

Abstract
Cushing’s disease (CD) requires accurate diagnosis, careful treatment selection, and long-term management to optimize patient outcomes. The Pituitary Society convened a Consensus Workshop comprising more than 50 academic researchers and clinical experts to discuss the application of recent evidence to clinical practice. In advance of the virtual meeting, recent data on screening and diagnosis; surgery, medical and radiation therapy; and disease- and treatment-related complications of CD were critically summarized in recorded lectures that were reviewed by all participants. During the meeting, concise summaries of the recorded lectures were presented, followed by small group breakout discussions. Consensus opinions from each group were collated into a draft document, which was reviewed and approved by all participants. Recommendations regarding use of laboratory tests, imaging, and treatment options are presented, along with algorithms for diagnosis of Cushing’s syndrome and management of CD. Topics considered most important to address in future research are also identified.
Keywords: Cushing’s disease, cortisol, transsphenoidal surgery, recurrence, medical therapy, Pituitary Tumor Centers of Excellence
INTRODUCTION
Cushing’s disease (CD), the most common cause of endogenous Cushing’s syndrome (CS), is caused by an adrenocorticotropin (ACTH)-secreting pituitary tumor.1 Optimal patient outcomes require accurate diagnosis, careful treatment selection, and management of the disease and its associated comorbidities to optimize patient outcomes.2 Notably, in comparison to patients with adrenal causes of CS, long-term quality of life (QoL) is worse for patients with CD.3 Since clinical guidelines published in 2003,4 2008,5,6 and 2015,7 novel screening and diagnostic modalities have been identified and new treatments approved for use. These new developments highlight the need for updates to clinical guidelines on this challenging disorder.
The Pituitary Society convened a 2-day virtual workshop in October 2020 to discuss management of CD, critically review the current literature, and provide recommendations for screening and diagnosis; optimal use of and monitoring outcomes from surgery, medical therapy, and radiation therapy; and identification and management of disease- and treatment-related complications. The focus was on pituitary, rather than adrenal or ectopic CS, and overlapping topics that had been recently covered in other consensus statements/reviews were not included.
We briefly review recent evidence and recommendations for clinical practice, grading the quality of the evidence and the strength of the consensus recommendations. Key considerations for use of different laboratory tests and medical therapies are presented in Tables 1 and 2. Consensus recommendations for management of CD complications and use of medical therapy for CD are presented in Panels 1 and 2. Evidence/recommendations grading schema8,9 are presented in the Appendix. Algorithms for diagnosis of CS and management of CD are presented in Figures 1 and 2. Topics that were rated the most important to address in future research are listed in Panel 3.
Table 1.
Laboratory Tests for CS Diagnosis and Monitoring for CD Recurrence
| Diagnosis | |||||
| Test | Cutoff level * | Sensitivity (%) | Specificity (%) | Advantages/Instructions for testing | Disadvantages/Pitfalls |
| 1-mg DST | 1.8 μg/dL (50 nmol/L) | 98 | 81 | • High negative predictive value • Easy for healthcare provider to administer |
• False positives common • Variable dexamethasone metabolism can confound results • Oral estrogen can increase CBG |
| 24-hr UFC | Assay-specific reference range | 91 | 81.5 | • Wide range for normal values | • Cumbersome for patient to undertake • Variability could be 50% between samples, thus 2–3 collections are needed |
| LNSC | Assay-specific reference range | 97 | 97.5 | • Easy for patient to perform • Patients should be cautioned not to eat, drink, smoke, or brush their teeth for 15 minutes prior to collecting saliva samples |
• Intra-patient variability • Cut-offs vary signficantly based on reference laboratory • Potential for contamination with topical hydrocortisone • Not available in all centers |
|
Clinical Considerations and Recommendations If CD is suspected: • Start with either UFC and/or LNSC; DST could also be an option if LNSC not feasible • Multiple LNSC may be easier for patient collection If confirming CD: • Use any test • UFC average 2–3 collections • LNSC ≥2 on consecutive days • DST useful in shift workers, not in women on estrogen-containing OC • Measuring dexamethasone level along with cortisol the morning after 1 mg dexamethasone ingestion improves test interpretability If CS due to adrenal tumor is suspected: • Start with DST • LNSC has lower specificity in these patients | |||||
| Monitoring for Recurrence | |||||
| Test | Cutoff level * | Sensitivity (%) | Specificity (%) | Advantages | Disadvantages |
| LNSC | 0.27 μg/dL (7.5 nmol/L) | 75–90 | 93–95 | • In most patients abnormal earlier than DST and UFC | • Intra-patient variability • May be normal despite recurrence |
| 24-hr UFC | 1.6 × ULN | 68 | 100 | • Direct reflection of bioavailable cortisol | • ∼50% intra-patient variability • Last test to become abnormal |
| Desmopressin | Absolute cortisol increments of 7.0–7.4 μg/dL from baseline** | 68 | 95 | • Earliest test to become positive in some studies • Predicts presence of corticotroph tumor • Can become positive before clinical adenoma recurrence |
• Dynamic labor-intensive testing |
| 1-mg DST | 1.8 μg/dL (50 nmol/L) | N/A | N/A | • Likely to be abnormal before 24-hr UFC | • Limited evidence specifically assessing utility for recurrence |
|
Clinical Considerations and Recommendations • LNSC most sensitive, should be done annually • DST and UFC usually become abnormal after LNSC • Consider which tests were abnormal at initial diagnosis | |||||
Based on Galm et al J Clin Endocrinol Metab 2020 and Hinojosa-Amaya et al Front Endocrinol (Lausanne) 2019. Sensitivity and specificity will vary based on assay used (Petersenn Best Pract Res Clin Endocrinol Metab 2021).
Cut-offs specified are for adults. Some experts recommend using the same cutoffs for initial diagnosis and recurrence.
Some studies use ACTH absolute cutoffs/increments.
Abbreviations: CD, Cushing’s disease; CS, Cushing’s syndrome; DST, dexamethasone suppression test; LNSC, late-night salivary cortisol; UFC, urinary free cortisol; ULN, upper level of normal.
Table 2.
Summary of Medical Therapies for CD
| Target | Drug | Commonly used doses | Efficacy | Adverse effects | Key considerations |
|---|---|---|---|---|---|
| Adrenal steroidogenesis | Ketoconazole | 400–1200 mg/d PO, dosing BID | Retrospective studies: ∼65% UFC normalization initially, but 15–25% escape | GI disturbances, ↑ liver enzymes, gynecomastia, skin rash, AI | • EMA approved for treatment of endogenous CS, off-label use in US • Increasing doses needed to counter escape • Needs gastric acid for absorption (avoid PPIs) • Decrease in testosterone would be preferred in women; men need follow-up for hypogonadism • Risk for serious hepatotoxicity; mostly transient but regular LFT monitoring required • Risk of QTc prolongation • Careful review of other medications for potential drug-drug interactions is essential |
| Osilodrostat | 2–7 mg/d BID PO as maintenance; 30 mg/d BID maximum | Phase 3 randomized withdrawal study: 86% UFC normalization | ↑ Androgenic and mineralocorticoid precursors (hirsutism, hypertension, hypokalemia), GI disturbances, asthenia, AI | FDA approved for patients with CD in whom pituitary surgery is not an option or has not been curative • EMA and Japan approved for treatment of endogenous CS • Not yet widely available • Rapid decrease in UFCHas • Risk for hypocortisolism, hypokalemia, and QTc prolongation • Cross-reaction in routine assays with 11-deoxycortisol • Careful monitoring for hyperandrogenism in women |
|
| Metyrapone | 500 mg/d to 6 g/d; dosing q 6–8 h | UFC normalization Retrospective studies: ∼70% Prospective study: 47% at week 12 |
↑ Androgenic and mineralocorticoid precursors (hirsutism, hypertension, hypokalemia), AI | • EMA approved for treatment of endogenous CS, off-label use in US • Rapid decrease in UFC, typically in first month • Possible cross reactivity with 11-deoxyxortisol in cortisol immunoassays • Hyperandrogenism needs to be monitored with long-term use in women |
|
| Mitotane | 250–500 mg/d PO up to 8 g/d | Retrospective studies: ∼80% UFC normalization | GI disturbances, dizziness, cognitive alterations, AI ↑ Liver enzymes; treatment should be stopped if elevations are >5 × ULN | • FDA and EMA approved for treatment of adrenal cancer with endogenous CS • Slow onset of action, highly variable bioavailability • Narrow therapeutic window (dose titration based on mitotane plasma levels) • Neurological toxicity could be a limiting factor • Mitotane is teratogenic and an abortifacient. Because of its long terminal half-life, this may limit its use in women who desire future pregnancy. • Cross-reaction in routine assays with 11-deoxycortisol |
|
| Etomidate | 0.04–1 mg/kg/h IV for patients in the ICU; 0.025 mg/kg/h for non-ICU patients | Retrospective studies: ∼100% serum cortisol control (10–20 μg/dL) | Sedation/anesthesia, AI Myoclonus, nausea, vomiting, and dystonic reactions at higher anesthetic doses | • Off-label use only Very rapid onset of action, appropriate for acute treatment of severe hypercortisolism • IV hydrocortisone required at high doses to avoid adrenal insufficiency |
|
| Somatostatin receptor | Pasireotide | 0.3–0.9 mg/mL BID SC | Phase 3 study: 15–26% UFC normalization | Hyperglycemia, T2DM, diarrhea, nausea, abdominal pain, cholelithiasis, fatigue | • Widely approved for patients with CD in whom pituitary surgery is not an option or has not been curative • Decreases tumor volume • High risk for hyperglycemia requires careful patient selection • Risk of QTc prolongation |
| Pasireotide LAR | 10–30 mg monthly IM | Phase 3 study: 40% UFC normalization Clinical signs and symptoms of hypercortisolism improved |
|||
| Dopamine receptor | Cabergoline | 0.5–7 mg weekly PO | Retrospective studies: ∼40% UFC normalization initially, but ∼25–40% escape Clinical signs and symptoms of hypercortisolism improved |
Headache, nasal congestion, hypotension, depression, dizziness | • Off-label use only for CD • Decreases tumor volume in up to 50% of the patients evaluated • Clinical signs and symptoms of hypercortisolism improved • Poor response may be due to under-titration • Risk for treatment-induced impulse-control disorder; unclear risk for cardiac valvulopathy |
| Glucocorticoid receptor | Mifepristone | 300–1200 mg/d PO | Open-label phase 3 study: significant improvement in glycemia (∼60%) and blood pressure Clinical signs and symptoms of hypercortisolism improved |
GI disturbances, headache, hypokalemia, arthralgia, peripheral edema, hypertension, vaginal bleeding, AI | • FDA approved for hyperglycemia associated with CS • No laboratory markers of efficacy • Challenging to use outside specialized clinical practice • Risk of hypokalemia and adrenal insufficiency; needs close monitoring • Careful review of other medications for potential drug-drug interactions is essential |
| Investigational drugs with completed phase 3 clinical trials | |||||
| Adrenal steroidogenesis | Levoketoconazole274,275 | 300–1200 mg/d, BID, PO | Phase 3 open label study: 31% UFC normalization primary end-point; 42% when using imputed data (comparable with other studies) Phase 3 randomized withdrawal study: 41% lost response with drug vs 96% with placebo Clinical signs and symptoms of hypercortisolism improved |
GI disturbances, headache, edema, ↑ liver enzymes, AI | • Investigational; FDA and EMA orphan drug status for treatment of endogenous CS • Possible lower risk for hepatotoxicity than with ketoconazole based on animal models, although no head to head studies in humans available • Needs gastric acid, avoid PPIs • Risk of QTc prolongation • Careful review of other medications for potential drug-drug interactions is essential |
Abbreviations: AI, adrenal insufficiency; BID, twice daily; CD, Cushing’s disease; CS, Cushing’s syndrome; EMA, European Medicines Agency; FDA, US Food and Drug Administration; GI, gastrointestinal; ICU, intensive care unit; IM, intramuscular; IV, intravenous; LAR, long-acting release; LFT, liver function test; PO, by mouth; PPI, proton pump inhibitor; q, every; SC, subcutaneous; UFC, urinary free cortisol.
Panel 1.
Complications of CD: Summary of Recommendations
| Hypercoagulation |
|
|
| • There is currently no standard practice for preoperative or postoperative thromboprophylaxis in patients with CD. Some experts hold estrogen therapy in women who are awaiting surgery, but care should be taken if it was being used as contraception, because pregnancy also is associated with increased risk of thrombosis (LQ, DR) |
| • Prophylactic anticoagulation should be considered for patients at risk for VTE, including history of embolism or abnormal coagulation testing; severe preoperative hypercortisolism; current use of estrogen or oral contraceptives; poor mobility; extended preoperative or postoperative hospital stay; and high postoperative cortisol levels or cortisol over-replacement in patients with AI (MQ, SR) |
| • Early postoperative ambulation and use of compression stockings should be encouraged for all patients (HQ, SR) |
| • If thromboprophylaxis is administered, there was strong consensus for preference of low molecular weight heparin over oral anticoagulants given the long half-life of the latter and the lack of therapy to reverse their effect, which may be especially concerning in the preoperative setting (LQ, DR) |
| • Anticoagulants may be discontinued before surgery to minimize intraoperative bleeding risk, but the timing of when to stop and when to reinitiate after surgery is unclear (LQ, DR) |
| • Among meeting participants, recommended anticoagulation duration ranged in the preoperative setting from 2–4 days to 1–2 weeks, and in the postoperative setting from 1–2 days of the hospital stay up to 2–4 weeks or even longer to 2–3 months (LQ, DR) |
| • Thromboprophylaxis should not be routinely used in pediatric patients due to bleeding risk but reserved for selected patients |
|
|
| Cardiovascular Disease |
|
|
| • Evaluate, monitor, and treat according to current guidelines for patients at high risk for cardiovascular disease (HQ, SR) |
| • Management approach should be individualized (HQ, SR) based on the complications present (e.g., hypertension or hyperlipidemia) and care should be coordinated with primary care and cardiology physicians as needed (VLQ, DR) |
|
|
| Bone Disease |
|
|
| • Risk assessment for bone loss and fracture recommended in all patients (HQ, SR) |
| • Given the risk for fracture even in patients without osteoporosis, standard DXA alone may not be sufficiently informative; bone quality (microscanner or trabecular bone score) or morphometric vertebral assessment is recommended where available (HQ, SR) and can be useful in detecting subclinical fractures (HQ, SR), but might not be practical for all patients. The FRAX tool to assess fracture risk is not validated for CD |
| • Monitor and follow-up as for all adult high-risk populations (HQ, SR) |
| • Consider conventional osteoporosis treatments, e.g., bisphosphonates, for patients with persistent CD even if BMD is normal because of increased fracture risk due to cortisol excess (HQ, SR) |
|
|
| GH Deficiency |
|
|
| • There is currently no standard practice for whether, when, and how to test for GHD in adults with CD. As postoperative HPA axis recovery is often delayed, we recommend waiting at least 6–12 months after surgery before considering GHD assessment (MQ, SR) |
| • Patients with macroadenomas and more aggressive surgical resection are at higher risk for hypopituitarism; patients with 3 or more pituitary hormone deficiencies are more likely to have GHD and do not need dynamic testing (HQ, SR) |
| • Serum IGF-I level alone is not likely to be a reliable indicator of GHD, as levels can be in the lower half of the normal range on dynamic tests |
| • Accessibility of GH replacement may be an important factor in determining testing and treatment considerations. If GH replacement is implemented earlier than 2 years after pituitary surgery, we recommend retesting periodically to determine whether GH secretion has normalized upon HPA axis recovery (MQ, SR) |
| • As CS-associated myopathy does not spontaneously resolve during remission, physical rehabilitation is recommended for all patients (HQ, SR). |
| • In children, evaluate for GHD 3–6 months after surgery and immediately initiate GH replacement if needed to ensure proper growth |
Abbreviations: AI, adrenal insufficiency; BMD, bone mineral density; CD, Cushing’s disease; DXA, dual x-ray absorptiometry; GHD, growth hormone deficiency; HPA, hypothalamus-pituitary-adrenal; VTE, venous thromboembolism.
Panel 2.
Medical Therapy for CD: Summary of Recommendations
| Which factors are helpful in selection of a medical therapy? |
|
|
| • If there is a need for rapid normalization of cortisol, we recommend an adrenal steroidogenesis inhibitor; osilodrostat and metyrapone have the fastest action and are orally available, while etomidate can be used intravenously in very severe cases (HQ, SR) |
| • In mild disease, if residual tumor is present and there is a potential for tumor shrinkage, consider pasireotide or cabergoline (MQ, SR) |
| • If there is a history of bipolar or impulse control disorder, consider avoiding cabergoline (MQ, SR) |
| • If an expert pituitary endocrinologist is not available to monitor treatment response, use mifepristone cautiously (LQ, DR); we recommend counseling patients that cortisol cannot be used to monitor treatment response or AI (SQ, SR). Drug-drug interactions must be considered when this medication is used. |
| • In pregnant women or those desiring pregnancy, consider cabergoline or metyrapone, although no CD medications are approved for use in pregnancy (LQ, DR) |
| • Drug intolerance or side effects as well as concomitant comorbidities such as T2DM and hypertension should further guide type of medication used (MQ, SR) |
| • Consider cost and estimated therapy duration, especially if definitive treatment (i.e., pituitary and adrenal surgery) is planned or while awaiting effects of radiotherapy (LQ, DR) |
|
|
| Which factors are used in selecting an adrenal steroidogenesis inhibitor? |
|
|
| • Rapidity of action, tolerability, ease-of-use, degree of likely biochemical normalization, and specific clinical improvement as well as local availability and cost of each drug should be considered at therapy start (MQ, SR) |
| • Ketoconazole may be favored for ease of dose titration; concern about inducing hepatotoxicity and the need to monitor liver enzymes may lead to under-dosing (MQ, SR). Drug-drug interactions must be considered and hypogonadism may occur in men |
| • Osilodrostat achieves high rates of cortisol normalization. Dosing schedule may be more convenient for patients compared with metyrapone, but neither metyrapone nor osilodrostat is limited by hypogonadism in men (HQ, SR) |
| • Mitotane is rarely used as monotherapy in CD in most centers (LQ, DR) |
|
|
| How is tumor growth monitored when using an adrenal steroidogenesis inhibitor or glucocorticoid receptor blocker? |
|
|
| • MRI is typically obtained 6–12 months after initiating treatment and repeated every few years depending on the clinical scenario (MQ, SR) |
| • It can be difficult to determine whether tumor progression is due to loss of cortisol feedback or reflects the underlying behavior of aggressive, recurrent disease (LQ, DR) |
| • We suggest monitoring ACTH levels, as progressive elevations in ACTH may be a sign of tumor growth and a need for MRI, although the half-life of ACTH is short, levels fluctuate and do not necessarily reflect tumor growth (LQ, DR) |
| • If progressive tumor growth is seen, medical treatment should be suspended and the management plan reassessed (MQ, SR) |
|
|
| When is preoperative medical therapy used? |
|
|
| • There are no rigorous data supporting use of preoperative medical therapy (MQ, SR) |
| • Most experts would consider use of adrenal steroidogenesis inhibitors if surgery is delayed, either because of scheduling or due to external factors (LQ, DR) |
| • Patients with severe CD who have potentially life-threatening metabolic, psychiatric, infectious, or cardiovascular/thromboembolic complications may benefit in select cases (LQ, DR) |
|
|
| How is treatment response monitored? Which factors are considered in deciding whether to use combination therapy or to switch to another therapy? |
|
|
| • Response should be defined based on a combination of clinical (improved phenotype, weight, hypertension, glucose metabolism, QoL) and biochemical endpoints or only clinical endpoints when glucocorticoid receptor blockers are used (MQ, SR) |
| • Cortisol levels are often measured by UFC (except when using mifepristone); UFC is not useful if AI is a concern (HQ, SR) |
| • Because of the loss of biologic circadian rhythm, it is unclear whether targeting diurnal secretion alone with morning cortisol and/or LNSC is meaningful (LQ, DR) |
| • Change in treatment should be considered if cortisol levels are persistently elevated after 2–3 months on maximum tolerated doses (MQ, SR) |
| • If cortisol does not normalize but is reduced and/or there is some clinical improvement, combination therapy can be considered (LQ, DR) |
| • If there is clear resistance to treatment despite dose escalation, we suggest switching to a different therapy (LQ, DR) |
|
|
| Which agents are used for optimal combination therapy? |
|
|
| • There are few rigorous data supporting specific regimens for combination therapy (HQ, SR) |
| • Many experts consider combining ketoconazole with metyrapone or potentially ketoconazole with osilodrostat to maximize adrenal blockade when monotherapy is not effective or to allow lower doses of both drugs (LQ, DR) |
| • Ketoconazole plus cabergoline or pasireotide, and pasireotide plus cabergoline may be rational combinations if there is visible tumor present (LQ, DR) |
| • Other combinations that may be used include triplets of cabergoline, pasireotide, plus ketoconazole, and ketoconazole, metyrapone, plus mitotane (LQ, DR) |
Abbreviations: ACTH, adrenocorticotropin; AI, adrenal insufficiency; CD, Cushing’s disease; LNSC, late-night salivary cortisol; MRI, magnetic resonance imaging; QoL, quality of life; UFC, urinary free cortisol.
Figure 1. Algorithm for diagnosis of Cushing’s syndrome.
