

Abstract
Mesenchymal stromal cell (MSC) transplantation has been a promising therapeutic strategy for repairing heart tissues post-myocardial infarction (MI). Nevertheless, its therapeutic efficacy remains low, which is mainly ascribed to the low viability of transplanted MSCs. Recently, long noncoding RNAs (lncRNAs) have been reported to participate in diverse physiological and pathological processes, but little is known about their role in MSC survival. Using unbiased transcriptome profiling of hypoxia-preconditioned MSCs (HP-MSCs) and normoxic MSCs (N-MSCs), we identified a lncRNA named lung cancer-associated transcript 1 (LUCAT1) under hypoxia. LUCAT1 knockdown reduced the survival of engrafted MSCs and decreased the MSC-based therapeutic potency, as shown by impaired cardiac function, reduced cardiomyocyte survival, and increased fibrosis post-MI. Conversely, LUCAT1 overexpression had the opposite results. Mechanistically, LUCAT1 bound with and recruited jumonji domain-containing 6 (JMJD6) to the promoter of forkhead box Q1 (FOXQ1), which demethylated FOXQ1 at H4R3me2(s) and H3R2me2(a), thus downregulating Bax expression and upregulating Bcl-2 expression to attenuate MSC apoptosis. Therefore, our findings revealed the protective effects of LUCAT1 on MSC apoptosis and demonstrated that the LUCAT1-mediated JMJD6-FOXQ1 pathway might represent a novel target to potentiate the therapeutic effect of MSC-based therapy for ischemic cardiovascular diseases.
Keywords: apoptosis, MSC, lncRNA, myocardial infarction, JMJD6, FOXQ1
Graphical Abstract
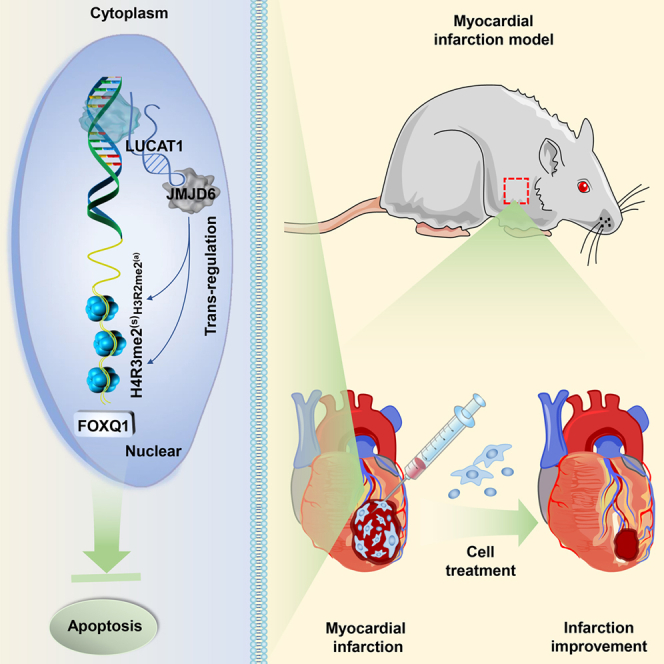
A novel lncRNA, LUCAT1, bound with and recruited JMJD6 to the promoter of FOXQ1, which demethylated FOXQ1 at H4R3me2(s) and H3R2me2(a), thus decreasing pro-apoptotic protein (Bax and Cleaved caspase-3) and increasing anti-apoptotic protein (Bcl-2) expression to attenuate MSC apoptosis. LUCAT1-mediated JMJD6-FOXQ1 pathway might be a promising pathway for improving MSCs’ therapeutic efficacy in MI.
Introduction
Myocardial infarction (MI), one of the most life-threatening diseases, is characterized by substantial cardiomyocyte death, which leads to myocardial remodeling, hypertrophy, and, eventually, heart failure. Over the past decades, mesenchymal stromal cell (MSC)-based cell therapy has emerged as a promising therapeutic strategy for MI due to its self-renewal, anti-inflammatory effects, neovascularization, and mild immunogenicity.1 However, this cell-based therapy is often inefficient, which is mainly ascribed to the low viability of transplanted MSCs. Most transplanted stromal cells die within 1 week.2 Therefore, the mechanism of MSC survival is still an important issue to be elucidated.
Previous studies by our group revealed that hypoxic preconditioning improved the therapeutic effect of MSCs for myocardial injury in mice and nonhuman primates by improving MSC tolerance to subsequent lethal injury, which was associated with increases in vascular density and myocardial glucose uptake and with declines in cardiac apoptosis.2,3 Therefore, it is important to investigate how hypoxic preconditioning improves MSC survival.
Long noncoding RNAs (lncRNAs), a class of noncoding RNAs with more than 200 nucleotides, regulate diverse physiological and pathological processes, including proliferation, differentiation, and apoptosis.4 lncRNAs can be involved in epigenetic regulation and directly interact with epigenetic modifiers,5, 6, 7, 8 thereby regulating the expression of protein-coding genes (PCGs). Previous studies have shown that lncRNAs play a crucial role in cell apoptosis. SNHG5 promoted colorectal cancer cell survival by counteracting STAU1-mediated mRNA instability.9 RBM7 activated P-TEFB to form a prosurvival transcriptional response to genotoxic stress.10 However, whether lncRNAs are involved in MSC apoptosis and the mechanisms of lncRNA-mediated apoptosis in MSCs remain unclear.
In this study, using unbiased sequencing of hypoxia-preconditioned MSCs (HP-MSCs) and normoxic MSCs (N-MSCs), we identified lung cancer-associated transcript 1 (LUCAT1) as the most significantly upregulated lncRNA under hypoxia. The role of LUCAT1 in MSC survival and the related mechanisms remain unknown. This paper proposes that MSCs with LUCAT1 knockdown have a decreased resistance to apoptosis and deteriorated cardiac function, accompanied by reduced cardiomyocyte survival and an augmented infarct size in infarcted mouse hearts, while MSCs with LUCAT1 overexpression have exhibited improved survival and cardiac repair. Mechanistically, LUCAT1 bound with jumonji domain-containing 6 (JMJD6), recruited it to the promoter of forkhead box Q1 (FOXQ1), and then demethylated FOXQ1 to inhibit the apoptotic response. Our study revealed that LUCAT1-mediated JMJD6-FOXQ1 activation is a novel pathway in MSC-mediated cardiac protection, which suggests a potential strategy for the effective therapy of MSCs for ischemic cardiovascular diseases.
Results
LUCAT1 is highly expressed in HP-MSCs
In our previous studies, we found that HP-MSCs exhibited significantly better survival than N-MSCs.2,3 However, it remains unclear whether lncRNAs affect the survival of MSCs. Therefore, we performed unbiased transcriptome profiling of HP-MSCs and N-MSCs (Figure 1A). Twenty-four lncRNAs had upregulated expression in HP-MSCs compared with N-MSCs (log2(fold change) > 2; adjusted p value < 0.05) (Figure 1B). Among them, LUCAT1 showed the most obviously upregulated expression in HP-MSCs, as confirmed by a quantitative reverse transcription-polymerase chain reaction (qRT-PCR) analysis in human samples (Figures 1C and 1D). As reported, LUCAT1 is located on chromosome 5 and consists of four exons and three introns. LUCAT1 promoted cell viability and proliferation in lung nonsmall cell cancers.11,12 However, the role of LUCAT1 in MSC survival is still unexplored. According to the annotations provided in the Ensembl database (GRCh38.p13), LUCAT1 had three different transcripts: LUCAT1-001 (ENST00000511918.5), 002 (ENST00000513626.3), and 003 (ENST00000513492.65). Among them, LUCAT1-002, which was the longest transcript, exhibited the highest expression in both N-MSCs and HP-MSCs (Figure S1I) and was amplified as a new 3,097-nt transcript by rapid amplification of cDNA ends (RACE) (Figures 1E, S1J, and S1K; Table S6). LUCAT1-001 was expressed at lower levels in MSCs and amplified as an 890-nt transcript by RACE approaches (Table S6). Compared with LUCAT1-001, LUCAT1-002 had a stronger effect on apoptosis (Figures S6A–S6D), so we focused on LUCAT1-002 in this study. Given that the cellular localization of lncRNAs determines their mechanisms, subcellular fractionation assays were performed and demonstrated that LUCAT1 was mainly distributed in the cell nucleus (Figure 1F), which was further supported by imaging LUCAT1 RNA through fluorescence in situ hybridization (FISH) in MSCs (Figure 1G).
Figure 1.

