

Abstract
Glioblastoma is the most frequently diagnosed type of primary brain tumour in adults. These aggressive tumours are characterised by inherent treatment resistance and disease progression, contributing to ~ 190 000 brain tumour‐related deaths globally each year. Current therapeutic interventions consist of surgical resection followed by radiotherapy and temozolomide chemotherapy, but average survival is typically around 1 year, with < 10% of patients surviving more than 5 years. Recently, a fourth treatment modality of intermediate‐frequency low‐intensity electric fields [called tumour‐treating fields (TTFields)] was clinically approved for glioblastoma in some countries after it was found to increase median overall survival rates by ~ 5 months in a phase III randomised clinical trial. However, beyond these treatments, attempts to establish more effective therapies have yielded little improvement in survival for patients over the last 50 years. This is in contrast to many other types of cancer and highlights glioblastoma as a recognised tumour of unmet clinical need. Previous work has revealed that glioblastomas contain stem cell‐like subpopulations that exhibit heightened expression of DNA damage response (DDR) factors, contributing to therapy resistance and disease relapse. Given that radiotherapy, chemotherapy and TTFields‐based therapies all impact DDR mechanisms, this Review will focus on our current knowledge of the role of the DDR in glioblastoma biology and treatment. We also discuss the potential of effective multimodal targeting of the DDR combined with standard‐of‐care therapies, as well as emerging therapeutic targets, in providing much‐needed improvements in survival rates for patients.
Keywords: chemotherapy, DNA damage response, glioblastoma, radiotherapy, synthetic lethality, tumour‐treating fields
Glioblastoma remains a cancer of unmet clinical need. Current treatment incorporates DNA damaging therapies which activate an intricate, multifaceted DNA damage response (DDR). This Review discusses the role of the DDR in the complex biology and therapeutic response of glioblastoma. Additionally, novel multimodal approaches to target the DDR alongside standard‐of‐care therapies are presented, towards the goal of improving patient survival.
Abbreviations
- ATM
ataxia telangiectasia mutated
- ATR
ataxia telangiectasia and Rad3‐related kinase
- BBB
blood–brain barrier
- BER
base excision repair
- BRCA
breast cancer gene
- CDK
cyclin‐dependent kinase
- cGAS
cyclic GMP‐AMP synthase
- CSCs
cancer stem cells
- DDR
DNA damage response
- DNA‐PK
DNA‐dependent protein kinase
- DSB
double‐strand break
- ERK5
extracellular‐related signalling kinase 5
- FA
Fanconi anaemia
- FDA
Food and Drug Administration (USA)
- GSCs
glioma stem‐like cells
- HGG
high‐grade glioma
- HR
homologous recombination
- ICL
interstrand crosslink
- IDH
isocitrate dehydrogenase
- IR
ionising radiation
- MGMT
methylguanine methyltransferase
- MMR
mismatch repair
- MRI
magnetic resonance imaging
- NER
nucleotide excision repair
- NHEJ
nonhomologous end joining
- O6MeG
O6‐methylguanine
- PARP
poly (ADP‐ribose) polymerase
- PIKK
phosphatidylinositol 3‐kinase‐related kinases
- ROS
reactive oxygen species
- RSS
replication stress signalling
- SSB
single‐strand break
- SSL
synthetic sensitivity or lethality
- STING
stimulator of interferon genes
- TMZ
temozolomide
- TTFields
tumour‐treating fields
- WHO
World Health Organization
1. Introduction
1.1. Glioblastoma and current treatment regimes
Brain tumours are globally responsible for around 190 000 deaths per year (around 5000 of which are in the UK) and are responsible for the greatest reduction in life expectancy of any cancer – around 20 years on average [1, 2]. Glioblastoma is ascribed the highest glioma grade designated by the World Health Organization (WHO grade IV glioma). It is the most frequently diagnosed primary brain tumour in adults and is associated with an exceptionally poor clinical course characterised by treatment resistance, rapid disease progression and dire patient survival rates of around 12–15 months following diagnosis in clinical studies [3]. However, average survival for unselected patients in the real‐world setting is typically closer to 8 months [4]. In most cases, glioblastoma arises rapidly without previous clinical presentation or histological confirmation of a less malignant precursor lesion, although, in a minority of cases, signs of progression from a lower grade diffuse (WHO grade II) or anaplastic (WHO grade III) astrocytoma are evident [5].
Despite some recent advances in the genetic, epigenetic and molecular subtyping of gliomas [6], the current standard‐of‐care treatment remains maximal safe surgical resection followed by a course of radiotherapy with concomitant and adjuvant chemotherapy [7, 8, 9]. The mainstay chemotherapeutic agent is oral delivery of the DNA alkylating agent temozolomide following successful clinical trial data evidencing a 2.5‐month increase in patient survival rates [7, 8]. However, despite this aggressive course of therapy, median patient survival was estimated at 14.6 months, with < 10% of patients surviving more than 5 years. Importantly, however, in the subset of patients with promoter methylation of the DNA repair gene MGMT (discussed below), the addition of temozolomide was associated with overall survival of 21.7 months, representing an increase of 6.4 months [10]. More recent studies corroborate the benefit of temozolomide in the context of methylated MGMT status [11, 12]. However, in contrast, efforts to improve the survival of patients with an unmethylated MGMT promoter have been less successful [13, 14].
Since the approval of temozolomide in 2005, no new approved treatments for glioblastoma were forthcoming until the recent development and clinical approval of tumour‐treating fields (TTFields) therapy, which uses locoregional delivery of alternating electrical fields within a narrow frequency range to specifically target rapidly dividing cancer cells within a defined brain region [15, 16, 17]. Akin to the introduction of temozolomide into existing radiotherapy regimes, the inclusion of TTFields to current temozolomide dosing schedules increased overall patient survival rates by approximately 5 months, leading to clinical approval for newly diagnosed glioblastoma by the Food and Drug Administration (FDA) in the United States [17] in 2015, following its initial approval for recurrent glioblastoma in 2011. However, the incorporation of TTFields into standard‐of‐care therapy for glioblastoma is not universally accepted for numerous reasons, including the lack of a placebo treatment, such as a sham TTFields device, in key phase 3 randomised trials [17, 18]; incomplete understanding of the mechanistic basis for TTFields therapeutic effects [16]; the challenge of maintaining high treatment compliance; and the current high financial costs associated with this treatment regime. Consequently, opportunities for patients to receive TTFields are not geographically equitable and are largely dependent on the country’s healthcare system approval structure for cancer therapies [16]. Importantly, and relevant to this Review, the current landscape of multimodal therapy used to treat glioblastoma – that is, chemotherapy, radiotherapy and TTFields – all induces DNA damage, replication stress and mitotic‐mediated cell death within tumour cells. As such, a deeper understanding of the innate DNA damage response (DDR) mechanisms and coordinated cell cycle regulation within these tumours offers opportunities to uncover exploitable tumour‐specific genetic and phenotypic vulnerabilities which could lead to more efficient targeting and effective treatment strategies in the clinic to confer much‐needed improvements in survival rates for patients.
1.2. Therapeutic challenges for glioblastoma
A cornerstone of glioblastoma treatment is effective surgical resection of the tumour bulk. However, clinical studies have suggested that neurosurgical removal of as much as 98% of the tumour volume may be required to provide an impact on median survival [19]. As such, a plethora of surgical innovations have been applied to help maximise surgical resection rates. Multimodal neuronavigation, intraoperative magnetic resonance imaging and ultrasound (MRI/US), and fluorescence‐guided surgery with 5‐aminolevulinic acid (5‐ALA) have demonstrated the potential to augment surgical reduction of tumour burden [20, 21, 22, 23, 24]. Additionally, forthcoming innovations, including the application of iKnife, which utilises rapid evaporative ionisation mass spectrometry (REIMS) to differentiate between tumour and healthy tissue [25, 26] or Raman spectroscopy, which is used to achieve similar tumour‐brain resolution in a nondestructive manner [27, 28, 29, 30], may yield further improvements in resection rates in the near future. However, given the highly infiltrative nature of glioblastoma and limited ability to resect infiltrated regions of eloquent cortex without unacceptable morbidity, it is unfortunately hard to envisage advances in surgical technology impacting patient survival outcomes beyond incremental gains.
In a similar manner to surgical interventions, radiotherapy has also seen technological advancements, including intensity‐modulated radiation therapy, stereotactic radiosurgery and proton beam therapy, which have the potential to lead to improved delivery of effective radiation doses to tumour sites whilst sparing the surrounding healthy brain tissue [31, 32].
Beyond the challenges associated with effective and maximal surgical resection and targeted radiotherapy regimes, delivery of a therapeutically effective dose of chemotherapy to postsurgical residual tumour cells is hampered by the presence of the blood–brain barrier/blood–tumour barrier (BBB) [3, 33], although there are several groups developing ways in which to disrupt or circumvent the BBB (discussed later).
The development of effective future therapies for glioblastoma will also need to tackle the biological mechanisms that help facilitate treatment resistance in these aggressive cancers. Unlike lower grade tumours, the majority of glioblastomas are classed as isocitrate dehydrogenase (IDH) wild‐type but often exhibit mutations and/or deletions in phosphatase and tensin homolog (PTEN), cyclin‐dependent kinase inhibitor 2A/B (CDKN2A/B), telomerase reverse transcriptase (TERT) promoter, tumour protein P53 (TP53), neurofibromin 1 (NF1), phosphatidylinositol‐4,5‐bisphosphate 3‐kinase catalytic subunit alpha (PIK3CA) and phosphoinositide‐3‐kinase regulatory subunit 1 (PIK3R1), as well as amplification of epidermal growth factor receptor (EGFR) and/or gain of chromosome 7, chromosome 10 monosomy and MGMT promoter methylation [5, 34, 35, 36]. However, at present, no targeted therapy designed to exploit such genetic and/or phenotypic traits has proven successful within the clinic [37]. One of the main reasons for this is that glioblastomas exhibit profound intratumoural heterogeneity, with spatiotemporally divergent subpopulations within a tumour displaying varying profiles of vulnerability and resistance [38, 39, 40, 41], which likely contributes to local disease recurrence. Additionally, increasing evidence supports the existence of a subpopulation of glioblastoma cells that possess an innate capacity for unlimited regeneration and self‐renewal, which are often described as cancer stem cells (CSCs) or glioblastoma stem cells (GSCs) [3, 42, 43, 44, 45, 46, 47]. Importantly, GSCs are often deemed responsible for resistance to conventional chemoradiotherapy treatment regimens through enhanced activation of DNA damage checkpoints and DNA repair capacity [48, 49, 50]. This will be highlighted with specific examples later within this Review.
1.3. Clinically relevant models of glioblastoma
Over the last decade, there has been a disappointing lack of success in translating promising novel agents or drug repurposing from preclinical studies into clinical survival benefit for patients with glioblastoma [51]. To some degree, this failure is representative of a lack of clinically and postsurgically relevant preclinical models of glioblastoma. In particular, the reported and further emerging intratumoural heterogeneity that exists within gliomas is a vital consideration for the development of future therapies. Variation in the genetic aberrations, phenotypic characteristics and clinical progression of individual patient tumours presupposes the need for personalised therapeutic strategies informed by a range of molecular biomarkers [52, 53, 54, 55, 56]. Furthermore, accumulating evidence suggests that a phylogenetic hierarchy of spatiotemporally divergent subclones exists within individual glioblastomas, each possessing a distinct collection of putative driver mutations and a characteristic transcriptome [38, 40, 53, 57, 58]. Creating appropriate preclinical models that are capable of recapitulating such profound intratumoural heterogeneity as well as GSC states represents a key challenge for the development of new therapeutic agents that can impact patient survival [3, 46, 59, 60]. A detailed discussion of the advantages and disadvantages of the various models is beyond the scope of this Review, but these currently consist of in vitro preclinical glioblastoma models that range from traditional commercially available immortalised cell lines or fresh patient‐derived primary cell cultures (‘bulk’ or stem cell enriched) which can all be maintained in either two‐ or three‐dimensional (2D or 3D) architectures, as well as more complex tumouroid and organoid cocultures and microfluidic ‘glioblastoma‐on‐a‐chip’ cultures [61, 62, 63, 64, 65, 66, 67, 68, 69, 70, 71, 72, 73]. Likewise, there are a plethora of in vivo preclinical glioblastoma models available, including subcutaneous, syngeneic, orthotopic and patient‐derived (PDX) xenografts as well as de novo genetically engineered rodent models [74, 75, 76, 77].
What is clear from the historic lack of newly approved therapies and limited improvement in patient survival rates over the last 50 years compared with other solid tumours is that glioblastoma represents a complex and difficult therapeutic challenge. However, recent work by numerous groups has started to reveal further genetic and molecular insights into glioblastoma biology, particularly within the area of DDR mechanisms that are triggered by current standard‐of‐care radiochemotherapy regimes used to treat these tumours. Furthermore, an evolving understanding of the GSC niche together with the development of novel preclinical models that better reflect postsurgical residual disease should hopefully begin to yield new therapeutic strategies over the next 5–10 years.
2. The DNA damage response and glioblastoma treatment
2.1. Cellular responses to DNA damage
The structural integrity of DNA is constantly threatened due to replication stress, telomere attrition and a multitude of endogenous and exogenous agents that generate high levels of varying DNA lesions. Such agents include metabolic by‐products, such as reactive oxygen species (ROS) and aldehydes, UV light, ionising radiation (IR) and chemical toxins [78, 79, 80]. Failure to repair DNA lesions induced by these processes can lead to mutagenesis, tumorigenesis or cell attrition. Given the potential deleterious effects on genome integrity induced by such lesions, it is perhaps not surprising that an intricate network of signalling pathways and reparative mechanisms have evolved to deal with a plethora of DNA lesions, preserving genomic architecture and integrity (genome stability). This network is collectively referred to as the DNA damage response (DDR), which encompasses a coordinated and interconnected network of pathways that regulate cell cycle progression/checkpoints, DNA repair mechanisms, DNA replication and mitotic progression, as well as transcriptional and cell death processes, in order to preserve the integrity of the genome [79, 81, 82, 83, 84, 85]. Importantly, given that radiation and chemotherapy treatments cause DNA lesions and affect cell cycle progression, heightened and/or dysregulated DDR mechanisms within tumour cells can often give rise to innate and/or acquired treatment resistance. However, such dysregulation to DDR mechanisms/processes during cancer development and progression can also offer vital cancer‐selective vulnerabilities that can be exploited for an improved therapeutic index as part of either monotherapy‐ or combination therapy‐targeted treatment strategies [82, 85, 86, 87, 88, 89].
The mechanisms associated with physically repairing DNA damage can be categorised into four main types based on the type of DNA lesion present: (a) base modifications (including alkylation damage) and mispaired bases; (b) intrastrand or interstrand DNA crosslinks; (c) single‐strand breaks (SSBs); and (d) double‐strand breaks (DSBs) [79, 81, 84, 85, 90]. Additionally, although DSBs are often considered amongst the most toxic of DNA lesions, it is worth considering that each type of lesion is not an enduringly separate entity. For example, radiation‐induced damage often leads to so‐called ‘complex lesions’ or ‘clustered damage’, in which multiple lesions are present within a few hundred base pairs of the DNA helix [91]. The collision of replication forks with SSBs or DNA interstrand crosslinks (ICLs), particularly as a consequence of oncogene activation, can result in the formation of DSBs and other deleterious lesions. Similarly, DNA : RNA hybrids known as R‐loops can be generated from replication forks colliding with transcription bubbles, which can have a plethora of deleterious effects if not adequately resolved [92, 93]. As such, the dynamic relationships that exist between various DNA lesions lead to functional redundancy within the reparative processes, which likely contributes to treatment resistance and forms the rationale for targeting multiple DDR pathways/processes to potentially overcome treatment resistance [78, 82, 83, 86, 87, 88, 89].