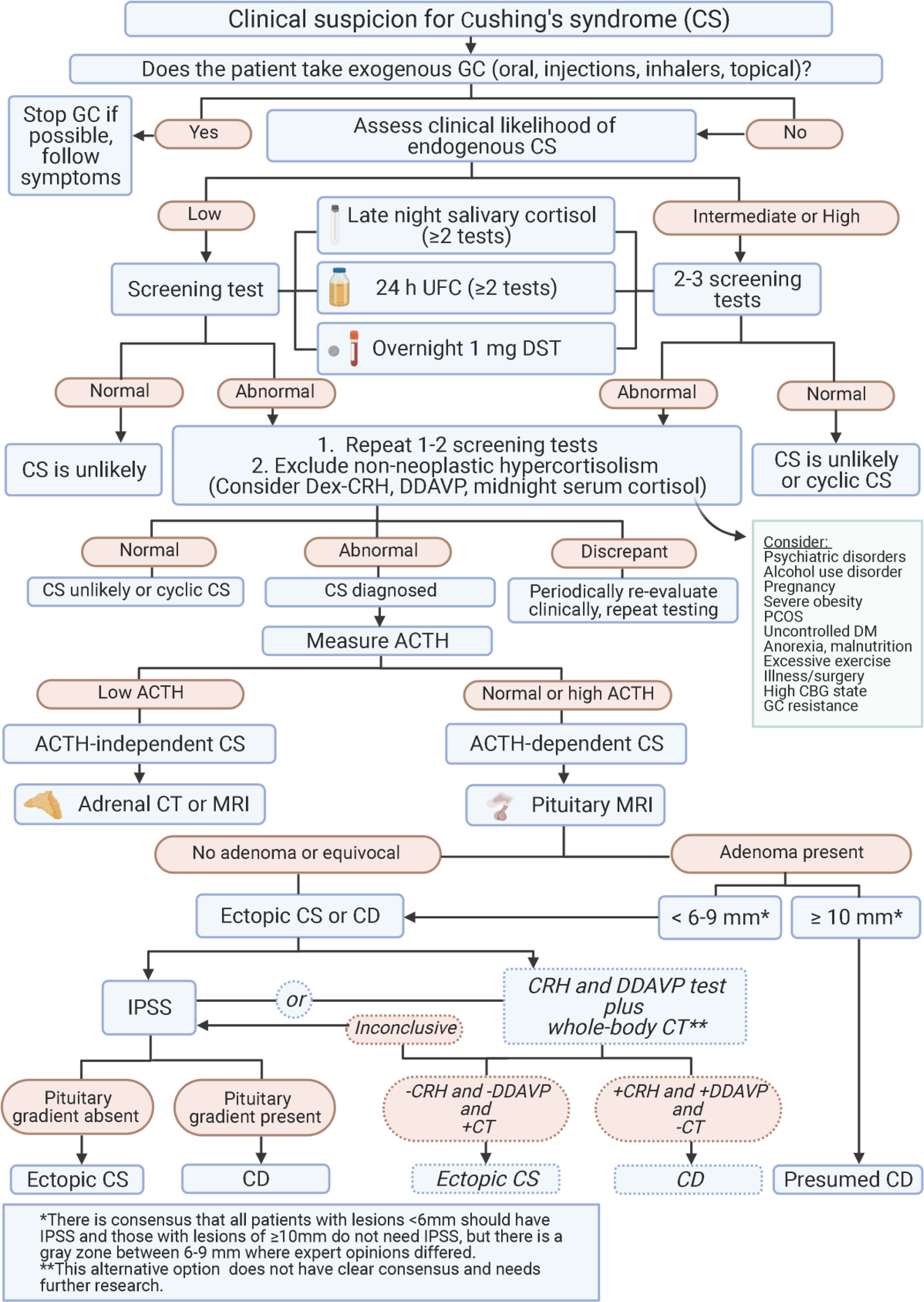
Abbreviations: ACTH, adrenocorticotropin; CBG, corticosteroid binding globulin; CD, Cushing’s disease; CRH, corticotropin stimulating hormone; CS, Cushing’s syndrome; CT, computed tomography; Dex, dexamethasone; DM, diabetes mellitus; DST, dexamethasone suppression test; GC, glucocorticoid; IPSS, inferior petrosal sinus sampling; MRI, magnetic resonance imaging; PCOS, polycystic ovary syndrome; UFC, urinary free cortisol.
Figure 2. Algorithm for management of Cushing’s disease.
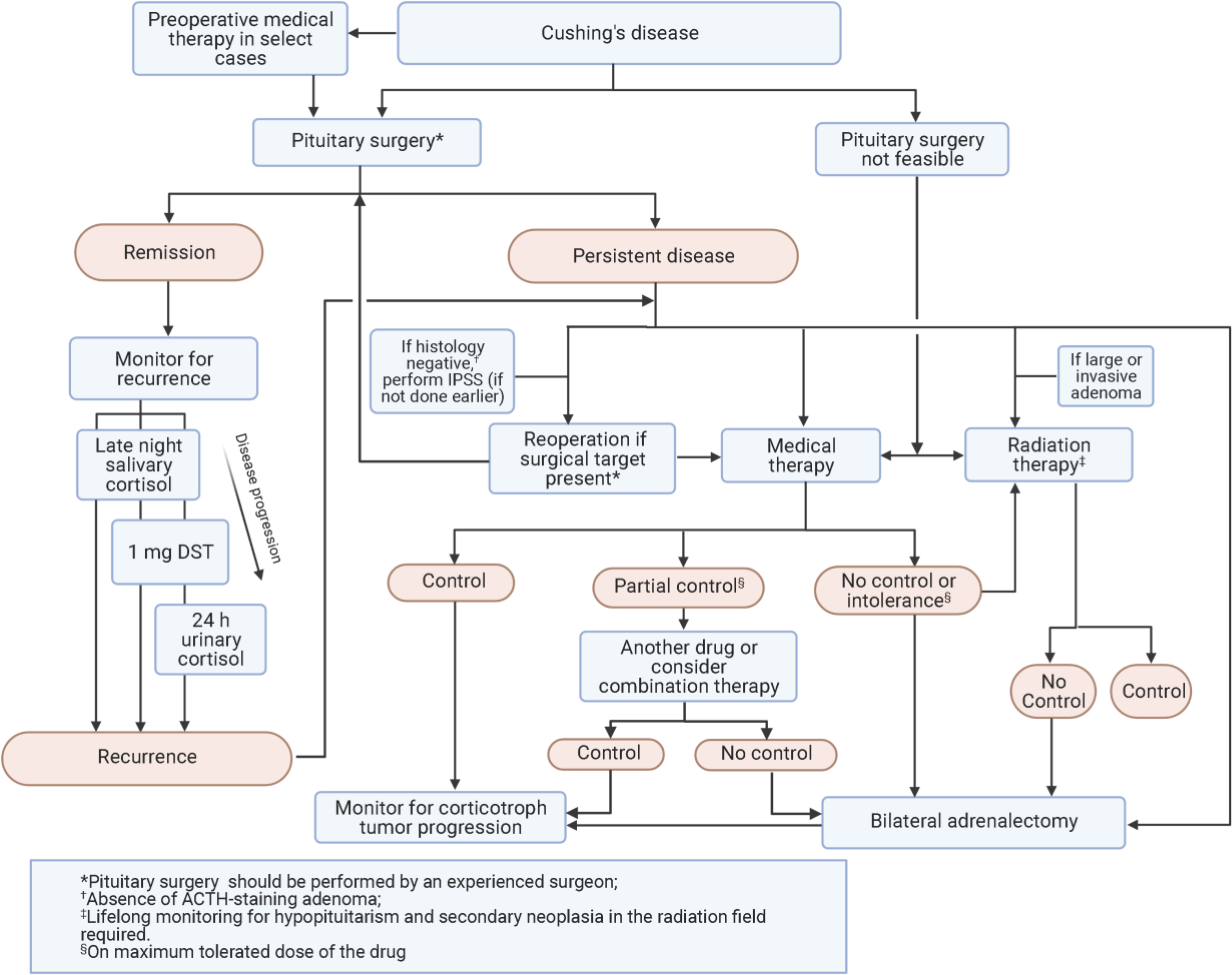
Abbreviations: ACTH, adrenocorticotropin; DST, dexamethasone suppression test; IPSS, inferior petrosal sinus sampling.
Panel 3.
Future Research Topics Ranked of Highest Importance
| Screening and diagnosis of CS |
|
|
| • Optimize pituitary MR and PET imaging using improved data acquisition and processing to improve microadenoma detection |
| • Compare diagnostic algorithms for the differential diagnosis using invasive versus non-invasive strategies |
| • Identify additional corticotroph adenoma mutations and development of a comprehensive panel of genomic/proteomic tests for CD diagnosis |
|
|
| Complications of CD |
|
|
| • Define use of anticoagulant prophylaxis and therapy in different populations and settings |
| • Optimize the approach in managing long-term complications |
|
|
| Treatment of CD |
|
|
| • Determine clinical benefit of restoring the circadian rhythm, potentially with a higher nighttime medication dose |
| • Identify better markers of disease activity and control |
| • Develop new, better tolerated, more effective medical therapies |
| • Define populations that might benefit from preoperative medical treatment |
Abbreviations: CD, Cushing’s disease; CS, Cushing’s syndrome; MR, magnetic resonance; PET, positron emission tomography.
Recommendations for adults with CD are presented here for use in clinical practice but should be considered alongside patient- and disease-specific factors for personalized care. A brief section regarding unique considerations in pediatric CD is also presented.
METHODS
Workshop co-chairs and steering committee members identified 28 discrete topics related to CD diagnosis, complications, and treatment to be addressed. Methods for critical review of the literature, pre-Workshop lectures, and Workshop discussions are described in the Appendix. A brief summary of the search strategy and selection criteria is given below.
DIAGNOSIS OF CS: SCREENING, CONFIRMATORY, AND LOCALIZATION MODALITIES
Laboratory Tests (Table 1)
Background
Diagnosis of CS is often delayed for years, partly due to lack of awareness of the insidious, progressive disease process and the testing complexity.10 Screening and diagnostic tests for CS assess cortisol secretory status: abnormal circadian rhythm with late night salivary cortisol (LNSC), impaired glucocorticoid feedback with overnight 1-mg dexamethasone suppression test (DST) or low dose 2-day dexamethasone test (LDDT), and increased bioavailable cortisol with 24-hour urinary free cortisol (UFC).5,6,11,12 In this setting, sensitivity of all tests is above 90%; the highest rates are seen with DST and LNSC and the lowest with UFC. Specificity is somewhat lower, with LNSC the most specific and DST and UFC the least.12,13
LNSC
The diagnostic utility of LNSC is based on the assumption that patients with CS lose the normal circadian nadir of cortisol secretion;14,15 at least two or three LNSC tests are recommended.5,16 Patients with mild CS may have LNSC just above the upper limit of normal (ULN). Sampling saliva at usual bedtime rather than at midnight could decrease false positive results,17 as cortisol nadir is tightly entrained to sleep onset. Although mass spectrometry can detect both cortisol and cortisone, therefore avoiding potential contamination from topical hydrocortisone preparations, sensitivity is better than with immunoassay, but at the expense of reduced specificity.18 Multiple, periodic, sequential LNSC are particularly useful for the longitudinal surveillance needed in distinguishing patients with cyclic CS who exhibit weeks to months of normal cortisol secretion interspersed with cortisol excess episodes.19 By contrast, this test should not be performed in patients with disruption of the normal day/night cycle, such as night-shift workers.14,15
Overnight 1-mg DST
In healthy individuals, a supraphysiologic dexamethasone dose inhibits vasopressin and ACTH secretion, thereby decreasing cortisol levels. Thus, a serum cortisol < 1.8 μg/dL (50 nmol/L) at 0800 h in the morning after 1 mg dexamethasone given between 2300 h and midnight is considered a normal response.5 A negative result strongly predicts CS absence. At higher cutoff points, e.g., 5 μg/dL (138 nmol/L), DST sensitivity is reduced.12 Cortisol values <1.8 μg/dL excludes dysregulated cortisol production from an adrenal incidentaloma;20 in this setting, values over 5 μg/dL generally identify patients with dysregulated cortisol secretion from an incidentaloma with overt CS. False positive results may be seen with rapid absorption/malabsorption of dexamethasone due to increased gut transit time, chronic diarrhea, or celiac disease; concomitant treatment with CYP3A4 inducers (e.g., phenobarbital, carbamazepine, St. John’s wort); and increased corticosteroid binding globulin (CBG) levels from oral estrogens, pregnancy, or chronic active hepatitis, which may increase total cortisol levels.21–23 Measuring dexamethasone concomitantly with cortisol, using laboratory-specific ranges of expected values, can reduce the risk for false-positive results.24,25 False negative results are less common, typically resulting from inhibition of dexamethasone metabolism by concomitant medications, such as fluoxetine, cimetidine, or diltiazem, leading to a higher biologically available dose. Decreased CBG and albumin levels, such as in patients with concurrent nephrotic syndrome, also might produce a falsely low value.26
UFC
At least two or three 24-hour urine collections are advised to measure UFC to account for intra-patient variability.5,27 One advantage with UFC over DST is that overall cortisol production is independent of CBG changes and dexamethasone compliance. However, although calculating the mean of several collections aids in correct interpretation, random variability can be as high as 50%.28 As with LNSC, UFC relies on accurate collection by the patient.
Sex, body mass index (BMI), age, very high or low urinary volume, and sodium intake can all influence UFC levels and should be taken into account for interpretation.29–33 As urine volume and glomerular filtration rate strongly predict UFC, other screening tests such as LNSC may be preferred for patients with renal impairment (CrCl <60mL/min) or significant polyuria (>5 L/24 h).34,35
Testing for non-neoplastic hypercortisolism (pseudo-CS)
Psychiatric disorders, alcohol use disorder, polycystic ovary syndrome, and obesity may activate the hypothalamic-pituitary-adrenal (HPA) axis.36,37 Such patients also may have concomitant features of CS that are common in the general population (e.g., weight gain) that lead to biochemical screening. DST, LNSC, and UFC may all show positive (abnormal) results in these patients with non-neoplastic clinical hypercortisolism, or so-called pseudo-CS.38 Furthermore, concomitant medications could result in steroid cross-reactivity or otherwise interfere with laboratory test results. However, these abnormal results tend to be mildly elevated; UFC is almost always within 3-fold of normal. The combined LDDT-CRH (Dex-CRH) test, LDDT, or the desmopressin test may be able to distinguish between ACTH-dependent CS and pseudo-CS.39–41 Utility of the Dex-CRH test in this setting is based on the assumption that only patients with ACTH-dependent CS will show a cortisol response to CRH after dexamethasone suppression.42 However, test reliability may differ due to different protocols, various ovine or human CRH doses, characteristics of cortisol and ACTH assays, and patients (e.g., degree of hypercortisolism, adrenal versus pituitary CS, and underlying conditions). Use of the desmopressin test is based on the finding that ACTH-secreting adenomas express vasopressin V1b (V3) receptors, producing a rise in plasma ACTH after desmopressin injection.43 The desmopressin test has a high specificity for CD44, is less complex and expensive than the Dex-CRH test, but both have shown good diagnostic performance in distinguishing CS from pseudo-CS in some studies; when both tests are done, they showed excellent agreement.45,46
Clinical Considerations and Recommendations
Screening and confirmatory testing for CS
There is no single preferred diagnostic test for CS, nor is there consensus on how to decide whether and when to test, despite attempts to develop a score for ease of diagnosis.47 Clinical judgment and index of suspicion for CS are very important48 and underscore the need to individualize decisions about timing and selection for diagnostic testing based on the clinical scenario (HQ, SR).
If CS is suspected, any of the diagnostic tests may be useful. We recommend starting with DST, UFC, and/or LNSC (HQ, SR) depending on local availability; multiple LNSCs may be easier for the patient to complete (HQ, SR). If an adrenal tumor is suspected, we recommend starting with DST (MQ, SR) and only using LNSC if cortisone levels can be also reported16,18 (MQ, SR).
DST may be the preferred test for shift workers and patients with disrupted circadian rhythm due to uneven sleep schedules, but may not be reliable in women treated with oral estrogen (HQ, SR). Measuring a dexamethasone level may be useful if a false-positive DST is suspected due to the clinical scenario (MQ, SR). If UFC is used, two or three collections should be obtained to evaluate variability (HQ, SR). If LNSC is used, we recommend at least two or three tests (HQ, SR). Although there were initial concerns about increased risk for infection from SARS-CoV-2 with LNSC,49 it remains safe for lab personnel when used with proper precautions.50 Bilateral inferior petrosal sinus sampling (IPSS) should not be used to diagnose hypercortisolism because the central-to-peripheral ACTH gradient in healthy controls and pseudo-CS overlaps that seen in patients with CD51 (HQ, SR). In classical cyclic CD or in patients with unpredictable fluctuating cortisol levels, dynamic testing and localization testing, including IPSS, should be preceded by a confirmatory LNSC, DST, or UFC to document that the patients are in the active phase.52
Currently, there is no preference for mass spectrometry over immunoassay in measuring cortisol level for diagnosis to ensure that patients with mild hypercortisolism are not excluded.18,27 However, normative data with modern assays are needed.
Ruling out pseudo-CS
Because the etiology of pseudo-CS can vary, there is no single approach to rule it out.53 We recommend considering the patient’s clinical history, particularly the duration of symptoms, and repeating testing to avoid implementing inappropriate treatment if CS is not present (LQ, DR). In most cases, patients have mild hypercortisolism and can be monitored for 3–6 months to see whether symptoms resolve; treatment of the underlying condition (such as depression) can restore normal HPA axis function and cortisol levels (LQ, DR). Standard diagnostic testing is unreliable in this population. LDDT or serial LNSCs over time correlate with the clinical picture (LQ, DR). Desmopressin is easy to use and easily administered in an outpatient setting. Dex-CRH in this setting could be valuable, but published diagnostic accuracy results have varied; use at an expert center with measurement of dexamethasone levels is advised (MQ, SR),54 as is cortisol cut-off adjustments in very obese patients. Ovine CRH is not presently available in the United States, Canada, Brazil, Argentina, Mexico and some other countries.
Imaging and Tumor Localization
Background
MRI is the imaging method of choice for detecting ACTH-secreting pituitary adenomas. However, as most lesions are very small, using standard 1.5T MRI, only approximately 50% of microadenomas are clearly depicted.55
Technical refinements including spoiled gradient–recalled (SPGR) acquisition echo with 1 mm slice intervals, fluid attenuation inversion recovery (FLAIR)56 and constructive interference in the steady state (CISS), may enhance detection, while variants of T1-weighted turbo spin echo (TSE) sequences and use of ultra high field 3T and 7T magnets allow improved localization of microadenomas.57–60 Nevertheless, approximately one-third of scans in patients with CD still remain negative,61 and higher resolution with 3T or 7T magnets can increase the risk of detecting incidentalomas potentially unrelated to the disorder.
Importantly, tumor size does not necessarily correlate with degree of hypercortisolism in CD. In fact, patients with larger adenomas frequently present with milder hypercortisolism.62
Positron emission tomography (PET) has been explored as an alternative to, or in combination with, MRI for localization of corticotroph adenomas. 18F-fluoro-deoxy-glucose (18F-FDG) PET/CT is largely comparable to standard fast spin echo MRI in detecting pituitary lesions in one series,63 while a separate study found both standard spin echo MRI and high resolution 18F-FDG PET were inferior to SPGR MRI.64 Prior ovine CRH stimulation can increase 18F-FDG uptake and thus increase detection.65 PET coregistration with volumetric MRI (PET/MRCR) combines functional and anatomical imaging, while 11C-methionine may permit more accurate localization of sites of radiotracer uptake.66 In one series, this technique correctly localized corticotroph adenomas in patients with de novo disease and persistent/recurrent hypercortisolism following primary surgery, most of whom had negative or equivocal standard spin echo MRI.67 However, this approach is not available or approved in most countries. Alternative strategies (e.g., targeting CRH-R1 expression on corticotroph tumors) have also recently been proposed, but require further study.68
Clinical Considerations and Recommendations
MRI remains the imaging modality of choice for ACTH-secreting pituitary adenomas (HQ, SR). We suggest 3T over 1.5T MRI where available (LQ, DR). 7T MRI is not widely available and there is currently no justification for re-imaging on 7T MRI if no tumor is detected on 1.5T/3T MRI.
It is likely that functional imaging will ultimately prove a better approach than MRI alone. However, more data are needed to define use of different ligands in various clinical settings. Although advanced imaging technologies may be available in some centers of excellence, the benefit of referring all patients for further imaging beyond 3T MRI remains unknown.
Distinguishing Between CD and Ectopic ACTH-dependent CS
Background
In patients with CD, glucocorticoid (GC) receptors typically retain the ability to inhibit ACTH secretion in the presence of high dexamethasone doses, and V2 and V1b (V3R), along with CRH receptor are all overexpressed. By contrast, most (but not all) ectopic ACTH-secreting do not express these receptors. Accordingly, desmopressin and CRH stimulation testing have proven useful in distinguishing between pituitary and ectopic tumors.69–71 Increased plasma ACTH and increased cortisol following CRH or desmopressin administration usually indicates CD.72–76 Using more than one dynamic test might further improve accuracy.77 Nevertheless, well-differentiated neuroendocrine tumors (NETs) may also express any or all of these receptors, potentially leading to false-positive results. High-dose DST, although it has low accuracy overall, is still used in some countries. None of the diagnostic tests reach 100% specificity and results may be discordant in up to one-third of patients;5,6 differences in type of ectopic tumor, as well as patient age, sex, and severity of hypercortisolism can all influence outcomes.
