

Abstract
A high-throughput method was developed for the automated enrichment of newly synthesized proteins (NSPs), which are labeled metabolically by substituting methionine with the “click-able” analogue azidohomoalanine (AHA). A suitable conjugate containing a dibenzocyclooctyne (DBCO) group allows the specific selection of NSPs by a fast 1 h click chemistry-based reaction with AHA. Through an automated pipetting platform, the samples are loaded into streptavidin cartridges for the selective binding of the NSPs by means of a biotin bait contained in the conjugate. The enriched proteins are eluted by a reproducible chemical cleavage of the 4,4-dimethyl-2,6-dioxocyclohexylidene (Dde) group in the conjugate, which increases selectivity. The NSPs can be collected and digested in the same well plate, and the resulting peptides can be subsequently loaded for automated cleanup, followed by mass spectrometry analysis. The proposed automated method allows for the robust and effective enrichment of samples in 96-well plates in a period of 3 h. Our developed enrichment method was comprehensively evaluated and then applied to the proteomics analysis of the melanoma A375 cell secretome, after treatment with the cytokines interferon α (IFN-α) and γ (IFN-γ), resulting in the quantification of 283 and 263 proteins, respectively, revealing intricate tumor growth-supportive and -suppressive effects.
Keywords: proteomics, mass spectrometry, secretome, newly synthesized proteins, Bravo AssayMAP, azidohomoalanine, protein enrichment, INF, melanoma
Introduction
Mass spectrometry (MS) is the key technology for protein analysis1 enabling a precise and deep coverage of the dynamic proteome.2,3 However, limitations still exist as, for example, small proteome changes, such as newly synthesized proteins (NSPs) in complex intracellular environments, which are still difficult to quantify accurately. The same holds true for secreted proteins, especially in the protein-rich culture media required for cell growth. The accurate study of these less abundant subsets of the proteome is crucial for our understanding of cellular communication and response to external stimuli. Improvements in this area will impact diverse research areas such as the field of cancer immunology and the tumor microenvironment, where secreted factors play essential roles.4 Until recently, a common approach for the identification of secreted proteins by MS was depletion of fetal bovine serum (FBS). However, cells depleted of serum activate many survival pathways and even apoptosis, independently of the experiment performed, which significantly affects the model system and related conclusions drawn.5,6
To deal with these challenges, bioorthogonal chemistry has been the tool of choice, specifically in the form of ‘click chemistry’.7,8 Several studies have focused on the use of the “click-able” analogue of methionine, azidohomoalanine (AHA),9−12 often combined with a version of stable isotope labeling by amino acids in cell culture (SILAC),13−16 to be compatible with the use of FBS, since AHA allows for protein enrichment by click chemistry and the pulsed version of SILAC (p-SILAC) marks NSPs for differential readout by MS.14 Several recent strategies have focused specifically on the improved detection of the actual biotinylated peptides using either biotin antibodies17,18 or alternative click-able phosphonate-handles.19 These methods are performed at the peptide level and provide an additional confirmation that detected peptides originate from actual newly synthesized proteins, at the cost of a reduction in protein identifications and unique peptides available for quantification. Moreover, recent developments in mammalian bioorthogonal labeling now enable the enrichment of cell-type-specific proteomes, including newly synthesized and secreted proteins.20
Overall, these approaches have many benefits; however, current variations also share several limitations, including limited throughput, long and complex sample preparation procedures, and limited flexibility. Therefore, here, we set out to develop an automated AHA-based protein enrichment method using a cleavable biotin probe, which deals with some of these limitations by improving simplicity of sample preparation and the overall speed and throughput, as well as providing flexibility for further adaptions, like the analysis of post-translational modifications. Central to these developments was the selection of a suitable conjugate that allowed (i) the robust enrichment of AHA-labeled proteins by click chemistry from highly complex backgrounds, (ii) in an automated manner, (iii) with increased reproducibility, and (iv) throughput. Specifically, we show that our method simplifies the sample preparation steps required, considerably reducing preparation time and increasing reproducibility through automation. This allows for high-throughput biological experiments, including multiple treatments and controls with a minimum increase in total enrichment time. We cross-validated identified proteins with complementary labeling strategies and enrichment controls. Combined, this work demonstrates the potential of our automated protocol for the identification of changes in the proteome in highly complex biological samples, which we showcase by analyzing the difference in melanoma cell secretomes upon IFN-α and IFN-γ stimulation.
Materials and Methods
Cell Culture
A375 cells were first grown in 10 cm dishes until 70% confluence at 37 °C in 5% CO2 in RPMI (Lonza) supplemented with 10% FBS, glutamine, penicillin, and streptomycin. Before the next step, medium was removed and the cells were washed once with warm PBS.
Pulse-Labeling with AHA and SILAC
Cells were incubated for 30 min in custom-made RPMI (Gibco) depleted of arginine, lysine, and methionine and supplemented with 10% dialyzed FBS (Gibco), glutamine, penicillin, and streptomycin. Next, the same medium was supplemented with 0.1 mM l-γ-azidohomoalanine (AHA) (Bachem) or l-methionine as control. Additionally, for p-SILAC experiments, the following reagents were used per labeling condition: intermediate (200 μg/mL [13C6] l-arginine, 40 μg/mL [4,4,5,5-D4] l-lysine (Cambridge Isotope Laboratories)) or heavy (200 μg/mL [13C6, 15N4] l-arginine, 40 μg/mL [13C6,15N2] l-lysine (Cambridge Isotope Laboratories)). For label-free experiments, 200 μg/mL l-arginine and 40 μg/mL l-lysine (Cambridge Isotope Laboratories) were used.
Treatment with IFN-γ and IFN-α
Cells were treated with 50 ng/mL of interferon γ (Peprotech) or α (Peprotech) in combination with the amino acids required for each experiment as described above. Otherwise, the cells were left untreated. For the protocol optimization, the cells were treated for 12 h. For the label-free experiments, secretome was collected after 24 h of treatment.
Sample Preparation for Method Development
Proteomes of melanoma cells were treated with interferon γ (see the above section) or left untreated as controls. The cells were labeled in culture with AHA and p-SILAC to mark NSPs during treatment. Samples were pooled to obtain enough material for technical replicates needed for optimization of the method parameters. To create these pooled samples, treated cells and controls were labeled with intermediate or heavy SILAC amino acids, and samples containing different labels were combined. Next, the samples were split in technical replicates and enriched.
Sample Collection
For secretome enrichment, the medium was collected after the indicated times and centrifuged for 5 min at 1000g to pellet remaining cells. After the supernatant was transferred to a new tube, protease inhibitors were added (Roche) and samples were frozen at −80 °C until further use. For cellular proteomes, the cells were washed three times with PBS and detached with trypsin (Gibco). The cells were spun down, PBS was removed, and the samples were frozen at −80 °C until further use.