Maintenance of genome integrity relies not only on accurate DNA repair, but also on the processes that detect DNA damage and co‐ordinate cell cycle checkpoints, allowing time for DNA to be repaired. The regulation of cell cycle checkpoints and subsequent DNA repair processes as part of the early DDR process is controlled by three main related protein kinases – ataxia telangiectasia mutated (ATM); ataxia telangiectasia and Rad3‐related kinase (ATR); and DNA‐dependent protein kinase (DNA‐PK) – along with the various cyclin‐dependent kinases (CDKs) and the central cell cycle regulator p53 and its associated factors [83, 90] (Fig. 1). ATM represents an apical, multifunctional kinase within the DDR and typically plays a key role in cellular responses to DNA DSBs, where it helps co‐ordinate all major cell cycle checkpoints via the ATM‐CHK2 axis [90, 94]. Through the ATR‐CHK1 signalling axis, ATR helps preserve DNA integrity in response to replication stress (perturbation to ongoing replication forks) [95], although it is important to note that there is significant crosstalk and some functional redundancy amongst the activities of these related kinases [90, 96]. However, due to the main differing functions of these kinases, they are considered as credible separate drug targets in cancer biology as part of either monotherapy or combinatorial (adjuvant) therapeutic approaches [88, 90, 97, 98].
Fig. 1.
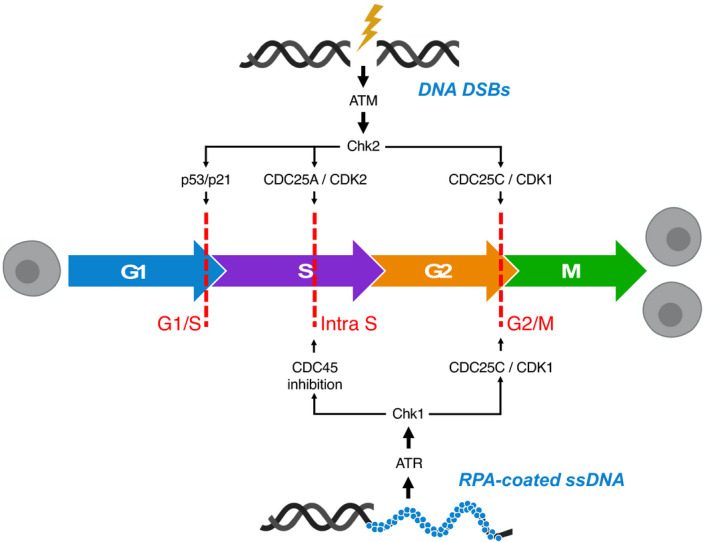
The role of ATM and ATR in cell cycle regulation following DNA damage. The processes of cell division (mitosis, M phase) and DNA synthesis (S phase) are separated by two important gap phases (G1 and G2). Progression of mitotic cells through the cell cycle is controlled by periodic accumulation and destruction of the aptly named cyclin‐dependent kinases (CDKs) and cyclins. Inappropriate progression through phases of the cell cycle is prevented by three main checkpoints (G1/S, intra‐S and G2/M checkpoints; dashed red lines). Following DNA damage, checkpoint activation is critical to provide ample time and recruit the necessary machinery required to maintain genomic integrity. Checkpoint activation: DNA double‐strand breaks (DSBs) activate the apical DNA damage response (DDR) kinase ataxia telangiectasia mutated (ATM), which can influence all three major cell cycle checkpoints via the phosphorylation of checkpoint kinase 2 (CHK2) and subsequent downstream signalling. In contrast, ataxia telangiectasia and Rad3‐related kinase (ATR) is activated by the presence of replication protein A (RPA)‐coated single‐stranded DNA (ssDNA) and contributes to maintenance of the intra‐S phase and G2/M checkpoints via phosphorylation of checkpoint kinase 1 (CHK1) and subsequent downstream signalling as indicated. G1/S checkpoint: Phosphorylation of p53 by CHK2 and ATM directly (arrow not shown) results in a reduction in the binding of mouse double minute 2 homolog (MDM2) to p53 and p53 activation, promoting its nuclear accumulation and stabilisation. Subsequently, elevated p53 levels promote increased transcription of p21, which inhibits CDK2–cyclin‐E activity, resulting in prevention of progression to S phase. Intra‐S checkpoint: Within S phase, the activation of cell division cycle 25 (CDC25) phosphatases predominantly by prevention of cell division cycle 45 (CDC45) loading onto replication origins (preventing subsequent DNA replication) primarily via the ATR–CHK1 axis, but also via ATM‐CHK2‐mediated phosphorylation of CDC25A, can instigate an intra‐S checkpoint in response to replication stress or other perturbations to optimal DNA synthesis, permitting a slowing of DNA replication. G2/M checkpoint: Both ATM‐ and ATR‐mediated phosphorylation of CHK2 and CHK1, respectively, lead to the phosphorylation of CDC25C phosphatases, which influence the G2/M checkpoint via interaction with the cyclinB1–CDK1 complex. This figure is adapted, with permission, from Ref. [227].
Beyond the initial DDR signalling and cell cycle checkpoint regulation, a key part of the cellular responses to DNA damage is obviously the physical repair of the vast array of DNA lesions that can be present within both heterochromatic and euchromatic regions of the genome [99, 100, 101, 102]. An exhaustive discussion of the vast array of DDR pathways is not possible within the confines of this Review; however, brief summaries of some of the DNA repair pathways most relevant to the therapy‐induced DNA damage within the context of glioblastoma treatment will be discussed in the following section.
2.2. Therapy‐induced DNA lesions and associated repair mechanisms
Abrogation of the mechanisms that glioblastoma cells use to repair the DNA lesions induced by chemo‐ and radiotherapy may represent a key to effective multimodal treatment approaches [97] (Fig. 2). The most effective chemotherapeutic agent in current standard‐of‐care clinical use for glioblastoma is the alkylating agent temozolomide [8]. Of the array of methylation lesions induced on both nitrogen and oxygen molecules within DNA nucleotides, O6‐methlyguanine (O6MeG) is considered the most toxic lesion induced by temozolomide once metabolised from its prodrug form to MTIC [3‐methyl‐(triazen‐1‐yl)imidazole‐4‐carboxamide] [103]. O6MeG can act as a miscoding base during DNA replication, leading to a corresponding C‐to‐T transversion within the complementary DNA strand. If O6MeG is not successfully excised by the mismatch repair (MMR) DNA repair machinery [104], it endures as a perpetually miscoding base, instigating ‘futile cycles’ of MMR with consequent stalling of DNA replication forks or double‐strand breakage [104, 105]. As such, MMR capacity within GSCs harbouring genetic or epigenetic defects that affect MMR gene expression can impact on temozolomide sensitivity as well as other phenotypic traits due to the well‐established hypermutation phenotype conferred by MMR defects [106, 107, 108] (Fig. 2). Additionally, the de‐alkylating enzyme methylguanine methyltransferase (MGMT) can sequester the methyl group from O6MeG to restore the guanine residue to its original state, but this leaves MGMT irreversibly inactivated and subject to ubiquitin‐mediated proteasomal degradation [105, 109]. Consequently, high expression levels of MGMT can contribute to temozolomide resistance in glioblastoma [109]. On the other hand, approximately 40% of IDH‐wild‐type glioblastomas exhibit transcriptional repression of MGMT expression due to hypermethylation of an MGMT‐associated 5’ CpG island, which confers a greater benefit from temozolomide therapy and prolonged patient survival [10].
Fig. 2.
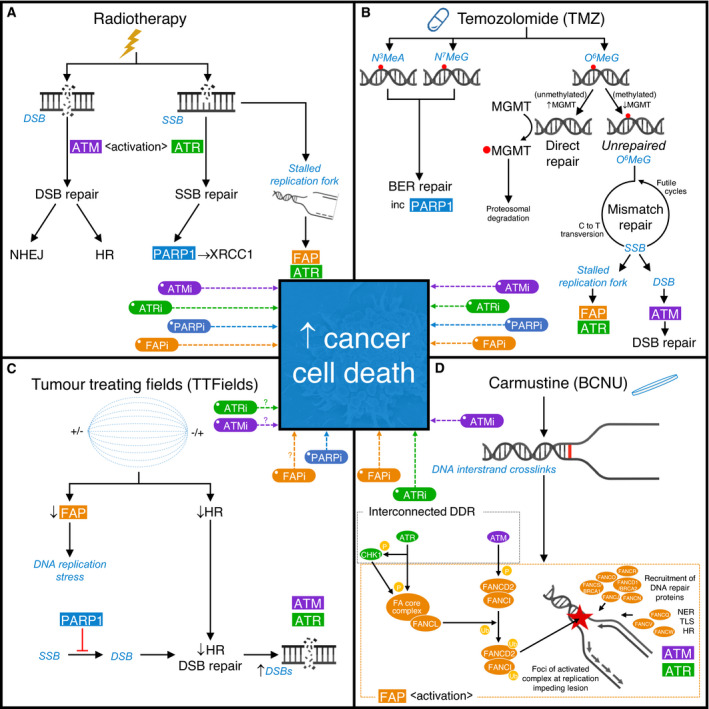
The effects of clinically approved therapies on the DNA damage response (DDR) and novel strategies to enhance efficacy of current standard‐of‐care treatments. Schematic representation of the main DNA damage lesions (in blue italic) induced by therapies approved for clinical use to treat glioblastoma and associated DDR mechanisms. For each approved treatment, putative strategies to enhance therapeutic efficacy through targeting relevant DDR mechanism(s) are indicated. (A) Radiotherapy: generates large amounts of DNA single‐strand breaks (SSBs) and double‐strand breaks (DSBs), which activate ATR and ATM, respectively. DSB repair is then predominantly undertaken by either nonhomologous end joining (NHEJ), which is available throughout the cell cycle but compromises fidelity, or homologous recombination (HR) DNA repair, which provides a high‐fidelity repair mechanism, but is only available during S and G2 phases of the cell cycle due to the requirement for a sister chromatid. SSB repair relies on PARP1 to detect SSBs and facilitate the recruitment of XRCC1. However, the presence of strand breaks also leads to stalling of DNA replication forks, which depend on the functions of ATR and proteins within the Fanconi anaemia pathway (FAP) for stability and replication restart. Consequently, a strong scientific rationale exists supporting inhibition of either ATM (ATMi), ATR (ATRi), PARP1 (PARPi) or the FAP (FAPi) to enhance the efficacy of radiotherapy. (B) Temozolomide: produces an array of methylation lesions including N3‐methyladenine (N3MeA) and N7‐methylguanine (N7MeG), which are substrates for effective removal via DNA base excision repair (BER), and O6‐methylguanine (O6MeG), which is removed directly by the enzyme MGMT in a suicide reaction. Hypermethylation of the MGMT gene promoter region leads to reduced MGMT expression, shifting the balance in favour of persistent O6MeG. O6MeG can act as a miscoding base during DNA replication, leading to a corresponding C‐to‐T transversion within the complementary DNA strand. If O6MeG is not successfully excised by the mismatch repair (MMR) DNA repair machinery, it endures as a perpetually miscoding base, instigating ‘futile cycles’ of MMR with consequent stalling of DNA replication forks or DSBs. (C) Tumour‐treating fields (TTFields): may negatively impact FAP and HR‐mediated DNA repair processes. TTFields‐induced ‘BRCAness’ (reflecting a relative HR deficiency) provides a compelling rationale to combine this therapeutic modality with PARPi, or potentially FAPi, ATRi or even ATMi. (D) Carmustine (BCNU) – Gliadel® wafers: provide local delivery of this bidirectional DNA alkylating agent, leading to the generation of DNA interstrand crosslinks which impede DNA replication during S phase. This leads to activation of the FAP, within which monoubiquitination of FANCD2 within the FANCD2‐I complex is a key quantifiable step. Activated FANCD2‐I coalesces as foci at sites of DNA damage and acts as a master regulator of downstream DNA repair, recruiting proteins involved in nucleotide excision repair (NER), translesion synthesis (TLS) and HR. Interplay with associated DDR mechanisms, for example ATM and ATR, leads to the phosphorylation of multiple FAP proteins (examples indicated), providing a rationale for the use of non‐FAP DDR inhibitors (e.g. ATRi or ATMi) to sensitise to crosslinking chemotherapy, and for the concept of combining multiple DDR inhibitors (including FAPi) to potentially maximise therapeutic enhancement.
The mere notion that a DNA repair mechanism such as MGMT expression can profoundly influence survival for some patients provides further evidence of the critical nature of DNA repair in glioblastoma. However, it is important to note that, although both MMR activity and MGMT expression can impact temozolomide sensitivity of glioblastomas, longitudinal genomic profiling has revealed that such defects do not account for the majority of therapy resistance exhibited within these tumours [106]. In addition to O6MeG, the more prevalent (but less toxic) temozolomide‐induced DNA lesions N3‐methyladenine and N7‐methylguanine are primarily removed via base excision repair (BER), leaving an intermediary abasic [or apurinic/apyrimidinic (AP)] site. The nucleotide gap is subsequently filled through triggered DNA synthesis processes and strand integrity restored through ligation of the DNA ends – a process involving an AP endonuclease (APEX1), DNA polymerase (pol‐β) and DNA ligases I and III, respectively [110, 111, 112]. Additionally, nucleotide excision repair (NER) is able to remove a wide variety of structurally unrelated DNA lesions that can be generated by both chemo‐ and radiotherapy. The process can be activated by DNA helix distortions associated with structural changes to nucleotides or by stalling of RNA polymerase II due to the presence of a DNA lesion during transcript elongation, and involves the DNA unwinding before removal of a section of DNA containing the lesion followed by resynthesis using the template strand and subsequent ligation [113].
Radiotherapy exerts cellular damage by inducing a wide range of DNA lesions, but it is particularly associated with the generation of large amounts of SSBs and DSBs (Fig. 2). Poly (ADP‐ribose) polymerase‐1 (PARP1) plays a pivotal role in detection of SSBs, facilitating the colocalisation of the single‐strand break repair (SSBR) polypeptide x‐ray repair cross‐complementing protein 1 (XRCC1) [114, 115]. DNA DSBs are predominantly repaired by the nonhomologous end joining (NHEJ) and homologous recombination (HR) DNA repair pathways. The HR pathway represents a complex, high‐fidelity mechanism of DNA DSB repair. However, given its reliance on a homologous DNA sequence (duplicate DNA strand on a sister chromatid) as a template for resynthesis of removed DNA sequences around the site of DNA damage, HR‐mediated repair mechanisms are only possible during S and G2 phases of the cell cycle, when such sister chromatids are available [116, 117]. Interestingly, although TTFields therapy does not directly induce DNA breaks (unlike IR), recent work has suggested that, in addition to its effects on mitotic cells, TTFields may negatively impact on HR‐mediated DNA repair processes [16, 118], which could be important for future DDR‐targeting strategies that combine with this recently approved glioblastoma therapy. In contrast to HR, the NHEJ DNA repair pathway provides rapid DSB repair capabilities throughout the cell cycle (since a homologous sister chromatid is not required), permitting repair of a range of DNA‐end configurations (Fig. 2). However, the flexibility and rapidity of NHEJ is provided at the expense of fidelity [119, 120]. As one might expect given the cell cycle regulated nature of these pathways, both HR and NHEJ are tightly regulated by CDK activity [120, 121], a family of kinases often dysregulated in human cancers [122, 123].
Finally, the Fanconi anaemia (FA or FA/BRCA) pathway is frequently activated in response to DNA strand breaks, alkylation damage and other cytotoxic DNA lesions induced by both alkylating and DNA crosslinking chemotherapeutic agents that impede ongoing DNA replication [124, 125, 126, 127, 128]. The FA pathway is also activated during normal S‐phase progression and is regulated by both ATM and ATR kinases [124, 125, 126, 127, 128, 129, 130, 131, 132, 133, 134]. The FA pathway consists of at least 22 proteins which can be broadly categorised into three distinct functional groups (core complex, ID complex and downstream effectors) that, through sequential interaction, facilitate lesion repair and the restart of replication forks via physical removal of lesions by NER and HR‐mediated processes, as well as interactions with components of the MMR system [135].