IPSS, which measures ACTH in pituitary vs peripheral venous drainage, has long been the gold standard to reliably exclude ectopic ACTH production78,79 and should preferably be performed in a specialized center due to potential patient risk. A central-to-peripheral ACTH gradient <2 before or <3 after stimulation suggests an ectopic tumor; however, both false negatives and positives have been reported. Prolactin measurement may improve diagnostic accuracy and it is essential that patient is hypercortisolemic at the time of IPSS.80
A non-invasive approach using a combination of three or four tests, specifically CRH and desmopressin stimulation plus MRI, followed by whole-body CT if diagnosis is equivocal, correctly diagnosed CD in approximately half of patients in one series, potentially eliminating the need for IPSS.81 Interestingly, a positive CT scan despite negative CRH/desmopressin stimulation and MRI had a negative predictive value of 100%. Currently, this combination of laboratory and imaging testing as a noninvasive approach to distinguish between pituitary and ectopic ACTH-secreting tumors is likely limited to specialized centers.82
68Ga-DOTATATE is a modified (Tyr3)-octreotide molecule covalently linked to 1,4,7,10-tetra-azacyclododecane-1,4,7,10-tetra-acetic acid (DOTA) combined with the radioactive 68Ga isotope. The radiopharmaceutical, with a half-life of approximately 1 hour, binds to somatostatin receptors with affinity similar to octreotide and can be used as a tracer in PET imaging of ectopic ACTH-secreting NETs.83 68Ga-DOTATATE localizes about 65% of these tumors,84 including those not seen or not definitively identified on cross-sectional imaging, and images are sharper than with single photon 111In-DTPA-pentetreotide, with greater sensitivity for small tumors.85,86 False positives can occur due to chronic inflammation, and a positive scan does not definitively prove that the NET is the source of ACTH, but 68Ga-DOTATATE imaging can be useful in guiding clinical management.87
The 68Ga isotope is typically derived from decaying 68Ge and the worldwide supply of 68Ge is being exhausted. The 68Ga isotope, if it can be generated locally via a cyclotron, or 64Cu, which has a longer 12.7-hour half-life and can be centrally produced, may be used as alternative DOTATATE, DOTATOC, or DOTANOC conjugates.88
Clinical Considerations and Recommendations
No single laboratory test or combination of tests can absolutely differentiate between pituitary and ectopic ACTH-secreting tumors (HQ, SR). We recommend using both the clinical context and test results to guide management (HQ, SR). When prompt access to brain MRI is not available, neck-to-pelvis thin-slice CT scan is useful if suspicion is high for ectopic ACTH syndrome, such as in a male with very high UFC and/or profound hypokalemia81 (LQ, DR).
If a pituitary tumor ≥10 mm is detected on MRI and dynamic testing results are consistent with CD, IPSS is not necessary for diagnosis (MQ, SR). As it is possible that a pituitary lesion seen on MRI is an incidental nonfunctioning adenoma or other sellar mass with an ectopic ACTH source, clinical presentation should always be considered. Some studies suggest this is true for lesions >6 mm, but not all expert centers use this lower cutoff. There was consensus that all patients with lesions <6 mm should have IPSS and those with lesions of ≥10 mm do not need IPSS (MQ, SR). Expert opinions differ regarding tumors 6–9 mm, but the majority recommended IPSS to confirm the diagnosis in this circumstance (MQ, DR). Notably, some differences between centers and countries are based on interventional radiology availability. Prolactin measurement can be useful in ruling out a false negative IPSS (MQ, DR). While IPSS has high diagnostic accuracy for localization to the pituitary gland, it is not sufficiently reliable for tumor lateralization to the right or left side of the gland (MQ, SR).
A noninvasive alternative using high-dose DST and CRH stimulation test predicts CD if both tests are positive.89 However, if tests are discordant, IPSS is necessary (LQ, DR). Emerging data suggest that CRH/desmopressin testing with pituitary MRI followed by whole-body CT scan might be a reliable alternative, if assessed by an experienced multidisciplinary team (VLQ, DR).
COMPLICATIONS OF CD
Strategies for CD management should consider how comorbidities and complications associated with CD may compromise patient health and QoL. Comorbidities should be addressed in many cases concomitant with or even before CD-specific treatments to restore normal cortisol levels. Clinical considerations and recommendations are summarized in Panel 1.
Hypercoagulability
Hypercoagulability in CS resulting in increased risk of thromboembolic events (TE) is paradoxically coupled with an increased bleeding tendency due to skin atrophy and capillary fragility.90,91 Most patients show an activated coagulation cascade, including shortened activated partial thromboplastin time and increased fibrinogen, von Willebrand factor, and factor VIII, as well as impaired fibrinolysis mediated by elevated plasminogen activator inhibitor-1 and antiplasmin. Increased thrombin, thromboxane 2, and platelets, with a compensatory increase in anti-coagulation factors such as protein C and S, have also been implicated.92,93
The incidence of venous thromboembolic events (VTE) in patients with endogenous CS is more than 10-fold higher versus those with nonfunctioning adenomas undergoing surgery94 and the odds-ratio is 18-fold higher compared with the healthy population.92 VTE risk persists in the first few months after CD surgery, indicating that hypercoagulability is not immediately reversible with cortisol normalization.92,95,96 At 30 days, VTE risk post adrenalectomy was 3.4 to 4.75%,96 and the odds ratio for TE after bilateral adrenalectomy (BLA) in a longer-term study was 3.74 (95% CI: 1.69–8.27).95 In a series of 17 patients, biochemical remission following short-term medical therapy (pasireotide ± cabergoline ± ketoconazole) also did not seem to reverse the risk or induce changes in pro-anticoagulation factors; pulmonary embolism occurred in two patients with a marked UFC decrease.90,97
Data from retrospective studies98,99 indicate that thromboprophylaxis can decrease the incidence of postoperative VTE, particularly when extended to 30 days. Surveys indicate increased awareness of the need for thromboprophylaxis and increased anticoagulation use in clinical practice,100 but strategies to identify patients most likely to benefit are still being developed.101
Cardiovascular Disease
Patients with CD show an adverse cardiovascular disease risk profile that may persist even after successful treatment.103–106 Visceral, subcutaneous, and total fat may decrease after remission, although most patients remain overweight or obese.107 Type 2 diabetes mellitus (T2DM) is present in up to 30% of patients, and dyslipidemia, with low high-density lipoprotein (HDL), high low-density lipoprotein (LDL), and high triglycerides, has been reported in 16–64% of cases at diagnosis. In many patients, but not all, T2DM resolves after remission.108 Structural cardiovascular changes improve, including left ventricular hypertrophy, concentric remodeling, dilated cardiomyopathy, increased intima media thickness, and increased formation of atherosclerotic plaques, as well as their clinical manifestations, including hypertension and heart failure, but may not fully resolve despite remission of hypercortisolism.109
Myocardial infarction, stroke,110,111 and other vascular events are a primary cause of increased standardized mortality ratio (SMR; 4.1 to 16) in patients with active/persistent CD.112 Most studies show these rates do not entirely normalize,111,113 but are lowered upon remission and patients in remission after a single pituitary surgery had normal SMR at 10 years in one study.114 Screening and risk assessment for cardiovascular risk factors before and after surgery is therefore essential.102
Bone Disease
Skeletal fragility is a frequent and early complication of hypercortisolism, and fractures may be the first clinical manifestation of the disease. Vertebral fractures occur in 30–50% of patients, largely correlating with hypercortisolism severity.115 Suppression of the growth hormone (GH)/insulin-like growth factor (IGF)-I and hypothalamic-pituitary-gonadal axes as well as altered parathyroid hormone pulsatility lead to decreased osteoblast number and function, as evidenced by decreased serum levels of bone formation markers including osteocalcin and alkaline phosphatase.116 Dual X-ray absorptiometry (DXA) of the lumbar spine may show low bone mineral density (BMD), but fractures may occur even in patients with BMD in the normal or osteopenic range.117 Although BMD increases were reported after hypercortisolism resolution, some patients show persistently high fracture risk, with men at higher risk compared with women. Conventional osteoporosis treatments, e.g., bisphosphonates, as well as supportive treatment with vitamin D and calcium may induce a more rapid improvement in BMD than cortisol normalization alone, and could be useful in patients with persistent postsurgical hypercortisolism to prevent further bone loss.118 Data on the role of specific bone treatments for patients with osteopenia who are in remission after CD treatment are lacking.
Growth Hormone Deficiency
GCs, both endogenous and exogenous, inhibit GH secretion, thereby decreasing IGF-I production by the liver in patients with CS.119,120 Although GH production can be fully restored in most patients after successful therapy and recovery of HPA axis, even years after remission,121 persistence of GH deficiency (GHD) can potentially worsen hypercortisolism complications such as bone loss, myopathy, and memory deficits.122 Using the insulin tolerance or glucagon stimulation test, GHD prevalence in adults varies with timing of the diagnosis, ranging from 50–60% when testing was performed within 2 years after surgery to 8–13% when done more than 2 years after surgery.121,123 A GHD rate of 65% was observed with the GHRH-arginine test after a median remission time of 3 years post-surgery,124 while 36% of patients were diagnosed with GHD at 99 months after remission post-radiotherapy.123 Prevalence using the newly approved macimorelin stimulation test is not known.120 Notably, IGF-I is an insensitive screening test for diagnosing GHD in adults.124
Compared with other GHD etiologies, GHD in patients with CS is more common in women and younger patients; generally, these patients exhibit higher rates of T2DM, hypertension, low bone mass, fractures, and worse QoL.125–127 Myopathy may be partially related to GHD among patients in remission. While preoperative IGF-I levels during active CS did not predict long-term myopathy risk, lower 6-month postoperative IGF-I levels strongly predicted more severe long-term muscle atrophy and weakness after CS remission.128
GH replacement ameliorates a number of complications associated with metabolic syndrome and risk for cardiovascular and cerebrovascular disease. Studies show decreased body weight, waist circumference, and total and LDL-cholesterol, as well as QoL and BMD improvement. Conversely, in patients with pre-existing glucose intolerance, it may worsen glucose metabolism.125–127,129–131 GH treatment has not yet been shown in randomized, prospective trials to reverse metabolic syndrome and cardiovascular or cerebrovascular complications.126
Other Complications
Increased risk for infection,102 dysfunction of one or more pituitary axes such as central hypothyroidism,133 gonadal function impairment, infertility, and other complications may be seen in patients with CD. Physical and psychological morbidity commonly affects QoL, even after successful treatment in some patients. Persistence of several features associated with prior hypercortisolism, including affective disorders, cognitive dysfunction, and negative illness perception can have a sustained impact on well-being.134 Proximal myopathy, with impaired stair climbing and straightening up, are characteristic of CS myopathy. The pathology is multifactorial, including protein degradation through the forkhead box O3 (FOXO3) pathway as well as accumulation of intramuscular fat and inactivity-associated muscle atrophy.135 Furthermore, hypercortisolism remission can induce exacerbation of pre-existing autoimmune disorders.
As these complications have been the subject of recent guidelines136 and reviews,102,134 they were not specifically addressed at the Workshop.
INITIAL TREATMENT OF CD AND MONITORING FOR RECURRENCE
Pituitary Surgery
Background
Transsphenoidal surgery (TSS) is recommended as first-line therapy for patients with CD.6,7 Remission, typically defined as postoperative serum cortisol <55 nmol/L (<2 μg/dL), is seen in approximately 80% of patients with microadenomas and 60% with macroadenomas if the procedure is performed by an experienced surgeon.137–140 Patients in remission require GC replacement until HPA axis recovery.7,136 As remission could be delayed, monitoring until postoperative cortisol nadir can usually identify such cases.141,142 Occasional patients with mild hypercortisolism, cyclic CD, or those treated medically prior to surgery may achieve remission without marked postoperative hypocortisolism. Treatment at a high-volume center by an experienced surgeon and tumor characteristics such as detection on MRI, noninvasiveness, and size <1 cm appear to correlate with higher remission rates;138,143 whether there is a potential incremental benefit with an endoscopic approach for macroadenomas remains unclear.144,145 Overall, complication rates are low, with more experienced surgeons having even lower rates.146,147 New-onset hypopituitarism, seen in approximately 10% of patients, as well as permanent diabetes insipidus (DI), cerebrospinal fluid (CSF) leak, and VTE seen in <5% of patients, are the most common complications; peri-operative mortality is <1%.143,144
How to measure surgical expertise for CD remains unclear. Hospitals that limit the number of neurosurgeons performing TSS show better outcomes and fewer complications, shorter postoperative length of stay, and lower costs. Survey data demonstrate that neurosurgeons who have performed more than 200 TSS have the lowest complication rates.148–151 Regionalized neurosurgery teams of 4–5 experts per 2.5–5 million inhabitants could potentially allow for optimal outcomes, reduced costs, and increased quality of care overall.149,152
Clinical Considerations and Recommendations
We recommend patients with CD undergo surgery in specialized Pituitary Tumor Centers of Excellence (PTCOE) wherever possible (HQ, SR).152 Surgery should be performed by an experienced pituitary neurosurgeon and follow-up conducted by a multidisciplinary team including a pituitary endocrinologist (HQ, SR). Outcomes of pituitary surgery and cost effectiveness (LQ, DR) should be reported and be made publicly available.
Monitoring for Recurrence (Table 1)
Background
Recurrence after successful pituitary surgery is characterized as the reappearance of clinical and biochemical features of hypercortisolism following initial remission. Low or undetectable cortisol in the immediate postoperative period is a defining criterion of remission, but does not necessarily predict lack of recurrence;153 some patients who show early remission with very low postoperative cortisol levels may experience later recurrence.154 Published recurrence rates vary between 5% and 35%, with half appearing within the first 5 years after surgery and half after up to 10 years or more.137,155–157
Lifelong monitoring for recurrence is required.158 In patients who responded preoperatively to desmopressin, early postoperative loss of response to desmopressin with/without dexamethasone or CRH may predict recurrence risk,70,159–165 but is not consistently used or recommended by most experts.
Compared to their use in the initial diagnosis of CS, LNSC, 1-mg DST, UFC, and desmopressin tests have a lower sensitivity for recurrence, but specificity is high, up to 95% or more.158 LNSC can detect postoperative elevated cortisol levels earlier than 1-mg DST, while UFC is usually the last test to become abnormal in patients who recur.166,167 Thus, LNSC may allow for earlier intervention, but serial tests are advised due to wide variability in results.167–170
Evaluation for recurrence should begin after HPA axis recovery, and then annually or sooner if clinical suspicion.171,172 In practice, however, clinical manifestations and biomarkers may be discordant. Moreover, diagnosis of early recurrence presents the additional challenge about when and how to intervene with treatment.171,172
Clinical Considerations and Recommendations
We recommend lifelong monitoring for recurrence of CD (MQ, SR). Postoperative dynamic testing can potentially predict recurrence (LQ, DR), but its utility in clinical practice remains to be established as some patients with low predicted likelihood of recurrence may recur many years later.
Among the tests available, LNSC is the most sensitive for detecting recurrence and should be done annually after HPA axis recovery postoperatively (MQ, SR). LNSC usually becomes abnormal before DST and UFC,166,167 although monitoring for recurrence should also take into consideration which specific tests were abnormal for an individual patient at initial diagnosis (MQ, SR). If only slight biochemical abnormalities are seen without clinical features of hypercortisolism, close monitoring with repeat testing and treatment of comorbidities rather than treatment of the underlying disorder per se can be considered (LQ, DR).
Repeat Pituitary Surgery
Background
Repeat TSS can be considered in patients with biochemical evidence of recurrent CD with visible tumor on MRI.139,173–176 At select expert centers where successful reoperation has been reported despite a lack of detectable adenoma on MRI, either ACTH-staining adenoma on pathology or a central ACTH gradient on IPSS at initial operation was often present.174,175
Tumor factors including size and presence of extra-sellar extension should be considered regarding eligibility for reoperation, and neurosurgeon experience likely plays a role in achieving good results.155,156,177 Remission rates after reoperation vary widely in the literature, ranging from 37% to 88%, at least in part due to different remission criteria and follow-up duration.174 Although some have reported a significantly higher incidence of both surgical (e.g., CSF leak, meningitis) and endocrinological complications (e.g., DI and hypopituitarism) with repeat versus initial surgery, significant deterioration of pituitary function or serious morbidity is less likely in experienced hands.155,156
Clinical Considerations and Recommendations
If there are no contraindications for surgery, we suggest repeat TSS in patients with biochemical evidence of recurrent CD if tumor is evident on MRI, especially if the first surgery was not done in a PTCOE (LQ, DR). If MRI does not show tumor presence, reoperation may be appropriate if an experienced surgeon at a high-volume center considers it feasible and positive pathology or a central gradient on IPSS was seen before initial operation (LQ, DR).
MEDICAL THERAPY FOR CD
Drugs used for treatment of CD target adrenal steroidogenesis, somatostatin and dopamine receptors in the pituitary, and GC receptors.6,7,178 They may be used to treat hypercortisolism in patients with persistent or recurrent CD and those who are not candidates or refuse surgery, and to control cortisol levels in patients undergoing radiation therapy (RT).139,179,180 Available medications and investigational drugs that reported phase 3 trial results are described in Table 2.
Medical Therapy: Targeting Adrenal Steroidogenesis
Background
Adrenal steroidogenesis inhibitors that have been available for many years, including ketoconazole, metyrapone, mitotane, and etomidate, as well as the recently approved osilodrostat, block one or more adrenal enzymes, decreasing GC synthesis and/or adrenal androgen production and secretion.181 They are effective in controlling cortisol excess, but do not directly target the pituitary ACTH-secreting adenoma, nor restore HPA axis circadian rhythm.182
When treatment is dose-titrated to achieve cortisol normalization, there is a risk of adrenal insufficiency (AI) with overtreatment. Alternatively, for patients treated with a block-and-replace regimen, there is a risk of inappropriate GC over-replacement if blockade is incomplete.180 Some adverse events (AEs) relate to ACTH increase in CD patients and buildup of adrenal hormones proximal to the blockade with mineralocorticoid or androgenic activity. Potential AEs related to drug-drug interactions are a key factor in treatment selection and use.183
Ketoconazole
Ketoconazole blocks multiple adrenal enzymes, including those involved early in the steroid biosynthetic pathway. This avoids excess circulation of androgen and mineralocorticoid precursors, but it may also decrease gonadal steroid synthesis; men may experience hypogonadism and gynecomastia, which can limit prolonged treatment.184 Review of 310 CS patients treated in 5 studies with a mean dose of 673.9 mg/d and followed for a mean of 12.6 months showed UFC normalization in 64.3% (median 50%; range 44.7–92.9%), but up to 23% of initially responsive patients lost biochemical control and escaped.179 Similarly, data derived from the largest retrospective study of 200 patients with CD who took ketoconazole showed that 64.7% of 51 patients treated for more than 24 months with a mean dose of 600 mg/d normalized UFC levels, but 15.4% escaped.185 Improvement in clinical features of CS has also been seen, including decreased body weight and blood pressure, improved glucose metabolism, and decreased muscle weakness.179
Hepatotoxicity, seen in 10–20% of patients, is mostly asymptomatic with mild or moderate increases in liver enzymes (≤5 × ULN)186 and typically appears within the first 6 months of treatment; these seem not to be dose-dependent and reverse within 2–12 weeks after dose decrease or discontinuation. However, as serious hepatotoxicity has been reported, in patients without obvious risk factors, the United States Food and Drug Administration (FDA) introduced a black-box warning and recommends weekly monitoring of liver function tests (LFTs) in patients with fungal infections treated with ketoconazole. Of note, ketoconazole use for CS is off-label in the US. Gastrointestinal disturbances and AI are also common, seen in 5–20% of patients, and skin rash is observed in approximately 5%.179 There are a number of drug-drug interactions with ketoconazole; careful review of the patient’s medication list for potentially problematic interactions is essential.
Metyrapone
Treatment with the 11β-hydroxylase inhibitor metyrapone in 120 CS patients (5 studies; mean dose 2127.5 mg/d, mean follow-up 8.7 months) showed UFC normalization in 71% (median 75.5%; range 45.4–100%), with up to 18% escaping after initial response.179 A subsequent retrospective multicenter study of 164 CS patients reported that 43% achieved biochemical control with a mean of 8 months monotherapy, at a mean starting dose of 1040 mg/d and escalating to 1425 mg/d.187 An observational study of 31 CS patients, including 20 with CD, demonstrated that a median dose of 1000 mg/d for 9 months induced a rapid decrease in both UFC and LNSC after the first month of treatment (−67 and −57%, respectively, from baseline), with sustained normalization in 70% and 37% of patients, respectively, at last visit.188 Three patients exhibited loss of control at 9 months despite normal UFC levels at 6 months and 2 patients also showed normal LNSC. Notably, 11-deoxycortisol may produce clinically relevant cross-reactivity with cortisol in both blood and urine immunoassays.189 A recently presented multicenter prospective study of 50 patients with CS showed 47% had UFC normalization at 12 weeks; median metyrapone dose was 1500 mg/day (250; 5750) and AI was reported in 12% of patients.190
Patients treated with metyrapone typically show a general improvement in clinical features of CS (66% in the prospective study), such as blood pressure, glucose metabolism, psychiatric disturbances, and muscle weakness.179
Hirsutism, dizziness, arthralgia, fatigue, hypokalemia, and nausea are the most commonly reported AEs with metyrapone; AI, abdominal pain, and atopic dermatitis are less frequently reported.179 AEs secondary to hyperandrogenism can limit prolonged treatment, especially in females.
