

Keywords: blood pressure, bosentan, endothelin-1, obesity, stress
Abstract
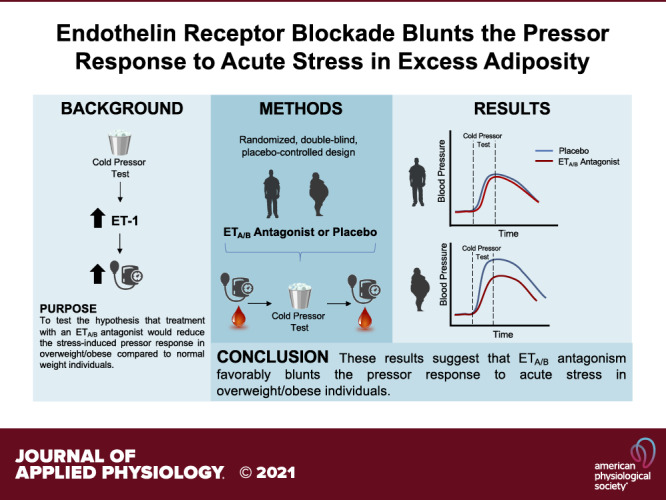
Obesity is associated with dysregulation of the endothelin system. In individuals with obesity, an exaggerated pressor response to acute stress is accompanied by increased circulating endothelin-1 (ET-1). The impact of combined endothelin A/B receptor (ETA/B) antagonism on the stress-induced pressor response in overweight/obese (OB) individuals is unknown. The objective of this study is to test the hypothesis that treatment with an ETA/B antagonist (bosentan) would reduce the stress-induced pressor response and arterial stiffness in overweight/obese compared with normal weight (NW) individuals. Forty participants [normal weight (NW): n = 20, body mass index (BMI): 21.7 ± 2.4 kg/m2 and overweight/obese (OB): n = 20, BMI: 33.8 ± 8.2 kg/m2] were randomized to placebo or 125 mg of bosentan twice a day (250 mg total) for 3 days. Hemodynamics were assessed before, during, and after a cold pressor test (CPT). Endothelin-1 was assessed at baseline and immediately after CPT. Following a washout period, the same protocol was repeated with the opposite treatment. The change from baseline in mean arterial pressure (MAP) during CPT following bosentan was significantly lower (P = 0.039) in the OB group than in the NW group (OB: 28 ± 12 vs. NW: 34 ± 15 mmHg). These results suggest that ETA/B antagonism favorably blunts the pressor response to acute stress in overweight/obese individuals.
NEW & NOTEWORTHY Findings from our current translational investigation demonstrate that dual endothelin A/B receptor antagonism blunts the pressor response to acute stress in overweight/obese individuals. These results suggest that modulation of the endothelin system may represent a novel therapeutic target to reduce cardiovascular disease (CVD) risk by blunting the stress response in overweight/obese individuals.
INTRODUCTION
The rate of overweight and obesity is rising and currently affects roughly 72% of the US population (1, 2). Increased adiposity intensifies vascular dysfunction, which contributes to a 3.5-fold increased risk for developing vascular diseases in individuals with obesity compared with normal weight (NW) individuals (3, 4). In fact, even in the absence of apparent vascular dysfunction, overweight/obese individuals have a 50% greater chance of developing cardiovascular disease (CVD) when compared with normal weight individuals (3, 5). Moreover, CVD-related mortality is almost 30% greater in overweight/obese than in normal weight individuals (5).
Growing evidence demonstrates that an exaggerated pressor response to acute stressors can increase the risk for developing CVD (6, 7). It is important to note that elevated adiposity is a major factor that can transiently increase arterial stiffness and exacerbate the pressor response to stress (8–11). Moreover, a heightened response to acute stress has been linked to increased long-term CVD risk (12, 13), and the cold pressor test (CPT) can predict CVD events in humans (14). Preclinical evidence indicates that endothelin-1 (ET-1) plays an important role in the pressor response to acute stress (15–17). ET-1 is the most abundant ligand of the endothelin system and is the most potent vasoconstrictor known (18) that is thought to contribute to the pathogenesis and progression of CVD (19). In addition, ET-1 is elevated with adiposity (20) and production of ET-1 is increased during pressor tests in animals and humans (21). Activation of vascular smooth muscle endothelin-A (ETA) receptors by ET-1 increases vascular tone (18) and exaggerates the pressor response to acute stress in animal models (15). Moreover, ET-1 binding to ETA increases central arterial stiffness, a clinical risk factor for the development of CVD (22). The actions of ETA are opposed by endothelin-B (ETB) receptors on endothelial cells (23). Activation of ETB not only elicits nitric oxide production and subsequent vasodilation but also aids in the clearance of ET-1 from circulation (23). However, in pathological states, such as CVD, dysregulation of the endothelin system, whether through alterations in ETA/B expression or receptor function, may contribute to the vasoconstrictive effect of ET-1, which plays a key role in the pathogenesis of CVD (24).
The concept of allostatic load suggests that the cumulative effects of repeated challenges may lead to chronic disease (25). Definition of the mechanistic response to an acute stressor may aid in understanding the chronic effects of stress. Despite the opposing actions of ETA and ETB, there are reports that dual ETA/B receptor blockade in animal models can be an effective method to alter the pressor response with few side effects (15, 26, 27). However, the impact of dual ETA/B antagonism on the pressor response to stress in overweight/obese individuals is unknown. Accordingly, the present investigation sought to test the hypothesis that subacute oral dual blockade of ETA/B receptors by bosentan (BOS) would blunt the pressor response to acute stress in overweight/obese than in normal weight individuals.
METHODS
Participant Characteristics
Forty healthy men and women aged 18–40 yr completed this investigation. Participants were excluded if they reported a history of cardiovascular disease, renal disease, metabolic disease, were on a sodium-restricted diet, a history of chronic pain, rheumatoid arthritis, hypertension, liver dysfunction, were using medications that are contraindicated with bosentan (i.e., glyburide, cyclosporine), or were taking any vasoactive medications (i.e., nitrates, β-blockers, ACE inhibitors, etc.). Of all the participants, seven were active smokers (3 NW and 4 OB) and were asked to refrain from smoking for at least 12 h before testing. All study protocols were approved by the Institutional Review Board at Augusta University. This study was registered at clinicaltrials.gov (NCT02116335).
Experimental Design and Clinical Laboratory Values
Figure 1 illustrates the clinical trial flowchart. All participants reported to the Laboratory of Integrative Vascular and Exercise Physiology (LIVEP) at the Georgia Prevention Institute for a preliminary visit that consisted of the informed consent process, anthropometric measures, and body composition assessment. On the preliminary day, a single stick venous blood draw was performed and a 3.5 mL of blood sample was collected. Hepatic enzymes, glycated hemoglobin (HbA1c), and concentrations of high-sensitivity C-reactive protein (hsCRP) were obtained using standard core laboratory techniques (Laboratory Corporation of American Holdings, Burlington, NC). Height and weight, determined using a stadiometer and standard platform scale (CN20, DETECTO, Webb City, MO), were used for calculation of body mass index (BMI). Total body fat, fat mass, and fat-free mass were determined using dual energy X-ray absorptiometry (QDR-4500W; Hologic, Waltham, MA).
Figure 1.

Clinical trial flowchart. n is the number of subjects in each group or category. CPT, cold pressor test.
Following the preliminary visit, all participants completed four experimental visits. For each experimental visit, participants reported to the LIVEP in the morning following an overnight fast and having abstained from moderate-to-vigorous physical activity for 24 h before investigation. On the first experimental day, an intravenous catheter was inserted and 24 mL of blood was collected for analysis of fasting concentrations of total cholesterol (TC), high-density lipoproteins (HDLs), low-density lipoproteins (LDLs), triglycerides (TG), and glucose using a Cholestech LDX point of care analyzer (Alere Inc., Scarborough, ME). Hemoglobin and hematocrit were determined using a HemoPoint H2 analyzer (Stanbio Laboratories, Boerne, TX). In addition, due to estrogen fluctuating across the menstrual cycle (28), estradiol was also measured in women at all testing visits. Resting systolic and diastolic blood pressures were evaluated using current American Heart Association recommendations (29). Briefly, participants were asked to sit with their backs supported for 10 min. Systolic and diastolic blood pressures were assessed using a digital vital signs monitor (Dinamap DPC200X, General Electric, Boston, MA) with an appropriately sized cuff. Demographic blood pressure values are reported as an average of duplicate measures taken 1-min apart. Based on BMI, participants were assigned to either a normal weight group (NW; BMI: 18.5–24.9 kg/m2) or an overweight/obese group (OB; BMI: 25.0–39.9 kg/m2).