3. Targeting functional interplay within the glioblastoma DDR network
3.1. Exploiting synthetic lethality and synthetic sensitivity strategies
As mentioned previously, there are a plethora of interconnected interactions and functional crosstalk between the various DDR, regulatory and DNA repair pathways that open up a multitude of potential therapeutic targeting strategies in heterogeneous tumours such as glioblastoma [88]. Perhaps most notable over the past 10 years or so is the concept of synthetic lethality: a concept that was originally described as a simultaneous genetic mutation in, or functional aberration of the product of, two genes which causes cell death; but in isolation, either change is survivable [136]. More recently, synthetic lethality in terms of oncology‐based therapeutic strategies has expanded to include scenarios in which specific phenotypic traits such as defective HR‐mediated DNA repair function, or aberration in the function of certain genes or pathways, result in impaired cell growth or proliferation, promoting lethal effects in the presence of additional insults such as IR or cytotoxic chemotherapy [137]. As such, the term ‘synthetic sensitivity or lethality’ (SSL) can be used to amalgamate these related concepts.
Although major advancements in the generation of RNAi and CRISPR/Cas9 libraries have undoubtedly advanced our ability to uncover potential SSL relationships at the genome scale, the most successful implementation of an SSL strategy to date in cancer treatment, the targeting of BRCA‐deficient cells with PARP1 inhibitors, was derived from hypothesis‐driven research rooted in a fundamental understanding of the specific molecular pathways concerned and relevant interplay between them, rather than broad screening approaches [138, 139]. This seminal work has led to a wave of clinical approvals for PARP1 inhibitors that are delivering survival benefits to patients with a range of cancers globally [140, 141, 142]. In the context of gliomas, tumours with IDH1/2 mutations may exhibit a ‘BRCAness’ phenotype as a consequence of inhibition of HR DNA repair processes by the enhanced levels of oncometabolites, which could explain why a subset of IDH‐mutant tumours respond well to conventional DNA damaging chemotherapies such as temozolomide as well as PARP1 and ATR inhibitors [141, 143, 144]. However, as highlighted earlier, the majority of high‐grade aggressive gliomas do not exhibit such defects in IDH genes and associated metabolism but, as discussed below, disruption to HR and associated DDR pathways may represent a credible mechanism to produce a similar phenotype in these tumours. Furthermore, challenges in efficient delivery of such compounds across the BBB are a major clinical consideration; however, at least for PARP1 inhibitors, early indications are encouraging in that therapeutically active doses can be achieved at the tumour site [145].
3.2. Single DDR inhibitor strategies to enhance therapeutic response
As mentioned previously, glioblastoma cells within a single tumour can demonstrate remarkable heterogeneity in the expression of DDR factors and consequently exhibit varying resistance profiles [146, 147], which likely plays a key role in treatment failure. Given the interconnected nature of the DDR pathways [88], it is perhaps not surprising that, although promising in preclinical models, targeting any single DDR pathway might not yield an effective therapeutic response clinically. The D’Andrea group were the first to demonstrate that components of the FA pathway could confer resistance to temozolomide in glioma cells [148], which was further corroborated by Kondo and colleagues [149]. Building upon these findings, we showed that the FA pathway is re‐expressed and active within high‐grade gliomas compared with low‐grade tumours as well as normal healthy tissue and that inhibition of the FA pathway in both established and primary glioma cells could confer an increased sensitivity to temozolomide [150]. These findings were recently confirmed by large‐scale CRISPR‐Cas9 screens that identified the FA and related HR pathways as key modulators of temozolomide resistance within glioma stem cells [108]. Importantly, we showed that disruption to FA pathway function was able to render glioma cells sensitive to temozolomide irrespective of MGMT status/expression levels [150], demonstrating a potential large scope for such an approach within the clinical setting. This is particularly important, as PARP1 inhibition (the most successful DDR‐targeting drug to date) may only confer a similar increased temozolomide sensitivity within cells that have MGMT promoter methylation [151], which represents ~ 40% of all patients diagnosed with glioblastoma and whose tumours are already intrinsically more sensitive to temozolomide. This is consistent with previous work by Gupta and colleagues, who demonstrated that siRNA‐mediated knockdown of either N‐methylpurine DNA glycosylase (MPG) or XRCC1 (two indispensable components of a functional BER pathway) did not confer temozolomide sensitivity in resistant glioma cell lines [152], even though PARP1 can act as a scaffold to promote BER‐mediated repair of alkylation damage [153].
Given that persistent bulky O6MeG lesions in the absence of MGMT result in elevated DNA replication stress [154, 155] and that replication fork stability and recovery is influenced by a number of DDR factors, such as PARP1, as well as the HR and FA proteins RAD51/FANCR, FANCD2, BRCA1/FANCS and BRCA2/FANCD1, the integrity of some of these interconnected DDR processes may underpin the heterogeneous preclinical success of PARP1 inhibitor‐mediated temozolomide sensitisation. At least theoretically, these data would support the use of adding an additional DDR inhibitor to maximise replication fork collapse and/or failure of resultant DNA DSB resolution, potentially resulting in more pronounced neoplastic cell death across a wider range of tumours. In this regard, it seems rational that the activity of apical DDR kinases, such as ATR and ATM, that regulate replication stress signalling and DNA break repair mechanisms might cooperate to limit the cytotoxicity of temozolomide in cancer cells. Interestingly, activation of both ATR and ATM following temozolomide chemotherapy occurs in an MMR‐dependent manner [156], which likely reflects the nature of the cytotoxicity associated with O6meG compared with N7‐meG and N3‐meA lesions, which do not activate MMR. Previous work by Eich and colleagues showed that siRNA‐mediated knockdown of either ATR or ATM sensitised an MGMT‐negative glioma cell line to temozolomide [157], with similar findings for ATM also reported by Nadkarni et al. [158]. Interestingly, the degree of sensitisation conferred by ATR knockdown in the Eich study was more than double that observed following ATM siRNA, indicating that impaired resolution of DNA replication stress generated from O6MeG lesions may be a key driver of sensitisation in this context, especially given that simultaneous ATR and ATM knockdown did not provide any additional sensitisation relative to ATR alone and that the enhanced temozolomide sensitisation could be rescued by ectopic expression of MGMT [157]. These findings were also recently corroborated by Jackson et al., who established that MGMT‐deficient glioma cells are profoundly susceptible to temozolomide sensitisation using small‐molecule ATR inhibitors both in vitro and in vivo [159].
3.3. The FA pathway as a foundation for future DDR‐centric combinatorial strategies
In addition to potential compounding genetic and/or epigenetic alterations within glioblastomas that might affect DDR‐targeting strategies, and the interplay within the BER, MMR, MGMT, FA and HR pathways (highlighted above), interactions between the FA pathway and other DDR elements have been characterised that are also important to consider with regard to potential FA‐based combination targeting strategies within glioblastomas [160]. For example, as mentioned previously, both ATM and ATR phosphorylate several proteins within the various FA subcomplexes [161, 162, 163] and components of the FA pathway have also been shown to promote activation of ATR and supress potentially deleterious repair of DNA ICLs by NHEJ [164, 165, 166]. Additionally, as the FA pathway promotes HR‐mediated DNA repair processes around stalled/collapsed replication forks [167], HR‐deficient cells have been shown to be sensitive to disruption of FA pathway function [168, 169]. Given this functional interplay, it has been postulated that FA dysfunctional tumours, or targeting of the FA pathway, may render them sensitive to PARP1 inhibitors [170, 171, 172], as well as a multitude of other drug targeting strategies within the FA and related DDR pathways [170]. Indeed, we have recently generated data in clinically relevant GSC 3D cell culture model systems highlighting profound radio‐ and chemosensitisation through the combined targeting of the FA pathway in combination with ATM, ATR or PARP1 inhibitors (Rominiyi et al, manuscript under preparation).
Unfortunately, there have been no reported rationally designed inhibitors of the FA pathway to date; however, recent structural insights into key regulatory components of the FA pathway together with current efforts by several groups to identify FA pathway inhibitors (including our own group) [173, 174, 175, 176, 177, 178] should hopefully lead to the emergence and further development of such compounds in the near future that could be of great clinical significance for glioblastoma treatment regimes. Apart from exhibiting high potency and specify, such compounds would also need to demonstrate high biological efficacy within the correct brain regions or be adaptable to novel delivery mechanisms in order to maximise their effectiveness in any such therapeutic regimes [3]. Encouragingly, tumour margin penetration within a biologically active drug concentration range has been recently reported for the PARP1 inhibitor olaparib (Lynparza) in combination with temozolomide chemotherapy as part of the OPARATIC trial [145], and it will be interesting to see the results from other DDR inhibitor trials for glioblastoma when they are reported (Table 1).
Table 1.
DNA damage response inhibitor trials in high‐grade glioma. AEs(G3‐4), grade 3–4 adverse events; AEs, adverse events; and WBRT, whole brain radiotherapy; CR, complete response; DIPG, diffuse intrinsic pontine glioma; DLTs, dose‐limiting toxicities; EFS, event‐free survival; F/U, follow‐up; IMRT, intensity‐modulated radiation therapy; IR, radiotherapy; MTD, maximum tolerated dose; nGBM, newly diagnosed GBM; NIRA, niraparib; OLAP, olaparib; ORR, overall response rate (proportion of patients with a PR or CR); OS, median overall survival; PAMI, pamiparib; PFS, median progression‐free survival; PR, partial response; QoL, quality of life; rGBM, recurrent GBM; rHGG, recurrent high‐grade glioma; RP2D, recommended phase II dose – highest dose with acceptable toxicity (producing a rate of around 20% DLTs); Rx, treatment; SAEs, severe adverse events; SD, stable disease; SoC, standard‐of‐care; TALA, talazoparib; TMZ, temozolomide; TTF, tumour‐treating fields; VELI, veliparib.
| Trial (reference) & indication | Design & n (rec dates) | Treatment(s) | 1° Endpoint | 2° Endpoint(s) | Results/remarks & conclusions |
|---|---|---|---|---|---|
| Summary of key PARP inhibitor in high‐grade glioma clinical trials – completed and ongoing | |||||
| Phase I studies | |||||
|
nGBM Veliparib (ABT‐888), radiation therapy, and temozolomide in treating patients with newly diagnosed glioblastoma multiforme |
Phase I Single arm 24 patients (2009–2012) |
VELI + IR + TMZ |
Phase I: VELI MTD Phase II: OS (with VELI MTD) |
A) Safety/toxicity B) Pharmacokinetics |
|
|
NCT01390571 [229, 230] rGBM Olaparib and temozolomide in treating patients with relapsed glioblastoma (OPARATIC) |
Phase 0/I Single arm 48 patients (2011–2017) |
Stage I: OLAP for 3/7 prior to surgery then usual Rx Stage II: Escalating OLAP 3/7 prior to surgery then OLAP + TMZ post‐op |
Phase 0: Tumour penetration via BBB/BTB Phase I: Safety |
A) BBB disruption/permeability B) Preliminary antitumour activity of OLAP + TMZ |
|
|
rGBM/rMelanoma/solid cancers Niraparib (MK‐4827) given with temozolomide in participants with advanced cancer |
Phase I Single arm 19 patients (2011–2012) |
NIRA + TMZ | No. of DLTs |
A) ORR within 30 days of last dose & 2m intervals B) PFS |
|
|
nGBM [231] Two parallel phase I studies of olaparib and radiotherapy or olaparib and radiotherapy plus temozolomide in patients with newly diagnosed glioblastoma, with treatment stratified by MGMT status (PARADIGM‐2) |
Phase I Parallel Estimated patients: 25–40 methylated; 19–28 unmethylated (2016–2021) |
Methylated: OLAP + IR + TMZ Unmethylated: OLAP + IR |
Safety/toxicity (MTD & optimum scheduling) | A) Define DLTs (+/− TMZ) |
|
| Phase I/II studies | |||||
|
rGBM A randomized phase I/II study of veliparib (ABT‐888) in combination with temozolomide in recurrent (temozolomide resistant) glioblastoma (RTOG0929) |
Phase I/II Randomised 225 patients: 151 BEV naïve (BEV‐N); 74 BEV refractory (BEV‐R) (2009–2017) |
Arm 1: VELI + TMZ 75 mg·m−2 (both 21/28 day cycle) Arm 2: VELI + TMZ 150 mg·m−2 (both 5/28 day cycle) |
Phase I: MTD. Phase II: PFS at 6m |
A) ORR B) OS |
|
|
NCT01514201 [214, 233] DIPG Veliparib, radiation therapy, and temozolomide in treating younger patients with newly diagnosed diffuse pontine gliomas: a paediatric brain tumor consortium study |
Phase I/II Single arm 65 patients (2012–2018) |
VELI + IR + TMZ |
Phase I: RP2D/DLTs. Phase II: OS |
A) PFS B) Pseudoprogression C) Pharmacokinetics |
|
|
Solid & haematological cancers Talazoparib and temozolomide in treating younger patients with refractory or recurrent malignancies |
Phase I/II Single arm 40 patients (2014–2018) |
TALA + TMZ |
Phase I: MTD/RP2D; Safety/toxicity; Pharmacokinetics Phase II: ORR (Ewing/PNET) |
A) ORR all solid tumours (RECIST) |
|
|
nGBM/rGBM Pamiparib (BGB‐290) with radiation and/or temozolomide (TMZ) in newly diagnosed or recurrent glioblastoma |
Phase Ib/II Parallel Estimated patients: 116 (2017–2021) |
nGBM (unmethylated) Arm 1: PAMI + IR Arm 2: PAMI + IR + TMZ rGBM (un‐ & methylated) Arm 3: PAMI + TMZ |
Phase I: Safety/toxicity Phase II: Disease response/control |
A) Pharmacokinetics B) PFS C) OS D) ORR |
|
|
Unresectable HGG Study of concomitant radiotherapy with olaparib and temozolomide in unresectable high‐grade gliomas patients (OLA‐TMZ‐RTE‐01) |
Phase I/IIa Sequential Estimated patients: 79 (2017–2022) |
OLAP + IR + TMZ |
Phase I: RP2D for both IR‐period and maintenance period. Phase II: OS |
A) PFS B) ORR C) Neurocognitive function D) Morphological and functional MRI findings |
|
| Phase II studies | |||||
|
rGBM Cediranib maleate and olaparib compared to bevacizumab in treating patients with recurrent glioblastoma |
Phase II Randomised Estimated patients: 70 (2017–2020) |
Arm 1: OLAP + cediranib Arm 2: BEV |
PFS |
A) OS B) Safety/toxicity C) Circulating biomarkers (inc DDR and cytokines) |
|
|
Solid tumours with DDR defects Phase 2 subprotocol of olaparib in patients with tumors harbouring defects in DNA damage repair genes (NCI‐COG Paediatric MATCH (Molecular Analysis for Therapy Choice)) |
Phase II Single group assignment Estimated patients: 49 (2017–2024) |
OLAP only | ORR |
A) PFS B) Safety/toxicity C) Pharmacokinetics |
|
|
rGlioma (WHO Grade II‐IV) / cholangiocarcinoma / solid tumours with IDH1/2 mutation Olaparib in treating patients with advanced glioma, cholangiocarcinoma, or solid tumours with IDH1 or IDH2 mutations |
Phase II Single arm Estimated patients: 145 (2018–2021) |
OLAP only |
ORR (3 cohorts) A) Glioma B) Cholangio C) Other solid tumours |
A) PFS B) OS C) Safety/toxicity D) Exploratory objectives inc correlation between baseline 2HG and response |
|
|
nHGG (H3K27M− BRAFV600−) Veliparib (ABT‐888), radiation therapy, and temozolomide in treating patients with newly diagnosed malignant glioma without H3 K27M or BRAFV600 mutations |
Phase II Single arm Estimated patients: 115 Age 3–25 (2018–2024) |
VELI + IR + TMZ | PFS |
A) ORR B) OS |
|
|
rGBM Evaluating the efficacy and safety of niraparib and tumor‐treating fields in recurrent glioblastoma (Niraparib/TTFields) [240] |
Phase II Parallel Estimated patients: 30 (2019–2025) |
All patients receive NIRA + TTFields Cohort 1: surgical resection indicated Cohort 2: resection not indicated |
Disease control (CR/PR or SD) |
A) Safety/toxicity B) ORR C) PFS D) OS |
|
|
rHGG (IDHmut) Olaparib in Recurrent IDH‐mutant High Grade Gliomas (OLAGLI) |
Phase II Single arm Estimated patients: 35 (2020–2021) |
OLAP only | PFS | n/a |
|
|
IDHmut solid tumours Olaparib and durvalumab in patients with IDH‐mutated solid tumors (MEDI 4736) |
Phase II Parallel Estimated patients: 78 (2020–2022) |
All pts receive OLAP + durvalumab Cohort 1: Glioma Cohort 2: Cholangio Cohort 3: Other solid tumours |
ORR |
A) PFS B) OS C) Safety/toxicity |
|
| Phase II/III studies | |||||
|
nGBM (MGMT promoter hypermethylated) Temozolomide with or without veliparib in treating patients with newly diagnosed glioblastoma multiforme |
Phase II/III RCT Randomised 447 patients (2014–2021) |
After SoC IR and concomitant TMZ: Arm 1: VELI + TMZ Arm 2: PLACEBO + TMZ |
OS |
A) ‘Interaction’ with TTFields (for pts receiving this) B) PFS C) ORR D) Safety/toxicity E) QoL |
|
| Summary of key ATM inhibitor in high‐grade glioma clinical trials – ongoing | |||||
| Phase I studies | |||||
|
NCT03423628 [241, 242] nGBM/rGBM/brain metastases Safety and tolerability of AZD1390 given with radiation therapy in patients with brain cancer |
Phase I Parallel Estimated patients: 132 (2018–2023) |
AZD1390 + SoC IR: nGBM: IMRT 60 Gy over 6 weeks rGBM: IMRT 35 Gy over 2 weeks Mets: WBRT 30 Gy over 2 weeks |
Safety/toxicity |
A) EFS B) ORR C) Pharmacokinetics |
|
| Summary of key DNA‐PK inhibitor in high‐grade glioma clinical trials – ongoing | |||||
| Phase II studies | |||||
|
nGBM INdividualized Screening Trial of Innovative Glioblastoma Therapy (INSIGhT) |
Phase II Parallel RCT Estimated patients: 250 (2017–2021) |
After SoC IR and concomitant TMZ: Arm 1: TMZ Arm 2: Neratinib + TMZ Arm 3: CC115 + TMZ Arm 4: Abemaciclib + TMZ |
OS |
A) Safety/toxicity B) PFS C) Biomarkers & survival associations |
|
| Summary of key WEE1 inhibitor in high‐grade glioma clinical trials – ongoing | |||||
| Phase I studies | |||||
|
rGBM A phase 0 study of AZD1775 in recurrent GBM patients |
Phase 0/I Single arm 20 patients (2015–2019) |
Single dose of AZD1775 (100mg, 200mg or 400mg) prior to surgery |
A) Plasma concentration B) Intratumoural concentration |
Tissue biomarker analysis |
|
|
nGBM/rGBM Adavosertib (AZD1775), radiation therapy, and temozolomide in treating patients with newly diagnosed or recurrent glioblastoma |
Phase I Nonrandomised Estimated patients: 114 (2013–2021) |
Arm 1: AZD1775 during initial IR + TMZ and maintenance TMZ Arm 2: AZD1775 during maintenance TMZ |
A) MTD B) Safety/toxicity |
A) OS B) PFS |
|
3.4. Expanding the preclinical evidence base for parallel targeting of the DDR
As highlighted previously, there has been intense research into the development of small‐molecule inhibitors of DDR proteins [82]; to date, however, only a handful of published preclinical studies have examined combined inhibition of multiple DDR elements simultaneously in glioblastoma. Given that ATM and PARP1 inhibition each individually possess some utility in sensitising GSCs to radiotherapy [179, 180], Ahmed et al. [181] investigated parallel inhibition of the DDR targets ATM, ATR, CHK1 and PARP1 in primary patient‐derived glioblastoma cell lines. Firstly, in agreement with previous findings by Bao et al. [48], this study demonstrated that these DDR factors were upregulated in the inherently treatment‐resistant subpopulation of GSCs compared to bulk populations. This further confirms the importance of targeting multiple DDR pathways and the potential for functional redundancy within the DDR to contribute to the treatment‐refractory nature of glioblastoma. Secondly, the study by Ahmed et al. demonstrated that combined inhibition of PARP1 and ATR resulted in a profound radiosensitisation of GSCs, with effects greater than any single inhibitor used in isolation [181].
