

Abstract
Disturbance of mitochondrial proteins by amyloid beta-protein (Aβ) that associates with mitochondrial stress responses (MSR) is one of the pathological mechanisms of Alzheimer’s disease (AD). This study tried to explore whether the axis of Jumonji domain-containing protein 3 (JMJD3)-trimethylated lysine 27 on histone H3 (H3K27me3)-brain derived neurotrophic factor (BDNF) is involved in the regulation of MSR which in turn intervenes in the process of AD, and whether curcumin (CUR) has a protective role against AD by influencing this axis, aiming to provide insights into AD treatment. AD mouse models presented a significant aggregation of Aβ, with conspicuous pathological changes in brain tissues and an increase in neuronal apoptosis. Moreover, the mRNA and protein levels of JMJD3 and BDNF were down-regulated, H3K27me3 methylation levels were increased, and the MSR markers (ClpP, HSP6, HSP-60, and ATFS-1) showed abnormal alterations. In in-vitro cellular models of AD, up-regulation of either JMJD3 or BDNF up-regulated BDNF levels, down-regulated H3K27me3 methylation levels, mitigated abnormalities of MSR markers and Aβ aggregation, and increased cell proliferation and inhibited apoptosis. JMJD3 was confirmed to regulate Aβ and MSR via BDNF. In addition, CUR was confirmed to modulate JMJD3-H3K27me3-BDNF axis. Furthermore, moderate and high doses of CUR could improve the morphology and histopathology of the brain, inhibit Aβ aggregation and cell apoptosis, and maintain MSR balance at least partly by modulating the JMJD3-H3K27me3-BDNF axis. To sum up, moderate and high doses of CUR regulate the progression of AD via MSR JMJD3-H3K27me3-BDNF axis.
Keywords: Curcumin, JMJD3, H3K27me3, BDNF, Alzheimer’s disease
Introduction
Alzheimer’s disease (AD), the most common neurodegenerative disease worldwide, has an increasing prevalence and is mainly manifested by dementia, therefore, it is also known as senile dementia [1,2]. The aggregation of amyloid-beta protein (Aβ), characterized by the production of Aβ1-40, may lead to dysfunction and even death of brain neurons which process is responsible for AD progression [3]. In addition, Aβ aggregation is associated with mitochondrial stress response (MSR), which is a pathological mechanism of AD and manifested mitochondrial dysfunction induced by disturbance of mitochondrial proteins [4]. The treatments of AD are still disappointing, failing to fundamentally increase the quality of life of patients and reduce their burdens [5,6]. Therefore, it is of great significance to understand the molecular mechanism of AD and develop novel therapeutic drugs.
Curcumin (CUR) is a natural polyphenol with pleiotropic pharmacological effects and plays a neuro-protective role in nervous system diseases [7,8]. It has been confirmed that CUR inhibits Aβ aggregation through anti-amyloidosis and restores mitochondrial dysfunction in AD and other neurodegenerative diseases [9,10]. We hypothesized that CUR might also play a neuro-protective role in AD by inhibiting Aβ aggregation and mitigating MSR. Still, the specific molecular mechanism remains to be explored.
In addition, the involvement of Jumonji domain-containing protein 3 (JMJD3)-trimethylated lysine 27 on histone H3 (H3K27me3)-brain derived neurotrophic factor (BDNF) in AD process and its association with CUR were investigated. It is known that JMJD3, a member of Utx/Uty subfamily, is an H3K27me3 demethylase whose knockdown induces the disturbance of mitochondrial proteins in nematodes and shortens their life span [11,12]. BDNF belongs to the neurotrophic factor family and is capable of regulating brain development, neuron regeneration and synaptic plasticity. Moreover, as an exogenous supplementation, BDNF is helpful to improve mitochondrial function [13,14]. Both increased levels of BDNF promoter methylation and decreased BDNF mRNA levels exist in the brain of patients with AD [15]. Based on the above findings, we inferred that JMJD3 might be a pivotal factor in the regulation of H3K27me3 demethylation at the BDNF promoter region, and its expression might be associated with methylation levels.
The innovation of this study lies in exploring the effects of CUR and JMJD3-H3K27me3-BDNF axis on AD in vivo and in vitro and the relationship between CUR and JMJD3-H3K27me3-BDNF axis, clarifying the anti-AD pathological mechanism of CUR more clearly, and providing a new molecular therapeutic strategy for the clinical treatment of AD.
Materials and methods
Animals
Ethical approval was granted by the Animal Care and Protection Committee (Second affiliated Hospital of Zhengzhou University) (PL-14-657), and all experimental procedures strictly followed the guidelines for animal care. APP/PS-1 double-transgenic mouse models of AD (n=40, male, aged 6 months, Institute of Laboratory Animal Science, CAMS & PUMC) served as APP/PS-1 group, whereas wild-type (WT) C57BL/6 mice (n=10, male, aged 6 months) served as WT group. With the aid of a stereotaxic apparatus, mice in the APP/PS-1 group were separately injected with low (100 mg/kg/d, low-dose group, n=10), moderate (200 mg/kg/d, moderate-dose group, n=10) and high (300 mg/kg/d, high-dose group, n=10) doses of CUR into the peritoneum (Baoman Biotech, Shanghai, China, C0190), for 2 consecutive weeks [16]. In addition, all animals were anesthetized with 30 g/L pentobarbital sodium (The BSZH Scientific Inc., Beijing, China, P3761) before operation to relieve pain with an intraperitoneal dose of 30 mg/kg, and mice were euthanized by intravenous injection of excessive pentobarbital (100-150 mg/kg).
Morris water maze (MWM) test
The MWM test [17] was used to evaluate the cognitive and motor function of mice. The test lasted for 5 days. A mouse was placed in the water facing the pool wall, and the time it took to find the platform was recorded. If the platform was not found within 2 min, the mouse was guided to stay on the platform for 30 s, and the time was recorded as 120 s. After the test, the dried mouse was reared in a cage and trained twice a day. We recorded the searching time in each quadrant and took the mean escape latency in four quadrants as the final latency of the day. On the sixth day, the spatial probe test was carried out, and the mouse was placed at the marked position in the quadrant. The number of platform crossings was recorded, and the test was carried out. After all the tests, the mouse was sacrificed, and the brain tissues were sampled.
