

Abstract
Background
The aim of this study was to assess the associations of modifiable lifestyle factors (smoking, coffee consumption, sleep, and physical activity) and cardiometabolic factors (body mass index, glycemic traits, type 2 diabetes, systolic and diastolic blood pressure, lipids, and inflammation and kidney function markers) with risks of any (ruptured or unruptured) intracranial aneurysm and aneurysmal subarachnoid hemorrhage using Mendelian randomization.
Methods and Results
Summary statistical data for the genetic associations with the modifiable risk factors and the outcomes were obtained from meta‐analyses of genome‐wide association studies. The inverse‐variance weighted method was used as the main Mendelian randomization analysis, with additional sensitivity analyses conducted using methods more robust to horizontal pleiotropy. Genetic predisposition to smoking, insomnia, and higher blood pressure was associated with an increased risk of both intracranial aneurysm and aneurysmal subarachnoid hemorrhage. For intracranial aneurysm, the odds ratios were 3.20 (95% CI, 1.93–5.29) per SD increase in smoking index, 1.24 (95% CI, 1.10–1.40) per unit increase in log‐odds of insomnia, and 2.92 (95% CI, 2.49–3.43) per 10 mm Hg increase in diastolic blood pressure. In addition, there was weak evidence for associations of genetically predicted decreased physical activity, higher triglyceride levels, higher body mass index, and lower low‐density lipoprotein cholesterol levels with higher risk of intracranial aneurysm and aneurysmal subarachnoid hemorrhage, with 95% CI overlapping the null for at least 1 of the outcomes. All results were consistent in sensitivity analyses.
Conclusions
This Mendelian randomization study suggests that smoking, insomnia, and high blood pressure are major risk factors for intracranial aneurysm and aneurysmal subarachnoid hemorrhage.
Keywords: intracranial aneurysm, lifestyle, Mendelian randomization, risk factors, single‐nucleotide polymorphisms, subarachnoid hemorrhage
Subject Categories: Lifestyle, Obesity, Risk Factors, High Blood Pressure, Genetics, Intracranial Hemorrhage
Nonstandard Abbreviations and Acronyms
- aSAH
aneurysmal subarachnoid hemorrhage
- IA
intracranial aneurysm
- IVW
inverse‐variance weighted
- MR
Mendelian randomization
Clinical Perspective
What Is New?
We performed Mendelian randomization to investigate the effect of modifiable lifestyle and cardiometabolic risk factors on risk of any (ruptured or unruptured) intracranial aneurysm (IA) and aneurysmal subarachnoid hemorrhage (aSAH).
Genetic predisposition to smoking, insomnia, and higher blood pressure were associated with an increased risk of both IA and aSAH.
In addition, there was weak evidence for associations of genetically predicted decreased physical activity, higher triglyceride levels, higher body mass index, and lower low‐density lipoprotein cholesterol levels with higher risk of IA and aSAH.
What Are the Clinical Implications?
Smoking, insomnia, and high blood pressure likely represent causal risk factors for IA and aSAH.
These results add to the body of evidence on causal risk factors for IA and aSAH, and warrant further investigation towards identifying preventative and therapeutic opportunities.
Aneurysmal subarachnoid hemorrhage (aSAH) is a type of stroke that is often caused by the rupture of an intracranial aneurysm (IA) and is associated with high mortality and morbidity. 1 , 2 In light of poor outcomes of aSAH, identification of modifiable risk factors for IA formation and rupture is of great importance. Suggested risk factors for aSAH include smoking, 3 , 4 , 5 heavy alcohol consumption, 3 , 6 , 7 hypertension, 3 , 4 and sleep apnea, 8 whereas coffee consumption, 9 regular physical activity, 10 , 11 , 12 high body mass index, 4 , 13 diabetes, 3 and hypercholesterolemia 3 , 14 have been proposed as risk‐reducing factors. In addition, impaired kidney function and chronic inflammation can damage the vascular endothelium, predisposing to IA and aSAH. 15 However, available data on risk factors for aSAH are mainly based on observational studies that are vulnerable to confounding and other biases, and the results are not conclusive. Hence, the causal associations of most modifiable risk factors other than smoking and high blood pressure with aSAH risk remain unestablished.
Mendelian randomization (MR) is an epidemiologic method that uses randomly allocated genetic variants related to the risk factor as instrumental variables to infer causality of the exposure–outcome relationship. 16 Here, we conducted a 2‐sample MR study to assess the associations of genetically predicted modifiable lifestyle factors (smoking, alcohol and coffee consumption, sleep, and physical activity) and cardiometabolic factors (body mass index, glycemic traits and type 2 diabetes, systolic and diastolic blood pressure, lipids, inflammation, and kidney function biomarkers) with risk of any (both ruptured and unruptured) IA and aSAH.
METHODS
Data Availability
We used summary data from published studies that had obtained participant content and ethical approval. The analysis scripts are available on request to the authors.
