

Abstract
Intestinal ischemia–reperfusion (IR) injury is a severe clinical condition, and unraveling its pathophysiology is crucial to improve therapeutic strategies and reduce the high morbidity and mortality rates. Here, we studied the dynamic proteome and phosphoproteome in the human intestine during ischemia and reperfusion, using liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis to gain quantitative information of thousands of proteins and phosphorylation sites, as well as mass spectrometry imaging (MSI) to obtain spatial information. We identified a significant decrease in abundance of proteins related to intestinal absorption, microvillus, and cell junction, whereas proteins involved in innate immunity, in particular the complement cascade, and extracellular matrix organization increased in abundance after IR. Differentially phosphorylated proteins were involved in RNA splicing events and cytoskeletal and cell junction organization. In addition, our analysis points to mitogen-activated protein kinase (MAPK) and cyclin-dependent kinase (CDK) families to be active kinases during IR. Finally, matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) MSI presented peptide alterations in abundance and distribution, which resulted, in combination with Fourier-transform ion cyclotron resonance (FTICR) MSI and LC-MS/MS, in the annotation of proteins related to RNA splicing, the complement cascade, and extracellular matrix organization. This study expanded our understanding of the molecular changes that occur during IR in the human intestine and highlights the value of the complementary use of different MS-based methodologies.
Keywords: human intestinal ischemia−reperfusion, proteomics, phosphoproteomics, mass spectrometry imaging, spatiotemporal data
Introduction
Intestinal ischemia–reperfusion (IR) is a clinical phenomenon carrying high morbidity and mortality and can occur in various conditions. Based on etiology, intestinal ischemia is classified into chronic ischemia, e.g., due to atherosclerosis, and acute ischemia. The latter is further divided into occlusive disease, caused by the obstruction of the mesenteric blood flow, or nonocclusive disease, caused by hypoperfusion, for example, due to major surgical procedures, trauma, hemorrhagic shock, or sepsis.1−3 The lack of oxygen during ischemia obviously leads to cell injury, and hence rapid reperfusion is crucial. However, reperfusion can also aggravate injury as the sudden oxygen supply to the ischemic intestine initiates a cascade of events, including Ca2+ influx and the production of reactive oxygen species (ROS), which damages cellular structures and activates an inflammatory response.4 The intestinal epithelium serves as an important barrier that protects the body from the hostile luminal environment. Disruption of this barrier, which can be caused by IR injury, allows entry of harmful luminal microorganisms and toxins into the sterile inner mucosa, which may cause a severe inflammatory response. Observations from a human experimental model of intestinal IR showed that the intestine was relatively resistant to short periods of ischemia,5,6 whereas prolonged ischemia (>45 min) followed by reperfusion disrupted the epithelial lining and induced inflammation.7 Cell death occurs initially at the villi tips and progresses toward the crypt with increasing duration of the ischemic period.7,8 Severe IR can eventually lead to bowel necrosis and severe systemic inflammation. The high mortality rates (60–80%) of acute intestinal ischemia1−3 can be attributed to the difficulty to diagnose acute mesenteric ischemia at an early stage, as well as the lack of effective therapeutic options.9−11 Further unraveling the molecular mechanisms underlying ischemia–reperfusion is crucial to improve therapeutic strategies and patient outcomes.
Identification of changes in (local) protein abundance that occur following ischemia and during reperfusion in the human experimental model is of great interest for understanding the biological processes involved in IR injury and tissue repair. As many cellular functions are regulated by the dynamic phosphorylation of proteins, we were interested in investigating changes in protein phosphorylation as well. Furthermore, given the aforementioned differences in function and susceptibility to IR across the layers and cell types of the intestinal wall, spatial information of protein changes is of particular interest in the context of intestinal IR injury.
Liquid chromatography coupled to mass spectrometry (LC-MS)-based proteomics has emerged as an important tool to study cell biology and disease mechanisms and enables untargeted identification and quantification of several thousand proteins.12 A multistep process, including protein extraction, digestion, and separation by LC, is currently used to obtain this thorough protein coverage and accuracy. However, with homogenization of the tissue during sample preparation, spatial information on protein distribution gets lost. A powerful technology to complement LC-MS-based proteomics is matrix-assisted laser desorption/ionization (MALDI) mass spectrometry imaging (MSI) of tissue sections. MALDI MSI allows analyzing hundreds of molecules simultaneously, providing spatial distribution and local abundance of these molecules in biological tissues in a label-free manner. Combining MALDI MSI with the histological information of these tissue enables a histology-driven analysis. Intact proteins can be detected by MSI; however, their detection is limited by low ionization efficiency that limits sensitivity in the high mass range of the instrumentation. Recent studies have shown imaging of proteins up to 200 kDa.13,14 MSI of trypsinized proteins can therefore—at least theoretically—increase the coverage of the proteome beyond this sensitivity limit. Moreover, the on-tissue digestion approach allows better integration of MSI data with liquid chromatography-tandem mass spectrometry (LC-MS/MS) of the same or an adjacent tissue section to enable the identification of the observed local peptide signals, especially when using high-mass resolution MSI instrumentation.15,16 However, these high-mass resolution MSI methods are limited by a long acquisition time and consequently suffer from a low throughput. This issue can be overcome by the use of high-speed MALDI-time-of-flight (TOF) MSI complemented by high-mass resolution MSI data,17 which is the approach that we applied in our study.
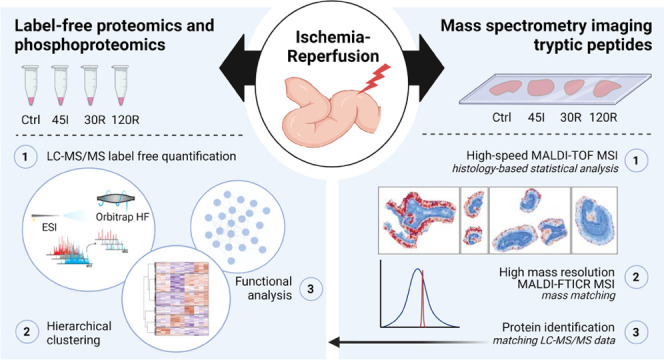
The main objective of this study is to analyze the dynamic proteome and phosphoproteome in the human intestine exposed to ischemia and reperfusion. To this end, we used two complementary MS-based technologies: LC-MS/MS (phospho)proteomics to gain in-depth quantitative information and MALDI MSI of tryptic peptides to obtain spatial information and study location-specific protein changes.
Experimental Section
Patients and Experimental Procedure
Human intestinal tissues exposed to ischemia and reperfusion were obtained using a controlled in vivo experimental model. The study was approved by the Medical Ethical Committee of the Maastricht University Medical Centre+ (Maastricht UMC+) and written informed consent was obtained from all patients. Nine patients (sex 3M/6F; median age 66 years, range 43–84 years) undergoing pancreatoduodenectomy were included in the study. The experimental procedure was performed as described previously.5 In short, a 6 cm jejunal segment, which is routinely resected as part of the surgical procedure, was isolated and subjected to ischemia by placing vascular clamps across the mesentery. After 45 min, one-third of the ischemic segment was resected (45I). Next, clamps were removed to start reperfusion. Another segment of isolated jejunum was removed after 30 min (30R) and 120 min of reperfusion (120R). Finally, a jejunal segment that was not exposed to IR but underwent similar surgical handling was resected (control, Ctrl). Jejunal tissue samples were immediately snap-frozen and stored at −80 °C.
LC-MS/MS Analysis
Cell Lysis and Protein Digestion
Tissue samples were treated with sodium deoxycholate (SDC) 1% to induce cell lysis. The buffer also contained 10 mM tris(2-carboxyethyl)-phosphine hydrochloride (TCEP), 40 mM chloroacetamide, 100 mM TRIS pH 8.0, further supplemented with a protease inhibitor (cOmplete mini ethylenediaminetetraacetic acid (EDTA)-free; Roche, Basel, Switzerland) and a phosphatase inhibitor (PhosSTOP, Roche). The samples were sonicated with a Bioruptor Plus (Diagenode, Liège, Belgium) for 15 cycles of 30 s. The protein amount in each sample was quantified by a Bradford protein assay. Next, proteins were digested overnight at 37 °C with Lys-C (FUJIFILM Wako pure chemical corporation, Osaka, Japan) and trypsin (Sigma-Aldrich, Zwijndrecht, The Netherlands), with enzyme to protein ratios of 1:75 and 1:50, respectively. SDC was precipitated by the addition of 2% formic acid (FA), and peptides were desalted using Sep Pak C18 cartridges (Waters Corporation, Milford, Massachusetts) to subsequently be dried down and stored at −80 °C.
Phosphopeptide Enrichment
Phosphopeptides were enriched by Ti(IV)-IMAC; 500 μg of beads were packed into microtip columns and washed with methanol and a loading buffer made of 80% acetonitrile (ACN) and 6% trifluoroacetic acid (TFA). Then, 200 μg of peptides per sample was dissolved in loading buffer and subsequently loaded into the columns. Peptides were washed with 50% ACN/0.5% TFA in 200 mM NaCl, followed by a second wash with 50% ACN/0.1% TFA. Phosphopeptides were eluted with 10% ammonia and 80% ACN/2% FA directly into 10% FA. The samples were dried down and stored at −80 °C until LC-MS/MS analysis. A detailed description of this protocol was published elsewhere.18
Data Acquisition by LC-MS/MS
The samples were analyzed using a UHPLC 1290 system (Agilent, Santa Clara, California) coupled to an Orbitrap Q Exactive HF (Thermo Fisher Scientific, Waltham, Massachusetts). The nanoflow rate (∼300 nL/min) was achieved by passively splitting the flow using an external valve.19 Peptides were first trapped into a precolumn (an inner diameter [ID] of 100 μm and 2 cm length; packed in-house with 3 μm C18 ReproSil particles [Dr. Maisch GmbH]) and eluted into an analytical column (ID of 75 μm and 50 cm length; packed in-house with 2.7 μm Poroshell EC-C18 particles [Agilent]). We used a two-system buffer consisting of solvent A (0.1% FA in water) and B (0.1% FA in 80% ACN). Peptides were trapped for 5 min at a 5 μL/min flow rate with solvent A before switching to a nanoflow of ∼300 nL/min. For the measurement of the full proteome, we used a 155 min gradient from 10 to 36% of solvent B. On the other hand, for the phosphoproteome, we used a 95 min gradient from 8 to 32% of solvent B. Both methods included a wash with 100% solvent B for 5 min followed by a column equilibration with 100% solvent A during the last 10 min.
The mass spectrometer was operated in a data-dependent acquisition mode. For the proteome analysis, full scan MS was acquired from m/z 375–1600 with a 60 000 resolution at m/z 200. The accumulation target value was set to 3 × 106 ions with a maximum injection time of 20 ms. Up to 15 of the most intense precursor ions were isolated (m/z 1.4 window) for fragmentation using high energy collision-induced dissociation (HCD) with a normalized collision energy of 27. For MS2 scans, an accumulation target value of 1 × 105 ions, a maximum injection time of 50 ms, and a dynamic exclusion time of 24 s were selected. Scans were acquired from m/z 200–2000 with a 30 000 resolution at m/z 200. For the phosphoproteome, the same settings were used with the exception of the dynamic exclusion window, which was set to 16 s. The electrospray voltage was set to 1.9 kV during the measurement of all samples.
