

Abstract
Metastasis formation is the major cause of death in most patients with cancer. Despite extensive research, targeting metastatic seeding and colonization is still an unresolved challenge. Only recently, attention has been drawn to the fact that metastasizing cancer cells selectively and dynamically adapt their metabolism at every step during the metastatic cascade. Moreover, many metastases display different metabolic traits compared with the tumours from which they originate, enabling survival and growth in the new environment. Consequently, the stage-dependent metabolic traits may provide therapeutic windows for preventing or reducing metastasis, and targeting the new metabolic traits arising in established metastases may allow their eradication.
The metastatic establishment of cancers at distant organs is largely uncurable and primarily contributes to the deaths of cancer patients. Nonetheless, meta stasis formation itself is a rare event in tumours because cancer cells need to overcome multiple environmental hurdles before they can successfully manifest themselves in other organs1,2. As a first step, cancer cells need to become motile, invasive and intravasate the tumour vasculature to enter the bloodstream, either directly or via the lymphatic system. Although numerous cancer cells can find their way into the circulation, the majority will succumb during their journey, with only few able to extravasate, expand and successfully colonize other organs3. Clinical observations and multiple experimental studies have led to several propositions that can explain the acquired metastatic traits of cancer cells (as described and summarized elsewhere2–7). Regardless, whether a sequential acquisition of random mutations in tumours may provide and select for metastatic traits in rare tumour clones8 or the metastatic predisposition of a tumour is already imprinted in the majority of cancer cells4–7, cancer cells need to be able to continuously adapt to changing environments during metastasis. Thus, only very few metastatic cancer cells with the appropriate adaptations will colonize at distant sites2,9. This knowledge supports the proposition that cancers do not randomly metastasize but, rather, dependent on the tumour type, seed to specific organs. Around 1900, James Ewing argued that tumour cells seed in a circulation-dependent manner10, while Stephen Paget proposed the ‘seed and soil’ hypothesis, stating that metastasizing cancer cells ‘seed’ only in certain especially hospitable tissues, akin to seeding in ‘fertile soil’11. Since then, subsequent studies have revealed several classes of metastasis genes whose altered activities are needed at individual steps along the metastatic process9. The expression of gene clusters associated with metastasis initiation and progression has been shown to confer a selective advantage to cancer cells both at primary and secondary sites as well as in the circulation, whereas protein products of metastatic virulence genes only promote survival and proliferation during colonization at the metastatic site, suggesting also a specific role in their mediation of organ-specific metastases12–14. Organotropism is facilitated by multiple factors including tumour-intrinsic factors, organ-specific niches and the interaction between tumour cells and the host microenvironment15,16. Furthermore, through the secretion of specific factors and extracellular vesicles, tumours are able to establish a pre-metastatic niche in distant organs17,18. These factors, for example, can activate distal lymphangiogenesis, recruit bone marrow-derived cells and promote extracellular matrix remodelling by activating stromal fibroblasts, which creates a permissive microenvironment for cancer cell colonization19–21.
Metastatic virulence genes.
Genes or factors that confer proliferation and/or survival advantages to metastasizing cancer cells at the secondary site without affecting primary tumours.
Only recently, attention has been drawn to the fact that metastatic cells also need to have or gain certain metabolic traits to be able to survive and grow in the new soil22–24, which may substantially vary in its nutrient and oxygen availability from the primary site. Such metabolic rewiring can be controlled transcriptionally, for example through epigenetic alterations, but also post-translationally or through metabolite availability to enzymes25. In this Review, we discuss growing evidence for the dynamic metabolic landscape metastasizing cells display to survive and propagate during the different steps of the metastatic cascade.
Metabolic adjustments during metastasis
Accumulating evidence supports the presence of dynamic changes in the metabolism of metastasizing cells, contributing to their ability to successfully transition through the changing microenvironments of the metastatic cascade. Here, we link the metabolic changes in cancer cells that traverse the metastatic cascade to the two intertwined concepts of metabolic plasticity and metabolic flexibility (FIG. 1). Similar to and extending beyond the previously described divergence in metabolism24,26, metabolite plasticity describes metastasizing cells that can use one metabolite to fuel the various metabolic requirements of different steps of the metastatic cascade. In contrast, metabolite flexibility builds on the well-described nutrient flexibility of cells27,28 and refers to metastasizing cancer cells that can use different metabolites to meet the same metabolic requirement imposed by a specific step of the metastatic cascade.
Fig. 1 |. Metabolite plasticity and flexibility in metastasizing cancer cells.
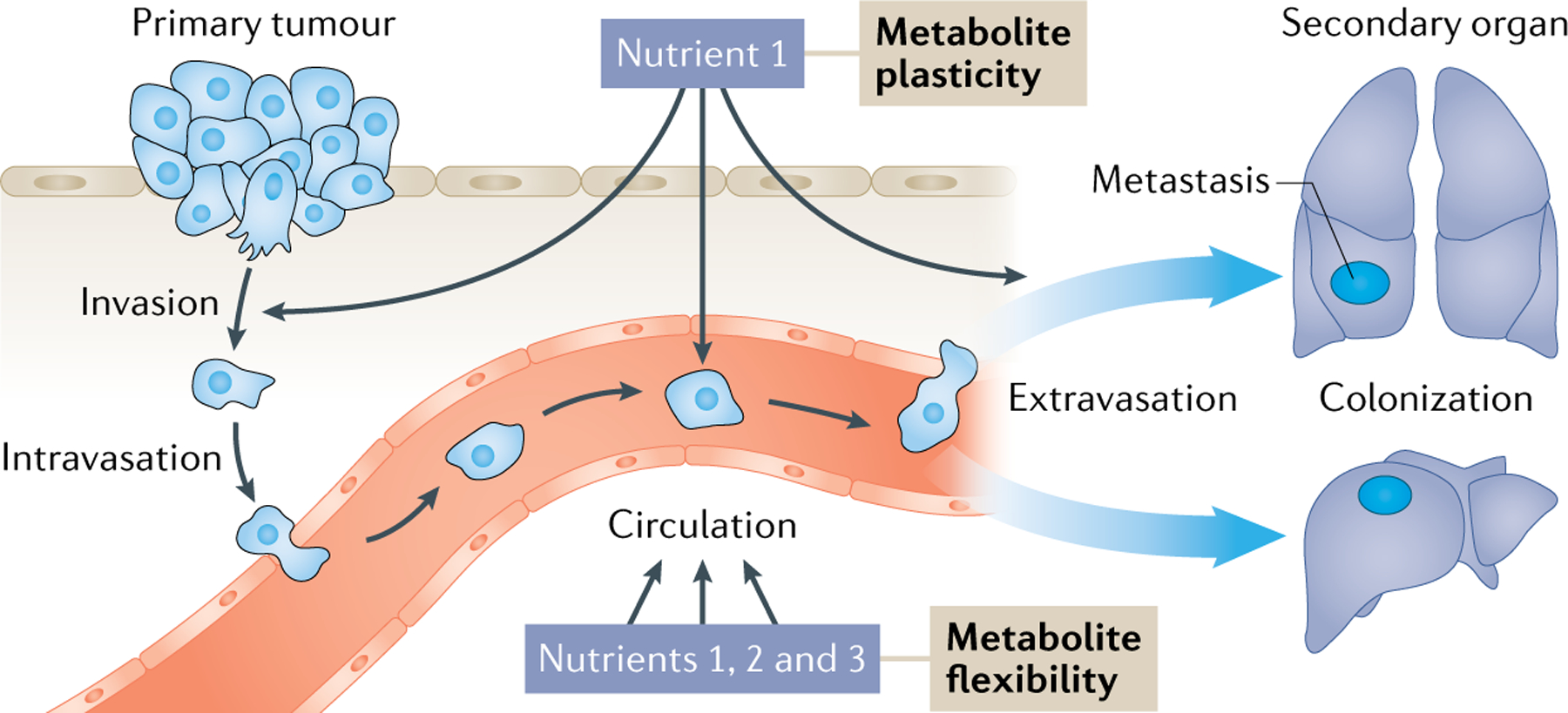
Metastasizing cells undergo dynamic metabolic changes to adjust to the differing microenvironments while travelling to distant organs. Thereby, metastasizing cells can exhibit metabolic plasticity in which they use one metabolite to fuel the various metabolic requirements of the different steps in the metastatic cascade. Alternatively, they can display nutrient flexibility by using multiple metabolites to meet the same metabolic requirement imposed by a specific step of the metastatic cascade. In cancer cells, both phenomena, metabolic plasticity and metabolic flexibility, contribute to metastasis formation and may be targeted for therapy.
Metabolic plasticity.
A metabolite can be used for multiple purposes.
Metabolic flexibility.
Several metabolites can be used for the same purpose.
Divergence in metabolism.
Divergent properties appear in distinct molecular subsets of cancer and contribute to metabolic heterogeneity.
Metabolite plasticity
It is conceivable that many nutrients can contribute to metabolite plasticity during metastasis formation. Yet the majority of metabolic research has focused on the early metastatic steps of tumour migration and invasion whereas only few nutrients have been analysed at several metastatic steps. Here, we discuss the implication of four such nutrients, namely lactate, pyruvate, glutamine and fatty acids, in regulating tumour invasion, survival in the circulation and colonization at a secondary site (FIGS 2,3; TABLE 1).
Fig. 2 |. The metabolism of invading and circulating (detached) cancer cells.
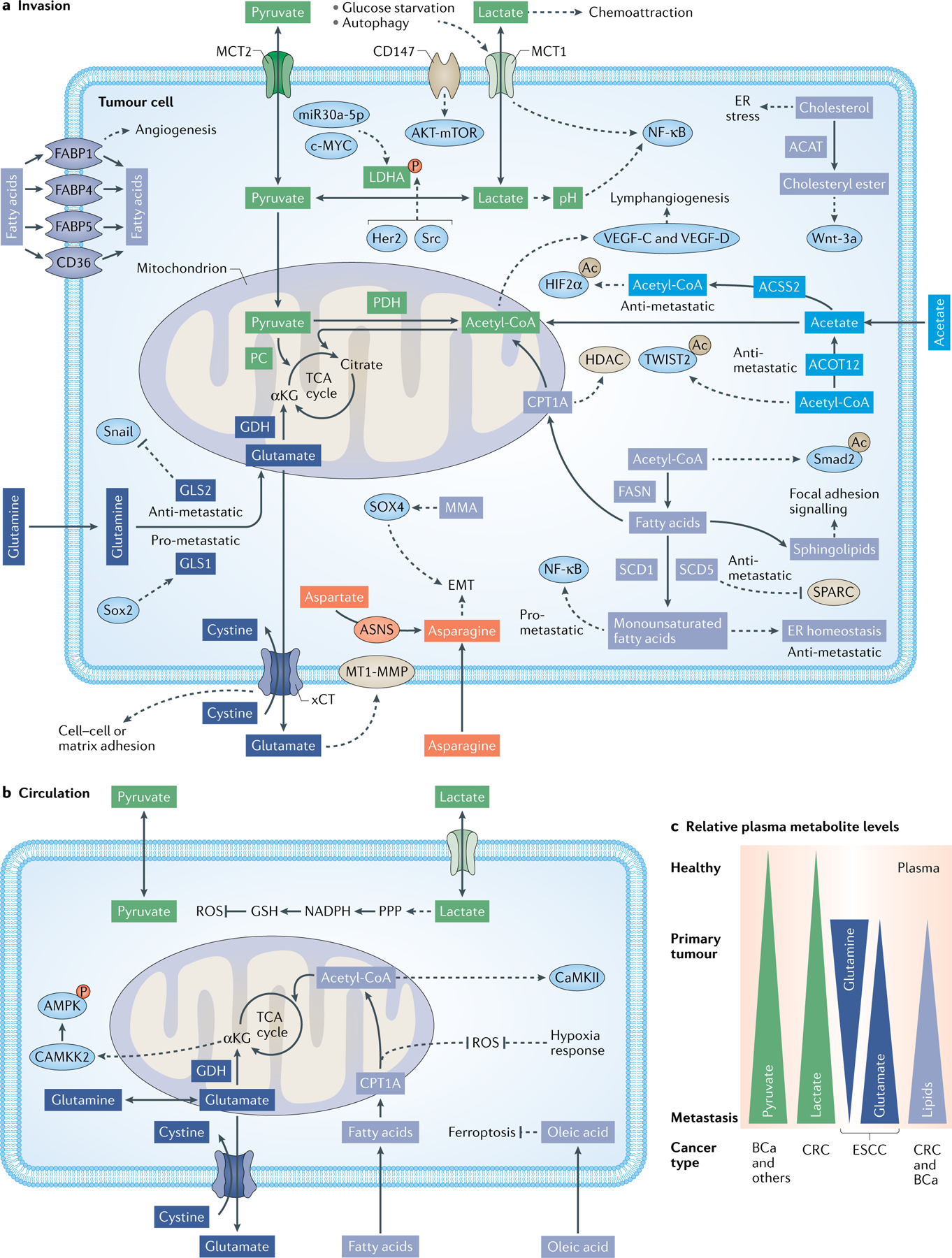
Multiple nutrients and metabolites facilitate the invasiveness and migratory abilities of cancer cells (part a) and their survival in the circulation (part b). Differential availability of nutrients and metabolites in the circulation of healthy individuals, patients with non-metastatic cancer (primary tumour) or patients with metastatic cancer have been reported. In the circulation, increased pyruvate levels have been observed in patients with non-metastatic versus metastatic breast cancer (BCa) and for patients with multiple types of cancer compared with healthy individuals. Increased lactate levels have been observed in patients with metastatic colorectal cancer (CRC). Low glutamine and high glutamate levels have been observed in patients with oesophageal squamous cell carcinoma (ESCC). Enhanced lipid levels have been observed in patients with colorectal and breast cancer, but not patients with oral cancer (part c). Fatty acid metabolism is depicted in light indigo. Lactate and pyruvate metabolism are depicted in green. Glutamine metabolism is depicted in dark indigo. Acetate metabolism is depicted in indigo. Asparagine metabolism is depicted in orange. Proteins whose function is regulated by metabolism pathways are shown in light blue. Enzymes depicted in box shape and membrane transporters not coloured in beige have been discussed as targets in the main text. Dashed lines indicate regulatory events. Multiple metabolic reactions are summarized for clarity reasons. Ac, acetylation; ACAT, acetyl-coenzyme A acetyltransferases; ACOT12, acyl-CoA thioesterase 12; ACSS2, acyl-coenzyme A synthetase short-chain family member 2; AMPK, adenosine monophosphate-activated protein kinase; αKG, α-ketoglutarate; ASNS, asparagine synthetase (glutamine-hydrolysing); CAMKK2, calcium/calmodulin-dependent protein kinase kinase 2; c-MYC, MYC proto-oncogene; CoA, coenzyme A; CPT1A, carnitine palmitoyltransferase 1; CTSB, cathepsin B; EMT, epithelial to mesenchymal transition; ER, endoplasmic reticulum; FABP1, fatty acid-binding protein 1; FASN, fatty acid synthase; GDH, glutamate dehydrogenase; GLS1, glutaminase 1; GSH, glutathione; HDAC, histone deacetylases; Her2, Erb-B2 receptor tyrosine kinase 2; HIF2α, hypoxia-inducible factor 2α; LDHA, lactate dehydrogenase A; MCT1, monocarboxylate transporter 1; miR-21, microRNA 21; MMA, methylmalonic acid; MT1-MMP, membrane type 1 matrix metalloproteinase; mTOR, mammalian target of rapamycin; NADPH, nicotinamide adenine dinucleotide phosphate; NF-κb, nuclear factor κ-light-chain-enhancer of activated B cells; P, phosphorylation; PC, pyruvate carboxylase; PDH, pyruvate dehydrogenase; PPP, pentose phosphate pathway; ROS, reactive oxygen species; SCD1, stearoyl-CoA desaturase 1; Smad2, mothers against decapentaplegic homologue 2; Sox2, SRY (sex determining region Y)-box 2; SPARC, secreted protein acidic and cysteine rich; TCA, tricarboxylic acid; TWIST2, twist family BHLH transcription factor 2; VEGF, vascular endothelial growth factor; Wnt-3a, wingless-type MMTV integration site family, member 3A; xCT, solute carrier family 7 member 11.
Fig. 3 |. The metabolism of cancer cells colonizing in distant organs.
