

Abstract
Activation of muscle-specific genes by the MEF2 transcription factor is inhibited by class II histone deacetylases (HDACs) 4 and 5, which contain carboxy-terminal deacetylase domains and amino-terminal extensions required for association with MEF2. The inhibitory action of HDACs is overcome by myogenic signals which disrupt MEF2-HDAC interactions and stimulate nuclear export of these transcriptional repressors. Nucleocytoplasmic trafficking of HDAC5 is mediated by binding of the chaperone protein 14-3-3 to two phosphoserine residues (Ser-259 and Ser-498) in its amino-terminal extension. Here we show that HDAC4 and -5 each contain a signal-responsive nuclear export sequence (NES) at their extreme carboxy termini. The NES is conserved in another class II HDAC, HDAC7, but is absent in class I HDACs and the HDAC-related corepressor, MEF2-interacting transcription repressor. Our results suggest that this conserved NES is inactive in unphosphorylated HDAC5, which is localized to the nucleus, and that calcium-calmodulin-dependent protein kinase (CaMK)-dependent binding of 14-3-3 to phosphoserines 259 and 498 activates the NES, with consequent export of the transcriptional repressor to the cytoplasm. A single amino acid substitution in this NES is sufficient to retain HDAC5 in the nucleus in the face of CaMK signaling. These findings provide molecular insight into the mechanism by which extracellular cues alter chromatin structure to promote muscle differentiation and other MEF2-regulated processes.
Whether or not a gene is transcribed is dependent on the packaging state of local chromatin, which is organized in nucleosomes. Chromosomal DNA present in condensed chromatin is generally inaccessible to high-molecular-weight transcriptional machinery and is thus transcriptionally silent. However, acetylation of lysine residues in the tails of nucleosomal histones relaxes chromatin structure and promotes gene expression. This posttranslational modification is catalyzed by histone acetyl transferases (HATs). Histone acetylation also provides a reinforcing mechanism for chromatin relaxation by creating binding sites for bromodomain-containing transcriptional activators, which typically possess HAT activity. The stimulatory effects of HATs on gene transcription are antagonized by histone deacetylases (HDACs) (reviewed in reference 30).
Skeletal muscle provides a tractable model system to study the dynamic interplay between HATs and HDACs in the control of cellular differentiation (reviewed in reference 23). During the formation of skeletal muscle, undifferentiated myoblasts irreversibly exit the cell cycle and fuse to form multinucleated myofibers with an organized contractile apparatus. Skeletal myogenesis is controlled by a highly specific transcriptional program, which regulates subordinate genes in either a positive or a negative manner. Members of the MEF2 family of transcription factors serve a central role in governing this program (reviewed in reference 1).
MEF2 belongs to the MADS-box superfamily of transcription factors and binds an A/T-rich element present in the regulatory regions of numerous muscle-specific genes. The four MEF2 factors, MEF2A, -B, -C, and -D, share homology in an amino-terminal MADS domain, which mediates DNA binding and dimerization, and an adjacent MEF2 domain, which regulates cofactor interactions. MEF2 can function as either a transcriptional activator or repressor, depending on whether it is physically associated with HATs or HDACs. Binding of the p300 coactivator, which possesses HAT activity, to the MADS-MEF2 domains of MEF2 results in activation of downstream target genes (28). Conversely, binding of class II HDAC4 and -5 to the same region of MEF2 converts the transcription factor into a potent repressor of gene expression (15, 16, 24, 33).
Class II HDACs contain a carboxy-terminal catalytic domain and an amino-terminal extension that mediates binding to MEF2 (4, 6, 31). HDAC4 and -5 act as potent repressors of skeletal myogenesis by virtue of their ability to associate with MEF2 (17). However, during myogenesis they dissociate from MEF2 and are exported to the cytoplasm, thereby freeing MEF2 to stimulate muscle gene expression (21). The effect of myogenic signals on HDAC localization can be mimicked by calcium-calmodulin-dependent protein kinase (CaMK) signaling (21). Previously, we showed that CaMK-mediated nuclear export of HDAC5 was dependent on phosphorylation of two serine residues in its amino-terminal extension, Ser-259 and Ser-498 (21). Phosphorylation at these residues creates docking sites for the intracellular chaperone protein 14-3-3 (22). Nuclear export of HDAC5 appears to require 14-3-3 binding, since replacement of Ser-259 and Ser-498 with alanine abolishes not only its association with 14-3-3 but also its nuclear export (21, 22). However, the existence of carboxy-terminal truncation mutants of HDAC5 that retain the ability to bind 14-3-3 but remain in the nucleus despite active CaMK signaling suggests that 14-3-3 binding alone is insufficient to drive HDAC5 out of the nucleus (22).
To further define the mechanism that regulates nuclear export of class II HDACs, we analyzed the behavior of a series of HDAC5 mutants. Here we describe a CaMK-responsive nuclear export sequence (NES) present near the extreme carboxy terminus of HDAC5. This regulatory sequence is conserved in HDAC4 and -7 but is absent in class I HDACs and the MEF2-interacting transcription repressor (MITR). Our studies indicate that phosphorylation-dependent binding of 14-3-3 to the amino-terminal extension of HDAC4 and -5 is required to activate the carboxy-terminal NES, culminating in shuttling of these transcriptional repressors to the cytoplasm. These results provide a molecular explanation for the signal-responsiveness of class II HDACs and the MEF2 target genes that are under their control.
MATERIALS AND METHODS
Cell culture and transfections.
COS cells were cultured in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum, 2 mM l-glutamine, and penicillin-streptomycin. Cells were plated at a density of 5 × 105 to 10 × 105 cells/35-mm-diameter dish 1 day prior to transfection with the lipid-based reagent Fugene 6 (Roche Molecular Biochemicals).
Plasmids.
Epitope-tagged derivatives of 14-3-3ɛ, HDAC4, and HDAC5 containing amino-terminal FLAG or Myc tags, MEF2C with a carboxy-terminal Myc tag, and CaMKI with a 3× hemagglutinin (HA) tag were generated using the pcDNA3.1 expression vector (Invitrogen). The cDNA encoding activated CaMKI contains a stop codon in place of isoleucine-294, thereby removing the carboxy-terminal autoinhibitory domain (8). This CaMK mutant functions constitutively without the requirement for calcium and calmodulin for activation. Mutagenesis was performed with the Quikchange kit (Stratagene). Internal deletion mutants of HDAC5 were generated by PCR with PFU Turbo polymerase (Stratagene).
Coimmunoprecipitation and immunoblotting.
COS cells were harvested 2 days posttransfection in phosphate-buffered saline containing 0.5% Triton X-100, 1 mM EDTA, 1 mM phenylmethylsulfonyl fluoride (PMSF), and protease inhibitors (Complete; Roche Molecular Biochemicals). After brief sonication and removal of cellular debris by centrifugation, FLAG-tagged proteins were immunoprecipitated from cell lysates using anti-FLAG affinity resin (Sigma) and were washed 5 times with lysis buffer. Precipitated proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, transferred to polyvinylidene difluoride membranes, and immunoblotted as indicated with either anti-Myc antibody (polyclonal, A-14; Santa Cruz), pan anti-14-3-3 antibody (polyclonal, A-19; Santa Cruz), or a monoclonal anti-FLAG antibody (M2; Sigma). Proteins were visualized with a chemiluminescence system (Santa Cruz).