LUCAT1 is highly expressed in HP-MSCs
(A) Schematic experimental design for screening specific lncRNAs. (B) lncRNA deep sequencing revealed long noncoding RNA expression profiles of HP-MSCs and N-MSCs from the three patient samples (fold change > 2; adjusted p value < 0.05). (C) The volcano map shows 24 differentially expressed lncRNAs (fold change > 2; adjusted p value < 0.05) in HP-MSCs, among which 5 lncRNAs showed upregulated expression in HP-MSCs. (D) qRT-PCR identified differentially expressed lncRNAs, with LUCAT1 having the highest fold change (n = 9). (E) RACE revealed that the genomic structure of LUCAT1-002 was a 3,097-nt transcript compared with the 2,627-nt transcript indexed in the Ensembl database. (F) Subcellular fractionation assays for evaluating the expression of LUCAT1 in MSC nuclei and cytoplasm (n = 3). (G) FISH for detecting the location of LUCAT1 in MSCs (bar, 20 μm). All data are presented as the mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
LUCAT1 knockdown increases the apoptosis of MSCs and impairs MSC-mediated cardioprotection
To investigate the role of LUCAT1 in MSC survival, we generated lentiviruses containing LUCAT1 small hairpin RNA (shRNA) and green fluorescent protein (GFP) reporters and transfected them into MSCs (Figure S7A) and then exposed the samples to 500-μM H2O2 with serum deprivation. LUCAT1-knockdown (sh-LUCAT1) MSCs exhibited deteriorated apoptosis, as shown by increased terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL)-positive cells compared with those of the control (sh-Ctr) MSCs (Figures 2A and 2B). Furthermore, more Annexin V- and propidium iodide (PI)-positive cells were detected by fluorescence-activated cell sorting (FACS) (Figures 2C and 2D) in the sh-LUCAT1 MSC group than in the control group. Western blotting showed that LUCAT1 knockdown increased Bax and Cleaved caspase-3 protein expression but decreased the expression of the anti-apoptotic protein Bcl-2 in MSCs (Figures 2E and 2F).
Figure 2.

LUCAT1 knockdown enhances the apoptosis of MSCs
(A) TUNEL staining with nuclei identified via DAPI staining (bar, 100 μm) of the control and LUCAT1-knockdown MSCs under 500-μM H2O2 for 1 h. (B) Quantification of the TUNEL-positive nuclei (n = 3). (C) Annexin V-APC/PI staining was used to assess cell apoptosis and was detected by FACS. (D) The late (Q2) apoptotic rates were quantified (n = 3). (E) Cleaved caspase-3, Bax, and Bcl-2 levels were measured by western blots. (F) Quantitative analysis of Cleaved caspase-3, Bax, and Bcl-2 protein expression (n = 3). The data are presented as the mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
It has been generally reported that MSCs may be immune privileged without eliciting immune responses due to a loss of immunological cell-surface markers.13,14 Human MSCs (hMSCs) with low levels of human leukocyte antigen (HLA) class I but without HLA class II escape T cell immune responses,15,16 which permits hMSCs to be applied for xenotransplantation.17 In our study, the quantification of immunofluorescence staining of CD68 and CD3 showed no significant difference among the Dulbecco’s modified Eagle medium (DMEM) and LUCAT1-knockdown or -overexpression MSC groups (Figures S4 and S5). Therefore, we used MSCs extracted from human bone marrow to explore the role of LUCAT1-engineered MSCs in the response to ischemic injury. MSCs were immediately implanted into the peri-infarct zone of mouse hearts post-MI surgery. At 3 days post-MI and cell injection, there were fewer GFP-positive MSCs in the sh-LUCAT1 MSC (MSCsh−LUCAT1) group than in the sh-Ctr MSC (MSCsh−Ctr) group in the peri-infarct area (Figures 3A and 3B). TUNEL staining of the infarct border area at 3 days post-MI showed that the survival of cardiac cells in the LUCAT1-knockdown MSC group was lower than that in the control group (Figures S2A and S2C). These results suggested that LUCAT1 knockdown increased MSC apoptosis, thus inhibiting the MSC-mediated protection of cardiac cells in the peri-infarct zone of mouse hearts post-MI surgery.
Figure 3.

LUCAT1 knockdown impairs MSC-mediated cardioprotection
(A) Representative images of MSCs in the hearts of the MSCsh−Ctr and MSCsh−LUCAT1 groups at 3 days post-MI. The heart tissue was immunostained for GFP (green), troponin T (red), and DAPI (blue) (bar, 100 μm; enlarged bar, 10 μm). (B) Quantification of MSCs in the hearts of the MSCsh−Ctr and MSCsh−LUCAT1 groups at 3 days post-MI was performed based on the number of GFP-positive cells per high-power field (HPF) (n = 5 per group). (C) Representative echocardiographic images demonstrated changes in cardiac function in each group, including the Sham, DMEM, MSCsh−Ctr, and MSCsh−LUCAT1 groups, at 28 days post-MI. (D and E) EF (D) and FS (E) were quantified by echocardiographic imaging (n = 5 per group). (F) Masson staining at 28 days post-MI indicated the scar size in MI hearts. (G) Quantitative analysis of the fibrotic size percentage at 28 days post-MI (n = 5 per group). All data are expressed as the mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
At 28 days post-MI and cell injection, the MSCsh−LUCAT1 group exhibited deteriorated cardiac function, as evidenced by the decreased ejection fraction (EF) and fractional shortening (FS) (Figures 3C–3E), consistent with the increased fibrotic size (Figures 3F and 3G) and fewer viable cardiomyocytes (Figures S3A and S3C) within MI segments than those in the MSCsh−Ctr group.
These results indicated that LUCAT1 is a prerequisite for the anti-apoptotic effect of MSCs and that LUCAT1 knockdown decreased the reparative potency of MSCs for MI.
LUCAT1 overexpression decreases MSC apoptosis and improves the effect of MSC-mediated cardioprotection
To further confirm the role of LUCAT1 in MSC survival, we generated a lentivirus containing the LUCAT1 plasmid with a GFP reporter (Figure S7B). The LUCAT1-overexpressing (Lv-LUCAT1) MSCs exhibited an increased survival potency when exposed to 500-μM H2O2 with serum deprivation compared with that of the vector (Lv-Ctr) group, as evidenced by decreased TUNEL-positive MSCs (Figures 4A and 4B) and Annexin V+-PI+ cells (Figures 4C and 4D), the decreased expression of Cleaved caspase-3 and Bax, and the increased expression of Bcl-2 (Figures 4E and 4F). In addition, we investigated the role of the LUCAT1-overexpressing MSCs in the response to MI. There were more GFP-positive MSCs in the peri-infarct area of the Lv-LUCAT1 MSC (MSCLv−LUCAT1) group than in the Lv-Ctr MSC (MSCLv−Ctr) group at 3 days post-MI (Figures 5A and 5B). TUNEL staining of the infarct border area at 3 days post-MI showed that the apoptotic level of cardiac cells was reduced in the MSCLv−LUCAT1 group (Figures S2B and S2D).
Figure 4.

LUCAT1 overexpression decreases MSC apoptosis
(A) TUNEL staining with nuclei identified via DAPI staining (bar, 100 μm) of the control and LUCAT1-overexpressing MSCs under 500-μM H2O2 for 1 h. (B) Quantification of the TUNEL-positive nuclei (n = 3). (C) Annexin V-APC/PI staining was used to assess cell apoptosis and was detected by FACS. (D) The late (Q2) apoptotic rates were quantified (n = 3). (E) Cleaved caspase-3, Bax, and Bcl-2 levels were measured by western blots. (F) Quantitative analysis of Cleaved caspase-3, Bax, and Bcl-2 protein expression (n = 3). The data are presented as the mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Figure 5.

LUCAT1 overexpression improves the effect of MSC-mediated cardioprotection
(A) Representative images of MSCs in the hearts of the MSCLv−Ctr and MSCLv−LUCAT1 groups at 3 days post-MI. The heart tissue was immunostained for GFP (green), troponin T (red), and DAPI (blue) (bar, 100 μm; enlarged bar, 10 μm). (B) Quantification of MSCs in the hearts of the MSCLv−Ctr and MSCLv−LUCAT1 groups at 3 days post-MI by the number of GFP-positive cells per HPF (n = 5 per group). (C) Representative echocardiographic images demonstrated changes in cardiac function in each group, including the Sham, DMEM, MSCLv−Ctr, and MSCLv−LUCAT1 groups, at 28 days post-MI. (D and E) EF (D) and FS (E) were quantified by echocardiographic imaging (n = 5 per group). (F) Masson staining at 28 days post-MI indicated the scar size in MI hearts. (G) Quantitative analysis of the percentage fibrotic size at 28 days post-MI (n = 5 per group). All data are expressed as the mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
At 28 days post-MI, the MSCLv−LUCAT1 group exhibited better preserved cardiac function (Figures 5C–5E), reduced fibrotic size (Figures 5F and 5G), and more surviving cardiomyocytes within the infarct area (Figures S3B and S3D). Thus, LUCAT1 overexpression decreased the apoptosis of MSCs both in vitro and in vivo, promoting the survival of cardiac cells in the infarct border area after MSC transplantation. LUCAT1 had a key role in MSC-mediated cardiac protection in response to ischemic injury and improved the prognosis after MI.