Preparation of AHA-Labeled Proteins for Enrichment
For secretome samples, the medium was thawed to room temperature and concentrated in 15 mL 3 kDa Amicon tubes (Millipore) to ∼2 mL (4 °C) following manufacturer instructions. Urea was added to a final concentration of 8 M. The samples were transfered to a new tube and sonicated in a water bath sonicator for 15 cycles of 1 min (30 s on/30 s off). The samples were maintained on ice until further use. For cellular proteomes, the cells were thawed to room temperature and resuspended in 8 M urea and 50 mM ammonium bicarbonate. Protease inhibitors were added, and the cells were sonicated for 15 cycles of 1 min (30 s on/30 s off) and kept on ice until further use.
Click Reaction
Cellular samples were transfered to 3 kDa Amicons, and secretome samples were kept in the same tubes. AHA-labeled proteins were “clicked” by adding 40 μM DBCO-Dde-(PEG-4)-Biotin conjugate (Jena Bioscience) and incubated in the dark while rotating the tubes for 1 h at room temperature. Next, the samples were buffer-exchanged three times with PBS (4 °C, 4000g) to eliminate excess of conjugate. Finally, the samples were concentrated to a volume of ∼250 μL or lower before being transfered to the automated platform. The samples grown with media containing methionine instead of AHA followed all steps as labeled samples.
Automated Enrichment of Clicked Proteins
Biotinylated proteins were enriched using streptavidin (SA-W) cartridges (Agilent Technologies) in the automated AssayMAP Bravo Platform (Agilent Technologies). The protocol for affinity purification included in the platform was used as a scaffold using the following settings. Cartridges were primed with 100 μL of PBS at 300 μL/min. Equilibration was done with 50 μL of PBS at 10 μL/min. The samples were loaded at 5 μL/min unless indicated otherwise. Next, loaded cartridges were first washed with 200 μL of buffer 1 (1 M NaCl in PBS) at 10 μL/min followed by a second wash with 50 μL of buffer 2 (100 mM PBS) at 10 μL/min. Cup washes 1 and 2 were done with 25 μL of the respective buffers. For the elution of the bound proteins, the Dde group was cleaved using a 2% hydrazine solution prepared in 100 mM PBS from a 35% stock (Sigma). The elution was performed at 0.4 μL/min with enough hydrazine solution to maintain the process for the desired reaction time, which was 90 min unless indicated otherwise (e.g., 36 μL for 90 min). Eluate was collected in 100 mM PBS. To elute the remaining proteins from the cartridges, three cycles of syringe washes were performed followed by a second elution with 25 μL of 5% acetic acid at 5 μL/min. The acid eluate was collected in 25 μL of 50 mM ammonium bicarbonate.
Digestion and Cleanup of Peptides
Eluted proteins were reduced, alkylated, and digested simultaneously by addition of 100 μL of buffer containing 10 mM tris(2-carboxyethyl)-phosphinehydrochloride, 40 mM chloroacetamide, 100 mM TRIS pH 8.5, 50 mM ammonium bicarbonate, and 1 μg trypsin (Gold, Promega). Plates were incubated overnight at 37 °C. Digestion was stopped with 2% formic acid (FA). The samples were cleaned using C18 cartridges (Agilent Technologies) in the automated AssayMAP Bravo Platform. The protocol for peptide cleanup included in the platform was used as scaffold using the following settings. Cartridges were primed with 100 μL of 80% acetonitrile (ACN)/0.1% FA at 300 μL/min. Equilibration was done with 50 μL of 0.1% FA at 10 μL/min. The sample was loaded at 5 μL/min. Cup wash (25 μL) and the internal cartridge wash (50 μL at 10 μL/min) were performed with 0.1% FA. Peptides were eluted with 50 μL of 80% ACN/0.1% FA at 5 μL/min. Samples were dried in vacuo and stored at −80 °C until further use.
Liquid Chromatography–Mass Spectrometry (LC-MS/MS)
Injections were randomized and the same proportion of each sample was analyzed, maintaining the same number of injection replicates intraexperiment. The samples were reconstituted in 10% formic acid and analyzed by nano-LC-MS/MS on an Orbitrap Q-Exactive HF or HF-X (Thermo Fisher Scientific) coupled to an Agilent 1290 Infinity System (Agilent Technologies) operating in reverse phase equipped with a Reprosil pur C18 (Dr. Maisch) trap column (100 μm x 2 cm, 3 μm) and a Poroshell 120 EC C18 (Agilent Technologies) analytical column (75 μm x 50 cm, 2.7 μm). After trapping with 100% solvent A (0.1% FA in H2O) for 10 min, peptides were eluted with a step gradient consisting of 95 min from 7% to 44% solvent B (0.1% FA, 80% ACN), 3 min from 44% to 100%, and 1 min at 100%. The mass spectrometer was operated in a data-dependent mode. Full-scan MS spectra with a mass range of 375–1600 m/z were acquired in profile mode with a resolution of 60 000. The filling time was set to a maximum of 20 ms with an AGC target of 3 × 106. The most intense ions (up to 15 for optimization experiments and up to 12 otherwise) were selected for fragmentation. A normalized collision energy of 27 was used and fragment spectra were recorded with a resolution of 30 000 in profile mode. The fragments were measured after reaching an AGC target of 1 × 105 or 50 ms accumulation time for the optimization experiments and 100 ms otherwise. The dynamic exclusion window was set at 16 s.
Data Analysis
Raw files were analyzed in one single search using the MaxQuant software package (version 1.6.3.3).21 Samples were grouped by type of experiment. The search was performed against the Uniprot database for Homo sapiens (20 409 sequences, downloaded October 17, 2018) using trypsin/P as enzyme and allowing maximum two miss cleavages. For all groups: cysteine carbamidomethylation was set as a fixed modification and oxidized methionine, protein N-terminal acetylation, and substitution of methionine for l-γ-azidohomoalanine were set as variable modifications. For SILAC experiments: intermediate and heavy arginine and lysine were added as medium and heavy labels. The protein and PSM FDR were set to 0.01. Protein quantification was set to use a minimum of two counts using only unique peptides. Match between runs and requantified were disabled in all groups. Potential contaminants suggested by the software were filtered out. Intensity values were log 2 transformed. Normalization was avoided to retain and evaluate the quantitative effect of the enrichment, and only raw intensity values were used.