Indirect immunofluorescence.
COS cells were grown on glass coverslips, fixed in 10% formalin and stained in phosphate-buffered saline containing 3% bovine serum albumin and 0.1% Nonidet-P40. Primary antibodies against FLAG (M2; Sigma), Myc (polyclonal, A-14; Santa Cruz), or HA (polyclonal, Y-11; Santa Cruz) were used at a dilution of 1:200. Secondary antibodies conjugated to either fluorescein or Texas Red (Vector Labs) were also used at a dilution of 1:200. All images were captured at a magnification of ×40.
RESULTS
Generation of HDAC5 mutants lacking internal amino acids.
In response to CaMK signaling, HDAC5 is efficiently exported from the nucleus to the cytoplasm (21). This process is dependent on phosphorylation of HDAC5 at two serine residues, Ser-259 and Ser-498, in its amino-terminal extension (21). These sites are conserved in HDAC4, which also displays CaMK responsiveness. However, HDAC4 tends to be less restricted to the nucleus than HDAC5 in the absence of CaMK signaling due to constitutive association of this repressor with the chaperone protein 14-3-3 (7, 22, 34). Nuclear-cytoplasmic trafficking of HDAC5 also requires carboxy-terminal sequences (22). Previously, we demonstrated that sequences between amino acids 767 and 921 in the catalytic domain of HDAC5 were capable of functioning as an autonomous NES when tethered to a heterologous protein (21). To further define the NES in HDAC5, we generated a series of HDAC5 deletion mutants lacking increasing amounts of carboxy-terminal sequence in the context of the full-length protein. Schematic depictions of these proteins are shown in Fig. 1A. Each mutant protein was expressed at the appropriate size and was efficiently associated with MEF2, as determined by sequential immunoprecipitation and immunoblotting (Fig. 1B).
FIG. 1.

Internal deletion mutants of HDAC5. (A) Schematic representations of HDAC5 deletion mutants containing amino acids 1 to 767 but lacking sequences in the HDAC domain from position 768 to 1080. The locations of the NLS, MEF2-binding domain, and CaMK phosphorylation sites (Ser-259 and Ser-498) are indicated. The right-hand panel summarizes localization data shown in Fig. 2. N, nuclear; C, cytoplasmic; N/C, nuclear and cytoplasmic; P, phosphorylation sites. (B) COS cells were cotransfected with expression plasmids encoding FLAG-tagged versions of the indicated HDAC5 protein and a vector for Myc-tagged MEF2C (1 μg each). HDAC5 was immunoprecipitated from cell lysates using a monoclonal anti-FLAG antibody, and associated MEF2 was detected by immunoblotting with polyclonal anti-Myc antibodies (bottom panel). The membrane was reprobed with anti-FLAG antibody to reveal HDAC5 protein (upper panel).
Subcellular distribution of HDAC5 deletion mutants.
Indirect immunofluorescence experiments were performed to determine the relative contribution of specific carboxy-terminal sequences in HDAC5 in governing its subcellular distribution. For these experiments, COS cells were transfected with FLAG epitope-tagged derivatives of the indicated HDAC5 mutants in the absence or presence of a plasmid for constitutively active CaMKI. Consistent with our previous finding (21), HDAC5 was localized exclusively to the nucleus of these cells and was efficiently transported to the cytoplasm in response to CaMK signaling (Fig. 2A and B). Surprisingly, deletion of HDAC5 sequences between amino acids 768 and 921 altered the localization of HDAC5 from the nucleus to a whole-cell pattern (Fig. 2C and E). Possible explanations for the presence of these mutants in the cytoplasm in the absence of CaMK signaling are that the deletions unmask an otherwise cryptic NES, remove sequences involved in nuclear retention, or hinder the function of the nuclear localization signal (NLS) between amino acids 260 and 304 (21). Despite this uncertainty, the deletion mutants clearly retained the capacity to undergo CaMK-dependent nuclear export (Fig. 2D and F).
FIG. 2.

Mapping of carboxy-terminal sequences in HDAC5 required for nuclear export. Plasmids encoding FLAG-tagged derivatives of the indicated HDAC5 proteins were transfected into COS cells in the absence (A, C, E, G, I, K, and M) or presence (B, D, F, H, J, L, and N) of a vector encoding constitutively active CaMKI (0.5 μg each). HDAC5 was detected by indirect immunofluorescence using primary antibodies against the FLAG tag and a fluorescein-conjugated secondary antibody. Magnification, ×40.
Deletion of additional carboxy-terminal sequences led to a redistribution of HDAC5 into the nucleus in the absence of active CaMK (Fig. 2G, I, and K). However, each mutant was efficiently exported to the cytoplasm in response to CaMK signaling (Fig. 2H, J, and L). A deletion mutant of HDAC5 containing amino acids 1 to 767 and no carboxy-terminal sequences was resistant to CaMK-mediated nuclear export, consistent with our prior work (Fig. 2M and N) (21). These results demonstrate that the region from 1081 to the carboxy terminus of HDAC5 is sufficient to confer proper nuclear export to the amino-terminal 767 amino acids of the protein, suggesting that these sequences may function as a CaMK-responsive NES.
Fusion of HDAC5 amino acids 1081 to 1122 to GFP.
NESs are often defined by their ability to confer nuclear export to heterologous proteins (reviewed in reference 5). To further define the properties of the putative NES in HDAC5, we fused it to green fluorescent protein (GFP) and assessed the localization of the resultant fusion protein. GFP alone was found in both the nucleus and the cytoplasm of transfected COS cells, with more prominent localization in the nucleus (Fig. 3A, a and b). Surprisingly, fusion of HDAC5 amino acids 1081 to 1122 to the carboxy terminus of GFP failed to alter the localization of the protein in either the absence or the presence of activated CaMK (Fig. 3A, c and d). Identical results were obtained when this HDAC5 sequence was fused to the amino terminus of GFP (data not shown). These findings raised the possibility that the HDAC5 NES might function in a protein context-dependent manner, possibly requiring additional sequences for full activity.
FIG. 3.

Subcellular localization of GFP-HDAC5 and MITR-HDAC5 fusion proteins. (A) COS cells were transfected with an expression vector for GFP or a GFP fusion protein containing amino acids 1081 to 1122 of HDAC5 fused to its carboxy terminus (GFP:1081-1122) in the absence (a and c) or presence (b and d) of a plasmid for constitutively active CaMKI (0.5 μg each). GFP-positive cells were photographed at a magnification of ×40. (B) COS cells were transfected with expression vectors for FLAG-tagged MITR or an MITR fusion protein containing amino acids 1081 to 1122 of HDAC5 fused to its carboxy terminus (MITR:1081-1122) in the absence (a and c) or presence (b and d) of a plasmid for constitutively active CaMKI (0.5 μg each). MITR proteins were detected by indirect immunofluorescence using a primary anti-FLAG antibody and a fluorescein-conjugated secondary antibody. Photographs were taken at a magnification of ×64 to reveal localization of MITR to discrete nuclear bodies in the absence of CaMK signaling.
Fusion of HDAC5 amino acids 1081 to 1122 to MITR.
