).
The previous gene expression results could be confounded by differences in subtype distribution across organs. To identify genes whose high expression was specific of metastatic site and independent of subtype, we performed adjusted logistic regression analysis for each individual gene. A total of 74 genes were identified (P < 0.05): 36 bone‐specific genes, 18 liver‐specific genes, 12 brain‐specific genes, and 8 skin‐specific genes (Table 2). Of note, we identified known organ‐specific genes such as the integrin‐binding sialoprotein (IBSP) for bone, the crystallin alpha B (CRYAB) for brain, the aldehyde dehydrogenase 1 family member A1 (ALDH1A1) for liver, or KRT14 for skin. In addition, we identified 3 genes found in the PAM50 gene list to be associated with bone (FOXC1) and skin (KRT14 and KRT5) metastasis.
Table 2.
Subtype‐independent organ‐specific genes.
| Gene | Gene description | Gene location | Metastatic site | P‐value | Lawler et al. |
|---|---|---|---|---|---|
| WIF1 | WNT Inhibitory Factor 1 | 12q14.3 | Bone | 8.94E‐07 | High expression in primary tumors associated with bone+visceral metastasis |
| IBSP | Integrin‐binding Sialoprotein | 4q22.1 | Bone | 1.37E‐06 | |
| MMP9 | Matrix Metallopeptidase | 20q13.12 | Bone | 2.44E‐06 | |
| ITGB3 | Integrin Subunit Beta 3 | 17q21.32 | Bone | 2.74E‐06 | |
| VIT | Vitrin | 2p22.2 | Bone | 3.25E‐06 | High expression in primary tumors associated with bone+visceral metastasis |
| HBB | Hemoglobin Subunit Beta | 11p15.4 | Bone | 1.40E‐05 | |
| WNT5B | Wnt Family Member 5B | 12p13.33 | Bone | 3.01E‐05 | High expression in primary tumors associated with visceral‐only |
| CHAD | Chondroadherin | 17q21.33 | Bone | 3.22E‐05 | High expression in primary tumors associated with bone‐only |
| BMP2 | Bone Morphogenetic Protein 2 | 20p12.3 | Bone | 3.38E‐05 | |
| EYA1 | EYA Transcriptional Coactivator And Phosphatase 1 | 8q13.3 | Bone | 5.05E‐05 | High expression in primary tumors associated with bone‐only |
| FOXC2 | Forkhead Box C2 | 16q24.1 | bone | 8.48E‐05 | High expression in primary tumors associated with bone+visceral metastasis |
| FZD8 | Frizzled Class Receptor 8 | 10p11.21 | bone | 0.0001 | |
| OLFML2B | Olfactomedin‐like 2B | 1q23.3 | bone | 0.0001 | |
| TGFB1 | Transforming Growth Factor Beta 1 | 19q13.2 | bone | 0.0004 | High expression in primary tumors associated with bone‐only |
| BMP5 | Bone Morphogenetic Protein 5 | 6p12.1 | bone | 0.0005 | |
| ENPP2 | Ectonucleotide Pyrophosphatase/Phosphodiesterase 2 | 8q24.12 | bone | 0.0006 | High expression in primary tumors associated with visceral‐only |
| NUDT1 | Nudix Hydrolase 1 | 7p22.3 | bone | 0.0014 | High expression in primary tumors associated with visceral‐only |
| FGF7 | Fibroblast Growth Factor 7 | 15q21.2 | bone | 0.0015 | |
| FOXC1 | Forkhead Box C1 | 6p25.3 | bone | 0.0024 | High expression in primary tumors associated with visceral‐only |
| BMP8A | Bone Morphogenetic Protein 8a | 1p34.3 | bone | 0.0044 | |
| EYA4 | EYA Transcriptional Coactivator And Phosphatase 4 | 6q23.2 | bone | 0.0045 | |
| RNASE2 | Ribonuclease A Family Member 2 | 14q11.2 | bone | 0.006 | |
| SRPX | Sushi‐repeat Containing Protein X‐linked | Xp11.4 | bone | 0.006 | |
| MME | Membrane Metalloendopeptidase | 3q25.2 | bone | 0.0143 | High expression in primary tumors associated with bone+visceral metastasis |
| LIFR | LIF Receptor Subunit Alpha | 5p13.1 | bone | 0.0146 | |
| BAX | BCL2‐associated X, Apoptosis Regulator | 19q13.33 | bone | 0.0192 | High expression in primary tumors associated with bone‐only |
| SCARA5 | Scavenger Receptor Class A Member 5 | 8p21.1 | bone | 0.0211 | |
| EYA2 | EYA Transcriptional Coactivator And Phosphatase 2 | 20q13.12 | bone | 0.0219 | High expression in primary tumors associated with visceral‐only |
| XRCC3 | X‐ray Repair Cross‐complementing 3 | 14q32.33 | bone | 0.0268 | |
| LEPR | Leptin Receptor | 1p31.3 | bone | 0.0281 | |
| BCL2L1 | BCL2 Like 1 | 20q11.21 | bone | 0.0325 | |
| NCAM1 | Neural Cell Adhesion Molecule 1 | 11q23.2 | bone | 0.0342 | |
| SMAD3 | SMAD Family Member 3 | 15q22.33 | bone | 0.0368 | |
| RAC2 | Rac Family Small GTPase 2 | 22q13.1 | bone | 0.0449 | High expression in primary tumors associated with visceral‐only |
| HOXA9 | Homeobox A9 | 7p15.2 | bone | 0.0483 | High expression in primary tumors associated with bone‐only |
| CKB | Creatine Kinase B | 14q32.33 | bone | 0.049 | High expression in primary tumors associated with visceral‐only |
| CRYAB | Crystallin Alpha B | 11q23.1 | brain | 0.0006 | |
| NRCAM | Neuronal Cell Adhesion Molecule | 7q31.1 | brain | 0.0007 | |
| FGF1 | Fibroblast Growth Factor 1 | 5q31.3 | brain | 0.0008 | High expression in primary tumors associated with bone+visceral metastasis |
| GDF15 | Growth Differentiation Factor 15 | 19p13.11 | brain | 0.0021 | |
| SOX2 | SRY‐Box Transcription Factor 2 | 3q26.33 | brain | 0.0049 | High expression in primary tumors associated with bone+visceral metastasis |
| GRIN1 | Glutamate Ionotropic Receptor NMDA Type Subunit 1 | 9q34.3 | brain | 0.0075 | |
| RASGRF1 | Ras Protein‐specific Guanine Nucleotide‐releasing Factor 1 | 15q25.1 | brain | 0.0103 | High expression in primary tumors associated with visceral‐only |