Participants were randomized to ingest either placebo (PLA) or 125 mg of oral bosentan (BOS) twice a day (250 mg total) for three consecutive days. This dose is prescribed clinically to patients with pulmonary artery hypertension. Bosentan is a black-boxed labeled drug that can lead to altered hepatic function and alteration in endothelin receptor expression with chronic use (30). To investigate a mechanism and minimize the risk of chronic treatment side effects on apparently healthy, normotensive participants, we minimized the treatment period to 3 days. In addition, 3 days of treatment minimizes the off-target effects of long-term treatment, and our 3-day treatment was effective in blocking the endothelin receptors as demonstrated by our data. A nonselective endothelin receptor antagonist, bosentan, was selected to effectively block the receptors and evaluate compliance. An increase in ET-1 following blockade of the ETB receptors demonstrates adherence and effective antagonism. Following bosentan/placebo treatment, participants returned to the LIVEP and completed a second experimental visit that was identical to the first. A washout period of at least 7 days was observed, and participants then returned to the LIVEP to complete a third identical experimental visit. Following completion of the third experimental visit, participants were assigned to the opposite treatment in a crossover fashion. Participants completed 3 days of the opposite treatment before returning to the LIVEP for a fourth and final experimental visit.
Arterial Stiffness Assessment
During each experimental visit, the SphygmoCor XCEL system (AtCor Medical, Itasca, IL) was used to obtain brachial mean arterial pressure (bMAP), to determine aortic augmentation index adjusted to a heart rate of 75 beats per minute (aAIx@75), aortic augmentation pressure (aAP), and aortic mean arterial pressure (aMAP) using pulse wave analysis (PWA) of the brachial artery in accordance with the manufacturer’s recommendations. Carotid-to-femoral pulse wave velocity (cfPWV) was determined following recommendations (31). Briefly, carotid (applanation tonometry) and femoral artery (oscillometry) waveforms were recorded simultaneously. Straight-line distances were measured from the suprasternal notch to the carotid sampling site and from the suprasternal notch to where the femoral artery waveform was measured. The cfPWV was calculated using the distance between the aforementioned points and the measured time delay between the carotid and femoral waveform (t) as follows: cfPWV = D/t (m/s). All values are reported from duplicate measurements with standard deviations <10%.
Hemodynamic Assessment
Mean arterial pressure (MAP), total peripheral resistance indexed (TPRi) to body size, heart rate (HR), stroke volume indexed (SVi) to body size, and cardiac output indexed (COi) to body size were obtained using the Finapres NOVA beat-to-beat finger photoplethysmography (Finapress Medical Systems B.V., Institutenweg, The Netherlands). Briefly, an appropriately sized finger cuff was placed on the middle or index finger of the left hand. A height correction unit was attached to the participant’s chest at the level of the heart. The readjusted finger measurements were used in the final analysis. Measurements were recorded throughout all phases of the cold pressor test (CPT).
Cold Pressor Test
The cold pressor test (CPT) activates the sympathetic nervous system (SNS), which evokes a strong acute hemodynamic and cardiovascular response (32). CPT procedures were explained to participants before initiation of the test. Briefly, participants were placed into a semi-Fowlers position and the CPT protocol consisted of a 5-min resting baseline. Participants were then provided a verbal countdown and instructed to immerse their right hand, up to the wrist, in ice water (∼4°C) for 3 min (33). Immediately following completion of the test, participants’ hands were removed and wrapped in a dry towel during a 5-min recovery period. In an effort to capture the hemodynamic response to increased sympathetic activity without the influence of an exaggerated heart rate response and norepinephrine (34), the first and third minutes of the CPT were excluded from analysis and 15-s averages during the second minute of the CPT were analyzed.
Endothelin-1
Venous blood samples were obtained at baseline and immediately following the CPT. Blood samples were separated by centrifugation and plasma was flash frozen in liquid nitrogen and stored at −80°C till further analysis. Plasma concentrations of ET-1, within the detection range of 0.250–1,000 pg/mL were determined using microfluidic ELISA Simple Plex cartridges run on the Ella platform (ProteinSimple, San Jose, CA) according to manufacturer’s instructions.
Statistical Methods
All statistical analyses were performed using SPSS version 25 (IBM Corporation, Somers, NY). Independent t tests were performed to identify group differences (i.e., overweight/obese vs. normal weight) in demographics and clinical laboratory markers. Chi-square tests were performed to assess equal distribution of sex and race between groups. The change in ET-1 during CPT was calculated by subtracting the baseline levels of ET-1 from the post measures (ΔET-1 = post-CPT – pre-CPT); this value was used in the final analysis. The changes in hemodynamic variables during CPT were all calculated the same for the final data analysis, by subtracting pre-CPT measures from those measured during the CPT (i.e., MAPCPT – MAPpre-CPT = ΔMAP). Baseline characteristics are presented as means ± standard deviation (SD), and all other values are presented as the posttreatment means ± SD unless otherwise noted. Baseline (pre-CPT), ΔCPT, and recovery (post-CPT) measures for all major variables are presented to provide the entire stress response. A mixed-effect linear model was performed to analyze the mean difference between primary outcome variables including the change in circulating ET-1 during CPT (ΔET-1), the change in COi (ΔCOi), SVi (ΔSVi), TPRi (ΔTPRi), MAP (ΔMAP), and HR (ΔHR) during CPT, and PWV after intervention. The tests of interest are treatment (bosentan vs. placebo), group (obese vs. normal weight), and the treatment × group interaction. If a treatment × group interaction was identified, further stratified analysis by group was conducted to test the treatment effect in each group. Due to the study design, a period effect was considered and reported, if significant, in the statistical model to control for any change that occurred independent of our primary outcomes. All variables were controlled for period, sex, race, age, and the preintervention values of the outcome variables. In addition, due to the impact of blood lipids (35, 36), systemic inflammation (37), and glycemic control (38) on blood pressure regulation, total cholesterol, CRP, and HbA1c were analyzed as covariates for all hemodynamic variables assessed during and immediately following the CPT. An α < 0.05 was considered statistically significant for all analyses.
RESULTS
Participant Characteristics and Laboratory Values
Participant characteristics and clinical laboratory values are presented in Table 1. As expected, significant differences between the overweight/obese compared with the normal weight group were observed in weight, BMI, body fat percentage, blood pressure, blood lipids, and HbA1c. Within the OB group, seven were considered overweight (BMI = 25.0–29.9 kg/m2) and 13 were obese (BMI > 30.0 kg/m2). All other characteristics and laboratory values were similar between groups. In women, estradiol did not significantly differ between experimental visits or between groups. Three women (2 NW and 1 OB) were on oral contraceptives (Sprintec, Yasmin, and Low-Ogesterel) and one woman (NW) had an intrauterine device (Mirena). Bosentan treatment was well tolerated with no adverse reactions reported from either group.
Table 1.