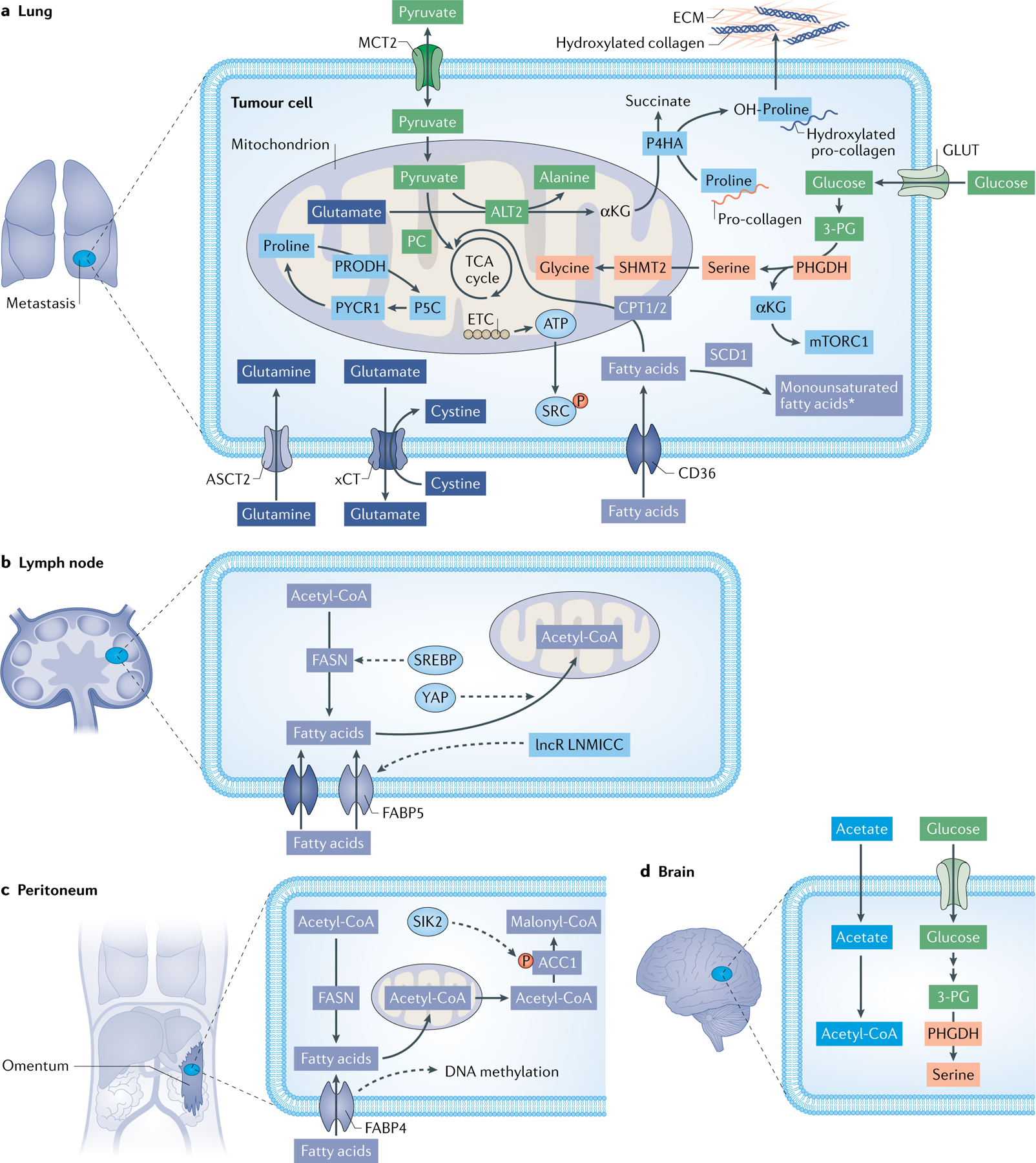
Different nutrients and metabolic pathways are supporting the nesting of metastasizing cancer cells in the lung (part a), lymph node (part b), peritoneum (part c) and brain (part d). In the lung, breast cancer cells take up extracellular pyruvate via monocarboxylate transporter 2 (MCT2) to generate α-ketoglutarate (αKG) from glutamate to activate the enzyme collagen prolyl-4-hydroxylase (P4HA), which is essential for extracellular matrix deposition and remodelling (part a). Uptake of glutamine via ASCT2 is important for the growth of prostate cancer cells and their metastatic seeding to the lung (part a). Elevated fatty acid metabolism is associated with a higher propensity of several cancer cell types to metastasize. Inhibition of CD36 and fatty acid binding proteins (FABPs), which both import fatty acids into the cell, as well as inhibition of fatty acid synthase (FASN) to block de novo fatty acid synthesis has been shown to restrain lung metastases (part a). In lymph nodes, melanoma and breast cancer cells undergo a metabolic shift towards fatty acid oxidation (FAO). Transcriptional co-activator yes-associated protein (YAP) is selectively activated in lymph node metastatic tumours, leading to the upregulation of genes in the FAO signalling pathway (part b). In the peritoneum, visceral adipocytes promote ovarian cancer progression and metastases in the peritoneal cavity by providing fatty acids and increasing CD36 expression in cancer cells (part c). Salt inducible kinase 2 (SIK2)-mediated ACC and SIK2 phosphorylation and overexpression promote fatty acid oxidation required for effective metastasis formation (part c). In the brain, acetate was discovered as an alternative energy source to glucose for human and mouse brain glioblastoma and lung and breast cancer-derived brain metastases (part d). As the brain exhibits low serine levels, inhibiting serine production by genetic phosphoglycerate dehydrogenase (PHGDH) silencing or PH-755 treatment in mice impairs breast cancer-derived brain metastases. *The metabolism of monounsaturated fatty acids can have a pro-metastatic or anti-metastatic function (part a). Fatty acid metabolism is depicted in light indigo. Lactate and pyruvate metabolism are depicted in green. Glutamine metabolism is depicted in dark indigo. Acetate metabolism is depicted in indigo. Serine metabolism is depicted in light orange. Proteins whose function is regulated by metabolism pathways are shown in light blue. Enzymes depicted in box shape and membrane transporters have been discussed as targets in the main text. Dashed lines indicate regulatory events. Multiple metabolic reactions are summarized for clarity reasons. ACC1, acetyl-CoA carboxylase 1; ALT2, alanine aminotransferase 2; CoA, coenzyme A; CPT1/2, carnitine palmitoyltransferase 1/2; ECM, extracellular matrix; ETC, electron transport chain; lncR LNMICC, link RNA LNMICC; P5C, 1-pyrroline-5-carboxylic acid; P, phosphorylation; PC, pyruvate carboxylase; PRODH, proline dehydrogenase; PYCR1, pyrroline-5-carboxylate reductase 1, mitochondrial; SCD1, stearoyl-CoA desaturase 1; SHMT2, serine hydroxymethyltransferase 2; TCA, tricarboxylic acid; xCT, solute carrier family 7 member 11.
Table 1 |.
Metabolic targets for metastasis prevention and treatment discussed in this Review
| Target | Cancer type | Investigated metastatic side | Model | Targeting strategy | Effect on primary tumour relative to control condition | Investigated therapeutic strategy | Refs |
|---|---|---|---|---|---|---|---|
| ACAT | Prostate, pancreas | Lung, liver, spleen, pancreas, lymph node | Human PC-3M, PC-3, MIA PaCa-2 cells | Avasimibe | Reduced invasion and proliferation | Metastasis prevention | 137,138 |
| ALT2 | Breast | Lung | Murine 4T1 cells, human MCF7 cells | shRNA knockdown | Reduced tumour size | Metastasis prevention | 69,175 |
| ASCT2 | Prostate | Liver (trend), lung (significant effect) | Human PC-3 cells | shRNA knockdown | Reduced tumour growth | Metastasis prevention | 92 |
| ASNS | Breast | Lung | Murine 4T1-T cells | shRNA knockdown, asparaginase, low-asparagine diet, combination of shRNA and drug or diet | No significant change in response to single treatments | Metastasis prevention | 177 |
| CD36 | Head and neck, breast, skin | Lymph node, lung, liver, bone | Patient-derived SCC tumours, murine SCC Ln-7 cells, human MCF7 cells, human 501mel cells | shRNA knockdown, neutralizing antibodies FA6.152, JC63.1 | No changes or slight reduction (one shRNA) of tumour growth | Metastasis prevention and treatment | 103 |
| CPT1A | Colon and/or rectum | Lung, liver | Human HCT15, HCT116 | Etomoxir, shRNA knockdown | n.d. | Metastasis prevention | 185 |
| FABP4 | Ovaries | Diaphragm, liver, spleen, lymph node, pelvic areas, omentum | Human HeyA8 MDR, Ovcar 5, OVCAR8 cells, murine ID8 cells | DOPC nanoliposomes containing siRNA injections, CRISPR knockout, BMS309403 | No changes (BMS309403) or reduced tumour weight | Metastasis prevention and treatment | 118,156 |
| FABP5 | Cervix, prostate | Lung, femur, lymph node | Human SiHa cells, PC-3 | siRNA, shRNA knockdown | Reduced tumour size | Metastasis prevention | 122,124 |
| FASN | Ovarian, skin, colorectal | Peritoneum, lung, liver, lymph node | Human SKOV-3, KM-20, HT29 cells, murine B16-F10, | shRNA knockdown, orlistat | Reduced tumour growth | Metastasis prevention | 127,128,157 |
| GDH | Lung | Liver, lung | Human A549, H460 | shRNA knockdown | n.d. | Metastasis prevention | 91 |
| GLS1 | Colorectal | Lung, liver, kidney | Human HT29 cells, murine VM-M3 cells | shRNA knockdown, 6-diazo-5-oxo-l-norleucine (DON) | Decreased tumour volume and weight | Metastasis prevention | 80,93 |
| LDHA | Breast | Lung | Human MDA-MB-231 cells | shRNA knockdown | Reduced tumour growth | Metastasis prevention | 37,39 |
| MCT1 | Skin | Lung | Patient-derived xenografts, murine YUMM1.7, YUMM3.3, YUMM5.2 cells | shRNA knockdown, AZD3965 | No change (AZD3965) or slight (one shRNA) reduction of tumour size | Metastasis prevention | 63 |
| MCT2 | Breast | Lung | Murine 4T1, EMT6.5 cells | CRISPR knockout, α-cyano-4-hydroxycinnamic acid | No (volume) to slight (weight) effect on tumour growth | Metastasis prevention | 69 |
| P4HA | Breast | Lung, lymph node | Human MDA-MB-231, MDA-MB-435 cells | shRNA knockdown, ethyl 3,4- dihydroxybenzoate | Reduced tumour volume | Metastasis prevention | 70 |
| PC | Breast | Lung | Murine D2A1 cells | shRNA knockdown | No significant effect | Metastasis prevention | 68 |
| PHGDH | Breast, kidney | Lung, brain | Human MDA-MB-231, MDA-MB-231 BrM cells, patient-derived clear cell renal cell carcinoma cells | shRNA knockdown, PH-755 | Similar to increased tumour mass | Metastasis prevention and treatment | 173,174 |
| PRODH | Breast | Lung | Murine 4T1, EMT6.5 cells | l-THFA | No significant effect | Metastasis prevention | 73 |
| SCD1 | Skin | Lung | Murine B16-F10 cells, MITF+ B16-F1 | shRNA knockdown, CAY10566, A939572 | n.d. | Metastasis prevention but also promotion | 135,159 |
| xCT | Breast, oesophagus, gastrointestinal tract | Lung | Murine 4T1, TUBO cells, human KYSE-150 cells | Anti-xCT vaccination, sulfasalazine | Reduced tumour size | Metastasis prevention and treatment | 86,87,94 |
ACAT, cholesterol acyltransferase; ALT2, alanine aminotransferase 2; ASNS, asparagine synthetase; CPT1A, carnitine palmitoyltransferase 1A; FABP4, fatty acid binding protein 4; FASN, fatty acid synthase; GDH, glutamate dehydrogenase; GLS1, glutaminase 1; LDHA, lactate dehydrogenase A; MCT1, monocarboxylate transporter 1; n.d., not determined in the referenced metastasis study; P4HA, prolyl-4-hydroxylase; PC, pyruvate carboxylase; PHGDH, phosphoglycerate dehydrogenase; PRODH, proline dehydrogenase; SCC, squamous cell carcinoma; SCD1, stearoyl-CoA desaturase 1; shRNA, short hairpin RNA; siRNA, small interfering RNA; xCT, solute carrier family 7 member 11.
Pyruvate and lactate metabolism
Pyruvate is produced from glucose and other nutrients that fuel the glycolytic pathway, and can give rise to lactate in a one-step reaction catalysed by lactate dehydrogenase (LDH). In addition, pyruvate and, consequently, lactate can be produced from glutamine and other amino acids that fuel the tricarboxylic acid (TCA) cycle via malic enzyme (ME) or pyruvate carboxy kinase (PCK), which funnel TCA cycle-derived carbon towards the lower part of glycolysis. Pyruvate (and lactate) can be reversibly converted to alanine, resulting either in the production of alanine from pyruvate or vice versa.
Additionally, pyruvate and, consequently, lactate can be oxidized in the mitochondria to acetyl-CoA by the enzyme pyruvate dehydrogenase (PDH) or converted by the enzyme pyruvate carboxylase (PC) to oxaloacetate; the latter replenishes the TCA cycle (anaplerosis). Yet both lactate and pyruvate are also nutrients that can be taken up by cells directly from the environment.
Anaplerosis.
Refilling of the tricarboxylic acid (TCA) cycle with carbon.
Pyruvate and lactate metabolism in invading and motile cancer cells.
One of the first metastatic attributes of cancer cells is their switch from a proliferative to a migrating phenotype. Multiple studies have provided evidence that metabolic changes in cancer cells modulate activity of signalling pathways and global gene expression programmes driving migration and invasion (for example, an epithelial to mesenchymal transition (EMT))23. These metabolic adaptations are not just consequential bystander effects because metabolites such as pyruvate and lactate can directly promote the invasion and migration ability of cancer cells (FIG. 2a). In breast cancer cells, the anaplerotic entry of pyruvate via PC into the TCA cycle promoted an invasive phenotype by increasing their motility, although the underlying molecular mechanism remains elusive29,30. Pyruvate oxidation requires PDH activity. The resulting acetyl-CoA production and further accumulation of acetyl-CoA, through phosphorylation-induced inhibition of its consuming enzyme acetyl-CoA carboxylase 1 (ACC1)31, led to the acetylation of the transcription factor Smad2, which is a known inducer of mesenchymal gene expression patterns31. Gene expression analysis of 20 different human cancers has shown that poor survival was associated with inhibition of at least one mitochondrial pathway. Oxidative phosphorylation was the most affected pathway in patients with a low versus high survival rate, and most frequently coincided with downregulated gene expression of the genes encoding the subunits of Complex I and IV of the respiratory chain32. Functionally, expression of genes contributing to oxidative phosphorylation showed a negative correlation with EMT32. Accordingly, metastasis-derived cell lines in vitro and metastases analysed ex vivo from a lung cancer mouse model had reduced mitochondrial functionality compared with non-metastatic primary tumours33. Although these changes in mitochondrial metabolism will have multiple effects, they may indicate a shift towards glycolytic energy production that requires lactate synthesis. Indeed, inhibition of lactate dehydrogenase A (LDHA) expression impaired invasion and migration in in vitro assays in renal cell carcinoma (RCC), pancreatic cancer and prostate cancer34–36 and decreased metastasis in an orthotopic renal xenograft model36. Moreover, downregulation of microRNA30a-5p suppressed LDHA expression and thereby inhibited gallbladder cancer progression as well as metastasis formation in breast cancer mouse models37,38, whereas knockdown of the oncogene c-MYC decreased LDHA expression in in vitro models of pancreatic cancer35. LDHA has also been found to be phosphorylated by HER2 and SRC39. Inhibition of this phosphorylation resulted in decreased invasiveness of head and neck as well as breast cancer cell lines in vitro and metastatic potential in breast cancer xenograft mouse models39. Interestingly, LDHA silencing could be rescued by lactate and the antioxidant N-acetylcysteine in in vitro assays, suggesting a redox metabolism-dependent mechanism39.
The transporters monocarboxylate transporter 1 (MCT1) and MCT4 preferentially exchange lactate between the extracellular and intracellular space40. Both proteins are independent prognostic markers for progression-free survival in clear cell RCC41, whereas high levels of MCT1 expression have been associated with a lower survival rate in patients with bladder cancer42. MCT1 expression also increased during glucose starvation in cervical cancer cell lines43 and during starvation-induced autophagy in hepatocellular carcinoma cell lines44, which are conditions that may occur in poorly perfused and hypoxic regions of the tumour microenvironment. Mechanistically, increased levels of MCT1 in cervical cancer cell lines can lead to the formation of a heterocomplex with CD147 (REF.43), a protein that stimulates secretion of matrix metalloproteinases (MMPs) and cytokines in many cancers45. This interaction was shown in oxidative phosphorylation-dependent tumour cells where it increased migratory capacities in vitro43. Notably, MCT1, independent of its lactate transport function, has been shown to activate NF-κB, which is an upstream regulator of a pro-invasive EMT in cervix squamous carcinoma cell lines and an experimental mammary carcinoma mouse model46, and promoted migration and invasion in osteosarcoma cell lines47. Interestingly, pH changes, which may result from lactate production, also activated NF-κB in a breast cancer cell line48. In addition, lactate has been shown to promote breast cancer progression by supporting chemoattraction, which stimulated cancer cell migration in a breast cancer cell line49. Accordingly, intraperitoneal administration of lactate to mice increased lung metastasis formation resulting from intravenously injected breast cancer cells49. Recently, lactate has also been identified as a precursor of histone lactylation50. It will be very interesting to reveal whether this post-translational histone modification provides another regulatory mechanism in tumour invasion.