| SOX10 | SRY‐Box Transcription Factor 10 | 22q13.1 | brain | 0.0199 | |
| CHI3L1 | Chitinase 3‐like 1 | 1q32.1 | brain | 0.0223 | High expression in primary tumors associated with visceral‐only |
| ZIC2 | Zic Family Member 2 | 13q32.3 | brain | 0.0276 | |
| NRXN1 | Neurexin 1 | 2p16.3 | brain | 0.0447 | |
| LEFTY2 | Left–Right Determination Factor 2 | 1q42.12 | brain | 0.0495 | |
| ALDH1A1 | Aldehyde Dehydrogenase 1 Family Member A1 | 9q21.13 | liver | 5.55E‐05 | |
| CYP4F3 | Cytochrome P450 Family 4 Subfamily F Member 3 | 19p13.12 | liver | 5.67E‐05 | High expression in primary tumors associated with bone‐only |
| PCK1 | Phosphoenolpyruvate Carboxykinase 1 | 20q13.31 | liver | 7.46E‐05 | |
| RELN | Reelin | 7q22.1 | liver | 0.0002 | |
| AGT | Angiotensinogen | 1q42.2 | liver | 0.0004 | |
| PPARGC1A | PPARG Coactivator 1 Alpha | 4p15.2 | liver | 0.0004 | |
| HNF1A | HNF1 Homeobox A | 12q24.31 | liver | 0.0009 | |
| CDH2 | Cadherin 2 | 18q12.1 | liver | 0.0029 | |
| APOE | Apolipoprotein E | 19q13.32 | liver | 0.0053 | |
| GGH | Gamma‐Glutamyl Hydrolase | 8q12.3 | liver | 0.0082 | High expression in primary tumors associated with visceral‐only |
| HGF | Hepatocyte Growth Factor | 7q21.11 | liver | 0.0159 | |
| MT1G | Metallothionein 1G | 16q13 | liver | 0.016 | |
| CLDN1 | Claudin 1 | 3q28 | liver | 0.017 | |
| UBB | Ubiquitin B | 17p11.2 | liver | 0.0173 | |
| HDAC1 | Histone Deacetylase 1 | 1p35.2‐p35.1 | liver | 0.0207 | |
| EDNRB | Endothelin Receptor Type B | 13q22.3 | liver | 0.0292 | |
| GATA4 | GATA‐binding Protein 4 | 8p23.1 | liver | 0.0444 | |
| MARCO | Macrophage Receptor With Collagenous Structure | 2q14.2 | liver | 0.0489 | High expression in primary tumors associated with visceral‐only |
| KRT14 | Keratin 14 | 17q21.2 | skin | 0.0005 | High expression in primary tumors associated with bone+visceral metastasis |
| KRT5 | Keratin 5 | 12q13.13 | skin | 0.0029 | |
| S100A7 | S100 Calcium‐binding Protein A7 | 1q21.3 | skin | 0.0044 | |
| SERPINB5 | Serpin Family B Member 5 | 18q21.33 | skin | 0.0069 | High expression in primary tumors associated with bone+visceral metastasis |
| MMP3 | Matrix Metallopeptidase 3 | 11q22.2 | skin | 0.0116 | |
| IL20RB | Interleukin 20 Receptor Subunit Beta | 3q22.3 | skin | 0.0133 | High expression in primary tumors associated with visceral‐only |
| SFN | Stratifin | 1p36.11 | skin | 0.021 | |
| TPSAB1 | Tryptase Alpha/Beta 1 | 16p13.3 | skin | 0.0333 |
Finally, we interrogated the 74 genes in 390 breast primary tumors from Lawler et al. [23] publicly available dataset, where 3 types of metastatic spread have been identified: bone and visceral metasynchronous spread, bone‐only spread, and visceral‐only metastasis. Among the 74 genes, 26 genes (35.1%) were found significantly associated with the type of metastatic spread, including 5 bone‐specific genes (CHAD, EYA1, TGFB1, BAX, and HOXA9) whose high expression was associated with bone‐only metastasis, 4 bone‐specific genes (WIF1, VIT, FOXC2, and MME) whose high expression was associated with bone and visceral metastasis, 2 brain‐specific genes (FGF1 and SOX2) whose high expression was associated with bone and visceral metastasis, 2 brain‐specific genes (RASGRF1 and CHI3L1) whose high expression was associated with visceral‐only metastasis, and 2 liver‐specific genes (GGH and MARCO) whose high expression was associated with visceral‐only metastasis (Table 2). This result suggests that particular metastatic organ‐specific genes might also be indicative of the type of metastatic spread when analyzed in primary tumors.
3.5. Immune expression profiles across organ sites
We then investigated differences in the expression of 95 immune genes across metastatic sites. The expression of 89 genes was found significantly different across metastatic sites (FDR < 5%) (Table S6 and Fig. S7A). Among them, we identified 18 genes of the tumor inflammation signature (TIS) (CCL5, CD27, CD276, CD274, CD8A, CMKLR1, CXCL9, CXCR6, HLA‐DQA1, HLA‐DRB1, HLA‐E, IDO1, LAG3, NKG7, PDCD1LG2, PSMB10, STAT1, and TIGIT) which has been previously associated with anti‐PD‐1/PD‐L1 response [28,29], 4 genes associated with CD8 T cells (CD8A, GZMM, CD8B, and PRF1), a marker of functional regulatory T cells (Treg) (FOXP3), a biomarker for B cells (CD19), and 4 macrophage‐related genes (C163, CD84, CD68, and CYBB) (Fig. 5). Lung and pleura were the sites with higher expression of immune genes, while brain had the lowest expression of immune genes. Moreover, we found 86 immune genes differentially expressed across the molecular subtypes (FDR < 5%) (Table S7). Basal‐like was the subtype with the highest expression of immune genes (Fig. S7B). Finally, to identify immune genes whose expression was specific of metastatic site and independent of subtype, we performed adjusted logistic regression analysis for each individual immune gene. Regardless of molecular subtype, we identified 27 highly expressed genes in lung, 23 highly expressed genes in pleura, 18 highly expressed and 9 lowly expressed genes in bone, 10 highly expressed and 21 lowly expressed genes in liver, or 7 highly expressed and 39 lowly expressed genes in brain (Table S8).