Data Processing
Raw files were processed with MaxQuant (version 1.6.17.0) using a false discovery rate (FDR) <0.01. The default settings were used, with the following exceptions: variable modifications, specifically methionine oxidation, protein N-term acetylation, and serine, threonine, and tyrosine phosphorylation were selected. Cysteine carbamidomethylation was selected as a fixed modification. Label-free quantification was performed, and we enabled the “match between runs” option with the default values. Database search was conducted against the human-reviewed Swiss-Prot database (October 2020).
The results were uploaded to Perseus (version 1.6.0.2) for subsequent analysis. For the proteome, only proteins identified by more than one unique peptide were considered, and for the phosphoproteome, only phosphosites with a localization probability score >0.75 were kept for further analysis. Decoys and potential contaminants were removed. The intensities were log 2 transformed and normalized by median subtraction. Finally, all values were filtered to keep only those proteins (or phosphosites) that were detected in a minimum of two out of three replicates for at least one condition. Missing values were replaced using a normal distribution applying a downshift of 1.8 times the standard deviation of the dataset and a width of 0.3 times the standard deviation.
An analysis of variance (ANOVA) test (P < 0.05) was used to keep only significantly changing proteins (or phosphosites) among the different conditions. Z-scored intensities were visualized using the Complex Heatmap package in R, applying a combination of k-means and hierarchical clustering. K-means clusters were set to 4 and 6 for proteome and phosphoproteome, respectively. This number was chosen based on the gap statistic method, which estimates the optimal number of groups for a given dataset.20 Next, we used the Pearson correlation distance with average linkage for clustering. Gene ontology (GO) analysis was done using the STRING web tool;21 enriched GO terms were filtered to keep only those with P < 0.01, fold enrichment (Log 10[observed/expected]) >5, and a minimum of five proteins per term. The list of terms was further condensed by the removal of redundant terms using the Revigo web tool.22
Sequence logos for motif analysis were obtained implementing previously described algorithms in R.23 Briefly, each sequence logo displays over- and underrepresented residues in each position of the sequence window centered on the phosphorylated residue. Calculations are based on the frequency change between a foreground (phosphosites from each cluster) and a background (all detected phosphosites). For kinase enrichment analysis, we used the online tool KEA2 to look for phosphosites previously linked to effector kinases.24 In addition, we applied post-translational modification-signature enrichment analysis (PTM-SEA), which looks for the enrichment of phosphosite-specific “signatures” related to specific kinases, signaling pathways, or perturbations previously reported in the literature.25
MALDI MSI Analysis
Tissue Preparation for MALDI MSI Analysis
Fresh frozen tissues were sectioned at 10 μm thickness at −20 °C using a cryostat (Leica, Leica CM1860, Leica Biosystems) and thaw-mounted onto clean indium tin oxide (ITO)-coated glass slides (Delta Technologies LTD, Loveland). A within-subjects experimental design was pursued, i.e., all four different tissue sections (Ctrl, 45I, 30R, and 120R) from one patient were always mounted on the same ITO slide. Mounted tissue sections were dried in a vacuum desiccator for 20 min, followed by three 2-min washes in 100% ethanol and then two 5 min washes in water. Fresh ethanol/water was used in every wash, and sections were not dried between steps. Antigen retrieval was performed in a 10 mM citric acid buffer (Sigma-Aldrich) (pH 6.0) for 20 min using the Antigen Retriever 2100 (Aptum Biologics, Rownhams, U.K.). Sections were allowed to cool down for 20 min, rinsed with water, and dried in a vacuum desiccator. Prior to trypsin digestion, 1 μL of 1 mg/mL cytochrome c (from equine heart, Sigma-Aldrich) was applied on the slide, away from the tissue, to evaluate digestion efficacy. Water-dissolved porcine trypsin (20 μg/mL) was sprayed onto the tissue samples using a SunCollect pneumatic sprayer device (Sunchrom GmbH, Friedrichsdorf, Germany) in 15 layers (flow rate: 10 μL/min, speed: 900 mm/min, track spacing: 1 mm, spray head distance: 25 mm). Afterward, the samples were incubated at 37 °C for 17 h in an airtight box containing 50% methanol. Finally, slides were coated with 5 mg/mL α-cyano-4-hydroxycinnamic acid (Sigma-Aldrich) in 50% acetonitrile and 0.2% trifluoroacetic acid using the SunCollect sprayer device. The matrix was applied in a series of seven layers with the increasing flow rate starting at 10 μL/min followed by 20, 30, and 40 μL/min for all subsequent layers (speed: 1390 mm/min, track spacing: 2 mm, spray head distance: 25 mm). Prior to matrix application, slides were scanned (Super Coolscan 5000 ED, Nikon) to obtain high-quality optical images.
MALDI MSI Data Acquisition
High-speed MALDI-TOF MSI analysis was performed on a RapifleX MALDI Tissuetyper (Bruker Daltonics GmbH, Bremen, Germany) equipped with a 10 kHz Nd:YAG (355 nm) laser. The instrument was operated in a positive ionization reflectron mode, and peptide spectra were acquired in a mass range m/z 620–3000 with a spatial raster of 50 μm and 500 averaged laser shots per pixel. An experimental mass resolution of 15 000 was achieved at m/z 1000. High mass resolution MALDI-Fourier-transform ion cyclotron resonance (FTICR) MSI experiments were performed with a Solarix 9.4 T (Bruker Daltonics), achieving an experimental mass resolution of 200 000 at m/z 1000. MSI data were acquired within a mass range of m/z 800–3000 (1 × 106 data points) in a positive ionization mode with a transient time of 2.94 s. The spatial raster width was 70 μm. At each pixel, 600 shots were accumulated with a laser frequency of 500 Hz. Data acquisition was controlled using ftmsControl and FlexImaging 4.1 (Bruker Daltonics). All MSI measurements were preceded by an instrument calibration using Red phosphorus. MALDI-TOF analysis was performed on 36 tissue samples from nine patients. MALDI-FTICR MSI measurements were performed on selected samples (four conditions from one patient) to improve the identification of the proteins behind the relevant peptides obtained from TOF analysis.
Histological Staining and Tissue Annotation
After MSI analysis, the matrix was removed by submersion in 70% ethanol and tissues were stained with hematoxylin and eosin (H&E). Optical images of H&E-stained tissues were obtained with a MIRAX desk scanner (Sysmex, Etten-Leur, The Netherlands). The MSI images were coregistered with the corresponding histological images in the FlexImaging software (v5.0, Bruker Daltonics), which allows the annotation of the histological regions of interest: mucosa, submucosa, and muscle layers.
MSI Data Preprocessing
MALDI-TOF MSI data were recalibrated using FlexAnalysis v3.4 (Bruker Daltonics). Cubic-enhanced calibration function was performed with a 500 ppm peak assignment tolerance and using m/z 868.5, 1138.6, 1562.8, 2115.2, 2567.3, and 2869.3 as calibrants. A total of 36 (nine patients with four conditions each) MALDI-TOF MSI datasets were imported into SCiLS 2019c (Bruker Daltonics), where mass spectra were normalized to their total ion count. Peak picking was performed on the overall mean spectrum in mMass26 using the following settings: 35 precision baseline correction, deisotoping with an isotope mass tolerance of m/z 0.1, isotope intensity tolerance of 50%, and a signal to noise (S/N) ratio of 7. The peak list was then imported back into SCiLS to create a data matrix containing every annotated region and sample the maximum intensity in each peak interval (m/z ±0.2).
The average spectra of the MALDI-FTICR MSI datasets were recalibrated in mMass with linear correction using the tryptic peptides of histone H2A (m/z 944.5312; pos. 22–30 “AGLQFPVGR”) and histone H4 (m/z 1325.7535; pos. 25–36 “DNIQGITKPAIR”) and known trypsin autolysis products at m/z 842.5094 (pos. 108–115 “VATVSLPR”) and m/z 1045.5637 (pos. 98–107 “LSSPATLNSR”). Peak picking on those recalibrated spectra was performed in MATLAB R2018 (Mathworks, Natick, Massachusetts) using the following settings: TopHat filter (window: 30 dp) for baseline correction, Gaussian smoothing (window: 20 dp), a minimum intensity for peak picking of 500, deisotoping with an isotope mass tolerance of 0.02 m/z and isotope intensity tolerance of 50%.
Statistical Analysis MALDI-TOF MSI Data
An outlier detection was performed based on a cytochrome c spot applied onto every slide prior to trypsin digestion. For this, peak picking was limited to known peptides of cytochrome c, which resulted in the consideration of four signals (HKTGPNLHGLFGR, m/z 1433.77; HKTGPNLHGLFGRK, m/z 1561.87; TGPNLHGLFGRK, m/z 1296.72; TGPNLHGLFGR, m/z 1168.62). Using these four features, principal component analysis (PCA) was performed on the average intensity in the cytochrome c spot from each slide (N = 9) in SCiLS. Any measurement outside the 95% confidence ellipse in the PC1–PC2 score plot was considered an outlier.
The data matrix from the included patients was exported for statistical analysis in R (v3.5.1). Trypsin-related peaks were determined by Pearson correlation >0.9 to the m/z 842.5 and excluded from analysis (removal of six peaks). Statistically significant differences in peak intensities between the conditions (Ctrl, 45I, 30R, 120R) were tested for each histological layer separately (mucosa, submucosa, muscle). The intensities for every peak were compared using a repeated-measurements ANOVA. P-values were corrected for multiple testing by Benjamini–Hochberg and P-values <0.01 were considered statistically significant. As several tissue sections lacked the muscle layer, statistical analysis could not be performed for the muscle layer.
Protein Identification Strategy
MALDI-FTICR MSI experiments were performed on selected samples to obtain high mass-resolution data of tryptic peptides, which was used to identify the proteins behind the significantly changed peptides from MALDI-TOF MSI statistics. These significantly changed m/z values were matched with a tolerance of ±80 ppm to the peaks in the MALDI-FTICR MSI measurements. Next, these accurate peptide masses were matched with the masses of the (phospho)peptides detected using LC-MS/MS and corresponding identified protein with a tolerance of ±6 ppm. As there is no alkylation and reduction step in the MSI workflow, the peptide masses of the LC-MS/MS were adapted by subtracting the mass shift (m/z 57.02146) caused by the formation of S-carboxyamidomethylcysteine for every cysteine in the peptide.
The matching and identification process was performed using four FTICR MSI datasets (data was obtained from one patient across all four conditions). The annotation of a protein was accepted if mass matching from TOF to FTICR (with ±80 ppm tolerance) and FTICR to LC-MS/MS (with ±5 ppm tolerance) resulted in one matching protein ID and this in at least three out of the four FTICR datasets. In cases where two FTICR peaks were detected in the analogous mass range in the TOF spectrum, the most intense FTICR peak (>10 fold higher) within this window was selected for mass matching with the LC-MS/MS data.
Data Availability
The mass spectrometry proteomics and phosphoproteomics datasets have been deposited to the ProteomeXchange Consortium via the PRIDE27 partner repository with the data set identifier PDX026076.
Results
Changes in protein expression and protein phosphorylation during IR of the human intestine were studied by combining a quantitative MS-based (phospho)proteomics approach with imaging MS. The dynamic (phospho)proteome during IR was investigated by analyzing tissue samples collected after 45 min of ischemia (45I), 30 and 120 min of reperfusion (30R and 120R), and in control tissue (Ctrl). A schematic of the experimental design and data analysis workflow are depicted in Figure 1.
Figure 1.