Osilodrostat
Proof-of-concept and phase 2 prospective studies showed that osilodrostat, an 11β-hydroxylase and aldosterone synthase inhibitor, was effective in reducing cortisol and was well-tolerated.191–193 This was further evaluated in 137 CD patients enrolled in a phase 3, prospective, multicenter, double-blind randomized withdrawal study.194 After 12 weeks of open-label dose-titrated and another 12 weeks of open-label dose-optimized treatment, 72 patients (53%) had maintained normal UFC and were eligible for randomization. By week 34, at the end of the randomized treatment period, 86% of those randomized to osilodrostat maintained normal UFC versus 29% of those randomized to placebo (OR 13.7 [95% CI: 3.7, 53.4]; p<0.0001).
Treatment with osilodrostat also yielded clinical improvements. By week 48, patients demonstrated significant decreases in body weight, blood pressure, total and LDL cholesterol, and decreased fasting serum glucose and HbA1c levels. QoL and depression scores also improved.194
Nausea, anemia, and headache were reported in 8–11% of patients, while AEs related to hypocortisolism were reported in about half of patients, mostly during the open-label dose-titration period. These were generally manageable with dose reductions or interruptions, although GC replacement was required in 25 of 70 (36%) patients with one or more hypocortisolism-related AE. In addition, 42% of treated patients in the phase 3 study showed effects from increased levels of adrenal steroid precursors, including hypokalemia and hypertension; 11% of women reported hirsutism.194 In another large prospective phase 3 study, a significantly greater proportion of patients receiving osilodrostat (77.1%) achieved mean UFC ≤ ULN after 12 weeks of treatment versus placebo (8.0%), with improvements seen in clinical features, cardiovascular disease markers, and QoL. Interestingly, hypocortisolism-related AEs occurred in 27.4% of patients, far fewer than in the prior study.195
Mitotane
Mitotane inhibits several steroidogenic enzymes and has a long-lasting adrenolytic action in steroid-secreting adrenocortical cells. It suppresses hypercortisolism in 80% of cases, but with a slow onset of action and highly variable bioavailability.180 Induction of CYP3A4-mediated rapid inactivation of cortisol leads to a requirement for a 2- to 3-fold increased GC replacement dose when treatment of AI is needed or with a block-and-replace strategy.196 It is rarely used for CD. Most participants considered that use of mitotane should be limited to patients with adrenal carcinoma.
Etomidate
Originally developed as an anesthetic, etomidate was shown to rapidly normalize cortisol levels, leading to use for acute control of severe hypercortisolism in hospitalized patients.198 Low-dose etomidate (0.04–0.05 mg/kg/h) produces partial blockade; a high-dose (0.5–1 mg/kg/h) provides for complete blockade, with IV hydrocortisone used to avoid etomidate-induced AI.199 Very low doses (0.025 mg/kg/h) may be used in hospitalized patients outside ICU,200 although this may depend on local practice.
Compared with the lipid formulation, the propylene glycol preparation is more frequently associated with thrombophlebitis and pain on injection, and also with additional AEs, such as hemolysis and renal tubular injury, as well as lactic acidosis at high doses.199
Medical Therapy: Targeting Pituitary Somatostatin and Dopamine Receptors
Background
Both the dopamine agonist cabergoline and the somatostatin receptor ligand pasireotide are used in CD patients with persistent or recurrent hypercortisolism,7,139,179 although only pasireotide is approved for use in this population.7,201,202 Tumor effect is clinically important for patients with a large residual tumor as well as for patients with corticotroph tumor progression, or Nelson’s syndrome.
Pasireotide
In a phase 3 study of 162 CD patients treated with SC pasireotide, UFC normalized at month 6 in 15–26% of without dose increases. Higher rates of UFC normalization were seen in those with baseline UFC <5 × ULN201 and significant clinical improvement was noted in most patients.202
A second phase 3 study treated 150 CD patients with 10 mg or 30 mg monthly IM pasireotide LAR. At month 7, 40% of patients in both groups showed normalized UFC regardless of dose titration, with higher response in those with baseline UFC <2 × ULN.203 At month 12, improvements in blood pressure were greater in those with normalized UFC; BMI, weight, waist circumference, and QoL were all improved regardless of UFC control.204 Long-term extension studies showed that biochemical and clinical improvements could be maintained for up to five years in select patients who continued the study.205,206 Of note, in real-life settings, limited data are available on long-term treatment compliance, and several studies show a high rate of treatment discontinuation. Treatment with pasireotide LAR also decreased median tumor volume by 17.8% on 10 mg and 16.3% on 30 mg, with 43% and 47% of patients, respectively, showing ≥20% reduction.203
Of note, a separate longitudinal study in CD patients with Nelson’s syndrome after BLA showed that pasireotide LAR rapidly suppressed ACTH levels and yielded sustained reductions over 24 weeks.207
Between one- and two-thirds of CD tumors harbor a mutation in USP8,208,209 and these mutated tumors may show higher SST5 expression compared with wild-type tumors.210,211 As pasireotide has a high affinity for this receptor, USP8 mutational status may prove a useful marker for predicting treatment response.
The risk for hyperglycemia is high with pasireotide.201,203,212 In the two phase 3 studies, approximately 70% of patients reported hyperglycemia-related AEs, with new antidiabetic medication initiation or dose adjustments required in approximately half of patients.201,203 The high rates of hyperglycemia are thought to result from inhibition of insulin and incretin secretion combined with a lesser degree of glucagon inhibition.213 Management with GLP-1 receptor agonists or DDP-4 inhibitors is therefore thought to be useful.214,215
Cabergoline
Available data in CD are derived mostly from small retrospective studies demonstrating biochemical normalization in 25–40% of patients, with loss of control in 20–40% initially normalized.216,217
A retrospective, multicenter cohort study of 53 patients treated with a median cabergoline dose of 2.3 mg/wk (range, 0.5–6.0) yielded normal UFC in 40% of patients during the first year, but only 23% of those showed sustained UFC normalization after a median 32.5 months follow-up.218 The lower control rate may be due to under-titration, as a smaller study of 20 patients on cabergoline titrated to maximum of 7 mg/wk (median 3.5 mg/wk) showed normalized UFC in 40% of patients at 24 months.219 Weight, glycemic control, and hypertension improved in 25–40% of complete responders,218 and tumor shrinkage was reported in 50%.219 Patients with Nelson’s syndrome may also respond to cabergoline, and both ACTH normalization and tumor shrinkage have been reported.220 Although not approved in this setting, cabergoline has been used in pregnant patients with prolactinomas and other pituitary adenomas, including CD.
Cabergoline-induced impulse-control disorder is likely under-reported, and can manifest as hypersexuality, pathological gambling, excessive alcohol consumption, overeating, and uncontrolled shopping.221 This behavior may occur within months of initiating cabergoline therapy, or may manifest later, and improves or resolves on treatment discontinuation.222,223
High cumulative doses of ergotamine-derived dopamine agonists used in patients with Parkinson’s disease were associated with risk for cardiac valve regurgitation.224 Although one study in prolactinomas found that moderate tricuspid regurgitation was more frequent with higher doses,225 a large multicenter study found no association between the cumulative cabergoline dose and age-corrected prevalence of any valvular abnormality.226 Furthermore, a meta-analysis showed that it remains an open question whether such echocardiographic findings are clinically significant.227
Medical Therapy: Targeting the Peripheral Tissue Glucocorticoid Receptor
Mifepristone
The glucocorticoid receptor blocker mifepristone is effective in controlling some effects of hypercortisolism regardless of etiology.
An open-label study of 50 patients with endogenous CS, including 43 with CD, showed that after 24 weeks of treatment, 60% with a concurrent diagnosis of T2DM or impaired glucose tolerance had a significant reduction of ≥25% from baseline in area under the curve for glucose during an oral glucose tolerance test, and 38% with hypertension showed a significant reduction of ≥5 mm Hg in diastolic blood pressure. Insulin resistance, weight, waist circumference, and QoL also improved.228
Twelve patients showed increased blood pressure, including 9 with hypokalemia who required spironolactone, consistent with mineralocorticoid receptor activation. Endometrial hypertrophy and irregular menstrual bleeding were also reported, consistent with the anti-progesterone activity of this medication. Dexamethasone was administered in 7 patients with signs and symptoms of AI, underscoring the need for careful monitoring.228 Importantly, cortisol levels remain high, and measures of low cortisol typically used to confirm AI due to overtreatment with other medical therapies cannot be used with mifepristone. Rather, only clinical features can be used.229
Continued mifepristone treatment of 27 patients with CD included in a long-term extension study showed sustained ≥2-fold ACTH elevations, but tumor volume progression, seen in 3 patients with macroadenomas up to 25 months from baseline, did not correlate with ACTH increases.230 Thyroid function should be closely monitored and thyroid hormone replacement adjusted as needed.231 All concomitant medications should be carefully reviewed given the potential for drug-drug interactions with mifepristone.
Medical Therapy: Clinical Considerations and Recommendations
We recommend individualizing medical therapy for all patients with CD based on the clinical scenario, including severity of hypercortisolism. Regulatory approvals, treatment availability, and drug costs vary between countries and determine treatment selection. However, where possible, it is important to consider balancing cost of treatment with the cost and significant adverse consequences of ineffective or insufficient treatment. In patients with severe disease, the primary goal is to treat aggressively to normalize cortisol levels (or cortisol action if using mifepristone). Multiple serial tests of both UFC and LNSC are used to monitor treatment outcomes.158,232,233
A brief summary of Workshop discussions about how to best incorporate each of the different treatment options is presented below and in Panel 2.
Initial treatment selection for medical therapy
Adrenal steroidogenesis inhibitors are usually used first given their reliable effectiveness. For patients with mild disease and no visible tumor on MRI, ketoconazole, osilodrostat, or metyrapone are typically preferred. Cabergoline also may be used for mild CD; it is less effective and has a slower onset of action, but requires less frequent dosing. For patients with mild-to-moderate disease and some residual tumor, there may be a preference for cabergoline or pasireotide because of the potential for tumor shrinkage. However, the high rate of hyperglycemia with pasireotide would make patient selection critical.
For patients with severe disease, rapid normalization of cortisol is the most important goal. With osilodrostat and metyrapone, response will typically be seen within hours, and with ketoconazole within a few days. Etomidate also works rapidly and could be used if the patient is hospitalized and cannot take oral medications. For patients with severe hypercortisolism, combinations of steroidogenesis inhibitors may be necessary. However, if hypercortisolism is very severe and not responsive to optimized medical therapy, including combinations, BLA should be considered to avoid worsening outcomes.
Other patient factors can be important for initial treatment selection. For example, cabergoline should not be used in patients with a history of bipolar or impulse control disorder, but may be preferred in a young woman desiring pregnancy. Although none of these drugs is specifically approved for use in pregnancy, metyrapone may be considered with precautions in selected women who are pregnant. In such cases, given the higher normal cortisol levels during pregnancy, a higher cut-off target for cortisol, such as 1.5 × ULN, is used.
Mifepristone improves key clinical features associated with hypercortisolism, specifically hyperglycemia and weight gain. However, it could be challenging to use in standard clinical practice, and often worsens hypokalemia. There are no reliable biochemical markers for monitoring cortisol levels, increasing the risk for AI due to overtreatment, and its long half-life requires several days of stress-dose GC replacement, preferably dexamethasone, if AI ensues. Because cortisol measurements not helpful for dosing or safety monitoring, mifepristone should be used only by clinicians with extensive experience in CD; counseling patients that cortisol levels monitoring is not reliable, especially for AI, is also important.
There are few rigorous data supporting specific regimens for combination therapy, but several have been described 234–236. Many experts consider combining ketoconazole with metyrapone to maximize adrenal blockade when monotherapy is not effective or to allow lower doses of both drugs, although a steroidogenesis inhibitor plus a tumor-targeting agent, such as ketoconazole plus cabergoline, is also a rational combination, especially if visible tumor is present. Other combinations that may be used include triplets of cabergoline, pasireotide, plus ketoconazole, and metyrapone, ketoconazole, plus mitotane. Risk for potentiating adverse effects with combination therapy, such as QTc prolongation, should also be considered.
Selecting an adrenal steroidogenesis inhibitor
The longest clinical experience for adrenal steroidogenesis inhibitors is with ketoconazole and metyrapone. These agents are approved for use in CD in Europe, but not in the United States (where only osilodrostat is approved in this category), and they may not be available in some countries. Ketoconazole may be favored for ease of dose titration, but it is often under-dosed for fear of inducing hepatotoxicity. LFTs should be regularly monitored, but treatment does not necessarily have to be discontinued if LFTs are mildly elevated, yet stable.237 Osilodrostat and metyrapone can induce rapid control in the majority of patients. They are not limited by monitoring of LFTs and hypogonadism does not occur in men. It is expected that osilodrostat will be increasingly used as it becomes widely available given its high efficacy and twice-daily dosing. It is necessary to monitor for AI and osilodrostat effects on androgens, but whether treatment selection should be based on patient sex in long-term treatment is not yet known. Mitotane, rarely used for patients with CD in most centers, has a slower onset of action.
A block-and-replace regimen may be considered for patients with severe disease, cyclical CS, and patients ineligible for surgery. This may be a particularly useful approach if monitoring visits are infrequent due to external factors such as pandemic, lack of transportation or other issues. Caution is needed to avoid GC over-replacement and inducing iatrogenic CS.
Monitoring response to medical therapy
For all patients, regular monitoring for treatment efficacy is required, including measures of cortisol (except with mifepristone) and patient symptoms and comorbidities, especially weight, glycemia, and blood pressure. In addition, QoL is important to take into account, preferably through patient-reported outcomes. Cortisol levels are often measured by UFC; notably, this test is not useful for AI diagnosis. Morning cortisol and/or LNSC may be used as an alternative, but because of the loss of circadian rhythm, it is unclear whether targeting diurnal secretion alone is meaningful. Nevertheless, morning cortisol values may be especially pertinent in patients taking higher medication doses in the evening versus morning.182 Patients who normalized both UFC and LNSC with pasireotide LAR showed better clinical outcomes than those who normalized UFC alone,232 and a higher treatment dose at bedtime for twice daily medications may help restore circadian rhythm patterns, but there is no rigorous evidence to support the latter approach.
As designs, medication up-titration schemes, comparator arms, inclusion/exclusion criteria, and primary endpoints differ even among prospective studies, it is difficult to directly compare treatment outcomes, either for efficacy or for adverse effects. Furthermore, some drugs have not been prospectively studied for CS. When using UFC normalization as a target, osilodrostat has the highest efficacy based on data from several prospective clinical trials, followed by metyrapone (retrospective and prospective data), ketoconazole (retrospective data), pasireotide (prospective), and cabergoline (retrospective and prospective). As improvement in clinical features of CS and diabetes are used as markers of mifepristone efficacy, it cannot be directly compared for biochemical efficacy with other available treatments.
Change in treatment should be considered if cortisol levels are persistently elevated after 2–3 months on maximum tolerated doses. If cortisol does not normalize but is reduced and/or there is some clinical improvement, combination therapy can be considered. If there is clear resistance to treatment, we suggest switching to a different therapy. However, it is important to ensure that insufficient disease control due to under-dosing is not misinterpreted as treatment resistance.
With adrenal-targeting agents, there may be concern for tumor growth due to ACTH-cortisol feedback interruption. However, it can be difficult to determine whether such tumor progression is due to this loss of feedback or reflects the underlying behavior of aggressive, recurrent disease. We suggest monitoring ACTH levels, as significant elevations may portend new tumor growth and a need for MRI, with the important caveats that ACTH has a short half-life and levels fluctuate and so may not necessarily reflect tumor growth. If progressive increase in tumor size is seen,238 treatment should be suspended and management reassessed. MRI is typically done 6–12 months after initiating treatment and repeated every few years depending on the clinical scenario.
With combination therapies, it is also important to monitor for potential overlapping toxicities, particularly QTc prolongation, as well as drug-drug interactions.
Primary and Preoperative Medical Therapy for De Novo CD
Primary medical therapy is used when successful adenoma resection is unlikely due to unfavorable localization, significant invasiveness, or lack of visualization on MRI. Recent double-blind randomized phase 3 studies evaluating the efficacy of several novel drugs included only a small percentage of patients with de novo CD, ranging from 0% to 28%.196 Further studies are needed to demonstrate utility of the different medical therapies in this setting, either as monotherapy or in combination, while also taking into account the potential effects of such treatment on adenoma size.
Published evidence regarding preoperative medical therapy in patients with CD is sparse, and it is not used in most patients, although there are regional variations. A meta-analysis showed no differences in cortisol normalization rate between those who received cortisol-lowering medications in the preoperative setting versus later use as adjuvant treatment.239 It may be an option in severely ill patients for whom surgery is contraindicated or if waiting time for surgery is long139 or in patients with life-threatening complications of hypercortisolism requiring rapid cortisol control.230,240 Physician surveys show that preoperative therapy, mostly with ketoconazole and/or metyrapone, is used in up to 20% of CD patients, especially those with more severe clinical features or nonvisible adenoma.241
Retrospective studies show preoperative steroidogenesis inhibitor therapy for a mean of 4 months yields cortisol normalization rates of 50–72%, although subjective symptom improvement was observed in only one-third of cases.185,187 Lower rates of postoperative hypoadrenalism from preoperative medical therapy could, in theory, protect against the occurrence of a proinflammatory and procoagulant state,94,241 but postsurgical complications, including VTE, are similar regardless of its use.241 If the HPA axis recovers during preoperative treatment, AI may not occur postoperatively, so it may be more difficult to determine whether remission is present.
Preoperative cabergoline likely has limited value, as a significant decrease in cortisol was seen in only one-fourth of patients in a cohort treated prospectively for 6 weeks.242
Clinical Considerations and Recommendations
There are no rigorous data supporting use of primary or preoperative medical therapy. Most experts would consider such an approach with adrenal steroidogenesis inhibitors if surgery is delayed, either because of scheduling or due to outside factors such as a pandemic (VLQ, DR).
Patients with severe CD who have potentially life-threatening metabolic, psychiatric, infectious, or cardiovascular/thromboembolic complications also may benefit from preoperative medical therapy in select cases (LQ, DR). Although this has not been clearly confirmed, some experts consider it may have a potentially favorable effect on glucose, cardiovascular, and coagulation parameters (VLQ, DR). Few use it to decrease the extent of postoperative cortisol withdrawal manifestations.
Monitoring and follow-up of patients treated with preoperative therapy can be challenging as postoperative cortisol assessments for surgical cure are not reliable. The patient’s perspective regarding this approach would be valuable to incorporate into future research studies (VLQ, DR).
RADIATION THERAPY
Background
RT is primarily used as adjuvant therapy for patients with persistent or recurrent disease after TSS 7,243 or for aggressive tumor growth. Approximately two-thirds of patients achieve biochemical remission during the years after treatment with conventional external-beam RT, typically 45–50 Gy administered in <2 Gy fractions, or stereotactic radiosurgery (SRS), which is administered as single dose or a few fractions of approximately 20 Gy.244 However, more recent series with SRS, including whole sellar RT,245 show higher biochemical remission rates. In a multicenter study of GammaKnife SRS in 278 subjects followed for a mean of 5.6 years, biochemical control was attained in 80% and durable hypercortisolism control was maintained in 57%.246 Tumor control rates are typically higher, with approximately 95% of patients treated with SRS showing decreased or stable tumor volume on MRI.244 A small single-center study of proton beam RT showed complete response (either cortisol or ACTH normalization) in patients with persistent corticotroph adenomas due to CD or Nelson’s syndrome, with low morbidity after a median follow-up of 62 months.247
SRS may also be used as primary therapy in patients with high surgical risk or who refuse surgery. In this setting, endocrine remission was attained in 81% of 46 patients at 5 years of follow-up.248 Long-term follow-up is needed as recurrence and tumor growth have been described post-RT.
Given the latency until post-RT remission, adjuvant medical therapy is needed to control hypercortisolism; periodic withdrawal allows cortisol secretion evaluation to assess treatment effect.7 Although data are mixed on whether ketoconazole246,249 or cabergoline250 treatment at the time of SRS limits efficacy, they are often withheld temporarily at the time of RT.
Hypopituitarism is the most common side effect of both conventional RT and SRS, seen in 25–50% of patients, and generally increases over time. Risk of secondary malignancy, cranial nerve damage, and stroke are low with SRS.251 In patients treated with SRS, distance of at least 3–5 mm between the tumor and the optic chiasm and a chiasm dose <8 Gy is recommended to limit treatment damage.251 Longer term data will help address whether use of different SRS modalities (GammaKnife, LINAC, proton beam) confers lower rates of stroke and hypopituitarism compared with conventional RT.252
Clinical Considerations and Recommendations
RT is most commonly used in cases of persistent hypercortisolism after incomplete corticotroph tumor resection, particularly if the tumor is aggressive or invasive and/or considered unresectable (HQ, SR). SRS is likely more convenient as few treatment sessions are required, but avoiding optic chiasm exposure is critical (HQ, SR). Lifelong monitoring for pituitary hormone deficiencies and recurrence is required in all patients undergoing RT (HQ, SR). Imaging for secondary neoplasia in the radiation field also should be considered (HQ, SR).