Taken together, the studies described above support the notion that lactate and pyruvate metabolism can directly promote cancer invasion by inducing various signalling pathways and molecules that drive and facilitate tumour cell migration and invasion.
Pyruvate and lactate metabolism in circulating tumour cells.
Cancer cells need to increase their antioxidant defence in the circulation51–53 to avoid matrix detachment-induced cell death (anoikis, reviewed elsewhere54,55). Lactate and pyruvate metabolism may potentially contribute to resistance of matrix-detached cells against reactive oxygen species (ROS) (FIG. 2b). Studies in vitro and in patients revealed that pyruvate concentrations correlate with matrix detachment and invasiveness. As such, pyruvate levels were increased intracellularly in matrix-detached 293T cells56, whereas highly invasive ovarian cancer cells exhibited an increased pyruvate uptake in matrix-detached conditions compared with less invasive ovarian cancer cells57. In patients, levels of pyruvate were elevated in the plasma of individuals with metastatic carcinoma of different origin compared with healthy individuals58. Congruently, pyruvate concentrations in serum of patients with aggressive metastatic breast cancer were higher compared with those of patients with early-stage breast cancer59. Interestingly, a study in non-malignant and malignant cells indicates that pyruvate itself can act as an antioxidant via a non-enzymatic reaction with hydrogen peroxide60. Moreover, it has been demonstrated that the serum lactate concentration was higher in patients with metastatic compared with non-metastatic colorectal cancer61. Thus, these observations suggest that pyruvate and lactate are available to circulating tumour cells and could support their successful transition through the blood circulation. Corroborating the idea that pyruvate and lactate metabolism can protect against ROS, the disruption of intracellular conversion of pyruvate via PC to oxaloacetate in cultured breast cancer cells resulted in a decreased ratio of NADPH/NADP+ and glutathione (GSH)/GSSG, indicating reduced ROS scavenging capacity and, consequently, causing elevated oxidative stress62. Striking evidence has been provided for an important role of lactate metabolism in circulating tumour cells. In particular, MCT1-mediated uptake of lactate facilitated melanoma metastasis in patient-derived xenografts by promoting pentose phosphate pathway flux and a consecutive ROS defence63.
Matrix-detached cells can induce hypoxia via cell clustering to reduce ROS56. Interestingly, intracellular pyruvate or lactate accumulation can induce a hypoxic response by stabilizing hypoxia inducible factor 1α (HIF1α)64–66. In the case of lactate, this can also occur via N-Myc Downstream-Regulated Gene 3 Protein (NDRG3) stabilization67. Thus, it is tempting to speculate that increased lactate and pyruvate concentrations in the blood circulation may support circulating tumour cells and cell clusters by facilitating a hypoxic response.
In conclusion, there is supporting evidence that lactate and pyruvate uptake can help circulating tumour cells to survive by enhancing their resistance against oxidative stress.
Pyruvate and lactate metabolism during metastatic colonization.
Once cancer cells reach a distant organ, they need to adapt to the new environment and also create a permissive niche in order to proliferate (FIG. 3; TABLE 1). Colonizing cancer cells are likely exposed to different types and/or levels of nutrients at the secondary site compared with the primary site. For example, pyruvate is enriched in lung interstitial fluid compared with plasma68, with the lung being an organ of frequent metastasis across several cancer types. Thus, it is conceivable that cancer cells utilize pyruvate when undergoing metastatic colonization in the lung. Indeed, there is evidence from mouse models that breast cancer cells colonizing in the lung require extracellular pyruvate, but not lactate, to create a permissive niche through extracellular matrix remodelling69. In particular, increased levels of pyruvate facilitated transamination between glutamate and pyruvate (catalysed by the mitochondrial enzyme alanine aminotransferase 2 (ALT2; also known as GPT2)), leading to the generation of α-ketoglutarate and alanine69. In turn, α-ketoglutarate boosted activity of the enzyme collagen prolyl-4-hydroxylase (P4HA)69, which independent studies have reported to be essential for collagen deposition and remodelling70. Yet, the role of pyruvate in the lung environment may go beyond collagen hydroxylation because expression of PC was required for breast cancer-derived lung metastases but not for extrapulmonary metastases in an experimental mouse model71. Moreover, pyruvate availability has been shown to increase activity of the serine biosynthesis pathway, which potentiated mTORC1 signalling in lung metastases but not primary breast tumours72.
Besides the above described findings, the number of studies that particularly investigate the impact of pyruvate and lactate metabolism in metastatic colonization in an in vivo setting is rather limited. However, supporting evidence for the importance of these two nutrients for metastatic colonization has been provided in vitro via colony formation assays, which may to some extent mirror a similar metabolic programme to that of in vivo metastatic colonization73. For example, pyruvate supplementation can stimulate in vitro colony formation in breast cancer cells by fuelling mitochondrial respiration74 and MCT1 inhibition with AR-C155858 had a cytostatic effect on colony growth in different cancer cell lines75. Similarly, blocking lactate and pyruvate entry into the mitochondria through inhibition of the mitochondrial pyruvate carrier (MPC) with 7ACC2 elicited cytotoxic effects on colony growth in different cell lines75. Whereas there is evidence that pyruvate and lactate fuel in vivo tumour proliferation68,76, the full extent to which lactate and pyruvate are important nutrients for cancer cells to seed and colonize in the metastatic niche is largely unknown.
Glutamine metabolism
Glutamine is the most abundant free amino acid in the plasma. Many cancer cells take up glutamine, which contributes to non-essential amino acid as well as nucleotide synthesis through carbon or nitrogen metabolism. Moreover, glutamine can be converted in the mitochondria to replenish the TCA cycle (anaplerosis) or can be fully oxidized to produce ATP (glutaminolysis).
Glutaminolysis.
Full oxidation of glutamine.
Glutamine metabolism in invading and motile cancer cells.
Glutamine metabolism has been extensively studied in proliferating cancer cells77. Emerging evidence suggest that glutamine metabolism is also important for invasion (FIG. 2a; TABLE 1). Invasive, but not non-invasive, ovarian cancer cells displayed a dependency on glutamine availability in vitro78, and metastatic melanoma cells exhibited elevated glutamine oxidation79. Moreover, mRNA expression levels of glutaminase 1 (GLS1), which catabolizes glutamine to glutamate, correlated with lymph node metastasis of colorectal cancer, and GLS1 expression was required for hypoxia-mediated cancer cell migration in vitro80. Moreover, SOX12 overexpression compared with control was found to promote colorectal cancer metastasis via GLS1 in experimental mouse models81. The isoenzyme GLS2 repressed hepatocellular carcinoma metastasis based on protein interactions with the small GTPase RAC1 (REF.82) and with Dicer83. The latter interaction inhibited the pro-invasive regulator and EMT transcription factor Snail83. Specifically, GLS2 binds to small GTPase RAC1 and inhibits its interaction with the RAC1 activators guanine-nucleotide exchange factors, which in turn inhibits RAC1 to suppress cancer metastasis82. Moreover, GLS2 also interacts with Dicer and stabilizes Dicer protein to facilitate miR-34a maturation, and subsequently represses Snail expression83. Both interactions are independent of the catalytic function of GLS2.
Glutamine-derived glutamate can have different fates, including secretion via the cystine antiporter xCT (also known as SLC7A11), expression of which has been identified as a predictive marker of recurrence, tumour invasion, lymph node metastasis and venous invasion in patients with colorectal cancer84. Mechanistically, secretion of glutamate via xCT can lead to paracrine activation of the metabotropic glutamate receptor GRM3, which in turn upregulates Rab27-dependent recycling of the transmembrane membrane type 1 (MT1)-MMP to promote the invasive behaviour of breast cancer cells85. Accordingly, xCT inhibition with sulfasalazine impaired lung metastasis through a ROS-dependent p38 MAPK activation in breast cancer and oesophageal squamous cell carcinoma mouse models86,87. In the latter scenario, disruption of xCT enhanced homotypic cell–cell adhesion and attenuated cell–extracellular matrix adhesion87.
Thus, cancer cells can rely on glutamine and cystine metabolism to alter cell adhesion and activate invasive cues.
Glutamine metabolism in circulating cancer cells.
So far, evidence of glutamine metabolism in supporting circulating tumour cells is sparse (FIG. 2b,c). Reduced glutamine concentrations and markedly elevated glutamate concentrations have been identified in the plasma of patients with oesophageal squamous cell carcinoma presenting lymph node metastasis compared with those who did not present with metastasis88. In line with this, circulating tumour cells with tumour-initiating capacity (CD44+/high versus CD44−/low) from patients with gastric cancer have increased expression of xCT89. Thus, it is conceivable that increased xCT expression may explain the above discussed elevated levels of glutamate in the plasma of patients with metastatic oesophageal squamous cell carcinoma because xCT overexpression may favour the secretion of glutamate. Yet, the intracellular metabolism of glutamate seems also to be linked to metastatic cancers. As such, glutamate dehydrogenase (GDH), which converts glutamate to α-ketoglutarate, has been identified as a prognostic marker of colorectal cancer metastasis90. Indeed, GDH knockdown or inhibition with R162 attenuated anoikis resistance and decreased tumour metastasis through CamKK2–AMPK signalling in an LKB1-deficient lung cancer mouse model91. Thus, different fates of glutamine may affect circulating tumour cells. For a broader understanding and interpretation of the above described observations, further investigations are required.
Glutamine metabolism during metastatic colonization.
Recent studies have described changes in glutamine metabolism of colonizing cancer cells (FIG. 3; TABLE 1). In this respect, inhibition of the glutamine transporter ASCT2 (also known as SLC1A5) with short hairpin RNA knockdown was shown to impair primary prostate cancer growth and lung, but not liver, metastasis in a mouse model92. Accordingly, treatment of the VM-M3 mouse model of systemic metastatic cancer with the glutamine analogue 6-diazo-5-oxo-l-norleucine (DON) reduced metastasis to the liver, lung and kidney93. Moreover, anti-xCT vaccination inhibited arising and established lung metastasis nodules in breast cancer mouse models94. On the molecular level, xCT activity regulated cancer stem cell self-renewal and the intracellular redox balance in breast cancer cells94. These data show that glutamine and cystine metabolism can support cancer cells to settle in distant organs.
Fatty acid metabolism
Fatty acids are an important fuel in anabolic and catabolic processes. Fatty acids can be synthesized de novo or taken up from the extracellular space. Newly synthesized fatty acids are often further desaturated to monounsaturated fatty acids, whereas the generation of most polyunsaturated fatty acids requires the uptake of essential fatty acids such as linoleic acid. Fatty acids are thereby important building blocks of lipids in cell membranes, and their desaturation status and double-bond position can define physical, chemical and biological properties such as membrane fluidity and peroxidation sensitivity95,96. Additionally, fatty acids can serve as important signalling molecules25,97. Moreover, many fatty acids can be oxidized in the mitochondria to acetyl-CoA, whereas specific long-chain and branched-chain fatty acids are oxidized in the peroxisomes.
Peroxidation.
A chemical reaction between mainly unsaturated fatty acids and the reactive forms of oxygen.
Fatty acid metabolism in invading and motile cancer cells.
Multiple studies have linked obesity to cancer progression, metastasis formation and mortality in several cancer types, including prostate cancer, melanoma and breast cancer98–101. Although several mechanisms can contribute to this correlation, lipids have been functionally implicated in several steps of the metastatic cascade (FIG. 2a; TABLE 1). Indeed, a metabolomics analysis of meta static versus non-metastatic oral squamous carcinoma cell lines showed differences in lipid metabolism102. In various cancers, increased fatty acid uptake, lipid accumulation and/or overexpression of genes encoding fatty acid transporters or other fatty acid metabolism genes induced invasive and migratory traits of cancer cells compared with control conditions, elevated the seeding capacity of tumour cells in distant organs in several mouse models103–106 and was associated with metastatic progression as well as poor prognosis in patients with various cancers105.
To explore a potential mechanistic link between fatty acid availability and metastasis formation, commonalities in gene expression across fatty acid-rich and cancer-associated environments have been analysed. Thereby, CD36, a transmembrane protein that facilitates the import of fatty acids into the cell, was found to be induced in ovarian cancer cells when co-cultured with adipocytes107, in cervical cancer of mice in response to a fat-enriched compared with control diet108 and in vitro in oral squamous carcinoma cells exposed to palmitate (compared with non-palmitate) supplementation in the culture media103. Congruently, the breast-associated adipocyte secretome enabled in vitro fatty acid uptake and invasiveness in breast cancer cells via induction of CD36 expression109. Moreover, CD36 was highly expressed in several metastasis-initiating compared with non-metastatic cancer cells103. Highlighting the relevance of these observations, elevated CD36 expression levels predicted poor prognosis in patients with clear cell RCC110 as well as glioblastoma111, and accelerated gastric cancer metastasis in experimental mouse models112,113. Conversely, CD36 inhibition (with Nobiletin, sulfo-N-succinimidyl oleate or CD36–short hairpin RNA-mediated knockdown) impaired angiogenesis as well as migration and invasion of breast cancer cell lines114,115. Silencing of CD36 in preclinical mouse models of prostate cancer also reduced fatty acid uptake, as well as the abundance of oncogenic signalling lipids, and slowed cancer progression compared with control conditions116. Different mechanisms have been identified as potentially responsible for the ability of CD36 to facilitate cancer cell invasion and motility. CD36-associated fatty acid uptake also promoted an EMT in hepatocellular carcinoma cell lines106, whereas the pro-metastatic effect of CD36 in cervical cancer was synergistic with a TGFβ-induced EMT in cell lines117.
Fatty acid binding proteins (FABPs) can likewise facilitate fatty acid uptake by cells. In this respect, expression of FABP4 (also known as A-FABP) was associated with metastatic potential, cancer progression and mortality of patients with ovarian cancer, and expression of FABP5 (also known as E-FABP) was accompanied by these parameters in patients with clear cell RCC and colorectal cancer118–120 (FIG. 2b). FABP5 induced a pro-metastatic EMT phenotype in hepatocellular carcinoma cell lines with FABP5 overexpression whereas knockdown cell lines showed the opposite phenotype121. Moreover, FABP5 silencing impaired MMP expression in cervical cancer cells in vitro and in vivo122, and decreased invasion and migration of gastric cancer cell lines in vitro123. Additionally, FABP5 expression was required in conjunction with fatty acid synthase (FASN) and monoacylglycerol lipase (MAGL) to support prostate cancer progression in mouse models124. Similarly, overexpression of the liver-specific FABP1 (also known as L-FABP) promoted angiogenic properties and migration in hepatocellular carcinoma cell lines and increased the number of liver metastases125. Further, FABP1 is highly expressed in 44% of patients with melanoma, enabling melanoma cells to increase their invasive potential through uptake of adipocyte-derived lipids126.
Cancer cells can also synthesize fatty acids de novo from various nutrients. In this respect, there is some evidence that de novo fatty acid synthesis contributes to the invasion and migration capacity of cancer cells. Inhibition of FASN with the compound orlistate impaired melanoma-induced metastases and angiogenesis in a mouse model127, and silencing of FASN attenuated CD44 expression-induced signalling and metastasis formation in colorectal cancer mouse models128. Mechanistically, FASN-mediated de novo lipogenesis regulated expression of CD44 at a post-transcriptional level, possibly by altering its palmitoylation128. In an independent study, the proliferation, adhesion and migration of colorectal cancer cells in vitro was promoted by FASN-driven sphingolipid metabolism modulating focal adhesion signalling129.
Fatty acids, regardless of whether they are taken up or synthetized de novo, can be further processed in the cell. This includes monodesaturation via the isoenzymes stearoyl-CoA desaturase 1 (SCD1) and SCD5, both producing the same fatty acids, for example palmitoleate, or via the enzyme fatty acid desaturase 2 (FADS2). Although FADS2 is mainly known for its function in polyunsaturation, it was recently discovered as an alternative metabolic route of monodesaturation in cancers leading to the production of the fatty acid sapienate130,131. SCD1 and SCD5 have been already studied in the context of cancer cell invasion and migration. High levels of SCD1 expression were prognostic in patients with colorectal cancer and supported cancer cell migration in vitro through monounsaturated fatty acid production compared with SCD1-silenced cancer cells132,133. In line with this, blocking fatty acid desaturation with the SCD inhibitor CAY10566 and the FADS2 inhibitor SC-26196 impaired NF-κB signalling, resulting in decreased stemness properties of ovarian cancer cells compared with vehicle-treated controls134. By contrast, blocking SCD1 with A939572 compared with vehicle treatment triggered endoplasmatic reticulum stress in several melanoma cells and ovarian cancer cells, which then led to enhanced invasion and metastatic dissemination to the lung of mice135. Thus, SCD1 inhibition has led to controversial results, either reducing or increasing metastasis formation in melanoma (TABLE 1), which may be dependent on the presence of an endoplasmatic reticulum stress response and the Melanocyte Inducing Transcription Factor (MITF) status. Expression of SCD5 was suggested to reduce melanoma-derived metastasis by impairing the secretion of the extracellular matrix-modifying protein SPARC136. To date, it remains to be determined to what extent FADS2-produced sapienate plays a role in metastasis formation.
