

SUMMARY
High-intensity transcription and replication supercoil DNA to levels that can impede or halt these processes. As a potent transcription amplifier and replication accelerator, the proto-oncogene MYC must manage this interfering torsional stress. By comparing gene expression with the recruitment of topoisomerases and MYC to promoters, we surmised a direct association of MYC with topoisomerase 1 (TOP1) and TOP2 that was confirmed in vitro and in cells. Beyond recruiting topoisomerases, MYC directly stimulates their activities. We identify a MYC-nucleated “topoisome” complex that unites TOP1 and TOP2 and increases their levels and activities at promoters, gene bodies, and enhancers. Whether TOP2A or TOP2B is included in the topoisome is dictated by the presence of MYC versus MYCN, respectively. Thus, in vitro and in cells, MYC assembles tools that simplify DNA topology and promote genome function under high output conditions.
In brief
Using biochemical and genomic approaches, Das et al. demonstrate that MYC or MYCN each unite TOP1 with TOP2A or TOP2B, respectively, in a “topoisome” in which their enzymatic activities are stimulated to resolve the local and global DNA topological issues associated with high output transcription.
Graphical Abstract
INTRODUCTION
The MYC “protooncogene” is central to most human cancers (Dang 2012). As a “universal amplifier” (Nie et al., 2012) or “super-transcription factor” (Kalkat et al., 2018), MYC regulates almost every cellular process. The MYC family includes pervasively expressed MYC, less broadly distributed MYCN, and more focused MYCL. MYC has 439 amino acids and includes four unstructured MYC boxes (MB I–IV) and a carboxyl-terminal basic helix-loop-helix leucine-zipper (bHLH-ZIP) domain that dimerizes with MAX (Blackwood and Eisenman, 1991) to bind E-boxes (CACGTG) (Guo et al., 2014). At RNA polymerase II (RNAPII) promoters, MYC-MAX increases gene expression by contributing to pause release (Rahl et al., 2010). MYC also stimulates RNAPI and RNAPIII transcription (Arabi et al., 2005; Gomez-Roman et al., 2003; Poortinga et al., 2004). MYC interacts with numerous factors and complexes that regulate transcription and chromatin (Baluapuri et al., 2020). MYC also associates with replication forks and contributes to origin of replication activity (Dominguez-Sola et al., 2007). The very number and diversity of MYC partner proteins and complexes have obscured the elucidation of its mechanism in genome biology.
MYC-driven transcription reorganizes chromatin and generates topological stress (Baranello et al., 2012). The positive and negative supercoils forming, respectively, upstream and downstream of an elongating RNAP (Wu et al., 1988) can impede or halt transcription (Ma et al., 2013). As a transcription amplifier, MYC would be impotent without removal of torsional stress. RNA-DNA hybrids, termed “R-loops,” promoted by negative supercoils behind RNAPII can disrupt transcription (Drolet et al., 1994; Aguilera and García-Muse 2012). The transcription of long genes is threatened by improper handling of their DNA topology (King et al., 2013; Joshi et al., 2012). Negative supercoiling transmitted through the DNA may promote non-B DNA structures that alter gene activity (Kouzine et al., 2017; Kouzine et al., 2008). Although many of the consequences of topological stress oppose MYC activity, the interplay of MYC and supercoiling has not been explored.
High levels of supercoiling and intertwining of chromatids also occur at replication forks, especially when two replisomes converge, provoking replication stress and inducing genome instability (Schalbetter et al., 2015). Nucleosome assembly involves DNA topology, with negative supercoiling increasing and positive decreasing nucleosome stability (Teves and Henikoff, 2014; Sheinin et al., 2013). To be mitogenic MYC must manage these stresses.
DNA topoisomerases (TOP1, TOP2A and TOP2B) remove supercoiling to regulate DNA conformation and to remove knots and tangles by passing DNA strands and fibers through each other while maintaining DNA integrity (Wang, 2002). TOP1 breaks one strand and rotates about the unbroken strand to change the DNA twist in steps of one before resealing. TOP2 passes an intact DNA helix through a transient double-strand break in a second helix to subtract or add two supercoils at a time. The facilitating role of topoisomerases in transcription is well-established. TOP1 is involved in pre-initiation complex formation (Merino et al., 1993; Shykind et al., 1997), elongation (Baranello et al., 2016), and enhancer activity (Puc et al., 2015). TOP2A favors transcription initiation of RNAPI genes (Ray et al., 2013), while TOP2B regulates stimulus-responsive genes (Ju et al., 2006; Madabhushi et al., 2015). TOP2A and TOP2B can also control the immediate early genes such as FOS (Herrero-Ruiz et al., 2021). Topoisomerases regulate supercoiling at replication forks; TOP1 and TOP2A resolve positive supercoils ahead of the forks (Kegel et al., 2011). TOP2A also resolves catenanes during the completion of replication (Le et al., 2019). TOP2B is ubiquitously expressed and is essential for neural differentiation and the expression of long neuronal genes (King et al., 2013).
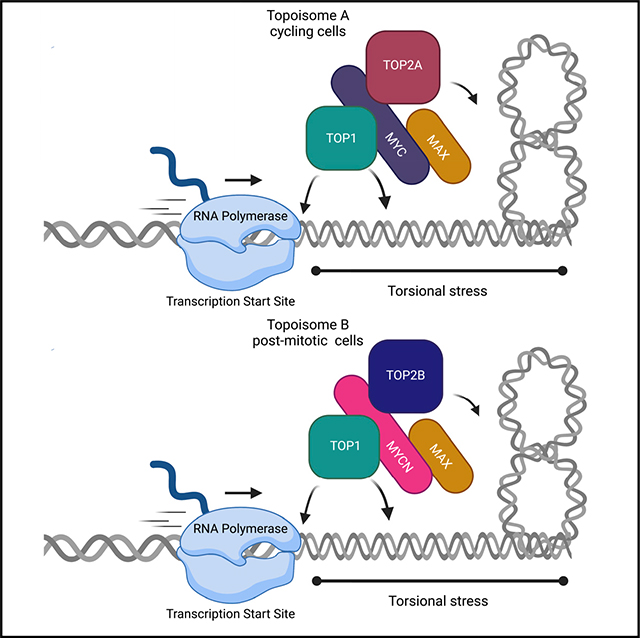
Whether MYC helps to resolve torsional stress during transcription or replication is unknown, although mass spectrometry and immunoprecipitation experiments have shown TOP1 and TOP2 to interact with MYC (Büchel et al., 2017; Kalkat et al., 2018; Koch et al., 2007). Here, we explore the association of MYC with topoisomerases, with topoisomerase delivery to sites of action, and MYC’s impact on the regulation of DNA topology by TOP1 or TOP2. We find that MYC assembles a novel complex, the “topoisome,” that includes both TOP1 and TOP2, and that in the topoisome, the activities of both enzymes are enhanced. The topoisome comes in two flavors: whereas MYC preferentially assembles with TOP1 and TOP2A, MYCN specifically joins with TOP1 and TOP2B, concordant with the high expression of MYCN in post-mitotic cells such as neurons. The MYC-induced topoisome controls DNA topology to sustain a high transcriptional output, allowing MYC-induced rapid growth under physiological and pathological conditions.
RESULTS
MYC binding parallels TOP1 and TOP2A at highly expressed genes
MYC associates with transcription regulators especially at highly expressed genes, where DNA becomes highly supercoiled (Kouzine et al., 2013). Unless dissipated, this torsional stress would oppose transcription amplification by MYC. To study MYC and topoisomerase recruitment at promoters, we performed chromatin immunoprecipitation sequencing (ChIP-seq) of MYC, TOP1 and TOP2A in the HCT116 colorectal cancer cell line, as TOP1 and TOP2A were previously detected in the MYC interactome (Koch et al., 2007; Büchel et al., 2017; Kalkat et al., 2018). Across the genome (Table S1), MYC, TOP1 and TOP2A mostly overlapped at promoters, co-localizing more than 60% (Figure 1A), as visualized at transcription start sites (TSSs) (Figures 1B, S1A, and S1B). If transcriptionally generated supercoiling increases the demand for topoisomerases, co-localization should be most prominent at highly expressed genes where MYC is preferentially enriched (Lorenzin et al., 2016; Nie et al., 2012). Genes were ranked by expression level, and binding of MYC, TOP1 and TOP2A at TSSs was assessed at the top 10,000 bound genes for each protein. MYC extensively overlapped with both TOP1 and TOP2A at the TSSs of bound genes (Figure 1C), especially at the top 5% expressed genes (Figure 1D). All three proteins were recruited to 38% of all expressed genes, rising to 62% for the top 5% (Figure 1E). Ranking MYC binding at TSSs by quartile, highest (100%–75%), high (75%–50%), medium (50%–25%), or low (25%–0%), and looking for the association of TOP1 (Figure S1C) or TOP2A (Figure S1D), MYC-binding paralleled the recruitment of both: the more MYC, the more TOP1 and TOP2. If transcription amplification by MYC provokes a higher demand for topoisomerase activity, decreasing MYC should decrease topoisomerase-DNA cleavage complexes (TOPccs). To detect TOP1ccs and TOP2Accs along the genome, we coupled Covalent Adduct Detection (CAD, a variant of RADAR assays [Kiianitsa and Maizels, 2013]) with ChIP (to immunoprecipitate only catalytically engaged topoisomerase) and sequencing (CAD-seq) or qPCR (CAD-qPCR). Auxin treatment rapidly degraded MYC in auxin-inducible K562 MYC_mAID cells (Muhar et al., 2018)(Figure 1F), whereas leaky degradation was prevented by the auxin competitor auxinole (Yesbolatova et al., 2019). Cells were treated sequentially with auxin, proteasome inhibitor MG132, and then TOP1 poison camptothecin (CPT) to trap and stabilize TOP1ccs (Pommier, 2013; Sciascia et al., 2020). In untreated cells, promoter-bound TOP1ccs were sparce but increased downstream of the TSSs, as expected (Baranello et al., 2016). With MYC degradation, TOP1ccs in the vicinity and downstream of TSSs were reduced (Figures 1G and 1H). In contrast, TOP2Accs trapped by MG132 and the TOP2 poison Etoposide (Eto) at selected highly transcribed TSSs, measured by CAD-qPCR, were increased upon MYC degradation (Figure S1E). This may reflect compensatory (Miao et al., 2007) TOP2A engagement with lingering supercoils (McClendon et al., 2005) at the TSS in the absence of TOP1 activity and MYC-driven pause release (Figure 1G). These results demonstrate that binding and catalytic engagement of TOP1 and TOP2A at TSSs are responsive to MYC.
Figure 1. TOP1, TOP2A and MYC are co-enriched at highly expressed genes.

(A) Genomic annotation of MYC, TOP1, and TOP2A peaks and their common peaks.
(B) MYC, TOP1, and TOP2A occupancy (reads per million [RPM]) at TSSs of all expressed genes.
(C and D) Number of genes where MYC, TOP1, and TOP2A were recruited at all TSSs ± 500 bps (C) and for the top 5% of expressed genes within the bound genes (D).
(E) Quantification of binding (%) in (C) and (D).
(F) MYC depletion in K562MYC_mAID cells.
(G) TOP1 CAD-seq profile (RPM) at all TSS of K562MYC_mAID cells treated with auxin, MG132 and CPT.
(H) Genome Browser of ENO1 and MAPK1 promoters from (G).
(I and J) HCT116 nuclear extracts immunoprecipitated (IP) with anti-MYC or immunoglobulin G (IgG) and probed for TOP1 (I) and TOP2A (J).
(K) Supercoiled plasmid was added to TOP1 alone or pre-incubated with increasing MYC, MYC-MAX or MAX and assessed by gel electrophoresis.
(L) TOP2A with or without pre-incubation of MYC, MYC-MAX or MAX was added to kDNA, and decatenation assay was analyzed by gel electrophoresis. Catenated and decatenated circles—nicked and intact—are indicated.
MYC physically interacts with topoisomerases
Coincident TSS-recruitment of TOP1 and TOP2A prompted the testing of their physical associations. To eliminate nucleic acid bridging, HCT116 cell lysates were treated with benzonase, a DNA/RNA nuclease. Endogenous MYC was immunoprecipitated from lysates, and immunocomplexes were interrogated for MYC, TOP1, and TOP2A by immunoblotting. MYC pulled down TOP1 (Figure 1I) and TOP2A (Figure 1J) and was immunoprecipitated by either anti-TOP1 or anti-TOP2A, indicating that MYC complexed with each topoisomerase. The impact of MYC upon the activity of purified enzymes was assayed. For TOP1, DNA relaxation yielded changes in the migration of a supercoiled plasmid DNA by agarose gel elecrophoresis. For TOP2A, decatenation assays displayed the unlinking of individual double-stranded DNA minicircles from a meshwork of interlinked circles (kinetoplast DNA [kDNA]) (Figure S1F). MYC or MYC-MAX greatly enhanced plasmid relaxation by TOP1 (note the intensity of the supercoiled DNA band in Figure 1K) and DNA decatenation by TOP2A (note nicked or intact minicircles in Figure 1L). MAX alone influenced neither TOP1 nor TOP2A activity, revealing MYC to be the topoisomerase stimulator. Besides decatenation, MYC stimulated relaxation by TOP2A, as the entire population of substrate plasmids appeared less negatively supercoiled in 2-dimensional gel electrophoresis (Figure S1G). The MYC and MAX used in relaxation and decatenation assays were free from contaminating nuclease and topoisomerase activity (Figure 1L).
We next examined the association of MYC with TOP1 and TOP2A. A panel of MYC proteins deleted of MYC boxes or other conserved domains (Figures 2A, S2A, and S2B) were compared with full-length MYC for co-immunoprecipitation with recombinant TOP1 or TOP2A. Equimolar MYC or MYC mutants were mixed together with TOP1 or TOP2A. While all of the MYC proteins bearing either the amino (N)- or the carboxyl (C)-terminal domain pulled down TOP1 from the mixtures of proteins (Figures 2B, S2C, and S2D), residues 200–320 (MB III and IV) did not (Figure 2B). In vitro immunoprecipitations using TOP2A showed that the panel of MYC mutants behaved similarly to TOP1 (Figures 2C and S2E–S2H). Reciprocally, full-length MYC was pulled down by either anti-TOP1 (Figure S1H) or anti-TOP2A (Figure S1I). Therefore, two discontiguous MYC domains physically associate with both TOP1 and TOP2A. Whereas the N-terminal transcription activation domain (TAD) of MYC and the C-terminal bHLH-ZIP domain each pull down TOP1 or TOP2A—although with reduced efficiency as compared with full-length MYC— the middle region of the protein does not (Figures 2B and 2C).
Figure 2. The N and C termini of MYC bind and stimulate TOP1 and TOP2A.

(A) Scheme of full-length MYC (WT) and truncations extending from the C terminus (1–199), N terminus (321–439), or from both ends (200–320).
(B) TOP1 was mixed with MYC truncations, IP with anti-TOP1 or IgG, and probed for MYC (as indicated) or TOP1.
(C) As in (B) but with TOP2A.
(D and E) MYC WT or truncations were pre-incubated with or without TOP1 (D) or TOP2A (E) before the addition of plasmid or kDNA, respectively, and assessed by gel electrophoresis. Chl, chloroquine.
(F and G) TOP1 (F) and TOP2A (G) were dephosphorylated and incubated with MYC and plasmid DNA (F) or kDNA (G). Relaxed and decatenated products were visualized on gels. The percentage of relaxed or decatenated product is indicated.
The N and C termini of MYC bind and stimulate TOP1 and TOP2A
To characterize the domain of MYC that stimulates TOP1, we conducted plasmid relaxation assays, analyzing the products on chloroquine gels. Chloroquine underwinds DNA, forcing relaxed plasmids to compensate by becoming positively supercoiled, more compact, and faster-migrating (Figures 2D, S3A, and S3C, left). We also performed decatenation assays for TOP2A, supplementing reactions with deleted or wild-type (WT) recombinant MYC (Figures 2E, S3B, and S3C, right). Whereas the activities of the C-terminal truncations were somewhat attenuated (Figure S3A, lanes 6, 8, 10, and 12), N-terminal MYC deletions retaining residues 144–439 stimulated TOP1 activity (Figures S3A, lanes 14,16, 18, and 20, and S3D, left). Even, the smallest construct— the C-terminal bHLH-ZIP domain (321–439 aa)—enhanced TOP1 activity, similar to full-length MYC. Stimulation of TOP2A by MYC truncations in decatenation assays paralleled the stimulation of TOP1 (Figure S3B and S3D, right), revealing the bHLH-ZIP region of MYC to strongly stimulate both TOP1 and TOP2A. Residues 200–320 had no effect on TOP1 or TOP2A activity, consistent with the pull-downs (Figures 2D, 2E, and S3C); note that this same region (MB III + IV) was recently found to dampen transcription amplification by MYC (Nie et al., 2020).
To ensure that the MYC stimulation of topoisomerases was direct and specific and not some arcane effect (perhaps DNA chirally wrapped around MYC complexes in the presence of topoisomerase), relaxation assays were conducted using E. coli TOP1A and analyzed by chloroquine gel. MYC had no impact on TOP1A activity (Figure S3E, compare lane 4 to lanes 8, 12, and 13; relaxed products are at the bottom). Relaxation assays also compared TOP1A (0.2 μM) and human TOP1 (0.01 μM) in the absence or presence of recombinant MYC (0.03–0.15 μM) (Figure S3E, compare lanes 1 with 4 and 3 with 8). MYC had no influence on bacterial topoisomerase. Certifying further that MYC’s effect on topoisomerase activity is specific, relaxation assays tested TATA-binding protein (TBP), positive cofactor 4 (PC4), and MAX, which are known DNA-binding factors. None of these affected the relaxation by TOP1 (Figure S3F), so topoisomerase stimulation seems a specific property of MYC.