ADRENALECTOMY
Background
BLA offers immediate control of cortisol excess in patients with persistent or recurrent CD not responsive to medical therapy,7,139,253 but is only considered for select patients due to the resultant AI and need for life-long GC and mineralocorticoid replacement therapy.254 Laparoscopic BLA using either a transperitoneal or posterior retroperitoneal approach is associated with a 10–18% complication rate in the largest series, and a mortality rate <1%.255,256 Long-term clinical relapse of hypercortisolism due to adrenal rest stimulation by high ACTH is uncommon (<10%), while clinical improvement in BMI, T2DM, hypertension, and muscle weakness is reported in more than 80%.257
Corticotroph tumor progression after BLA is a long-term concern in 25–40% of patients after 5 to 10 years.257–259 Most cases can be managed with surgery, RT, or medical therapy. However, a subset of aggressive tumors will continue to grow and long-term monitoring is required. A European consensus focused on management of these patients was recently published.260
Corticotroph tumor progression after BLA does not seem to be influenced by pregnancy.261 This may make BLA a preferred option in female patients with an immediate pregnancy plan. In most cases, however, BLA is rarely performed as the first-line treatment after failure of initial pituitary surgery, and duration of disease before adrenal surgery is typically 3 to 4 years or more.256 Whether and how this might impact long-term treatment outcomes remains unknown.
Clinical Considerations and Recommendations
In patients with CD, BLA is often considered a treatment of last resort in most centers after all other options have failed (MQ, SR). However, BLA may be warranted earlier in patients with severe hypercortisolism in whom a rapid, definitive effect on cortisol is needed to avoid prolonged systemic effects of uncontrolled disease (MQ, SR). Many expert centers recommend BLA earlier in the course of the disease for females with CD desiring pregnancy (MQ, SR).
After BLA, plasma ACTH and serial pituitary imaging are used for monitoring at intervals dictated by the clinical scenario, usually starting 6 months after surgery (HQ, SR). More frequent evaluation may be necessary if there is a clinical suspicion of corticotroph tumor progression (HQ, SR).
ADDITIONAL CONSIDERATIONS
Genetics of CD
Corticotroph adenomas are predominantly of sporadic origin, based on a monoclonal expansion of a singular mutated cell.262 These adenomas abundantly express EGFR, which signals to induce ACTH production.263 Somatic activating driver mutations in USP8 are present in 36–60% of corticotroph adenomas.209 These mutations lead to persistent overexpression of EGFR, thereby perpetuating the hyper-synthesis of ACTH. Rarely, mutations in the glucocorticoid receptor NR3C1, the BRAF oncogene, the deubiquitinase USP48, and TP53 are encountered.262 Patients with familial tumor syndromes, such as MEN1, FIPA, and DICER1 rarely develop corticotroph adenomas. It has been proposed that corticotroph tumors may be sub-classified based on USP8 driver mutations and clinical behavior.264 As USP8 mutational status may predict recurrence after TSS,265 such genomic classifications may open new avenues for more targeted, personalized treatment modalities in the future.
Diagnosis and Management of CS in Children
Endogenous CS is extraordinarily rare before age 18. Germline mutations in MEN1, RET, AIP, PRKAR1A, CDKN1B, DICER1, SDHx, and CABLES1 may all predispose children to CD, although screening is usually reserved for cases in which there is either family history or other signs suggestive of a genetic syndrome.266
Lack of height gain concomitant with weight gain is the most common CS presentation in children, making the disorder somewhat easier to detect compared with post-pubertal adolescents or adults. Using the insulin tolerance or glucagon stimulation test, prevalence of severe GHD (< 9 mU/L) and partial GHD (<30 mU/L) is estimated at 31% and 54%, respectively.267
Documentation of hypercortisolism with 24-hour UFC, LNSC, or overnight 1 mg DST are all used to confirm diagnosis. The diagnostic approach and test performances are slightly different from adults, as recently extensively reviewed.268 The Dex-CRH test is not useful in children. In children over age 6, CD is the most common cause of CS, while adrenal causes are more common in younger children. Algorithms for testing to distinguish ACTH-dependent from ACTH-independent CS are available. Notably IPSS role in children is more limited compared with adults.269
As in adults, surgical resection of the ACTH-secreting tumor is the first-line treatment. However, unlike in adults, thromboprophylaxis should not be routinely used due to bleeding risk, but reserved for selected pediatric patients. With successful treatment, adrenal function typically recovers within approximately 12 months.270 Evaluation for GHD should be done by 3–6 months postoperatively and immediate GH replacement given if needed to ensure proper growth; GH replacement ensures adequate final height, but obesity is not fully reversible.271 For those requiring medical therapy, ketoconazole or metyrapone is typically used with morning cortisol for monitoring response. Pasireotide is not recommended and clinical trials of osilodrostat in children are underway. Block-and-replace regimens with metyrapone also may be considered.
Early diagnosis and expert management are critical given the potential for long-term adverse health outcomes from prolonged hypercortisolism as well as from morbidity associated with TSS or RT. Children with CS should be referred to multidisciplinary centers of excellence with pediatric endocrinologists expert in managing disorders of the pituitary, and with specialized neurosurgery units. If an underlying genetic syndrome is present, genetic counseling for the child and family members as well as investigations into other disorders associated with the syndrome are necessary.268,272,273
Supplementary Material
Acknowledgments
The authors thank Professors Annamaria Colao, Richard Feelders, and A.J. van der Lely for their contributions to the meeting. The authors thank Shira Berman of Cedars-Sinai Medical Center for editorial assistance.
Declaration of Interests
MF has received grants to the institution from Novartis, Strongbridge, Novo Nordisk, Crinetics, Millendo, Ascendis, and Pfizer, and personal honoraria for consulting and advisory boards from Crinetics, HRA Pharma, Novartis, Recordati, Strongbridge, Sparrow, Ascendis, Novo Nordisk, and Pfizer, and has served on the Board for Pituitary Society.
RA has received grants to the institution from Strongbridge Biopharma, Novartis Pharmaceuticals, and Corcept Therapeutics, and personal honoraria for consulting and advisory boards from Strongbridge Biopharma, Recordati Rare Diseases, Corcept Therapeutics, and Novartis Pharmaceuticals.
IB has received grants and fees for consulting, advisory boards, and authorship to the institution from the National Institutes of Health, Strongbridge, Corcept, HRA Pharma, Sparrow Pharmaceutics, Adrenas Pharmaceutics, and Elsevier, and non-financial support to the institution from HRA Pharma.
AB-S has received personal honoraria for advisory boards from Recordati.
JB has received grants to the institution from Novartis, HRA Pharma, and Recordati, and personal honoraria for consulting, lectures, and meeting attendance from Novartis, HRA Pharma, Ipsen, and Recordati.
NB has served on the Board or as an advisor for European Neuroendocrine Association and the European Reference Network on Rare Endocrine Conditions.
CLB has received grants and personal honoraria for lectures from Novartis and served on the Board or as an advisor for Sociedade Brasileira de Endocrinologia e Metabologia, Endocrine Society, and European Society Of Endocrinology.
MDB has nothing to declare.
MB has nothing to declare.
JDC has received grants to the institution from Novartis, Strongbridge, and Crinetics, personal honoraria for consulting and authorship from Recordati, Novo Nordisk, Corcept, and Merck Manual, and served on the Board or as an advisor for Pituitary Society, Endocrine Society, and American Association of Clinical Endocrinologists.
FFC has served on the Board for Pituitary Society.
FC has received personal honoraria for consulting, lectures, and support for meeting attendance from Recordati Rare Diseases, Ipsen, and HRA Pharma.
PC has received grants and honoraria to the institution for consulting and lectures from Novartis and Recordati.
JF has received grants to the institution from Novartis and personal honoraria for consulting and advisory boards from Corcept, Recordati, and Novartis.
MG has received non-financial support from Novartis and Recordati and served on the Board or as an advisor for Pituitary Society and Brazilian Society of Endocrinology and Metabolism.
EBG has received grants and personal honoraria for consulting, lectures, and advisory boards from Novartis, Corcept, Strongbridge, Bristol-Myers Squibb, Recordati, and HRA Pharma, and has served as an advisor for Cushing’s Support & Research Foundation.
AG has received grants to the institution from Pfizer and personal honoraria for consulting and advisory boards from Abiogen, Novo Nordisk, and Recordati, and has served on the Board or as an advisor to European Society of Endocrinology and Glucocorticoid Induced Osteoporosis Skeletal Endocrinology Group.
AG has served as an advisor to Novartis and as an editor for Neuroendocrinology and Journal of Neuroendocrinology.
MG has received personal honoraria for consulting and lectures from Recordati Rare Diseases UK, HRA Pharma, and Ipsen, and is a Board member or advisor for UK Society for Endocrinology and European Society of Endocrinology.
KH has nothing to declare.
AGI has received grants to the institution from Recordati, Novartis, and Strongbridge, and personal honoraria for consulting from Recordati and Strongbridge.
UBK has received grants as co-investigator from Corcept, and personal honoraria for consulting, advisory board, and lectures from Acerus Pharmaceuticals and Novo Nordisk, and serves as editor for Journal of Clinical Endocrinology and Metabolism and as Board member for Endocrine Society.
NK has received personal honoraria for consulting from Recordati Rare Diseases and HRA Pharma, and has served as Board member or advisor for Pituitary Society, European Neuroendocrine Association, Endocrine Society, and European Society of Endocrinology.
LK has received personal honoraria for consulting from Strongbridge and Recordati.
DFK has received royalties from Mizuho, Inc..
AL has received grants from Recordati and Corcept, personal honoraria for lectures, support for meeting attendance, and advisory boards from Pfizer, Ipsen, Corcept, and the European Journal of Endocrinology, and royalties from UpToDate Endocrinology, and served as a Board member for International Society of Endocrinology.
AM has received grants and personal honoraria for lectures/presentations and support for meeting attendance from Pfizer, Ipsen, Novartis, and Novo Nordisk, and served as a Council member for Endocrine Society of Australia.
SM has received grants to the institution from US Food and Drug Administration and nonfinancial support from Cyclacel.
MM has received grants to the institution from Corcept, Novartis, and Strongbridge, fees for expert testimony from Janssen and Corcept, personal honoraria for advisory boards for Merck, Pfizer, Recordati, and Strongbridge.
PM has nothing to declare.
JN-P has received grants and honoraria to the institution for consulting and advisory boards from Diurnal Ltd and HRA Pharma, Recordati, and Novartis, and served as a Board member or advisor for Pituitary Foundation and Endocrine Society.
LN has received royalties from UpToDate and support for meeting attendance from the National Institutes of Health, and has served as a Board member for Endocrine Society.
AMP has received grants to the institution from HRA Pharma and served as advisor for European Reference Network on rare Endocrine Conditions and European Endocrine Society.
SP has received personal honoraria for lectures and advisory boards from HRA Pharma, Recordati Pharma, Novartis Pharma, and Crinetics Pharmaceuticals.
RP has received grants to the institution from Novartis, Pfizer, Ipsen, Shire, IBSA Farmaceutici, HRA Pharma, Cortendo AB, Corcept Therapeutics, and Merck Serono, personal honoraria for consulting, lectures, support for meeting attendance, and advisory boards from Novartis, Shire, HRA Pharma, Cortendo AB, Pfizer, Recordati, IBSA Farmaceutici, and Crinetics Pharmaceuticals.
HR has received personal honoraria for consulting from Cerium and non-financial support from Corcept.
MR has received personal honoraria for consulting, lectures, and advisory boards from Novartis, Recordati, HRA Pharma, and Ipsen.
RS has received grants to the institution from Corcept and Crinetics, and personal honoraria for consulting from Strongbridge, Corcept, HRA Pharma, and Sterotherapeutics.
CS has received grants and personal honoraria for lectures from Pfizer, Novartis, Recordati Rare Diseases, and HRA Pharma.
IL has received personal honoraria for consulting from Medison Pharma.
CAS has received grants from Pfizer, personal honoraria for consulting and support for meeting attendance from Lundbeck Pharma and the National Institutes of Health, hold patents on the genetics of PRKAR1A, PDE11A, and GPR101, and has served as a Board member or advisor for Society for Pediatric Research, Children’s Inn at NIH, and Cushing’s Foundation.
BS has nothing to declare.
AT has received grants to the institution from HRA Pharma, personal honoraria for consulting, lectures, and support for meeting attendance from HRA Pharma, Recordati Rare Diseases, and Ipsen.
YT has received personal honoraria for consulting and lectures from Novo Nordisk, Recordati Rare Diseases, Bohringer Ingelheim, and Sumitomo Dainippon Pharma.
MT has served as an advisor to European Society of Endocrinology.
ST has received grants to the institution from Strongbridge, Crinetics, and Novartis, and personal honoraria for lectures, advisory boards, and support for meeting attendance from Recordati, Pfizer, and Ipsen.
EV has received from grants through the European Society of Endocrinology from HRA Pharma, and from Novartis, Recordati, and Corcept, and received personal honoraria for consulting, lectures, and advisory boards from HRA Pharma, HRA Pharma Spain, and Recordati Rare Diseases.
EVV has nothing to declare.
GV has received grants to the institution from Novartis, Recordati, and Corcept, and personal honoraria and honoraria to the institution for consulting and lectures from HRA Pharma and Recordati, and served as an advisor for Cushing’s Hub.
JW has nothing to declare.
SMW has received from grants through the European Society of Endocrinology from HRA Pharma, and from Novartis, Recordati, and Corcept, and received personal honoraria for consulting and lectures from HRA Pharma and Recordati Rare Diseases.
MCZ has served as a Board member for European Neuroendocrine Association.
BMKB has received grants to the institution from Novartis, Opko, Strongbridge, and Millendo, personal honoraria for consulting from Aeterna Zentaris, Ascendis, Crinetics, Merck Serono, Novartis, Novo Nordisk, Recordati, Strongbridge, and Sparrow, and served as an advisor for Endocrine Society.
Funding Statement
Virtual meeting logistics were supported by unrestricted educational grants to the Pituitary Society from Ascendis Pharma, Crinetics Pharmaceuticals, HRA Pharma, Novo Nordisk, Recordati Rare Diseases, and Strongbridge Biopharma. Supporters were invited to observe the highlight summaries presented during the meeting, but did not participate in the small group discussions, had no role in the development of consensus recommendations or topics for future research, and did not review the manuscript prior to publication.
Footnotes
Search Strategy and Selection Criteria
References for this review were identified through searches of PubMed for articles published from January, 2015, to April, 2021, by use of the terms “diagnosis,” “urinary free cortisol,” “salivary cortisol,” “screening tests,” “confirmatory testing,” “differential diagnosis,” “localization testing,” “genetics,” “surgery,” “radiation therapy,” “medical therapy,” “biochemical treatment goals,” “tumor shrinkage,” “clinical outcomes,” “adrenal steroidogenesis inhibitors,” “glucocorticoid receptor blockers,” “somatostatin receptor ligands,” “dopamine agonists,” “mortality,” “comorbidities,” “quality of life,” “preoperative treatment,” “combination therapy,” and “guidelines” in combination with the terms “Cushing’s disease” and “ectopic Cushing’s”. English-language articles resulting from these searches and relevant references cited in those articles were reviewed.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Data Availability
Data sharing is not applicable to this article as no datasets were generated or analyzed.