Beyond desaturation, other fatty acid modifications can occur that foster cancer progression. For example, there is evidence that inhibition of cholesterol esterification through targeting cholesterol acyltransferase (ACAT) suppressed the development and growth of metastatic lesions in prostate cancer137 as well as pancreatic cancer138 mouse models and reduced in vitro cell migration in Lewis lung carcinoma cells139. In prostate cancer cell lines, inhibition of cholesterol esterification blocked secretion of Wnt3a through reduction of mono unsaturated fatty acids, which limited Wnt3a acylation and, consequently, cancer cell migration137, whereas in pancreatic cancer cell lines it induced endoplasmatic reticulum stress through cholesterol accumulation138.
Fatty acids can be oxidized to generate acetyl-CoA. Major fatty acid oxidizing organelles are the mitochondria into which many long-chain fatty acids, such as palmitate, are transported depending on carnitine palmitoyltransferase 1 (CPT1). Elevated fatty acid oxidation has been observed in breast and ovarian cancer cells in co-culture with adipocytes compared with sole monocultures140,141, whereas CPT1A-silenced breast cancer cells lacked the ability to effectively drive tumour-associated lymphangiogenesis because of decreased VEGF-C and VEGF-D expression142. Further, a splice variant of CPT1A supported cancer cell survival and invasiveness by promoting histone deacetylase (HDAC) activity in the nucleus through protein–protein interaction with HDAC1 (REF.143). In addition, fatty acid oxidation can generate mitochondrial ROS, which is known to facilitate an EMT in different cancer cell lines144. Finally, the breakdown of odd-chain fatty acids, cholesterol and certain amino acids can lead to the production of methyl malonic acid (MMA). This metabolite was recently found to be elevated in the serum of old compared with young human individuals and to drive cancer progression in breast cancer mouse models due to the induction of a Sox4-mediated EMT145.
In summary, fatty acid metabolism can increase tumour cell migration and invasion by altering cellular signalling cues and regulation of epigenetic modifiers.
Fatty acid metabolism in circulating cancer cells.
Higher lipid serum concentrations have been detected in colorectal and breast cancer patients with distant metastases compared with patients without metastases146,147. There was, however, no correlation between lipid serum levels and metastases in patients with oral squamous cell carcinoma148. Thus, it appears that, at least in certain tumour types, metastasizing cancer cells can be exposed to higher lipid levels in the blood circulation (FIG. 2b,c). Given that exogenous lipids could be metabolized by circulating cancer cells, this could suggest that their metabolism can potentially support survival in the circulation. Congruently, blocking fatty acid oxidation by CPT1A silencing led to ROS accumulation in matrix-detached colorectal cancer cell lines149. The accumulation of ROS has been shown to be detrimental for matrix-detached cells in vitro150 and circulating tumour cells in vivo51,52,151. In line, fatty acid oxidation-generated acetyl-CoA supported calcium/calmodulin-dependent kinase II (CaMKII) activity in prostate cancer cells that resulted in reduced anoikis and cell migration152. Thereby, the presumed mechanism may depend on acetyl-CoA-derived CoA, generated locally by a yet to be identified acetyl-CoA hydrolysing reaction, and consecutive binding to the calmodulin (CaM)-binding domain of CaMKII to promote CaM binding and activation of CaMKII at basal calcium concentrations153. Moreover, a recent study demonstrated in mice bearing patient-derived melanomas that cancer cells metastasizing through the lymphatic system conveyed reduced levels of ROS-induced ferroptosis compared with cells metastasizing through the blood vascular system154. This was based on increased availability of oleic acid-containing vesicles in the lymph compared with blood that allowed circulating cancer cells to take up oleate, resulting in less desaturated cell membranes and reduced sensitivity to lipid peroxidation154. Interestingly, cancer cells that disseminated through the lymph and then entered the blood circulation were protected from ferroptosis compared with cancer cells directly disseminating into the blood circulation154. Thus, fatty acids may be available to and protective of certain circulating cancer cells, depending on the metastatic route.
Fatty acid metabolism during metastatic colonization.
Little is known about the availability of fatty acids in the different sites of metastasis relative to the primary tumour tissues but there is ample evidence that fatty acid uptake and metabolism can boost the nesting of metastasizing cancer cells in multiple organs (FIG. 3; TABLE 1). Whereas targeting CD36 had only minor to no effects on primary tumour growth, it dramatically impaired metastasis formation in the lungs and lymph nodes (and likely other organs) of mice bearing oral carcinomas103 (FIG. 3a,b). In line with this observation, long non-coding RNA LNMICC promoted lymph node metastasis through FABP5-mediated fatty acid metabolism in cervical cancer mouse models155, whereas FABP4 inhibition increased α-ketoglutarate concentrations and, consequently, increased DNA demethylation through regulation of ten–eleven translocase (TET) enzymes in vitro and reduced omental colonization in a mouse model of ovarian cancer156 (FIG. 3c).
In addition to fatty acid uptake, de novo fatty acid synthesis has also been implicated in the ability of cancer cells to colonize a distant organ. Accordingly, FASN overexpression resulted in increased peritoneal metastasis of ovarian cancers in mice, and promoted cellular colony formation and metastatic ability in vitro157 (FIG. 3c). CD147 knockout in hepatocellular carcinoma cells decreased fatty acid synthesis by impairing the Akt/mTOR signalling pathway and upregulated peroxisome proliferator-activated receptor-α (PPARα), resulting in increased proliferation and metastasis formation compared with control in cell lines and a mouse model158. Similarly, hyperactivation of sterol regulatory element-binding protein (SREBP), a downstream target of mTOR, by Pten and Pml double-null compared with Pten-null genetic modification of mouse prostates was capable of promoting prostate cancer metastasis by upregulating de novo fatty acid synthesis98 (FIG. 3b). In addition, the fatty acid monodesaturating enzyme SCD1 was induced in melanoma cells when they were co-cultured with lung fibroblasts, and genetically silencing SCD1 in cancer cells impaired metastasis formation and prolonged their survival of mice injected with melanoma cells159. Yet it has also been shown that an increased ratio of monounsaturated to saturated fatty acids can result in mitochondrial dysfunction and, consequently, reduced breast cancer metastasis to the lung160 (FIG. 3a). In line with this and as discussed above, elevation of saturated fatty acids, for example through SCD1 inhibition, increased melanoma-derived lung metastasis through inducing invasion in a mouse model135.
Little is known about the role of fatty acid oxidation in metastatic colonization. In a mouse model of ovarian cancer, salt-inducible kinase 2 (SIK2)-phosphorylated ACC and SIK2 overexpression compared with control promoted fatty acid oxidation required for effective metastasis formation141 (FIG. 3c). Moreover, inducible silencing of YAP in melanoma cells in mice showed that cancer cells were critically dependent on YAP-induced fatty acid oxidation to seed in the lymph node, but not in the lung161 (FIG. 3b).
In summary, fatty acid uptake, synthesis and modification fosters the colonization of the metastatic niche through multiple mechanisms.
Additional metabolic rewiring
The studies described above infer several metabolic liabilities that can be potentially targeted while cancer cells transition through the metastatic cascade (TABLE 1). Notably, there are additional metabolic vulnerabilities that have been evaluated only for a limited number of metastatic steps, and thus were not possible to include as examples for metabolite plasticity. This includes, but is not limited to, acetate, serine, alanine, proline and asparagine metabolism, which are highlighted below (FIGS 2,3; TABLE 1). We do not address the metabolic rewiring of glucose metabolism during metastasis formation, which is covered in other recent reviews22,162,163.
Acetate metabolism.
Acetate is a nutrient that can be converted to acetyl-CoA or produced from acetyl-CoA via different enzymes. Acetyl-CoA concentrations can be increased through downregulation of the enzyme acyl-CoA thioesterase 12 (ACOT12), which converts acetyl-CoA to acetate. Consequently, ACOT12 downregulation by genetic silencing and subsequent acetyl-CoA accumulation in hepatocellular carcinoma cell lines facilitated histone acetylation-induced activation of Twist2 (REF.164), a known inducer of EMT. Phenotypically, elevated acetyl-CoA concentrations led to increased invasive properties of hepatocellular carcinoma cells164, and similar observations have been made in breast cancer cells31,164 (FIG. 2a). By contrast, inhibition of the cytosolic enzyme acetyl-CoA synthase 2 (ACSS2; which converts acetate to acetyl-CoA), resulted in increased invasiveness and migration of hepatocellular carcinoma cells due to a HIF2α-dependent induction of an EMT165 (FIG. 2a). Acetate is available to tumours via the circulation and, potentially, through production by other organs such as the liver166,167. As such, through conversion to acetyl-CoA and contribution to the TCA cycle, acetate can serve as a bioenergetic fuel for breast cancer, non-small cell lung cancer, clear cell RCC, melanoma and endometrial cancer-derived brain metastases168 (FIG. 3d). Thus, further studies are needed to evaluate the role of acetate metabolism and acetyl-CoA concentrations in metastasis formation.
Serine, alanine, proline and asparagine metabolism.
Serine, alanine, proline and asparagine are non-essential amino acids that can be produced by cells but are also available to differing extents in body fluids. Serine is produced from the glycolytic intermediate 3-phosphoglycerate, alanine is made from pyruvate, and proline and asparagine are often generated from glutamine. Serine, alanine and proline, but not asparagine, can be catabolized by human cells, resulting in pyruvate production (serine, alanine) and TCA and/or urea cycle fuelling and/or glutamate/glutamine production (proline).
It has been reported that the expression levels of all serine biosynthesis enzymes were elevated in MDA-MB-231 breast cancer cells with enhanced bone metastatic abilities compared with the parental cell lines169. Expression levels of the first enzyme of the serine biosynthesis pathway, namely phosphoglycerate dehydrogenase (PHGDH), has been associated with lymph node metastasis in patients with non-small cell lung cancer or pancreatic cancer170,171 and high expression levels of PHGDH have been linked to shorter overall survival of patients with breast cancer-derived liver metastasis172. Serine conversion to glycine (indicated by high levels of serine hydroxymethyltransferase 2 (SHMT2) expression) has been prognostic in patients with breast cancer-derived lung metastasis172. In mice, PHGDH activity facilitated breast cancer-derived lung metastasis formation through maintaining mitochondrial redox homeostasis173 (FIG. 3a). Moreover, PHGDH inhibition by genetic silencing or PH-755 treatment in mice impaired breast cancer-derived brain metastases due to low availability of serine in the brain174 (FIG. 3d) and it decreased mTORC1 signalling in lung meta stases but not in primary breast cancers72 (FIG. 3a). These observations indicate a role of serine metabolism in metastasis formation.
The biosynthesis and catabolism of alanine depends on the reversible reaction catalysed by ALT1 (cytosolic) or ALT2 (mitochondrial), which convert alanine and α-ketoglutarate to pyruvate and glutamate or vice versa. In a mouse model of primary breast cancer, inhibition of alanine catabolism by silencing ALT2 impaired primary tumour growth, in particular, leading to elevated α-ketoglutarate concentrations, resulting in HIF1α degradation and subsequent reduced sonic hedgehog signalling175. Conversely, ALT2 silencing compared with control led to inhibition of alanine synthesis from pyruvate in breast cancer-derived lung metastasis, thereby reducing the metastatic burden in an experimental mouse model, based on decreased α-ketoglutarate concentrations impairing P4HA activity and, consequently, collagen hydroxylation69 (FIG. 3a). Interestingly, these data suggest that primary breast cancers rely on alanine catabolism, whereas breast cancer-derived lung metastases rely on alanine biosynthesis.
Asparagine and glutamine metabolism are intertwined because limited glutamine availability makes asparagine an essential amino acid176. Interestingly, reducing the bioavailability of asparagine to cancer cells through knockdown of asparagine synthetase (ASNS), treatment with l-asparaginase or dietary asparagine restriction reduced lung metastasis without affecting the growth of the primary breast tumour in mice177. Mechanistically, ASNS silencing impaired the number of circulating tumour cells as well as invasiveness by preventing the induction of mesenchymal gene expression patterns177 (FIG. 2a).
Proline is synthesized and catabolized by different enzymes coupled to different cofactors, namely proline dehydrogenase (PRODH) and pyrroline-5-carboxylate reductase 1 (PYCR1)178. PYCR1 is highly expressed in patients with invasive ductal breast carcinoma179 and its levels have been predictive for lymph node metastasis in patients with non-small cell lung cancer180. Moreover, PYCR1 silencing reduced invasiveness in neuroblastoma cell lines with overexpression of MZF1-AS1 in vitro181, whereas proline starvation impaired clonicity in different cancer cell lines182. Accordingly, the proline cycle consisting of catabolism via PRODH and synthesis via PYCR1 is required for breast cancer-derived lung metastasis in mice73 (FIG. 3a).
Taken together, nutrients such as lactate, pyruvate, glutamine and lipids (and likely others) appear to be crucial metabolites in many steps of the metastatic cascade — that is, their use is plastic. Although the evidence presented here supports the concept of metabolite plasticity, several questions arise. For example, it remains elusive as to how the cancer cell origin impacts the different aspects of metabolite plasticity-driven metastasis formation. The largest body of evidence for metabolite plasticity is based on a wide range of different in vitro and in vivo models across multiple cancer types. Thus, additional studies are required that follow metabolite plasticity within the same cancer model to link changes in the metabolization of a particular nutrient to phenotypic switches of the cancer cells. Moreover, it will be important to understand how concentration changes of one and the same metabolite can arise across different organelles, tissues, tumours and patients and how these contribute to the ability of cancer cells to leverage these metabolites during metastasis formation. Interestingly, some of the targets discussed above also affect primary tumours, whereas others only affect metastasis formation (BOX 1; TABLE 1). The latter supports the proposition that the metastatic phenotype and/or the site of metastasis defines the metabolic vulnerabilities of cancer cells.
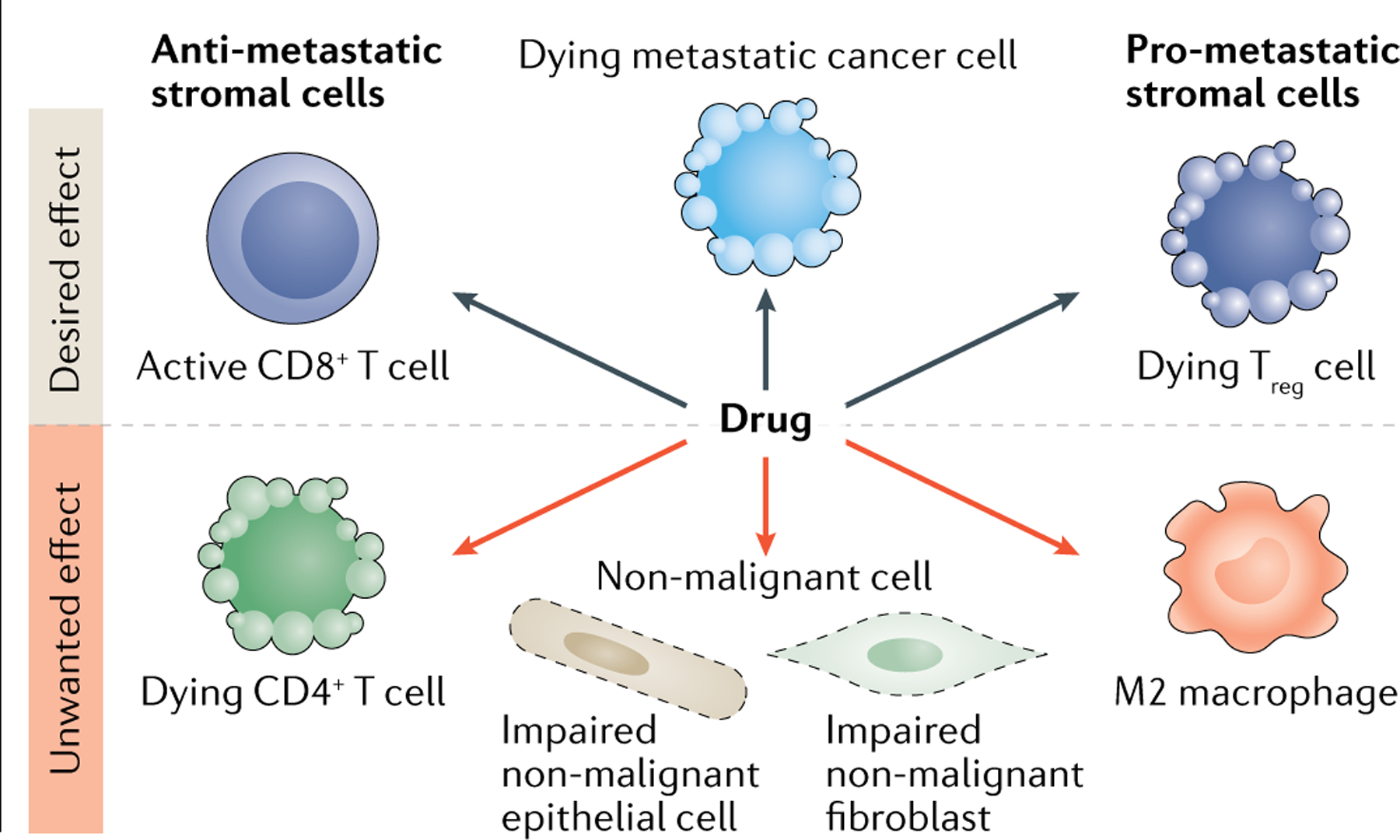
Box 1 |. Targeting metabolism in the context of the tumour microenvironment.