Pathological evaluation
Brain tissue morphology was assessed by hematoxylin and eosin (HE) staining (Baiaolaibo, Beijing, China, ZN1970-KPC). The sections were then observed under an optical microscope (Pooher Optoelectronic Technology Co., Ltd., Shanghai, China, OLYMPUS), and five visual fields were randomized for quantitative analysis by evaluating the number of pyloric nerve cells. Neuronal apoptosis was tested by terminal deoxynucleotidyl transferase (TdT)-mediated deoxyuridine triphosphate (dUTP)-biotin nick end-labeling (TUNEL; Qiming Biotechnology Co., Ltd., Shanghai, China, OX02752). The accumulation and clearance of Aβ1-40 were determined by immunohistochemistry.
qPCR
Total RNA was extracted by a high-purity extraction kit (Angfei, Guangzhou, China, RZ102). cDNA synthesized with Super M-MLV reverse transcriptase (Bio Teke, Beijing, China, PR6502) was amplified using SYBR Green reaction mixture (Think-Far, Beijing, China, 4913914001), followed by a PCR (Image Trading, Beijing, China, 100131). The data were determined by 2-ΔΔCT method. The sequences of primers are shown in Table 1.
Table 1.
Primer sequence
| Genes | Upstream primer | Downstream primer |
|---|---|---|
| JMJD3 | 5’-TCAGGAGAGGAAGGCCTCAG-3’ | 5’-AGCTGGGTATGGATGAGGGT-3’ |
| BDNF | 5’-CCAGGAGCGUGACAACAAUTT-3’ | 5’-AUUGUUGUCACGCUCCUGGTT-3’ |
| GAPDH | 5’-ACCACAGTCCATGCCATCAC-3’ | 5’-TTTAATG TCACGCACGATTTC-3’ |
Western blot (WB)
The treated hippocampal tissues and cells were homogenized in RIPA buffer (Runwell, Shanghai, China, N653-100ML), followed by centrifugation at 1500×g and 4°C for 10 min. Afterwards, the supernatant was collected, and the protein concentration was measured by a BCA kit (Baiaolaibo, Beijing, China, GL1484). After SDS-PAGE separation (Neobio, Beijing, China, WB1102), proteins were blotted onto a polyvinylidene fluoride (PVDF) membrane (WeGene Shanghai, China, IPVH00010). It was then blocked with 5% nonfat milk powder (Xinfan Bio, Shanghai, China, XF-P438) and reacted with primary antibodies (dilution concentration 1:1000) at 4°C for 12 h, including JMJD3 (Fine Biotech Co., Ltd., Wuhan, China, FNab04440) and BDNF (Fine Biotech Co., Ltd., Wuhan, China, FNab10014), ClpP (Fine Biotech Co., Ltd., Wuhan, China, FNab01772), HSP6 (Chundu Bio, Wuhan, China, CD-B106978A-KT), HSP-60 (Fine Biotech Co., Ltd., Wuhan, China, FNab10250), ATFS-1 (Fine Biotech Co., Ltd., Wuhan, China, FNab02597), PINK1 (Fine Biotech Co., Ltd., Wuhan, China, FNab09992), PARK2 (Yipu Biotechnology Co., Ltd., ATA35111), BNIP3 (Fantai Bio-technology Co. Ltd., Shanghai, China, FT-B3764S), P62 (Fine Biotech Co., Ltd., Wuhan, China, FNab06087), LC3(I) (Fine Biotech Co., Ltd., Wuhan, China, FNab04717), Cox5a (Fine Biotech Co., Ltd., Wuhan, China, FNab01900), Cox2 (Fine Biotech Co., Ltd., Wuhan, China, FNab10407), Nd1 (Fine Biotech Co., Ltd., Wuhan, China, FNab05597), Sdhc (Fine Biotech Co., Ltd., Wuhan, China, FNab07671), and GAPDH (Fine Biotech Co., Ltd., Wuhan, China, FNab03343). The washed samples were incubated with HRP-conjugated secondary antibodies (SHR Biotech, Nanjing, China, 330) (dilution ratio 1:12000) for 1 h at room temperature. Enhanced chemiluminescence (ECL) (HZ Industrial, Shanghai, China, HZ-F252), chemiluminescence imaging system (Xinyu Biotech, Shanghai, China, XY-I600C) and Quantity One software were used to visualize and quantify the proteins.
Chromatin immunoprecipitation with qPCR (ChIP-qPCR)
First, DNA-protein complexes were allowed to react with 1% formaldehyde for 15 min. Immune complexes were formed with nonspecific IgGs targeting BDNF, H3K27me3 and JMJD3. Next, the DNA in the complexes was eluted and purified. At last, the BDNF promoter was amplified by PCR and qPCR was performed to verify the effect of JMJD3 on H3K27me expression at the BDNF promoter region.
Cellular models of AD and drug intervention
APPswe-SH-SY5 cells bought from iPhase Pharmaceutical Services were allocated into in-vitro AD group, and SH-SY5Y cells without mutation were used as control group. The cells were placed in DMEM/F-12 (Gaochuang Chemical, Shanghai, China) containing 10% FBS (Lianshuo Biotech, Shanghai, China, A3160802, 10×50 ml/box) and penicillin-streptomycin (Sciencell Biotech, Shanghai, China, 0513-2). Before the test, the cells were passaged for three generations with geneticin (Yaji Biotech, Shanghai, China, YS-10555R) selective antibiotic at 4 μg/ml. Cur (20 μm) was used to intervene the modeling of AD cells [18].
Over-expression and knockout of JMJD3 and BNDF
Adeno-associated virus (AAV) system was used to construct the AD cell model transfected with JMJD3 over-expression plasmid (JMJD3) and BDNF over-expression plasmid (BDNF) in AD group for 48 h. RNA interference technique (RNAi) was used to construct the AD cell model transfected with JMJD3 knockout plasmid (JMJD3 RNAi) for 48 h in AD group. Biotechnology was completed by Shanghai Hanheng Biological Engineering Co., Ltd., China, and their empty vectors were used as negative control (NC). At last, the success of the construction was verified by qPCR and WB. The sequences of transfected vectors are shown in Table 2.
Table 2.
