

Abstract
The somatic mutations in each cancer genome are caused by multiple mutational processes, each of which leaves a characteristic imprint (or “signature”), potentially caused by specific etiologies or exposures. Deconvolution of these signatures offers a glimpse into the evolutionary history of individual tumors. Recent work has shown that mutational signatures may also yield therapeutic and prognostic insights, including the identification of cell-intrinsic signatures as biomarkers of drug response and prognosis. For example, mutational signatures indicating homologous recombination deficiency are associated with PARP inhibitor sensitivity, while APOBEC-associated signatures are associated with ATR inhibitor sensitivity. Further, therapy-induced mutational signatures implicated in cancer progression have also been uncovered, including the identification of thiopurine-induced TP53 mutations in leukemia. In this review, we explore the various ways mutational signatures can reveal new therapeutic and prognostic insights, thus extending their traditional role in identifying disease etiology.
Introduction
Mutational signature analysis has become a powerful approach for unveiling the mutagenic processes that shape the landscape of cancer genomes. Mutational signatures can be characterized into single-nucleotide variants (SNVs; See Glossary), doublet (dinucleotide) substitutions, small insertion-deletions (indels), and structural variations (SVs) (Fig. 1). Over 60 SNV mutational signatures have been identified across cancers based on 96 possible variant types which consider the SNV’s trinucleotide context (including the base 5’ and the base 3’ of the mutated position; Fig. 1) [1]. Eleven doublet and 18 indel signatures have also been identified, the latter of which also rely on sequence context [1]. These signatures are included in the COSMIC database [2], which is regularly updated as new signatures get discovered. For example, the COSMIC v2.0 SNV signature set contained 30 signatures, which was expanded to ~60 signatures in the COSMIC v3.0 update in 2019 due largely to new signatures identified through the Pan-Cancer Analysis of Whole Genomes (PCAWG) initiative [1], with v3.2 recently released with several additions. Finally, SV signatures have been identified based on adjacent copy number patterns, breakpoint orientation, the presence of clustered SVs, and the nearby sequence context [3–5]; these signatures are not currently included in the COSMIC database.
Figure 1. Features of mutational signatures among various classes of genetic alterations.
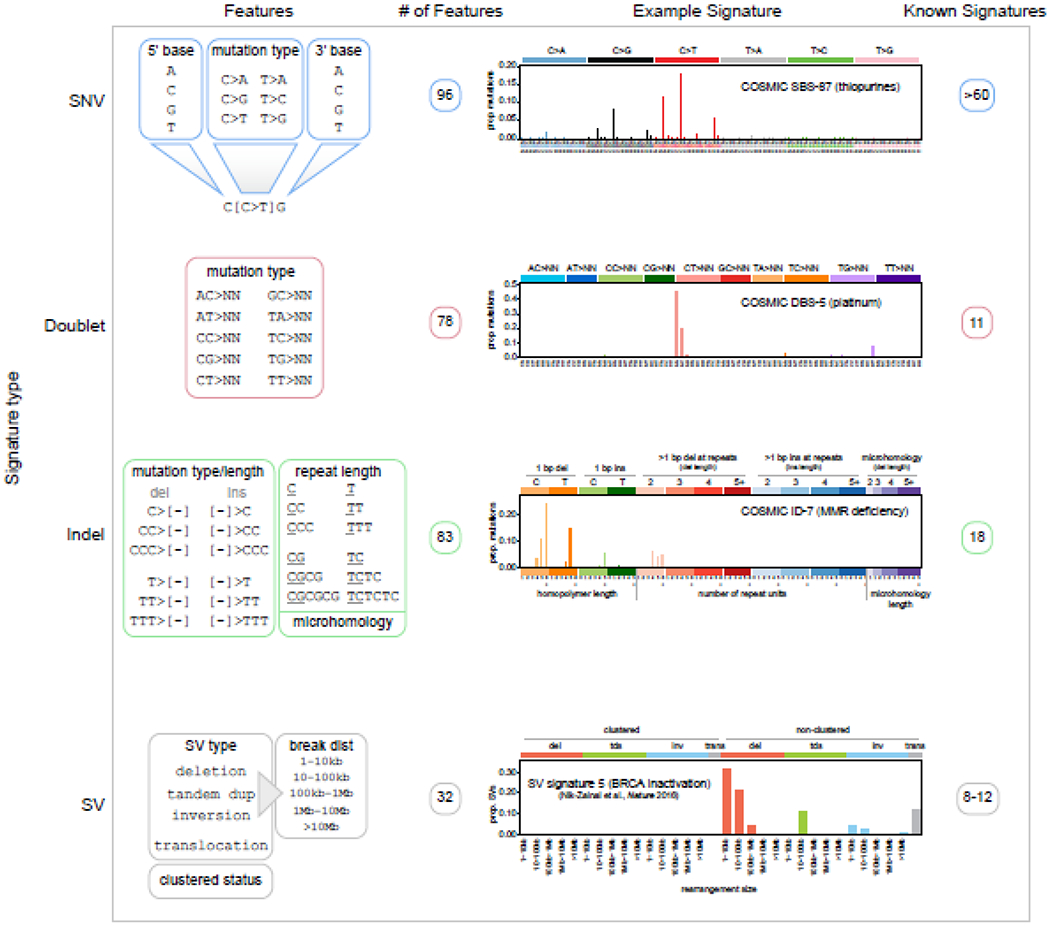
Features of SNV, doublet, insertion-deletion (indel), and SV signatures. SV signatures are derived from published studies [3,23]; others are based on COSMIC v3 classification. The first column shows the genomic features of each signature type. For SNVs, the trinucleotide context is considered, including bases 5’ and 3’ of the mutated site, along with the variant type (e.g. C>T). For doublets, only reference and altered alleles are considered (N indicates any base). For indels, the mutation type and length are considered, where “[−]” indicates sequences deleted (del) or inserted (ins). The length of local repeats is also considered (e.g. 3 for 3 C’s if GCCCG becomes GCCG), as well as whether microhomology was present adjacent to the deleted region. SVs are classified into the four variant types shown, and intrachromosomal SVs (tandem duplications, deletions, and inversions), are further subclassified based on the distance between the two breakpoints (break dist). Whether similar SVs are clustered in a specific genomic region is also considered. The second column indicates the number of features used to classify each signature; for example, SNV signatures have 4 possible 5’ bases, 6 possible mutation types, and 4 possible 3’ bases, making 96 features. The third column indicates example signatures, with the y-axis indicating the proportion of mutations falling into each feature; text indicates COSMIC signature identifiers (if available) and signature etiology. The fourth column indicates the number of signatures reported in COSMIC or published studies.
The notion of a mutational signature encapsulates both a biological process giving rise to mutagenesis, together with a mathematical vector representing the probability of inducing specific variant types. The mathematical vectors for mutational signatures have been discovered using non-negative matrix factorization (NMF) approaches such as SigProfiler [1] or SignatureAnalyzer (a Bayesian variant of NMF) [6–8] applied to catalogs of mutations from cancer patients. For most mutational signatures, the specific biological process causing the signature is well-established or inferred, based on experimental [9–13] or correlative data [1,14,15]. However, some signatures are induced by unknown etiologies which remain to be explored [1]. The duration of a signature can be constant or episodic. For example, “clock-like” signatures are constantly active in virtually all normal and malignant cell types, such as COSMIC signature 1 (caused by 5-methylcytosine deamination) and COSMIC signature 5 (unknown etiology) [16], and thus offer windows into the evolutionary timing of tumorigenesis [17]. By contrast, APOBEC-induced COSMIC signatures 2 and 13 are intermittently triggered and then turned off by unknown mechanisms [18].
