

Summary
The GroEL/GroES chaperonin system assists the folding of many proteins, through conformational transitions driven by ATP hydrolysis. Although structural information about bullet-shaped GroEL:ES1 complexes has been extensively reported, the substrate interactions of another functional complex, the football-shaped GroEL:ES2, remain elusive. Here, we report single-particle cryo-EM structures of reconstituted wild-type GroEL:ES2 complexes with a chemically denatured substrate, ribulose-1,5-bisphosphate carboxylase oxygenase (RuBisCO). Our structures demonstrate that native-like folded RuBisCO density is captured at the lower part of the GroEL chamber and that GroEL's bulky hydrophobic residues Phe281, Tyr360, and Phe44 contribute to direct contact with RuBisCO density. In addition, our analysis found that GroEL:ES2 can be occupied by two substrates simultaneously, one in each chamber. Together, these observations provide insights to the football-shaped GroEL:ES2 complex as a functional state to assist the substrate folding with visualization.
Subject areas: Biochemistry, Biological sciences, Structural biology
Graphical abstract
Highlights
-
•
Presenting cryo-EM structures of football-shaped GroEL:ES2 encapsulating RuBisCO
-
•
Visualizing direct contacts between the chaperone and an encapsulated substrate
-
•
Visualizing native-like folded substrate inside the chaperone chamber
Biochemistry; Biological sciences; Structural biology
Introduction
Protein misfolding interferes with cellular proteostasis and causes cell death and diseases such as Alzheimer's, Parkinson's, and the prion diseases (Balch et al., 2008). Ensuring correct protein folding in the highly crowded cellular environment depends on an elaborate network of molecular chaperones (Balchin et al., 2016). Chaperonins are double-ring-architecture chaperones that are essential for the efficient folding of numerous proteins that mediate vital cellular functions (Hartl and Hayer-Hartl, 2013). Chaperonins act as catalytic unfolding enzymes, or unfoldases, that drive iterative cycles of unfolding/pulling (Hammarström et al., 2000). Chaperonins also function as an assembly station for regulatory protein complexes (Roh et al., 2016). The best studied example is the bacterial chaperonin GroEL and its co-chaperone GroES, which together comprise a classic ATP-dependent type I co-chaperonin system that is required for bacterial viability. The 14 subunits of GroEL form the barrel-shaped chaperonin, and the 7 subunits of GroES form its lid-like cofactor (Lorimer, 1994). These oligomers assemble into large (>800 kDa) double-ring complexes, with an internal chamber in each ring to provide a folding cage for nascent protein molecules. Current understanding of how the GroEL:ES system promotes protein folding has been obtained from structural studies, presenting the structures of bullet-shaped GroEL:ES chaperone containing substrate inside its chamber (Chen et al., 2013; Clare et al., 2009).
Mechanistically, apical domains of GroEL bind and encapsulate the nonnative substrate protein upon ATP hydrolysis. GroES then seals the cavity, and the substrate moves into a GroEL:ES chamber to continue the folding process (Hayer-Hartl et al., 2016). GroEL:ES systems operate via dynamic assemblies between asymmetric and symmetric states, driven by cycles of ATP hydrolysis and substrate interaction (Ye and Lorimer, 2013). GroEL with one GroES molecule forms an asymmetric bullet-shaped GroEL:ES1 complex, while the complex of one GroEL with two GroES units forms a symmetric football-shaped GroEL:ES2 complex, depending on the association and hydrolysis of ATP in the two GroEL rings. The bullet-shaped GroEL:ES1 has been extensively characterized as a predominant conformation to assist the protein folding process with inter-ring negative cooperativity in an asymmetric manner (Yan et al., 2018), whereas the role of the football-shaped GroEL:ES2 complex has been less studied. The football-shaped complex is proposed to be a transient functional state that is promoted by the substrate (Ye and Lorimer, 2013). Binding of unfolded substrate to GroEL accelerates the rate of ADP/ATP exchange and shifts the equilibrium between footballs and bullets (Ye and Lorimer, 2013). The 3D structure of a bullet-shaped chaperonin, GroEL, and its co-chaperone gp31 (a phage-encoded homologue of GroES) with a partially/native folded substrate, the T4 bacteriophage capsid protein gp23, in the folding chamber has been described (Clare et al., 2009). The structure of RuBisCO in complex with GroEL:ES has also been studied, with the substrate bound to both the lower part of the apical domains and the extended C termini of GroEL (Chen et al., 2013). The question remains as to how the double-cage GroEL:ES2 complex encapsulates and assists obligate substrate folding. In the present study, we use single-particle cryo-electron microscopy (cryo-EM) to visualize the folding process of Rhodospirillum rubrum RuBisCO trapped within a GroEL:ES2 chamber, at 6- to 9-Å resolution. Our structures demonstrate that native-like folded RuBisCO density is captured at the lower part of the cis chamber and that GroEL's bulky hydrophobic residues Phe281, Tyr360, and Phe44 make direct contact with RuBisCO density. In addition, our analysis found that ∼13% of GroEL:ES2-RuBisCO complexes have two substrates bound simultaneously, one in each chamber. This observation suggests that substrate binding to each ring of GroEL:ES2 can be stochastic under our experimental conditions.
