

Abstract
The proximal tubule relies on oxidative mitochondrial metabolism to meet its energy needs and has limited capacity for glycolysis, which makes it uniquely susceptible to damage during AKI, especially after ischemia and anoxia. Under these conditions, mitochondrial ATP production is initially decreased by several mechanisms, including fatty acid–induced uncoupling and inhibition of respiration related to changes in the shape and volume of mitochondria. Glycolysis is initially insufficient as a source of ATP to protect the cells and mitochondrial function, but supplementation of tricarboxylic acid cycle intermediates augments anaerobic ATP production, and improves recovery of mitochondrial oxidative metabolism. Incomplete recovery is characterized by defects of respiratory enzymes and lipid metabolism. During the transition to CKD, tubular cells atrophy but maintain high expression of glycolytic enzymes, and there is decreased fatty acid oxidation. These metabolic changes may be amenable to a number of therapeutic interventions.
Keywords: acute kidney injury and ICU nephrology, AKI, aTP, basic science, CKD, glycolysis, metabolism, mitochondria, tricarboxylic acid cycle
Introduction
AKI is one of the most common problems encountered by nephrologists, yet we lack efficacious therapies. The proximal tubule of the nephron is a prime site for tubular injury, which is partially due to its high energy requirements to sustain solute transport and its dependence on oxidative metabolism to meet its energy needs. Our understanding of the central role of mitochondrial abnormalities and alterations in metabolism in both acute and chronic kidney injury has steadily improved over the last 20 years. There are now potential therapeutic targets to improve mitochondrial function in AKI that positively affect the metabolic derangements that occur in AKI (1–3).
These promising possibilities increase the need for nephrologists to understand energy metabolism in the proximal tubule and alterations that occur during AKI. The purpose of this review is to briefly examine the basics of energy metabolism in the kidney under normal circumstances, and consider some of the adaptions of oxidative metabolism during two stages of injury, which we have investigated in detail along with related work by others. First, we will address the contribution of glycolysis and anaerobic pathways of mitochondrial ATP production in survival and protection of tubule cell mitochondrial function during primary injury, and, second, the initial metabolic shift that occurs during the AKI to CKD transition.
Modes of ATP Production in the Kidney
Tubule cells utilize several different pathways and sources of fuel to produce ATP. When glucose is used as the primary energy source, one molecule of glucose is converted to two pyruvates via glycolysis (Figure 1). Under aerobic conditions, the pyruvate formed will then be shuttled into the tricarboxylic acid (TCA) cycle, and the reduced NAD (NADH) generated will be used to drive mitochondrial oxidative metabolism (Figure 1). During aerobic metabolism of one molecule of glucose, two ATPs are generated in the cytosol via glycolysis (4). Subsequent mitochondrial oxidative metabolism by pyruvate dehydrogenase and the TCA cycle generates an additional 30 ATPs, or roughly five ATPs per carbon of the glucose molecule (5). If fatty acids are used as the fuel source, they undergo beta-oxidation to generate acetyl-CoA, which is then shuttled into the TCA cycle to undergo mitochondrial oxidation (6,7). In contrast, if mitochondrial oxidative metabolism is absent, the anaerobic metabolism of glucose yields just the net of two ATPs available at the end of the glycolysis pathway, and pyruvate is subsequently converted to lactate by lactate dehydrogenase. Converting pyruvate to lactate serves to remove the proton from NADH, allowing NAD to recycle and continue the process (Figure 2) (4). Less well known as a potential source of anaerobic ATP production than anaerobic glycolysis is a mitochondrial pathway for substrate level phosphorylation via α-ketoglutarate (αKG)-dehydrogenase and succinyl-CoA synthetase in the TCA cycle (Figure 2) (8,9). Although not readily amenable to interventions in vivo (10,11), manipulating this pathway in isolated tubules has proven to be very informative for understanding mitochondrial sensitivity to injury (12–14).
Figure 1.

Pathways of aerobic tubule ATP production. Glycolysis of one glucose molecule yields two pyruvates and two molecules of ATP. In the TCA cycle, further aerobic metabolism of these pyruvates yields an additional 30–32 ATPs or five ATPs/carbon. Amino acids (via transamination) and carboxylic acids are aerobically metabolized in this fashion. Fatty acids enter the TCA cycle via β-oxidation and yield a net of 6–8 ATPs per carbon. ATP yields are corrected for estimated efficiency as discussed in refs. (4–7). CoA, coenzyme A; FADH2, reduced flavin adenine dinucleotide; NADH, reduced NAD; TCA, tricarboxylic acid.
Figure 2.

Pathways of anaerobic tubule ATP production. Under anaerobic conditions, pyruvate undergoes anaerobic fermentation and generates lactate and only two ATPs. A limited amount of ATP can also be generated anaerobically during TCA cycle substrate level phosphorylation. ADP, adenosine diphosphate; αKG, α-ketoglutarate; ASP, aspartate; CoA, Coenzyme A; FADH2, reduced flavin adenine dinucleotide; FUM, fumarate; GDP, guanosine diphosphate; GTP, guanosine triphosphate; MAL, malate; NADH, reduced NAD; OAA, oxaloacetate; Pi, inorganic phosphate; SUCC, succinate; TCA, tricarboxylic acid.
Proximal Tubule Cells Rely on Mitochondria and Oxidative Metabolism to Meet Their Energy Needs
Oxidative metabolism mediated by abundant mitochondria is essential to proximal tubule function because high amounts of ATP are needed for their high rates of transport (15). During mitochondrial oxidative metabolism, substrates that are transported across the outer and inner mitochondrial membranes into the matrix are metabolized in the TCA cycle to produce NADH and reduced flavin adenine dinucleotide that, in turn, delivers electrons to the electron transport chain on the membrane, where they are progressively transferred to oxygen (Figure 3). In this process, hydrogen ions are pumped out of the matrix at three different points along the electron transport chain, to generate an electrochemical potential across the membrane that consists of both a hydrogen ion gradient and a membrane potential. That potential is used via the F1F0-ATPAse on the inner mitochondrial membrane to drive phosphorylation of ADP to ATP (16,17).
Figure 3.

Mitochondrial energy transduction. NADH and FADH2 deliver electrons to the electron transport chain on the inner mitochondrial membrane where they are progressively transferred to oxygen. Hydrogen ions are pumped out of the mitochondrial matrix to generate an electrochemical potential, which powers the F1F0-ATPAse on the inner mitochondrial membrane to drive phosphorylation of ADP to ATP. ADP, adenosine diphosphate; FADH2, reduced flavin adenine dinucleotide; IMM, inner mitochondrial membrane; NADH, reduced NAD; TCA, tricarboxylic acid. Adapted from ref. (17).