Mutational signature analysis generally involves three analytical steps. First, somatic variants are called. As different SNV callers are generally concordant in variant calls [19,20], downstream SNV signature results are likewise similar [21]. However, indel callers are less concordant [19,20], suggesting the need to intersect multiple callers [1,22], although Pindel has been used alone [9]. SV signatures have been identified using BRASS [4,5] and MANTA [23]. Second (and optionally), to discover novel mutational signatures, NMF-based tools such as SigProfiler [24], SignatureAnalyzer [6–8], or MutationalPatterns [25], are fed a catalog of mutations from multiple samples. The extracted signatures can be compared to COSMIC signatures to determine which, if any, are novel. This step is most effective with large sample sizes which helps to crisply resolve non-ubiquitous signatures. Third, to determine the activity level of signatures in individual tumor samples, tools such as those noted above are used to measure a user-selected set of signatures (COSMIC and/or user-detected signatures from the second step) in mutation catalogs from any number of samples. Interestingly, two frequently-used signature extraction tools (second step), SigProfiler and SignatureAnalyzer, often extract equivalent signatures [1,26], suggesting that signature extraction often functions similarly between tools given similar NMF-related algorithms. By contrast, signature application results (third step) differ between tools, as some algorithms “overfit” by applying an excessive number of signatures to individual samples to achieve a better mathematical fit, whereas others are conservative. We suggest using tools that allow tuning of this sensitivity, such as SigProfilerSingleSample (by adjusting the cosine increase threshold for signature inclusion) and careful testing to verify results are biologically plausible (e.g. no chemotherapy-induced signatures in diagnosis samples). The burgeoning number of mutational signatures tools and their varying mathematical approaches have been reviewed elsewhere [27].
Importantly, the identification of a mutational signature within a tumor sample provides insight into the underlying cancer etiology. This is exemplified in melanoma and lung cancer, where mutational signature analysis demonstrates the presence of ultraviolet-light and smoking-induced mutations respectively [28]. Further, probabilistic approaches have been developed that can indicate the nature of driver mutations likely induced by specific mutational signatures [29,30], thereby unveiling the biological processes causing driver mutations. For instance, if three SNV signatures (A, B, and C) are detected in a sample, and only signature A frequently induces C>G SNVs at a CCG trinucleotide context, then signature A most likely caused any given C>G at CCG variant. This is illustrated in Fig. 2 for a specific EZH2 mutation that was likely induced by thiopurine treatment in a relapsed acute lymphoblastic leukemia (ALL) patient from a previous study [12]. This analysis, while straightforward, has remarkable implications for understanding the causes of specific driver mutations.
Figure 2. Calculating the probability a putative driver mutation was caused by a mutational signature.
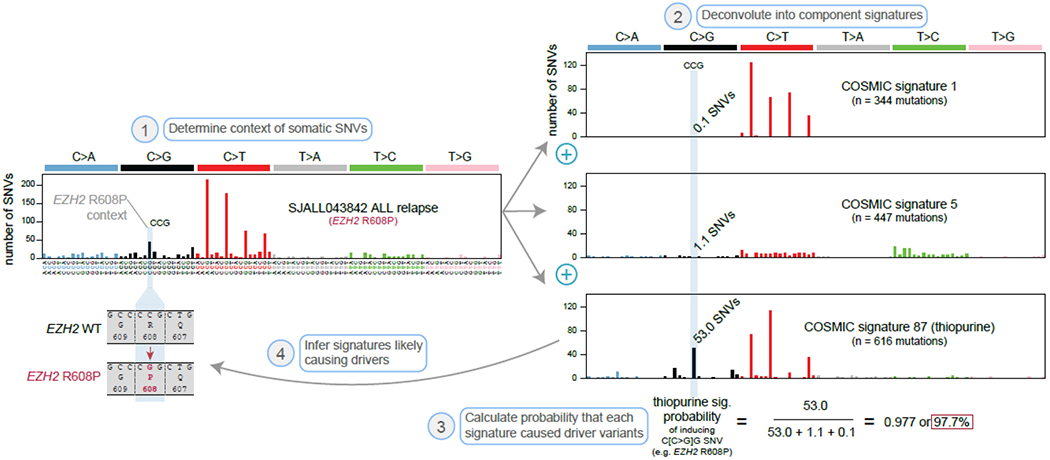
Example calculation showing an EZH2 mutation was likely induced by thiopurine treatment. First, the trinucleotide context of somatic SNVs are determined. The example shown includes the relapse-specific somatic SNVs in a pediatric ALL patient, SJALL043842, sequenced previously by WGS at diagnosis and relapse [12]. A relapse-specific EZH2 R608P mutation occurred in this patient (a C>G at the center of a CCG context). Second, the mutational profile is deconvoluted into signatures, including COSMIC signature 1 (5-mC deamination), signature 5 (clock-like), and signature 87 (thiopurine-induced). When these 3 signatures are added together at the dosages indicated (“n = X mutations”), they have a cosine similarity of 0.99 to the actual sample profile, indicating the signatures effectively explain the mutational processes in the relapsed ALL. The three mutation spectra (shown at right) represent the absolute numbers of mutations of each SNV mutation type which are predicted to have been caused by each mutational signature. The C>G (CCG) channel had 53.0 mutations predicted to have been caused by the thiopurine mutational signature, while the other signatures caused a predicted 0.1 (signature 1) or 1.1 (signature 5) mutations. Third, we can therefore calculate a 97.7% probability that the EZH2 R608P was caused by thiopurines, as the thiopurine signature contributed that percentage of mutations occurring at the EZH2 R608P channel (C>G at CCG). Fourth and finally, we can infer that thiopurine treatment most likely caused the EZH2 variant.
In addition to identifying cancer etiologies and the causes of driver mutations, analysis of mutational signatures can also lead to direct therapeutic and prognostic insights. In this review, we discuss several areas in which knowledge of mutational signatures have yielded insights that directly affect therapeutic decision-making and prognosis of cancer (Fig. 3). We then discuss possible future uses of mutational signatures in research and clinical care of cancer patients.
Figure 3. Three ways mutational signatures can be incorporated into clinical decision-making.
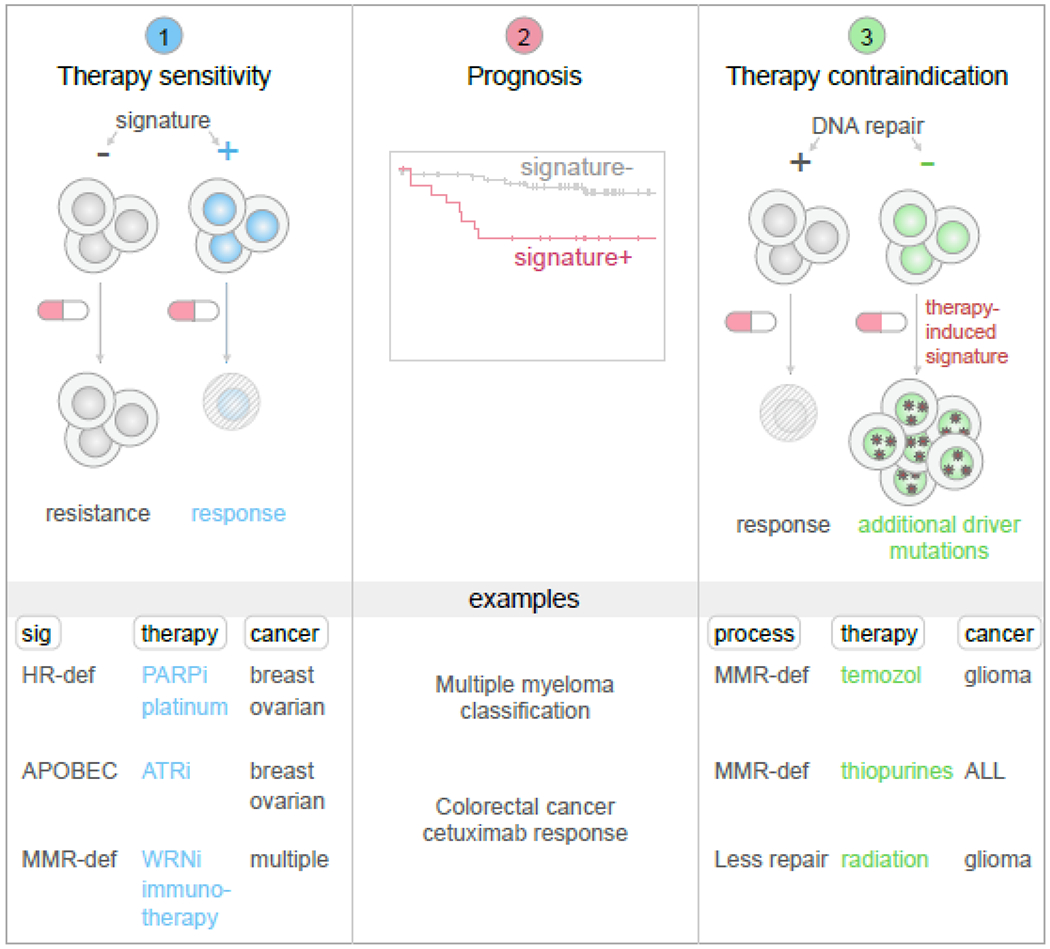
Left, mutational signatures are sometimes associated with sensitivity to a specific anticancer drug (shown as a red-and-white pill), while signature-negative samples lack responsiveness. Middle, mutational signatures may also be associated with clinical outcomes, such as worse survival, which may require more aggressive therapy or monitoring. Right, DNA-damaging chemotherapies may be associated with an enhanced rate of mutagenesis in cancers with decreased activity of specific DNA repair pathways, such as through MMR gene mutations, which may increase the possibility of therapy inducing driver mutations and which may thus represent a contraindication. This process has been discovered by identifying specific therapy-induced mutational signatures which were only detected in tumors lacking certain DNA repair pathways. At bottom, several specific examples are shown in each category.
