

Abstract
The accumulation of an excessive amount of body fat can cause type 2 diabetes, and the risk of type 2 diabetes increases linearly with an increase in body mass index. Accordingly, the worldwide increase in the prevalence of obesity has led to a concomitant increase in the prevalence of type 2 diabetes. The cellular and physiological mechanisms responsible for the link between obesity and type 2 diabetes are complex and involve adiposity-induced alterations in β-cell function, adipose tissue biology, and multi-organ insulin resistance, which are often ameliorated and can even be normalized with adequate weight loss.
Introduction
Adipose tissue is the body’s major fuel reserve and provides an important transportable energy source that is critical for survival when food is scarce. The high energy density and hydrophobic nature of triglyceride make it a five-fold better fuel per unit mass than glycogen; triglycerides are compactly stored as an oil inside adipocytes and produce ~9.3 kcal per gram when oxidized, whereas glycogen is stored intracellularly as a gel, containing approximately 2 grams of water for every gram of glycogen, and produces only 4.1 kcal per gram when oxidized (Klein et al., 2002). During starvation, the duration of survival is determined by the size of the adipose tissue mass. Men who are lean die after approximately 60 days of starvation when more than 35% of body weight is lost (Leiter and Marliss, 1982), whereas people with extreme obesity can tolerate long-term fasting; the longest known duration of fasting was reported in a man with extreme obesity who fasted for 382 days, consuming only acaloric fluids, vitamins and minerals, resulting in a 60% loss in body weight without adverse effects (Stewart and Fleming, 1973). Adipose tissue also produces and secretes adipokines and exosomes, which are involved in the regulation of important physiological functions, such as appetite, reproductive function and insulin action (Funcke and Scherer, 2019).
The accumulation of an excessive amount of body fat induces a constellation of metabolic abnormalities and diseases, including insulin resistance, atherogenic dyslipidemia (high plasma triglyceride and low plasma HDL-cholesterol concentrations), nonalcoholic fatty liver disease (NAFLD), β-cell dysfunction, prediabetes, and type 2 diabetes. In general, a progressive increase in body mass index (BMI), which provides an index of adiposity, is associated with a progressive increase in the risk of developing type 2 diabetes (Colditz et al., 1995). However, fat and triglyceride distribution modify the risk of adiposity-induced metabolic dysfunction (Klein et al., 2002). People who are obese with a predominant increase in upper body fat (abdominal subcutaneous and intra-abdominal fat), intrahepatic triglyceride content, intramyocelluar lipid content and pancreatic fat) are at higher risk of developing type 2 diabetes than those with a lower body (gluteofemoral) fat phenotype. In fact, increased gluteofemoral body fat mass is associated with decreased plasma triglyceride and increased HDL-cholesterol concentrations, decreased fasting blood glucose and insulin concentrations, increased oral glucose tolerance and insulin sensitivity, and decreased risk of type 2 diabetes in people who are lean, overweight or obese (Manolopoulos et al., 2010).
Type 2 diabetes is caused by multi-organ insulin resistance, in conjunction with a decline in β-cell insulin secretory function (Bogardus and Tataranni, 2002). The worldwide increase in the prevalence of obesity is likely responsible for the recent increase in the prevalence of type 2 diabetes because obesity influences both insulin action and β-cell function. In this article, we will review the cellular and physiological mechanisms responsible for the link between excess adiposity and type 2 diabetes and the therapeutic metabolic effects of weight (fat) loss.
β-cell physiology and insulin kinetics
Pancreatic β-cells secrete insulin directly into the portal vein for delivery to the liver, which is the major site for insulin clearance. Plasma insulin concentration is determined by the balance between the rate of insulin secretion and the rate of insulin removal by the liver and extrahepatic tissues. A large portion (~50%) of the insulin secreted from β-cells and delivered to the liver is cleared during first pass transit and an additional 20% is cleared through subsequent passes; the remaining 30% of secreted insulin is primarily removed by the kidneys (about 20%) and skeletal muscle (about 10%) (Duckworth et al., 1998). The increase in both basal and postprandial plasma insulin concentrations observed in people with obesity is caused by both increased pancreatic insulin secretion and decreased fractional extraction and clearance of portal and peripheral plasma insulin (Gastaldelli et al., 2021; Kotronen et al., 2008; Kotronen et al., 2007; Smith et al., 2020a).
Pancreatic β-cell function is a critical determinant of whether people with obesity develop type 2 diabetes. Plasma insulin concentrations and the rate of insulin secretion during basal conditions and after glucose ingestion is typically greater in people with obesity who do not have type 2 diabetes than people who are lean (van Vliet et al., 2020). This increase in insulin secretion rate and plasma insulin concentration is often able to overcome the resistance to insulin action, so that fasting blood glucose concentration and oral glucose tolerance are normal. However, a progressive decline in β-cell function causes a progressive decline in glycemic control, resulting in prediabetes and ultimately type 2 diabetes (Gastaldelli et al., 2004; Weyer et al., 1999).
It is likely that the number of pancreatic β-cells, per se, influences the secretion of insulin during both basal and postprandial conditions. Pancreatic β-cell mass, often expressed as the relative volume (ratio of the β-cell area to exocrine area assessed at autopsy), is about 50% greater in people with obesity than in people who are lean. However the relative β-cell volume in those with impaired fasting glucose or type 2 diabetes is about 50% lower than the relative volume in lean people because of β-cell apoptosis (Butler et al., 2003). It is unlikely that the increase in β-cell mass associated with obesity is simply caused by insulin resistance, because weight gain in mice fed a high-fat diet is associated with an increased proliferation in β-cell mass before the development of insulin resistance (Mosser et al., 2015) and both basal and postprandial insulin secretion rates are greater in people with obesity than in lean people when both groups are matched on insulin sensitivity (van Vliet et al., 2020). The mechanism(s) responsible for β-cell hyperplasia in people with obesity is not known, but could be related to stimulation by specific nutrients, insulin, incretins and growth factors associated with high-calorie diet consumption and obesity (Linnemann et al., 2014).
Increased plasma free fatty acid (FFA) concentrations associated with obesity and type 2 diabetes can have adverse effects on β-cells. During basal conditions, circulating FFAs are responsible for about 30% of insulin secretion in people with or without diabetes (Boden et al., 1998). An acute (1.5 to 6 hours) increase in plasma FFAs, induced by infusing a lipid emulsion, increases glucose-stimulated insulin secretion, whereas a more prolonged increase in FFAs for 24 to 48 hours decreases glucose-stimulated insulin secretion in relationship to insulin sensitivity (Carpentier et al., 1999; Paolisso et al., 1995).. However, it has been argued that the results from studies that infused a lipid emulsion are not clinically relevant, because the plasma FFA concentrations achieved during those studies were much higher than the usual range found in people with obesity or type 2 diabetes and the composition of the plasma FFA profile generated by hydrolysis of lipid emulsion triglycerides is not physiologic (Weir, 2020). In addition, increases in plasma glucose are probably more harmful to β-cells than FFAs; mild increases in plasma glucose (11 mg/dL) causes phenotypic changes in gene expression that adversely affect β-cells function, growth, and survival (Ebrahimi et al., 2020). Nonetheless, this does not mean that the concept of β-cell “lipotoxicity” is incorrect, but that other lipid mediators, such as increased β-cell production of long-chain acyl-CoA esters, ceramides, phosphatidic acid and diacylglycerides, rather than plasma FFAs, are involved (Weir, 2020). It is likely that increases in plasma FFAs, triglycerides and glucose act synergistically, known as “glucolipotoxicity” (Poitout and Robertson, 2008), or possibly in combination with excess plasma amino acids, known as “nutrient-induced metabolic stress,” (Prentki et al., 2020) cause β-cell dysfunction and death, mediated by oxidative, mitochondrial, and endoplasmic reticulum stress, and β-cell dedifferentiation.
Adipose tissue biology and insulin resistance
Adipose tissue must have considerable metabolic flexibility to be able to cope with the large and rapid changes in energy balance during feeding and fasting throughout the day and to adjust to more long term changes in energy balance that cause adipose tissue expansion or reduction. The increase in adipose tissue mass after chronic positive energy balance is caused by an accumulation of triglycerides in adipocytes, which increases adipocyte size and requires structural remodeling to provide the scaffolding needed to support the expanded adipocyte mass. The specific adaptive responses of adipose tissue to its expansion are an important determinant of adipose tissue health and systemic metabolic homeostasis, and differences in responses likely contribute to the heterogeneity in metabolic health observed in people with obesity (Crewe et al., 2017; Scherer, 2016; Smith et al., 2019).
Studies conducted in a variety of mouse models of obesity demonstrate a series of complex but interactive biological processes in adipose tissue that contribute to whole-body insulin resistance, including: 1) adipocyte hypoxia, due to inadequate oxygen delivery and increased adipocyte oxygen demand (Seo et al., 2019; Sun et al., 2012), which stimulates adipose tissue fibrogenesis and macrophage chemotaxis (Halberg et al., 2009; Khan et al., 2009) and can suppress adipose tissue branched-chain amino acid catabolism thereby increasing plasma branched-chain amino acid concentrations (Bianchi et al., 2003; Lo et al., 2013); 2) increased number and relative proportion of adipose tissue proinflammatory immune cells (macrophages and T cells) and the expression of genes that encode proinflammatory proteins (Crewe et al., 2017); 3) decreased adipose tissue production and secretion of adiponectin, an insulin-sensitizing hormone (Straub and Scherer, 2019); 4) increased adipose tissue lipolytic activity and the release of FFAs into the circulation (Petersen and Shulman, 2018); and 5) metabolically-deleterious alterations in the cargo content of exosomes derived from adipose tissue macrophages (Liu et al., 2019; Thomou et al., 2017; Ying et al., 2017). Although each of these factors have been shown to cause insulin resistance in mouse models and reversing their effects improve insulin action, their importance in the pathogenesis of insulin resistance in people is less clear or not yet validated.