Mitochondrial oxidative metabolism is also required for the utilization of fatty acids, which are a high-yield source of fuel in the proximal tubule (18–21). The ATP yield from beta oxidation and TCA cycle metabolism of the acetyl CoA formed is roughly 6–8 ATPs per carbon or 106–129 ATPs per molecule of the typical fatty acid, palmitate (7).
Role of Anaerobic Glycolysis in Proximal Tubular Cells
Although quantitatively much less than oxidative metabolism, ATP production by anaerobic glycolysis can be critically important in the cellular response to injury because it can provide the very small amounts of ATP necessary to preserve viability during the primary insult and permit recovery afterwards (22). Glycolytic metabolism is limited in fully differentiated proximal tubule cells, because the enzymes needed for it are present at much lower levels than in distal segments, to favor the gluconeogenic function of the tubules (Figure 4) (23). In the proximal tubule, S3 segments in the outer medulla have higher glycolytic capacity than the S1 and S2 segments in the cortex (24,25).
Figure 4.
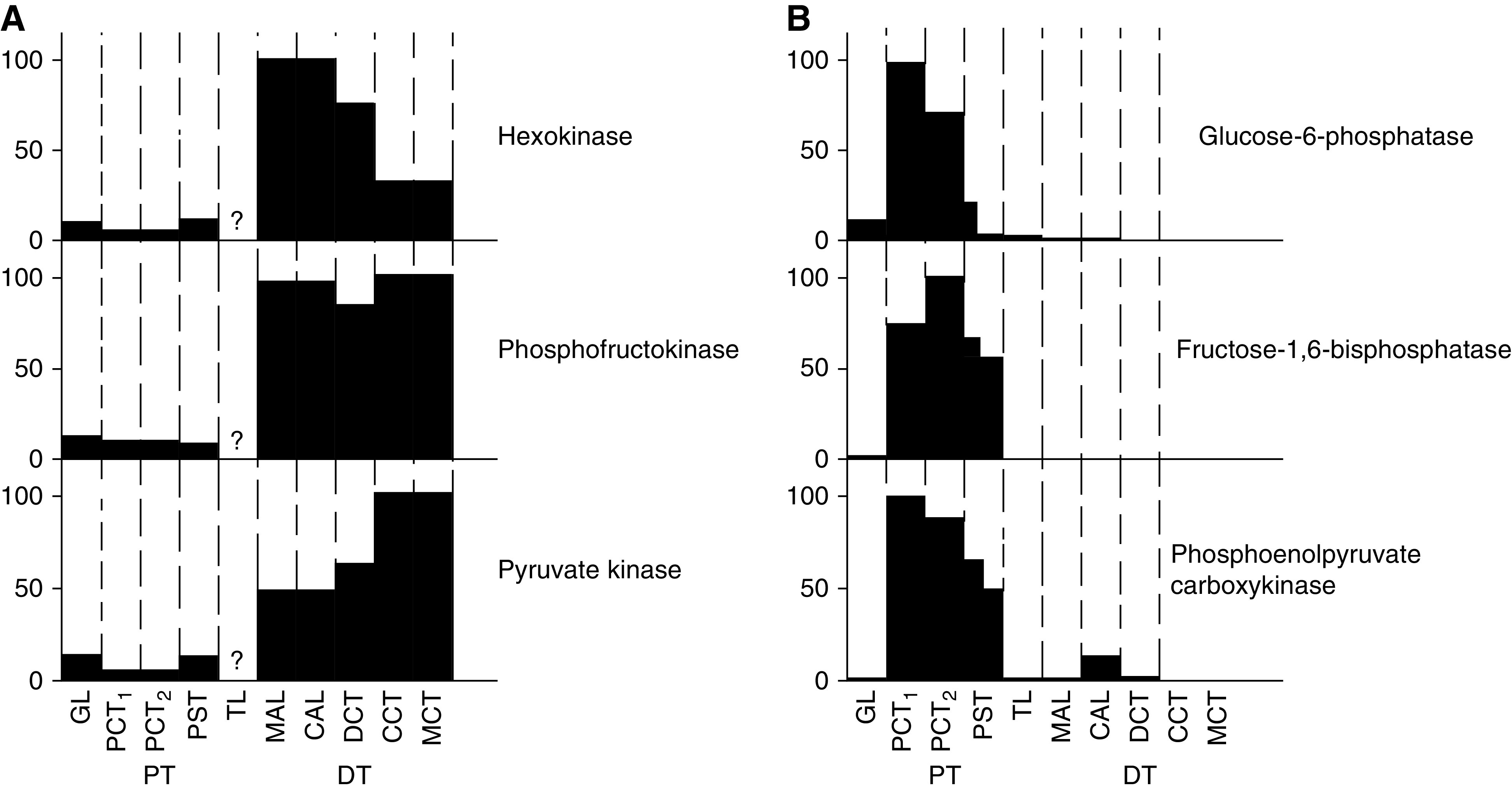
Differential expression of glycolytic and gluconeogenic enzymes along the nephron. (A) Expression of enzymes involved in glycolysis. (B) Expression of enzymes involved in gluconeogenesis. GL, glomerulus; PCT, early proximal convoluted tubule; PCT2, late proximal convoluted tubule; PST, proximal straight tubule; TL, loop of Henle; MAL, medullary thick ascending limb; CAL, cortical ascending limb; CT, distal convoluted tubule; CCT, cortical collecting tubule; MCT, medullary collecting tubule. Figure is from ref. (23) with permission.
Proximal tubules microdissected from snap-frozen kidney after short periods of ischemia had the most ATP depletion during inhibition of mitochondrial oxidative metabolism compared with other nephron segments, and had the lowest amount of lactate production, suggesting there was less flux through the glycolytic pathways in the proximal tubule compared with other nephron segments (26). Other investigators have shown that there is essentially no lactate production in the proximal tubule under complete inhibition of electron transport (27). More recent imaging studies have demonstrated better preservation of mitochondrial energization in distal than proximal tubules in intact kidney tissue treated with cyanide, which is attributable to the use of glycolytic ATP in the distal segments to support energization (28).
Whether the small amount of glycolysis possible in proximal tubules modifies ATP depletion and resulting injury has been debated. In isolated tubule suspensions, addition of glucose provided a small benefit against cell killing during anoxia, but it did not translate into improved respiratory function during reoxygenation of the more sensitive proximal convoluted tubules (24). Despite the presence of glucose, isolated proximal tubules subject to anoxia undergo very rapid ATP depletion and extensive cell killing, more so than in vivo (29). Even if this heightened in vitro sensitivity to injury is corrected by replacement of the cytoprotective amino acid, glycine, which delays ATP depletion-induced lethal plasma membrane damage, recovery of energetic function during reoxygenation is still impaired (29). Reoxygenated tubules have markedly decreased mitochondrial membrane potential (13,16,30).