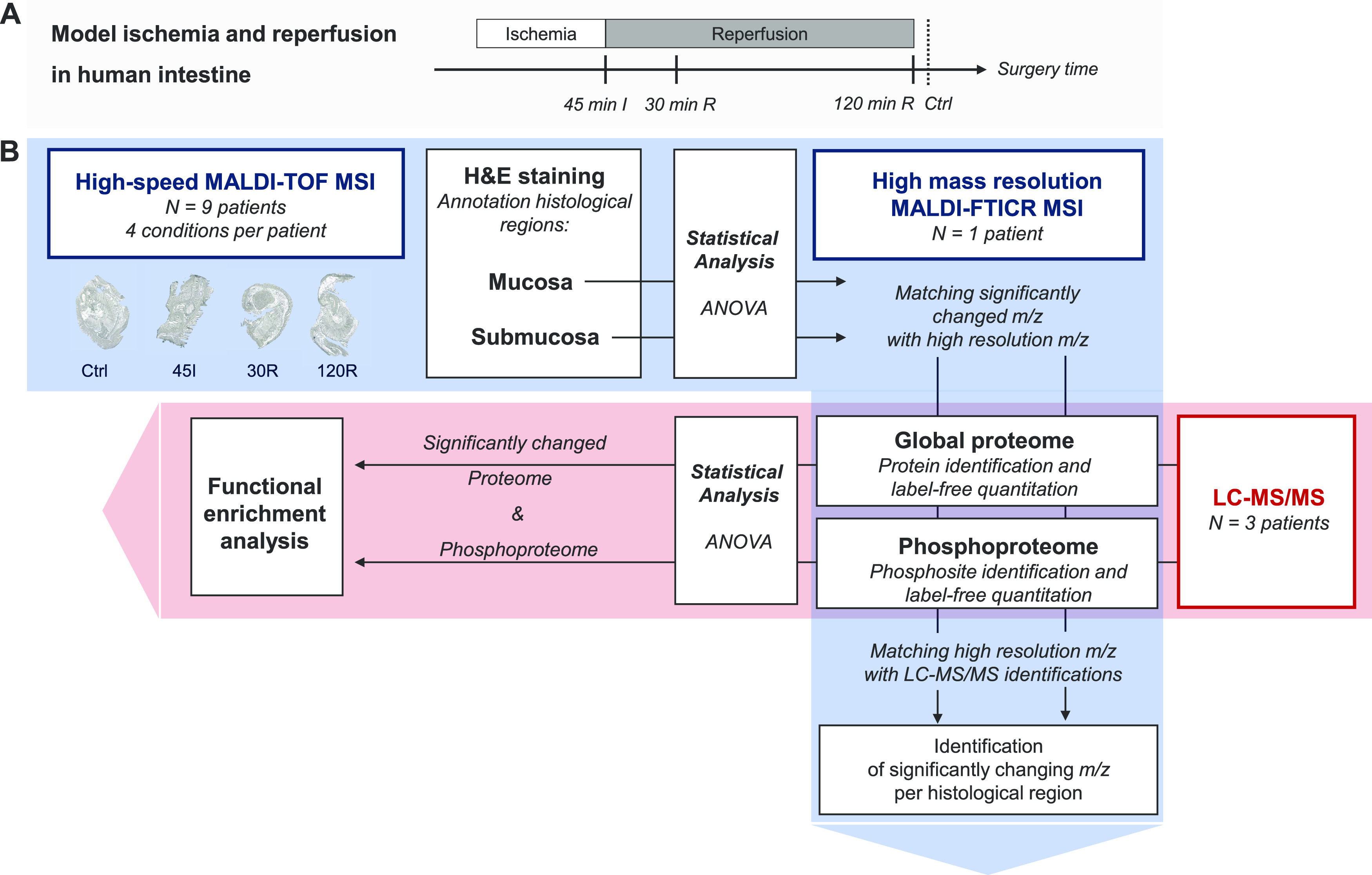
Experimental design and data analysis workflow. (A) Experimental model of ischemia–reperfusion in the human intestine, with tissue collection after 45 min of ischemia (45I), 30 min of reperfusion (30R), and 120 min of reperfusion (120R), and tissue not exposed to ischemia–reperfusion (Ctrl). (B) Mass spectrometry and data analysis workflow. The red area shows the workflow of LC-MS/MS measurement and subsequent data analysis. The blue area shows the MS imaging (MSI) measurements.
Proteomics
LC-MS/MS analysis resulted in the identification and quantification of 2562 proteins. The correlation between biological replicates was high (Pearson r > 0.9), with the exception of one 30R sample (r < 0.85), which was therefore excluded from further analysis (Figure S1). Cluster analysis of the complete proteome showed that the two main clusters were represented by 45I and Ctrl samples, on the one hand, and 30R and 120R samples, on the other hand (Figure S2). Analysis of dynamic changes in the proteome revealed that the abundance of 239 proteins was significantly altered during IR (Table S1). Hierarchical clustering of these differentially expressed proteins resulted in four main clusters reflecting distinct temporal expression profiles (Figure 2A) and clearly distinguished the proteins decreasing (clusters 1 and 2) and proteins increasing in abundance (clusters 3 and 4) during reperfusion.
Figure 2.

Dynamic proteome during ischemia–reperfusion in the human intestine. (A) Heatmap visualizing clustering of differentially expressed proteins. Hierarchical clustering was based on the Z scores of the log 2 values of differentially expressed proteins. The ANOVA test was performed and P < 0.05 was considered statistically significant. Average temporal profiles are shown for every cluster (a gray area represents CI). (B) Functional enrichment analysis of differentially expressed proteins. Over-represented gene ontology (GO) terms are shown (P < 0.05). GO term fold enrichment (log 10[observed/expected]) is plotted against P-value (−log 10). The size of the dot correlates with the number of proteins linked to that GO term, as indicated in the legend. Red dot, GO Biological Processes; blue dot, GO cellular component, gray dot, GO molecular function. (C) Dynamic profile for proteins in clusters 2 (blue) and 3 (red) is shown, as well as corresponding overrepresented GO terms. (D, E) Protein networks showing interacting proteins. Networks were generated using STRING. The color of the circle indicates the temporal profile (cluster) of the protein. Blue line, cluster 1; blue fill, cluster 2; red fill, cluster 3, red line, cluster 4. Color cubes below each protein indicate the Z-score intensity in Ctrl, 45I, 30R, and 120R, respectively. The majority of proteins in the networks in (D) are located in cluster 2 (blue fill). The majority of proteins in the network in (E) are located in cluster 3 (red fill).
Functional Enrichment Analysis of the Dynamic Proteome
To gain global functional insight into the changing proteome during IR, we performed GO term enrichment analysis of differentially expressed proteins. These proteins were predominantly involved in processes such as intestinal absorption and digestion, cell junction organization, and innate immune responses (Figure 2B). In line with this observation, overrepresented cellular component GO terms included brush border and microvillus, actin filament, and cell–cell contact zone. Proteins in cluster 1 of the changing proteome, showing a decrease in abundance at 30R, which restored at 120R, were related to various processes such as protein translation, including EIF2S2, EIF4EBP1, EIF5B, and the cellular response to stress, such as HSPA4, TNIK, Nup50, and LAMTOR1.
Protein abundances in cluster 2 decreased during reperfusion (Figure 2C) and were significantly enriched for GO terms related to intestinal digestion/absorption, microvillus, and cell junction (Table S2). Moreover, network analysis of the total changing proteome showed interactions of proteins involved in these GO terms. The majority of interacting proteins in these networks exhibited the same temporal profile (cluster 2; Figure 2D, blue fill). Proteins involved in digestion and absorption included IFABP, LCT, ANPEP, NAALADL1, and SLC5A1 (Figure 2D, left). Proteins associated with microvillus organization included actin-bundling proteins such as VIL1, ESPN, and PLS1, motor protein MYO1A, anchoring protein EZR, and microvillus–microvillus adhesion molecules CDHR2 and MYO7B. Other proteins playing a role in actin filament organization were COBL and MAP7. In addition, various proteins playing an important role in cell–cell junction organization were decreased, such as CDH1, CDH17, CDHR5, CDHR2, AFDN, NECTIN-1, F11R, EpCAM, and CD2AP and CGN (Figure 2D, right; Table S1).
Cluster 3 showed an opposite expression profile with increased protein levels during reperfusion. Over-represented GO terms in this cluster were predominantly associated with the innate immune response, in particular the complement pathway (Figure 2C). Both regulatory and effector proteins of the complement cascade were significantly increased at reperfusion and included C3, C5, C6, C8B, PROS1, and F2, which were all among the top 20 proteins showing the highest fold change amongst conditions, and exhibited very similar temporal profiles. In addition, various proteins in cluster 3 were involved in an extracellular matrix organization, including COL15A1, COL18A1, FBN1, NID2, ITGA, and JAM3. One of the protein interaction networks, resulting from network analysis of the complete changing proteome, contained mostly proteins of cluster 3 and showed interactions of complement proteins and proteins related to extracellular matrix organization (Figure 2E, red fill).
Proteins in cluster 4 gradually increased in abundance during ischemia and reperfusion and were involved in a variety of biological processes without a clear overrepresentation, and included the metabolism of amino acids, metabolism of nucleotides, post-translational protein modification, and cellular response to stress. Interesting proteins in this cluster include CYGB, SQSTM1, and CASP1.
In addition to the protein interaction networks that could be linked to clusters 2 and 3, another network showed interactions of proteins located in all four clusters and thus showing distinct temporal profiles (Figure S3). These proteins were associated with the cellular response to stress, protein, and RNA metabolism.
Phosphoproteomics
LC-MS/MS analysis resulted in the identification and quantification of 1802 phosphosites derived from 1214 proteins (whole phosphoproteome). The observed distribution of phosphosites was 90% phosphoserine, 9.8% phosphothreonine, and 0.2% phosphotyrosine (Figure S4A). The majority of these phosphosites had a high localization probability score, indicative of accurate localization of the phosphorylated residue in the peptide backbone (Figure S4B). In total, 305 phosphosites on 162 proteins showed a significant change during IR (dynamic phosphoproteome) (Table S3)
Prediction of the Kinases Responsible for Detected Phosphorylation
A kinase enrichment analysis for all detected phosphosites was performed to get an overview of which kinases were potentially active during IR. Here, 279 detected phosphosites were mapped to putative effector kinases (Figure S4C). Some of these kinases, such as GSK3B and casein kinase II, are constitutively active and participate in a myriad of cellular processes. We also predicted the activity of cyclin-dependent kinases (CDK), namely, CDK1 and CDK2, both involved in cell cycle control. Finally, we detected putative targets of the mitogen-activated protein (MAP) kinases MAPK9 (JNK2) and MAPK10 (JNK3), part of the JNK signaling pathway, and MAPK14 (p38α), part of the p38 MAP kinase pathway, which are both activated in response to cellular stress. The full list of targets related to each of these kinases are listed in Table S4.
Kinase enrichment analysis that focused on the 305 significantly regulated phosphosites (Table S3) and mapped 47 phosphosites to potential effector kinases (Figure S4D). Notably, putative substrates of several MAP kinases, like MAPK9 (JNK2), MAPK3 (ERK1), MAPK13 (p38δ), and MAPK8 (JNK1) were present. Dynamic phosphosites linked to casein kinase II and CDKs were detected as well. The full list of phosphosites and predicted effector kinases can be found in Table S5.