The rationale for a combination DDR inhibitory strategy in glioblastoma is also supported by the work of Signore and colleagues, who performed a simultaneous multipathway approach with subsequent reverse‐phase protein microarrays and kinase inhibitor library screening to identify dual inhibition of CHK1 and PDK1, resulted in profound retardation of GSC growth in both in vitro and in vivo (subcutaneous and intracranial mouse) models [182]. Although this study is highly informative, the lack of target specificity with use of a drug such as UCN‐01 severely limits potential clinical utility due to the high likelihood of dose‐limiting toxicities mediated by known off‐target effects associated with this compound. Nevertheless, this work provides further preclinical proof‐of‐concept data in support of such combinatorial approaches within heterogeneous glioblastoma tumour populations. More recently, Rasmussen et al. [183] demonstrated that reduced DDR capacity through PARP1 inhibition (olaparib) in conjunction with epigenetic‐downregulation‐induced oxidative stress through histone deacetylase (HDAC) inhibition (vorinostat) led to reduced glioblastoma cell survival, induced apoptosis and impaired cell cycle progression.
As demonstrated through these examples, the principle of multimodal targeting within the DDR network based on an in‐depth mechanistic understanding of functional interplay and regulatory mechanisms offers a potentially powerful approach to combat biological complexity and functional redundancy within the DDR, as well as intratumour heterogeneity within glioblastoma tumour subpopulations (Fig. 3). Indeed, preclinical research investigating such dual DDR inhibition approaches outside of glioblastoma research is supportive of this concept, for example CHK1 and PARP1 inhibition in pancreatic cancer [184], CHK2 and PARP1 inhibition in lymphoma [185], CHK1 and PARP1 inhibition in breast cancer [186], ATR and PARP1 inhibition in breast and ovarian cancer [187], and ATM and PARP1 inhibition in lung cancer [188].
Fig. 3.
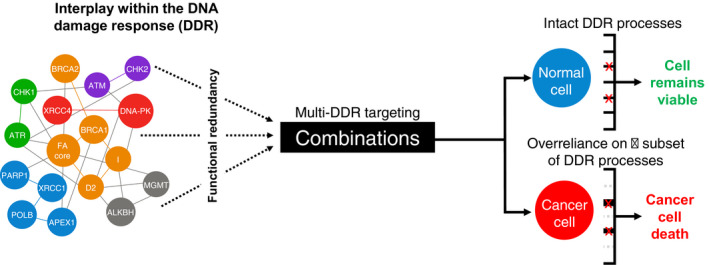
An approach for cancer‐selective killing through multimodality targeting of interconnected DNA damage response (DDR) pathways. A schematic representation of simultaneous targeting of multiple interconnected DDR processes to achieve cancer‐selective killing. Left – a simplified network schematic of key DDR proteins illustrating the complexity of intra‐ and interpathway protein–protein interactions within the global DDR. This complexity provides a degree of functional redundancy in DDR processes, which is likely to afford therapeutic resistance to current DNA damaging therapies. Right – due to the loss of functionality within some DDR pathways during carcinogenesis, cancerous cells often demonstrate overreliance on a reduced subset of DDR processes for cell survival. Where inhibition of a single DDR pathway may not be sufficient to provide synthetic lethality or substantial cancer cell killing, targeting multiple DDR processes simultaneously may overwhelm the remaining functional DDR leading to exquisitely potent cancer cell killing. However, by virtue of their complete repertoire of fully functional DDR processes, normal cells might continue to avoid significant toxicity associated with multi‐DDR‐targeting strategies (e.g. PARPi in noncancerous breast tissue that exhibits normal BRCA1/2 expression/function).
3.5. Indirect DDR‐targeting strategies in glioblastoma
In addition to direct combination targeting of DDR network factors, a comparable approach can be to target other signalling pathways that impact on DDR activity and/or capacity. For example, Gomez‐Roman et al. [64] recently demonstrated in 3D GSC models that disruption to functional VEGF and AKT signalling pathways impacted the balance between NHEJ and HR DNA DSB break repair activities to increase radiation sensitivity. This is particularly interesting given that targeted therapies against VEGF (such as bevacizumab) have generally failed to improve patient overall survival in large clinical trials [51].
In a similar manner to identifying nonclassical DDR pathway targeting strategies, we recently identified ERK5/MAPK5 through a kinome‐wide RNAi screen as a novel temozolomide resistance factor, with abrogation of ERK5 activity in glioma cells leading to defective DNA repair capacity, likely through inappropriate NHEJ activity prior to mitosis [189]. Interestingly, ERK5 has recently been identified as a key factor in promoting cell growth and cell survival in the aggressive diffuse intrinsic pontine gliomas [190], supporting recent evidence around ERK5 as an emerging novel oncology drug target [191, 192, 193]. As such, we are currently further assessing the potential of ERK5 targeting in glioblastomas as part of various combinatorial approaches together with current standard‐of‐care therapy.
On the topic on non‐DDR signalling kinases that impact the DDR, Riess et al. [194] recently assessed targeting of the CDK family of signalling kinases given their common dysregulation in glioblastoma and the recent advancement of a new generation of clinically approved compounds. Using a CDK‐based monotherapy approach in various 3D glioblastoma preclinical models, they showed that CDK inhibitors could negatively affect tumour growth, but also that some CDK inhibitors were able to effectively combine with DNA damaging regimes such as radiation and temozolomide treatments. However, they also showed that not all tested CDK inhibitors behaved in the same way, with some conferring antagonistic properties when combined with temozolomide, potentially through differential effects on global gene expression patterns [194]. This study further highlights the need to understand the intricate functional interplay and regulatory mechanisms within the DDR as part of preclinical studies to help maximise the therapeutic potential of such new combinatorial regimes within the clinic.
In addition to CDK dysregulation, another common feature of cancer cells is a heightened level of replication stress due to the activation of oncogenes [92, 195]. Oncogene‐induced replication stress has been shown to be present in GSCs [49] and is capable of triggering the DDR during early tumorigenesis [196]. Recent work from Ning et al. [197] revealed that heightened MYC activity in GSCs can lead to suppression of ATR‐mediated replication stress signalling through transcriptional repression of CDK18. This is consistent with recent findings from our laboratory that identified CDK18 as a novel component of the ATR‐mediated replication stress signalling module that promotes cellular resistance to a variety of replication stress‐inducing chemotherapeutic agents [198, 199]. In keeping with the aforementioned interplay between ATR signalling and PARP1 activity (see above), Ning et al. [197] further showed that GSCs with reduced CDK18 expression and subsequent retarded ATR activity, as a consequence of oncogenic MYC action, were rendered sensitive to PARP1 inhibitors. In keeping with a heightened S‐phase fraction within a subpopulation of GSCs, a recent study by Zhou et al. [200] showed that purine metabolism was increased in aggressive/high‐grade tumours and represents a potential target to improve the effectiveness of radiotherapy regimes. This is especially compelling given that, as highlighted within this study, there are currently FDA‐approved inhibitors of GTP synthesis, although obviously the efficient delivery of therapeutic doses within the brain will be key to the success of such strategies.
Finally, another common feature of cancer cells, particularly solid tumours, is an imbalance between oxygen supply and demand from active aerobic metabolism, causing regional hypoxia defined as regions of reduced oxygen concentration. This presents both challenges in terms of cell death mechanisms, which are less effective in the context of hypoxia, such as those elicited by radiotherapy, but also potentially exploitable therapeutic opportunities given the effects hypoxia has on several DDR factors [201, 202, 203, 204]. Recent discoveries outside of glioblastoma have revealed key molecular and functional links between the DDR, replication stress signalling and the cGAS‐STING immune pathways [205, 206], and that targeting of replication stress signalling may synergise with immuno‐oncology (IO) therapies [207, 208]. However, the propensity of glioblastoma to escape immunosurveillance, potentially poor receptor expression and anatomical considerations have so far limited progress in the development of effective immunotherapies for glioblastoma compared to other cancers [209, 210, 211, 212]. However, strategies to circumvent such immunosurveillance escape in gliomas have recently been reported [213] and, as more mechanistic understanding around how these pathways interact becomes available, further therapeutic opportunities for gliomas will hopefully be developed. Together, these studies raise the possibility that oncogene‐induced replication stress within residual GSC populations following surgical resection may be targeted with agents that exploit such defective ATR signalling, for example. However, as is unfortunately all too common in glioblastoma research, promising preclinical studies do not necessarily translate into clinical benefit for patients [3, 214] so expectations in this regard need to be measured.
4. Conclusions and future perspectives
As highlighted in this Review, there is a critical need for new and more effective treatment strategies to combat the long‐standing dismal survival rates experienced by patients diagnosed with high‐grade brain tumours such as glioblastoma. Through the combined acquisition of fundamental biological insights into DDR mechanisms and interplay within this network and associated pathways, together with continued progress in imaging, surgical technologies and radiochemotherapy delivery within the clinic, it is expected that targeting of the DDR has a potential to tackle the historic lack of treatment options for these tumours. It will be particularly interesting to see the results of DDR inhibitor clinical trials currently in various phases around the world (Table 1), as this will give an indication of how successful such approaches may or may not be [145, 215]. There is, of course, the risk, given the extensive intratumoural heterogeneity of glioblastoma and inherently treatment‐resistant GSCs within these tumours, that any single targeted therapy may in fact be ‘too targeted’, leading to inevitable resurgence of resistant subclones and tumour repopulation. In a similar manner to other difficult‐to‐treat diseases such as HIV and multidrug‐resistant tuberculosis [216], novel drug combinations may be required to overcome the extensive genetic heterogeneity and resistance mechanisms observed in glioblastoma. Consequently, as highlighted within this Review, targeting multiple DDR constituents in parallel has the potential to provide new effective treatment paradigms that might help prevent disease progression by counteracting complex and overarching phenotypic delinquency within the DDR of cancerous cells (Fig. 3).
In addition to the aforementioned needed improvement in preclinical models that better reflect postsurgical residual disease, an important factor alongside the development of such multimodal strategies will be further improvements in the efficient delivery of therapeutically active doses of drugs beyond the BBB, which remains a significant challenge in glioblastoma therapy [3]. Potential innovations on the horizon include the use of MRI‐directed magnetic nanoparticles [217, 218], surgical delivery of in situ gelling agents [219, 220] and enhanced intrathecal/cerebrospinal fluid delivery using novel viral vectors, antibody ligands or exosomes [221, 222, 223]. Such approaches, in addition to traditional oral or intravenous delivery approaches, coupled with novel ways to disrupt the BBB, such as ultrasound [224, 225] or TTFields [226]‐based approaches, will hopefully provide the best chance for DDR‐targeting approaches to provide much‐needed clinical benefit to patients and families faced with the devastating diagnosis of a high‐grade glioma.
Conflict of interest
OR and SJC have received research funding from the funding bodies acknowledged below and are recipients of an Inovitro™ TTFields preclinical research system (on loan from Novocure) and take part in the annual Inovitro™ Users Meeting hosted by Novocure.
Author contributions
OR and SJC conceived and wrote the manuscript following an invitation from Prof. Kevin Ryan (co‐EIC).
Acknowledgements
OR is funded by an NIHR Clinical Lectureship. OR and SJC acknowledge funding support from The Brain Tumour Charity, NC3Rs, Royal College of Surgeons, Neurocare, Yorkshire’s Brain Tumour Charity (formerly BTRS), Sheffield Hospitals Charity, Weston Park Cancer Charity, American Association for Cancer Research and the NIHR Sheffield Biomedical Research Centre/NIHR Sheffield Clinical Research Facility.
Contributor Information
Ola Rominiyi, Email: o.rominiyi@sheffield.ac.uk.
Spencer J. Collis, Email: s.collis@sheffield.ac.uk.