It is important to note that targeting metabolism in cancer in general, or metastasis formation in particular, can lead to (un)wanted systemic effects of a drug on stromal cells within the tumour microenvironment. Notwithstanding, targeting similar metabolic traits in the tumour microenvironment (for example, the tumour vasculature, immunosuppressive cells) and cancer cells can also lead to improved beneficial effects (see the figure). For example, many enzymes implicated in fatty acid uptake are active in stromal cells207. Immunosuppressive regulatory CD4 T cells (Treg cells) have been shown to take up fatty acids208. Accordingly, genetic ablation of the fatty acid transporter cD36 in Treg cells suppressed tumour growth and enhanced antitumour activity in tumour-infiltrating lymphocytes without disrupting immune homeostasis209. Moreover, tumour-associated compared with non-tumour-associated macrophages showed upregulation of cD36 expression210 and lipid accumulation in natural killer cells limited their antitumour response211,212. Thus, targeting lipid uptake through cD36 inhibition may have positive effects on stromal cells during cancer therapy. In contrast, lipid uptake activity through fatty acid binding protein 4 (FaBP4) and FaBP5 was required for the long-term survival of memory cD8+ T cells213. It is not known how FaBP4 and FaBP5 inhibition impacted memory cD8+ T cells in a tumour context, but double knockouts were less effective at protecting mice from cutaneous viral infection213, which may suggest undesired side effects in cancer treatment. This example highlights the challenge but also the potential benefit of targeting metabolic traits in metastasizing cancer cells in the context of the tumour microenvironment. Yet our current knowledge regarding stromal cells is often limited to observations in primary tumours or even non-cancerous situations, making it difficult to predict the overall effects in the metastatic setting. In addition, it remains elusive to which extent immune cell populations differ between metastatic sites. For example, distinct immune profiles have been documented between brain and extracranial metastases200, whereas lung metastases shared a common immune profile regardless of tumour origin214. Therefore, further studies are urgently needed that consider the effect of metabolism targeting drugs on metastasizing cancer and stromal cells.
Metabolite (in)flexibility
Multiple studies demonstrate that cancer cells rely on different nutrients to fuel the same metabolic requirements, a phenomenon termed metabolite flexibility, which might allow them to overcome the hurdle of a specific step in the metastatic cascade. Here, we will discuss metabolite flexibility in light of circulating and colonizing tumour cells. We decided to cover these two specific steps in the metastatic cascade because there is evidence that circulating cancer cells require certain metabolic products (that is, NADPH, glutathione) to control ROS levels, whereas cancer cells that seed and colonize the metastatic niche have an increased need for the metabolic product ATP compared with proliferating cancer cells in the primary tumour23,24,183,184. ATP and NADPH can be produced from various nutrients; however, ATP and NADPH in metastasizing cancer cells seem to be produced from only a limited range of nutrients (nutrient inflexibility).
Nutrient inflexibility.
A dependence on one nutrient despite the fact that multiple nutrients can lead to the production of a certain metabolite.
ROS defense in circulating tumour cells
Antioxidant metabolism and resistance to ROS is essential for the survival of cancer cells in the circulation23,24. Cancer cells can rely on different nutrients to avoid ROS-induced cell deaths such as ferroptosis (FIG. 4). As described above, pyruvate can act as an extracellular antioxidant60 whereas lactate-driven pentose phosphate pathway activity63 and fatty acid oxidation185 generate NADPH. NADPH is required for the recovery of the ROS scavenger GSH in melanoma and colorectal cancer, and potentially other cancers. As mentioned above, oleic acid uptake reduced the susceptibility of lymph-circulating melanoma cells to ferroptosis in blood circulation through the reduction of peroxidation-sensitive double bounds in lipids154. In addition, glutamine reductive carboxylation and glucose fermentation induced through cell detachment protected cells from anoikis by NADPH regeneration in the mitochondria and reduced mitochondrial ROS production in various cancer cell lines150,186. Moreover, glucose-fuelled folate metabolism reduced oxidative stress in circulating melanoma cells based on low-dose methotrexate treatment, ALDH1L2 knockdown or MTHFD1 knockdown inhibiting distant melanoma metastasis but not primary tumours in mouse51. These findings highlight that numerous nutrients, including pyruvate, lactate, fatty acids, glutamine and glucose, can contribute to ROS resistance in circulating tumour cells.
Fig. 4 |. Nutrient inflexibility during metastasis formation.
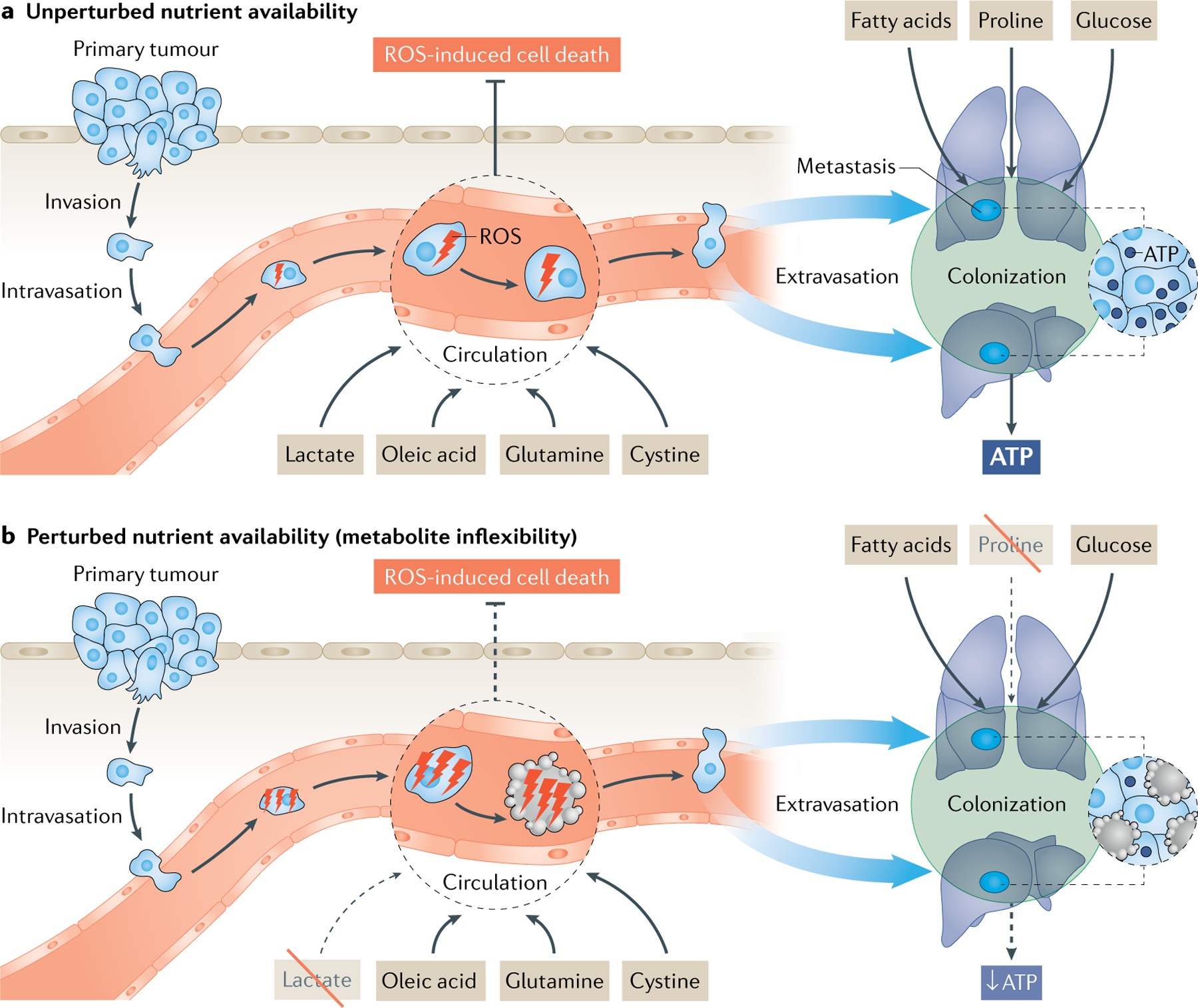
a | Cancer cells can use multiple nutrients to support the specific metabolic needs that arise when transitioning through the metastatic cascade. One example are circulating tumour cells that rely, for example, on lactate, glutamine, oleic acid and cystine to avoid reactive oxygen species (ROS)-induced cell death. Another example are colonizing cancer cells that rely, for example, on fatty acids, proline and glucose to generate sufficient ATP. b | Despite the fact that cancer cells can use multiple nutrients for this purpose, a therapeutically relevant nutrient inflexibility arises. For example, inhibition of lactate or proline uptake is sufficient to impair circulating and colonizing cancer cells, respectively by either inducing cell death or preventing cell division. Consequently, metastasis formation is reduced and may be prevented. Dashed lines in (b) indicate a decrease relative to (a).
Reductive carboxylation.
A metabolic pathway in which α-ketoglutarate is converted to citrate through a reaction with carbon dioxide.
Glucose fermentation.
A biological process in which glucose is converted to lactate.
ATP production in seeding tumour cells
Energy generation has emerged as an important metabolic output for cancer cells establishing tumours in distant organs23. The reason for increased energy requirement of cancer cells colonizing in distant organs is unknown; however, it is conceivable that this could relate to protein as well as extracellular matrix production and trafficking, which are both highly energy-demanding processes187 and required to form a permissive metastatic niche. Cancer cells can rely on different nutrients to increase their ATP availability during metastatic colonization (FIG. 4). Fatty acids have been identified as important nutrients in this respect. In breast cancer, metastatic triple-negative breast cancer cells compared with benign cell lines required fatty acid-driven energy metabolism for the formation of distant metastasis based on the inhibition of fatty acid oxidation with etomoxir or the perturbance of fatty acid metabolism through inhibition of CUB-domain containing protein 1 (CDCP1) activity with an engineered blocking fragment in experimental mouse models188,189. The rewiring of energy metabolism yielded more ATP that was needed to activate Src through autophosphorylation188. Moreover, both proline catabolism and glucose metabolism-derived ATP contributed to energy production during metastatic colonization of breast cancer cells in the lung of mice68,190. Interestingly, breast cancer-derived lung metastases used glucose to fuel glycolysis and mitochondrial metabolism to account for their energy needs190, whereas breast cancer-derived liver metastases only relied on glycolytic energy production191. In mouse models of colon cancer, colon cancer-derived liver metastases scavenged ATP from the extracellular space192. In particular, colon cancer cells released creatine kinase brain-type into the liver extracellular space, which converted creatine into phosphocreatine in an ATP-driven reaction192,193. Phosphocreatine was then taken up by the colonizing colon cancer cells and used for intracellular phosphorylation of ADP to ATP. In summary, there is evidence that cancer cells, in principle, can rely on several metabolites (for example, fatty acids, proline, glucose) to accommodate their energy needs when they colonize distant organs.
The fact that cancer cells rely on multiple metabolites and nutrients to produce NADPH, GSH and ATP is not surprising, especially as these metabolic products are substrates and products of multiple reactions across the entire metabolic network. It is, however, surprising that targeting only one of the nutrients producing these molecules is sufficient to exhibit therapeutic efficacy in mouse models; for example, leading to reduction of the number of circulating tumour cells or reduced metastasis formation (FIG. 4). Thus, metastasizing cancer cells entail a certain metabolic rigidity, that is, nutrient inflexibility that seems to depend on the organ of metastasis, cell state, cell of origin, microenvironment or (epi) genetic landscape, which opens a therapeutic window to target these metabolic vulnerabilities. Accordingly, proline catabolism inhibition through blocking PRODH or inhibition of lactate uptake through MCT1 did not impair primary tumour growth, whereas it had dramatic effects on metastasis formation due to the different cell state and/or microenvironment during metastasis63,73.
Consequently, interesting questions arise such as which patients would benefit most from such treatments; and whether an anti-metabolic therapy would be more effective and eventually less toxic than a standard chemopreventative strategy in counteracting the metastatic seeding, or would also be considered for patients who already have developed metastatic disease. If organ-specific metastases may indeed demonstrate specific metabolic vulnerabilities that are targetable, anti-metabolic therapy of patients with established metastases might become an achievable goal. Considering the possibility that some of the vulnerabilities of metastasizing cancer cells, such as increased energy needs during seeding or sensitivity of circulating tumour cells to oxidative stress, may be lost in established metastases further argues for distinct therapeutic windows in which metabolic targeting therapies would be most effective. Clinical trials in the neoadjuvant setting may thus be a promising strategy to evaluate metabolic therapeutics given that tumour spread has become an accepted clinical trial end point194.
Metabolic evolution of metastases
Once cancer cells have successfully metastasized to a distant organ, the secondary tumour shows a similar behaviour (that is, phenotype) to the primary tumour — both grow, proliferate and can reseed2,9. One could then argue that primary tumours and established metastases are metabolically similar, or at least depend on the same metabolic pathways or enzymes. Some examples discussed above are in line with this proposition because, in some cases, disrupting metabolic activity leads to effects in cancer cells regardless of whether they grow as metastases or primary tumours (TABLE 1). However, there is an increasing body of evidence that suggests a metabolic evolution from primary tumours to metastases in different organs. Accordingly, some metabolic vulnerabilities discussed above only target metastases (sometimes only in specific organs) but not primary tumours (TABLE 1). Here, we summarize the current evidence suggesting that primary cancers and their metastatic progenies differ in their metabolic profiles.
Over the last decade, expression analysis of a limited number of metabolic genes has revealed distinct expression profiles between primary and secondary lesions. For example, a study on patients with pancreatic adenocarcinoma and corresponding metastatic lesions demonstrated common but also highly distinct metabolic gene expression profiles between primary tumours and metastases195. A recent single-cell RNA-sequencing study of metastatic patient-derived xenograft tumours confirmed a clear intra-tumour and inter-tumour heterogeneity in metabolic expression profiles in distinct cancer cell populations in primary breast tumours, and lung and lymph node micrometastases196. Additionally, single-cell transcriptional profiling of tumour tissue samples from six patients with triple-negative breast cancer showed subclonal heterogeneity of malignant cells shared by various tumours with multiple signatures of treatment resistance and metastasis as characterized by elevation of glycosphingolipid metabolism and associated innate immunity pathways197. Moreover, lung micrometastases from breast cancer patient-derived xenograft mouse models displayed a distinct metabolic gene expression signature of mitochondrial oxidative metabolism compared with the corresponding primary breast cancers196. These results support the notion that metastases can be metabolically different compared with their corresponding primary tumours. Notably, whereas the evidence presented here and below is mainly based on transcriptional changes, post-translational modifications and metabolite concentration-driven metabolic rewiring in primary tumours versus metastasis is likely to occur, yet is less demonstrated due to technological limitations.
Another layer of complexity is observed by inter-metastatic metabolic heterogeneity in relation to their metastatic site. Data from Flura-seq (fluorouracil-labelled RNA sequencing), a technique that determines which genes are active in small clusters of cells in a tissue, showed that lung micrometastases had differential transcriptional activity compared with brain micrometastases and primary mammary tumours in mice198. This may suggest that metabolic requirements exist in the lung microenvironment that are distinct from brain or breast tissue. Accordingly, mitochondrial electron transport chain genes were higher expressed in lung metastases compared with both brain metastases and orthotopic mammary tumours198. In addition, a proteomics study on patient-derived breast cancer cells observed a unique metabolic protein profile of brain metastasis compared with bone metastasis in mice, which suggests either a selection of predisposed cells or bioenergetic adaptation of the tumour cells to the brain environment199. In line with this, melanoma-derived brain metastases displayed an enrichment for oxidative phosphorylation-associated gene expression patterns in comparison with patient-matched extracranial metastases200.