MITR (29), also referred to as HDRP (HDAC-related protein) (38), shares high homology with the amino-terminal extensions of HDAC4 and -5 and interacts with MEF2 but lacks an HDAC catalytic domain (29, 38). Nevertheless, MITR inhibits MEF2-dependent reporter genes by recruiting other HDACs and the CtBP corepressor (29, 36, 38). CaMK signaling stimulates binding of 14-3-3 to MITR, and this binding is dependent on phosphorylation of two serines in the protein, Ser-218 and Ser-448, that are analogous to Ser-259 and Ser-498 of HDAC5 (37). However, unlike HDAC4 and -5, MITR remains in the nucleus of CaMK-expressing cells, presumably due to the lack of a carboxy-terminal NES. Experiments were next performed to determine whether amino acids 1081 to 1122 of HDAC5 could function as a constitutive or signal-dependent NES when fused to MITR.
MITR was localized in discrete nuclear bodies in transfected COS cells (Fig. 3B, a). A similar expression pattern was observed in other cell types, including C2 myoblasts and 10T1/2 fibroblasts (data not shown). In response to CaMK signaling, MITR remained in the nucleus, although the protein was evenly distributed and no longer localized to nuclear speckles (Fig. 3B, b). This redistribution of MITR is dependent on CaMK-dependent binding of 14-3-3 to Ser-218 and Ser-448 (37). MITR remained nuclear when amino acids 1081 to 1122 of HDAC5 were fused to its carboxy terminus, although the protein was typically found in fewer foci than wild-type MITR (Fig. 3B, c). However, in response to CaMK, the MITR-HDAC5 fusion protein was efficiently transported from the nucleus to the cytoplasm. Translocation of the MITR fusion protein was a result of active nuclear export, since the process was blocked by leptomycin B, a fungal toxin that inhibits the exportin protein CRM1 (data not shown). These findings suggest that amino acids 1081 to 1122 of HDAC5 function as a signal-dependent NES.
Fine mapping the HDAC5 NES.
HDAC5 nuclear export is dependent on the CRM1 exportin protein (21). A consensus CRM1-dependent NES has been established (Leu-X-X-X-Leu-X-X-Leu-X-Leu) (2). However, in several known CRM1 substrates, isoleucine and/or valine substitute for leucine at one or more positions (5). We analyzed HDAC5 amino acids 1081 to 1122 for the presence of a consensus NES and found none (Fig. 4A). However, this region of human and mouse HDAC5 contains two leucines and two valines that are conserved in HDAC4 and -7. In this regard, HDAC4 also undergoes CaMK-mediated nuclear export (22) and HDAC7 is likely to be subject to similar control on the basis of its homology to HDAC4 and -5 (12).
FIG. 4.

Identification of hydrophobic residues that regulate nuclear export of HDAC5Δ768-1080. (A) Alignment of the carboxy termini of class II HDACs (h, human; m, mouse). Conserved leucine (L) and valine (V) residues are highlighted. The arrow at 1108 indicates a consensus CaMK phosphorylation site in HDAC5. (B) COS cells were transfected with expression vectors for FLAG-tagged forms of HDAC5Δ768-1080 (see Fig. 1A) or point mutants of HDAC5Δ768-1080 containing an alanine substitution at valine 1086 (V1086A) or alanine in place of both leucine 1091 and 1092 (L1091/1092A) in the absence (a, c, e, and g) or presence (b, d, f, and h) of a plasmid for constitutively active CaMKI (0.5 μg each). HDAC5 proteins were visualized by indirect immunofluorescence with an anti-FLAG primary antibody and a fluorescein-conjugated secondary antibody. Magnification, ×40.
To address the possible roles of these residues in nuclear export of HDAC5, we replaced Val-1086, Leu-1091, and Leu-1092 with alanine. These mutants were initially generated in the context of the HDAC5Δ768-1080 protein (Fig. 1A) to circumvent potential complications in interpretation of the data due to the presence of redundant NESs in the carboxy terminus of HDAC5 (see below). The HDAC5Δ768-1080 protein was efficiently transported from the nucleus to the cytoplasm in response to CaMK signaling (Fig. 4B, a and b). In contrast, when Val-1086 was converted to alanine (mutant Δ768-1080 V1086A), CaMK-mediated nuclear export was significantly impaired, although it was not eliminated (Fig. 4B, c and d). Simultaneous conversion of Leu-1091 and Leu-1092 to alanine (mutant Δ768-1080 L1091/1092A) completely abolished nuclear export of the protein (Fig. 4B, e and f).
A consensus CaMK phosphorylation site (Arg-X-X-Ser) is present within HDAC5 amino acids 1081 to 1122 (Fig. 4A). To address its possible involvement in CaMK-mediated nuclear export of HDAC5, we generated a mutant protein containing alanine in place of the potential phosphoacceptor at position 1108. However, as shown in Fig. 4B (g and h), this substitution had no effect on export of the HDAC5Δ768-1080 protein.
Identical results to those presented in Fig. 4B were obtained when Val-1086, Leu-1091, Leu-1092, or Ser-1108 was converted to alanine in the context of the MITR-HDAC5 fusion protein described in Fig. 3B (data not shown). Taken together, these results suggest that Val-1086, Leu-1091, and Leu-1092 are critical elements of the HDAC5 NES and that the CaMK signal for nuclear export is transmitted to these sequences via cross-talk with amino-terminal residues in HDAC5 rather than by phosphorylation of an adjacent site (e.g., Ser-1108).
A single NES regulates nuclear export of HDAC5.
Our previous demonstration that amino acids 768 to 921 can function as an NES when fused to GFP and that a deletion mutant of HDAC5 lacking 101 carboxy-terminal amino acids undergoes CaMK-dependent nuclear export raised the possibility that HDAC5 contains redundant NESs (21). Indeed, there is precedent for multiple NESs in other transcriptional regulators (39). To further address this issue, we generated an HDAC5 deletion mutant lacking only amino acids 1081 to 1122 (mutant 1-1080). In addition, changes of Val-1086 to alanine and Leu-1092 to alanine were generated in the context of full-length HDAC5 (mutants V1086A and L1092A, respectively). As shown in Fig. 5A (a and b), full-length HDAC5 was efficiently exported to the cytoplasm in response to CaMK signaling. In contrast, the 1-1080 and V1086A mutants of HDAC5 were largely resistant to CaMK-mediated nuclear export (Fig. 5A, c through f), and replacement of Leu-1092 with alanine resulted in a complete block to HDAC5 nuclear export (Fig. 5A, g and h). The relative importance of Val-1094 in governing nuclear export of HDAC5 was not addressed. However, given its proximity to Leu-1092, it is also likely to play an important role in the HDAC5 NES. Of note, this experiment was performed with a mutant in which only Leu-1092 was converted to alanine, while other experiments in this study employed the double Leu-1091 and Leu-1092 to alanine mutant. The double-alanine mutant was originally constructed to avoid complications due to possible redundant use of the adjacent leucines in the NES. However, the results presented here demonstrate that the conserved leucine at position 1092 is a critical regulator of HDAC5 nuclear export.
FIG. 5.