Data Sources
Genetic associations for the lifestyle and cardiometabolic factors were obtained from summary statistics of large‐scale genome‐wide association studies (GWAS) comprising individuals of European ancestry. 17 , 18 , 19 , 20 , 21 , 22 , 23 , 24 , 25 , 26 , 27 , 28 , 29 , 30 , 31 Alcohol was omitted from the list of exposures because the principal single‐nucleotide variation (SNV; formerly SNP) affecting alcohol consumption in individuals of European ancestry (ie, rs1229984 in ADH1B) 27 was not available in the outcome data sets and no suitable proxy SNV was available. The number of individuals included in each exposure GWAS are shown in Table 1.
Table 1.
Data Sources for the Genetically Predicted Modifiable Risk Factors
| Trait | Sample size | Number of variants | Unit | Variance explained (%) |
|---|---|---|---|---|
| Lifestyle exposures | ||||
| Caffeine consumption 17 | 47 341 | 2 | SD | 0.29 |
| Coffee consumption 24 | 375 833 | 10 | 50% increase | 0.36 |
| Insomnia*, 25 | 397 959 cases; 933 051 controls | 143 | Log‐odds | 0.53 † |
| Long sleep duration 26 | 34 184 cases; 305 742 controls | 4 | Log‐odds (≥9 h/d, compared with 7–8 h/d) | 0.10 † |
| Physical activity 22 | 377 234 | 6 | SD (MET‐minutes per week of moderate‐to‐vigorous physical activity) | 0.08 |
| Short sleep duration 26 | 106 192 cases; 305 742 controls | 19 | Log‐odds (<7 h/d, compared with 7–8 h/d) | 0.20 † |
| Sleep duration 26 | 446 118 | 54 | Hours per day | 0.49 |
| Smoking index 28 | 462 690 | 85 | SD (continuous lifetime smoking measure) | 0.90 |
| Smoking initiation 27 | 1 232 091 | 235 | SD (prevalence of smoking initiation, ie, ever smoker) | 0.88 † |
| Cardiometabolic exposures | ||||
| Body mass index 21 | 806 834 | 967 | SD | 8.1 |
| HDL‐C 19 | 188 577 | 113 | SD | 7.8 |
| LDL‐C 19 | 188 577 | 88 | SD | 9.0 |
| Systolic blood pressure 23 | 318 417 | 214 | 10 mm Hg | 3.4 |
| Diastolic blood pressure 23 | 318 417 | 721 | 10 mm Hg | 6.2 |
| Triglycerides 19 | 188 577 | 60 | SD | 5.6 |
| Type 2 diabetes 29 | 148 726 cases; 965 732 controls | 422 | Log‐odds | 0.90 † |
| Fasting glucose 18 | 133 010 | 32 | 1 mmol/L | 2.8 |
| Fasting insulin 18 | 133 010 | 9 | 1 pmol/L (log‐transformed) | 0.27 |
| HbA1c 20 | 123 665 | 34 | Percentage point | 1.9 |
| Interleukin‐6 receptor 31 | 343 524 | 2 | SD C‐reactive protein levels | 0.46 |
| Chronic kidney disease 30 | 41 395 cases; 439 303 controls | 21 | Log‐odds | 0.29 † |
| Blood urea nitrogen 30 | 243 029 | 67 | 1 mg/dL | 2.0 |
| eGFR 30 | 567 460 | 235 | (mL×min−1)/(1.73 m2) (log‐transformed) | 2.9 |
eGFR indicates estimated glomerular filtration rate; HbA1c, hemoglobin A1c; HDL‐C, high‐density lipoprotein cholesterol; LDL‐C, low‐density lipoprotein cholesterol; and MET, metabolic equivalent of task.
In UK Biobank, insomnia cases were defined as participants who answered "usually" on the question "Do you have trouble falling asleep at night or do you wake up in the middle of the night?" Participants who answered "never/rarely" or "sometimes" were defined as controls. In 23andMe, insomnia cases were defined as participants who affirmed at least 1 of the following questions: "Have you ever been diagnosed with, or treated for insomnia, insomnia but not narcolepsy, sleep apnea or restless leg syndrome?"; "Has a doctor ever told you that you have any of these conditions: insomnia?"; "Have you ever been diagnosed by a doctor with sleep disturbance?"; "Do you routinely have trouble getting to sleep at night?"; "What sleep disorders have you been diagnosed with? Please select all that apply: insomnia, trouble falling or staying asleep"; "In the last 2 years, have you taken prescription sleep aids?".
Calculated assuming a logistic distribution for the liability.
For the outcomes (any [ruptured or unruptured] IA and aSAH), the genetic associations were taken from the International Stroke Genetics Consortium GWAS meta‐analysis of individuals of European ancestry. 32 To avoid bias because of sample overlap in the summary statistics with the exposures, individuals from UK Biobank were excluded, resulting in 6252 cases and 59 544 controls for any (ruptured or unruptured) IA and 4196 cases and 59 544 controls for aSAH. For all exposures and outcomes, participant consent and ethical approval were obtained in the original studies.
Selection of Genetic Instrumental Variables
We selected SNVs that associated with the corresponding modifiable risk factor at P<5×10−8 as instrumental variables for the risk factor. For interleukin‐6 receptor (IL6R), we considered genetic variants only within ±300 kb of the IL6R gene. The independence of the variants was ensured by clumping them so that variants with r 2>0.01 (based on European ancestry reference in 1000 Genomes Project) with the lead SNV within ±10 000 kb window were excluded.