Clustering and Motif Enrichment of the Dynamic Phosphoproteome
Hierarchical clustering of altered phosphosites resulted in six distinct groups and revealed a highly dynamic regulation of protein phosphorylation during the course of ischemia and reperfusion (Figure 3A). In contrast to the global proteome, phosphorylation changes occur rapidly, already following ischemia. Kinases recognize their substrate partly through certain sequence motifs near the phosphorylation site, and some of these motifs are associated with specific kinases. Motif analysis revealed differences in its composition amongst clusters (Figure S5). First, we observed that proline-directed phosphorylation comprised almost half of the significantly changed phosphosites (144 out of 305). These were spread across all six clusters. Amongst the nonproline-directed motifs, the clusters 1, 2, 5, and 6 showed predominantly acidic motifs characterized by the presence of aspartic and glutamic acid. Cluster 3 showed basic residues upstream of the phosphorylation site, and cluster 4 was mostly comprised of proline-directed phosphosites. These results show how the activity of different kinases changes during the different stages of IR injury.
Figure 3.

Dynamic phosphoproteome during ischemia–reperfusion in the human intestine. (A) Heatmap visualizing clustering of changing phosphosites and average temporal profiles for every cluster (a gray area represents CI). Hierarchical clustering was based on Z scores of the log 2 values of differentially expressed proteins. The ANOVA test was performed, and P < 0.05 was considered statistically significant. (B) Functional enrichment analysis of differentially phosphorylated proteins. Over-represented gene ontology (GO) terms are shown (P < 0.05). The size of the dot correlates with the number of proteins linked to that GO term, as indicated in the legend. Red, GO biological processes; blue, GO cellular component, gray, GO molecular function. (C) Networks of highly interconnected phosphoproteins related to overrepresented terms of Cell junction and RNA splicing. (D) Heat maps of single phosphosites related to different biological processes. Showing the Z scores of the averaged log 2 intensities for each condition.
Functional Enrichment Analysis of the Proteins with Significantly Regulated Phosphorylation
In general, differentially phosphorylated proteins were involved in the regulation of mRNA processing and RNA splicing, supramolecular fiber organization/cytoskeleton, and cell junction organization (Figure 3B). Related molecular functions—e.g., RNA polymerase binding and actin binding—and cellular components—e.g., spliceosomal complex, actin cytoskeleton, and adherence junction—were overrepresented as well. In contrast to the changing proteome, the GO analysis per cluster revealed that differentially phosphorylated proteins related to the same biological process appeared in different clusters (Table S6). Moreover, changing phosphosites from the same protein were represented in different clusters, for instance, phosphosites on MISP (clusters 2–5) or SRRM2 (clusters 1, 3, 5) (Table S3).
Next, similar to the proteome data, we explored the connectivity and association of dynamically phosphorylated proteins. The network analysis showed that many of the phosphorylated proteins were related to either cell junctions or RNA splicing, forming discrete networks of highly interconnected proteins (Figure 3C). This highlights that proteins related to these two biological processes are highly regulated by phosphorylation during IR.
Altered phosphorylation of RNA splicing factors was observed for various serine/arginine proteins, including SRRM2, SRRM1, and SRSF2 (Figure 3D). In addition, several heterogeneous nuclear ribonucleoproteins were found to show changes in phosphorylation and included HNRNPK and HNRNPC (Figure 3D). Differentially phosphorylated proteins related to cell junction organization included cadherin-associated CTNND1, scaffolding proteins such as ZO-1 (TJP1), and other proteins contributing to cell adhesion and related cytoskeleton organization, including CTTN, DSP, and SYNPO2 (Figure 3D). Phosphorylated proteins associated with supramolecular fiber organization included STMN1 and ESPN.
Exploration of Specific Phosphosites with Known Functionality
After focusing on the proteins that displayed dynamic phosphorylation during IR, we next explored our data for phosphosites with a previously studied functionality. By applying PTM-SEA to all of the phosphosites detected, various signatures were found to be regulated during IR injury (Figure S4E). Overall, control and ischemic samples showed a lower expression of phosphosites related to the growth factor response (e.g., EGF treatment and ERK2/MAPK1 signature) and cell division (e.g., CDK1 and CDK2 signatures), with the lowest intensities observed during ischemia. In contrast, 30 min reperfusion presented a high intensity of phosphosites related to growth factor stimulus. A similar trend was observed for CDK targets, which were highly phosphorylated during the reperfusion conditions when compared to the control and ischemic samples.
When looking at the individual trend of biologically relevant phosphosites belonging to these signatures, we encountered phosphorylation on transcription factor ATF2, namely, T69 and T71, to be upregulated upon ischemia, peaking at 30 min reperfusion and dropping at 120 min (Figure 3D). On the other hand, phosphorylation on the ribosomal protein RPS6, a known marker of active translation, increased drastically at 30 min reperfusion. Potential targets of MAP kinases and CDKs were upregulated during reperfusion times, such as S405 and S418 of CTTN and S25 and S38 of STMN1 (Figure 3D). The latter two proteins are related to cytoskeleton organization.
Proteins Showing Regulated Phosphosites as well as Significant Alterations in Abundance
When looking at the overlap between the proteome and the phosphoproteome, we found that twenty-two of the differentially phosphorylated proteins were also found to be significantly changed in abundance at the protein level. Among these overlapping proteins, a major part (16/22) was located in cluster 2 of the changing proteome, and hence these proteins showed a decrease in abundance during reperfusion and the majority was related to cell junction and cytoskeleton (e.g., CDHR5, CTTN, CGN, MYO7B, and ESPN) and digestion/absorption (e.g., LCT, SLC9A3R). For these 16 proteins, almost half of the phosphorylation changes occurred already at 45I (27 changing phosphosites in clusters 1, 2, 5, and 6 versus 29 altered phosphosites in clusters 3 and 4), indicating that alterations in phosphorylation preceded a decrease in their abundance.
On-Tissue Imaging of Tryptic Peptides Using MALDI-TOF MSI
Next to IR-induced proteomic changes in whole tissues, we explored histological region-specific protein changes. High-speed MALDI-TOF MSI enabling bottom-up tissue proteomics experiments were performed on a total of 36 tissue sections belonging to nine patients (four experimental conditions per patient: Ctrl, 45I, 30R, 120R). Prior to imaging, proteins underwent tryptic on-tissue proteolysis. As digestion efficiency greatly influences signal intensities of tryptic peptides, a spot of cytochrome c was added to each slide as a quality control,28 and its digestion profile was used to detect outliers. Based on PCA analysis of the average cytochrome c mass spectrum, one out of nine patient datasets was excluded from analysis (Figure S6). Figure 4A shows the average peptide spectrum across all control tissues.
Figure 4.

MALDI-TOF MSI of distinct histological structures in the human small intestine. (A) Average mass spectrum of whole tissues (Ctrl). (B) H&E staining showing annotation of histological layers, mucosa, submucosa, and muscle. See also Figure S7 for region annotations in all tissues. (C) Heatmap visualizing intensity differences for average mass spectra obtained from mucosa, submucosa, and muscle layers of the small intestine. Individual peptide images of m/z values with specific localization in (D) mucosa (m/z 886.5 ± 0.2 Da), (E) muscle (m/z 1198.7 ± 0.2 Da), (F) submucosa (m/z 840.5 ± 0.2 Da), and blood vessels (m/z 1274.6 ± 0.2 Da). All peptide images were generated from TOF-MSI data.
MALDI-TOF MSI of Distinct Histological Structures in the Human Small Intestine
We first compared peptide distributions with the tissue’s histology to evaluate the potential of MALDI MSI to detect region-specific IR-induced protein changes. After MALDI MSI, tissue sections were H&E-stained, and optical scans were coregistered to the MSI images. Histological regions were annotated in the H&E images (Figure 4B and S7) and heat maps of average mass spectra acquired from mucosa, submucosa, and muscle regions showed distinct peptide profiles for the different histological regions (Figure 4C). In addition, individual peptide images showed specific localization in distinct intestinal tissue structures. For example, m/z 866.5 was located in the mucosa region (Figure 4D) and m/z 1198.7 in muscle (Figure 4E). In addition, m/z 840.5 was found to be associated with connective tissues of the submucosa layer but also surrounding muscle tissue (Figure 4F), and m/z 1274.6 was specifically located in blood vessels (Figure 4G). To identify region-specific IR-induced protein changes, we performed subsequent analysis for each histological layer separately. Since 10 out of 32 tissues did not contain muscle in the analyzed section (Figure S7), statistical analysis could not be performed for the muscle layer.
IR-Induced Changes in Mucosa and Submucosa
In the context of IR injury, we are particularly interested in the mucosa layer as this is the most susceptible to damage. PCA analysis of mucosa regions revealed the highest similarity between Ctrl and 45I conditions, on the one hand, and reperfusion conditions (30R, 120R) on the other hand (Figure 5A), which is congruent with the clustering of the LC-MS/MS proteomics data. Peak picking resulted in 319 peptide signals to be included for statistical analysis, of which 154 m/z values were found to be significantly changing in intensity during IR (Table S7). In general, signal intensities were either gradually correlated or anticorrelated to the IR sequence (Ctrl-45I-30R-120R). Remarkably, a decreasing intensity gradient was observed predominantly for peptides in the lower mass range (m/z < 1500), whereas peptides in the higher mass range (m/z > 1500) showed an increasing intensity gradient (Table S7). Images of the peptides with the 10 highest fold changes showed mucosa-specific localization and a decreasing abundance during IR in most of the patients (Figure 5C-II, all tissues in Figure S8A–D). Two of these were among the top 10 peaks with the highest intensity (Figure S8C,D). Interestingly, some peptides show a distribution shift from the whole mucosa in Ctrl toward the villus tips after 120R, as shown in Figure 5C-III (Figure S8A). The peaks with an increasing intensity gradient appeared to be mostly low-intensity peptides, expressed in all histological layers.
Figure 5.

Region-specific changes in response to ischemia–reperfusion. Principle component analysis of (A) mucosa regions and (B) submucosa regions. (C) H&E staining (I) and peptide images showing high-fold change peptides in the mucosa (II, III) and submucosa (IV). Corresponding graphs show intensity data for all patients (presented peptide images correspond to a red line in the graph). All peptide images were generated from TOF-MSI data for the indicated m/z values ±0.2 Da. See also Figures S8 and S9 for peptide images from all samples.
PCA analysis of submucosa regions resulted in a slightly different grouping of samples compared to the mucosa (Figure 5B). In total, 185 peptide signals were found to be significantly changed between IR conditions, and all showed a decreasing intensity gradient (Table S8). Looking at peptides exhibiting a high-fold change, only a few showed specific localization in the submucosa layer (Figure 5C-IV, all tissues in Figure S9A,B). Images of high-intensity peptides better represented submucosa-specific peptide changes (Figure S9C,D).
Protein Identification of Relevant Peptides Obtained from MALDI-TOF MSI Analysis
To link the MALDI-TOF MSI data (R = 15 000 at m/z 1000) of peptides to the LC-MS/MS protein identifications, we used MALDI-FTICR MSI measurements (R = 200 000 at m/z 1000) as an intermediate step to obtain more accurate mass descriptions of the peptides of interest. With regard to the mucosa, 75 out of 154 significantly changing m/z values from TOF-MSI could be matched to one or more peaks in the high mass resolution FTICR data. Matching these accurate m/z values to the LC-MS/MS (phospho)-proteome data enabled annotation of 10 proteins (Table S9). Performing the same analysis for the submucosa data resulted in 96 matched FTICR m/z values and in the annotation of 11 proteins (Table S9). None of the annotations were based on matched phosphopeptides. We next highlight a selection of proteins of interest, showing layer-specific localization (peptide images can be found in Figure S10).