REFERENCES
- 1.Cushing H The basophil adenomas of the pituitary body and their clinical manifestation. Bull Johns Hopkins Hosp 1932; 50: 137–95. [Google Scholar]
- 2.Melmed S Pituitary-tumor endocrinopathies. N Engl J Med 2020; 382: 937–50. [DOI] [PubMed] [Google Scholar]
- 3.Valassi E, Feelders R, Maiter D, et al. Worse health-related quality of life at long-term follow-up in patients with Cushing’s disease than patients with cortisol producing adenoma: data from the ERCUSYN. Clin Endocrinol (Oxf) 2018; 88: 787–98. [DOI] [PubMed] [Google Scholar]
- 4.Arnaldi G, Angeli A, Atkinson AB, et al. Diagnosis and complications of Cushing’s syndrome: a consensus statement. J Clin Endocrinol Metab 2003; 88: 5593–602. [DOI] [PubMed] [Google Scholar]
- 5.Nieman LK, Biller BM, Findling JW, et al. The diagnosis of Cushing’s syndrome: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2008; 93: 1526–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Biller BM, Grossman AB, Stewart PM, et al. Treatment of adrenocorticotropin-dependent Cushing’s syndrome: a consensus statement. J Clin Endocrinol Metab 2008; 93: 2454–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nieman LK, Biller BM, Findling JW, et al. Treatment of Cushing’s syndrome: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2015; 100: 2807–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guyatt GH, Oxman AD, Vist GE, et al. GRADE: an emerging consensus on rating quality of evidence and strength of recommendations. BMJ 2008; 336: 924–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Giustina A, Chanson P, Kleinberg D, et al. Expert consensus document: A consensus on the medical treatment of acromegaly. Nat Rev Endocrinol 2014; 10: 243–8. [DOI] [PubMed] [Google Scholar]
- 10.Rubinstein G, Osswald A, Hoster E, et al. Time to diagnosis in Cushing’s syndrome: a meta-analysis based on 5367 patients. J Clin Endocrinol Metab 2020; 105: e12–e22. [DOI] [PubMed] [Google Scholar]
- 11.Raff H, Carroll T. Cushing’s syndrome: from physiological principles to diagnosis and clinical care. J Physiol 2015; 593: 493–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Galm BP, Qiao N, Klibanski A, Biller BMK, Tritos NA. Accuracy of laboratory tests for the diagnosis of Cushing syndrome. J Clin Endocrinol Metab 2020; 105: 2081–94. [DOI] [PubMed] [Google Scholar]
- 13.Petersenn S Biochemical diagnosis of Cushing’s disease: screening and confirmatory testing. Best Pract Res Clin Endocrinol Metab 2021: 101519. [DOI] [PubMed] [Google Scholar]
- 14.Putignano P, Toja P, Dubini A, Pecori Giraldi F, Corsello SM, Cavagnini F. Midnight salivary cortisol versus urinary free and midnight serum cortisol as screening tests for Cushing’s syndrome. J Clin Endocrinol Metab 2003; 88: 4153–7. [DOI] [PubMed] [Google Scholar]
- 15.Raff H Cushing’s syndrome: diagnosis and surveillance using salivary cortisol. Pituitary 2012; 15: 64–70. [DOI] [PubMed] [Google Scholar]
- 16.Carroll T, Raff H, Findling JW. Late-night salivary cortisol for the diagnosis of Cushing syndrome: a meta-analysis. Endocr Pract 2009; 15: 335–42. [DOI] [PubMed] [Google Scholar]
- 17.Raff H, Phillips JM. Bedtime salivary cortisol and cortisone by LC-MS/MS in healthy adult subjects: evaluation of sampling time. J Endocr Soc 2019; 3: 1631–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kannankeril J, Carroll T, Findling JW, et al. Prospective evaluation of late-night salivary cortisol and cortisone by EIA and LC-MS/MS in suspected Cushing syndrome. J Endocr Soc 2020; 4: bvaa107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jahandideh D, Swearingen B, Nachtigall LB, Klibanski A, Biller BMK, Tritos NA. Characterization of cyclic Cushing’s disease using late night salivary cortisol testing. Clin Endocrinol (Oxf) 2018; 89: 336–45. [DOI] [PubMed] [Google Scholar]
- 20.Debono M, Bradburn M, Bull M, Harrison B, Ross RJ, Newell-Price J. Cortisol as a marker for increased mortality in patients with incidental adrenocortical adenomas. J Clin Endocrinol Metab 2014; 99: 4462–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ueland GA, Methlie P, Kellmann R, et al. Simultaneous assay of cortisol and dexamethasone improved diagnostic accuracy of the dexamethasone suppression test. Eur J Endocrinol 2017; 176: 705–13. [DOI] [PubMed] [Google Scholar]
- 22.Orbach O, Schussler GC. Increased serum cortisol binding in chronic active hepatitis. Am J Med 1989; 86: 39–42. [DOI] [PubMed] [Google Scholar]
- 23.Fleseriu M, Hamrahian AH, Hoffman AR, et al. American Association of Clinical Endocrinologists and American College of Endocrinology disease state clinical review: diagnosis of recurrence in Cushing disease. Endocr Pract 2016; 22: 1436–48. [DOI] [PubMed] [Google Scholar]
- 24.Ceccato F, Artusi C, Barbot M, et al. Dexamethasone measurement during low-dose suppression test for suspected hypercortisolism: threshold development with and validation. J Endocrinol Invest 2020; 43: 1105–13. [DOI] [PubMed] [Google Scholar]
- 25.Roper SM. Yield of serum dexamethasone measurement for reducing false-positive results of low-dose dexamethasone suppression testing. J Appl Lab Med 2021. [DOI] [PubMed] [Google Scholar]
- 26.Findling JW, Raff H, Aron DC. The low-dose dexamethasone suppression test: a reevaluation in patients with Cushing’s syndrome. J Clin Endocrinol Metab 2004; 89: 1222–6. [DOI] [PubMed] [Google Scholar]
- 27.Raff H, Auchus RJ, Findling JW, Nieman LK. Urine free cortisol in the diagnosis of Cushing’s syndrome: is it worth doing and, if so, how? J Clin Endocrinol Metab 2015; 100: 395–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Petersenn S, Newell-Price J, Findling JW, et al. High variability in baseline urinary free cortisol values in patients with Cushing’s disease. Clin Endocrinol (Oxf) 2014; 80: 261–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen AX, Haas AV, Williams GH, Vaidya A. Dietary sodium intake and cortisol measurements. Clin Endocrinol (Oxf) 2020; 93: 539–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moffat SD, An Y, Resnick SM, Diamond MP, Ferrucci L. Longitudinal change in cortisol levels across the adult life span. J Gerontol A Biol Sci Med Sci 2020; 75: 394–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schorr M, Lawson EA, Dichtel LE, Klibanski A, Miller KK. Cortisol measures across the weight spectrum. J Clin Endocrinol Metab 2015; 100: 3313–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Deutschbein T, Broecker-Preuss M, Hartmann MF, et al. Measurement of urinary free cortisol by current immunoassays: need for sex-dependent reference ranges to define hypercortisolism. Horm Metab Res 2011; 43: 714–9. [DOI] [PubMed] [Google Scholar]
- 33.Mericq MV, Cutler GB Jr. High fluid intake increases urine free cortisol excretion in normal subjects. J Clin Endocrinol Metab 1998; 83: 682–4. [DOI] [PubMed] [Google Scholar]
- 34.Rosmalen JG, Kema IP, Wust S, et al. 24 h urinary free cortisol in large-scale epidemiological studies: short-term and long-term stability and sources of variability. Psychoneuroendocrinology 2014; 47: 10–6. [DOI] [PubMed] [Google Scholar]
- 35.Raff H, Cohen EP, Findling JW. A commentary on diagnosing Cushing’s disease in the context of renal failure. Eur J Endocrinol 2019; 181: C9–C11. [DOI] [PubMed] [Google Scholar]
- 36.Carroll TB, Findling JW. The diagnosis of Cushing’s syndrome. Rev Endocr Metab Disord 2010; 11: 147–53. [DOI] [PubMed] [Google Scholar]
- 37.Fukuoka H, Shichi H, Yamamoto M, Takahashi Y. The mechanisms underlying autonomous adrenocorticotropic hormone secretion in Cushing’s disease. Int J Mol Sci 2020; 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Scaroni C, Albiger NM, Palmieri S, et al. Approach to patients with pseudo-Cushing’s states. Endocr Connect 2020; 9: R1–R13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Alwani RA, Schmit Jongbloed LW, de Jong FH, van der Lely AJ, de Herder WW, Feelders RA. Differentiating between Cushing’s disease and pseudo-Cushing’s syndrome: comparison of four tests. Eur J Endocrinol 2014; 170: 477–86. [DOI] [PubMed] [Google Scholar]
- 40.Pecori Giraldi F, Pivonello R, Ambrogio AG, et al. The dexamethasone-suppressed corticotropin-releasing hormone stimulation test and the desmopressin test to distinguish Cushing’s syndrome from pseudo-Cushing’s states. Clin Endocrinol (Oxf) 2007; 66: 251–7. [DOI] [PubMed] [Google Scholar]
- 41.Tirabassi G, Faloia E, Papa R, Furlani G, Boscaro M, Arnaldi G. Use of the desmopressin test in the differential diagnosis of pseudo-Cushing state from Cushing’s disease. J Clin Endocrinol Metab 2010; 95: 1115–22. [DOI] [PubMed] [Google Scholar]
- 42.Martin NM, Dhillo WS, Banerjee A, et al. Comparison of the dexamethasone-suppressed corticotropin-releasing hormone test and low-dose dexamethasone suppression test in the diagnosis of Cushing’s syndrome. J Clin Endocrinol Metab 2006; 91: 2582–6. [DOI] [PubMed] [Google Scholar]
- 43.Moro M, Putignano P, Losa M, Invitti C, Maraschini C, Cavagnini F. The desmopressin test in the differential diagnosis between Cushing’s disease and pseudo-Cushing states. J Clin Endocrinol Metab 2000; 85: 3569–74. [DOI] [PubMed] [Google Scholar]
- 44.Rollin GA, Costenaro F, Gerchman F, Rodrigues TC, Czepielewski MA. Evaluation of the DDAVP test in the diagnosis of Cushing’s Disease. Clin Endocrinol (Oxf) 2015; 82: 793–800. [DOI] [PubMed] [Google Scholar]
- 45.Tirabassi G, Papa R, Faloia E, Boscaro M, Arnaldi G. Corticotrophin-releasing hormone and desmopressin tests in the differential diagnosis between Cushing’s disease and pseudo-Cushing state: a comparative study. Clin Endocrinol (Oxf) 2011; 75: 666–72. [DOI] [PubMed] [Google Scholar]
- 46.Ceccato F, Tizianel I, Vedolin CK, Boscaro M, Barbot M, Scaroni C. Human corticotropin-releasing hormone tests: 10 years of real-life experience in pituitary and adrenal disease. J Clin Endocrinol Metab 2020; 105: e3938–e49. [DOI] [PubMed] [Google Scholar]
- 47.Braun LT, Riester A, Osswald-Kopp A, et al. Toward a diagnostic score in Cushing’s syndrome. Front Endocrinol (Lausanne) 2019; 10: 766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nieman LK. Cushing’s syndrome: update on signs, symptoms and biochemical screening. Eur J Endocrinol 2015; 173: M33–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Newell-Price J, Nieman LK, Reincke M, Tabarin A. ENDOCRINOLOGY IN THE TIME OF COVID-19: Management of Cushing’s syndrome. Eur J Endocrinol 2020; 183: G1–G7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fleseriu M, Buchfelder M, Cetas JS, et al. Pituitary Society guidance: pituitary disease management and patient care recommendations during the COVID-19 pandemic-an international perspective. Pituitary 2020; 23: 327–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yanovski JA, Cutler GB Jr., Doppman JL, et al. The limited ability of inferior petrosal sinus sampling with corticotropin-releasing hormone to distinguish Cushing’s disease from pseudo-Cushing states or normal physiology. J Clin Endocrinol Metab 1993; 77: 503–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Alexandraki KI, Kaltsas GA, Isidori AM, et al. The prevalence and characteristic features of cyclicity and variability in Cushing’s disease. Eur J Endocrinol 2009; 160: 1011–8. [DOI] [PubMed] [Google Scholar]
- 53.Findling JW, Raff H. DIAGNOSIS OF ENDOCRINE DISEASE: Differentiation of pathologic/neoplastic hypercortisolism (Cushing’s syndrome) from physiologic/non-neoplastic hypercortisolism (formerly known as pseudo-Cushing’s syndrome). Eur J Endocrinol 2017; 176: R205–R16. [DOI] [PubMed] [Google Scholar]
- 54.Valassi E, Swearingen B, Lee H, et al. Concomitant medication use can confound interpretation of the combined dexamethasone-corticotropin releasing hormone test in Cushing’s syndrome. J Clin Endocrinol Metab 2009; 94: 4851–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Buchfelder M, Nistor R, Fahlbusch R, Huk WJ. The accuracy of CT and MR evaluation of the sella turcica for detection of adrenocorticotropic hormone-secreting adenomas in Cushing disease. AJNR Am J Neuroradiol 1993; 14: 1183–90. [PMC free article] [PubMed] [Google Scholar]
- 56.Chatain GP, Patronas N, Smirniotopoulos JG, et al. Potential utility of FLAIR in MRI-negative Cushing’s disease. J Neurosurg 2018; 129: 620–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Portocarrero-Ortiz L, Bonifacio-Delgadillo D, Sotomayor-Gonzalez A, Garcia-Marquez A, Lopez-Serna R. A modified protocol using half-dose gadolinium in dynamic 3-Tesla magnetic resonance imaging for detection of ACTH-secreting pituitary tumors. Pituitary 2010; 13: 230–5. [DOI] [PubMed] [Google Scholar]
- 58.Patel V, Liu CJ, Shiroishi MS, et al. Ultra-high field magnetic resonance imaging for localization of corticotropin-secreting pituitary adenomas. Neuroradiology 2020; 62: 1051–4. [DOI] [PubMed] [Google Scholar]
- 59.Chowdhury IN, Sinaii N, Oldfield EH, Patronas N, Nieman LK. A change in pituitary magnetic resonance imaging protocol detects ACTH-secreting tumours in patients with previously negative results. Clin Endocrinol (Oxf) 2010; 72: 502–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.MacFarlane J, Bashari WA, Senanayake R, et al. Advances in the Imaging of Pituitary Tumors. Endocrinol Metab Clin North Am 2020; 49: 357–73. [DOI] [PubMed] [Google Scholar]
- 61.Grober Y, Grober H, Wintermark M, Jane JA, Oldfield EH. Comparison of MRI techniques for detecting microadenomas in Cushing’s disease. J Neurosurg 2018; 128: 1051–7. [DOI] [PubMed] [Google Scholar]
- 62.Mathioudakis N, Pendleton C, Quinones-Hinojosa A, Wand GS, Salvatori R. ACTH-secreting pituitary adenomas: size does not correlate with hormonal activity. Pituitary 2012; 15: 526–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Alzahrani AS, Farhat R, Al-Arifi A, Al-Kahtani N, Kanaan I, Abouzied M. The diagnostic value of fused positron emission tomography/computed tomography in the localization of adrenocorticotropin-secreting pituitary adenoma in Cushing’s disease. Pituitary 2009; 12: 309–14. [DOI] [PubMed] [Google Scholar]
- 64.Chittiboina P, Montgomery BK, Millo C, Herscovitch P, Lonser RR. High-resolution(18)F-fluorodeoxyglucose positron emission tomography and magnetic resonance imaging for pituitary adenoma detection in Cushing disease. J Neurosurg 2015; 122: 791–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Boyle J, Patronas NJ, Smirniotopoulos J, et al. CRH stimulation improves (18)F-FDG-PET detection of pituitary adenomas in Cushing’s disease. Endocrine 2019; 65: 155–65. [DOI] [PubMed] [Google Scholar]
- 66.Ikeda H, Abe T, Watanabe K. Usefulness of composite methionine-positron emission tomography/3.0-tesla magnetic resonance imaging to detect the localization and extent of early-stage Cushing adenoma. J Neurosurg 2010; 112: 750–5. [DOI] [PubMed] [Google Scholar]
- 67.Koulouri O, Steuwe A, Gillett D, et al. A role for 11C-methionine PET imaging in ACTH-dependent Cushing’s syndrome. Eur J Endocrinol 2015; 173: M107–20. [DOI] [PubMed] [Google Scholar]
- 68.Walia R, Gupta R, Bhansali A, et al. Molecular imaging targeting corticotropin-releasing hormone receptor for corticotropinoma: a changing paradigm. J Clin Endocrinol Metab 2021; 106: e1816–e26. [DOI] [PubMed] [Google Scholar]
- 69.Newell-Price J, Perry L, Medbak S, et al. A combined test using desmopressin and corticotropin-releasing hormone in the differential diagnosis of Cushing’s syndrome. J Clin Endocrinol Metab 1997; 82: 176–81. [DOI] [PubMed] [Google Scholar]
- 70.Vassiliadi DA, Tsagarakis S. DIAGNOSIS OF ENDOCRINE DISEASE: The role of the desmopressin test in the diagnosis and follow-up of Cushing’s syndrome. Eur J Endocrinol 2018; 178: R201–R14. [DOI] [PubMed] [Google Scholar]
- 71.Lacroix A, Feelders RA, Stratakis CA, Nieman LK. Cushing’s syndrome. Lancet 2015; 386: 913–27. [DOI] [PubMed] [Google Scholar]
- 72.Messager M, Carriere C, Bertagna X, de Keyzer Y. RT-PCR analysis of corticotroph-associated genes expression in carcinoid tumours in the ectopic-ACTH syndrome. Eur J Endocrinol 2006; 154: 159–66. [DOI] [PubMed] [Google Scholar]
- 73.Tsagarakis S, Tsigos C, Vasiliou V, et al. The desmopressin and combined CRH-desmopressin tests in the differential diagnosis of ACTH-dependent Cushing’s syndrome: constraints imposed by the expression of V2 vasopressin receptors in tumors with ectopic ACTH secretion. J Clin Endocrinol Metab 2002; 87: 1646–53. [DOI] [PubMed] [Google Scholar]
- 74.Ritzel K, Beuschlein F, Berr C, et al. ACTH after 15 min distinguishes between Cushing’s disease and ectopic Cushing’s syndrome: a proposal for a short and simple CRH test. Eur J Endocrinol 2015; 173: 197–204. [DOI] [PubMed] [Google Scholar]
- 75.Reimondo G, Paccotti P, Minetto M, et al. The corticotrophin-releasing hormone test is the most reliable noninvasive method to differentiate pituitary from ectopic ACTH secretion in Cushing’s syndrome. Clin Endocrinol (Oxf) 2003; 58: 718–24. [DOI] [PubMed] [Google Scholar]
- 76.Newell-Price J, Morris DG, Drake WM, et al. Optimal response criteria for the human CRH test in the differential diagnosis of ACTH-dependent Cushing’s syndrome. J Clin Endocrinol Metab 2002; 87: 1640–5. [DOI] [PubMed] [Google Scholar]
- 77.Barbot M, Trementino L, Zilio M, et al. Second-line tests in the differential diagnosis of ACTH-dependent Cushing’s syndrome. Pituitary 2016; 19: 488–95. [DOI] [PubMed] [Google Scholar]
- 78.Oldfield EH, Chrousos GP, Schulte HM, et al. Preoperative lateralization of ACTH-secreting pituitary microadenomas by bilateral and simultaneous inferior petrosal venous sinus sampling. N Engl J Med 1985; 312: 100–3. [DOI] [PubMed] [Google Scholar]
- 79.Loriaux DL. Diagnosis and differential diagnosis of Cushing’s syndrome. N Engl J Med 2017; 376: 1451–9. [DOI] [PubMed] [Google Scholar]
- 80.Sharma ST, Raff H, Nieman LK. Prolactin as a marker of successful catheterization during IPSS in patients with ACTH-dependent Cushing’s syndrome. J Clin Endocrinol Metab 2011; 96: 3687–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Frete C, Corcuff JB, Kuhn E, et al. Non-invasive diagnostic strategy in ACTH-dependent Cushing’s syndrome. J Clin Endocrinol Metab 2020; 105: 3273–84. [DOI] [PubMed] [Google Scholar]
- 82.Nieman LK. Is it time for a new approach to the differential diagnosis of ACTH-dependent Cushing syndrome? J Clin Endocrinol Metab 2020; 105: e4964–e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hofman MS, Lau WF, Hicks RJ. Somatostatin receptor imaging with 68Ga DOTATATE PET/CT: clinical utility, normal patterns, pearls, and pitfalls in interpretation. Radiographics 2015; 35: 500–16. [DOI] [PubMed] [Google Scholar]
- 84.Varlamov E, Hinojosa-Amaya JM, Stack M, Fleseriu M. Diagnostic utility of Gallium-68-somatostatin receptor PET/CT in ectopic ACTH-secreting tumors: a systematic literature review and single-center clinical experience. Pituitary 2019; 22: 445–55. [DOI] [PubMed] [Google Scholar]
- 85.Deppen SA, Blume J, Bobbey AJ, et al. 68Ga-DOTATATE compared with 111In-DTPA-octreotide and conventional imaging for pulmonary and gastroenteropancreatic neuroendocrine tumors: a systematic review and meta-analysis. J Nucl Med 2016; 57: 872–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Isidori AM, Sbardella E, Zatelli MC, et al. Conventional and nuclear medicine imaging in ectopic Cushing’s syndrome: a systematic review. J Clin Endocrinol Metab 2015; 100: 3231–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wannachalee T, Turcu AF, Bancos I, et al. The clinical impact of [(68) Ga]-DOTATATE PET/CT for the diagnosis and management of ectopic adrenocorticotropic hormone - secreting tumours. Clin Endocrinol (Oxf) 2019; 91: 288–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Senanayake R, Gillett D, MacFarlane J, et al. New types of localization methods for adrenocorticotropic hormone-dependent Cushing’s syndrome. Best Pract Res Clin Endocrinol Metab 2021: 101513. [DOI] [PubMed] [Google Scholar]
- 89.Isidori AM, Kaltsas GA, Mohammed S, et al. Discriminatory value of the low-dose dexamethasone suppression test in establishing the diagnosis and differential diagnosis of Cushing’s syndrome. J Clin Endocrinol Metab 2003; 88: 5299–306. [DOI] [PubMed] [Google Scholar]
- 90.van der Pas R, de Bruin C, Leebeek FW, et al. The hypercoagulable state in Cushing’s disease is associated with increased levels of procoagulant factors and impaired fibrinolysis, but is not reversible after short-term biochemical remission induced by medical therapy. J Clin Endocrinol Metab 2012; 97: 1303–10. [DOI] [PubMed] [Google Scholar]
- 91.Van Zaane B, Nur E, Squizzato A, et al. Hypercoagulable state in Cushing’s syndrome: a systematic review. J Clin Endocrinol Metab 2009; 94: 2743–50. [DOI] [PubMed] [Google Scholar]