Statistical Analysis
To evaluate statistical power, we calculated the minimum detectable odds ratios (ORs) for the continuous exposures with 80% power and α=0.05, based on the exposure GWAS sample size and the sum of the variance explained by the individual genetic instruments. For the main MR analysis, we used the multiplicative random‐effects inverse‐variance weighted method. This method provides consistent causal estimates when all genetic variants used are valid instrumental variables. We used MR‐Egger, 33 weighted median, 34 and weighted mode 35 methods as sensitivity analyses, all of which are more robust to inclusions of invalid instrumental variables, with the trade‐off of decreased statistical power. MR‐Egger is robust to invalid instrumental variables, provided that the pleiotropic effects of the instruments are independent of the instrument strengths. The presence of horizontal pleiotropy was evaluated by the MR‐Egger intercept test. 33 The weighted median method provides robust causal estimates if more than half of the weights are provided by valid instrumental variables. 34 The weighted mode provides robust causal estimates if the weights associated with valid instrumental variables are the largest among homogeneous subsets of instruments. 35 Finally, to further investigate the exposures with strong evidence for association on IA and aSAH risk in the main MR analysis, we conducted multivariable MR 36 to explore the mutually adjusted direct effects of (1) insomnia liability and sleep apnea (proxied by snoring liability 37 ), (2) insomnia liability and systolic blood pressure, and (3) smoking index and systolic blood pressure. For multivariable MR, the considered instruments were SNVs that both associated at P<5×10−8 with either exposure under consideration, and were available in the outcome GWAS data set. The variants were clumped at r 2>0.01 as in the univariable MR described above, based on the lower SNV‐wise P value with the considered exposures.
The results are reported as ORs for the outcomes per unit increase in the exposure (Table 1). Insomnia, long and short sleep duration, type 2 diabetes, and chronic kidney disease were treated as binary exposures, and the ORs are per unit increase in the log‐odds of the exposure. For smoking initiation as the exposure, the ORs are per SD increase in the prevalence of smoking initiation. For coffee consumption as the exposure, the ORs are per 50% increase in the exposure. For sleep duration, the ORs are per 1‐hour increase of sleep per day. For systolic and diastolic blood pressure, the ORs are per 10 mm Hg increase. For hemoglobin A1c, the ORs are per percentage point increase in the exposure. For fasting glucose, the ORs are per mmol/L increase in glucose levels, and for fasting insulin, the ORs are per unit increase in log(insulin[pmol/L]). For blood urea nitrogen, the ORs are per mg/dL increase, and for estimated glomerular filtration rate (eGFR), the ORs are per unit increase in log(eGFR). For the rest of the exposures, the ORs are per 1‐SD increase in the exposure (Table 1).
Strength of Evidence
As a reference value, the Bonferroni‐corrected significance level for 23 exposures is 0.05/23=0.0022. However, we interpret the evidence based on the effect size, the consistency of the results (both in the sensitivity analyses and between the outcomes), and the statistical evidence as a continuous measure, and we refrain from dichotomous decisions based on any P value threshold. 38
RESULTS
The minimum detectable ORs for the continuous exposures are given in Table S1. Of the 18 continuous exposures considered, we had adequate power to detect ORs at least ≥1.5 (or ≤0.67) per unit change in the exposure for 12 and 10 exposures for any IA and aSAH, respectively.
The MR estimates for the associations of the modifiable lifestyle and cardiometabolic factors with any IA and aSAH are presented in Figure. We found strong evidence for associations between genetically proxied smoking, insomnia liability, and blood pressure with increased risk of both any IA (OR [95% CI] per 1‐SD increase in smoking index 3.20 [1.93–5.29], P=5.8×10−6; OR per 1‐SD increase in the prevalence of smoking initiation 1.85 [1.50–2.28], P=1.4×10−8; OR per unit increase in log‐odds of insomnia liability 1.24 [1.10–1.40], P=5.0×10−4; OR per 10 mm Hg increase in diastolic blood pressure 2.92 [2.49–3.43], P=8.4×10−40; OR per 10 mm Hg increase in systolic blood pressure 1.87 [1.61–2.17], P=1.4×10−16) and aSAH (OR per 1‐SD increase in smoking index 3.00 [1.55–5.74], P=0.0010; OR per 1‐SD increase in the prevalence of smoking initiation 1.62 [1.24–2.12], P=4.6×10−4; OR per unit increase in log‐odds of insomnia liability 1.20 [1.03–1.40], P=0.023; OR per 10 mm Hg increase in diastolic and systolic blood pressure 3.21 [2.66–3.87], P=2.1×10−34 and 2.17 [1.82–2.58], P=2.4×10−18, respectively). There was also weak evidence of association for genetically predicted decreased physical activity, higher triglyceride levels, higher body mass index, and lower low‐density lipoprotein cholesterol levels with increased risk of both outcomes, with 95% CI overlapping the null for at least 1 of the outcomes. Increased fasting glucose levels were associated with lower risk of any IA (OR per unit increase in fasting glucose levels 0.64 [0.43–0.95], P=0.029) and aSAH (OR, 0.57 [0.34–0.94], P=0.029); however, these results were not supported by the point estimates from sensitivity analysis more robust to horizontal pleiotropy (Figure S1).