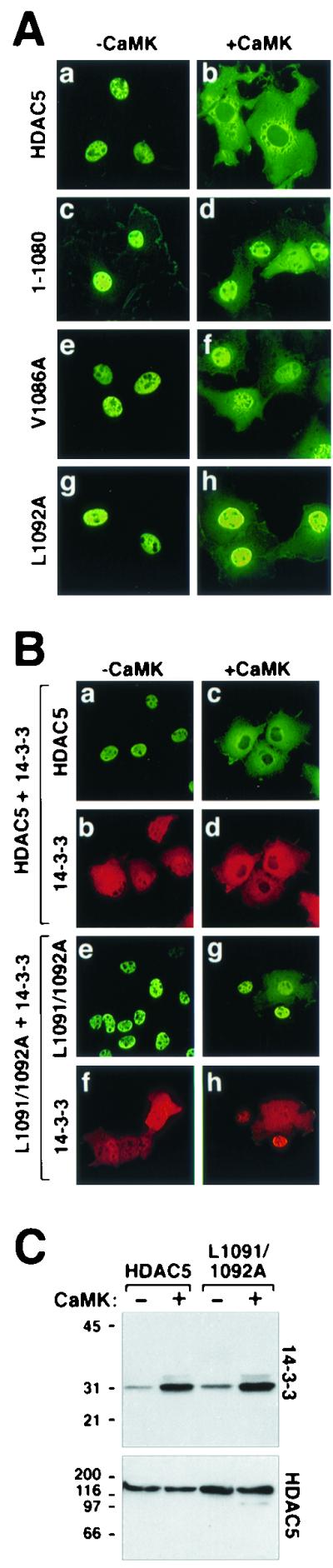
Subcellular localization of NES mutants of HDAC5. (A) COS cells were transfected with expression vectors for FLAG-tagged derivatives of full-length HDAC5 (a and b), a deletion mutant of HDAC5 lacking amino acids 1081 to 1122 (1-1080), or point mutants of HDAC5 containing an alanine substitution at valine 1086 (V1086A) or leucine 1092 (L1092A) in the absence (a, c, e, and g) or presence (b, d, f, and h) of a plasmid for constitutively active CaMKI (0.5 μg each). HDAC5 proteins were visualized by indirect immunofluorescence with an anti-FLAG antibody and a fluorescein-conjugated secondary antibody. (B) CaMK-dependent colocalization of HDAC5 and 14-3-3. COS cells were cotransfected with expression vectors for FLAG-tagged wild-type HDAC5 (a through d) or HDAC5 L1091/1092A (e through h) and Myc-tagged 14-3-3 in the absence (a, b, e, and f) or presence (c, d, g, and h) of a plasmid for constitutively active CaMK (0.5 μg each). HDAC5 and 14-3-3 were costained with anti-FLAG and anti-Myc primary antibodies and fluorescein (HDAC5)- and Texas Red (14-3-3)-conjugated secondary antibodies. All images were taken at a magnification of ×40. (C) Association of HDAC5 with endogenous 14-3-3. COS cells were transfected with expression vectors for FLAG-tagged HDAC5 or HDAC5 L1091/1092A in the absence or presence of a plasmid for activated CaMKI (1 μg). To compensate for CaMK-mediated increases in expression from the cytomegalovirus-driven expression plasmids, cells receiving CaMKI were transfected with 0.5 μg of HDAC5 plasmid, compared to 1 μg in those lacking CaMKI. Ectopic HDAC5 was immunoprecipitated from cell lysates with an anti-FLAG antibody, and associated endogenous 14-3-3 was detected by immunoblotting with a pan anti-14-3-3 antibody (top panel). The membrane was reprobed with anti-FLAG antibody to reveal total immunoprecipitated HDAC5 protein (bottom panel).
Block to HDAC5 nuclear export despite efficient binding to 14-3-3.
The above results strongly suggest that HDAC5 contains a single NES near its extreme carboxy terminus and that Val-1086 and Leu-1092 serve critical functions within this NES. However, one explanation for the inability of these mutants to undergo nuclear export is that the amino acid substitutions inhibit binding of 14-3-3 to HDAC5, which is required for nuclear-cytoplasmic trafficking of this repressor (22). Indirect immunofluorescence and coimmunoprecipitation experiments were performed to address this possibility. As shown in Fig. 5B (a and b), in cells coexpressing HDAC5 and 14-3-3, HDAC5 was exclusively nuclear, while 14-3-3 was found in both the nucleus and the cytoplasm. This localization pattern is identical to that seen when either protein is expressed individually, suggesting that the proteins fail to interact in the absence of a signal (22). In contrast, in the presence of activated CaMK both HDAC5 and 14-3-3 were colocalized exclusively in the cytoplasm, consistent with our prior findings showing that CaMK signaling promotes association of the two proteins (Fig. 5B, c and d) (22). Likewise, in response to CaMK signaling, 14-3-3 efficiently associated with the L1091A-L1092A mutant of HDAC5, as indicated by the colocalization of these two proteins in the nuclear compartment (Fig. 5B, g and h). These results were further supported by coimmunoprecipitation experiments, which revealed CaMK-inducible binding of endogenous 14-3-3 to both wild-type HDAC5 and the L1091-L1092A mutant (Fig. 5C). Together, these results suggest that the failure of the L1091-L1092A mutant to undergo nuclear export is due to a loss of NES function rather than a block to 14-3-3 binding to the amino terminus of HDAC5.
A conserved NES in HDAC4.
HDAC4 and -5 share 54% amino acid identity. Consistent with this sequence homology, both proteins are potent inhibitors of MEF2-dependent transcription and skeletal myogenesis (15, 16, 17, 24, 33). Furthermore, like HDAC5, HDAC4 is efficiently exported from the nucleus to the cytoplasm in response to CaMK signaling, and this process is dependent on 14-3-3 binding (22). However, there is evidence to suggest that HDAC4 and -5 are differentially regulated in vivo. For example, ectopic expression of HDAC4 in various cell types has revealed that the protein is constitutively localized to the cytoplasm in 50 to 80% of the cells in which it is expressed, while HDAC5 is exclusively nuclear in the same cells under the same conditions (Fig. 5 and 6) (22, 24).
FIG. 6.
