

Abstract
Remibrutinib, a novel oral Bruton’s Tyrosine Kinase inhibitor (BTKi) is highly selective for BTK, potentially mitigating the side effects of other BTKis. Enzyme phenotyping identified CYP3A4 to be the predominant elimination pathway of remibrutinib. The impact of concomitant treatment with CYP3A4 inhibitors, grapefruit juice and ritonavir (RTV), was investigated in this study in combination with an intravenous microtracer approach. Pharmacokinetic (PK) parameters, including the fraction absorbed, the fractions escaping intestinal and hepatic first‐pass metabolism, the absolute bioavailability, systemic clearance, volume of distribution at steady‐state, and the fraction metabolized via CYP3A4 were evaluated. Oral remibrutinib exposure increased in the presence of RTV 4.27‐fold, suggesting that remibrutinib is not a sensitive CYP3A4 substrate. The rich PK dataset supported the building of a robust physiologically‐based pharmacokinetic (PBPK) model, which well‐described the therapeutic dose range of 25–100 mg. Simulations of untested scenarios revealed an absence of drug‐drug interaction (DDI) risk between remibrutinib and the weak CYP3A4 inhibitor fluvoxamine (area under the concentration‐time curve ratio [AUCR] <1.25), and a moderate effect with the CYP3A4 inhibitor erythromycin (AUCR: 2.71). Predictions with the moderate and strong CYP3A4 inducers efavirenz and rifampicin, suggested a distinct remibrutinib exposure decrease of 64% and 89%. Oral bioavailability of remibrutinib was 34%. The inclusion of an intravenous microtracer allowed the determination of all relevant remibrutinib PK parameters, which facilitated construction of the PBPK model. This will provide guidance on the selection or restriction of comedications and prediction of DDI risks.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Remibrutinib is an irreversible Bruton’s Tyrosine Kinase inhibitor and moderate CYP3A4 substrate to be administered with caution with strong inhibitors.
WHAT QUESTION DID THIS STUDY ADDRESS?
This study explored the drug‐drug interaction (DDI) risk of remibrutinib as a victim with CYP3A4 inhibitors.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
Using a specifically tailored study design, most model‐relevant pharmacokinetic (PK) parameters were determined, including fractions of the dose escaping intestinal and hepatic first‐pass metabolism, absolute bioavailability, systemic drug clearance, apparent volume of distribution at steady‐state, and fraction of the drug metabolized via CYP3A4.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
The clinical study design offers opportunities to obtain important PK parameters, which enabled physiologically‐based pharmacokinetic (PBPK) model building of complex PK compounds. Taken together, the PK parameters and the PBPK simulations allowed robust DDI predictions of untested scenarios.
INTRODUCTION
Remibrutinib is a highly selective, potent, oral, covalent Bruton’s Tyrosine Kinase inhibitor (BTKi) that is in development for the treatment of autoimmune disorders, including chronic spontaneous urticaria. 1 , 2 It is hypothesized that signaling pathways downstream of BTK, relevant to the activation and function of innate and adaptive immune system cells, could be modulated by remibrutinib. 3 , 4 Regarding drug‐drug interactions (DDIs), the effects as “victim” and “perpetrator” must be considered. 5 , 6 The impact of remibrutinib as perpetrator of cytochrome (CYP) enzymes was recently investigated in a separate clinical DDI study, demonstrating weak inhibition effects on CYP2C9 and CYP3A4/5 (unpublished data, Schiller, Huth, Schuhler, Drollmann, Kaul, Woessner, et al. 2021).
In vitro data identified CYP3A4 as the major metabolizing enzyme of remibrutinib. Comedications affecting CYP3A4 by inhibition or induction may influence remibrutinib exposure. Therefore, an investigation regarding the DDI potential risk between remibrutinib and CYP3A4 perpetrators should be considered, as recommended by the European Medicines Agency (EMA) and US Food and Drug Administration (FDA). 5 , 7 , 8 , 9 , 10 , 11 , 12
In this study, an integrated approach was used to evaluate remibrutinib as victim of DDI by concomitant medications. We present the in vitro assessments of intestinal transport, cellular uptake, and enzyme kinetic parameters. Next, a clinical study was conducted to assess the effects of the CYP3A4 inhibitors, ritonavir (RTV) and grapefruit juice, on remibrutinib. RTV, an antiretroviral drug, is a strong inhibitor of intestinal and hepatic CYP3A4/5 and gut P‐glycoprotein (P‐gp), and is used to evaluate the maximal CYP3A inhibition potential of substrates in clinical DDI studies. 13 , 14 Grapefruit juice is a strong inhibitor of intestinal but not hepatic CYP3A4/5, allowing differentiation when compared to the RTV results. 15 , 16 Remibrutinib was given orally; an intravenous microtracer was administered 2 h post‐remibrutinib in the form of a stable isotope labeled remibrutinib (SIL‐remibrutinib).
Secondary objectives were to investigate absolute bioavailability (F) and safety and tolerability of remibrutinib when administered alone or concomitantly with RTV or grapefruit juice. Based on in vitro results and the outcome of the clinical DDI study, a physiologically‐based pharmacokinetic (PBPK) model was established to predict DDI effects mediated via CYP3A4 for a set of known perpetrator drugs. 17 , 18 , 19
METHODS
In vitro assessment of remibrutinib as transporter and metabolizing enzyme substrate
To assess remibrutinib as substrate of adenosine triphosphate (ATP)‐binding cassette efflux transporters, Caco‐2 cells were utilized. Human hepatocytes were used to assess whether remibrutinib is a substrate of hepatic uptake transporters. 20 The rates and pathways of remibrutinib metabolism were investigated by using recombinant human CYPs (rCYP) and human liver microsomes (HLMs). Method details are specified in the Supplementary Document.
Clinical DDI study
This was a single‐center, open‐label, three‐period, single sequence, cross‐over phase I study to investigate the effect of RTV and grapefruit juice on the pharmacokinetics (PKs) of oral (p.o.) remibrutinib in healthy adult participants (Table S3, NCT03918980). Two hours post p.o. administration, an intravenous (i.v.) microtracer dose of SIL‐remibrutinib was administered. Remibrutinib doses (50 mg and 20 mg p.o. and 100 µg i.v.) were selected to ensure remibrutinib levels were above the lower limit of quantification (LLOQ) to reliably characterize the PKs of remibrutinib. This study consisted of a 21‐day screening period followed by 9 days of treatment with the treatment periods 1 to 3 and the end of study (EOS) assessment, which was performed 5 days after the last dose on day 14 (Figure 1).
FIGURE 1.
