

Abstract
We conducted cohort- and race-specific epigenome-wide association analyses of mitochondrial deoxyribonucleic acid (mtDNA) copy number (mtDNA CN) measured in whole blood from participants of African and European origins in five cohorts (n = 6182, mean age = 57–67 years, 65% women). In the meta-analysis of all the participants, we discovered 21 mtDNA CN-associated DNA methylation sites (CpG) (P < 1 × 10−7), with a 0.7–3.0 standard deviation increase (3 CpGs) or decrease (18 CpGs) in mtDNA CN corresponding to a 1% increase in DNA methylation. Several significant CpGs have been reported to be associated with at least two risk factors (e.g. chronological age or smoking) for cardiovascular disease (CVD). Five genes [PR/SET domain 16, nuclear receptor subfamily 1 group H member 3 (NR1H3), DNA repair protein, DNA polymerase kappa and decaprenyl-diphosphate synthase subunit 2], which harbor nine significant CpGs, are known to be involved in mitochondrial biosynthesis and functions. For example, NR1H3 encodes a transcription factor that is differentially expressed during an adipose tissue transition. The methylation level of cg09548275 in NR1H3 was negatively associated with mtDNA CN (effect size = −1.71, P = 4 × 10−8) and was positively associated with the NR1H3 expression level (effect size = 0.43, P = 0.0003), which indicates that the methylation level in NR1H3 may underlie the relationship between mtDNA CN, the NR1H3 transcription factor and energy expenditure. In summary, the study results suggest that mtDNA CN variation in whole blood is associated with DNA methylation levels in genes that are involved in a wide range of mitochondrial activities. These findings will help reveal molecular mechanisms between mtDNA CN and CVD.
Introduction
Mitochondria are power houses which generate adenosine triphosphate (ATP) through oxidative phosphorylation (OXPHOS) (1). ATP is the molecular energy for normal cellular activities. Mitochondria are also centrally involved in a wide range of fundamental biochemical processes, including intermediate metabolite synthesis, ion homeostasis, oxidative stress and programmed apoptosis (2,3). Furthermore, mitochondria uniquely harbor their own nuclear double-stranded circular genome, i.e. mitochondrial DNA (mtDNA). This mtDNA is made up of 16 569 base pairs and contains 37 genes, which code for 13 essential protein subunits of the OXPHOS complexes as well as two rRNAs and 22 tRNAs required for protein synthesis within the mitochondrial matrix (4). In addition to the 37 genes in the mtDNA, over 1000 genes in the nuclear genome also code for mitochondria-related proteins, including those that are responsible for mtDNA replication and transcription (5). Unlike the diploid nuclear DNA, individual cells typically contain hundreds, even thousands of mtDNA copies (6). The replication of mtDNA is tightly coupled with the transcription and subsequent expression of mtDNA genes (7), and thus, mtDNA copy number (mtDNA CN) is strictly regulated to meet the energy needs of diverse cell types (8). This is critical for the maintenance of mitochondrial functions and cellular homeostasis. Human mtDNA CN generally declines after middle age, and this decline progresses at different rates across tissues in mice (9) and in humans (10,11). Reduced mtDNA CN has also been reported in several complex diseases, including cancers (12), cardiovascular diseases (CVDs) (13), metabolic traits (14,15), kidney diseases (16) and neurodegenerative disorders (17–19). These observations suggest a link between mtDNA CN, mitochondrial dysfunction and impaired energy metabolism in the pathogenesis of these disorders.
DNA methylation, the addition of methyl groups to the DNA molecule, is by far its most commonly characterized epigenetic modification (20). DNA methylation regulates gene expression (20). Aberrant nuclear DNA methylation levels are associated with many complex diseases (21,22). Intriguingly, a recent study found different nuclear DNA methylation patterns in the brain tissues of a hybrid mouse model that contained identical nuclear genomes but different mtDNA backgrounds (23). Experimental studies also revealed that nuclear DNA methylation and gene expression levels changed after experimentally induced mtDNA depletion and restoration in human cells (24,25). Conversely, previous studies have also reported that altering DNA methylation levels in the nuclear-encoded DNA polymerase gamma catalytic subunit (POLGA) influenced the mtDNA CN levels in a cell-specific manner (26). These findings suggest the existence of a dynamic ‘cross-regulation’ between nuclear and mitochondrial genomes, and this ‘cross-regulation’ is at least partially regulated through the methylation of the nuclear genome.
To that end, we hypothesized that mtDNA CN variation is associated with DNA methylation changes in the nuclear genome with downstream transcriptomic features that can be characterized in whole blood. To map the epigenetic links between nuclear DNA and mtDNA CN, we conducted cohort- and race-specific epigenome-wide association studies (EWAS) of mtDNA CN in whole blood, followed by a meta-analysis of all participants (n = 6182). In addition, we performed Mendelian randomization (MR) analyses to infer possible causal effects of identified DNA methylation sites (CpG) on mtDNA CN variation. In the whole blood samples from Framingham Heart Study (FHS) participants, we examined the associations of the identified CpG sites with the expression levels of the cis-genes. We further integrated the findings with known biological pathways and their molecular functions to explore the functional relevance of the mtDNA CN-associated CpG sites (Fig. 1).
Figure 1.
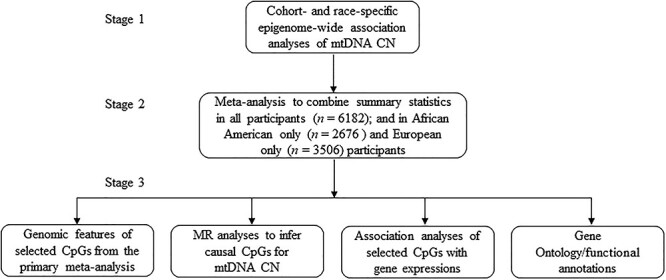
Flowchart: study design and statistical analysis.
Results
Participant characteristics
This study included a total of 6182 participants (3506 participants of European origin and 2676 African origin) from five cohorts (Supplementary Material, Table S1). The participants were mostly middle-aged or older (mean age = 57–67 years across cohorts), and about 65% of them were women. Except for the Women’s Health Initiative (WHI) study, which recruited only women, the other four cohorts had both men and women. Most (n = 4808, n = 78%) participants’ DNA methylation was measured using HumanMethylation 450K BeadChip arrays, and 1374 (22%) participants’ DNA methylation was measured with MethylationEPIC BeadChip arrays. We prioritized the meta-analysis results of the common CpGs (n ~ 430 000) between two platforms from 6182 participants for subsequent statistical analyses and functional inference. The results in meta-analysis of 1374 participants with additional CpGs that were only measured with MethylationEPIC BeadChip arrays are presented in Supplementary Results.
Significant CpGs identified by meta-analyses in all participants
No substantial inflation was observed in the test statistics in cohort-specific EWASs (Supplementary Material, Table S2). In the primary meta-analysis of all cohort participants, 21 CpGs at 13 genetic loci were significantly associated with mtDNA at P < 1 × 10−7 (Table 1, Fig. 2), and 285 CpGs showed evidence of association with mtDNA CN at P < 1 × 10−4 (Supplementary Material, Table S3). A slightly larger proportion of epigenome-wide CpGs (56%) were negatively associated with mtDNA CN (Fig. 3). Of these 21 CpGs, 18 (86%) were negatively associated with mtDNA CN, with a 1.3–2.6 standard deviation (s.d.) decrease in mtDNA CN corresponding to 1% increase in DNA methylation (Table 1). For example, a 1% increase in methylation level of cg03732020 in the nuclear receptor subfamily 1 group H member 3 (NR1H3) was significantly associated with a 2.2 s.d. decrease in the mtDNA CN level in the meta-analysis across all participants (P = 2.2 × 10−8). Three significant CpGs showed positive association with mtDNA CN. For example, a 1% increase in methylation level of cg05673882 in DNA polymerase kappa (POLK) was significantly associated with a 2.2 s.d. increase in mtDNA CN level in the meta-analysis across all participants (P = 2.3 × 10−8).
Table 1.