LUCAT1 decreases MSC apoptosis through FOXQ1
To investigate the mechanisms of LUCAT1 in regulating MSC apoptosis, we performed transcriptome sequencing to analyze PCG expression in the MSCs with LUCAT1 knockdown (Figure 6A). A total of 518 genes (adjusted p value < 0.05) showed downregulated expression (Figure 6B) and participated in the regulation of aging, oxidative stress, growth, external stimulus, metabolism, and apoptosis (Figure 6C), while 594 genes showed upregulated expression (Figure 6B) and were found to be mainly associated with cell-junction assembly, wound healing, and angiogenesis (Figure S7C) in Gene Ontology (GO) analysis. Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis revealed that the PCGs with downregulated expression in MSCs with LUCAT1 knockdown were enriched in the advanced glycation endproduct (AGE)-receptor for AGE (RAGE), mitogen-activated protein kinase (MAPK), and FoxO signaling pathways related to cell survival regulation (Figure 6D), and the PCGs with upregulated expression were enriched in the tight junction and thyroid hormone pathways (Figure S7D).
Figure 6.

LUCAT1 decreases MSC apoptosis through FOXQ1
(A) Heatmap of the LUCAT1-knockdown and control groups for 3 samples per group. (B) Volcano map revealed that 518 genes showed downregulated expression and 594 genes showed upregulated expression (adjusted p value < 0.05). (C) Gene Ontology (GO) terms for the protein-coding gene (PCG) cohort with downregulated expression in the LUCAT1-knockdown group. (D) Functional enrichment analysis was linked to biological pathways of the PCGs with downregulated expression in the LUCAT1-knockdown group. (E) The expression of relevant apoptosis-related genes was analyzed through qRT-PCR either for LUCAT1 knockdown or overexpression. (F–I) Western blot analysis for FOXQ1 in LUCAT1-knockdown (F and G) or -overexpression (H and I) MSCs (n = 3). (J–M) Cleaved caspase-3 levels after FOXQ1 knockdown (J and K) or overexpression (L and M) were assessed under 500-μM H2O2 for 1 h (n = 3). (N) Representative images of TUNEL staining with nuclei identified through DAPI staining in the control and LUCAT1-knockdown MSC groups with or without FOXQ1 overexpression under the above treatments (bar, 100 μm). (O) Apoptotic cells were quantified by TUNEL-positive nuclei in the total nuclei and analyzed via one-way ANOVA (n = 3). (P) Representative images of TUNEL staining with nuclei identified using DAPI staining in the control and LUCAT1-overexpression MSC groups with or without FOXQ1 knockdown as described above (bar, 100 μm). (Q) Apoptotic cells were quantified by TUNEL-positive nuclei in the total nuclei and analyzed via one-way ANOVA (n = 3). All data are presented as the mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
We screened the differentially expressed PCGs (log2(fold change) > 1; adjusted p value < 0.05) and the apoptosis-related biological processes from these enriched GO biological processes and KEGG pathways, and we obtained 12 anti-apoptotic genes with downregulated expression (for example, FOXQ1 and CCL7) and 14 pro-apoptotic molecules (for example, RARB and EROB1) with upregulated expression in the LUCAT1-knockdown MSCs. The PCGs with downregulated and upregulated expression were quantitatively analyzed by qRT-PCR, and only three genes, FOXQ1, CCL7, and ALPL, showed expression trends consistent with that of LUCAT1 expression (Figures 6E and S7E). Among them, FOXQ1 expression decreased most obviously when LUCAT1 was knocked down in MSCs but increased most significantly when LUCAT1 was overexpressed (Figure 6E). FOXQ1 protein expression was also changed following LUCAT1 knockdown or overexpression (Figures 6F–6I), suggesting that FOXQ1 may be the target of LUCAT1.
To investigate the role of FOXQ1 in MSC apoptosis, we constructed FOXQ1 siRNA and lentivirus-containing FOXQ1 cDNA. The efficiency of the FOXQ1 siRNA and the overexpression lentivirus is shown in Figures S8A–S8D, and the expression levels of LUCAT1 and FOXQ1 in rescue experiments are shown in Figures S8E and S8F. FOXQ1 knockdown enhanced MSC apoptosis, as evidenced by the higher level of Cleaved caspase-3 (Figures 6J and 6K), while FOXQ1 overexpression decreased Cleaved caspase-3 expression (Figures 6L and 6M). Furthermore, we found that FOXQ1 attenuated LUCAT1-knockdown-induced apoptosis, as detected by TUNEL staining (Figures 6N and 6O). However, FOXQ1 knockdown abolished the LUCAT1-mediated protection against apoptosis (Figures 6P and 6Q). These findings indicated that LUCAT1-regulated MSC apoptosis was at least partially dependent on FOXQ1.
LUCAT1 regulates FOXQ1 expression in a JMJD6-dependent manner
lncRNAs have been reported to interact with chromatin-modulating proteins, facilitating their recruitment to chromatin, thereby regulating transcriptional activity.18, 19, 20, 21 Cis-regulation has been extensively studied in the regulatory function of lncRNAs on nearby genes to govern cell fate.22, 23, 24 Therefore, we explored the nearest PCG, ARRDC3, which was 54 kilobytes away from LUCAT1. However, ARRDC3 expression had no change when LUCAT1 was knocked down or overexpressed (Figures S6E–S6H), which suggested that LUCAT1 might not regulate the expression of apoptosis-related proteins through the cis-pathway. To further characterize the LUCAT1-FOXQ1 interaction at the molecular level, we identified intracellular LUCAT1-interacting proteins using an unbiased RNA pull-down approach. Biotinylated full-length anti-sense and sense LUCAT1 (3,097 bp) sequences transcribed in vitro were incubated with total protein extracts from MSCs and pulled down by streptavidin magnetic beads. The binding proteins were analyzed by mass spectrometry (MS) analysis (Figure 7A; Table S4). As LUCAT1 was located in the nucleus, we focused on nuclear regulatory proteins and found that several proteins, including JMJD6, KDM5B, RU2A, PGAM5, MED30, MPH6, FEN1, DDB2, and MCA3, had a strong binding ability with LUCAT1. Interestingly, JMJD6, as a protein hydroxylase or histone demethylase in the nucleus, is ubiquitously expressed in bone marrow and associated with cell apoptosis.25, 26, 27, 28 Therefore, we selected JMJD6 as the potential LUCAT1-interacting protein, and the binding activity of LUCAT1 with JMJD6 was confirmed by western blot analysis using the retrieved proteins from the RNA pull-down assays (Figure 7B) and RNA-binding protein immunoprecipitation (RIP) assays (Figure 7C). Furthermore, to investigate the roles of JMJD6 in the LUCAT1-FOXQ1 interaction, we constructed JMJD6 siRNA to knock down JMJD6 expression and a lentivirus containing JMJD6 cDNA to overexpress JMJD6. The efficiency of JMJD6 siRNA and a lentivirus was detected by qRT-PCR (Figures S9A–S9D). The mRNA level of FOXQ1 was decreased following JMJD6 knockdown, while FOXQ1 had no changes after JMJD6 overexpression (Figures S9E and S9F). Western blotting analysis revealed that JMJD6 knockdown increased Cleaved caspase-3 expression (Figures 7D and 7E) and decreased the protein level of FOXQ1 (Figures 7F and 7G). These results indicate that JMJD6 played an indispensable role in the LUCAT1-FOXQ1 interaction.
Figure 7.

LUCAT1 regulates FOXQ1 expression dependent on JMJD6
(A) Representative image of SDS-PAGE gels showing proteins pulled down by biotin-labeled LUCAT1 and LUCAT1 anti-sense antibodies (anti-sense). (B) Western blot analysis was used to detect JMJD6 expression in proteins retrieved from LUCAT1 and anti-sense pull-down assays. (C) RNA immunoprecipitation using an anti-JMJD6 antibody to assess endogenous JMJD6 binding with LUCAT1 in MSCs. Immunoglobulin G (IgG) was used as the control. LUCAT1 was detected by qRT-PCR products with specific primers at sites 2726–2886, and H2O was used as the negative control. The results are shown by nucleic acid gel electrophoresis. (D and E) With JMJD6-knockdown treatment, the expression of Cleaved caspase-3 was assessed (n = 3). (F and G) With JMJD6-knockdown treatment, the expression of FOXQ1 was assessed (n = 3). All data are presented as the mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
LUCAT1 regulates FOXQ1 through JMJD6-mediated demethylation of H4R3me2(s) and H3R2me2(a) at the FOXQ1 promoter
As previous studies showed, the chromatin-modulating factor JMJD6 demethylated histone H3 at arginine 2 (H3R2) and histone H4 at arginine 3 (H4R3),27 and we hypothesized that JMJD6 recruited by LUCAT1 might regulate the target FOXQ1 by demethylation. Chromatin immunoprecipitation (ChIP) assays with an anti-JMJD6 antibody revealed that the two regions (−1065∼–1281, −1619∼–2019) of the FOXQ1 promoter were enriched upon JMJD6 immunoprecipitation (Figure 8A), indicating that JMJD6 could physiologically bind with the promoter of FOXQ1 and might regulate FOXQ1 expression. Luciferase assays independently confirmed the binding of JMJD6 with the FOXQ1 promoter (Figure 8B). Next, we used ChIP assays to explore the roles of LUCAT1 in the binding efficiency between JMJD6 and FOXQ1. LUCAT1 knockdown remarkably decreased the enrichment of the FOXQ1 promoter in four regions (−144∼–488, −399∼–766, −757∼–1156, −1056∼–1281) (Figure 8C), while LUCAT1 overexpression increased the enrichment in the same regions (Figure 8D). These findings demonstrate that LUCAT1 knockdown decreased JMJD6’s binding efficiency to the FOXQ1 promoter, while LUCAT1 overexpression enhanced the efficiency.