Mutational signatures associated with therapy sensitivity
Mutational signatures offer indications of the DNA repair capacity of cancer cells, and as such may predict response to DNA-damaging or other therapies (Fig. 3, left). Signatures of homologous recombination (HR) deficiency are among the first mutational signatures applied to therapeutic decision-making. Indeed, signature analysis integrating multiple variant types can predict HR deficiency associated with mutations in the homologous repair genes BRCA1 or BRCA2 in breast cancer, and can also remarkably identify HR-deficient cancers without known BRCA1 or BRCA2 alterations [4]. HR deficiency is indicated by the presence of an SNV signature (COSMIC signature 3), a distinctive indel signature with microhomology at deletions junctions, and two specific SV signatures, which can be computationally integrated to robustly identify HR-deficient cancers using the “HRDetect” algorithm [4]. More recently, the “CHORD” classifier has also been developed to identify HR-deficient cancers using features similar to HRDetect [31]. Importantly, HR-deficient cancers respond more robustly to certain DNA-damaging agents due to decreased ability to repair therapy-induced DNA damage [32], making these algorithms clinically valuable. Indeed, HRDetect effectively predicts breast cancer response to adjuvant chemotherapy [33] and sensitivity to platinum treatment [34,35]. Further, preliminary clinical trial data suggest that HRDetect can detect PARP inhibitor sensitivity in breast cancer [36]. Mechanistically, the PARP1 protein aids in the resolution of single-stranded DNA breaks, which progress to double-stranded breaks (DSBs) when PARP1 is pharmacologically inhibited; these DSBs cannot be resolved in HR-deficient cancers and lead to cell death, which may explain the clinical sensitivity observed [37]. The detection of HR deficiency represents the most mature example of mutational signature analysis being integrated into clinical treatment decisions.
A study by Secrier and colleagues presents another example of using mutational signatures to predict therapy responsiveness [38]. This study classified esophageal cancers into three therapeutically relevant groups based on SNV mutational signature analysis. The first group was defined, as in the breast cancer example above, by the HR deficiency signature, and the authors hypothesized that PARP inhibitors combined with DNA-damaging agents may be effective in this group. The second group was defined by T>G-dominated COSMIC signature 17b, a signature detected primarily in esophageal and stomach cancers plus a subset of colorectal cancers, and possibly related to gastric acid reflux exposure. This group had the highest mutation burden and was therefore hypothesized to be more sensitive to immunotherapy. Additionally, an esophageal cell line within this second group was more sensitive to inhibitors of the cell cycle checkpoint regulators WEE1 and CHK1/2, as compared to cell lines representing other groups—although only a few cell lines were tested and the mechanism of sensitivity was unclear. The third group was dominated by COSMIC signature 1 (caused by 5-methylcytosine deamination) and 18 (caused by reactive oxygen species (ROS)) which do not have specific therapeutic indications currently. Notably, these three signature-defined groups did not differ by clinical features (such as survival, smoking status, and/or age) nor by specific genetic alterations, indicating that mutational signatures offered orthogonal information that may inform esophageal cancer response to specific treatments [38]. In summary, this study suggests that mutational signatures in esophageal cancer may predict subgroup-specific responsiveness to PARP inhibitors, DNA-damaging agents, immunotherapies, and specific cell-cycle inhibitors, although follow-up clinical studies are needed to validate these hypotheses.
APOBEC-associated SNV signatures (COSMIC signatures 2 and 13) are also associated with sensitivity to certain therapies. The APOBEC family of enzymes provides defense against viruses by inducing mutations in single-stranded DNA (ssDNA). However, a subset of APOBEC enzymes, including APOBEC3A [39] and APOBEC3B [40], can also be activated in cancer to act upon endogenous ssDNA [41], and induce C>T and C>G mutations at TCN trinucleotide contexts. Interestingly, the APOBEC signatures are associated with sensitivity to ATR inhibition in breast, ovarian, and other cancer cell lines [42]. Given that APOBEC enzymes induce mutations in ssDNA at replication forks, while ATR stabilizes DNA at stalled replication forks [43], it was hypothesized that the sensitivity of APOBEC-activated cancers to ATR inhibition was due to catastrophic abasic site accumulation leading to replication catastrophe [42]. This demonstrates how understanding mutational signature mechanisms may lead directly to therapeutic insights. Notably, ATR is a ubiquitously essential gene required for survival of both normal and cancer cells and its inhibition may cause toxicity to normal cells [44]. Thus, careful usage of ATR inhibitors in APOBEC-positive cancers, including chemotherapy-like intermittent dosage rather than constant daily dosage, may be needed to achieve a successful therapeutic index for these patients [44]. Further, APOBEC activity may be intermittent [18,45], suggesting that only tumors with current APOBEC activity may respond to ATR inhibition.
COSMIC signature 18, which is dominated by C>A mutations caused by ROS-induced guanine oxidation [9,46], is another signature with potential therapeutic relevance. Although ROS are metabolic byproducts of virtually all cell types [47], signature 18 is strikingly enriched in neuroblastoma [28]. It is mechanistically unclear why most neuroblastomas bear this ROS-related signature, which is absent in most other cancers. A recent study compared mutational and gene expression profiles between signature 18-bearing vs. -lacking neuroblastomas, and found that 17q gain and increased expression of mitochondrial genes on 17q were positively associated with signature 18 [48]. As most ROS are generated in the mitochondria due to the electron transport chain [49], these results suggest that signature 18 may be induced by copy gains that promote ROS through increasing electron transport chain activity. MYCN amplification was also associated with increased signature 18 [48] and ROS production [50] in neuroblastoma, which may explain the sensitivity of MYCN-amplified neuroblastoma to mitochondria-perturbing therapies such as MCT1 inhibitors and electron transport chain complex I inhibitors [51]. As measuring ROS requires complex biochemical assays, the presence of signature 18 offers a convenient readout for possible ROS activity that can be measured in existing genomic datasets, pointing to the benefit of mutational signatures for biomarker development. Other cancers with signature 18, such as colon cancers [1], pediatric leukemias [52], or rhabdomyosarcomas [48], may also benefit from therapies exploiting ROS overabundance, particularly if follow-up studies can identify the mechanistic causes of ROS/signature 18 enrichment in these cancers.
MMR deficiency-associated SNV signatures (including COSMIC signatures 6, 15, 26, and 44) occur in cancers with inactivation of MSH2, MSH6, PMS2, or MLH1, a link shown to be causal using experimental models [53]. Two general therapeutic vulnerabilities are observed in MMR-deficient cancers: (1) hypersensitivity to loss of the WRN DNA helicase gene through mechanisms still being explored [54,55], and (2) increased sensitivity to immunotherapies related to an increased mutation burden [56]. However, MMR deficiency can be inferred through a variety of assays independent of mutational signature analysis, including mutational analysis of MMR genes (MSH2, MSH6, PMS2, andMLH1), total mutation burden measurement, and microsatellite instability assays [57]. However, these assays may miss cryptic causes of MMR inactivation, such as promoter hypermethylation which is common in colorectal cancer [58] or focal deletions of MMR genes [12] which may be missed by exome sequencing. Thus, mutational signatures may provide orthogonal or confirmatory information pointing to MMR deficiency.