Results
Cryo-EM structures of GroEL:ES reconstituted in vitro with RuBisCO
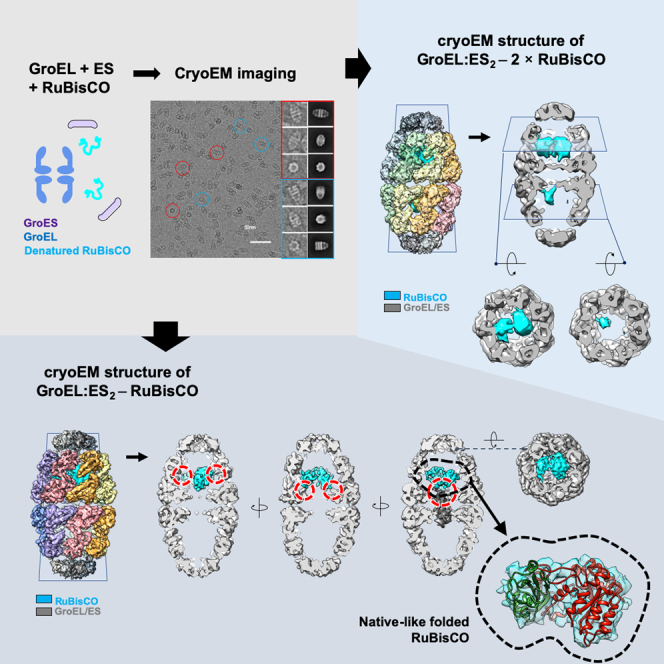
It has been demonstrated in a number of different structural studies that folding intermediates can be captured by rapid mixing of denatured substrate proteins with GroEL (Yan et al., 2018). To investigate how molecular chaperonin assists substrate folding, we reconstituted the wild-type GroEL and GroES with their obligate substrate RuBisCO. To accomplish this, chemically denatured RuBisCO was rapidly diluted into a buffer containing purified GroEL and GroES, resulting in a 10:1 molar ratio of substrate to GroEL:ES2. We added ATP with metal fluoride, BeFx, to coordinate hydrolysis transition states (pre-hydrolytic state) of the ATP γ-phosphate and to trap the step in the GroEL:ES cycle at which GroEL forms a football-shaped structure with two GroES molecules (Figure 1A) (Yan et al., 2018; Taguchi et al., 2004). After incubation, the reaction mixture was vitrified and imaged by cryo-EM. About 350K particles from 925 cryo-EM movies were picked and subjected to 2D reference-free classification and 3D reconstruction protocols. Although ATP + BeFx treatment provided the majority of the population in football shape, we also found bullet-shaped GroEL:ES1 particles in the 2D class averages. Particles were further processed to classify various subpopulations according to 3D conformational and compositional states. After multiple rounds of structure processing, three major classes of the chaperonin complex were clustered: an empty football complex, a football complex containing significant density in a GroEL chamber, and a GroEL:ES1 bullet complex with additional density around its trans ring (Figures 1C and S2). Each class was reconstructed to produce 7- to 9-Å maps with no symmetry to conserve the conformation of the substrate density inside the chambers. It should be noted that we performed the same reconstitution procedure with GroEL and ES without RuBisCO. The collected cryo-EM images for these control samples yielded two major populations of football-shaped GroEL:ES2 (approximately 24K particles) and bullet-shaped GroEL:ES1 (6K particles) without any notable density inside the GroEL chambers. Each class was finally reconstructed at 4.5 and 6.0 Å with applied 7-fold symmetry (Figures 1B and S1). The modest resolution of the structures may be based on the limitations of the instrument (McMullan et al., 2014) and the strong preferred orientation of the GroEL:ES2 complex in vitreous ice. Therefore, the sample was treated with detergent, (0.1% of n-octyl-β-D-glucopyranoside) to obtain more distributed angular views of particles in thicker ice layer, which leads to successful 3D reconstruction but less prominent high-frequency signal.
Figure 1.

Cryo-EM structures of reconstituted GroEL:ES2 with RuBisCO
(A) Experimental design ATP + BeFx to obtain reconstituted GroEL:ES2 containing RuBisCO protein.
(B) Cryo-EM structures of GroEL + ES and (C) GroEL + ES + RuBisCO. Representative micrographs and selected particles are shown. 2D reference-free class averages indicate conformational heterogeneity with bullet and football-shaped particles in each dataset. Various classes of the GroEL:ES complex were reconstructed at 4- to 9-Å maps. RuBisCO density (cyan color) is found in GroEL:ES1 and GroEL:ES2 complexes, marked with red circles.
Approximately 58% of the population of football images in the RuBisCO dataset was devoid of any density within cavities. This map was highly symmetric and very similar to that of the control sample footballs. On the other hand, 28% of the population of football particle images displayed strong density within one of the chambers, most likely representing a RuBisCO monomer trapped in the stalled complex. The estimated mass of the visible RuBisCO monomer, at a contour level of 1.0 σ, was ∼40 kDa, representing roughly 80% of the mass of the RuBisCO monomer. Therefore, we concluded that this extra density in the single chamber can be attributed to RuBisCO and that our experiment successfully captured the structural state of the wild-type GroEL:ES2 complex with RuBisCO encapsulated in the chamber. Of note, ∼14% of the total population of particle images yielded the bullet-shaped GroEL:ES1, and its map showed significant density around the apical domain of the trans chamber (Figure 1C). We consider that the bullet state observed here is the substrate-bound conformation before encapsulation in an asymmetric conformation of GroEL:ES1 because we drove the directional reaction with BeFx (Taguchi et al., 2004). Because molecular contact of GroEL:ES1 with RuBisCO has been described in a previous study (Chen et al., 2013), hereafter we will focus our analysis and discussion on the structure of football-shaped GroEL:ES2.
Molecular contacts of RuBisCO inside the GroEL:ES2 chamber
Denatured RuBisCO was previously shown to be aggregation-prone when diluted from chemical denaturant into solution without chaperonin (van der Vies et al., 1992). The structure of GroEL-RuBisCO binary complexes was previously obtained by the same approach of rapid dilution of denatured RuBisCO (Natesh et al., 2018). In the binary structure, extra density of the RuBisCO was observed in contact with the apical domains of GroEL, indicating a folding state of initial binding. Subsequent recruitment of ATP and GroES would be expected to cause encapsulation of the substrate protein, through GroEL's conformational change.