Epigenome-wide association and meta-analysis of mtDNA CN (P < 1 × 10−7) in 6182 participants
| IlmnID | Effect size | SE | P | Chr | MAPINFO | Gene name | CpG island | Relation CpG island |
|---|---|---|---|---|---|---|---|---|
| cg21848084 | −2.158 | 0.391 | 3.4E-08 | 1 | 3 264 381 | PRDM16 | ||
| cg27187555 | −1.478 | 0.266 | 2.9E-08 | 1 | 3 269 252 | PRDM16 | ||
| cg21393163 | 3.029 | 0.474 | 1.7E-10 | 1 | 12 217 629 | |||
| cg00988037 | −1.548 | 0.276 | 2.0E-08 | 1 | 54 869 007 | SSBP | chr1:54870853–54 872 476 | N_Shore |
| cg05673882 | 1.456 | 0.261 | 2.3E-08 | 5 | 74 862 702 | POLK | ||
| cg04368724 | −1.749 | 0.294 | 2.8E-09 | 6 | 31 760 593 | VARS | chr6:31763240–31 763 905 | N_Shelf |
| cg04018738 | −1.446 | 0.256 | 1.6E-08 | 6 | 31 760 616 | VARS | chr6:31763240–31 763 905 | N_Shelf |
| cg17619755 | −1.479 | 0.219 | 1.4E-11 | 6 | 31 760 629 | VARS | chr6:31763240–31 763 905 | N_Shelf |
| cg02980249 | −2.219 | 0.317 | 2.7E-12 | 6 | 31 760 762 | VARS | chr6:31763240–31 763 905 | N_Shelf |
| cg02597894 | −2.586 | 0.356 | 3.7E-13 | 6 | 31 760 796 | VARS | chr6:31763240–31 763 905 | N_Shelf |
| cg08899667 | −2.333 | 0.315 | 1.3E-13 | 6 | 31 761 055 | VARS | chr6:31763240–31 763 905 | N_Shelf |
| cg22761205 | −2.205 | 0.404 | 5.0E-08 | 11 | 457 256 | PTDSS2 | chr11:459088–459 345 | N_Shore |
| cg24420089 | −1.784 | 0.309 | 7.6E-09 | 11 | 457 304 | PTDSS2 | chr11:459088–459 345 | N_Shore |
| cg03732020 | −2.246 | 0.401 | 2.2E-08 | 11 | 47 282 968 | NR1H3 | ||
| cg09548275 | −1.715 | 0.312 | 3.8E-08 | 11 | 47 282 999 | NR1H3 | ||
| cg10713715 | −2.567 | 0.451 | 1.3E-08 | 11 | 63 533 656 | C11orf95 | chr11:63535652–63 537 435 | N_Shore |
| cg02194129 | −2.237 | 0.360 | 5.3E-10 | 14 | 1.04E+08 | XRCC3 | ||
| cg27192248 | 0.712 | 0.120 | 3.1E-09 | 15 | 65 285 669 | chr15:65281928–65 282 375 | S_Shelf | |
| cg20507228 | −1.329 | 0.231 | 9.1E-09 | 15 | 91 460 071 | MAN2A2 | ||
| cg04983687 | −1.624 | 0.294 | 3.4E-08 | 16 | 88 558 223 | ZFPM1 | chr16:88558051–88 558 329 | Island |
| cg26094004 | −2.331 | 0.238 | 1.3E-22 | 17 | 42 075 116 | PYY | chr17:42072138–42 072 444 | S_Shelf |
We performed cohort- and race-specific association analysis of mtDNA CN as the outcome variable with DNA methylation. Inverse variance-weighted fixed effect model was used in all meta-analyses.
Figure 2.
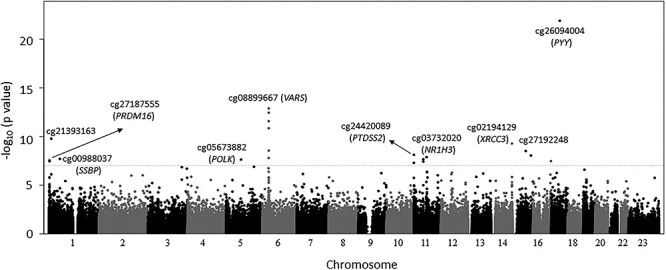
Epigenome-wise association and meta- analysis of mtDNA CN. Cohort- and race-specific association analysis of mtDNA CN with DNA methylation was performed, followed by meta-analysis to combine summary statistics (n = 6182). 23, X-chromosome.
Figure 3.
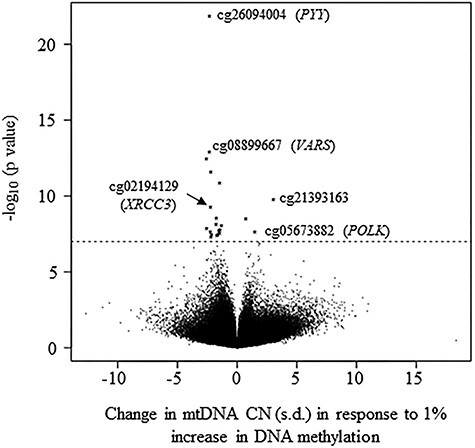
Volcano plot. A 1-s.d. change in mtDNA CN corresponds to a 1% increase in DNA methylation difference in the meta-analysis of all participants.
Comparison of effect sizes between participants of European and African origins
Six CpGs were significantly (P < 1 × 10−7) associated with mtDNA CN in the meta-analysis of participants of European origin (n = 3506), and a total of 183 CpGs displayed evidence of association with mtDNA CN (P < 1 × 10−4) (Supplementary Material, Table S4). Three CpGs were found to be significantly associated with mtDNA CN in the meta-analysis of participants of African origin (n = 2676), and 72 CpGs displayed evidence of association with mtDNA CN (P < 1 × 10−4) (Supplementary Material, Table S5). We compared the effect sizes of the 21 (P < 1 × 10−7 in the meta-analysis of all participants) and 285 (P < 1 × 10−4 in the meta-analysis of all participants) CpGs in the meta-analyses of participants of European origin to that of African origin. As expected, these 21 significant CpGs and 285 CpGs showed highly consistent effect sizes, Pearson r = 0.91 and 0.89, respectively, in their associations with mtDNA CN between participants of the two races (Supplementary Material, Fig. S1).
The previously identified CpG sites
Six CpGs (cg21051031, cg26563141, cg08899667, cg26094004, cg14575356 and cg23513930) were recently reported in a study that used a discovery-validation study design followed by meta-analysis (27). Of those, four CpGs, cg21051031 (effect size = 4.5, P = 2.5 × 10−17), cg26563141 (effect size = −2.4, P = 9.1 × 10−22), cg08899667 (effect size = −2.3, P = 1.3 × 10−13) and cg26094004 (effect size = −2.3, P = 1.3 × 10−22) were also found to be significant (P < 1 × 10−7) in the meta-analysis of all the participants in the present study. However, cg21051031 and cg26563141 were among the list of cross-reactive probes that were mapped to multiple locations (28,29), and therefore, we did not include them in the significant CpG list. The two CpGs, cg14575356 and cg23513930, were not significant in the present study.
No clear causal effects of validated CpG sites on mtDNA CN
To investigate the potential causal effects of the 21 mtDNA CN-associated CpG sites, we browsed the previously established whole blood meQTL [i.e. single nucleotide polymorphisms (SNPs) that were associated with DNA methylation] database in the FHS (30). We found that 19 significant CpGs had at least one independent cis-meQTL (P < 5 × 10−8) as instrument variables (IVs) for MR after pruning at linkage disequilibrium (LD) r2 < 0.01. However, none of these CpGs showed a significant causal effect on mtDNA CN variation (MR P < 0.05/20 = 0.0025). One CpG (cg10713715; C11orf95) showed an evidence, albeit not significant, that it may have a causal effect on mtDNA CN (P = 0.005) (Supplementary Material, Table S6).
Genomic features of the top CpGs in the meta-analysis of all participants
Eleven of the 21 significant CpGs in meta-analysis of all participants were located in the shelves or shores of CpG islands, which was not significantly different from expected (Fisher’s test, P = 1). We further tested possible enrichment in epigenetic features of the 285 CpGs with P < 1 × 10−4 in pooled data. Based on their annotated genomic features, the 285 CpG sites contained a slightly lower proportion of CpG that were located in CpG island (P = 0.001), while they contained similar proportions of CpGs that were located in shelves or shores of CpG islands (P > 0.1) as those observed in the whole methylation sites (Supplementary Material, Table S7). These 285 CpG sites were significantly overlapped with the three H3 markers and overlap with DNase I hypersensitive sites (DHSs) across various tissue types, such as blood, digestive, lung, placenta and digestive and heart tissues (31) (Supplementary Material, Tables S8 and S9).
Significant mtDNA CN-associated CpG sites and their associations with other traits
By querying the EWAS catalog database, we linked the significant 21 CpGs (Table 1) to previous publications of EWAS with various traits. We made two important observations. First, 20 significant mtDNA CN-associated CpGs have been associated with traits that are related to aging. These traits included chronological age, gestational age and or age from birth to late adolescence. Second, in previous EWASs, multiple significant mtDNA CN-associated CpGs have been significantly associated with at least two traits (Supplementary Material, Table S10). For example, the DNA methylation level at cg05673882 in POLK was positively associated with lung function (forced expiratory volume), while it was negatively associated with chronological age (adulthood aging, fetal age and brain development and the age from birth to late adolescence), birth weight, C-reactive protein and smoking (and prenatal smoke exposure in newborn) and forced expiratory volume (Fig. 4, Supplementary Material, Table S10). Similarly, the DNA methylation level at cg04983687 in zinc finger protein member 1 (ZFPM1) was positively associated with age from birth to late adolescence, while it was negatively associated with childhood asthma, birth weight and atopy—a disease condition with the tendency to produce an exaggerated immunoglobulin E (IgE) immune response and with tumor necrosis factor receptor 2 (Fig. 4, Supplementary Material, Table S10).