Participant demographics and clinical laboratory values
| Variable | NW | OB | P Value |
|---|---|---|---|
| n | 20 | 20 | |
| Sex (M/F) | 10/10 | 12/8 | 0.525 |
| Race | |||
| Caucasian (M/F) | 11 (4/7) | 7 (6/1) | 0.096 |
| African America (M/F) | 7 (5/2) | 13 (6/7) | |
| Asian (M/F) | 2 (1/1) | 0 | |
| Age, yr | 27 ± 7 | 31 ± 6 | 0.070 |
| Height, cm | 172.0 ± 8.9 | 171.3 ± 9.8 | 0.866 |
| Weight, kg | 64.4 ± 9.7 | 101.0 ± 18.3 | <0.001* |
| BMI, kg/m2 | 21.7 ± 2.4 | 33.8 ± 8.2 | <0.001* |
| Body fat, % | 25.8 ± 7.9 | 38.5 ± 7.9 | <0.001* |
| SBP, mmHg | 113 ± 8 | 126 ± 9 | <0.001* |
| DBP, mmHg | 66 ± 13 | 74 ± 9 | 0.015* |
| MAP, mmHg | 81 ± 9 | 92 ± 8 | 0.001* |
| Heart Rate, beats/min | 64 ± 3 | 65 ± 1 | 0.600 |
| Clinical laboratory markers | |||
| TC, mg/dL | 151 ± 27 | 170 ± 29 | 0.037* |
| HDL, mg/dL | 59 ± 14 | 49 ± 14 | 0.023* |
| LDL, mg/dL | 74 ± 26 | 102 ± 28 | 0.002* |
| Triglycerides, mg/dL | 62 ± 22 | 93 ± 45 | 0.008* |
| TC:HDL | 2.6 ± 0.6 | 3.7 ± 1.3 | 0.002* |
| Glucose, mg/dL | 84 ± 6 | 89 ± 8 | 0.062 |
| HbA1c, % | 5.2 ± 0.2 | 5.5 ± 0.4 | 0.018* |
| Hemoglobin, g/dL | 14.0 ± 1.5 | 13.9 ± 1.5 | 0.408 |
| Hematocrit, % | 41.6 ± 3.8 | 41.3 ± 4.1 | 0.617 |
| C-reactive protein, mg/L | 3.2 ± 11.0 | 5.7 ± 12.9 | 0.514 |
| Estradiol, pg/mL | 42.2 ± 25.5 | 61.5 ± 36.9 | 0.157 |
Baseline participant characteristics for normal weight (NW) and obese (OB) groups. Data presented as means ± SD; *P < 0.05 between groups; Chi-square test performed for sex and race; independent samples t test. Estradiol was only measured in women. Bold type indicates significance. BMI, body mass index; DBP, diastolic blood pressure; HbA1c, glycated hemoglobin; HDL, high-density lipoprotein; LDL, low-density lipoprotein; MAP, mean arterial pressure; SBP, systolic blood pressure; TC, total cholesterol.
Endothelin-1 following Acute Stress
The main findings for ET-1 data are presented in Table 2. Circulating ET-1 before CPT significantly (P < 0.001) increased in both NW and OB groups following treatment with bosentan. During the CPT, the change in ET-1 significantly (P = 0.019) increased with treatment of bosentan regardless of BMI, no group-by-treatment effect was observed.
Table 2.
Concentrations of endothelin-1
| Treatment |
Obesity |
|||||
|---|---|---|---|---|---|---|
| Placebo | Bosentan | P Value | NW | OB | P Values | |
| Pre-CPT ET-1, pg/mL | 1.92 ± 0.70 | 2.75 ± 1.32 | <0.001 | 2.26 ± 1.18 | 2.42 ± 1.09 | 0.174 |
| ΔET-1, pg/mL | 0.37 ± 0.54 | 0.69 ± 1.06 | 0.019 | 0.50 ± 0.77 | 0.56 ± 0.94 | 0.391 |
| Post-CPT ET-1, pg/mL | 2.30 ± 0.65 | 3.44 ± 1.22 | <0.001 | 2.77 ± 1.07 | 2.98 ± 1.19 | 0.615 |
Means ± SD for the average circulating ET-1 following treatment with placebo or bosentan (treatment effect) regardless of adiposity or between the NW and OB groups (group effect) regardless of treatment. When a group-by-treatment interaction was observed, the means ± SD of the ET-1 following treatment in the placebo or bosentan are presented separately for the NW and OB group. ΔET-1 = the change in ET-1 (post-pre). P values were obtained from linear mixed model with age, sex, race, and pretreatment ET-1 included as covariates in the analysis. Bold type indicates significance. CPT, cold pressor test; ET-1, endothelin-1; NW, normal weight; OB, obese.
Hemodynamic Response to Acute Stress
Hemodynamic variables during CPT are presented in Table 3. There was a significant group-by-treatment interaction on ΔMAP (Fig. 2, P = 0.014). Specifically, with bosentan, the OB group had a significantly lower (P = 0.039) change in MAP during CPT than NW individuals. Given differences in baseline MAP, a secondary analysis was performed to control for differences in starting MAP (pre-CPT). Even when covarying for pre-CPT MAP, a significant group-by-treatment interaction was still observed for ΔMAP (β= −7.5 mmHg; P = 0.013). Specifically, the change in MAP during CPT (P < 0.001) was significantly lower following treatment with bosentan within the OB group compared with NW individuals.
Table 3.
Hemodynamics
| Treatment |
Obesity |
|||||
|---|---|---|---|---|---|---|
| Placebo | Bosentan | P Value | NW | OB | P Values | |
| Pre-CPT MAP, mmHg | 99 ± 10 | 96 ± 10 | 0.202 | 93 ± 8 | 101 ± 10 | 0.010 |
| ΔMAP, mmHg | ||||||
| NW | 31 ± 13 | 34 ± 15 | 0.071 | |||
| OB | 29 ± 14 | 28 ± 12 | 0.113 | |||
| Post-CPT MAP, mmHg | 102 ± 11 | 100 ± 11 | 0.353 | 98 ± 10 | 105 ± 11 | 0.094 |
| Pre-CPT HR, beats/min | 66 ± 9 | 68 ± 9 | 0.029 | 64 ± 9 | 70 ± 7 | 0.016 |
| ΔHR, beats/min | 16 ± 10 | 16 ± 8 | 0.315 | 17 ± 9 | 15 ± 9 | 0.283 |
| Post-CPT HR, beats/min | ||||||
| NW | 18 ± 10 | 16 ± 7 | 0.495 | |||
| OB | 14 ± 9 | 16 ± 9 | 0.003 | |||
| Pre-CPT SVi, mL/m2 | 51.4 ± 9.5 | 52.9 ± 9.9 | 0.406 | 51.3 ± 8.9 | 53.0 ± 10.5 | 0.999 |
| ΔSVi, mL/m2 | −4.2 ± 6.7 | −5.4 ± 4.8 | 0.173 | −6.2 ± 5.4 | −3.6 ± 5.9 | <0.001 |
| Post-CPT SVi, mL/m2 | 52.7 ± 10.7 | 53.9 ± 10.5 | 0.664 | 52.6 ± 9.3 | 54.1 ± 11.6 | 0.308 |
| Pre-CPT TPRi, dyn/s/cm5/m2 | ||||||
| NW | 0.62 ± 0.25 | 0.51 ± 0.20 | 0.002 | |||
| OB | 0.36 ± 0.15 | 0.35 ± 0.22 | 0.806 | |||
| ΔTPRi, dyn/s/cm5/m2 | 0.13 ± 0.13 | 0.12 ± 0.09 | 0.564 | 0.17 ± 0.13 | 0.08 ± 0.07 | <0.001 |
| Post-CPT TPRi, dyn/s/cm5/m2 | ||||||
| NW | 0.69 ± 0.29 | 0.56 ± 0.21 | 0.002 | |||
| OB | 0.38 ± 0.18 | 0.37 ± 0.25 | 0.824 | |||
| Pre-CPT COi, L/min/m2 | 6.7 ± 1.8 | 7.0 ± 2.0 | 0.089 | 5.8 ± 1.3 | 8.0 ± 1.8 | 0.015 |
| ΔCOi, L/min/m2 | 1.3 ± 1.1 | 1.3 ± 0.9 | 0.812 | 1.2 ± 0.8 | 1.4 ± 1.1 | 0.038 |
| Post-CPT COi, L/min/m2 | 6.7 ± 1.9 | 7.0 ± 2.1 | 0.093 | 5.7 ± 1.2 | 8.1 ± 1.9 | 0.006 |
Means ± SD for the average hemodynamics following treatment with placebo or bosentan (treatment effect) regardless of adiposity or between the NW and OB groups (group effect) regardless of treatment. When a group-by-treatment interaction was observed, the means ± SD of the variable following treatment in the placebo or bosentan are presented separately for the NW and OB groups. Δ = change in variable during CPT. P values were obtained from linear mixed model with age, sex, race, total cholesterol, HbA1c, CRP, and the pretreatment change as covariates in the analysis. Bold type indicates significance. COi, cardiac output indexed; CPT, cold pressor test; CRP, C-reactive protein; HbA1c, glycated hemoglobin; HR, heart rate; MAP, mean arterial pressure; NW, normal weight; OB, obese; SVi, stroke volume indexed; TPRi, total peripheral resistance indexed.
Figure 2.

The means ± SD and individual change from pre-CPT baseline during CPT between pre-to-post placebo (PLA), pre-to-post bosentan (BOS) visits in mean arterial pressure (MAP) in normal weight (NW) and overweight/obese (OB) groups. Bars represent the mean. Group × treatment interaction P = 0.0031; linear mixed-model. *P < 0.05 vs. NW BOS; linear mixed-model. CPT, cold pressor test.