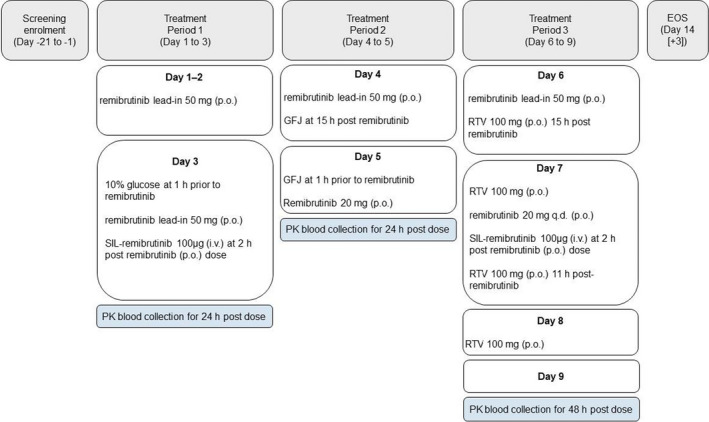
Study design. A single‐center, open‐label, single sequence, cross‐over study to investigate the effect of ritonavir and grapefruit juice on the pharmacokinetics of intravenous and oral remibrutinib. EOS, end of study; GFJ, grapefruit juice; PK, pharmacokinetic; RTV, ritonavir; SIL‐remibrutinib, stable isotope labeled remibrutinib
Treatment period 1 (remibrutinib): A single dose of remibrutinib 50 mg p.o. was administered on the morning of days 1 and 2 after an overnight fast of greater than or equal to 10 h. On day 3, participants received 240 ml of 10% glucose (to mimic grapefruit juice calorific content) after an overnight fast of greater than or equal to 9 h. A single dose of remibrutinib 50 mg p.o. was administered 1 h later. SIL‐remibrutinib 100 μg i.v. was administered 2 h later.
Treatment period 2 (remibrutinib and grapefruit juice): On day 4, a single dose of remibrutinib 50 mg p.o. was administered after an overnight fast of greater than or equal to 10 h. Participants received grapefruit juice 240 ml p.o. 15 h post‐remibrutinib intake. On day 5, participants were given a further 240 ml of grapefruit juice after an overnight fast of greater than or equal to 9 h, followed by a single dose of remibrutinib 20 mg p.o. 1 h later. Blood samples were collected for up to 24 h after the remibrutinib p.o. dosing for remibrutinib measurements.
Treatment period 3 (remibrutinib and RTV): On day 6, a single dose of remibrutinib 50 mg p.o. was administered after an overnight fast of greater than or equal to 9 h. Participants received a single dose of RTV 100 mg p.o. 15 h post‐remibrutinib intake. A second lead‐in dose of RTV 100 mg p.o. was administered on the morning of day 7 after an overnight fast of greater than or equal to 9 h. A single dose of remibrutinib 20 mg p.o. was administered 1 h later. SIL‐remibrutinib 100 μg i.v. was administered 2 h later. A third dose of RTV 100 mg p.o. was administered on day 7, 11 h post‐remibrutinib dose, and a fourth dose of RTV 100 mg p.o. was administered on day 8 after an overnight fast of greater than or equal to 9 h. Blood samples were collected up to 48 h after the p.o. and i.v. remibrutinib dosing, for remibrutinib and SIL‐remibrutinib measurements.
Pharmacokinetic assessments
A validated liquid chromatography tandem mass spectrometry method was used for determining the remibrutinib and SIL‐remibrutinib concentrations in human blood. The primary variables to evaluate the effect of RTV and grapefruit juice on the PKs of oral remibrutinib were the maximal concentration (Cmax) and the area under the curve (to 24 h, [AUC0–24 h], last quantifiable concentration [AUClast], and to infinity [AUCinf]), or remibrutinib. The secondary variables to evaluate the effect of RTV and grapefruit juice on the PKs of oral remibrutinib were the apparent total clearance (CL/F), systemic clearance (CL), apparent volume of distribution (Vz/F), volume of distribution at steady‐state (V ss), and terminal half‐life (t 1/2).
Remibrutinib is cleared by oxidative metabolism and hydrolysis. As only the oxidative metabolism, which is almost exclusively driven by CYP3A4, contributes to first‐pass metabolism, the following calculations can be applied for the fraction of the dose escaping intestinal first‐pass metabolism (Fg ), Equation 1:
| (1) |
with AUCpo,without inhibitor representing the remibrutinib AUC after oral administration without inhibitor and AUCpo,with grapefruit juice representing the remibrutinib AUC after oral administration with grapefruit juice.
The fraction of the dose escaping hepatic first‐pass metabolism (Fh ) value for remibrutinib was calculated by Equation 2:
| (2) |
with AUC ratioiv,RTV and AUC ratiopo,RTV representing the respective remibrutinib AUC ratios after i.v. and p.o. administration with RTV.
The hepatic fraction of the drug metabolized (fm[CYP3A4]) for remibrutinib was calculated by Equation 3:
| (3) |
with AUCiv,without inhibitor representing the remibrutinib AUC after i.v. administration without inhibitor and AUCiv,with RTV representing the remibrutinib AUC after i.v administration with RTV.
The absolute bioavailability (F) of remibrutinib was calculated by Equation 4:
| (4) |
Estimation of the remibrutinib systemic PK parameters total CL and V ss, and calculation of fraction of the dose absorbed from gastrointestinal tract (Fa ) was calculated by Equation 5:
| (5) |
Ethics
The trial was approved by the institutional review board, and written informed consent was obtained from all participants before any assessment was performed. The study was designed and implemented in accordance with the International Conference on Harmonization (ICH) Harmonized Tripartite Guidelines for Good Clinical Practice (GCP), with applicable local regulations, and with the ethical principles laid down in the Declaration of Helsinki.
Safety
Safety was monitored throughout the study and the safety analysis set included all participants who received any study drug. Safety assessments consisted of collecting all adverse events (AEs) and serious adverse events (SAEs); physical examinations; vital signs measurement (pulse rate, blood pressure, height and weight); clinical laboratory evaluations (hematology, blood chemistry, urinalysis, coagulation, hepatitis, and HIV markers); and electrocardiogram (ECG).
Statistical analysis
The effect of RTV and grapefruit juice on the PKs of oral remibrutinib drug exposure was analyzed using an analysis of variance model for the primary PK parameters (Cmax, AUC0–24 h, AUClast, and AUCinf) that included the clinical trial treatment as a fixed effect, and the participant as a random effect. Concentrations below the LLOQ were treated as zero in summary statistics and for PK parameter calculations. No formal statistical analysis was done for safety and tolerability evaluation.