Many of the abnormalities in adipose tissue observed in obese rodents have also been identified in subcutaneous adipose tissue in people with obesity, including decreased interstitial adipose tissue partial pressure of oxygen (Cifarelli et al., 2020), increased rates of fibrogenesis and expression of genes involved in extracellular matrix formation (Beals et al., 2021), increased proinflammatory macrophage and T cell content and expression of genes that encode proinflammatory proteins (Fuchs et al., 2021), production of exosomes that can induce insulin resistance (Fuchs et al., 2021), and alterations in expression of genes involved in branched-chain amino acid catabolism (Cifarelli et al., 2020). However, the mean differences in many of these outcome measures between people with obesity who were “insulin-sensitive” and those who were “insulin-resistant” were often small, albeit statistically significant, and there was often considerable overlap in individual values between the two obese groups. Although these findings suggest many of these alterations are more closely associated with excess adiposity than insulin resistance, it is possible that these factors in aggregate induce insulin resistance.
Since the discovery in mice that adipose tissue produces proinflammatory cytokines that cause insulin resistance (Hotamisligil et al., 1993) and the finding that obesity in people is associated with an accumulation of adipose tissue macrophages (Weisberg et al., 2003), it has been proposed that adipose inflammation is a major driver of insulin resistance in people with obesity. Although inflammatory macrophages and the expression of genes that encode inflammatory proteins are increased in subcutaneous abdominal adipose tissue in people with “metabolically unhealthy obesity” compared with those with “metabolically healthy obesity” (Fuchs et al., 2021), it is difficult to determine whether these associations are a cause or an effect of insulin resistance. In fact, studies conducted in mouse models have found: i) insulin resistance causes an infiltration of proinflammatory macrophages in adipose tissue (Shimobayashi et al., 2018); ii) adipose tissue inflammation is part of the normal remodeling that occurs with an increase in body fat (Wernstedt Asterholm et al., 2014); and iii) suppressing adipose tissue inflammation causes insulin resistance (Zhu et al., 2020). The importance of adipose tissue immune cells and inflammation as a cause of insulin resistance in people with obesity is not resolved and requires further study.
If adipose tissue regulates metabolic function in other organs, it must somehow communicate with these organs. Several potential signaling mechanisms have been proposed that involve the secretion of adipose tissue products into the circulation that are then delivered to target tissues. These products include proinflammatory proteins, adiponectin, FFAs and exosomes, which are discussed below.
Chronic, low-grade, inflammation, manifested by increased proinflammatory immune cells and expression of genes that encode proinflammatory proteins, is present in people with obesity. However, the increase in adipose tissue gene expression often does not translate to an increase in plasma concentrations of the encoded proteins, and many of these cytokines act locally. A recent study that evaluated plasma cytokines every hour for 24 hours in people with obesity who were insulin-sensitive and those who were insulin-resistant found no difference in 24-hour plasma concentration area-under-the-curves for a battery of cytokines, with the exception of plasminogen activator-1 (PAI-1) (Fuchs et al., 2021). Plasma PAI-1 concentration was much greater in the obese insulin-resistant group than the obese insulin-sensitive group, and plasma PAI-1 was inversely correlated with measures of both hepatic and skeletal muscle insulin sensitivity. PAI-1 is produced by adipocytes and adipose tissue macrophages (encoded by SERPINE1). In rodent models, adipocyte-specific PAI-1 overexpression causes insulin resistance whereas whole-body and adipocyte-specific knockouts of PAI-1 improve insulin action (Alessi et al., 2007). These results suggest adipose tissue production of most cytokines, with the exception of PAI-1, have local paracrine effects, but do not have direct effects on systemic metabolic function.
Plasma adiponectin concentrations are often inversely associated with percent body fat, directly associated with insulin sensitivity (Straub and Scherer, 2019; Tschritter et al., 2003), and can be considered a biomarker of adipose tissue health (Li et al., 2021; Scherer, 2019). In rodents, adiponectin has anti-inflammatory, anti-fibrotic and insulin-sensitizing effects and increases pancreatic β cell survival and regeneration (Li et al., 2021; Rutkowski et al., 2013; Ye et al., 2015). The mechanism responsible for the pleiotropic therapeutic effects of adiponectin is likely mediated, at least in part, by increasing ceramidase activity and decreasing intracellular ceramide levels, which occurs when adiponectin binds to adiponectin cell surface receptors 1 and 2 (Holland et al., 2011).
Obesity is associated with an increased rate of release of FFAs into the bloodstream and delivery of FFAs to body tissues (Mittendorfer et al., 2009). Although there is a widespread belief that increased plasma FFA concentrations are an important cause of liver and muscle insulin resistance in people with obesity, this notion has been questioned because of conflicting data from different studies and the questionable extrapolation of data from experimental manipulations to real world conditions (Karpe et al., 2011). During basal conditions, the rate of release of FFAs into the bloodstream in relation to body fat mass is lower in people with obesity than in people who are lean; however, because of the large volume of body fat, the rate of release of FFAs in relation to body fat-free mass is greater in people with obesity than in people who are lean (Mittendorfer et al., 2009). Lipolysis of adipose tissue triglycerides is very sensitive to insulin (Conte et al., 2012; van Vliet et al., 2020), so the postprandial suppression of lipolysis and plasma FFA concentrations is often the same in people who are lean or obese because the greater postprandial increase in plasma insulin in people with obesity can compensate for their increase in fat mass (McQuaid et al., 2011; van Vliet et al., 2020). Experimental increases in plasma FFAs, induced by infusing a lipid emulsion and heparin, impairs, whereas suppression of plasma FFAs by treatment with acipimox, a nicotinic acid derivative that blocks adipose tissue hormone sensitive lipase, increases insulin-stimulated glucose disposal and insulin-mediated suppression of endogenous glucose production (Boden et al., 1994; Santomauro et al., 1999). However, the changes in plasma FFA concentrations in these studies were much greater than the usual small differences (or no differences) in fasting plasma FFA concentrations observed between lean people and people with obesity and between people with obesity who are insulin-sensitive and those who are insulin-resistant (Arner and Ryden, 2015; Fabbrini et al., 2009; Gastaldelli et al., 2017; Karpe et al., 2011). Nonetheless, it is possible that the specific intracellular metabolism of fatty acids, which can produce toxic products (e.g. reactive oxygen species, diacylglycerol and ceramides), rather than the amount of fatty acids delivered to a tissue, determines whether plasma FFAs have adverse effects on metabolic function.
The correlation between visceral (intra-peritoneal) adipose tissue mass, insulin resistance and risk of developing type 2 diabetes (Kissebah et al., 1982) has led to the belief that an increase in visceral fat is a major contributor to insulin resistance because of an increased release of FFAs directly into the portal circulation. However, only about 20% of FFAs delivered to the liver are derived from lipolysis of visceral fat in people with obesity, whereas about 80% are derived from lipolysis of subcutaneous fat (Nielsen et al., 2004). But there was a large range in values among individuals and the percentage of FFAs delivered to the liver from visceral adipose tissue lipolysis was as high as 50% in some participants, suggesting visceral fat might be an important contributor to hepatic insulin resistance and steatosis in some people. Only about 14% of total FFAs that appeared in the systemic circulation are derived from splanchnic sources (comprised of FFAs released from both subcutaneous and visceral fat). In addition, removal of about one-third of visceral fat by laparoscopic omentectomy does not affect insulin sensitivity in people with type 2 diabetes (Fabbrini et al., 2010b). Therefore, although FFAs released from visceral fat could influence hepatic insulin sensitivity, they are unlikely to be an important cause of insulin resistance in other tissues.
Recently, it has been shown that adipose tissue derived exosomes, which are small membrane bound extracellular vesicles that contain microRNAs, bioactive lipids and regulatory proteins, regulate metabolic function in other organs. Injecting exosomes isolated from adipose tissue macrophages obtained from lean mice into obese mice improves liver and muscle insulin sensitivity, whereas injecting exosomes isolated from adipose tissue macrophages obtained from obese mice induces liver and muscle insulin resistance in lean mice (Ying et al., 2017). This study also showed the effect of exosomes on insulin action was mediated by the transfer of specific microRNAs. People with “metabolically-unhealthy obesity” (defined as prediabetes and high intrahepatic triglyceride content) have a much greater number of exosomes in plasma than people who are lean or those with “metabolically-healthy obesity” (defined as normal glucose tolerance and normal intrahepatic triglyceride content) (Freeman et al., 2018; Fuchs et al., 2021). Moreover, plasma and adipose tissue-derived exosomes obtained from people with metabolically-unhealthy obesity, but not from people who are lean or those with metabolically-healthy obesity, decrease insulin-signaling in muscle myotubes and hepatocytes (Fuchs et al., 2021).