Regardless of BMI, the ΔHR during CPT was not significantly different following treatment with bosentan compared with placebo (P = 0.315). A significant period effect was observed for ΔHR (P = 0.010). There was a significant group-by-treatment interaction on HR recovery following CPT (P = 0.048). Specifically, HR significantly increased in the OB group following treatment with bosentan (P = 0.003), whereas HR recovery was not different in the NW group (P = 0.495).
Treatment with bosentan did not impact the ΔSVi during CPT overall (P = 0.173). However, independent of treatment, the ΔSVi was significantly greater in the OB group than in the NW group (P < 0.001). There were no observed differences following treatment overall (P = 0.664) or between NW and OB (P = 0.308) on the recovery of SVi after CPT.
A significant group-by-treatment interaction was observed for baseline TPRi before the CPT test (P = 0.028). Specifically, the NW group experienced a significant decrease in baseline TPRi following treatment with bosentan (P = 0.002), whereas no change was observed in the OB after treatment (P = 0.806). Treatment with bosentan did not change the ΔTPRi response during CPT (P = 0.564). However, the NW group had a significantly greater ΔTPRi during CPT than the OB group (P < 0.001). A significant group-by-treatment interaction for TPRi recovery after CPT was observed (P = 0.033). Specifically, the NW group experienced a significant decrease in TPRi during recovery from CPT following treatment with bosentan (P = 0.002), whereas the OB group did not have any change in TPRi during recovery after treatment with bosentan (P = 0.824).
Treatment with bosentan did not change the overall ΔCOi during CPT (P = 0.089); however, the OB group had a greater baseline COi before CPT (P = 0.015) and ΔCOi during CPT than the NW group, independent of treatment (P = 0.038). A significant period effect was also observed (P = 0.010). The OB group had significantly greater COi during recovery than the NW group regardless of treatment (P = 0.006).
Arterial Stiffness
Indices of arterial stiffness are presented in Table 4. A significant group-by-treatment interaction was observed for bMAP (P < 0.001). Specifically, NW individuals experienced a significant decrease in bMAP following treatment with bosentan compared with placebo (P < 0.001), whereas no change in bMAP following treatment with bosentan compared with placebo was observed in the OB group (P = 0.423). In this line, a similar interaction was observed for aMAP (group × treatment interaction, P < 0.001). A significant decline in aMAP after treatment with bosentan compared with placebo (P < 0.001) was observed in the NW group, whereas no change in aMAP was observed in the OB group following treatment with bosentan (P = 0.417). There was no observed effect of bosentan on aAP or aAIx@75; however, treatment with bosentan significantly reduced overall cfPWV, independent of BMI (P = 0.002). This finding held true when adjusted for the change in MAP after treatment with bosentan (P = 0.003). No period effect was observed for any arterial stiffness measures.
Table 4.
Arterial stiffness
| Treatment |
Obesity |
|||||
|---|---|---|---|---|---|---|
| Placebo | Bosentan | P Value | NW | OB | P Value | |
| bMAP, mmHg | ||||||
| NW | 85 ± 7 | 80 ± 5 | <0.001 | |||
| OB | 91 ± 9 | 89 ± 8 | 0.423 | |||
| aMAP, mmHg | ||||||
| NW | 83 ± 7 | 78 ± 9 | <0.001 | |||
| OB | 91 ± 10 | 88 ± 9 | 0.417 | |||
| aAP, mmHg | 7 ± 6 | 7 + 6 | 0.707 | 5 ± 5 | 9 ± 7 | 0.536 |
| aAIx@75, % | 14 ± 16 | 14 + 15 | 0.658 | 8 ± 15 | 19 ± 14 | 0.114 |
| cfPWV, m/s | 6.0 ± 0.8 | 5.7 + 0.7 | 0.002 | 5.7 ± 0.9 | 6.0 ± 0.6 | 0.725 |
Means ± SD for the average arterial stiffness variables following treatment with placebo or bosentan (treatment effect) regardless of adiposity or between the NW and OB groups (group effect) regardless of treatment. When a group-by-treatment interaction was observed, the means ± SD of the variable are presented separately for the NW and OB group. P values were obtained from linear mixed model with age, sex, race, and pretreatment values were included as covariates in the analysis. Bold type indicates significance. aAP, aortic augmentation pressure; aAIx@75, aortic augmentation index adjusted for heart rate of 75; aMAP, aortic mean arterial pressure; bMAP, brachial mean arterial pressure; cfPWV, carotid-to-femoral pulse wave velocity; NW, normal weight; OB, obese.
DISCUSSION
Exaggerated pressor responses to stress and arterial stiffness can each increase the risk of CVD. Increased adiposity has been associated with increased arterial stiffness and an elevation in the pressor response to stress. In addition, acute stress increases endothelin-1, which directly increases arterial stiffness and further exacerbates the pressor responses to stress. Accordingly, this investigation sought to determine the impact of endothelin A/B receptor blockade on the acute stress-induced pressor response in overweight/obese compared with normal weight individuals. To our knowledge, this is the first study to demonstrate that nonselective ETA/B receptor antagonism reduces the pressor response to stress in overweight/obese individuals.
Efficacy of Treatment
In addition to its vasodilatory actions, ETB has been shown to be responsible for clearance of ET-1 from circulation (23). Consistent with previously published data (23, 39), basal plasma concentrations of ET-1 in the current investigation were significantly elevated in both groups following bosentan treatment. Although bosentan has a 20-fold selectivity for ETA receptors (40), which can change under in vivo conditions (40), bosentan’s impact on the ETB receptor is likely responsible for the observed increase in plasma ET-1 and demonstrates the effectiveness of our 3-day treatment to inhibit both ET receptors. Furthermore, acute stress has been shown to significantly increase plasma concentrations of ET-1 (15, 41). Although ET-1 increased in response to acute stress in both groups, the increase in ET-1 in response to CPT was greater following bosentan treatment. These data suggest that bosentan reduced ET-1 clearance during acute stress in both NW and OB groups. Although ETB antagonism reduces ETB receptor-mediated nitric oxide production by vascular endothelial cells, no significant changes were observed in the basal blood pressure of either group, which is consistent with previous bosentan studies in apparently healthy individuals (39, 42). Taken together, these data suggest that 3 days of treatment with bosentan was an effective endothelin receptor antagonist.
Stress-Induced Pressor Response
An exaggerated stress-induced pressor response has been linked with increased CVD risk (12, 43). Perhaps even more important, the cumulative effects of repeated stressors over time may accelerate both the development of hypertension and the atherosclerotic disease process (25). Circulating ET-1 is increased in response to acute stress (15, 16), and ETA/B receptor blockade has been shown to reduce the pressor response to acute stress in prehypertensive rats (15). As expected, the CPT increased MAP in both groups. However, when modulating endothelin activity with bosentan, a significant decrease in MAP to the CPT was observed following treatment with bosentan only in the OB group. These findings support the mechanistic involvement of ET-1 in mediating the pressure response to acute stress in individuals with elevated adiposity. Although participants in the current investigation had blood pressure within normal limits, it is important to note that baseline systolic blood pressure (SBP), diastolic blood pressure (DBP), and MAP were higher in the OB group. Although we cannot rule out that greater sympathetic nervous activity (SNA) could alter the pressor response to stress, when statistically controlling for baseline MAP, the results remained the same. Nonetheless, previous studies have shown that individuals with greater stress-induced pressor responses have a 52% greater risk of developing hypertension (44) and that the stress pressor response is positively associated with the progression of carotid atherosclerosis over a 2-year period (45). The primary purpose of the investigation was to understand the pressor response; however, the recovery from CPT was included and evaluated to provide the complete effects of the stressor. The poststress recovery response is a valuable contributor to the physiological harm that the actual stress (i.e., CPT) exerts. Specifically, a quicker recovery immediately after a stressor is associated with lower risk of CVD events (46). In this line, we observed a slower recovery in blood pressure or longer time to return to baseline after a stress event in the OB group, which results in an increase in exposure time to the harmful effects of stress events. Activation of ETA receptors on vascular smooth muscle induces vasoconstriction, which is normally opposed by ETB-mediated nitric oxide (NO) production by endothelial cells and ETB-mediated clearance of ET-1 from circulation. In humans, renal artery ETA expression is nine times greater than ETB expression (47), and greater concentrations of circulating ET-1 have been shown to contribute to increased vascular tone and higher basal blood pressure (19). In the current investigation, basal systolic, diastolic, and mean arterial pressures were significantly greater in the OB group than the NW group, despite similar basal concentrations of circulating ET-1. Furthermore, the change in the ET-1 response to stress was not significantly different between the OB and NW groups and no significant differences were observed between groups in post-CPT absolute concentrations of plasma ET-1 following treatments. However, although not different between the groups, the stress-induced change in ET-1 was significantly greater following bosentan treatment. These data suggest that both ET-1 production and clearance may be similar between apparently healthy normal weight individuals and individuals with obesity.