Remarkably, among annotated proteins significantly changing in the mucosa, three proteins (ELAV1, SNRNP70, and HNRNPC) were associated with mRNA splicing and processing. Peptide signals belonging to these proteins were mucosa-specific and had the highest intensity in the control sample, which decreased during the IR sequence (Figures 6-II and A–C). Images of complement C5 showed localization specifically in the mucosa layer. The decreasing intensity during IR was accompanied by a distribution shift toward the villus tips (Figure 6-III) and expression in villus debris in reperfusion samples (Figure S10D). Interestingly, for complement C7, a decreasing intensity (P < 0.01) in submucosa was accompanied by an increase in the mucosa layer (P = 0.02). Peptide images confirm this distribution shift (Figures 6-IV and S10E). The collagen-α-2(I) chain, a structural constituent of the extracellular matrix, exhibited a significant downregulation in the submucosa (Figures 6-V and S10F).
Figure 6.

Selection of annotated proteins using MALDI-TOF MSI in combination with FTICR and LC-MS/MS. H&E staining (I) and peptide images from one patient are shown for indicated m/z values, which were annotated as ELAV-like protein (II), complement C5 (III), complement C7 (IV), and collagen α2(I)chain (V). Graphs show intensities for all patients in the mucosa (left) and/or submucosa (right) in case the m/z was differentially expressed in the respective layer. Intensities corresponding to the presented peptide images are shown in red. All peptide images were generated from TOF-MSI data for the indicated m/z values ±0.2 Da. See also Figure S10 for peptide images from all patient samples.
Imaging MS and Quantitative MS-Based Proteomics are Complementary Methods
When comparing imaging MS with LC-MS/MS proteomics results, only two (Nup50, complement C5) of the 22 annotated proteins overlapped with the list of significantly changed proteins based on LC-MS/MS analysis. Nevertheless, the proteins found to be changed in MSI experiments were involved in processes that were over-represented among differentially expressed proteins in LC-MS/MS analysis. Figure 7 summarizes and connects the most important results of this study.
Figure 7.

Summary of MS-based proteome and phosphoproteome functional enrichment analysis and imaging MS data showing that these methods complement as well as support each other. Overview summarizing regulated processes during IR based on the dynamic proteome (blue) and phosphoproteome (red) or both (blue-red). Processes that were shown to be significantly enriched are indicated in bold. Peptide images show significantly changing peptides, specifically located in the mucosa (red arrow) or submucosa (blue arrow). The annotated proteins were related to indicated processes (arrow). The overview image was adjusted from ReacFoam format (Reactome.org). C5, complement 5; C7, complement 7; SNRNP70, small nuclear ribonucleoprotein U1 subunit 70; HNRNPC, heterogeneous nuclear ribonucleoprotein C; ELAV1, ELAV-like protein 1; and COL1A1, collagen 1 α2 (I) chain.
Over-represented processes in the changing proteome (blue), phosphoproteome (red), or both (blue-red) are highlighted, and images of significantly changed peptides and corresponding annotated proteins are shown with arrows pointing toward the corresponding process (red, mucosa; blue, submucosa). The overview shows that the applied methods complement each other; insight into the different regulated processes in the global (phospho)proteome is accompanied by partially overlapping spatial information provided by MSI.
Discussion
Here, we present a comprehensive study of the proteome and phosphoproteome in combination with MALDI MSI to unravel protein alterations during IR of the human intestine. LC-MS/MS-based (phospho)proteomics resulted in the identification and quantification of thousands of proteins and phosphosites and enabled thorough functional enrichment and interaction network analyses. We showed that proteins related to the intestinal absorption, microvillus structure, and cell junction were decreased in abundance after IR, whereas proteins involved in innate immunity were increased in abundance. Phosphoproteome analysis revealed regulation of RNA splicing events and cytoskeletal/cell junction organization and suggested MAPK and CDK families to be active kinases during IR. In addition, MALDI MSI enabled the identification of mucosa-specific protein changes as well as alterations in protein distribution, for instance, a shift in localization of complement C5 during the course of IR.
Functional Interpretation of the Dynamic Proteome in Intestinal IR
Functional enrichment analysis of the dynamic proteome during IR revealed that proteins showing a decrease in abundance during reperfusion were overrepresented for GO terms related to the microvillus/cell junction/cytoskeleton. Proteins showing an increase during reperfusion were related to the innate immune response. Functional interpretation of the changing proteome will be discussed per cluster.
The downregulation of various translation initiation factors (EIF2S2, EIF4EBP1, EIF5B) suggests that protein translation is inhibited in the early reperfusion phase. Inhibition of translation initiation is one of the cytoprotective mechanisms of the unfolded protein response,29 which is induced in response to proteotoxic stress in the ER and known to play an important role in IR injury.8 Interestingly, we observed a decrease in abundance of HspA4, a member of the Hsp70 family, which act as chaperones and is known to be induced in response to proteotoxic stress and protect cells from its harmful effects.30 We speculate that acute IR-induced oxidative stress depleted the Hsp70 protein at 30R, which was rapidly recovered by IR stress-induced transcription of Hsp70s.31 Another stress-response related protein showing decreased abundance was Nup50, which has a direct role in nuclear transport and is known to be sensitive to different stressors, including oxidative stress.32 A reduction in TNIK, which acts as a critical activator of Wnt targets,33 suggests that this kinase may play a role in the inhibition of proliferation during IR-induced cellular stress.
The decreased abundance of proteins involved in intestinal digestion/absorption and related to microvillus and cell junction organization likely reflects the loss of villus tips as a consequence of reperfusion injury. This group of proteins included many structural proteins of intestinal microvilli and also brush border enzymes and transporters for nutrient absorption. Our group has previously described how IR-induced destruction of the intestinal epithelium led to the contraction of the epithelial sheets and shedding of the damaged villus tips, which resulted in reduced length of the villi.6 Reorganization of the actin cytoskeleton and accumulation of F-actin at the basal side of enterocytes enabled this protective mechanism. The actin cytoskeleton is tightly anchored to the lateral membranes by cell–cell junction complexes. This interaction between the cytoskeleton and the cell junction is crucial for the integrity of the epithelial barrier and changes in the organization of either one affect the other and may contribute to gut barrier disruption,34 which is known to occur in inflamed and injured intestine35 and cardiac IR.36 A decreased abundance of the cytosolic IFABP protein in the villus is a well-known consequence of intestinal IR.5 The loss of the enterocyte membrane integrity results in the release of IFABP into the circulation and has been shown to be a useful serological biomarker for intestinal IR injury.37−39
Functional enrichment analysis of the cluster of proteins exhibiting increased expression during reperfusion (cluster 3) strongly indicates activation of the innate immune response upon reperfusion of the ischemically damaged intestine and points in particular to a crucial role of the complement system, as also demonstrated by the presented network of interconnected proteins in this cluster. Activation of the complement system has been well-documented in animal models of intestinal IR.40−43 In addition, our group has previously reported complement activation after IR in the human intestine.7 In that study, high amounts of complement activation product C3c were detected in the luminal debris of shed enterocytes but not in mucosal tissue, whereas native C3 was present in the tissue.7 Interestingly, our MSI data show that C5 expression shifted toward the villus tips and that C5 was almost absent in the mucosal tissue itself during reperfusion. The decrease of complement C5 in the mucosa layer seems contradictory to the quantitative proteomics data, showing an overall increase in complement proteins. This discrepancy may be explained by homogenization of whole tissue, including the luminal debris that may contain complement proteins, for proteomics analysis. Images of C7, on the other hand, showed increased abundance in the mucosa layer together with a decrease in submucosal expression. It should be noted that we cannot verdict on actual complement activation, as our proteomics analysis could not distinguish the native and active forms of complement factors. Our findings shed new light on the importance of the complement system in IR of the human intestine and could give rise to further studies investigating the activation of complement and its role in human intestinal IR. To date, the potentially protective effects of complement inhibition during intestinal IR have been investigated in animal models only.41−43 Moreover, these data underline the strength of combining LC-MS/MS data giving robust and reliable quantitative data and MS images exposing changes in protein localization. An alternative spatial proteomics approach that could be very useful to identify and quantify peptides in a specific tissue area is laser capture microdissection of the area of interest followed by LC-MS/MS analysis.44,45
Furthermore, both LC-MS/MS and MSI approaches point to changing ECM organization during IR, which is in accordance with our previous proteomics analysis of hypoxia-reoxygenation in a human intestinal organoid model.46 Interestingly, network analysis indicated interconnection of proteins directly related to innate immunity and ECM proteins. Unbalanced ECM remodeling, and associated altered expression of ECM proteins, are a well-known feature in inflammatory bowel disease (IBD).47 Immune activation and inflammation are known to induce both the degradation and synthesis of the ECM. The interplay between inflammation and the ECM is a dynamic process in which ECM alterations can also actively promote inflammation and contribute to disease progression in IBD.48 The observed alterations in the expression of ECM proteins, both quantitatively and in peptide images, may reflect remodeling of the ECM as a result of IR-induced inflammation.
The small cluster of proteins that gradually increased during IR (cluster 4) contained proteins involved in a variety of processes. Cytoglobin, which has an important role in oxygen transport, was significantly increasing during IR and has been shown to be protective against IR injury in other organs.49,50 Interestingly, the increase in the caspase-1 protein may point to the promotion of pyroptosis, a pro-inflammatory form of programmed cell death, which is initiated by caspase-1, and has recently been shown to play a role in murine intestinal IR injury.51
Dynamic Phosphoproteome
Protein phosphorylation and its regulation by kinases and phosphatases play a key role in the regulation of cellular functions, and changes in phosphorylation can be a cause as well as a consequence of a variety of diseases.52 By analyzing our data with a combination of phosphosite- and protein-centric approaches, we identified dynamic protein phosphorylation events that regulate specific biological processes, and hence are expected to play an important role in the cellular response to IR.
Hierarchical clustering accompanied with motif analysis per cluster revealed the dynamic nature of protein phosphorylation during IR. The majority of phosphosites are proline-directed, which suggests that they are potential targets of a variety of kinases, from CDKs to MAP kinases.53,54 In the case of IR, it is very likely that many of the regulated phosphosites are targets of MAP kinases such as JNK and p38, which are activated by cellular stress and thus their targets are expected to locate to clusters showing upregulation after IR (clusters 1, 3, 4, 5). Indeed, phosphosites of STMN1 (S25, S38), ATF2 (T69, T71), and CTTN (S405, S418), which are proposed targets of either JNK or p38 (Table S5), locate to clusters 3 and 4, which contain phosphosites that appear upregulated during reperfusion. Proline-directed phosphosites could also be targets of MAP kinases related to growth and survival, like ERK1/ERK2. We found that phosphorylation of ERK2 on T185 and Y187, which are indicative of kinase activation,55 were located in cluster 4, which suggests that ERK2 is highly active shortly after reperfusion. Cluster 5, which exhibits upregulated phosphosites at 45I, is predominantly comprised of nonproline-directed phosphosites, which suggests that kinases other than MAPK are likely to be active during ischemia. Overall, the differences in dynamics and motif composition amongst clusters suggest that protein phosphorylation response to IR is complex and comprises different effector kinases acting at different stages.