Sequence of transfected vector
| Genes | Target sequences |
|---|---|
| JMJD3 | 5’-CCGGCCTGTTCGTTACAAGTGAGAACTCGAGTTCTCACTTGTAACGAACAGGTTTTTG-3’ |
| BDNF | 5’-CCAGCACUCUCGUAAUGAUTT-3’ |
| JMJD3 RNAi | 5’-GCCUUCAUGCGAGUAACAUTT-3’ |
| AAV-NC | 5’-UUCUCCGAA CGUGUCACGUTT-3’ |
CCK-8 assay
We measured cell proliferation with the cell counting kit 8 (CCK-8) (Jingke Chemica, Shanghai, China, CK04-2) assay. First, logarithmic phase cells seeded in a 96-well plate (6×103 cells/well) were cultured in 10% CCK-8 for 1 h. Absorbances were measured at 450 nm by a microplate reader (Yanhui Biotech, Shanghai, China, HBS-1096A).
Flow cytometry
The Annexin V-FITC/PI Apoptosis Kit (Sanshu Biotech, Shanghai, China, BYT0035) was adopted in the test. The washed and digested APPswe/SH-SY5Y cells were centrifuged at 1500×g and 4°C for 10 min, then suspended in 100 μL binding buffer (1×105 cells). Afterwards, cell apoptosis was quantitatively analyzed with FACSCalibur flow cytometry (Shiwei Tech, Shanghai, China) within 1 h.
Statistical analysis
All tests were carried out independently for at least 3 times, and the values were expressed as mean ± standard deviation (SD). GraphPad Prism 6 was employed for data processing and graphing. Multi-group comparisons were conducted with ANOVA and Dunnett’s post-test, or Bonferroni’s post-test and two-way ANOVA as appropriate. Between-group comparisons were performed with t-test. Values of P<0.05 were considered statistically significant.
Results
Behavioral and pathological assessments
APP/PS-1 double-transgenic mouse models of AD and control WT C57BL/6 mice were classified into APP/PS-1 and WT groups, respectively. First, the MWM test indicated that compared with WT group, the APP/PS-1 group showed prolonged escape latency (Figure 1A), decreased number of platform crossings (Figure 1B), shortened time spent in the target quadrant (Figure 1C), as well slower swimming speed (Figure 1D) (P<0.05). After behavioral assessment, we collected the brain tissues of euthanized mice for pathological assessment. As shown by HE staining findings, mice in the APP/PS-1 group presented typical pathological features of AD in the cortex and hippocampus, and the quantitative results were consistent with the pathological conditions (Figure 1E, 1F), enhanced apoptosis of TUNEL positive cells (Figure 1G), and aggregated Aβ (Figure 1H) (P<0.05). Therefore, those rats had impaired cognitive and motor functions, the pathological features conforming to AD, indicating successful modeling.
Figure 1.

Verification of AD modeling in mice. APP/PS-1 transgenic AD mice were used to induce mouse models of AD (APP/PS-1 group, n=10) and wild C57BL/6 mice served as controls (WT group, n=10). The escape latency (A), the number of platform crossings (B), the time spent in the target quadrant (C) and swimming speed (D) of mice were recorded in MWM test to assess their cognitive and motor functions. Pathological features of the cortex and hippocampus were visualized by HE staining with qualitative and quantitative analysis (scale bar =30 μm, magnification: ×200) (E, F). Apoptosis of brain tissues was measured by TUNEL (×200) (scale bar =30 µm) (G). Aβ aggregation of brain tissues was measured by immunohistochemistry (×200) (scale bar =30 µm) (H). Note: *P<0.05 and **P<0.01 vs. WT group.
Abnormalities of JMJD3-H3K27me3-BDNF axis in AD models
The expression of JMJD3-H3K27me3-BDNF axis in mouse models of AD was quantified. Compared with WT group, mRNA and protein levels of JMJD3 (Figure 2A and 2B) and BDNF (Figure 2C and 2D) in APP/PS-1 group were lower, while H3K27me levels at the BDNF promoter region were higher (Figure 2E) (P<0.05). Therefore, JMJD3-H3K27me3-BDNF axis may be associated with pathological changes of AD model mice.
Figure 2.

Abnormalities of JMJD3-H3K27me3-BDNF axis in AD models. We examined the abnormality of JMJD3-H3K27me3-BDNF axis in APP/PS-1 (n=10) and WT (n=10) groups. mRNA and protein levels of JMJD3 (A, B) and BDNF (C, D) were determined by qPCR and WB, and H3K27me3 levels at the BDNF promoter region were determined by ChIP-qPCR (E). Note: *P<0.05 and **P<0.01 vs. WT group.
MSR markers were inhibited in AD models
The levels of MSR markers in AD models were quantified. Compared with WT group, the protein levels of UPRmt markers (ClpP, HSP6, HSP-60, ATFS-1) (Figure 3A), mitochondrial autophagy markers (PINK1, PARK2, BNIP3, P62, LC3) (Figure 3B), and OXPHOS markers (Cox5a, Cox2, Nd1, Sdhc) (Figure 3C) decreased in APP/PS-1 group (P<0.05). These results suggested that the pathological changes of AD model mice were associated with abnormal MSR, which was mainly reflected in the inhibition of MSR markers.
Figure 3.
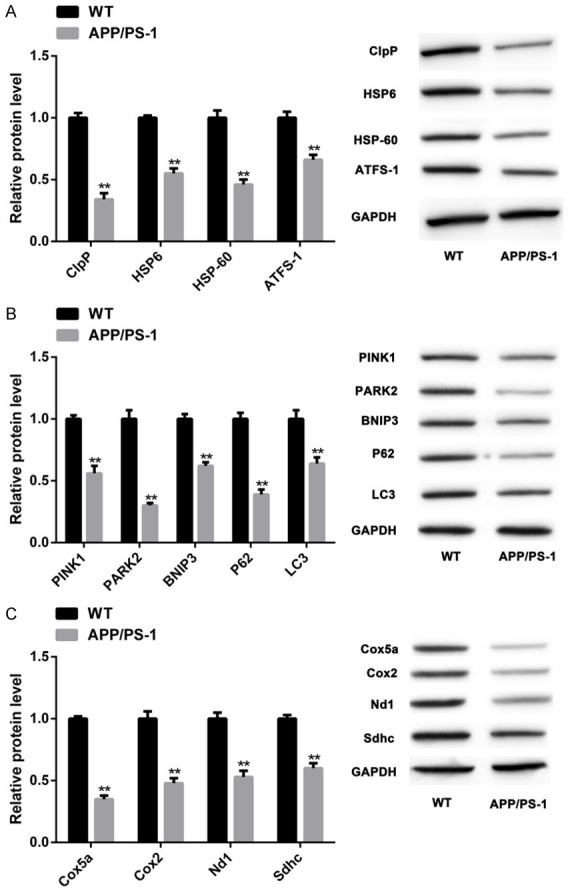
MSR markers were inhibited in AD models. The protein levels of UPRmt markers in two groups of mice (both n=10) (ClpP, HSP6, HSP-60, ATFS-1) (A), mitochondrial autophagy markers (PINK1, PARK2, BNIP3, P62, LC3) (B), and OXPHOS markers (Cox5a, Cox2, Nd1, Sdhc) (C) were quantified by WB. Note: **P<0.01 vs. WT group.