In summary, the data from studies conducted in animal models and in people demonstrate that obesity-induced alterations in adipose tissue metabolism, extracellular matrix formation, immune cells (primarily macrophages) and inflammation (primarily SERPINE1) are involved in regulating metabolic function in other organs (Figure 1). Differences in these factors among individuals likely contribute to the heterogeneity in metabolic health associated with obesity in people.
Figure 1.
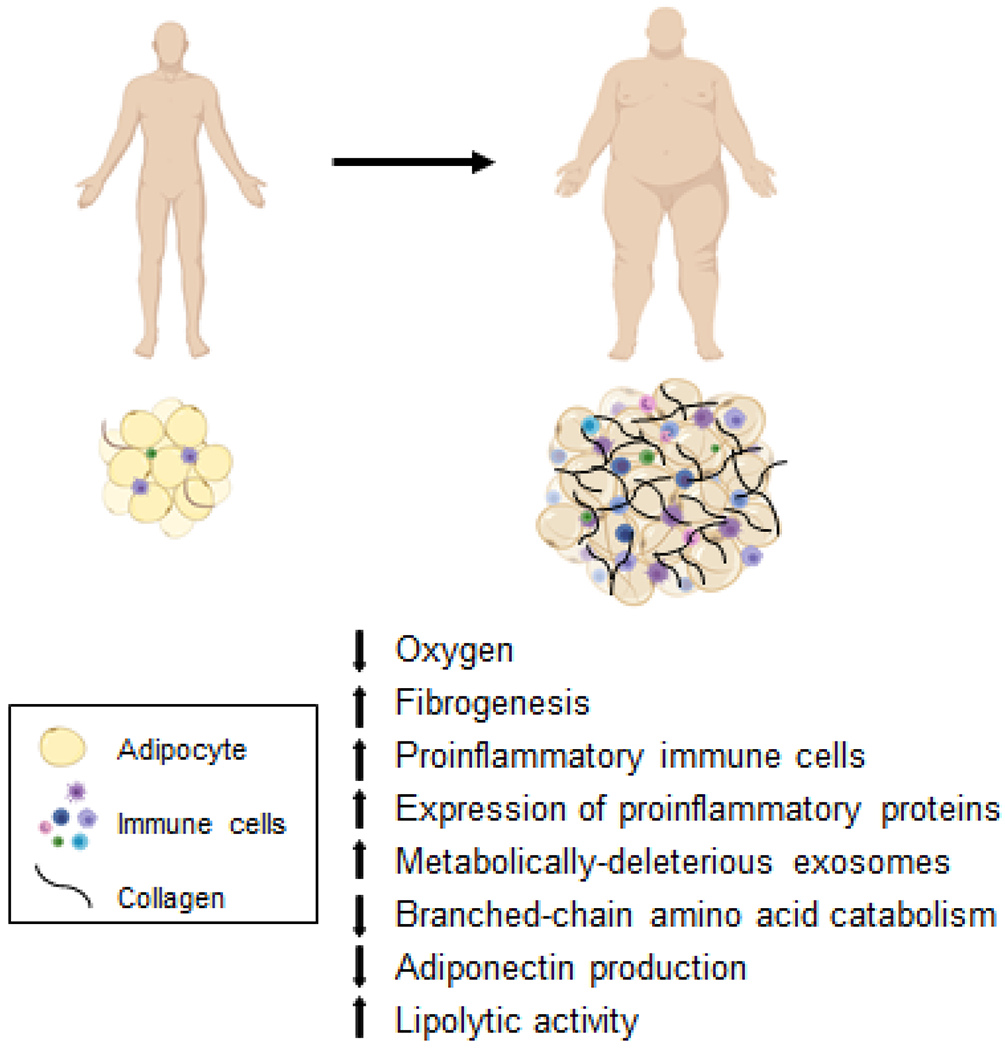
Alterations in adipose tissue biology associated with metabolic dysfunction in persons with obesity.
Hepatic glucose and lipid metabolism
The liver is the major source of endogenous glucose production. During basal conditions about 80% of endogenous glucose production is derived from hepatic glycogenolysis (glucose produced from the breakdown of liver glycogen) and gluconeogenesis (glucose produced from precursors such as lactate, glycerol and amino acids), and about 20% is derived from gluconeogenesis by the kidneys (Alsahli and Gerich, 2017). In lean people, glycogenolysis and gluconeogenesis contribute equally to total endogenous glucose production, but the contribution of gluconeogenesis is much higher in people with obesity or type 2 diabetes (Gastaldelli et al., 2000). An increase in gluconeogenesis is responsible for fasting hyperglycemia, and impaired suppression of endogenous glucose production and gluconeogenesis after meal ingestion contributes to postprandial hyperglycemia in people with prediabetes and type 2 diabetes (Gastaldelli et al., 2007).
Insulin in the portal vein is the major regulator of hepatic glucose production. During basal conditions, about 70% of the blood delivered to the liver comes from the portal vein and about 30% from the hepatic artery. Moreover, the concentration of insulin in the portal vein is about three times higher than the concentration in the hepatic artery and portal blood flow nearly doubles after ingestion of a mixed-meal (Dauzat et al., 1994). People with obesity typically have an impairment in the ability of insulin to suppress hepatic glucose production, but often have normal basal and postprandial hepatic glucose production rates because of increased insulin secretion (Smith et al., 2020a). However, increased hepatic glucose production occurs when the increased secretion of insulin is not adequate to compensate for insulin resistance as in people with impaired fasting glucose or when the secretion of insulin decreases as in people with type 2 diabetes (Groop et al., 1989; Natali et al., 2000). Insulin resistance in adipose tissue has indirect effects on hepatic glucose metabolism because impaired suppression of adipose tissue lipolysis increases the release of fatty acids that are delivered to the liver, which increases hepatic gluconeogenesis (Ader and Bergman, 1990).
Obesity and type 2 diabetes also have adverse effects on intrahepatic lipid metabolism and are a major cause of nonalcoholic fatty liver disease (NAFLD); about two-thirds of adults with obesity or type 2 diabetes have NAFLD (Browning et al., 2004; Leite et al., 2009). Hepatic steatosis is primarily caused by an increased production of triglycerides, not a decrease in either the oxidation of fatty acids or the export of triglycerides through very-low-density-lipoprotein (VLDL) secretion (Fabbrini et al., 2008; Iozzo et al., 2010). Insulin resistance and chronic hyperinsulinemia increase hepatic de novo lipogenesis (fatty acid synthesis from glucose) and the delivery of lipogenic substrates to the liver (i.e. glucose, fatty acids released from hydrolysis of subcutaneous and intra-peritoneal adipose tissue triglycerides, fatty acids released from hepatic hydrolysis of plasma triglycerides, and fatty acids that spill over into the systemic circulation during postprandial lipolysis of triglycerides in chylomicrons) (Donnelly et al., 2005; Fabbrini et al., 2010a; Smith et al., 2020b; Ter Horst et al., 2021). In fact, hepatic steatosis is unlikely to occur in people who do not have significant insulin resistance (Fabbrini et al., 2009; Ter Horst et al., 2017). Even people with a genetic variant of the patatin-like phospholipase domain-containing 3 gene (PNPLA3), which markedly increases the risk of NAFLD, are unlikely to develop hepatic steatosis in the absence of obesity and concomitant insulin resistance (Stender et al., 2017). In addition, both insulin resistance and increased intrahepatic triglyceride content are likely responsible for the increase in hepatic VLDL-triglyceride secretion rate and hypertriglyceridemia observed in people with obesity and NAFLD and those with type 2 diabetes (Packard et al., 2020).
The molecular mechanisms responsible for hepatic insulin-resistant glucose metabolism in people with obesity are not completely clear. Data from genetically modified rodent models and the limited data from studies conducted in humans demonstrate a decrease in hepatic insulin receptor number and function, in conjunction with proximal insulin signaling defects, cause a decrease in downstream insulin signaling pathways (Petersen and Shulman, 2018; Ter Horst et al., 2021). Although intrahepatic triglycerides per se are inert and do not directly impair insulin action, their production is associated with the formation of metabolically bioactive lipids, namely ceramides and diacylglycerols (DAGs) that can cause insulin resistance. Studies conducted in genetically modified rodents have shown that both ceramides, specifically those containing saturated C16 and C18 acyl chains (C16:0 and C18:0), and DAGs, specifically the sn-1,2-DAG stereoisomer, can inhibit proximal insulin signaling by affecting phosphorylation of insulin receptor substrate (IRS), phosphoinositide-3-kinase (PI3K), protein kinase C-theta (PKC-Θ), and protein kinase B (PKB also known as AKT) (Petersen and Shulman, 2018). The potential clinical relevance of these findings is supported by studies showing alterations in both intrahepatic ceramides and DAGs in people with NAFLD (Apostolopoulou et al., 2018; Luukkonen et al., 2016; Ter Horst et al., 2017).