References
- 1. Burnet NG, Jefferies SJ, Benson RJ, Hunt DP & Treasure FP (2005) Years of life lost (YLL) from cancer is an important measure of population burden–and should be considered when allocating research funds. B J Cancer 92, 241–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Patel AP, Fisher JL, Nichols E, Abd‐Allah F, Abdela J, Abdelalim A, Abraha HN, Agius D, Alahdab F, Alam T et al. (2019) Global, regional, and national burden of brain and other CNS cancer, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol 18, 376–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Aldape K, Brindle KM, Chesler L, Chopra R, Gajjar A, Gilbert MR, Gottardo N, Gutmann DH, Hargrave D, Holland EC et al. (2019) Challenges to curing primary brain tumours. Nat Rev Clin Oncol 16, 509–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ostrom QT, Patil N, Cioffi G, Waite K, Kruchko C & Barnholtz‐Sloan JS (2020) CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2013–2017. Neuro Oncol 22, iv1–iv96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Reifenberger G, Wirsching HG, Knobbe‐Thomsen CB & Weller M (2017) Advances in the molecular genetics of gliomas – implications for classification and therapy. Nat Rev Clin Oncol 14, 434–452. [DOI] [PubMed] [Google Scholar]
- 6. Bi J, Chowdhry S, Wu S, Zhang W, Masui K & Mischel PS (2020) Altered cellular metabolism in gliomas – an emerging landscape of actionable co‐dependency targets. Nat Rev Cancer 20, 57–70. [DOI] [PubMed] [Google Scholar]
- 7. Stupp R, Hegi ME, Mason WP, van den Bent MJ , Taphoorn MJB, Janzer RC, Ludwin SK, Allgeier A, Fisher B, Belanger K et al. (2009) Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5‐year analysis of the EORTC‐NCIC trial. Lancet Oncol 10, 459–466. [DOI] [PubMed] [Google Scholar]
- 8. Stupp R, Mason WP, van den Bent MJ , Weller M, Fisher B, Taphoorn MJB, Belanger K, Brandes AA, Marosi C, Bogdahn U et al. (2005) Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 352, 987–996. [DOI] [PubMed] [Google Scholar]
- 9. Weller M, van den Bent M , Preusser M, Le Rhun E, Tonn JC, Minniti G, Bendszus M, Balana C, Chinot O, Dirven L et al. (2021) EANO guidelines on the diagnosis and treatment of diffuse gliomas of adulthood. Nat Rev Clin Oncol 18, 170–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N , Weller M, Kros JM, Hainfellner JA, Mason W, Mariani L et al. (2005) MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med 352, 997–1003. [DOI] [PubMed] [Google Scholar]
- 11. Chinot OL, Wick W, Mason W, Henriksson R, Saran F, Nishikawa R, Carpentier AF, Hoang‐Xuan K, Kavan P, Cernea D et al. (2014) Bevacizumab plus radiotherapy‐temozolomide for newly diagnosed glioblastoma. N Engl J Med 370, 709–722. [DOI] [PubMed] [Google Scholar]
- 12. Gilbert MR, Wang M, Aldape KD, Stupp R, Hegi ME, Jaeckle KA, Armstrong TS, Wefel JS, Won M, Blumenthal DT et al. (2013) Dose‐dense temozolomide for newly diagnosed glioblastoma: a randomized phase III clinical trial. J Clin Oncol 31, 4085–4091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Khasraw M, McDonald KL, Rosenthal M, Lwin Z, Ashley DM, Wheeler H, Barnes E, Foote MC, Koh E‐S, Sulman EP et al. (2019) A randomized phase II trial of veliparib (V), radiotherapy (RT) and temozolomide (TMZ) in patients (pts) with unmethylated MGMT (uMGMT) glioblastoma (GBM). J Clin Oncol 37, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Oronsky B, Reid TR, Oronsky A, Sandhu N & Knox SJ (2020) A review of newly diagnosed glioblastoma. Front Oncol 10, 574012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chaudhry A, Benson L, Varshaver M, Farber O, Weinberg U, Kirson E & Palti Y (2015) NovoTTF‐100A system (Tumor Treating Fields) transducer array layout planning for glioblastoma: a NovoTAL system user study. World J Surg Oncol 13, 316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rominiyi O, Vanderlinden A, Clenton SJ, Bridgewater C, Al‐Tamimi Y & Collis SJ (2021) Tumour treating fields therapy for glioblastoma: current advances and future directions. Br J Cancer 124, 697–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Stupp R, Taillibert S, Kanner A, Read W, Steinberg D, Lhermitte B, Toms S, Idbaih A, Ahluwalia MS, Fink K et al. (2017) Effect of tumor‐treating fields plus maintenance temozolomide vs maintenance temozolomide alone on survival in patients with glioblastoma: a randomized clinical trial. JAMA 318, 2306–2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Stupp R, Wong ET, Kanner AA, Steinberg D, Engelhard H, Heidecke V, Kirson ED, Taillibert S, Liebermann F, Dbaly V et al. (2012) NovoTTF‐100A versus physician's choice chemotherapy in recurrent glioblastoma: a randomised phase III trial of a novel treatment modality. Eur J Cancer 48, 2192–2202. [DOI] [PubMed] [Google Scholar]
- 19. Lacroix M, Abi‐Said D, Fourney DR, Gokaslan ZL, Shi W, DeMonte F, Lang FF, McCutcheon IE, Hassenbusch SJ, Holland E et al. (2001) A multivariate analysis of 416 patients with glioblastoma multiforme: prognosis, extent of resection, and survival. J Neurosurg 95, 190–198. [DOI] [PubMed] [Google Scholar]
- 20. Ferraro N, Barbarite E, Albert TR, Berchmans E, Shah AH, Bregy A, Ivan ME, Brown T & Komotar RJ (2016) The role of 5‐aminolevulinic acid in brain tumor surgery: a systematic review. Neurosurg Rev 39, 545–555. [DOI] [PubMed] [Google Scholar]
- 21. Kuhnt D, Becker A, Ganslandt O, Bauer M, Buchfelder M & Nimsky C (2011) Correlation of the extent of tumor volume resection and patient survival in surgery of glioblastoma multiforme with high‐field intraoperative MRI guidance. Neuro Oncol 13, 1339–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Prada F, Bene MD, Fornaro R, Vetrano IG, Martegani A, Aiani L, Sconfienza LM, Mauri G, Solbiati L, Pollo B et al. (2016) Identification of residual tumor with intraoperative contrast‐enhanced ultrasound during glioblastoma resection. Neurosurg Focus 40, E7. [DOI] [PubMed] [Google Scholar]
- 23. Stummer W, Novotny A, Stepp H, Goetz C, Bise K & Reulen HJ (2000) Fluorescence‐guided resection of glioblastoma multiforme by using 5‐aminolevulinic acid‐induced porphyrins: a prospective study in 52 consecutive patients. J Neurosurg 93, 1003–1013. [DOI] [PubMed] [Google Scholar]
- 24. Stummer W, Pichlmeier U, Meinel T, Wiestler OD, Zanella F, Reulen HJ & Group AL‐GS (2006) Fluorescence‐guided surgery with 5‐aminolevulinic acid for resection of malignant glioma: a randomised controlled multicentre phase III trial. Lancet Oncol 7, 392–401. [DOI] [PubMed] [Google Scholar]
- 25. Alexander J, Gildea L, Balog J, Speller A, McKenzie J, Muirhead L, Scott A, Kontovounisios C, Rasheed S, Teare J et al. (2017) A novel methodology for in vivo endoscopic phenotyping of colorectal cancer based on real‐time analysis of the mucosal lipidome: a prospective observational study of the iKnife. Surg Endosc 31, 1361–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Balog J, Sasi‐Szabo L, Kinross J, Lewis MR, Muirhead LJ, Veselkov K, Mirnezami R, Dezso B, Damjanovich L, Darzi A et al. (2013) Intraoperative tissue identification using rapid evaporative ionization mass spectrometry. Sci Transl Med 5, 194ra93. [DOI] [PubMed] [Google Scholar]
- 27. Depciuch J, Tolpa B, Witek P, Szmuc K, Kaznowska E, Osuchowski M, Krol P & Cebulski J (2020) Raman and FTIR spectroscopy in determining the chemical changes in healthy brain tissues and glioblastoma tumor tissues. Spectrochim Acta A Mol Biomol Spectrosc 225, 117526. [DOI] [PubMed] [Google Scholar]
- 28. Livermore LJ, Isabelle M, Bell IM, Edgar O, Voets NL, Stacey R, Ansorge O, Vallance C & Plaha P (2020) Raman spectroscopy to differentiate between fresh tissue samples of glioma and normal brain: a comparison with 5‐ALA‐induced fluorescence‐guided surgery. J Neurosurg 1–11. 10.3171/2020.5.JNS20376 [DOI] [PubMed] [Google Scholar]
- 29. Livermore LJ, Isabelle M, Bell IM, Scott C, Walsby‐Tickle J, Gannon J, Plaha P, Vallance C & Ansorge O (2019) Rapid intraoperative molecular genetic classification of gliomas using Raman spectroscopy. Neurooncol Adv 1, vdz008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Riva M, Sciortino T, Secoli R, D'Amico E, Moccia S, Fernandes B, Conti Nibali M, Gay L, Rossi M, De Momi E et al. (2021) Glioma biopsies classification using Raman spectroscopy and machine learning models on fresh tissue samples. Cancers 13, 1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ludmir EB, Mahajan A, Ahern V, Ajithkumar T, Alapetite C, Bernier‐Chastagner V, Bindra RS, Bishop AJ, Bolle S, Brown PD et al. (2019) Assembling the brain trust: the multidisciplinary imperative in neuro‐oncology. Nat Rev Clin Oncol 16, 521–522. [DOI] [PubMed] [Google Scholar]
- 32. Thompson MK, Poortmans P, Chalmers AJ, Faivre‐Finn C, Hall E, Huddart RA, Lievens Y, Sebag‐Montefiore D & Coles CE (2018) Practice‐changing radiation therapy trials for the treatment of cancer: where are we 150 years after the birth of Marie Curie? Br J Cancer 119, 389–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Arvanitis CD, Ferraro GB & Jain RK (2020) The blood–brain barrier and blood–tumour barrier in brain tumours and metastases. Nat Rev Cancer 20, 26–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, Berman SH et al. (2013) The somatic genomic landscape of glioblastoma. Cell 155, 462–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Neftel C, Laffy J, Filbin MG, Hara T, Shore ME, Rahme GJ, Richman AR, Silverbush D, Shaw ML, Hebert CM et al. (2019) An integrative model of cellular states, plasticity, and genetics for glioblastoma. Cell 178, 835–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ohgaki H & Kleihues P (2013) The definition of primary and secondary glioblastoma. Clin Cancer Res 19, 764–772. [DOI] [PubMed] [Google Scholar]
- 37. Le Rhun E, Preusser M, Roth P, Reardon DA, van den Bent M , Wen P, Reifenberger G & Weller M (2019) Molecular targeted therapy of glioblastoma. Cancer Treat Rev 80, 101896. [DOI] [PubMed] [Google Scholar]
- 38. Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, Cahill DP, Nahed BV, Curry WT, Martuza RL et al. (2014) Single‐cell RNA‐seq highlights intratumoral heterogeneity in primary glioblastoma. Science 344, 1396–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Piccirillo SGM, Colman S, Potter NE, van Delft FW , Lillis S, Carnicer MJ, Kearney L, Watts C & Greaves M (2015) Genetic and functional diversity of propagating cells in glioblastoma. Stem Cell Rep 4, 7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sottoriva A, Spiteri I, Piccirillo SG, Touloumis A, Collins VP, Marioni JC, Curtis C, Watts C & Tavare S (2013) Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc Natl Acad Sci USA 110, 4009–4014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Spiteri I, Caravagna G, Cresswell GD, Vatsiou A, Nichol D, Acar A, Ermini L, Chkhaidze K, Werner B, Mair R et al. (2019) Evolutionary dynamics of residual disease in human glioblastoma. Ann Oncol 30, 456–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Castellan M, Guarnieri A, Fujimura A, Zanconato F, Battilana G, Panciera T, Sladitschek HL, Contessotto P, Citron A, Grilli A et al. (2021) Single‐cell analyses reveal YAP/TAZ as regulators of stemness and cell plasticity in Glioblastoma. Nat Cancer 2, 174–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Galli R, Binda E, Orfanelli U, Cipelletti B, Gritti A, De Vitis S, Fiocco R, Foroni C, Dimeco F & Vescovi A (2004) Isolation and characterization of tumorigenic, stem‐like neural precursors from human glioblastoma. Cancer Res 64, 7011–7021. [DOI] [PubMed] [Google Scholar]
- 44. Lathia JD, Mack SC, Mulkearns‐Hubert EE, Valentim CL & Rich JN (2015) Cancer stem cells in glioblastoma. Genes Dev 29, 1203–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mitchell K, Troike K, Silver DJ & Lathia JD (2021) The evolution of the cancer stem cell state in glioblastoma: emerging insights into the next generation of functional interactions. Neuro Oncol 23, 199–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Prager BC, Bhargava S, Mahadev V, Hubert CG & Rich JN (2020) Glioblastoma stem cells: driving resilience through chaos. Trends Cancer 6, 223–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD & Dirks PB (2004) Identification of human brain tumour initiating cells. Nature 432, 396–401. [DOI] [PubMed] [Google Scholar]
- 48. Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD & Rich JN (2006) Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 444, 756–760. [DOI] [PubMed] [Google Scholar]
- 49. Carruthers RD, Ahmed SU, Ramachandran S, Strathdee K, Kurian KM, Hedley A, Gomez‐Roman N, Kalna G, Neilson M, Gilmour L et al. (2018) Replication stress drives constitutive activation of the DNA damage response and radioresistance in glioblastoma stem‐like cells. Cancer Res 78, 5060–5071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chen J, Li Y, Yu TS, McKay RM, Burns DK, Kernie SG & Parada LF (2012) A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 488, 522–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Stupp R, Lukas RV & Hegi ME (2019) Improving survival in molecularly selected glioblastoma. Lancet 393, 615–617. [DOI] [PubMed] [Google Scholar]
- 52. Bedard PL, Hansen AR, Ratain MJ & Siu LL (2013) Tumour heterogeneity in the clinic. Nature 501, 355–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Johansson P, Krona C, Kundu S, Doroszko M, Baskaran S, Schmidt L, Vinel C, Almstedt E, Elgendy R, Elfineh L et al. (2020) A patient‐derived cell atlas informs precision targeting of glioblastoma. Cell Rep 32, 107897. [DOI] [PubMed] [Google Scholar]
- 54. Phillips HS, Kharbanda S, Chen R, Forrest WF, Soriano RH, Wu TD, Misra A, Nigro JM, Colman H, Soroceanu L et al. (2006) Molecular subclasses of high‐grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 9, 157–173. [DOI] [PubMed] [Google Scholar]
- 55. Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP et al. (2010) Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17, 98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wang Q, Hu B, Hu X, Kim H, Squatrito M, Scarpace L, deCarvalho AC, Lyu S, Li P, Li Y et al. (2017) Tumor evolution of glioma‐intrinsic gene expression subtypes associates with immunological changes in the microenvironment. Cancer Cell 32, 42–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Soeda A, Hara A, Kunisada T, Yoshimura S, Iwama T & Park DM (2015) The evidence of glioblastoma heterogeneity. Sci Rep 5, 7979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Vartanian A, Singh SK, Agnihotri S, Jalali S, Burrell K, Aldape KD & Zadeh G (2014) GBM's multifaceted landscape: highlighting regional and microenvironmental heterogeneity. Neuro Oncol 16, 1167–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Caragher S, Chalmers AJ & Gomez‐Roman N (2019) Glioblastoma's next top model: novel culture systems for brain cancer radiotherapy research. Cancers 11, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Rominiyi O, Al‐Tamimi Y & Collis SJ (2019) The 'Ins and Outs' of early preclinical models for brain tumor research: are they valuable and have we been doing it wrong? Cancers 11, 426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. De Witt Hamer PC, Van Tilborg AA, Eijk PP, Sminia P, Troost D, Van Noorden CJ, Ylstra B & Leenstra S (2008) The genomic profile of human malignant glioma is altered early in primary cell culture and preserved in spheroids. Oncogene 27, 2091–2096. [DOI] [PubMed] [Google Scholar]