In our study, we mimicked the transient state of GroEL:ES2 with an encapsulated substrate, before ATP hydrolysis, using ATP + BeFx. Our structure of the football-shaped GroEL:ES2 with RuBisCO displayed aspects of the interaction between RuBisCO and the GroEL ring, but no direct contact with GroES. Of importance, the observed RuBisCO density is linked directly to the density of specific GroEL subunits at several sites that involve hydrophobic residues Phe281, Tyr360, and residues of the S-loop (Figure 2A) according to the rigid-body fitting and Molecular Dynamics Flexible Fitting (MDFF) applied model of GroEL:ES2 from crystal structure (PDB code: 4PKO). Phe281 and Tyr360 reside on helices of the apical domain and interact with the upper part of RuBisCO, whereas the S-loop is located on the equatorial domain, suitably positioned to hold the lower part of the substrate (Figure 2B). This observation accords with the previous results for GroEL:ES1 showing that Phe281, Tyr360, and S-loop were in direct contact with the substrate and contributed to its encapsulation (Chen et al., 2013; Weaver and Rye, 2014). In particular, the hydrophobic residue Phe44 resides on the S-loop, as do other hydrophobic residues, which suggests that hydrophobic interactions between substrate and cavity help to settle the substrate during the refolding process. Together, these hydrophobic residues protrude toward the center and align along prominent hydrophobicity belts at the middle and lower levels of the ring, which enables the ready interaction with substrate. Apart from these hydrophobicity belts and areas of partial positive charge, the electrostatic charge inside the GroEL cavity is mostly negative, an arrangement that was proposed to accelerate the folding process (Figure 2C) (Motojima et al., 2012). In addition to the direct physical contacts with RuBisCO in the football-shaped complex, we have observed additional density from the base of the GroEL cavity (Figure 2A). This density was located principally in the region of the unstructured C-terminal tails of the GroEL subunit and was linked to the density of the substrate, which was not evident in the control map without RuBisCO (Figure S3). Therefore, we concluded that the map represents a heterogeneous interaction between RuBisCO and the C-terminal tails. Contact between nonnative substrate and C termini of GroEL:ES1 was reported previously, and several studies have suggested that the unstructured C-terminal tails of the GroEL subunits play important roles in substrate encapsulation (Weaver and Rye, 2014) and protein folding (Chen et al., 2013).
Figure 2.

Molecular contact of RuBisCO inside the GroEL:ES2 chamber
(A) Connected density of RuBisCO to GroEL is identified in mostly three locations inside of the chamber and shown in red circles; bulky hydrophobic residues (Phe281, Tyr360), S-loop, and around C-terminal tails (C-terminal density is segmented and shown at 0.5 contour level for clear display).
(B) Direct contact of the GroEL chamber to RuBisCO. Phe281 and Tyr360 reside on helices on the apical domain and interact with the upper part of RuBisCO. The S-loop is located on the equatorial domain to hold the lower part of the substrate.
(C) Electrostatic surface representations of the inside of the GroEL chamber.
Unwrapped views of the density map displayed clearly asymmetric binding, with mainly three GroEL subunits contributing to direct contacts with RuBisCO via hydrophobic interaction with Phe281 or Tyr360, while the S-loops protruding from two GroEL subunits mainly held the substrate (Figure 3A). Previous studies of GroEL:ES1 with RuBisCO reported chamber expansion and disruption of symmetry, showing that folding substrates directly and indirectly affect the conformation of the chamber (Elad et al., 2007; Chen et al., 2013). To examine the alteration in symmetry of the GroEL chamber upon binding of RuBisCO, we compared the density maps of the substrate-bound chamber and empty chamber of the football-shaped complex. Specific subunits without direct contact with RuBisCO draw closer to the next one, which were not observed in the structure of the GroEL:ES2 complex without RuBisCO. The empty chamber of the football-shaped structure with or without RuBisCO did not exhibit such differences. Of note, C1 symmetry is applied for the structure of GroEL:ES2 without RuBisCO for this comparison, to avoid the misleading from different symmetry applied for the two structures (Figure 3B). Rotational correlation coefficients of apical domains of the RuBisCO-bound chamber show that two GroEL subunits of the substrate-bound chamber are shifted around ∼3°, compared with those of the empty GroEL chamber (Figure 3C). Possibly, this feature allows GroEL subunits to provide space for substrate binding by the others.
Figure 3.

Asymmetric interaction of RuBisCO inside the GroEL:ES2 chamber
(A) Unwrapped maps indicate asymmetric contact between RuBisCO and GroEL residues. Contacting GroEL subunits by Phe281/Tyr360 are indicated by magenta circles. Contacting the GroEL subunit via S-loop are indicated by blue circles.
(B) Altered contacts of GroEL subunits (black arrowheads) upon substrate binding.
(C) Correlation coefficients of apical domains of substrate-bound or empty GroEL chamber compared with that of control map. Rotational degree has been shifted (red circles) for specific subunits of the substrate-bound chamber.
In the previous report, density corresponding to RuBisCO was seen in contact with multiple apical domains of bullet-shaped GroEL (Natesh et al., 2018). That group identified two distinct densities of a presumed partially folded RuBisCO domain contacting an apical domain of the GroEL ring. Therefore, we also examined whether the encapsulated density in our structures had domain-like features. In its crystal structure, RuBisCO is mainly composed of two domains: the smaller N-terminal domain (residues 1–135) and the bigger C-terminal domain (residues 136–466) (Mizohata et al., 2002). A flexible loop linking the two domains may allow their different orientations when RuBisCO exists in the monomeric state. Our RuBisCO density possessed two clearly separate domains with different volumes, which were linked by a narrow density. The volume of the larger density at a contour level of σ ∼ 1.0 was approximately 30 ų, which corresponds to the size of the C-terminal domain of RuBisCO. The volume of the smaller density was approximately 18 Å3, which may be attributed to the N-terminal domain of RuBisCO. To explore potential heterogeneity in this RuBisCO density, we performed focused classification inside the GroEL cavity without additional orientation searching (Figure 4A) (Roh et al., 2017). The resulting classes after 3D clustering exhibit heterogeneity, but all display two clear segments, which superimposed well with one another with minor structural variation. To identify compatibility between the crystal structure and the cryo-EM-observed RuBisCO density inside the chaperonin, we fitted the crystal structure of RuBisCO (PDB code: 9RUB) (Mizohata et al., 2002) into the cryo-EM map, using either rigid-body fitting or the MDFF method. The two domains of RuBisCO monomer from the crystal structure fitted into each subregion of the cryo-EM density map fairly well after the flexible fitting, but the relative orientation of the two domains had changed. Each domain of the MDFF applied model becomes closer to each other in comparison with the model of RuBisCO from crystal structure, due to this altered relative orientation (Figure 4B). This was consistent with preliminary fluorescence resonance energy transfer (FRET) data showing decreased distance between Cys58 and Ala454 in monomeric RuBisCO inside the GroEL ring, which implies that the substrate encapsulated inside the GroEL cavity was partially folded and spatially confined (Fei et al., 2014; Lin and Rye, 2004). Together with the previous study, our structure provides insight into the state of native-like folded substrates inside of the GroEL:ES2 chamber.