Skeletal muscle glucose metabolism
During basal conditions, plasma free fatty acids serve as the primarily fuel for skeletal muscle. After glucose or mixed-meal ingestion, the increase in plasma insulin suppresses the lipolysis of adipose tissue triglycerides, decreases plasma free fatty acid concentrations and stimulates muscle glucose uptake, which causes a switch in the predominant muscle fuel from fatty acids to glucose. Insulin-stimulated muscle glucose uptake involves the binding of insulin to myocyte insulin receptors, which initiates a cascade of intracellular signaling events that result in the translocation of glucose transporter 4 (GLUT4) to the plasma membrane, which is necessary for glucose transport into the cell. After entering the myocyte glucose is immediately phosphorylated and can be oxidized for fuel via glycolysis or stored as glycogen (i.e. non-oxidative glucose disposal). Normally, approximately 30% of ingested glucose is taken up by skeletal muscle, of which 50% is oxidized, 35% is stored as glycogen and about 15% undergoes non-oxidative glycolysis, which produces lactate, alanine, and pyruvate (Kelley et al., 1988; Woerle et al., 2003). People with obesity and people with type 2 diabetes typically have an impairment in both insulin-stimulated muscle glucose oxidation and glycogen synthesis caused by a downregulation in the number and function of insulin receptors and multiple defects in post-receptor insulin signaling (Mitrakou et al., 1990; Petersen and Shulman, 2018). In addition, increases in intramyocellular lipid and intermyocellular adipose tissue are associated with obesity, type 2 diabetes, and impaired skeletal muscle insulin signaling (Kiefer et al., 2021; Virkamaki et al., 2001), suggesting alterations in muscle lipid distribution and metabolism contribute to the pathogenesis of insulin resistance (Kiefer et al., 2021; Virkamaki et al., 2001).
Effect of weight (fat) loss
Weight loss can have profound therapeutic effects on metabolic function, type 2 diabetes and diabetes comorbidities (Cohen et al., 2020; Gomez-Ambrosi et al., 2017; Klein et al., 2002) (Figure 2). Moderate 5%-10% weight loss improves glycemic control, plasma triglyceride and HDL-cholesterol concentrations and blood pressure (Wing et al., 2011). Greater weight loss can achieve diabetes remission, but the rate of remission depends primarily on the duration of diabetes, the ability of weight loss to improve β-cell function, and the criteria used to define remission (Camastra et al., 2011; Taylor et al., 2018). The ability of weight loss to induce diabetes remission has been demonstrated in randomized controlled trials that compared bariatric surgery with medical therapy: i) 73% of patients who had diabetes for less than 2 years achieved diabetes remission (defined as glycated hemoglobin [HbA1c] <6.2% without diabetes medications) at 24 months after laparoscopic adjustable gastric banding and 20% weight loss (Dixon et al., 2008); ii) 42% of patients who had diabetes for a mean of 8 years achieved diabetes remission (defined as HbA1c ≤6.0% without diabetes medications) at 12 months after Roux-en-Y gastric bypass surgery and 28% weight loss (Schauer et al., 2012); and iii) 75% of patients who had diabetes for a mean of 6 years achieved diabetes remission (defined as HbA1c <6.5% without diabetes medications) at 24 months after Roux-en-Y gastric bypass surgery and 33% weight loss (Mingrone et al., 2012). However, the durability of diabetes remission after bariatric surgery decreases over time and is associated with weight regain; only about 50% of patients maintain remission of diabetes (defined as no diabetes medications and either fasting plasma glucose <100 mg/dL or <126 mg/dL, or HbA1c <6.0%) 5 to 10 years after surgery (Arterburn et al., 2013; Brethauer et al., 2013; Sjostrom et al., 2004).
Figure 2.

Multi-organ therapeutic effects of weight loss
Recently, the ability to induce diabetes remission with a structured weight management program incorporated into a medical care setting was demonstrated by the DiRECT trial (Lean et al., 2018; Lean et al., 2019). This cluster-randomized trial was conducted within primary care practices in the United Kingdom in patients with type 2 diabetes diagnosed in the previous 6 years (mean 3 years) and not being treated with insulin. Diabetes remission (defined as HbA1c <6.5% [<48 mmol/mol] without diabetes medications) was achieved in 46% of patients at the end of 12 months and an average weight loss of approximately 10% (10 kg). Remission varied with the amount of weight loss and occurred in 7% of participants who lost 0–5 kg, 34% who lost 5–10 kg, 57% of participants who lost 10–15 kg, and 86% of participants who lost 15 kg or more.
In contrast to weight (fat) loss achieved by negative energy balance induced by bariatric surgery, diet therapy, or pharmacotherapy, surgical removal of adipose tissue does not result in metabolic benefits. For example, the removal of large amounts of subcutaneous abdominal adipose tissue (10 kg of adipose tissue which was equal to a 12% body weight loss and a decrease in 20% of total body fat mass) by using liposuction does not improve adipose tissue, liver or skeletal muscle insulin sensitivity or plasma lipids in women with obesity or obesity with type 2 diabetes (Klein et al., 2004), and removing approximately 30% of intra-abdominal adipose tissue by laparoscopic omentectomy does not improve whole-body insulin sensitivity in people with obesity and type 2 diabetes (Fabbrini et al., 2010b). These results demonstrate that fat loss must be induced by negative energy balance to achieve metabolic benefits.
Weight loss has potent effects on insulin action and even 5% weight loss improves multiorgan (adipose tissue, liver and skeletal muscle) insulin sensitivity (Lim et al., 2011; Magkos et al., 2016). However, the degree of functional improvement in response to a given amount of weight loss is not the same among all organs, and the amount of weight loss needed to achieve maximum metabolic benefits among different organs systems is not clear, and will likely differ from person-to-person based on the severity and duration of the metabolic abnormality. In general, insulin sensitivity in the liver (insulin-mediated suppression of glucose production) and adipose tissue (insulin-mediated suppression of lipolysis) are probably maximally improved with 5%-8% weight loss, whereas greater amounts of weight loss cause a further increase in skeletal muscle insulin sensitivity (insulin-mediated increase in glucose disposal) (Magkos et al., 2016; Petersen et al., 2005). Large weight loss can achieve remission of type 2 diabetes. For example, in the DiRECT trial, 57% of participants who lost 10%-15% body weight and 86% of participants who lost ≥15% body weight achieved remission of their diabetes (Lean et al., 2018).
The cellular mechanisms responsible for the therapeutic effects of weight loss on metabolic function are not clear. Adipose tissue is very responsive to negative energy balance and a decrease in body weight. A progressive increase in diet-induced weight loss causes a progressive decrease in whole-body, subcutaneous and intra-abdominal adipose tissue masses due primarily to a decrease in adipocyte size. Progressive weight loss also causes progressive changes in adipose tissue biology, manifested as a stepwise down-regulation of metabolic pathways and genes involved in lipid synthesis, extracellular matrix remodeling and collagen synthesis, plasminogen activator inhibitor-1 (PAI-1) production and oxidative stress (Magkos et al., 2016). However, the improvement in multi-organ insulin sensitivity after 5% weight loss is usually not associated with a decline in either circulating or subcutaneous adipose tissue markers of inflammation, suggesting the beneficial effect of 5% weight loss on insulin action is not mediated by a reduction in systemic or adipose tissue markers of inflammation. Progressive weight loss also causes a progressive decline in intrahepatic triglyceride content and improvement in liver histological features of NAFLD. In fact, intrahepatic triglyceride is particularly sensitive to negative energy balance; even 48 hours of treatment with a low-calorie diet causes a large decrease in intrahepatic triglyceride content (Kirk et al., 2009). The sensitivity of adipose tissue and the liver to small decreases in body weight suggest the therapeutic effects of weight loss are mediated, at least in part, by alterations in adipose tissue and liver physiology and the effect of adipose tissue on systemic signaling mechanisms, such as adiponectin, PAI-1 and exosomes released from adipose tissue.
Conclusions
Obesity, particularly when associated with increased abdominal and intra-abdominal fat distribution and increased intrahepatic and intramuscular triglyceride content, is a major risk factor for prediabetes and type 2 diabetes because it causes both insulin resistance and β-cell dysfunction. Accordingly, the worldwide increase in the prevalence of obesity has led to the concomitant increase in the prevalence of type 2 diabetes. A better understanding of the mechanisms responsible for the adverse effects of excess body fat on the factors involved in the pathogenesis of type 2 diabetes can lead to novel therapeutic interventions to prevent and treat this debilitating disease. A series of studies conducted in mouse models and in people have demonstrated alterations in adipose tissue biology that link obesity with insulin resistance and β-cell dysfunction. These alterations include adipose tissue fibrosis (increased rates of fibrogenesis and expression of genes involved in extracellular matrix formation), inflammation (increased proinflammatory macrophage and T cell content and the production of PAI-1), and the production of exosomes that can induce insulin resistance. However, none of these factors can influence systemic metabolic function without a mechanism for adipose tissue communication with other organs. It is possible that several adipose tissue secretory products that are released into the bloodstream, including PAI-1, adiponectin, FFAs and exosomes, are involved in this signaling process, but additional research is needed to fully assess their clinical importance. In addition, it is also likely that cross-talk among adipose tissue, the liver, muscle and pancreatic islets contribute to insulin resistance and hepatic steatosis (Figure 3). Decreasing body fat mass by inducing a negative energy balance, not by surgical removal, can ameliorate or normalize obesity-induced metabolic dysfunction and can even achieve diabetes remission if there is adequate restoration of β-cell function.
Figure 3.