Stress Hemodynamics
Heightened hemodynamic responses to stress have been implicated as a cause of vascular damage that contributes to the pathogenesis and progression of CVD (6). TPR, which is the ratio of CO to blood pressure (48), has a strong positive association with CVD risk and has been suggested to be a primary factor in CVD progression (49). In addition, both normotensive individuals and individuals with hypertension with elevated blood pressure maintained by increased TPR relative to CO have an increased risk of cardiovascular events and death (50). In the current investigation, TPRi at baseline and during recovery was significantly lower following bosentan treatment compared with placebo treatment in the NW group. In the NW group, lower TPRi was also accompanied by greater COi during baseline and recovery following bosentan treatment compared with placebo. These responses are likely due to reduced vascular tone elicited by ETA antagonism (51). Interestingly, bosentan treatment did not impact the hemodynamic response to stress in the OB group. It is possible that endothelin receptor antagonism was not enough to significantly reduce vascular resistance during CPT in the OB group. There are a number of factors that can contribute to vascular resistance, particularly in a multifactorial disease such as obesity or elevated adiposity (52). Although not measured in this study, inflammation (53) and elevated ROS (24) can contribute to systemic vascular resistance or endothelial dysfunction—all of which are commonly observed in people with elevated adiposity (53). Nonetheless, bosentan only reduced pre- and post-CPT TRPi in the NW group, whereas the change in TPRi to the CPT, the primary variable of interest, was similar in both groups following treatment with bosentan. Taken together, these data suggest that ET-1 receptor antagonism reduces vascular resistance in normal weight individuals and does not negatively impact the hemodynamic response to acute stress in individuals with obesity.
Arterial Stiffness
Carotid-to-femoral pulse wave velocity is a noninvasive assessment of central arterial stiffness. Increased arterial stiffness has been associated with accelerated blood pressure progression and increased incidence of cardiovascular events in normotensive individuals (54). For the first time, we demonstrate that nonselective ETA/B receptor antagonism significantly reduces cfPWV in nonhypertensive, normal weight, and overweight/obese individuals. These findings are in agreement with previous studies that demonstrate that ET-1 directly increases arterial stiffness (55) and that ETA blockade mitigates these effects (22). Interestingly, the magnitude of the reduction in arterial stiffness following bosentan was similar between individuals with obesity and normal weight individuals. Although ETA activation has been implicated in increasing arterial stiffness, the mechanisms responsible are unknown. Both groups exhibited similar reductions in cfPWV following bosentan treatment. Therefore, it is likely that the mechanisms by with the endothelin system regulates arterial stiffness are similar between normal weight individuals and individuals with obesity, such as endothelin-mediated arterial compliance (56, 57). In addition, both groups had similar baseline endothelin activity (circulating ET-1) and similar baseline arterial stiffness, which could explain the similar change observed following treatment with bosentan. Recognizing that blood pressure can affect arterial stiffness, when the change in MAP following bosentan was statistically controlled for, a significant reduction in cfPWV continued to be observed. Moreover, the change in both bMAP and aMAP following bosentan was significantly reduced in the NW group, potentially contributing to the decline in arterial stiffness in that group. These findings suggest that bosentan elicits more favorable hemodynamics at rest compared with placebo in normal weight individuals. Nonetheless, these data suggest that blockade of ETA/B receptors reduces arterial stiffness in both overweight/obese and normal weight individuals.
Experimental Considerations
Both endothelin receptor expression and circulating concentrations of ET-1 have been demonstrated to be transiently altered by smoke inhalation and exposure (58). The number of active smokers were equally distributed in each group, three NW and four OB, and all were instructed to refrain from smoking for at least 12 h before their visits to reduce short-term effects. In addition, a secondary analysis excluding the seven smokers was performed and the overall findings held true with or without smokers included.
Although both, visceral (VAT) and subcutaneous (SAT) adipose tissue are correlated with metabolic risk factors, VAT has been demonstrated to be more strongly associated with adverse cardiovascular risk (59). Although the primary focus of this investigation was on adiposity in general, future research should focus on determining the impact that regional distribution of body fat (central vs. peripheral) may have on the endothelin system and pressor response to stress.
Circulating sex-steroid hormones can play a role on the endothelin system (28, 60, 61). Although the exact phase of the menstrual cycle during testing is unknown, estradiol was measured at each experimental visit in all women to ensure that testing was performed in a similar phase of the menstrual cycle. Four women, three NW and one OB, were on an oral contraceptive (n = 3) or had an intrauterine device (n = 1). To the best of our knowledge, contraceptive pills do not cause major changes in circulating concentrations of ET-1 (62, 63). Nonetheless, sex was used as a variable in our statistical analysis and our results remained the same; a significant treatment by group interaction (P = 0.010) was observed for ΔMAP during CPT.
ET receptors are heterotrimeric G protein-coupled receptors, which stimulate multiple signaling pathways and regulate diverse cellular functions (64). Therefore, activation of the ET receptors can result in a multitude of downstream effects, one important downstream consequence is its proinflammatory properties (65). Regulation of cyclooxygenase (COX)-dependent vasoconstriction by ETA receptors has been reported by several groups (65–68). Evidence also indicates that ET stimulates phospholipase A2 activity, ultimately increasing arachidonic acid-derived mediators such as prostaglandins and thromboxane (69). Furthermore, ET-induced vasoconstriction is dependent on the presence of COX (67). Not only is obesity associated with enhanced ET-1 contractility, COX-dependent prostanoids are also upregulated with augmented sensitivity (65, 67). It is possible that blockade of the ET receptors with bosentan could reduce the expression of COX-induced prostanoids and contribute to the observed reductions in the pressor response. However, COX-inhibitors were not administered in the present study and we cannot rule out the potential effects of COX-induced vasoconstriction. Future investigations should consider the interaction of ET and COX during the stress response.
Although endothelin receptor function was not directly assessed in the current investigation, adiposity has been shown to alter endothelin system signaling including enhanced ETA-mediated vasoconstriction (70) in humans and ETB-induced vasoconstriction in mice (71). Moreover, animal models have demonstrated significantly lower renal expression of ETA (72) and adipose tissue ET-1 (73) in normal weight than in obese, a characteristic that may have impacted the pressor response to stress. In addition, although ETA/B expression has been shown to be altered in response to chronic bosentan treatment (30), the current study only examined 3 days of bosentan administration. Therefore, investigation into the long-term efficacy of bosentan on the stress-induced pressor response in healthy individuals with increased adiposity may produce different findings. It is also important to note that there are other assessments that can induce a stress response similar to the CPT (i.e., mental stress, isometric handgrip, or postexercise ischemia). The CPT has been heavily studied in relationship to CVD risk and is easily assessable, affordable, and validated in cardiovascular research. Nonetheless, future studies are needed to evaluate if our findings are consistent with other forms of physiological and biological stressors. In a similar line, CPT-mediated blood pressure alterations are, in part, mediated by alterations in the sympathetic nervous system (SNS). However, an increase in ET-1 has been observed in preclinical models and a number of clinical populations in response to CPT. More importantly, the acute stress-mediated increase in ET-1 has been observed in both animals and humans without activation of SNS activity (14). Before our investigation, the direct contributions of ET-1 to the hemodynamic changes during/after CPT were unclear. Although we cannot rule out the presence of SNS activity, our findings suggest that the stress-mediated increases in ET-1 contribute, in part, to the alterations in hemodynamics. Given that activation of endothelial ETB receptors mediates NO production (23) and ETA receptor activation modulates an increase in oxidative stress (74), it is plausible that receptor antagonism could alter the NO-oxidative stress balance. We recognize that additional assays could have provided useful insight; however, future studies should consider evaluating markers of NO or oxidative stress following endothelin receptor modulation. In addition, the actions of ETA are opposed by ETB receptors and to some extent, macrophages (23). Although not directly related to the scope of the present investigation, assessments of macrophages and even the polarization of M1- and M2-like macrophages could help provide mechanistic insight warranting future investigations.