Supplementation of TCA Cycle Intermediates can Improve Survival during Anoxia
Although there is little or no benefit of anaerobic glycolysis for fully differentiated proximal tubule cells subjected to anoxia, they can be salvaged by supplementing TCA cycle intermediates that enable anaerobic ATP generation. As discussed above and shown in Figure 2, substrate level phosphorylation via αKG-dehydrogenase and succinyl-CoA synthetase in the TCA cycle results in direct phosphorylation of GDP, and the newly formed GTP can generate ATP by transphosphorylation. However, this process cannot continue unless NAD can be regenerated anaerobically from the NADH formed. This can be promoted by additionally providing aspartate, which oxidizes the NADH back to NAD, and produces malate, then fumarate (Figure 2) (8,9,12). Additionally, the fumarate formed can theoretically drive energization via an anaerobic cycle that can occur between complexes I and II of the electron transport chain, although the presence of this second pathway in mammalian cells has been debated (31).
Consistent with operation of these pathways, supplementing intact tubules with αKG + aspartate supports both ATP production and energization, with strong effects to increase ATP levels and mitochondrial membrane potential when provided during reoxygenation after anoxia (Figure 5) (12). It is also of benefit when provided during anoxia alone and is best when present during both anoxia and reoxygenation (12).
Figure 5.
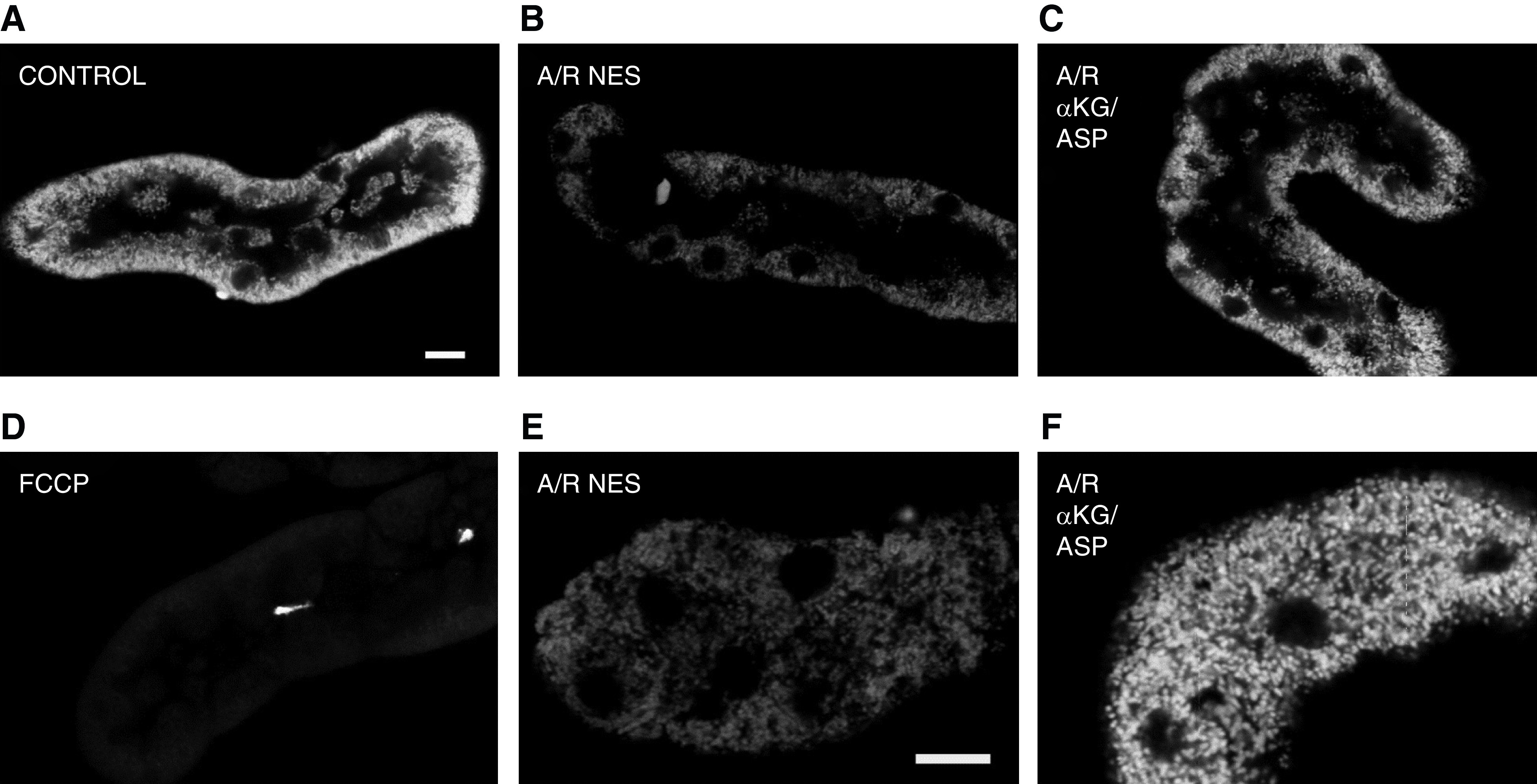
Tricarboxylic acid cycle intermediate supplementation improves mitochondrial membrane potential of isolated proximal tubules. Isolated proximal tubules showing mitochondrial tetramethylrhodamine methyl ester (TMRM) fluorescence, which detects changes in mitochondrial membrane potential. (A) In control samples, mitochondria in the basal portion of tubule cells are brightly stained, indicating high membrane potential. (B) After anoxia/reperfusion (A/R) with no extra substrates (NES), TMRM fluorescence is decreased indicating decreased mitochondrial membrane potential. (C) Supplementation of α-ketoglutarate + aspartate (αKG/ASP) during only reoxygenation improves mitochondrial fluorescence. (D) In the presence of carbonyl fluoride-m-chlorophenylhydrazone (FCCP), a mitochondrial uncoupler that completely eliminates membrane potential, there is virtually no fluorescence. (E) High magnification image of A/R with NES. (F) High magnification image of A/R with αKG/ASP. Sizing bars are 10 μm. Figure is from from ref. (13) with permission.
Further studies have shown that benefit of TCA cycle substrate supplementation is not limited to αKG + aspartate, but is seen with other metabolites and combinations, such as malate, succinate, citrate, and fumarate (13). Some of these are only effective during reoxygenation, so they may be working by overcoming substrate delivery issues at that time in addition to feeding the anaerobic path5ways.