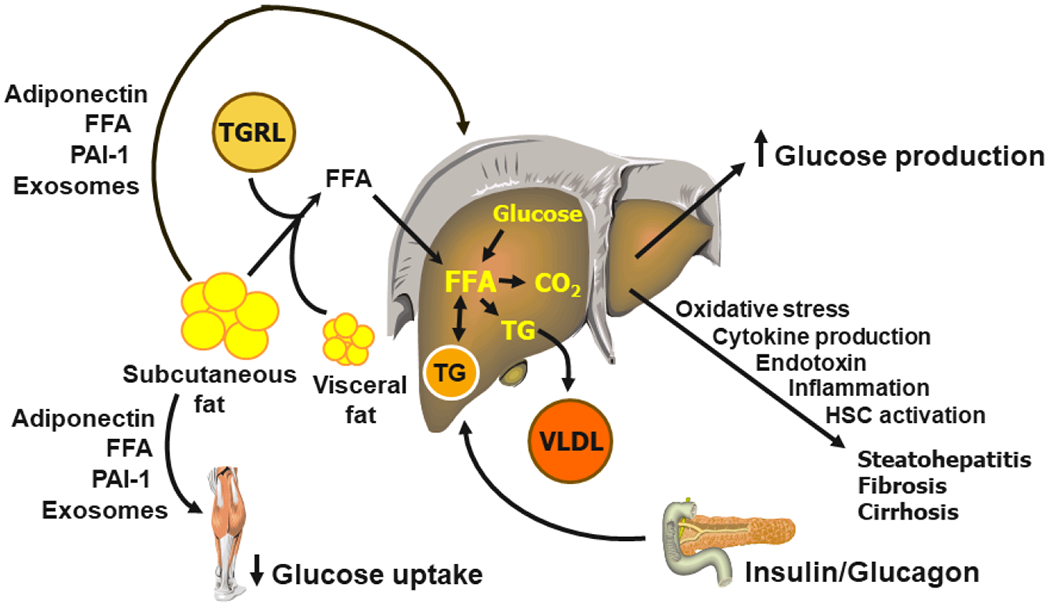
Inter-organ cross-talk in the pathogenesis of metabolic dysfunction in people with obesity. CO2 = carbon dioxide, HSC = hepatic stellate cell, FFA = free fatty acid, PAI-1 = plasminogen activator inhibitor-1, TG = triglyceride, TGRL = triglyceride-rich lipoprotein, VLDL = very-low-density lipoprotein.
Klein et al. examine the cellular and physiological mechanisms that explain why people with obesity develop metabolic dysfunction and type 2 diabetes. They also review the effects of weight loss, induced by negative energy balance, on adipose tissue biology and multi-organ system function that can cause remission of diabetes.
Funding:
Supported by National Institutes of Health grants DK56341 (Nutrition Obesity Research Center), DK20579 (Diabetes Research Center), UL1TR002345 (Clinical and Translational Science Award), RC2-DK118620, R01-DK55758 and R01-DK099110, the Academy of Finland, and support from Merck, Novo Nordisk and the Sigrid Jusélius Foundation.
Declaration of interests:
SK serves as a scientific consultant for Janssen and Altimmune and has a sponsored research agreement with Janssen. AG serves as a scientific consultant for Eli Lilly, Gilead, Inventiva, and Boehringer. HY-J serves as a scientific consultant for Merck, Novo Nordisk, Eli Lilly and Hamni Pharmaceuticals. PES has sponsored research agreements with Merck and Novo Nordisk.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ader M, and Bergman RN (1990). Peripheral effects of insulin dominate suppression of fasting hepatic glucose production. Am J Physiol 258, E1020–1032. 10.1152/ajpendo.1990.258.6.E1020 [DOI] [PubMed] [Google Scholar]
- Alessi MC, Poggi M, and Juhan-Vague I (2007). Plasminogen activator inhibitor-1, adipose tissue and insulin resistance. Curr Opin Lipidol 18, 240–245. 10.1097/MOL.0b013e32814e6d29 [DOI] [PubMed] [Google Scholar]
- Alsahli M, and Gerich JE (2017). Renal glucose metabolism in normal physiological conditions and in diabetes. Diabetes Res Clin Pract 133, 1–9. 10.1016/j.diabres.2017.07.033 [DOI] [PubMed] [Google Scholar]
- Apostolopoulou M, Gordillo R, Koliaki C, Gancheva S, Jelenik T, De Filippo E, Herder C, Markgraf D, Jankowiak F, Esposito I, et al. (2018). Specific Hepatic Sphingolipids Relate to Insulin Resistance, Oxidative Stress, and Inflammation in Nonalcoholic Steatohepatitis. Diabetes Care 41, 1235–1243. 10.2337/dc17-1318 [DOI] [PubMed] [Google Scholar]
- Arner P, and Ryden M (2015). Fatty Acids, Obesity and Insulin Resistance. Obes Facts 8, 147–155. 10.1159/000381224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arterburn DE, Bogart A, Sherwood NE, Sidney S, Coleman KJ, Haneuse S, O’Connor PJ, Theis MK, Campos GM, McCulloch D, et al. (2013). A multisite study of long-term remission and relapse of type 2 diabetes mellitus following gastric bypass. Obes Surg 23, 93–102. 10.1007/s11695-012-0802-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beals JW, Smith GI, Shankaran M, Fuchs A, Schweitzer GG, Yoshino J, Field T, Matthews M, Nyangau E, Morozov D, et al. (2021). Increased Adipose Tissue Fibrogenesis, Not Impaired Expandability, Is Associated With Nonalcoholic Fatty Liver Disease. Hepatology 74, 1287–1299. 10.1002/hep.31822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi G, Marchesini G, Brunetti N, Manicardi E, Montuschi F, Chianese R, and Zoli M (2003). Impaired insulin-mediated amino acid plasma disappearance in non-alcoholic fatty liver disease: a feature of insulin resistance. Dig Liver Dis 35, 722–727. [DOI] [PubMed] [Google Scholar]
- Boden G, Chen X, and Iqbal N (1998). Acute lowering of plasma fatty acids lowers basal insulin secretion in diabetic and nondiabetic subjects. Diabetes 47, 1609–1612. 10.2337/diabetes.47.10.1609 [DOI] [PubMed] [Google Scholar]
- Boden G, Chen X, Ruiz J, White JV, and Rossetti L (1994). Mechanisms of fatty acid-induced inhibition of glucose uptake. J Clin Invest 93, 2438–2446. 10.1172/JCI117252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogardus C, and Tataranni PA (2002). Reduced early insulin secretion in the etiology of type 2 diabetes mellitus in Pima Indians. Diabetes 51 Suppl 1, S262–264. 10.2337/diabetes.51.2007.s262 [DOI] [PubMed] [Google Scholar]
- Brethauer SA, Aminian A, Romero-Talamas H, Batayyah E, Mackey J, Kennedy L, Kashyap SR, Kirwan JP, Rogula T, Kroh M, et al. (2013). Can diabetes be surgically cured? Long-term metabolic effects of bariatric surgery in obese patients with type 2 diabetes mellitus. Ann Surg 258, 628–636. 10.1097/SLA.0b013e3182a5034b [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning JD, Szczepaniak LS, Dobbins R, Nuremberg P, Horton JD, Cohen JC, Grundy SM, and Hobbs HH (2004). Prevalence of hepatic steatosis in an urban population in the United States: impact of ethnicity. Hepatology 40, 1387–1395. 10.1002/hep.20466 [DOI] [PubMed] [Google Scholar]
- Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, and Butler PC (2003). Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes 52, 102–110. 10.2337/diabetes.52.1.102 [DOI] [PubMed] [Google Scholar]
- Camastra S, Gastaldelli A, Mari A, Bonuccelli S, Scartabelli G, Frascerra S, Baldi S, Nannipieri M, Rebelos E, Anselmino M, et al. (2011). Early and longer term effects of gastric bypass surgery on tissue-specific insulin sensitivity and beta cell function in morbidly obese patients with and without type 2 diabetes. Diabetologia 54, 2093–2102. 10.1007/s00125-011 [DOI] [PubMed] [Google Scholar]
- Carpentier A, Mittelman SD, Lamarche B, Bergman RN, Giacca A, and Lewis GF (1999). Acute enhancement of insulin secretion by FFA in humans is lost with prolonged FFA elevation. Am J Physiol 276, E1055–1066. 10.1152/ajpendo.1999.276.6.E1055 [DOI] [PubMed] [Google Scholar]
- Cifarelli V, Beeman SC, Smith GI, Yoshino J, Morozov D, Beals JW, Kayser BD, Watrous JD, Jain M, Patterson BW, et al. (2020). Decreased adipose tissue oxygenation associates with insulin resistance in individuals with obesity. J Clin Invest 130, 6688–6699. 10.1172/JCI141828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen RV, Pereira TV, Aboud CM, Petry TBZ, Lopes Correa JL, Schiavon CA, Pompilio CE, Pechy FNQ, da Costa Silva ACC, de Melo FLG, et al. (2020). Effect of Gastric Bypass vs Best Medical Treatment on Early-Stage Chronic Kidney Disease in Patients With Type 2 Diabetes and Obesity: A Randomized Clinical Trial. JAMA Surg 155, e200420. 10.1001/jamasurg.2020.0420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colditz GA, Willett WC, Rotnitzky A, and Manson JE (1995). Weight gain as a risk factor for clinical diabetes mellitus in women. Ann Intern Med 122, 481–486. 10.7326/0003-4819-122-7-199504010-00001 [DOI] [PubMed] [Google Scholar]
- Conte C, Fabbrini E, Kars M, Mittendorfer B, Patterson BW, and Klein S (2012). Multiorgan insulin sensitivity in lean and obese subjects. Diabetes Care 35, 1316–1321. 10.2337/dc11-1951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crewe C, An YA, and Scherer PE (2017). The ominous triad of adipose tissue dysfunction: inflammation, fibrosis, and impaired angiogenesis. J Clin Invest 127, 74–82. 10.1172/JCI88883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dauzat M, Lafortune M, Patriquin H, and Pomier-Layrargues G (1994). Meal induced changes in hepatic and splanchnic circulation: a noninvasive Doppler study in normal humans. Eur J Appl Physiol Occup Physiol 68, 373–380. 10.1007/BF00843732 [DOI] [PubMed] [Google Scholar]
- Dixon JB, O’Brien PE, Playfair J, Chapman L, Schachter LM, Skinner S, Proietto J, Bailey M, and Anderson M (2008). Adjustable gastric banding and conventional therapy for type 2 diabetes: a randomized controlled trial. JAMA 299, 316–323. 10.1001/jama.299.3.316 [DOI] [PubMed] [Google Scholar]
- Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, and Parks EJ (2005). Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest 115, 1343–1351. 10.1172/JCI23621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duckworth WC, Bennett RG, and Hamel FG (1998). Insulin degradation: progress and potential. Endocr Rev 19, 608–624. 10.1210/edrv.19.5.0349 [DOI] [PubMed] [Google Scholar]