Volcano plots were generated after a two-sided t-test, using Perseus (version 1.6.10.0).22 Statistical tests were performed with GraphPad Prism 8, and an adjusted p-value of 0.05 was considered significant when pertinent. Graphs were generated with GraphPad Prism and BioVenn.23 Gene ontology enrichments were performed with GOrilla,24 and the totality of protein identifications obtained in the search was used as a background set.
Results and Discussion
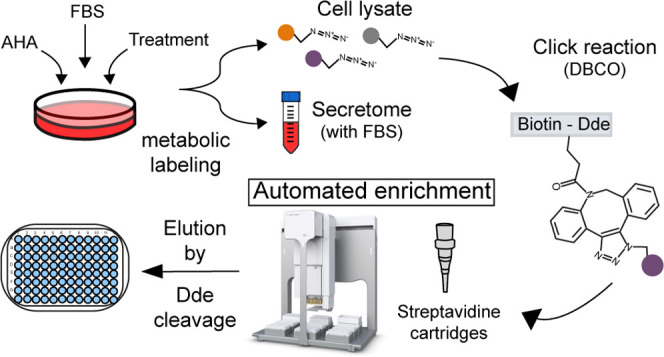
To align the existing methodology for MS-based analysis of newly synthesized and secreted proteins with the sample size and throughput required in biomedical and translational research, we developed an automated enrichment strategy with simplified sample preparation. For this, we selected a molecule for chemoselective ligation that allowed us to robustly automate the enrichment procedure and reduce sample preparation time. To fit with these requirements, we opted for a commercial conjugate containing four regions (Figure 1A): a dibenzocyclooctyne (DBCO), which allows for a fast copper-free alkyne-azide click chemistry reaction, thereby reducing reaction times up to 18-fold compared to a regular alkyne; a 4,4-dimethyl-2,6-dioxocyclohexylidene (Dde) group, which provides a cleavage site to elute the proteins from the enrichment matrix;25 biotin for quick capture of the proteins bound to the DBCO by streptavidin; and finally, a chain of polyethylene glycol (PEG) for increased hydrophilicity, beneficial in water-based solvents used in common proteomics sample preparation protocols. Moreover, the (PEG-4) chain provides distance between the protein and the streptavidin–biotin complex, increasing the efficiency of the affinity capture. The use of biotin provides the flexibility to select highly efficient automation tools based on streptavidin capture. Other alkynes could benefit from the automated protocol described here; however, further optimization of the binding and elution steps would be required.
Figure 1.

Enrichment platform for AHA-labeled proteins. (A) Chemical structure of the conjugate employed to capture AHA-labeled proteins with the four functional regions that facilitate the success of the highlighted protocol. Red arrows indicate point of chemical cleavage. (B) Schematic representation of the enrichment protocol showing the main steps from cell culture treatment to data analysis. For a more detailed protocol, see the Materials and Methods section.
Automated Protocol for Enrichment of NSPs
Newly synthesized proteins can be enriched from both intracellular and extracellular sources. Here, to set up our automated method and test its reproducibility and robustness, we created a large stock of labeled newly synthesized protein, which allowed us to perform initial experiments under controlled conditions. As shown in Figure 1B, the first steps are performed manually. After cell lysis, secretome samples are filter-concentrated by centrifugation, after which AHA-labeled proteins are clicked to the DBCO-Dde-(PEG-4)-biotin conjugate during a 1 h incubation at room temperature. We chose 1 h as the optimal time point after we tested the conjugate reaction time from 30 min to 1.5 h, as part of the process of optimization. Next, samples are buffer-exchanged and filter-concentrated by centrifugation to ≤250 μL, after which they are ready for enrichment. For the automated enrichment, we chose the streptavidin cartridges for the AssayMap Bravo platform as capturing matrix for the biotinylated proteins. The cartridges allow for batch-consistent capture of biotinylated proteins and are capable of handling up to 100 μg of bound protein in various pH ranges. The chosen platform is a solution-handling robot, capable of handling small sample volumes, and it allows for a custom setup of solvent use and drawing speed, down to <1 μL per minute. Moreover, the platform allows in-plate protein digestion and desalting and can be combined with other automated protocols like phosphopeptide enrichment.26 Overall, the enrichment protocol developed here enables the simultaneous processing of 96 samples in an automated and reproducible manner in just 3 h and consists of the following steps: First, clicked cell lysates are loaded onto the streptavidin cartridges followed by a series of automated washing steps for the removal of possible contaminants and nonspecific binding proteins. Then, proteins are eluted from the resin using a chemical cleavage reaction with hydrazine, which targets specifically the Dde group of the conjugate. This minimizes elution of nonspecific binders and allows for reproducible release of the specifically captured proteins. Each of these steps have been optimized carefully and are described in the following sections. The protocol is finalized with the digestion, automated desalting, and reconstitution of the enriched proteins for MS analysis, all in the same sample plate to reduce sample loss.
Loading Speed of the AHA-Labeled Proteins
The first important optimization step was the loading speed onto the streptavidin-embedded cartridges since proteins need sufficient time of contact to be captured, and this should be in balance with the required sample throughput. This is especially relevant in complex samples like FBS-embedded secretomes, where the actual proteins of interest are considerably less abundant than the complex background. Here, the speed at which this high amount of protein passes the matrix of the cartridge may decrease the interaction efficiency and thus the chance of capture. A slower speed increase time of contact, however, increases the total enrichment duration. Therefore, we set out to test the optimal speed for efficient protein capture based on manufacturer experience with similar samples. We tested both 2 and 5 μL/min loading speed and enrichment efficiency was determined by comparing technical triplicates of AHA-labeled p-SILAC cell lysates. For this, two samples with either medium or heavy SILAC labels were pooled and split in technical replicates, one triplicate was used to test each speed. In the absence of FBS, as any intracellular sample, this resulted in the identification and quantification of more than 964 NSPs in at least one sample, of which 81% were shared between the two loading speeds (Figure 2A). The unique proteins found were 8 and 12% of the total at 2 and 5 μL/min, respectively. Because of the comparable results, we chose 5 μL/min as the loading speed for further experiments to keep overall analysis time as short as possible without compromising the enrichment capacity. Sample loading now takes a mere 50 min for a standard 250 μL sample.
Figure 2.

Optimization of the automated protocol. (A) Comparison of two different loading speeds based on the number of identified NSPs after loading three sample replicates. Sample is a cell lysate. (B) Effect of increasing sample complexity, achieved by the addition of FBS to the cell lysate, to imitate a secretome sample obtained in complete cell culture media. Newly synthesized proteins from three sample replicates loaded at 5 μL/min are shown. (C) Elution of captured proteins by either chemical cleavage of the Dde group present in the conjugate through incubation with 2% hydrazine, PBS wash, or denaturing biotin with 5% acetic acid. Sample is a cell lysate. The error bar represents the standard error of the mean of four replicates. The solid red filling represents NSPs enriched consistently in three or more replicates of the same condition. (D) NSPs obtained in C organized to showcase the first elution step in which they eluted consistently in three replicates.