Figure 8.

LUCAT1 regulates FOXQ1 through JMJD6-mediated demethylation of H4R3me2(s) and H3R2me2(a) at the FOXQ1 promoter
(A) ChIP assays were performed with an anti-JMJD6 antibody. IgG was used as the control. Fold enrichment of the indicated regions of the FOXQ1 promoter was examined through qPCR (n = 3). (B) Luciferase assays were used to measure the trigger of JMJD6 at the FOXQ1 promoter (n = 3). (C and D) ChIP-qPCR assays were performed with an anti-JMJD6 antibody and revealed the enrichment of the indicated regions at the FOXQ1 promoter in the LUCAT1-knockdown or -overexpression MSCs compared with the control (n = 3). (E and F) The ChIP-qPCR assay with the anti-H4R3me2(s) and anti-H3R2me2(a) antibodies indicated region enrichment of the FOXQ1 promoter in the LUCAT1-knockdown MSCs compared with the control (n = 3). (G and H) The ChIP-qPCR assay with the anti-H4R3me2(s) and anti-H3R2me2(a) antibodies indicated enrichment of the FOXQ1 promoter in the LUCAT1-overexpressing MSCs compared with the control (n = 3). (I) A schematic illustration of the overall process. Data are presented as the mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
To explore whether LUCAT1 affected FOXQ1 expression through JMJD6-mediated histone demethylation of the FOXQ1 promoter, we performed ChIP assays using anti-H4R3me2(s) and anti-H3R2me2(a) antibodies. ChIP-qPCR following LUCAT1 knockdown demonstrated that the H4R3me2(s) (regions: −144∼–488, −399∼–766, −757∼–1156, −1056∼–1281, −1619∼–2019) and H3R2me2(a) (regions: −144∼–488, −399∼–766, −757∼–1156, −1056∼–1281, −1216∼–1643, −1619∼–2019) methylation levels in the FOXQ1 promoter region were significantly increased (Figures 8E and 8F). However, with LUCAT1 overexpression, the H4R3me2(s) methylation level (regions: −399∼–766, −1056∼–1281) of FOXQ1 was decreased (Figure 8G), while the H3R2me2(a) methylation level had no significant changes (Figure 8H). These results indicated that LUCAT1 mainly mediated the demethylation of H4R3me2(s) and partially demethylated H3R2me2(a) at the FOXQ1 promoter. Thus, LUCAT1 recruited JMJD6 to FOXQ1, thereby regulating FOXQ1 expression by JMJD6-mediated demethylation of H4R3me2(s) and H3R2me2(a) in the FOXQ1 promoter (Figure 8I).
Discussion
Although the cardioprotective effect of MSC transplantation after MI has been well-documented, the low survival rate of engrafted MSCs remains a major challenge. In this study, we discovered for the first time that the LUCAT1-mediated JMJD6-FOXQ1 pathway protected MSCs from H2O2-induced apoptosis and promoted the survival of implanted MSCs in infarcted hearts. MSCLv−LUCAT1 implantation decreased cardiac cell apoptosis in the infarcted area at the early stage of MI and preserved heart function with a reduced fibrotic size at 28 days post-MI, while the MSCsh−LUCAT1 group exhibited poor results. Therefore, LUCAT1-mediated cardiac protection may reinforce MSC-based therapy to treat post-MI hearts in the clinic. Mechanistically, we found that LUCAT1 directly bound with JMJD6 and recruited JMJD6 to the promoter of the anti-apoptotic protein FOXQ1. LUCAT1 mediated JMJD6 to ensure the symmetric demethylation of H4R3 (di-H4R3me2(s)) and might also influence the asymmetric demethylation of H3R2 (di-H3R2me2(a)) of FOXQ1, thereby protecting MSCs from apoptosis. These findings provide new insights into the mechanisms of MSC survival and provide novel avenues to potentiate the therapeutic effect of MSC-based therapy for MI.
Emerging studies have shown that lncRNAs play an important role in cell biological functions and act as signals, decoys, guides, and scaffolds in cell-specific responses.29, 30, 31 However, the roles of lncRNAs in MSC apoptosis and the related mechanisms remain unclear. In our study, we found that LUCAT1 showed the most significantly upregulated expression among the lncRNAs in HP-MSCs. LUCAT1 was first identified in smoking-related lung cancer and termed smoke- and cancer-associated lncRNA-1 (SCAL1), which is involved in multiple types of cancers and influences cell proliferation, migration, and tumorigenesis.12,32,33 Recently, some reports have shown the influence of LUCAT1 on cell apoptosis. Although few studies have found that LUCAT1 acts as a microRNA sponge,34,35 more studies have demonstrated that LUCAT1, like other epigenetic regulatory lncRNAs, binds with proteins to regulate downstream responses.11,33,36 LUCAT1 was reported to modulate cancer cell viability by physically interacting with PTBP1 to modulate the alternative splicing of a set of DNA-damage-related genes.36 Here, we found that the LUCAT1-mediated JMJD6-FOXQ1 pathway protected MSCs from H2O2-induced apoptosis and promoted the survival of implanted MSCs in infarcted hearts, thus preserving cardiac function and reducing fibrotic size. Therefore, our study has provided additional novel insight into the regulation of lncRNAs that drive MSC protection, suggesting that lncRNA LUCAT1 may be a potential candidate for reinforcing MSC-based therapy to treat post-MI hearts.
Understanding the molecular basis of lncRNAs is important for exploring their function.37 Three different transcripts of LUCAT1 were found in the Ensembl database. In this study, we found that LUCAT1-002, which was the most highly expressed and the longest transcript in MSCs, played a stronger protective role in the apoptosis of MSCs than did LUCAT1-001, so we focused on LUCAT1-002 in subsequent studies. However, the role of LUCAT1-001 needs further study. In addition, we found that LUCAT1 directly bound with JMJD6, a JmjC-containing iron- and 2-oxoglutarate-dependent dioxygenase that demethylates H3R2 and H4R3 in both biochemical and cell-based assays.38, 39, 40 The LUCAT1-JMJD6 interaction ensured a specific demethylation of H4R3me2(s) and H3R2me2(a) at the promoter of FOXQ1. H4R3me2(s) and H3R2me2(a) were reported to act as repressive histone marks, and their transcriptional inhibition could be attenuated by demethylation.41 Our findings showed that the LUCAT1-mediated suppression of the apoptotic response, at least in part, was due to a direct interaction with JMJD6. However, elucidating the structure of LUCAT1’s functional motif and its complex interaction with JMJD6 or other transcriptional regulators requires further exploration.
FOXQ1 is a member of the FOX gene family, which is characterized by a conserved 110-amino acid DNA-binding motif and is widely involved in embryonic development, cell-cycle regulation, and cell apoptosis.42, 43, 44 FOXQ1 was reported to functionally interact with lncRNAs, act as a direct target of miRNAs, or competitively bind miRNA sites with circRNA.45,46 Our findings reveal that lncRNA LUCAT1-mediated regulation of FOXQ1 is a novel mechanism to specifically induce demethylation of H4R3me2(s) and H3R2me2(a) at the FOXQ1 promoter. FOXQ1 was reported to reduce the apoptosis of NKTCL cells (SNK-6) with the downregulation of the protein expression of pro-apoptotic Bax and Cleaved caspase-3 and the upregulation of the protein expression of anti-apoptotic Bcl-2,43,44 and in human renal cell carcinoma, FOXQ1 is potentially linked to the activation of the caspase cascade and the deregulation of the GLI1/Bcl-2 pathway.47 Notably, GLI1 elevated Bcl-2 expression and decreased the expression of Bax, which acts as a transcription factor to mediate Sonic hedgehog (Shh) signaling.48 Our findings revealed that knockdown of FOXQ1 significantly increased MSC apoptosis, while overexpression of FOXQ1 reversed this cell fate. When we overexpressed FOXQ1 following LUCAT1 knockdown, the deteriorated apoptosis of MSCs was attenuated. However, when FOXQ1 was knocked down after LUCAT1 overexpression, MSC apoptosis was enhanced. Here, we found that LUCAT1 can change the expression of FOXQ1 by recruiting JMJD6 and can affect the expression of the downstream apoptosis-related proteins Bcl-2 and Bax to mediate MSC apoptotic progression.
Thus, we found that LUCAT1 can change the expression of FOXQ1 by recruiting JMJD6 and can affect the expression of downstream apoptosis-related proteins Bcl-2 and Bax, indicating that the LUCAT1-JMJD6-mediated regulation of FOXQ1 is a novel mechanism to confer anti-apoptotic effects.
In summary, we demonstrated a vital role of LUCAT1 in MSC apoptosis and its therapeutic effect in lethal environments post-MI. Mechanistically, the anti-apoptotic effects of LUCAT1 were based on recruiting the epigenetic modifier JMJD6 to induce promoter activity of FOXQ1 by demethylating H4R3me2(s) and H3R2me2(a). These results revealed that LUCAT1-JMJD6-FOXQ1 might be a promising pathway for improving MSC therapeutic efficacy in MI.