PBPK model
A PBPK model was established using Simcyp (version 18; Simcyp Limited, a CERTARA company, Princeton, NJ), with White healthy volunteers population‐based on remibrutinib’s physico‐chemical parameters, in vitro absorption, distribution, metabolism, and excretion data, and clinical PK parameters from a single and multiple ascending dose study (Table S6) and the DDI trial with RTV and grapefruit juice (NCT03918980). The model was verified by predicting remibrutinib exposure of a separate clinical study (DDI trial with CYP1A2, CYP2C9, and CYP3A4 substrates, unpublished data, Schiller, Huth, Schuhler, Drollmann, Kaul, Woessner et al. 2021) and the DDI impact of RTV on remibrutinib administrated i.v. illustrated by the model development scheme (Figure S2). A detailed description on model building, used compound files, and simulation details is provided in the Supplementary Document. For DDI simulations, the different CYP3A4/5 inhibitors were administered from days 1–8, with remibrutinib dosed 100 mg p.o. either as a single dose on day 5 or once daily (q.d.) for 8 days. CYP3A4/5 inducers were administered for 14 days, with a single dose of remibrutinib at day 11, or from days 1–14 q.d. Unexplored scenarios were predicted by simulating the impact of the weak, moderate, and strong CYP3A4 inhibitors fluvoxamine (100 mg q.d.), erythromycin (500 mg twice daily [b.i.d.]), and ketoconazole (200 mg b.i.d.), as well as the impact of the moderate and strong CYP3A4 inducers efavirenz (600 mg q.d.) and rifampicin (600 mg q.d.) on remibrutinib.
Prediction accuracy (PE) for the PK parameters (Cmax and AUC) was calculated by %PE = [predicted−observed]/observed*100, with a value of within −50% to +100% considered as acceptable prediction. 21 , 22 The accuracy of DDI predictions was assessed by comparing the simulated versus the observed DDI ratio using the Guest criteria, by calculating the limits according to: Upper limit: , Lower limit: , with R obs: observed DDI ratio. Average fold error (AFE) was calculated to compare the quality of the PBPK model with and without target‐mediated drug disposition (TMDD), where n is the sample number. 23 , 24 , 25 , 26
RESULTS
Determination of kinetic transport parameters for remibrutinib and identification of human enzymes involved in the oxidative liver metabolism of remibrutinib
Results of experiments using Caco‐2 cells predicted Fa of about 89% based on passive permeability (P app: 16.5 × 10−6 cm/s), and an absorption rate constant (ka ) of 0.8 h−1 was estimated with Simcyp to match time to maximum concentration (Tmax). Remibrutinib was identified as a P‐gp substrate. Based on its low apparent Km value (13.8 μM), efflux activity is likely saturated in the intestine at clinical doses greater than or equal to 25 mg (gut lumen concentration greater than or equal to 200 μM). The uptake clearance of remibrutinib was not affected by inhibitors of hepatic uptake transporters; thus, its uptake is driven by passive diffusion. In pooled HLM, the metabolic rates of remibrutinib were characterized by an apparent intrinsic affinity (Kmapp) of 5.43 μM and a maximal rate of metabolism (Vmax) of 560.7 pmol/min/mg protein, suggesting a high metabolic clearance of remibrutinib in the liver. The selective CYP3A4 inhibitors ketoconazole and azamulin inhibited remibrutinib metabolism up to 100%. For all other inhibitors tested, either no or less than 30% inhibition was observed. Using recombinant CYP450 enzymes, CYP3A4 was identified as the major metabolizing enzyme contributing to oxidative hepatic metabolism with 97.9% attributed to this enzyme. Minor enzymes involved were CYP2C19 (1%), CYP3A5 (0.8%), and CYP2B6 (0.3%). The CYP3A4 Km,u and Vmax values were 1.51 μM and 16.3 pmol/min/pmol CYP, respectively (Table S2). In addition, the metabolism rates using HLM from 16 individual donors correlated strongly with the activity of CYP3A4/5 (testosterone R: 0.986 and midazolam R: 0.989).
Clinical DDI study
Of the 17 participants enrolled, 16 (94.1%) completed treatment period 1 to treatment period 3 (TP1–TP3) and one participant was discontinued in TP1 due to an AE. All participants received at least one dose of trial drug and were included in the safety and PK analysis sets. There were no major protocol deviations and 17 minor deviations; no participants were excluded from the analysis due to protocol deviations. Participant demographics are outlined in Table S3.
Blood PKs of remibrutinib was compared from participants following the administration of remibrutinib alone, in combination with grapefruit juice, and in combination with RTV. Different remibrutinib doses were administered during TP1 (50 mg), TP2 (20 mg), and TP3 (20 mg). Hence, the actual Cmax and AUC values were dose‐normalized. Arithmetic mean remibrutinib Cmax increased from 2.77 ng/ml/mg following administration of remibrutinib alone, to 9.00 ng/ml/mg for remibrutinib with ritonavir (Table S4). Arithmetic mean dose‐normalized AUC0–24 h values were 4.89 ng*h/ml/mg for remibrutinib alone, and 20.3 ng*h/ml/mg for remibrutinib with RTV. Arithmetic mean dose‐normalized remibrutinib Cmax and AUC0–24 h values increased following administration of remibrutinib with grapefruit juice to values of 3.27 ng/ml/mg and 6.28 ng*h/ml/mg, respectively.
The geometric mean ratios of the dose‐normalized values of Cmax and AUC0–24 h for remibrutinib, comparing administration with grapefruit juice to remibrutinib alone, indicate a 1.24‐fold and 1.29‐fold increase, respectively (Table 1). The geometric mean ratios of the dose‐normalized values of Cmax and AUC0–24 h for remibrutinib, comparing administration with RTV to remibrutinib alone, indicate a 3.32‐fold and 4.27‐fold increase, respectively. Remibrutinib i.v. showed an AUC0–24 h increase in total remibrutinib exposure in the presence of RTV of 1.67‐fold (Table 1).
TABLE 1.