Over-expression of JMJD3 or BNDF mitigated Aβ aggregation and MSR
To figure out the specific mechanism underlying the regulation of AD process by JMJD3-H3K27me3-BDNF axis, we constructed the overexpression vectors of JMJD3 or BNDF in AD models (APPswe SH-SY5Y) through the AAV system (Figure 4A, 4B). Compared with negative controls (AAV-NC group), Aβ clearance occurred in the AD cells with upregulated JMJD3 (Figure 4C), cell proliferation was enhanced (Figure 4D), apoptosis was restricted (Figure 4E), and levels of MSR markers were increased (Figure 4F-H). Identical results were achieved after BNDF up-regulation. In addition, overexpression of JMJD3 increased H3K27me3 levels at the BDNF promoter region and increased the protein levels of BNDF (Figure 4I) (P<0.05). These findings indicated that overexpression of JMJD3 or BNDF was helpful to mitigate Aβ aggregation and MSR in AD models.
Figure 4.

Overexpression of JMJD3 and BNDF mitigated Aβ aggregation and MSR. The establishment of JMJD3 and BNDF overexpression vectors in cellular models of AD was confirmed by WB (A, B). Aβ levels were determined by WB (C). Cell proliferation was measured by CCK-8 assay (D). Cell apoptosis was detected by flow cytometry (E). Levels of MSR markers were determined by WB (F-H). H3K27me3 levels at the BDNF promoter region were measured by ChIP-qPCR (I). Note: *P<0.05, **P<0.01 vs. AAV-NC group, a P<0.05 vs. JMJD3.
JMJD3 regulated Aβ and MSR through BDNF
We also constructed a JMJD3 knockdown cell model by RNAi technology (JMJD3 RNAi group), and the AAV system was used to upregulate BNDF expression (JMJD3 RNAi+BNDF group). Compared with AAV-NC group, mRNA and protein levels of JMJD3 (Figure 5A) and BNDF (Figure 5B) in JMJD3 RNAi group decreased, Aβ aggregated (Figure 5C), cell proliferation decreased (Figure 5D), and apoptosis increased (Figure 5E). Moreover, levels of MSR markers were suppressed (Figure 5F-H), and H3K27me3 level at the BDNF promoter region increased (Figure 5I). However, the above indexes of JMJD3 RNAi+BNDF group were not different from AAV-NC group. All the results were statistically significant (P<0.05), suggesting that the knockdown of JMJD3 downregulated BNDF levels and thereby offset the anti-AD effect of BNDF overexpression. Therefore, JMJD3 can regulate Aβ and MSR in AD models through BDNF.
Figure 5.
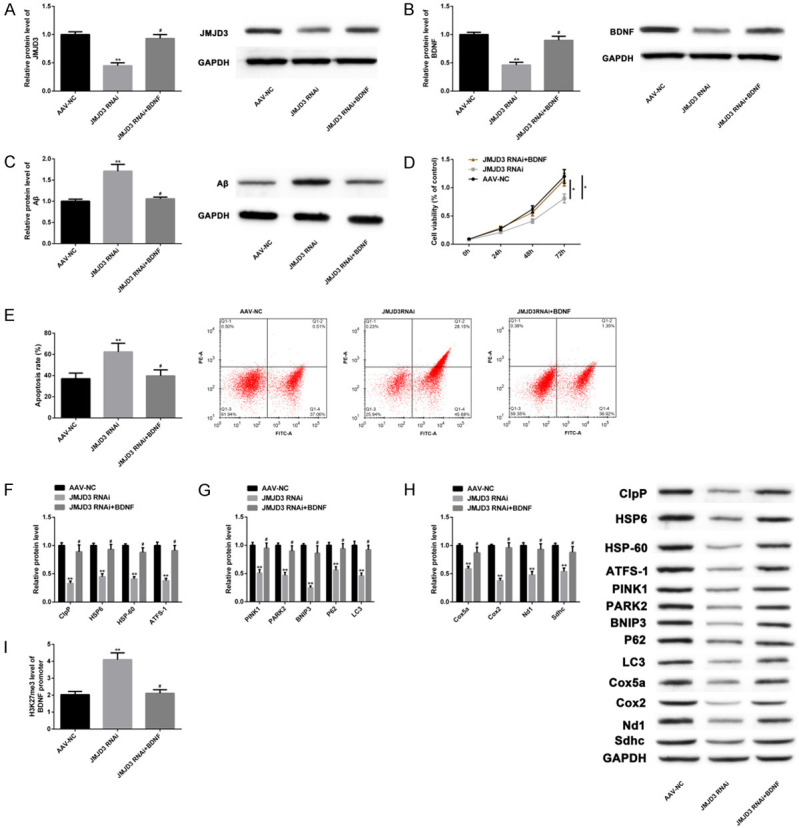
JMJD3 regulated Aβ and MSR through BDNF. The effects of JMJD3 RNAi and JMJD3 RNAi+BNDF on protein levels of JMJD3 and BNDF were analyzed by WB (A, B). Their effects on Aβ levels were analyzed by WB (C). Their effects on cell proliferation were analyzed by CCK-8 assay (D). Their effects on apoptosis were analyzed by Flow cytometry (E). Their effects on MSR markers were analyzed by WB (F-H). Their effects on H3K27me3 levels at the BDNF promoter region were analyzed by ChIP-qPCR (I). Note: *P<0.05, **P<0.01 vs. AAV-NC group, #P<0.05 vs. JMJD3 RNAi.