Materials and methods
A more detailed description of the materials and methods is presented in the supplemental information.
Animals and ethics statement
All animals used were in accordance with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH publication no. 85-23, revised 1996) and approved by the Animal Use Committee of Zhejiang University. Mice were fed a standard laboratory diet and maintained with a 12:12-h light/dark cycle.
Isolation and culture of MSCs
Human bone-marrow-derived MSCs were isolated from patients who underwent hip replacements, and cells at passages 4–8 were used. Informed consent was obtained from the donors, and all studies were approved by the Human Ethics Committee of the Second Affiliated Hospital of Zhejiang University and complied with the edicts of the 1975 Declaration of Helsinki. MSCs were isolated and harvested as previously described.49,50 Briefly, the bone marrow was obtained by a posterior iliac bone puncture, and then the aspirate was centrifuged at 1,000 RPM for 5 min. The cell-containing precipitate was resuspended and added to DMEM (Corning, NY, USA) containing 10% fetal bovine serum (FBS; Corning), which was placed in a 10-cm Petri dish and incubated in an incubator at 37°C for 24 h. The cells that did not adhere to the wall were washed with phosphate-buffered saline (PBS; Corning) 3 times, and then adherent cells were cultured in complete medium (DMEM + 10% FBS). The complete culture medium was refreshed every 3–4 days. After 1 week of culture, bone marrow MSCs covered the entire 10-cm culture dish, were digested by trypsin, and were cultured for passage.
Flow cytometry and characterization of MSCs
Passage 6 MSCs were characterized and incubated with specific surface molecular antibodies (mesenchymal surface markers: PE-CD105, PE-CD29, and APC-CD90; endothelial cell surface marker: FITC-CD34; hematopoietic surface marker: FITC-CD45 and isotype control) in the dark for 30 min at room temperature. The expression of cell-surface markers was quantified using a FACSCanto II Flow Cytometer (BD Bioscience, San Jose, CA, USA). The staining for the surface markers CD29, CD90, and CD105 of the MSCs we used was positive, while that for the hematopoietic surface marker CD45 and endothelial cell-surface marker CD34 was negative (Figures S1A–S1E).
The differentiation of MSCs into osteocytes, chondrocytes, and adipocytes was based on different differentiation protocols as previously described.2 MSCs were inoculated in 6-well plates at a density of 2 × 105 cells/well, cultured to the maximum degree of fusion, and then photographed under an optical microscope after the differentiation process. The MSCs we used can be differentiated into osteocytes, adipocytes, and chondrocytes (Figures S1F–S1H).
Recombinant lentivirus construction and transfection
The full-length cDNAs of human LUCAT1, JMJD6, and FOXQ1 were cloned and recombined into the lentivirus expression vector with a GFP reporter provided by GeneChem (Shanghai, China). A lentivirus containing an empty plasmid served as a control. A lentivirus encoding the shRNA targeting human LUCAT1 (sh-LUCAT1) and its nonspecific shRNA were recombined with a GFP reporter (GeneChem, Shanghai, China).
MSCs were transfected with a lentivirus at a multiplicity of infection (MOI) of 50 overnight, and then fresh medium was added. After 72 h of infection, qRT-PCR and western blot analyses were utilized to determine the effect of infection.
MI model and transplantation of MSCs
The MI model was established in 8- to 10-week-old male C57BL/6 J mice (Zhejiang Chinese Medical University, Zhejiang, China) by ligation of the left anterior descending (LAD) coronary artery as previously reported.49
Human bone-marrow-derived MSCs were infected by a lentivirus labeled with GFP reporter. After ligation, MSCs were resuspended in 20 μL of DMEM at a concentration of 1 × 104 cells/μL and injected into 5 sites: 4 sites at the border of the infarct, and 1 site at the apex of the mouse heart. After surgery and cell transplantation, heart tissue was harvested post-MI when needed.
Hypoxic preconditioning culture and H2O2-induced apoptosis assay
HP-MSCs were cultured in complete medium under well-controlled hypoxia in a ProOx-C-chamber system (Biospherix, Redfield, NY, USA) for 24 h. The oxygen concentration of the incubator was sustained at 0.5%, and the remaining gas mixture consisted of 5% CO2 and of equilibrium N2. N-MSCs were cultured in complete medium under conditions of 21% O2 and 5% CO2 as previously reported.2
For the H2O2-induced apoptosis model, MSCs were exposed to 500-μM H2O2 treatment without serum for 1 h as previously described.51
siRNA transfection
JMJD6 siRNA (si-JMJD6), FOXQ1 siRNA (si-FOXQ1), and control siRNA (si-Ctr) were purchased from GenePharma (Shanghai, China). The nucleotide sequences used are presented in the supplemental information (Table S1). MSCs were transfected with 50-nM siRNAs in serum-free DMEM containing Lipofectamine RNAi MAX (Invitrogen, Carlsbad, CA, USA). Twelve hours later, the medium was replaced with fresh complete medium, and siRNA efficiency was determined after 48 h.
Transcriptome sequencing
Total RNA was extracted from MSCs using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. Library construction and sequencing were performed in Novogene (Beijing, China). First, RNA integrity and DNA contamination of samples were analyzed by agarose gel electrophoresis. RNA purity (OD260/280 and OD260/230 ratio) was determined by a NanoPhotometer NP80 (Implen, Munich, Germany), and RNA integrity was examined accurately by a Gilent 2100 BioAnalyzer (Invitrogen, Carlsbad, CA, USA). The preparation of whole-transcriptome libraries was conducted in Novogene (Beijing, China). Illumina sequencing was performed after pooling different libraries according to the effective concentration and the requirement of target data volume, and 150-bp paired terminal readings were generated. In the sequencing of the flow cell with four kinds of fluorescence-labeled deoxyribonucleotide triphosphates (dNTPs), primers for amplification, DNA polymerase, and sequence complementary chain clusters, each to join a fluorescently labeled dNTP, can release the corresponding fluorescence, and a sequencing machine was used to capture the fluorescent signal. Software was used to convert optical signals to the sequencing peaks, thereby identifying fragments with sequence information. FastQC (v.0.11.8) was adopted to assess the quality of the raw data. Moreover, Fastp (v.0.19.5) was used to remove low-quality data, adapters, and poly-N sequences. Transcript expression was analyzed using Hisat5 and quantified as FPKM. FPKM, the expected number of fragments per kilobase of transcript sequence per million base pairs sequenced, considers the effect of sequencing depth and gene length on the read count at the same time and is currently the most commonly used method for estimating gene expression levels. The RNA sequencing data were deposited in the Gene Expression Omnibus (GEO) repository.
lncRNA deep sequencing
Total RNA was extracted from cells using TRIzol reagent. The next-generation sequencing libraries were sequenced using the HiSeq6000 platform at Novogene Bioinformatics Technology (Beijing, China), and a total of 150-bp paired single-end reads were generated as per Illumina’s protocol description. The expression of RNAs was normalized using reads mapped to the reference genome. The transcripts of single exons, length ≤200, fragments per kilobase per million (FPKM reads) <0.5, and transcripts per kilobase million (TPM) <0.5 were filtered by StringTie when merging the file of transcripts. The remaining transcripts that passed the above filtered steps were assessed by their coding potential by the coding potential calculator 2 (CPC2), coding−noncoding index (CNCI), and Pfam-scan, and the thresholds of these software programs were scores <0 and were tagged as “noncoding.” HTSeq v.0.6.0 was used to count the read numbers mapped to each gene. Gene expression was analyzed by StringTie (version 1.2.3) software and quantified with FPKM. The DESeq2 software package was used to determine differentially expressed lncRNAs (DELs and differentially expressed mRNAs [DEMs]). DESeq2 provided statistical routines for determining differential expression in digital gene expression data using a model-based negative binomial distribution. The resulting p values were adjusted using Benjamini and Hochberg’s approach for controlling the false discovery rate. We used padj (adjusted p value) <0.05 found by DESeq2 and FPKM log2(fold change) >1 as thresholds to determine the differentially expressed lncRNAs (and mRNAs). The raw data have been shown on the National Center for Biotechnology Information (NCBI) databases as BioProject PRJNA744741, and project information is accessible with the following link: http://www.ncbi.nlm.nih.gov/bioproject/744741.
Quantitative reverse transcription polymerase chain reaction (qRT-PCR) analysis
Total RNA was extracted and reverse-transcribed using the PrimeScript RT reagent Kit with gDNA Eraser (TaKaRa, Kyoto, Japan). Quantitative real-time PCR was performed utilizing SYBR Green Master Mix (TaKaRa, Kyoto, Japan) on an iCycleriQ system (Bio-Rad, Berkeley, CA, USA). The PCR primer sequences used for quantitative real-time PCR are listed in Table S2. RNA expression levels were normalized to that of beta-actin (β-actin).