Anaerobic Mitochondrial ATP Generation Limits Nonesterified Fatty Acid Accumulation and Maintains Mitochondrial Respiratory Function
The mechanism by which anaerobic mitochondrial ATP generation helps foster recovery of effective oxidative metabolism and cellular energetics was initially unclear. Investigations subsequently demonstrated that supplementation of TCA cycle intermediates alters nonesterified fatty acid (NEFA) accumulation that occurs during anoxia (32). These fatty acids can enter the mitochondrial matrix via nonionic diffusion, which delivers their protons to the matrix, and, thus, short-circuits the normal path of proton entry through the ATPase to drive ATP generation and uncouples oxidative phosphorylation (Figure 6). The process only requires small catalytic amounts of NEFA because the fatty acid ions can then be recycled out of the matrix by the ADP/ATP carrier and other carriers, thus making it self-sustaining (33,34). The net leak of protons into the matrix decreases membrane potential, the driving force for ATP synthesis by the mitochondrial F1F0-ATPase (16,17). Isolated proximal tubules that are provided with protective TCA cycle substrates during anoxia and reoxygenation experiments have strikingly reduced NEFA accumulation during both anoxia and reoxygenation (32). The role of decreased fatty acid accumulation is supported by other studies showing the substrate effects can be enhanced by further sequestering NEFA with delipidated albumin, which strongly restores membrane potential and ATP. Addition of delipidated albumin to permeabilized tubules rapidly and strongly restores membrane potential (32).
Figure 6.

Fatty acid cycling across the inner mitochondrial membrane decrease membrane potential and uncouples oxidative phosphorylation. Fatty acids (FA) enter the mitochondrial matrix via nonionic diffusion and deliver their protons to the matrix, which short-circuits the normal path of proton re-entry via the ATPase. Fatty acid ions can then be recycled out of the matrix by membrane carriers including ADP/ATP carrier (AAC), glutamate-aspartate transport (AGC) and uncoupling proteins (UCP). Figure is from ref. (34) with permission.
Respiration of both intact and permeabilized tubules is profoundly impaired during reoxygenation, but, unlike membrane potential, respiratory function is not substantially improved by the addition of albumin to permeabilized tubules, indicating this abnormality is not being driven by a continued presence of NEFA. Respiratory dysfunction is only improved when tubules are substrate supplemented for 60 minutes of reoxygenation before sampling and permeabilization for the assay (14).
These data indicate there are two components to the failure of oxidative phosphorylation. One is a readily reversible NEFA-induced uncoupling, but the other is a severe impairment of intrinsic respiratory function that can be at least partially repaired in the cells during an hour of reoxygenation if tubules are supplemented with protective substrates to anaerobically restore ATP. The respiratory effects for intact mitochondria are most severe for Complex 1–dependent substrates, but if the tubules and their mitochondria are disrupted, the core enzymatic function of Complex I measured as NADH:CoQ reductase activity is not markedly decreased by anoxia, or changed by protective substrates (35). TCA cycle activity and complex I in particular require NAD, which is rapidly degraded during anoxia and only partially recovers during unprotected reoxygenation (35). This degradation during anoxia is strongly alleviated by the presence of protective substrates with improvement of subsequent recovery of NAD during reoxygenation (35). So, it is likely that preservation of NAD by protective substrates contributes to their benefit for respiratory function. Other more recent studies have emphasized the important role played by NAD and its applicability to injury in vivo (1,36).
The isolated tubule changes are not sex specific. The published studies for rabbits used females, those for mice used males (12–14,16,29,30,32,34–38), but both sexes have been tested in each species, and showed similar behavior with regard to the metabolic changes, just with a tendency to overall less injury in females (JMW, unpublished).
The nature of the continuing mitochondrial defect has not been further studied in isolated tubule models, but recent work in other systems has suggested it also likely involves mitochondrial shape and volume changes that remodel cristae, which causes disruption of respiratory chain supramolecular complexes (37). The volume changes may also affect metabolite access to the respiratory chain (38). Consistent with this possibility, several mitochondria-targeted agents have shown strong benefit in intact tissue models, which has been associated with preservation of matrix volume and structure of cristae (39–41).
Metabolic Switch to Glycolysis during AKI to CKD Transition
In circumstances where damaged proximal tubular cells are unable to fully recover, there is a metabolic switch that occurs during the AKI to CKD transition in vivo. During ischemia reperfusion, there is downregulation of respiratory enzyme mRNA and protein 24 hours after reperfusion (42). There is also substantial downregulation of enzymes of fatty acid oxidation (18,19). Additionally, glycolytic enzymes are rapidly upregulated during the first 24 hours after ischemia and enzymes necessary for gluconeogenesis are downregulated (43). Metabolically during the first 18 hours of reperfusion, tissue pyruvate was low whereas lactate was unchanged, consistent with the shunting of available pyruvate to lactate. Subsequently, levels of both metabolites increased in a sustained way and hexokinase activity was substantially increased (44). Interestingly, however, in a recent study using a murine model of septic AKI, it was shown that, despite increased expression of rate-limiting enzymes of glycolysis, glycolytic capacity remained limited (45).
By 2 weeks after an ischemic insult, many tubules appear fully recovered, but a subpopulation has an atrophic simplified morphology, and these tubules are surrounded by areas of fibrosis. The atrophic tubules are sites of production of cytokines such as TGF-β and PDGF, and autacoids such as lysophosphatidic acid that drive both the maladaptive tubule phenotype and the development of the surrounding fibrosis (46). These atrophic tubules have greatly simplified cells and far fewer mitochondria, as seen by both ultrastructure and staining for the mitochondrial protein TOM 20 (Figure 7). There is evidence of ongoing mitochondrial damage as indicated by the ultrastructural presence of autophagolysosomes (Figure 7) (46). All of these changes were mitigated by a TGF-β antagonist, SD208. Rate-limiting enzymes of glycolysis, hexokinase, phosphofructokinase platelet isoform, phosphofructokinase B3, and pyruvate kinase M2 were all increased (Figure 8A). Conversely, PKLR, the pyruvate kinase isoform that mediates gluconeogenesis and is most strongly expressed under preischemic control conditions, decreases. SD208, the TGF-β antagonist, mitigated all these changes. The increases of glycolytic enzymes occurred to some extent in all proximal tubules but were greatest in the atrophic tubules. The atrophic tubules also show increased inhibitory phosphorylation of the E1-α isoform of pyruvate dehydrogenase, which will serve to limit entry of pyruvate into the TCA cycle and its oxidative metabolism, thus favoring conversion to lactate (46).
Figure 7.