Regulatory effect of CUR on JMJD3-H3K27me3-BDNF axis
We tried to explore the influence of CUR on the JMJD3-H3K27me3-BDNF axis to verify the relationship between them in the pathological process of AD. It turned out that CUR upregulated the mRNA and protein levels of JMJD3 and BDNF (Figure 6A-D), and inhibited H3K27me3 levels at the BDNF promoter region (Figure 6E) (P<0.05). Thus, CUR might play a protective role in AD by regulating JMJD3-H3K27me3-BDNF axis.
Figure 6.
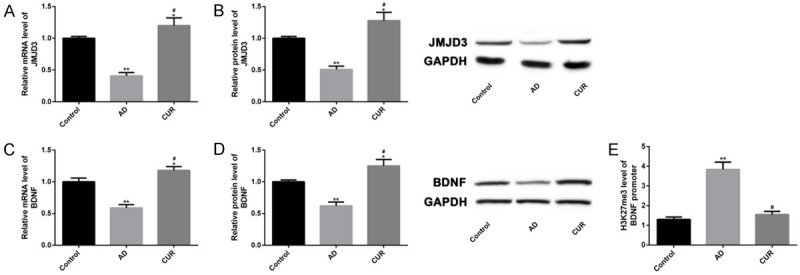
Regulatory effect of CUR on JMJD3-H3K27me3-BDNF axis. The effects of JMJD3 RNAi and JMJD3 RNAi+BNDF on mRNA and protein levels of JMJD3 and BNDF were analyzed by qPCR and WB (A-D). Their effects on H3K27me3 levels at the BDNF promoter region were analyzed by ChIP-qPCR (E). Note: *P<0.05, **P<0.01 vs. Control group, a P<0.05 vs. AD group.
Therapeutic effect of different doses of CUR
To estimate the therapeutic effect of CUR in AD mouse models, we divided CUR group into low (100 mg/kg/d), moderate (200 mg/kg/d) and high (300 mg/kg/d) dose subgroups. Compared with low dose group, the mice in moderate and high dose groups had shortened escape latency (Figure 7A), increased number of platform crossings (Figure 7B), longer time spent in the target quadrant (Figure 7C), and a faster swimming speed (Figure 7D). Pathological evaluation revealed that the improvement of pathological characteristics of cortex and hippocampus in the middle and high dose groups was better than that in the low dose group, and the quantitative results were consistent with the pathological condition (Figure 7E, 7F), Aβ levels were evidently reduced (Figure 7G), and the apoptosis in brain tissue was restricted (Figure 8) (P<0.05 for each comparison). These results suggested that moderate and high doses of CUR shortened the course of the treatment of AD in mice.
Figure 7.

Therapeutic effects of different doses of CUR in AD mouse models. We evaluated the cognitive and motor functions of mice in APP/PS-1 group (n=10), low dose group (n=10), middle dose group (n=10) and high dose group (n=10), escape latency (A), platform crossing times (B), target quadrant stay time (C), and swimming speed (D) by MWM test. The effects of different doses of CUR on the pathological characteristics of cortex and hippocampus of mice (scale bar =30 µm) (E, F) were analyzed qualitatively and quantitatively by HE staining (×200), and the effects of different doses of CUR on the accumulation of Aβ in brain tissue of mice were detected by immunohistochemistry (×200) (scale bar =30 µm) (G). Note: *P<0.05, **P<0.01 vs. AD group, a P<0.05 vs. low-dose group, b P<0.05 vs. moderate-dose group.
Figure 8.
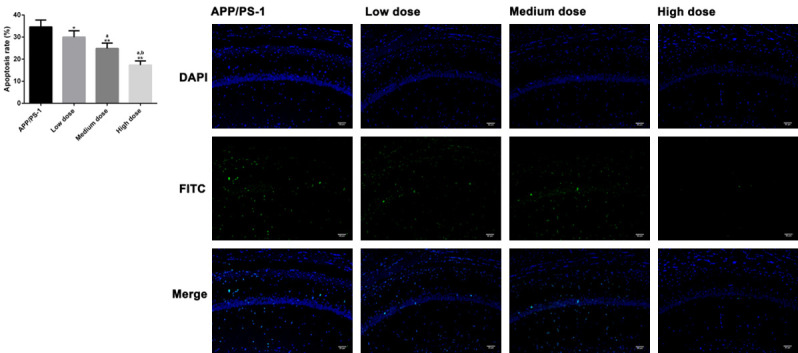
Effects of different doses of CUR on apoptosis. TUNEL (×200) analyzed the effects of different doses of CUR on apoptosis in brain tissue (scale bar =30 µm). Note: *P<0.05, **P<0.01 vs. AD group, a P<0.05 vs. low-dose group, b P<0.05 vs. moderate-dose group.
Regulatory effect of different doses of CUR on JMJD3-H3K27me3-BDNF axis
Serial explorations were performed to verify whether different doses of CUR had dose-dependent regulation on JMJD3-H3K27me3-BDNF axis. Moderate- and high-dose CUR elevated mRNA and protein levels of JMJD3 and BDNF (Figure 9A-D), and suppressed H3K27me3 levels at the BDNF promoter region (Figure 9E); moreover, high dose CUR had a more marked effect on JMJD3-H3K27me3-BDNF axis (P<0.05). Therefore, CUR had a dose-dependent effect on the regulation of JMJD3-H3K27me3-BDNF axis, and significant effect was achieved at least at moderate doses. Based on all the above results, a schematic diagram of the anti-AD mechanism of CUR was drawn (Figure 10).
Figure 9.

Regulatory effect of different doses of CUR on JMJD3-H3K27me3-BDNF axis. The mRNA and protein expressions of JMJD3 and BDNF in APP/PS-1 group (n=10), low dose group (n=10), middle dose group (n=10) and high dose group (n=10) were analyzed by qPCR and WB (A-D), and the level of H3K27me3 in BDNF promoter region (E) was detected by ChIP-qPCR. Note: *P<0.05, **P<0.01 vs. AD group, a P<0.05 vs. low-dose group, b P<0.05 vs. moderate-dose group.
Figure 10.