Effect of ritonavir and grapefruit juice on the PKs of oral and intravenous remibrutinib
| p.o. | Period 1 remibrutinib alone (Geo‐mean, 90% CI) | Period 2 remibrutinib + grapefruit juice (Geo‐mean, 90% CI) | Period 3 remibrutinib + ritonavir (Geo‐mean, 90% CI) | Ratio period 2 / period 1 (Geo mean ratio, 90% CI) | Ratio period 3 / period 1 (Geo‐mean ratio, 90% CI) |
|---|---|---|---|---|---|
| Cmax norm by dose (ng/ml/mg) | 2.45 (1.99, 3.02) | 3.03 (2.45, 3.75) | 8.15 (6.59, 10.1) | 1.24 (1.05, 1.46) | 3.32 (2.81, 3.93) |
| AUC0–24 h norm by dose (h*ng/ml/mg) | 4.49 (3.83, 5.26) | 5.80 (4.95, 6.80) | 19.2 (16.4, 22.5) | 1.29 (1.16, 1.43) | 4.27 (3.84, 4.74) |
| AUClast norm by dose (h*ng/ml/mg) | 4.37 (3.73, 5.13) | 5.80 (4.93, 6.81) | 19.7 (16.7, 23.1) | 1.33 (1.19, 1.47) | 4.50 (4.05, 4.99) |
| AUCinf norm by dose (h*ng/ml/mg) | 4.66 (3.92, 5.54) | 5.98 (4.97, 7.19) | 20.1 (16.9, 23.8) | 1.28 (1.09, 1.51) | 4.31 (3.76, 4.94) |
| i.v. | Period 1 remibrutinib alone day 3 Geo‐mean (%CV) | Period 3 remibrutinib + ritonavir day 3 Geo‐mean (%CV) | Ratio period 3 / period 1 day 3 |
|---|---|---|---|
| Cmax norm by dose (ng/ml/mg) | 0.0326 (71.3%) | 0.0291 (81.6%) | 0.89 |
| AUC0–24 h norm by dose (h*ng/ml/mg) | 14.2 (21.8%) | 23.8 (31.0%) | 1.68 |
| AUClast norm by dose (h*ng/ml/mg) | 13.9 (22.3%) | 23.5 (31.0%) | 1.68 |
| AUCinf norm by dose (h*ng/ml/mg) | 14.5 (17.9%) | 24.8 (33.3%) | 1.70 |
Dose‐normalized PK parameters were used. For p.o. statistics were adjusted geometric mean and 90% CI. Model was mixed ANOVA model with a fixed term for treatment and a random effect for subject.
Abbreviations: %CV, percent coefficient of variation; AUC0–24 h, area under the curve during 24 h; AUCinf, area under the blood concentration–time curve from time zero to infinity; AUClast, area under the blood concentration–time curve from time zero to the time of the last quantifiable concentration; CI, confidence interval; Cmax, maximum (peak) blood drug concentration.
Median (minimum‐maximum) time to reach peak blood concentration following drug administration (Tmax) values of remibrutinib reduced slightly from 1.00 h (0.5–2.5 h) for remibrutinib alone, to 0.73 h (0.5–2.0 h) for remibrutinib with grapefruit juice and 0.72 h (0.5–1.5 h) for remibrutinib with RTV. Arithmetic means (coefficient of variation [CV%]) of t 1/2 values were comparable for remibrutinib alone (6.58 h [26.9%]) and when administered with grapefruit juice (7.13 h [15.0%]) but increased to 12.1 h (23.3%) when remibrutinib was administered with RTV. The CV%, CL/F, and Vz/F values decreased following administration of remibrutinib with RTV compared to administration of remibrutinib alone or with grapefruit juice.
Absolute bioavailability of remibrutinib, absorption in the gut, and metabolism by CYP3A
Applying Equations (1), (2), (3), (4), (5) above, the remibrutinib PK parameters Fa, Fg, Fh, F, F inhibited, and FmCYP3A4 were calculated using the results of the DDI study with RTV and grapefruit juice (Table 2). The calculated PK parameters based on observed data were consistent with the PBPK parameters.
TABLE 2.
Summary of observed and simulated remibrutinib parameters using the established PBPK model
| PK parameters | Observed mean value | Simulated mean value |
|---|---|---|
| CL (L/h) | 70.1 ± 12.8a | 73.1 ± 14.1c |
| Fa (%) | 86.9e | 88.9 ± 9.70 |
| Fg (%) | 78.9 ± 15.6 | 55.4 ± 14.6 |
| Fh (%)d | 49.3d | 62.3 ± 10.7 |
| F (%) | 33.8 ± 11.5 | 30.6 ± 10.1 |
| F inhibited (%)b | 80.9 ± 11.5 | 70.8g |
| fm (CYP3A4) (%) | 40.0 | 43.0 ± 10.8 |
| V ss (Subs) (L/Kg) | 0.81 ± 0.22f | 0.812 ± 0.228 |
After target saturation (third dose 0.1 mg i.v.), due to short t 1/2 no accumulation.
Bioavailability in the presence of ritonavir.
Clearance after target saturation (third dose) in ritonavir DDI study simulation with 20 mg remibrutinib at day 3.
Observed first‐pass metabolism (Fh ) calculated:
Fh = AUCratio,iv/(AUCratio,po * Fg ) where AUCratio of the ritonavir DDI study were used.
Observed Fa calculated: Fa = F/Fg /Fh ).
Observed value, calculated from mean V ss: 63.1 L divided by mean body weight of 77.8 kg.
Calculated based on PBPK simulations: remibrutinib p.o. AUCinhibited divided by i.v. AUCinhibited by ritonavir.
Abbreviations: AUCratio, area under the blood concentration–time; CL, systemic drug clearance; DDI, drug‐drug interaction; F, absolute bioavailability; Fa , fraction of the dose absorbed from gastrointestinal tract; Fg , fraction of the dose escaping intestinal first‐pass metabolism; Fh , fraction of the dose escaping hepatic first‐pass metabolism; F inhibited, absolute bioavailability when inhibiting CYP3A4; fm (CYP3A4), fraction of the drug metabolized via CYP3A4; PBPK, physiologically‐based pharmacokinetic; PK, pharmacokinetic; t 1/2, terminal elimination half‐life; V ss, apparent volume of distribution at steady‐state.
Safety
Remibrutinib was well‐tolerated by participants in this study when administered alone, with grapefruit juice, or with RTV. No SAEs, deaths, or AEs of high‐grade severity were reported during the trial. Almost all the AEs reported were rated grade 1 (13/14 occurring in 5 [29.4%] participants) in severity. One grade 2 AE of toothache was reported in TP3, which was not considered to be treatment related. There were no clinically relevant changes in clinical laboratory evaluations, vital sign measurements, and ECG results, except for the isolated AE of mildly increased white blood cell count in one patient who discontinued the study.
Remibrutinib pharmacokinetics and DDI effects predicted by PBPK modeling
The established PBPK model predicted remibrutinib exposure when including TMDD with residual values ranging from −46.6% to +113% for the parameters Cmax and AUC for the dose range of 15–400 mg at single dose or multiple q.d. doses, or b.i.d. doses of 100–200 mg (Figure [Link], [Link] and [Link], [Link], Table S6). Without considering TMDD, the model performed with less accuracy with residual values ranging from −41.6% to +155%. Moreover, the prediction error plots (Figure S1) demonstrate a better Cmax and AUC accuracy for single dose when including TMDD. The AFE was also higher for the model without TMDD, especially for the first dose PKs (Tables S5 and S6). The overprediction at 400–600 mg is based on solubility limitations at these doses, which were not accounted for by the model. As these doses are outside the foreseen therapeutic dose range of 25–100 mg, the model liability is considered acceptable.