Figure 4.
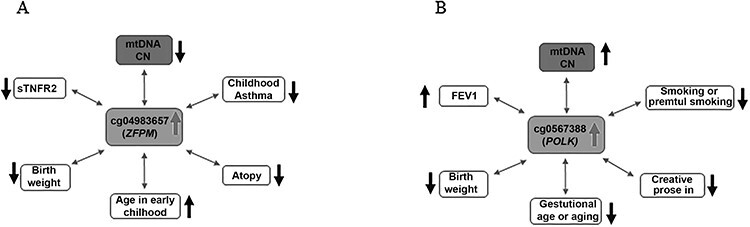
Examples that mtDNA CN-associated CpGs were also associated with aging and multiple CVD risk factors: (A) cg04983687 (ZFPM1) cg04983687 in ZFPM1; (B) cg0567388 in POLK.
Transcriptomic implications of mtDNA CN-associated CpG sites
A total of 431 cis-genes were located within ±1 Mb windows around the 21 CpG sites, which established 922 CpG-gene pairs. Of those, 30 CpG-gene pairs (19 distinct cis-genes with 10 distinct CpGs) exhibited P ≤ 0.005, and 120 CpG-gene pairs (73 distinct cis-genes with 21 CpGs) exhibited P < 0.05 (Supplementary Material, Table S11). Three CpG-gene pairs showed significant associations (P < 0.05/923 = 5.4 × 10−5): the DNA methylation levels of two CpGs (cg03732020 and cg09548275) in the NR1H3 gene at chromosome 11 were negatively associated with the expression levels of protein tyrosine phosphatase receptor type J (PTPRJ). The NR1H3 gene is about 720 kb upstream from the PTPRJ gene. The third pair was between a CpG in valyl-tRNA synthetase (VARS) and the transcript of general transcription factor IIH subunit 4 (GTF2H4A) which was 884 kb downstream. Several CpGs in the VARS genes also showed small P-values (P < 0.005) for their associations with several surrounding cis-genes that were involved in immune responses (Supplementary Material, Table S11). We further performed Go Ontology enrichment analyses for these 73 distinct genes, which were nominally associated with the 21 CpGs. These 73 genes were not enriched in Gene Ontology (GO) biological processes (FDR > 0.05), while they were enriched in peptide antigen binding for molecular processes and in MHC protein complex for cellular components (FDR ≤ 0.05) (Supplementary Material, Table S12).
Discussion
To test the hypothesis that mtDNA CN variation was associated with DNA methylation changes in the nuclear genome, we conducted a large epigenome-wide association and meta-analysis to investigate mtDNA CN in association with nuclear DNA methylation levels. We discovered that mtDNA CN were significantly associated with 21 CpG sites at P < 1 × 10−7 in the meta-analysis of 6182 participants of European and African origins from five cohorts. The effect sizes of 21 CpGs-mtDNA CN associations were consistent between the two races. Several significant CpGs have been reported to be associated with at least two CVD risk factors, e.g. chronological age, alcohol consumption, cigarette smoking, lung function and inflammation-related proteins. These results suggest that epigenetic regulation pathways underlie mtDNA CN and CVD risk.
Several significant CpGs are located in genes known to be involved in mitochondrial biosynthesis and mtDNA replication: cg21848084 and cg27187555 in PR/SET domain 16 (PRDM16) (32); cg03732020 and cg09548275 in NR1H3 (33); cg02194129 in DNA repair protein XRCC3 (34); cg05673882 in POLK (35) and cg22761205 and cg24420089 in decaprenyl-diphosphate synthase subunit 2 (PDSS2) (36). PRDM16 encodes a transcription coregulator that controls the development of brown adipocytes in brown adipose tissue that contains densely packed mitochondria. The expression of PRDM16 elevates the mRNA levels of many genes involved in OXPHOS and also stimulates mitochondrial biogenesis (32). The transcription factor encoding by NR1H3 was found to be differentially expressed during the adipose tissue transition from brown adipose to white adipose and was also linked to energy expenditure, lipolysis and glucose transport (37). mtDNA CN was previously reported to drastically decline during brown-to-white adipose transformation (37). XRCC3, a part of the mitochondrial nucleoid, facilitates mtDNA replication and maintains the integrity of the mitochondrial genome (34). A previous study showed that XRCC3 is localized to mitochondria and participates in the maintenance of mtDNA integrity during oxidative stress (38). POLK encodes the nuclear POLK. A previous study found that POLK localizes to mitochondria in the protozoan parasite Trypanosoma cruzi (35). The protein encoded by PDSS2 is an enzyme that synthesizes the prenyl side-chain of coenzyme Q, or ubiquinone, one of the key elements in the mitochondrial respiratory chain (36). Previous studies have shown that coenzyme Q deficiency triggers mitochondria degradation by mitophagy (39), and PDSS2 deficiency induces hepatocarcinogenesis by decreasing mitochondrial respiration and reprogramming glucose metabolism (40). The peptide YY (PYY) gene harbors the most significant CpG, cg26094004, with mtDNA CN. The PYY gene encodes peptide tyrosine tyrosine. The primary function of the PYY peptide is to slow gastric emptying, and obese people secrete less PYY peptide than non-obese people (41). Therefore, PYY peptide has been used directly as a weight-loss drug with some success (42). Although the mechanism of the PYY peptide action has not been fully established, previous studies have demonstrated that the PYY receptor stimulation increases protein kinase C activity, which couples to inhibit apoptosis (43) via mitochondrial pathways.
At the Bonferroni correction P < 5.4 × 10−5, the methylation levels of the significant CpGs in PRDM16, NR1H3, XRCC3, POLK, PDSS2 and PYY were not significantly associated with the transcription levels of these genes. The Bonferroni correction may be too conservative because some of these CpGs or genes were expected to be correlated (e.g. see Supplementary Material, Fig. S2). Nevertheless, the DNA methylation levels of cg09548275 and cg03732020 in the NR1H3 were positively correlated with the expression level of NR1H3 at P = 0.0003 and 0.0046, respectively. Along with the finding that the methylation levels of cg09548275 and cg03732020 were negatively associated with mtDNA CN, one might expect that the increase in DNA methylation levels in cg09548275 and cg03732020 may underlie the drastic decline of mtDNA CN in brown-to-white adipose transformation (37). In addition, at P < 5.4 × 10−5, cg09548275 and cg03732020 were negatively associated with the expression levels of the PTPRJ gene that is located 720 kb away. The PTPRJ gene is involved in dephosphorylation of many proteins in cell adhesion, migration, proliferation and differentiation (44). A previous study in lung cancer patients found that PTPRJ negatively modulated several proteins that are related to mitochondrial functions (45). Six CpGs in the VAR gene showed small P < 0.005 with surrounding gene expression levels that are involved in immune functions. mtDNA CN was significantly associated with white blood cell counts and differential counts, indicating that mtDNA CN is involved in inflammation. Future studies are warranted to study the roles of DNA methylation levels in the VAR gene that underlies mtDNA CN and inflammation.
Strengths and weaknesses
We explored the relationship of mtDNA CN with nDNA methylation in a large human population. The estimation of mtDNA CN using whole genome sequencing (WGS) has proven to be more accurate than mtDNA CN obtained from other methods (46). In addition, we applied careful consideration to account for batch effects and unobserved confounders, although residual confounding might still remain. Despite these strengths, several limitations should be noted in the present study. Previous studies have showed that mtDNA CN are associated with a range of pathologies, including CVD (13) and metabolic traits (14,15). Future studies may be needed to adjust for comorbidities in EWAS of mtDNA CN to understand the interplay between mtDNA CN, these comorbidities and DNA methylation. DNA methylation and mtDNA CN were measured in peripheral whole blood samples, and thus, our findings may not be generalizable to other tissues. However, whole blood samples circulate to all parts of the body. Additionally, the procurement of specific tissues (e.g. adipose or arterial tissue) is not feasible in large-scale cohort studies. For this reason, mtDNA CN variation and DNA methylation changes in readily accessible whole blood may reflect the metabolic health status across multiple systems. Indeed, a recent study showed that blood-derived mtDNA CN is associated with gene expression across various tissues (47). In addition, this study showed that mtDNA CN derived in whole blood is predictive for incident neurodegenerative disease (47). These observations provide further evidence supporting the use of blood-derived measures in association analyses to reveal mechanisms which underlie mtDNA CN, DNA methylation and human disease. The MR analyses did not find sufficient evidence that the newly discovered CpG sites might be causally linked to mtDNA CN. A recent genome-wide association analysis discovered 96 independent loci (P < 5e-8), and these loci jointly explain a small proportion of the variance (~1%) (48). However, there was a great degree of pleiotropy for the identified SNPs, making it challenging to use MR approaches to establish causality of mtDNA CN with DNA methylation. In summary, the results in the present study suggest that mtDNA CN variation in peripheral blood cells is associated with DNA methylation levels in genes related to a wide range of mitochondrial activities. These findings, if confirmed in future studies, may contribute to understanding the molecular mechanisms of mitochondria-related diseases.