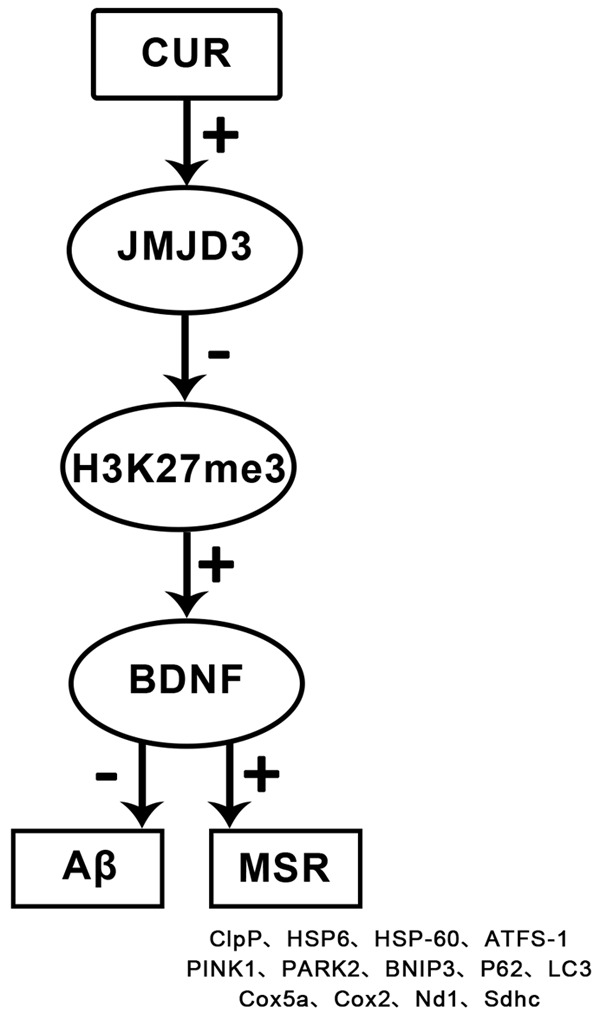
Schematic diagram of related mechanism of CUR. Note: CUR up-regulates the level of BDNF by up-regulating JMJD3 and suppressing the level of H3K27me3 in the BDNF promoter region, thereby reducing the level of Aβ and improving MSR. In short, CUR exerts its anti-AD effect by regulating the JMJD3-H3K27me3-BDNF axis.
Discussion
AD is a brain-specific disease that induces symptoms of memory loss, mental disorder and cognitive impairment [19]. CUR has been reported to be effective in improving memory impairment with in vivo experiments, and in restoring cognitive function partially by increasing lactate content and monocarboxylic acid transporter 2 level in APP/PS1 mouse model of AD [20,21].
Aβ has been confirmed to be a pathogenic factor of AD in animal models in vivo and in vitro, and its aggregation may lead to neuronal apoptosis and mitochondrial dysfunction, thereby aggravating the disease [22,23]. It is known that UPRmt, mitochondrial autophagy and OXPHOS can synergistically alleviate MSR and improve mitochondrial function, which is manifested in the remarkable upregulation of the expression of related biomarkers [24-26]. In the present study, we used APP/PS-1 transgenic AD mice and APPswe SH-SY5Y as AD models in vivo and in vitro, respectively. Mice in APP/PS-1 group presented cognitive and motor dysfunction, pathologically manifested by aggregated Aβ, pathological alterations and increased neuronal apoptosis in brain tissue, conforming to characteristics of AD. Next, we tried to explore the molecular mechanism. The mRNA and protein levels of JMJD3 and BDNF in APP/PS-1 group were lower than those in WT group, and H3K27me3 methylation levels were higher, suggesting that there were abnormalities in the JMJD3-H3K27me3-BDNF axis in in vivo AD models. Also, Hu et al. reported that JMJD3 actively regulated H3K27me3 demethylation at the BDNF promoter region, which means that up-regulating JMJD3 inhibited H3K27me3 methylation levels, promoted BDNF transcription, and maintained the function of the central nervous system [27].
JMJD3 and BDNF have been found to be related to AD process. JMJD3 is capable of mediating the progression of AD through indirect connection with antisense noncoding RNA in the INK4 locus (ANRIL) in INK4b-ARF-INK4a locus [28]. Abnormally low serum BDNF levels in AD patients indicate that BDNF may be a specific biological indicator of AD process [29]. There is evidence that after overexpressing BDNF in human umbilical mesenchymal stem cells, the spatial learning and memory are greatly improved in AD rat models, and the recovery of neural function is promoted by inhibiting Aβ production and neuronal apoptosis [30]. On the basis of the above, we hypothesized that JMJD3-H3K27me3-BDNF axis may be a new target for AD therapy, and a promoter for JMJD3 expression may have an inhibitory effect on AD process. In addition, compared with WT group, UPRmt, mitochondrial autophagy and OXPHOS of MSR markers in APP/PS-1 group were all abnormally downregulated, indicating inhibited MSR and disturbance of mitochondrial proteins. The relationship between JMJD3-H3K27me3-BDNF axis and MSR has not been dissected, so we made some explorations from the molecular level.
In cellular models of AD, we found that up-regulating either JMJD3 or BDNF reduced Aβ aggregation, enhanced cell proliferation, restricted apoptosis, and maintained MSR balance; the former elevated BDNF levels by down-regulating H3K27me3 methylation levels. Afterwards, we constructed JMJD3 knockdown models, in which significantly opposite results were obtained. Once again, it was confirmed that the JMJD3 partially manipulated AD process, and that up-regulation of JMJD3 had a positive therapeutic effect on AD. On the basis of JMJD3 knockdown, we up-regulated BDNF levels. It turned out that the favorable effect of up-regulated BDNF in AD was offset, which may be related to the decrease of BDNF levels caused by the increase of H3K27me3 methylation at the BDNF promoter region after JMJD3 knockdown, indicating that JMJD3 is able to mediate Aβ and MSR through BDNF. Moreover, CUR was found to regulate JMJD3-H3K27me3-BDNF axis in in vitro AD models. Moderate and high doses of CUR could improve the morphology and histopathology of the brain, inhibit Aβ aggregation and cell apoptosis, and maintain MSR balance at least partly by modulating the JMJD3-H3K27me3-BDNF axis. The above findings confirmed the therapeutic potential and protective role of CUR in AD. The association between CUR and JMJD3-H3K27me3-BDNF axis has been previously reported. For example, Zhu revealed that CUR relieved neuropathic pain in rats by regulating BDNF levels [31]. Other studies demonstrated that CUR affected H3K27me3 methylation levels at the promoter region [32,33].