Fig. 5.
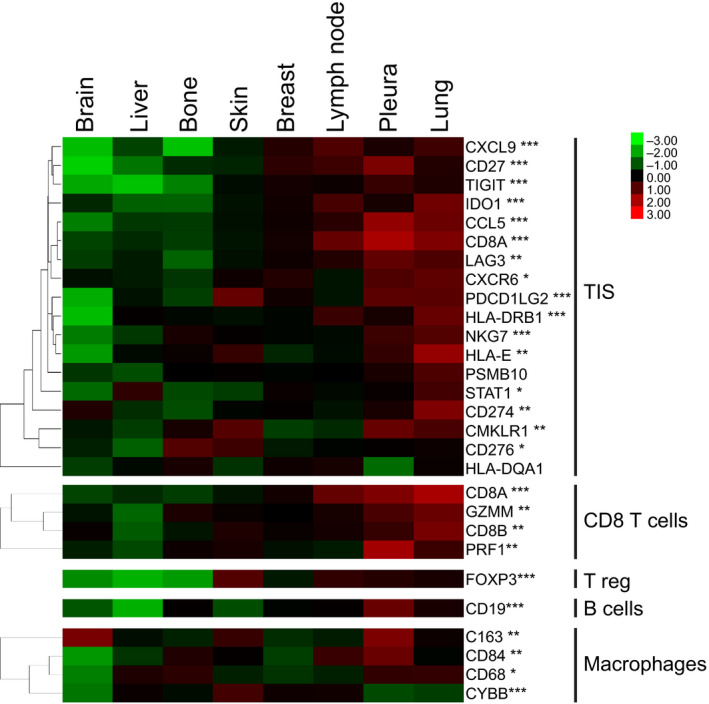
Differential expression of immune genes across metastatic sites. Expression of genes comprised in immune signatures across metastatic sites. The heatmap shows high (red) to low (green) expression of mRNAs in each metastatic site. Significant changes across metastatic sites by multiclass SAM analysis are indicated: *FDR < 5%, **FDR < 1%, and ***FDR < 0.1%.
3.6. Associations with overall survival
We evaluated the prognostic ability of the PAM50 subtypes and the site of metastasis. PAM50 molecular subtypes were associated with OS (P < 0.001) and better discriminated prognosis than site of metastasis (Fig. 6A). Median OS was 99.7 months for luminal A, 63.6 for luminal B, 34.7 months for HER2‐enriched, and 22.4 months for basal‐like.
Fig. 6.
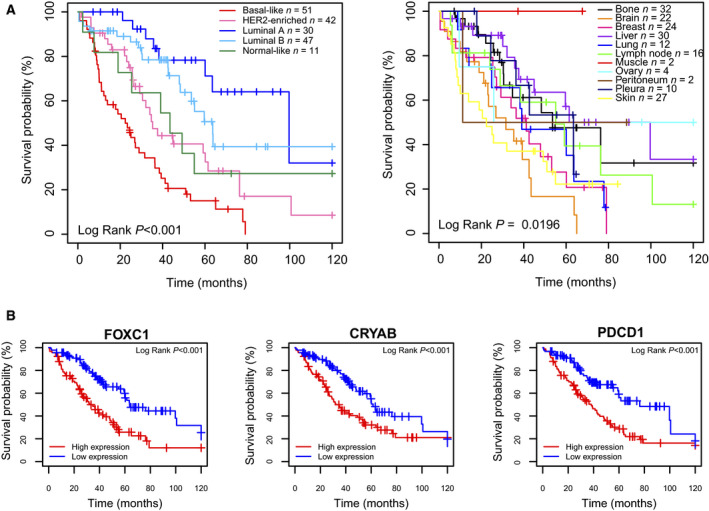
Associations with overall survival. (A) Kaplan–Meier curves of 10‐year OS (log‐rank test) according to PAM50 molecular subtype and metastatic site. (B) Examples genes associated with poor OS, including two organ‐specific genes (FOXC1 and CRYAB) and the immune gene PDCD1. Kaplan–Meier curves of 10‐year OS (log‐rank test) according to median gene expression of the selected genes.
We then explored the association of 771 individual genes and 9 signatures with OS. We identified 1 signature score (i.e., luminal A signature score) and 51 genes whose high expression was significantly associated with better OS, and 2 signature scores (i.e., basal‐like signature score and PAM50MET signature score [27]) and 25 genes whose high expression was significantly associated with worse OS (Table S9). When adjusting for PAM50 subtype, 45 of the 771 genes (5.8%) were significantly associated with OS (Fig. S8 and Table S10), of which the high expression of 9 genes was associated with worse OS (ENO1, CDCA5, FAM83D, ANLN, MMP7, CRYAB, FOXC1, E2F1, and PDCD1) including the bone‐specific gene FOXC1, the brain‐specific genes CRYAB, or the immune‐related gene PDCD1 (Fig. 6B). Finally, we performed a multivariate analysis adjusting for clinicopathological variables including menopausal status, type of metastasis (de novo or relapsed), number of metastatic sites, metastatic site of the biopsy, PAM50 molecular subtype, and number of lines of therapy and found that 5 of the 9 genes were still significantly associated with worse OS (FAM83D, ANLN, CRYAB, FOXC1, and E2F1) (Table S10).