- 62. Fael Al‐Mayhani TM, Ball SL, Zhao JW, Fawcett J, Ichimura K, Collins PV & Watts C (2009) An efficient method for derivation and propagation of glioblastoma cell lines that conserves the molecular profile of their original tumours. J Neurosci Methods 176, 192–199. [DOI] [PubMed] [Google Scholar]
- 63. Gomez‐Roman N & Chalmers AJ (2019) Patient‐specific 3D‐printed glioblastomas. Nat Biomed Eng 3, 498–499. [DOI] [PubMed] [Google Scholar]
- 64. Gomez‐Roman N, Chong MY, Chahal SK, Caragher SP, Jackson MR, Stevenson KH, Dongre SA & Chalmers AJ (2020) Radiation responses of 2D and 3D glioblastoma cells: a novel, 3D‐specific radioprotective role of VEGF/Akt signaling through functional activation of NHEJ. Mol Cancer Ther 19, 575–589. [DOI] [PubMed] [Google Scholar]
- 65. Gomez‐Roman N, Stevenson K, Gilmour L, Hamilton G & Chalmers AJ (2017) A novel 3D human glioblastoma cell culture system for modeling drug and radiation responses. Neuro Oncol 19, 229–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Hubert CG, Rivera M, Spangler LC, Wu Q, Mack SC, Prager BC, Couce M, McLendon RE, Sloan AE & Rich JN (2016) A three‐dimensional organoid culture system derived from human glioblastomas recapitulates the hypoxic gradients and cancer stem cell heterogeneity of tumors found in vivo. Cancer Res 76, 2465–2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Iorio F, Knijnenburg TA, Vis DJ, Bignell GR, Menden MP, Schubert M, Aben N, Goncalves E, Barthorpe S, Lightfoot H et al. (2016) A landscape of pharmacogenomic interactions in cancer. Cell 166, 740–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Linkous A, Balamatsias D, Snuderl M, Edwards L, Miyaguchi K, Milner T, Reich B, Cohen‐Gould L, Storaska A, Nakayama Y et al. (2019) Modeling patient‐derived glioblastoma with cerebral organoids. Cell Rep 26, 3203–3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Ogawa J, Pao GM, Shokhirev MN & Verma IM (2018) Glioblastoma model using human cerebral organoids. Cell Rep 23, 1220–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Pine AR, Cirigliano SM, Nicholson JG, Hu Y, Linkous A, Miyaguchi K, Edwards L, Singhania R, Schwartz TH, Ramakrishna R et al. (2020) Tumor microenvironment is critical for the maintenance of cellular states found in primary glioblastomas. Cancer Discov 10, 964–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Pollard SM, Yoshikawa K, Clarke ID, Danovi D, Stricker S, Russell R, Bayani J, Head R, Lee M, Bernstein M et al. (2009) Glioma stem cell lines expanded in adherent culture have tumor‐specific phenotypes and are suitable for chemical and genetic screens. Cell Stem Cell 4, 568–580. [DOI] [PubMed] [Google Scholar]
- 72. Rahman M, Reyner K, Deleyrolle L, Millette S, Azari H, Day BW, Stringer BW, Boyd AW, Johns TG, Blot V et al. (2015) Neurosphere and adherent culture conditions are equivalent for malignant glioma stem cell lines. Anat Cell Biol 48, 25–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Yi HG, Jeong YH, Kim Y, Choi YJ, Moon HE, Park SH, Kang KS, Bae M, Jang J, Youn H et al. (2019) A bioprinted human‐glioblastoma‐on‐a‐chip for the identification of patient‐specific responses to chemoradiotherapy. Nat Biomed Eng 3, 509–519. [DOI] [PubMed] [Google Scholar]
- 74. Ben‐David U, Ha G, Tseng YY, Greenwald NF, Oh C, Shih J, McFarland JM, Wong B, Boehm JS, Beroukhim R et al. (2017) Patient‐derived xenografts undergo mouse‐specific tumor evolution. Nat Genet 49, 1567–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Holland EC (2000) A mouse model for glioma: biology, pathology, and therapeutic opportunities. Toxicol Pathol 28, 171–177. [DOI] [PubMed] [Google Scholar]
- 76. Lenting K, Verhaak R, Ter Laan M, Wesseling P & Leenders W (2017) Glioma: experimental models and reality. Acta Neuropathol 133, 263–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Oh T, Fakurnejad S, Sayegh ET, Clark AJ, Ivan ME, Sun MZ, Safaee M, Bloch O, James CD & Parsa AT (2014) Immunocompetent murine models for the study of glioblastoma immunotherapy. J Transl Med 12, 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Goldstein M & Kastan MB (2015) The DNA damage response: implications for tumor responses to radiation and chemotherapy. Annu Rev Med 66, 129–143. [DOI] [PubMed] [Google Scholar]
- 79. Hoeijmakers JH (2001) Genome maintenance mechanisms for preventing cancer. Nature 411, 366–374. [DOI] [PubMed] [Google Scholar]
- 80. Lindahl T (1993) Instability and decay of the primary structure of DNA. Nature 362, 709–715. [DOI] [PubMed] [Google Scholar]
- 81. Ciccia A & Elledge SJ (2010) The DNA damage response: making it safe to play with knives. Mol Cell 40, 179–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Curtin NJ (2012) DNA repair dysregulation from cancer driver to therapeutic target. Nat Rev Cancer 12, 801–817. [DOI] [PubMed] [Google Scholar]
- 83. Jeggo PA, Pearl LH & Carr AM (2016) DNA repair, genome stability and cancer: a historical perspective. Nat Rev Cancer 16, 35–42. [DOI] [PubMed] [Google Scholar]
- 84. Olivieri M, Cho T, Alvarez‐Quilon A, Li K, Schellenberg MJ, Zimmermann M, Hustedt N, Rossi SE, Adam S, Melo H et al. (2020) A genetic map of the response to DNA damage in human cells. Cell 182, 481–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Smith HL, Southgate H, Tweddle DA & Curtin NJ (2020) DNA damage checkpoint kinases in cancer. Expert Rev Mol Med 22, e2. [DOI] [PubMed] [Google Scholar]
- 86. Gourley C, Balmana J, Ledermann JA, Serra V, Dent R, Loibl S, Pujade‐Lauraine E & Boulton SJ (2019) Moving from poly (ADP‐ribose) polymerase inhibition to targeting DNA repair and DNA damage response in cancer therapy. J Clin Oncol 37, 2257–2269. [DOI] [PubMed] [Google Scholar]
- 87. Jackson SP & Helleday T (2016) Drugging DNA repair. Science 352, 1178–1179. [DOI] [PubMed] [Google Scholar]
- 88. Pearl LH, Schierz AC, Ward SE, Al‐Lazikani B & Pearl FM (2015) Therapeutic opportunities within the DNA damage response. Nat Rev Cancer 15, 166–180. [DOI] [PubMed] [Google Scholar]
- 89. Pilie PG, Tang C, Mills GB & Yap TA (2019) State‐of‐the‐art strategies for targeting the DNA damage response in cancer. Nat Rev Clin Oncol 16, 81–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Blackford AN & Jackson SP (2017) ATM, ATR, and DNA‐PK: the trinity at the heart of the DNA damage response. Mol Cell 66, 801–817. [DOI] [PubMed] [Google Scholar]
- 91. Mavragani IV, Nikitaki Z, Kalospyros SA & Georgakilas AG (2019) Ionizing radiation and complex DNA damage: from prediction to detection challenges and biological significance. Cancers 11, 1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Kotsantis P, Petermann E & Boulton SJ (2018) Mechanisms of oncogene‐induced replication stress: jigsaw falling into place. Cancer Discov 8, 537–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Primo LMF & Teixeira LK (2019) DNA replication stress: oncogenes in the spotlight. Genet Mol Biol 43, e20190138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Shiloh Y & Ziv Y (2013) The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol 14, 197–210. [PubMed] [Google Scholar]
- 95. Saldivar JC, Cortez D & Cimprich KA (2017) The essential kinase ATR: ensuring faithful duplication of a challenging genome. Nat Rev Mol Cell Biol 18, 622–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Marechal A & Zou L (2013) DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb Perspect Biol 5, a012716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Beckta JM, Bindra RS & Chalmers AJ (2019) Targeting DNA repair in gliomas. Curr Opin Neurol 32, 878–885. [DOI] [PubMed] [Google Scholar]
- 98. Bradbury A, Hall S, Curtin N & Drew Y (2020) Targeting ATR as cancer therapy: a new era for synthetic lethality and synergistic combinations? Pharmacol Ther 207, 107450. [DOI] [PubMed] [Google Scholar]
- 99. Chapman JR, Taylor MR & Boulton SJ (2012) Playing the end game: DNA double‐strand break repair pathway choice. Mol Cell 47, 497–510. [DOI] [PubMed] [Google Scholar]
- 100. Jeggo PA & Downs JA (2014) Roles of chromatin remodellers in DNA double strand break repair. Exp Cell Res 329, 69–77. [DOI] [PubMed] [Google Scholar]
- 101. Jeggo PA, Downs JA & Gasser SM (2017) Chromatin modifiers and remodellers in DNA repair and signalling. Philos Trans R Soc Lond B Biol Sci 372, 20160279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Panier S & Boulton SJ (2014) Double‐strand break repair: 53BP1 comes into focus. Nat Rev Mol Cell Biol 15, 7–18. [DOI] [PubMed] [Google Scholar]
- 103. Kaina B, Christmann M, Naumann S & Roos WP (2007) MGMT: key node in the battle against genotoxicity, carcinogenicity and apoptosis induced by alkylating agents. DNA Repair 6, 1079–1099. [DOI] [PubMed] [Google Scholar]
- 104. Jiricny J (2013) Postreplicative mismatch repair. Cold Spring Harb Perspect Biol 5, a012633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Fu D, Calvo JA & Samson LD (2012) Balancing repair and tolerance of DNA damage caused by alkylating agents. Nature Rev Cancer 12, 104–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Barthel FP, Johnson KC, Varn FS, Moskalik AD, Tanner G, Kocakavuk E, Anderson KJ, Abiola O, Aldape K, Alfaro KD et al. (2019) Longitudinal molecular trajectories of diffuse glioma in adults. Nature 576, 112–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Daniel P, Sabri S, Chaddad A, Meehan B, Jean‐Claude B, Rak J & Abdulkarim BS (2019) Temozolomide induced hypermutation in glioma: evolutionary mechanisms and therapeutic opportunities. Front Oncol 9, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. MacLeod G, Bozek DA, Rajakulendran N, Monteiro V, Ahmadi M, Steinhart Z, Kushida MM, Yu H, Coutinho FJ, Cavalli FMG et al. (2019) Genome‐wide CRISPR‐Cas9 screens expose genetic vulnerabilities and mechanisms of temozolomide sensitivity in glioblastoma stem cells. Cell Rep 27, 971–986. [DOI] [PubMed] [Google Scholar]
- 109. Wick W, Weller M, van den Bent M , Sanson M, Weiler M, von Deimling A , Plass C, Hegi M, Platten M & Reifenberger G (2014) MGMT testing–the challenges for biomarker‐based glioma treatment. Nat Rev Neurol 10, 372–385. [DOI] [PubMed] [Google Scholar]
- 110. Frosina G (2000) Overexpression of enzymes that repair endogenous damage to DNA. Eur J Biochem 267, 2135–2149. [DOI] [PubMed] [Google Scholar]
- 111. Johannessen TC, Bjerkvig R & Tysnes BB (2008) DNA repair and cancer stem‐like cells–potential partners in glioma drug resistance? Cancer Treat Rev 34, 558–567. [DOI] [PubMed] [Google Scholar]
- 112. Trivedi RN, Almeida KH, Fornsaglio JL, Schamus S & Sobol RW (2005) The role of base excision repair in the sensitivity and resistance to temozolomide‐mediated cell death. Cancer Res 65, 6394–6400. [DOI] [PubMed] [Google Scholar]
- 113. Marteijn JA, Lans H, Vermeulen W & Hoeijmakers JH (2014) Understanding nucleotide excision repair and its roles in cancer and ageing. Nat Rev Mol Cell Biol 15, 465–481. [DOI] [PubMed] [Google Scholar]
- 114. Harrision D, Gravells P, Thompson R & Bryant HE (2020) Poly(ADP‐ribose) glycohydrolase (PARG) vs. Poly(ADP‐ribose) polymerase (PARP) – function in genome maintenance and relevance of inhibitors for anti‐cancer therapy. Front Mol Biosci 7, 191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Liao H, Ji F, Helleday T & Ying S (2018) Mechanisms for stalled replication fork stabilization: new targets for synthetic lethality strategies in cancer treatments. EMBO Rep 19, e46263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Lord CJ & Ashworth A (2016) BRCAness revisited. Nat Rev Cancer 16, 110–120. [DOI] [PubMed] [Google Scholar]
- 117. Sung P & Klein H (2006) Mechanism of homologous recombination: mediators and helicases take on regulatory functions. Nat Rev Mol Cell Biol 7, 739–750. [DOI] [PubMed] [Google Scholar]
- 118. Giladi M, Munster M, Schneiderman RS, Voloshin T, Porat Y, Blat R, Zielinska‐Chomej K, Haag P, Bomzon Z, Kirson ED et al. (2017) Tumor treating fields (TTFields) delay DNA damage repair following radiation treatment of glioma cells. Radiat Oncol 12, 206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Chang HHY, Pannunzio NR, Adachi N & Lieber MR (2017) Non‐homologous DNA end joining and alternative pathways to double‐strand break repair. Nat Rev Mol Cell Biol 18, 495–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Scully R, Panday A, Elango R & Willis NA (2019) DNA double‐strand break repair‐pathway choice in somatic mammalian cells. Nat Rev Mol Cell Biol 20, 698–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Yata K & Esashi F (2009) Dual role of CDKs in DNA repair: to be, or not to be. DNA Repair 8, 6–18. [DOI] [PubMed] [Google Scholar]
- 122. Malumbres M & Barbacid M (2009) Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer 9, 153–166. [DOI] [PubMed] [Google Scholar]
- 123. Vijayaraghavan S, Moulder S, Keyomarsi K & Layman RM (2018) Inhibiting CDK in cancer therapy: current evidence and future directions. Target Oncol 13, 21–38. [DOI] [PubMed] [Google Scholar]
- 124. Degan P, Cappelli E, Regis S & Ravera S (2019) New insights and perspectives in Fanconi Anemia research. Trends Mol Med 25, 167–170. [DOI] [PubMed] [Google Scholar]
- 125. Kennedy RD & D'Andrea AD (2005) The Fanconi Anemia/BRCA pathway: new faces in the crowd. Genes Dev 19, 2925–2940. [DOI] [PubMed] [Google Scholar]
- 126. Nalepa G & Clapp DW (2018) Fanconi anaemia and cancer: an intricate relationship. Nat Rev Cancer 18, 168–185. [DOI] [PubMed] [Google Scholar]
- 127. Niraj J, Farkkila A & D'Andrea AD (2019) The Fanconi anemia pathway in cancer. Annu Rev Cancer Biol 3, 457–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Wang AT & Smogorzewska A (2015) SnapShot: Fanconi anemia and associated proteins. Cell 160, 354–354.e1. [DOI] [PubMed] [Google Scholar]