While the present study proposed that CUR mitigates MSR and inhibits AD progress via JMJD3-H3K27me3-BDNF axis, there are several limitations. First of all, it may be feasible to explore the potential upstream targets of this axis from miRNA or lncRNA levels, so as to expand molecular networks associated with AD; then, the influence of MSR-related signal pathways in AD process and their connection with JMJD3-H3K27me3-BDNF axis can be supplemented.
In brief, CUR mitigates MSR by modulating JMJD3-H3K27me3-BDNF axis and thereby slows the progression of AD. The present study not only clarifies the molecular mechanism underlying protection against AD by JMJD3-H3K27me3-BDNF axis, but also uncovers the molecular mechanism of CUR in AD, which may contribute to the development of therapies for AD.
Acknowledgements
Natural Fund of Shandong Province (ZR2019MH065).
Disclosure of conflict of interest
None.
References
- 1.Ke S, Yang Z, Yang F, Wang X, Tan J, Liao B. Long noncoding RNA NEAT1 aggravates abeta-induced neuronal damage by targeting miR-107 in Alzheimer’s disease. Yonsei Med J. 2019;60:640–650. doi: 10.3349/ymj.2019.60.7.640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gu C, Chen C, Wu R, Dong T, Hu X, Yao Y, Zhang Y. Long noncoding RNA EBF3-AS promotes neuron apoptosis in Alzheimer’s disease. DNA Cell Biol. 2018;37:220–226. doi: 10.1089/dna.2017.4012. [DOI] [PubMed] [Google Scholar]
- 3.Li F, Wang Y, Yang H, Xu Y, Zhou X, Zhang X, Xie Z, Bi J. The effect of BACE1-AS on beta-amyloid generation by regulating BACE1 mRNA expression. BMC Mol Biol. 2019;20:23. doi: 10.1186/s12867-019-0140-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Poirier Y, Grimm A, Schmitt K, Eckert A. Link between the unfolded protein response and dysregulation of mitochondrial bioenergetics in Alzheimer’s disease. Cell Mol Life Sci. 2019;76:1419–1431. doi: 10.1007/s00018-019-03009-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stypa V, Haussermann P, Fleiner T, Neumann S. Validity and reliability of the german quality of life-Alzheimer’s disease (QoL-AD) self-report scale. J Alzheimers Dis. 2020;77:581–590. doi: 10.3233/JAD-200400. [DOI] [PubMed] [Google Scholar]
- 6.Enette L, Vogel T, Merle S, Valard-Guiguet AG, Ozier-Lafontaine N, Neviere R, Leuly-Joncart C, Fanon JL, Lang PO. Effect of 9 weeks continuous vs. interval aerobic training on plasma BDNF levels, aerobic fitness, cognitive capacity and quality of life among seniors with mild to moderate Alzheimer’s disease: a randomized controlled trial. Eur Rev Aging Phys Act. 2020;17:2. doi: 10.1186/s11556-019-0234-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu Y, Wang X, Zeng S, Zhang X, Zhao J, Zhang X, Chen X, Yang W, Yang Y, Dong Z, Zhu J, Xu X, Tian F. The natural polyphenol curcumin induces apoptosis by suppressing STAT3 signaling in esophageal squamous cell carcinoma. J Exp Clin Cancer Res. 2018;37:303. doi: 10.1186/s13046-018-0959-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang L, Chen C, Zhang X, Li X, Chen Z, Yang C, Liang X, Zhu G, Xu Z. Neuroprotective effect of curcumin against cerebral ischemia-reperfusion via mediating autophagy and inflammation. J Mol Neurosci. 2018;64:129–139. doi: 10.1007/s12031-017-1006-x. [DOI] [PubMed] [Google Scholar]
- 9.Teter B, Morihara T, Lim GP, Chu T, Jones MR, Zuo X, Paul RM, Frautschy SA, Cole GM. Curcumin restores innate immune Alzheimer’s disease risk gene expression to ameliorate Alzheimer pathogenesis. Neurobiol Dis. 2019;127:432–448. doi: 10.1016/j.nbd.2019.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bagheri H, Ghasemi F, Barreto GE, Rafiee R, Sathyapalan T, Sahebkar A. Effects of curcumin on mitochondria in neurodegenerative diseases. Biofactors. 2020;46:5–20. doi: 10.1002/biof.1566. [DOI] [PubMed] [Google Scholar]
- 11.De Santa F, Totaro MG, Prosperini E, Notarbartolo S, Testa G, Natoli G. The histone H3 lysine-27 demethylase Jmjd3 links inflammation to inhibition of polycomb-mediated gene silencing. Cell. 2007;130:1083–1094. doi: 10.1016/j.cell.2007.08.019. [DOI] [PubMed] [Google Scholar]
- 12.Merkwirth C, Jovaisaite V, Durieux J, Matilainen O, Jordan SD, Quiros PM, Steffen KK, Williams EG, Mouchiroud L, Tronnes SU, Murillo V, Wolff SC, Shaw RJ, Auwerx J, Dillin A. Two conserved histone demethylases regulate mitochondrial stress-induced longevity. Cell. 2016;165:1209–1223. doi: 10.1016/j.cell.2016.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen KW, Chen L. Epigenetic regulation of BDNF gene during development and diseases. Int J Mol Sci. 2017;18:571. doi: 10.3390/ijms18030571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nguyen Nguyen HT, Kato H, Sato H, Yamaza H, Sakai Y, Ohga S, Nonaka K, Masuda K. Positive effect of exogenous brain-derived neurotrophic factor on impaired neurite development and mitochondrial function in dopaminergic neurons derived from dental pulp stem cells from children with attention deficit hyperactivity disorder. Biochem Biophys Res Commun. 2019;513:1048–1054. doi: 10.1016/j.bbrc.2019.04.084. [DOI] [PubMed] [Google Scholar]