Effect of Increased Sample Complexity
To assess the performance of our approach when handling the complexity typically observed when analyzing secretomes, we spiked technical sample replicates in an equal volume of FBS (1:1). Triplicates were analyzed with and without FBS, and their content of NSPs was compared. As can be seen in Figure 2B, from 979 newly synthesized proteins, 66% was shared between both conditions in at least one replicate, despite the significant increase in matrix complexity. As expected, this increase in complexity does lower the total number of identifications. However, with 735 unique NSPs identified, only 18% less than the condition without FBS, our method demonstrated to be compatible with typical conditions expected for secretome analysis. Moreover, the reproducibility observed between the two conditions is also very comparable, with 61% overlap among replicates of the same condition in the absence of FBS and 59% when FBS is present (data not shown). It is clear from this experiment that, despite the challenge of adding a matrix of highly abundant proteins like FBS, our automated approach can handle such complex samples, while maintaining reproducibility.
Selective Elution of Enriched Proteins
Next, we set out to characterize and optimize the elution of the captured proteins from the streptavidin cartridges by chemical cleavage of the Dde group. Before this step, the cartridges were washed with 1 M NaCl solutions to remove unspecific binders. Although the biotin–streptavidin interaction allows for more stringent washing, we chose milder conditions to secure the integrity of the cartridges. It is of interest to test more washing conditions in the future. For the elution, we chose a cleavable molecule to add flexibility and versatility to the elution, since this allows the controlled release of the proteins,25 and it leaves most of the click-able molecule on the column, reducing background. The Dde group was chosen for its known resistance to acidic conditions,27 which is common in protein preparations, and the convenient possibility to be cleaved by hydrazine in PBS, both compatible with further preparation steps (e.g., tryptic digestion) and common proteomics setups. To evaluate the cleavage of the Dde, we divided a pool of labeled cells in four technical replicates and measured the released proteins after different incubation times. As in previous experiments, we used the output of SILAC-labeled proteins as a measure for the capacity of each step to elute specifically enriched proteins. Each replicate was loaded onto the streptavidin cartridges, washed, and eluted using 2% hydrazine. We sampled after 60, 90, and 120 min elution, using a constant unidirectional flow of 0.4 μL/min of 2% hydrazine, and inspected the acquired data for three characteristics: (1) overall protein identifications; (2) reproducibility of identifications based on their detection in at least three replicates per elution step; and (3) the first time of appearance of each detected protein (Figure 2C,D). An average of 1010 NSPs were released from the cartridges in the first 60 min of elution, with 91% identified in triplicate (Figure 2C). After collection of the first eluate, two additional cleavage steps of 30 min were added to complete 90 and 120 min. Each of these subsequent steps released a decreasing number of proteins, while maintaining high reproducibility, with 86% of proteins identified in three replicates in the second elution step and 81% in the third. Importantly, as can be seen from Figure 2D, most of the total number of proteins identified were already observed in the first elution (93%). Additionally, we tested whether the dead volume in the cartridges still contain protein, for this, one wash with PBS was performed after the 120 min time point. This wash resulted in an average of 51 protein identifications, including only one unique protein (Figure 2D). Finally, we performed an acetic acid-based elution, to elute all proteins remaining on the cartridge after the previous steps. This final step resulted in the identification of an average of 288 proteins (92% previously eluted) but showed high inter-replicate variability. These data show that longer hydrazine incubation, PBS wash, and acetic acid elution hardly contributed to the total number of reproducible identifications. After 90 min of elution with hydrazine, 98% of the proteins are identified in triplicate in at least one step (Figure 2D). Overall, these results show that the elution of enriched proteins based on the cleavage of Dde is highly reproducible and consistent over time. Moreover, most proteins are released consistently over the first hour. Based on these results, 90 min was chosen as the default cleavage time for further experiments.
Label-Free Identification of Enriched Proteins
Taking advantage of the observed reproducibility of our automated method, we next set out to test the performance of a label-free secretome enrichment. In all previous optimization steps, we made use of an AHA-labeled intracellular proteome in which NSPs were additionally labeled using p-SILAC. Although SILAC labeling is the ideal approach to simultaneously validate the identifications as newly synthesized, we reasoned that for many experimental approaches, the use of p-SILAC can be cumbersome and providing an alternative could be beneficial. Since our method enables the highly reproducible and selective enrichment of proteins that contain SILAC labels, we here wanted to test the strength of our method in a label-free quantification approach and simultaneously benchmark this method to a p-SILAC counterpart. For this, we set up an experiment where we stimulated melanoma cells with IFN-α and IFN-γ in the presence of AHA and included unstimulated cells as treatment control and methionine in place of AHA to produce enrichment controls (Figure 3A). Furthermore, we took along a p-SILAC-labeled version of the IFN-γ stimulation. This labeled counterpart was necessary to validate the proteins we identify as newly synthesized since label-free methods cannot do this distinction on their own. All samples were prepared in quadruplicate, making a total of 28 samples, from which the supernatant was concentrated and secreted proteins were enriched using our optimized protocol.
Figure 3.

Performance of the automated method in a label-free approach to study melanoma secretory response to IFN treatment. (A) Schematic representation of the workflow for label-free identification of NSPs. The plates were seeded, treated, and collected simultaneously in one experiment and secretomes were enriched with the workflow shown in Figure 1B. The control in the metabolic labeling section consists of the substitution of AHA with methionine in the culture media. The controls in the 24 h treatment section consist of untreated samples. The secretomes correspond to the collection of the complete media (including FBS) after the treatment, without detachment of the cells. (B) Proteins obtained by label-free identification in the presence or absence of AHA are compared to the p-SILAC proteins filtered by the presence of the label. The asterisk (*) area is shared between p-SILAC and methionine control. (C) Linear correlation between 240 mutual proteins obtained after AHA enrichment by the label-free and p-SILAC approaches. Proteins shown have at least two values per condition. A linear fit has been applied (red line) with an R2 = 0.85 and a slope of 0.98. (D) Statistical comparison of 144 label-free proteins identified in both the AHA-labeled proteins and methionine controls (unspecific binding), in at least three replicates. Proteins above the curve are significantly over-represented in the presence of AHA (right) or methionine (left). Proteins in red are cross-validated as newly synthesized by the results in the p-SILAC dataset. Curve represents FDR = 0.05.