- 129. Andreassen PR, D'Andrea AD & Taniguchi T (2004) ATR couples FANCD2 monoubiquitination to the DNA‐damage response. Genes Dev 18, 1958–1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Elia AE, Wang DC, Willis NA, Boardman AP, Hajdu I, Adeyemi RO, Lowry E, Gygi SP, Scully R & Elledge SJ (2015) RFWD3‐dependent ubiquitination of RPA regulates repair at stalled replication forks. Mol Cell 60, 280–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Feeney L, Munoz IM, Lachaud C, Toth R, Appleton PL, Schindler D & Rouse J (2017) RPA‐mediated recruitment of the E3 ligase RFWD3 is vital for interstrand crosslink repair and human health. Mol Cell 66, 610–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Inano S, Sato K, Katsuki Y, Kobayashi W, Tanaka H, Nakajima K, Nakada S, Miyoshi H, Knies K, Takaori‐Kondo A et al. (2017) RFWD3‐mediated ubiquitination promotes timely removal of both RPA and RAD51 from DNA damage sites to facilitate homologous recombination. Mol Cell 66, 622–634. [DOI] [PubMed] [Google Scholar]
- 133. Ishiai M, Kitao H, Smogorzewska A, Tomida J, Kinomura A, Uchida E, Saberi A, Kinoshita E, Kinoshita‐Kikuta E, Koike T et al. (2008) FANCI phosphorylation functions as a molecular switch to turn on the Fanconi anemia pathway. Nat Struct Mol Biol 15, 1138–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Lossaint G, Larroque M, Ribeyre C, Bec N, Larroque C, Decaillet C, Gari K & Constantinou A (2013) FANCD2 binds MCM proteins and controls replisome function upon activation of s phase checkpoint signaling. Mol Cell 51, 678–690. [DOI] [PubMed] [Google Scholar]
- 135. Walden H & Deans AJ (2014) The Fanconi anemia DNA repair pathway: structural and functional insights into a complex disorder. Annu Rev Biophys 43, 257–278. [DOI] [PubMed] [Google Scholar]
- 136. Kaelin WG Jr (2005) The concept of synthetic lethality in the context of anticancer therapy. Nat Rev Cancer 5, 689–698. [DOI] [PubMed] [Google Scholar]
- 137. Ashworth A & Lord CJ (2018) Synthetic lethal therapies for cancer: what's next after PARP inhibitors? Nat Rev Clin Oncol 15, 564–576. [DOI] [PubMed] [Google Scholar]
- 138. Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ & Helleday T (2005) Specific killing of BRCA2‐deficient tumours with inhibitors of poly(ADP‐ribose) polymerase. Nature 434, 913–917. [DOI] [PubMed] [Google Scholar]
- 139. Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C et al. (2005) Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434, 917–921. [DOI] [PubMed] [Google Scholar]
- 140. Mateo J, Lord CJ, Serra V, Tutt A, Balmana J, Castroviejo‐Bermejo M, Cruz C, Oaknin A, Kaye SB & de Bono JS (2019) A decade of clinical development of PARP inhibitors in perspective. Ann Oncol 30, 1437–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Ning J & Wakimoto H (2020) Therapeutic application of PARP inhibitors in neuro‐oncology. Trends Cancer 6, 147–159. [DOI] [PubMed] [Google Scholar]
- 142. Yap TA, Plummer R, Azad NS & Helleday T (2019) The DNA damaging revolution: PARP inhibitors and beyond. Am Soc Clin Oncol Educ Book 39, 185–195. [DOI] [PubMed] [Google Scholar]
- 143. Sulkowski PL, Oeck S, Dow J, Economos NG, Mirfakhraie L, Liu Y, Noronha K, Bao X, Li J, Shuch BM et al. (2020) Oncometabolites suppress DNA repair by disrupting local chromatin signalling. Nature 582, 586–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Wang Y, Wild AT, Turcan S, Wu WH, Sigel C, Klimstra DS, Ma X, Gong Y, Holland EC, Huse JT et al. (2020) Targeting therapeutic vulnerabilities with PARP inhibition and radiation in IDH‐mutant gliomas and cholangiocarcinomas. Sci Adv 6, eaaz3221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Hanna C, Kurian KM, Williams K, Watts C, Jackson A, Carruthers R, Strathdee K, Cruickshank G, Dunn L, Erridge S et al. (2020) Pharmacokinetics, safety, and tolerability of olaparib and temozolomide for recurrent glioblastoma: results of the phase I OPARATIC trial. Neuro Oncol 22, 1840–1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Meyer M, Reimand J, Lan X, Head R, Zhu X, Kushida M, Bayani J, Pressey JC, Lionel AC, Clarke ID et al. (2015) Single cell‐derived clonal analysis of human glioblastoma links functional and genomic heterogeneity. Proc Natl Acad Sci USA 112, 851–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Xie Y, Bergstrom T, Jiang Y, Johansson P, Marinescu VD, Lindberg N, Segerman A, Wicher G, Niklasson M, Baskaran S et al. (2015) The human glioblastoma cell culture resource: validated cell models representing all molecular subtypes. EBioMedicine 2, 1351–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Chen CC, Taniguchi T & D'Andrea A (2007) The Fanconi anemia (FA) pathway confers glioma resistance to DNA alkylating agents. J Mol Med 85, 497–509. [DOI] [PubMed] [Google Scholar]
- 149. Kondo N, Takahashi A, Mori E, Noda T, Zdzienicka MZ, Thompson LH, Helleday T, Suzuki M, Kinashi Y, Masunaga S et al. (2011) FANCD1/BRCA2 plays predominant role in the repair of DNA damage induced by ACNU or TMZ. PLoS One 6, e19659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Patil AA, Sayal P, Depondt ML, Beveridge RD, Roylance A, Kriplani DH, Myers KN, Cox A, Jellinek D, Fernando M et al. (2014) FANCD2 re‐expression is associated with glioma grade and chemical inhibition of the Fanconi Anaemia pathway sensitises gliomas to chemotherapeutic agents. Oncotarget 5, 6414–6424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Gupta SK, Kizilbash SH, Carlson BL, Mladek AC, Boakye‐Agyeman F, Bakken KK, Pokorny JL, Schroeder MA, Decker PA, Cen L et al. (2016) Delineation of MGMT hypermethylation as a biomarker for veliparib‐mediated temozolomide‐sensitizing therapy of glioblastoma. J Natl Cancer Inst 108, djv369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Gupta SK, Smith EJ, Mladek AC, Tian S, Decker PA, Kizilbash SH, Kitange GJ & Sarkaria JN (2018) PARP inhibitors for sensitization of alkylation chemotherapy in glioblastoma: impact of blood‐brain barrier and molecular heterogeneity. Front Oncol 8, 670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Helleday T, Petermann E, Lundin C, Hodgson B & Sharma RA (2008) DNA repair pathways as targets for cancer therapy. Nat Rev Cancer 8, 193–204. [DOI] [PubMed] [Google Scholar]
- 154. D'Atri S, Tentori L, Lacal PM, Graziani G, Pagani E, Benincasa E, Zambruno G, Bonmassar E & Jiricny J (1998) Involvement of the mismatch repair system in temozolomide‐induced apoptosis. Mol Pharmacol 54, 334–341. [DOI] [PubMed] [Google Scholar]
- 155. Sarkaria JN, Kitange GJ, James CD, Plummer R, Calvert H, Weller M & Wick W (2008) Mechanisms of chemoresistance to alkylating agents in malignant glioma. Clin Cancer Res 14, 2900–2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Caporali S, Falcinelli S, Starace G, Russo MT, Bonmassar E, Jiricny J & D'Atri S (2004) DNA damage induced by temozolomide signals to both ATM and ATR: role of the mismatch repair system. Mol Pharmacol 66, 478–491. [DOI] [PubMed] [Google Scholar]
- 157. Eich M, Roos WP, Nikolova T & Kaina B (2013) Contribution of ATM and ATR to the resistance of glioblastoma and malignant melanoma cells to the methylating anticancer drug temozolomide. Mol Cancer Ther 12, 2529–2540. [DOI] [PubMed] [Google Scholar]
- 158. Nadkarni A, Shrivastav M, Mladek AC, Schwingler PM, Grogan PT, Chen J & Sarkaria JN (2012) ATM inhibitor KU‐55933 increases the TMZ responsiveness of only inherently TMZ sensitive GBM cells. J Neuro Oncol 110, 349–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Jackson CB, Noorbakhsh SI, Sundaram RK, Kalathil AN, Ganesa S, Jia L, Breslin H, Burgenske DM, Gilad O, Sarkaria JN et al. (2019) Temozolomide sensitizes MGMT‐deficient tumor cells to ATR inhibitors. Cancer Res 79, 4331–4338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Lopez‐Martinez D, Liang CC & Cohn MA (2016) Cellular response to DNA interstrand crosslinks: the Fanconi anemia pathway. Cell Mol Life Sci 73, 3097–3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Boisvert RA & Howlett NG (2014) The Fanconi anemia ID2 complex: dueling saxes at the crossroads. Cell Cycle 13, 2999–3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Ceccaldi R, Sarangi P & D'Andrea AD (2016) The Fanconi anaemia pathway: new players and new functions. Nat Rev Mol Cell Biol 17, 337–349. [DOI] [PubMed] [Google Scholar]
- 163. Kim H & D'Andrea AD (2012) Regulation of DNA cross‐link repair by the Fanconi anemia/BRCA pathway. Genes Dev 26, 1393–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Adamo A, Collis SJ, Adelman CA, Silva N, Horejsi Z, Ward JD, Martinez‐Perez E, Boulton SJ & La Volpe A (2010) Preventing nonhomologous end joining suppresses DNA repair defects of Fanconi anemia. Mol Cell 39, 25–35. [DOI] [PubMed] [Google Scholar]
- 165. Collis SJ, Ciccia A, Deans AJ, Horejsi Z, Martin JS, Maslen SL, Skehel JM, Elledge SJ, West SC & Boulton SJ (2008) FANCM and FAAP24 function in ATR‐mediated checkpoint signaling independently of the Fanconi anemia core complex. Mol Cell 32, 313–324. [DOI] [PubMed] [Google Scholar]
- 166. Pace P, Mosedale G, Hodskinson MR, Rosado IV, Sivasubramaniam M & Patel KJ (2010) Ku70 corrupts DNA repair in the absence of the Fanconi anemia pathway. Science 329, 219–223. [DOI] [PubMed] [Google Scholar]
- 167. Eccles LJ, Bell AC & Powell SN (2018) Inhibition of non‐homologous end joining in Fanconi Anemia cells results in rescue of survival after interstrand crosslinks but sensitization to replication associated double‐strand breaks. DNA Repair 64, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Kais Z, Rondinelli B, Holmes A, O'Leary C, Kozono D, D'Andrea AD & Ceccaldi R (2016) FANCD2 maintains fork stability in BRCA1/2‐deficient tumors and promotes alternative end‐joining DNA repair. Cell Rep 15, 2488–2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Michl J, Zimmer J, Buffa FM, McDermott U & Tarsounas M (2016) FANCD2 limits replication stress and genome instability in cells lacking BRCA2. Nat Struct Mol Biol 23, 755–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Cleary JM, Aguirre AJ, Shapiro GI & D'Andrea AD (2020) Biomarker‐guided development of DNA repair inhibitors. Mol Cell 78, 1070–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. D'Andrea AD (2018) Mechanisms of PARP inhibitor sensitivity and resistance. DNA Repair 71, 172–176. [DOI] [PubMed] [Google Scholar]
- 172. Zimmermann M, Murina O, Reijns MAM, Agathanggelou A, Challis R, Tarnauskaite Z, Muir M, Fluteau A, Aregger M, McEwan A et al. (2018) CRISPR screens identify genomic ribonucleotides as a source of PARP‐trapping lesions. Nature 559, 285–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Alcón P, Shakeel S, Chen ZA, Rappsilber J, Patel KJ & Passmore LA (2020) FANCD2‐FANCI is a clamp stabilized on DNA by monoubiquitination of FANCD2 during DNA repair. Nat Struct Mol Biol 27, 240–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Rennie ML, Lemonidis K, Arkinson C, Chaugule VK, Clarke M, Streetley J, Spagnolo L & Walden H (2020) Differential functions of FANCI and FANCD2 ubiquitination stabilize ID2 complex on DNA. EMBO Rep 21, e50133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Sharp MF, Murphy VJ, Twest SV, Tan W, Lui J, Simpson KJ, Deans AJ & Crismani W (2020) Methodology for the identification of small molecule inhibitors of the Fanconi Anaemia ubiquitin E3 ligase complex. Sci Rep 10, 7959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Tan W, van Twest S , Leis A, Bythell‐Douglas R, Murphy VJ, Sharp M, Parker MW, Crismani W & Deans AJ (2020) Monoubiquitination by the human Fanconi anemia core complex clamps FANCI:FANCD2 on DNA in filamentous arrays. eLife 9, e54128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. van Twest S , Murphy VJ, Hodson C, Tan W, Swuec P, O'Rourke JJ, Heierhorst J, Crismani W & Deans AJ (2017) Mechanism of ubiquitination and deubiquitination in the Fanconi anemia pathway. Mol Cell 65, 247–259. [DOI] [PubMed] [Google Scholar]
- 178. Wang S, Wang R, Peralta C, Yaseen A & Pavletich NP (2021) Structure of the FA core ubiquitin ligase closing the ID clamp on DNA. Nat Struct Mol Biol 28, 300–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Carruthers R, Ahmed SU, Strathdee K, Gomez‐Roman N, Amoah‐Buahin E, Watts C & Chalmers AJ (2015) Abrogation of radioresistance in glioblastoma stem‐like cells by inhibition of ATM kinase. Mol Oncol 9, 192–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Venere M, Hamerlik P, Wu Q, Rasmussen RD, Song LA, Vasanji A, Tenley N, Flavahan WA, Hjelmeland AB, Bartek J et al. (2014) Therapeutic targeting of constitutive PARP activation compromises stem cell phenotype and survival of glioblastoma‐initiating cells. Cell Death Differ 21, 258–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Ahmed SU, Carruthers R, Gilmour L, Yildirim S, Watts C & Chalmers AJ (2015) Selective inhibition of parallel DNA damage response pathways optimizes radiosensitization of glioblastoma stem‐like cells. Cancer Res 75, 4416–4428. [DOI] [PubMed] [Google Scholar]
- 182. Signore M, Pelacchi F, di Martino S , Runci D, Biffoni M, Giannetti S, Morgante L, De Majo M, Petricoin EF, Stancato L et al. (2014) Combined PDK1 and CHK1 inhibition is required to kill glioblastoma stem‐like cells in vitro and in vivo. Cell Death Differ 5, e1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Rasmussen RD, Gajjar MK, Jensen KE & Hamerlik P (2016) Enhanced efficacy of combined HDAC and PARP targeting in glioblastoma. Mol Oncol 10, 751–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Vance S, Liu E, Zhao L, Parsels JD, Parsels LA, Brown JL, Maybaum J, Lawrence TS & Morgan MA (2011) Selective radiosensitization of p53 mutant pancreatic cancer cells by combined inhibition of Chk1 and PARP1. Cell Cycle 10, 4321–4329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Hoglund A, Stromvall K, Li Y, Forshell LP & Nilsson JA (2011) Chk2 deficiency in Myc overexpressing lymphoma cells elicits a synergistic lethal response in combination with PARP inhibition. Cell Cycle 10, 3598–3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Booth L, Cruickshanks N, Ridder T, Dai Y, Grant S & Dent P (2013) PARP and CHK inhibitors interact to cause DNA damage and cell death in mammary carcinoma cells. Cancer Biol Ther 14, 458–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Peasland A, Wang LZ, Rowling E, Kyle S, Chen T, Hopkins A, Cliby WA, Sarkaria J, Beale G, Edmondson RJ et al. (2011) Identification and evaluation of a potent novel ATR inhibitor, NU6027, in breast and ovarian cancer cell lines. Br J Cancer 105, 372–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Riches LC, Trinidad AG, Hughes G, Jones GN, Hughes AM, Thomason AG, Gavine P, Cui A, Ling S, Stott J et al. (2020) Pharmacology of the ATM inhibitor AZD0156: potentiation of irradiation and olaparib responses preclinically. Mol Cancer Ther 19, 13–25. [DOI] [PubMed] [Google Scholar]