Although these observations reiterate the existence of differential metabolic traits between primary tumours and metastases, and across metastases, the need for these metabolic alterations are less understood. At least two fundamental principles could lay the foundation for these alterations (FIG. 5). Either the (epi)genetically or metabolically heterogeneous tumour cell pool in a primary tumour provides a selection for a distinct cancer cell subpopulation that is optimally suited to flourish in a specific organ environment, or several cancer cell subpopulations may be able to adapt to a certain organ environment. It is conceivable that both selective and adaptive processes occur, which may again depend on the tumour origin and metastatic site.
Fig. 5 |. Selection and adaptation processes contributing to the metabolic differences between primary tumours and metastases.
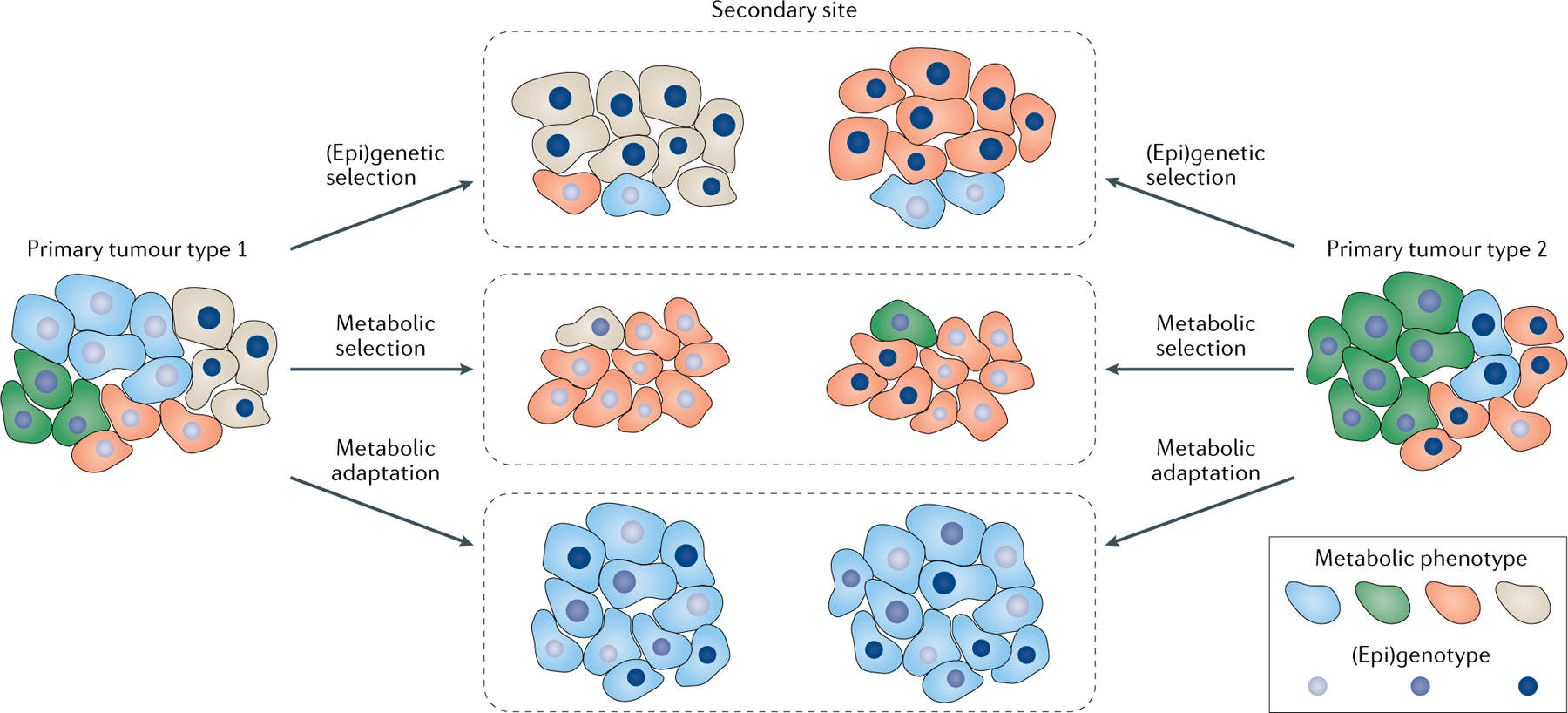
Cancer cells growing as a secondary tumour are challenged with a different environment. Either (epi) genetic or metabolic subpopulations of cancer cells from the primary tumour have a selective growth advantage in the new soil, or cancer cells may have the ability to adapt to the new soil. In the case of adaptation, the metabolic make-up of secondary tumours depends on the organ of metastasis rather than the cancer cell origin. In the case of (epi)genetic selection, a subpopulation of cancer cells selected by their (epi)genetic features grows in the secondary site, resulting in a different metabolism depending on the cancer origin. In the case of metabolic selection, a subpopulation of cancer cells selected by their metabolic features grows in the secondary site, resulting in the same metabolism regardless of the cancer origin. It is conceivable that likely a combination of these processes may occur in vivo.
Regardless of whether selection or adaptation is the determinant factor for successful growth of secondary tumours, emerging evidence shows that the (metabolic) environment matters. An interesting experiment compared the influence of in vitro and in vivo environments. Thereby, brain and lung metastases as well as mammary tumours from mice were directly analysed after harvesting the tissue198. In parallel, an aliquot of these tissue samples was dissociated into single cells, cultured for 1–2 weeks and subjected to RNA sequencing. Strikingly, several thousand genes were distinctly expressed across the different tissues whereas the same cells showed differential expression of only a few hundred genes when cultured in vitro198. Moreover, in vitro culture of lung cancer cells versus in vivo growth of the same tumour cells resulted in a different metabolic phenotype201. Moreover, microenvironmental differences in per-fusion also correlated with intra-tumour metabolic differences in patients with lung cancer202. Such experiments demonstrate the environmental dependency of metabolism.
In addition, there is also circumstantial evidence that the nutrient availability in the environment is important. For example, breast cancer-derived brain metastases, similar to glioblastoma, can use acetate for propagation168,203 (FIG. 3d). This observation was based on 13C tracer infusions in humans and mice, which assess in vivo nutrient contribution to metabolism204,205. Moreover, there is evidence that secreted factors from primary breast tumours increased the glucose availability in the metastatic niche, which elevated the effectiveness of metastatic seeding in the lungs of mice206 (FIG. 3a).
Thus, nutrient availability in the metastatic niche is important and can be altered to increase the permissiveness of the niche. Data from 13C-glucose infusions in mice harbouring metastatic primary breast cancer demonstrated an increase in PC-dependent anaplerosis in lung metastases compared with primary tumours, which was recapitulated in vitro by adding pyruvate to the media68. Additionally, the dependence of breast cancer-derived brain metastases on PHGDH activity can be explained through the very low serine availability in the brain environment174 (FIG. 3d). Finally, there is also some evidence that the microenvironment may prepare cancer cells en route metabolically for another environment because melanoma cells that traversed through the oleate-enriched environment of the lymph system before entering the blood circulation were better prepared to avoid cell death, and consequently more successful in seeding in distant organs of mice154 (FIG. 2b).
The studies described above support the notion that primary tumours and their metastases differ in their metabolic attributes because metastases adapt or have growth advantages for specific environments (FIG. 5). Although limited evidence exists, metabolic priming of the metastatic niche206 and by a certain environment154 may also exist. Whereas the here presented evidence suggests a role of the nutrient environment, additional factors of the microenvironment such as the presence of stromal cells may also contribute to environment-dependent metabolic requiring. Thus, it is tempting to speculate that metastases of differing origin growing in the same organ or taking the same route of metastasis may metabolically be more similar to each other than metastases of the same origin growing in different organs.
It is important to note that the environment is certainly not the only determinant of metabolic rewiring and that other factors such as the cancer cell origin, that is the cell lineage from which the cancer cell arises, may override the influence of nutrient availability (as suggested for PCG1α in breast versus prostate cancer23). Moreover, some metastatic niches may be more permissive for cancer cells with different genetically embedded metabolic programmes to successfully proliferate. Finally, some metabolic traits such as the reliance on CD36 activity in metastasis-initiating cells seem to be essential across multiple cancers and metastatic sites103, making them very attractive targets for cancer therapy.
Further studies that dissect the metabolic dependencies of metastases will be instrumental in determining and mechanistically understanding when the influence of the microenvironment outweighs the cellular origin, cell state (that is, phenotype) and (epi)genetic landscape in imposing metabolic constrains on metastasis growing in different organs.
Conclusion
Emerging evidence depicts specific metabolic vulnerabilities in cancer cells during their metastatic journey that could be exploited to potentially halt metastatic growth and even prevent successful seeding. These vulnerabilities are not just manifested intrinsically in a tumour type-specific manner but, rather, dynamically dependent on the stage of and location during their metastatic journey. Importantly, the success of metastatic manifestation may be influenced by the nutrient composition of the respective organ to which metastatic tumour cells need to adapt. These observations fit well with the ‘seed and soil’ hypothesis in which tumour cells require an appropriate soil with the necessary metabolites and nutrients in order to expand successfully. Nonetheless, the complexity of metabolic traits and their necessities in maintenance and propagation, not only of tumour cells but of every normal cell, makes therapeutic targeting of metabolic pathways challenging and requires a more detailed understanding of the effects of metabolite inhibition in the context of the heterogeneous cell types in metastases and the organ in which they seed.
Acknowledgements
The authors regret the inability to cite all studies that have shaped the understanding of cancer metastasis metabolism. S.-M.F. acknowledges funding from the European Research Council under the ERC Consolidator Grant Agreement n. 771486 — MetaRegulation, FWO — Odysseus II, FWO research projects (G098120N, G088318N), KU Leuven — Methusalem Co-Funding and Fonds Baillet Latour. G.B. acknowledges funding from the Flemish cancer society Stichting tegen Kanker (STK 1303), the Flemish government FWO (G0A0818N) and the National Institutes of Health (NIH)/National Cancer Institute (NCI) (R01CA201537).
Competing interests
S.-M.F. has received funding from Bayer, Merck and BlackBelt Therapeutics and has consulted for Fund+. G.B. declares no competing interests.
References
- 1.Fares J, Fares MY, Khachfe HH, Salhab HA & Fares Y Molecular principles of metastasis: a hallmark of cancer revisited. Sig. Transduct Target Ther 5, 28 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lambert AW, Pattabiraman DR & Weinberg RA Emerging biological principles of metastasis. Cell 168, 670–691 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vanharanta S & Massagué J Origins of metastatic traits. Cancer Cell 24, 410–421 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Steeg PS Targeting metastasis. Nat. Rev. Cancer 16, 201–218 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Turajlic S & Swanton C Metastasis as an evolutionary process. Science 352, 169–175 (2016). [DOI] [PubMed] [Google Scholar]
- 6.Plaks V, Kong N & Werb Z The cancer stem cell niche: how essential is the niche in regulating stemness of tumor cells? Cell Stem Cell 16, 225–238 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gonzalez H, Hagerling C & Werb Z Roles of the immune system in cancer: from tumor initiation to metastatic progression. Genes Dev. 32, 1267–1284 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fidler IJ & Kripke ML Metastasis results from preexisting variant cells within a malignant tumor. Science 197, 893 (1977). [DOI] [PubMed] [Google Scholar]
- 9.Valastyan S & Weinberg RA Tumor metastasis: molecular insights and evolving paradigms. Cell 147, 275–292 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ewing J Neoplastic Diseases: A Treatise on Tumors (Saunders WB, 1928). [Google Scholar]
- 11.Paget S The distribution of secondary growths in cancer of the breast. Lancet 133, 571–573 (1889). [PubMed] [Google Scholar]
- 12.Nguyen DX, Bos PD & Massagué J Metastasis: from dissemination to organ-specific colonization. Nat. Rev. Cancer 9, 274–284 (2009). [DOI] [PubMed] [Google Scholar]
- 13.Pavlovic M et al. Enhanced MAF oncogene expression and breast cancer bone metastasis. J. Natl Cancer Inst 107, djv256 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ganesh K et al. L1CAM defines the regenerative origin of metastasis-initiating cells in colorectal cancer. Nat. Cancer 1, 28–45 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gao Y et al. Metastasis organotropism: redefining the congenial soil. Dev. Cell 49, 375–391 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tian X et al. Organ-specific metastases obtained by culturing colorectal cancer cells on tissue-specific decellularized scaffolds. Nat. Biomed. Eng 2, 443–452 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Peinado H et al. Pre-metastatic niches: organ-specific homes for metastases. Nat. Rev. Cancer 17, 302–317 (2017). [DOI] [PubMed] [Google Scholar]
- 18.Doglioni G, Parik S & Fendt S-M Interactions in the (pre)metastatic niche support metastasis formation. Front. Oncol 9, 219–219 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaplan RN et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature 438, 820–827 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hoshino A et al. Tumour exosome integrins determine organotropic metastasis. Nature 527, 329–335 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Olmeda D et al. Whole-body imaging of lymphovascular niches identifies pre-metastatic roles of midkine. Nature 546, 676–680 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Teoh Shao T & Lunt Sophia Y Metabolism in cancer metastasis: bioenergetics, biosynthesis, and beyond. Wiley Interdiscip. Rev. Syst. Biol. Med 10, e1406 (2018). [DOI] [PubMed] [Google Scholar]
- 23.Elia I, Doglioni G & Fendt S-M Metabolic hallmarks of metastasis formation. Trends Cell Biol. 28, 673–687 (2018). [DOI] [PubMed] [Google Scholar]
- 24.Faubert B, Solmonson A & DeBerardinis RJ Metabolic reprogramming and cancer progression. Science 368, eaaw5473 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lorendeau D, Christen S, Rinaldi G & Fendt S-M Metabolic control of signaling pathways and metabolic auto-regulation. Biol. Cell 107, 251–272 (2015). [DOI] [PubMed] [Google Scholar]
- 26.Kim J & DeBerardinis RJ Mechanisms and implications of metabolic heterogeneity in cancer. Cell Metab. 30, 434–446 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kelley DE & Mandarino LJ Fuel selection in human skeletal muscle in insulin resistance: a reexamination. Diabetes 49, 677–683 (2000). [DOI] [PubMed] [Google Scholar]
- 28.Randle PJ, Garland PB, Hales CN & Newsholme EA The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet 1, 785–789 (1963). [DOI] [PubMed] [Google Scholar]
- 29.Phannasil P et al. Mass spectrometry analysis shows the biosynthetic pathways supported by pyruvate carboxylase in highly invasive breast cancer cells. Biochim. Biophys. Acta Mol. Basis Dis 1863, 537–551 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Phannasil P et al. Pyruvate carboxylase is up-regulated in breast cancer and essential to support growth and invasion of MDA-MB-231 cells. PLoS ONE 10, e0129848 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rios Garcia M et al. Acetyl-CoA carboxylase 1-dependent protein acetylation controls breast cancer metastasis and recurrence. Cell Metab. 26, 842–855.e5 (2017). [DOI] [PubMed] [Google Scholar]
- 32.Gaude E & Frezza C Tissue-specific and convergent metabolic transformation of cancer correlates with metastatic potential and patient survival. Nat. Commun 7, 13041 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chuang C-H et al. Altered mitochondria functionality defines a metastatic cell state in lung cancer and creates an exploitable vulnerability. Cancer Res. 10.1158/0008-5472.CAN-20-1865 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xian Z-Y et al. Inhibition of LDHA suppresses tumor progression in prostate cancer. Tumor Biol. 36, 8093–8100 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.He T-L et al. The c-Myc–LDHA axis positively regulates aerobic glycolysis and promotes tumor progression in pancreatic cancer. Med. Oncol 32, 187 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhao J et al. LDHA promotes tumor metastasis by facilitating epithelial–mesenchymal transition in renal cell carcinoma. Mol. Med. Rep 16, 8335–8344 (2017). [DOI] [PubMed] [Google Scholar]
- 37.Li L et al. miR-30a-5p suppresses breast tumor growth and metastasis through inhibition of LDHA-mediated Warburg effect. Cancer Lett. 400, 89–98 (2017). [DOI] [PubMed] [Google Scholar]
- 38.He Y et al. LDHA is a direct target of miR-30d-5p and contributes to aggressive progression of gallbladder carcinoma. Mol. Carcinog 57, 772–783 (2018). [DOI] [PubMed] [Google Scholar]
- 39.Jin L et al. Phosphorylation-mediated activation of LDHA promotes cancer cell invasion and tumour metastasis. Oncogene 36, 3797 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pérez-Escuredo J et al. Monocarboxylate transporters in the brain and in cancer. Biochim. Biophys. Acta 1863, 2481–2497 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cao Y-W et al. Monocarboxylate transporters MCT1 and MCT4 are independent prognostic biomarkers for the survival of patients with clear cell renal cell carcinoma and those receiving therapy targeting angiogenesis. Urol. Oncol 36, 311.e315–311.e25 (2018). [DOI] [PubMed] [Google Scholar]
- 42.Zhang G et al. MCT1 regulates aggressive and metabolic phenotypes in bladder cancer. J. Cancer 9, 2492–2501 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.De Saedeleer CJ et al. Glucose deprivation increases monocarboxylate transporter 1 (MCT1) expression and MCT1-dependent tumor cell migration. Oncogene 33, 4060 (2013). [DOI] [PubMed] [Google Scholar]
- 44.Fan Q et al. Autophagy promotes metastasis and glycolysis by upregulating MCT1 expression and Wnt/β-catenin signaling pathway activation in hepatocellular carcinoma cells. J. Exp. Clin. Cancer Res 37, 9 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xiong L, Edwards CK 3rd & Zhou L The biological function and clinical utilization of CD147 in human diseases: a review of the current scientific literature. Int. J. Mol. Sci 15, 17411–17441 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Payen VL et al. Monocarboxylate transporter MCT1 promotes tumor metastasis independently of its activity as a lactate transporter. Cancer Res. 77, 5591 (2017). [DOI] [PubMed] [Google Scholar]
- 47.Zhao Z et al. Downregulation of MCT1 inhibits tumor growth, metastasis and enhances chemotherapeutic efficacy in osteosarcoma through regulation of the NF-κB pathway. Cancer Lett. 342, 150–158 (2014). [DOI] [PubMed] [Google Scholar]
- 48.Gupta SC, Singh R, Pochampally R, Watabe K & Mo Y-Y Acidosis promotes invasiveness of breast cancer cells through ROS–AKT–NF-κB pathway. Oncotarget 5, 12070–12082 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bonuccelli G et al. Ketones and lactate “fuel” tumor growth and metastasis. Cell Cycle 9, 3506–3514 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang D et al. Metabolic regulation of gene expression by histone lactylation. Nature 574, 575–580 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Piskounova E et al. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 12, 186–191 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; This article provides evidence that antioxidants increase the survival of melanoma cells in the circulation.