MYC stimulation of topoisomerases is independent of phosphorylation
Post-translational modifications of human topoisomerase influence activity, stability and localization. Phosphorylation has been shown to impact topoisomerase activity and cell-cycle checkpoint control (Bedez et al., 2018). As dephosphorylation has been associated with the loss of enzyme activity (Pommier et al., 1990), we asked if topoisomerase stimulation by MYC depends on the phosphorylation status. The recombinant TOP1 and TOP2A used were expressed in insect cells or in S. cerevisiae, respectively, and so are likely to carry eukaryotic post-translational modifications—including phopshorylation— that support activity. Following dephosphorylation (Figure S3G), TOP1 and TOP2A, and their phosphorylated counterparts, were tested for MYC binding and stimulation. Despite diminished activity (Figure 2F), phosphatase-treated TOP1 remained responsive to MYC. MYC activation of TOP2A was also independent of TOP2A’s phosphorylation status (Figure 2G). Because MYC-stimulated topoisomerase activity is independent of the latter’s phosphorylation, MYC may potentially substitute for direct enzymatic activation by signaling or other kinases. That dephosphorylated topoisomerases interact at least as well (TOP1) (Figure S3H) or better (TOP2A) (Figures S3I) with MYC than their phosphorylated counterparts suggests that signal transduction may modulate MYC-topoisomerase interactions.
MYC stimulates topoisomerases activity ex vivo
To test if MYC could activate topoisomerases in a complex environment, plasmid relaxation assays were performed using extracts from U2OS cells harboring lentivirus-encoded doxycycline (Dox)-inducible MYC-EGFP (Nie et al., 2020). Western blot (WB) confirmed the expression of native TOP1 and TOP2A, as well as of MYC-EGFP (Figure 3A). Besides MYC-EGFP, the anti-MYC antibody detected endogenous MYC in the extracts. MYC-EGFP increased according to the inducing dose of Dox, while TOP1 and TOP2A levels remained constant (Figure 3A). To investigate the influence of MYC-EGFP on cellular TOP1 activity, extracts from cells induced or not with 0.6 μg/ml Dox were assayed for TOP1 activity, and products were run on a chloroquine gel (Figure 3B). Plasmid relaxation was greatest when MYC-EGFP expression was highest (0.6 μg/ml Dox) and was lowest with the least MYC-EGFP (untreated); thus, MYC stimulates cellular TOP1 activity ex vivo (Figures 3B and S4A). The influence of MYC-EGFP upon cellular TOP2A in extracts treated or not with Dox was similarly tested using decatenation assays. DNA decatenation was greater when extracts had more MYC, indicating that MYC stimulates TOP2A activity ex vivo (Figures 3C and S4B). As a global amplifier of transcription, MYC might alter the cellular milieu, indirectly conditioning the cell to sustain higher levels of topoisomerase activity to relax supercoils generated during transcription. To assess whether cells must be primed to support MYC-stimulated topoisomerase activity, cellular extracts (treated or not with Dox) were tested for topoisomerase activity in the presence/absence of full-length recombinant MYC. Supplementing untreated extracts with MYC or MYC-MAX at levels comparable to those in treated cells, replicated the activation of TOP1 by cellularly expressed MYC (Figures 3D, compare lane 5 or 13 with lane 7, and S4C). Thus, the enhancement of cellular TOP1 activity requires neither post-translational MYC modifications nor upregulation by MYC of other components of the transcription or chromatin machineries.
Figure 3. MYC increases DNA engagement of topoisomerases in cells.

(A) Scheme of U2OS-MYC-EGFP cells harboring Dox-inducible MYC-EGFP. Cells were checked for the indicated proteins.
(B) TOP1 relaxation assay using untreated or Dox-induced U2OS-MYC-EGFP lysates.
(C) As in (B) but decatenation assay with TOP2A.
(D) Relaxation assay using untreated or Dox-induced U2OS-MYC-EGFP lysates, with/without purified MYC or MYC-MAX.
(E) Scheme of HO15.19-MYC-ER cells devoid of endogenous MYC but expressing MYC-ER protein that translocates to the nucleus upon Tam.
(E and F) After treatments (as indicated), CAD extracts were probed with anti-TOP1 (E) or anti-TOP2A (F).
(G and I) PLAs using anti-MYC and anti-TOP1 (left) or anti-TOP2A (right) on U2OS-MYC-EGFP cells induced with Dox (G) or HO15.19-MYC-ER cells treated with Tam (I) (scale bars, 25 μm).
(H and J) Quantification of (G) and (I) for 100 cells. Significance by 2-way ANOVA using Dunnett’s correction for multiple comparisons (n = 5; mean ± SEM; t test) (****: p < 0.0001).
See also Figure S4.
MYC increases topoisomerase-DNA complexes in cells
If MYC stimulates topoisomerase activity in cells, then increased MYC should increase TOPccs. To detect MYC-induced TOPccs in cells, we employed the CAD assay (Gittens et al., 2019). U2OS-MYC-EGFP cells were treated with the indicated amount of Dox and then incubated with MG132 and the TOP1 poison CPT before the CAD assay was performed (Figure S4D). TOP1ccs were strongly elevated following Dox treatment compared to untreated cells (Figures S4D, S4E, and S4A), showing that MYC induced more TOP1ccs in cells. Increased TOP1cc was most likely due to the direct activation of TOP1 by MYC, but transcription amplification by MYC could provoke a greater genomic demand for TOP1 action. In yet another scenario, expression of an immediate-early target of MYC might be the agent that activates TOP1. Reasoning that direct activation of TOP1 by MYC would increase TOP1ccs more swiftly than indirect action, CAD assays were done in HO15.19-MYCER12 cells (MYC-ER cells) (O’Connell et al., 2003). This cell line is devoid of endogenous MYC but expresses a MYC-ER protein that immediately translocates to the nucleus upon tamoxifen (Tam) treatment. MYC-ER cells were briefly treated with Tam in the presence of MG132 and CPT, and TOP1ccs were analyzed by CAD assay. TOP1ccs were elevated ~22-fold by Tam+MG132+CPT compared to Tam-untreated cells (Figures 3E, and S4F, left). Similarly, to detect MYC-induced TOP2Acc, MYC-ER cells were treated with Tam in the presence of the TOP2 poison Eto. TOP2Accs were increased by Eto ~30-fold in Tam-treated cells compared to untreated cells (Figures 3F and S4F, right). To ensure that MYC stimulation of topoisomerase activity was not an indirect result of ongoing transcription, MYC-ER cells were treated with triptolide, a transcription inhibitor, and MG132+CPT and examined by CAD assay. The levels of TOP1cc in Tam+CPT+MG132-treated cells were insensitive to triptolide treatment (Figures S4G and S4H), confirming that MYC-induced TOP1cc formation was independent of transcription. Thus, MYC directly sponsors the catalytic engagement of TOP1 and TOP2A with DNA in cells.
MYC and topoisomerases are closely associated inside cells
Previous in vitro high-throughput studies have indicated an uncharacterized association of MYC with TOP1 and TOP2A (Büchel et al., 2017; Kalkat et al., 2018; Koch et al., 2007); we sought to further study these interactions in cells. Proximity ligation assay (PLA) was performed in U2OS-MYC-EGFP cells that upregulate MYC-EGFP with Dox treatment. Successful interactions were detected using mouse anti-MYC and either rabbit anti-TOP1 or rabbit anti-TOP2A, as monitored by confocal microscopy. Quantification of the PLA dots due to the proximity of MYC with TOP1 (Figures 3G, left, and S4I) or MYC with TOP2A (Figure 3G, right) revealed that the association of MYC with either TOP1 or TOP2A increased as the MYC-EGFP was induced. The number of PLA dots/cell increased from ~60 for untreated cells to more than ~80 dots/cell with Dox (Figures 3G and 3H), documenting a dose-dependent interaction of MYC with TOP1 or TOP2A. The PLA dots in untreated cells were presumed to be due to an interaction of TOP1 or TOP2A with endogenous MYC. The PLA signal between nucleolin and TOP1, which are not known to associate, documented the background. We next examined the proximity of TOP1 and TOP2A to MYC in MYC-ER cells. In the absence of Tam, MYC-ER is cytoplasmic, complexed with HSP90, and held away from nuclear TOP1 or TOP2A. Indeed, the PLA signal between MYC and the topoisomerases was very low. With a pulse of Tam, MYC-ER engaged with TOP1 (Figure 3I, left) and with TOP2A (Figure 3I, right). The average number of PLA dots increased from ~20 for untreated cells to more than ~80 with Tam treatment (Figures 3I and 3J), suggesting that, as in vitro, the interaction of MYC with TOP1 or TOP2A is rapid and likely to be independent of other nuclear processes.
MYC assembles a multi-component "topoisome" complex
By PLA, both topoisomerases displayed very similar patterns, prompting speculation that the MYC-TOP1 and MYC-TOP2A complexes were connected. Recombinant TOP1 and TOP2A were mixed together in the presence or absence of recombinant MYC or MYC-MAX and were pulled down with either anti-TOP1 or anti-TOP2A. TOP2A co-precipitated with TOP1 only in the presence of MYC or MYC-MAX, and the co-precipitation of TOP1 with TOP2A also required MYC or MYC-MAX, yielding at least a ternary or quaternary complex, respectively (Figures 4A and 4B). To ask whether MYC nucleates TOP1 with TOP2A in cells, U2OS-MYC-EGFP cells were induced with Dox, and cellular extracts were immunoprecipitated with anti-TOP1 or anti-TOP2A. The resulting complexes were analyzed by WB for co-precipitating proteins (Figure 4C). Consistent with the formation of a TOP1-TOP2A-MYC complex, the co-precipitation of TOP1 with TOP2A was enhanced by increasing cellular MYC (Figure 4C, compare pellet with supernatant). While in uninduced U2OS cells the fraction of co-associating TOP1 and TOP2A was ~3%, upon MYC induction it rose up to ~15% (Table S3). This supports the notion that MYC bridges the interaction of TOP1 with TOP2A. To investigate whether the MYC-induced association of TOP1 and TOP2A required transcription, MYC-ER cells were treated with or without Tam to drive MYC into the nucleus, and cellular extracts were immunoprecipitated using anti-TOP1 or anti-TOP2A. Co-precipitation of TOP1 with TOP2A was dramatically increased by Tam (Figures 4D and 4E), suggesting that nuclear MYC rapidly unites TOP1 with TOP2A.
Figure 4. MYC joins TOP1 and TOP2A in a complex.

(A and B) Recombinant proteins were mixed as indicated, IP with either anti-TOP1 (A) or anti-TOP2A (B) and probed for TOP1 and TOP2A.
(C) Untreated or Dox-treated U2OS-MYC-EGFP cells were IP with anti-TOP1 (left) or anti-TOP2A (right). The IP material (Pellet), unbound (Sup) and nuclear extract prior to IP (Input) were probed for TOP1 or TOP2A. Quantitation in Table S3.
(D and E) Tam-induced or not (Un) HO15.19-MYC-ER cells were IP with anti-TOP1 (D) or anti-TOP2A (E) and probed for TOP1 and TOP2A.
(F and G) Indicated proteins were mixed, IP with anti-TOP1 (F) or anti-TOP2A (G), and probed for TOP1 and TOP2A.
(H) Scheme of EGFP-tagged full-length MYC (1–439 aa) or mutant MYC (200–320). EGFP-MYC constructs were expressed in HCT116 cells, IP with anti-GFP, and probed for TOP1, TOP2A, or GFP.
(I) PLAs (red) of anti-TOP1 with anti-TOP2A in Dox-induced U2OS-MYC-EGFP cells (left) or in Tam-treated HO15.19-MYC-ER cells (right) (scale bars, 25 μm).
(J) Quantification of (I) for 100 cells. Significance by 2-way ANOVA using Dunnett’s correction for multiple comparisons (n = 5; mean ± SEM; t test) (****: p < 0.0001).
To define the minimal MYC region necessary for unifying TOP1 and TOP2A in a complex, in vitro pull-downs used TOP1, TOP2A and the panel of MYC proteins deleted of their N-, N+C - or C-terminal domains (Figures S5A, S5B, 4F, and 4G). While separated N- or C-terminal regions alone could nucleate the TOP1-TOP2A complex, TOP1 and TOP2A did not co-precipitate in presence of MYC mutant 200–320 (Figures 4F, 4G, S5A, and S5B). Further, this mutant suppressed the association of TOP1 with TOP2A by the N terminus mutant 1–199, acting as a dominant negative in vitro. Similarly, transient expression of the EGFP-tagged 200–320 in HCT116 cells abrogated the WT-MYC-EGFP promotion of TOP1 interaction with TOP2A (Figure 4H). Thus, beyond not forming a complex with TOP1+TOP2A, MYC residues 200–320 can disrupt this complex.
We also examined the proximity of TOP1 with TOP2A by PLA in U2OS-MYC-EGFP cells either induced or not with Dox. Rabbit anti-TOP1 and mouse anti-TOP2A were used along with the negative control anti-nucleolin. MYC boosted the interaction of TOP1 with TOP2A (Figures 4I and 4J, left). PLA dots were ~85 in cells treated with Dox compared to ~60 in untreated cells, suggesting that MYC promoted TOP1-TOP2A association inside cells. The basal interaction of TOP1 with TOP2A seemed likely to be due to high levels of endogenous MYC in U2OS (Lorenzin et al., 2016). To prove that the TOP1-TOP2A interaction in cells was promoted by MYC, PLA of TOP1 with TOP2A was done in MYC-ER cells with or without Tam. Without Tam, very little TOP1 was juxtaposed with TOP2A, as the average number of PLA dots was very low. Brief treatment with Tam dramatically increased the PLA signal (Figures 4I and 4J, right), indicating that MYC rapidly complexes with TOP1 and TOP2A. We designate this complex of TOP1, TOP2 and MYC the “topoisome.”
The highly stable topoisome has specific composition and stochiometry
How big and stable is the topoisome formed in cells, and what are the stoichiometries of its components? HeLa cell nuclear extracts (Dignam et al., 1983) were fractionated by P11 phospho-cellulose column chromatography using step elutions of increasing salt. These fractions were examined by WB for coelution of MYC (or MYCN), TOP1, TOP2 (including both isoforms A and B), and MAX. Only the 1 M salt eluate contained MYC, TOP1, TOP2A, TOP2B and MAX (Figure 5A), suggesting the topoisome eluted as a complex in the high salt. This fraction contained ~45% of the total MYC in the nuclear extract. To estimate the size of the native topoisome complex, the P11 (1 M) eluate was further fractionated by size-exclusion chromatography (SEC; Superose 6) and analyzed by WB for the coelution of TOP1, TOP2A, TOP2B, MYC and MAX. TOP1, TOP2A, MYC and MAX were present in fractions 18–28, and TOP1 and TOP2A were most prominent in fractions 18–26, indicative of a complex size of ~450 to ~800 kDa relative to calibrating size markers (Figures 5B, S5C, and S5D), confirming the existence of a high molecular topoisome in cellular extracts. TOP2B was found only in fractions 26–28, suggesting that the main isoform participating in the topoisome is TOP2A. Although SEC lacks sufficient resolution to fix the precise stoichiometry of the topoisome, a 1–1-1–1 combination of native MYC-MAX, TOP1 and TOP2 would weigh a total of ~500 kDa. Although the breadth of the peak suggests some variation in topoisome composition, we speculate that this size range can accommodate no more than a single TOP2 dimer (~350 kDa) and still include one or two molecules each of TOP1 (~90 kDa) and MYC (~50 kDa) or MYC-MAX (~70 kDa). To interrogate other molecular species in this region of the SEC that interact with TOP1, we performed native mass spectrometry of material immunoprecipitated with anti-TOP1 from fraction 22 (a region of the SEC between TOP2A and TOP2B). A comparison with the BioGrid database (Stark et al., 2006) revealed that the immunoprecipitated material was enriched for known TOP1 interaction partners such as casein kinase II, XRCC5, and PARP1 (Stark et al., 2006)(Table S4). MYC, TOP2A and, although to a lesser extent, TOP2B were among the proteins passing the selection threshold. Therefore, the analysis further supported the notion that TOP1 is associated with TOP2A and MYC. Other TOP1-containing complexes included cohesin, condensin, SWI/SNF, SMC5/6, PAF1, GTFIIIC, and RNAPIII (Table S4), suggesting that TOP1 physically associates with other machines that mechanically stress the DNA (Neguembor et al., 2021; Uusküla-Reimand et al., 2016). The poorer recovery of TOP2B peptides by mass spectrometry of the same SEC fractions that included TOP2A, TOP1 and MYC was puzzling, as TOP2B was present in the 1 M P11 eluate subjected to SEC (Figure 5A). This suggested that TOP2B was more weakly incorporated into the topoisome than TOP2A. Testing combinations of recombinant TOP1, TOP2A, TOP2B and MYC by anti-MYC pull down and WB, showed that TOP2B was weakly incorporated in the topoisome only when TOP2A was absent; when TOP2A was present, TOP2B was displaced from the topoisome (Figure 5C). TOP2A is expressed only in cycling cells (Thakurela et al., 2013), while TOP2B expression is ubiquitous and persists in post-mitotic cells such as neurons (Tiwari et al., 2012) where MYCN is the preferred MYC isoform (Rickman et al., 2018; Liu et al., 2021). Might TOP2B preferentially join a MYCN-driven topoisome? Different mixtures of TOP1, TOP2A and TOP2B were incubated with recombinant MYCN, immunoprecipitated with anti-MYCN, and analyzed by WB for the presence of each protein. MYCN forms a topoisome with TOP1 and TOP2B that displaces weakly incorporated TOP2A (Figure 5D). The absence of MYCN in the 1 M P11 eluate (in contrast to the presence of MYC) (Figure 5A) accounts for the failure to find a TOP2B-containing topoisome in this fraction. That MYC and MYCN are highly homologous (47% identical, BLASTP) and TOP2A versus TOP2B are even more so (69% identical, BLASTP), testifies to the fine specificity of topoisome assembly. Thus, topoisomes come in two flavors: A (including MYC, TOP1 and TOP2A) and B (including MYCN, TOP1 and TOP2B). We compared the stimulation of the catalytic activities of TOP1, TOP2A and TOP2B by MYC versus MYCN. Whereas TOP1 was equivalently activated by both MYC and MYCN (Figure S5E), TOP2A was stimulated only by MYC but not MYCN (Figure 5E), while TOP2B was stimulated only by MYCN but not MYC (Figures 5F and S5F). Thus, the specificity of topoisome A versus B assembly is congruent with catalytic upregulation of TOP2A versus TOP2B.