Mitochondrial pathology in proximal tubules during the AKI to CKD transition. Electron micrographs showing (A) normal recovery, (B) atrophy with loss and simplification of mitochondria (C) autophagolysosome associated with atrophy and (D) TGF-β receptor antagonist, SD-208, improves mitochondrial number and morphology. Figure is from ref. (46) with permission.
Figure 8.

Changes in glycolysis enzymes and hypoxia markers during transition to CKD after ischemia-reperfusion injury. (A) Western blot showing increases in glycolytic enzyme protein expression during progression to CKD after ischemia/reperfusion injury (IRI). (B) Western blot showing increases in hypoxia markers after IRI. HK2, Hexokinase 2; PFKP, Phosphofructokinase platelet isoform; PFKFB3, 6-phosphofructo-2-kinase/fructose-2,6-bisphosphate-3; PKM2, Pyruvate kinase M2; PKLR, pyruvate kinase liver/RBC isoform; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; CA9, carbonic anhydrase 9; SD, SD208, a TGF-β receptor antagonist. Nephrectomy control 14 days after sham left-kidney ischemia (14 days neph ctrl). Figure is from ref. (46) with permission.
Hypoxia-Inducible Factors as Critical Regulators of the Metabolic Switch after AKI
Hypoxia inducible factor-1α (HIF-1α) is a key transcription factor regulating the cellular response to hypoxia, and has far-reaching effects on energy metabolism, including increasing expression of enzymes involved in glycolysis, and is one of the primary transcription factors regulating the switch from oxidative metabolism to anaerobic metabolism (47–49). Regulation of HIF-1α and its downstream genes in the kidney is compartment specific and likely cell-type specific, and its effects on kidney tissue vary by cell type, injury type, and stage (50–57).
In cell culture, HIF-1α–deficient proximal tubule epithelial cells that were exposed to hypoxia had decreased expression of glycolytic genes and decreased utilization of glucose in culture medium (58). Stabilization of HIFs in the thick ascending limb resulted in preserved expression of glycolytic enzymes and attenuated the severity of proximal tubular injury in a murine model of ischemia-reperfusion injury (59). During the AKI to CKD transition, HIF-1α expression is strongly increased as is expression of carbonic anhydrase-9 (CA9), a hypoxia marker whose transcription is directed by HIF-1α. CA9 increases were seen in all tubules, but most strongly in the atrophic ones. TGF-β inhibition decreased but did not eliminate the changes of HIF-1α and CA9 (Figure 8B) (46).
Lipid Metabolism Abnormalities during the AKI to CKD Transition
A growing and important body of information indicates that the transition to CKD is accompanied by persistent decreases in fatty acid oxidation, which has been considered the primary source of fuel for proximal tubular epithelial cells (18–21). In a murine ischemia-reperfusion model of AKI, isolated tubular cells showed downexpression of genes involved in fatty acid oxidation and increased expression of genes involved in lipid storage (60). Similarly, downregulation of carnitine palmitoyltransferase-1, a rate-limiting enzyme of fatty acid oxidation, is seen in septic models of AKI (45). Moreover, proximal tubule specific upregulation of peroxisome proliferator activated receptors, transcription factors that activate fatty acid oxidation, mitigates the severity of AKI in murine ischemia-reperfusion and cisplatin models of AKI (61). The severity of AKI is also mitigated when fenofibrate is administered, which is a known peroxisome proliferator activated receptor activator (62–64), and when the diminished activity of carnitine palmitoyltransferase-1 is pharmacologically stimulated (65). Taken together, these data implicate sustained, decreased fatty acid oxidation as a process that worsens and perpetuates AKI. Decreased expression of fatty acid oxidation enzymes in tubular cells has been documented in kidney samples from patients with CKD (18).
Discussion
The past two decades of research have improved the understanding of metabolic regulation during AKI and the AKI to CKD transition. These efforts have culminated in initial studies manipulating these pathways as potential therapeutics for AKI (1–3). We have presented here a detailed description of the biochemistry to improve understanding. In summary, during the initial oxygen deprivation insult, very small amounts of ATP are required to maintain cell viability and enable recovery of mitochondrial oxidative metabolism via effects to limit fatty acid uncoupling, increase NAD, and likely preserve respiratory complex organization via regulation of mitochondrial volume and cristae structure. These levels of ATP cannot be provided by glycolysis in proximal tubules, but can be produced by anaerobic mitochondrial metabolism.
Later during the transition to CKD, a metabolic switch to favor glycolysis occurs, driven in large part by HIF-1α and TGF-β. The switch is prevented or reversed in proximal tubules that recover normally, but persists and becomes progressively more severe in atrophied tubules that are associated with areas of fibrosis and exhibit extensive loss of mitochondria. Abnormalities of fatty acid oxidation occur early during AKI, persist during CKD, and are promising therapeutic targets. Although there are multiple cellular processes that contribute to the AKI to CKD transition (66), the totality of these data suggests changes in energy metabolism are a critical component of the AKI to CKD transition.
Disclosures
M.A. Venkatachalam reports honoraria from Surrozen. All remaining authors have nothing to disclose.
Funding
J. Schaub was supported by National Institutes of Health (NIH) National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) grant K08 DK124449. Original work summarized in this paper from J.M. Weinberg was supported by NIH NIDDK grant DK34275 and the Veteran’s Administration. M.A. Venkatachalam was supported by NIH NIDDK grants DK104128 and DK119693.
Author Contributions
J.M. Weinberg and M.A. Venkatachalam were responsible for investigation; and all authors were responsible for the conceptualization, wrote the original draft, and reviewed and edited the writing.