Phosphorylation Dynamics and Its Relation with Signaling Pathways
We further examined which kinases may be responsible for the protein phosphorylations by analyzing how phosphosite dynamics relate to signatures of specific kinases or signaling pathways. As expected, we observed that most potential MAPK and CDK targets tend to decrease during ischemic conditions, followed by a drastic increase during reperfusion. However, when looking into specific phosphosites with known biological functions, we highlighted some examples displaying interesting trends. One of these is the phosphorylation of ATF2, a transcription factor regulating cell growth and survival, on T69 and T71, which are known targets of MAP kinases in response to both cellular stress and growth factors.56 Both phosphosites showed increased intensity upon ischemia and peaked at 30R, which equals induction of ATF2 transcription activity.57−59 This is in line with previously reported increased ATF2 binding activity in renal IR.60 It is conceivable that stress-responsive MAP kinases (JNK and/or p38) induce phosphorylation during ischemia, which is subsequently boosted by other MAP kinase activity (e.g., ERK1/ERK2) in response to growth factors upon reperfusion.61
In addition, we identified protein phosphorylation events known to regulate protein translation, namely, S235 and S236 phosphorylation on RPS6, an important ribosomal protein that is regulated by kinases responsive to growth factors.62,63 As these phosphosites on RPS6 promote assembly of the preinitiation complex,64 our data indicate translation seemed to be inhibited during ischemia and resumed upon reperfusion. The latter supports our findings in the proteome data, where proteins involved in translation appeared to decrease in abundance early in reperfusion and then recover after 120R.
Functional Interpretation of Changes in the Phosphoproteome
Through GO enrichment analysis, we found that most of the phosphorylation-regulated proteins were involved in RNA splicing and cell junction/cytoskeleton organization. This is consistent with a previous phosphoproteome study in a swine model for cardiac IR, reporting that the majority of phosphoprotein alterations were involved in RNA processing and cell junction.65
Pre-mRNA splicing is executed by the spliceosome. The phosphorylation state of splicing factors is crucial for the correct regulation of their function and organization and for the formation of the spliceosome complex.66 Splicing factors exhibited increasing as well as decreasing phosphorylation (clusters 1 and 5), suggesting that both phosphorylation and dephosphorylation events play a role in the regulation of RNA splicing, which has been shown previously.67 In addition to the role in constitutive splicing, phosphorylation acts as a major player in the regulation of alternative splicing,68 a process that 95% of human genes undergo, and is considered an important mechanism in pathological cellular processes.69 Accumulating evidence indicates that pre-mRNA splicing plays an essential role in the adaptation to hypoxic stress.70−72 Hypoxia-induced alternative splicing of, for example, VEGF, Bcl-x, BNIP3, and CAIX, changes gene expression patterns to enhance proliferation and survival.71 This may explain the ischemia-induced changes in phosphorylation of splicing-related proteins in our model. Furthermore, stress-activated MAP kinases such as JNK and p38, which are activated during IR, can indirectly modulate splicing by phosphorylation of selected splicing factors.73 In addition to the phosphorylation changes in splicing factors, MSI results showed a locally decreased mucosal intensity of peptides annotated as spliceosome component SNRNP70 and RNA binding proteins ELAV1 and HNRNPC, which play a role in RNA splicing. This further supports the regulation of splicing events during IR.
A substantial part of phosphorylation-regulated proteins was related to cell junction and cytoskeletal organization. In contrast to the decrease in abundance of these proteins following reperfusion, changes in phosphorylation exhibited various temporal profiles, including phosphosites that were regulated immediately after ischemia. Regulation of cell junction proteins by phosphorylation plays an important role in the assembly and disassembly of adherence junctions.74 The latter are known to interact with the actin cytoskeleton of adjacent cells, suggesting that the dynamic phosphorylation of junctional and cytoskeletal proteins during IR likely affects its organization and integrity. Interestingly, we detected some specific phosphosites with a known function in the regulation of cytoskeleton organization, which showed an increase during reperfusion. Cortactin (CTTN) phosphorylation on S405 and S418 is associated with cytoskeleton reorganization,75,76 and STMN1 phosphosites S16, S25, and S38 are linked to polymerization of the microtubule cytoskeleton.77 Together, these findings suggest that increased phosphorylation of junctional proteins upon reperfusion may be related to the reorganization of the cytoskeleton during IR of the human intestine.6
MSI Data Interpretation and Limitations
The clustering of control and ischemia samples, on the one hand, and reperfusion samples, on the other hand, was comparable for MSI and proteome data. However, the fact that for MSI, the peptides in the lower mass range exhibited a decreasing gradient, and those in the higher mass range an increasing intensity gradient may suggest that enzymatic digestion efficiency was affected negatively by ischemia and an increasing reperfusion time. While these results have to be interpreted with care, we can speculate that IR-induced changes in endogenous proteolytic enzymes may affect digestion efficiency. This is supported by animal studies which have shown that during IR, pancreatic enzymes from the intestinal lumen leak into the intestinal wall, resulting in self-digestion.78,79
Where LC-MS/MS proteomics is a well-established and robust technology, MSI methodologies are evolving rapidly. One of the existing challenges of tryptic peptide MSI is the identification of the corresponding proteins. Here, we assigned MSI-detected peptides to protein data from LC-MS/MS analysis of the same tissue samples. A trade-off between speed, sensitivity, and mass resolution made us decide to use MALDI-TOF MSI for the screening of the 36 tissue samples and to perform additional MALDI-FTICR measurements only as an intermediate step to aid in the identification process because of their higher mass accuracy. When compared to phospho- and global proteomics, MSI identification results were sparse. There are several explanations for this. First, the number of peptide signals is relatively low in MSI analysis due to the mass resolution of the TOF system and the lack of an additional dimension of separation. This low mass resolution was also the major limiting factor for the identification of the peptide signals since about 30% of the TOF peaks could be further resolved into at least two peptide signals in the FTICR spectrum. We therefore assume that the use of high-resolution MALDI MSI for all tissue sections would have significantly increased the number of identified (phospho)peptides, although it is more time-consuming. Another factor that limits identification is the use of different ionization methods (ESI for LC-MS/MS versus MALDI in MSI), which inherently limits the overlap between the detected peptides. Despite these detrimental factors, the proteins annotated to significantly changing peptides with our MSI analysis were related to the same processes that were shown to be altered in our quantitative proteomics analysis, supporting the coherence between the different types of data used. For further studies, MSI- and/or morphology-guided laser microdissection could be performed and analyzed by LC-MS/MS.44
Conclusions
Altogether, we identified IR-induced alterations in abundance, phosphorylation, and distribution of proteins, which expanded our understanding of the molecular events that occur during IR in the human intestine. In addition, the study highlights the strength of the complementary use of different MS-based methodologies.
Acknowledgments
This work was funded by NUTRIM, Maastricht University (NUTRIM Graduate Program grant to A.M.K.), NWO (Aspasia grant 015.010.046 to K.L.), NWO (project 184.034.019), and was part of the Netherlands X-omics Initiative. J.M.V. was supported by scholarships from the Ministry of Science and Technology of Costa Rica (MICITT) and the University of Costa Rica (UCR). We thank the surgical team of Maastricht UMC+ for their excellent surgical assistance. Table of Contents graphic was created with BioRender.com.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jproteome.1c00447.
Correlation plots (phospho)proteome (Figure S1); hierarchical clustering of the proteome (Figure S2); protein interaction network (Figure S3); global overview of the phosphoproteome (Figure S4); sequence motif logos (Figure S5); PCA analysis of the cytochrome c mass spectrum (Figure S6); annotation histological regions in tissue samples (Figure S7); peptide images significantly changed m/z mucosa (Figure S8); peptide images significantly changed m/z submucosa (Figure S9); peptide images for a selection of annotated proteins (Figure S10) (PDF)
Differentially expressed proteins during ischemia–reperfusion (Table S1); GO term enrichment analysis of the dynamic proteome per cluster (Table S2); changing phosphosites during ischemia–reperfusion (Table S3); kinase enrichment analysis in whole phosphoproteome (Table S4); kinase enrichment analysis in dynamic phosphoproteome (Table S5); GO term enrichment analysis of the dynamic phosphoproteome per cluster (Table S6); significantly changed m/z in mucosa layer (Table S7); significantly changed m/z in the submucosa layer (Table S8); annotated proteins using MALDI-TOF MSI of tryptic peptides combined with MALDI-FTICR MSI and LC-MS/MS (Table S9) (XLSX)
The authors declare no competing financial interest.