The simulation of the DDI trial design with ritonavir, administering remibrutinib i.v. or p.o. resulted in the AUC ratios 1.51 and 4.27 (Table 3), respectively, which were close to the respective observed AUC ratios of 1.66 and 4.27 (Table 1). The predicted values were within the criteria ranges of 1.18–2.28 for the i.v. AUC ratio, and 2.42–7.54 for the p.o. AUC ratio. 24 As further model verification, the remibrutinib PK parameters and concentration time curve in the DDI trial with the CYP450 enzyme substrate cocktail (unpublished data, Schiller, Huth, Schuhler, Drollmann, Kaul, Woessner, et al. 2021) were well‐predicted with residual values of 23% and 36% for Cmax,ss and AUC0–12 h simulated AUCtau/Cmax values: 587ng*h/ml/258 ng/ml, observed AUCtau/Cmax: 916 ng*h/ml/334 ng/ml (Figure 2 and Table 3).
TABLE 3.
Remibrutinib pharmacokinetics after single or multiple oral administration of 100 mg remibrutinib (with TMDD) with CYP3A4 inhibitors or inducers
| Perpetrator, schedule | Perpetrator present | Remibrutinib | Predicted Cmax (ng/ml) | Predicted AUC (ng*h/ml) | Cmax ratio | AUC ratio |
|---|---|---|---|---|---|---|
| Ritonavir, 100 mg b.i.d. |
No Yes |
20 mg s.d. p.o. |
30.8 (28.5–3.3) 105 (99.1–112) |
70.2 (64.8–76.0) 300 (282–318) |
3.41 (3.22–3.62) | 4.27 (4.00–4.55) |
| Ritonavir, 100 mg b.i.d. |
No Yes |
20 mg s.d. i.v. |
1089 (960–1234) 1056 (935–1193) |
286 (277–295) 435 (416–455) |
0.97 (0.97–0.97) | 1.51 (1.48–1.56) |
| Ketoconazole, 200 mg b.i.d. |
No Yes |
100 mg s.d. p.o. |
136 (125–148) 542 (515–570) |
329 (304–357) 1522 (1452–1596) |
3.98 (3.72–4.26) |
4.62 (4.32–4.94) |
| Ketoconazole, 200 mg b.i.d. |
No Yes |
100 mg q.d. p.o. |
188 (176–202) 648 (620–678) |
432 (402–465) 1803 (1723–1887) |
3.44 (3.24–3.66) | 4.17 (3.90–4.46) |
| Erythromycin, 500 mg b.i.d. |
No Yes |
100 mg s.d. p.o. |
136 (125–148) 343 (323–365) |
329 (304–357) 893 (842–947) |
2.52 (2.38–2.67) | 2.71 (2.56–2.87) |
| Erythromycin, 500 mg b.i.d. |
No Yes |
100 mg q.d. p.o. |
188 (176–202) 405 (384–427) |
432 (402–465) 1037 (981–1097) |
2.15 (2.05–2.26) | 2.40 (2.27–2.53) |
| Fluvoxamine, 50 mg q.d. |
No Yes |
100 mg s.d. p.o. |
136 (125–148) 162 (150–176) |
329 (304–357) 379 (351–409) |
1.19 (1.18–1.21) | 1.15 (1.14–1.16) |
| Fluvoxamine, 50 mg q.d. |
No Yes |
100 mg q.d. p.o. |
188 (176–202) 224 (210–239) |
432 (402–465) 500 (466–535) |
1.15 (1.14–1.17) | 1.13 (1.12–1.14) |
| Rifampicin, 600 mg q.d. |
No Yes |
100 mg s.d. p.o. |
143 (131–155) 18.9 (16.5–21.8) |
341 (316–368) 38.7 (33.7–44.6) |
0.13 (0.12–0.15) | 0.11 (0.10–0.13) |
| Rifampicin, 600 mg q.d. |
No Yes |
100 mg q.d. p.o. |
198 (185–212) 25.4 (22.0–29.2) |
451 (421–482) 50.1 (43.5–57.8) |
0.13 (0.11–0.14) | 0.11 (0.10–0.13) |
| Rifampicin, 600 mg q.d. |
No Yes |
100 mg s.d. i.v. |
1103 (1070–1137) 850 (830–870) |
1358 (1315–1402) 976 (952–1000) |
0.77 (0.76–0.78) | 0.72 (0.71–0.73) |
| Efavirenz, 600 mg q.d. |
No Yes |
100 mg s.d. p.o. |
136 (125–148) 53.6 (48.1–59.8) |
329 (304–356) 120 (107–133) |
0.39 (0.37–0.42) | 0.36 (0.34–0.39) |
| Efavirenz, 600 mg q.d. |
No Yes |
100 mg q.d. p.o. |
189 (176–202) 74.5 (67.3–82.6) |
433 (402–466) 156 (140–174) |
0.40 (0.37–0.43) | 0.36 (0.33–0.39) |
Remibrutinib pharmacokinetics and DDI effects predicted by PBPK modeling. Pharmacokinetic data are presented as geometric means with 90% CI range in parentheses, AUC reported as AUCinf for single dose and AUCtau for multiple dose remibrutinib.
Abbreviations: AUC, area under the blood concentration‐time; CI, confidence interval; Cmax, maximum (peak) blood drug concentration; Cmax ratio, maximum (peak) blood drug concentration ratio; DDI, drug‐drug interaction; s.d, single‐dose; TMDD, target‐mediated drug disposition.
FIGURE 2.