Figure 5. The stable topoisome has specific composition and stoicheometry.

(A) HeLa cell nuclear extracts was eluted stepwise with an increasing KCl concentration and examined for the indicated proteins. A mix of recombinant proteins identified the migration of each protein.
(B) The P11 1 M eluate was further fractionated, and every other fraction from 16–44 co-elution was examined. Protein mix as in (A).
(C) Proteins were mixed as shown, IP with anti-MYC, and probed for TOP2A, TOP2B, TOP1 and MYC.
(D) Indicated proteins were mixed, IP with anti-MYCN, and probed as in (C).
(E and F) Full-length MYC or MYCN were pre-incubated with TOP2A (E) or TOP2B (F) before the addition of kDNA and decatenation assay.
MYC increases topoisome-DNA covalent adducts in cells
Reasoning that when one topoisomerase in the topoisome is DNA-engaged, the co-tethered second topoisomerase may engage that same DNA segment more frequently than if independently diffusing, cells were treated with CPT to lock TOP1 onto DNA. If accompanied by TOP2A, within the topoisome, this TOP1 inhibition should increase the frequency of TOP2Accs, an experiment we call “cross-CAD” (Figure 6A). Similar logic dictates that trapping TOP2Accs with Eto should augment co-tethered TOP1ccs (MG132 was included to prevent TOPcc degradation [Sciascia et al., 2020]). Cross-CAD failed with either CPT or Eto in the HO15.19-MYC-ER cells in the absence of Tam (Figures 6B and S6A), indicating that in the absence of nuclear MYC, TOP1 and TOP2A separately engage the DNA. Upon the addition of Tam, cross-CAD showed the enhancement of TOP2Acc by CPT-mediated topoisome tethering through TOP1 (Figures S6A and S6B, left) and enhancement of TOP1cc by Eto-mediated topoisome tethering through TOP2 (Figures 6B and S6B, right). Similar cross-CAD results were obtained in U2OS-MYC-EGFP cells when incubated with MG132 and Eto, and TOP1ccs were increased after Dox (Figures 6C and S6D). Note that the relatively short treatment with MG132 (45 min) only marginally increased topoisome-MYC levels (Figure S6C), suggesting that MG132 does not promote topoisome hyper-ubiquitylation.
Figure 6. The topoisome engages promoters and enhancers to favor transcription.

(A) Scheme of “cross-CAD” assay. CPT increased engagement of TOP2A as well as TOP1 in the presence of MYC, while Eto increased engagement of TOP1 as well as TOP2A. Red dots with Y indicate the active site of TOP1 (Y723) or TOP2 (Y805).
(B) Detection of MYC-tethered TOP1 along with Eto-trapped TOP2Acc by cross-CAD assays in HO15.19-MYC-ER cells with or without Tam with anti-TOP1.
(C) As in (B) but in Dox-induced U2OS-MYC-EGFP cells.
(D) Genome Browser of TOP1 cross-CAD-seq (RPM) showing two pairs of divergent promoters (dashed boxes).
(E and F) Average TOP1 cross-CAD-seq (RPM) at all RNAPII genes (between TSS and transcription end site [TES]) (E) or at highly (75%–100%; Q, quartile) expressed genes (F) and at active enhancers (G) in U2OS cells.
The topoisome engages promoters and enhancers to favor transcription
To map catalytically engaged topoisomes on the genome, we developed “cross-CAD-seq,” a modification of the cross-CAD assay. After Dox induction, U2OS-MYC-EGFP cells were treated with MG132 and Eto to trap both TOP2Accs and TOP2Bccs. By immunoprecipitaing lysates with anti-TOP1 (not anti-TOP2) to target the common subunit (TOP1) shared by both topoisomes A and B, we recovered all the bound DNA, enabling topoisome mapping by DNA sequencing. Note that cross-CAD-seq relies upon topoisomerase-DNA-adducts, as no formaldehyde is used. In the absence of Eto, MG132, or Dox, the only DNA recovered must represent either TOP1 that is catalytically engaged at the moment of SDS-lysis or non-specific background (Input). Browser shots and metagene analysis of this control revealed an apparent consistent “background” throughout the genome (Figures 6D and 6E). Yet, at most genes, a sharp valley fell to baseline precisely at TSSs, suggesting that the steady background in fact represented widely dispersed, genomically engaged TOP1. That such engagement was inhibited at TSSs is in accord with previous work (Baranello et al., 2016) showing TOP1cc formation to be suppressed until after pause release when the phosphorylated-CTD of RNAPII stimulates TOP1 activity. Using MG132+Eto (“Untreated+Eto+MG132” in Figures 6D and 6E), to stabilize topoisomes, endogenous MYC promoted their covalent engagement at TSSs, transforming the TSS valleys into peaks. Upon Dox induction, a portion of the topoisomes (engaged through Eto+MG132) were released from the TSS into gene bodies (Figure 6F), as expected (Rahl et al., 2010). To test whether topoisome engagement with DNA is MYC-dependent, we identified MYC-binding sites in MYC ChIP-seq from U2OS (Lorenzin et al., 2016) and analyzed these sites in our cross-CAD-seq data for TOP1 binding. In untreated cells, valleys of TOP1 activity were centered around MYC binding. Treatment with Dox, MG132, or Eto+MG132 turned the valleys into topoisome peaks that strongly co-localized with MYC peaks (Figure S6E). We also used the MYC ChIP-seq to sort TSSs in deciles of decreasing MYC binding and profiled the TOP1 cross-CAD-seq in untreated and Dox-induced U2OS cells in each decile. In the absence of treatment, cross-CAD-seq signals did not differ across the TSS deciles (Figure S6F). Following Dox induction and stabilization with Eto+MG132, topoisome binding paralleled MYC recruitment. TSSs with high MYC carried more topoisomes than TSSs with low MYC (Figure S6G), indicating that MYC fundamentally alters topoisomerase engagement at genes, increasing catalytic activity near TSSs, where supercoils otherwise accumulate (Ma et al., 2013; Wu et al., 1988). At enhancers, in the presence of Eto+MG132, “enhancer invasion” driven by MYC induction with Dox was accompanied by an increase in engaged TOP1, consistent with topoisome activation (Rahl et al., 2010; Lin et al., 2012) (Figure 6G). These results bolster the notion that MYC-mediated topoisomes focus and enhance topoisomerase action at sites of intense gene activity.
The topoisome facilitates RNAPII transcription and MYC recruitment at promoters
If the topoisome manages supercoiling that opposes high output transcription, then blocking topoisome assembly should impede RNAPII translocation. We generated auxin-inducible degron cell lines (HCT116TOP1_mAID and HCT116TOP2A_mAID [Nielsen et al., 2020]) to rapidly deplete TOP1 and TOP2A in cells and assessed the RNAPII and MYC recruitment to genes. In these systems, the degradation of mAID-tagged TOP1 and TOP2A is achieved within 2 h of auxin treatment (Figures 7A and 7B). RNAPII occupancy at TSSs increased upon acute depletion of TOP1 and TOP2A, as measured by spike-in normalized ChIP-seq (Figure 7C; Table S2). This effect was more dramatic at highly expressed genes and at genes bearing transcriptionally engaged RNAPII, where supercoiling not efficiently removed by topoisomes might accumulate. Indeed, the magnitude of the defect correlated with the level of gene expression, as the top 5% of expressed TSSs showed stronger RNAPII accumulation upon topoisomerases depletion (Figure 7C). We also identified elongating genes by pausing index and compared RNAPII levels with or without auxin. TOP1 or TOP2A degradation provoked a strong accumulation of RNAPII (Figure S7A), likely due to the buildup of supercoiling, whether positive or negative, opposing RNAPII translocation (Ma et al., 2013; Joshi et al., 2012). The accumulation of RNAPII at TSSs upon topoisomerase depeletion mirrors the increase of RNAPII seen upon acute MYC depletion (Muhar et al., 2018). We next measured MYC binding at TSSs and found its levels reduced without TOP1 or TOP2A (Figure 7D). The reduction in MYC binding was more pronounced at the top 5% of expressed TSSs upon TOP1 or TOP2A depletion compared to the whole TSS distribution (Figure 7D). Taken together, these and previous results (Figures 6D–6G) indicate that MYC controls topoisomerase engagement to DNA and that increasing topoisomerase levels favor MYC recruitment at genes.
Figure 7. The topoisome favors RNAPII transcription and MYC recruitment at TSSs.

(A and B) TOP1 (A) and TOP2A (B) depletion in HCT116TOP1_mAID and HCT116TOP2A_ mAID cells with auxin.
(C and D) Average RNAPII (C) and MYC (D) occupancy at TSS (spike-in normalized signal) of all and the top 5% of expressed genes. Significance between auxin and auxinole samples for RNAPII and MYC were calculated using a Wilcoxon signed-rank test (p < e-100) comparing means of average signal ± 500bp window around TSS.
(E) A translocating polymerase unwinds DNA, generating torque transmitted through the chromatin fiber that forms supercoils. By nucleating a topisome, MYC (with TOP2A) and MYCN (with TOP2B) controls DNA topology regulating DNA over- or under-twisting as well as supercoiling that may otherwise halt ongoing transcription or replication. Created with BioRender.com.
DISCUSSION
Topoisomes A and B are highly specific complexes
MYC and MYCN are largely disordered despite bHLH-ZIP dimerization with MAX. The C termini of TOP2A and TOP2B and the N terminus of TOP1 are also disordered (Jumper et al., 2021). All four proteins bind DNA. Their interaction, though, is neither non-specific or mediated by nucleic acid bridging because: (1) There are two types of topoisomes (Figure 7E). The A and B topoisomes, with their distinct activation profiles, are veritably the definition of specificity. (2) Topoisomes from cells or in vitro are resistant to nucleases or high ionic strength. Topoisome formation and activation by MYC versus MYC-MAX are equivalent, though only the latter binds DNA. (3) Other DNA-binding proteins did not stimulate topoisomerase activity. (4) PLA and immunoprecipitation from cell extracts are concordant with the activity and composition of reconstituted topoisomes. Thus, topoisomes A and B are real, discrete biochemical entities. The molecular discriminants of topoisome A versus B assembly (MYC versus MYCN; TOP2A versus TOP2B) remain to be defined; whether subtle amino acid substitutions in the conserved regions versus coarse changes in less well-conserved protein segments—or specific modifications—guide their interactions is yet unknown. Topoisomes A versus B may prove functionally distinct. MYC and TOP2A are associated with proliferation; if TOP2A and MYC are high, and TOP2B and MYCN are low, full topoisome A activity would maximize transcription amplification by MYC. High TOP2B, in the absence of MYCN, would modulate the dynamic range of the MYC amplifier (and vice versa, where TOP2A modulates TOP2B when MYCN exceeds MYC). Thus, the amounts and ratios of TOP2A and TOP2B, or MYC and MYCN, control amplifier “gain” and determine promoter saturation. By tuning torsional stress, topoisomes A or B would adjust elongation rate (Singh and Padgett, 2009) and promoter burst-size.
Why tether TOP1 with TOP2?
MYC influences transcription by RNAPI, RNAPII and RNAPIII (Poortinga et al., 2004; Arabi et al., 2005; Gomez-Roman et al., 2003; Nie et al., 2012) and promotes replication via interactions with the replisome (Dominguez-Sola et al., 2007). Common to these processes are enzymes that unwind and translocate along the DNA, generating over- or under-twisting DNA, supercoils or alternative DNA structures. All these DNA conformations may directly stall a translocating enzyme. Whatever else MYC does to increase gene expression would be wasted if topological impedances are not resolved. Torsional stress is partitioned between twist (Tw) and writhe (Wr). TOP1s are “twist-ases” that remove excess ± Tw; for TOP1 to remove supercoils, Wr must repartition into Tw. In contrast, TOP2s are “writhe-ases” directly removing ± supercoils (Wr); for TOP2 to remove excess ± Tw, it must first repartition into Wr (Baranello et al., 2013). TOP1 and TOP2 are largely redundant when Tw and Wr are in equilbrium and freely interconvert, as in most in vitro reactions. Within the mammalian nucleus, the topoisomerases encounter chemically and physically complex conditions that may slow equilibration between Tw and Wr. Entangling chromatin, protein-protein looping, tethering of transcribed regions to static structures such as nuclear pores, and situating active genes in viscous condensates (Sabari et al., 2018) may all delay the interconversion of Tw and Wr. When promoter firing and pause release are sporadic, the repartitioning of Tw and Wr may be resolved by any topoisomerase. At high output promoters–the main targets of MYC–where bursts of transcription are frequent (Rodriguez and Larson, 2020), topoisomes may ensure the immediate removal of Tw or Wr, to sustain transcription. Unlike other transcription factors, MYC prolongs bursts. Perhaps similar to prokaryotes (Chong et al., 2014), transcription bursts terminate when opposing torsional stress spikes; if spikes are suppressed by topoisomes, the burst may endure.
Binding to multiple partners dictates MYC function
The peak of topoisome components suggests a 1:1:1:1 TOP2-dimer:TOP1:MYC or MYC:MAX. Topoisomes can form in vitro without MAX. MAX-independent MYC function has been noted in Drosophila (Gallant and Steiger, 2009) and in some pheochromocytomas (Hopewell and Ziff, 1995). Although incorporating ~10%–15% of cellular TOP1, TOP2, and MYC, topoisome formation is likely to have a major impact on total cellular topoisomerase activity. Because MYC boosts TOP1 and TOP2A catalytic activities 5- to 10-fold, net cellular topoisomerase activity may be doubled, while still leaving components available to other complexes.
The power of MYC as an oncogenic driver derives from its assembly into a broad array of complexes that regulate transcription, chromatin remodeling (Knoepfler et al., 2006) and replication. Mass spectrometry identified more than 300 partners of MYC, including over 100 transcription factors (Kalkat et al., 2018). The scope of these interactions indicates that MYC function may vary depending on partner availability and E-boxes in target promoters. For example, the PAF complex binds with MYC and promotes transactivation activity in a predominantly MAX-dependent manner (Gerlach et al., 2017), while the trans-activation domain of MYC can bind to TBP and facilitate DNA engagement independently of E-boxes (Wei et al., 2019). The diversity of MYC partners likely allows MYC to switch between its gene-specific, E-box-dependent role as an upregulator of specific target genes or to play a more expansive role as an E-box-independent expression amplifier. MYC’s influence has been thought to rest on targeting complexes to strategic locations through heterodimerization with MAX and binding to E-boxes (Fernandez et al., 2003) or by bringing common components of the transcription apparatus to all active genes to amplify global promoter output (Lin et al., 2012; Nie et al., 2012). Beyond this “general delivery service”, our results indicate that MYC directly manages catalytic effector functions of partner proteins, e.g., MYC inhibits KDM5-H3K4 demethylase activity (Secombe et al., 2007). MYC also regulates DNA replication independent of MAX by recruiting CDC45 to replication origins (Dominguez-Sola et al., 2007). This suggests that via topoisomes (e.g., topoisome A), MYC may help to resolve topological issues during replication. Elucidating partitioning of MYC between its partners and its effects on their activities represents a largely unaddressed challenge.
Topoisome formation and MYC regulation via other binding partners need not be mutually exclusive. MYC is recruited to promoters either directly by DNA binding via its MAX-dimerized bHLH-ZIP domain or indirectly by the N-terminal TAD interacting with promoter-bound complexes. Redundant topoisome assembly via either terminus bypasses whatever steric hinderances occur upon DNA binding or by engaging large complexes (transcription and epigenetic co-regulators) to ensure that the topoisome is available throughout transcription to swiftly resolve topological issues. It has been hypothesized that MYC amplifies transcription via the acceleration of multiple steps of the transcription cycle–i.e., kinetic synergy (Nie et al., 2020). As many events during transcription are accompanied by changes in DNA topology (Baranello et al., 2013), the amplifier capacity of MYC is contingent upon its ability to control torsional stress.
Targeting an oncogene as polyamorous as MYC may seem to be impossible; however, the DNA supercoiling produced by almost all DNA transactions may create a bottleneck for gene activity. The novel coupling of TOP1 and TOP2 sponsored by MYC (or MYCN) creates a tool to sustain accelerated cellular growth. Without topoisomes, tumor growth may be compromised compared to low MYC cells (Zhang et al., 2016). MYC-driven oncogenic transcription and replication may create a vulnerability that can be therapeutically exploited through strategies that target the topoisome.
Limitation of the study
Topoisomes may include other topoisomerase subspecies. TOP2A/B heterodimers have been detected in HeLa cells (Gromova et al., 1998; Biersack et al., 1996). Although less than 5% of the total TOP2A heterodimerizes with TOP2B and less than 25% of the less abundant (in HeLa cells) TOP2B heterodimerizes with TOP2A (and so TOP2A/B heterodimers area small fraction of the total TOP2), we cannot exclude that topoisomes form with these heterodimers, and we cannot predict their response to MYC or MYCN. Techniques such as ChIP-seq do not provide the kinetic information required to understand the dynamics of genetic processes. Further characterization of topoisome activity at genes requires systems using single-molecule tracking to reveal the mechanical coupling between DNA and protein in vitro and in cells.