- 15.Rao JS, Keleshian VL, Klein S, Rapoport SI. Epigenetic modifications in frontal cortex from Alzheimer’s disease and bipolar disorder patients. Transl Psychiatry. 2012;2:e132. doi: 10.1038/tp.2012.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Feng HL, Dang HZ, Fan H, Chen XP, Rao YX, Ren Y, Yang JD, Shi J, Wang PW, Tian JZ. Curcumin ameliorates insulin signalling pathway in brain of Alzheimer’s disease transgenic mice. Int J Immunopathol Pharmacol. 2016;29:734–741. doi: 10.1177/0394632016659494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dinel AL, Lucas C, Guillemet D, Laye S, Pallet V, Joffre C. Chronic supplementation with a mix of salvia officinalis and salvia lavandulaefolia improves morris water maze learning in normal adult C57Bl/6J mice. Nutrients. 2020;12:6. doi: 10.3390/nu12061777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Qian W, Li H, Pan N, Zhang C. Curcumin treatment is associated with increased expression of the N-Methyl-D-Aspartate Receptor (NMDAR) subunit, NR2A, in a Rat PC12 cell line model of Alzheimer’s disease treated with the acetyl amyloid-beta peptide, abeta(25-35) Med Sci Monit. 2018;24:2693–2699. doi: 10.12659/MSM.906933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tublin JM, Adelstein JM, Del Monte F, Combs CK, Wold LE. Getting to the Heart of Alzheimer Disease. Circ Res. 2019;124:142–149. doi: 10.1161/CIRCRESAHA.118.313563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lu WT, Sun SQ, Li Y, Xu SY, Gan SW, Xu J, Qiu GP, Zhuo F, Huang SQ, Jiang XL, Huang J. Curcumin ameliorates memory deficits by enhancing lactate content and MCT2 expression in APP/PS1 transgenic mouse model of Alzheimer’s disease. Anat Rec (Hoboken) 2019;302:332–338. doi: 10.1002/ar.23969. [DOI] [PubMed] [Google Scholar]
- 21.Bisceglia F, Seghetti F, Serra M, Zusso M, Gervasoni S, Verga L, Vistoli G, Lanni C, Catanzaro M, De Lorenzi E, Belluti F. Prenylated curcumin analogues as multipotent tools to tackle Alzheimer’s disease. ACS Chem Neurosci. 2019;10:1420–1433. doi: 10.1021/acschemneuro.8b00463. [DOI] [PubMed] [Google Scholar]
- 22.Resende R, Marques SC, Ferreiro E, Simoes I, Oliveira CR, Pereira CM. Effect of alpha-synuclein on amyloid beta-induced toxicity: relevance to Lewy body variant of Alzheimer disease. Neurochem Res. 2013;38:797–806. doi: 10.1007/s11064-013-0982-7. [DOI] [PubMed] [Google Scholar]
- 23.Zhao N, Yan QW, Xia J, Zhang XL, Li BX, Yin LY, Xu B. Treadmill exercise attenuates abeta-induced mitochondrial dysfunction and enhances mitophagy activity in APP/PS1 transgenic mice. Neurochem Res. 2020;45:1202–1214. doi: 10.1007/s11064-020-03003-4. [DOI] [PubMed] [Google Scholar]
- 24.Martinez BA, Petersen DA, Gaeta AL, Stanley SP, Caldwell GA, Caldwell KA. Dysregulation of the mitochondrial unfolded protein response induces non-apoptotic dopaminergic neurodegeneration in C. elegans models of Parkinson’s disease. J Neurosci. 2017;37:11085–11100. doi: 10.1523/JNEUROSCI.1294-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kang MH, Das J, Gurunathan S, Park HW, Song H, Park C, Kim JH. The cytotoxic effects of dimethyl sulfoxide in mouse preimplantation embryos: a mechanistic study. Theranostics. 2017;7:4735–4752. doi: 10.7150/thno.21662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sorrentino V, Romani M, Mouchiroud L, Beck JS, Zhang H, D’Amico D, Moullan N, Potenza F, Schmid AW, Rietsch S, Counts SE, Auwerx J. Enhancing mitochondrial proteostasis reduces amyloid-beta proteotoxicity. Nature. 2017;552:187–193. doi: 10.1038/nature25143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hu E, Du H, Zhu X, Wang L, Shang S, Wu X, Lu H, Lu X. Beta-hydroxybutyrate promotes the expression of BDNF in hippocampal neurons under adequate glucose supply. Neuroscience. 2018;386:315–325. doi: 10.1016/j.neuroscience.2018.06.036. [DOI] [PubMed] [Google Scholar]
- 28.Popov N, Gil J. Epigenetic regulation of the INK4b-ARF-INK4a locus: in sickness and in health. Epigenetics. 2010;5:685–690. doi: 10.4161/epi.5.8.12996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ng TKS, Ho CSH, Tam WWS, Kua EH, Ho RC. Decreased serum Brain-Derived Neurotrophic Factor (BDNF) levels in patients with Alzheimer’s disease (AD): a systematic review and meta-analysis. Int J Mol Sci. 2019;20:257. doi: 10.3390/ijms20020257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hu W, Feng Z, Xu J, Jiang Z, Feng M. Brain-derived neurotrophic factor modified human umbilical cord mesenchymal stem cells-derived cholinergic-like neurons improve spatial learning and memory ability in Alzheimer’s disease rats. Brain Res. 2019;1710:61–73. doi: 10.1016/j.brainres.2018.12.034. [DOI] [PubMed] [Google Scholar]
- 31.Zhu X, Li Q, Chang R, Yang D, Song Z, Guo Q, Huang C. Curcumin alleviates neuropathic pain by inhibiting p300/CBP histone acetyltransferase activity-regulated expression of BDNF and cox-2 in a rat model. PLoS One. 2014;9:e91303. doi: 10.1371/journal.pone.0091303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shu L, Khor TO, Lee JH, Boyanapalli SS, Huang Y, Wu TY, Saw CL, Cheung KL, Kong AN. Epigenetic CpG demethylation of the promoter and reactivation of the expression of Neurog1 by curcumin in prostate LNCaP cells. AAPS J. 2011;13:606–614. doi: 10.1208/s12248-011-9300-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ma L, Zhang X, Wang Z, Huang L, Meng F, Hu L, Chen Y, Wei J. Anti-cancer effects of curcumin on myelodysplastic syndrome through the inhibition of Enhancer of Zeste Homolog-2 (EZH2) Curr Cancer Drug Targets. 2019;19:729–741. doi: 10.2174/1568009619666190212121735. [DOI] [PubMed] [Google Scholar]