Western blot analysis
As previously reported, cells were harvested with a RIPA buffer containing a phosphatase inhibitor cocktail and a protease inhibitor cocktail (Roche, Basel, Switzerland), and the protein concentration was determined by a Pierce BCA Protein Assay Kit (Thermo Fisher Scientific, Waltham, MA, USA).49 Proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred to polyvinylidene difluoride (PVDF) membranes (Merck Millipore, Darmstadt, Germany). After they were blocked with 5% milk for 1 h, the membranes were incubated overnight at 4°C with primary antibodies at appropriate concentrations. Then, the PVDF membranes were incubated with the corresponding horseradish peroxidase (HRP)-conjugated secondary antibodies (1:3,000) for 1 h at room temperature and analyzed using the Gel Doc EZ Imaging System (Bio-Rad, Berkeley, CA, USA) with an ECL kit (Merck Millipore, Darmstadt, Germany). All the antibodies used are listed in Table S3.
Masson’s trichrome staining
At 28 days post-MI, hearts were excised, dehydrated in 30% sucrose solution, and embedded in Tissue Tek O.C.T. compound. The tissue sections (6.0-μm thick) were stained with Masson’s trichrome kit (Sigma, St. Louis, MO, USA) to measure the fibrotic area. The fibrotic area of the left ventricle (LV) was calculated by Image-Pro Plus 6.0 and evaluated by the ratio of the fibrotic area in the LV area.
Immunofluorescence assay
For immunofluorescence, myocardial tissue (6.0-μm thick) was fixed in 4% paraformaldehyde and permeabilized with 0.2% Triton X-100. After the samples were blocked with 5% BSA (BSA, Gibco, Grand Island, NY, USA) for 1 h, sections were incubated with a primary antibody overnight at 4°C. The primary antibodies anti-GFP and anti-troponin I (TnI) were used to observe the remaining MSCs at the border zone of mouse hearts post-MI. Anti-CD68 was used to mark macrophages, and anti-CD3 was used to observe T cells in border areas. Then, the cells were incubated with the corresponding fluorescent dye-conjugated secondary antibodies for 1 h at room temperature, and nuclei were counterstained with DAPI (Vector, Silicon Valley, CA, USA) according to the manufacturer’s instructions.
Wheat germ agglutinin (WGA) and anti-TnI were used to examine viable myocardium in infarcted areas. The tissue sections were first stained with primary anti-TnI overnight and incubated with corresponding fluorescent secondary antibodies for 1 h at room temperature. Then, tissue sections were placed in WGA (100 μg/mL), labeled with immune markers, and incubated at room temperature for 30 min. Finally, DAPI staining was performed.
At least five randomly selected fields in each heart were analyzed, and five hearts were evaluated per group. The primary and corresponding fluorescent dye-conjugated secondary antibodies are listed in Table S3.
TUNEL assay
Cell samples were fixed in 4% paraformaldehyde, permeabilized with 0.2% Triton X-100, incubated with a TUNEL reaction mixture (Roche, Basel, Switzerland) for 1.5 h at 37°C in the dark, and stained with DAPI for 15 min. The apoptotic ratio was calculated by measuring the TUNEL-positive cells in total cells/nuclei. Images were obtained from 6–9 randomly selected fields per well. We also used a TUNEL kit to evaluate cell apoptosis at the border zone of mouse hearts with MI. The TUNEL-positive cardiac cell percentage was calculated by the ratio of the TUNEL-positive nuclei to the total nuclei in at least six randomly selected fields in each heart, and five hearts were evaluated per group.
Echocardiography
Echocardiography was performed at days 0, 3, 7, 14, and 28 post-MI surgery as previously described.52 Two-dimensional and M-mode images were used to capture and analyze cardiac morphology and function with a Vevo 2100 system (VisualSonics, Toronto, ON, Canada). The EF and FS were measured for at least three interval cardiac cycles.
Subcellular fractionation assay
According to the manufacturer’s instructions, a cytoplasmic and nuclear RNA purification kit (Norgen, Thorold, ON, Canada) was used. Briefly, cells were incubated with a lysis solution to disrupt the cell membrane and centrifuged at 4°C and 1,200 × g to separate cytoplasmic and nuclear fractions. The RNA from both fractions was extracted and purified with columns in the kit and detected with qRT-PCR.
FISH assay
FISH procedures were performed according to the product manual of the Fluorescent In Situ Hybridization Kit (RiboBio, Guangzhou, China). LUCAT1 FISH Probe Mix and control FISH probes 18S and U6 were designed and synthesized at RiboBio. Cells were washed with PBS, fixed in 4% paraformaldehyde, and then permeabilized using 0.5% Triton X-100. After protease reagent treatment, the cells were incubated with 200-μL prehybridization buffer at 37°C for 30 min and discarded. Then, 2.5 μL of 20 μM LUCAT1 FISH Probe Mix and control FISH probes 18S and U6 were separately added to the cells with 100-μL hybridization buffer at 37°C overnight. All procedures were performed away from light after adding the probes. After the samples were washed with 3 different washing buffers, they were incubated with DAPI at 37°C for 10 min and photographed by a confocal microscope.
RNA pull-down assay
The sense and anti-sense sequences of full-length LUCAT1 were amplified in accordance with the Biotin RNA Labeling Mix and T7 RNA polymerase (Roche, Basel, Switzerland) protocols and purified with the RNeasy Mini Kit (Qiagen, Dusseldorf, Germany). Then, purified biotin-labeled LUCAT1 was incubated with the total lysates of MSCs, and eluted proteins were detected using western blots. The whole band pulled down from the sense or anti-sense sequence was excised from the gel, analyzed by MS, and retrieved in the human proteomic library. Table S4 shows the list of those proteins predicted to be pulled down only by the LUCAT1 sense sequence compared with the proteins predicted to be pulled down by the anti-sense sequence.
The 5’ and 3’ RACE assays
Total RNA was extracted from MSCs with TRIzol reagent, as described above. Then, 5’ and 3′ RACE were performed using the GeneRacer Kit (Invitrogen, Carlsbad, CA, USA) as previously reported.53 First-strand cDNA was synthesized using the MMuLV First-Strand cDNA Synthesis Kit (BBI, Toronto, ON, Canada), and PCR was performed using LA Taq polymerase (TaKaRa, Kyoto, Japan). The 5ʹ and 3ʹ RACE PCR primers were designed and synthesized for nested PCR, and PCR products were separated on a 1.5% agarose gel. The electrophoresis results were confirmed, and the amplified bands were sequenced. All the sequences were spliced together to obtain the whole sequence. The 5’ and 3’ amplification primers are shown in Table S5, and full-length LUCAT1-001 and LUCAT1-002 are presented in Table S6.
RIP assay
The RIP assay was performed using the Magna RIP RNA-Binding Protein Immunoprecipitation Kit (Millipore, Billerica, MA, USA). Cells were harvested, lysed in polysome lysis buffer, immunoprecipitated overnight at 4°C using anti-JMJD6 magnetic beads, and immobilized in a purification column. The immunoprecipitated RNAs were extracted and measured by qRT-PCR to confirm the binding relationship to LUCAT1.
ChIP assay
The ChIP assay was performed using the SimpleChIP Plus Sonication Chromatin IP Kit (CST, Boston, MA, USA) according to the manufacturer’s guidelines. Specifically, cells were immersed in 4% formaldehyde to fix proteins (both histone and nonhistone) to the DNA. Chromatin was sheared with sonication into 200–1000 bp, added with specific target antibodies (anti-JMJD6, anti-H4R3me2(s) and anti-H3R2me2(a)), and captured by Protein G beads. Then, the protein-DNA crosslinks were reversed, and the DNA was purified. The enrichment of target DNA sequences was detected via qRT-PCR analysis. The antibodies and primers used in ChIP are illustrated in Tables S3, S4, and S5.
Dual-luciferase reporter assay
The luciferase reporter assay was conducted by the Dual-Glo Luciferase Assay System (Promega, Madison, WI, USA) based on the manufacturer’s instructions. Briefly, JMJD6 plasmids were cotransfected with a FOXQ1 promoter-luciferase vector and a pRL-TK vector. In the other group, LUCAT1-overexpressing or LUCAT1-knockdown lentiviruses were cotransfected with the FOXQ1 promoter-luciferase vector and the pRL-TK vector. The pGL3-basic vector was transfected as a negative control. After 48 h, the cell extracts were used to calculate the respective luciferase activity on a Spark multimode microplate reader (Tecan, Mannedorf, Switzerland).
Statistical analysis
All data are presented as the mean ± SEM. The group sizes were determined based on previously published experiments and previous experience in which differences were observed. Student’s t test was used to compare two groups. Comparisons among three or more groups were performed by one-way analysis of variance (ANOVA), and we chose the Tukey post hoc test when comparing every mean with every other mean, while the Sidak post hoc test was used when comparing selected pairs of means, with the selection based on experimental design. Data analysis was performed using SPSS 20.0 and GraphPad Prism 5. p < 0.05 was considered statistically significant (∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001).
Acknowledgments
This work was supported by the National Key R&D Program of China (2019YFA0110400 and 2016YFC1301204 to J.W. and 2017YFA0103700 to X.H.; grants from the National Natural Science Foundation of China (nos. 81870292 to J.W., 81622006 and 81670261 to X.H., 81800351 to K.W.); Key R & D Projects of Zhejiang Province (nos. 2015C03028to J.W. and 2018C03014 to X.H.); and the Fundamental Research Funds for the Central Universities (2020FZZX003-02-11 to R.W.).