STAR*METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact Laura Baranello (laura.baranello@ki.se).
Materials availability
The material generated in this study is available upon request from the lead contact.
Data and code availability
The deep sequencing data reported in this paper were deposited in the GEO database and are publicly available as of the date of publication. Previously published RNA-seq data, RNAPII and MYC ChIP-seq data from HCT116 and U2OS cells (Lorenzin et al., 2016; Baranello et al., 2016; Ibarra et al., 2016) are publicly available in GEO. Accession numbers of all aforementioned datasets are listed in the Key resource table.
This paper does not report original code since it is based on implementation of publicly available software. Published software and code used in this study are listed in the Key resource table. Any additional information required to reanalyze the data reported in this paper is available from the lead contacts upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell cultures, drug treatments and transfection
Human colon carcinoma HCT116 expressing FLAG-TOP2A were generated as described. The HCT116 FLAG-TOP2A cells were made by CRISPR-Cas9 gene editing. The 3xFLAG sequence (DYKDHDGDYKDHDIDYKDDDDKL) was knocked-in at the N-terminal of TOP2A and digital droplet PCR was used to determine efficiency. Initially, cells with heterozygous FLAG knock-in were isolated by single cell cloning and validated by Sanger sequencing. This process was repeated to obtain cells with homozygous FLAG-TOP2A. HCT116 FLAG-TOP2A and HCT116 cells were grown in high glucose (4.5g/L) DMEM (Thermo Fisher, 11965092) containing 10% FBS (Thermo Fisher, 26170043) in a 37°C incubator supplied with 5% CO2. HCT116CMVOsTIR1mAIDTop1 (HCT116TOP1_mAID) (Wiegard et al., 2021) and HCT116CMVOsTIR1Top2A mAID (HCT116TOP2A_mAID) (Nielsen et al., 2020) were grown in media containing 1μg/ml puromycin (Thermo Fisher, A11138–03) and 125μg/ml hygromycin B (Thermo Fisher, 10687010). The K562MYCmAIDTir1-eBFP2 (K562MYC_mAID) cells (Muhar et al., 2018) were grown in RPMI (Thermo Fisher, 21875034) media containing FBS 10%, 2mM glutamate and 1mM sodium pyruvate. For MYC, TOP1 or TOP2A degradation, cells were incubated in the presence of 500 μM auxin (Sigma, I3750–100MG-A) at 37°C. To suppress basal degradation of MYC, TOP1 and TOP2A auxinole (Medchem Express, HY-111444) treatment (final concentration of 100 μM) was used as a control. The bone osteosarcoma epithelial cells U2OS expressing a Dox inducible lentivirus- MYC-EGFP (Nie et al., 2020) were grown in DMEM containing 10% Tet approved FBS (Takara, 631106), MEM Nonessential Amino Acids (Corning, 25–025-Cl), 50 U/ml penicillin G and 50 μg/ml streptomycin sulfate (GIBCO, 15140122). Indicated dose of Dox treatment (0.6 μg/ml) was executed for 7 h. CPT or Eto treatments were performed for 15 min on U2OS-MYC-EGFP cells at 37°C with a final concentration of 10 μM or 25 μM, respectively. HO15.19-MYC-ER cells were grown with DMEM and 10% of calf serum (GIBCO, 26170–043) as previously described (Nie et al., 2020). 4-OH Tam treatment (200 nM final concentration) was performed for 15 min on HO15.19-MYC-ER cells to express a retrovirally encoded MYC-ER chimeric protein that immediately translocate to the nucleus. MG132 (Cayman chemicals, 13697) treatment was performed for 1 h on HO15.19-MYC-ER and U2OS-MYC-EGFP cells at 37°C with a final concentration of 10 μM. In K562MYC_mAID cells, MG132 treatment (10 μM) was performed for 30 min after auxin treatment (30 min, 500 μM). This MG132 treatment did not affect MYC levels (data not shown). CPT (Sigma, C9911) (20 μM) and Eto (Sigma, E1383) (100 μM) were added in the last 5 min of MG132. HCT116 cells were transfected for 24 h with the indicated plasmid DNAs using Opti-MEMTM I Reduced Serum medium (Thermo Fisher, 31985062) and Lipofectamine 2000 (Thermo Fisher, 11668019) according to the manufacturer’s protocol.
Generation of the HCT116 CMV-OsTIR1 mAID-Top1
Plasmid construction. To insert mAID at the N terminus of the TOP1 gene using CRISPR/Cas, the sgRNA sequence (5'-CCCCACT CATGTCGGCCCGG-3') targeting near the start codon of the gene was inserted into pX330-U6-Chimeric_BB-CBh-hSpCas9 (Cong et al., 2013) (Addgene plasmid #42230) to give pX330-TOP1-N. The donor plasmid was constructed as described previously (Yesbolatova et al., 2019). Briefly, the homology arm sequence was cloned into pBluescript II after being amplified from genomic DNA using the primer set: TOP1-N_HA_50_SacI 5'-ATGCgagctcGTTCCGAGAAAAAGCGTCTGGAGAG-3' and TOP1-N_HA_3'_KpnI 5'-ATGCggtaccCCTTCCCTCTCTGGTGAAGTATGTG-3'. Using this plasmid as a template, inverse PCR was performed using the primer set: TOP1-N_HA_INV_F_BamHI 5'-ATGCggatccATGAGTGGGGACCACCTCCACAACG-3' and TOP1-N_HA_INV_R_EcoRV 5'-ATGCAgatatcGTCGGCCCGGAGGGACGAGC-3'. The EcoRV-BamHI fragment containing Hygro-P2A-mAID was excised from pMK344 and ligated to the above inverse PCR product to give Hygro-P2A-mAID-TOP1 donor. HCT116 cells expressing OsTIR1 under control of the CMV promoter from the AAVS1 locus was described previously (Natsume et al., 2016). HCT116mAID-TOP1 cells were generated by co-transfecting HCT116 CMV-OsTIR1 cells with pX330-TOP1-N and Hygro-P2A-mAID-TOP1 donor using Fu-GENE HD (Promega, E2311). After selection with 100 μg/ml hygromycin B Gold (Invivogen, ant-hg-5), single colonies were isolated and homologous recombination mediated knock-in of mAID at both TOP1 alleles was confirmed by genomic PCR and western blotting. The detailed protocol is described in (Yesbolatova et al., 2019). The HCT116CMVOsTIR1mAIDTop2 (HCT116TOP2_mAID) cell line was a kind gift from Dr. D. Hudson (MCRI, Australia). The K562MYC_mAID cell line was a kind gift from Dr. J. Zuber (IMP, Austria).
METHOD DETAILS
Drug and antibodies
Camptothecin (CPT), etoposide (Eto), 4-OH tamoxifen (Tam), doxycycline (Dox), triptolide (Tript), and auxin were purchased from Sigma. Auxinole was from MedChemexpress (HY-111444), puromycin (A11138–03) and hygromycin B (10687010) were purchased from Thermo Fisher. MG132 was purchased from Cayman Chemical. Mouse monoclonal anti-TOP1 (C-21), anti-c-MYC (9E10), anti-MAX (H-2), anti-TOP2A (E-10), anti-N-MYC (B8.4.B), anti-nucleolin (C-23) and anti-IgG [mouse (Sc-2025), rabbit (Sc-2027)] antibodies were from Santa Cruz Biotechnology. Rabbit monoclonal anti-c-MYC antibody (N-ter) (Y69) (ab32072), anti-TOP1 (N-ter) (ab109374), anti-TOP2A (ab52934) and mouse monoclonal anti-c-MYC (C-ter) (ab56), anti-GAPDH (ab9484), anti-RNAPII (ab5408) and anti-Histone H3 (ab1791) were from Abcam. Rabbit polyclonal anti-c-MYC (mid-ter) was from Novus (NBP2–49201). Anti-FLAG antibody (F1804) was from Sigma. Anti-KAP1 antibody (A300–274A) was from Bethyl. Spike-in antibody (61686) was purchased from Active motif. Rabbit polyclonal anti-GFP (A-11122) was from Thermo Fisher. Rabbit polyclonal anti-TOP2B antibody (20549–1-AP) was from Proteintech (Rosemont, IL, USA). Secondary antibodies like horseradish peroxidase conjugated anti-rabbit IgG (ab205718) or anti-mouse IgG (ab205719) were purchased from Abcam.
Plasmids and generation of recombinant MYC and heterodimer MYC-MAX
Construction of full length and truncated c-MYC in pET-28a (+) has been carried out as described. Briefly, PCR was performed to amplify the full-length MYC using forward primer 5'-CGATTCGATcatatgCCCCTCAACGTTAGCTTC and reverse primer 5'- GGTA CAATCctcgag TTACGCACAAGAGTTCCGTAG. The forward primer was designed with a NdeI recognition site and the reverse primer with a XhoI site. Reverse primer was also designed with stop codon to remove the adjacent His tag of the pET-28a (+) vector. This PCR amplified fragment was cloned in NdeI/XhoI site of bacterial expression vector pET-28a (+) and confirmed by restriction enzyme digestion and sequencing. The newly generated construct pET28a(+)-MYC full length was transformed in One Shot BL21(DE3) Chemically Competent E. coli (Thermo Fisher, C601003), expressed by 1 mM IPTG for 4 h in a 37°C shaker and purified using Ni-Sepharose™ 6 Fast Flow (Sigma, GE17–5318-01) as described in (Guo et al., 2014). In summary, the cell pellet was lysed and sonicated in UHSW buffer (7 M urea, 10 mM HEPES pH 7.5, 500 mM NaCl, 25 mM imidazole, 1% Triton X-100, 5% glycerol and protease inhibitor cocktail) and centrifuged at 13000 rpm for 20 min at 4°C. Supernatant was passed through pre-equilibrated Ni-Sepharose™ 6 Fast Flow column (UHSW buffer). The column was washed with ULSW buffer (containing 100 mM KCl) and was eluted by UE buffer containing 250 mM imidazole. To eliminate non-specific proteins, the urea eluted fraction was further purified through MONO-Q and dialysis. Next, the remaining His tag was removed by thrombin cleavage using the Thrombin cleavage kit (ab207000). The reaction product was passed through Ni-Sepharose™ 6 Fast Flow and the flow-through containing recombinant MYC protein without His tag was collected. Purified untagged MYC protein was further mixed with recombinant His-tagged MAX protein in equimolar ratio and dialysis was performed in HGKEDP buffer (25 μM HEPES pH 7.5, 15% glycerol, 100 mM KCl, 0.1 mM EDTA, 1 mM DTT and protease inhibitor cocktail) for 4 h at 4°C (Das et al., 2004). The dialyzed mixture was further purified through Ni-Sepharose™ 6 Fast Flow column with urea free buffer. The eluted fraction contained purified heterodimer MYC-MAX protein as confirmed by 4%–12% SDS-PAGE gel with Coomassie blue staining. The EGFP-tagged human MYC full length and truncated constructs were generated as described. Briefly, PCR was performed to amplify the mid term (200–320 aa) truncated MYC constructs using forward primer 5'-CGATTCGATgaattcATGGACAGCAGCTCGCCCAAG and reverse primer 5'- GGTACAATCggatccAGCAGGATAGTCCTTCCG. This PCR amplified fragment was cloned in EcoRI/BamHI site of bacterial EGFP expression vector Pd4 EGFP-N1 (Clontech, 6085–1) using One shot™ TOP10 Chemically Competent E. coli (Thermo Fisher, C404010) and confirmed by restriction enzyme digestion and sequencing (NCI core).
Expression and purification of recombinant protein
The recombinant human TOP1 was kindly gifted by Dr Y. Pommier (NIH/USA). Recombinant human TOP2B was a kind gift from Dr. J. Berger (Johns Hopkins School of Medicine/USA). Recombinant human TOP2A and E. coli TOP1 were purchased from Topogen (Buena Vista, CO, USA). Recombinant MYCN were purchased from abcam (ab241520). Recombinant TBP and PC4 were purified as described previously (Maldonado et al., 1996; Ge et al., 1996). Recombinant TOP2A was also produced using the protocol published in (Lee et al., 2017) with slight modifications. The Gal1/10 His6 TEV Ura S. cerevisiae expression vector (12URA-B) was a gift from Scott Gradia (Addgene plasmid # 48304) and the plasmid expressing human TOP2A was kindly provided by Dr. J. Berger. URA-deficient yeast transformed with the expression plasmid was incubated in uracil-deficient media for 24 h, followed by YPLG (1% yeast extract, 2% peptone, 2% sodium DL-lactate, 1.5% glycerol) for 24 h, then expression of TOP2A was induced by 2% galactose for 4 h. Protein was extracted from the snap-frozen yeast using a cryo-mill, the lysate was filtered, and the tagged TOP2A was enriched and eluted from HisTrap Excel nickel and HiTrap CP cation exchange columns (GE Healthcare) before overnight incubation with His-tagged TEV protease. A HisTrap column was used to remove the cleaved His tag and the TEV protease, and the TOP2A was purified further with a Superdex 200 16/60 column, before storing at −80°C. TOP2A purity and activity was confirmed by immunoblotting.
MYC and RNAPII Chromatin Immunoprecipitation coupled to sequencing (ChIP-seq)
MYC and RNAPII ChIP was performed on HCT116 cells as described previously (Baranello et al., 2016) with minor modifications (Cameron et al., 2021). Briefly, cells were crosslinked with 1% formaldehyde (Thermo Fisher, 28906) for 5 min. Cross-linking was stopped by the addition of glycine (125 mM) and cells were washed twice with cold PBS. After harvesting cells by scraping, the pellet was washed once with PBS plus 0.5% BSA and resuspended in RIPA buffer (10 mM Tris-HCl pH 8.0, 1 mM EDTA pH 8.0, 1% Triton X-100, 0.1% Na-Deoxycholate, 0.1% SDS, 200 mM NaCl, with the addition of protease inhibitor cocktail) to a final concentration of 1x107 cells/ml. Samples were sonicated with a Bandelin probe sonicator to produce chromatin fragments of 400 bps on average. After centrifugation, extracts were immunoprecipitated. 2 μg of anti-MYC antibody (N-ter) (Y69) (ab32072) or anti-RNAPII (ab5408) were mixed with 35 μL Protein A/G magnetic beads (Pierce, 88803) and incubated at 4°C for 6 h with controlled rotation. For ChIP normalization, 1 μg of spike-in antibody (Active Motif, 61686) was added to the antibody-Protein A/G beads mix. Chromatin from 1x107 cells was added together with 20 ng of spike-in chromatin (Active Motif, 53083) to the Protein A/G-antibody complexes and incubated with rotation overnight at 4°C. Samples were washed twice with RIPA buffer, twice with RIPA buffer containing 300 mM NaCl, twice with LiCl buffer (10 mM Tris-HCl pH 8.0, 1 mM EDTA pH 8.0, 250 mM LiCl, 0.5% NP40, 0.5% Na-Deoxycholate) and twice with TE (10 mM Tris-HCl pH 8.0, 1 mM EDTA pH 8.0). The beads were then resuspended in 125 μL TE plus 0.25% SDS supplemented with proteinase K (500 μg/ml, NEB, P8107S) and incubated overnight at 65°C. DNA was recovered from the elute by phenol:chloroform (25:24:1) (Sigma, P2069) extraction followed by ethanol (100%) precipitation in the presence of 20 μg of GlycoBlue (Thermo Fisher, AM9515) and dissolved in Tris-HCl pH 8.5. All ChIP-seq experiments were performed in biological duplicates.
TOP2A ChIP-seq
ChIP (in biological duplicates) was performed as described above with some modifications. Briefly, cells were treated with 100 μM Etoposide for 4 min and crosslinked with 1% formaldehyde for 5 min. Cross-linking was stopped by the addition of glycine as described above. TE-SDS 0.25% was added directly after removal of media with glycine and cells were collected to reach a final concentration of 1x107 cells/ml. Samples were sonicated with a Bandelin probe sonicator to produce chromatin fragments of 400 bp on average. After clarification by centrifugation, sonicated extracts were immunoprecipitated. 2 μg of anti-FLAG (F1804) and 1 μg of anti-TOP2A (ab52934) were mixed with 35 μl Protein A/G magnetic beads and incubated at 4°C for 6 h with rotation. Chromatin from 1 × 107 cells was added to the Protein A/G-antibody complexes and incubated overnight at 4°C with rotation. Washing, precipitation and elution was performed as described above.
Library preparation and sequencing of ChIP-seq
DNA from ChIP was quantified with the Qubit dsDNA HS Assay Kit (Thermo Fisher, Q33230). To cleave off covalently bound tyrosyl residues from TOP2A, the samples were additionally treated with ExoVII (NEB, M0379S) (0.5–1U per 10 ng of DNA) and purified by PCR purification Kit (QIAGEN, 28106). Sequencing libraries were created according to the ThruPLEX DNA-seq kit protocol (Takara, R400676). Size selection was performed in the range of 200–700bp with AMPure XP beads (Beckman, A63880) and confirmed using the Agilent High Sensitivity DNA Kit (Agilent, 5067–4626) on the Agilent 2100 Bioanalyzer. Libraries were pooled and sequenced using the NextSeq 500/550 High Output Kit v2.5 (Illumina, 20024906). The sequencing run was Single End and Dual Index with 75 bp reads.
Dephosphorylation of topoisomerases
Recombinant TOP1 and TOP2A proteins were dephosphorylated by incubation with lambda protein phosphatase (NEB, P0753) for either 16 h at 4°C or 1 h at 30°C. Dephosphorylation was visualized by transferring the protein onto PVDF membrane by western blotting, and probing the membrane for phosphoproteins using the Pro-Q Diamond Phosphoprotein Blot Stain Kit (Thermo Fisher, P33356) and counterstaining for total protein using Ponceau stain. Experiments performed in 3 independent replicas.