- 92.Wagner J, Langlois F, Lim DST, McCartney S, Fleseriu M. Hypercoagulability and risk of venous thromboembolic events in endogenous cushing’s syndrome: a systematic meta-analysis. Front Endocrinol (Lausanne) 2018; 9: 805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Coelho MC, Vieira Neto L, Kasuki L, et al. Rotation thromboelastometry and the hypercoagulable state in Cushing’s syndrome. Clin Endocrinol (Oxf) 2014; 81: 657–64. [DOI] [PubMed] [Google Scholar]
- 94.Stuijver DJ, van Zaane B, Feelders RA, et al. Incidence of venous thromboembolism in patients with Cushing’s syndrome: a multicenter cohort study. J Clin Endocrinol Metab 2011; 96: 3525–32. [DOI] [PubMed] [Google Scholar]
- 95.Suarez MG, Stack M, Hinojosa-Amaya JM, et al. Hypercoagulability in Cushing syndrome, prevalence of thrombotic events: a large, single-center, retrospective study. J Endocr Soc 2020; 4: bvz033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Babic B, De Roulet A, Volpe A, Nilubol N. Is VTE prophylaxis necessary on discharge for patients undergoing adrenalectomy for Cushing syndrome? J Endocr Soc 2019; 3: 304–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Barbot M, Guarnotta V, Zilio M, et al. Effects of pasireotide treatment on coagulative profile: a prospective study in patients with Cushing’s disease. Endocrine 2018; 62: 207–14. [DOI] [PubMed] [Google Scholar]
- 98.Boscaro M, Sonino N, Scarda A, et al. Anticoagulant prophylaxis markedly reduces thromboembolic complications in Cushing’s syndrome. J Clin Endocrinol Metab 2002; 87: 3662–6. [DOI] [PubMed] [Google Scholar]
- 99.Barbot M, Daidone V, Zilio M, et al. Perioperative thromboprophylaxis in Cushing’s disease: What we did and what we are doing? Pituitary 2015; 18: 487–93. [DOI] [PubMed] [Google Scholar]
- 100.Fleseriu M, Biller B, Grossman A, Swearingen B, Melmed S, on behalf of the Pituitary Society International Cushing Disease Workshop Task Force. Hypercoagulability in Cushing disease: a risk awareness and prophylaxis survey on behalf of the Pituitary Society. 15th International Pituitary Congress; March 29–31, 2017; Orlando, Florida. Abstract P7. [Google Scholar]
- 101.Zilio M, Mazzai L, Sartori MT, et al. A venous thromboembolism risk assessment model for patients with Cushing’s syndrome. Endocrine 2016; 52: 322–32. [DOI] [PubMed] [Google Scholar]
- 102.Varlamov EV, Langlois F, Vila G, Fleseriu M. Cardiovascular risk assessment, thromboembolism and infection prevention in Cushing’s syndrome - a practical approach. Eur J Endocrinol 2021. [DOI] [PubMed] [Google Scholar]
- 103.Pivonello R, Isidori AM, De Martino MC, Newell-Price J, Biller BM, Colao A. Complications of Cushing’s syndrome: state of the art. Lancet Diabetes Endocrinol 2016; 4: 611–29. [DOI] [PubMed] [Google Scholar]
- 104.Ntali G, Asimakopoulou A, Siamatras T, et al. Mortality in Cushing’s syndrome: systematic analysis of a large series with prolonged follow-up. Eur J Endocrinol 2013; 169: 715–23. [DOI] [PubMed] [Google Scholar]
- 105.Roldan-Sarmiento P, Lam-Chung CE, Hinojosa-Amaya JM, et al. Diabetes, active disease, and afternoon serum cortisol levels predict Cushing’s disease mortality: a cohort study. J Clin Endocrinol Metab 2021; 106: e103–e11. [DOI] [PubMed] [Google Scholar]
- 106.Schernthaner-Reiter MH, Siess C, Micko A, et al. Acute and life-threatening complications in cushing syndrome: prevalence, predictors, and mortality. J Clin Endocrinol Metab 2021; 106: e2035–e46. [DOI] [PubMed] [Google Scholar]
- 107.Geer EB, Shen W, Strohmayer E, Post KD, Freda PU. Body composition and cardiovascular risk markers after remission of Cushing’s disease: a prospective study using whole-body MRI. J Clin Endocrinol Metab 2012; 97: 1702–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Giordano R, Picu A, Marinazzo E, et al. Metabolic and cardiovascular outcomes in patients with Cushing’s syndrome of different aetiologies during active disease and 1 year after remission. Clin Endocrinol (Oxf) 2011; 75: 354–60. [DOI] [PubMed] [Google Scholar]
- 109.Toja PM, Branzi G, Ciambellotti F, et al. Clinical relevance of cardiac structure and function abnormalities in patients with Cushing’s syndrome before and after cure. Clin Endocrinol (Oxf) 2012; 76: 332–8. [DOI] [PubMed] [Google Scholar]
- 110.Dekkers OM, Horvath-Puho E, Jorgensen JO, et al. Multisystem morbidity and mortality in Cushing’s syndrome: a cohort study. J Clin Endocrinol Metab 2013; 98: 2277–84. [DOI] [PubMed] [Google Scholar]
- 111.Papakokkinou E, Olsson DS, Chantzichristos D, et al. Excess morbidity persists in patients with Cushing’s disease during long-term remission: a Swedish nationwide study. J Clin Endocrinol Metab 2020; 105: 2616–24. [DOI] [PubMed] [Google Scholar]
- 112.Ntali G, Hakami O, Wattegama M, Ahmed S, Karavitaki N. Mortality of patients with Cushing’s disease. Exp Clin Endocrinol Diabetes 2021; 129: 203–7. [DOI] [PubMed] [Google Scholar]
- 113.Ragnarsson O, Olsson DS, Papakokkinou E, et al. Overall and disease-specific mortality in patients with Cushing disease: a Swedish nationwide study. J Clin Endocrinol Metab 2019; 104: 2375–84. [DOI] [PubMed] [Google Scholar]
- 114.Clayton RN, Jones PW, Reulen RC, et al. Mortality in patients with Cushing’s disease more than 10 years after remission: a multicentre, multinational, retrospective cohort study. Lancet Diabetes Endocrinol 2016; 4: 569–76. [DOI] [PubMed] [Google Scholar]
- 115.Mazziotti G, Frara S, Giustina A. Pituitary diseases and bone. Endocr Rev 2018; 39: 440–88. [DOI] [PubMed] [Google Scholar]
- 116.Braun LT, Fazel J, Zopp S, et al. The effect of biochemical remission on bone metabolism in Cushing’s syndrome: a 2-year follow-up study. J Bone Miner Res 2020; 35: 1711–7. [DOI] [PubMed] [Google Scholar]
- 117.Scillitani A, Mazziotti G, Di Somma C, et al. Treatment of skeletal impairment in patients with endogenous hypercortisolism: when and how? Osteoporos Int 2014; 25: 441–6. [DOI] [PubMed] [Google Scholar]
- 118.Di Somma C, Colao A, Pivonello R, et al. Effectiveness of chronic treatment with alendronate in the osteoporosis of Cushing’s disease. Clin Endocrinol (Oxf) 1998; 48: 655–62. [DOI] [PubMed] [Google Scholar]
- 119.Mazziotti G, Giustina A. Glucocorticoids and the regulation of growth hormone secretion. Nat Rev Endocrinol 2013; 9: 265–76. [DOI] [PubMed] [Google Scholar]
- 120.Tritos NA. Growth hormone deficiency in adults with Cushing’s disease. Best Pract Res Clin Endocrinol Metab 2021; 35: 101474. [DOI] [PubMed] [Google Scholar]
- 121.Tzanela M, Karavitaki N, Stylianidou C, Tsagarakis S, Thalassinos NC. Assessment of GH reserve before and after successful treatment of adult patients with Cushing’s syndrome. Clin Endocrinol (Oxf) 2004; 60: 309–14. [DOI] [PubMed] [Google Scholar]
- 122.Formenti AM, Maffezzoni F, Doga M, Mazziotti G, Giustina A. Growth hormone deficiency in treated acromegaly and active Cushing’s syndrome. Best Pract Res Clin Endocrinol Metab 2017; 31: 79–90. [DOI] [PubMed] [Google Scholar]
- 123.Hughes NR, Lissett CA, Shalet SM. Growth hormone status following treatment for Cushing’s syndrome. Clin Endocrinol (Oxf) 1999; 51: 61–6. [DOI] [PubMed] [Google Scholar]
- 124.Pecori Giraldi F, Andrioli M, De Marinis L, et al. Significant GH deficiency after long-term cure by surgery in adult patients with Cushing’s disease. Eur J Endocrinol 2007; 156: 233–9. [DOI] [PubMed] [Google Scholar]
- 125.Feldt-Rasmussen U, Abs R, Bengtsson BA, et al. Growth hormone deficiency and replacement in hypopituitary patients previously treated for acromegaly or Cushing’s disease. Eur J Endocrinol 2002; 146: 67–74. [DOI] [PubMed] [Google Scholar]
- 126.Webb SM, Mo D, Lamberts SW, et al. Metabolic, cardiovascular, and cerebrovascular outcomes in growth hormone-deficient subjects with previous cushing’s disease or non-functioning pituitary adenoma. J Clin Endocrinol Metab 2010; 95: 630–8. [DOI] [PubMed] [Google Scholar]
- 127.Hoybye C, Ragnarsson O, Jonsson PJ, et al. Clinical features of GH deficiency and effects of 3 years of GH replacement in adults with controlled Cushing’s disease. Eur J Endocrinol 2010; 162: 677–84. [DOI] [PubMed] [Google Scholar]
- 128.Vogel F, Braun L, Rubinstein G, et al. Patients with low IGF-I after curative surgery for Cushing’s syndrome have an adverse long-term outcome of hypercortisolism-induced myopathy. Eur J Endocrinol 2021; 184: 813–21. [DOI] [PubMed] [Google Scholar]
- 129.Colson A, Brooke AM, Walker D, et al. Growth hormone deficiency and replacement in patients with treated Cushing’s Disease, prolactinomas and non-functioning pituitary adenomas: effects on body composition, glucose metabolism, lipid status and bone mineral density. Horm Res 2006; 66: 257–67. [DOI] [PubMed] [Google Scholar]
- 130.Johannsson G, Sunnerhagen KS, Svensson J. Baseline characteristics and the effects of two years of growth hormone replacement therapy in adults with growth hormone deficiency previously treated for Cushing’s disease. Clin Endocrinol (Oxf) 2004; 60: 550–9. [DOI] [PubMed] [Google Scholar]
- 131.Ragnarsson O, Hoybye C, Jonsson PJ, et al. Comorbidity and cardiovascular risk factors in adult GH deficiency following treatment for Cushing’s disease or non-functioning pituitary adenomas during childhood. Eur J Endocrinol 2012; 166: 593–600. [DOI] [PubMed] [Google Scholar]
- 132.Vogel F, Braun LT, Rubinstein G, et al. Persisting muscle dysfunction in Cushing’s syndrome despite biochemical remission. J Clin Endocrinol Metab 2020; 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Shekhar S, McGlotten R, Auh S, Rother KI, Nieman LK. The hypothalamic-pituitary-thyroid axis in Cushing syndrome before and after curative surgery. J Clin Endocrinol Metab 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.McBride M, Crespo I, Webb SM, Valassi E. Quality of life in Cushing’s syndrome. Best Pract Res Clin Endocrinol Metab 2021: 101505. [DOI] [PubMed] [Google Scholar]
- 135.Reincke M Cushing syndrome associated myopathy: it is time for a change. Endocrinol Metab (Seoul) 2021; 36: 564–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Fleseriu M, Hashim IA, Karavitaki N, et al. Hormonal replacement in hypopituitarism in adults: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2016; 101: 3888–921. [DOI] [PubMed] [Google Scholar]
- 137.Alexandraki KI, Kaltsas GA, Isidori AM, et al. Long-term remission and recurrence rates in Cushing’s disease: predictive factors in a single-centre study. Eur J Endocrinol 2013; 168: 639–48. [DOI] [PubMed] [Google Scholar]
- 138.Ciric I, Zhao JC, Du H, et al. Transsphenoidal surgery for Cushing disease: experience with 136 patients. Neurosurgery 2012; 70: 70–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Capatina C, Hinojosa-Amaya JM, Poiana C, Fleseriu M. Management of patients with persistent or recurrent Cushing’s disease after initial pituitary surgery. Expert Rev Endocrinol Metab 2020; 15: 321–39. [DOI] [PubMed] [Google Scholar]
- 140.Stroud A, Dhaliwal P, Alvarado R, et al. Outcomes of pituitary surgery for Cushing’s disease: a systematic review and meta-analysis. Pituitary 2020; 23: 595–609. [DOI] [PubMed] [Google Scholar]
- 141.Valassi E, Biller BM, Swearingen B, et al. Delayed remission after transsphenoidal surgery in patients with Cushing’s disease. J Clin Endocrinol Metab 2010; 95: 601–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Fan Y, Li Y, Bao X, et al. Development of machine learning models for predicting postoperative delayed remission of patients with Cushing’s disease. J Clin Endocrinol Metab 2020. [DOI] [PubMed] [Google Scholar]
- 143.Petersenn S, Beckers A, Ferone D, et al. Therapy of endocrine disease: outcomes in patients with Cushing’s disease undergoing transsphenoidal surgery: systematic review assessing criteria used to define remission and recurrence. Eur J Endocrinol 2015; 172: R227–39. [DOI] [PubMed] [Google Scholar]
- 144.Broersen LHA, Biermasz NR, van Furth WR, et al. Endoscopic vs. microscopic transsphenoidal surgery for Cushing’s disease: a systematic review and meta-analysis. Pituitary 2018; 21: 524–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Hattori Y, Tahara S, Aso S, et al. Pituitary surgery’s epidemiology using a national inpatient database in Japan. Acta Neurochir (Wien) 2020; 162: 1317–23. [DOI] [PubMed] [Google Scholar]
- 146.Honegger J, Grimm F. The experience with transsphenoidal surgery and its importance to outcomes. Pituitary 2018; 21: 545–55. [DOI] [PubMed] [Google Scholar]
- 147.de Vries F, Lobatto DJ, Verstegen MJT, et al. Outcome squares integrating efficacy and safety, as applied to functioning pituitary adenoma surgery. J Clin Endocrinol Metab 2021: dgab138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Ciric I, Ragin A, Baumgartner C, Pierce D. Complications of transsphenoidal surgery: results of a national survey, review of the literature, and personal experience. Neurosurgery 1997; 40: 225–37. [DOI] [PubMed] [Google Scholar]
- 149.Mortini P, Nocera G, Roncelli F, Losa M, Formenti AM, Giustina A. The optimal numerosity of the referral population of pituitary tumors centers of excellence (PTCOE): A surgical perspective. Rev Endocr Metab Disord 2020; 21: 527–36. [DOI] [PubMed] [Google Scholar]
- 150.Mortini P, Losa M, Barzaghi R, Boari N, Giovanelli M. Results of transsphenoidal surgery in a large series of patients with pituitary adenoma. Neurosurgery 2005; 56: 1222–33. [DOI] [PubMed] [Google Scholar]
- 151.Barker FG 2nd, Curry WT Jr., Carter BS. Surgery for primary supratentorial brain tumors in the United States, 1988 to 2000: the effect of provider caseload and centralization of care. Neuro Oncol 2005; 7: 49–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Casanueva FF, Barkan AL, Buchfelder M, et al. Criteria for the definition of Pituitary Tumor Centers of Excellence (PTCOE): A Pituitary Society Statement. Pituitary 2017; 20: 489–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Pendharkar AV, Sussman ES, Ho AL, Hayden Gephart MG, Katznelson L. Cushing’s disease: predicting long-term remission after surgical treatment. Neurosurg Focus 2015; 38: E13. [DOI] [PubMed] [Google Scholar]
- 154.Patil CG, Prevedello DM, Lad SP, et al. Late recurrences of Cushing’s disease after initial successful transsphenoidal surgery. J Clin Endocrinol Metab 2008; 93: 358–62. [DOI] [PubMed] [Google Scholar]
- 155.Hofmann BM, Hlavac M, Martinez R, Buchfelder M, Muller OA, Fahlbusch R. Long-term results after microsurgery for Cushing disease: experience with 426 primary operations over 35 years. J Neurosurg 2008; 108: 9–18. [DOI] [PubMed] [Google Scholar]
- 156.Mortini P, Barzaghi LR, Albano L, Panni P, Losa M. Microsurgical therapy of pituitary adenomas. Endocrine 2018; 59: 72–81. [DOI] [PubMed] [Google Scholar]
- 157.Braun LT, Rubinstein G, Zopp S, et al. Recurrence after pituitary surgery in adult Cushing’s disease: a systematic review on diagnosis and treatment. Endocrine 2020; 70: 218–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Hinojosa-Amaya JM, Varlamov EV, McCartney S, Fleseriu M. Hypercortisolemia recurrence in Cushing’s disease; a diagnostic challenge. Front Endocrinol (Lausanne) 2019; 10: 740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Valero R, Vallette-Kasic S, Conte-Devolx B, Jaquet P, Brue T. The desmopressin test as a predictive factor of outcome after pituitary surgery for Cushing’s disease. Eur J Endocrinol 2004; 151: 727–33. [DOI] [PubMed] [Google Scholar]
- 160.Le Marc’hadour P, Muller M, Albarel F, et al. Postoperative follow-up of Cushing’s disease using cortisol, desmopressin and coupled dexamethasone-desmopressin tests: a head-to-head comparison. Clin Endocrinol (Oxf) 2015; 83: 216–22. [DOI] [PubMed] [Google Scholar]
- 161.Lindsay JR, Oldfield EH, Stratakis CA, Nieman LK. The postoperative basal cortisol and CRH tests for prediction of long-term remission from Cushing’s disease after transsphenoidal surgery. J Clin Endocrinol Metab 2011; 96: 2057–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Castinetti F, Martinie M, Morange I, et al. A combined dexamethasone desmopressin test as an early marker of postsurgical recurrence in Cushing’s disease. J Clin Endocrinol Metab 2009; 94: 1897–903. [DOI] [PubMed] [Google Scholar]
- 163.Ambrogio AG, Andrioli M, De Martin M, Cavagnini F, Pecori Giraldi F. Usefulness of desmopressin testing to predict relapse during long-term follow-up in patients in remission from Cushing’s disease. Endocr Connect 2017; 6: 791–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Vassiliadi DA, Balomenaki M, Asimakopoulou A, Botoula E, Tzanela M, Tsagarakis S. The desmopressin test predicts better than basal cortisol the long-term surgical outcome of Cushing’s disease. J Clin Endocrinol Metab 2016; 101: 4878–85. [DOI] [PubMed] [Google Scholar]
- 165.Cambos S, Mohammedi K, Castinetti F, et al. Persistent cortisol response to desmopressin predicts recurrence of Cushing’s disease in patients with post-operative corticotropic insufficiency. Eur J Endocrinol 2020; 182: 489–98. [DOI] [PubMed] [Google Scholar]
- 166.Amlashi FG, Swearingen B, Faje AT, et al. Accuracy of late-night salivary cortisol in evaluating postoperative remission and recurrence in Cushing’s disease. J Clin Endocrinol Metab 2015; 100: 3770–7. [DOI] [PubMed] [Google Scholar]
- 167.Carroll TB, Javorsky BR, Findling JW. Postsurgical recurrent Cushing disease: clinical benefit of early intervention in patients with normal urinary free cortisol. Endocr Pract 2016; 22: 1216–23. [DOI] [PubMed] [Google Scholar]
- 168.Bou Khalil R, Baudry C, Guignat L, et al. Sequential hormonal changes in 21 patients with recurrent Cushing’s disease after successful pituitary surgery. Eur J Endocrinol 2011; 165: 729–37. [DOI] [PubMed] [Google Scholar]
- 169.Sandouk Z, Johnston P, Bunch D, et al. Variability of late-night salivary cortisol in Cushing disease: a prospective study. J Clin Endocrinol Metab 2018; 103: 983–90. [DOI] [PubMed] [Google Scholar]
- 170.Danet-Lamasou M, Asselineau J, Perez P, et al. Accuracy of repeated measurements of late-night salivary cortisol to screen for early-stage recurrence of Cushing’s disease following pituitary surgery. Clin Endocrinol (Oxf) 2015; 82: 260–6. [DOI] [PubMed] [Google Scholar]
- 171.Geer EB, Shafiq I, Gordon MB, et al. Biochemical control during long-term follow-up of 230 adult patients with Cushing disease: a multicenter retrospective study. Endocr Pract 2017; 23: 962–70. [DOI] [PubMed] [Google Scholar]
- 172.Geer EB, Ayala A, Bonert V, et al. Follow-up intervals in patients with Cushing’s disease: recommendations from a panel of experienced pituitary clinicians. Pituitary 2017; 20: 422–9. [DOI] [PubMed] [Google Scholar]
- 173.Locatelli M, Vance ML, Laws ER. Clinical review: the strategy of immediate reoperation for transsphenoidal surgery for Cushing’s disease. J Clin Endocrinol Metab 2005; 90: 5478–82. [DOI] [PubMed] [Google Scholar]
- 174.Rutkowski MJ, Flanigan PM, Aghi MK. Update on the management of recurrent Cushing’s disease. Neurosurg Focus 2015; 38: E16. [DOI] [PubMed] [Google Scholar]
- 175.Patil CG, Veeravagu A, Prevedello DM, Katznelson L, Vance ML, Laws ER Jr. Outcomes after repeat transsphenoidal surgery for recurrent Cushing’s disease. Neurosurgery 2008; 63: 266–70; discussion 70–1. [DOI] [PubMed] [Google Scholar]
- 176.Ram Z, Nieman LK, Cutler GB Jr., Chrousos GP, Doppman JL, Oldfield EH. Early repeat surgery for persistent Cushing’s disease. J Neurosurg 1994; 80: 37–45. [DOI] [PubMed] [Google Scholar]
- 177.Hameed N, Yedinak CG, Brzana J, et al. Remission rate after transsphenoidal surgery in patients with pathologically confirmed Cushing’s disease, the role of cortisol, ACTH assessment and immediate reoperation: a large single center experience. Pituitary 2013; 16: 452–8. [DOI] [PubMed] [Google Scholar]
- 178.Cuevas-Ramos D, Fleseriu M. Treatment of Cushing’s disease: a mechanistic update. J Endocrinol 2014; 223: R19–39. [DOI] [PubMed] [Google Scholar]
- 179.Pivonello R, De Leo M, Cozzolino A, Colao A. The treatment of Cushing’s disease. Endocr Rev 2015; 36: 385–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Tritos NA, Biller BMK. Advances in the medical treatment of Cushing disease. Endocrinol Metab Clin North Am 2020; 49: 401–12. [DOI] [PubMed] [Google Scholar]
- 181.Pivonello R, Ferrigno R, De Martino MC, et al. Medical treatment of Cushing’s disease: an overview of the current and recent clinical trials. Front Endocrinol (Lausanne) 2020; 11: 648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Castinetti F, Nieman LK, Reincke M, Newell-Price J. Approach to the patient treated with steroidogenesis inhibitors. J Clin Endocrinol Metab 2021; 106: 2114–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Varlamov EV, Han AJ, Fleseriu M. Updates in adrenal steroidogenesis inhibitors for Cushing’s syndrome - a practical guide. Best Pract Res Clin Endocrinol Metab 2021; 35: 101490. [DOI] [PubMed] [Google Scholar]
- 184.Feelders RA, Hofland LJ, de Herder WW. Medical treatment of Cushing’s syndrome: adrenal-blocking drugs and ketaconazole. Neuroendocrinology 2010; 92 Suppl 1: 111–5. [DOI] [PubMed] [Google Scholar]