Conclusions
To the best of our knowledge, for the first time, findings from the present investigation demonstrate that nonselective endothelin A/B receptor antagonism blunts the pressor response to acute stress in people with elevated adiposity. These results extend preclinical data and suggest that modulation of the endothelin system may represent a novel therapeutic approach to reduce CVD risk by blunting the stress response in overweight/obese individuals.
GRANTS
This study was supported by Grant 5P01HL069999 from NIH/National Heart, Lung, and Blood Institute (NHLBI).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
D.M.P. and R.A.H. conceived and designed research; A.M.B., M.A.T., J.H.J., P.R-M, J.L., and J.T. performed experiments; C.C.D., A.M.B., X.W., M.A.T., C.H., J.H.J., and R.A.H. analyzed data; C.C.D., A.M.B., X.W., M.A.T., and R.A.H. interpreted results of experiments; C.C.D. and A.M.B. prepared figures; A.M.B. drafted manuscript; C.C.D., X.W., M.A.T., C.H., J.H.J., P.R-M., J.L., D.M.P., and R.A.H. edited and revised manuscript; C.C.D., A.M.B., X.W., M.A.T., C.H., J.H.J., P.R-M, J.L. J.T., D.M.P., and R.A.H approved final version of manuscript.
REFERENCES
- 1.Mitchell NS, Catenacci VA, Wyatt HR, Hill JO. Obesity: overview of an epidemic. Psychiatr Clin North Am 34: 717–732, 2011. doi: 10.1016/j.psc.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Centers for Disease Control and Prevention. Data and Statistics. Obesity and Overweight. National Center for Health Statistics Trend Tables, 2020. https://www.cdc.gov/obesity/index.html.
- 3.Shimbo D, Muntner P, Mann D, Viera AJ, Homma S, Polak JF, Barr RG, Herrington D, Shea S. Endothelial dysfunction and the risk of hypertension: the multi-ethnic study of atherosclerosis. Hypertension 55: 1210–1216, 2010. doi: 10.1161/HYPERTENSIONAHA.109.143123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kotchen TA. Obesity-related hypertension: epidemiology, pathophysiology, and clinical management. Am J Hypertens 23: 1170–1178, 2010. doi: 10.1038/ajh.2010.172. [DOI] [PubMed] [Google Scholar]
- 5.Jiang J, Ahn J, Huang WY, Hayes RB. Association of obesity with cardiovascular disease mortality in the PLCO trial. Prev Med 57: 60–64, 2013. doi: 10.1016/j.ypmed.2013.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van der Valk ES, Savas M, van Rossum EFC. Stress and obesity: are there more susceptible individuals? Curr Obes Rep 7: 193–203, 2018. doi: 10.1007/s13679-018-0306-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.D’Angelo G, Mintz JD, Tidwell JE, Schreihofer AM, Pollock DM, Stepp DW. Exaggerated cardiovascular stress responses and impaired β-adrenergic-mediated pressor recovery in obese Zucker rats. Hypertension 48: 1109–1115, 2006. doi: 10.1161/01.HYP.0000247306.53547.d4. [DOI] [PubMed] [Google Scholar]
- 8.Barnes VA, Treiber FA, Davis H, Kelley TR, Strong WB. Central adiposity and hemodynamic functioning at rest and during stress in adolescents. Int J Obes Relat Metab Disord 22: 1079–1083, 1998. doi: 10.1038/sj.ijo.0800730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sung BH, Wilson MF, Izzo JL Jr, Ramirez L, Dandona P. Moderately obese, insulin-resistant women exhibit abnormal vascular reactivity to stress. Hypertension 30: 848–853, 1997. doi: 10.1161/01.HYP.30.4.848. [DOI] [PubMed] [Google Scholar]
- 10.Ribeiro MM, Silva AG, Santos NS, Guazzelle I, Matos LN, Trombetta IC, Halpern A, Negrão CE, Villares SM. Diet and exercise training restore blood pressure and vasodilatory responses during physiological maneuvers in obese children. Circulation 111: 1915–1923, 2005. doi: 10.1161/01.CIR.0000161959.04675.5A. [DOI] [PubMed] [Google Scholar]
- 11.Tabara Y, Kohara K, Nakagawa S, Handa J, Hayashi M, Hamada C, Miyaguchi M, Shigemi Y, Miki T, Konishi M. Effects of obesity and smoking on mental stress-induced blood pressure and augmentation index responses in normotensive young males: the J-SHIPP study. Hypertens Res 31: 1219–1224, 2008. doi: 10.1291/hypres.31.1219. [DOI] [PubMed] [Google Scholar]
- 12.Chida Y, Steptoe A. Greater cardiovascular responses to laboratory mental stress are associated with poor subsequent cardiovascular risk status: a meta-analysis of prospective evidence. Hypertension 55: 1026–1032, 2010. doi: 10.1161/HYPERTENSIONAHA.109.146621. [DOI] [PubMed] [Google Scholar]
- 13.Dimsdale JE. Psychological stress and cardiovascular disease. J Am Coll Cardiol 51: 1237–1246, 2008. doi: 10.1016/j.jacc.2007.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pouwels S, Van Genderen ME, Kreeftenberg HG, Ribeiro R, Parmar C, Topal B, Celik A, Ugale S. Utility of the cold pressor test to predict future cardiovascular events. Expert Rev Cardiovasc Ther 17: 305–318, 2019. doi: 10.1080/14779072.2019.1598262. [DOI] [PubMed] [Google Scholar]
- 15.D'Angelo G, Loria AS, Pollock DM, Pollock JS. Endothelin activation of reactive oxygen species mediates stress-induced pressor response in Dahl salt-sensitive prehypertensive rats. Hypertension 56: 282–289, 2010. doi: 10.1161/HYPERTENSIONAHA.110.152629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fox BM, Becker BK, Loria AS, Hyndman KA, Jin C, Clark H, Johns R, Yanagisawa M, Pollock DM, Pollock JS. Acute pressor response to psychosocial stress is dependent on endothelium-derived endothelin-1. J Am Heart Assoc 7: e007863, 2018. doi: 10.1161/JAHA.117.007863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Speed JS, D'Angelo G, Wach PA, Sullivan JC, Pollock JS, Pollock DM. High salt diet increases the pressor response to stress in female, but not male ETB-receptor-deficient rats. Physiol Rep 3: e12326, 2015. doi: 10.14814/phy2.12326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kowalczyk A, Kleniewska P, Kolodziejczyk M, Skibska B, Goraca A. The role of endothelin-1 and endothelin receptor antagonists in inflammatory response and sepsis. Arch Immunol Ther Exp (Warsz) 63: 41–52, 2015. doi: 10.1007/s00005-014-0310-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mathew V, Hasdai D, Lerman A. The role of endothelin in coronary atherosclerosis. Mayo Clin Proc 71: 769–777, 1996. doi: 10.1016/S0025-6196(11)64842-8. [DOI] [PubMed] [Google Scholar]
- 20.Ferri C, Bellini C, Desideri G, Di Francesco L, Baldoncini R, Santucci A, De Mattia G. Plasma endothelin-1 levels in obese hypertensive and normotensive men. Diabetes 44: 431–436, 1995. doi: 10.2337/diabetes.44.4.431. [DOI] [PubMed] [Google Scholar]
- 21.Danese C, Parlapiano C, Zavattaro E, Di Prima M, Campana E, Rota C, Tonnarini G, Di Siena G, Borgia MC. ET-1 plasma levels during cold stress test in sclerodermic patients. Angiology 48: 965–968, 1997. doi: 10.1177/000331979704801105. [DOI] [PubMed] [Google Scholar]
- 22.McEniery CM, Qasem A, Schmitt M, Avolio AP, Cockcroft JR, Wilkinson IB. Endothelin-1 regulates arterial pulse wave velocity in vivo. J Am Coll Cardiol 42: 1975–1981, 2003. doi: 10.1016/j.jacc.2003.06.016. [DOI] [PubMed] [Google Scholar]
- 23.Fukuroda T, Fujikawa T, Ozaki S, Ishikawa K, Yano M, Nishikibe M. Clearance of circulating endothelin-1 by ETB receptors in rats. Biochem Biophys Res Commun 199: 1461–1465, 1994. doi: 10.1006/bbrc.1994.1395. [DOI] [PubMed] [Google Scholar]