- 52.Le Gal K et al. Antioxidants can increase melanoma metastasis in mice. Sci. Transl Med 7, 308re308 (2015). [DOI] [PubMed] [Google Scholar]; This article provides evidence that antioxidants and the glutathione synthesis can increase lymph node metastasis formation of melanoma.
- 53.Wiel C et al. BACH1 stabilization by antioxidants stimulates lung cancer metastasis. Cell 178, 330–345.e22 (2019). [DOI] [PubMed] [Google Scholar]
- 54.Guadamillas MC, Cerezo A & del Pozo MA Overcoming anoikis — pathways to anchorage-independent growth in cancer. J. Cell Sci 124, 3189 (2011). [DOI] [PubMed] [Google Scholar]
- 55.Grossmann J Molecular mechanisms of “detachment-induced apoptosis — anoikis”. Apoptosis 7, 247–260 (2002). [DOI] [PubMed] [Google Scholar]
- 56.Labuschagne CF, Cheung EC, Blagih J, Domart M-C & Vousden KH Cell clustering promotes a metabolic switch that supports metastatic colonization. Cell Metab. 30, 720–734.e725 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; This work reveals hypoxia-induced mitophagy of clustering tumour cells in the circulation that limits mitochondrial ROS production, and promotes survival and metastasis formation.
- 57.Caneba CA, Bellance N, Yang L, Pabst L & Nagrath D Pyruvate uptake is increased in highly invasive ovarian cancer cells under anoikis conditions for anaplerosis, mitochondrial function, and migration. Am. J. Physiol. Endocrinol. Metab 303, E1036–E1052 (2012). [DOI] [PubMed] [Google Scholar]
- 58.Maneche HC Blood pyruvate in malignant neoplastic disorders. Clin. Chem 12, 158–164 (1966). [PubMed] [Google Scholar]
- 59.Jobard E et al. A serum nuclear magnetic resonance-based metabolomic signature of advanced metastatic human breast cancer. Cancer Lett. 343, 33–41 (2014). [DOI] [PubMed] [Google Scholar]
- 60.Donnell-Tormey J, Nathan CF, Lanks K, DeBoer CJ & de la Harpe J Secretion of pyruvate. An antioxidant defense of mammalian cells. J. Exp. Med 165, 500 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wei Y et al. Prognostic significance of serum lactic acid, lactate dehydrogenase, and albumin levels in patients with metastatic colorectal cancer. BioMed. Res. Int 2018, 1804086 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wilmanski T et al. Inhibition of pyruvate carboxylase by 1α,25-dihydroxyvitamin D promotes oxidative stress in early breast cancer progression. Cancer Lett. 411, 171–181 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tasdogan A et al. Metabolic heterogeneity confers differences in melanoma metastatic potential. Nature 577, 115–120 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study shows that MCT1-dependent lactate uptake increases melanoma metastasis by elevating the survival of cancer cells in the circulation.
- 64.Vande Voorde J et al. Improving the metabolic fidelity of cancer models with a physiological cell culture medium. Sci. Adv 5, eaau7314 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zera K & Zastre J Stabilization of the hypoxia-inducible transcription factor-1α (HIF-1α) in thiamine deficiency is mediated by pyruvate accumulation. Toxicol. Appl. Pharmacol 355, 180–188 (2018). [DOI] [PubMed] [Google Scholar]
- 66.Saedeleer CJ et al. Lactate activates HIF-1 in oxidative but not in Warburg-phenotype human tumor cells. PloS ONE 7, e46571 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lee DC et al. A lactate-induced response to hypoxia. Cell 161, 595–609 (2015). [DOI] [PubMed] [Google Scholar]
- 68.Christen S et al. Breast cancer-derived lung metastasis show increased pyruvate carboxylase-dependent anaplerosis. Cell Rep. 17, 837–848 (2016). [DOI] [PubMed] [Google Scholar]
- 69.Elia I et al. Breast cancer cells rely on environmental pyruvate to shape the metastatic niche. Nature 568, 117–121 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; This article demonstrates that breast cancer cells are dependent on pyruvate uptake to initiate collagen-based extracellular matrix remodelling for the establishment of a metastatic niche and subsequent metastatic growth in the lung.
- 70.Gilkes DM et al. Collagen prolyl hydroxylases are essential for breast cancer metastasis. Cancer Res. 73, 3285–3296 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 71.Shinde A, Wilmanski T, Chen H, Teegarden D & Wendt MK Pyruvate carboxylase supports the pulmonary tropism of metastatic breast cancer. Breast Cancer Res. 20, 76–76 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rinaldi G et al. In vivo evidence for serine biosynthesis-defined sensitivity of lung metastasis, but not of primary breast tumors, to mTORC1 inhibition. Mol. Cell 10.1016/j.molcel.2020.11.027 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Elia I et al. Proline metabolism supports metastasis formation and could be inhibited to selectively target metastasizing cancer cells. Nat. Commun 8, 15267 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Diers AR, Broniowska KA, Chang CF & Hogg N Pyruvate fuels mitochondrial respiration and proliferation of breast cancer cells: effect of monocarboxylate transporter inhibition. Biochem. J 444, 561–571 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Corbet C et al. Interruption of lactate uptake by inhibiting mitochondrial pyruvate transport unravels direct antitumor and radiosensitizing effects. Nat. Commun 9, 1208 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Faubert B et al. Lactate metabolism in human lung tumors. Cell 171, 358–371.e9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Elia I, Schmieder R, Christen S & Fendt S-M Organ-specific cancer metabolism and its potential for therapy. Handb. Exp. Pharmacol 233, 321–353 (2016). [DOI] [PubMed] [Google Scholar]
- 78.Yang L et al. Metabolic shifts toward glutamine regulate tumor growth, invasion and bioenergetics in ovarian cancer. Mol. Syst. Biol 10, 728 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rodrigues MF et al. Enhanced OXPHOS, glutaminolysis and β-oxidation constitute the metastatic phenotype of melanoma cells. Biochem. J 473, 703 (2016). [DOI] [PubMed] [Google Scholar]
- 80.Xiang L et al. Glutaminase 1 expression in colorectal cancer cells is induced by hypoxia and required for tumor growth, invasion, and metastatic colonization. Cell Death Dis. 10, 40 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Du F et al. SOX12 promotes colorectal cancer cell proliferation and metastasis by regulating asparagine synthesis. Cell Death Dis. 10, 239 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhang C et al. Glutaminase 2 is a novel negative regulator of small GTPase Rac1 and mediates p53 function in suppressing metastasis. eLife 5, e10727–e10727 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kuo T-C et al. Glutaminase 2 stabilizes Dicer to repress Snail and metastasis in hepatocellular carcinoma cells. Cancer Lett. 383, 282–294 (2016). [DOI] [PubMed] [Google Scholar]
- 84.Sugano KMK, Ohtani H, Nagahara H, Shibutani M & Hirakawa K Expression of xCT as a predictor of disease recurrence in patients with colorectal cancer. Anticancer Res. 35, 677–682 (2015). [PubMed] [Google Scholar]
- 85.Dornier E et al. Glutaminolysis drives membrane trafficking to promote invasiveness of breast cancer cells. Nat. Commun 8, 2255 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yae T et al. Alternative splicing of CD44 mRNA by ESRP1 enhances lung colonization of metastatic cancer cell. Nat. Commun 3, 883 (2012). [DOI] [PubMed] [Google Scholar]
- 87.Chen RS et al. Disruption of xCT inhibits cancer cell metastasis via the caveolin-1/β-catenin pathway. Oncogene 28, 599 (2008). [DOI] [PubMed] [Google Scholar]
- 88.Jin H et al. Serum metabolomic signatures of lymph node metastasis of esophageal squamous cell carcinoma. J. Proteome Res 13, 4091–4103 (2014). [DOI] [PubMed] [Google Scholar]
- 89.Toyoshima K et al. Analysis of circulating tumor cells derived from advanced gastric cancer. Int. J. Cancer 137, 991–998 (2015). [DOI] [PubMed] [Google Scholar]
- 90.Liu G et al. Glutamate dehydrogenase is a novel prognostic marker and predicts metastases in colorectal cancer patients. J. Transl Med 13, 144 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jin L et al. The PLAG1–GDH1 axis promotes anoikis resistance and tumor metastasis through CamKK2–AMPK signaling in LKB1-deficient lung cancer. Mol. Cell 69, 87–99.e7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wang Q et al. Targeting ASCT2-mediated glutamine uptake blocks prostate cancer growth and tumour development. J. Pathol 236, 278–289 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Shelton LM, Huysentruyt LC & Seyfried TN Glutamine targeting inhibits systemic metastasis in the VM-M3 murine tumor model. Int. J. Cancer 127, 2478–2485 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lanzardo S et al. Immunotargeting of antigen xCT attenuates stem-like cell behavior and metastatic progression in breast cancer. Cancer Res. 76, 62 (2016). [DOI] [PubMed] [Google Scholar]
- 95.Gaschler MM & Stockwell BR Lipid peroxidation in cell death. Biochem. Biophys. Res. Commun 482, 419–425 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Peck B & Schulze A Lipid desaturation — the next step in targeting lipogenesis in cancer? FEBS J. 283, 2767–2778 (2016). [DOI] [PubMed] [Google Scholar]
- 97.Rohrig F & Schulze A The multifaceted roles of fatty acid synthesis in cancer. Nat. Rev. Cancer 16, 732–749 (2016). [DOI] [PubMed] [Google Scholar]
- 98.Chen M et al. An aberrant SREBP-dependent lipogenic program promotes metastatic prostate cancer. Nat. Genet 50, 206–218 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Pandey V, Vijayakumar MV, Ajay AK, Malvi P & Bhat MK Diet-induced obesity increases melanoma progression: involvement of Cav-1 and FASN. Int. J. Cancer 130, 497–508 (2012). [DOI] [PubMed] [Google Scholar]
- 100.Jiralerspong S & Goodwin PJ Obesity and breast cancer prognosis: evidence, challenges, and opportunities. J. Clin. Oncol 34, 4203–4216 (2016). [DOI] [PubMed] [Google Scholar]
- 101.O’Flanagan CH et al. Metabolic reprogramming underlies metastatic potential in an obesity-responsive murine model of metastatic triple negative breast cancer. NPJ Breast Cancer 3, 26 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sant’Anna-Silva ACB et al. Metabolic profile of oral squamous carcinoma cell lines relies on a higher demand of lipid metabolism in metastatic cells. Front. Oncol 8, 13 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Pascual G et al. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature 541, 41–45 (2017). [DOI] [PubMed] [Google Scholar]; This article identifies CD36+ metastasis-initiating cells in human oral carcinoma and other human cancer types and shows that blocking CD36 inhibits metastasis formation.
- 104.Antalis CJ, Uchida A, Buhman KK & Siddiqui RA Migration of MDA-MB-231 breast cancer cells depends on the availability of exogenous lipids and cholesterol esterification. Clin. Exp. Metastasis 28, 733–741 (2011). [DOI] [PubMed] [Google Scholar]
- 105.Nath A & Chan C Genetic alterations in fatty acid transport and metabolism genes are associated with metastatic progression and poor prognosis of human cancers. Sci. Rep 6, 18669 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Nath A, Li I, Roberts LR & Chan C Elevated free fatty acid uptake via CD36 promotes epithelial–mesenchymal transition in hepatocellular carcinoma. Sci. Rep 5, 14752 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ladanyi A et al. Adipocyte-induced CD36 expression drives ovarian cancer progression and metastasis. Oncogene 37, 2285–2301 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Yang P et al. Dietary oleic acid-induced CD36 promotes cervical cancer cell growth and metastasis via up-regulation Src/ERK pathway. Cancer Lett. 438, 76–85 (2018). [DOI] [PubMed] [Google Scholar]
- 109.Zaoui M et al. Breast-associated adipocytes secretome induce fatty acid uptake and invasiveness in breast cancer cells via CD36 independently of body mass index, menopausal status and mammary density. Cancers 11, 2012 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Xu WH et al. Elevated CD36 expression correlates with increased visceral adipose tissue and predicts poor prognosis in ccRCC patients. J. Cancer 10, 4522–4531 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hale JS et al. Cancer stem cell-specific scavenger receptor CD36 drives glioblastoma progression. Stem Cell 32, 1746–1758 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wang R et al. Fatty-acid receptor CD36 functions as a hydrogen sulfide-targeted receptor with its Cys333–Cys272 disulfide bond serving as a specific molecular switch to accelerate gastric cancer metastasis. EBioMedicine 45, 108–123 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Pan J et al. CD36 mediates palmitate acid-induced metastasis of gastric cancer via AKT/GSK-3β/β-catenin pathway. J. Exp. Clin. Cancer Res 38, 52–52 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Sp N et al. Nobiletin inhibits CD36-dependent tumor angiogenesis, migration, invasion, and sphere formation through the CD36/STAT3/NF-κB signaling axis. Nutrients 10, 772 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Casciano JC et al. MYC regulates fatty acid metabolism through a multigenic program in claudin-low triple negative breast cancer. Br. J. Cancer 122, 868–884 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Watt MJ et al. Suppressing fatty acid uptake has therapeutic effects in preclinical models of prostate cancer. Sci. Transl Med 11, eaau5758 (2019). [DOI] [PubMed] [Google Scholar]
- 117.Deng M et al. CD36 promotes the epithelial–mesenchymal transition and metastasis in cervical cancer by interacting with TGF-β. J. Transl Med 17, 352 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Gharpure KM et al. FABP4 as a key determinant of metastatic potential of ovarian cancer. Nat. Commun 9, 2923 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kawaguchi K et al. High expression of fatty acid-binding protein 5 promotes cell growth and metastatic potential of colorectal cancer cells. FEBS Open Bio 6, 190–199 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Wu G et al. FABP5 is correlated with poor prognosis and promotes tumour cell growth and metastasis in clear cell renal cell carcinoma. Eur. J. Pharmacol 862, 172637 (2019). [DOI] [PubMed] [Google Scholar]
- 121.Ohata T et al. Fatty acid-binding protein 5 function in hepatocellular carcinoma through induction of epithelial–mesenchymal transition. Cancer Med. 6, 1049–1061 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wang W et al. FABP5 correlates with poor prognosis and promotes tumor cell growth and metastasis in cervical cancer. Tumor Biol. 37, 14873–14883 (2016). [DOI] [PubMed] [Google Scholar]
- 123.Pan J, Dai Q, Zhang T & Li C Palmitate acid promotes gastric cancer metastasis via FABP5/SP1/UCA1 pathway. Cancer Cell Int. 19, 69 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Carbonetti G et al. FABP5 coordinates lipid signaling that promotes prostate cancer metastasis. Sci. Rep 9, 18944 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Ku C-Y, Liu Y-H, Lin H-Y, Lu S-C & Lin J-Y Liver fatty acid-binding protein (L-FABP) promotes cellular angiogenesis and migration in hepatocellular carcinoma. Oncotarget 7, 18229–18246 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Zhang M et al. Adipocyte-derived lipids mediate melanoma progression via FATP proteins. Cancer Discov. 8, 1006–1025 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Seguin F et al. The fatty acid synthase inhibitor orlistat reduces experimental metastases and angiogenesis in B16-F10 melanomas. Br. J. Cancer 107, 977 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zaytseva YY et al. Inhibition of fatty acid synthase attenuates CD44-associated signaling and reduces metastasis in colorectal cancer. Cancer Res. 72, 1504 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Jafari N et al. De novo fatty acid synthesis-driven sphingolipid metabolism promotes metastatic potential of colorectal cancer. Mol. Cancer Res 17, 140–152 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Vriens K et al. Evidence for an alternative fatty acid desaturation pathway increasing cancer plasticity. Nature 566, 403–406 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Triki M et al. mTOR signaling and SREBP activity increase FADS2 expression and can activate sapienate biosynthesis. Cell Rep. 31, 107806 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Ran H et al. Stearoyl-CoA desaturase-1 promotes colorectal cancer metastasis in response to glucose by suppressing PTEN. J. Exp. Clin. Cancer Res 37, 54 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Zhao J et al. Exogenous lipids promote the growth of breast cancer cells via CD36. Oncol. Rep 38, 2105–2115 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Li J et al. Lipid desaturation is a metabolic marker and therapeutic target of ovarian cancer stem cells. Cell Stem Cell 20, 303–314.e5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Vivas-García Y et al. Lineage-restricted regulation of SCD and fatty acid saturation by MITF controls melanoma phenotypic plasticity. Mol. Cell 77, 120–137.e9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; This work reveals that SCD1 is regulated by MITF being implicated in melanoma proliferation. SCD1 inhibition leads to endoplasmic reticulum stress response and enhanced invasion and metastasis formation.