Figure 1. Associations of genetically predicted lifestyle and cardiometabolic factors with risk of any (ruptured or unruptured) IA and aSAH, using multiplicative random‐effects inverse‐variance weighted method.
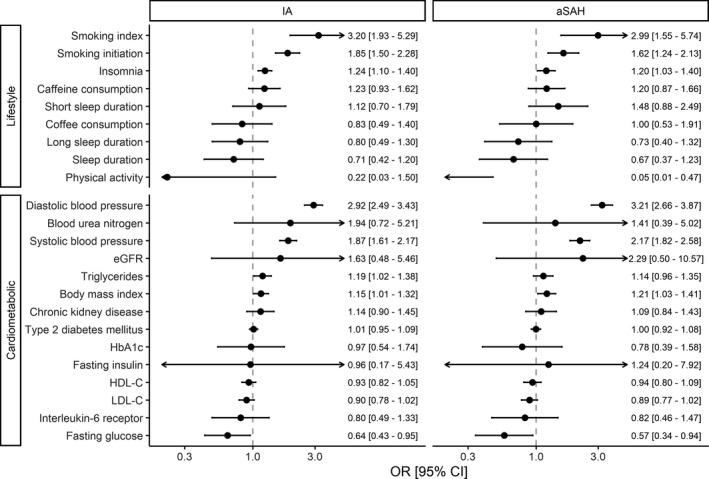
aSAH indicates aneurysmal subarachnoid hemorrhage; eGFR estimated glomerular filtration rate; HDL‐C, high‐density lipoprotein cholesterol; IA, intracranial aneurysm; LDL‐C, low‐density lipoprotein cholesterol; and OR, odds ratio.
For the exposures that showed evidence for association in inverse‐variance weighted results, the MR‐Egger intercept test indicated evidence for horizontal pleiotropy with genetically predicted systolic and diastolic blood pressure and risk of both IA (P=0.004 for systolic, P=0.001 for diastolic) and aSAH (P=0.008 for systolic, P=2×10−4 for diastolic), and weaker evidence for horizontal pleiotropy of genetically predicted smoking initiation liability (P=0.03) with aSAH risk (Table S2). However, all point estimates in sensitivity analyses by weighted median and weighted mode methods for these exposures were consistent with the main inverse‐variance weighted analysis (Table S2; Figure S1). In the multivariable MR investigation of direct effects adjusted for genetically predicted effects of other relevant exposures, there was evidence for direct effects of smoking (independent of blood pressure), blood pressure (independent of insomnia liability or smoking), and insomnia liability (independent of sleep apnea or blood pressure), with point estimates concordant with those in univariable MR (Table 2).
Table 2.
Multivariable Mendelian Randomization Results of Mutually Adjusted Direct Effects on Risk of Any (Ruptured or Unruptured) IA and aSAH for Exposures That Showed Evidence for Association in the Main Mendelian Randomization
| Trait | IA | aSAH | ||||
|---|---|---|---|---|---|---|
| Number of variants | OR (95% CI) | P value | Number of variants | OR (95% CI) | P value | |
| Insomnia | 269 | 1.29 (1.06–1.58) | 0.012 | 230 | 1.30 (1.03–1.65) | 0.030 |
| SBP | 1.87 (1.62–2.17) | 2.8×10−15 | 2.13 (1.80–2.53) | 6.9×10−16 | ||
| Insomnia | 104 | 1.27 (1.07–1.50) | 0.006 | 88 | 1.28 (1.05–1.55) | 0.017 |
| Sleep apnea | 3.43 (0.89–13.24) | 0.077 | 3.01 (0.64–14.24) | 0.17 | ||
| Smoking | 330 | 5.78 (3.14–10.62) | 3.5×10−8 | 295 | 4.91 (2.42–9.94) | 1.4×10−5 |
| SBP | 1.82 (1.59–2.09) | 5.6×10−16 | 2.15 (1.83–2.52) | 4.3×10−18 | ||
aSAH indicates aneurysmal subarachnoid hemorrhage; IA, intracranial aneurysm; OR, odds ratio; and SBP, systolic blood pressure.
DISCUSSION
The present MR study found further evidence for smoking and high blood pressure as the strongest risk factors for IA and aSAH. This study additionally found evidence that insomnia may be a novel risk factor for IA and aSAH. These results were consistent in multivariable MR, indicating direct effects of these exposures on IA and aSAH risk. Weak evidence of possible associations was found for higher triglyceride levels and body mass index with increased risk of both IA and aSAH and for higher levels of moderate‐to‐vigorous physical activity and low‐density lipoprotein cholesterol with decreased risk of the outcomes. Other lifestyle and cardiometabolic factors showed no strong and consistent associations with either IA or aSAH.