Another study in pancreatic cancer identified increased antitumor immune activation in tumors with mutational signatures of MMR or HR deficiency, including increased CD8+ T cell infiltration [59]. Twelve of 28 tumors with the HR deficiency mutational signature lacked a detectable alteration in BRCA1, BRCA2, or PALB2, reaffirming that mutational signatures have predictive value independent of individual gene alterations. While MMR deficiency may increase immune infiltration by generating neoantigens, HR deficiency likely increases immune infiltration by permitting DSBs which lead to micronuclei formation, activation of STING-induced interferon secretion, and hence the recruitment and activation of T cells in the tumor microenvironment [60,61].
Levatić colleagues recently performed a systematic correlation between SNV mutational signatures in cell lines (under basal culture conditions) and drug response using molecular data from the Genomics of Drug Sensitivity in Cancer (GDSC) [62,63]. Mutational signature analysis generally requires matched germline tissue, in addition to tumor tissue, so that somatic mutations can be identified; however, germline tissue is usually not available for cancer cell lines. Therefore, Levatić and colleagues performed a sophisticated ancestry-matching analysis to remove known and likely germline variants from cell line exome sequencing data, thus recapitulating mutational signatures derived from patient tissues. With somatic mutational signatures thus inferred, the authors then correlated mutational signatures with drug response in 930 cell lines spanning 29 cancer types treated with 518 drugs. Remarkably, mutational signatures predicted global drug response more effectively than oncogenic mutations, copy number alterations, and DNA hypermethylation. While gene expression predicted drug response better than mutational signatures or other features alone, mutational signatures combined with gene expression or other molecular features improved the prediction of drug response, indicating that mutational signatures offer complementary predictive value rather than simply recapitulating known variables. In all, over 500 significant mutational-signature/drug-sensitivity correlations were observed, including signature 26 (MMR deficiency) associated with camptothecin sensitivity in colorectal cancer cell lines; signature 20 (POLD1 mutation and MMR deficiency) associated with topotecan sensitivity in skin cancer and HDAC inhibitor sensitivity in multiple cancers; and signature 36 (ROS-induced) associated with sensitivity to the kinase inhibitor cabozantinib in several cancers. This study thus reveals a plethora of mutational signature biomarkers that might be used to inform precision cancer therapy [62].
Together, these examples show that mutational signatures can serve as a unique class of biomarkers of therapy sensitivity in multiple cancer types. We have summarized several of these examples in Fig. 3.
Mutational signatures associated with cancer prognosis
Mutational signatures may also be associated with general or treatment-specific prognosis (Fig. 3, middle column). In a study by Woolston et al., COSMIC signature 17b (possibly acid reflux-associated, as noted above) was associated with worse progression-free survival in colorectal cancers treated with the EGFR inhibitor cetuximab [64]. Mechanistically, signature 17b likely induces mutations in KRAS, NRAS, and EGFR, which cause resistance to cetuximab [65], indicating the prognostic value of understanding a cancer’s “evolvability” based on the intrinsic mutational processes at its disposal.
Further, a sophisticated prognostic scheme for multiple myeloma, based solely on mutational signatures, has been developed by Hoang and colleagues, which integrates SNV and SV signatures to separate myelomas into seven subgroups (Fig. 4) [23]. Although some mutational signatures recapitulate known prognostic markers in multiple myeloma, such as the APOBEC signature ,which is associated with known high-risk translocations [66], the Hoang et al. scheme also captured prognostic information independent of known risk factors. For example, signature cluster F samples in Hoang et al. lacked APOBEC-induced signatures but harbored an intermediate level of COSMIC SNV signature 9, associated with DNA polymerase eta-induced somatic hypermutation, and a non-clustered SV pattern (SV signature 4; see Fig. 4). Cluster F was associated with poor progression-free and overall survival, independent of known high-risk molecular features [23], again highlighting the value of mutational signatures in defining cancer prognosis.
Figure 4. Multiple myeloma prognostic classification based on SV and SNV mutational signatures.
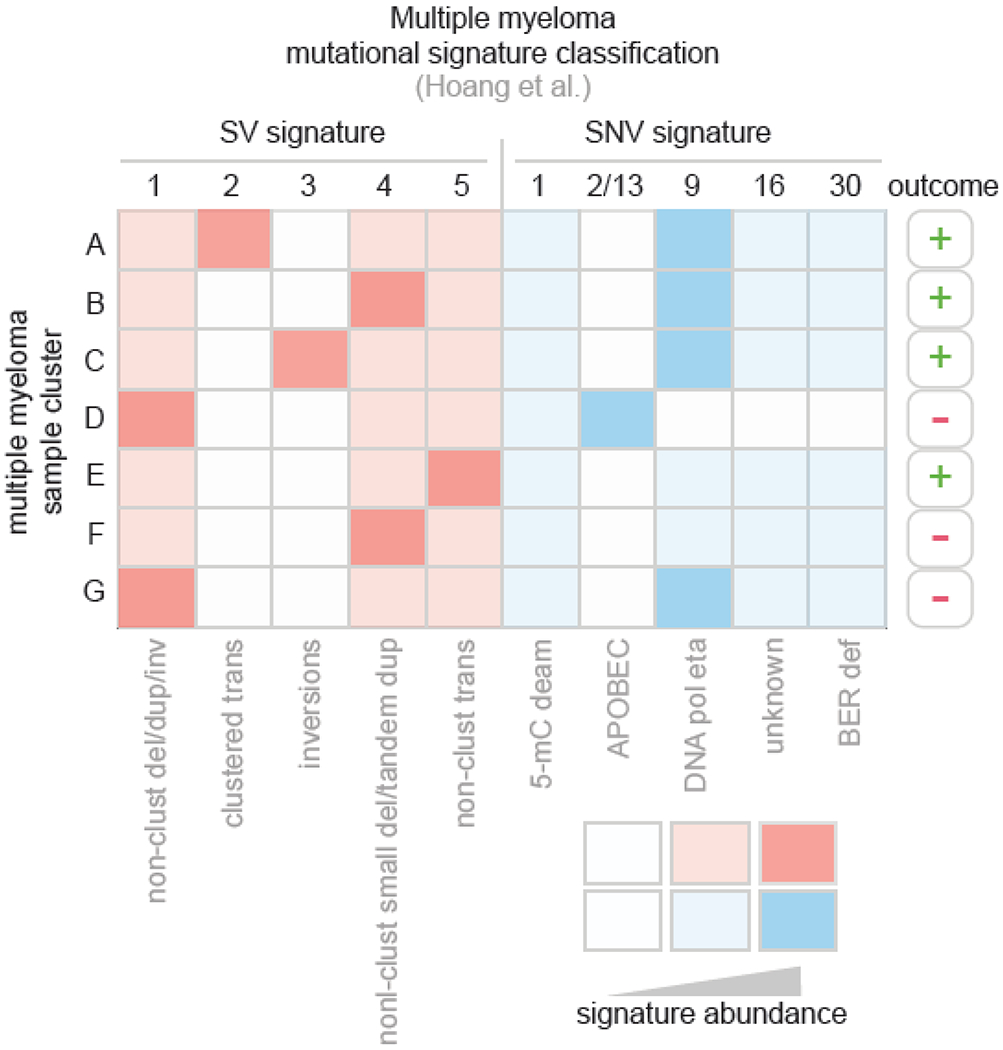
This figure summarizes the work of Hoang and colleagues [23], which extracted SV signatures de novo from multiple myeloma (left) and analyzed known COSMIC SNV signatures (right) in this disease. Each row represents one group or cluster of multiple myeloma samples, and each column represents one signature. The presence of a signature is indicated by red (SVs) or blue (SNVs) color, with lighter color indicating intermediate signature abundance. White color indicates absence or paucity of the mutational signature. For SV signatures, the signature’s characteristics are shown at bottom, where “del” indicates deletion; “dup”, tandem duplication; “inv”, inversion; and clustered status (multiple similar SVs nearby) indicated by “clust” For SNV signatures, the signature’s etiology if known, is indicated at bottom At right, the outcome, as measured by progression-free and overall survival, is indicated. “+” indicates relatively good outcome, while “−” indicates poor outcome.