- 189. Carmell N, Rominiyi O, Myers KN, McGarrity‐Cottrell C, Vanderlinden A, Lad N, Perroux‐David E, El‐Khamisy SF, Fernando M, Finegan KG et al. (2021) Identification and validation of ERK5 as a DNA damage modulating drug target in glioblastoma. Cancers 13, 944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Koncar RF, Dey BR, Stanton AJ, Agrawal N, Wassell ML, McCarl LH, Locke AL, Sanders L, Morozova‐Vaske O, Myers MI et al. (2019) Identification of novel RAS signaling therapeutic vulnerabilities in diffuse intrinsic pontine gliomas. Cancer Res 79, 4026–4041. [DOI] [PubMed] [Google Scholar]
- 191. Pereira DM & Rodrigues CMP (2020) Targeted avenues for cancer treatment: the MEK5‐ERK5 signaling pathway. Trends Mol Med 26, 394–407. [DOI] [PubMed] [Google Scholar]
- 192. Simoes AE, Rodrigues CM & Borralho PM (2016) The MEK5/ERK5 signalling pathway in cancer: a promising novel therapeutic target. Drug Discov Today 21, 1654–1663. [DOI] [PubMed] [Google Scholar]
- 193. Stecca B & Rovida E (2019) Impact of ERK5 on the hallmarks of cancer. Int J Mol Sci 20, 1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Riess C, Koczan D, Schneider B, Linke C, Del Moral K, Classen CF & Maletzki C (2021) Cyclin‐dependent kinase inhibitors exert distinct effects on patient‐derived 2D and 3D glioblastoma cell culture models. Cell Death Discov 7, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Gaillard H, Garcia‐Muse T & Aguilera A (2015) Replication stress and cancer. Nat Rev Cancer 15, 276–289. [DOI] [PubMed] [Google Scholar]
- 196. Bartkova J, Hamerlik P, Stockhausen MT, Ehrmann J, Hlobilkova A, Laursen H, Kalita O, Kolar Z, Poulsen HS, Broholm H et al. (2010) Replication stress and oxidative damage contribute to aberrant constitutive activation of DNA damage signalling in human gliomas. Oncogene 29, 5095–5102. [DOI] [PubMed] [Google Scholar]
- 197. Ning JF, Stanciu M, Humphrey MR, Gorham J, Wakimoto H, Nishihara R, Lees J, Zou L, Martuza RL, Wakimoto H et al. (2019) Myc targeted CDK18 promotes ATR and homologous recombination to mediate PARP inhibitor resistance in glioblastoma. Nat Commun 10, 2910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Barone G, Arora A, Ganesh A, Abdel‐Fatah T, Moseley P, Ali R, Chan SY, Savva C, Schiavone K, Carmell N et al. (2018) The relationship of CDK18 expression in breast cancer to clinicopathological parameters and therapeutic response. Oncotarget 9, 29508–29524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Barone G, Staples CJ, Ganesh A, Patterson KW, Bryne DP, Myers KN, Patil AA, Eyers CE, Maslen S, Skehel JM et al. (2016) Human CDK18 promotes replication stress signaling and genome stability. Nucleic Acids Res 44, 8772–8785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Zhou W, Yao Y, Scott AJ, Wilder‐Romans K, Dresser JJ, Werner CK, Sun H, Pratt D, Sajjakulnukit P, Zhao SG et al. (2020) Purine metabolism regulates DNA repair and therapy resistance in glioblastoma. Nat Commun 11, 3811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Bader SB, Dewhirst MW & Hammond EM (2020) Cyclic hypoxia: an update on its characteristics, methods to measure it and biological implications in cancer. Cancers 13, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Bindra RS, Chalmers AJ, Evans S & Dewhirst M (2017) GBM radiosensitizers: dead in the water..or just the beginning? J Neurooncol 134, 513–521. [DOI] [PubMed] [Google Scholar]
- 203. Chedeville AL & Madureira PA (2021) The role of hypoxia in glioblastoma radiotherapy resistance. Cancers 13, 542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Ng N, Purshouse K, Foskolou IP, Olcina MM & Hammond EM (2018) Challenges to DNA replication in hypoxic conditions. FEBS J 285, 1563–1571. [DOI] [PubMed] [Google Scholar]
- 205. Feng X, Tubbs A, Zhang C, Tang M, Sridharan S, Wang C, Jiang D, Su D, Zhang H, Chen Z et al. (2020) ATR inhibition potentiates ionizing radiation‐induced interferon response via cytosolic nucleic acid‐sensing pathways. EMBO J 39, e104036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Motwani M, Pesiridis S & Fitzgerald KA (2019) DNA sensing by the cGAS‐STING pathway in health and disease. Nat Rev Genet 20, 657–674. [DOI] [PubMed] [Google Scholar]
- 207. Forment JV & O'Connor MJ (2018) Targeting the replication stress response in cancer. Pharmacol Ther 188, 155–167. [DOI] [PubMed] [Google Scholar]
- 208. Sheridan C (2019) Drug developers switch gears to inhibit STING. Nat Biotechnol 37, 199–201. [DOI] [PubMed] [Google Scholar]
- 209. Akhavan D, Alizadeh D, Wang D, Weist MR, Shepphird JK & Brown CE (2019) CAR T cells for brain tumors: lessons learned and road ahead. Immunol Rev 290, 60–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Chan HY, Choi J, Jackson C & Lim M (2021) Combination immunotherapy strategies for glioblastoma. J Neurooncol 151, 375–391. [DOI] [PubMed] [Google Scholar]
- 211. Chuntova P, Chow F, Watchmaker P, Galvez M, Heimberger AB, Newell EW, Diaz A, DePinho RA, Li MO, Wherry EJ et al. (2021) Unique challenges for glioblastoma immunotherapy – discussions across neuro‐oncology and non‐neuro‐oncology experts in cancer immunology. Neuro Oncol 23, 356–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Jackson CM, Choi J & Lim M (2019) Mechanisms of immunotherapy resistance: lessons from glioblastoma. Nat Immunol 20, 1100–1109. [DOI] [PubMed] [Google Scholar]
- 213. von Roemeling CA , Wang Y, Qie Y, Yuan H, Zhao H, Liu X, Yang Z, Yang M, Deng W, Bruno KA et al. (2020) Therapeutic modulation of phagocytosis in glioblastoma can activate both innate and adaptive antitumour immunity. Nat Commun 11, 1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Baxter PA, Su JM, Onar‐Thomas A, Billups CA, Li XN, Poussaint TY, Smith ER, Thompson P, Adesina A, Ansell P et al. (2020) A phase I/II study of veliparib (ABT‐888) with radiation and temozolomide in newly diagnosed diffuse pontine glioma: a Pediatric Brain Tumor Consortium study. Neuro Oncol 22, 875–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Sanai N, Li J, Boerner J, Stark K, Wu J, Kim S, Derogatis A, Mehta S, Dhruv HD, Heilbrun LK et al. (2018) Phase 0 trial of AZD1775 in first‐recurrence glioblastoma patients. Clin Cancer Res 24, 3820–3828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Calcagno A, Di Perri G & Bonora S (2017) Treating HIV infection in the Central Nervous System. Drugs 77, 145–157. [DOI] [PubMed] [Google Scholar]
- 217. Sur‐Erdem I, Muslu K, Pinarbasi N, Altunbek M, Seker‐Polat F, Cingoz A, Aydin SO, Kahraman M, Culha M, Solaroglu I et al. (2020) TRAIL‐conjugated silver nanoparticles sensitize glioblastoma cells to TRAIL by regulating CHK1 in the DNA repair pathway. Neurol Res 42, 1061–1069. [DOI] [PubMed] [Google Scholar]
- 218. Zhang H, van Os WL , Tian X, Zu G, Ribovski L, Bron R, Bussmann J, Kros A, Liu Y & Zuhorn IS (2021) Development of curcumin‐loaded zein nanoparticles for transport across the blood‐brain barrier and inhibition of glioblastoma cell growth. Biomater Sci. 10.1039/d0bm01536a [DOI] [PubMed] [Google Scholar]
- 219. McCrorie P, Vasey CE, Smith SJ, Marlow M, Alexander C & Rahman R (2020) Biomedical engineering approaches to enhance therapeutic delivery for malignant glioma. J Control Release 328, 917–931. [DOI] [PubMed] [Google Scholar]
- 220. Vasey CE, Cavanagh RJ, Taresco V, Moloney C, Smith S, Rahman R & Alexander C (2021) Polymer pro‐drug nanoparticles for sustained release of cytotoxic drugs evaluated in patient‐derived glioblastoma cell lines and in situ gelling formulations. Pharmaceutics 13, 208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. D'Amico RS, Aghi MK, Vogelbaum MA & Bruce JN (2021) Convection‐enhanced drug delivery for glioblastoma: a review. J Neurooncol 151, 415–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Mozhei O, G. Teschemacher A & Kasparov S (2020) Viral vectors as gene therapy agents for treatment of glioblastoma. Cancers 12, 3724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Romano E, Netti PA & Torino E (2020) Exosomes in gliomas: biogenesis, isolation, and preliminary applications in nanomedicine. Pharmaceuticals 13, 319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Etame AB, Diaz RJ, Smith CA, Mainprize TG, Hynynen K & Rutka JT (2012) Focused ultrasound disruption of the blood‐brain barrier: a new frontier for therapeutic delivery in molecular neurooncology. Neurosurg Focus 32, E3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Xie F, Boska MD, Lof J, Uberti MG, Tsutsui JM & Porter TR (2008) Effects of transcranial ultrasound and intravenous microbubbles on blood brain barrier permeability in a large animal model. Ultrasound Med Biol 34, 2028–2034. [DOI] [PubMed] [Google Scholar]
- 226. Chang E, Patel CB, Pohling C, Young C, Song J, Flores TA, Zeng Y, Joubert LM, Arami H, Natarajan A et al. (2018) Tumor treating fields increases membrane permeability in glioblastoma cells. Cell Death Discov 4, 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Carruthers RD & Chalmers AJ (2019) DNA repair in Radiation Oncology. Radiat Oncol 1–16. Available at: https://link.springer.com/referenceworkentry/10.1007/978‐3‐319‐52619‐5_111‐1 [Google Scholar]
- 228. Kleinberg L, Supko JG, Mikkelsen T, Blakeley JON, Stevens G, Ye X, Desideri S, Ryu S, Desai B, Giranda VL et al. (2013) Phase I adult brain tumor consortium (ABTC) trial of ABT‐888 (veliparib), temozolomide (TMZ), and radiotherapy (RT) for newly diagnosed glioblastoma multiforme (GBM) including pharmacokinetic (PK) data. J Clin Oncol 31, 2065.23650415 [Google Scholar]
- 229. Halford SER, Cruickshank G, Dunn L, Erridge S, Godfrey L, Herbert C, Jefferies S, Lopez JS, McBain C, Pittman M et al. (2017) Results of the OPARATIC trial: A phase I dose escalation study of olaparib in combination with temozolomide (TMZ) in patients with relapsed glioblastoma (GBM). J Clin Oncol 35, 2022. [Google Scholar]
- 230. Kurzrock R, Galanis E, Johnson DR, Kansra V, Wilcoxen K, McClure T, Martell RE & Agarwal S (2014) A phase I study of niraparib in combination with temozolomide (TMZ) in patients with advanced cancer. J Clin Oncol 32, 2092. [Google Scholar]
- 231. Fulton B, Short SC, James A, Nowicki S, McBain C, Jefferies S, Kelly C, Stobo J, Morris A, Williamson A et al. (2017) PARADIGM‐2: Two parallel phase I studies of olaparib and radiotherapy or olaparib and radiotherapy plus temozolomide in patients with newly diagnosed glioblastoma, with treatment stratified by MGMT status. Clin Transl Radiat Oncol 8, 12–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Robins HI, Zhang P, Gilbert M, Chakravarti A, deGroot J, Grimm S, Wang F, Lieberman F, Krauze A, Sharma A et al. (2015) ATCT‐27: NRG oncology/RTOG 0929: a randomized phase I/II study of ABT‐888 in combination with temozolomide in recurrent temozolomide resistant glioblastoma. Neuro Oncol 17, v7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Baxter PA, Su JM, Li X‐N, Onar‐Thomas A, Billups CA, Thompson PA, Goldman S, Gururangan S, Young‐Poussaint T, McKeegan EM et al. (2015) A phase I/II clinical trial of veliparib (ABT‐888) and radiation followed by maintenance therapy with veliparib and temozolomide in patients with newly diagnosed diffuse intrinsic pontine glioma (DIPG): A Pediatric Brain Tumor Consortium Interim Report of Phase I Study. J Clin Oncol 33, 10053. [Google Scholar]
- 234. Schafer ES, Rau RE, Berg SL, Liu X, Minard CG, Bishop AJR, Romero JC, Hicks MJ, Nelson MD Jr et al. (2020) Phase 1/2 trial of talazoparib in combination with temozolomide in children and adolescents with refractory/recurrent solid tumors including Ewing sarcoma: A Children's Oncology Group Phase 1 Consortium study (ADVL1411). Pediatr Blood Cancer 67, e28073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Piotrowski A, Puduvalli V, Wen P, Campian J, Colman H, Pearlman M, Butowski N, Battiste J, Glass J, Cloughesy T et al. (2019) ACTR‐39. pamiparib in combination with radiation therapy (RT) and/or temozolomide (TMZ) in patients with newly diagnosed or recurrent/refractory (R/R) glioblastoma (GBM); phase 1b/2 study update. Neuro Oncol 21, vi21–vi22. [Google Scholar]
- 236. Lesueur P, Lequesne J, Grellard JM, Dugue A, Coquan E, Brachet PE, Geffrelot J, Kao W, Emery E, Berro DH et al. (2019) Phase I/IIa study of concomitant radiotherapy with olaparib and temozolomide in unresectable or partially resectable glioblastoma: OLA‐TMZ‐RTE‐01 trial protocol. BMC Cancer 19, 198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Parsons DW, Janeway KA, Patton D, Coffey B, Williams PM, Hamilton SR, Purkayastha A, Tsongalis GJ, Routbort M, Gastier‐Foster JM et al. (2019) Identification of targetable molecular alterations in the NCI‐COG Pediatric MATCH trial. J Clin Oncol 37, 10011. [Google Scholar]
- 238. Sulkowski PL, Corso CD, Robinson ND, Scanlon SE, Purshouse KR, Bai H, Liu Y, Sundaram RK, Hegan DC, Fons NR et al. (2017) 2‐Hydroxyglutarate produced by neomorphic IDH mutations suppresses homologous recombination and induces PARP inhibitor sensitivity. Sci Transl Med 9, eaal2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. Karanam NK, Ding L, Aroumougame A & Story MD (2020) Tumor treating fields cause replication stress and interfere with DNA replication fork maintenance: Implications for cancer therapy. Transl Res 217, 33–46. [DOI] [PubMed] [Google Scholar]
- 240. Karanam NK, Srinivasan K, Ding L, Sishc B, Saha D & Story MD (2017) Tumor‐treating fields elicit a conditional vulnerability to ionizing radiation via the downregulation of BRCA1 signaling and reduced DNA double‐strand break repair capacity in non‐small cell lung cancer cell lines. Cell Death Dis 8, e2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241. Cancer Research UK (2020) A trial of AZD1390 and radiotherapy for some types of brain tumour. Available at: https://www.cancerresearchuk.org/about‐cancer/find‐a‐clinical‐trial/a‐trial‐of‐azd1390‐and‐radiotherapy‐for‐some‐types‐of‐brain‐tumour.
- 242. Reddy VP, Sykes A, Colclough N, Durant ST, Connor LO, Hoch M, Bruna NB, Vita SD, Merchant M & Pass M (2019) A preclinical PK/PD model based on a mouse glioblastoma survival model for AZD1390, a novel, brain‐penetrant ATM kinase inhibitor, to predict the inhibition of DNA damage response induced by radiation and the human efficacious dose. Cancer Res 79, 4868. [Google Scholar]
- 243. Durant ST, Zheng L, Wang Y, Chen K, Zhang L, Zhang T, Yang Z, Riches L, Trinidad AG, Fok JHL et al. (2018) The brain‐penetrant clinical ATM inhibitor AZD1390 radiosensitizes and improves survival of preclinical brain tumor models. Sci Adv 4, eaat1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244. Alexander B, Supko J, Agar N, Ahluwalia M, Desai A, Dietrich J, Kaley T, Peereboom D, Takebe N, Desideri S et al. (2018) ACTR‐14. Phase I study of AZD1775 with radiation therapy (RT) and temozolomide (TMZ) in patients with newly diagnosed glioblastoma (GBM) and evaluation of intratumoral drug distribution in patients with recurrent GBM. Neuro Oncol 20, vi13–vi14. [Google Scholar]