To benchmark our label-free quantification, we compared the results of the label-free melanoma secretome against the p-SILAC-based method, both AHA-labeled and IFN-γ-treated, and simultaneously tested the usefulness of the methionine control. We identified 568 proteins in the label-free AHA-labeled (LF-AHA) IFN-γ-treated samples and 339 in the p-SILAC (pS-AHA) counterpart, which we compared to 282 proteins identified in the methionine control. As can be seen from Figure 3B, using the SILAC label as a marker for NSPs results in less background proteins, although still 33% of pS-AHA overlap with the methionine control. This result also reaffirms the need to use better ways to question the background proteome, instead of the common practice to filter them out as contaminants. Reassuringly, the majority (87%) of the pS-AHA identified proteins were present in the LF-AHA dataset.
Next, we compared the shared identifications between LF-AHA and pS-AHA and assessed the quality of the quantitative component in the label-free dataset. We took the proteins present with at least two values in both groups and analyzed the linearity, correlation, and CV of LF-AHA using the pS-AHA as a reference. The analysis revealed a linearity of 0.85 with a slope of 0.98 (Figure 3C), a significant correlation between mean intensities (Pearson = 0.92, p-value <0.0001), and no significant difference between their CV (p-value = 0.86, Supporting Table S1). This detailed comparison confirmed the reproducibility and robustness of our method, and the reliability of the quantitative findings in the label-free approach.
Given the overlap of proteins identified in pS-AHA and the methionine control, we reasoned a protein can be both an unspecific binder and truly enriched under the condition tested. Therefore, we did not want to simply exclude all proteins identified in the methionine control but rather use the methionine controls to filter proteins found significantly more abundant in the LF-AHA condition. First, we selected the proteins present in LF-AHA and methionine control in at least three replicates per dataset, resulting in 144 shared proteins. Then, to identify which proteins have a significant enrichment in the AHA dataset, we applied a two-sided T-test with an FDR of 0.05 as a significance threshold. As shown in Figure 3D, 78 proteins are significantly enriched under the LF-AHA condition compared to their presence in the methionine control, and importantly, from those, 60 proteins (77%) are also identified in the p-SILAC method. From these cross-verified proteins, 92% (55/60) are associated with the gene ontology (GO) term “extracellular” (Supporting Table S1). Most secreted proteins are expected to be associated with this term and its variants, although the secretome also includes all secretory vesicles and their content, which can be cytoplasmic in origin.
Next to the pS-AHA shared proteins, there were 18 proteins significantly enriched versus the methionine control that were unique to our LF-AHA method, which brings the total of uniquely identified LF-AHA proteins to 153, which is 3.5 times more than our pS-AHA experiment (Figure 3B). These results show that the use of a specific control for unspecific binding (e.g., methionine control) can substantially increase the number of newly enriched proteins that can be identified. This, however, is often avoided due to the increase in the number of samples to be processed. Our automated method allows processing of multiple types of control samples to guide data analysis and filter the resulting complex datasets with no increment on the enrichment time and minimum increase in total processing time. Overall, our analysis confidently showcased the compatibility of our automated enrichment protocol of AHA-labeled proteins with label-free protein quantification.
Melanoma-Secreted Response to IFN
After validating our label-free quantitative approach, we set to use the label-free dataset to study the secretome response of melanoma cells to IFN-α and IFN-γ stimulation. This comparison is relevant since these interferons have emerged as central regulators of interactions between tumors and the immune system.30 Their effect on tumor cells and use as clinical treatments has been extensively studied,31,32 however, mostly based on the intracellular response of the cancer cells, which omits the protein-mediated influence of the tumor on its environment.
Our here described automated protocol enables the handling of IFN stimulated and control secretomes, produced under FBS-rich culture conditions, together with their respective enrichment controls in one single batch, with minimum manual manipulation, minimizing error and increasing reproducibility. We performed a stringent, filtering procedure, specifically (i) we selected the proteins significantly enriched in the AHA samples compared to their methionine controls, (ii) we added the unique identifications present in each LF-AHA dataset, and finally, (iii) from this dataset, we selected the proteins present in at least three replicates (Supporting Table S1). This resulted in the selection of 283 high-quality identifications for the IFN-α, 263 for IFN-γ, and 195 for the untreated control. GO enrichment showed terms associated with the extracellular space, lumen of vesicles, or membranes, but no cytosolic or nuclear markers (Figure 4A and Supporting Table S1).
Figure 4.

Melanoma secretory response to IFN-α and IFN-γ measured by our label-free automated enrichment approach. Proteins analyzed were significantly enriched against an enrichment control and selected by their presence in at least three replicates. (A) Functional enrichment analysis for the secretome of IFN-α-treated A375 melanoma cells. For the comparable results of IFN-γ and control, see Supporting Table S1. The circle size reflects the statistical significance of a given term. Distance between terms represents similarity of the terms. Analysis was performed with GOrilla, and the plot was made with REViGO28 using p-values and similarity set to small (0.5). (B) Statistical comparison of 213 label-free proteins identified in both IFN treatments. Proteins above the curve are significantly upregulated after treatment with IFN-α (right, red) or IFN-γ (left, purple). Curve represents FDR = 0.05. (C) Schematic summary of the major biological processes upregulated and downregulated by the IFN treatments, in comparison to untreated cells. Proteins not connected by lines contribute to the biological process but are not known to be connected. Network relationships and analysis of GO enrichment were retrieved from STRING.29 Terms represented have FDR <0.05.
To study the changes on the secretome after the IFN treatments, we first tested for differential regulation between the shared proteins of the IFN treatments (Figure 4B), resulting in eight upregulated proteins in the IFN-γ and 20 in the IFN-α treatment. Then, we identified treatment-specific up- and downregulated proteins compared to the untreated melanoma cells. This resulted in 34 up- and 16 downregulated proteins for the IFN-γ treatment and 54 up- and 16 downregulated proteins for the IFN-α treatment (Supporting Table S1).