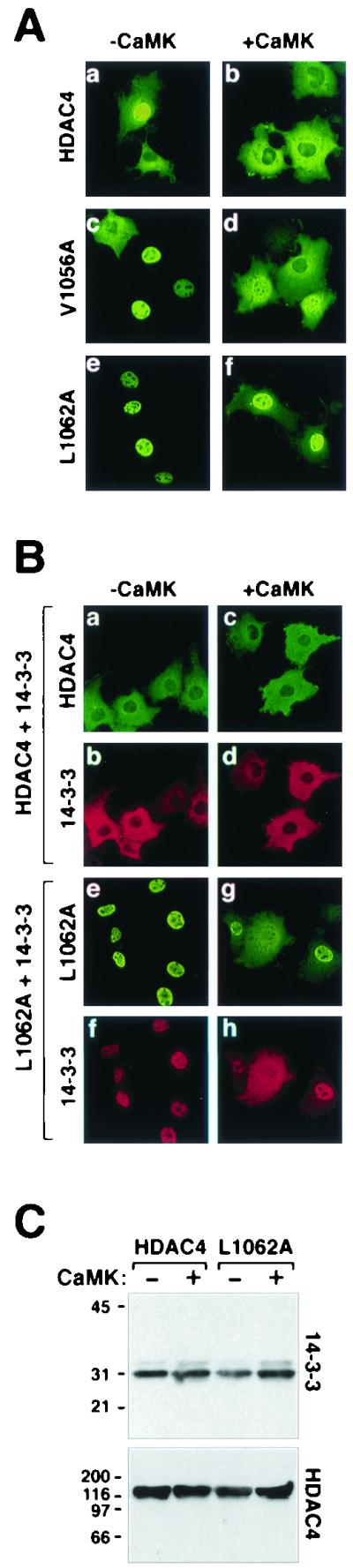
Mutagenesis of a conserved NES in HDAC4. (A) COS cells were transfected with expression vectors for FLAG-tagged derivatives of full-length HDAC4 or point mutants of HDAC4 containing an alanine substitution at leucine 1056 (L1056A) or alanine in place of leucine 1062 (L1062A) in the absence or presence of a plasmid for constitutively active CaMKI (0.5 μg each). HDAC4 proteins were visualized by indirect immunofluorescence with anti-FLAG antibody and a fluorescein-conjugated secondary antibody. (B) Constitutive colocalization of HDAC4 and 14-3-3. COS cells were cotransfected with expression vectors for FLAG-tagged wild-type HDAC4 (a through d) or HDAC4 L1062A (e through h) and Myc-tagged 14-3-3 in the absence (a, b, e, and f) or presence (c, d, g, and h) of a plasmid for constitutively active CaMK (0.5 μg each). HDAC4 and 14-3-3 were costained with anti-FLAG and anti-Myc primary antibodies and fluorescein (HDAC5)- and Texas Red (14-3-3)-conjugated secondary antibodies. All images were taken at a magnification of ×40. (C) Association of HDAC4 with endogenous 14-3-3. COS cells were transfected with expression vectors for FLAG-tagged HDAC4 or HDAC4 L1062A in the absence or presence of a plasmid for activated CaMKI (1 μg). To compensate for CaMK-mediated increases in expression from the cytomegalovirus-driven expression plasmids, cells receiving CaMKI were transfected with 0.5 μg of HDAC4 plasmid, compared to 1 μg in those lacking CaMKI. Ectopic HDAC4 was immunoprecipitated from cell lysates with anti-FLAG antibody, and associated endogenous 14-3-3 was detected by immunoblotting with a pan anti-14-3-3 antibody (top panel). The membrane was reprobed with anti-FLAG antibody to reveal total immunoprecipitated HDAC4 protein (bottom panel).
To determine whether the NES in HDAC5 is functionally conserved in HDAC4, mutants of HDAC4 were generated in which the residues analogous to Val-1086 or Leu-1092 in HDAC5 (Val-1056 and Leu-1062) were converted to alanine. As shown in Fig. 6A, HDAC4 was localized to either the nucleus or the cytoplasm of transfected COS cells and was efficiently exported to the cytoplasm in response to CaMK signaling (a and b). HDAC4 was cytoplasmic in 100% of the cells expressing activated CaMK (Fig. 6A, b). Unlike wild-type HDAC4, a mutant of HDAC4 containing alanine in place of leucine at position 1056 (mutant L1056A) was exclusively nuclear in the majority of cells in which it was expressed, and CaMK-mediated nuclear export of this mutant was severely impaired (Fig. 6A, c and d). The block to nuclear export was even more pronounced when alanine was substituted for Leu-1062 in HDAC4 (mutant L1062A) (Fig. 6A, e and f). These results demonstrate that the NESs in HDAC4 and -5 are structurally and functionally conserved.
NES mutants of HDAC4 retain the capacity to bind 14-3-3.
While HDAC5 binding to 14-3-3 is largely dependent on CaMK-mediated phosphorylation, HDAC4 binds constitutively to 14-3-3 in yeast and mammalian cells, presumably due to the basal activity of an as-yet unidentified kinase (7, 22, 34). Binding of 14-3-3 to HDAC4 is dependent on phosphorylation at three serine residues: Ser-246, Ser-467, and Ser-632 (7, 22, 34). Ser-246 and Ser-467 in HDAC4 are analogous to Ser-259 and Ser-498, respectively, in HDAC5. We performed experiments to determine whether sequences in the NES of HDAC4 influenced association with 14-3-3. Consistent with our prior findings, HDAC4 was predominantly cytoplasmic in COS cells coexpressing 14-3-3 (Fig. 6B, a) (22). Ectopic 14-3-3, which localizes to both the nucleus and the cytoplasm in the absence of HDAC4 (22), was exclusively cytoplasmic in the presence of HDAC4 in either the absence or the presence of activated CaMK (Fig. 6B, b and d). The NES mutant of HDAC4 (L1062A) retained the capacity to bind 14-3-3, as evidenced by the nuclear accumulation of both proteins in the absence of CaMK signaling (Fig. 6B, e and f). The HDAC4 mutant and 14-3-3 also colocalized in the presence of activated CaMK (Fig. 6B, g and h). Of note, increased cytoplasmic staining of the HDAC4 L1062A mutant was evident in cells overexpressing 14-3-3 (compare Fig. 6A, f, and B, g). This enhanced cytoplasmic localization could be due to the ability of 14-3-3 to block binding of the nuclear import factor, importin α, to HDAC4 (7) (see Discussion). Nevertheless, these data clearly demonstrate that nuclear export-resistant mutants of HDAC4 retain the capacity to constitutively associate with 14-3-3 and suggest that the failure of these mutants to localize in the cytoplasm is due to an NES-autonomous defect.
DISCUSSION
The results of this study demonstrate that class II HDACs contain conserved NESs located at their extreme carboxy termini. These NESs function in a signal-dependent manner requiring phosphorylation and subsequent binding of 14-3-3 to serine residues near the amino termini. These results provide a potential molecular explanation for the dynamic nucleocytoplasmic shuttling of HDACs during skeletal myogenesis and in response to CaMK signaling (21).
CRM1-dependent nuclear export.
A major pathway for nuclear export involves the exportin protein CRM1 (5). CRM1 has been implicated in the regulation of several transcription factors, including NFAT and STAT1 (20, 39). A role for CRM1 in nuclear export of HDACs is strongly supported by the ability of the CRM1 antagonist, leptomycin B, to block constitutive nuclear export of HDAC4 and CaMK-mediated nuclear export of HDAC5 (21, 24).
A consensus leucine-rich binding site for CRM1 has been defined (Leu-X-X-X-Leu-X-X-Leu-X-Leu) (2), although CRM1-dependent NESs that diverge from this consensus on the basis of amino acid composition and spacing have also been identified, with isoleucine and valine being commonly substituted for leucine among these divergent NESs. Sequence analysis of class II HDACs failed to reveal a consensus NES. However, by examining the subcellular localization of a panel of deletion and point mutants, we have defined an NES (Val-X-X-X-X-X-Leu-X-Val) present in HDAC4, -5, and -7 but absent in class I HDACs and MITR. To our knowledge, this regulatory motif has not been previously described, and thus it represents a novel CRM1-dependent NES.
HDAC6 is unique among the class II HDACs in that it contains two tandem deacetylase domains and lacks a MEF2-binding domain (6). Furthermore, HDAC6 appears to translocate to the nucleus in the absence of mitogens (32). Consistent with these differences in regulation and function relative to other class II HDACs, HDAC6 lacks the carboxy-terminal NES described here but contains an amino-terminal NES not found in HDAC4, -5, and -7 (32).