- Ebrahimi AG, Hollister-Lock J, Sullivan BA, Tsuchida R, Bonner-Weir S, and Weir GC (2020). Beta cell identity changes with mild hyperglycemia: Implications for function, growth, and vulnerability. Mol Metab 35, 100959. 10.1016/j.molmet.2020.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabbrini E, Magkos F, Mohammed BS, Pietka T, Abumrad NA, Patterson BW, Okunade A, and Klein S (2009). Intrahepatic fat, not visceral fat, is linked with metabolic complications of obesity. Proc Natl Acad Sci U S A 106, 15430–15435. 10.1073/pnas.0904944106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabbrini E, Mohammed BS, Magkos F, Korenblat KM, Patterson BW, and Klein S (2008). Alterations in adipose tissue and hepatic lipid kinetics in obese men and women with nonalcoholic fatty liver disease. Gastroenterology 134, 424–431. 10.1053/j.gastro.2007.11.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabbrini E, Sullivan S, and Klein S (2010a). Obesity and nonalcoholic fatty liver disease: biochemical, metabolic, and clinical implications. Hepatology 51, 679–689. 10.1002/hep.23280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabbrini E, Tamboli RA, Magkos F, Marks-Shulman PA, Eckhauser AW, Richards WO, Klein S, and Abumrad NN (2010b). Surgical removal of omental fat does not improve insulin sensitivity and cardiovascular risk factors in obese adults. Gastroenterology 139, 448–455. 10.1053/j.gastro.2010.04.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman DW, Noren Hooten N, Eitan E, Green J, Mode NA, Bodogai M, Zhang Y, Lehrmann E, Zonderman AB, Biragyn A, et al. (2018). Altered Extracellular Vesicle Concentration, Cargo, and Function in Diabetes. Diabetes 67, 2377–2388. 10.2337/db17-1308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs A, Samovski D, Smith GI, Cifarelli V, Farabi SS, Yoshino J, Pietka T, Chang SW, Ghosh S, Myckatyn TM, et al. (2021). Associations Among Adipose Tissue Immunology, Inflammation, Exosomes and Insulin Sensitivity in People With Obesity and Nonalcoholic Fatty Liver Disease. Gastroenterology 161, 968–981 e912. 10.1053/j.gastro.2021.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funcke JB, and Scherer PE (2019). Beyond adiponectin and leptin: adipose tissue-derived mediators of inter-organ communication. J Lipid Res 60, 1648–1684. 10.1194/jlr.R094060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gastaldelli A, Abdul Ghani M, and DeFronzo RA (2021). Adaptation of Insulin Clearance to Metabolic Demand Is a Key Determinant of Glucose Tolerance. Diabetes 70, 377–385. 10.2337/db19-1152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gastaldelli A, Baldi S, Pettiti M, Toschi E, Camastra S, Natali A, Landau BR, and Ferrannini E (2000). Influence of obesity and type 2 diabetes on gluconeogenesis and glucose output in humans: a quantitative study. Diabetes 49, 1367–1373. 10.2337/diabetes.49.8.1367 [DOI] [PubMed] [Google Scholar]
- Gastaldelli A, Casolaro A, Pettiti M, Nannipieri M, Ciociaro D, Frascerra S, Buzzigoli E, Baldi S, Mari A, and Ferrannini E (2007). Effect of pioglitazone on the metabolic and hormonal response to a mixed meal in type II diabetes. Clin Pharmacol Ther 81, 205–212. 10.1038/sj.clpt.6100034 [DOI] [PubMed] [Google Scholar]
- Gastaldelli A, Ferrannini E, Miyazaki Y, Matsuda M, DeFronzo RA, and San Antonio metabolism study (2004). Beta-cell dysfunction and glucose intolerance: results from the San Antonio metabolism (SAM) study. Diabetologia 47, 31–39. 10.1007/s00125-003-1263-9 [DOI] [PubMed] [Google Scholar]
- Gastaldelli A, Gaggini M, and DeFronzo RA (2017). Role of Adipose Tissue Insulin Resistance in the Natural History of Type 2 Diabetes: Results From the San Antonio Metabolism Study. Diabetes 66, 815–822. 10.2337/db16-1167 [DOI] [PubMed] [Google Scholar]
- Gomez-Ambrosi J, Andrada P, Valenti V, Rotellar F, Silva C, Catalan V, Rodriguez A, Ramirez B, Moncada R, Escalada J, et al. (2017). Dissociation of body mass index, excess weight loss and body fat percentage trajectories after 3 years of gastric bypass: relationship with metabolic outcomes. Int J Obes (Lond) 41, 1379–1387. 10.1038/ijo.2017.134 [DOI] [PubMed] [Google Scholar]
- Groop LC, Bonadonna RC, DelPrato S, Ratheiser K, Zyck K, Ferrannini E, and DeFronzo RA (1989). Glucose and free fatty acid metabolism in non-insulin-dependent diabetes mellitus. Evidence for multiple sites of insulin resistance. J Clin Invest 84, 205–213. 10.1172/JCI114142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halberg N, Khan T, Trujillo ME, Wernstedt-Asterholm I, Attie AD, Sherwani S, Wang ZV, Landskroner-Eiger S, Dineen S, Magalang UJ, et al. (2009). Hypoxia-inducible factor 1alpha induces fibrosis and insulin resistance in white adipose tissue. Mol Cell Biol 29, 4467–4483. 10.1128/MCB.00192-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland WL, Miller RA, Wang ZV, Sun K, Barth BM, Bui HH, Davis KE, Bikman BT, Halberg N, Rutkowski JM, et al. (2011). Receptor-mediated activation of ceramidase activity initiates the pleiotropic actions of adiponectin. Nat Med 17, 55–63. 10.1038/nm.2277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotamisligil GS, Shargill NS, and Spiegelman BM (1993). Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science 259, 87–91. [DOI] [PubMed] [Google Scholar]
- Iozzo P, Bucci M, Roivainen A, Nagren K, Jarvisalo MJ, Kiss J, Guiducci L, Fielding B, Naum AG, Borra R, et al. (2010). Fatty acid metabolism in the liver, measured by positron emission tomography, is increased in obese individuals. Gastroenterology 139, 846–856. 10.1053/j.gastro.2010.05.039 [DOI] [PubMed] [Google Scholar]
- Karpe F, Dickmann JR, and Frayn KN (2011). Fatty acids, obesity, and insulin resistance: time for a reevaluation. Diabetes 60, 2441–2449. 10.2337/db11-0425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley D, Mitrakou A, Marsh H, Schwenk F, Benn J, Sonnenberg G, Arcangeli M, Aoki T, Sorensen J, Berger M, et al. (1988). Skeletal muscle glycolysis, oxidation, and storage of an oral glucose load. J Clin Invest 81, 1563–1571. 10.1172/JCI113489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan T, Muise ES, Iyengar P, Wang ZV, Chandalia M, Abate N, Zhang BB, Bonaldo P, Chua S, and Scherer PE (2009). Metabolic dysregulation and adipose tissue fibrosis: role of collagen VI. Mol Cell Biol 29, 1575–1591. 10.1128/MCB.01300-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiefer LS, Fabian J, Rospleszcz S, Lorbeer R, Machann J, Kraus MS, Roemer F, Rathmann W, Meisinger C, Heier M, et al. (2021). Distribution patterns of intramyocellular and extramyocellular fat by magnetic resonance imaging in subjects with diabetes, prediabetes and normoglycaemic controls. Diabetes Obes Metab 23, 1868–1878. 10.1111/dom.14413 [DOI] [PubMed] [Google Scholar]
- Kirk E, Reeds DN, Finck BN, Mayurranjan SM, Patterson BW, and Klein S (2009). Dietary fat and carbohydrates differentially alter insulin sensitivity during caloric restriction. Gastroenterology 136, 1552–1560. 10.1053/j.gastro.2009.01.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kissebah AH, Vydelingum N, Murray R, Evans DJ, Hartz AJ, Kalkhoff RK, and Adams PW (1982). Relation of body fat distribution to metabolic complications of obesity. J Clin Endocrinol Metab 54, 254–260. 10.1210/jcem-54-2-254 [DOI] [PubMed] [Google Scholar]
- Klein S, Fontana L, Young VL, Coggan AR, Kilo C, Patterson BW, and Mohammed BS (2004). Absence of an effect of liposuction on insulin action and risk factors for coronary heart disease. N Engl J Med 350, 2549–2557. 10.1056/NEJMoa033179 [DOI] [PubMed] [Google Scholar]
- Klein S, Wadden T, and Sugerman HJ (2002). AGA technical review on obesity. Gastroenterology 123, 882–932. [DOI] [PubMed] [Google Scholar]