Materials and Methods
Study design and study participants
This study was carried out in three major stages (Fig. 1). First, we performed cohort- and race- specific epigenome-wide association analyses in several studies, including the Atherosclerosis Risk in Communities (ARIC) study (n = 2027, 75.6% African origin) (49), the FHS (n = 1952, 100% European origins) (50,51), the Genetic Epidemiology Network of Arteriopathy (GENOA, n = 797, 100% African origin) (52), the Multi-Ethnic Study of Atherosclerosis (MESA, n = 577, 31.5% African origin) (17) and the WHI (n = 829, 20.4% African origins). To maximize the power, we performed a meta-analysis in all participants (n = 6182) of both races. We also performed race-specific meta-analysis in participants of European origin (n = 3506) or African origin (n = 2676). Fixed effects inverse variance method was conducted to combine summary statistics from individual cohorts. Detailed information on all participating studies (FHS, WHI, GENOA, MESA and ARIC) has been described previously (49–54) (Supplementary Materials). Lastly, we performed analyses to infer the biological functions of the selected CpG sites from the primary meta-analyses. The study-specific protocols were approved by the institutional review board of each study. Written informed consent for genetic research was obtained for all participants.
DNA methylation measurement in whole blood and quality control
Fasting morning peripheral whole blood samples were obtained during routine research clinic visits. Genomic DNA was extracted from whole blood samples and bisulfite-converted for methylation profiling. DNA samples then underwent whole genome amplification, fragmentation, array hybridization and single-base pair extension following the manufacturer’s protocols (55,56). In FHS, WHI and ARIC, DNA methylation levels were measured using the Infinium HumanMethylation 450K BeadChip array (Illumina, Inc., San Diego, CA), which simultaneously queries the methylation state of over 480 000 CpG sites in the nuclear genome. In GENOA and MESA, DNA methylation profiling was performed using the Infinium MethylationEPIC BeadChip array (Illumina, Inc.), which covers more than 850 000 CpG sites across the genome. Detailed information about DNA extraction, bisulfite conversion, DNA methylation profiling, normalization and quality control (QC) procedures in each cohort are described in the Supplementary Methods. In brief, we excluded cross-reactive probes that mapped to multiple locations (28,29). We also excluded low-quality probes if these probes with high missing rate (>20%), with detection P-value > 0.01 (57), and those with SNPs at CpG sites or ≤10 bp of single base extension. After QC procedures, we excluded bad samples if the samples were multi-dimensional scaling (MDS) outliers (58), high missing rate (>1%) and poor matching to SNP genotype (Supplementary Methods).
Measurement of mtDNA CN in whole blood
mtDNA CN estimation in WGS
mtDNA CN was estimated by a ratio of two times of average coverage of mtDNA to average coverage of nDNA from WGS data (~30×) through the NHLBI Trans-Omics for Precision Medicine (TOPMed) Consortium (Supplementary Material, Table S1). The coverage was defined as the number of reads that were mapped to a given nucleotide in the reconstructed sequence. Samples from ARIC were sequenced at the Human Genome Sequencing Center at Baylor College of Medicine and at the Broad Institute; FHS, WHI and MESA were sequenced at the Broad Institute of MIT and Harvard, while samples from the GENOA study were sequenced at the University of Washington Northwest Genomics Center. Detailed methods of DNA sample handling, library construction, data acquisition, processing and QC were described previously (59). Briefly, after sequencing, the reads were aligned to human genome build GRCh37 at each sequencing center, and the resulting BAM files were transferred from all centers to the TOPMed’s Informatics Research Center (IRC) where they were re-aligned to build GRCh37 using a common pipeline to produce a set of ‘harmonized’ BAM files (60).
mtDNA CN estimation by low-pass WGS in ARIC
Low-pass WGS data (>4-fold) for ARIC was generated at the Baylor College of Medicine Human Genome Sequencing Center using Nano or PCR-free DNA libraries on the Illumina HiSeq 2000 (Supplementary Material, Table S1). Sequence reads were mapped to the human genome build GRCh37 using BWA (61). QC was performed as previously described (62). A count for the total number of reads in a sample was scraped from the NCBI sequence read archive using the R package RCurl, while reads aligned to the mitochondrial genome were downloaded directly through Samtools (version 1.3.1). mtDNA CN estimation from low-pass WGS was calculated as the ratio of mitochondrial reads to the number of total aligned reads.
Gene expression profiling
Details of gene expression profiling in FHS were described previously (63). In brief, fasting peripheral whole blood samples were collected in PAXgene blood tubes (PreAnalytiX, Hombrechtikon, Switzerland) at the same time as DNA methylation profiling. Gene expression was assayed using the Affymetrix Human Exon 1.0 ST GeneChip platform (Affymetrix Inc., Santa Clara, CA, USA), which contained > 5.5 million probes covering 17 873 distinct genes. Collected gene expression values were normalized using the robust multi-array average (RMA) method and were adjusted for chip batch effects, first principal component and several technical factors (64).
Statistical analyses
Association analysis and meta-analysis of mtDNA CN with epigenome-wide DNA methylation
Outcome and predictor variables: DNA methylation residuals (predictors) were obtained by regressing beta values on age, sex, imputed white blood cell count and fractions, surrogate variables (SVs) and technical covariates (plate/row/column numbers on the methylation chip). The residuals of mtDNA CN were obtained by adjusting for age, age2, sex, imputed white blood cell count and fractions and platelet count and the year of blood collection (as a cluster variable to account for additional batch effect). The mtDNA CN residuals were then standardized to have a mean of 0 and a s.d. of 1 (outcome). The mtDNA CN standardized residuals were used as the outcome variables in all models. All clinical covariates were contemporaneous to blood collection for DNA methylation and WGS. The white blood cell fractions were imputed using the Houseman method (65) via a reference panel, which included CD8+ T cells, CD4+ T cells, natural killer cells, B cells, monocytes and granulocytes. SVs were imputed to account for hidden confounders (66,67). The mtDNA CN variable was put in the model so it was ‘protected’ from being treated as unexplained heterogeneity in SV analysis.
Association analyses
We performed power estimation (Supplementary Methods). We performed cohort- and race-specific association analysis of mtDNA CN with DNA methylation. In all models across studies, the residuals of CpG levels at each CpG site were used as the independent variable and the standardized residuals of mtDNA CN as the dependent variable. Linear regression models or linear mixed effects models were used for cohorts with unrelated or families, respectively. The genomic control factor (λ) was used to evaluate any possible inflation (68). All association analyses were performed using the ‘lme’ function within the R package ‘nlme’ (69).
Meta-analyses
We performed meta-analysis to combine summary statistics from individual studies rather than a direct analysis of pooled individual-level data. The primary results were obtained by meta-analysis in participants of both African and European origins for common DNA methylation CpGs between MethylationEPIC BeadChip and HumanMethylation 450K BeadChip arrays. We also applied race-specific meta-analysis. We compared effect sizes of significant associations in pooled meta-analyses between participants of African origin and European origins. Inverse variance weighted fixed effect models were used in all meta-analyses with the ‘metagen’ function of the R package ‘meta’ (70). About 430 000 CpGs were common between MethylationEPIC BeadChip and HumanMethylation 450K BeadChip arrays. Therefore, we prioritized these common CpGs in reporting meta-analysis results. To correct for multiple testing, we applied Bonferroni correction (P < 1 × 10−7 = 0.05/430 000) for significance in the primary meta-analysis. In Supplementary file, we reported CpGs with P < 1 × 10−4 from the primary meta-analysis and meta-analysis for the CpGs that were only covered by MethylationEPIC BeadChip array (Supplementary Material, Table S13).
Furthermore, we investigated the results of the six CpGs (cg21051031, cg26563141, cg08899667, cg26094004, cg14575356 and cg23513930) in this study. These six CpGs were recently reported from a meta-analysis (27). Of note, mtDNA-CN in ARIC cohort was estimated from Affymetrix Human SNP 6.0 arrays in the previous study (27).