NESs are commonly defined by their ability to confer cytoplasmic localization to heterologous proteins. However, the localization of chimeric proteins containing the HDAC5 NES fused to either GFP or the MITR corepressor was unaltered relative to the wild-type proteins (Fig. 3A and B). In contrast, an MITR-HDAC5 fusion protein was efficiently translocated to the cytoplasm in response to CaMK signaling, whereas wild-type MITR remained nuclear (Fig. 3). This signal-dependent nuclear export was specific for the MITR fusion protein, as the localization of the GFP chimera was unaltered by activated CaMK. Thus, while the HDAC5 NES fails to act as a constitutive export signal, it is capable of functioning in a signal-dependent manner when tethered to CaMK response elements present in the amino-terminal extensions of HDAC4, -5, and -7.
Role of other carboxy-terminal residues in the regulation of HDAC5 localization.
We previously analyzed the localization of a panel of carboxy-terminal deletion mutants of HDAC5 (21). Removal of approximately 100 carboxy-terminal amino acids from HDAC5 led to a diffuse, whole-cell pattern of HDAC5 localization, and a mutant lacking an additional 100 amino acids was completely excluded from the nucleus. Further deletion of HDAC5 carboxy-terminal sequences up to residue 767 resulted in a nuclear pattern of HDAC5 localization. We concluded from these results that sequences of HDAC5 between 768 and 921 function to localize the protein in the cytoplasm (21). Indeed, a GFP fusion protein containing these sequences was efficiently targeted to the cytoplasm. However, as shown here, deletion of amino acids 768 to 921 in the context of full-length HDAC5 had no effect on CaMK-mediated nuclear export of this protein (Fig. 2E and F). Furthermore, single amino acid substitutions in the carboxy-terminal NES of HDAC5 are sufficient to block nuclear export (Fig. 5A, e and f). Therefore, the bona fide NES in HDAC5 is located near the extreme carboxy terminus of the protein. The basis for the altered localization of the carboxy-terminal truncation mutants of HDAC5 remains unclear, but several possible explanations exist. The amino acid deletions may (i) expose a cryptic NES in HDAC5 that is normally masked, (ii) block the function of the NLS in HDAC5 located between amino acids 260 and 304, (iii) expose sequences in HDAC5 that serve to dock the protein in the cytoplasm, or (iv) remove sequences in HDAC5 involved in nuclear retention. Resolution of these issues awaits further experimentation.
Cross-talk between the amino and carboxy termini of class II HDACs?
How do signals received within the amino-terminal extensions of class II HDACs communicate with the carboxy-terminal NES? CaMK-mediated nuclear export of HDAC5 appears to require binding of 14-3-3 to Ser-259 and Ser-498 in its amino terminus. Indeed, substitution of these serines with alanines blocks both 14-3-3 binding and nuclear export of HDAC5 (22). However, association with 14-3-3 alone is not sufficient to localize HDAC5 in the cytoplasm, since NES mutants of HDAC5 associate with 14-3-3 in a CaMK-dependent manner but remain in the nucleus (Fig. 5 and 6). The simplest interpretation of these findings is schematized in Fig. 7. According to this model, amino-terminal sequences in HDAC5 mask the carboxy-terminal NES to retain HDAC5 in the nucleus. Upon 14-3-3 binding to phosphorylated Ser-259 and Ser-498, the conformation of HDAC5 is altered such that the carboxy-terminal NES becomes exposed, allowing export of the repressor to the cytoplasm. Based on the sensitivity of HDAC5 nuclear export to leptomycin B, this process likely involves binding of CRM1 to the NES, although we have not demonstrated this interaction directly.
FIG. 7.

A model for signal-dependent nuclear export of HDAC5. In the unphosphorylated state, the carboxy-terminal NES in HDAC5 is inactive, perhaps due to masking by sequences in the amino terminus of the protein. HDAC5 translocates into the nucleus via binding of importin α to the NLS positioned between amino acids 260 and 304. In the nucleus, HDAC5 associates with MEF2 to block expression of MEF2 target genes. In response to CaMK signaling, Ser-259 and Ser-498 in HDAC5 are phosphorylated and subsequently bound by 14-3-3. Association of 14-3-3 with HDAC5 disrupts MEF2-HDAC5 complexes, activates the carboxy-terminal NES in HDAC5, and stimulates nuclear export of the transcriptional repressor through a CRM1 exportin-dependent mechanism. Reentry of HDAC5 into the nucleus presumably requires the action of a protein phosphatase (PPase) which targets Ser-259 and Ser-498.
Of note, association of 14-3-3 with HDAC4 has been shown to decrease binding of the nuclear import factor, importin α, to this transcriptional repressor (7). Consistent with this, we previously mapped the NLS in HDAC5 to sequences that are flanked by Ser-259 and Ser-498 (21). Thus, 14-3-3 may serve a dual role in the regulation of HDAC localization by functioning to expose the NES and mask the NLS.
The model in Fig. 7 predicts that nuclear entry of HDAC5 requires dephosphorylation of Ser-259 and Ser-498 by a phosphatase. Indeed, treatment of cells with the phosphatase inhibitor calyculin A has been shown to increase association of 14-3-3 with HDAC4 (7). Calyculin A is a broad-specificity phosphatase inhibitor that blocks the activities of protein phosphatase 1 and protein phosphatase 2A. Thus, the exact identity of the HDAC phosphatase remains unknown. This phosphatase is of particular interest, because it would be expected to antagonize CaMK signaling to MEF2-dependent genes by enhancing the repressive activity of class II HDACs.
Class II HDAC–SMRT–N-CoR interactions.
Class II HDACs were recently shown to associate with nuclear receptor corepressor (N-CoR) and silencing mediator for retinoid and thyroid receptors (SMRT) (10, 12). Interestingly, the binding site for SMRT–N-CoR on class II HDACs overlaps with the NES we describe here, suggesting that these corepressors may regulate the function of the HDAC NES. Indeed, a recent report demonstrated that SMRT is capable of driving HDAC4 from the cytoplasm to the nucleus (35). It will be interesting to determine whether or not SMRT–N-CoR masks the carboxy-terminal NESs in HDAC4 and -5 and, if so, whether CaMK signaling alters this inhibitory activity.
MITR: a constitutively nuclear repressor of MEF2-dependent transcription.
MITR is highly homologous to the amino-terminal extensions of class II HDACs but lacks a carboxy-terminal catalytic domain (29, 38). Like HDAC4 and -5, MITR is a repressor of skeletal myogenesis and contains two regulatory serine residues that are targets for CaMK signaling (37). Phosphorylation of these sites releases MITR from MEF2 through a 14-3-3-dependent mechanism. However, unlike HDAC4 and -5, MITR remains in the nucleus following phosphorylation by CaMK, although the subnuclear distribution of the protein is altered (Fig. 3B, b). These results demonstrate that MITR is a nuclear export-resistant inhibitor of MEF2 and suggest that MITR may perform functions in the nucleus that distinguish it from HDAC4 and -5. Of note, analysis of the human genome sequence reveals the presence of a coding region for a putative HDAC domain approximately 50 kb downstream of the MITR sequence on chromosome 7, and this putative MITR HDAC domain possesses the conserved leucines and valines present in the class II HDAC-specific NES. The sequence of this putative NES is as follows: Val-Ser-Ala-Leu-Ala-Ser-Leu-Thr-Val.
Nuclear export of HDACs in the control of muscle differentiation.