Figure 4.

Native-like folded RuBisCO in the GroEL:ES2 chamber
(A) Focused clustering on RuBisCO density in GroEL chamber (K = 5). All the five classes present distinct two domains, which are superimposed together well in minor structural variation.
(B) The crystal structure of RuBisCO (PDB code: 9RUB) is fitted into the cryo-EM map. Two domains of the RuBisCO monomer fitted into each subregion of the cryo-EM density map fairly well after MDFF, whose relative orientation has been changed.
Visualizing a two-substrate-bound state of GroEL:ES2
Previously, mass spectrometry analysis suggested a stoichiometry of one RuBisCO per GroEL oligomer, whereas smaller substrates, such as gp23 and malate dehydrogenase (MDH), can bind at up to 2:1 ratio (Duijn et al., 2007). To see whether this biochemical stoichiometry was consistent with our imaging data, we analyzed individual GroEL:ES2 particles to investigate binding of multiple RuBisCO molecules, which may be related to potential cooperativity between the two GroEL:ES chambers. Contribution of the signal from RuBisCO may be too weak for global 3D classification to define substrate binding, because GroEL:ES2 signal dominates in particle orientation searching. Therefore, we took advantage of the signal subtraction procedure to reduce the contribution of GroEL:ES2 in order to analyze RuBisCO binding more precisely (Roh et al., 2017). The overall workflow for signal subtraction and localized reconstruction of a single ring of GroEL:ES2 is illustrated in Figure 5A. Signal subtraction was applied for each chamber of GroEL:ES2 and yielded two sub-particles, for the top and for the bottom single-ring GroEL:ES sub-particles. We were then able to perform 3D classification based on the substrate-bound state of single-ring GroEL:ES2. We initially classified them into five classes and then clustered them into two classes depending on whether the sub-particle had substrate or not. After establishing the presence of RuBisCO in each chamber, we traced the information for each ring back to the original double-ring GroEL:ES2 particles. We pooled only particles occupied by two substrates and then refined the map with two substrates per GroEL:ES2 football at a resolution of 8.7 Å (Figure 5B). From this analysis, we found that the number of substrates varied from particle to particle. Almost all of the individual GroEL:ES2 particles had one or no substrate molecule present, but a significant proportion of particles (14.9%) were found to have two substrates simultaneously, one occupying each chamber (Figure 5C). Of note, although we found varied stoichiometry per single GroEL:ES complex, overall substrate binding per GroEL:ES complex in this dataset was still less than 1. This suggests that the previous mass spectroscopy analysis (Duijn et al., 2007) may represent the global average of substrate binding under the experimental condition.
Figure 5.

Two-substrates-bound state of GroEL:ES2
(A) Signal subtraction and 3D refinement workflow to analyze RuBisCO binding to each GroEL ring.
(B) Two RuBisCO-bound particles are pooled and reconstructed at 8.7 Å.
(C) Histogram shows distribution of varied number of substrates per GroEL:ES2 particle. Each particle was divided into two sub-particles, particle pooled, and reconstituted again.
Discussion
The bacterial GroEL/ES system is the archetype chaperonin and the most extensively studied member of the cellular folding nanomachines (Hartl and Hayer-Hartl, 2013). These chamber-forming oligomers have the ability to form dynamic assembly states driven by cycles of ATP hydrolysis and substrate interaction. Structures of GroEL with various types of substrates have been reported from enzymes such as RuBisCO, MDH, or PepQ to a viral capsid protein gp23 (Chen et al., 2013; Clare et al., 2009; Elad et al., 2007; Weaver et al., 2017). Our resulting structures further visualize the native-like substrate and the proximal contacts of GroEL:ES2 residues interacting with the RuBisCO. Previous studies provide insights as to how the bullet-shaped GroEL:ES1 complex functions in an asymmetric manner (Chen et al., 2013; Clare et al., 2009), whereas this study provides a notable view of the symmetric football-shaped GroEL:ES2 as it functions allosterically to encapsulate and fold a physiological substrate protein.
The domain-level definition of RuBisCO density in the GroEL:ES2 chamber suggests that this physiological substrate protein is trapped in a unique position and orientation in the chaperonin chamber. The combination of size and shape of the chamber and of RuBisCO results in the major domain of the substrate being hinged into the chamber in a constrained position. A previous FRET study measured the distances between Ala58 and Cys454 of RuBisCO-bound GroEL and suggested that the GroEL ring can confine the RuBisCO monomer and the RuBisCO inside the GroEL ring is likely to be partially folded rather than randomly positioned (Lin and Rye, 2004). A cryo-EM study of GroEL:gp31-encapsulated gp23 also reported native folded substrate within the upper part of a bullet-shaped chaperonin (Figure S4) (Clare et al., 2009). It is also interesting to compare our structure with one of RuBisCO in a complex with a chemically modified mutant of GroEL (EL43Py):ES1 that is trapped at a normally transient stage (Chen et al., 2013). In both studies, the interactions between GroEL/ES and encapsulated RuBisCO are consistent mainly via hydrophobic residues on the apical domain or S-loop. Distinctively, our structures show that the folded substrate is notably shifted to the apical domains, whereas GroEL (EL43Py) anchors the transient substrate at the lower part of the chamber (Figure S4). It is tempting to speculate that the relocation of substrate inside of the chamber is related to the protein folding process. Further high-resolution experiments will be required to uncover the physiological role of the interaction of GroEL with substrates and whether the observations obtained here for the archetype chaperonin system are conserved in higher organisms.