The IFN-γ treatment seems to generate a mixed profile with strong tendency toward tumor growth progression. The upregulated proteins quantified show the enhancement of three processes associated with tumor mobility and recruitment of the immune system for its negative modulation (Figure 4C). Representative of those processes, MMP1 is associated with rapid tumor growth progression in melanoma patients,33 CSF1 is known to induce pro-tumorigenic modulation of macrophages in melanoma,34 and A2M, part of the network of MMP1, has been associated with immunomodulation in cancer.35 Moreover, the secretion of ICAM1 by melanoma cells has been found to inhibit non-MHC-restricted cytotoxicity.36 This treatment also upregulates the protein C1R and induces the production of C1S, both reported to degrade collagen and correlate with cancer progression and metastasis.37 Contrary to this pro-tumor growth profile, IFN-γ treatment downregulated four proteins active in a process that has been associated with inducing metastasis in BRAFV600E melanoma cells, the “glycosaminoglycan catabolic process” (Figure 4C).38
In the case of the IFN-α treatment, we identified a mixed profile with strong tendency toward tumor growth suppression. To start, among the proteins specifically secreted in this treatment, we found IL24, which inhibits tumor growth and metastasis in melanoma and other cancers.39 In addition, 12 proteins found upregulated upon the IFN-α treatment, are associated with antigen presentation via the major histocompatibility complex I (MHC I) (Figure 4C). The enhancement of antigen presentation is part of the tumor-suppressive effects associated with IFN-α.40 A key player in this process is the protein calnexin (CANX), found specific to the IFN-α treatment, which is a chaperone responsible for folding of glycosylated proteins that are crucial in the maturation of MHC I.41 IFN-α also downregulated the expression of proteins like MIA,42 and IGFBP2,43 known to be associated with metastasis and poor prognosis in melanoma patients. Further supporting the suppressive effect of IFN-α, we found biological processes that have been reported to correlate with invasiveness of melanoma cells downregulated, like “peptidyl-proline hydroxylation” and “positive regulation of locomotion”44,45 (Figure 4C). Conversely, several upregulated proteins are also associated with tumor growth, like those involved in the “carbohydrate catabolic process”. This process has been connected to metastatic behavior of melanoma and other cancer cells,46 and although it is mainly a cytosolic process, proteins like ENO1 are also known to have extracellular metastatic functions.46
The mixed profiles identified with our secretome analysis are common in cancer research and are a reflection of the complexity of studying the tumor–immune system interactions. Secretome research is especially difficult to interpret since, until recently, most studies were performed on intracellular proteins and when secreted proteins were studied, these were either overexpressed, studied as individual cases, or from cells grown under low or starved FBS conditions.
Conclusions
Our optimized automated enrichment protocol allows for the high-throughput generation of rich and confident datasets, like the one described above. The quality and high reproducibility of the generated data supports confident biological interpretations and careful selection of candidates for follow-up studies, while minimizing analysis time (3 h of total enrichment time). With the ever-increasing accessibility to automated sample-handling platforms, we expect the methodology described here to become a powerful approach for quantitative proteomic analysis of newly synthesized and secreted proteins.
Acknowledgments
The authors thank Dr. Barbara Steigenberger for helpful discussion during the process of selecting the conjugate used in their studies and Agilent for providing the Streptavidin (SA-W) cartridges. This research was part of the Netherlands X-omics Initiative and partially funded by NWO (Project 184.034.019) and the Horizon 2020 program INFRAIA project Epic-XS (Project 823839). They also thank the University of Costa Rica for partial support to D.V.-D.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jproteome.1c00743.
Detailed description of materials in XSLX file (PDF)
Extracted datasets used in Figures 2A–D, 3B–D, and 4A–C (Table S1); coefficient of variation data for Figure 3C (Figure 3C + CV); results of the GO enrichment used in Figure 4A and additional results for IFN γ treatment and control (GO F4A + IFNγ and control); complete list of secreted proteins changing after treatment with IFN α and γ (treatment-specific changes) (XLSX)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Author Contributions
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE47 partner repository with the dataset identifier “PXD025255”.
The authors declare no competing financial interest.
Supplementary Material
References
- Altelaar A. F. M.; Munoz J.; Heck A. J. R. Next-Generation Proteomics: Towards an Integrative View of Proteome Dynamics. Nat. Rev. Genet. 2013, 14, 35–48. 10.1038/nrg3356. [DOI] [PubMed] [Google Scholar]
- Richards A. L.; Merrill A. E.; Coon J. J. Proteome Sequencing Goes Deep. Curr. Opin. Chem. Biol. 2015, 24, 11–17. 10.1016/j.cbpa.2014.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger T.; Wehner A.; Schaab C.; Cox J.; Mann M. Comparative Proteomic Analysis of Eleven Common Cell Lines Reveals Ubiquitous but Varying Expression of Most Proteins. Mol. Cell. Proteomics 2012, 11, M111.014050 10.1074/mcp.M111.014050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makridakis M.; Vlahou A. Secretome Proteomics for Discovery of Cancer Biomarkers. J. Proteomics 2010, 73, 2291–2305. 10.1016/j.jprot.2010.07.001. [DOI] [PubMed] [Google Scholar]
- Pirkmajer S.; Chibalin A. V. Serum Starvation: Caveat Emptor. Am. J. Physiol.: Cell Physiol. 2011, 301, C272–C279. 10.1152/ajpcell.00091.2011. [DOI] [PubMed] [Google Scholar]