Author contributions
J.W. and X.H. conceived of and designed the study. Y.T. designed the experiments and analyzed data. Q.L. and R.W. drafted and revised the manuscript. C.X., C.N., K.W., and W.H. acquired in vitro and in vivo data. Z.Z., J.Z., Q.L., D.Z., and S.Z. analyzed and interpreted the data. H.Y., W.Z., and J.C. revised the manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2021.12.006.
Contributor Information
Xinyang Hu, Email: hxy0507@zju.edu.cn.
Jian'an Wang, Email: wangjianan111@zju.edu.cn.
Supplemental information
References
- 1.Galipeau J., Sensebe L. Mesenchymal stromal cells: clinical challenges and therapeutic opportunities. Cell Stem Cell. 2018;22:824–833. doi: 10.1016/j.stem.2018.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hu X., Xu Y., Zhong Z., Wu Y., Zhao J., Wang Y., Cheng H., Kong M., Zhang F., Chen Q., et al. A large-scale investigation of hypoxia-preconditioned allogeneic mesenchymal stem cells for myocardial repair in nonhuman primates: paracrine activity without remuscularization. Circ. Res. 2016;118:970–983. doi: 10.1161/CIRCRESAHA.115.307516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hu X., Yu S.P., Fraser J.L., Lu Z., Ogle M.E., Wang J.A., Wei L. Transplantation of hypoxia-preconditioned mesenchymal stem cells improves infarcted heart function via enhanced survival of implanted cells and angiogenesis. J. Thorac. Cardiovasc. Surg. 2008;135:799–808. doi: 10.1016/j.jtcvs.2007.07.071. [DOI] [PubMed] [Google Scholar]
- 4.Yao R.W., Wang Y., Chen L.L. Cellular functions of long noncoding RNAs. Nat. Cell Biol. 2019;21:542–551. doi: 10.1038/s41556-019-0311-8. [DOI] [PubMed] [Google Scholar]
- 5.Wang Z., Wang Y. Dawn of the epi-LncRNAs: new path from myheart. Circ. Res. 2015;116:235–236. doi: 10.1161/CIRCRESAHA.114.305490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Klattenhoff C.A., Scheuermann J.C., Surface L.E., Bradley R.K., Fields P.A., Steinhauser M.L., Ding H., Butty V.L., Torrey L., Haas S., et al. Braveheart, a long noncoding RNA required for cardiovascular lineage commitment. Cell. 2013;152:570–583. doi: 10.1016/j.cell.2013.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li L., Liu B., Wapinski O.L., Tsai M.C., Qu K., Zhang J., Carlson J.C., Lin M., Fang F., Gupta R.A., et al. Targeted disruption of Hotair leads to homeotic transformation and gene derepression. Cell Rep. 2013;5:3–12. doi: 10.1016/j.celrep.2013.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang K.C., Yang Y.W., Liu B., Sanyal A., Corces-Zimmerman R., Chen Y., Lajoie B.R., Protacio A., Flynn R.A., Gupta R.A., et al. A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature. 2011;472:120–124. doi: 10.1038/nature09819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Damas N.D., Marcatti M., Come C., Christensen L.L., Nielsen M.M., Baumgartner R., Gylling H.M., Maglieri G., Rundsten C.F., Seemann S.E., et al. SNHG5 promotes colorectal cancer cell survival by counteracting STAU1-mediated mRNA destabilization. Nat. Commun. 2016;7:13875. doi: 10.1038/ncomms13875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bugai A., Quaresma A.J.C., Friedel C.C., Lenasi T., Duster R., Sibley C.R., Fujinaga K., Kukanja P., Hennig T., Blasius M., et al. P-TEFb activation by RBM7 shapes a pro-survival transcriptional response to genotoxic stress. Mol. Cell. 2019;74:254–267.e10. doi: 10.1016/j.molcel.2019.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yoon J.H., You B.H., Park C.H., Kim Y.J., Nam J.W., Lee S.K. The long noncoding RNA LUCAT1 promotes tumorigenesis by controlling ubiquitination and stability of DNA methyltransferase 1 in esophageal squamous cell carcinoma. Cancer Lett. 2018;417:47–57. doi: 10.1016/j.canlet.2017.12.016. [DOI] [PubMed] [Google Scholar]
- 12.Thai P., Statt S., Chen C.H., Liang E., Campbell C., Wu R. Characterization of a novel long noncoding RNA, SCAL1, induced by cigarette smoke and elevated in lung cancer cell lines. Am. J. Respir. Cell Mol. Biol. 2013;49:204–211. doi: 10.1165/rcmb.2013-0159RC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grinnemo K.H., Mansson A., Dellgren G., Klingberg D., Wardell E., Drvota V., Tammik C., Holgersson J., Ringden O., Sylven C., et al. Xenoreactivity and engraftment of human mesenchymal stem cells transplanted into infarcted rat myocardium. J. Thorac. Cardiovasc. Surg. 2004;127:1293–1300. doi: 10.1016/j.jtcvs.2003.07.037. [DOI] [PubMed] [Google Scholar]
- 14.Wang Y., Chen X., Armstrong M.A., Li G. Survival of bone marrow-derived mesenchymal stem cells in a xenotransplantation model. J. Orthop. Res. 2007;25:926–932. doi: 10.1002/jor.20385. [DOI] [PubMed] [Google Scholar]
- 15.Devaney J., Horie S., Masterson C., Elliman S., Barry F., O'Brien T., Curley G.F., O'Toole D., Laffey J.G. Human mesenchymal stromal cells decrease the severity of acute lung injury induced by E. coli in the rat. Thorax. 2015;70:625–635. doi: 10.1136/thoraxjnl-2015-206813. [DOI] [PubMed] [Google Scholar]
- 16.Rossignol J., Boyer C., Thinard R., Remy S., Dugast A.S., Dubayle D., Dey N.D., Boeffard F., Delecrin J., Heymann D., et al. Mesenchymal stem cells induce a weak immune response in the rat striatum after allo or xenotransplantation. J. Cell Mol. Med. 2009;13:2547–2558. doi: 10.1111/j.1582-4934.2008.00657.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang F., Wu R., Jiang Z., Chen J., Nan J., Su S., Zhang N., Wang C., Zhao J., Ni C., et al. Leptin increases mitochondrial OPA1 via GSK3-mediated OMA1 ubiquitination to enhance therapeutic effects of mesenchymal stem cell transplantation. Cell Death Dis. 2018;9:556. doi: 10.1038/s41419-018-0579-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Almeida M., Pintacuda G., Masui O., Koseki Y., Gdula M., Cerase A., Brown D., Mould A., Innocent C., Nakayama M., et al. PCGF3/5-PRC1 initiates Polycomb recruitment in X chromosome inactivation. Science. 2017;356:1081–1084. doi: 10.1126/science.aal2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Joung J., Engreitz J.M., Konermann S., Abudayyeh O.O., Verdine V.K., Aguet F., Gootenberg J.S., Sanjana N.E., Wright J.B., Fulco C.P., et al. Genome-scale activation screen identifies a lncRNA locus regulating a gene neighbourhood. Nature. 2017;548:343–346. doi: 10.1038/nature23451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee J.T. Epigenetic regulation by long noncoding RNAs. Science. 2012;338:1435–1439. doi: 10.1126/science.1231776. [DOI] [PubMed] [Google Scholar]
- 21.Munschauer M., Nguyen C.T., Sirokman K., Hartigan C.R., Hogstrom L., Engreitz J.M., Ulirsch J.C., Fulco C.P., Subramanian V., Chen J., et al. The NORAD lncRNA assembles a topoisomerase complex critical for genome stability. Nature. 2018;561:132–136. doi: 10.1038/s41586-018-0453-z. [DOI] [PubMed] [Google Scholar]
- 22.Barakat T.S., Loos F., van Staveren S., Myronova E., Ghazvini M., Grootegoed J.A., Gribnau J. The trans-activator RNF12 and cis-acting elements effectuate X chromosome inactivation independent of X-pairing. Mol. Cell. 2014;53:965–978. doi: 10.1016/j.molcel.2014.02.006. [DOI] [PubMed] [Google Scholar]
- 23.Fang H., Bonora G., Lewandowski J.P., Thakur J., Filippova G.N., Henikoff S., Shendure J., Duan Z., Rinn J.L., Deng X., et al. Trans- and cis-acting effects of Firre on epigenetic features of the inactive X chromosome. Nat. Commun. 2020;11:6053. doi: 10.1038/s41467-020-19879-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rinn J.L., Chang H.Y. Long noncoding RNAs: molecular modalities to organismal functions. Annu. Rev. Biochem. 2020;89:283–308. doi: 10.1146/annurev-biochem-062917-012708. [DOI] [PubMed] [Google Scholar]