Results of this MR study support the findings of previous studies, 3 , 4 , 5 which have shown that smoking is a strong risk factor for aSAH. Tobacco smoke contains nicotine and numerous other substances that could promote vascular endothelial dysfunction and IA rupture. 39 , 40 Accumulated evidence indicates that endothelial dysfunction, hemodynamic stress, and inflammatory responses play a central role in IA formation, growth, and rupture. 41 Mechanistic evidence indicates that nicotine exposure increases IA rupture risk through actions on the vascular cell nicotinic acetylcholine receptors containing α7 subunits, leading to increased levels of vascular endothelial growth factor, platelet‐derived growth factor‐B, and inflammatory cytokines. 39
Studies of coffee consumption and risk of aSAH are limited and results are inconsistent, with an inverse 9 and a neutral 42 association observed in cohorts of Swedish women and Finnish male smokers, respectively. We observed no association of genetically predicted coffee or caffeine intake with IA or aSAH. However, the CIs were broad, and weak associations in either direction cannot be ruled out.
Data on the role of sleep in the development of IA formation and rupture are scarce, but a previous observational study found an increased risk for aSAH in patients with sleep apnea. 8 No association between short or long sleep duration and subarachnoid hemorrhage was found in a cohort of Swedish adults. 43 Here, we found evidence to support associations of insomnia with increased risk of IA and aSAH. The results were similar in multivariable MR, where we found evidence for a direct effect of insomnia liability, after adjusting for either blood pressure or sleep apnea. The point estimates for other sleep‐related traits (total sleep duration, short sleep, and long sleep) were consistent with our finding for insomnia liability, albeit with large uncertainty in the estimates. Given the limited data, whether lack of sleep is an etiological risk factor for both IA and aSAH risk merits further study.
Regular physical activity was associated with a reduced risk of aSAH in a cohort of 8006 men of Japanese ancestry, 11 a cohort of 65 521 Finnish adults, 10 and a cohort of >1 million UK women. 12 Our MR results provide tentative support for a causal association between physical activity and decreased risk of aSAH; however, caution should be warranted because of the large uncertainty in our estimates. Physical activity may reduce the risk of aSAH by improving endothelial function, 44 lowering blood pressure, 45 and decreasing systemic inflammation. 46
Among cardiometabolic risk factors, previous studies have conclusively revealed that hypertension is associated with an increased risk of aSAH, 3 , 4 , 32 supported by our MR results for systolic and diastolic blood pressure, but have yielded conflicting results for body mass index 4 , 13 , 47 and diabetes. 3 , 4 Body mass index was inversely associated with risk of aSAH in women but was not associated with aSAH in men in a pooled analysis of 21 Swedish cohort studies. 4 An inverse association between body mass index and risk of aSAH was also observed in a cohort of 1.3 million UK women. 13 In contrast, a borderline significant positive association between body mass index and aSAH was observed in a previous MR study in UK women and men 47 and the present MR study in another population. High body mass index is a strong risk factor for type 2 diabetes, which was found to be inversely associated with aSAH in a meta‐analysis of case–control studies 3 and nonsignificantly inversely associated with aSAH in a pooled analysis of cohort studies. 4 In our analysis, there was no evidence for association of type 2 diabetes with IA or aSAH, and further research on the causal associations of adiposity and type 2 diabetes with risk of aSAH is necessary. With regard to lipids, limited observational data, mainly case–control studies, suggest that hypercholesterolemia 3 and high levels of high‐density lipoprotein 14 are associated with a lower risk of aSAH. 3 Our MR findings provided weak evidence for positive and negative associations for triglyceride levels and low‐density lipoprotein cholesterol levels, respectively, with only modest effect sizes.
Major strengths of this study include the MR design, which reduced confounding and reverse causality. We were able to utilize the summary statistics from the largest GWAS on IA and aSAH to date, which ensured maximal statistical power. Population stratification bias was minimized by restricting the analyses to individuals of European descent. With regard to limitations, the statistical power was low in some analyses as demonstrated in Table S1, because of instrumental variables only explaining a small proportion of variance in the exposure, particularly for physical activity, and coffee and caffeine consumption. Another limitation is that because we used summarized data, we could not assess nonlinear associations. Finally, our analyses were restricted to individuals of European ancestry and may therefore not be generalizable to other populations.
In conclusion, this MR study found that genetic predisposition to smoking, insomnia, and high blood pressure was robustly associated with increased risk of IA and aSAH. Physical activity, body mass index, triglyceride levels, and low‐density lipoprotein cholesterol levels may also affect the risk of both IA and aSAH. These results add to the triangulation of evidence on risk factors of IA and aSAH, and warrant further investigation in future large MR and other epidemiological studies.
Sources of Funding
Gill and Karhunen are supported by the British Heart Foundation Centre of Research Excellence (RE/18/4/34215) at Imperial College London. Karhunen is supported by the Academy of Finland Project 312123, and European Union’s Horizon 2020 research and innovation programme under Grant Agreement No 848158. Gill is supported by a National Institute for Health Research Clinical Lectureship at St. George's, University of London (CL‐2020‐16‐001). Larsson acknowledges research support from the Swedish Research Council for Health, Working Life and Welfare (Forte, 2018‐00123), the Swedish Heart‐Lung Foundation (Hjärt‐Lungfonden, 20190247), and the Swedish Research Council (Vetenskapsrådet, 2019‐00977). Bakker was supported by the Netherlands Cardiovascular Research Initiative: An initiative with support of the Dutch Heart Foundation, CVON2015‐08 ERASE. Ruigrok received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (PRYSM, grant agreement No. 852173).