MR analysis
We previously conducted methylation quantitative trait loci (meQTL) mapping in FHS (30). SNPs were imputed from the 1000 Genomes Project panel (Phase 3, version 5) using MaCH/Minimac software (71). SNPs (meQTLs) with minor allele frequency (MAF) > 0.01 and imputation quality ratio > 0.5 were used to identify instrument variables (IVs) for MR analysis. We selected the independent cis-meQTL (residing within 1 Mb of the CpG sites with LD r2 < 0.01) with the lowest SNP-CpG P-value using the ‘clump_data’ function within the R package ‘TwoSampleMR’ (72). The LD proxies were defined using the 1000 Genomes European samples (73). Two-step MR, with CpGs as the exposure and mtDNA CN as the outcome, was performed to test for the significant CpGs (P < 1 × 10−7) selected from the primary meta-analysis.
Association analysis of selected CpG sites with expression levels of cis-gene
To explore the possible roles of DNA methylation on regulating of gene expression, we performed association analyses between the significant CpG sites and the transcript expression levels of the genes residing within ±1 Mb of each of the CpG sites (cis-genes) for the significant CpGs (P < 1 × 10−7). The residuals of methylation levels at each CpG site were used as the independent variables in the models. The residuals of expression levels (dependent variable) of each transcript were obtained by regressing on age, sex, imputed white blood cell fractions based on Houseman method (65) and cohort index. Linear mixed effects models were applied to account for family structure in the FHS data. Statistically significant associations were define as P < 0.05/n, where n was the number of CpG-gene pairs. CpG-cis-gene association analyses were performed using the ‘lme’ function within the R package ‘nlme’.
Functional inference analyses
We performed several analyses to infer functional relevance for mtDNA CN-associated CpG sites. For mtDNA CN-associated CpG sites (P < 1 × 10−7), genomic features (74) of CpG sites were annotated. Hypergeometric tests were used to evaluate if the identified CpG sites were enriched with a genomic feature. We also used eFORGE (v2.0) (31) to determine if two set of selected CpG sites (P < 1 × 10−7 and P < 1 × 10−4) were enriched across 15 chromatin states and overrepresented at loci with overlapping histone modifications (H3K4me1, H3K4me3, H3K9me3, H3K27me3 and H3K36me3) across multiple cell lines and tissues from the Roadmap Epigenomics Project (75), BLUEPRINT Epigenome (76) and Encyclopedia of DNA Elements (ENCODE) (77) data. Furthermore, we searched the EWAS Catalog database (78) to link the validated CpG sites with specific disease/trait phenotypes in previously published EWAS of DNA methylation. For cis-genes that were associated with mtDNA CN-associated CpG sites, we used the PANTHER (79) overrepresentation test (release: 14 February 2020) to assess whether the identified cis-genes were overrepresented in specific GO biological processes, molecular functions and cellular component pathways with Fisher’s exact test. We used FDR-corrected P < 0.05 (i.e. FDR < 0.05) for significance.
Data Availability
We describe data availability in the following: In ARIC, DNA methylation data are available upon request at https://sites.cscc.unc.edu/aric/distribution-agreements. The mtDNA CN data were calculated using TOPMed sequencing data on the Database of Genotypes and Phenotypes (dbGaP): phs000993. In FHS, the methylation data can be downloaded at the dbGaP: phs000724. The mtDNA CN data were calculated using TOPMed sequencing data on the dbGaP: phs000974. In GENOA, the methylation data from the EPIC array are on Gene Expression Omnibus (GEO): GSE157131. The mtDNA CN data were estimated using the GENOA TOPMed sequencing data on the dbGaP: phs001345. Owing to IRB restriction, mapping of the sample IDs between genotype data in dbGaP and methylation data in GEO cannot be provided publicly but are available upon written request to J.A.S. and Sharon LR Kardia. In MESA, the individual-level genotype, phenotype and methylation data are available on dbGaP (https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000209.v13.p3). The mtDNA CN data were calculated using TOPMed sequence data. In WHI, a part of DNA methylation data are under phs000200 and phs001335. Because DNA methylation data were not covered by the genetic data rules when it was generated, users should contact C.K. for requesting DNA methylation. The mtDNA CN data can be accessed using the TOPMed sequence data on dbGaP: phs001237.
URLs
TwoSampleMR: https://github.com/MRCIEU/TwoSampleMR
eFORGE: https://eforge.altiusinstitute.org/
Gene Ontology: http://geneontology.org/
EWAS Catalog database: http://www.ewascatalog.org/
Supplementary Material
Acknowledgements
We are very grateful to Cynthia A. Korhonen for her reviewing and editing this manuscript. Molecular data for the Trans-Omics in Precision Medicine (TOPMed) program was supported by the National Heart, Lung and Blood Institute (NHLBI). See the TOPMed Omics Support Table in the Supplementary Materials for study specific omics support information. We gratefully acknowledge the studies and participants who provided biological samples and data for TOPMed. Additional study-specific acknowledgements are included in Supplementary Materials.
Conflict of Interest Statement. Ryan Longchamps is a full-time employee of Deep Genomics Inc. and is entitled to a stock option. All other authors reported no conflict of interest.
Funding supports
Core support, including centralized genomic read mapping and genotype calling, along with variant quality metrics and filtering were provided by the IRC (3R01HL-117626-02S1; contract HHSN268201800002I). Core support, including phenotype harmonization, data management, sample-identity QC and general program coordination, were provided by the TOPMed Data Coordinating Center (R01HL-120393; U01HL-120393; contract HHSN268201800001I). Infrastructure for the CHARGE Consortium is supported in part by the National Heart, Lung, and Blood Institute grant R01HL105756. Statistical analyses in this study are partly supported by supported by R01HL155569.
Authors contributed equally to the work.
Contributor Information
Penglong Wang, Population Sciences Branch, National Heart, Lung, and Blood Institute, National Institutes of Health, Bethesda, MD 20892, USA.
Christina A Castellani, McKusick-Nathans Department of Genetic Medicine, Johns Hopkins University School of Medicine, Baltimore, MD 21287, USA; Department of Pathology and Laboratory Medicine, Western University, London, Ontario N6A 5C1, Canada.
Jie Yao, Department of Pediatrics, The Institute for Translational Genomics and Population Sciences, The Lundquist Institute for Biomedical Innovation at Harbor-UCLA Medical Center, Torrance, CA 90502, USA.
Tianxiao Huan, Population Sciences Branch, National Heart, Lung, and Blood Institute, National Institutes of Health, Bethesda, MD 20892, USA.
Lawrence F Bielak, Department of Epidemiology, School of Public Health, University of Michigan, Ann Arbor, MI 48109, USA.
Wei Zhao, Department of Epidemiology, School of Public Health, University of Michigan, Ann Arbor, MI 48109, USA.
Jeffrey Haessler, Division of Public Health Science, Fred Hutchinson Cancer Research Center, Seattle, WA 98109, USA.
Roby Joehanes, Population Sciences Branch, National Heart, Lung, and Blood Institute, National Institutes of Health, Bethesda, MD 20892, USA.
Xianbang Sun, Department of Biostatistics, Boston University, Boston, MA 02118, USA.
Xiuqing Guo, Department of Pediatrics, The Institute for Translational Genomics and Population Sciences, The Lundquist Institute for Biomedical Innovation at Harbor-UCLA Medical Center, Torrance, CA 90502, USA.
Ryan J Longchamps, McKusick-Nathans Department of Genetic Medicine, Johns Hopkins University School of Medicine, Baltimore, MD 21287, USA.
JoAnn E Manson, Department of Epidemiology, Harvard T.H. Chan School of Public Health, Boston, MA 02115, USA.
Megan L Grove, Human Genetics Center, Department of Epidemiology, Human Genetics and Environmental Sciences, School of Public Health, The University of Texas Health Science Center at Houston, Houston, TX 77030, USA.
Jan Bressler, Human Genetics Center, Department of Epidemiology, Human Genetics and Environmental Sciences, School of Public Health, The University of Texas Health Science Center at Houston, Houston, TX 77030, USA.
Kent D Taylor, Department of Pathology and Laboratory Medicine, Western University, London, Ontario N6A 5C1, Canada.
Tuuli Lappalainen, New York Genome Center, New York, NY 10013, USA; Department of Systems Biology, Columbia University, New York, NY 10034, USA.
Silva Kasela, New York Genome Center, New York, NY 10013, USA; Department of Systems Biology, Columbia University, New York, NY 10034, USA.
David J Van Den Berg, Department of Population and Public Health Sciences, Center for Genetic Epidemiology, Keck School of Medicine of USC, University of Southern California, Los Angeles, CA 90033, USA.
Lifang Hou, Feinberg School of Medicine, Northwestern University, Chicago, IL 60611, USA.