References
- 1.Mehr A, Tran MT, Ralto KM, Leaf DE, Washco V, Messmer J, Lerner A, Kher A, Kim SH, Khoury CC, Herzig SJ, Trovato ME, Simon-Tillaux N, Lynch MR, Thadhani RI, Clish CB, Khabbaz KR, Rhee EP, Waikar SS, Berg AH, Parikh SM: De novo NAD+ biosynthetic impairment in acute kidney injury in humans. Nat Med 24: 1351–1359, 2018. 10.1038/s41591-018-0138-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Szeto HH: Pharmacologic approaches to improve mitochondrial function in AKI and CKD. J Am Soc Nephrol 28: 2856–2865, 2017. 10.1681/ASN.2017030247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cameron RB, Beeson CC, Schnellmann RG: Development of therapeutics that induce mitochondrial biogenesis for the treatment of acute and chronic degenerative diseases. J Med Chem 59: 10411–10234, 2016. 10.1021/acs.jmedchem.6b00669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Romano AH, Conway T: Evolution of carbohydrate metabolic pathways. Res Microbiol 147: 448–455, 1996. 10.1016/0923-2508(96)83998-2 [DOI] [PubMed] [Google Scholar]
- 5.Schmidt-Rohr K: Oxygen is the high-energy molecule powering complex multicellular life: Fundamental corrections to traditional bioenergetics. ACS Omega 5: 2221–2233, 2020. 10.1021/acsomega.9b03352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stahl A: A current review of fatty acid transport proteins (SLC27). Pflugers Arch 447: 722–727, 2004. 10.1007/s00424-003-1106-z [DOI] [PubMed] [Google Scholar]
- 7.Houten SM, Wanders RJA: A general introduction to the biochemistry of mitochondrial fatty acid β-oxidation. J Inherit Metab Dis 33: 469–477, 2010. 10.1007/s10545-010-9061-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hochachka PW, Owen TG, Allen JF, Whittow GC: Multiple end products of anaerobiosis in diving vertebrates. Comp Biochem Physiol B 50: 17–22, 1975. 10.1016/0305-0491(75)90292-8 [DOI] [PubMed] [Google Scholar]
- 9.Gronow GH, Cohen JJ: Substrate support for renal functions during hypoxia in the perfused rat kidney. Am J Physiol 247: F618–F631, 1984. 10.1152/ajprenal.1984.247.4.F618 [DOI] [PubMed] [Google Scholar]
- 10.Bienholz A, Petrat F, Wenzel P, Ickerott P, Weinberg JM, Witzke O, Kribben A, de Groot H, Feldkamp T: Adverse effects of α-ketoglutarate/malate in a rat model of acute kidney injury. Am J Physiol Renal Physiol 303: F56–F63, 2012. 10.1152/ajprenal.00070.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bienholz A, Reis J, Sanli P, de Groot H, Petrat F, Guberina H, Wilde B, Witzke O, Saner FH, Kribben A, Weinberg JM, Feldkamp T: Citrate shows protective effects on cardiovascular and renal function in ischemia-induced acute kidney injury. BMC Nephrol 18: 130, 2017. 10.1186/s12882-017-0546-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weinberg JM, Venkatachalam MA, Roeser NF, Nissim I: Mitochondrial dysfunction during hypoxia/reoxygenation and its correction by anaerobic metabolism of citric acid cycle intermediates. Proc Natl Acad Sci U S A 97:2826–2831, 2000. 10.1073/pnas.97.6.2826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weinberg JM, Venkatachalam MA, Roeser NF, Saikumar P, Dong Z, Senter RA, Nissim I: Anaerobic and aerobic pathways for salvage of proximal tubules from hypoxia-induced mitochondrial injury. Am J Physiol Renal Physiol 279: F927–F943, 2000. 10.1152/ajprenal.2000.279.5.F927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bienholz A, Al-Taweel A, Roeser NF, Kribben A, Feldkamp T, Weinberg JM: Substrate modulation of fatty acid effects on energization and respiration of kidney proximal tubules during hypoxia/reoxygenation. PLoS One 9: e94584, 2014. 10.1371/journal.pone.0094584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pfaller W, Rittinger M: Quantitative morphology of the rat kidney. Int J Biochem 12: 17–22, 1980. 10.1016/0020-711X(80)90035-X [DOI] [PubMed] [Google Scholar]
- 16.Feldkamp T, Kribben A, Weinberg JM: F1FO-ATPase activity and ATP dependence of mitochondrial energization in proximal tubules after hypoxia/reoxygenation. J Am Soc Nephrol 16: 1742–1751, 2005. 10.1681/ASN.2005010053 [DOI] [PubMed] [Google Scholar]
- 17.Nicholls DG, Ferguson SJ: Bioenergetics, 4th Ed., San Diego, CA, Academic Press, 2013 [Google Scholar]
- 18.Kang HM, Ahn SH, Choi P, Ko YA, Han SH, Chinga F, Park ASD, Tao J, Sharma K, Pullman J, Bottinger EP, Goldberg IJ, Susztak K: Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nature Med 21: 37–46, 2015. 10.1038/nm.3762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Simon N, Hertig A: Alteration of fatty acid oxidation in tubular epithelial cells: From acute kidney injury to renal fibrogenesis. Front Med (Lausanne) 2: 52, 2015. 10.3389/fmed.2015.00052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weidemann MJ, Krebs HA: The fuel of respiration of rat kidney cortex. Biochem J 112: 149–166, 1969. 10.1042/bj1120149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wirthensohn G, Guder WG: Renal substrate metabolism. Physiol Rev 66: 469–497, 1986. 10.1152/physrev.1986.66.2.469 [DOI] [PubMed] [Google Scholar]
- 22.Venkatachalam MA, Patel YJ, Kreisberg JI, Weinberg JM: Energy thresholds that determine membrane integrity and injury in a renal epithelial cell line (LLC-PK1). Relationships to phospholipid degradation and unesterified fatty acid accumulation. J Clin Invest 81: 745–758, 1988. 10.1172/JCI113380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guder WG, Ross BD: Enzyme distribution along the nephron. Kidney Int 26: 101–111, 1984. 10.1038/ki.1984.143 [DOI] [PubMed] [Google Scholar]
- 24.Ruegg CE, Mandel LJ: Bulk isolation of renal PCT and PST. I. Glucose-dependent metabolic differences. Am J Physiol 259: F164–F175, 1990. 10.1152/ajprenal.1990.259.1.F164 [DOI] [PubMed] [Google Scholar]
- 25.Uchida S, Endou H: Substrate specificity to maintain cellular ATP along the mouse nephron. Am J Physiol 255: F977–F983, 1988. 10.1152/ajprenal.1988.255.5.F977 [DOI] [PubMed] [Google Scholar]