Mutational signatures indicating therapy contraindication due to therapy-induced mutagenesis
DNA-damaging therapies, such as chemotherapy and radiation treatment, often induce characteristic mutational signatures which can be detected by whole-genome sequencing of post-treatment (relapsed) cancers. This treatment-induced mutagenesis may be accelerated in patients deficient in certain DNA repair processes, suggesting a possible treatment contraindication (Fig. 3, right). For example, temozolomide induces an SNV signature dominated by C>T mutations at specific contexts (COSMIC signature 11), which was first identified in post-treatment melanomas and glioblastomas [28]. Importantly, temozolomide likely induces specific driver mutations in glioblastoma, including CDKN2A and PI3K pathway mutations [67], which may accelerate disease progression. Later it was recognized that only cancers and experimental models with mismatch repair (MMR) deficiency acquired signature 11 after temozolomide treatment [68], indicating that therapy-induced signatures are modulated by the genetic background. This suggests that avoiding (or “contraindicating”) temozolomide treatment in MMR-deficient cancers may prevent the development of additional driver mutations induced by temozolomide.
Further, whole-genome sequencing of relapsed ALL has recently revealed that thiopurine treatment may lead to drug resistance mutations leading to relapse [12]. Thiopurines primarily induced C>T mutations at the center of NCG trinucleotides in relapsed ALL samples and experimental models (COSMIC signature 87). Notably, relapse-specific NR3C1, TP53, and NT5C2 mutations were preferentially of these mutation types, suggesting that thiopurines directly induced mutations leading to glucocorticoid resistance (NR3C1 mutations), cytotoxic chemotherapy resistance (TP53 mutations), and even thiopurine resistance itself (NT5C2 mutations). Subsequent experimental work has confirmed that thiopurines can indeed directly induce TP53 drug resistance mutations [69], and that thiopurine-induced mutagenesis is accelerated by MMR deficiency, similar to temozolomide, which increases the likelihood of TP53 mutagenesis (Fig. 5). This is likely because thiopurine incorporation into DNA leads to DNA mismatches which cannot be resolved in the context of MMR deficiency, resulting in mutation fixation after subsequent DNA replication [70]. Thus, patients with MMR-deficient clones should perhaps not be subjected to thiopurine treatment, as this would predispose to TP53 mutations and relapse. Indeed, one randomized clinical trial of pediatric ALL compared higher doses of thiopurine treatment to standard doses, and reported an increased risk of relapse in the group receiving higher dosage (where relapse risk as measured by Cox regression was the primary endpoint) [71]. This suggests that excess thiopurine usage may enrich for drug resistance mutations, and indicates the need for careful titration of thiopurine dosage to minimize undesired mutagenesis in these patients. Importantly, the findings of these studies offer a paradigm shift in the understanding of how drug resistance mutations develop, as these mutations have often been considered to simply pre-exist within subclones at diagnosis, only arising to prominence via selection during treatment [72–74]. Whereas these studies indicate that some drug resistance mutations may in fact be preventable by adjusting treatment regimens.
Figure 5. Example of how thiopurine treatment and MMR deficiency cooperate to induce a TP53 driver mutation.
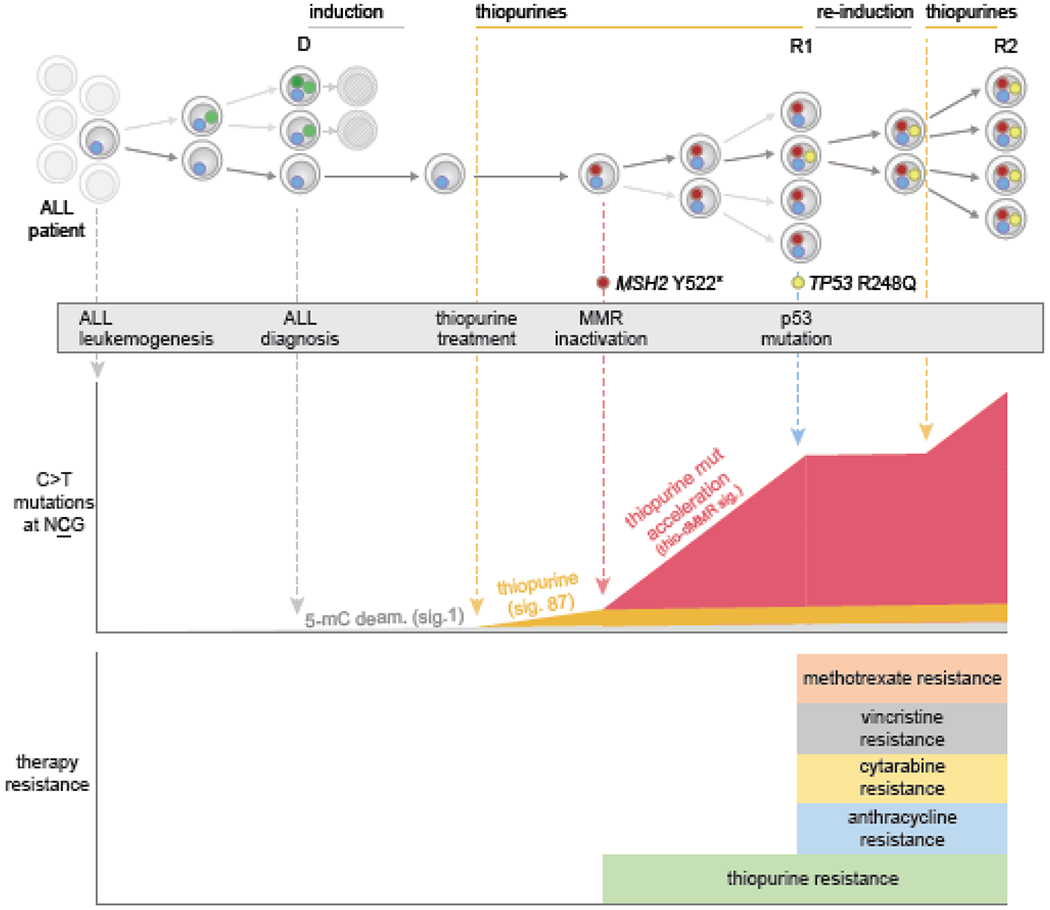
This shows a specific ALL patient analyzed by WGS at diagnosis and through multiple relapses, from a previous study [69]. Individual ALL clones are shown as cells harboring specific mutations (small colored circles), and time goes from left (ALL initiation) to right (through to second relapse). Initial therapy eliminated multiple clones, but the first relapse (R1) acquired MSH2 mutation leading to MMR deficiency. The resulting MMR deficiency enhanced the rate of mutation by thiopurine treatment approximately 10-fold (orange signature originally, in middle panel, to red signature with enhanced rate). Thiopurines induce C>T mutations at NCG contexts, increasing the probability of a TP53 R248Q mutation, which is of this C>T at NCG mutation class. Indeed, a subclonal TP53 R248Q mutation was present at a low level at R1, which was likely induced by thiopurines after the MSH2 mutation occurred (based on experimental and patient analysis). TP53 mutations cause resistance to induction and re-induction therapies, including resistance to anthracyclines, vincristine, and other drugs (see bottom), resulting in a multi-drug resistant clone which became dominant at second relapse (R2). Thus, thiopurine treatment can induce important drug resistance mutations that lead to more aggressive disease, and this process is more likely in MMR-deficient clones which are unable to repair thiopurine mismatches incorporated into DNA.
Platinum-based therapies (including cisplatin and carboplatin) induce SNVs at a high rate [11] and are represented by COSMIC signatures 31 and 35 [1]. Platinum-induced driver or drug resistance SNVs were rare in studies of post-treatment ovarian cancer [75] and post-treatment osteosarcoma [76]. However, the induction of drug resistance mutations by therapy is influenced by the combination of the drug’s mutational signature, the tumor’s DNA repair capacity, and selection. Thus, the absence of platinum-induced drug resistance mutations in these studies could be due to limitations in the drug’s mutational capability (the signature) and/or a paucity of SNVs genome-wide which are capable of inducing resistance to platinum therapy (lack of selection). In addition to SNVs, cisplatin can also induce single-base indels (both insertions and deletions) [11,77], including variants of the type that are known to revert BRCA2 frameshifts back to wild-type [77]. Such BRCA2 reversions are known to confer resistance to platinum therapy in BRCA2-mutant cancers [78], suggesting that platinum therapy may limit its own efficacy by causing drug resistance mutations [77], a hypothesis that requires experimental validation. Interestingly, the platinum signature was observed to appear at relapse in an Asian hepatoblastoma patient which harbored COSMIC SNV signature 22 (thought to be caused by aristolochic acid, a compound found in Chinese medicinal herbs, based on associational [79] and experimental [80] studies) at diagnosis. Both signature 22 and the cisplatin signature were present together at relapse in this patient [52], indicating that multiple signatures can evolve and accumulate through different exposures over time within the same tumor.