Having obtained evidence of native-like structural features in RuBisCO density, we identified specific residues of the GroEL chamber that contributed to the direct contact with this density: Phe281, Tyr360, and residues 38–50 on the S-loop. Although a negatively charged surface inside the GroEL cavity was proposed to be involved in the function of protein folding (Motojima et al., 2012), distinct surface hydrophobicity at a certain level of the ring seems to be critical to orient the folded substrate in this study. In particular, Phe281 and Tyr360 on helices of the apical domain interact with the upper part of RuBisCO, whereas the S-loop located on an equatorial domain holds the lower part of the substrate, and the highly dynamic C-terminal tail of GroEL also makes direct contact. Of note, residues from only specific subunits participated in direct contact with RuBisCO, which suggests that the orientation of the folding substrate is guided and maintained inside the chamber at a specific stage of its encapsulation. This observation is consistent with the previous observation in bullet-shaped GroEL:ES1 that Phe281, Tyr360, or S-loop residues contribute to substrate encapsulation and folding rates (Chen et al., 2013). This conserved mode of contact of bullet and football complexes suggests that each chamber can function independently to fold the substrate. In addition, previous studies of GroEL-gp23 and of GroEL-RuBisCO contacts in bullet-shaped GroEL:ES1 reported significant expansion of the folding chamber (Clare et al., 2009; Chen et al., 2013). However, structural changes are not very distinct at the resolution in this study, and near-atomic resolution will be needed for the further investigation of this phenomenon.
The allosteric behavior of GroEL:ES2 involves positively cooperative interactions among the subunits of one ring (Roh et al., 2017) and negatively cooperative long-range allosteric communication between the GroEL chambers (Yan et al., 2018). Negative cooperativity has been described both for nucleotide and for substrate binding; thus, bullet-shaped GroEL:ES1 has been considered as a major active functional conformation (Yan et al., 2018). Football-shaped GroEL:ES2 is recognized as a transient functional conformation, supported by thermodynamically plausible scenarios in which ATP can be bound simultaneously to both rings of GroEL; that, in turn, enables GroES to bind both rings simultaneously (Fei et al., 2014). Therefore, symmetric GroEL:ES2 and asymmetric GroEL:ES1 particles exist in a dynamic, substrate protein-dependent equilibrium (Fei et al., 2014; Ye and Lorimer, 2013). For the particular conditions chosen for our study, we mixed GroEL, GroES, and ATP in the presence of BeFx, leading to the formation of symmetrical GroEL:ES2 particles and mimicking the directional events of chamber closing and substrate encapsulation (Taguchi et al., 2004). Our structure hints that closing of a single chamber provides a sufficient environment to support substrate folding, which suggests that GroEL:ES1 may operate passively as an environment provider for RuBisCO folding, rather than an active, mechanistic folding machine. We adopted a signal subtraction strategy (Roh et al., 2017) to analyze substrate binding in each chamber of the football independently and revealed that the football state can encapsulate two substrates simultaneously. The population of two-substrate-containing GroEL:ES2 particles was significant, which is also consistent with a previous biochemical study that found that the football-shaped complex can provide both chambers for substrate binding simultaneously (Fei et al., 2014). This observation implies that the symmetric football, in dynamic assembly equilibrium with asymmetric bullets, works as a functional state by holding two substrates. When we refined the structure for two-substrate-bound footballs, we observed a well-defined substrate in one chamber and a less structured density in the other chamber. This observation may imply that the two bound substrates are in different folded states, which would support an asymmetric transition of football to bullet, based on the folding state of the substrate (Ye and Lorimer, 2013). However, we cannot rule out the possibility that observation of a less defined substrate is the result of a structural average of symmetry mismatch between two substrates in each chamber. Further high-resolution experiments will be required to uncover whether our in vitro observations of football-shaped GroEL:ES2 complexes with substrates represent real events in cellular systems and their physiological meaning.
Limitations of the study
This study visualizes the football-shaped GroEL:ES2 complex encapsulating RuBisCO and the direct interaction between the chaperone and the substrate by using cryo-EM. However, the limited resolution of the presented structure makes further analyses difficult. Further analyses with improved resolution of chaperone structures encapsulating folding intermediate will allow much intensive investigation on the chaperonin-assisted substrate folding.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| JM105 Escherichia coli cells | ATCC | ATCC-47016 |
| Chemicals, peptides, and recombinant proteins | ||
| Beryllium Sulfate tetrahydrate | Alfa Aesar | Q05F063 |
| Potassium Fluoride | Sigma-Aldrich | 60238-100G-F |
| Adenosine 50-triphosphate (ATP) disodium salt hydrate | Sigma-Aldrich | A1852 |
| Ni-nitrilotriacetate resin | Qiagen | Cat No. 30210 |
| SP Sepharose HP column | Amersham | 17-1137-01 |
| G25M column | Amersham | 17-0851-01 |
| DEAE Sepharose Fast Flow column | Amersham | 17-5154-01 |
| Deposited data | ||
| Football-shaped GroEL:ES complex encapsulating native-like folded substrate RuBisCO, cryo-EM map | This paper | EMD: 32164 |
| cryoEM structure of football-shaped GroEL:ES2 with model of native-like folded substrate, model | This paper | PDB: 7VWX |
| Experimental models: Cell lines | ||
| Not applicable to this article | ||
| Recombinant DNA | ||
| Plasmid containing His-tagged RuBisCO | Fei et al., 2014 | N/A |
| Plasmid containing GroEL | Grason et al., 2008 | N/A |
| Plasmid containing GroES | Grason et al., 2008 | N/A |
| Software and algorithms | ||
| DE_process_frames.py | Direct-Electron open source software scripts | https://www.directelectron.com/support/ |
| cryoSPARC ver 3.1 | Punjani et al., 2017 | https://cryosparc.com/ |
| Gctf | Zhang, 2016 | https://www2.mrc-lmb.cam.ac.uk/research/locally-developed-software/zhang-software/#gctf |
| UCSF Chimera | Pettersen et al., 2004 | https://www.cgl.ucsf.edu/chimera/ |
| e2spt_rotationalplot.py | Galaz-Montoya et al., 2015 | https://github.com/jgalaz84/eman2/blob/master/examples/e2spt_rotationalplot.py |
| Isolde | Croll, 2018 | https://isolde.cimr.cam.ac.uk/ |
| Phenix | Afonine et al., 2018 | https://phenix-online.org/documentation/reference/real_space_refine.html |
| Coot | Emsley et al., 2010 | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| relion 3.1 | Zivanov et al., 2018 | https://www3.mrc-lmb.cam.ac.uk/relion/index.php/Main_Page |
| Other | ||
| Quantifoil R1.2/1.3 holey carbon grids | SPI supplies | 4220C-XA |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Soung-Hun Roh (shroh@snu.ac.kr).