- 26.Bastin J, Cambon N, Thompson M, Lowry OH, Burch HB: Change in energy reserves in different segments of the nephron during brief ischemia. Kidney Int 31: 1239–1247, 1987. 10.1038/ki.1987.137 [DOI] [PubMed] [Google Scholar]
- 27.Bagnasco S, Good D, Balaban R, Burg M: Lactate production in isolated segments of the rat nephron. Am J Physiol 248: F522–F526, 1985. 10.1152/ajprenal.1985.248.4.F522 [DOI] [PubMed] [Google Scholar]
- 28.Hall AM, Unwin RJ, Parker N, Duchen MR: Multiphoton imaging reveals differences in mitochondrial function between nephron segments. J Am Soc Nephrol 20: 1293–1302, 2009. 10.1681/ASN.2008070759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weinberg JM, Roeser NF, Davis JA, Venkatachalam MA: Glycine-protected, hypoxic, proximal tubules develop severely compromised energetic function. Kidney Int 52: 140–151, 1997. 10.1038/ki.1997.313 [DOI] [PubMed] [Google Scholar]
- 30.Feldkamp T, Kribben A, Weinberg JM: Assessment of mitochondrial membrane potential in proximal tubules after hypoxia-reoxygenation. Am J Physiol Renal Physiol 288: F1092–F1102, 2005. 10.1152/ajprenal.00443.2004 [DOI] [PubMed] [Google Scholar]
- 31.Chinopoulos C: Succinate in ischemia: Where does it come from? Int J Biochem Cell Biol 115: 105580, 2019. 10.1016/j.biocel.2019.105580 [DOI] [PubMed] [Google Scholar]
- 32.Feldkamp T, Kribben A, Roeser NF, Senter RA, Weinberg JM: Accumulation of nonesterified fatty acids causes the sustained energetic deficit in kidney proximal tubules after hypoxia-reoxygenation. Am J Physiol Renal Physiol 290: F465–F477, 2006. 10.1152/ajprenal.00305.2005 [DOI] [PubMed] [Google Scholar]
- 33.Schönfeld P, Wojtczak AB, Geelen MJ, Kunz W, Wojtczak L: On the mechanism of the so-called uncoupling effect of medium- and short-chain fatty acids. Biochim Biophys Acta 936: 280–288, 1988. 10.1016/0005-2728(88)90003-5 [DOI] [PubMed] [Google Scholar]
- 34.Feldkamp T, Kribben A, Roeser NF, Ostrowski T, Weinberg JM: Alleviation of fatty acid and hypoxia-reoxygenation-induced proximal tubule deenergization by ADP/ATP carrier inhibition and glutamate. Am J Physiol Renal Physiol 292: F1606–F1616, 2007. 10.1152/ajprenal.00476.2006 [DOI] [PubMed] [Google Scholar]
- 35.Feldkamp T, Kribben A, Roeser NF, Senter RA, Kemner S, Venkatachalam MA, Nissim I, Weinberg JM: Preservation of complex I function during hypoxia-reoxygenation-induced mitochondrial injury in proximal tubules. Am J Physiol Renal Physiol 286: F749–F759, 2004. 10.1152/ajprenal.00276.2003 [DOI] [PubMed] [Google Scholar]
- 36.Zhou HL, Zhang R, Anand P, Stomberski CT, Qian Z, Hausladen A, Wang L, Rhee EP, Parikh SM, Karumanchi SA, Stamler JS: Metabolic reprogramming by the S-nitroso-CoA reductase system protects against kidney injury [published correction appears in Nature 570: E23, 2019 10.1038/s41586-019-1225-0]. Nature 565: 96–100, 2019. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cogliati S, Frezza C, Soriano ME, Varanita T, Quintana-Cabrera R, Corrado M, Cipolat S, Costa V, Casarin A, Gomes LC, Perales-Clemente E, Salviati L, Fernandez-Silva P, Enriquez JA, Scorrano L: Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell 155: 160–171, 2013. 10.1016/j.cell.2013.08.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Halestrap AP: Regulation of mitochondrial metabolism through changes in matrix volume. Biochem Soc Trans 22: 522–529, 1994. 10.1042/bst0220522 [DOI] [PubMed] [Google Scholar]
- 39.Birk AV, Liu S, Soong Y, Mills W, Singh P, Warren JD, Seshan SV, Pardee JD, Szeto HH: The mitochondrial-targeted compound SS-31 re-energizes ischemic mitochondria by interacting with cardiolipin. J Am Soc Nephrol 24: 1250–1261, 2013. 10.1681/ASN.2012121216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Suzuki T, Yamaguchi H, Kikusato M, Hashizume O, Nagatoishi S, Matsuo A, Sato T, Kudo T, Matsuhashi T, Murayama K, Ohba Y, Watanabe S, Kanno SI, Minaki D, Saigusa D, Shinbo H, Mori N, Yuri A, Yokoro M, Mishima E, Shima H, Akiyama Y, Takeuchi Y, Kikuchi K, Toyohara T, Suzuki C, Ichimura T, Anzai JI, Kohzuki M, Mano N, Kure S, Yanagisawa T, Tomioka Y, Toyomizu M, Tsumoto K, Nakada K, Bonventre JV, Ito S, Osaka H, Hayashi KI, Abe T: Mitochonic acid 5 binds mitochondria and ameliorates renal tubular and cardiac myocyte damage. J Am Soc Nephrol 27: 1925–1932, 2016. 10.1681/ASN.2015060623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Szeto HH, Liu S, Soong Y, Birk AV: Improving mitochondrial bioenergetics under ischemic conditions increases warm ischemia tolerance in the kidney. Am J Physiol Renal Physiol 308: F11–F21, 2015. 10.1152/ajprenal.00366.2014 [DOI] [PubMed] [Google Scholar]
- 42.Funk JA, Schnellmann RG: Persistent disruption of mitochondrial homeostasis after acute kidney injury. Am J Physiol Renal Physiol 302: F853–F864, 2012. 10.1152/ajprenal.00035.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fukuhara Y, Yamamoto S, Yano F, Orita Y, Fujiwara Y, Ueda N, Kamada T, Noguchi T, Tanaka T: Changes in activities and mRNA levels of glycolytic enzymes of ischemia-reperfused rat kidney. Contrib Nephrol 95: 222–228, 1991. 10.1159/000420663 [DOI] [PubMed] [Google Scholar]
- 44.Zager RA, Johnson AC, Becker K: Renal cortical pyruvate depletion during AKI. J Am Soc Nephrol 25: 998–1012, 2014. 10.1681/ASN.2013070791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li Y, Nourbakhsh N, Pham H, Tham R, Zuckerman JE, Singh P: Evolution of altered tubular metabolism and mitochondrial function in sepsis-associated acute kidney injury. Am J Physiol Renal Physiol 319: F229–F244, 2020. 10.1152/ajprenal.00390.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lan R, Geng H, Singha PK, Saikumar P, Bottinger EP, Weinberg JM, Venkatachalam MA: Mitochondrial pathology and glycolytic shift during proximal tubule atrophy after ischemic AKI. J Am Soc Nephrol 27: 3356–3367, 2016. 10.1681/ASN.2015020177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.de Laplanche E, Gouget K, Cléris G, Dragounoff F, Demont J, Morales A, Bezin L, Godinot C, Perrière G, Mouchiroud D, Simonnet H: Physiological oxygenation status is required for fully differentiated phenotype in kidney cortex proximal tubules. Am J Physiol Renal Physiol 291: F750–F760, 2006. 10.1152/ajprenal.00022.2006 [DOI] [PubMed] [Google Scholar]