Supplementary Material
References
- American Gastroenterological Association Medical Position Statement: guidelines on intestinal ischemia. Gastroenterology 2000, 118, 951−953 10.1016/S0016-5085(00)70182-X. [DOI] [PubMed] [Google Scholar]
- Bala M.; Kashuk J.; Moore E. E.; Kluger Y.; Biffl W.; Gomes C. A.; Ben-Ishay O.; Rubinstein C.; Balogh Z. J.; Civil I.; Coccolini F.; Leppaniemi A.; Peitzman A.; Ansaloni L.; Sugrue M.; Sartelli M.; Di Saverio S.; Fraga G. P.; Catena F. Acute mesenteric ischemia: guidelines of the World Society of Emergency Surgery. World J. Emerg. Surg. 2017, 12, 38 10.1186/s13017-017-0150-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldenburg W. A.; Lau L. L.; Rodenberg T. J.; Edmonds H. J.; Burger C. D. Acute mesenteric ischemia: a clinical review. Arch. Intern. Med. 2004, 164, 1054–1062. 10.1001/archinte.164.10.1054. [DOI] [PubMed] [Google Scholar]
- Kalogeris T.; Baines C. P.; Krenz M.; Korthuis R. J. Ischemia/Reperfusion. Compr. Physiol. 2016, 7, 113–170. 10.1002/cphy.c160006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derikx J. P.; Matthijsen R. A.; de Bruine A. P.; van Bijnen A. A.; Heineman E.; van Dam R. M.; Dejong C. H.; Buurman W. A. Rapid reversal of human intestinal ischemia-reperfusion induced damage by shedding of injured enterocytes and reepithelialisation. PLoS One 2008, 3, e3428 10.1371/journal.pone.0003428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grootjans J.; Thuijls G.; Derikx J. P.; van Dam R. M.; Dejong C. H.; Buurman W. A. Rapid lamina propria retraction and zipper-like constriction of the epithelium preserves the epithelial lining in human small intestine exposed to ischaemia-reperfusion. J. Pathol. 2011, 224, 411–419. 10.1002/path.2882. [DOI] [PubMed] [Google Scholar]
- Grootjans J.; Lenaerts K.; Derikx J. P.; Matthijsen R. A.; de Bruine A. P.; van Bijnen A. A.; van Dam R. M.; Dejong C. H.; Buurman W. A. Human intestinal ischemia-reperfusion-induced inflammation characterized: experiences from a new translational model. Am. J. Pathol. 2010, 176, 2283–2291. 10.2353/ajpath.2010.091069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grootjans J.; Hodin C. M.; de Haan J. J.; Derikx J. P.; Rouschop K. M.; Verheyen F. K.; van Dam R. M.; Dejong C. H.; Buurman W. A.; Lenaerts K. Level of activation of the unfolded protein response correlates with Paneth cell apoptosis in human small intestine exposed to ischemia/reperfusion. Gastroenterology 2011, 140, 529.e3–539.e3. 10.1053/j.gastro.2010.10.040. [DOI] [PubMed] [Google Scholar]
- Kassahun W. T.; Schulz T.; Richter O.; Hauss J. Unchanged high mortality rates from acute occlusive intestinal ischemia: six year review. Langenbeck’s Arch. Surg. 2008, 393, 163–171. 10.1007/s00423-007-0263-5. [DOI] [PubMed] [Google Scholar]
- Mastoraki A.; Mastoraki S.; Tziava E.; Touloumi S.; Krinos N.; Danias N.; Lazaris A.; Arkadopoulos N. Mesenteric ischemia: Pathogenesis and challenging diagnostic and therapeutic modalities. World J. Gastrointest. Pathophysiol. 2016, 7, 125–130. 10.4291/wjgp.v7.i1.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kougias P.; Lau D.; El Sayed H. F.; Zhou W.; Huynh T. T.; Lin P. H. Determinants of mortality and treatment outcome following surgical interventions for acute mesenteric ischemia. J. Vasc. Surg. 2007, 46, 467–474. 10.1016/j.jvs.2007.04.045. [DOI] [PubMed] [Google Scholar]
- Altelaar A. F.; Munoz J.; Heck A. J. Next-generation proteomics: towards an integrative view of proteome dynamics. Nat. Rev. Genet. 2013, 14, 35–48. 10.1038/nrg3356. [DOI] [PubMed] [Google Scholar]
- Liu H.; Han M.; Li J.; Qin L.; Chen L.; Hao Q.; Jiang D.; Chen D.; Ji Y.; Han H.; Long C.; Zhou Y.; Feng J.; Wang X. A Caffeic Acid Matrix Improves In Situ Detection and Imaging of Proteins with High Molecular Weight Close to 200,000 Da in Tissues by Matrix-Assisted Laser Desorption/Ionization Mass Spectrometry Imaging. Anal. Chem. 2021, 93, 11920–11928. 10.1021/acs.analchem.0c05480. [DOI] [PubMed] [Google Scholar]
- Han J.; Permentier H.; Bischoff R.; Groothuis G.; Casini A.; Horvatovich P. Imaging of protein distribution in tissues using mass spectrometry: An interdisciplinary challenge. TrAC, Trends Anal. Chem. 2019, 112, 13–28. 10.1016/j.trac.2018.12.016. [DOI] [Google Scholar]
- Huber K.; Khamehgir-Silz P.; Schramm T.; Gorshkov V.; Spengler B.; Rompp A. Approaching cellular resolution and reliable identification in mass spectrometry imaging of tryptic peptides. Anal. Bioanal. Chem. 2018, 410, 5825–5837. 10.1007/s00216-018-1199-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schober Y.; Schramm T.; Spengler B.; Rompp A. Protein identification by accurate mass matrix-assisted laser desorption/ionization imaging of tryptic peptides. Rapid Commun. Mass Spectrom. 2011, 25, 2475–2483. 10.1002/rcm.5135. [DOI] [PubMed] [Google Scholar]
- Spraggins J. M.; Rizzo D. G.; Moore J. L.; Noto M. J.; Skaar E. P.; Caprioli R. M. Next-generation technologies for spatial proteomics: Integrating ultra-high speed MALDI-TOF and high mass resolution MALDI FTICR imaging mass spectrometry for protein analysis. Proteomics 2016, 16, 1678–1689. 10.1002/pmic.201600003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H.; Ye M.; Dong J.; Corradini E.; Cristobal A.; Heck A. J.; Zou H.; Mohammed S. Robust phosphoproteome enrichment using monodisperse microsphere-based immobilized titanium (IV) ion affinity chromatography. Nat. Protoc. 2013, 8, 461–480. 10.1038/nprot.2013.010. [DOI] [PubMed] [Google Scholar]
- Meiring H. D.; van der Heeft E.; ten Hove G. J.; de Jong A. P. J. M. Nanoscale LC-MS(n): technical design and applications to peptide and protein analysis. J. Sep. Sci. 2002, 25, 557–568. . [DOI] [Google Scholar]
- Tibshirani R.; Walther G.; Hastie T. Estimating the number of clusters in a data set via the gap statistic. J. Royal Stat. Soc.: Ser. B (Stat. Methodol.) 2001, 63, 411–423. 10.1111/1467-9868.00293. [DOI] [Google Scholar]
- Szklarczyk D.; Gable A. L.; Lyon D.; Junge A.; Wyder S.; Huerta-Cepas J.; Simonovic M.; Doncheva N. T.; Morris J. H.; Bork P.; Jensen L. J.; Mering C. V. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. 10.1093/nar/gky1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supek F.; Bošnjak M.; Škunca N.; Šmuc T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS One 2011, 6, e21800 10.1371/journal.pone.0021800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vacic V.; Iakoucheva L. M.; Radivojac P. Two Sample Logo: a graphical representation of the differences between two sets of sequence alignments. Bioinformatics 2006, 22, 1536–1537. 10.1093/bioinformatics/btl151. [DOI] [PubMed] [Google Scholar]
- Lachmann A.; Ma’ayan A. KEA: kinase enrichment analysis. Bioinformatics 2009, 25, 684–686. 10.1093/bioinformatics/btp026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krug K.; Mertins P.; Zhang B.; Hornbeck P.; Raju R.; Ahmad R.; Szucs M.; Mundt F.; Forestier D.; Jane-Valbuena J.; Keshishian H.; Gillette M. A.; Tamayo P.; Mesirov J. P.; Jaffe J. D.; Carr S.; Mani D. R. A Curated Resource for Phosphosite-specific Signature Analysis*[S]. Mol. Cell. Proteomics 2019, 18, 576–593. 10.1074/mcp.TIR118.000943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strohalm M.; Hassman M.; Kosata B.; Kodicek M. mMass data miner: an open source alternative for mass spectrometric data analysis. Rapid Commun. Mass Spectrom. 2008, 22, 905–908. 10.1002/rcm.3444. [DOI] [PubMed] [Google Scholar]
- Perez-Riverol Y.; Csordas A.; Bai J.; Bernal-Llinares M.; Hewapathirana S.; Kundu D. J.; Inuganti A.; Griss J.; Mayer G.; Eisenacher M.; Pérez E.; Uszkoreit J.; Pfeuffer J.; Sachsenberg T.; Yilmaz S.; Tiwary S.; Cox J.; Audain E.; Walzer M.; Jarnuczak A. F.; Ternent T.; Brazma A.; Vizcaíno J. A. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. 10.1093/nar/gky1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vos D. R. N.; Jansen I.; Lucas M.; Paine M. R. L.; de Boer O. J.; Meijer S. L.; Savci-Heijink C. D.; Marquering H. A.; de Bruin D. M.; Heeren R. M. A.; Ellis S. R.; Balluff B. Strategies for managing multi-patient 3D mass spectrometry imaging data. J. Proteomics 2019, 193, 184–191. 10.1016/j.jprot.2018.10.008. [DOI] [PubMed] [Google Scholar]
- Pavitt G. D.; Ron D. New insights into translational regulation in the endoplasmic reticulum unfolded protein response. Cold Spring Harbor Perspect. Biol. 2012, 4, a012278 10.1101/cshperspect.a012278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenzweig R.; Nillegoda N. B.; Mayer M. P.; Bukau B. The Hsp70 chaperone network. Nat. Rev. Mol. Cell Biol. 2019, 20, 665–680. 10.1038/s41580-019-0133-3. [DOI] [PubMed] [Google Scholar]
- Kip A. M.; Grootjans J.; Manca M.; Hadfoune M.; Boonen B.; Derikx J. P. M.; Biessen E. A. L.; Olde Damink S. W. M.; Dejong C. H. C.; Buurman W. A.; Lenaerts K. Temporal transcript profiling identifies a role for unfolded protein stress in human gut ischemia-reperfusion injury. Cell. Mol. Gastroenterol. Hepatol. 2021, 10.1016/j.jcmgh.2021.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodiha M.; Chu A.; Matusiewicz N.; Stochaj U. Multiple mechanisms promote the inhibition of classical nuclear import upon exposure to severe oxidative stress. Cell Death Differ. 2004, 11, 862–874. 10.1038/sj.cdd.4401432. [DOI] [PubMed] [Google Scholar]
- Mahmoudi T.; Li V. S.; Ng S. S.; Taouatas N.; Vries R. G.; Mohammed S.; Heck A. J.; Clevers H. The kinase TNIK is an essential activator of Wnt target genes. EMBO J. 2009, 28, 3329–3340. 10.1038/emboj.2009.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delacour D.; Salomon J.; Robine S.; Louvard D. Plasticity of the brush border - the yin and yang of intestinal homeostasis. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 161–174. 10.1038/nrgastro.2016.5. [DOI] [PubMed] [Google Scholar]
- Lechuga S.; Ivanov A. I. Disruption of the epithelial barrier during intestinal inflammation: Quest for new molecules and mechanisms. Biochim. Biophys. Acta, Mol. Cell Res. 2017, 1864, 1183–1194. 10.1016/j.bbamcr.2017.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tansey E. E.; Kwaku K. F.; Hammer P. E.; Cowan D. B.; Federman M.; Levitsky S.; McCully J. D. Reduction and redistribution of gap and adherens junction proteins after ischemia and reperfusion. Ann. Thorac. Surg. 2006, 82, 1472–1479. 10.1016/j.athoracsur.2006.04.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanda T.; Nakatomi Y.; Ishikawa H.; Hitomi M.; Matsubara Y.; Ono T.; Muto T. Intestinal fatty acid-binding protein as a sensitive marker of intestinal ischemia. Dig. Dis. Sci. 1992, 37, 1362–1367. 10.1007/BF01296004. [DOI] [PubMed] [Google Scholar]
- Kanda T.; Tsukahara A.; Ueki K.; Sakai Y.; Tani T.; Nishimura A.; Yamazaki T.; Tamiya Y.; Tada T.; Hirota M.; Hasegawa J.; Funaoka H.; Fujii H.; Hatakeyama K. Diagnosis of ischemic small bowel disease by measurement of serum intestinal fatty acid-binding protein in patients with acute abdomen: a multicenter, observer-blinded validation study. J. Gastroenterol. 2011, 46, 492–500. 10.1007/s00535-011-0373-2. [DOI] [PubMed] [Google Scholar]