- 25.Liu W., Ma Q., Wong K., Li W., Ohgi K., Zhang J., Aggarwal A., Rosenfeld M.G. Brd4 and JMJD6-associated anti-pause enhancers in regulation of transcriptional pause release. Cell. 2013;155:1581–1595. doi: 10.1016/j.cell.2013.10.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tsai W.C., Reineke L.C., Jain A., Jung S.Y., Lloyd R.E. Histone arginine demethylase JMJD6 is linked to stress granule assembly through demethylation of the stress granule-nucleating protein G3BP1. J. Biol. Chem. 2017;292:18886–18896. doi: 10.1074/jbc.M117.800706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yi J., Shen H.F., Qiu J.S., Huang M.F., Zhang W.J., Ding J.C., Zhu X.Y., Zhou Y., Fu X.D., Liu W. JMJD6 and U2AF65 co-regulate alternative splicing in both JMJD6 enzymatic activity dependent and independent manner. Nucleic Acids Res. 2017;45:3503–3518. doi: 10.1093/nar/gkw1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chang B., Chen Y., Zhao Y., Bruick R.K. JMJD6 is a histone arginine demethylase. Science. 2007;318:444–447. doi: 10.1126/science.1145801. [DOI] [PubMed] [Google Scholar]
- 29.Uchida S., Dimmeler S. Long noncoding RNAs in cardiovascular diseases. Circ. Res. 2015;116:737–750. doi: 10.1161/CIRCRESAHA.116.302521. [DOI] [PubMed] [Google Scholar]
- 30.Wapinski O., Chang H.Y. Long noncoding RNAs and human disease. Trends Cell Biol. 2011;21:354–361. doi: 10.1016/j.tcb.2011.04.001. [DOI] [PubMed] [Google Scholar]
- 31.Wang K.C., Chang H.Y. Molecular mechanisms of long noncoding RNAs. Mol. Cell. 2011;43:904–914. doi: 10.1016/j.molcel.2011.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang L., Liu S.K., Song L., Yao H.R. SP1-induced up-regulation of lncRNA LUCAT1 promotes proliferation, migration and invasion of cervical cancer by sponging miR-181a. Artif. Cells Nanomed. Biotechnol. 2019;47:556–564. doi: 10.1080/21691401.2019.1575840. [DOI] [PubMed] [Google Scholar]
- 33.Zhou Q., Hou Z., Zuo S., Zhou X., Feng Y., Sun Y., Yuan X. LUCAT1 promotes colorectal cancer tumorigenesis by targeting the ribosomal protein L40-MDM2-p53 pathway through binding with UBA52. Cancer Sci. 2019;110:1194–1207. doi: 10.1111/cas.13951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gao Y.S., Liu X.Z., Zhang Y.G., Liu X.J., Li L.Z. Knockdown of long noncoding RNA LUCAT1 inhibits cell viability and invasion by regulating miR-375 in glioma. Oncol. Res. 2018;26:307–313. doi: 10.3727/096504017X15088061795756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zheng A., Song X., Zhang L., Zhao L., Mao X., Wei M., Jin F. Long non-coding RNA LUCAT1/miR-5582-3p/TCF7L2 axis regulates breast cancer stemness via Wnt/beta-catenin pathway. J. Exp. Clin. Cancer Res. 2019;38:305. doi: 10.1186/s13046-019-1315-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huan L., Guo T., Wu Y., Xu L., Huang S., Xu Y., Liang L., He X. Hypoxia induced LUCAT1/PTBP1 axis modulates cancer cell viability and chemotherapy response. Mol. Cancer. 2020;19:11. doi: 10.1186/s12943-019-1122-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang Z., Zhang X.J., Ji Y.X., Zhang P., Deng K.Q., Gong J., Ren S., Wang X., Chen I., Wang H., et al. The long noncoding RNA Chaer defines an epigenetic checkpoint in cardiac hypertrophy. Nat. Med. 2016;22:1131–1139. doi: 10.1038/nm.4179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cui P., Qin B., Liu N., Pan G., Pei D. Nuclear localization of the phosphatidylserine receptor protein via multiple nuclear localization signals. Exp. Cell Res. 2004;293:154–163. doi: 10.1016/j.yexcr.2003.09.023. [DOI] [PubMed] [Google Scholar]
- 39.Tibrewal N., Liu T., Li H., Birge R.B. Characterization of the biochemical and biophysical properties of the phosphatidylserine receptor (PS-R) gene product. Mol. Cell Biochem. 2007;304:119–125. doi: 10.1007/s11010-007-9492-8. [DOI] [PubMed] [Google Scholar]
- 40.Webby C.J., Wolf A., Gromak N., Dreger M., Kramer H., Kessler B., Nielsen M.L., Schmitz C., Butler D.S., Yates J.R., 3rd, et al. Jmjd6 catalyses lysyl-hydroxylation of U2AF65, a protein associated with RNA splicing. Science. 2009;325:90–93. doi: 10.1126/science.1175865. [DOI] [PubMed] [Google Scholar]
- 41.Blanc R.S., Richard S. Arginine methylation: the coming of age. Mol. Cell. 2017;65:8–24. doi: 10.1016/j.molcel.2016.11.003. [DOI] [PubMed] [Google Scholar]
- 42.Wei C., Yang C., Wang S., Shi D., Zhang C., Lin X., Liu Q., Dou R., Xiong B. Crosstalk between cancer cells and tumor associated macrophages is required for mesenchymal circulating tumor cell-mediated colorectal cancer metastasis. Mol. Cancer. 2019;18:64. doi: 10.1186/s12943-019-0976-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu P., Chen L. Inhibition of sonic hedgehog signaling blocks cell migration and growth but induces apoptosis via suppression of FOXQ1 in natural killer/T-cell lymphoma. Leuk. Res. 2018;64:1–9. doi: 10.1016/j.leukres.2017.11.001. [DOI] [PubMed] [Google Scholar]
- 44.Zhang X., Wang L., Wang Y., Shi S., Zhu H., Xiao F., Yang J., Yang A., Hao X. Inhibition of FOXQ1 induces apoptosis and suppresses proliferation in prostate cancer cells by controlling BCL11A/MDM2 expression. Oncol. Rep. 2016;36:2349–2356. doi: 10.3892/or.2016.5018. [DOI] [PubMed] [Google Scholar]
- 45.Peng X.H., Huang H.R., Lu J., Liu X., Zhao F.P., Zhang B., Lin S.X., Wang L., Chen H.H., Xu X., et al. MiR-124 suppresses tumor growth and metastasis by targeting Foxq1 in nasopharyngeal carcinoma. Mol. Cancer. 2014;13:186. doi: 10.1186/1476-4598-13-186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hong X., Liu N., Liang Y., He Q., Yang X., Lei Y., Zhang P., Zhao Y., He S., Wang Y., et al. Circular RNA CRIM1 functions as a ceRNA to promote nasopharyngeal carcinoma metastasis and docetaxel chemoresistance through upregulating FOXQ1. Mol. Cancer. 2020;19:33. doi: 10.1186/s12943-020-01149-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sun Y., Sheng Q., Cheng Y., Xu Y., Han Y., Wang J., Shi L., Zhao H., Du C. Zerumbone induces apoptosis in human renal cell carcinoma via Gli-1/Bcl-2 pathway. Pharmazie. 2013;68:141–145. [PubMed] [Google Scholar]
- 48.Gai X., Tu K., Li C., Lu Z., Roberts L.R., Zheng X. Histone acetyltransferase PCAF accelerates apoptosis by repressing a GLI1/BCL-2/BAX axis in hepatocellular carcinoma. Cell Death Dis. 2015;6:e1712. doi: 10.1038/cddis.2015.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hu X., Wu R., Jiang Z., Wang L., Chen P., Zhang L., Yang L., Wu Y., Chen H., Chen H., et al. Leptin signaling is required for augmented therapeutic properties of mesenchymal stem cells conferred by hypoxia preconditioning. Stem Cells. 2014;32:2702–2713. doi: 10.1002/stem.1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Soleimani M., Nadri S. A protocol for isolation and culture of mesenchymal stem cells from mouse bone marrow. Nat. Protoc. 2009;4:102–106. doi: 10.1038/nprot.2008.221. [DOI] [PubMed] [Google Scholar]
- 51.Liu X., Hu D., Zeng Z., Zhu W., Zhang N., Yu H., Chen H., Wang K., Wang Y., Wang L., et al. SRT1720 promotes survival of aged human mesenchymal stem cells via FAIM: a pharmacological strategy to improve stem cell-based therapy for rat myocardial infarction. Cell Death Dis. 2017;8:e2731. doi: 10.1038/cddis.2017.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xiao C., Wang K., Xu Y., Hu H., Zhang N., Wang Y., Zhong Z., Zhao J., Li Q., Zhu D., et al. Transplanted mesenchymal stem cells reduce autophagic flux in infarcted hearts via the exosomal transfer of miR-125b. Circ. Res. 2018;123:564–578. doi: 10.1161/CIRCRESAHA.118.312758. [DOI] [PubMed] [Google Scholar]
- 53.Hu Y.P., Jin Y.P., Wu X.S., Yang Y., Li Y.S., Li H.F., Xiang S.S., Song X.L., Jiang L., Zhang Y.J., et al. LncRNA-HGBC stabilized by HuR promotes gallbladder cancer progression by regulating miR-502-3p/SET/AKT axis. Mol. Cancer. 2019;18:167. doi: 10.1186/s12943-019-1097-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.