- Kotronen A, Juurinen L, Tiikkainen M, Vehkavaara S, and Yki-Jarvinen H (2008). Increased liver fat, impaired insulin clearance, and hepatic and adipose tissue insulin resistance in type 2 diabetes. Gastroenterology 135, 122–130. 10.1053/j.gastro.2008.03.021 [DOI] [PubMed] [Google Scholar]
- Kotronen A, Vehkavaara S, Seppala-Lindroos A, Bergholm R, and Yki-Jarvinen H (2007). Effect of liver fat on insulin clearance. Am J Physiol Endocrinol Metab 293, E1709–1715. 10.1152/ajpendo.00444.2007 [DOI] [PubMed] [Google Scholar]
- Lean ME, Leslie WS, Barnes AC, Brosnahan N, Thom G, McCombie L, Peters C, Zhyzhneuskaya S, Al-Mrabeh A, Hollingsworth KG, et al. (2018). Primary care-led weight management for remission of type 2 diabetes (DiRECT): an open-label, cluster-randomised trial. Lancet 391, 541–551. 10.1016/S0140-6736(17)33102-1 [DOI] [PubMed] [Google Scholar]
- Lean MEJ, Leslie WS, Barnes AC, Brosnahan N, Thom G, McCombie L, Peters C, Zhyzhneuskaya S, Al-Mrabeh A, Hollingsworth KG, et al. (2019). Durability of a primary care-led weight-management intervention for remission of type 2 diabetes: 2-year results of the DiRECT open-label, cluster-randomised trial. Lancet Diabetes Endocrinol 7, 344–355. 10.1016/S2213-8587(19)30068-3 [DOI] [PubMed] [Google Scholar]
- Leite NC, Salles GF, Araujo AL, Villela-Nogueira CA, and Cardoso CR (2009). Prevalence and associated factors of non-alcoholic fatty liver disease in patients with type-2 diabetes mellitus. Liver Int 29, 113–119. 10.1111/j.1478-3231.2008.01718.x [DOI] [PubMed] [Google Scholar]
- Leiter LA, and Marliss EB (1982). Survival during fasting may depend on fat as well as protein stores. JAMA 248, 2306–2307. [PubMed] [Google Scholar]
- Li N, Zhao S, Zhang Z, Zhu Y, Gliniak CM, Vishvanath L, An YA, Wang MY, Deng Y, Zhu Q, et al. (2021). Adiponectin preserves metabolic fitness during aging. Elife 10, e65108. 10.7554/eLife.65108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim EL, Hollingsworth KG, Aribisala BS, Chen MJ, Mathers JC, and Taylor R (2011). Reversal of type 2 diabetes: normalisation of beta cell function in association with decreased pancreas and liver triacylglycerol. Diabetologia 54, 2506–2514. 10.1007/s00125-011-2204-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linnemann AK, Baan M, and Davis DB (2014). Pancreatic β-cell proliferation in obesity. Adv Nutr 5, 278–288. 10.3945/an.113.005488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, Sun YC, Cheng P, and Shao HG (2019). Adipose tissue macrophage-derived exosomal miR-29a regulates obesity-associated insulin resistance. Biochem Biophys Res Commun 515, 352–358. 10.1016/j.bbrc.2019.05.113 [DOI] [PubMed] [Google Scholar]
- Lo KA, Labadorf A, Kennedy NJ, Han MS, Yap YS, Matthews B, Xin X, Sun L, Davis RJ, Lodish HF, et al. (2013). Analysis of in vitro insulin-resistance models and their physiological relevance to in vivo diet-induced adipose insulin resistance. Cell Rep 5, 259–270. 10.1016/j.celrep.2013.08.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luukkonen PK, Zhou Y, Sadevirta S, Leivonen M, Arola J, Oresic M, Hyotylainen T, and Yki-Jarvinen H (2016). Hepatic ceramides dissociate steatosis and insulin resistance in patients with non-alcoholic fatty liver disease. J Hepatol 64, 1167–1175. 10.1016/j.jhep.2016.01.002 [DOI] [PubMed] [Google Scholar]
- Magkos F, Fraterrigo G, Yoshino J, Luecking C, Kirbach K, Kelly SC, de Las Fuentes L, He S, Okunade AL, Patterson BW, et al. (2016). Effects of moderate and subsequent progressive weight loss on metabolic function and adipose tissue biology in humans with obesity. Cell Metab 23, 591–601. 10.1016/j.cmet.2016.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manolopoulos KN, Karpe F, and Frayn KN (2010). Gluteofemoral body fat as a determinant of metabolic health. Int J Obes (Lond) 34, 949–959. 10.1038/ijo.2009.286 [DOI] [PubMed] [Google Scholar]
- McQuaid SE, Hodson L, Neville MJ, Dennis AL, Cheeseman J, Humphreys SM, Ruge T, Gilbert M, Fielding BA, Frayn KN, et al. (2011). Downregulation of adipose tissue fatty acid trafficking in obesity: a driver for ectopic fat deposition? Diabetes 60, 47–55. 10.2337/db10-0867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mingrone G, Panunzi S, De Gaetano A, Guidone C, laconelli A, Leccesi L, Nanni G, Pomp A, Castagneto M, Ghirlanda G, et al. (2012). Bariatric surgery versus conventional medical therapy for type 2 diabetes. N Engl J Med 366, 1577–1585. 10.1056/NEJMoa1200111 [DOI] [PubMed] [Google Scholar]
- Mitrakou A, Kelley D, Veneman T, Jenssen T, Pangburn T, Reilly J, and Gerich J (1990). Contribution of abnormal muscle and liver glucose metabolism to postprandial hyperglycemia in NIDDM. Diabetes 39, 1381–1390. 10.2337/diab.39.11.1381 [DOI] [PubMed] [Google Scholar]
- Mittendorfer B, Magkos F, Fabbrini E, Mohammed BS, and Klein S (2009). Relationship between body fat mass and free fatty acid kinetics in men and women. Obesity (Silver Spring) 17, 1872–1877. 10.1038/oby.2009.224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosser RE, Maulis MF, Moulle VS, Dunn JC, Carboneau BA, Arasi K, Pappan K, Poitout V, and Gannon M (2015). High-fat diet-induced beta-cell proliferation occurs prior to insulin resistance in C57Bl/6J male mice. Am J Physiol Endocrinol Metab 308, E573–582. 10.1152/ajpendo.00460.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natali A, Toschi E, Camastra S, Gastaldelli A, Groop L, and Ferrannini E (2000). Determinants of postabsorptive endogenous glucose output in non-diabetic subjects. European Group for the Study of Insulin Resistance (EGIR). Diabetologia 43, 1266–1272. 10.1007/s001250051522 [DOI] [PubMed] [Google Scholar]
- Nielsen S, Guo Z, Johnson CM, Hensrud DD, and Jensen MD (2004). Splanchnic lipolysis in human obesity. J Clin Invest 113, 1582–1588. 10.1172/JCI21047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Packard CJ, Boren J, and Taskinen MR (2020). Causes and Consequences of Hypertriglyceridemia. Front Endocrinol (Lausanne) 11, 252. 10.3389/fendo.2020.00252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paolisso G, Gambardella A, Amato L, Tortoriello R, D’Amore A, Varricchio M, and D’Onofrio F (1995). Opposite effects of short- and long-term fatty acid infusion on insulin secretion in healthy subjects. Diabetologia 38, 1295–1299. 10.1007/BF00401761 [DOI] [PubMed] [Google Scholar]
- Petersen KF, Dufour S, Befroy D, Lehrke M, Hendler RE, and Shulman GI (2005). Reversal of nonalcoholic hepatic steatosis, hepatic insulin resistance, and hyperglycemia by moderate weight reduction in patients with type 2 diabetes. Diabetes 54, 603–608. 10.2337/diabetes.54.3.603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen MC, and Shulman GI (2018). Mechanisms of Insulin Action and Insulin Resistance. Physiol Rev 98, 2133–2223. 10.1152/physrev.00063.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poitout V, and Robertson RP (2008). Glucolipotoxicity: fuel excess and beta-cell dysfunction. Endocr Rev 29, 351–366. 10.1210/er.2007-0023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prentki M, Peyot ML, Masiello P, and Madiraju SRM (2020). Nutrient-Induced Metabolic Stress, Adaptation, Detoxification, and Toxicity in the Pancreatic beta-Cell. Diabetes 69, 279–290. 10.2337/dbi19-0014 [DOI] [PubMed] [Google Scholar]
- Rutkowski JM, Wang ZV, Park AS, Zhang J, Zhang D, Hu MC, Moe OW, Susztak K, and Scherer PE (2013). Adiponectin promotes functional recovery after podocyte ablation. J Am Soc Nephrol 24, 268–282. 10.1681/ASN.2012040414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santomauro AT, Boden G, Silva ME, Rocha DM, Santos RF, Ursich MJ, Strassmann PG, and Wajchenberg BL (1999). Overnight lowering of free fatty acids with Acipimox improves insulin resistance and glucose tolerance in obese diabetic and nondiabetic subjects. Diabetes 48, 1836–1841. 10.2337/diabetes.48.9.1836 [DOI] [PubMed] [Google Scholar]