Materials availability
This study did not generate new unique reagents.
Method details
Purification of GroEL
Proteins used for the imaging are expressed and purified by F. Xue and G.H.Lorimer as described previously (Grason et al., 2008). Briefly, JM105 Escherichia coli cells were used to overexpress the GroEL and were harvested and resuspended in buffer containing 50 mM Tris (pH 8), 1 mM EDTA, 5 mM MgCl2, 1 mM DTT, 5 mM Ɛ-amino-n-caproic acid, and 1 mM benzamidine. After sonication, streptomycin sulfate at a final concentration of 6 mg/mL was added into cell lysates and the supernatant was applied onto DEAE Sepharose Fast Flow column (Amersham). GroEL was eluted at a conductivity of ∼28 mS employing Tris (pH 8), 1 mM EDTA, 5 mM MgCl2, 1 mM DTT and corresponding concentration of NaCl. Eluted fraction was supplemented with (NH4)2SO4 up to 65%, and the precipitate was centrifuged, resuspended in buffer (50 mM Tris (pH 7.5), 1 mM EDTA, 10 mM MgCl2, and 1 mM DTT), and desalted. Collected GroEL was concentrated to ∼10 mg/mL and supplemented with acetone, and the precipitate was centrifuged and resuspended in a buffer containing 10 mM Tris (pH 7.5), 10 mM magnesium acetate, and 1 mM DTT. The supernatant was treated with saturated (NH4)2SO4 to a final concentration of 65%. After centrifugation, pellet was collected, dissolved in 10 mM Tris (pH 7.5), 10 mM magnesium acetate, and desalted.
Purification of GroES
GroES was expressed and lysed as for GroEL, following the process described in a previous paper (Grason et al., 2008). Briefly, cleared cell lysates after centrifugation were treated with saturated (NH4)2SO4 to 65% and stirred overnight at 4°C. The precipitate was recovered by centrifugation and resuspended in a buffer containing 10 mM Tris (pH 7.5), 0.1 mM EDTA, 0.1 mM DTT. After desalting, pH of the solution was adjusted to 5.11 by adding 50 mM of NaOAc (pH 5). The fraction was further applied onto an SP Sepharose HP column (Amersham) and eluted with a buffer containing 50 mM NaOAc, 0.1 mM EDTA, 0.1 mM DTT, and 200 mM NaCl. Eluted GroES was supplemented with saturated (NH4)2SO4 to 65% and stirred overnight at 4°C. The precipitate was recovered by centrifugation, resuspended in a buffer of 10 mM Tris (pH 7.5).
Preparation of RuBisCO
Purification and denaturation of RuBisCO follows the process as described in previous paper (Fei et al., 2014). Briefly, RuBisCO from Rhodospirillum rubrum constructed with His-tag was overexpressed and lysed as for GroEL and GroES. The crude lysate was mixed with Ni-nitrilotriacetate resin (Qiagen), equilibrated with buffer of 10 mM imidazole, 20 mM Tris (pH 8), 300 mM NaCl, and incubated for 30 min on ice. After washing with a buffer containing 60 mM imidazole, 20 mM Tris (pH 8), 300 mM NaCl, RuBisCO was eluted with 250 mM imidazole and 20 mM Tris (pH 8). Eluted protein was buffer-changed to remove imidazole. After purification, 20-30 μM of RuBisCO solution was incubated with 8 M acid urea (20 mM glycine-HCl, pH 2.5) to be denatured.
Reconstitution of GroEL:ES2 and of GroEL:ES2 with RuBisCO
Reconstitution of GroEL:ES2 complex with RuBisCO follows the protocol described in the preliminary paper (Fei et al., 2014). Briefly, chemically denatured RuBisCO in 8 M urea was diluted 100-fold into the buffer containing 50 mM Tris-Acetate (pH 7.5), 20 mM MgCl2, 200 mM KCl with 1 μM purified GroEL and 2 μM GroES, resulting in a 10:1 molar ratio of substrate to GroEL:ES2. After a 30 min incubation at room temperature, 1 mM ATP, 5 mM BeCl2, and 10 mM KF were added into the reaction mixture and the mixture was vitrified.
CryoEM image acquisition
For cryoEM specimen preparation and imaging, we applied 3-μL aliquots of empty GroEL:ES2 and RuBisCO-GroEL:ES2 to glow-discharged holey-carbon Quantifoil R1.2/1.3 grids, blotted them for 2–3 s, and then plunge-froze them in liquid ethane using a Leica EM GP plunge freezer. The sample was treated with detergent, (0.1% of n-Octyl-β-D-Glucopyranoside) to obtain more distributed angular views. We transferred the grids into cartridges and loaded them into a 300 KeV JEM 3200FSC (JEOL) electron microscope with an in-column Ω filter (25 eV energy slit). We recorded images at 0.7–2.5 μm underfocus on a DE20 direct electron detector (Direct Electron) at nominal 30K magnification, corresponding to a sampling of 1.97 Å/pixel.
CryoEM map reconstruction
We corrected for drift and radiation damage using DE_process_frames.py (Direct Election Ltd.). Rest of image processing was performed in cryosparc2 (Punjani et al., 2017). Briefly, we imported corrected micrographs and performed contrast transfer function parameters estimation using Gctf (Zhang, 2016). Manually picked particles were averaged and served as template for automatic particle picking. We then performed 2D reference-free averaging and then converged particles were subjected to further 3D classification. Since maps of empty GroEL:ES2 observed empty in both chambers, we applied D7 symmetry for final reconstruction and Global non-uniform refinement resulted in maps at 4.8 and 6.6 Å resolution for bullet and football, respectively, based on the gold-standard Fourier shell correlation (FSC) at 0.143. RuBisCO-GroEL:ES2 displayed asymmetric features on substrate after 3D classification in the chamber, and thus we reconstructed multiple maps with C1 at the range of resolution between 4 and 9 Å.
Correlation coefficient calculation
To calculate the correlation coefficient of the density map of GroEL:ES2 containing RuBisCO, we used e2spt_rotationalplot.py in EMAN2 program with 1 degree of azimuth. Rotational correlation plot of either substrate-bound chamber or empty chamber was computed against that of the control map (GroEL:ES2 without RuBisCO) and compared to each other.