- 24.Dhalla NS, Temsah RM, Netticadan T. Role of oxidative stress in cardiovascular diseases. J Hypertens 18: 655–673, 2000. doi: 10.1097/00004872-200018060-00002. [DOI] [PubMed] [Google Scholar]
- 25.McEwen BS, Seeman T. Protective and damaging effects of mediators of stress. Elaborating and testing the concepts of allostasis and allostatic load. Ann N Y Acad Sci 896: 30–47, 1999. doi: 10.1111/j.1749-6632.1999.tb08103.x. [DOI] [PubMed] [Google Scholar]
- 26.Jasmin JF, Lucas M, Cernacek P, Dupuis J. Effectiveness of a nonselective ETA/B and a selective ETA antagonist in rats with monocrotaline-induced pulmonary hypertension. Circulation 103: 314–318, 2001. doi: 10.1161/01.CIR.103.2.314. [DOI] [PubMed] [Google Scholar]
- 27.Iglarz M, Steiner P, Wanner D, Rey M, Hess P, Clozel M. Vascular effects of endothelin receptor antagonists depends on their selectivity for ETA versus ETB receptors and on the functionality of endothelial ETB receptors. J Cardiovasc Pharmacol 66: 332–337, 2015. doi: 10.1097/FJC.0000000000000283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Merki-Feld GS, Imthurn B, Keller PJ. The effect of the menstrual cycle and of ethinylestradiol on nitric oxide, endothelin-1 and homocysteine plasma levels. Horm Metab Res 32: 288–293, 2000. doi: 10.1055/s-2007-978638. [DOI] [PubMed] [Google Scholar]
- 29.Muntner P, Shimbo D, Carey RM, Charleston JB, Gaillard T, Misra S, Myers MG, Ogedegbe G, Schwartz JE, Townsend RR, Urbina EM, Viera AJ, White WB, Wright JT Jr. Measurement of blood pressure in humans: a scientific statement from the American Heart Association. Hypertension 73: e35–e66, 2019. doi: 10.1161/HYP.0000000000000087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sauvageau S, Thorin E, Villeneuve L, Dupuis J. Change in pharmacological effect of endothelin receptor antagonists in rats with pulmonary hypertension: role of ETB-receptor expression levels. Pulm Pharmacol Ther 22: 311–317, 2009. doi: 10.1016/j.pupt.2009.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Laurent S, Cockcroft J, Van Bortel L, Boutouyrie P, Giannattasio C, Hayoz D, Pannier B, Vlachopoulos C, Wilkinson I, Struijker-Boudier H; European Network for Non-invasive Investigation of Large Arteries. Expert consensus document on arterial stiffness: methodological issues and clinical applications. Eur Heart J 27: 2588–2605, 2006. doi: 10.1093/eurheartj/ehl254. [DOI] [PubMed] [Google Scholar]
- 32.Kelsey RM, Patterson SM, Barnard M, Alpert BS. Consistency of hemodynamic responses to cold stress in adolescents. Hypertension 36: 1013–1017, 2000. doi: 10.1161/01.HYP.36.6.1013. [DOI] [PubMed] [Google Scholar]
- 33.Mizushima T, Tajima F, Okawa H, Umezu Y, Furusawa K, Ogata H. Cardiovascular and endocrine responses during the cold pressor test in subjects with cervical spinal cord injuries. Arch Phys Med Rehabil 84: 112–118, 2003. doi: 10.1053/apmr.2003.50072. [DOI] [PubMed] [Google Scholar]
- 34.Victor RG, Leimbach WN Jr, Seals DR, Wallin BG, Mark AL. Effects of the cold pressor test on muscle sympathetic nerve activity in humans. Hypertension 9: 429–436, 1987. doi: 10.1161/01.hyp.9.5.429. [DOI] [PubMed] [Google Scholar]
- 35.Drexler H, Zeiher AM. Endothelial function in human coronary arteries in vivo. Focus on hypercholesterolemia. Hypertension 18: II90–99, 1991. doi: 10.1161/01.hyp.18.4_suppl.ii90. [DOI] [PubMed] [Google Scholar]
- 36.Sung BH, Izzo JL Jr, Wilson MF. Effects of cholesterol reduction on BP response to mental stress in patients with high cholesterol. Am J Hypertens 10: 592–599, 1997. doi: 10.1016/S0895-7061(97)00050-2. [DOI] [PubMed] [Google Scholar]
- 37.Yudkin JS, Stehouwer CD, Emeis JJ, Coppack SW. C-reactive protein in healthy subjects: associations with obesity, insulin resistance, and endothelial dysfunction: a potential role for cytokines originating from adipose tissue? Arterioscler Thromb Vasc Biol 19: 972–978, 1999. doi: 10.1161/01.ATV.19.4.972. [DOI] [PubMed] [Google Scholar]
- 38.Bower JK, Appel LJ, Matsushita K, Young JH, Alonso A, Brancati FL, Selvin E. Glycated hemoglobin and risk of hypertension in the atherosclerosis risk in communities study. Diabetes Care 35: 1031–1037, 2012. doi: 10.2337/dc11-2248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weber C, Schmitt R, Birnboeck H, Hopfgartner G, van Marle SP, Peeters PA, Jonkman JH, Jones CR. Pharmacokinetics and pharmacodynamics of the endothelin-receptor antagonist bosentan in healthy human subjects. Clin Pharmacol Ther 60: 124–137, 1996. doi: 10.1016/S0009-9236(96)90127-7. [DOI] [PubMed] [Google Scholar]
- 40.Maguire JJ, Kuc RE, Davenport AP. Defining the affinity and receptor sub-type selectivity of four classes of endothelin antagonists in clinically relevant human cardiovascular tissues. Life Sci 91: 681–686, 2012. doi: 10.1016/j.lfs.2012.05.008. [DOI] [PubMed] [Google Scholar]
- 41.Heimlich JB, Speed JS, Bloom CJ, O'Connor PM, Pollock JS, Pollock DM. ET-1 increases reactive oxygen species following hypoxia and high-salt diet in the mouse glomerulus. Acta Physiol (Oxf) 213: 722–730, 2015. doi: 10.1111/apha.12397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.MacIntyre IM, Dhaun N, Lilitkarntakul P, Melville V, Goddard J, Webb DJ. Greater functional ETB receptor antagonism with bosentan than sitaxsentan in healthy men. Hypertension 55: 1406–1411, 2010. doi: 10.1161/HYPERTENSIONAHA.109.148569. [DOI] [PubMed] [Google Scholar]
- 43.Carroll D, Smith GD, Shipley MJ, Steptoe A, Brunner EJ, Marmot MG. Blood pressure reactions to acute psychological stress and future blood pressure status: a 10-year follow-up of men in the Whitehall II study. Psychosom Med 63: 737–743, 2001. doi: 10.1097/00006842-200109000-00006. [DOI] [PubMed] [Google Scholar]
- 44.Wood DL, Sheps SG, Elveback LR, Schirger A. Cold pressor test as a predictor of hypertension. Hypertension 6: 301–306, 1984. doi: 10.1161/01.hyp.6.3.301. [DOI] [PubMed] [Google Scholar]
- 45.Matthews KA, Owens JF, Kuller LH, Sutton-Tyrrell K, Lassila HC, Wolfson SK. Stress-induced pulse pressure change predicts women's carotid atherosclerosis. Stroke 29: 1525–1530, 1998. doi: 10.1161/01.STR.29.8.1525. [DOI] [PubMed] [Google Scholar]
- 46.Morshedi-Meibodi A, Larson MG, Levy D, O'Donnell CJ, Vasan RS. Heart rate recovery after treadmill exercise testing and risk of cardiovascular disease events (The Framingham Heart Study). Am J Cardiol 90: 848–852, 2002. doi: 10.1016/S0002-9149(02)02706-6. [DOI] [PubMed] [Google Scholar]
- 47.Davenport AP, Kuc RE, Maguire JJ, Harland SP. ETA receptors predominate in the human vasculature and mediate constriction. J Cardiovasc Pharmacol 26, Suppl 3: S265–S267, 1995. [PubMed] [Google Scholar]
- 48.Laughlin MH. Cardiovascular response to exercise. Am J Physiol 277: S244–S259, 1999. doi: 10.1152/advances.1999.277.6.S244. [DOI] [PubMed] [Google Scholar]
- 49.Julius S. Transition from high cardiac output to elevated vascular resistance in hypertension. Am Heart J 116: 600–606, 1988. doi: 10.1016/0002-8703(88)90557-1. [DOI] [PubMed] [Google Scholar]