- Schauer PR, Kashyap SR, Wolski K, Brethauer SA, Kirwan JP, Pothier CE, Thomas S, Abood B, Nissen SE, and Bhatt DL (2012). Bariatric surgery versus intensive medical therapy in obese patients with diabetes. N Engl J Med 366, 1567–1576. 10.1056/NEJMoa1200225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherer PE (2016). The Multifaceted Roles of Adipose Tissue-Therapeutic Targets for Diabetes and Beyond: The 2015 Banting Lecture. Diabetes 65, 1452–1461. 10.2337/db16-0339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherer PE (2019). The many secret lives of adipocytes: implications for diabetes. Diabetologia 62, 223–232. 10.1007/s00125-018-4777-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo JB, Riopel M, Cabrales P, Huh JY, Bandyopadhyay GK, Andreyev AY, Murphy AN, Beeman SC, Smith GI, Klein S, et al. (2019). Knockdown of ANT2 reduces adipocyte hypoxia and improves insulin resistance in obesity. Nature Metabolism 1, 86–97. 10.1038/s42255-018-0003-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimobayashi M, Albert V, Woelnerhanssen B, Frei IC, Weissenberger D, Meyer-Gerspach AC, Clement N, Moes S, Colombi M, Meier JA, et al. (2018). Insulin resistance causes inflammation in adipose tissue. J Clin Invest 128, 1538–1550. 10.1172/JCI96139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjostrom L, Lindroos AK, Peltonen M, Torgerson J, Bouchard C, Carlsson B, Dahlgren S, Larsson B, Narbro K, Sjostrom CD, et al. (2004). Lifestyle, diabetes, and cardiovascular risk factors 10 years after bariatric surgery. N Engl J Med 351, 2683–2693. 10.1056/NEJMoa035622 [DOI] [PubMed] [Google Scholar]
- Smith GI, Mittendorfer B, and Klein S (2019). Metabolically healthy obesity: facts and fantasies. J Clin Invest 129, 3978–3989. 10.1172/JCI129186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith GI, Polidori DC, Yoshino M, Kearney ML, Patterson BW, Mittendorfer B, and Klein S (2020a). Influence of adiposity, insulin resistance, and intrahepatic triglyceride content on insulin kinetics. J Clin Invest 130, 3305–3314. 10.1172/JCI136756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith GI, Shankaran M, Yoshino M, Schweitzer GG, Chondronikola M, Beals JW, Okunade AL, Patterson BW, Nyangau E, Field T, et al. (2020b). Insulin resistance drives hepatic de novo lipogenesis in nonalcoholic fatty liver disease. J Clin Invest 130, 1453–1460. 10.1172/JCI134165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stender S, Kozlitina J, Nordestgaard BG, Tybjaerg-Hansen A, Hobbs HH, and Cohen JC (2017). Adiposity amplifies the genetic risk of fatty liver disease conferred by multiple loci. Nat Genet 49, 842–847. 10.1038/ng.3855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart WK, and Fleming LW (1973). Features of a successful therapeutic fast of 382 days’ duration. Postgrad Med J 49, 203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straub LG, and Scherer PE (2019). Metabolic Messengers: Adiponectin. Nat Metab 1, 334–339. 10.1038/s42255-019-0041-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun K, Wernstedt Asterholm I, Kusminski CM, Bueno AC, Wang ZV, Pollard JW, Brekken RA, and Scherer PE (2012). Dichotomous effects of VEGF-A on adipose tissue dysfunction. Proc Natl Acad Sci U S A 109, 5874–5879. 10.1073/pnas.1200447109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor R, Al-Mrabeh A, Zhyzhneuskaya S, Peters C, Barnes AC, Aribisala BS, Hollingsworth KG, Mathers JC, Sattar N, and Lean MEJ (2018). Remission of Human Type 2 Diabetes Requires Decrease in Liver and Pancreas Fat Content but Is Dependent upon Capacity for beta Cell Recovery. Cell Metab 28, 547–556 e543. 10.1016/j.cmet.2018.07.003 [DOI] [PubMed] [Google Scholar]
- Ter Horst KW, Gilijamse PW, Versteeg RI, Ackermans MT, Nederveen AJ, la Fleur SE, Romijn JA, Nieuwdorp M, Zhang D, Samuel VT, et al. (2017). Hepatic Diacylglycerol-Associated Protein Kinase Cepsilon Translocation Links Hepatic Steatosis to Hepatic Insulin Resistance in Humans. Cell Rep 19, 1997–2004. 10.1016/j.celrep.2017.05.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ter Horst KW, Vatner DF, Zhang D, Cline GW, Ackermans MT, Nederveen AJ, Verheij J, Demirkiran A, van Wagensveld BA, Dallinga-Thie GM, et al. (2021). Hepatic Insulin Resistance Is Not Pathway Selective in Humans With Nonalcoholic Fatty Liver Disease. Diabetes Care 44, 489–498. 10.2337/dc20-1644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomou T, Mori MA, Dreyfuss JM, Konishi M, Sakaguchi M, Wolfrum C, Rao TN, Winnay JN, Garcia-Martin R, Grinspoon SK, et al. (2017). Adipose-derived circulating miRNAs regulate gene expression in other tissues. Nature 542, 450–455. 10.1038/nature21365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tschritter O, Fritsche A, Thamer C, Haap M, Shirkavand F, Rahe S, Staiger H, Maerker E, Haring H, and Stumvoll M (2003). Plasma adiponectin concentrations predict insulin sensitivity of both glucose and lipid metabolism. Diabetes 52, 239–243. 10.2337/diabetes.52.2.239 [DOI] [PubMed] [Google Scholar]
- van Vliet S, Koh HE, Patterson BW, Yoshino M, LaForest R, Gropler RJ, Klein S, and Mittendorfer B (2020). Obesity Is Associated With Increased Basal and Postprandial beta-Cell Insulin Secretion Even in the Absence of Insulin Resistance. Diabetes 69, 2112–2119. 10.2337/db20-0377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virkamaki A, Korsheninnikova E, Seppala-Lindroos A, Vehkavaara S, Goto T, Halavaara J, Hakkinen AM, and Yki-Jarvinen H (2001). Intramyocellular lipid is associated with resistance to in vivo insulin actions on glucose uptake, antilipolysis, and early insulin signaling pathways in human skeletal muscle. Diabetes 50, 2337–2343. 10.2337/diabetes.50.10.2337 [DOI] [PubMed] [Google Scholar]
- Weir GC (2020). Glucolipotoxicity, beta-Cells, and Diabetes: The Emperor Has No Clothes. Diabetes 69, 273–278. 10.2337/db19-0138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, and Ferrante AW Jr. (2003). Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest 112, 1796–1808. 10.1172/JCI19246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wernstedt Asterholm I, Tao C, Morley TS, Wang QA, Delgado-Lopez F, Wang ZV, and Scherer PE (2014). Adipocyte inflammation is essential for healthy adipose tissue expansion and remodeling. Cell Metab 20, 103–118. 10.1016/j.cmet.2014.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weyer C, Bogardus C, Mott DM, and Pratley RE (1999). The natural history of insulin secretory dysfunction and insulin resistance in the pathogenesis of type 2 diabetes mellitus. J Clin Invest 104, 787–794. 10.1172/JCI7231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wing RR, Lang W, Wadden TA, Safford M, Knowler WC, Bertoni AG, Hill JO, Brancati FL, Peters A, Wagenknecht L, et al. (2011). Benefits of modest weight loss in improving cardiovascular risk factors in overweight and obese individuals with type 2 diabetes. Diabetes Care 34, 1481–1486. 10.2337/dc10-2415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woerle HJ, Meyer C, Dostou JM, Gosmanov NR, Islam N, Popa E, Wittlin SD, Welle SL, and Gerich JE (2003). Pathways for glucose disposal after meal ingestion in humans. Am J Physiol Endocrinol Metab 284, E716–725. 10.1152/ajpendo.00365.2002 [DOI] [PubMed] [Google Scholar]
- Ye R, Wang M, Wang QA, and Scherer PE (2015). Adiponectin-mediated antilipotoxic effects in regenerating pancreatic islets. Endocrinology 156, 2019–2028. 10.1210/en.2015-1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying W, Riopel M, Bandyopadhyay G, Dong Y, Birmingham A, Seo JB, Ofrecio JM, Wollam J, Hernandez-Carretero A, Fu W, et al. (2017). Adipose Tissue Macrophage-Derived Exosomal miRNAs Can Modulate In Vivo and In Vitro Insulin Sensitivity. Cell 171, 372–384 e312. 10.1016/j.cell.2017.08.035 [DOI] [PubMed] [Google Scholar]
- Zhu Q, An YA, Kim M, Zhang Z, Zhao S, Zhu Y, Asterholm IW, Kusminski CM, and Scherer PE (2020). Suppressing adipocyte inflammation promotes insulin resistance in mice. Mol Metab 39, 101010. 10.1016/j.molmet.2020.101010 [DOI] [PMC free article] [PubMed] [Google Scholar]