Disclosures
Gill is employed part‐time by Novo Nordisk. The other authors have no conflicts of interest to disclose.
Supporting information
Tables S1–S2
Figure S1
Acknowledgments
The authors would like to thank the International Stroke Genetics Consortium (ISGC) Intracranial Aneurysm working group. Data on glycemic traits have been contributed by the Meta‐Analyses of Glucose and Insulin‐related traits Consortium investigators and have been downloaded from www.magicinvestigators.org. We acknowledge the Chronic Kidney Disease Genetics (CKDGen) Consortium for releasing GWAS summary data.
For Sources of Funding and Disclosures, see page 7.
References
- 1. Macdonald RL, Schweizer TA. Spontaneous subarachnoid haemorrhage. Lancet. 2017;389:655–666. doi: 10.1016/S0140-6736(16)30668-7 [DOI] [PubMed] [Google Scholar]
- 2. Lawton MT, Vates GE. Subarachnoid hemorrhage. N Engl J Med. 2017;377:257–266. doi: 10.1056/NEJMcp1605827 [DOI] [PubMed] [Google Scholar]
- 3. Feigin VL, Rinkel GJE, Lawes CMM, Algra A, Bennett DA, van Gijn J, Anderson CS. Risk factors for subarachnoid hemorrhage: an updated systematic review of epidemiological studies. Stroke. 2005;36:2773–2780. doi: 10.1161/01.STR.0000190838.02954.e8 [DOI] [PubMed] [Google Scholar]
- 4. Sundström J, Söderholm M, Söderberg S, Alfredsson L, Andersson M, Bellocco R, Björck M, Broberg P, Eriksson M, Eriksson M, et al. Risk factors for subarachnoid haemorrhage: a nationwide cohort of 950 000 adults. Int J Epidemiol. 2019;48:2018–2025. doi: 10.1093/ije/dyz163 [DOI] [PubMed] [Google Scholar]
- 5. Larsson SC, Mason AM, Bäck M, Klarin D, Damrauer SM, Program MV, Michaëlsson K, Burgess S. Genetic predisposition to smoking in relation to 14 cardiovascular diseases. Eur Heart J. 2020;41:3304–3310. doi: 10.1093/eurheartj/ehaa193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Larsson SC, Wallin A, Wolk A, Markus HS. Differing association of alcohol consumption with different stroke types: a systematic review and meta‐analysis. BMC Med. 2016;14:178. doi: 10.1186/s12916-016-0721-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Larsson SC, Burgess S, Mason AM, Michaëlsson K. Alcohol consumption and cardiovascular disease: a Mendelian randomization study. Circ Genom Precis Med. 2020;13:e002814. doi: 10.1161/CIRCGEN.119.002814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zaremba S, Albus L, Schuss P, Vatter H, Klockgether T, Güresir E. Increased risk for subarachnoid hemorrhage in patients with sleep apnea. J Neurol. 2019;266:1351–1357. doi: 10.1007/s00415-019-09265-5 [DOI] [PubMed] [Google Scholar]
- 9. Larsson SC, Virtamo J, Wolk A. Coffee consumption and risk of stroke in women. Stroke. 2011;42:908–912. doi: 10.1161/STROKEAHA.110.603787 [DOI] [PubMed] [Google Scholar]
- 10. Lindbohm JV, Rautalin I, Jousilahti P, Salomaa V, Kaprio J, Korja M. Physical activity associates with subarachnoid hemorrhage risk—a population‐based long‐term cohort study. Sci Rep. 2019;9:9219. doi: 10.1038/s41598-019-45614-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Abbott RD, Rodriguez BL, Burchfiel CM, Curb JD. Physical activity in older middle‐aged men and reduced risk of stroke: the Honolulu Heart Program. Am J Epidemiol. 1994;139:881–893. doi: 10.1093/oxfordjournals.aje.a117094 [DOI] [PubMed] [Google Scholar]
- 12. Armstrong MEG, Green J, Reeves GK, Beral V, Cairns BJ. Frequent physical activity may not reduce vascular disease risk as much as moderate activity: large prospective study of women in the United Kingdom. Circulation. 2015;131:721–729. doi: 10.1161/CIRCULATIONAHA.114.010296 [DOI] [PubMed] [Google Scholar]
- 13. Kroll ME, Green J, Beral V, Sudlow CLM, Brown A, Kirichek O, Price A, Yang TO, Reeves GK; For the Million Women Study Collaborators . Adiposity and ischemic and hemorrhagic stroke: prospective study in women and meta‐analysis. Neurology. 2016;87:1473–1481. doi: 10.1212/WNL.0000000000003171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Can A, Castro VM, Dligach D, Finan S, Yu S, Gainer V, Shadick NA, Savova G, Murphy S, Cai T, et al. Lipid‐lowering agents and high HDL (high‐density lipoprotein) are inversely associated with intracranial aneurysm rupture. Stroke. 2018;49:1148–1154. doi: 10.1161/STROKEAHA.117.019972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Diaz‐Ricart M, Torramade‐Moix S, Pascual G, Palomo M, Moreno‐Castaño AB, Martinez‐Sanchez J, Vera M, Cases A, Escolar G. Endothelial damage, inflammation and immunity in chronic kidney disease. Toxins. 2020;12:361. doi: 10.3390/toxins12060361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Davey Smith G, Ebrahim S. ‘Mendelian randomization’: can genetic epidemiology contribute to understanding environmental determinants of disease? Int J Epidemiol. 2003;32:1–22. doi: 10.1093/ije/dyg070 [DOI] [PubMed] [Google Scholar]