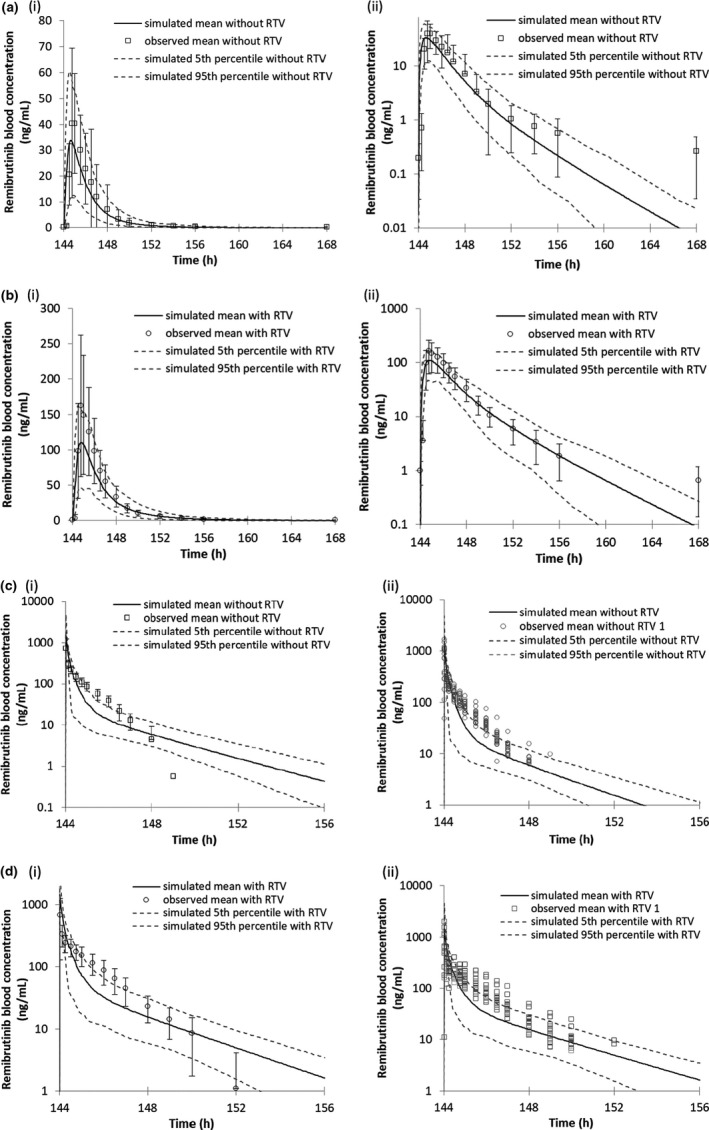
PBPK model predicted and observed remibrutinib blood concentrations when co‐administered with the strong CYP3A4 inhibitor ritonavir. All graphs (i) are linear and (ii) logarithmic. Black lines represent the simulated mean remibrutinib blood concentrations after multiple daily oral doses of 20 mg at days 5 and 7, 50 mg at days 1–3 and 6 without ritonavir. (a) Remibrutinib 20 mg p.o. day 7 with ritonavir 100 mg b.i.d. (b) Remibrutinib 20 mg p.o. day 7 without ritonavir 100 mg b.i.d. (c) Remibrutinib 20 mg i.v. day 7 with ritonavir 100 mg b.i.d. (d) Remibrutinib 20 mg i.v. day 7 without ritonavir 100 mg b.i.d. PBPK, physiologically‐based pharmacokinetic; RTV, ritonavir
The DDI simulations with the weak, moderate, and strong CYP3A4 inhibitors fluvoxamine, erythromycin, and ketoconazole with single dose remibrutinib resulted in remibrutinib exposure increases of 1.15, 2.71, and 4.62, respectively. At steady‐state, remibrutinib AUCtau increased to 1.13, 2.40, and 4.17‐fold, respectively. The exposure of a single remibrutinib 100 mg i.v. dose administered at day 11 in a simulated DDI trial with rifampicin (600 mg q.d., day 1–14) was reduced by 28% (Table 3). For single and multiple oral administration of remibrutinib, an AUC decrease of 0.11‐fold and 0.36‐fold was predicted for the strong CYP3A4 inducer rifampicin and the moderate inducer efavirenz. The anticipated disposition scheme of remibrutinib is shown in Figure 3.
FIGURE 3.
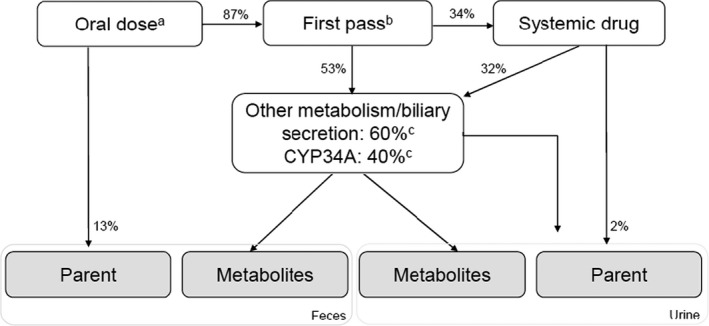
Potential disposition pathways of remibrutinib. aOral dose equals 100%. bFirst pass for intestine and liver, calculated using PBPK modeling. cFractional contribution of CYP3A4 based on clinical DDI results and sum of minor oxidative pathways (<2%), hydrolysis, GSH conjugates, and biliary secretion. Disposition values are derived from observed PK parameters listed in Table 1. DDI, drug‐drug interaction; GSH, glutathione; PBPK, physiologically‐based pharmacokinetic model, PK, pharmacokinetic
DISCUSSION
In this study, an integrated approach was applied to evaluate the DDI potential of remibrutinib as a victim drug. DDI data were generated from both an in vitro study and a clinical interaction study, including an i.v. microtracer treatment arm that allowed for robust and reliable DDI predictions using sophisticated PBPK modeling. In addition to standard PK parameters, such as the absolute bioavailability (F), total systemic CL and V ss, the clinical DDI study design enabled the determination of Fg , Fh , Fa , the fractional contribution of CYP3A4 to its clearance and the inhibition potential with a strong CYP3A4 inhibitor.
To avoid a potential confounding impact of TMDD on the PK parameters, the remibrutinib exposure increase was determined at steady‐state conditions. Administering remibrutinib p.o. with the strong CYP3A4/5 inhibitor RTV resulted in an exposure increase of 4.27‐fold. Although this is a substantial increase, remibrutinib does not meet the criteria for a sensitive CYP3A4 substrate (AUC ratio <5‐fold). When administering remibrutinib i.v. with ritonavir, the AUC increased 1.67‐fold compared to remibrutinib i.v. alone, indicating substantial CYP3A4 contribution to first‐pass metabolism after oral administration. Fg of remibrutinib escaping intestinal CYP3A4 metabolism was estimated to be 78.9%, by the selective inhibition of intestinal CYP3A4 using grapefruit juice (Table 2). In consequence, CYP3A4‐mediated hepatic first‐pass metabolism was calculated to transform 50.7% of remibrutinib (100%‐Fh ). However, intestinal inhibition by grapefruit juice may have been incomplete, resulting in an overestimation of Fg . The PBPK model Fg and Fh values were 55.4% and 62.3%, respectively, based on CL, fractional CYP3A4 contribution of CYP3A4 and Q gut. The product of Fg ⋅Fh is comparable with 39% (calculated values) and 34.5% (PBPK values). The Fg and Fh parameters established in the PBPK model are considered more conservative, because lower Fg value react more sensitive toward weak/moderate inhibitors or inducers. F of remibrutinib of 33.8% was determined, which increased to 80.9% under co‐administration of RTV. Fa was calculated using the measured Fg , F values and the estimated Fh , resulting in a value of 86.9%, which was in line with the predicted value of 88.9% based on Caco‐2 permeability data, characterizing remibrutinib as a highly permeable compound. Assuming complete absorption, the established Fa value of 86.9% would reflect additional first‐pass metabolism (13.1%), not inhibited by RTV. Considering the fact that remibrutinib CL is mainly driven by CYP3A4 metabolism and hydrolysis, additional first‐pass effects and complete absorption are unlikely. Based on the remibrutinib exposure increase of 1.66‐fold following i.v. administration in the presence of RTV, a 40% fraction metabolized via CYP3A4 was calculated.