4. Discussion
To our knowledge, this is the first study to evaluate gene expression profiles of breast cancer across metastatic organs. In particular, we explored genomic differences between sites of metastatic disease and made the following observations: (a) All intrinsic molecular subtypes are identified within IHC groups; (b) HER2‐low disease is identified in all metastatic sites; (c) intrinsic molecular subtypes determined in the metastatic site are associated with OS regardless of where biopsy was performed; (d) lung and pleural metastases have the highest expression of immune genes, while brain and liver have the lowest; and (e) the expression of individual genes is organ‐specific and is associated with OS.
Previously, we reported that approximately 15% of primary luminal A and B HR+/HER2‐negative tumors become HER2‐enriched once they metastasize, regardless of HER2 status [11, 17]. Concordant with this observation, here we observed a higher frequency of HER2‐enriched and basal‐like subtypes in HR+/HER2‐negative metastasis compared to primary tumors [17]. On the other side, in primary HER2+ disease we have previously reported 47% of HER2‐enriched, 24% of luminal A, 20% of luminal B, and 9% of basal‐like tumors. Here, despite the small HER2+ metastatic sample size, we did not detect luminal A tumors, while 70% were HER2‐enriched. Finally, approximately 60–80% of TNBC primary tumors have been reported to be basal‐like and 9% HER2‐enriched [32, 33] and our results showed similar distribution of molecular subtypes in metastatic TNBC.
The acquisition of more aggressive molecular subtypes in the metastatic setting, such as HER2‐enriched and basal‐like [34], especially in HR+/HER2‐negative disease, may be due to patient selection, changes in the tumor biology due to its inherent evolution, the effects of therapies, or a combination of all. Recently, comprehensive genomic studies of metastatic breast cancers linked an increase in APOBEC genetic signatures with metastatic HR+/HER2‐negative breast cancer [14, 35]. Interestingly, high frequency of APOBEC3B‐associated mutations occurs in HER2‐enriched subtype [36] which is consistent with the increase in this subtype observed in the metastatic setting.
Here, we also report HER2‐low disease in all metastatic sites, including bone. Bone metastases are usually not accepted for inclusion in clinical trials due to decalcification procedures related to IHC. Here, we show that ERBB2 mRNA is highly correlated with HER2 IHC in bone metastasis, suggesting that bone metastasis might be a reliable organ to detect HER2 expression. Nonetheless, alternative quantitative measurements of HER2 (i.e., ERBB2 mRNA) may help better identify patients who might benefit from potent anti‐HER2 antibody–drug conjugates, like T‐DXd [31, 37].
Interestingly, we have previously observed in a wide population of almost 1600 HER2‐negative tumors that HER2‐low disease was enriched in luminal molecular subtypes (about 80%), especially when compared to HER2‐0 (about 50%) [29]. In the present study on a smaller sample size (80 HER2‐low specimens), luminal subtypes accounted for roughly half of the total and no difference in subtypes distribution was observed between HER2‐low and HER2‐0 tumors. However, in our previous study, only 2.4% of HER2‐low tumors were metastatic. A potential shift in molecular subtype distribution between primary and metastatic tumors might thus merit a more careful evaluation, along with its potential prognostic and therapeutic implications.
Our study identified particular genes differentially expressed across metastatic sites, suggesting a potential role of the tumor microenvironment. Indeed, our functional enrichment analysis of upregulated genes in each metastatic site identified biological processes and pathways related with the organ where metastasis was seeded. Moreover, we have validated some previously reported overexpressed genes such as TGFB1, IBSP, MMP9, or ITGB3 in bone metastasis [4, 38, 39]; CRYAB, NRCAM, and SOX2 in brain metastasis [40, 41, 42]; VEGF and IL6 in lung metastasis [4, 43]; and CYP4F3 in liver metastasis [44]. ALDH1A1, PCK1, and APOE were previously described to be upregulated in liver metastases of colorectal cancer [45, 46, 47]. Further studies are required to understand whether these genes could be used as therapeutic targets or biomarkers of response.
Our data indicate that lung and pleura are the sites of metastasis with higher expression of immune genes, while brain and liver have the lowest expression of immune genes. This is consistent with the findings of a recently published study of over 400 metastatic samples which indicate that lung metastasis has the highest TIS compared to other metastatic sites regardless of the cancer of origin [48]. Notably, high TIS is a biomarker of response to immunotherapy [49, 50]. Taken together, these data suggest that patients with lung and pleural metastasis might benefit from immune checkpoint blockade, while other treatment approaches could be more suitable for liver and brain metastases.
Concordant with early‐stage breast cancer [51], PAM50 subtypes in metastatic tissues were found highly prognostic. At the same time, we identified 45 genes whose expression provides prognostic information beyond PAM50 subtypes. For example, we identified high PD1 expression as being associated with poor prognosis. Functional studies are needed to better understand whether the genes associated with worse OS could also be therapeutic targets. Indeed, high PD1 mRNA might be a tumor‐agnostic biomarker of benefit from anti‐PD1 therapy [52].
Our study has some limitations worth noting. First, this is a retrospective study using the available metastatic tumor samples at Hospital Clinic of Barcelona and a set of 23 TNBC samples from Hospital Universitario 12 de Octubre; therefore, selection bias is likely. For instance, we had a very small HER2+ sample size. However, the distribution of IHC groups was very similar to the seminal work by Bertucci and colleagues [14]. On the other hand, the distribution of the organs of the selected biopsies may not reflect the actual frequency of breast cancer metastatic sites due to the accessibility to the organs of metastasis. Second, our cohort is very heterogeneous in terms of systemic therapies received. Thus, we could not link the biological findings with treatment benefit. Third, our dataset surprisingly had longer median OS than expected, possibly because those patients that have biopsies are the more likely to survive longer. Indeed, having a biopsy upon recurrence has been associated with longer survival [53]. Fourth, our analyses are limited to 771 genes; whether different results might be obtained with more genes is unknown. Fifth, we did not explore the biological differences across metastatic sites within a single patient.
5. Conclusion
In summary, although main molecular features from primary tumors are known to be maintained in advanced disease [9, 10], here we report higher proportion of aggressive molecular subtypes in the metastatic setting, especially in HR+/HER2‐negative disease, and unique biological features of each metastatic site, indicating a role of the tumor microenvironment and the need to biopsy metastatic disease in patients with advanced breast cancer to better select the treatment strategy for each patient. Understanding the biology of each metastatic site can potentially impact the design of new therapies and ultimately improve patient outcomes. Finally, our study provides a precious dataset of cancer metastasis that can be further exploited.