- 17. Cornelis MC, Monda KL, Yu K, Paynter N, Azzato EM, Bennett SN, Berndt SI, Boerwinkle E, Chanock S, Chatterjee N, et al. Genome‐wide meta‐analysis identifies regions on 7p21 (AHR) and 15q24 (CYP1A2) as determinants of habitual caffeine consumption. PLoS Genet. 2011;7:e1002033. doi: 10.1371/journal.pgen.1002033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Scott RA, Lagou V, Welch RP, Wheeler E, Montasser ME, Luan J, Mägi R, Strawbridge RJ, Rehnberg E, Gustafsson S, et al. Large‐scale association analyses identify new loci influencing glycemic traits and provide insight into the underlying biological pathways. Nat Genet. 2012;44:991–1005. doi: 10.1038/ng.2385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Willer CJ, Schmidt EM, Sengupta S, Peloso GM, Gustafsson S, Kanoni S, Ganna A, Chen J, Buchkovich ML, Mora S, et al. Discovery and refinement of loci associated with lipid levels. Nat Genet. 2013;45:1274–1283. doi: 10.1038/ng.2797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wheeler E, Leong A, Liu C‐T, Hivert M‐F, Strawbridge RJ, Podmore C, Li M, Yao J, Sim X, Hong J, et al. Impact of common genetic determinants of hemoglobin A1c on type 2 diabetes risk and diagnosis in ancestrally diverse populations: a transethnic genome‐wide meta‐analysis. PLoS Med. 2017;14:e1002383. doi: 10.1371/journal.pmed.1002383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pulit SL, Stoneman C, Morris AP, Wood AR, Glastonbury CA, Tyrrell J, Yengo L, Ferreira T, Marouli E, Ji Y, et al. Meta‐analysis of genome‐wide association studies for body fat distribution in 694 649 individuals of European ancestry. Hum Mol Genet. 2018;28:166–174. doi: 10.1093/hmg/ddy327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Klimentidis YC, Raichlen DA, Bea J, Garcia DO, Wineinger NE, Mandarino LJ, Alexander GE, Chen Z, Going SB. Genome‐wide association study of habitual physical activity in over 377,000 UK Biobank participants identifies multiple variants including CADM2 and APOE. Int J Obes. 2018;42:1161–1176. doi: 10.1038/s41366-018-0120-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Carter AR, Gill D, Davies NM, Taylor AE, Tillmann T, Vaucher J, Wootton RE, Munafò MR, Hemani G, Malik R, et al. Understanding the consequences of education inequality on cardiovascular disease: Mendelian randomisation study. BMJ. 2019;365:l1855. doi: 10.1136/bmj.l1855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhong VW, Kuang A, Danning RD, Kraft P, van Dam RM, Chasman DI, Cornelis MC. A genome‐wide association study of bitter and sweet beverage consumption. Hum Mol Genet. 2019;28:2449–2457. doi: 10.1093/hmg/ddz061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jansen PR, Watanabe K, Stringer S, Skene N, Bryois J, Hammerschlag AR, de Leeuw CA, Benjamins JS, Muñoz‐Manchado AB, Nagel M, et al. Genome‐wide analysis of insomnia in 1,331,010 individuals identifies new risk loci and functional pathways. Nat Genet. 2019;51:394–403. doi: 10.1038/s41588-018-0333-3 [DOI] [PubMed] [Google Scholar]
- 26. Dashti HS, Jones SE, Wood AR, Lane JM, van Hees VT, Wang H, Rhodes JA, Song Y, Patel K, Anderson SG, et al. Genome‐wide association study identifies genetic loci for self‐reported habitual sleep duration supported by accelerometer‐derived estimates. Nat Commun. 2019;10:1100. doi: 10.1038/s41467-019-08917-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liu M, Jiang YU, Wedow R, Li Y, Brazel DM, Chen F, Datta G, Davila‐Velderrain J, McGuire D, Tian C, et al. Association studies of up to 1.2 million individuals yield new insights into the genetic etiology of tobacco and alcohol use. Nat Genet. 2019;51:237–244. doi: 10.1038/s41588-018-0307-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wootton RE, Richmond RC, Stuijfzand BG, Lawn RB, Sallis HM, Taylor GMJ, Hemani G, Jones HJ, Zammit S, Davey Smith G, et al. Evidence for causal effects of lifetime smoking on risk for depression and schizophrenia: a Mendelian randomisation study. Psychol Med. 2020;50:2435–2443. doi: 10.1017/S0033291719002678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Vujkovic M, Keaton JM, Lynch JA, Miller DR, Zhou J, Tcheandjieu C, Huffman JE, Assimes TL, Lorenz K, Zhu X, et al. Discovery of 318 new risk loci for type 2 diabetes and related vascular outcomes among 1.4 million participants in a multi‐ancestry meta‐analysis. Nat Genet. 2020;52:680–691. doi: 10.1038/s41588-020-0637-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wuttke M, Li Y, Li M, Sieber KB, Feitosa MF, Gorski M, Tin A, Wang L, Chu AY, Hoppmann A, et al. A catalog of genetic loci associated with kidney function from analyses of a million individuals. Nat Genet. 2019;51:957. doi: 10.1038/s41588-019-0407-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. The Neale Lab . Updated GWAS Analysis of the UK Biobank. Published 2018. Available at: http://www.nealelab.is/uk‐biobank. Accessed February 3, 2021.