- 48.Seagroves TN, Ryan HE, Lu H, Wouters BG, Knapp M, Thibault P, Laderoute K, Johnson RS: Transcription factor HIF-1 is a necessary mediator of the pasteur effect in mammalian cells. Mol Cell Biol 21: 3436–3444, 2001. 10.1128/MCB.21.10.3436-3444.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Semenza GL, Roth PH, Fang HM, Wang GL: Transcriptional regulation of genes encoding glycolytic enzymes by hypoxia-inducible factor 1. J Biol Chem 269: 23757–23763, 1994 [PubMed] [Google Scholar]
- 50.Bernhardt WM, Câmpean V, Kany S, Jürgensen JS, Weidemann A, Warnecke C, Arend M, Klaus S, Günzler V, Amann K, Willam C, Wiesener MS, Eckardt KU: Preconditional activation of hypoxia-inducible factors ameliorates ischemic acute renal failure. J Am Soc Nephrol 17: 1970–1978, 2006. 10.1681/ASN.2005121302 [DOI] [PubMed] [Google Scholar]
- 51.Haase VH: Pathophysiological consequences of HIF activation: HIF as a modulator of fibrosis. Ann N Y Acad Sci 1177: 57–65, 2009. 10.1111/j.1749-6632.2009.05030.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hill P, Shukla D, Tran MG, Aragones J, Cook HT, Carmeliet P, Maxwell PH: Inhibition of hypoxia inducible factor hydroxylases protects against renal ischemia-reperfusion injury. J Am Soc Nephrol 19: 39–46, 2008. 10.1681/ASN.2006090998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kapitsinou PP, Jaffe J, Michael M, Swan CE, Duffy KJ, Erickson-Miller CL, Haase VH: Preischemic targeting of HIF prolyl hydroxylation inhibits fibrosis associated with acute kidney injury. Am J Physiol Renal Physiol 302: F1172–F1179, 2012. 10.1152/ajprenal.00667.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kimura K, Iwano M, Higgins DF, Yamaguchi Y, Nakatani K, Harada K, Kubo A, Akai Y, Rankin EB, Neilson EG, Haase VH, Saito Y: Stable expression of HIF-1alpha in tubular epithelial cells promotes interstitial fibrosis. Am J Physiol Renal Physiol. 295: F1023–F1029, 2008. 10.1152/ajprenal.90209.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rosenberger C, Mandriota S, Jürgensen JS, Wiesener MS, Hörstrup JH, Frei U, Ratcliffe PJ, Maxwell PH, Bachmann S, Eckardt KU: Expression of hypoxia-inducible factor-1alpha and -2alpha in hypoxic and ischemic rat kidneys. J Am Soc Nephrol 13: 1721–1732, 2002. 10.1097/01.ASN.0000017223.49823.2A [DOI] [PubMed] [Google Scholar]
- 56.Shved N, Warsow G, Eichinger F, Hoogewijs D, Brandt S, Wild P, Kretzler M, Cohen CD, Lindenmeyer MT: Transcriptome-based network analysis reveals renal cell type-specific dysregulation of hypoxia-associated transcripts. Sci Rep 7:8576, 2017. 10.1038/s41598-017-08492-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tanaka T, Nangaku M: The role of hypoxia, increased oxygen consumption, and hypoxia-inducible factor-1 alpha in progression of chronic kidney disease. Curr Opin Nephrol Hypertens 19: 43–50, 2010. 10.1097/MNH.0b013e3283328eed [DOI] [PubMed] [Google Scholar]
- 58.Biju MP, Akai Y, Shrimanker N, Haase VH: Protection of HIF-1-deficient primary renal tubular epithelial cells from hypoxia-induced cell death is glucose dependent. Am J Physiol Renal Physiol 289: F1217–F1226, 2005. 10.1152/ajprenal.00233.2005 [DOI] [PubMed] [Google Scholar]
- 59.Schley G, Klanke B, Schödel J, Forstreuter F, Shukla D, Kurtz A, Amann K, Wiesener MS, Rosen S, Eckdart KU, Maxwell PH, Willam C: Hypoxia-inducible transcription factors stabilization in the thick ascending limb protects against ischemic acute kidney injury. J Am Soc Nephrol 22: 2004–2015, 2011. 10.1681/ASN.2010121249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang D, Xing Y, Li W, Yang F, Lang Y, Yang J, Liu Z: Renal tubules transcriptome reveals metabolic maladaption during the progression of ischemia-induced acute kidney injury. Biochem Biophys Res Commun 505: 432–438, 2018. 10.1016/j.bbrc.2018.08.111 [DOI] [PubMed] [Google Scholar]
- 61.Li S, Nagothu KK, Desai V, Lee T, Branham W, Moland C, Megyesi JK, Crew MD, Portilla D: Transgenic expression of proximal tubule peroxisome proliferator-activated receptor-alpha in mice confers protection during acute kidney injury. Kidney Int 76: 1049–1062, 2009. 10.1038/ki.2009.330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li S, Basnakian A, Bhatt R, Megyesi J, Gokden N, Shah SV, Portilla D: PPAR-alpha ligand ameliorates acute renal failure by reducing cisplatin-induced increased expression of renal endonuclease G. Am J Physiol Renal Physiol 287: F990–F998, 2004. 10.1152/ajprenal.00206.2004 [DOI] [PubMed] [Google Scholar]
- 63.Li S, Gokden N, Okusa MD, Bhatt R, Portilla D: Anti-inflammatory effect of fibrate protects from cisplatin-induced ARF. Am J Physiol Renal Physiol 289: F469–F480, 2005. 10.1152/ajprenal.00038.2005 [DOI] [PubMed] [Google Scholar]
- 64.Nagothu KK, Bhatt R, Kaushal GP, Portilla D: Fibrate prevents cisplatin-induced proximal tubule cell death. Kidney Int 68: 2680–2693, 2005. 10.1111/j.1523-1755.2005.00739.x [DOI] [PubMed] [Google Scholar]
- 65.Idrovo JP, Yang WL, Nicastro J, Coppa GF, Wang P: Stimulation of carnitine palmitoyltransferase 1 improves renal function and attenuates tissue damage after ischemia/reperfusion. J Surg Res 177: 157–164, 2012. 10.1016/j.jss.2012.05.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liu J, Kumar S, Dolzhenko E, Alvarado GF, Guo J, Lu C, Chen Y, Li M, Dessing MC, Parvez RK, Cippà PE, Krautzberger AM, Saribekyan G, Smith AD, McMahon AP: Molecular characterization of the transition from acute to chronic kidney injury following ischemia/reperfusion. JCI Insight 2: e94716, 2017. 10.1172/jci.insight.94716 [DOI] [PMC free article] [PubMed] [Google Scholar]