Conflict of interest
Potential conflicts of interest are the following: A.P. reports advisory and consulting fees from Roche, Pfizer, Novartis, Amgen, BMS, Puma, Oncolytics Biotech, MSD, Guardant Health, Peptomyc, and Lilly, lecture fees from Roche, Pfizer, Novartis, Amgen, BMS, NanoString Technologies, and Daiichi Sankyo, institutional financial interests from Boehringer, Novartis, Roche, NanoString, Sysmex Europe GmbH, Medica Scientia inno. Research, SL, Celgene, Astellas, and Pfizer; a leadership role in Reveal Genomics, SL; and a patent PCT/EP2016/080056.
Author contributions
FB‐M and AP performed experimental study design. FB‐M, NC, OM‐S, TP, MM‐L, BG‐F, ES, DM, PG, EB, PT, ECi, ECa, BA, RM, MV, MM, and AP acquired the data. FB‐M, LP, FS, BG‐F, ES, DM, PG, EB, and AP analyzed the data. FB‐M, NC, OM‐S, TP, MM‐L, BG‐F, ES, PT, ECi, MM, and AP interpreted the data. FB‐M and AP wrote the manuscript. All authors reviewed the manuscript.
Peer Review
The peer review history for this article is available at https://publons.com/publon/10.1002/1878‐0261.13021.
Supporting information
Fig. S1. Correlation between ESR1 mRNA and % ER protein expression. Pearson correlation between ESR1 mRNA and ER protein expression across all metastatic sites (n = 148) and across bone metastasis (n = 29) with coloring of PAM50 molecular subtype.
Fig. S2. PCA in 184 metastatic tumors. Unsupervised PCA of 181 metastatic tumors with coloring of samples included in the gene expression analyses (grey) and the outliers, excluded from gene expression analyses (red).
Fig. S3. Gene expression features. Unsupervised hierarchical clustering of 181 metastatic samples. Heatmaps show high (red) to low (green) expression of mRNAs in each sample. The organ of biopsy, IHC and PAM50 molecular subtype of each sample are shown.
Fig. S4. PCA in primary and metastatic tumors. Unsupervised PCA of 186 primary and 181 metastatic tumors with coloring of type of biopsy (primary [P] vs metastatic [M]) and PAM50 molecular subtypes.
Fig. S5. Differential gene expression across metastatic sites. (A) Volcano plots showing differentially expressed genes in each organ vs others (B) Venn diagram showing common significantly upregulated genes in all metastatic sites. Each circle includes the number of genes upregulated and the sites where these genes are upregulated.
Fig. S6. Functional enrichement analysis of upregulated genes. Gene ontology (GO) and KEGG pathway analysis were performed using DAVID. Significantly enriched (p < 0.05) biological processes and pathways are presented. Functional enrichement analysis of upregulated genes. Gene ontology (GO) and KEGG pathway analysis were performed using DAVID. Significantly enriched (p < 0.05) biological processes and pathways are presented.
Fig. S7. Expression of immune genes. Differential expression of immune genes across (A) metastatic sites and (B) PAM50 molecular subtypes. Heatmaps show high (red) to low (green) expression of RNAs in each sample. Significantly different gene expression were identified using multiclass SAM (*FDR<5%, **FDR<1%, ***FDR<0.01%).
Fig. S8. Associations with overall survival. Forest plot showing genes and signatures associated with OS.
Table S1. Distribution of PAM50 subtypes and IHC groups across metastatic sites.
Table S2. Distribution of PAM50 subtypes in each metastatic site according to IHC group.
Table S3. Multiclass SAM of 771 genes between metastatic sites.
Table S4. Unpaired SAM between each metastatic site and others.
Table S5. Functional enrichment GO analyses of the up‐regulated genes in each metastatic site.
Table S6. Multiclass SAM of 95 immune genes between metastatic sites.
Table S7. Multiclass SAM of 95 immune genes between molecular subtypes.
Table S8. Adjusted logistic regression analysis for 95 immune genes.
Table S9. Univariate analysis to investigate the association of 771 individual genes and 9 signatures with OS.
Table S10. Bivariate analysis (adjusted for PAM50) and multivariate analysis (adjusted for menopausal status, type of metastasis, number of metastatic sites, metastatic site of the biopsy, PAM50 subtype, number of lines of therapy) to investigate the association of 771 individual genes and 9 signatures with OS.
Acknowledgements
This study has received funding from Instituto de Salud Carlos III—PI16/00904 and PI19/01846 (to A.P.), Breast Cancer Now—2018NOVPCC1294 (to A.P.), Breast Cancer Research Foundation‐AACR Career Development Awards for Translational Breast Cancer Research 19‐20‐26‐PRAT (to A.P.), Fundació La Marató TV3 201935‐30 (to A.P.), the RESCUER, funded by European Union's Horizon 2020 Research and Innovation Programme under Grant Agreement No. 847912 (to A.P.) Pas a Pas (to A.P.), Save the Mama (to A.P.), Fundación Científica Asociación Española Contra el Cáncer AECC_Postdoctoral17‐1062 (to F.B‐M.), Generalitat de Catalunya Peris PhD4MD 2019 SLT008/18/00122 (to N.C.), Fundación SEOM, Becas FSEOM para Formación en Investigación en Centros de Referencia en el Extranjero 2018 (to T.P.), and Fundación Científica Asociación Española Contra el Cáncer AECC_PLCA18 (M. M‐L.). [Correction added on 28 September 2021, after first online publication: the phrase ‘European Union’s Horizon 2020 research and innovation programme H2020‐SC1‐BHC‐2018‐2020 (to A.P.)’ has been modified to ‘RESCUER, funded by European Union's Horizon 2020 Research and Innovation Programme under Grant Agreement No. 847912 (to A.P.)’.]
Data accessibility
The data that support this study are available in Tables S1–10 and are derived from the gene expression data deposited in the Gene Expression Omnibus under the accession number GSE175692.