As with the above-mentioned chemotherapeutic drugs, radiation therapy can also induce driver mutations leading to tumor progression. This is exemplified in a study by Kocakavuk and colleagues who analyzed 190 glioma patients with paired primary and recurrent sample, and detected mutational signatures induced by radiation therapy in recurrent samples [81]. Radiation induced characteristic copy number changes and a specific indel signature, including small deletions of 5-15 bp or less, and CDKN2A deletions (possibly radiation-induced) were enriched in recurrent disease. Strikingly, patients with enrichment of radiation-induced indels had worse survival than patients with fewer radiation-induced indels [81]. This suggests that tumors lacking the ability to repair radiation-induced damage may evolve more rapidly and thus resist treatment. Thus, the radiation-induced indel signature may be a biomarker of prognosis if it can be assessed prospectively, prior to recurrence—a challenge given the difficulties of repeated biopsies of brain tumors. Alternatively, if the mechanism of dampened repair in a subset of gliomas can be identified and prospectively measured, this would provide valuable prognostic information.
In addition to inducing drug resistance mutations, DNA-damaging chemotherapies are also associated with secondary cancer initiation, potentially through inducing driver variants or chromosomal aberrations. Indeed, radiation [82], anthracyclines [83], epipodophyllotoxins [84], platinums [85], and thiopurines [86] are each associated with a risk of secondary cancer. These therapies can leave their imprint on the genomes of the secondary cancers, and the resulting signatures can suggest which driver variants were likely therapy-induced. For example, Behjati and colleagues identified a radiation-induced mutational signature in twelve secondary cancers (including breast cancer and sarcoma) which developed after radiation treatment for a primary malignancy (including lymphoma, breast, and ovarian cancers) [87]. In addition to detection of small deletions, similar to the analysis of Kocakavuk et al. in relapsed glioma [81], they also identified balanced inversions as variant types induced by radiation. These radiation-associated balanced inversions frequently disrupted genes at inversion junctions, including TP53, while radiation-associated indels affected both TP53 and CASP8. This indicates that radiation can induce driver alterations that may initiate secondary cancersand suggests further consideration of how to optimize treatment to prevent these driver alterations.
Recently, a mutational signature analysis on whole-genome sequencing of 84 therapy-related myeloid pediatric neoplasms (tMNs, including acute myeloid leukemia and myelodysplasia) occurring after treatment of a primary neoplasm, was performed [88]. Eight patients showed evidence of cisplatin- or thiopurine-induced mutational signatures, including several driver mutations that were mathematically likely to have been induced by these therapies as they appeared at genomic contexts frequently mutated by these therapies. One tMN patient had a germline mutation in PMS2 (inactivating MMR) and their tMN had acquired RUNX1 and KRAS mutations that were likely induced by thiopurine treatment given during primary leukemia treatment. Thus, thiopurine treatment may be contraindicated in patients with germline MMR inactivation, as it may increase the likelihood of secondary malignancies. Together, these studies suggest that the epidemiological evidence associating DNA-damaging therapies with secondary cancers may be, in part, explained by their direct induction of driver SNVs. Thus, further investigation is required to determine whether primary cancer treatment regimens can be modified to reduce the induction of such driver mutations.
Future directions
The above-mentioned studies demonstrate that mutational signatures can have both prognostic and therapeutic significance in cancer. For the remainder of this review, we focus on how future studies can build on these findings (see Outstanding Questions Box).
Outstanding Questions Box.
Can therapy-induced mutational signatures be measured on-treatment using cfDNA or minimal residual disease-derived DNA in a cost-effective manner for clinical implementation? Can this information effectively guide clinical decision-making?
Why do certain tumors acquire therapy-induced mutagenesis (e.g. from radiation and thiopurine treatment) while others do not?
What are the mechanisms underlying cell-intrinsic mutational signatures with potential therapeutic relevance?
For example, in the study of thiopurine-induced mutagenesis in ALL [12], it is interesting to note that only a subset of relapsed ALL samples (~15%) acquired the thiopurine-induced signature even though all had received thiopurine treatment. Notably, thiopurine signature-positive relapses had lower expression of thiopurine methyltransferase (TPMT), an enzyme which inactivates thiopurines [89], than relapse samples lacking the signature. This suggests that the presence of a mutational signature may be an early biomarker of sensitivity to a specific therapy, and thus may have utility, if it can be measured, during therapy such as by circulating cell-free DNA (cfDNA) or in the case of leukemias, via minimal residual disease analysis of bone marrow DNA. Indeed, genomic analysis of cfDNA from breast cancer patients has revealed that it is possible to detect mutational signatures from cfDNA [90]. Therefore, it is possible that cfDNA analysis could provide an early indication of responsiveness to individual drugs. This would be particularly useful for drugs that are given in drug combinations where it is difficult to deconvolute the tumor’s responsiveness to each individual drug. We hypothesize that drug signature-positive cfDNA would be indicative of therapy response, based on the TPMT example above, and that signature-negative cases would be predictive of non-response. One recently developed technology, mutREAD, has also shown the ability to detect mutational signatures in small quantities of DNA based on reduced representation sequencing [91], further demonstrating the feasibility of analyzing mutational signatures in small-quantity samples such as cfDNA for prognostic purposes.
Further, as noted above, thiopurine [69] and temozolomide [68] treatment may induce bursts of mutagenesis in MMR-deficient cancers, which may cause drug resistance mutations. It thus may prove valuable to assess the presence of MMR-deficient clones during treatment with these drugs by testing for mutations in the MSH2, MSH6, PMS2, or MLH1 MMR genes within cfDNA or bone marrow (for leukemia). This would require high-depth sequencing and detection of subclonal mutations with high confidence, which has been facilitated by recent computational algorithms that suppress the error rate in high-depth sequencing data [92,93]. Studies are thus needed to test the feasibility of detecting of MMR-deficiency mutations before treatment begins or during treatment—and could for instance begin with the retrospective analysis of plasma samples acquired from patients who later relapsed with new MMR-deficiency mutations.
In the above-mentioned pediatric osteosarcoma study, cisplatin treatment was able to double the mutation burden in some patients [76], Importantly, this process may make relapsed cancers susceptible to immunotherapies if new neoantigens are produced, even in cancers where the primary tumors normally do not bear a sufficient mutation burden to respond to immunotherapy. This is particularly evident in thiopurine- or temozolomide-treated relapsed cancers with MMR-deficient subclones. In this instance, we propose that the burst of mutations may provide large numbers of neoantigens for on which immunotherapies may act.
The further development of prognostic and drug response-predictive schemes incorporating mutational signatures, similar to those noted above [4,23,36,38,59,64], is also of value for future clinical research. Whole-genome or -exome sequencing is often required to detect mutational signatures, and most cancer centers are not able to perform these assays for each patient. However, the emergence of reduced-representation approaches such as mutREAD [91] that substantially reduce cost, protocols for high-intensity sequencing of specific genomic regions [90], and the continual decrease of sequencing costs overall, may make signature analysis within the clinic more commonplace in the future. Further, it will also be of value to gain a mechanistic understanding of why certain mutational signatures are associated with different prognoses—for example, whether the response to a specific therapy is affected by the biological process underlying a certain mutational signature. This endeavor will therefore potentially enable more tailored therapy approaches in addition to the prognostic value added.
The remarkable progress in understanding cancer mutational signatures thus has the capability to transfer a fundamental aspect of cancer cells—their aberrant mutational processes and genomic instability—into a mathematical construct that is predictive of treatment response and prognosis. We therefore envision mutational signatures to become an increasingly valuable analysis tool for both research and clinical decision-making that should complement the use of more traditional molecular features for improved clinical care of cancer patients.
Highlights.
Mutational signatures have revolutionized our understanding of cancer etiology, by identifying biological processes underlying somatic mutagenesis.
Mutational signatures can also represent biomarkers indicative of therapy sensitivity, prognosis, and therapy contraindications. Importantly, mutational signatures provide predictive information independent of known clinical and molecular biomarkers.
New technologies have increased the feasibility of measuring mutational signatures in a cost-effective manner, which may facilitate integration of signature analysis into clinical decision-making.
Acknowledgments
J.Z. is funded in part by NCI grants R01CA216354 and R01CA216391. This work was also supported by and the American Lebanese Syrian Associated Charities of St. Jude Children’s Research Hospital.