Alexander Reiner, Division of Public Health Science, Fred Hutchinson Cancer Research Center, Seattle, WA 98109, USA.
Yongmei Liu, Department of Medicine, Divisions of Cardiology and Neurology, Duke University Medical Center, Durham, NC 27704, USA.
Eric Boerwinkle, Human Genetics Center, Department of Epidemiology, Human Genetics and Environmental Sciences, School of Public Health, The University of Texas Health Science Center at Houston, Houston, TX 77030, USA; Human Genome Sequencing Center, Baylor College of Medicine, Houston, TX 77030, USA.
Jennifer A Smith, Department of Epidemiology, School of Public Health, University of Michigan, Ann Arbor, MI 48109, USA.
Patricia A Peyser, Department of Epidemiology, School of Public Health, University of Michigan, Ann Arbor, MI 48109, USA.
Myriam Fornage, Human Genetics Center, Department of Epidemiology, Human Genetics and Environmental Sciences, School of Public Health, The University of Texas Health Science Center at Houston, Houston, TX 77030, USA; Brown Foundation Institute of Molecular Medicine, McGovern Medical School, University of Texas Health Science Center at Houston, Houston, TX 77030, USA.
Stephen S Rich, Center for Public Health Genomics, University of Virginia, Charlottesville, VA 22903, USA.
Jerome I Rotter, Department of Pediatrics, The Institute for Translational Genomics and Population Sciences, The Lundquist Institute for Biomedical Innovation at Harbor-UCLA Medical Center, Torrance, CA 90502, USA.
Charles Kooperberg, Division of Public Health Science, Fred Hutchinson Cancer Research Center, Seattle, WA 98109, USA.
Dan E Arking, McKusick-Nathans Department of Genetic Medicine, Johns Hopkins University School of Medicine, Baltimore, MD 21287, USA.
Daniel Levy, Population Sciences Branch, National Heart, Lung, and Blood Institute, National Institutes of Health, Bethesda, MD 20892, USA; Framingham Heart Study, National Heart, Lung, and Blood Institute (NHLBI), Framingham, MA 01702, USA.
Chunyu Liu, Department of Biostatistics, Boston University, Boston, MA 02118, USA; Framingham Heart Study, National Heart, Lung, and Blood Institute (NHLBI), Framingham, MA 01702, USA.
References
- 1. Sherratt, H.S. (1991) Mitochondria: structure and function. Rev. Neurol. (Paris), 147, 417–430. [PubMed] [Google Scholar]
- 2. Nunnari, J. and Suomalainen, A. (2012) Mitochondria: in sickness and in health. Cell, 148, 1145–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pagliarini, D.J. and Rutter, J. (2013) Hallmarks of a new era in mitochondrial biochemistry. Genes Dev., 27, 2615–2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Taylor, R.W. and Turnbull, D.M. (2005) Mitochondrial DNA mutations in human disease. Nat. Rev. Genet., 6, 389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pagliarini, D.J., Calvo, S.E., Chang, B., Sheth, S.A., Vafai, S.B., Ong, S.E., Walford, G.A., Sugiana, C., Boneh, A., Chen, W.K. et al. (2008) A mitochondrial protein compendium elucidates complex I disease biology. Cell, 134, 112–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. D'Erchia, A.M., Atlante, A., Gadaleta, G., Pavesi, G., Chiara, M., De Virgilio, C., Manzari, C., Mastropasqua, F., Prazzoli, G.M., Picardi, E. et al. (2015) Tissue-specific mtDNA abundance from exome data and its correlation with mitochondrial transcription, mass and respiratory activity. Mitochondrion, 20, 13–21. [DOI] [PubMed] [Google Scholar]
- 7. Kasiviswanathan, R., Collins, T.R. and Copeland, W.C. (2012) The interface of transcription and DNA replication in the mitochondria. Biochim. Biophys. Acta, 1819, 970–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Clay Montier, L.L., Deng, J.J. and Bai, Y. (2009) Number matters: control of mammalian mitochondrial DNA copy number. J. Genet. Genomics, 36, 125–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Barazzoni, R., Short, K.R. and Nair, K.S. (2000) Effects of aging on mitochondrial DNA copy number and cytochrome c oxidase gene expression in rat skeletal muscle, liver, and heart. J. Biol. Chem., 275, 3343–3347. [DOI] [PubMed] [Google Scholar]
- 10. Short, K.R., Bigelow, M.L., Kahl, J., Singh, R., Coenen-Schimke, J., Raghavakaimal, S. and Nair, K.S. (2005) Decline in skeletal muscle mitochondrial function with aging in humans. Proc. Natl. Acad. Sci. U. S. A., 102, 5618–5623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mengel-From, J., Thinggaard, M., Dalgard, C., Kyvik, K.O., Christensen, K. and Christiansen, L. (2014) Mitochondrial DNA copy number in peripheral blood cells declines with age and is associated with general health among elderly. Hum. Genet., 133, 1149–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Reznik, E., Miller, M.L., Senbabaoglu, Y., Riaz, N., Sarungbam, J., Tickoo, S.K., Al-Ahmadie, H.A., Lee, W., Seshan, V.E., Hakimi, A.A. et al. (2016) Mitochondrial DNA copy number variation across human cancers. elife, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ashar, F.N., Zhang, Y., Longchamps, R.J., Lane, J., Moes, A., Grove, M.L., Mychaleckyj, J.C., Taylor, K.D., Coresh, J., Rotter, J.I. et al. (2017) Association of mitochondrial DNA copy number with cardiovascular disease. JAMA Cardiol., 2, 1247–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Monickaraj, F., Aravind, S., Gokulakrishnan, K., Sathishkumar, C., Prabu, P., Prabu, D., Mohan, V. and Balasubramanyam, M. (2012) Accelerated aging as evidenced by increased telomere shortening and mitochondrial DNA depletion in patients with type 2 diabetes. Mol. Cell. Biochem., 365, 343–350. [DOI] [PubMed] [Google Scholar]
- 15. Liu, X., Longchamps, R.J., Wiggins, K., Raffield, L., Bielak, L., Zhao, W., Pitsillides, A.N., Blackwell, T., Yao, J., Guo, X. et al. (2020) Association of mitochondrial DNA copy number with cardiometabolic diseases in a large cross-sectional study of multiple ancestries. medRxiv, in press, April 24, 2020, doi 10.1101/2020.04.20.20016337, preprint: not peer reviewed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tin, A., Grams, M.E., Ashar, F.N., Lane, J.A., Rosenberg, A.Z., Grove, M.L., Boerwinkle, E., Selvin, E., Coresh, J., Pankratz, N. et al. (2016) Association between mitochondrial DNA copy number in peripheral blood and incident CKD in the Atherosclerosis Risk in Communities study. J. Am. Soc. Nephrol., 27, 2467–2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Blokhin, A., Vyshkina, T., Komoly, S. and Kalman, B. (2008) Variations in mitochondrial DNA copy numbers in MS brains. J. Mol. Neurosci., 35, 283–287. [DOI] [PubMed] [Google Scholar]
- 18. Petersen, M.H., Budtz-Jørgensen, E., Sørensen, S.A., Nielsen, J.E., Hjermind, L.E., Vinther-Jensen, T., Nielsen, S.M. and Nørremølle, A. (2014) Reduction in mitochondrial DNA copy number in peripheral leukocytes after onset of Huntington’s disease. Mitochondrion, 17, 14–21. [DOI] [PubMed] [Google Scholar]
- 19. Pyle, A., Anugrha, H., Kurzawa-Akanbi, M., Yarnall, A., Burn, D. and Hudson, G. (2016) Reduced mitochondrial DNA copy number is a biomarker of Parkinson’s disease. Neurobiol. Aging, 38, 216.e217–216.e210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Moore, L.D., Le, T. and Fan, G. (2013) DNA methylation and its basic function. Neuropsychopharmacology, 38, 23–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Portela, A. and Esteller, M. (2010) Epigenetic modifications and human disease. Nat. Biotechnol., 28, 1057–1068. [DOI] [PubMed] [Google Scholar]
- 22. Bergman, Y. and Cedar, H. (2013) DNA methylation dynamics in health and disease. Nat. Struct. Mol. Biol., 20, 274–281. [DOI] [PubMed] [Google Scholar]
- 23. Vivian, C.J., Brinker, A.E., Graw, S., Koestler, D.C., Legendre, C., Gooden, G.C., Salhia, B. and Welch, D.R. (2017) Mitochondrial genomic backgrounds affect nuclear DNA methylation and gene expression. Cancer Res., 77, 6202–6214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bellizzi, D., D'Aquila, P., Giordano, M., Montesanto, A. and Passarino, G. (2012) Global DNA methylation levels are modulated by mitochondrial DNA variants. Epigenomics, 4, 17–27. [DOI] [PubMed] [Google Scholar]