- 185.Castinetti F, Guignat L, Giraud P, et al. Ketoconazole in Cushing’s disease: is it worth a try? J Clin Endocrinol Metab 2014; 99: 1623–30. [DOI] [PubMed] [Google Scholar]
- 186.Young J, Bertherat J, Vantyghem MC, et al. Hepatic safety of ketoconazole in Cushing’s syndrome: results of a Compassionate Use Programme in France. Eur J Endocrinol 2018; 178: 447–58. [DOI] [PubMed] [Google Scholar]
- 187.Daniel E, Aylwin S, Mustafa O, et al. Effectiveness of metyrapone in treating Cushing’s syndrome: a retrospective multicenter study in 195 patients. J Clin Endocrinol Metab 2015; 100: 4146–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Ceccato F, Zilio M, Barbot M, et al. Metyrapone treatment in Cushing’s syndrome: a real-life study. Endocrine 2018; 62: 701–11. [DOI] [PubMed] [Google Scholar]
- 189.Krasowski MD, Drees D, Morris CS, Maakestad J, Blau JL, Ekins S. Cross-reactivity of steroid hormone immunoassays: clinical significance and two-dimensional molecular similarity prediction. BMC Clin Pathol 2014; 14: 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Nieman L, Boscaro M, Scaroni C, et al. Metyrapone treatment in endogenous Cushing’s syndrome: results at week 12 from PROMPT, a prospective international multicenter, open-label, phase III/IV study. 2021 Virtual Meeting of the Endocrine Society (ENDO 2021); March 20–23, 2021. Abstract OR14–2. [Google Scholar]
- 191.Bertagna X, Pivonello R, Fleseriu M, et al. LCI699, a potent 11beta-hydroxylase inhibitor, normalizes urinary cortisol in patients with Cushing’s disease: results from a multicenter, proof-of-concept study. J Clin Endocrinol Metab 2014; 99: 1375–83. [DOI] [PubMed] [Google Scholar]
- 192.Fleseriu M, Pivonello R, Young J, et al. Osilodrostat, a potent oral 11beta-hydroxylase inhibitor: 22-week, prospective, phase II study in Cushing’s disease. Pituitary 2016; 19: 138–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Tanaka T, Satoh F, Ujihara M, et al. A multicenter, phase 2 study to evaluate the efficacy and safety of osilodrostat, a new 11beta-hydroxylase inhibitor, in Japanese patients with endogenous Cushing’s syndrome other than Cushing’s disease. Endocr J 2020; 67: 841–52. [DOI] [PubMed] [Google Scholar]
- 194.Pivonello R, Fleseriu M, Newell-Price J, et al. Efficacy and safety of osilodrostat in patients with Cushing’s disease (LINC 3): a multicentre phase III study with a double-blind, randomised withdrawal phase. Lancet Diabetes Endocrinol 2020; 8: 748–61. [DOI] [PubMed] [Google Scholar]
- 195.Gadelha M, Bex M, Feelders R, et al. Osilodrostat is an effective and well-tolerated treatment for Cushing’s disease: results from a phase III study with an upfront, randomized, double-blind, placebo-controlled phase (LINC 4). 2021 Virtual Meeting of the Endocrine Society (ENDO 2021); March 20–23, 2021. Abstract OR14–1. [Google Scholar]
- 196.Hinojosa-Amaya JM, Cuevas-Ramos D, Fleseriu M. Medical management of Cushing’s syndrome: current and emerging treatments. Drugs 2019; 79: 935–56. [DOI] [PubMed] [Google Scholar]
- 197.Baudry C, Coste J, Bou Khalil R, et al. Efficiency and tolerance of mitotane in Cushing’s disease in 76 patients from a single center. Eur J Endocrinol 2012; 167: 473–81. [DOI] [PubMed] [Google Scholar]
- 198.Carroll TB, Peppard WJ, Herrmann DJ, et al. Continuous etomidate infusion for the management of severe Cushing syndrome: validation of a standard protocol. J Endocr Soc 2018; 3: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Preda VA, Sen J, Karavitaki N, Grossman AB. Etomidate in the management of hypercortisolaemia in Cushing’s syndrome: a review. Eur J Endocrinol 2012; 167: 137–43. [DOI] [PubMed] [Google Scholar]
- 200.Constantinescu SM, Driessens N, Lefebvre A, Furnica RM, Corvilain B, Maiter D. Etomidate infusion at low doses is an effective and safe treatment for severe Cushing’s syndrome outside intensive care. Eur J Endocrinol 2020; 183: 161–7. [DOI] [PubMed] [Google Scholar]
- 201.Colao A, Petersenn S, Newell-Price J, et al. A 12-month phase 3 study of pasireotide in Cushing’s disease. N Engl J Med 2012; 366: 914–24. [DOI] [PubMed] [Google Scholar]
- 202.Pivonello R, Petersenn S, Newell-Price J, et al. Pasireotide treatment significantly improves clinical signs and symptoms in patients with Cushing’s disease: results from a phase III study. Clin Endocrinol (Oxf) 2014; 81: 408–17. [DOI] [PubMed] [Google Scholar]
- 203.Lacroix A, Gu F, Gallardo W, et al. Efficacy and safety of once-monthly pasireotide in Cushing’s disease: a 12 month clinical trial. Lancet Diabetes Endocrinol 2018; 6: 17–26. [DOI] [PubMed] [Google Scholar]
- 204.Lacroix A, Bronstein MD, Schopohl J, et al. Long-acting pasireotide improves clinical signs and quality of life in Cushing’s disease: results from a phase III study. J Endocrinol Invest 2020; 43: 1613–22. [DOI] [PubMed] [Google Scholar]
- 205.Fleseriu M, Petersenn S, Biller BMK, et al. Long-term efficacy and safety of once-monthly pasireotide in Cushing’s disease: a phase III extension study. Clin Endocrinol (Oxf) 2019; 91: 776–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Petersenn S, Salgado LR, Schopohl J, et al. Long-term treatment of Cushing’s disease with pasireotide: 5-year results from an open-label extension study of a Phase III trial. Endocrine 2017; 57: 156–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Daniel E, Debono M, Caunt S, et al. A prospective longitudinal study of pasireotide in Nelson’s syndrome. Pituitary 2018; 21: 247–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Ma ZY, Song ZJ, Chen JH, et al. Recurrent gain-of-function USP8 mutations in Cushing’s disease. Cell Res 2015; 25: 306–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Reincke M, Sbiera S, Hayakawa A, et al. Mutations in the deubiquitinase gene USP8 cause Cushing’s disease. Nat Genet 2015; 47: 31–8. [DOI] [PubMed] [Google Scholar]
- 210.Castellnou S, Vasiljevic A, Lapras V, et al. SST5 expression and USP8 mutation in functioning and silent corticotroph pituitary tumors. Endocr Connect 2020; 9: 243–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Hayashi K, Inoshita N, Kawaguchi K, et al. The USP8 mutational status may predict drug susceptibility in corticotroph adenomas of Cushing’s disease. Eur J Endocrinol 2016; 174: 213–26. [DOI] [PubMed] [Google Scholar]
- 212.Fleseriu M, Iweha C, Salgado L, et al. Safety and efficacy of subcutaneous pasireotide in patients with Cushing’s disease: results from an open-label, multicenter, single-arm, multinational, expanded-access study. Front Endocrinol (Lausanne) 2019; 10: 436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Henry RR, Ciaraldi TP, Armstrong D, Burke P, Ligueros-Saylan M, Mudaliar S. Hyperglycemia associated with pasireotide: results from a mechanistic study in healthy volunteers. J Clin Endocrinol Metab 2013; 98: 3446–53. [DOI] [PubMed] [Google Scholar]
- 214.Colao A, De Block C, Gaztambide MS, Kumar S, Seufert J, Casanueva FF. Managing hyperglycemia in patients with Cushing’s disease treated with pasireotide: medical expert recommendations. Pituitary 2014; 17: 180–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Reznik Y, Bertherat J, Borson-Chazot F, et al. Management of hyperglycaemia in Cushing’s disease: experts’ proposals on the use of pasireotide. Diabetes Metab 2013; 39: 34–41. [DOI] [PubMed] [Google Scholar]
- 216.Godbout A, Manavela M, Danilowicz K, Beauregard H, Bruno OD, Lacroix A. Cabergoline monotherapy in the long-term treatment of Cushing’s disease. Eur J Endocrinol 2010; 163: 709–16. [DOI] [PubMed] [Google Scholar]
- 217.Petersenn S, Fleseriu M. Pituitary-directed medical therapy in Cushing’s disease. Pituitary 2015; 18: 238–44. [DOI] [PubMed] [Google Scholar]
- 218.Ferriere A, Cortet C, Chanson P, et al. Cabergoline for Cushing’s disease: a large retrospective multicenter study. Eur J Endocrinol 2017; 176: 305–14. [DOI] [PubMed] [Google Scholar]
- 219.Pivonello R, De Martino MC, Cappabianca P, et al. The medical treatment of Cushing’s disease: effectiveness of chronic treatment with the dopamine agonist cabergoline in patients unsuccessfully treated by surgery. J Clin Endocrinol Metab 2009; 94: 223–30. [DOI] [PubMed] [Google Scholar]
- 220.Casulari LA, Naves LA, Mello PA, Pereira Neto A, Papadia C. Nelson’s syndrome: complete remission with cabergoline but not with bromocriptine or cyproheptadine treatment. Horm Res 2004; 62: 300–5. [DOI] [PubMed] [Google Scholar]
- 221.Hinojosa-Amaya JM, Johnson N, Gonzalez-Torres C, et al. Depression and impulsivity self-assessment tools to identify dopamine agonist side effects in patients with pituitary adenomas. Front Endocrinol (Lausanne) 2020; 11: 579606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Klos KJ, Bower JH, Josephs KA, Matsumoto JY, Ahlskog JE. Pathological hypersexuality predominantly linked to adjuvant dopamine agonist therapy in Parkinson’s disease and multiple system atrophy. Parkinsonism Relat Disord 2005; 11: 381–6. [DOI] [PubMed] [Google Scholar]
- 223.Dodd ML, Klos KJ, Bower JH, Geda YE, Josephs KA, Ahlskog JE. Pathological gambling caused by drugs used to treat Parkinson disease. Arch Neurol 2005; 62: 1377–81. [DOI] [PubMed] [Google Scholar]
- 224.Zanettini R, Antonini A, Gatto G, Gentile R, Tesei S, Pezzoli G. Valvular heart disease and the use of dopamine agonists for Parkinson’s disease. N Engl J Med 2007; 356: 39–46. [DOI] [PubMed] [Google Scholar]
- 225.Colao A, Galderisi M, Di Sarno A, et al. Increased prevalence of tricuspid regurgitation in patients with prolactinomas chronically treated with cabergoline. J Clin Endocrinol Metab 2008; 93: 3777–84. [DOI] [PubMed] [Google Scholar]
- 226.Drake WM, Stiles CE, Bevan JS, et al. A follow-up study of the prevalence of valvular heart abnormalities in hyperprolactinemic patients treated with cabergoline. J Clin Endocrinol Metab 2016; 101: 4189–94. [DOI] [PubMed] [Google Scholar]
- 227.Stiles CE, Tetteh-Wayoe ET, Bestwick J, Steeds RP, Drake WM. A meta-analysis of the prevalence of cardiac valvulopathy in hyperprolactinemic patients treated with cabergoline. J Clin Endocrinol Metab 2019; 104: 523–38. [DOI] [PubMed] [Google Scholar]
- 228.Fleseriu M, Biller BM, Findling JW, et al. Mifepristone, a glucocorticoid receptor antagonist, produces clinical and metabolic benefits in patients with Cushing’s syndrome. J Clin Endocrinol Metab 2012; 97: 2039–49. [DOI] [PubMed] [Google Scholar]
- 229.Fleseriu M, Molitch ME, Gross C, Schteingart DE, Vaughan TB, 3rd, Biller BM. A new therapeutic approach in the medical treatment of Cushing’s syndrome: glucocorticoid receptor blockade with mifepristone. Endocr Pract 2013; 19: 313–26. [DOI] [PubMed] [Google Scholar]
- 230.Fleseriu M, Findling JW, Koch CA, Schlaffer SM, Buchfelder M, Gross C. Changes in plasma ACTH levels and corticotroph tumor size in patients with Cushing’s disease during long-term treatment with the glucocorticoid receptor antagonist mifepristone. J Clin Endocrinol Metab 2014; 99: 3718–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Guarda FJ, Findling J, Yuen KCJ, Fleseriu M, Nachtigall LB. Mifepristone increases thyroid hormone requirements in patients with central hypothyroidism: a multicenter study. J Endocr Soc 2019; 3: 1707–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Newell-Price J, Pivonello R, Tabarin A, et al. Use of late-night salivary cortisol to monitor response to medical treatment in Cushing’s disease. Eur J Endocrinol 2020; 182: 207–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Marques JVO, Boguszewski CL. Medical therapy in severe hypercortisolism. Best Pract Res Clin Endocrinol Metab 2021; 35: 101487. [DOI] [PubMed] [Google Scholar]
- 234.Barbot M, Albiger N, Ceccato F, et al. Combination therapy for Cushing’s disease: effectiveness of two schedules of treatment: should we start with cabergoline or ketoconazole? Pituitary 2014; 17: 109–17. [DOI] [PubMed] [Google Scholar]
- 235.Feelders RA, de Bruin C, Pereira AM, et al. Pasireotide alone or with cabergoline and ketoconazole in Cushing’s disease. N Engl J Med 2010; 362: 1846–8. [DOI] [PubMed] [Google Scholar]
- 236.Kamenicky P, Droumaguet C, Salenave S, et al. Mitotane, metyrapone, and ketoconazole combination therapy as an alternative to rescue adrenalectomy for severe ACTH-dependent Cushing’s syndrome. J Clin Endocrinol Metab 2011; 96: 2796–804. [DOI] [PubMed] [Google Scholar]
- 237.Ollivier M, Haissaguerre M, Ferriere A, Tabarin A. Should we avoid using ketoconazole in patients with severe Cushing’s syndrome and increased levels of liver enzymes? Eur J Endocrinol 2018; 179: L1–L2. [DOI] [PubMed] [Google Scholar]
- 238.Fontaine-Sylvestre C, Letourneau-Guillon L, Moumdjian RA, Berthelet F, Lacroix A. Corticotroph tumor progression during long-term therapy with osilodrostat in a patient with persistent Cushing’s disease. Pituitary 2021; 24: 207–15. [DOI] [PubMed] [Google Scholar]
- 239.Broersen LHA, Jha M, Biermasz NR, Pereira AM, Dekkers OM. Effectiveness of medical treatment for Cushing’s syndrome: a systematic review and meta-analysis. Pituitary 2018; 21: 631–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Feelders RA, Newell-Price J, Pivonello R, Nieman LK, Hofland LJ, Lacroix A. Advances in the medical treatment of Cushing’s syndrome. Lancet Diabetes Endocrinol 2019; 7: 300–12. [DOI] [PubMed] [Google Scholar]
- 241.Valassi E, Franz H, Brue T, et al. Preoperative medical treatment in Cushing’s syndrome: frequency of use and its impact on postoperative assessment: data from ERCUSYN. Eur J Endocrinol 2018; 178: 399–409. [DOI] [PubMed] [Google Scholar]
- 242.Burman P, Eden-Engstrom B, Ekman B, Karlsson FA, Schwarcz E, Wahlberg J. Limited value of cabergoline in Cushing’s disease: a prospective study of a 6-week treatment in 20 patients. Eur J Endocrinol 2016; 174: 17–24. [DOI] [PubMed] [Google Scholar]
- 243.Hughes JD, Young WF, Chang AY, et al. Radiosurgical management of patients with persistent or recurrent Cushing disease after prior transsphenoidal surgery: a management algorithm based on a 25-year experience. Neurosurgery 2020; 86: 557–64. [DOI] [PubMed] [Google Scholar]
- 244.Ironside N, Chen CJ, Lee CC, Trifiletti DM, Vance ML, Sheehan JP. Outcomes of pituitary radiation for Cushing’s disease. Endocrinol Metab Clin North Am 2018; 47: 349–65. [DOI] [PubMed] [Google Scholar]
- 245.Shepard MJ, Mehta GU, Xu Z, et al. Technique of whole-sellar stereotactic radiosurgery for Cushing disease: results from a multicenter, international cohort study. World Neurosurg 2018; 116: e670–e9. [DOI] [PubMed] [Google Scholar]
- 246.Mehta GU, Ding D, Patibandla MR, et al. Stereotactic radiosurgery for Cushing disease: results of an international, multicenter study. J Clin Endocrinol Metab 2017; 102: 4284–91. [DOI] [PubMed] [Google Scholar]
- 247.Petit JH, Biller BM, Yock TI, et al. Proton stereotactic radiotherapy for persistent adrenocorticotropin-producing adenomas. J Clin Endocrinol Metab 2008; 93: 393–9. [DOI] [PubMed] [Google Scholar]
- 248.Gupta A, Xu Z, Kano H, et al. Upfront Gamma Knife radiosurgery for Cushing’s disease and acromegaly: a multicenter, international study. J Neurosurg 2018; 131: 532–8. [DOI] [PubMed] [Google Scholar]
- 249.Castinetti F, Nagai M, Dufour H, et al. Gamma knife radiosurgery is a successful adjunctive treatment in Cushing’s disease. Eur J Endocrinol 2007; 156: 91–8. [DOI] [PubMed] [Google Scholar]
- 250.Thakkar K, Lila A, Sarathi V, et al. Cabergoline may act as a radioprotective agent in Cushing’s disease. Clin Endocrinol (Oxf) 2020; 92: 55–62. [DOI] [PubMed] [Google Scholar]
- 251.Starke RM, Williams BJ, Vance ML, Sheehan JP. Radiation therapy and stereotactic radiosurgery for the treatment of Cushing’s disease: an evidence-based review. Curr Opin Endocrinol Diabetes Obes 2010; 17: 356–64. [DOI] [PubMed] [Google Scholar]
- 252.Sims-Williams HP, Rajapaksa K, Yianni J, et al. Long-term safety of gamma knife radiosurgery (SRS) for acromegaly. Pituitary 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Katznelson L Bilateral adrenalectomy for Cushing’s disease. Pituitary 2015; 18: 269–73. [DOI] [PubMed] [Google Scholar]
- 254.Guerin C, Taieb D, Treglia G, et al. Bilateral adrenalectomy in the 21st century: when to use it for hypercortisolism? Endocr Relat Cancer 2016; 23: R131–42. [DOI] [PubMed] [Google Scholar]
- 255.Ritzel K, Beuschlein F, Mickisch A, et al. Clinical review: Outcome of bilateral adrenalectomy in Cushing’s syndrome: a systematic review. J Clin Endocrinol Metab 2013; 98: 3939–48. [DOI] [PubMed] [Google Scholar]
- 256.Reincke M, Ritzel K, Osswald A, et al. A critical reappraisal of bilateral adrenalectomy for ACTH-dependent Cushing’s syndrome. Eur J Endocrinol 2015; 173: M23–32. [DOI] [PubMed] [Google Scholar]
- 257.Osswald A, Plomer E, Dimopoulou C, et al. Favorable long-term outcomes of bilateral adrenalectomy in Cushing’s disease. Eur J Endocrinol 2014; 171: 209–15. [DOI] [PubMed] [Google Scholar]
- 258.Assie G, Bahurel H, Coste J, et al. Corticotroph tumor progression after adrenalectomy in Cushing’s disease: a reappraisal of Nelson’s syndrome. J Clin Endocrinol Metab 2007; 92: 172–9. [DOI] [PubMed] [Google Scholar]
- 259.Fountas A, Lim ES, Drake WM, et al. Outcomes of patients with Nelson’s syndrome after primary treatment: a multicenter study from 13 UK pituitary centers. J Clin Endocrinol Metab 2020; 105: dgz200. [DOI] [PubMed] [Google Scholar]
- 260.Reincke M, Albani A, Assie G, et al. Corticotroph tumor progression after bilateral adrenalectomy (Nelson’s syndrome): systematic review and expert consensus recommendations. Eur J Endocrinol 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Jornayvaz FR, Assie G, Bienvenu-Perrard M, et al. Pregnancy does not accelerate corticotroph tumor progression in Nelson’s syndrome. J Clin Endocrinol Metab 2011; 96: E658–62. [DOI] [PubMed] [Google Scholar]
- 262.Reincke M, Theodoropoulou M. Genomics in Cushing’s disease: the dawn of a new era. J Clin Endocrinol Metab 2021; 106: e2455–e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Fukuoka H, Cooper O, Ben-Shlomo A, et al. EGFR as a therapeutic target for human, canine, and mouse ACTH-secreting pituitary adenomas. J Clin Invest 2011; 121: 4712–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Neou M, Villa C, Armignacco R, et al. Pangenomic classification of pituitary neuroendocrine tumors. Cancer Cell 2020; 37: 123–34 e5. [DOI] [PubMed] [Google Scholar]
- 265.Albani A, Perez-Rivas LG, Dimopoulou C, et al. The USP8 mutational status may predict long-term remission in patients with Cushing’s disease. Clin Endocrinol (Oxf) 2018; 89: 454–8. [DOI] [PubMed] [Google Scholar]
- 266.Tatsi C, Flippo C, Stratakis CA. Cushing syndrome: old and new genes. Best Pract Res Clin Endocrinol Metab 2020; 34: 101418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Carroll PV, Monson JP, Grossman AB, et al. Successful treatment of childhood-onset Cushing’s disease is associated with persistent reduction in growth hormone secretion. Clin Endocrinol (Oxf) 2004; 60: 169–74. [DOI] [PubMed] [Google Scholar]
- 268.Ferrigno R, Hasenmajer V, Caiulo S, et al. Paediatric Cushing’s disease: epidemiology, pathogenesis, clinical management and outcome. Rev Endocr Metab Disord 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Lodish MB, Keil MF, Stratakis CA. Cushing’s Syndrome in Pediatrics: An Update. Endocrinol Metab Clin North Am 2018; 47: 451–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Tatsi C, Neely M, Flippo C, Bompou ME, Keil M, Stratakis CA. Recovery of hypothalamic-pituitary-adrenal axis in paediatric Cushing disease. Clin Endocrinol (Oxf) 2021; 94: 40–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Davies JH, Storr HL, Davies K, et al. Final adult height and body mass index after cure of paediatric Cushing’s disease. Clin Endocrinol (Oxf) 2005; 62: 466–72. [DOI] [PubMed] [Google Scholar]
- 272.Makri A, Akshintala S, Derse-Anthony C, et al. Multiple endocrine neoplasia type 2B presents early in childhood but often is undiagnosed for years. J Pediatr 2018; 203: 447–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Makri A, Bonella MB, Keil MF, et al. Children with MEN1 gene mutations may present first (and at a young age) with Cushing disease. Clin Endocrinol (Oxf) 2018; 89: 437–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Fleseriu M, Pivonello R, Elenkova A, et al. Efficacy and safety of levoketoconazole in the treatment of endogenous Cushing’s syndrome (SONICS): a phase 3, multicentre, open-label, single-arm trial. Lancet Diabetes Endocrinol 2019; 7: 855–65. [DOI] [PubMed] [Google Scholar]
- 275.Zacharieva S, Pivonello R, Elenkova A, et al. Safety and efficacy of levoketoconazole in the treatment of endogenous Cushing’s syndrome (LOGICS): results from a double-blind, placebo-controlled, randomized withdrawal stud y. 2021 Virtual Meeting of the Endocrine Society (ENDO 2021); March 20–23, 2021. Abstract P30–1. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data sharing is not applicable to this article as no datasets were generated or analyzed.