Previously, we performed chromatin immunoprecipitation assays to examine the acetylation state of nucleosomal histones surrounding MEF2 binding sites in the regulatory regions of muscle-specific genes (17). Our results demonstrated that the degree of histone acetylation associated with these MEF2 response elements was significantly higher in differentiated myotubes than in undifferentiated myoblasts, supporting the notion that repression of the skeletal muscle differentiation program is coupled to histone deacetylation. Consistent with this, we have shown that HDAC5, which is a potent inhibitor of MEF2-dependent transcription, resides in the nucleus of proliferating, undifferentiated myoblasts and shuttles to the cytoplasm when cells are triggered to differentiate (21). Recently, HDAC7 was also shown to undergo nuclear export in response to myogenic cues (3). The data presented here provide a molecular explanation for these findings (see Fig. 7).
Our studies demonstrate that CaMK acts as a potent export kinase for class II HDACs. However, the existence of other HDAC kinases remains likely. Furthermore, it is conceivable that different class II HDACs are regulated by distinct kinases. Indeed, we have previously shown that HDAC4 and -5 are subject to differential regulation in yeast and mammalian cells (22).
The MEF2-HDAC axis in the control of diverse biological processes.
Class II HDACs are expressed at highest levels in skeletal muscle, heart, and brain (4, 6, 31), the same tissues in which MEF2 is most abundant (1). These overlapping expression patterns suggest roles for MEF2-HDAC interactions that extend beyond regulation of skeletal muscle differentiation. In neurons, MEF2 has been implicated in calcium-dependent survival pathways (18, 25), and CaMK signaling has been shown to affect learning and memory (9, 19, 27). In cardiac myocytes, the activities of MEF2 and CaMK are upregulated in response to stimuli that trigger cardiac hypertrophy and heart failure (11, 13, 14, 26, 40). As such, it is intriguing to speculate that MEF2-HDAC complexes serve to integrate normal and pathological signals in diverse organ systems.
ACKNOWLEDGMENTS
We thank S. Schreiber and A. Means for expression constructs. We are grateful to J. Page and W. Simpson for editorial assistance, A. Tizenor for graphics, and S. Bezprozvannaya for technical support.
This work was supported by grants from the National Institutes of Health, The D. W. Reynolds Center for Clinical Cardiovascular Research, and The Robert A. Welch Foundation to E.N.O. T.A.M. is a Pfizer fellow of the Life Sciences Research Foundation.
REFERENCES
- 1.Black B L, Olson E N. Transcriptional control of muscle development by myocyte enhancer factor-2 (MEF2) proteins. Annu Rev Cell Dev Biol. 1998;14:167–196. doi: 10.1146/annurev.cellbio.14.1.167. [DOI] [PubMed] [Google Scholar]
- 2.Bogerd H P, Fridell R A, Benson R E, Hua J, Cullen B R. Protein sequence requirements for function of the human T-cell leukemia virus type 1 Rex nuclear export signal delineated by a novel in vivo randomization-selection assay. Mol Cell Biol. 1996;16:4207–4214. doi: 10.1128/mcb.16.8.4207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dressel U, Bailey P J, Wang S C, Downes M, Evans R M, Muscat G E. A dynamic role for HDAC7 in MEF2-mediated muscle differentiation. J Biol Chem. 2001;276:17007–17013. doi: 10.1074/jbc.M101508200. [DOI] [PubMed] [Google Scholar]
- 4.Fischle W, Emiliani S, Hendzel M J, Nagase T, Nomura N, Voelter W, Verdin E. A new family of human histone deacetylases related to Saccharomyces cerevisiae HDA1p. J Biol Chem. 1999;274:11713–11720. doi: 10.1074/jbc.274.17.11713. [DOI] [PubMed] [Google Scholar]
- 5.Gorlich D, Kutay U. Transport between the cell nucleus and the cytoplasm. Annu Rev Cell Dev Biol. 1999;15:607–660. doi: 10.1146/annurev.cellbio.15.1.607. [DOI] [PubMed] [Google Scholar]
- 6.Grozinger C M, Hassig C A, Schreiber S L. Three proteins define a class of human histone deacetylases related to yeast Hda1p. Proc Natl Acad Sci USA. 1999;96:4868–4873. doi: 10.1073/pnas.96.9.4868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grozinger C M, Schreiber S L. Regulation of histone deacetylase 4 and 5 and transcriptional activity by 14-3-3-dependent cellular localization. Proc Natl Acad Sci USA. 2000;97:7835–7840. doi: 10.1073/pnas.140199597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Haribabu B, Hook S S, Selbert M A, Goldstein E G, Tomhave E D, Edelman A M, Snyderman R, Means A R. Human calcium-calmodulin dependent protein kinase I: cDNA cloning, domain structure and activation by phosphorylation at threonine-177 by calcium-calmodulin-dependent protein kinase I kinase. EMBO J. 1995;14:3679–3686. doi: 10.1002/j.1460-2075.1995.tb00037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ho N, Liauw J A, Blaeser F, Wei F, Hanissian S, Muglia L M, Wozniak D F, Nardi A, Arvin K L, Holtzman D M, Linden D J, Zhuo M, Muglia L J, Chatila T A. Impaired synaptic plasticity and cAMP response element-binding protein activation in Ca2+/calmodulin-dependent protein kinase type IV/Gr-deficient mice. J Neurosci. 2000;20:6459–6472. doi: 10.1523/JNEUROSCI.20-17-06459.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang E Y, Zhang J, Miska E A, Guenther M G, Kouzarides T, Lazar M A. Nuclear receptor corepressors partner with class II histone deacetylases in a Sin3-independent repression pathway. Genes Dev. 2000;14:45–54. [PMC free article] [PubMed] [Google Scholar]
- 11.Irons C E, Sei C A, Hidaka H, Glembotski C C. Protein kinase C and calmodulin kinase are required for endothelin-stimulated atrial natriuretic factor secretion from primary atrial myocytes. J Biol Chem. 1992;267:5211–5216. [PubMed] [Google Scholar]
- 12.Kao H Y, Downes M, Ordentlich P, Evans R M. Isolation of a novel histone deacetylase reveals that class I and class II deacetylases promote SMRT-mediated repression. Genes Dev. 2000;14:55–66. [PMC free article] [PubMed] [Google Scholar]
- 13.Kirchhefer U, Schmitz W, Scholz H, Neumann J. Activity of cAMP-dependent protein kinase and Ca2+/calmodulin-dependent protein kinase in failing and nonfailing human hearts. Cardiovasc Res. 1999;42:254–261. doi: 10.1016/s0008-6363(98)00296-x. [DOI] [PubMed] [Google Scholar]
- 14.Kolodziejczyk S M, Wang L, Balazsi K, DeRepentigny Y, Kothary R, Megeney L A. MEF2 is upregulated during cardiac hypertrophy and is required for normal post-natal growth of the myocardium. Curr Biol. 1999;9:1203–1206. doi: 10.1016/S0960-9822(00)80027-5. [DOI] [PubMed] [Google Scholar]