- 136.Bellenghi M et al. SCD5-induced oleic acid production reduces melanoma malignancy by intracellular retention of SPARC and cathepsin B. J. Pathol 236, 315–325 (2015). [DOI] [PubMed] [Google Scholar]
- 137.Lee HJ et al. Cholesterol esterification inhibition suppresses prostate cancer metastasis by impairing the Wnt/β-catenin pathway. Mol. Cancer Res 16, 974 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Li J et al. Abrogating cholesterol esterification suppresses growth and metastasis of pancreatic cancer. Oncogene 35, 6378–6388 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Bi M et al. Effect of inhibiting ACAT-1 expression on the growth and metastasis of Lewis lung carcinoma. Oncol. Lett 18, 1548–1556 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Wang YY et al. Mammary adipocytes stimulate breast cancer invasion through metabolic remodeling of tumor cells. JCI Insight 2, e87489 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Miranda F et al. Salt-inducible kinase 2 couples ovarian cancer cell metabolism with survival at the adipocyte-rich metastatic niche. Cancer Cell 30, 273–289 (2016). [DOI] [PubMed] [Google Scholar]
- 142.Xiong Y et al. CPT1A regulates breast cancer-associated lymphangiogenesis via VEGF signaling. Biomed. Pharmacother 106, 1–7 (2018). [DOI] [PubMed] [Google Scholar]
- 143.Pucci S et al. Carnitine palmitoyl transferase-1A (CPT1A): a new tumor specific target in human breast cancer. Oncotarget 7, 19982–19996 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Wang C et al. Elevated level of mitochondrial reactive oxygen species via fatty acid β-oxidation in cancer stem cells promotes cancer metastasis by inducing epithelial–mesenchymal transition. Stem Cell Res. Ther 10, 175 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Gomes AP et al. Age-induced accumulation of methylmalonic acid promotes tumour progression. Nature 585, 283–287 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study shows that age-dependent metabolic change, that is, methylmalonic acid accumulation in the blood, promotes cancer progression by inducing Sox4.
- 146.Notarnicola M et al. Serum lipid profile in colorectal cancer patients with and without synchronous distant metastases. Oncology 68, 371–374 (2005). [DOI] [PubMed] [Google Scholar]
- 147.Liu Y-L et al. Association of serum lipid profile with distant metastasis in breast cancer patients. Zhonghua Zhong Liu Za Zhi 34, 129–131 (2012). [DOI] [PubMed] [Google Scholar]
- 148.Acharya S, Rai P, Hallikeri K, Anehosur V & Kale J Serum lipid profile in oral squamous cell carcinoma: alterations and association with some clinicopathological parameters and tobacco use. Int. J. Oral. Maxillofac. Surg 45, 713–720 (2016). [DOI] [PubMed] [Google Scholar]
- 149.Wang YN et al. CPT1A-mediated fatty acid oxidation promotes colorectal cancer cell metastasis by inhibiting anoikis. Oncogene 37, 6025–6040 (2018). [DOI] [PubMed] [Google Scholar]
- 150.Jiang L et al. Reductive carboxylation supports redox homeostasis during anchorage-independent growth. Nature 532, 255–258 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Lignitto L et al. Nrf2 activation promotes lung cancer metastasis by inhibiting the degradation of Bach1. Cell 178, 316–329.e18 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Yu G et al. Organelle-derived acetyl-CoA promotes prostate cancer cell survival, migration, and metastasis via activation of calmodulin kinase II. Cancer Res. 78, 2490 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.McCoy F et al. Metabolic activation of CaMKII by coenzyme A. Mol. Cell 52, 325–339 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Ubellacker JM et al. Metastasis through lymph protects melanoma cells from ferroptosis. Nature 158, 113–118 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; This work reveals that metastasizing melanoma cells in lymphatic vessels experience less oxidative stress than cells in the blood because they are protected from ferroptosis, and thus are more efficient to form metastases.
- 155.Shang C et al. LNMICC promotes nodal metastasis of cervical cancer by reprogramming fatty acid metabolism. Cancer Res. 78, 877 (2018). [DOI] [PubMed] [Google Scholar]
- 156.Mukherjee A et al. Adipocyte-induced FABP4 expression in ovarian cancer cells promotes metastasis and mediates carboplatin resistance. Cancer Res. 80, 1748–1761 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Jiang L et al. Up-regulated FASN expression promotes transcoelomic metastasis of ovarian cancer cell through epithelial–mesenchymal transition. Int. J. Mol. Sci 15, 11539–11554 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Li J et al. CD147 reprograms fatty acid metabolism in hepatocellular carcinoma cells through Akt/mTOR/SREBP1c and P38/PPARα pathways. J. Hepatol 63, 1378–1389 (2015). [DOI] [PubMed] [Google Scholar]
- 159.Liu G et al. Lung fibroblasts promote metastatic colonization through upregulation of stearoyl-CoA desaturase 1 in tumor cells. Oncogene 37, 1519–1533 (2018). [DOI] [PubMed] [Google Scholar]
- 160.Blomme A et al. Myoferlin regulates cellular lipid metabolism and promotes metastases in triple-negative breast cancer. Oncogene 36, 2116 (2016). [DOI] [PubMed] [Google Scholar]
- 161.Lee C-K et al. Tumor metastasis to lymph nodes requires YAP-dependent metabolic adaptation. Science 363, 644 (2019). [DOI] [PubMed] [Google Scholar]
- 162.Lu J The Warburg metabolism fuels tumor metastasis. Cancer Metastasis Rev. 38, 157–164 (2019). [DOI] [PubMed] [Google Scholar]
- 163.Lehuédé C, Dupuy F, Rabinovitch R, Jones RG & Siegel PM Metabolic plasticity as a determinant of tumor growth and metastasis. Cancer Res. 76, 5201 (2016). [DOI] [PubMed] [Google Scholar]
- 164.Lu M et al. ACOT12-dependent alteration of acetyl-CoA drives hepatocellular carcinoma metastasis by epigenetic induction of epithelial–mesenchymal transition. Cell Metab. 29, 886–900.e5 (2019). [DOI] [PubMed] [Google Scholar]
- 165.Sun L et al. Decreased expression of acetyl-CoA synthase 2 promotes metastasis and predicts poor prognosis in hepatocellular carcinoma. Cancer Sci. 108, 1338–1346 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Comerford SA et al. Acetate dependence of tumors. Cell 159, 1591–1602 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Schug ZT et al. Acetyl-CoA synthetase 2 promotes acetate utilization and maintains cancer cell growth under metabolic stress. Cancer Cell 27, 57–71 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Mashimo T et al. Acetate is a bioenergetic substrate for human glioblastoma and brain metastases. Cell 159, 1603–1614 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Pollari S et al. Enhanced serine production by bone metastatic breast cancer cells stimulates osteoclastogenesis. Breast Cancer Res. Treat 125, 421–430 (2011). [DOI] [PubMed] [Google Scholar]
- 170.Zhu J et al. High expression of PHGDH predicts poor prognosis in non-small cell lung cancer. Transl Oncol. 9, 592–599 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Song Z, Feng C, Lu Y, Lin Y & Dong C PHGDH is an independent prognosis marker and contributes cell proliferation, migration and invasion in human pancreatic cancer. Gene 642, 43–50 (2018). [DOI] [PubMed] [Google Scholar]
- 172.Kim HM, Jung WH & Koo JS Site-specific metabolic phenotypes in metastatic breast cancer. J. Transl Med 12, 354 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Samanta D et al. PHGDH expression is required for mitochondrial redox homeostasis, breast cancer stem cell maintenance, and lung metastasis. Cancer Res. 76, 4430 (2016). [DOI] [PubMed] [Google Scholar]
- 174.Ngo B et al. Limited environmental serine and glycine confer brain metastasis sensitivity to PHGDH inhibition. Cancer Discov. 12, 1352–1373 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Cao Y et al. Glutamic pyruvate transaminase GPT2 promotes tumorigenesis of breast cancer cells by activating sonic hedgehog signaling. Theranostics 7, 3021–3033 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Pavlova NN et al. As extracellular glutamine levels decline, asparagine becomes an essential amino acid. Cell Metab. 27, 428–438.e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Knott SRV et al. Asparagine bioavailability governs metastasis in a model of breast cancer. Nature 554, 378 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; This work finds limiting asparagine bioavailability by silencing ASNS or treatment with L-asparaginase, or dietary asparagine restriction, reduces metastasis formation without affecting primary tumour growth.
- 178.Tanner JJ, Fendt S-M & Becker DF The proline cycle as a potential cancer therapy target. Biochemistry 57, 3433–3444 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Loayza-Puch F et al. Tumour-specific proline vulnerability uncovered by differential ribosome codon reading. Nature 530, 490 (2016). [DOI] [PubMed] [Google Scholar]
- 180.Wang D et al. PYCR1 promotes the progression of non-small-cell lung cancer under the negative regulation of miR-488. Biomed. Pharmacother 111, 588–595 (2019). [DOI] [PubMed] [Google Scholar]
- 181.Fang E et al. Therapeutic targeting of MZF1-AS1/PARP1/E2F1 axis inhibits proline synthesis and neuroblastoma progression. Adv. Sci 0, 1900581 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Sahu N et al. Proline starvation induces unresolved ER stress and hinders mTORC1-dependent tumorigenesis. Cell Metab. 24, 753–761 (2016). [DOI] [PubMed] [Google Scholar]
- 183.Fendt S-M Metabolic vulnerabilities of metastasizing cancer cells. BMC Biol. 17, 54 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Lunt SY & Fendt S-M Metabolism — a cornerstone of cancer initiation, progression, immune evasion and treatment response. Curr. Opin. Syst. Biol 8, 67–72 (2018). [Google Scholar]
- 185.Wang Y. n. et al. CPT1A-mediated fatty acid oxidation promotes colorectal cancer cell metastasis by inhibiting anoikis. Oncogene 37, 6025–6040 (2018). [DOI] [PubMed] [Google Scholar]
- 186.Kamarajugadda S et al. Glucose oxidation modulates anoikis and tumor metastasis. Mol. Cell. Biol 32, 1893–1907 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Kruse NJ & Bornstein P The metabolic requirements for transcellular movement and secretion of collagen. J. Biol. Chem 250, 4841–4847 (1975). [PubMed] [Google Scholar]
- 188.Park JH et al. Fatty acid oxidation-driven Src links mitochondrial energy reprogramming and oncogenic properties in triple-negative breast cancer. Cell Rep. 14, 2154–2165 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Wright HJ et al. CDCP1 drives triple-negative breast cancer metastasis through reduction of lipid-droplet abundance and stimulation of fatty acid oxidation. Proc. Natl Acad. Sci. USA 114, E6556 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Andrzejewski S et al. PGC-1α promotes breast cancer metastasis and confers bioenergetic flexibility against metabolic drugs. Cell Metab.26, 778–787 (2017). [DOI] [PubMed] [Google Scholar]
- 191.Dupuy F et al. PDK1-dependent metabolic reprogramming dictates metastatic potential in breast cancer. Cell Metab. 22, 577–589 (2015). [DOI] [PubMed] [Google Scholar]
- 192.Loo JM et al. Extracellular metabolic energetics can promote cancer progression. Cell 160, 393–406 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Payne CE et al. A novel selective and orally bioavailable Nav 1.8 channel blocker, PF-01247324, attenuates nociception and sensory neuron excitability. Br. J. Pharmacol 172, 2654–2670 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Fernandes M, Rosel D & Brábek J Solid cancer: the new tumour spread endpoint opens novel opportunities. Br. J. Cancer 121, 513–514 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Chaika NV et al. Differential expression of metabolic genes in tumor and stromal components of primary and metastatic loci in pancreatic adenocarcinoma. PLoS ONE 7, e32996 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Davis RT et al. Transcriptional diversity and bioenergetic shift in human breast cancer metastasis revealed by single-cell RNA sequencing. Nat. Cell Biol 22, 310–320 (2020). [DOI] [PubMed] [Google Scholar]
- 197.Karaayvaz M et al. Unravelling subclonal heterogeneity and aggressive disease states in TNBC through single-cell RNA-seq. Nat. Commun 9, 3588 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Basnet H et al. Flura-seq identifies organ-specific metabolic adaptations during early metastatic colonization. eLife 8, e43627 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Chen EI et al. Adaptation of energy metabolism in breast cancer brain metastases. Cancer Res. 67, 1472 (2007). [DOI] [PubMed] [Google Scholar]
- 200.Fischer GM et al. Molecular profiling reveals unique immune and metabolic features of melanoma brain metastases. Cancer Discov. 9, 628–645 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Davidson SM et al. Environment impacts the metabolic dependencies of Ras-driven non-small cell lung cancer. Cell Metab. 23, 517–528 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Hensley CT et al. Metabolic heterogeneity in human lung tumors. Cell 164, 681–694 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Maher EA et al. Metabolism of [U-13C]glucose in human brain tumors in vivo. NMR Biomed. 25, 1234–1244 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Fernández-García J, Altea-Manzano P, Pranzini E & Fendt S-M Stable isotopes for tracing mammalian-cell metabolism in vivo. Trends Biochem. Sci 45, 185–201 (2020). [DOI] [PubMed] [Google Scholar]
- 205.Buescher JM et al. A roadmap for interpreting 13C metabolite labeling patterns from cells. Curr. Opin. Biotechnol 34, 189–201 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Fong MY et al. Breast-cancer-secreted miR-122 reprograms glucose metabolism in premetastatic niche to promote metastasis. Nat. Cell Biol 17, 183–194 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Hao Y et al. Investigation of lipid metabolism dysregulation and the effects on immune microenvironments in pan-cancer using multiple omics data. BMC Bioinf. 20, 195 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Berod L et al. De novo fatty acid synthesis controls the fate between regulatory T and T helper 17 cells. Nat. Med 20, 1327 (2014). [DOI] [PubMed] [Google Scholar]
- 209.Wang H et al. CD36-mediated metabolic adaptation supports regulatory T cell survival and function in tumors. Nat. Immunol 21, 298–308 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Su P et al. Enhanced lipid accumulation and metabolism are required for the differentiation and activation of tumor-associated macrophages. Cancer Res. 80, 1438–1450 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Niavarani SR et al. Lipid accumulation impairs natural killer cell cytotoxicity and tumor control in the postoperative period. BMC Cancer 19, 823 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Michelet X et al. Metabolic reprogramming of natural killer cells in obesity limits antitumor responses. Nat. Immunol 19, 1330–1340 (2018). [DOI] [PubMed] [Google Scholar]
- 213.Pan Y et al. Survival of tissue-resident memory T cells requires exogenous lipid uptake and metabolism. Nature 543, 252 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.García-Mulero S et al. Lung metastases share common immune features regardless of primary tumor origin. J. Immunother. Cancer 8, e000491 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]