- 25. Sun, X. and St John, J.C. (2018) Modulation of mitochondrial DNA copy number in a model of glioblastoma induces changes to DNA methylation and gene expression of the nuclear genome in tumours. Epigenetics Chromatin, 11, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lee, W., Johnson, J., Gough, D.J., Donoghue, J., Cagnone, G.L., Vaghjiani, V., Brown, K.A., Johns, T.G. and St John, J.C. (2015) Mitochondrial DNA copy number is regulated by DNA methylation and demethylation of POLGA in stem and cancer cells and their differentiated progeny. Cell Death Dis., 6, e1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Castellani, C.A., Longchamps, R.J., Sumpter, J.A., Newcomb, C.E., Lane, J.A., Grove, M.L., Bressler, J., Brody, J.A., Floyd, J.S., Bartz, T.M. et al. (2020) Mitochondrial DNA copy number can influence mortality and cardiovascular disease via methylation of nuclear DNA CpGs. Genome Med., 12, 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chen, Y.A., Lemire, M., Choufani, S., Butcher, D.T., Grafodatskaya, D., Zanke, B.W., Gallinger, S., Hudson, T.J. and Weksberg, R. (2013) Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics, 8, 203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Benton, M.C., Johnstone, A., Eccles, D., Harmon, B., Hayes, M.T., Lea, R.A., Griffiths, L., Hoffman, E.P., Stubbs, R.S. and Macartney-Coxson, D. (2015) An analysis of DNA methylation in human adipose tissue reveals differential modification of obesity genes before and after gastric bypass and weight loss. Genome Biol., 16, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Huan, T., Joehanes, R., Song, C., Peng, F., Guo, Y., Mendelson, M., Yao, C., Liu, C., Ma, J., Richard, M. et al. (2019) Genome-wide identification of DNA methylation QTLs in whole blood highlights pathways for cardiovascular disease. Nat. Commun., 10, 4267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Breeze, C.E., Reynolds, A.P., van Dongen, J., Dunham, I., Lazar, J., Neph, S., Vierstra, J., Bourque, G., Teschendorff, A.E., Stamatoyannopoulos, J.A. et al. (2019) eFORGE v2.0: updated analysis of cell type-specific signal in epigenomic data. Bioinformatics, 35, 4767–4769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Seale, P., Kajimura, S., Yang, W., Chin, S., Rohas, L.M., Uldry, M., Tavernier, G., Langin, D. and Spiegelman, B.M. (2007) Transcriptional control of brown fat determination by PRDM16. Cell Metab., 6, 38–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Permuth-Wey, J., Chen, Y.A., Tsai, Y.Y., Chen, Z., Qu, X., Lancaster, J.M., Stockwell, H., Dagne, G., Iversen, E., Risch, H. et al. (2011) Inherited variants in mitochondrial biogenesis genes may influence epithelial ovarian cancer risk. Cancer Epidemiol. Biomark. Prev., 20, 1131–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mishra, A., Saxena, S., Kaushal, A. and Nagaraju, G. (2018) RAD51C/XRCC3 facilitates mitochondrial DNA replication and maintains integrity of the mitochondrial genome. Mol. Cell. Biol., 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rajao, M.A., Passos-Silva, D.G., DaRocha, W.D., Franco, G.R., Macedo, A.M., Pena, S.D., Teixeira, S.M. and Machado, C.R. (2009) DNA polymerase kappa from Trypanosoma cruzi localizes to the mitochondria, bypasses 8-oxoguanine lesions and performs DNA synthesis in a recombination intermediate. Mol. Microbiol., 71, 185–197. [DOI] [PubMed] [Google Scholar]
- 36. Peng, M., Falk, M.J., Haase, V.H., King, R., Polyak, E., Selak, M., Yudkoff, M., Hancock, W.W., Meade, R., Saiki, R. et al. (2008) Primary coenzyme Q deficiency in Pdss2 mutant mice causes isolated renal disease. PLoS Genet., 4, e1000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Basse, A.L., Dixen, K., Yadav, R., Tygesen, M.P., Qvortrup, K., Kristiansen, K., Quistorff, B., Gupta, R., Wang, J. and Hansen, J.B. (2015) Global gene expression profiling of brown to white adipose tissue transformation in sheep reveals novel transcriptional components linked to adipose remodeling. BMC Genomics, 16, 215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sage, J.M., Gildemeister, O.S. and Knight, K.L. (2010) Discovery of a novel function for human Rad51: maintenance of the mitochondrial genome. J. Biol. Chem., 285, 18984–18990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rodriguez-Hernandez, A., Cordero, M.D., Salviati, L., Artuch, R., Pineda, M., Briones, P., Gomez Izquierdo, L., Cotan, D., Navas, P. and Sanchez-Alcazar, J.A. (2009) Coenzyme Q deficiency triggers mitochondria degradation by mitophagy. Autophagy, 5, 19–32. [DOI] [PubMed] [Google Scholar]
- 40. Li, Y., Lin, S., Li, L., Tang, Z., Hu, Y., Ban, X., Zeng, T., Zhou, Y., Zhu, Y., Gao, S. et al. (2018) PDSS2 deficiency induces hepatocarcinogenesis by decreasing mitochondrial respiration and reprogramming glucose metabolism. Cancer Res., 78, 4471–4481. [DOI] [PubMed] [Google Scholar]
- 41. Batterham, R.L., Cowley, M.A., Small, C.J., Herzog, H., Cohen, M.A., Dakin, C.L., Wren, A.M., Brynes, A.E., Low, M.J., Ghatei, M.A. et al. (2002) Gut hormone PYY(3-36) physiologically inhibits food intake. Nature, 418, 650–654. [DOI] [PubMed] [Google Scholar]
- 42. Alvarez Bartolome, M., Borque, M., Martinez-Sarmiento, J., Aparicio, E., Hernandez, C., Cabrerizo, L. and Fernandez-Represa, J.A. (2002) Peptide YY secretion in morbidly obese patients before and after vertical banded gastroplasty. Obes. Surg., 12, 324–327. [DOI] [PubMed] [Google Scholar]
- 43. Basu, A. and Sivaprasad, U. (2007) Protein kinase cepsilon makes the life and death decision. Cell. Signal., 19, 1633–1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Honda, H., Inazawa, J., Nishida, J., Yazaki, Y. and Hirai, H. (1994) Molecular cloning, characterization, and chromosomal localization of a novel protein-tyrosine phosphatase, HPTP eta. Blood, 84, 4186–4194. [PubMed] [Google Scholar]
- 45. D'Agostino, S., Lanzillotta, D., Varano, M., Botta, C., Baldrini, A., Bilotta, A., Scalise, S., Dattilo, V., Amato, R., Gaudio, E. et al. (2018) The receptor protein tyrosine phosphatase PTPRJ negatively modulates the CD98hc oncoprotein in lung cancer cells. Oncotarget, 9, 23334–23348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Longchamps, R.J., Castellani, C.A., Yang, S.Y., Newcomb, C.E., Sumpter, J.A., Lane, J., Grove, M.L., Guallar, E., Pankratz, N., Taylor, K.D. et al. (2020) Evaluation of mitochondrial DNA copy number estimation techniques. PLoS One, 15, e0228166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yang, S.Y., Castellani, C.A., Longchamps, R.J., Pillalamarri, V.K., O’Rourke, B., Guallar, E. and Arking, D.E. (2020) Blood-derived mitochondrial DNA copy number is associated with gene expression across multiple tissues and is predictive for incident neurodegenerative disease (2021). Genome Res., 31, 349–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Longchamps, R.J., Yang, S.Y., Castellani, C.A., Shi, W., Lane, J., Grove, M.L., Bartz, T.M., Sarnowski, C., Burrows, K., Guyatt, A.L. et al. (2021) Genome-wide analysis of mitochondrial DNA copy number reveals multiple loci implicated in nucleotide metabolism, platelet activation, and megakaryocyte proliferation. BioRxiv, in press, January 28, 2021, doi 10.1101/2021.01.25.428086, preprint: not peer reviewed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. The ARIC Investigators (1989) The Atherosclerosis Risk in Communities (ARIC) study: design and objectives. Am. J. Epidemiol., 129, 687–702. [PubMed] [Google Scholar]
- 50. Feinleib, M., Kannel, W.B., Garrison, R.J., McNamara, P.M. and Castelli, W.P. (1975) The Framingham offspring study. Design and preliminary data. Prev. Med., 4, 518–525. [DOI] [PubMed] [Google Scholar]
- 51. Splansky, G.L., Corey, D., Yang, Q., Atwood, L.D., Cupples, L.A., Benjamin, E.J., D'Agostino, R.B., Sr., Fox, C.S., Larson, M.G., Murabito, J.M. et al. (2007) The third generation cohort of the National Heart, Lung, and Blood Institute’s Framingham Heart Study: design, recruitment, and initial examination. Am. J. Epidemiol., 165, 1328–1335. [DOI] [PubMed] [Google Scholar]