References
- 1. Harbeck N, Penault‐Llorca F, Cortes J, Gnant M, Houssami N, Poortmans P, Ruddy K, Tsang J & Cardoso F (2019) Breast cancer. Nat Rev Dis Prim 5, 1–31. [DOI] [PubMed] [Google Scholar]
- 2. Spratt DE, Spratt EAG, Wu S, De Rosa A, Lee NY, Lacouture ME & Barker CA (2014) Efficacy of skin‐directed therapy for cutaneous metastases from advanced cancer: a meta‐analysis. J Clin Oncol 32, 3144–3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Schwarz C, Lübbert H, Rahn W, Schönfeld N, Serke M & Loddenkemper R (2004) Medical thoracoscopy: hormone receptor content in pleural metastases due to breast cancer. Eur Respir J 24, 728–730. [DOI] [PubMed] [Google Scholar]
- 4. Chen W, Hoffmann AD, Liu H & Liu X (2018) Organotropism: new insights into molecular mechanisms of breast cancer metastasis. NPJ Precis Oncol 2, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kotecki N, Lefranc F, Devriendt D & Awada A (2018) Therapy of breast cancer brain metastases: challenges, emerging treatments and perspectives. Ther Adv Med Oncol 10, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bale R, Putzer D & Schullian P (2019) Local treatment of breast cancer liver metastasis. Cancers 11, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wu Q, Li J, Zhu S, Wu J, Chen C, Liu Q, Wei W, Zhang Y & Sun S (2017) Breast cancer subtypes predict the preferential site of distant metastases: a SEER based study. Oncotarget 8, 27990–27996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Paget S (1889) The distribution of secondary growths in cancer of the breast. Lancet 133, 571–573. [PubMed] [Google Scholar]
- 9. Weigelt B, Hu Z, He X, Livasy C, Carey LA, Ewend MG, Glas AM, Perou CM & van't Veer LJ (2005) Molecular portraits and 70‐gene prognosis signature are preserved throughout the metastatic process of breast cancer. Cancer Res 65, 9155–9158. [DOI] [PubMed] [Google Scholar]
- 10. De Dueñas EM, Hernández AL, Zotano ÁG, Carrión RMP, López‐Muñiz JIC, Novoa SA, Rodríguez ÁL, Fidalgo JAP, Lozano JF, Gasión OB et al. (2014) Prospective evaluation of the conversion rate in the receptor status between primary breast cancer and metastasis: results from the GEICAM 2009–03 ConvertHER study. Breast Cancer Res Treat 143, 507–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cejalvo JM, Martínez de Dueñas E, Galván P, García‐Recio S, Burgués Gasión O, Paré L, Antolín S, Martinello R, Blancas I, Adamo B et al. (2017) Intrinsic subtypes and gene expression profiles in primary and metastatic breast cancer. Cancer Res 77, 2213–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Osborne CK & Schiff R (2011) Mechanisms of endocrine resistance in breast cancer. Annu Rev Med 62, 233–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fabi A, Di Benedetto A, Metro G, Perracchio L, Nisticò C, Di Filippo F, Ercolani C, Ferretti G, Melucci E, Buglioni S et al. (2011) HER2 protein and gene variation between primary and metastatic breast cancer: significance and impact on patient care. Clin Cancer Res 17, 2055–2064. [DOI] [PubMed] [Google Scholar]
- 14. Bertucci F, Ng CKY, Patsouris A, Droin N, Piscuoglio S, Carbuccia N, Soria JC, Dien AT, Adnani Y, Kamal M et al. (2019) Genomic characterization of metastatic breast cancers. Nature 569, 560–564. [DOI] [PubMed] [Google Scholar]
- 15. O’Leary B, Finn RS & Turner NC (2016) Treating cancer with selective CDK4/6 inhibitors. Nat Rev Clin Oncol 13, 417–430. [DOI] [PubMed] [Google Scholar]
- 16. Nayar U, Cohen O, Kapstad C, Cuoco MS, Waks AG, Wander SA, Painter C, Freeman S, Persky NS, Marini L et al. (2019) Acquired HER2 mutations in ER + metastatic breast cancer confer resistance to estrogen receptor–directed therapies. Nat Genet 51, 207–216. [DOI] [PubMed] [Google Scholar]
- 17. Cejalvo JM, Pascual T, Fernández‐Martínez A, Brasó‐Maristany F, Gomis RR, Perou CM, Muñoz M & Prat A (2018) Clinical implications of the non‐luminal intrinsic subtypes in hormone receptor‐positive breast cancer. Cancer Treat Rev 67, 63–70. [DOI] [PubMed] [Google Scholar]
- 18. Brasó‐Maristany F, Griguolo G, Pascual T, Paré L, Nuciforo P, Llombart‐Cussac A, Bermejo B, Oliveira M, Morales S, Martínez N et al. (2020) Phenotypic changes of HER2‐positive breast cancer during and after dual HER2 blockade. Nat Commun 11, 385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yu J, Green MD, Li S, Sun Y, Journey SN, Choi JE, Rizvi SM, Qin A, Waninger JJ, Lang X et al. (2021) Liver metastasis restrains immunotherapy efficacy via macrophage‐mediated T cell elimination. Nat Med 27, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li Y, Chang C‐W, Tran D, Denker M, Hegde P & Molinero L (2018) Abstract PD6‐01: Prevalence of PDL1 and tumor infiltrating lymphocytes (TILs) in primary and metastatic TNBC. In Cancer Research pp. PD6‐01‐PD6‐01. American Association for Cancer Research (AACR).