- 50.Fagard RH, Pardaens K, Staessen JA, Thijs L. Prognostic value of invasive hemodynamic measurements at rest and during exercise in hypertensive men. Hypertension 28: 31–36, 1996. doi: 10.1161/01.HYP.28.1.31. [DOI] [PubMed] [Google Scholar]
- 51.Bruno RM, Sudano I, Ghiadoni L, Masi L, Taddei S. Interactions between sympathetic nervous system and endogenous endothelin in patients with essential hypertension. Hypertension 57: 79–84, 2011. doi: 10.1161/HYPERTENSIONAHA.110.163584. [DOI] [PubMed] [Google Scholar]
- 52.Rohde K, Keller M, la Cour Poulsen L, Blüher M, Kovacs P, Böttcher Y. Genetics and epigenetics in obesity. Metabolism 92: 37–50, 2019. doi: 10.1016/j.metabol.2018.10.007. [DOI] [PubMed] [Google Scholar]
- 53.Fernández-Sánchez A, Madrigal-Santillán E, Bautista M, Esquivel-Soto J, Morales-González A, Esquivel-Chirino C, Durante-Montiel I, Sánchez-Rivera G, Valadez-Vega C, Morales-González JA. Inflammation, oxidative stress, and obesity. Int J Mol Sci 12: 3117–3132, 2011. doi: 10.3390/ijms12053117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mitchell GF, Hwang SJ, Vasan RS, Larson MG, Pencina MJ, Hamburg NM, Vita JA, Levy D, Benjamin EJ. Arterial stiffness and cardiovascular events: the Framingham Heart Study. Circulation 121: 505–511, 2010. doi: 10.1161/CIRCULATIONAHA.109.886655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vuurmans TJ, Boer P, Koomans HA. Effects of endothelin-1 and endothelin-1 receptor blockade on cardiac output, aortic pressure, and pulse wave velocity in humans. Hypertension 41: 1253–1258, 2003. doi: 10.1161/01.HYP.0000072982.70666.E8. [DOI] [PubMed] [Google Scholar]
- 56.Marano G, Grigioni M, Palazzesi S, Ferrari AU. Endothelin and mechanical properties of the carotid artery in Wistar-Kyoto and spontaneously hypertensive rats. Cardiovasc Res 41: 701–707, 1999. doi: 10.1016/s0008-6363(98)00240-5. [DOI] [PubMed] [Google Scholar]
- 57.Corden B, Keenan NG, de Marvao AS, Dawes TJ, Decesare A, Diamond T, Durighel G, Hughes AD, Cook SA, O'Regan DP. Body fat is associated with reduced aortic stiffness until middle age. Hypertension 61: 1322–1327, 2013. doi: 10.1161/HYPERTENSIONAHA.113.01177. [DOI] [PubMed] [Google Scholar]
- 58.Haak T, Jungmann E, Raab C, Usadel KH. Elevated endothelin-1 levels after cigarette smoking. Metabolism 43: 267–269, 1994. doi: 10.1016/0026-0495(94)90091-4. [DOI] [PubMed] [Google Scholar]
- 59.Fox CS, Massaro JM, Hoffmann U, Pou KM, Maurovich-Horvat P, Liu CY, Vasan RS, Murabito JM, Meigs JB, Cupples LA, D'Agostino RB Sr, O'Donnell CJ. Abdominal visceral and subcutaneous adipose tissue compartments: association with metabolic risk factors in the Framingham Heart Study. Circulation 116: 39–48, 2007. doi: 10.1161/CIRCULATIONAHA.106.675355. [DOI] [PubMed] [Google Scholar]
- 60.Kellogg DL, Liu Y, Pérgola PE. Selected contribution: gender differences in the endothelin-B receptor contribution to basal cutaneous vascular tone in humans. J Appl Physiol (1985) 91: 2407–2411, 2001.doi: 10.1152/jappl.2001.91.5.2407. [DOI] [PubMed] [Google Scholar]
- 61.Kuczmarski AV, Shoemaker LN, Hobson JC, Edwards DG, Wenner MM. Altered endothelial ETB receptor expression in postmenopausal women. Am J Physiol Heart Circ Physiol 319: H242–H247, 2020. doi: 10.1152/ajpheart.00342.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Merki-Feld GS, Rosselli M, Dubey RK, Jäger AW, Keller PJ. Long-term effects of combined oral contraceptives on markers of endothelial function and lipids in healthy premenopausal women. Contraception 65: 231–236, 2002. doi: 10.1016/s0010-7824(01)00312-2. [DOI] [PubMed] [Google Scholar]
- 63.Eagan LE, Chesney CA, Mascone SE, Shenouda N, Ranadive SM. Interleukin-6 is higher in naturally menstruating women compared with oral contraceptive pill users during the low-hormone phase. J Appl Physiol (1985) 131: 544–552, 2021. doi: 10.1152/japplphysiol.00921.2020. [DOI] [PubMed] [Google Scholar]
- 64.Davenport AP, Hyndman KA, Dhaun N, Southan C, Kohan DE, Pollock JS, Pollock DM, Webb DJ, Maguire JJ. Endothelin. Pharmacol Rev 68: 357–418, 2016. doi: 10.1124/pr.115.011833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Félétou M, Huang Y, Vanhoutte PM. Endothelium-mediated control of vascular tone: COX-1 and COX-2 products. Br J Pharmacol 164: 894–912, 2011. doi: 10.1111/j.1476-5381.2011.01276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Traupe T, D'Uscio LV, Muenter K, Morawietz H, Vetter W, Barton M. Effects of obesity on endothelium-dependent reactivity during acute nitric oxide synthase inhibition: modulatory role of endothelin. Clin Sci (Lond) 103, Suppl 48: 13S–15S, 2002. doi: 10.1042/CS103S013S. [DOI] [PubMed] [Google Scholar]
- 67.Traupe T, Lang M, Goettsch W, Münter K, Morawietz H, Vetter W, Barton M. Obesity increases prostanoid-mediated vasoconstriction and vascular thromboxane receptor gene expression. J Hypertens 20: 2239–2245, 2002. doi: 10.1097/00004872-200211000-00024. [DOI] [PubMed] [Google Scholar]
- 68.Martínez-Revelles S, Caracuel L, Márquez-Martín A, Dantas A, Oliver E, D'Ocon P, Vila E. Increased endothelin-1 vasoconstriction in mesenteric resistance arteries after superior mesenteric ischaemia-reperfusion. Br J Pharmacol 165: 937–950, 2012. doi: 10.1111/j.1476-5381.2011.01617.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Simonson MS. Endothelins: multifunctional renal peptides. Physiol Rev 73: 375–411, 1993. doi: 10.1152/physrev.1993.73.2.375. [DOI] [PubMed] [Google Scholar]
- 70.Weil BR, Westby CM, Van Guilder GP, Greiner JJ, Stauffer BL, DeSouza CA. Enhanced endothelin-1 system activity with overweight and obesity. Am J Physiol Heart Circ Physiol 301: H689–H695, 2011. doi: 10.1152/ajpheart.00206.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Contreras C, Sánchez A, Martínez P, Climent B, Benedito S, García-Sacristán A, Hernández M, Prieto D. Impaired endothelin calcium signaling coupled to endothelin type B receptors in penile arteries from insulin-resistant obese Zucker rats. J Sex Med 10: 2141–2153, 2013. doi: 10.1111/jsm.12234. [DOI] [PubMed] [Google Scholar]
- 72.Baretella O, Chung SK, Xu A, Vanhoutte PM. Endothelial overexpression of endothelin-1 modulates aortic, carotid, iliac and renal arterial responses in obese mice. Acta Pharmacol Sin 38: 498–512, 2017. doi: 10.1038/aps.2016.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Eriksson AK, van Harmelen V, Stenson BM, Aström G, Wåhlén K, Laurencikiene J, Rydén M. Endothelin-1 stimulates human adipocyte lipolysis through the ETA receptor. Int J Obes (Lond) 33: 67–74, 2009. doi: 10.1038/ijo.2008.212. [DOI] [PubMed] [Google Scholar]
- 74.Montezano AC, Burger D, Paravicini TM, Chignalia AZ, Yusuf H, Almasri M, He Y, Callera GE, He G, Krause KH, Lambeth D, Quinn MT, Touyz RM. Nicotinamide adenine dinucleotide phosphate reduced oxidase 5 (Nox5) regulation by angiotensin II and endothelin-1 is mediated via calcium/calmodulin-dependent, rac-1-independent pathways in human endothelial cells. Circ Res 106: 1363–1373, 2010. doi: 10.1161/CIRCRESAHA.109.216036. [DOI] [PMC free article] [PubMed] [Google Scholar]