Immunoprecipitation and western blotting
Cells were washed twice with PBS, scraped off and lysed in RIPA buffer (50 mM Tris-HCl pH 8.0, 50 mM NaCl, 1 mM EDTA, 0.1% sodium dodecyl sulfate, 1% NP40, 0.1% Na-deoxycholate supplemented with complete protease inhibitors). After vortexing and incubation at 4°C for 1 h, lysates were centrifuged at 13000 rpm for 10 min at 4°C. Supernatants were collected and stored at −80°C. Nuclear extract was prepared by washing the cells with PBS and scrape off cells. Cells were spin down, resuspend in HP buffer (50 mM HEPES pH 7.5, plus protease inhibitors), incubated on ice for 5 min, passed 10 times through a 20 G needle (to disrupt the cell membrane) and then centrifuged for 10 min 4000 rpm at 4°C. The pellet was resuspended in 300 μL HLSP buffer (50 mM Tris-HCl pH 7.5, 100 mM LiCl, 0.2% sarkosyl and protease inhibitors), treated with 1 μL benzonase (250 units/μl) for 1 h at 4°C and further dissolved through sonication and passing through a 25 G needle. The sample was centrifuged at 15000 rpm for 15 min and the supernatant was collected and quantified. For the immunoprecipitation of nuclear extract, 1 mg of protein was pre-cleared with 25 μL of Protein A/G for 1 h. The precleared supernatant was incubated overnight with 2 μg of specific antibody and 30 μL of Protein A/G magnetic beads (blocked by 3% BSA) at 4°C. The immunocomplexes were washed four times with HLSP buffer and 2 times with RIPA buffer with 250 mM NaCl and 0.2% sarkosyl. The immunoprecipitated proteins were eluted by heating in 40 μL of 1 x NuPAGE LDS loading dye (Thermo Fisher, NP0007) for 10 min at high temp. Protein samples were examined by western blotting. Briefly, samples were run on 4%–12% Bis-Tris NuPAGE protein gel (Thermo Fisher, NP0323), transferred onto nitrocellulose blots, probed with antibodies against the desired protein of interest, and imaged by chemiluminescence using ECL chemiluminescence reaction (Thermo Fisher, 34095) under the Odyssey infrared scanner (Li-Cor). All densitometric quantifications of immunoblots were carried out using ImageJ software (Das et al., 2016). Experiments performed in 3 independent replicas. Immunoprecipitations with recombinant proteins were carried out by mixing equimolar concentrations of proteins in PDB buffer (10 mM Tris-HCl pH 7.5, 100 M KCl, 0.5% NP40, 7.5% Glycerol, 200 μM EDTA, 250 μg/ml BSA and protease inhibitors) on ice for 1 h and 500 ng of anti-MYC or IgG was added to the mixture followed by incubation in ice for 1 h. For each sample, 10 mL Protein A/G beads were blocked in PDB buffer with 5% skim milk powder for 1 h at 4°C, washed in PDB and added to the protein solution in 100 μL PDB. Elution was performed by heating samples in presence of protein 1 x NuPAGE LDS loading dye. Protein samples were subjected to standard western blotting protocol as discussed before. Experiments performed in 3 independent replicas.
Topoisomerase I relaxation assay
The activity of type I DNA topoisomerases was assayed by analyzing the mobility of relaxed isomers of supercoiled plasmid DNA (FLIP-FUSE plasmid) on agarose gel (Das et al., 2018; Kundu et al., 2019). Briefly, recombinant human TOP1 (0.23 ng) was incubated with increasing amount of recombinant full length MYC-MAX, MYC or truncated MYC mutants (0.4 to 2 ng) for 10 min in ice in the TOP1 buffer (10 mM Tris-HCl pH 7.5, 50 mM KCl, 5 mM MgCl2, 0.1 mM EDTA, 50 μg/ml). Supercoiled plasmid DNA (200 ng) was added to the mixture, incubated at 37°C for 15 min and reactions were terminated by adding TE-SDS 1% plus proteinase K (200 μg/ml). Reaction products were purified by phenol: chloroform and analyzed on 1.4% (w/v) agarose gel electrophoresis in TAE buffer pH 7.6 containing 15 μM chloroquine. To visualize the relaxed species, gels were stained with Diamond™ Nucleic acid dye (Promega, H1181) and imaged under the Odyssey infrared scanner (Li-Cor). All the factors used in this work have been tested for the absence of nuclease and topoisomerase contamination using the same conditions described above but excluding TOP1. Experiments performed at least in 3 independent replicas.
Ex-vivo relaxation assays were carried out with whole cell extracts from Dox inducible lentivirus-expressed U2OS-MYC-EGFP cells as described (Nie et al., 2020; Das et al., 2016). Briefly, the indicated amount of whole cell extracts (equivalent to purified recombinant protein amount as compared by western blot of specific protein) from untreated or Dox-induced (0.6 μg/ml) U2OS-MYC-EGFP cells were incubated with supercoiled DNA (200 ng) in TOP1 buffer at 37°C for 15 min, terminated with TE-SDS 1% plus proteinase K (200 μg/ml) and purified with phenol: chloroform. Purified products from relaxation assay were analyzed by agarose gel electrophoresis in presence of chloroquine. All densitometric quantifications of relaxation gels were carried out using ImageJ software. Experiments performed at least in 3 independent replicas.
Topoisomerase II decatenation assay
TOP2A activity was assayed by decatenation assay of kinetoplast DNA. Briefly, recombinant human TOP2 (0.23 ng) was incubated with increasing amount of recombinant full length MYC-MAX, MYC, MYCN or truncated MYC mutants (0.4 to 2 ng) for 10 min in ice in the TOP2 reaction buffer (50 mM Tris-HCl pH 8.0, 10 mM MgCl2, 2 mM ATP, 0.5 mM DTT, 30 μg/ml BSA) before addition of 100–200 ng of kinetoplast DNA (Topogen, TG2013–3) and was incubated at 37°C for 10 min. The reaction was stopped by adding TE-SDS 1% plus proteinase K (200 μg/ml) and reaction products were purified by phenol: chloroform and run on a 1% agarose gel containing 0.5 μg/ml SYBR Green (Thermo Fisher, S7563) or Ethidium Bromide (Thermo Fisher, 15585011). Image was detected under the ChemiDoc System (Bio-Rad). All the factors used in this work have been tested for the absence of nuclease and topoisomerase contamination using the same conditions described above but excluding TOP2. Experiments performed at least in 3 independent replicas.
Nuclear extracts were harvested for the TOP2A activity assay with a modified version of a previously described protocol (Nitiss et al., 2012). Briefly, U2OS-MYC-EGFP cells were collected in PBS by scraping, resuspended in 1 ml LSB (20 mM Tris-HCl pH 7.5, 5 mM KCl, 1 mM MgCl2, 10% glycerol, 1 mM DTT, plus protease inhibitors), passed through a 20 G needle 10 times and incubated on ice for 10 min. The nuclei were pelleted, resuspended in HSB (LSB but with 350 mM KCl) and incubated on ice for 30 min. For the ex vivo decatenation assay, the indicated amount of nuclear extracts from untreated or Dox-induced (0.6 μg/ml) U2OS-MYC-EGFP cells were incubated with 100–200 ng of kinetoplast DNA in TOP2 reaction buffer (50 mM Tris pH 8.0, 10 mM MgCl2, 2 mM ATP, 0.5 mM DTT, 30 μg/ml BSA) at 37°C for 10 min. Reactions were stopped with the addition of adding TE-SDS 1% plus proteinase K (200 μg/ml). After phenol: chloroform extraction, samples were run on agarose gel containing SYBR Green or Ethidium Bromide and image was detected under the ChemiDoc System (Bio-Rad). All densitometric quantifications of relaxation gels were carried out using ImageJ software. Experiments performed at least in 3 independent replicas.
Two-dimensional (2-D) Gel Electrophoresis
To resolve the full range of DNA topoisomers that might simultaneously include both positively and negatively supercoiled topoisomers of varying densities, 2-D agarose gel electrophoresis is used. A mixture of DNA topoisomers is first separated in an 1.8% (w/v) agarose gel in TAE buffer pH 7.6. using a low concentration of chloroquine (3 μM). To better resolve the topoisomers, electrophoresis in a second, perpendicular, is perfomed in the presence of an increased concentration of chloroquine (20μM). The distribution of topoisomers was visualized by staining with Sybr Green. Experiments performed in 2 independent replicas.
Covalant adduct detection (CAD) IN living cells
U2OS-MYC-EGFP or HO15.19-MYC-ER cells (1.5 x107) were treated as indicated in the scheme on top of each figure, CPT 10 μM and Eto 25 μM. Cells were washed with 15 ml ice-cold PBS and 10 mL of STE buffer (2% SDS, 0.5 M Tris-HCl pH 8.1, 10 mM EDTA) was added to the cells before scraping. Lysates were incubated at 65°C for 10 min and were disrupted using a Dounce homogenizer tight pestle shearing apparatus. Phenol: chloroform was added to the samples and centrifugated at 15000 rpm for 15 min to induce separation of the phases. After centrifugation, the aqueous phase containing the protein peptides covalently linked to DNA, was precipitated by adding 2.5 volume ethanol 100% and 1/10 volume sodium acetate (3 M, pH 5.2), incubated for 1 h at room temperature and centrifugated at 15000 rpm for 30 min. DNA pellet was washed by ethanol 70%, dried and finally dissolved in elution buffer (10 mM Tris pH 8.0) by overnight incubation at 4°C. Samples were further purified through QIAquick PCR purification kit (QIAGEN, 28106) using columns (provided in the kit) such that the total DNA bound per column was not exceeded 10 μg and was eluted in 100 μL of elution buffer. To quantify the purified DNA, 20 μL sample was treated with 0.2 mg/ml RNase A (Thermo Fisher, EN0531) at 37°C for 30 min before addition of 1 μl proteinase K (1U) at 55°C for 1.5 h. The reaction products were further purified through Minielute PCR purification kit (QIAGEN, 28004) and quantified by nanodrop. Each CAD sample was prepared by diluting 1–3 μg of sample DNA into elution buffer (total volume 20 μl), was applied to nitrocellulose membrane (Bio-Rad) using a vacuum slot-blot (Minifold II), blocked for 1 h in 3% BSA prepared in TBS (50 mM Tris-HCl pH 7.5, 150 mM NaCl) containing 0.1% Tween 20 (TBST) and incubated with specific primary and secondary antibodies. The peptide linked DNA was detected using ECL chemiluminescence reaction under the Odyssey infrared scanner (Li-Cor). Experiments performed in 3 independent replicas.
Covalent adduct detection coupled to ChIP-seq for TOP1 and ChIP-qPCR for TOP2 (TOP1 CAD-seq and TOP2 CAD-qPCR)
1x107 (K562MYC_mAID) cells were treated (in biological duplicates for the sequencing assays and in quadruplicates for the qPCRs) with auxin 500 μM for 30 min followed by MG132 10 μM for 25 min and addition of ETO 100 μM or CPT 20 μM for 5 min to trap TOP2cc or TOP1cc, respectively. The MG132 treatment did not affect MYC levels (data not shown). Cells in suspension were collected by centrifugation, resupended in 2 ml of M buffer (9.3 mM Tris-HCl pH 6.5, 18.6 mM EDTA, 5.59 M guanidine thiocyanate, 0.93% DTT, 0.93% Sarcosyl, 3.72% Triton X-100) and briefly sonicated with Bandelin probe sonicator at 20% amplitude for 3 cycles with 30 s ON, 30 s OFF. DNA covalent adducts were precipitated with 50% EtOH at −20°C, centrifuged at 14000 rpm and pellets were washed thrice in wash buffer (20 mM Tris-HCl pH 7.5, 50 mM NaCl, 1 mM EDTA, 50% EtOH). Pellets were dried for 5 min and resuspended in TE-SDS 0.1% (10 mM Tris-HCl pH 8.0, 1 mM EDTA pH 8.0, 0.1% SDS). After 30 min incubation by gentle agitation samples were further sonicated with Covaris ME220 sonicator for 5 min at High Cell protocol in milliTUBE–1 ml with AFA Fiber to produce fragments of about 1kb. For the immunoprecipitation, 2 μg anti-TOP1 (ab109374) or 3 μg of 1:1 mixture of anti-TOP2A (sc166934 and ab52934) antibodies were mixed with 30 μl Protein A/G magnetic beads (Pierce, 88803) and incubated at 4°C for 6 h with rotation. Beads were washed once with ice-cold PBS and DNA covalent adducts from 1 × 107 cells were added to the Protein A/G-antibody complexes and incubated overnight at 4°C with rotation. Washing was performed as described for the ChIP protocol but only once with every buffer and always in presence of 0.1% SDS. The beads were then resuspended in 100 μl TE plus 0.5% SDS supplemented with proteinase K (500 μg/ml) and incubated for 4 h at 65°C. Samples were then purified by QIAquick PCR purification Kit. Enrichment of specific DNA loci was determined by qPCR with the following primers: KRT19 promoter GCTCAGATATC CGCCCCTGACA and CCCTCACCTGGCGCCTTTTATG; EEF1A1 promoter CCTGCGAGTGTGTGTGTG and GCAAGTGTTGGGGT TAGGAA; MYC promoter GGACTCAGTCTGGGTGGAAGG and AAGGAGGAAAACGATGCCTAGA; UQCRQ promoter GCTGAGGA GAAGTGTGAGC and GGATGACGCCTTTGTCC; RPL4 promoter GCTTCCCGGCGCGTCCTGTGC and GGTGTGGAACTGG GATGTGCGGCG; α-sat: CTTTTTCATCATAGGCCTCAA and AGCTCACAGAGCTGAAACATT. Serial dilution if Input (non immunoprecipitated) material were used to for relative normalization of samples.
In situ proximity ligation assay
Proximity ligation assay (PLA) was performed using the Duolink In Situ PLAKit (Sigma-Millipore, DUO92101–1KT) according to the manufacturer’s protocol. In brief, approximately 1 x 104 cells of U2OS-MYC- EGFP or HO15.19-MYC-ER cells were plated on Ibidi 15 μ-slide angiogenesis slide (ibidi-treat, 81506) for 30–45 min, treated with the indicated concentration of Dox or Tam to induce MYC and utilized for PLA according to the manufacturer’s protocol. The primary antibodies used were: anti-c-MYC (9E10) mouse monoclonal antibody (sc-40) (1:100 dilution), anti-nucleolin (sc-8031) (1:100 dilution), anti-TOP1 rabbit monoclonal antibody (ab109374, abcam) (1:300 dilution), anti-TOP2A (E-10) mouse monoclonal antibody (sc-365799) (1:100 dilution) and anti-c-MYC (Y69) rabbit monoclonal antibody (ab168727) (1:100 dilution). Cells were observed with Zeiss LSM880 Multi-Photon Confocal Microscope. The distinct red PLA dots were generated due to close association of two protein of interest. The number of PLA dots were quantified using Macro for ImageJ (Schindelin et al., 2012) software (available at https://sites.imageJ.net/Janek/). Macro software counts PLA dots and measures the area of each nucleus present in the image. The output consists of composite images allowing efficient quality control of detection parameters and a spreadsheet with measurements. For the quantification plot, the number of PLA dots were quantified for more than 100 cells (nuclei) using macro software. Data was plotted using Graphpad Prism version 9.1.0. Statistical analysis was carried out using GraphPad Prism 9.1.0. The number of replicates for each experiment are reported in the figure legends.
TOP1 cross covalent adduct detection and ChIP for TOP1 (TOP1 Cross-CAD-seq)
We performed the TOP1 Cross-CAD-seq in the U2OS due to the extensive avaliability of genomic data. U2OS-MYC-EGFP cells were treated (in biological duplicates) with Dox (0.6 μg/ml), Eto (25 μM) and MG132 (10 μM) as indicated in the figure scheme. Cells were processed for harvesting covalent DNA adducts as discussed previously (see section Covalent adduct detection (CAD) in living cells). CAD samples were sonicated with a Bandelin probe sonicator to produce chromatin fragments of about 1 kb on average, were further purified through QIAquick PCR purification kit using columns (provided in the kit) such that the total DNA bound per column was not exceeded 10 μg (each sample were divided into 3 columns) and was eluted in elution buffer (Tris-HCl pH 8.5). 2 μg of anti-TOP1 antibody (ab109374) were mixed with 40 μL Protein A/G magnetic beads (Pierce, 88803) and incubated at 4°C for 6 h with controlled rotation. Eluted CAD samples from 1 × 107 cells were added to the Protein A/G-antibody complexes and incubated with rotation overnight at 4°C. Washing was performed as described for the ChIP protocol but twice with every buffer and always in presence of 0.1% SDS. The beads were then resuspended in 100 μl TE plus 0.5% SDS supplemented with proteinase K (500 μg/ml) and incubated for 4 h at 65°C. Samples were then either purified by QIAquick PCR purification Kit or extracted with Phenol: Chloroform and precipitated with EtOH (1/10 volume of sodium acetate 3 M pH 5.2, 2.5 volumes of 100% EtOH and 1.5 μl Glycogen).
Library preparation and sequencing of CAD-seq and Cross-CAD-seq
To cleave off covalently bound tyrosyl residues from immunoprecipitated DNA samples, the samples were additionally treated with ExoVII (0.5–1U per 10 ng of DNA) and purified by QIAquick PCR purification Kit. CAD-seq libraries were prepared using the protocol described (above) for ChIP-seq libraries. Cross-CAD libraries were prepared with AccelNGS 2S Plus DNA kit (Swift Bioscience, 21024) by sequencing facility at Fredrick National Laboratory for Cancer Research and sequenced on NovaSeq in Single End mode with 101 bp reads.