- 21. Wolff AC, Elizabeth Hale Hammond M, Allison KH, Harvey BE, Mangu PB, Bartlett JMS, Bilous M, Ellis IO, Fitzgibbons P, Hanna W et al. (2018) Human epidermal growth factor receptor 2 testing in breast cancer: American society of clinical oncology/ college of American pathologists clinical practice guideline focused update. J Clin Oncol 36, 2105–2122. [DOI] [PubMed] [Google Scholar]
- 22. Allison KH, Hammond MEH, Dowsett M, McKernin SE, Carey LA, Fitzgibbons PL, Hayes DF, Lakhani SR, Chavez‐MacGregor M, Perlmutter J et al. (2020) Estrogen and progesterone receptor testing in breast cancer: ASCO/CAP guideline update. J Clin Oncol 38, 1346–1366. [DOI] [PubMed] [Google Scholar]
- 23. Lawler K, Papouli E, Naceur‐Lombardelli C, Mera A, Ougham K, Tutt A, Kimbung S, Hedenfalk I, Zhan J, Zhang H et al. (2017) Gene expression modules in primary breast cancers as risk factors for organotropic patterns of first metastatic spread: a case control study. Breast Cancer Res 19, 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Adamo B, Bellet M, Paré L, Pascual T, Vidal M, Pérez Fidalgo JA, Blanch S, Martinez N, Murillo L, Gómez‐Pardo P et al. (2019) Oral metronomic vinorelbine combined with endocrine therapy in hormone receptor‐positive HER2‐negative breast cancer: SOLTI‐1501 VENTANA window of opportunity trial. Breast Cancer Res 21, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Parker JS, Mullins M, Cheang MCU, Leung S, Voduc D, Vickery T, Davies S, Fauron C, He X, Hu Z et al. (2009) Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol 27, 1160–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nielsen TO, Parker JS, Leung S, Voduc D, Ebbert M, Vickery T, Davies SR, Snider J, Stijleman IJ, Reed J et al. (2010) A comparison of PAM50 intrinsic subtyping with immunohistochemistry and clinical prognostic factors in tamoxifen‐treated estrogen receptor‐positive breast cancer. Clin Cancer Res 16, 5222–5232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Prat A, Tsai Y‐H, Pascual T, Paré L, Adamo B, Vidal M, Brasó‐Maristany F, Galván P, Brase JC, Rodrik‐Outmezguine V et al. (2020) A prognostic model based on PAM50 and clinical variables (PAM50MET) for metastatic hormone‐receptor‐positive HER2‐negative breast cancer. Clin Cancer Res 26, 6141–6148. [DOI] [PubMed] [Google Scholar]
- 28. Tusher VG, Tibshirani R & Chu G (2001) Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci USA 98, 5116–5121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Schettini F, Chic N, Brasó‐Maristany F, Paré L, Pascual T, Conte B, Martínez‐Sáez O, Adamo B, Vidal M, Barnadas E et al. (2021) Clinical, pathological, and PAM50 gene expression features of HER2‐low breast cancer. NPJ Breast Cancer 7, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dennis G, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC & Lempicki RA (2003) DAVID: database for annotation, visualization, and integrated discovery. Genome Biol 4, P3. [PubMed] [Google Scholar]
- 31. Modi S, Park H, Murthy RK, Iwata H, Tamura K, Tsurutani J, Moreno‐Aspitia A, Doi T, Sagara Y, Redfern C et al. (2020) Antitumor activity and safety of trastuzumab deruxtecan in patients with HER2‐low‐expressing advanced breast cancer: results from a phase Ib study. J Clin Oncol 38, 1887–1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Perou CM (2010) Molecular stratification of triple‐negative breast cancers. Oncologist 15, 39–48. [DOI] [PubMed] [Google Scholar]
- 33. Koboldt DC, Fulton RS, McLellan MD, Schmidt H, Kalicki‐Veizer J, McMichael JF, Fulton LL, Dooling DJ, Ding L, Mardis ER et al. (2012) Comprehensive molecular portraits of human breast tumours. Nature 490, 61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Prat A, Chaudhury A, Solovieff N, Paré L, Martinez D, Chic N, Martínez‐Sáez O, Brasó‐Maristany F, Lteif A, Taran T et al. (2021) Correlative biomarker analysis of intrinsic subtypes and efficacy across the MONALEESA phase III studies. J Clin Oncol 39, 1458–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lefebvre C, Bachelot T, Filleron T, Pedrero M, Campone M, Soria J‐C, Massard C, Lévy C, Arnedos M, Lacroix‐Triki M et al. (2016) Mutational profile of metastatic breast cancers: a retrospective analysis. PLoS Medicine 13, e1002201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Roberts SA, Lawrence MS, Klimczak LJ, Grimm SA, Fargo D, Stojanov P, Kiezun A, Kryukov GV, Carter SL, Saksena G et al. (2013) An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat Genet 45, 970–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Griguolo G, Brasó‐Maristany F, González‐Farré B, Pascual T, Chic N, Saurí T, Kates R, Gluz O, Martínez D, Paré L et al. (2020) ERBB2 mRNA expression and response to Ado‐Trastuzumab Emtansine (T‐DM1) in HER2‐positive breast cancer. Cancers (Basel) 12, 1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhang Y, He W & Zhang S (2019) Seeking for correlative genes and signaling pathways with bone metastasis from breast cancer by integrated analysis. Front Oncol 9, 138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sloan EK, Pouliot N, Stanley KL, Chia J, Moseley JM, Hards DK & Anderson RL (2006) Tumor‐specific expression of αvβ3 integrin promotes spontaneous metastasis of breast cancer to bone. Breast Cancer Res 8, R20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Voduc KD, Nielsen TO, Perou CM, Harrell JC, Fan C, Kennecke H, Minn AJ, Cryns VL & Cheang MCU (2015) αB‐crystallin expression in breast cancer is associated with brain metastasis. NPJ Breast Cancer 1, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sehgal A, Boynton AL, Young RF, Vermeulen SS, Yonemura KS, Kohler EP, Aldape HC, Simrell CR & Murphy GP (1998) Cell adhesion molecule Nr‐CAM is over‐expressed in human brain tumors. Int J Cancer 76, 451–458. [DOI] [PubMed] [Google Scholar]