- Shin J.; Rhim J.; Kwon Y.; Choi S. Y.; Shin S.; Ha C.-W.; Lee C. Comparative Analysis of Differentially Secreted Proteins in Serum-Free and Serum-Containing Media by Using BONCAT and Pulsed SILAC. Sci. Rep. 2019, 9, 3096 10.1038/s41598-019-39650-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sletten E. M.; Bertozzi C. R. Bioorthogonal Chemistry: Fishing for Selectivity in a Sea of Functionality. Angew. Chem., Int. Ed. 2009, 48, 6974–6998. 10.1002/anie.200900942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker C. G.; Pratt M. R. Click Chemistry in Proteomic Investigations. Cell 2020, 180, 605–632. 10.1016/j.cell.2020.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieterich D. C.; Link A. J.; Graumann J.; Tirrell D. A.; Schuman E. M. Selective Identification of Newly Synthesized Proteins in Mammalian Cells Using Bioorthogonal Noncanonical Amino Acid Tagging (BONCAT). Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 9482–9487. 10.1073/pnas.0601637103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieterich D. C.; Lee J. J.; Link A. J.; Graumann J.; Tirrell D. A.; Schuman E. M. Labeling, Detection and Identification of Newly Synthesized Proteomes with Bioorthogonal Non-Canonical Amino-Acid Tagging. Nat. Protoc. 2007, 2, 532–540. 10.1038/nprot.2007.52. [DOI] [PubMed] [Google Scholar]
- Schiapparelli L. M.; McClatchy D. B.; Liu H. H.; Sharma P.; Yates J. R.; Cline H. T. Direct Detection of Biotinylated Proteins by Mass Spectrometry. J. Proteome Res. 2014, 13, 3966–3978. 10.1021/pr5002862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y.; McClatchy D. B.; Barkallah S.; Wood W. W.; Yates J. R. Quantitative Analysis of Newly Synthesized Proteins. Nat. Protoc. 2018, 13, 1744–1762. 10.1038/s41596-018-0012-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwanhäusser B.; Gossen M.; Dittmar G.; Selbach M. Global Analysis of Cellular Protein Translation by Pulsed SILAC. Proteomics 2009, 9, 205–209. 10.1002/pmic.200800275. [DOI] [PubMed] [Google Scholar]
- Eichelbaum K.; Winter M.; Diaz M. B.; Herzig S.; Krijgsveld J. Selective Enrichment of Newly Synthesized Proteins for Quantitative Secretome Analysis. Nat. Biotechnol. 2012, 30, 984–990. 10.1038/nbt.2356. [DOI] [PubMed] [Google Scholar]
- Howden A. J. M.; Geoghegan V.; Katsch K.; Efstathiou G.; Bhushan B.; Boutureira O.; Thomas B.; Trudgian D. C.; Kessler B. M.; Dieterich D. C.; Davis B. G.; Acuto O. QuaNCAT: Quantitating Proteome Dynamics in Primary Cells. Nat. Methods 2013, 10, 343–346. 10.1038/nmeth.2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichelbaum K.; Krijgsveld J. Combining Pulsed SILAC Labeling and Click-Chemistry for Quantitative Secretome Analysis. Methods Mol. Biol. 2014, 1174, 101–114. 10.1007/978-1-4939-0944-5_7. [DOI] [PubMed] [Google Scholar]
- Udeshi N. D.; Pedram K.; Svinkina T.; Fereshetian S.; Myers S. A.; Aygun O.; Krug K.; Clauser K.; Ryan D.; Ast T.; Mootha V. K.; Ting A. Y.; Carr S. A. Antibodies to Biotin Enable Large-Scale Detection of Biotinylation Sites on Proteins. Nat. Methods 2017, 14, 1167–1170. 10.1038/nmeth.4465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D. I.; Cutler J. A.; Na C. H.; Reckel S.; Renuse S.; Madugundu A. K.; Tahir R.; Goldschmidt H. L.; Reddy K. L.; Huganir R. L.; Wu X.; Zachara N. E.; Hantschel O.; Pandey A. BioSITe: A Method for Direct Detection and Quantitation of Site-Specific Biotinylation. J. Proteome Res. 2018, 17, 759–769. 10.1021/acs.jproteome.7b00775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinpenning F.; Steigenberger B.; Wu W.; Heck A. J. R. Fishing for Newly Synthesized Proteins with Phosphonate-Handles. Nat. Commun. 2020, 11, 3244 10.1038/s41467-020-17010-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang A. C.; du Bois H.; Olsson N.; Gate D.; Lehallier B.; Berdnik D.; Brewer K. D.; Bertozzi C. R.; Elias J. E.; Wyss-Coray T. Multiple Click-Selective TRNA Synthetases Expand Mammalian Cell-Specific Proteomics. J. Am. Chem. Soc. 2018, 140, 7046–7051. 10.1021/jacs.8b03074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J.; Mann M. MaxQuant Enables High Peptide Identification Rates, Individualized p.p.b.-Range Mass Accuracies and Proteome-Wide Protein Quantification. Nat. Biotechnol. 2008, 26, 1367–1372. 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- Tyanova S.; Temu T.; Sinitcyn P.; Carlson A.; Hein M. Y.; Geiger T.; Mann M.; Cox J. The Perseus Computational Platform for Comprehensive Analysis of (Prote)Omics Data. Nat. Methods 2016, 13, 731–740. 10.1038/nmeth.3901. [DOI] [PubMed] [Google Scholar]
- Hulsen T.; de Vlieg J.; Alkema W. BioVenn - A Web Application for the Comparison and Visualization of Biological Lists Using Area-Proportional Venn Diagrams. BMC Genomics 2008, 9, 488 10.1186/1471-2164-9-488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eden E.; Navon R.; Steinfeld I.; Lipson D.; Yakhini Z. GOrilla: A Tool for Discovery and Visualization of Enriched GO Terms in Ranked Gene Lists. BMC Bioinformatics 2009, 10, 48 10.1186/1471-2105-10-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y.; Verhelst S. H. L. Cleavable Trifunctional Biotin Reagents for Protein Labelling, Capture and Release. Chem. Commun. 2013, 49, 5366–5368. 10.1039/c3cc42076k. [DOI] [PubMed] [Google Scholar]
- Post H.; Penning R.; Fitzpatrick M. A.; Garrigues L. B.; Wu W.; Macgillavry H. D.; Hoogenraad C. C.; Heck A. J. R.; Altelaar A. F. M. Robust, Sensitive, and Automated Phosphopeptide Enrichment Optimized for Low Sample Amounts Applied to Primary Hippocampal Neurons. J. Proteome Res. 2017, 16, 728–737. 10.1021/acs.jproteome.6b00753. [DOI] [PubMed] [Google Scholar]
- Bycroft B. W.; Chan W. C.; Chhabra S. R.; Hone N. D. A Novel Lysine-Protecting Procedure for Continuous Flow Solid Phase Synthesis of Branched Peptides. J. Chem. Soc. Chem. Commun. 1993, 53, 778–779. 10.1039/c39930000778. [DOI] [Google Scholar]