Biliary clearance of unchanged remibrutinib is unlikely a major pathway, as an extended clearance classification concept system (EC3S) class 2b compound like remibrutinib (high permeability and high intrinsic metabolic clearance) is dominantly eliminated via oxidative metabolism and hydrolysis with minor contributions of biliary secretion. 27 This was confirmed by rat in vivo data, showing that excretion of unchanged remibrutinib into the bile comprised only 0.4% of the dose (data on file, Novartis). However, in rats, a significant amount of glutathione adducts were excreted via bile, indicating that glutathione conjugation may also be a relevant elimination pathway in humans. In humans, renal excretion was identified as a minor elimination pathway with a fractional contribution of about 1.8%.
The rich remibrutinib PK parameter set determined in this study supported the development of a robust PBPK model, which predicted the single dose and steady‐state PK parameters Cmax and AUC within twofold across the dose range of 15–200 mg. In addition, the DDI effect of RTV on remibrutinib i.v. or p.o. was predicted within the range calculated based on Guest et al., confirming a good model prediction. 24 On day 1, TMDD substantially contributed to the compound clearance, indicated by higher day 12 AUC, most likely due to BTK target saturation. The TMDD effect was dose‐dependent, described by the AUC ratio day 12 to day 1, which declined from 4.86 to 1.14 for the dose range 10–600 mg (Supplementary Table S6). To account for the TMDD process, metabolism by an additional clearance pathway together with an auto‐inhibition mechanism was added to the PBPK model. Hence, complete inhibition of the potential TMDD after several doses and/or high doses was achieved. Besides the remibrutinib dose and dosing frequency, the saturation of the target is dependent on the BTK abundance and re‐synthesis rate, which may not be fully reflected by the introduced auto‐inhibition mechanism of CYP2J2. 28 , 29 However, the applied k deg,CYPJ2 (0.0194 h−1, Simcyp V18) may differ from the human BTK k deg value (value unknown), which may result in a different time to develop full target saturation. The unknown abundance for whole body BTK expression was addressed by optimizing the CYP2J2 KI/k inact parameters accounting for the dose‐dependent TMDD effect. Exploring the impact of weak, moderate, and strong CYP3A4/5 inhibitors on the exposure of remibrutinib at steady‐state, PBPK simulations revealed no DDI effect by fluvoxamine, and moderate AUC ratios in the presence of erythromycin (AUC ratio: 2.40) and ketoconazole (AUC ratio: 4.17; Table 3).
Remibrutinib mean systemic CL is high (70.1 L/h), which is ~ 80% of hepatic blood flow (87 L/h). 30 For a high clearance drug, no major exposure decrease is expected when co‐administering strong CYP3A4 inducers, as clearance is considered blood flow‐limited. However, based on the significant first‐pass metabolism mediated mainly by CYP3A4, remibrutinib exposure might be sensitive to strong CYP3A4 inducers, which was confirmed by PBPK simulation with rifampicin predicting an 89% exposure loss. Co‐administration of remibrutinib with the moderate inducer efavirenz predicted an AUC reduction of 66%. As expected, a PBPK simulation of a single i.v. remibrutinib dose co‐administered with rifampicin resulted in a modest exposure decrease of 0.72‐fold. Thus, co‐administration of moderate or strong CYP3A4 inducers with p.o. remibrutinib should be considered carefully.
In summary, applying a specifically tailored clinical design can yield a rich set of PK parameters, which enabled building a robust PBPK model. The combination of clinical DDI trial results with PBPK predictions enabled conclusions on comedication with CYP3A4 perpetrators. Remibrutinib was identified as moderate sensitive CYP3A4 substrate when co‐administered with strong CYP3A4/5 inhibitors; this suggests weak CYP3A4 inhibitors may be used without restrictions. Based on PBPK simulations, remibrutinib exposure is predicted to be decreased by moderate and strong CYP3A4 inducers greater than 50%. These conclusions will aid in selection or restriction of comedications. Finally, for drugs with rather complex disposition based on nonclinical information, the current applied DDI study design (in particular, the inclusion of an i.v. microtracer arm) reveals a powerful method to determine key PK parameters.
CONFLICTS OF INTEREST
F.H., H.S., Y.J., B.P., W.W., R.W., A.D., and P.E. are full‐time employees of Novartis Institutes for BioMedical Research and hold shares of Novartis. C.S. is a full‐time employee of GCE Solutions, the Clinical Research Organization sponsored by Novartis for the analysis of the study data.
AUTHORS CONTRIBUTIONS
F.H., H.S., P.E., and H.F. wrote the manuscript. F.H., H.S., Y.J., B.P., C.S., W.W., R.W., A.D., and P.E. designed the research. F.H., H.S., Y.J., and B.P. performed the research. F.H., H.S., Y.J., B.P., C.S., R.W., A.D., and P.E. analyzed the data.
Supporting information
Fig S1
Fig S2
Fig S3
Supplementary Material
Supplementary Material
ACKNOWLEDGEMENTS
All the authors collaborated on writing the manuscript (with the assistance of a professional medical writer funded by Novartis) and made the decision to submit the manuscript for publication. Data analysis was conducted by PAREXEL, under supervision by Novartis. The authors thank Hayley Furlong, PhD, of Novartis Ireland Ltd., for providing medical writing assistance in accordance with Good Publication Practice guidelines (www.ismpp.org/gpp3).
Huth F, Schiller H, Jin Y, et al. Novel Bruton’s Tyrosine Kinase inhibitor remibrutinib: Drug‐drug interaction potential as a victim of CYP3A4 inhibitors based on clinical data and PBPK modeling. Clin Transl Sci. 2022;15:118–129. 10.1111/cts.13126
Funding information
This investigation was sponsored by Novartis Pharma AG, Basel, Switzerland.