- 32. Bakker MK, van der Spek RAA, van Rheenen W, Morel S, Bourcier R, Hostettler IC, Alg VS, van Eijk KR, Koido M, Akiyama M, et al. Genome‐wide association study of intracranial aneurysms identifies 17 risk loci and genetic overlap with clinical risk factors. Nat Genet. 2020;52:1303–1313. doi: 10.1038/s41588-020-00725-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol. 2015;44:512–525. doi: 10.1093/ije/dyv080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bowden J, Davey Smith G, Haycock PC, Burgess S. Consistent estimation in Mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol. 2016;40:304–314. doi: 10.1002/gepi.21965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hartwig FP, Davey Smith G, Bowden J. Robust inference in summary data Mendelian randomization via the zero modal pleiotropy assumption. Int J Epidemiol. 2017;46:1985–1998. doi: 10.1093/ije/dyx102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Burgess S, Thompson SG. Multivariable Mendelian randomization: the use of pleiotropic genetic variants to estimate causal effects. Am J Epidemiol. 2015;181:251–260. doi: 10.1093/aje/kwu283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Campos AI, García‐Marín LM, Byrne EM, Martin NG, Cuéllar‐Partida G, Rentería ME. Insights into the aetiology of snoring from observational and genetic investigations in the UK Biobank. Nat Commun. 2020;11:817. doi: 10.1038/s41467-020-14625-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wasserstein RL, Schirm AL, Lazar NA. Moving to a world beyond “p < 0.05”. Am Stat. 2019;73(sup1):1–19. doi: 10.1080/00031305.2019.1583913 [DOI] [Google Scholar]
- 39. Kamio Y, Miyamoto T, Kimura T, Mitsui K, Furukawa H, Zhang D, Yokosuka K, Korai M, Kudo D, Lukas RJ, et al. Roles of nicotine in the development of intracranial aneurysm rupture. Stroke. 2018;49:2445–2452. doi: 10.1161/STROKEAHA.118.021706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Messner B, Bernhard D. Smoking and cardiovascular disease: mechanisms of endothelial dysfunction and early atherogenesis. Arterioscler Thromb Vasc Biol. 2014;34:509–515. doi: 10.1161/ATVBAHA.113.300156 [DOI] [PubMed] [Google Scholar]
- 41. Chalouhi N, Hoh BL, Hasan D. Review of cerebral aneurysm formation, growth, and rupture. Stroke. 2013;44:3613–3622. doi: 10.1161/STROKEAHA.113.002390 [DOI] [PubMed] [Google Scholar]
- 42. Larsson SC, Männistö S, Virtanen MJ, Kontto J, Albanes D, Virtamo J. Coffee and tea consumption and risk of stroke subtypes in male smokers. Stroke. 2008;39:1681–1687. doi: 10.1161/STROKEAHA.107.504183 [DOI] [PubMed] [Google Scholar]
- 43. Titova OE, Michaëlsson K, Larsson SC. Sleep duration and stroke: prospective cohort study and mendelian randomization analysis. Stroke. 2020;51:3279–3285. doi: 10.1161/STROKEAHA.120.029902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sherman DL. Exercise and endothelial function. Coron Artery Dis. 2000;11:117–122. doi: 10.1097/00019501-200003000-00005 [DOI] [PubMed] [Google Scholar]
- 45. Fagard RH, Cornelissen VA. Effect of exercise on blood pressure control in hypertensive patients. Eur J Cardiovasc Prev Rehabil. 2007;14:12–17. doi: 10.1097/HJR.0b013e3280128bbb [DOI] [PubMed] [Google Scholar]
- 46. Ford ES. Does exercise reduce inflammation? Physical activity and C‐reactive protein among U.S. adults. Epidemiology. 2002;13:561–568. doi: 10.1097/00001648-200209000-00012 [DOI] [PubMed] [Google Scholar]
- 47. Larsson SC, Bäck M, Rees JMB, Mason AM, Burgess S. Body mass index and body composition in relation to 14 cardiovascular conditions in UK Biobank: a Mendelian randomization study. Eur Heart J. 2020;41:221–226. doi: 10.1093/eurheartj/ehz388 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tables S1–S2
Figure S1
Data Availability Statement
We used summary data from published studies that had obtained participant content and ethical approval. The analysis scripts are available on request to the authors.