DATA ANALYSIS
The generated fastq files were quality controlled with FastQC and MultiQC (Ewels et al., 2016), trimmed with cutadapt, aligned to hg38 reference genome (Drosophila melanogaster Release 6 –dm6–in case of spike-in normalization) with bowtie2 (Langmead and Salzberg, 2012), deduplicated, sorted and indexed using samtools (Li et al., 2009) and Picard. BigWig files for visualization were generated using Deeptools (Ramírez and Dündar, 2014) from combined reads of two replicas. The profiles of short reads average distribution near TSSs and along normalized gene bodies were generated by ngs.plot (Shen et al., 2014), custom R scripts, Bioconductor (Huber et al., 2015) packages and ggplot2. Normalization was performed based on library size (RPM) or based on spike-in in individual replicas by multiplying the total number of reads by a factor inversely proportional to Drosophila spike-in unique deduplicated reads. Reads from two merged replicas were used for average profile generation except for Figures 1B, 7C and 7D where replicas were first normalized and then averaged.. Protein coding genes (n=19791) from Ensembl 76 (Fliceket al., 2014) database were used for generating the profiles at genes. The profiles were smoothed using a moving average algorithm. To select the most TOP1, TOP2A and MYC bound genes, RPM values were calculated ± 500 bps around the TSSs and 10,000 genes with highest values were taken. The expression of genes was determined based on previously published RNA-seq in HCT116 (Baranello et al., 2016) and U2OS (Ibarra et al., 2016) cells. Venn diagrams and boxplots were generated in R. TOP1, TOP2A and MYC peaks (shown in Table S1) were called on combined reads from two replicas with a random background model in TOP1 and MYC ChIP-seq data using MACS3 (Zhang et al., 2008) and in TOP2A ChIP-seq data using EPIC2 (Stovnerand Sætrom, 2019), which uses SICER algorithm proven efficient for finding diffuse binding sites. To determined MYC binding sites in U2OS cells (n=23475), a publicly available dataset (Lorenzin et al., 2016) was taken and MACS3 was used to determine peak summits for further analysis. Bedtools (Quinlanand Hall, 2010) package was used to filter peaks for hg38 blacklisted regions (Amemiyaet al., 2019) and find common peaks. ChIPseeker (Yu et al., 2015) R package was used for peak annotation and calculation of peak distances to TSS. Active enhancers in U2OS cells (n=6886) were determined as Enhancer Atlas 2.0 (Gao and Qian, 2020) U2OS enhancers not overlapping with any TSSs (+1 kb and −1 kb). Pausing index calculations to determine elongating genes (TOP1_mAID, n=406; TOP2A_mAID, n=636) were performed in python with custom scripts based on modified PIC software and in R.
Pausing Index (PI) of RNAPII
RNAPII pausing index is calculated to classify genes according to RNAPII recruitment (Zeitlinger et al., 2007)(Muse et al., 2007). The pausing index is the ratio of RNAPII reads density at the TSS over the average reads density in the gene body. At genes where the rate of promoter proximal pause release is similar to the rate of initiation, the PI is close to 1. At genes where promoter proximal pause release is lower than the initiation rate, the PI is higher than 1. The definition and thresholds used in this work were as in (Schones et al., 2008).
Gel filtration size exclusion chromatography
HeLa nuclear extracts were made according to (Dignam et al., 1983) and were fractionated at 4°C on a P11 column by step elution using BC buffers (20 mM Tris, pH.7.9, 10% glycerol, 0.2 mM EDTA, 0.2 mM DTT) containing 0.1 M, 0.3 M, 0.5 M and 1 M KCl. Pooled peak fractions were then dialyzed back into BC100 (BC buffer with 100 mM KCl). These four fractions were analyzed by westernblot for MYC, MYCN, TOP1, TOP2A, TOP2B and MAX. The 1 M fraction was then injected onto a Superose 6 Increase 10/300 column (Cytiva) and isocractically fractionated using BC100 using an AKTA Pure system. Calibration of Superose 6 Increase 10/300 column was performed with high molecular weight marker (calibration each fraction volume was 1 ml). Fractions were collected at 1% of the total column volume (each fraction volume was 500 μl), and were analyzed by western blot for the co-elution of MYC, TOP1, TOP2A, TOP2B and MAX.
Trypsin/LysC digestion
On-bead digestion was carried out using the EasyPep Mini MS Sample Prep Kit (Thermo Fisher, PN A40006). Beads were resuspended in 100 μl of EasyPep lysis buffer and treated with 50 μl of reduction and alkylation solutions provided with the kit and heated at 95°C for 10 min. Samples were allowed to cool to room temperature and then treated with 40 μl of 0.1 μg/μl trypsin/LysC (Pierce, A41007) and incubated at 37°C with shaking for 4 h. 50 μl of Stop Solution was added and samples were cleaned using EasyPep cleanup column and eluting in 300 μl of Elution Solution. Eluted sample were dried in speed-vac.
Liquid chromatography-mass spectrometry (LC/MS) analysis
Digested TOP1-IP peptides were resuspended in 30 μL of 0.1% formic acid and 6 μl was analyzed in triplicate on a Dionex U3000 RSLC in front of a Orbitrap Eclipse (Thermo Fisher) equipped with an EasySpray ion source and FAIMSTM. Peptides were first loaded onto the trap column (Specs) at a flow rate of 7 μl/min for 5 min then eluted from the analytical column (Specs) using a flow rate of 300 nl/min and a linear gradient of 5%–7% B for 1min, 7%–30% B for 34min, 30%–50% B for 15min, 50%–95% B for 4 min, holding at 95% B for 7min, then re-equilibration of analytical column at 5% B for 17min. Three injections were made utilizing different FAIMS compensation voltages (CVs) in each method: (−45, −60, −75), (−50, −65, −80), (−55, −70, −85). Each FAIMS voltage had a cycle time of 1 s and consisted of an MS1 scan at 120,000 resolution from 350–1600 m/z, with automatic gain control (AGC) of 4e5 ions and max injection time of 50ms. Each FAIMS voltage was operated using the TopSpeed method for MS2 acquisition which was carried out using High Collision induced Dissociation (HCD) fragmentation and rapid acquisition in the Ion Trap using a quadrupole isolation window of 1.6, an AGC of 1e4 ions, a max injection time of 35 ms. For all cycles Easy-IC was enabled for the MS1 scan, Monoisotopic Precursor Selection was enabled, HCD energy was set at 30%, Advanced Peak Determination was enabled, charge state selection was set from 2–5, dynamic exclusion was set to a count of 1 for 15 sec, and intensity threshold was set to 1e4 to trigger MS2 scan. Database search: the three acquisitions were searched together in Proteome Discoverer 2.4 using the Sequest node with Fixed Value PSM Validator. Data was searched against the Uniprot Human database from Feb 2020 using a fmll tryptic digest, 2 max missed cleavages, minimum peptide length of 6 amino acids, an MS1 mass tolerance of 10 ppm, MS2 mass tolerance of 0.6 Da, variable oxidation on methionine (+15.995 Da), and fixed carbamidomethyl on cysteine (+57.021 Da).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical testing
All statistical tests were performed using Graphpad Prism version 8 or 9 or statistical functions in R using the tests described for each experiment. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; *****p < 0.00001. Detailed information about statistical tests is provided in figure legends for respective figures.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Rabbit monoclonal Anti-Topoisomerase I antibody [EPR5375] | Abcam | Cat# ab109374; RRID: AB_10861978 |
| Mouse monoclonal Anti-Topo I Antibody (C-21) | Santa cruz Biotechnology | Cat# sc-32736; RRID: AB_628382 |
| Rabbit monoclonal Anti-Topoisomerase II alpha antibody [EP1102Y] | Abcam | Cat# ab52934; RRID: AB_883143 |
| Mouse monoclonal Anti-Topo IIα Antibody (E-10) | Santa cruz Biotechnology | Cat# sc-365799; RRID: AB_10842163 |
| Rabbit monoclonal Recombinant Anti-c-Myc antibody [Y69] | Abcam | Cat# ab32072; RRID: AB_731658 |
| Mouse monoclonal c-Myc antibody [9E11] - ChIP Grade | Abcam | Cat# ab56; RRID: AB_304976 |
| Mouse monoclonal c-Myc Antibody (9E10) | Santa Cruz Biotechnology | Cat# sc-40 AC; RRID: AB_2857941 |
| Rabbit c-Myc polyclonal Antibody | Novus biologicals | cat# NBP2–49201 |
| Mouse Anti-Human Max (H-2) Monoclonal, Unconjugated, Clone H-2 antibody | Santa Cruz Biotechnology | Cat# sc-8011; RRID: AB_627913 |
| Rabbit GFP Polyclonal Antibody | Thermo Fisher Scientific | Cat# A-11122; RRID: AB_221569 |
| Anti-C23 (Nucleolin) (MS-3) Antibody | Santa Cruz Biotechnology | Cat# sc-8031; RRID: AB_670271 |
| Mouse Anti-Human N-Myc(B8.4.B) Monoclonal, Unconjugated, Clone B8.4.b antibody | Santa Cruz Biotechnology | Cat# sc-53993; RRID: AB_831602 |
| Rabbit TOP2B Polyclonal antibody | Proteintech | Cat# 20549–1-AP; RRID: AB_10700004 |
| Anti-Ubiquitin Antibody (P4D1) | Santa Cruz Biotechnology | Cat# sc-8017; RRID: AB_628423 |
| Monoclonal ANTI-FLAG® M2 antibody | Sigma-Aldrich | Cat# F1804; RRID: AB_262044 |
| Anti-RNA polymerase II CTD repeat YSPTSPS (phospho S5) antibody [4H8] - ChIP Grade (ab5408) | Abcam | Cat# ab5408; RRID: AB_304868 |
| Mouse Anti-GAPDH Monoclonal Antibody | Abcam | Cat# ab9484; RRID: AB_307274 |
| normal mouse IgG antibody | Santa Cruz Biotechnology | Cat# sc-2025; RRID: AB_737182 |
| normal rabbit IgG antibody | Santa Cruz Biotechnology | Cat# sc-2027; RRID: AB_737197 |
| Goat Anti-Rabbit IgG H&L (HRP) antibody | Abcam | Cat# ab205718; RRID: AB_2819160 |
| Goat Anti-Mouse IgG H&L (HRP) antibody | Abcam | Cat# ab205719; RRID: AB_2755049 |
| Anti-Histone H3 antibody - Nuclear Loading Control and ChIP Grade, Abcam | Abcam | Cat# ab1791; RRID: AB_302613 |
| Rabbit anti-KAP-1 Antibody, Affinity Purified, Bethyl | Bethyl | Cat# A300–274A; RRID: AB_185559 |
| Spike-in antibody | Active motif | Cat# 61686; RRID: AB_2737370 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| One Shot TOP10 Chemically Competent E. coli | Thermo Fisher Scientific | Cat# C404010 |
| One Shot BL21 Star (DE3) Chemically Competent E. coli | Thermo Fisher Scientific | Cat# C601003 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| DMEM, high glucose | Thermo Fisher Scientific | Cat# 11965092 |
| RPMI 1640 Medium | Thermo Fisher Scientific | Cat# 21875034 |
| Opti-MEM I Reduced Serum Medium | Thermo Fisher Scientific | Cat# 31985062 |
| Fetal bovine serum for cell culture (tetracycline-free) | Takara | Cat# 631106 |
| Bovine Serum, heat inactivated, New Zealand origin | Thermo Fisher Scientific | Cat# 26170043 |
| Hygromycin B (50 mg/mL) | Thermo Fisher Scientific | Cat# 10687010 |
| Hygromycin B Gold | Invivogen | Cat# ant-hg-5 |
| Puromycin Dihydrochloride | Thermo Fisher Scientific | Cat# A1113803 |
| Penicillin-Streptomycin (10,000 U/mL) | GIBCO | Cat# 15140122 |
| Corning® 100 mL MEM Nonessential Amino Acids, 100x | Corning | Cat# 25–025-CI |
| cOmplete, Mini, EDTA-free Protease Inhibitor Cocktail | Milipore-Sigma | Cat# 11836170001 |
| 3-Indoleacetic acid (Auxin) | Milipore-Sigma | Cat# I3750–100MG-A |
| Auxinole CAS:86445–22–9 Purity: > 98% | Medchemexpress | Cat# HY11144410MG |
| Tamoxifen | Milipore-Sigma | Cat# T5648–1G |
| (R)-MG132 | Cayman chemicals | Cat# 13697 |
| Doxycycline hydrochloride | Milipore-Sigma | Cat# D3447–500MG |
| Benzonase® Nuclease, ultrapure ≥ 250 units/μL, ≥ 99% (SDS-PAGE) | Milipore-Sigma | Cat# E8263–25KU |
| Ni Sepharose® 6Fast Flow | Milipore-Sigma | Cat# GE17–5318-01 |
| Pierce Protein A/G Magnetic Beads | Thermo Fisher Scientific | Cat# 88803 |
| UltraPure Agarose | Thermo Fisher Scientific | Cat# 16500500 |
| Camptothecin | Milipore-Sigma | Cat# C9911–250MG |
| Etoposide synthetic, 98.0–105.0%, powder | Milipore-Sigma | Cat# E1383–250MG |
| FuGENE® HD Transfection Reagent | Promega | Cat# E2311 |
| Lambda Protein Phosphatase (Lambda PP) | New England Biolabs Inc. | Cat# P0753S |
| Triptolide from Tripterygium wilfordii, ≥ 98% (HPLC), solid | Milipore-Sigma | Cat# T3652–5MG |
| VECTASHIELD® Antifade Mounting Medium with DAPI | Vector laboratories | Cat# H-1200–10 |
| Diamond Nucleic Acid Dye | Promega | Cat# H1181 |
| Lipofectamine 2000 Transfection Reagent | Thermo Fisher Scientific | Cat# 11668019 |
| RNase A, DNase and protease-free (10 mg/mL) | Thermo Fisher Scientific | Cat# EN0531 |
| Proteinase K, Molecular Biology Grade | New England Biolabs Inc. | Cat# P8107S |
| Phenol:Chloroform:Isoamyl Alcohol 25:24:1 Saturated with 10 mM Tris, pH 8.0, 1 mM EDTA | Milipore-Sigma | Cat# P2069–400ML |
| Sodium dodecyl sulfate | Milipore-Sigma | Cat# L3771–100G |
| Lithium chloride anhydrous, free-flowing, Redi-Dri, ReagentPlus®, 99% | Milipore-Sigma | Cat# 793620–100G |
| Glycerol for molecular biology, ≥ 99.0% | Milipore-Sigma | Cat# G5516–1L |
| TWEEN® 20 for molecular biology, viscous liquid | Milipore-Sigma | Cat# P9416–50ML |
| Triton X-100 for molecular biology | Milipore-Sigma | Cat# T8787–50ML |
| N-Lauroylsarcosine sodium salt BioXtra, ≥ 97% (TLC) | Milipore-Sigma | Cat# L5777–50G |
| Sodium deoxycholate monohydrate BioXtra, ≥ 99.0% (titration) | Milipore-Sigma | Cat# D5670–25G |
| Urea ACS reagent, 99.0–100.5% | Milipore-Sigma | Cat# U5128–5KG |
| DTT 1,4-Dithiothreitol | Milipore-Sigma | Cat# 11583786001 |
| Nonidet P 40 Substitute | Milipore-Sigma | Cat# 74385 |
| Chloroquine diphosphate salt | Milipore-Sigma | Cat# C6628–25G |
| Ethylenediaminetetraacetic acid disodium salt dihydrate | Milipore-Sigma | Cat# 03685–500G |
| Potassium chloride for molecular biology, ≥ 99.0% | Milipore-Sigma | Cat# P9541–500G |
| AcquaStain Protein Gel Stain (Coomassie) | Bulldog Bio | Cat# AS001000 |
| UltraPure Ethidium Bromide, 10 mg/mL | Thermo Fisher Scientific | Cat# 15585011 |
| Exonuclease VII | New England Biolabs Inc. | Cat# M0379S |
| XhoI | New England Biolabs Inc. | Cat# R0146L |
| NdeI | New England Biolabs Inc. | Cat# R0111L |
| EcoRI | New England Biolabs Inc. | Cat# R0101L |
| BamHI | New England Biolabs Inc. | Cat# R0136L |
| IPTG ≥ 99% (TLC), ≤ 0.1% Dioxane | Milipore-Sigma | Cat# I6758–5G |
| Recombinant Human n-Myc/MYCN protein (His tag) | abcam | Cat# ab241520 |
| Recombinant Human Topoisomerase IIα | Topogen | Cat# TG2000H-4 |
| Recombinat Human TOPIIβ protein | This paper | N/A |
| Recombinant Human c-MYC/MYC protein | This paper | N/A |
| Recombinant Human c-MYC-MAX protein | This paper | N/A |
| Recombinat Human TOP1 protein | NCI core facility | N/A |
| Pro-Q Diamond Phosphoprotein Blot Stain Kit | Thermo Fisher Scientific | Cat# P33356 |
| Spike-in chromatin | Active Motif | Cat# 53083 |
| AMPure XP | Beckman | Cat# A63880 |
| Pierce Trypsin/Lys-C Protease Mix | Pierce | Cat# A41007 |
| GlycoBlue Coprecipitant | Thermo Fisher Scientific | Cat# AM9515 |
| Ibidi 15 m-slide angiogenesis slide | Ibidi-treat | Cat# 81506 |
| Pierce 16% Formaldehyde (w/v), Methanol-free | Thermo Fisher Scientific | Cat# 28906 |
| 4–12% Bis-Tris NuPAGE protein gel | Thermo Fisher Scientific | Cat# NP0323 |
| NuPAGE LDS Sample Buffer (4X) | Thermo Fisher Scientific | Cat# NP0007 |
| SYBR Green I Nucleic Acid Gel Stain - 10,000X concentrate in DMSO | Thermo Fisher Scientific | Cat# S7563 |
| SuperSignal West Femto Maximum Sensitivity Substrate | Thermo Fisher Scientific | Cat# 34095 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Duolink® In Situ Red Starter Kit Mouse/Rabbit | Milipore-Sigma | Cat# DUO92101–1KT |
| Thrombin cleavage kit | Abcam | Cat# ab207000 |
| Agilent High Sensitivity DNA Kit | Agilent | Cat# 5067–4626 |
| NextSeq 500/550 High Output Kit v2.5 (75 Cycles) | Illumina | Cat# 20024906 |
| QIAquick PCR Purification Kit | QIAGEN | Cat# 28106 |
| ThruPLEX DNA-seq kit | Takara | Cat# R400676 |
| Qubit 1X dsDNA HS Assay Kit | Thermo Fisher | Cat# Q33230 |
| Accel-NGS 2S Plus DNA Library Kit | Swift Biosciences | Cat# 21024 |
| Minielute PCR purification kit | QIAGEN | Cat# 28004 |
| EasyPep Mini MS Sample Prep Kit | Thermo Fisher Scientific | Cat# A40006 |