Model building
To derive our pseudo atomic model, we used Chimera's Fit in Map tool (Solar et al., 2019) to perform rigid-body fitting of a previously published model (PDB code: 4PKO for GroEL:ES2, (Fei et al., 2014), PDB code: 1PCQ for GroEL:ES, and PDB code: 9RUB for RuBisCO (Lundqvist and Schneider, 1993) into the cryoEM density map. We then performed flexible fitting using MDFF to improve correlation between map and model (Adams et al., 2010). To optimize the final model, we used Phenix.real_space_refine with default parameters, which quickly adjusted the fit-to-density for the subunit. We then used Coot to manually adjust loop regions that did not converge into the density (Emsley et al., 2010). We also adjusted Ramachandran outliers and amino acids with distortions in their bond lengths and angles. A final round of model optimization was performed with Phenix.real_space_refine using new parameters: global minimization, morphing, and atomic displacement (Adams et al., 2010).
Particle signal subtraction for a single ring GroELS analysis
The overall workflow for signal subtraction and localized reconstruction of a single ring of GroEL:ES is described in Figure 4 (Roh et al., 2017). The mask for signal subtraction on the single ring GroEL:ES was generated by color zone in Chimera (UCSF) (Solar et al., 2019). Subtraction of the signal for single ring GroEL:ES from the whole GroEL:ES2 particles yielded two sub-particles from one original particle. Therefore, we performed 3D classification on the set of sub-particles. Classification into 5 classes clustered two major classes whether the sub-particle has substrate or not. Then the substrate information in a single ring reconstituted to original particles for clustering one or two substrate bindings in GroEL:ES2 chambers. Then, we performed homogeneous refinement for two substrates bound GroEL:ES2 which yielded a 8.7 Å map.
Quantification and statistical analysis
Quantification, statistical analysis and the validation are implemented in the software packages used for 3-D classification, reconstruction and model refinement.
Acknowledgement
Purified and reconstituted GroEL, ES, and RuBisCO are generous gifts from F. Xue and G. H. Lorimer (University of Maryland). This work has been supported by the Korean National Research Foundation (NRF-2019M3E5D6063871, 2019R1C1C1004598, 2020R1A5A1018081, 2021M3A9I4021220, and 2020R1A6C101A183), SUHF foundation, and the Creative-Pioneering Researchers Program of Seoul National University to S.-H.R. The cryo-EM data were collected at the National Center for Macromolecular Imaging at Baylor College of Medicine (P41GM103832, R01GM079429, and S10OD021600).
Author contributions
Conceptualization, S.-H.R.; investigation, H.K., J.P., S.L., S.-H.J., M.J., and S.-H.R.; writing, H.K., J.P., S.L., and S.-H.R.; funding acquisition, S.-H.R.
Declaration of interest
The authors claim no competing interests.
Published: January 21, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.103704.
Supplemental information
Data and code availability
-
•
Cryo-EM density maps and model of GroEL:ES2 encapsulating RuBisCO are deposited in the EMDB and PDB with the accession codes PDB: 7VWX and EMD: 32164, respectively.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Adams P.D., Afonine P.V., Bunkóczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.-W., Kapral G.J., Grosse-Kunstleve R.W., et al. PHENIX: a comprehensive python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afonine P.V., Poon B.K., Read R.J., Sobolev O.V., Terwilliger T.C., Urzhumtsev A., Adams P.D. Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr. D Struct. Biol. 2018;74:531–544. doi: 10.1107/S2059798318006551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balch W.E., Morimoto R.I., Dillin A., Kelly J.W. Adapting proteostasis for disease intervention. Science. 2008;319:916–919. doi: 10.1126/science.1141448. [DOI] [PubMed] [Google Scholar]
- Balchin D., Hayer-Hartl M., Ulrich Hartl F. In vivo aspects of protein folding and quality control. Science. 2016;353:aac4354. doi: 10.1126/science.aac4354. [DOI] [PubMed] [Google Scholar]
- Chen D.-H., Madan D., Weaver J., Lin Z., Schröder G.F., Chiu W., Rye H.S. Visualizing GroEL/ES in the act of encapsulating a folding protein. Cell. 2013;153:1354–1365. doi: 10.1016/j.cell.2013.04.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clare D.K., Bakkes P.J., van Heerikhuizen H., van der Vies S.M., Saibil H.R. Chaperonin complex with a newly folded protein encapsulated in the folding chamber. Nature. 2009;457:107–110. doi: 10.1038/nature07479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croll T.I. ISOLDE: a physically realistic environment for model building into low-resolution electron-density maps. Acta Crystallogr. D Struct. Biol. 2018;74:519–530. doi: 10.1107/S2059798318002425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duijn E.V., Heck A.J.R., van der Vies S.M. Inter-ring communication allows the GroEL chaperonin complex to distinguish between different substrates. Protein Sci. 2007;16:956–965. doi: 10.1110/ps.062713607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elad N., Farr G.W., Clare D.K., Orlova E.V., Horwich A.L., Saibil H.R. Topologies of a substrate protein bound to the chaperonin GroEL. Mol. Cell. 2007;26:415–426. doi: 10.1016/j.molcel.2007.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P., Lohkamp B., Scott W.G., Cowtan K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fei X., Xiang Y., LaRonde N.A., Lorimer G.H. Formation and structures of GroEL:GroES2 chaperonin footballs, the protein-folding functional form. Proc. Natl. Acad. Sci. U S A. 2014;111:12775–12780. doi: 10.1073/pnas.1412922111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galaz-Montoya J.G., Flanagan J., Schmid M.F., Ludtke S.J. Single particle tomography in EMAN2. J. Struct. Biol. 2015;190:279–290. doi: 10.1016/j.jsb.2015.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grason J.P., Gresham J.S., Lorimer G.H. Setting the chaperonin timer: a two-stroke, two-speed, protein machine. Proc. Natl. Acad. Sci. U S A. 2008;105:17339–17344. doi: 10.1073/pnas.0807418105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammarström P., Persson M., Owenius R., Lindgren M., Carlsson U. Protein substrate binding induces conformational changes in the chaperonin GroEL. A suggested mechanism for unfoldase activity. J. Biol. Chem. 2000;275:22832–22838. doi: 10.1074/jbc.M000649200. [DOI] [PubMed] [Google Scholar]