- Supek F.; Bošnjak M.; Škunca N.; Šmuc T. REVIGO Summarizes and Visualizes Long Lists of Gene Ontology Terms. PLoS One 2011, 6, e21800 10.1371/journal.pone.0021800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D.; Gable A. L.; Lyon D.; Junge A.; Wyder S.; Huerta-Cepas J.; Simonovic M.; Doncheva N. T.; Morris J. H.; Bork P.; Jensen L. J.; Mering C. von. STRING V11: Protein–Protein Association Networks with Increased Coverage, Supporting Functional Discovery in Genome-Wide Experimental Datasets. Nucleic Acids Res. 2019, 47, D607–D613. 10.1093/nar/gky1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn G. P.; Koebel C. M.; Schreiber R. D. Interferons, Immunity and Cancer Immunoediting. Nat. Rev. Immunol. 2006, 6, 836–848. 10.1038/nri1961. [DOI] [PubMed] [Google Scholar]
- Tarhini A. A.; Gogas H.; Kirkwood J. M. IFN-α in the Treatment of Melanoma. J. Immunol. 2012, 189, 3789–3793. 10.4049/jimmunol.1290060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro F.; Cardoso A. P.; Gonçalves R. M.; Serre K.; Oliveira M. J. Interferon-Gamma at the Crossroads of Tumor Immune Surveillance or Evasion. Front. Immunol. 2018, 9, 847 10.3389/fimmu.2018.00847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikkola J.; Vihinen P.; Vuoristo M. S.; Kellokumpu-Lehtinen P.; Kähäri V. M.; Pyrhönen S. High Serum Levels of Matrix Metalloproteinase-9 and Matrix Metalloproteinase-1 Are Associated with Rapid Progression in Patients with Metastatic Melanoma. Clin. Cancer Res. 2005, 11, 5158–5166. 10.1158/1078-0432.CCR-04-2478. [DOI] [PubMed] [Google Scholar]
- Neubert N. J.; Schmittnaegel M.; Bordry N.; Nassiri S.; Wald N.; Martignier C.; Tillé L.; Homicsko K.; Damsky W.; Maby-El Hajjami H.; Klaman I.; Danenberg E.; Ioannidou K.; Kandalaft L.; Coukos G.; Hoves S.; Ries C. H.; Fuertes Marraco S. A.; Foukas P. G.; De Palma M.; Speiser D. E. T Cell-Induced CSF1 Promotes Melanoma Resistance to PD1 Blockade. Sci. Transl. Med. 2018, 10, eaan3311 10.1126/scitranslmed.aan3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Businaro R.; Fabrizi C.; Fumagalli L.; Lauro G. M. Synthesis and Secretion of A2-Macroglobulin by Human Glioma Established Cell Lines. Exp. Brain Res. 1992, 88, 213–218. 10.1007/BF02259144. [DOI] [PubMed] [Google Scholar]
- Becker J. C.; Dummer R.; Hartmann A. A.; Burg G.; Schmidt R. E. Shedding of ICAM-1 from Human Melanoma Cell Lines Induced by IFN-Gamma and Tumor Necrosis Factor-Alpha. Functional Consequences on Cell-Mediated Cytotoxicity. J. Immunol. 1991, 147, 4398–4401. [PubMed] [Google Scholar]
- Yamaguchi K.; Sakiyama H.; Matsumoto M.; Moriya H.; Sakiyama S. Degradation of Type I and II Collagen by Human C1-S. FEBS Lett. 1990, 268, 206–208. 10.1016/0014-5793(90)81009-D. [DOI] [PubMed] [Google Scholar]
- Price M. A.; Colvin Wanshura L. E.; Yang J.; Carlson J.; Xiang B.; Li G.; Ferrone S.; Dudek A. Z.; Turley E. A.; McCarthy J. B. CSPG4, a Potential Therapeutic Target, Facilitates Malignant Progression of Melanoma. Pigm. Cell Melanoma Res. 2011, 24, 1148–1157. 10.1111/j.1755-148X.2011.00929.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menezes M. E.; Bhatia S.; Bhoopathi P.; Das S. K.; Emdad L.; Dasgupta S.; Dent P.; Wang X.-Y.; Sarkar D.; Fisher P. B.. MDA-7/IL-24: Multifunctional Cancer Killing Cytokine. In Anticancer Genes; Grimm S., Ed.; Springer London: London, 2014; Vol. 818, pp 127–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer K. J.; Harries M.; Gore M. E.; Collins M. K. L. Interferon-Alpha (IFN- α) Stimulates Anti-Melanoma Cytotoxic T Lymphocyte (CTL) Generation in Mixed Lymphocyte Tumour Cultures (MLTC). Clin. Exp. Immunol. 2000, 119, 412–418. 10.1046/j.1365-2249.2000.01159.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsson K.; Wang P. Chaperones and Folding of MHC Class I Molecules in the Endoplasmic Reticulum. Biochim. Biophys. Acta, Mol. Cell Res. 2003, 1641, 1–12. 10.1016/S0167-4889(03)00048-X. [DOI] [PubMed] [Google Scholar]
- Reiniger I. W.; Schaller U. C.; Haritoglou C.; Hein R.; Bosserhoff A. K.; Kampik A.; Mueller A. J. “Melanoma Inhibitory Activity” (MIA): A Promising Serological Tumour Marker in Metastatic Uveal Melanoma. Graefe’s Arch. Clin. Exp. Ophthalmol. 2005, 243, 1161–1166. 10.1007/s00417-005-1171-4. [DOI] [PubMed] [Google Scholar]
- Wang H.; Shen S. S.; Wang H.; Diwan A. H.; Zhang W.; Fuller G. N.; Prieto V. G. Expression of Insulin-like Growth Factor-Binding Protein 2 in Melanocytic Lesions. J. Cutaneous Pathol. 2003, 30, 599–605. 10.1034/j.1600-0560.2003.00120.x. [DOI] [PubMed] [Google Scholar]
- Atkinson A.; Renziehausen A.; Wang H.; Lo Nigro C.; Lattanzio L.; Merlano M.; Rao B.; Weir L.; Evans A.; Matin R.; Harwood C.; Szlosarek P.; Pickering J. G.; Fleming C.; Sim V. R.; Li S.; Vasta J. T.; Raines R. T.; Boniol M.; Thompson A.; Proby C.; Crook T.; Syed N. Collagen Prolyl Hydroxylases Are Bifunctional Growth Regulators in Melanoma. J. Invest. Dermatol. 2019, 139, 1118–1126. 10.1016/j.jid.2018.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S. B.; Schramme A.; Doberstein K.; Dummer R.; Abdel-Bakky M. S.; Keller S.; Altevogt P.; Oh S. T.; Reichrath J.; Oxmann D.; Pfeilschifter J.; Mihic-Probst D.; Gutwein P. ADAM10 Is Upregulated in Melanoma Metastasis Compared with Primary Melanoma. J. Invest. Dermatol. 2010, 130, 763–773. 10.1038/jid.2009.335. [DOI] [PubMed] [Google Scholar]
- Liberato T.; Pessotti D. S.; Fukushima I.; Kitano E. S.; Serrano S. M. T.; Zelanis A. Signatures of Protein Expression Revealed by Secretome Analyses of Cancer Associated Fibroblasts and Melanoma Cell Lines. J. Proteomics 2018, 174, 1–8. 10.1016/j.jprot.2017.12.013. [DOI] [PubMed] [Google Scholar]
- Perez-Riverol Y.; Csordas A.; Bai J.; Bernal-Llinares M.; Hewapathirana S.; Kundu D. J.; Inuganti A.; Griss J.; Mayer G.; Eisenacher M.; Pérez E.; Uszkoreit J.; Pfeuffer J.; Sachsenberg T.; Yilmaz S.; Tiwary S.; Cox J.; Audain E.; Walzer M.; Jarnuczak A. F.; Ternent T.; Brazma A.; Vizcaíno J. A. The PRIDE Database and Related Tools and Resources in 2019: Improving Support for Quantification Data. Nucleic Acids Res. 2019, 47, D442–D450. 10.1093/nar/gky1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.