REFERENCES
- 1. Kocatürk E, Zuberbier T. New biologics in the treatment of urticaria. Curr Opin Allergy Clin Immunol. 2018;18:425‐431. [DOI] [PubMed] [Google Scholar]
- 2. Volpe DA. Permeability classification of representative fluoroquinolones by a cell culture method. AAPS J. 2004;6:1‐6. [DOI] [PubMed] [Google Scholar]
- 3. Crawford JJ, Johnson AR, Misner DL, et al. Discovery of GDC‐0853: a potent, selective, and noncovalent Bruton’s tyrosine kinase inhibitor in early clinical development. J Med Chem. 2018;61:2227‐2245. [DOI] [PubMed] [Google Scholar]
- 4. Regan JA, Cao Y, Dispenza MC, et al. Ibrutinib, a Brutons tyrosine kinase inhibitor used for treatment of lymphoproliferative disorders, eliminates both aeroallergen skin test and basophil activation test reactivity. J Allergy Clin Immunol. 2017;140:875‐79.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Park MH, Shin SH, Byeon JJ, Lee GH, Yu BY, Shin YG. Prediction of pharmacokinetics and drug‐drug interaction potential using physiologically based pharmacokinetic (PBPK) modeling approach: a case study of caffeine and ciprofloxacin. Korean J Physiol Pharmacol. 2017;21:107‐115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lu C, Di L. In vitro and in vivo methods to assess pharmacokinetic drug– drug interactions in drug discovery and development. Biopharm Drug Dispos. 2020;41:3‐31. [DOI] [PubMed] [Google Scholar]
- 7. Bjornsson TD, Callaghan JT, Einolf HJ, et al. The conduct of in vitro and in vivo drug‐drug interaction studies: a Pharmaceutical Research and Manufacturers of America (PhRMA) perspective. Drug Metab Dispos. 2003;31:815‐832. [DOI] [PubMed] [Google Scholar]
- 8. Shitara Y. Clinical importance of OATP1B1 and OATP1B3 in drug‐drug interactions. Drug Metab Pharmacokinet. 2011;26:220‐227. [DOI] [PubMed] [Google Scholar]
- 9. Wittwer MB, Zur AA, Khuri N, et al. Discovery of potent, selective multidrug and toxin extrusion transporter 1 (MATE1, SLC47A1) inhibitors through prescription drug profiling and computational modeling. J Med Chem. 2013;56:781‐795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. CDER . FDA guidance for Industry (2020) in vitro drug interaction studies — cytochrome P450 enzyme‐ and transporter‐mediated drug interactions. 2020.
- 11. CDER . FDA guidance for Industry (2020) clinical drug interaction studies — cytochrome P450 enzyme‐ and transporter‐mediated drug interactions. 2020.
- 12. EMA . Guideline on the investigation of drug interactions. European Medicines Agency. 2015.
- 13. Adeloye T, Sahgal O, Puri A, et al. Amenamevir: studies of potential CYP3A‐mediated pharmacokinetic interactions with midazolam, cyclosporine, and ritonavir in healthy volunteers. Clin Pharmacol Drug Dev. 2018;7(8):844‐859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Umehara K‐I, Huth F, Won CS, Heimbach T, He H. Verification of a physiologically based pharmacokinetic model of ritonavir to estimate drug–drug interaction potential of CYP3A4 substrates. Biopharm Drug Dispos. 2018;39:152‐163. [DOI] [PubMed] [Google Scholar]
- 15. Bailey DG, Malcolm J, Arnold O, David Spence J. Grapefruit juice–drug interactions. Br J Clin Pharmacol. 1998;46:101‐110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Won CS, Oberlies NH, Paine MF. Mechanisms underlying food–drug interactions: inhibition of intestinal metabolism and transport. Pharmacol Ther. 2012;136:186‐201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rostami‐Hodjegan A, Tucker GT. Simulation and prediction of in vivo drug metabolism in human populations from in vitro data. Nat Rev Drug Discovery. 2007;6:140‐148. [DOI] [PubMed] [Google Scholar]
- 18. Luzon E, Blake K, Cole S, Nordmark A, Versantvoort C, Berglund EG. Physiologically based pharmacokinetic modeling in regulatory decision‐making at the European Medicines Agency. Clin Pharmacol Ther. 2017;102:98‐105. [DOI] [PubMed] [Google Scholar]
- 19. Huang WN, Tso TK, Kuo YC, Tsay GJ. Distinct impacts of syndesmophyte formation on male and female patients with ankylosing spondylitis. Int J Rheum Dis. 2012;15:163‐168. [DOI] [PubMed] [Google Scholar]
- 20. Research. Guidance for industry. Waiver of in vivo bioavailability and bioequivalence studies for immediate release solid oral dosage forms based on a biopharmaceutics classification system. Food and Drug Administration 2000.
- 21. Zhou W, Johnson TN, Xu H, et al. Predictive performance of physiologically based pharmacokinetic and population pharmacokinetic modeling of renally cleared drugs in children. CPT Pharmacometrics Syst Pharmacol. 2016;5:475‐483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jones H, Chen Y, Gibson C, et al. Physiologically based pharmacokinetic modeling in drug discovery and development: a pharmaceutical industry perspective. Clin Pharmacol Ther. 2015;97:247‐262. [DOI] [PubMed] [Google Scholar]
- 23. Coutant DE, Kulanthaivel P, Turner PK, et al. Understanding disease‐drug interactions in cancer patients: implications for dosing within the therapeutic window. Clin Pharmacol Ther. 2015;98:76‐86. [DOI] [PubMed] [Google Scholar]
- 24. Guest EJ, Aarons L, Houston JB, Rostami‐Hodjegan A, Galetin A. Critique of the two‐fold measure of prediction success for ratios: application for the assessment of drug‐drug interactions. Drug Metab Dispos. 2011;39:170‐173. [DOI] [PubMed] [Google Scholar]
- 25. Cheeti S, Budha NR, Rajan S, Dresser MJ, Jin JY. A physiologically based pharmacokinetic (PBPK) approach to evaluate pharmacokinetics in patients with cancer. Biopharm Drug Dispos. 2013;34:141‐154. [DOI] [PubMed] [Google Scholar]
- 26. Schwenger E, Reddy VP, Moorthy G, et al. Harnessing meta‐analysis to refine an oncology patient population for physiology‐based pharmacokinetic modeling of drugs. Clin Pharmacol Ther. 2018;103:271‐280. [DOI] [PubMed] [Google Scholar]
- 27. Camenisch GP. Drug disposition classification systems in discovery and development: a comparative review of the BDDCS, ECCS and ECCCS concepts. Pharm Res. 2016;33:2583‐2593. [DOI] [PubMed] [Google Scholar]
- 28. Pal Singh S, Dammeijer F, Hendriks RW. Role of Bruton’s Tyrosine Kinase in B cells and malignancies. Mol Cancer. 2018;17:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Alsadhan AA, Cheung J, Gulrajani M, et al. Variable Bruton Tyrosine Kinase (BTK) Resynthesis across patients with Chronic Lymphocytic Leukemia (CLL) on acalabrutinib therapy affect target occupancy and reactivation of B‐Cell Receptor (BCR) signaling. Blood. 2018;132:4401‐4501. [Google Scholar]
- 30. Davies B, Morris T. Physiological parameters in laboratory animals and humans. Pharm Res. 1993;10:1093‐1095. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S2
Fig S3
Supplementary Material
Supplementary Material