Glossary
- Contraindication
A situation in which a certain therapy should be avoided due to adverse risks.
- Doublet substitution
A mutation changing the DNA sequence of two adjacent bases, such as a cytosine-guanine sequence being mutated to thymine-adenine (CG>TA).
- Homologous recombination
A DNA repair pathway which resolves double-stranded DNA breaks, involving a suite of genes including BRCA1 and BRCA2. The pathway uses the intact (unbroken) sister chromatid as a template to correctly repair the break. Specific mutational signatures are indicative of loss of this pathway’s activity.
- Mismatch repair
A DNA repair pathway which corrects single-base mismatches (for example, a guanine mispaired with a thymine), involving several genes including MSH2, MSH6, PMS2, MLH1, This pathway prevents excess mutagenesis, and its inactivation can lead to resistance to certain chemotherapies. Specific mutational signatures are indicative of loss of this pathway’s activity.
- Mutational signature
A mathematical construct associated with a specific mutagenic process, which indicates the relative predilection to cause various mutation types.
- Single-nucleotide variant
A DNA mutation affecting a single base, such as cytosine to thymine (C>T).
- Structural variation
A DNA rearrangement resulting from an interchromosomal translocation, large genomic deletion, inversion, or duplication of a specific region.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing interests
The authors declare no competing interests.
References
- 1.Alexandrov LB et al. (2020) The repertoire of mutational signatures in human cancer. Nature 578, 94–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Forbes SA et al. (2015) COSMIC: exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res. 43, D805–D811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nik-Zainal S et al. (2016) Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 534, 47–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Davies H et al. (2017) HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat. Med 23, 517–525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Papaemmanuil E et al. (2014) RAG-mediated recombination is the predominant driver of oncogenic rearrangement in ETV6-RUNX1 acute lymphoblastic leukemia. Nat. Genet 46, 116–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim J et al. (2016) Somatic ERCC2 mutations are associated with a distinct genomic signature in urothelial tumors. Nat. Genet. 2016 486 48, 600–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kasar S et al. (2015) Whole-genome sequencing reveals activation-induced cytidine deaminase signatures during indolent chronic lymphocytic leukaemia evolution. Nat. Commun 6, 8866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tan VYF and Févotte C (2013) Automatic relevance determination in nonnegative matrix factorization with the (β)-divergence. IEEE Trans. Pattern Anal. Mach. Intell 35, 1592–1605 [DOI] [PubMed] [Google Scholar]
- 9.Kucab JE et al. (2019) A Compendium of Mutational Signatures of Environmental Agents. Cell 177, 821–836.e16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zou X et al. (2018) Validating the concept of mutational signatures with isogenic cell models. Nat. Commun 9, 1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boot A et al. (2018) In-depth characterization of the cisplatin mutational signature in human cell lines and in esophageal and liver tumors. Genome Res 28, 654–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li B et al. (2020) Therapy-induced mutations drive the genomic landscape of relapsed acute lymphoblastic leukemia. Blood 135, 41–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koh G et al. Mutational signatures: Experimental design and analytical framework. , Genome Biology, 21. 14-Feb-(2020), BioMed Central Ltd., 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Inman GJ et al. (2018) The genomic landscape of cutaneous SCC reveals drivers and a novel azathioprine associated mutational signature. Nat. Commun 9, 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Helleday T et al. (2014) Mechanisms underlying mutational signatures in human cancers. Nat. Rev. Genet 15, 585–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alexandrov LB et al. (2015) Clock-like mutational processes in human somatic cells. Nat. Genet 47, 1402–1407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gerstung M et al. (2020) The evolutionary history of 2,658 cancers. Nature 578, 122–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Petljak M et al. (2019) Characterizing Mutational Signatures in Human Cancer Cell Lines Reveals Episodic APOBEC Mutagenesis. Cell 176, 1282–1294.e20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kishikawa T et al. (2019) Empirical evaluation of variant calling accuracy using ultra-deep whole-genome sequencing data. Sci. Reports 2019 91 9, 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Supernat A et al. (2018) Comparison of three variant callers for human whole genome sequencing. Sci. Reports 2018 81 8, 1–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.García-Prieto C et al. (2020) The consequences of variant calling decisions in secondary analyses of cancer genomics data. bioRxiv DOI: 10.1101/2020.01.29.924860 [DOI] [Google Scholar]
- 22.Campbell PJ et al. (2020) Pan-cancer analysis of whole genomes. Nature 578, 82–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hoang PH et al. (2019) Mutational processes contributing to the development of multiple myeloma. Blood Cancer J. 9, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Islam SMA et al. (2021) Uncovering novel mutational signatures by de novo extraction with SigProfilerExtractor. bioRxiv DOI: 10.1101/2020.12.13.422570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Blokzijl F et al. (2017) MutationalPatterns: comprehensive genome-wide analysis of mutational processes. bioRxiv DOI: 10.1101/071761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pich O et al. (2019) The mutational footprints of cancer therapies. Nat. Genet. 2019 5112 51, 1732–1740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim Y-A et al. (2021) Mutational Signatures: From Methods to Mechanisms. 10.1146/annurev-biodatasci-122320-120920 4, 189–206 [DOI] [PubMed] [Google Scholar]
- 28.Alexandrov LB et al. (2013) Signatures of mutational processes in human cancer. Nature 500, 415–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morganella S et al. (2016) The topography of mutational processes in breast cancer genomes. Nat. Commun 7, 11383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Giacomelli AO et al. (2018) Mutational processes shape the landscape of TP53 mutations in human cancer. Nat. Genet 50, 1381–1387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nguyen L et al. (2020) Pan-cancer landscape of homologous recombination deficiency. Nat. Commun. 2020 111 11, 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tumiati M et al. (2018) A Functional Homologous Recombination Assay Predicts Primary Chemotherapy Response and Long-Term Survival in Ovarian Cancer Patients. Clin. Cancer Res 24, 4482–4493 [DOI] [PubMed] [Google Scholar]
- 33.Staaf J et al. (2019) Whole-genome sequencing of triple-negative breast cancers in a population-based clinical study. Nat. Med. 2019 2510 25, 1526–1533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhao EY et al. (2017) Homologous recombination deficiency and platinum-based therapy outcomes in advanced breast cancer. Clin. Cancer Res 23, 7521–7530 [DOI] [PubMed] [Google Scholar]
- 35.Koh G et al. (2021) Mutational signatures: emerging concepts, caveats and clinical applications. Nat. Rev. Cancer 2021 DOI: 10.1038/s41568-021-00377-7 [DOI] [PubMed] [Google Scholar]
- 36.Chopra N et al. (2020) Homologous recombination DNA repair deficiency and PARP inhibition activity in primary triple negative breast cancer. Nat. Commun 11, 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mateo J et al. (2019) A decade of clinical development of PARP inhibitors in perspective. Ann. Oncol 30, 1437–1447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Secrier M et al. (2016) Mutational signatures in esophageal adenocarcinoma define etiologically distinct subgroups with therapeutic relevance. Nat. Genet 48, 1131–1141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Langenbucher A et al. (2021) An extended APOBEC3A mutation signature in cancer. Nat. Commun 12, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chan K et al. (2015) An APOBEC3A hypermutation signature is distinguishable from the signature of background mutagenesis by APOBEC3B in human cancers. Nat. Genet 47, 1067–1072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kazanov MD et al. (2015) APOBEC-Induced Cancer Mutations Are Uniquely Enriched in Early-Replicating, Gene-Dense, and Active Chromatin Regions. Cell Rep. 13, 1103–1109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Buisson R et al. (2017) APOBEC3A and APOBEC3B activities render cancer cells susceptible to ATR inhibition. Cancer Res. 77, 4567–4578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liao H et al. (2018) Mechanisms for stalled replication fork stabilization: new targets for synthetic lethality strategies in cancer treatments. EMBO Rep. 19, e46263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chang L et al. Targeting pan-essential genes in cancer: Challenges and opportunities., Cancer Cell, 39. 12-Apr-(2021) , Cell Press, 466–479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Van Hoeck A et al. Portrait of a cancer: Mutational signature analyses for cancer diagnostics., BMC Cancer, 19. 15-May-(2019), BioMed Central Ltd., 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Viel A et al. (2017) A Specific Mutational Signature Associated with DNA 8-Oxoguanine Persistence in MUTYH-defective Colorectal Cancer. EBioMedicine 20, 39–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hamanaka RB and Chandel NS Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes., Trends in Biochemical Sciences, 35. 01-Sep-(2010), Elsevier Current Trends, 505–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brady SW et al. (2020) Pan-neuroblastoma analysis reveals age- and signature-associated driver alterations. Nat. Commun 11, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Balaban RS et al. Mitochondria, oxidants, and aging., Cell, 120. 25-Feb-(2005), Cell Press, 483–495 [DOI] [PubMed] [Google Scholar]