- 52. Daniels, P.R., Kardia, S.L., Hanis, C.L., Brown, C.A., Hutchinson, R., Boerwinkle, E., Turner, S.T. and Genetic Epidemiology Network of Arteriopathy, s (2004) Familial aggregation of hypertension treatment and control in the Genetic Epidemiology Network of Arteriopathy (GENOA) study. Am. J. Med., 116, 676–681. [DOI] [PubMed] [Google Scholar]
- 53. The Women’s Health Initiative Study Group (1998) Design of the women’s health initiative clinical trial and observational study. Control. Clin. Trials, 19, 61–109. [DOI] [PubMed] [Google Scholar]
- 54. Bild, D.E., Bluemke, D.A., Burke, G.L., Detrano, R., Diez Roux, A.V., Folsom, A.R., Greenland, P., Jacob, D.R., Kronmal, R., Liu, K. et al. (2002) Multi-Ethnic Study of Atherosclerosis: objectives and design. Am. J. Epidemiol., 156, 871–881. [DOI] [PubMed] [Google Scholar]
- 55. Bibikova, M., Barnes, B., Tsan, C., Ho, V., Klotzle, B., Le, J.M., Delano, D., Zhang, L., Schroth, G.P., Gunderson, K.L. et al. (2011) High density DNA methylation array with single CpG site resolution. Genomics, 98, 288–295. [DOI] [PubMed] [Google Scholar]
- 56. Bibikova, M., Le, J., Barnes, B., Saedinia-Melnyk, S., Zhou, L., Shen, R. and Gunderson, K.L. (2009) Genome-wide DNA methylation profiling using Infinium® assay. Epigenomics, 1, 177–200. [DOI] [PubMed] [Google Scholar]
- 57. Kuan, P.F., Wang, S., Zhou, X. and Chu, H. (2010) A statistical framework for Illumina DNA methylation arrays. Bioinformatics, 26, 2849–2855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Taguchi, Y.H. and Oono, Y. (2005) Relational patterns of gene expression via non-metric multidimensional scaling analysis. Bioinformatics, 21, 730–740. [DOI] [PubMed] [Google Scholar]
- 59. Taliun, D., Harris, D.N., Kessler, M.D., Carlson, J., Szpiech, Z.A., Torres, R., Taliun, S.A.G., Corvelo, A., Gogarten, S.M., Kang, H.M. et al. (2021) Sequencing of 53,831 diverse genomes from the NHLBI TOPMed program. Nature, 590, 290–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ding, J., Sidore, C., Butler, T.J., Wing, M.K., Qian, Y., Meirelles, O., Busonero, F., Tsoi, L.C., Maschio, A., Angius, A. et al. (2015) Assessing mitochondrial DNA variation and copy number in lymphocytes of ~2,000 Sardinians using tailored sequencing analysis tools. PLoS Genet., 11, e1005306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Li, H. and Durbin, R. (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics, 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Morrison, A.C., Voorman, A., Johnson, A.D., Liu, X., Yu, J., Li, A., Muzny, D., Yu, F., Rice, K., Zhu, C. et al. (2013) Whole-genome sequence-based analysis of high-density lipoprotein cholesterol. Nat. Genet., 45, 899–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Joehanes, R., Zhang, X., Huan, T., Yao, C., Ying, S.X., Nguyen, Q.T., Demirkale, C.Y., Feolo, M.L., Sharopova, N.R., Sturcke, A. et al. (2017) Integrated genome-wide analysis of expression quantitative trait loci aids interpretation of genomic association studies. Genome Biol., 18, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Joehanes, R., Ying, S., Huan, T., Johnson, A.D., Raghavachari, N., Wang, R., Liu, P., Woodhouse, K.A., Sen, S.K., Tanriverdi, K. et al. (2013) Gene expression signatures of coronary heart disease. Arterioscler. Thromb. Vasc. Biol., 33, 1418–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Houseman, E.A., Accomando, W.P., Koestler, D.C., Christensen, B.C., Marsit, C.J., Nelson, H.H., Wiencke, J.K. and Kelsey, K.T. (2012) DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics, 13, 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Leek, J.T., Johnson, W.E., Parker, H.S., Jaffe, A.E. and Storey, J.D. (2012) The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics, 28, 882–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Leek, J.T. and Storey, J.D. (2007) Capturing heterogeneity in gene expression studies by surrogate variable analysis. PLoS Genet., 3, 1724–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Devlin, B. and Roeder, K. (1999) Genomic control for association studies. Biometrics, 55, 997–1004. [DOI] [PubMed] [Google Scholar]
- 69. Pinheiro, J., Bates, D., DebRoy, S., Sarkar, D., Authors, E., Heisterkamp, S., Willigen, B.V. and Ranke, J. (2007) Linear and Nonlinear Mixed Effects Models. CRAN. https://cran.r-project.org/web/packages/nlme/nlme.pdf.
- 70. Schwarze, G. (2015) General Package for Meta-Analysis. CRAN. https://cran.r-project.org/web/packages/meta/meta.pdf.
- 71. Li, Y., Willer, C.J., Ding, J., Scheet, P. and Abecasis, G.R. (2010) MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet. Epidemiol., 34, 816–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Hemani, G., Haycock, P., Zheng, J., Gaunt, T., Elsworth, B. and Tom Palmer, T. (2020) Mendelian Randomization with GWAS Summary Data. https://mrcieu.github.io/TwoSampleMR/.
- 73. Zheng, J., Erzurumluoglu, A.M., Elsworth, B.L., Kemp, J.P., Howe, L., Haycock, P.C., Hemani, G., Tansey, K., Laurin, C., Pourcain, B.S. et al. (2017) LD Hub: a centralized database and web interface to perform LD score regression that maximizes the potential of summary level GWAS data for SNP heritability and genetic correlation analysis. Bioinformatics, 33, 272–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Price, M.E., Cotton, A.M., Lam, L.L., Farré, P., Emberly, E., Brown, C.J., Robinson, W.P. and Kobor, M.S. (2013) Additional annotation enhances potential for biologically-relevant analysis of the Illumina Infinium HumanMethylation450 BeadChip array. Epigenetics Chromatin, 6, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Bernstein, B.E., Stamatoyannopoulos, J.A., Costello, J.F., Ren, B., Milosavljevic, A., Meissner, A., Kellis, M., Marra, M.A., Beaudet, A.L., Ecker, J.R. et al. (2010) The NIH roadmap epigenomics mapping consortium. Nat. Biotechnol., 28, 1045–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Martens, J.H. and Stunnenberg, H.G. (2013) BLUEPRINT: mapping human blood cell epigenomes. Haematologica, 98, 1487–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Consortium, E.P (2012) An integrated encyclopedia of DNA elements in the human genome. Nature, 489, 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Battram, T., Yousefi, P., Crawford, G., Prince, C., Babaei, M. S., Sharp, G., Hatcher, C., Vega-Salas, M. J., Khodabakhsh, S., Whitehurst, O. et al. (2021) The EWAS Catalog: A Database of Epigenome-Wide As-sociation Studies. in press. OSF Preprint, https://osf.io/837wn [DOI] [PMC free article] [PubMed]
- 79. Mi, H., Muruganujan, A., Ebert, D., Huang, X. and Thomas, P.D. (2019) PANTHER version 14: more genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res., 47, D419–D426. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
We describe data availability in the following: In ARIC, DNA methylation data are available upon request at https://sites.cscc.unc.edu/aric/distribution-agreements. The mtDNA CN data were calculated using TOPMed sequencing data on the Database of Genotypes and Phenotypes (dbGaP): phs000993. In FHS, the methylation data can be downloaded at the dbGaP: phs000724. The mtDNA CN data were calculated using TOPMed sequencing data on the dbGaP: phs000974. In GENOA, the methylation data from the EPIC array are on Gene Expression Omnibus (GEO): GSE157131. The mtDNA CN data were estimated using the GENOA TOPMed sequencing data on the dbGaP: phs001345. Owing to IRB restriction, mapping of the sample IDs between genotype data in dbGaP and methylation data in GEO cannot be provided publicly but are available upon written request to J.A.S. and Sharon LR Kardia. In MESA, the individual-level genotype, phenotype and methylation data are available on dbGaP (https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000209.v13.p3). The mtDNA CN data were calculated using TOPMed sequence data. In WHI, a part of DNA methylation data are under phs000200 and phs001335. Because DNA methylation data were not covered by the genetic data rules when it was generated, users should contact C.K. for requesting DNA methylation. The mtDNA CN data can be accessed using the TOPMed sequence data on dbGaP: phs001237.