- 42. Lee JY, Park K, Lee E, Ahn TJ, Jung HH, Lim SH, Hong M, Do IG, Cho EY, Kim DH et al. (2016) Gene expression profiling of breast cancer brain metastasis. Sci Rep 6, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jayatilaka H, Tyle P, Chen JJ, Kwak M, Ju J, Kim HJ, Lee JSH, Wu PH, Gilkes DM, Fan R et al. (2017) Synergistic IL‐6 and IL‐8 paracrine signalling pathway infers a strategy to inhibit tumour cell migration. Nat Commun 8, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kimbung S, Johansson I, Danielsson A, Veerla S, Brage SE, Stolt MF, Skoog L, Carlsson L, Einbeigi Z, Lidbrink E et al. (2016) Transcriptional profiling of breast cancer metastases identifies liver metastasis‐selective genes associated with adverse outcome in luminal a primary breast cancer. Clin Cancer Res 22, 146–157. [DOI] [PubMed] [Google Scholar]
- 45. van der Waals LM , Borel Rinkes IHM & Kranenburg O (2018) ALDH1A1 expression is associated with poor differentiation, ‘right‐sidedness’ and poor survival in human colorectal cancer. PLoS One 13, e0205536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yamaguchi N, Weinberg EM, Nguyen A, Liberti MV, Goodarzi H, Janjigian YY, Paty PB, Saltz LB, Kingham TP, Loo J et al. (2019) PCK1 and DHODH drive colorectal cancer liver metastatic colonization and hypoxic growth by promoting nucleotide synthesis. Elife 8, e52135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhao Z, Zou S, Guan X, Wang M, Jiang Z, Liu Z, Li C, Lin H, Liu X, Yang R et al. (2018) Apolipoprotein E overexpression is associated with tumor progression and poor survival in colorectal cancer. Front Genet 9, 650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. García‐Mulero S, Alonso MH, Pardo J, Santos C, Sanjuan X, Salazar R, Moreno V, Piulats JM & Sanz‐Pamplona R (2020) Lung metastases share common immune features regardless of primary tumor origin. J Immunother Cancer 8, e000491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR, Albright A, Cheng JD, Kang SP, Shankaran V et al. (2017) IFN‐γ‐related mRNA profile predicts clinical response to PD‐1 blockade. J Clin Invest 127, 2930–2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Danaher P, Warren S, Lu R, Samayoa J, Sullivan A, Pekker I, Wallden B, Marincola FM & Cesano A (2018) Pan‐cancer adaptive immune resistance as defined by the Tumor Inflammation Signature (TIS): Results from The Cancer Genome Atlas (TCGA). J Immunother Cancer 6, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Prat A & Perou CM (2011) Deconstructing the molecular portraits of breast cancer. Mol Oncol 5, 5–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Paré L, Pascual T, Seguí E, Teixidó C, Gonzalez‐Cao M, Galván P, Rodríguez A, González B, Cuatrecasas M, Pineda E et al. (2018) Association between PD1 mRNA and response to anti‐PD1 monotherapy across multiple cancer types. Ann Oncol 29, 2121–2128. [DOI] [PubMed] [Google Scholar]
- 53. Shachar SS, Mashiach T, Fried G, Drumea K, Shafran N, Muss HB & Bar‐Sela G (2017) Biopsy of breast cancer metastases: patient characteristics and survival. BMC Cancer 17, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Correlation between ESR1 mRNA and % ER protein expression. Pearson correlation between ESR1 mRNA and ER protein expression across all metastatic sites (n = 148) and across bone metastasis (n = 29) with coloring of PAM50 molecular subtype.
Fig. S2. PCA in 184 metastatic tumors. Unsupervised PCA of 181 metastatic tumors with coloring of samples included in the gene expression analyses (grey) and the outliers, excluded from gene expression analyses (red).
Fig. S3. Gene expression features. Unsupervised hierarchical clustering of 181 metastatic samples. Heatmaps show high (red) to low (green) expression of mRNAs in each sample. The organ of biopsy, IHC and PAM50 molecular subtype of each sample are shown.
Fig. S4. PCA in primary and metastatic tumors. Unsupervised PCA of 186 primary and 181 metastatic tumors with coloring of type of biopsy (primary [P] vs metastatic [M]) and PAM50 molecular subtypes.
Fig. S5. Differential gene expression across metastatic sites. (A) Volcano plots showing differentially expressed genes in each organ vs others (B) Venn diagram showing common significantly upregulated genes in all metastatic sites. Each circle includes the number of genes upregulated and the sites where these genes are upregulated.
Fig. S6. Functional enrichement analysis of upregulated genes. Gene ontology (GO) and KEGG pathway analysis were performed using DAVID. Significantly enriched (p < 0.05) biological processes and pathways are presented. Functional enrichement analysis of upregulated genes. Gene ontology (GO) and KEGG pathway analysis were performed using DAVID. Significantly enriched (p < 0.05) biological processes and pathways are presented.
Fig. S7. Expression of immune genes. Differential expression of immune genes across (A) metastatic sites and (B) PAM50 molecular subtypes. Heatmaps show high (red) to low (green) expression of RNAs in each sample. Significantly different gene expression were identified using multiclass SAM (*FDR<5%, **FDR<1%, ***FDR<0.01%).
Fig. S8. Associations with overall survival. Forest plot showing genes and signatures associated with OS.
Table S1. Distribution of PAM50 subtypes and IHC groups across metastatic sites.
Table S2. Distribution of PAM50 subtypes in each metastatic site according to IHC group.
Table S3. Multiclass SAM of 771 genes between metastatic sites.
Table S4. Unpaired SAM between each metastatic site and others.
Table S5. Functional enrichment GO analyses of the up‐regulated genes in each metastatic site.
Table S6. Multiclass SAM of 95 immune genes between metastatic sites.
Table S7. Multiclass SAM of 95 immune genes between molecular subtypes.
Table S8. Adjusted logistic regression analysis for 95 immune genes.
Table S9. Univariate analysis to investigate the association of 771 individual genes and 9 signatures with OS.
Table S10. Bivariate analysis (adjusted for PAM50) and multivariate analysis (adjusted for menopausal status, type of metastasis, number of metastatic sites, metastatic site of the biopsy, PAM50 subtype, number of lines of therapy) to investigate the association of 771 individual genes and 9 signatures with OS.
Data Availability Statement
The data that support this study are available in Tables S1–10 and are derived from the gene expression data deposited in the Gene Expression Omnibus under the accession number GSE175692.