| NovaSeq 6000 S1 Reagent Kit v1.5 (100 cycles) | Illumina | Cat# 20028319 |
|
| ||
| Deposited data | ||
|
| ||
| Raw and processed sequencing data | This paper | GEO: GSE181450 |
| Previously published RNA sequencing data in U2OS cells | Ibarra et al., 2016 | GEO: GSE87831 |
| Previously published MYC ChIP-seq data from U2OS cells | Lorenzin et al., 2016 | GEO: GSM2050639 |
| Previously published RNA sequencing data from HCT116 cells | Baranello et al., 2016 | GEO: GSE57628 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| HCT116 | Developmental Therapeutics Programme | N/A |
| HCT116 FLAG-TOP2A | This paper | N/A |
| HCT116TOP1_mAID | (Wiegard et al., 2021) | N/A |
| HCT116TOP2A_mAID | gift from Dr. D.Hudson (MCRI, Australia) (Nielsen et al., 2020) | N/A |
| U2OS | Developmental Therapeutics Programme | N/A |
| U2OS-MYC-EGFP | Nie et al., 2020 | N/A |
| Rat HO15.19-MYC-ER | Nie et al., 2020 | N/A |
| K562 MYCmAIDTir1-eBFP2 (K562MYC_mAID) | gift from Dr. J. Zuber (IMP, Austria) (Muhar et al., 2018) | N/A |
|
| ||
| Oligonucleotides | ||
|
| ||
| Full-length MYC forward primer for cloning into pET-28a(+): 5′- CGATTCGATCATAT GCCCCTCAACGTTAGCTTC-3′ | IDT oligo | N/A |
| Full-length MYC reverse primer for cloning into pET-28a(+):5′- GGTACAATCCTCGAG TTACGCACAAGAGTTCCGTAG-3′ | IDT oligo | N/A |
| MYC (321–439) forward primer for cloning into pET-28a(+):5′-CGATTCGATCATAT GGCCAAGAGGGTCAAGTTG-3′ | IDT oligo | N/A |
| MYC (200–320) forward primer for cloning into Pd4 EGFP-N1: 5′-CGATTCGATGAATT CATGGACAGCAGCTCGCCCAAG-3′ | IDT oligo | N/A |
| MYC (200–320) reverse primer for cloning into Pd4 EGFP-N1: 5′- GGTACAATCGGAT CCAGCAGGATAGTCCTTCCG-3′ | IDT oligo | N/A |
| MYC (200–320) forward primer for cloning into pET-28a(+): 5′-CGATTCGATCATATG GACAGCAGCTCGCCCAAG-3′ | IDT oligo | N/A |
| MYC (200–320) reverse primer for cloning into pET-28a(+): 5′-GGTACAATCCTCG AGAGCAGGATAGTCCTTCCG-3′ | IDT oligo | N/A |
| 3xFLAG sequence (DYKDHDGDYK DHDIDYKDDDDKL) | IDT oligo | N/A |
| To insert mAID at the N terminus of the TOP1 gene using CRISPR/Cas, the sgRNA sequence (5′-CCCCACTCATGTCGGCCCGG-3′) | IDT oligo | N/A |
| TOP1-N_HA_5′_SacI for cloning into pBluescript II forward primer: 5′-ATGCGAGCTCGTTCCGAG AAAAAGCGTCTGGAGAG-3′ | IDT oligo | N/A |
| TOP1-N_HA_5′_KpnI for cloning into pBluescript II reverse primer:5′-ATGCGGTACCCCTTCC CTCTCTGGTGAAGTATGTG-3′ | IDT oligo | N/A |
| For inverse PCR using the forward primer: TOP1- N_HA_INV_F_BamHI: 5′-ATGCGGATCCATGAGTGGG GACCACCTCCACAACG-3′ | IDT oligo | N/A |
| For inverse PCR using the reverse primer:TOP1-N_HA_INV_R_EcoRV: 5′-ATGCAGATATCGTCGGCCC GGAGGGACGAGC-3′ | IDT oligo | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| pET-28 a (+) | Addgene (lab stock) | Cat# 69864–3 (EMD Biosciences) |
| Pd4 EGFP-N1 | Addgene (lab stock) | Cat# 6085–1 (Clontech) |
| pET-28 a(+)-c-MYC (FL) | This paper | N/A |
| pET-28 a(+)-c-MYC (200–320) | This paper | N/A |
| Pd4 EGFP-N1-c-MYC (FL) | Nie etal., 2012 | N/A |
| Pd4 EGFP-N1-c-MYC (200–320) | This paper | N/A |
| Kinetoplast DNA (kDNA) | Topogen | Cat# TG2013–3 |
| Gal1/10 His6 TEV Ura S. cerevisiae expression vector (12URA-B) | Gift from Scott Gradia (Addgene plasmid) | Cat#: 48304(Addgene); RRID: Addgene_48304 |
| pX330-U6-Chimeric_BB-CBh-hSpCas9 | Cong et al., 2013; PMID: 23287718 | Cat#: 42230(Addgene); RRID: Addgene_42230 |
|
| ||
| Software and algorithms | ||
|
| ||
| GraphPad Prism 9.1.0 (software for graph and statistics analysis | GraphPad | https://www.graphpad.com/scientific-software/prism/; RRID: SCR_002798 |
| Adobe acrobat (illustrator for preparation of manuscript) | Adobe | https://www.adobe.com/products/illustrator; RRID: SCR_010279 |
| Fiji | Schindelin et al., 2012 | https://fiji.sc/; RRID: SCR_002285 |
| ImageJ Fiji macros for quantification of PLA dots | This paper | https://sites.imageJ.net/Janek/ |
| Bowtie2 2.3.5.1 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml; RRID: SCR_016368 |
| Deeptools 2.5.0 | (Ramirez and Dundar, 2014) | https://deeptools.readthedocs.io/en/develop/; RRID: SCR_016366 |
| FastQC 0.11.9 | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ | https://github.com/s-andrews/FastQC; RRID: SCR_014583 |
| MultiQC 1.10 | Ewels et al., 2016 | https://multiqc.info/; RRID: SCR_014982 |
| Picard tools 2.10.3 | Broad Institute | https://broadinstitute.github.io/picard/; RRID: SCR_006525 |
| Samtools 1.8 | Li et al., 2009 | http://www.htslib.org/; RRID: SCR_002105 |
| R 4.1.0 | R Core Team (2021). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. | http://www.r-project.org/; RRID: SCR_001905 |
| Bioconductor | Huber et al., 2015 | https://www.bioconductor.org/; RRID: SCR_006442 |
| Cutadapt 3.1 | http://journal.embnet.org/index.php/embnetjournal/article/view/200 | https://cutadapt.readthedocs.io/en/stable/; RRID: SCR_011841 |
| UCSC Genome Browser | (Kent and Sugnet, 2002) | http://genome.ucsc.edu/; RRID: SCR_005780 |
| Python 2.7.18 | G. van Rossum, Python tutorial, Technical Report CS-R9526, Centrum voor Wiskunde en Informatica (CWI), Amsterdam, May 1995. | https://www.python.org/; RRID: SCR_008394 |
| PIC 1.0 | (Day and Zhang, 2016) | https://github.com/MiMiroot/PIC |
| ggplot2 | https://wires.onlinelibrary.wNey.com/doi/10.1002/wics.147 | https://cran.r-project.org/web/packages/ggplot2/index.html; RRID: SCR_014601 |
| ngsplot 2.61 | Shen et al., 2014 | https://github.com/shenlab-sinai/ngsplot; RRID: SCR_011795 |
| MACS3 3.0.0a6 | Zhang et al., 2008 | https://github.com/macs3-project/MACS; RRID: SCR_013291 |
| EPIC2 0.0.44 | Stovner and Ŝtrom, 2019 | https://github.com/biocore-ntnu/epic2 |
| Bedtools 2.27.1 | (Quinlan and Hall, 2010) | https://bedtools.readthedocs.io/en/latest/index.html; RRID: SCR_006646 |
| ChIPseeker 1.28.3 | Yu etal., 2015 | https://bioconductor.org/packages/release/bioc/html/ChIPseeker.html; RRID: SCR_021322 |
| Enhancer Atlas 2.0 | (Gao and Qian, 2020) | http://www.enhanceratlas.org/ |
| Proteome Discoverer 2.4 | Thermo fisher |
https://www.thermofisher.com/se/en/home/industrial/mass-spectrometry/liquid-chromatography-mass-spectrometry-lc-ms/lc-ms-software/multi-omics-data-analysis/proteome-discoverer-software.html; RRID: SCR_014477 |
Highlights.
MYC manages high levels of torsional stress during transcription and replication
Both topoisomerase 1 and 2 activities are stimulated by MYC inside cells
The MYC nucleated "topoisome" harbors TOP1 and TOP2 and is activated at promoters
Inclusion of TOP2A versus TOP2B in a topoisome is decided by MYC versus MYCN, respectively
ACKNOWLEDGMENTS
This work was supported by grants from the Wallenberg foundation (KAW 2016.0161) (L.B.), the Swedish Research Council (2016-02610 VR) (L.B.), the Swedish Governmental Agency for Innovation Systems (2016-02055 VINNOVA) (L.B.), Cancerfonden (180608) (L.B.), and the Intramural Research Programs of the National Cancer Institute (Center for Cancer Research) (D.L.). We thank Prof. Eilers and Dr. Zuber for sharing reagents and helpful advice. We thank Dr. Hudson, Dr. Nielsen and Dr. Canela for sharing reagents and protocols. We thank Dr. Ballachanda and Dr. Lamour for helpful advice and sharing protocols and Dr. Wiegard for help in some experiments. We thank Dr. Kutter and Ms. Geng for technical expertise with the sequencing machine. The computations and data storage were enabled by resources in project [SNIC 2018/8-390], provided by the Swedish National Infrastructure for Computing (SNIC) at UPPMAX, partially funded by the Swedish Research Council through grant agreement no. 2018-05973. Part of this work was facilitated by the Protein Science Facility at Karolinska Institutet/SciLifeLab (https://ki.se/psf) and the Karolinska Genome Engineering Facility (KGE). Part of this work was carried out at the KGE. Cross-CAD-seq libraries were prepared and/or sequenced by the NCI Sequencing Facility and Genomics Core. The graphical abstract was created with BioRender.com.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.molcel.2021.11.016.
INCLUSION AND DIVERSITY
While citing references scientifically relevant for this work, we also actively worked to promote gender balance in our reference list.
REFERENCES
- Aguilera A, and García-Muse T (2012). R loops: from transcription byproducts to threats to genome stability. Mol. Cell 46, 115–124. [DOI] [PubMed] [Google Scholar]
- Amemiya HM, Kundaje A, and Boyle AP (2019). The ENCODE Blacklist: Identification of Problematic Regions of the Genome. Sci. Rep 9, 9354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arabi A, Wu S, Ridderstråle K, Bierhoff H, Shiue C, Fatyol K, Fahlén S, Hydbring P, Söderberg O, Grummt I, et al. (2005). c-Myc associates with ribosomal DNA and activates RNA polymerase I transcription. Nat. Cell Biol 7, 303–310. [DOI] [PubMed] [Google Scholar]
- Baluapuri A, Wolf E, and Eilers M (2020). Target gene-independent functions of MYC oncoproteins. Nat. Rev. Mol. Cell Biol 21, 255–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baranello L, Levens D, Gupta A, and Kouzine F (2012). The importance of being supercoiled: how DNA mechanics regulate dynamic processes. Biochim. Biophys. Acta 1819, 632–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baranello L, Kouzine F, and Levens D (2013). DNA topoisomerases beyond the standard role. Transcription 4, 232–237. [DOI] [PubMed] [Google Scholar]
- Baranello L, Wojtowicz D, Cui K, Devaiah BN, Chung HJ, Chan-Salis KY, Guha R, Wilson K, Zhang X, Zhang H, et al. (2016). RNA Polymerase II Regulates Topoisomerase 1 Activity to Favor Efficient Transcription. Cell 165, 357–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedez C, Lotz C, Batisse C, Broeck AV, Stote RH, Howard E, Pradeau-Aubreton K, Ruff M, and Lamour V (2018). Post-translational modifications in DNA topoisomerase 2α highlight the role of a eukaryote-specific residue in the ATPase domain. Sci. Rep 8, 9272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biersack H, Jensen S, Gromova I, Nielsen IS, Westergaard O, and Andersen AH (1996). Active heterodimers are formed from human DNA topoisomerase II alpha and II beta isoforms. Proc. Natl. Acad. Sci. USA 93, 8288–8293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackwood EM, and Eisenman RN (1991). Max: a helix-loop-helix zipper protein that forms a sequence-specific DNA-binding complex with Myc. Science 251, 1211–1217. [DOI] [PubMed] [Google Scholar]
- Büchel G, Carstensen A, Mak KY, Roeschert I, Leen E, Sumara O, Hofstetter J, Herold S, Kalb J, Baluapuri A, et al. (2017). Association with Aurora-A Controls N-MYC-Dependent Promoter Escape and Pause Release of RNA Polymerase II during the Cell Cycle. Cell Rep 21, 3483–3497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron DP, Kuzin V, and Baranello L (2021). Analysis of Myc Chromatin Binding by Calibrated ChIP-Seq Approach. Methods Mol. Biol 2318, 161–185. [DOI] [PubMed] [Google Scholar]
- Chong S, Chen C, Ge H, and Xie XS (2014). Mechanism of transcriptional bursting in bacteria. Cell 158, 314–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, and Zhang F (2013). Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang CV (2012). MYC on the path to cancer. Cell 149, 22–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das BB, Sen N, Ganguly A, and Majumder HK (2004). Reconstitution and functional characterization of the unusual bi-subunit type I DNA topoisomerase from Leishmania donovani. FEBS Lett 565, 81–88. [DOI] [PubMed] [Google Scholar]
- Das SK, Rehman I, Ghosh A, Sengupta S, Majumdar P, Jana B, and Das BB (2016). Poly(ADP-ribose) polymers regulate DNA topoisomerase I (Top1) nuclear dynamics and camptothecin sensitivity in living cells. Nucleic Acids Res 44, 8363–8375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das SK, Ghosh A, Paul Chowdhuri S, Halder N, Rehman I, Sengupta S, Sahoo KC, Rath H, and Das BB (2018). Neutral Porphyrin Derivative Exerts Anticancer Activity by Targeting Cellular Topoisomerase I (Top1) and Promotes Apoptotic Cell Death without Stabilizing Top1-DNA Cleavage Complexes. J. Med. Chem 61, 804–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day D, Zhang B, et al. (2016). Comprehensive analysis of promoter-proximal RNA polymerase II pausing across mammalian cell types. Genome Biology 17 (120). 10.1186/s13059-016-0984-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dignam JD, Lebovitz RM, and Roeder RG (1983). Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res 11, 1475–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez-Sola D, Ying CY, Grandori C, Ruggiero L, Chen B, Li M, Galloway DA, Gu W, Gautier J, and Dalla-Favera R (2007). Non-transcriptional control of DNA replication by c-Myc. Nature 448, 445–451. [DOI] [PubMed] [Google Scholar]
- Drolet M, Bi X, and Liu LF (1994). Hypernegative supercoiling of the DNA template during transcription elongation in vitro. J. Biol. Chem 269, 2068–2074. [PubMed] [Google Scholar]
- Ewels P, Magnusson M, Lundin S, and Käller M(2016). MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32, 3047–3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez PC, Frank SR, Wang L, Schroeder M, Liu S, Greene J, Cocito A, and Amati B (2003). Genomic targets of the human c-Myc protein. Genes Dev 17, 1115–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flicek P, Amode MR, Barrell D, Beal K, Billis K, Brent S, Carvalho-Silva D, Clapham P, Coates G, Fitzgerald S, et al. (2014). Ensembl 2014. Nucleic Acids Res 42, D749–D755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallant P, and Steiger D (2009). Myc’s secret life without Max. Cell Cycle 8, 3848–3853. [DOI] [PubMed] [Google Scholar]
- Gao T, and Qian J (2020). EnhancerAtlas 2.0: an updated resource with enhancer annotation in 586 tissue/cell types across nine species. Nucleic Acids Res 48 (D1), D58–D64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge H, Martinez E, Chiang CM, and Roeder RG (1996). Activator-dependent transcription by mammalian RNA polymerase II: in vitro reconstitution with general transcription factors and cofactors. Methods Enzymol 274, 57–71. [DOI] [PubMed] [Google Scholar]
- Gerlach JM, Furrer M, Gallant M, Birkel D, Baluapuri A, Wolf E, and Gallant P (2017). PAF1 complex component Leo1 helps recruit Drosophila Myc to promoters. Proc. Natl. Acad. Sci. USA 114, E9224–E9232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gittens WH, Johnson DJ, Allison RM, Cooper TJ, Thomas H, and Neale MJ (2019). A nucleotide resolution map of Top2-linked DNA breaks in the yeast and human genome. Nat. Commun 10, 4846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Roman N, Grandori C, Eisenman RN, and White RJ (2003). Direct activation of RNA polymerase III transcription by c-Myc. Nature 421, 290–294. [DOI] [PubMed] [Google Scholar]
- Gromova I, Biersack H, Jensen S, Nielsen OF, Westergaard O, and Andersen AH (1998). Characterization of DNA topoisomerase II alpha/beta heterodimers in HeLa cells. Biochemistry 37, 16645–16652. [DOI] [PubMed] [Google Scholar]
- Guo J, Li T, Schipper J, Nilson KA, Fordjour FK, Cooper JJ, Gordân R, and Price DH (2014). Sequence specificity incompletely defines the genome-wide occupancy of Myc. Genome Biol 15, 482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrero-Ruiz A, Martínez-García PM, Terroón-Bautista J, Millán-Zambrano G, Lieberman JA, Jimeno-González S, and Cortés-Ledesma F (2021). Topoisomerase IIa represses transcription by enforcing promoter-proximal pausing. Cell Rep 35, 108977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopewell R, and Ziff EB (1995). The nerve growth factor-responsive PC12 cell line does not express the Myc dimerization partner Max. Mol. Cell. Biol 15, 3470–3478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber W, Carey VJ, Gentleman R, Anders S, Carlson M, Carvalho BS, Bravo HC, Davis S, Gatto L, Girke T, et al. (2015). Orchestrating high-throughput genomic analysis with Bioconductor. Nat. Methods 12, 115–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibarra A, Benner C, Tyagi S, Cool J, and Hetzer MW (2016). Nucleoporin-mediated regulation of cell identity genes. Genes Dev 30, 2253–2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi RS, Piña B, and Roca J (2012). Topoisomerase II is required for the production of long Pol II gene transcripts in yeast. Nucleic Acids Res 40, 7907–7915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju BG, Lunyak VV, Perissi V, Garcia-Bassets I, Rose DW, Glass CK, and Rosenfeld MG (2006). A topoisomerase IIbeta-mediated dsDNA break required for regulated transcription. Science 312, 1798–1802. [DOI] [PubMed] [Google Scholar]
- Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Žídek A, Potapenko A, et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalkat M, Resetca D, Lourenco C, Chan PK, Wei Y, Shiah YJ, Vitkin N, Tong Y, Sunnerhagen M, Done SJ, et al. (2018). MYC Protein Interactome Profiling Reveals Functionally Distinct Regions that Cooperate to Drive Tumorigenesis. Mol. Cell 72, 836–848.e7. [DOI] [PubMed] [Google Scholar]
- Kegel A, Betts-Lindroos H, Kanno T, Jeppsson K, Ström L, Katou Y, Itoh T, Shirahige K, and Sjögren C (2011). Chromosome length influences replication-induced topological stress. Nature 471, 392–396. [DOI] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, et al. (2002). The Human Genome Browser at UCSC. Genome Research 12, 996–1006. 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiianitsa K, and Maizels N (2013). A rapid and sensitive assay for DNA-protein covalent complexes in living cells. Nucleic Acids Res 41, e104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King IF, Yandava CN, Mabb AM, Hsiao JS, Huang HS, Pearson BL, Calabrese JM, Starmer J, Parker JS, Magnuson T, et al. (2013). Topoisomerases facilitate transcription of long genes linked to autism. Nature 501, 58–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoepfler PS, Zhang XY, Cheng PF, Gafken PR, McMahon SB, and Eisenman RN (2006). Myc influences global chromatin structure. EMBO J 25, 2723–2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch HB, Zhang R, Verdoodt B, Bailey A, Zhang CD, Yates JR 3rd, Menssen A, and Hermeking H (2007). Large-scale identification of c-MYC-associated proteins using a combined TAP/MudPIT approach. Cell Cycle 6, 205–217. [DOI] [PubMed] [Google Scholar]
- Kouzine F, Sanford S, Elisha-Feil Z, and Levens D (2008). The functional response of upstream DNA to dynamic supercoiling in vivo. Nat. Struct. Mol. Biol 15, 146–154. [DOI] [PubMed] [Google Scholar]
- Kouzine F, Gupta A, Baranello L, Wojtowicz D, Ben-Aissa K, Liu J, Przytycka TM, and Levens D (2013). Transcription-dependent dynamic supercoiling is a short-range genomic force. Nat. Struct. Mol. Biol 20, 396–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzine F, Wojtowicz D, Baranello L, Yamane A, Nelson S, Resch W, Kieffer-Kwon KR, Benham CJ, Casellas R, Przytycka TM, and Levens D (2017). Permanganate/S1 Nuclease Footprinting Reveals Non-B DNA Structures with Regulatory Potential across a Mammalian Genome. Cell Syst 4, 344–356.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundu B, Das SK, Paul Chowdhuri S, Pal S, Sarkar D, Ghosh A, Mukherjee A, Bhattacharya D, Das BB, and Talukdar A (2019). Discovery and Mechanistic Study of Tailor-Made Quinoline Derivatives as Topoisomerase 1 Poison with Potent Anticancer Activity. J. Med. Chem 62, 3428–3446. [DOI] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le TT, Gao X, Park SH, Lee J, Inman JT, Lee JH, Killian JL, Badman RP, Berger JM, and Wang MD (2019). Synergistic Coordination of Chromatin Torsional Mechanics and Topoisomerase Activity. Cell 179, 619–631.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Wendorff TJ, and Berger JM (2017). Resveratrol: A novel type of topoisomerase II inhibitor. J. Biol. Chem 292, 21011–21022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CY, Lovén J, Rahl PB, Paranal RM, Burge CB, Bradner JE, Lee TI, and Young RA (2012). Transcriptional amplification in tumor cells with elevated c-Myc. Cell 151, 56–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Chen SS, Clarke S, Veschi V, and Thiele CJ (2021). Targeting MYCN in Pediatric and Adult Cancers. Front. Oncol 10, 623679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenzin F, Benary U, Baluapuri A, Walz S, Jung LA, von Eyss B, Kisker C, Wolf J, Eilers M, and Wolf E (2016). Different promoter affinities account for specificity in MYC-dependent gene regulation. eLife 5, e15161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Bai L, and Wang MD (2013). Transcription under torsion. Science 340, 1580–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madabhushi R, Gao F, Pfenning AR, Pan L, Yamakawa S, Seo J, Rueda R, Phan TX, Yamakawa H, Pao PC, et al. (2015). Activity-Induced DNA Breaks Govern the Expression of Neuronal Early-Response Genes. Cell 161, 1592–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldonado E, Drapkin R, and Reinberg D (1996). Purification of human RNA polymerase II and general transcription factors. Methods Enzymol 274, 72–100. [DOI] [PubMed] [Google Scholar]
- McClendon AK, Rodriguez AC, and Osheroff N (2005). Human topoisomerase IIalpha rapidly relaxes positively supercoiled DNA: implications for enzyme action ahead of replication forks. J. Biol. Chem 280, 39337–39345. [DOI] [PubMed] [Google Scholar]
- Merino A, Madden KR, Lane WS, Champoux JJ, and Reinberg D (1993). DNA topoisomerase I is involved in both repression and activation of transcription. Nature 365, 227–232. [DOI] [PubMed] [Google Scholar]
- Miao ZH, Player A, Shankavaram U, Wang YH, Zimonjic DB, Lorenzi PL, Liao ZY, Liu H, Shimura T, Zhang HL, et al. (2007). Nonclassic functions of human topoisomerase I: genome-wide and pharmacologic analyses. Cancer Res 67, 8752–8761. [DOI] [PubMed] [Google Scholar]
- Muhar M, Ebert A, Neumann T, Umkehrer C, Jude J, Wieshofer C, Rescheneder P, Lipp JJ, Herzog VA, Reichholf B, et al. (2018). SLAM-seq defines direct gene-regulatory functions of the BRD4-MYC axis. Science 360, 800–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muse GW, Gilchrist DA, Nechaev S, Shah R, Parker JS, Grissom SF, Zeitlinger J, and Adelman K (2007). RNA polymerase is poised for activation across the genome. Nat. Genet 39, 1507–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natsume T, Kiyomitsu T, Saga Y, and Kanemaki MT (2016). Rapid Protein Depletion in Human Cells by Auxin-Inducible Degron Tagging with Short Homology Donors. Cell Rep 15, 210–218. [DOI] [PubMed] [Google Scholar]
- Neguembor MV, Martin L, Castells-García Á, Gómez-Garciía PA, Vicario C, Carnevali D, AlHaj Abed J, Granados A, Sebastian-Perez R, Sottile F, et al. (2021). Transcription-mediated supercoiling regulates genome folding and loop formation. Mol. Cell 81, 3065–3081.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie Z, Hu G, Wei G, Cui K, Yamane A, Resch W, Wang R, Green DR, Tessarollo L, Casellas R, et al. (2012). c-Myc is a universal amplifier of expressed genes in lymphocytes and embryonic stem cells. Cell 151, 68–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie Z, Guo C, Das SK, Chow CC, Batchelor E, Simons SS Jnr, and Levens D (2020). Dissecting transcriptional amplification by MYC. eLife 9, e52483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen CF, Zhang T, Barisic M, Kalitsis P, and Hudson DF (2020). Topoisomerase IIa is essential for maintenance of mitotic chromosome structure. Proc. Natl. Acad. Sci. USA 117, 12131–12142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitiss JL, Soans E, Rogojina A, Seth A, and Mishina M (2012). Topoisomerase assays. Curr. Protoc. Pharmacol. Chapter 3. Unit 3.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connell BC, Cheung AF, Simkevich CP, Tam W, Ren X, Mateyak MK, and Sedivy JM (2003). A large scale genetic analysis of c-Myc-regulated gene expression patterns. J. Biol. Chem 278, 12563–12573. [DOI] [PubMed] [Google Scholar]
- Pommier Y (2013). Drugging topoisomerases: lessons and challenges. ACS Chem. Biol 8, 82–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pommier Y, Kerrigan D, Hartman KD, and Glazer RI (1990). Phosphorylation of mammalian DNA topoisomerase I and activation by protein kinase C. J. Biol. Chem 265, 9418–9422. [PubMed] [Google Scholar]
- Poortinga G, Hannan KM, Snelling H, Walkley CR, Jenkins A, Sharkey K, Wall M, Brandenburger Y, Palatsides M, Pearson RB, et al. (2004). MAD1 and c-MYC regulate UBF and rDNA transcription during granulocyte differentiation. EMBO J 23, 3325–3335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puc J, Kozbial P, Li W, Tan Y, Liu Z, Suter T, Ohgi KA, Zhang J, Aggarwal AK, and Rosenfeld MG (2015). Ligand-dependent enhancer activation regulated by topoisomerase-I activity. Cell 160, 367–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahl PB, Lin CY, Seila AC, Flynn RA, McCuine S, Burge CB, Sharp PA, and Young RA (2010). c-Myc regulates transcriptional pause release. Cell 141, 432–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F, Dündar F, et al. (2014). deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res 42 (W1), 187–191. 10.1093/nar/gku365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray S, Panova T, Miller G, Volkov A, Porter AC, Russell J, Panov KI, and Zomerdijk JC (2013). Topoisomerase IIa promotes activation of RNA polymerase I transcription by facilitating pre-initiation complex formation. Nat. Commun 4, 1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickman DS, Schulte JH, and Eilers M (2018). The Expanding World of N-MYC-Driven Tumors. Cancer Discov 8, 150–163. [DOI] [PubMed] [Google Scholar]
- Rodriguez J, and Larson DR (2020). Transcription in Living Cells: Molecular Mechanisms of Bursting. Annu. Rev. Biochem 89, 189–212. [DOI] [PubMed] [Google Scholar]
- Sabari BR, Dall’Agnese A, Boija A, Klein IA, Coffey EL, Shrinivas K, Abraham BJ, Hannett NM, Zamudio AV, Manteiga JC, et al. (2018). Coactivator condensation at super-enhancers links phase separation and gene control. Science 361, eaar3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schalbetter SA, Mansoubi S, Chambers AL, Downs JA, and Baxter J (2015). Fork rotation and DNA precatenation are restricted during DNA replication to prevent chromosomal instability. Proc. Natl. Acad. Sci. USA 112, E4565–E4570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schones DE, Cui K, Cuddapah S, Roh TY, Barski A, Wang Z, Wei G, and Zhao K (2008). Dynamic regulation of nucleosome positioning in the human genome. Cell 132, 887–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciascia N, Wu W, Zong D, Sun Y, Wong N, John S, Wangsa D, Ried T, Bunting SF, Pommier Y, and Nussenzweig A (2020). Suppressing proteasome mediated processing of topoisomerase II DNA-protein complexes preserves genome integrity. eLife 9, e53447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Secombe J, Li L, Carlos L, and Eisenman RN (2007). The Trithorax group protein Lid is a trimethyl histone H3K4 demethylase required for dMyc-induced cell growth. Genes Dev 21, 537–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheinin MY, Li M, Soltani M, Luger K, and Wang MD (2013). Torque modulates nucleosome stability and facilitates H2A/H2B dimer loss. Nat. Commun 4, 2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L, Shao N, Liu X, and Nestler E (2014). ngs.plot: Quick mining and visualization of next-generation sequencing data by integrating genomic databases. BMC Genomics 15, 284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shykind BM, Kim J, Stewart L, Champoux JJ, and Sharp PA (1997). Topoisomerase I enhances TFIID-TFIIA complex assembly during activation of transcription. Genes Dev 11, 397–407. [DOI] [PubMed] [Google Scholar]
- Singh J, and Padgett RA (2009). Rates of in situ transcription and splicing in large human genes. Nat. Struct. Mol. Biol 16, 1128–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark C, Breitkreutz BJ, Reguly T, Boucher L, Breitkreutz A, and Tyers M (2006). BioGRID: a general repository for interaction datasets. Nucleic Acids Res 34, D535–D539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stovner EB, and Sætrom P (2019). epic2 efficiently finds diffuse domains in ChIP-seq data. Bioinformatics 35, 4392–4393. [DOI] [PubMed] [Google Scholar]
- Teves SS, and Henikoff S (2014). Transcription-generated torsional stress destabilizes nucleosomes. Nat. Struct. Mol. Biol 21, 88–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakurela S, Garding A, Jung J, Schübeler D, Burger L, and Tiwari VK (2013). Gene regulation and priming by topoisomerase IIa in embryonic stem cells. Nat. Commun 4, 2478. [DOI] [PubMed] [Google Scholar]
- Tiwari VK, Burger L, Nikoletopoulou V, Deogracias R, Thakurela S, Wirbelauer C, Kaut J, Terranova R, Hoerner L, Mielke C, et al. (2012). Target genes of Topoisomerase IIb regulate neuronal survival and are defined by their chromatin state. Proc. Natl. Acad. Sci. USA 109, E934–E943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uusküla-Reimand L, Hou H, Samavarchi-Tehrani P, Rudan MV, Liang M, Medina-Rivera A, Mohammed H, Schmidt D, Schwalie P, Young EJ, et al. (2016). Topoisomerase II beta interacts with cohesin and CTCF at topological domain borders. Genome Biol 17, 182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JC (2002). Cellular roles of DNA topoisomerases: a molecular perspective. Nat. Rev. Mol. Cell Biol 3, 430–440. [DOI] [PubMed] [Google Scholar]
- Wei Y, Resetca D, Li Z, Johansson-Åkhe I, Ahlner A, Helander S, Wallenhammar A, Morad V, Raught B, Wallner B, et al. (2019). Multiple direct interactions of TBP with the MYC oncoprotein. Nat. Struct. Mol. Biol 26, 1035–1043. [DOI] [PubMed] [Google Scholar]
- Wiegard A, Kuzin V, Cameron DP, et al. (2021). Topoisomerase 1 activity during mitotic transcription favors the transition from mitosis to G1. Mol Cell 10.1016/j.molcel.2021.10.015. [DOI] [PubMed] [Google Scholar]
- Wu HY, Shyy SH, Wang JC, and Liu LF (1988). Transcription generates positively and negatively supercoiled domains in the template. Cell 53, 433–440. [DOI] [PubMed] [Google Scholar]
- Yesbolatova A, Natsume T, Hayashi KI, and Kanemaki MT (2019). Generation of conditional auxin-inducible degron (AID) cells and tight control of degron-fused proteins using the degradation inhibitor auxinole. Methods 164–165, 73–80. [DOI] [PubMed] [Google Scholar]
- Yu G, Wang LG, and He QY (2015). ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 31, 2382–2383. [DOI] [PubMed] [Google Scholar]
- Zeitlinger J, Stark A, Kellis M, Hong JW, Nechaev S, Adelman K, Levine M, and Young RA (2007). RNA polymerase stalling at developmental control genes in the Drosophila melanogaster embryo. Nat. Genet 39, 1512–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Chambers W, Sciascia S, and Cuadrado MJ (2016). Emerging therapies in systemic lupus erythematous: from clinical trial to the real life. Expert Rev. Clin. Pharmacol 9, 681–694. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The deep sequencing data reported in this paper were deposited in the GEO database and are publicly available as of the date of publication. Previously published RNA-seq data, RNAPII and MYC ChIP-seq data from HCT116 and U2OS cells (Lorenzin et al., 2016; Baranello et al., 2016; Ibarra et al., 2016) are publicly available in GEO. Accession numbers of all aforementioned datasets are listed in the Key resource table.
This paper does not report original code since it is based on implementation of publicly available software. Published software and code used in this study are listed in the Key resource table. Any additional information required to reanalyze the data reported in this paper is available from the lead contacts upon request.