- 50.Zeineldin M et al. (2020) MYCN amplification and ATRX mutations are incompatible in neuroblastoma. Nat. Commun 11, 913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dalton KM et al. (2021) Catastrophic ATP loss underlies a metabolic combination therapy tailored for MYCN-amplified neuroblastoma. Proc. Natl. Acad. Sci. U. S. A 118, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McLeod C et al. (2021) St. Jude Cloud-a Pediatric Cancer Genomic Data Sharing Ecosystem. Cancer Discov. DOI: 10.1158/2159-8290.cd-20-1230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zou X et al. (2021) A systematic CRISPR screen defines mutational mechanisms underpinning signatures caused by replication errors and endogenous DNA damage. Nat. Cancer 2021 26 2, 643–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Picco G et al. (2021) Werner helicase is a synthetic-lethal vulnerability in Mismatch Repair-Deficient Colorectal Cancer Refractory to Targeted Therapies, Chemotherapy and Immunotherapy. Cancer Discov. DOI: 10.1158/2159-8290.cd-20-1508 [DOI] [PubMed] [Google Scholar]
- 55.Chan EM et al. (2019) WRN helicase is a synthetic lethal target in microsatellite unstable cancers. Nature 568, 551–556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Le DT et al. (2017) Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 357, 409–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nowak JA et al. (2017) Detection of Mismatch Repair Deficiency and Microsatellite Instability in Colorectal Adenocarcinoma by Targeted Next-Generation Sequencing. J. Mol. Diagnostics 19, 84–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Esteller M et al. (1998) MLH1 promoter hypermethylation is associated with the microsatellite instability phenotype in sporadic endometrial carcinomas. Oncogene 17, 2413–2417 [DOI] [PubMed] [Google Scholar]
- 59.Connor AA et al. (2017) Association of distinct mutational signatures with correlates of increased immune activity in pancreatic ductal adenocarcinoma. JAMA Oncol. 3, 774–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ma J et al. The therapeutic significance of mutational signatures from DNA repair deficiency in cancer., Nature Communications, 9. 01-Dec-(2018), Nature Publishing Group, 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Weiss JM et al. (2017) The STING agonist DMXAA triggers a cooperation between T lymphocytes and myeloid cells that leads to tumor regression. Oncoimmunology 6, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Levatić J et al. (2021) Mutational signatures are markers of drug sensitivity of cancer cells. bioRxiv DOI: 10.1101/2021.05.19.444811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yang W et al. (2013) Genomics of Drug Sensitivity in Cancer (GDSC): A resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 41, D955–D961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Woolston A et al. (2021) Mutational signatures impact the evolution of anti-EGFR antibody resistance in colorectal cancer. Nat. Ecol. Evol DOI: 10.1038/s41559-021-01470-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Misale S et al. (2012) Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature 486, 532–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Walker BA et al. (2015) APOBEC family mutational signatures are associated with poor prognosis translocations in multiple myeloma. Nat. Commun. 6, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Johnson BE et al. (2014) Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science 343, 189–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Touat M et al. (2020) Mechanisms and therapeutic implications of hypermutation in gliomas. Nature 580, 517–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yang F et al. (2021) Chemotherapy and mismatch repair deficiency cooperate to fuel TP53 mutagenesis and ALL relapse. Nat. Cancer 2021 DOI: 10.1038/s43018-021-00230-8 [DOI] [PubMed] [Google Scholar]
- 70.Uribe-Luna S et al. (1997) Mutagenic consequences of the incorporation of 6-thioguanine into DNA. Biochem. Pharmacol. 54, 419–424 [DOI] [PubMed] [Google Scholar]
- 71.Schmiegelow K et al. (2003) Intensification of mercaptopurine/methotrexate maintenance chemotherapy may increase the risk of relapse for some children with acute lymphoblastic leukemia. J. Clin. Oncol 21, 1332–1339 [DOI] [PubMed] [Google Scholar]
- 72.Diaz LA et al. (2012) The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature 486, 537–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Turke AB et al. (2010) Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer Cell 17, 77–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pfeifer H et al. (2007) Kinase domain mutations of BCR-ABL frequently precede imatinib-based therapy and give rise to relapse in patients with de novo Philadelphia-positive acute lymphoblastic leukemia (Ph+ ALL). Blood 110, 727–734 [DOI] [PubMed] [Google Scholar]
- 75.Patch A-M et al. (2015) Whole-genome characterization of chemoresistant ovarian cancer. Nature 521, 489–94 [DOI] [PubMed] [Google Scholar]
- 76.Brady SW et al. (2019) The Clonal Evolution of Metastatic Osteosarcoma as Shaped by Cisplatin Treatment. Mol. Cancer Res DOI: 10.1158/1541-7786.MCR-18-0620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Szikriszt B et al. (2016) A comprehensive survey of the mutagenic impact of common cancer cytotoxics. Genome Biol. 17, 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Waks AG et al. (2020) Reversion and non-reversion mechanisms of resistance to PARP inhibitor or platinum chemotherapy in BRCA1/2-mutant metastatic breast cancer. Ann. Oncol 31, 590–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Poon SL et al. (2013) Genome-Wide Mutational Signatures of Aristolochic Acid and Its Application as a Screening Tool. Sci. Transl. Med 5, 197ra101–197ra101 [DOI] [PubMed] [Google Scholar]
- 80.Nik-Zainal S et al. (2015) The genome as a record of environmental exposure. Mutagenesis 30, 763–770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kocakavuk E et al. (2021) Radiotherapy is associated with a deletion signature that contributes to poor outcomes in patients with cancer. Nat. Genet DOI: 10.1038/s41588-021-00874-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Armstrong GT et al. Long-term effects of radiation exposure among adult survivors of childhood cancer: Results from the childhood cancer survivor study. Radiation Research, 174. 01-Dec-(2010), Allen Press, 840–850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Henderson TO et al. (2016) Breast cancer risk in childhood cancer survivors without a history of chest radiotherapy: A report from the childhood cancer survivor study. J. Clin. Oncol 34, 910–918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Neglia JP et al. (2001) Second malignant neoplasms in five-year survivors of childhood cancer: Childhood cancer survivor study. J. Natl. Cancer Inst 93, 618–629 [DOI] [PubMed] [Google Scholar]
- 85.Travis LB et al. (2000) Treatment-associated leukemia following testicular cancer. J. Natl. Cancer Inst 92, 1165–1171 [DOI] [PubMed] [Google Scholar]
- 86.Schmiegelow K et al. (2009) Methotrexate/6-mercaptopurine maintenance therapy influences the risk of a second malignant neoplasm after childhood acute lymphoblastic leukemia: Results from the NOPHO ALL-92 study. Blood 113, 6077–6084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Behjati S et al. (2016) Mutational signatures of ionizing radiation in second malignancies. Nat. Commun 7, 1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Schwartz JR et al. (2021) The acquisition of molecular drivers in pediatric therapy-related myeloid neoplasms. Nat. Commun 12, 985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Krynetski EY et al. Genetic polymorphism of thiopurine S-methyltransferase: Clinical importance and molecular mechanisms. Pharmacogenetics, 6. 01-Aug-(1996) , Lippincott Williams and Wilkins, 279–290 [DOI] [PubMed] [Google Scholar]
- 90.Razavi P et al. (2019) High-intensity sequencing reveals the sources of plasma circulating cell-free DNA variants. Nat. Med 25, 1928–1937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Perner J et al. (2020) The mutREAD method detects mutational signatures from low quantities of cancer DNA. Nat. Commun 11, 1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ma X et al. (2019) Analysis of error profiles in deep next-generation sequencing data. Genome Biol. 20, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Davis EM et al. (2021) SequencErr: measuring and suppressing sequencer errors in next-generation sequencing data. Genome Biol. 22, 1–18 [DOI] [PMC free article] [PubMed] [Google Scholar]