- Hartl F.U., Hayer-Hartl M. The first chaperonin. Nat. Rev. Mol. Cell Biol. 2013;14:611. doi: 10.1038/nrm3665. [DOI] [PubMed] [Google Scholar]
- Hayer-Hartl M., Bracher A., Ulrich Hartl F. The GroEL–GroES chaperonin machine: a nano-cage for protein folding. Trends Biochem. Sci. 2016;41:62–76. doi: 10.1016/j.tibs.2015.07.009. [DOI] [PubMed] [Google Scholar]
- Lin Z., Rye H.S. Expansion and compression of a protein folding intermediate by GroEL. Mol. Cell. 2004;16:23–34. doi: 10.1016/j.molcel.2004.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorimer G.H. GroEL structure: a new chapter on assisted folding. Structure. 1994;2:1125–1128. doi: 10.1016/s0969-2126(94)00114-6. [DOI] [PubMed] [Google Scholar]
- Lundqvist T., Schneider G. Crystal structure of activated ribulose-1,5-bisphosphate carboxylase complexed with its substrate, ribulose-1,5-bisphosphate. J. Biol. Chem. 1993;266:12604–12611. doi: 10.1016/S0021-9258(18)98942-8. [DOI] [PubMed] [Google Scholar]
- McMullan G., Faruqi A.R., Clare D., Henderson R. Comparison of optimal performance at 300keV of three direct electron detectors for use in low dose electron microscopy. Ultramicroscopy. 2014;147:156–163. doi: 10.1016/j.ultramic.2014.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizohata E., Matsumura H., Okano Y., Kumei M., Takuma H., Onodera J., Kato K., Shibata N., Inoue T., Yokota A., Kai Y. Crystal structure of activated ribulose-1,5-bisphosphate carboxylase/oxygenase from green alga chlamydomonas reinhardtii complexed with 2-carboxyarabinitol-1,5-bisphosphate. J. Mol. Biol. 2002;316:679–691. doi: 10.1006/jmbi.2001.5381. [DOI] [PubMed] [Google Scholar]
- Motojima F., Motojima-Miyazaki Y., Yoshida M. Revisiting the contribution of negative charges on the chaperonin cage wall to the acceleration of protein folding. Proc. Natl. Acad. Sci. U S A. 2012;109:15740–15745. doi: 10.1073/pnas.1204547109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natesh R., Clare D.K., Farr G.W., Horwich A.L., Saibil H.R. A two-domain folding intermediate of RuBisCO in complex with the GroEL chaperonin. Int. J. Biol. Macromol. 2018;118:671–675. doi: 10.1016/j.ijbiomac.2018.06.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Punjani A., Rubinstein J.L., Fleet D.J., Brubaker M.A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods. 2017;14:290–296. doi: 10.1038/nmeth.4169. [DOI] [PubMed] [Google Scholar]
- Roh S.-H., Kasembeli M., Galaz-Montoya J.G., Trnka M., Lau W.C.-Y., Burlingame A., Chiu W., Tweardy D.J. Chaperonin TRiC/CCT modulates the folding and activity of leukemogenic fusion oncoprotein AML1-ETO. J. Biol. Chem. 2016;291:4732–4741. doi: 10.1074/jbc.M115.684878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roh S.-H., Hryc C.F., Jeong H.-H., Xue F., Jakana J., Lorimer G.H., Chiu W. Subunit conformational variation within individual GroEL oligomers resolved by cryo-EM. Proc. Natl. Acad. Sci. U S A. 2017;114:8259–8264. doi: 10.1073/pnas.1704725114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solar P., Chacon P., Lopez-Blanco J.R. Easing exhaustive rigid-body and flexible fitting in UCSF Chimera. Biophys. J. 2019;116 doi: 10.1016/j.bpj.2018.11.3081. [DOI] [Google Scholar]
- Taguchi H., Tsukuda K., Motojima F., Koike-Takeshita A., Yoshida M. BeF stops the chaperonin cycle of GroEL-GroES and generates a complex with double folding chambers. J. Biol. Chem. 2004;279:45737–45743. doi: 10.1074/jbc.m406795200. [DOI] [PubMed] [Google Scholar]
- van der Vies S.M., Viitanen P.V., Gatenby A.A., Lorimer G.H., Jaenicke R. Conformational states of ribulosebisphosphate carboxylase and their interaction with chaperonin 60. Biochemistry. 1992;31:3635–3644. doi: 10.1021/bi00129a012. [DOI] [PubMed] [Google Scholar]
- Weaver J., Jiang M., Roth A., Puchalla J., Zhang J., Rye H.S. GroEL actively stimulates folding of the endogenous substrate protein PepQ. Nat Commun. 2017;8 doi: 10.1038/ncomms15934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver J., Rye H.S. The C-terminal tails of the bacterial chaperonin GroEL stimulate protein folding by directly altering the conformation of a substrate protein. J. Biol. Chem. 2014;289:23219–23232. doi: 10.1074/jbc.M114.577205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan X., Shi Q., Bracher A., Miličić G., Singh A.K., Hartl F.U., Hayer-Hartl M. GroEL ring separation and exchange in the chaperonin reaction. Cell. 2018;172:605–617.e11. doi: 10.1016/j.cell.2017.12.010. [DOI] [PubMed] [Google Scholar]
- Ye X., Lorimer G.H. Substrate protein switches GroE chaperonins from asymmetric to symmetric cycling by catalyzing nucleotide exchange. Proc. Natl. Acad. Sci. U S A. 2013;110:E4289–E4297. doi: 10.1073/pnas.1317702110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K. Gctf: real-time CTF determination and correction. J. Struct. Biol. 2016;193:1–12. doi: 10.1016/j.jsb.2015.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zivanov J., Nakane T., Forsberg B.O., Kimanius D., Hagen W.J., Lindahl E., Scheres S.H. New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife. 2018;7:e42166. doi: 10.7554/eLife.42166. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Cryo-EM density maps and model of GroEL:ES2 encapsulating RuBisCO are deposited in the EMDB and PDB with the accession codes PDB: 7VWX and EMD: 32164, respectively.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.