- Schellekens D. H.; Grootjans J.; Dello S. A.; van Bijnen A. A.; van Dam R. M.; Dejong C. H.; Derikx J. P.; Buurman W. A. Plasma intestinal fatty acid-binding protein levels correlate with morphologic epithelial intestinal damage in a human translational ischemia-reperfusion model. J. Clin. Gastroenterol. 2014, 48, 253–260. 10.1097/MCG.0b013e3182a87e3e. [DOI] [PubMed] [Google Scholar]
- Arumugam T. V.; Shiels I. A.; Woodruff T. M.; Granger D. N.; Taylor S. M. The role of the complement system in ischemia-reperfusion injury. Shock 2004, 21, 401–409. 10.1097/00024382-200405000-00002. [DOI] [PubMed] [Google Scholar]
- Wada K.; Montalto M. C.; Stahl G. L. Inhibition of complement C5 reduces local and remote organ injury after intestinal ischemia/reperfusion in the rat. Gastroenterology 2001, 120, 126–133. 10.1053/gast.2001.20873. [DOI] [PubMed] [Google Scholar]
- Fleming S. D.; Mastellos D.; Karpel-Massler G.; Shea-Donohue T.; Lambris J. D.; Tsokos G. C. C5a causes limited, polymorphonuclear cell-independent, mesenteric ischemia/reperfusion-induced injury. Clin. Immunol. 2003, 108, 263–273. 10.1016/S1521-6616(03)00160-8. [DOI] [PubMed] [Google Scholar]
- Arumugam T. V.; Shiels I. A.; Woodruff T. M.; Reid R. C.; Fairlie D. P.; Taylor S. M. Protective effect of a new C5a receptor antagonist against ischemia-reperfusion injury in the rat small intestine. J. Surg. Res. 2002, 103, 260–267. 10.1006/jsre.2002.6369. [DOI] [PubMed] [Google Scholar]
- Dewez F.; Oejten J.; Henkel C.; Hebeler R.; Neuweger H.; De Pauw E.; Heeren R. M. A.; Balluff B. MS Imaging-Guided Microproteomics for Spatial Omics on a Single Instrument. Proteomics 2020, 20, 1900369 10.1002/pmic.201900369. [DOI] [PubMed] [Google Scholar]
- Mezger S. T. P.; Mingels A. M. A.; Bekers O.; Heeren R. M. A.; Cillero-Pastor B. Mass Spectrometry Spatial-Omics on a Single Conductive Slide. Anal. Chem. 2021, 93, 2527–2533. 10.1021/acs.analchem.0c04572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kip A. M.; Soons Z.; Mohren R.; Duivenvoorden A. A. M.; Röth A. A. J.; Cillero-Pastor B.; Neumann U. P.; Dejong C. H. C.; Heeren R. M. A.; Olde Damink S. W. M.; Lenaerts K. Proteomics analysis of human intestinal organoids during hypoxia and reoxygenation as a model to study ischemia-reperfusion injury. Cell Death Dis. 2021, 12, 95 10.1038/s41419-020-03379-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortensen J. H.; Lindholm M.; Langholm L. L.; Kjeldsen J.; Bay-Jensen A. C.; Karsdal M. A.; Manon-Jensen T. The intestinal tissue homeostasis - the role of extracellular matrix remodeling in inflammatory bowel disease. Expert Rev. Gastroenterol. Hepatol. 2019, 13, 977–993. 10.1080/17474124.2019.1673729. [DOI] [PubMed] [Google Scholar]
- Petrey A. C.; de la Motte C. A. The extracellular matrix in IBD: a dynamic mediator of inflammation. Curr. Opin. Gastroenterol. 2017, 33, 234–238. 10.1097/MOG.0000000000000368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian S. F.; Yang H. H.; Xiao D. P.; Huang Y. J.; He G. Y.; Ma H. R.; Xia F.; Shi X. C. Mechanisms of neuroprotection from hypoxia-ischemia (HI) brain injury by up-regulation of cytoglobin (CYGB) in a neonatal rat model. J. Biol. Chem. 2013, 288, 15988–16003. 10.1074/jbc.M112.428789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishi H.; Inagi R.; Kawada N.; Yoshizato K.; Mimura I.; Fujita T.; Nangaku M. Cytoglobin, a novel member of the globin family, protects kidney fibroblasts against oxidative stress under ischemic conditions. Am. J. Pathol. 2011, 178, 128–139. 10.1016/j.ajpath.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia Y.; Cui R.; Wang C.; Feng Y.; Li Z.; Tong Y.; Qu K.; Liu C.; Zhang J. Metformin protects against intestinal ischemia-reperfusion injury and cell pyroptosis via TXNIP-NLRP3-GSDMD pathway. Redox Biol. 2020, 32, 101534 10.1016/j.redox.2020.101534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen P. The role of protein phosphorylation in human health and disease. The Sir Hans Krebs Medal Lecture. Eur. J. Biochem. 2001, 268, 5001–5010. 10.1046/j.0014-2956.2001.02473.x. [DOI] [PubMed] [Google Scholar]
- Malumbres M. Cyclin-dependent kinases. Genome Biol. 2014, 15, 122 10.1186/gb4184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z.; Gibson T. B.; Robinson F.; Silvestro L.; Pearson G.; Xu B.; Wright A.; Vanderbilt C.; Cobb M. H. MAP kinases. Chem. Rev. 2001, 101, 2449–2476. 10.1021/cr000241p. [DOI] [PubMed] [Google Scholar]
- Roskoski R. Jr. ERK1/2 MAP kinases: structure, function, and regulation. Pharmacol. Res. 2012, 66, 105–143. 10.1016/j.phrs.2012.04.005. [DOI] [PubMed] [Google Scholar]
- Morton S.; Davis R. J.; Cohen P. Signalling pathways involved in multisite phosphorylation of the transcription factor ATF-2. FEBS Lett. 2004, 572, 177–183. 10.1016/j.febslet.2004.07.031. [DOI] [PubMed] [Google Scholar]
- Gupta S.; Campbell D.; Dérijard B.; Davis R. J. Transcription factor ATF2 regulation by the JNK signal transduction pathway. Science 1995, 267, 389–393. 10.1126/science.7824938. [DOI] [PubMed] [Google Scholar]
- Livingstone C.; Patel G.; Jones N. ATF-2 contains a phosphorylation-dependent transcriptional activation domain. EMBO J. 1995, 14, 1785–1797. 10.1002/j.1460-2075.1995.tb07167.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dam H.; Wilhelm D.; Herr I.; Steffen A.; Herrlich P.; Angel P. ATF-2 is preferentially activated by stress-activated protein kinases to mediate c-jun induction in response to genotoxic agents. EMBO J. 1995, 14, 1798–1811. 10.1002/j.1460-2075.1995.tb07168.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morooka H.; Bonventre J. V.; Pombo C. M.; Kyriakis J. M.; Force T. Ischemia and reperfusion enhance ATF-2 and c-Jun binding to cAMP response elements and to an AP-1 binding site from the c-jun promoter. J. Biol. Chem. 1995, 270, 30084–30092. 10.1074/jbc.270.50.30084. [DOI] [PubMed] [Google Scholar]
- Ouwens D. M.; de Ruiter N. D.; van der Zon G. C.; Carter A. P.; Schouten J.; van der Burgt C.; Kooistra K.; Bos J. L.; Maassen J. A.; van Dam H. Growth factors can activate ATF2 via a two-step mechanism: phosphorylation of Thr71 through the Ras-MEK-ERK pathway and of Thr69 through RalGDS-Src-p38. EMBO J. 2002, 21, 3782–3793. 10.1093/emboj/cdf361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari S.; Bandi H. R.; Hofsteenge J.; Bussian B. M.; Thomas G. Mitogen-activated 70K S6 kinase. Identification of in vitro 40 S ribosomal S6 phosphorylation sites. J. Biol. Chem. 1991, 266, 22770–22775. 10.1016/S0021-9258(18)54634-2. [DOI] [PubMed] [Google Scholar]
- Bandi H. R.; Ferrari S.; Krieg J.; Meyer H. E.; Thomas G. Identification of 40 S ribosomal protein S6 phosphorylation sites in Swiss mouse 3T3 fibroblasts stimulated with serum. J. Biol. Chem. 1993, 268, 4530–4533. 10.1016/S0021-9258(18)53642-5. [DOI] [PubMed] [Google Scholar]
- Roux P. P.; Shahbazian D.; Vu H.; Holz M. K.; Cohen M. S.; Taunton J.; Sonenberg N.; Blenis J. RAS/ERK signaling promotes site-specific ribosomal protein S6 phosphorylation via RSK and stimulates cap-dependent translation. J. Biol. Chem. 2007, 282, 14056–14064. 10.1074/jbc.M700906200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledee D.; Kang M. A.; Kajimoto M.; Purvine S.; Brewer H.; Pasa-Tolic L.; Portman M. A. Quantitative cardiac phosphoproteomics profiling during ischemia-reperfusion in an immature swine model. Am. J. Physiol.: Heart Circ. Physiol. 2017, 313, H125–H137. 10.1152/ajpheart.00842.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misteli T. RNA splicing: What has phosphorylation got to do with it?. Curr. Biol. 1999, 9, R198–R200. 10.1016/S0960-9822(99)80128-6. [DOI] [PubMed] [Google Scholar]
- Prasad J.; Colwill K.; Pawson T.; Manley J. L. The protein kinase Clk/Sty directly modulates SR protein activity: both hyper- and hypophosphorylation inhibit splicing. Mol. Cell. Biol. 1999, 19, 6991–7000. 10.1128/MCB.19.10.6991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naro C.; Sette C. Phosphorylation-mediated regulation of alternative splicing in cancer. Int. J. Cell Biol. 2013, 2013, 151839 10.1155/2013/151839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang E. T.; Sandberg R.; Luo S.; Khrebtukova I.; Zhang L.; Mayr C.; Kingsmore S. F.; Schroth G. P.; Burge C. B. Alternative isoform regulation in human tissue transcriptomes. Nature 2008, 456, 470–476. 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natua S.; Ashok C.; Shukla S. Hypoxia-induced alternative splicing in human diseases: the pledge, the turn, and the prestige. Cell. Mol. Life Sci. 2021, 78, 2729–2747. 10.1007/s00018-020-03727-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanopka A. Cell survival: Interplay between hypoxia and pre-mRNA splicing. Exp. Cell Res. 2017, 356, 187–191. 10.1016/j.yexcr.2017.03.018. [DOI] [PubMed] [Google Scholar]
- Nakayama K.; Kataoka N. Regulation of Gene Expression under Hypoxic Conditions. Int. J. Mol. Sci. 2019, 20, 3278 10.3390/ijms20133278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Ayoubi A. M.; Zheng H.; Liu Y.; Bai T.; Eblen S. T. Mitogen-activated protein kinase phosphorylation of splicing factor 45 (SPF45) regulates SPF45 alternative splicing site utilization, proliferation, and cell adhesion. Mol. Cell. Biol. 2012, 32, 2880–2893. 10.1128/MCB.06327-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertocchi C.; Vaman Rao M.; Zaidel-Bar R. Regulation of adherens junction dynamics by phosphorylation switches. J. Signal Transduction 2012, 2012, 125295 10.1155/2012/125295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Quiles N.; Ho H. Y.; Kirschner M. W.; Ramesh N.; Geha R. S. Erk/Src phosphorylation of cortactin acts as a switch on-switch off mechanism that controls its ability to activate N-WASP. Mol. Cell. Biol. 2004, 24, 5269–5280. 10.1128/MCB.24.12.5269-5280.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley L. C.; Hayes K. E.; Ammer A. G.; Martin K. H.; Weed S. A. Cortactin phosphorylated by ERK1/2 localizes to sites of dynamic actin regulation and is required for carcinoma lamellipodia persistence. PLoS One 2010, 5, e13847 10.1371/journal.pone.0013847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson N.; Marklund U.; Gradin H. M.; Brattsand G.; Gullberg M. Control of microtubule dynamics by oncoprotein 18: dissection of the regulatory role of multisite phosphorylation during mitosis. Mol. Cell. Biol. 1997, 17, 5530–5539. 10.1128/MCB.17.9.5530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang M.; Kistler E. B.; Schmid-Schönbein G. W. Disruption of the mucosal barrier during gut ischemia allows entry of digestive enzymes into the intestinal wall. Shock 2012, 37, 297–305. 10.1097/SHK.0b013e318240b59b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang M.; Alsaigh T.; Kistler E. B.; Schmid-Schönbein G. W. Breakdown of mucin as barrier to digestive enzymes in the ischemic rat small intestine. PLoS One 2012, 7, e40087 10.1371/journal.pone.0040087. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry proteomics and phosphoproteomics datasets have been deposited to the ProteomeXchange Consortium via the PRIDE27 partner repository with the data set identifier PDX026076.