- 15.Lemercier C, Verdel A, Galloo B, Curtet S, Brocard M P, Khochbin S. mHDA1/HDAC5 histone deacetylase interacts with and represses MEF2A transcriptional activity. J Biol Chem. 2000;275:15594–15599. doi: 10.1074/jbc.M908437199. [DOI] [PubMed] [Google Scholar]
- 16.Lu J, McKinsey T A, Nicol R L, Olson E N. Signal-dependent activation of the MEF2 transcription factor by dissociation from histone deacetylases. Proc Natl Acad Sci USA. 2000;97:4070–4075. doi: 10.1073/pnas.080064097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lu J, McKinsey T A, Zhang C L, Olson E N. Regulation of skeletal myogenesis by association of the MEF2 transcription factor with class II histone deacetylases. Mol Cell. 2000;6:233–244. doi: 10.1016/s1097-2765(00)00025-3. [DOI] [PubMed] [Google Scholar]
- 18.Mao Z, Bonni A, Xia F, Nadal-Vicens M, Greenberg M E. Neuronal activity-dependent cell survival mediated by transcription factor MEF2. Science. 1999;286:785–790. doi: 10.1126/science.286.5440.785. [DOI] [PubMed] [Google Scholar]
- 19.Mayford M, Bach M E, Huang Y Y, Wang L, Hawkins R D, Kandel E R. Control of memory formation through regulated expression of a CaMKII transgene. Science. 1996;274:1678–1683. doi: 10.1126/science.274.5293.1678. [DOI] [PubMed] [Google Scholar]
- 20.McBride K M, McDonald C, Reich N C. Nuclear export signal located within theDNA-binding domain of the STAT1 transcription factor. EMBO J. 2000;19:6196–6206. doi: 10.1093/emboj/19.22.6196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McKinsey T A, Zhang C L, Lu J, Olson E N. Signal-dependent nuclear export of a histone deacetylase regulates muscle differentiation. Nature. 2000;408:106–111. doi: 10.1038/35040593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McKinsey T A, Zhang C L, Olson E N. Activation of the myocyte enhancer factor-2 transcription factor by calcium/calmodulin-dependent protein kinase-stimulated binding of 14-3-3 to histone deacetylase 5. Proc Natl Acad Sci USA. 2000;97:14400–14405. doi: 10.1073/pnas.260501497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McKinsey, T. A., C. L. Zhang, and E. N. Olson. Control of muscle development by dueling HATs and HDACs. Curr. Opin. Genet. Dev., in press. [DOI] [PubMed]
- 24.Miska E A, Karlsson C, Langley E, Nielsen S J, Pines J, Kouzarides T. HDAC4 deacetylase associates with and represses the MEF2 transcription factor. EMBO J. 1999;18:5099–5107. doi: 10.1093/emboj/18.18.5099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Okamoto S, Krainc D, Sherman K, Lipton S A. Antiapoptotic role of the p38 mitogen-activated protein kinase-myocyte enhancer factor 2 transcription factor pathway during neuronal differentiation. Proc Natl Acad Sci USA. 2000;97:7561–7566. doi: 10.1073/pnas.130502697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Passier R, Zeng H, Frey N, Naya F J, Nicol R L, McKinsey T A, Overbeek P, Richardson J A, Grant S R, Olson E N. CaM kinase signaling induces cardiac hypertrophy and activates the MEF2 transcription factor in vivo. J Clin Investig. 2000;105:1395–1406. doi: 10.1172/JCI8551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rongo C, Kaplan J M. CaMKII regulates the density of central glutamatergic synapses in vivo. Nature. 1999;402:195–199. doi: 10.1038/46065. [DOI] [PubMed] [Google Scholar]
- 28.Sartorelli V, Huang J, Hamamori Y, Kedes L. Molecular mechanisms of myogenic coactivation by p300: direct interaction with the activation domain of MyoD and with the MADS box of MEF2C. Mol Cell Biol. 1997;17:1010–1026. doi: 10.1128/mcb.17.2.1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sparrow D B, Miska E A, Langley E, Reynaud-Deonauth S, Kotecha S, Towers N, Spohr G, Kouzarides T, Mohun T J. MEF-2 function is modified by a novel co-repressor, MITR. EMBO J. 1999;18:5085–5098. doi: 10.1093/emboj/18.18.5085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Strahl B D, Allis C D. The language of covalent histone modifications. Nature. 2000;403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 31.Verdel A, Khochbin S. Identification of a new family of higher eukaryotic histone deacetylases. Coordinate expression of differentiation-dependent chromatin modifiers. J Biol Chem. 1999;274:2440–2445. doi: 10.1074/jbc.274.4.2440. [DOI] [PubMed] [Google Scholar]
- 32.Verdel A, Curtet S, Brocard M P, Rousseaux S, Lemercier C, Yoshida M, Khochbin S. Active maintenance of mHDA2/mHDAC6 histone-deacetylase in the cytoplasm. Curr Biol. 2000;10:747–749. doi: 10.1016/s0960-9822(00)00542-x. [DOI] [PubMed] [Google Scholar]
- 33.Wang A H, Bertos N R, Vezmar M, Pelletier N, Crosato M, Heng H H, Th'ng J, Han J, Yang X J. HDAC4, a human histone deacetylase related to yeast HDA1, is a transcriptional corepressor. Mol Cell Biol. 1999;19:7816–7827. doi: 10.1128/mcb.19.11.7816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang A H, Kruhlak M J, Wu J, Bertos N R, Vezmar M, Posner B I, Bazett-Jones D P, Yang X J. Regulation of histone deacetylase 4 by binding of 14-3-3 proteins. Mol Cell Biol. 2000;20:6904–6912. doi: 10.1128/mcb.20.18.6904-6912.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu X Y, Li H, Park E J, Chen J D. SMRTe inhibits MEF2C transcriptional activation by targeting HDAC4 and 5 to nuclear domains. J Biol Chem. 2001;276:24177–24185. doi: 10.1074/jbc.M100412200. [DOI] [PubMed] [Google Scholar]
- 36.Zhang C L, McKinsey T A, Lu J R, Olson E N. Association of COOH-terminal-binding protein (CtBP) and MEF2-interacting transcription repressor (MITR) contributes to transcriptional repression of the MEF2 transcription factor. J Biol Chem. 2001;276:35–39. doi: 10.1074/jbc.M007364200. [DOI] [PubMed] [Google Scholar]
- 37.Zhang C L, McKinsey T A, Olson E N. The transcriptional corepressor MITR is a signal-responsive inhibitor of myogenesis. Proc Natl Acad Sci USA. 2001;98:7354–7359. doi: 10.1073/pnas.131198498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou X, Richon V M, Rifkind R A, Marks P A. Identification of a transcriptional repressor related to the noncatalytic domain of histone deacetylases 4 and 5. Proc Natl Acad Sci USA. 2000;97:1056–1061. doi: 10.1073/pnas.97.3.1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhu J, McKeon F. NF-AT activation requires suppression of Crm1-dependent export by calcineurin. Nature. 1999;398:256–260. doi: 10.1038/18473. [DOI] [PubMed] [Google Scholar]
- 40.Zhu W, Zou Y, Shiojima I, Kudoh S, Aikawa R, Hayashi D, Mizukami M, Toko H, Shibasaki F, Yazaki Y, Nagai R, Komuro I. Ca2+/calmodulin-dependent kinase II and calcineurin play critical roles in endothelin-1-induced cardiomyocyte hypertrophy. J Biol Chem. 2000;275:15239–15245. doi: 10.1074/jbc.275.20.15239. [DOI] [PubMed] [Google Scholar]